
miR23b* targets proline oxidase, a novel tumor suppressor protein in renal cancer
11
ORIGINAL ARTICLE miR-23b* targets proline oxidase, a novel tumor suppressor protein in renal cancer W Liu 1 , O Zabirnyk 1 , H Wang 2 , Y-H Shiao 1 and ML Nickerson 3 , S Khalil 1 , LM Anderson 1 , AO Perantoni 2 and JM Phang 1 1 Laboratory of Comparative Carcinogenesis, Center for Cancer Research, National Cancer Institute at Frederick, National Institutes of Health, Frederick, MD, USA; 2 Laboratory of Cancer and Developmental Biology, Center for Cancer Research, National Cancer Institute at Frederick, National Institutes of Health, Frederick, MD, USA and 3 Cancer and Inflammation Program, Center for Cancer Research, National Cancer Institute at Frederick, National Institutes of Health, Frederick, MD, USA Proline oxidase (POX) is a novel mitochondrial tumor suppressor that can suppress proliferation and induce apoptosis through the generation of reactive oxygen species (ROS) and decreasing hypoxia-inducible factor (HIF) signaling. Recent studies have shown the absence of expression of POX in human cancer tissues, including renal cancer. However, the mechanism for the loss of POX remains obscure. No genetic or epigenetic variation of POX gene was found. In this study, we identified the upregulated miR-23b* in renal cancer as an important regulator of POX. Ectopic overexpression of miR-23b* in normal renal cells resulted in striking downregulation of POX, whereas POX expression increased markedly when endogenous miR-23b* was knocked down by its anta- gomirs in renal cancer cells. Consistent with the POX- mediated tumor suppression pathway, these antagomirs induced ROS, inhibited HIF signaling and increased apoptosis. Furthermore, we confirmed the regulation of miR-23b* on POX and its function in the DLD1 Tet-off POX cell system. Using a luciferase reporter system, we verified the direct binding of miR-23b* to the POX mRNA 3 0 -untranslated region. In addition, pairs of human renal carcinoma and normal tissues showed a negative correlation between miR-23b* and POX protein expres- sion, providing its clinical corroboration. Taken together, our results suggested that miR-23b*, by targeting POX, could function as an oncogene; decreasing miR-23b* expression may prove to be an effective way of inhibiting kidney tumor growth. Oncogene advance online publication, 21 June 2010; doi:10.1038/onc.2010.237 Keywords: proline oxidase; miR-23b*; renal cancer; ROS; tumor suppressor Introduction During the last decade, the important functions of proline metabolism in human health and disease have received increasing attention. Proline, together with hydroxypro- line, constitutes more than 25% of incorporated residues of collagen, the most abundant protein in the human body. Proline oxidase (POX) is a mitochondrial inner- membrane enzyme involved in the metabolism of proline to pyrroline-5-carboxylate. This reaction, when coupled with the conversion of pyrroline-5-carboxylate to proline by pyrroline-5-carboxylate reductase, mediates the pro- line cycle to shuttle redox equivalents between mitochon- dria and the cytosol (Phang, 1985; Liu et al., 2005; Phang et al., 2008). Pyrroline-5-carboxylate provides a direct carbon bridge connecting proline metabolism with the tricarboxylic cycle through glutamate and a-ketoglutarate (Phang, 1985). Changes in the cycling of proline through POX may alter the redox balance, critical for regulation of cell growth and apoptosis. POX has been identified as one of a few mitochon- drial tumor suppressors. It can induce apoptosis through the generation of reactive oxygen species (ROS) and also reduce hypoxia-inducible factor (HIF) signaling by elevating cellular a-ketoglutarate, a critical substrate for prolyl hydroxylase-catalyzed hydroxyla- tion of HIF-1a and degradation (Liu et al., 2005, 2006, 2008, 2009). POX participates in the apoptosis induced by p53, cytotoxic agent adriamycin and peroxisome proliferator-activated receptor-g ligands, in a variety of cancer cell types (Polyak et al., 1997; Donald et al., 2001; Liu et al., 2005, 2008; Pandhare et al., 2006). Its upregulation inhibits growth of various cultured tumor cells and suppresses tumor formation in a xenograft model (Liu et al., 2008, 2009). Recent studies have shown the absence or reduction of POX in a variety of human tumor tissues as compared with their normal tissue counterparts, including kidney, colon, stomach, liver and pancreas (Maxwell and Rivera, 2003; Liu et al., 2008, 2009). However, we did not find any genetic or epigenetic variation of the POX gene and its promoter in human tumor panels (COSMIC database and our unpublished data). The mechanism for differential POX expression and its contribution to tumor develop- ment is still not clearly defined. Received 18 December 2009; revised 22 March 2010; accepted 16 May 2010 Correspondence: Dr W Liu, NCI-Frederick, National Institutes of Health, Building 538, Room 144, Frederick, MD 21702, USA E-mail: [email protected] or Dr JM Phang, NCI-Frederick, National Institutes of Health, Building 538, Room 115, Frederick, MD 21702, USA. E-mail: [email protected] Oncogene (2010), 1–11 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of miR23b* targets proline oxidase, a novel tumor suppressor protein in renal cancer

ORIGINAL ARTICLE
miR-23b* targets proline oxidase, a novel tumor suppressor protein in renal
cancer
W Liu1, O Zabirnyk1, H Wang2, Y-H Shiao1 and ML Nickerson3, S Khalil1, LM Anderson1,AO Perantoni2 and JM Phang1
1Laboratory of Comparative Carcinogenesis, Center for Cancer Research, National Cancer Institute at Frederick, National Institutesof Health, Frederick, MD, USA; 2Laboratory of Cancer and Developmental Biology, Center for Cancer Research, National CancerInstitute at Frederick, National Institutes of Health, Frederick, MD, USA and 3Cancer and Inflammation Program, Center for CancerResearch, National Cancer Institute at Frederick, National Institutes of Health, Frederick, MD, USA
Proline oxidase (POX) is a novel mitochondrial tumorsuppressor that can suppress proliferation and induceapoptosis through the generation of reactive oxygenspecies (ROS) and decreasing hypoxia-inducible factor(HIF) signaling. Recent studies have shown the absence ofexpression of POX in human cancer tissues, includingrenal cancer. However, the mechanism for the loss ofPOX remains obscure. No genetic or epigenetic variationof POX gene was found. In this study, we identified theupregulated miR-23b* in renal cancer as an importantregulator of POX. Ectopic overexpression of miR-23b* innormal renal cells resulted in striking downregulation ofPOX, whereas POX expression increased markedly whenendogenous miR-23b* was knocked down by its anta-gomirs in renal cancer cells. Consistent with the POX-mediated tumor suppression pathway, these antagomirsinduced ROS, inhibited HIF signaling and increasedapoptosis. Furthermore, we confirmed the regulation ofmiR-23b* on POX and its function in the DLD1 Tet-offPOX cell system. Using a luciferase reporter system, weverified the direct binding of miR-23b* to the POXmRNA 30-untranslated region. In addition, pairs of humanrenal carcinoma and normal tissues showed a negativecorrelation between miR-23b* and POX protein expres-sion, providing its clinical corroboration. Taken together,our results suggested that miR-23b*, by targeting POX,could function as an oncogene; decreasing miR-23b*expression may prove to be an effective way of inhibitingkidney tumor growth.Oncogene advance online publication, 21 June 2010;doi:10.1038/onc.2010.237
Keywords: proline oxidase; miR-23b*; renal cancer;ROS; tumor suppressor
Introduction
During the last decade, the important functions of prolinemetabolism in human health and disease have receivedincreasing attention. Proline, together with hydroxypro-line, constitutes more than 25% of incorporated residuesof collagen, the most abundant protein in the humanbody. Proline oxidase (POX) is a mitochondrial inner-membrane enzyme involved in the metabolism of prolineto pyrroline-5-carboxylate. This reaction, when coupledwith the conversion of pyrroline-5-carboxylate to prolineby pyrroline-5-carboxylate reductase, mediates the pro-line cycle to shuttle redox equivalents between mitochon-dria and the cytosol (Phang, 1985; Liu et al., 2005; Phanget al., 2008). Pyrroline-5-carboxylate provides a directcarbon bridge connecting proline metabolism with thetricarboxylic cycle through glutamate and a-ketoglutarate(Phang, 1985). Changes in the cycling of proline throughPOX may alter the redox balance, critical for regulationof cell growth and apoptosis.
POX has been identified as one of a few mitochon-drial tumor suppressors. It can induce apoptosisthrough the generation of reactive oxygen species(ROS) and also reduce hypoxia-inducible factor (HIF)signaling by elevating cellular a-ketoglutarate, a criticalsubstrate for prolyl hydroxylase-catalyzed hydroxyla-tion of HIF-1a and degradation (Liu et al., 2005, 2006,2008, 2009). POX participates in the apoptosis inducedby p53, cytotoxic agent adriamycin and peroxisomeproliferator-activated receptor-g ligands, in a variety ofcancer cell types (Polyak et al., 1997; Donald et al.,2001; Liu et al., 2005, 2008; Pandhare et al., 2006). Itsupregulation inhibits growth of various cultured tumorcells and suppresses tumor formation in a xenograftmodel (Liu et al., 2008, 2009). Recent studies haveshown the absence or reduction of POX in a variety ofhuman tumor tissues as compared with their normaltissue counterparts, including kidney, colon, stomach,liver and pancreas (Maxwell and Rivera, 2003; Liu et al.,2008, 2009). However, we did not find any genetic orepigenetic variation of the POX gene and its promoter inhuman tumor panels (COSMIC database and ourunpublished data). The mechanism for differentialPOX expression and its contribution to tumor develop-ment is still not clearly defined.
Received 18 December 2009; revised 22 March 2010; accepted 16 May2010
Correspondence: Dr W Liu, NCI-Frederick, National Institutes ofHealth, Building 538, Room 144, Frederick, MD 21702, USAE-mail: [email protected] or Dr JM Phang, NCI-Frederick,National Institutes of Health, Building 538, Room 115, Frederick,MD 21702, USA.E-mail: [email protected]
Oncogene (2010), 1–11& 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10
www.nature.com/onc

Recently, with the discovery of microRNAs (miR-NAs), a new mechanism to regulate protein expressionhas been revealed. miRNAs are a class of conserved,endogenously expressed, noncoding small RNAs (18–25nucleotides). They can negatively regulate gene expres-sion at the posttranscriptional level by cleavage and/ortranslational repression of their mRNA targets throughspecific, although imperfect, base pairing with the 30-untranslated region (30UTR) of target mRNAs (Bartel,2004; Chen and Rajewsky, 2007). This imperfect andlimited complementarity to target sites enables indivi-dual miRNAs to target a large number of mRNAs andalso hampers the identification of the specific genes itregulates. Early analyses estimated that B30% ofknown human genes are under miRNA control (Lewiset al., 2005), whereas later reports increased this numberto 490% (Miranda et al., 2006). However, althoughhundreds of miRNAs have been identified to date, theelucidation of their specific targets and functions hasbeen limited.
miRNAs have been shown to modulate a variety ofbiological processes, including cellular differentiationand proliferation, metabolic signaling and apoptosis. Inrecent years, growing evidence has strongly implicatedthe involvement of miRNAs in carcinogenesis (Iorioet al., 2005; Ma et al., 2007). Dysregulated miRNAsmay function as oncogenes, such as miR-17-92 clusterand miR-214 (Yang et al., 2008; Northcott et al., 2009),or tumor suppressor genes, such as miR-205 and let-7(Johnson et al., 2005; Gandellini et al., 2009), dependingon the targets they regulate. Although some targets ofthese miRNAs have been identified, miRNA regulatorsof critical cancer proteins and pathways remain largelyunknown, including the POX-mediated pathways des-cribed above. Therefore, we proposed that the dysregu-lation of candidate miRNAs could be a possiblemechanism for POX downregulation in tumors. In thisstudy, we examined miRNAs to identify a novelmechanism resulting in POX downregulation in renalcancer.
Results
POX expression is markedly reduced in renal cancer celllines and human renal carcinoma tissue samplesWe measured POX mRNA and protein expression byreal-time reverse transcriptase PCR (RT–PCR) andwestern blot in several renal cancer and normal celllines. Figures 1a and b showed that POX mRNA levelsin the clear cell renal carcinoma cell lines, including 786-0, TK10 and UO31, were about 50% of those in normalrenal cell lines HREpC and HRCEpC, whereas POXprotein levels were only 4% of those in normal renalcells.
To confirm the expression levels of POX in vivo, weobtained 16 paired renal cell carcinomas (clear cellsubtype) and the corresponding normal renal tissues toanalyze its protein expression by immunohistochemicalstaining. We found that in 13 out of 16 pairs of renaltissues, the expression of POX was strikingly decreased
in carcinoma tissues compared with their normalcounterparts. The representative images from five pairsof normal/cancer tissues are shown in Figure 1c.
Overexpressed miRNAs potentially related to POXin renal cancer cell linesTo identify possible miRNAs negatively regulating POXexpression, we performed a bioinformatic analysis usingthree public databases, miRBase, TargetScan andMicroinspector. We then integrated the projected resultsfrom these three databases and identified 91 potentialmiRNAs targeting POX mRNA 30UTR totally (datanot shown).
To investigate the involvement of miRNAs in thereduced expression of POX in renal cancer, wecompared miRNA expression profiles of three renalcancer cell lines (786-0, TK10 and UO31) with twonormal renal cell lines (HREpC and HRCEpC) bymiRNA microarrays. We analyzed the changes of 91putative miRNAs related to POX from 723 testedhuman miRNAs. In all, 10 miRNAs showed anincreased expression in renal cancer cells relative tonormal cells (Table 1). Among the 10 miRNAs, miR-23b* (predicted by Microinspector), miR-151-5p (pre-dicted by miRBase and Microinspector), miR-30b*(predicted by miRBase and Microinspector), miR-595(by miRBase, TargetScan and Microinspector) andmiR-766 (by TargetScan) showed an over 1.5-fold(0.58 in log2-transformed value) increase in cancer cellscompared with nontransformed cell lines, which wereselected for further study.
Negative regulation of miR-23b* on POX expressionTo assess whether the identified miRNAs could modu-late the expression of POX, we transfected the normalrenal cell line HREpC, which expressed relatively lowlevels of those miRNAs, with 100 nM synthesized mimicmiRNAs. The overexpression of each miRNA aftertransfection was confirmed by real-time RT–PCR usingU6 as an internal control (Supplementary Figure S1a).POX mRNA and protein levels were observed by real-time RT–PCR and western blot, respectively. As shownin Figures 2a and b, ectopic expression of miR-151-5pand miR-23b*, especially the latter, decreased POXprotein expression significantly (about 65% calculatedby densitometry), whereas POX mRNA level wasdecreased by only 19% (Po0.05).
We further showed the inhibitory effect of miR-23b*on POX protein level in DLD1 Tet-off POX cells, acolon cancer cell line stably transfected with full-lengthPOX complementary DNA containing 30UTR. POX isoverexpressed when doxycycline is removed. MimicmiR-151-5p and miR-23b* were transfected into thosecells overexpressing POX. The scrambled negative smallinterference RNA (siRNA) had no effects either onPOX mRNA or protein expression. miR-23b* but notmiR-151-5p inhibited POX mRNA (15% decrease,Po0.05), and especially POX protein expression sig-nificantly (53% decrease calculated by densitometry),which is comparable to those transfected with a
miR-23b* targets proline oxidaseW Liu et al
2
Oncogene

Figure 1 Reduced POX expression in renal cancer cell lines and human renal carcinoma tissue samples. (a) POX mRNA levels wereevaluated by real-time RT–PCR in normal renal cell lines (HREpC and HRCEpC) and renal cancer cell lines (786-0, TK10 and UO31).b-actin was used as an internal control. Data are reported as mean±s.e.m. (b) POX protein expression was tested by western blot.b-actin served as loading control. All results are representative of three independent experiments. (c) 16 paired renal carcinoma/normaltissues were stained by immunohistochemistry for the expression of POX. The representative images from five pairs of normal/cancertissues are shown (40� ).
Table 1 Upregulated miRNAs related to POX in renal cancer cells
Upregulated miRNA potentially targeting POX Sequence of target POX mRNA 30UTR Log2 (Tumor/Normal cells)
P(t-test)
Hsa-miR-23b*(uggguuccuggcaugcugauuu) 50yCTGCCAAGGCCAGCCCACACAGCCCGAGCCCC 1.79 o0.01Hsa-miR-151-5p (ucgaggagcucacagucuagu) 50yACTTTTGGGAACTCTCCTCGA 1.75 o0.01Hsa-miR-30b*(cugggagguggauguuuacuuc) 50yGAGGTGAGGTCAGGTGCCTCCCAG 1.20 o0.01Hsa-miR-595 (gaagugugccguggugugucu) 50yCACCUUUUUUCACCCCACACUUG 1.00 o0.01Hsa-miR-766 (acuccagccccacagccucagc) 50yCACACUUGCAGAGCUGCUGGAGG 0.58 o0.05Hsa-miR-296-5p (agggcccccccucaauccugu) 50yUCUCCUCGAAUGUGUGGGCCCAA 0.48 o0.05Hsa-miR-299-5p (ugguuuaccgucccacauacau) 50yGGCCUGCCUGGUCAAUAAACCAC 0.36 o0.05Hsa-miR-342-3p (ucucacacagaaaucgcacccgu) 50yAACTTTTGGGAACTCTCCTCGAATGTGTGGGC 0.34 o0.05Hsa-miR-940 (aaggcagggcccccgcucccc) 50yGGCACTCAGGTGTGGGCCGAACCTGATACCTG 0.30 o0.05Hsa-miR-30e*(cuuucagucggauguuuacagc) 50yCCTGGGACAGCCACTGGAAA 0.26 o0.05
Abbreviations: miRNA, microRNA; POX, proline oxidase; UTR, untranslated region.miRNA expression profiling was performed using human miRNA microarray chips in renal tumor cell lines (786-0, TK10 and UO31) andnormal renal cell lines (HREpC and HRCEpC). A total of 91 putative miRNAs related to POX were analyzed through the average log2 ratios(tumor to normal cells) calculated based on the two measurements of each miRNA and t-test. Upregluated miRNAs potentially related to POX areshown in the table.
miR-23b* targets proline oxidaseW Liu et al
3
Oncogene
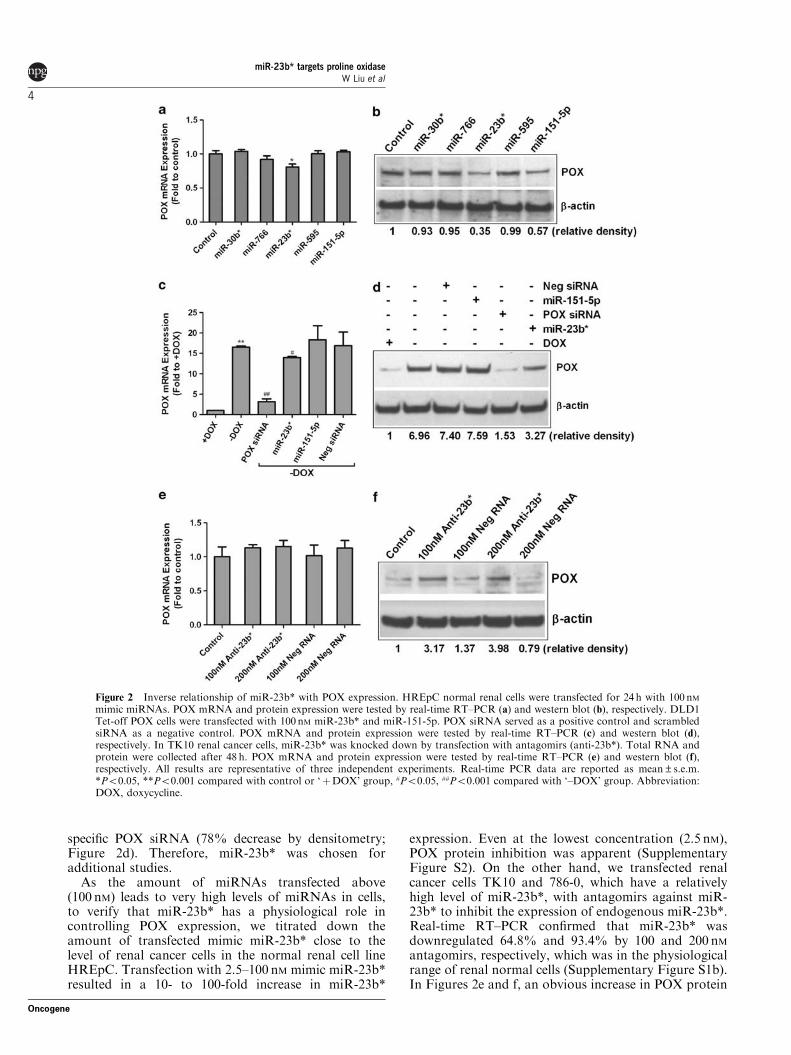
specific POX siRNA (78% decrease by densitometry;Figure 2d). Therefore, miR-23b* was chosen foradditional studies.
As the amount of miRNAs transfected above(100 nM) leads to very high levels of miRNAs in cells,to verify that miR-23b* has a physiological role incontrolling POX expression, we titrated down theamount of transfected mimic miR-23b* close to thelevel of renal cancer cells in the normal renal cell lineHREpC. Transfection with 2.5–100 nM mimic miR-23b*resulted in a 10- to 100-fold increase in miR-23b*
expression. Even at the lowest concentration (2.5 nM),POX protein inhibition was apparent (SupplementaryFigure S2). On the other hand, we transfected renalcancer cells TK10 and 786-0, which have a relativelyhigh level of miR-23b*, with antagomirs against miR-23b* to inhibit the expression of endogenous miR-23b*.Real-time RT–PCR confirmed that miR-23b* wasdownregulated 64.8% and 93.4% by 100 and 200 nMantagomirs, respectively, which was in the physiologicalrange of renal normal cells (Supplementary Figure S1b).In Figures 2e and f, an obvious increase in POX protein
Figure 2 Inverse relationship of miR-23b* with POX expression. HREpC normal renal cells were transfected for 24 h with 100 nMmimic miRNAs. POX mRNA and protein expression were tested by real-time RT–PCR (a) and western blot (b), respectively. DLD1Tet-off POX cells were transfected with 100 nM miR-23b* and miR-151-5p. POX siRNA served as a positive control and scrambledsiRNA as a negative control. POX mRNA and protein expression were tested by real-time RT–PCR (c) and western blot (d),respectively. In TK10 renal cancer cells, miR-23b* was knocked down by transfection with antagomirs (anti-23b*). Total RNA andprotein were collected after 48 h. POX mRNA and protein expression were tested by real-time RT–PCR (e) and western blot (f),respectively. All results are representative of three independent experiments. Real-time PCR data are reported as mean±s.e.m.*Po0.05, **Po0.001 compared with control or ‘þDOX’ group, #Po0.05, ##Po0.001 compared with ‘–DOX’ group. Abbreviation:DOX, doxycycline.
miR-23b* targets proline oxidaseW Liu et al
4
Oncogene
(2.2- to 2.9-fold), but not mRNA expression (only 12%increase, no statistical significance), was observed inTK10 cells on transfection with those two concentra-tions of antagomirs, suggesting that the identifiedmiRNA physiologically regulates POX expression.Similar changes were obtained in the 786-0 cell line(data not shown).
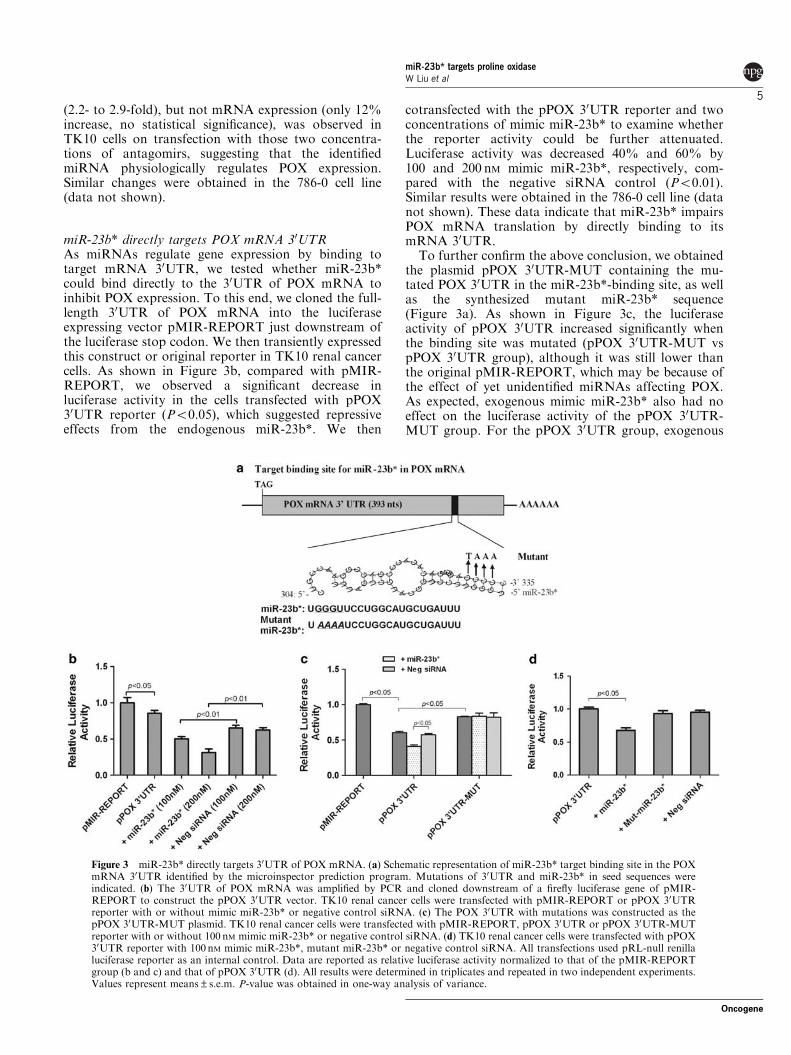
miR-23b* directly targets POX mRNA 30UTRAs miRNAs regulate gene expression by binding totarget mRNA 30UTR, we tested whether miR-23b*could bind directly to the 30UTR of POX mRNA toinhibit POX expression. To this end, we cloned the full-length 30UTR of POX mRNA into the luciferaseexpressing vector pMIR-REPORT just downstream ofthe luciferase stop codon. We then transiently expressedthis construct or original reporter in TK10 renal cancercells. As shown in Figure 3b, compared with pMIR-REPORT, we observed a significant decrease inluciferase activity in the cells transfected with pPOX30UTR reporter (Po0.05), which suggested repressiveeffects from the endogenous miR-23b*. We then
cotransfected with the pPOX 30UTR reporter and twoconcentrations of mimic miR-23b* to examine whetherthe reporter activity could be further attenuated.Luciferase activity was decreased 40% and 60% by100 and 200 nM mimic miR-23b*, respectively, com-pared with the negative siRNA control (Po0.01).Similar results were obtained in the 786-0 cell line (datanot shown). These data indicate that miR-23b* impairsPOX mRNA translation by directly binding to itsmRNA 30UTR.
To further confirm the above conclusion, we obtainedthe plasmid pPOX 30UTR-MUT containing the mu-tated POX 30UTR in the miR-23b*-binding site, as wellas the synthesized mutant miR-23b* sequence(Figure 3a). As shown in Figure 3c, the luciferaseactivity of pPOX 30UTR increased significantly whenthe binding site was mutated (pPOX 30UTR-MUT vspPOX 30UTR group), although it was still lower thanthe original pMIR-REPORT, which may be because ofthe effect of yet unidentified miRNAs affecting POX.As expected, exogenous mimic miR-23b* also had noeffect on the luciferase activity of the pPOX 30UTR-MUT group. For the pPOX 30UTR group, exogenous
Figure 3 miR-23b* directly targets 30UTR of POX mRNA. (a) Schematic representation of miR-23b* target binding site in the POXmRNA 30UTR identified by the microinspector prediction program. Mutations of 30UTR and miR-23b* in seed sequences wereindicated. (b) The 30UTR of POX mRNA was amplified by PCR and cloned downstream of a firefly luciferase gene of pMIR-REPORT to construct the pPOX 30UTR vector. TK10 renal cancer cells were transfected with pMIR-REPORT or pPOX 30UTRreporter with or without mimic miR-23b* or negative control siRNA. (c) The POX 30UTR with mutations was constructed as thepPOX 30UTR-MUT plasmid. TK10 renal cancer cells were transfected with pMIR-REPORT, pPOX 30UTR or pPOX 30UTR-MUTreporter with or without 100 nM mimic miR-23b* or negative control siRNA. (d) TK10 renal cancer cells were transfected with pPOX30UTR reporter with 100 nM mimic miR-23b*, mutant miR-23b* or negative control siRNA. All transfections used pRL-null renillaluciferase reporter as an internal control. Data are reported as relative luciferase activity normalized to that of the pMIR-REPORTgroup (b and c) and that of pPOX 30UTR (d). All results were determined in triplicates and repeated in two independent experiments.Values represent means±s.e.m. P-value was obtained in one-way analysis of variance.
miR-23b* targets proline oxidaseW Liu et al
5
Oncogene
mimic miR-23b* decreased its luciferase activity,whereas mutant miR-23b* did not (Figure 3d).
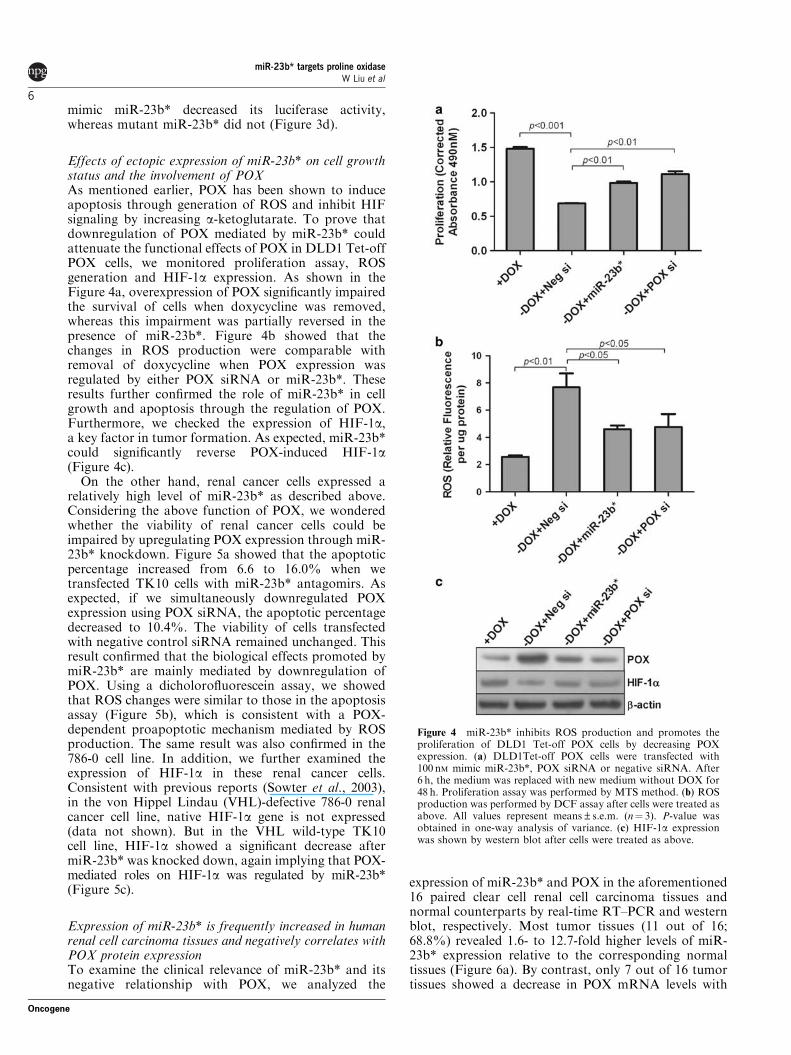
Effects of ectopic expression of miR-23b* on cell growthstatus and the involvement of POXAs mentioned earlier, POX has been shown to induceapoptosis through generation of ROS and inhibit HIFsignaling by increasing a-ketoglutarate. To prove thatdownregulation of POX mediated by miR-23b* couldattenuate the functional effects of POX in DLD1 Tet-offPOX cells, we monitored proliferation assay, ROSgeneration and HIF-1a expression. As shown in theFigure 4a, overexpression of POX significantly impairedthe survival of cells when doxycycline was removed,whereas this impairment was partially reversed in thepresence of miR-23b*. Figure 4b showed that thechanges in ROS production were comparable withremoval of doxycycline when POX expression wasregulated by either POX siRNA or miR-23b*. Theseresults further confirmed the role of miR-23b* in cellgrowth and apoptosis through the regulation of POX.Furthermore, we checked the expression of HIF-1a,a key factor in tumor formation. As expected, miR-23b*could significantly reverse POX-induced HIF-1a(Figure 4c).
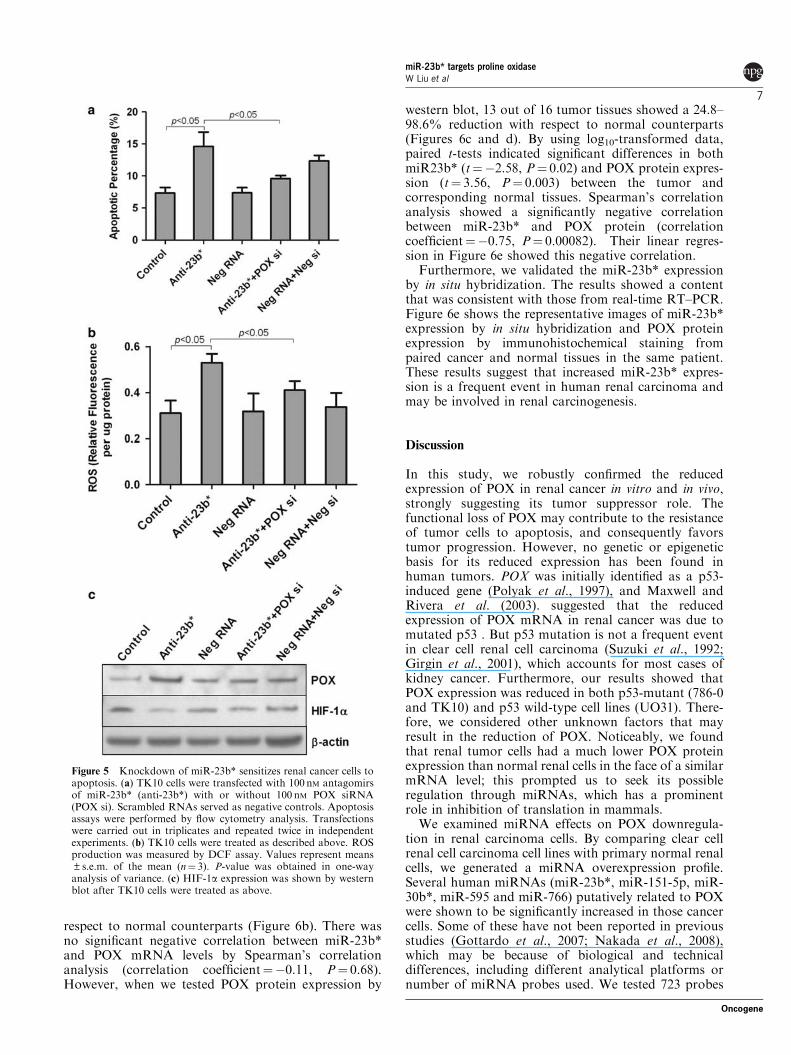
On the other hand, renal cancer cells expressed arelatively high level of miR-23b* as described above.Considering the above function of POX, we wonderedwhether the viability of renal cancer cells could beimpaired by upregulating POX expression through miR-23b* knockdown. Figure 5a showed that the apoptoticpercentage increased from 6.6 to 16.0% when wetransfected TK10 cells with miR-23b* antagomirs. Asexpected, if we simultaneously downregulated POXexpression using POX siRNA, the apoptotic percentagedecreased to 10.4%. The viability of cells transfectedwith negative control siRNA remained unchanged. Thisresult confirmed that the biological effects promoted bymiR-23b* are mainly mediated by downregulation ofPOX. Using a dicholorofluorescein assay, we showedthat ROS changes were similar to those in the apoptosisassay (Figure 5b), which is consistent with a POX-dependent proapoptotic mechanism mediated by ROSproduction. The same result was also confirmed in the786-0 cell line. In addition, we further examined theexpression of HIF-1a in these renal cancer cells.Consistent with previous reports (Sowter et al., 2003),in the von Hippel Lindau (VHL)-defective 786-0 renalcancer cell line, native HIF-1a gene is not expressed(data not shown). But in the VHL wild-type TK10cell line, HIF-1a showed a significant decrease aftermiR-23b* was knocked down, again implying that POX-mediated roles on HIF-1a was regulated by miR-23b*(Figure 5c).
Expression of miR-23b* is frequently increased in humanrenal cell carcinoma tissues and negatively correlates withPOX protein expressionTo examine the clinical relevance of miR-23b* and itsnegative relationship with POX, we analyzed the
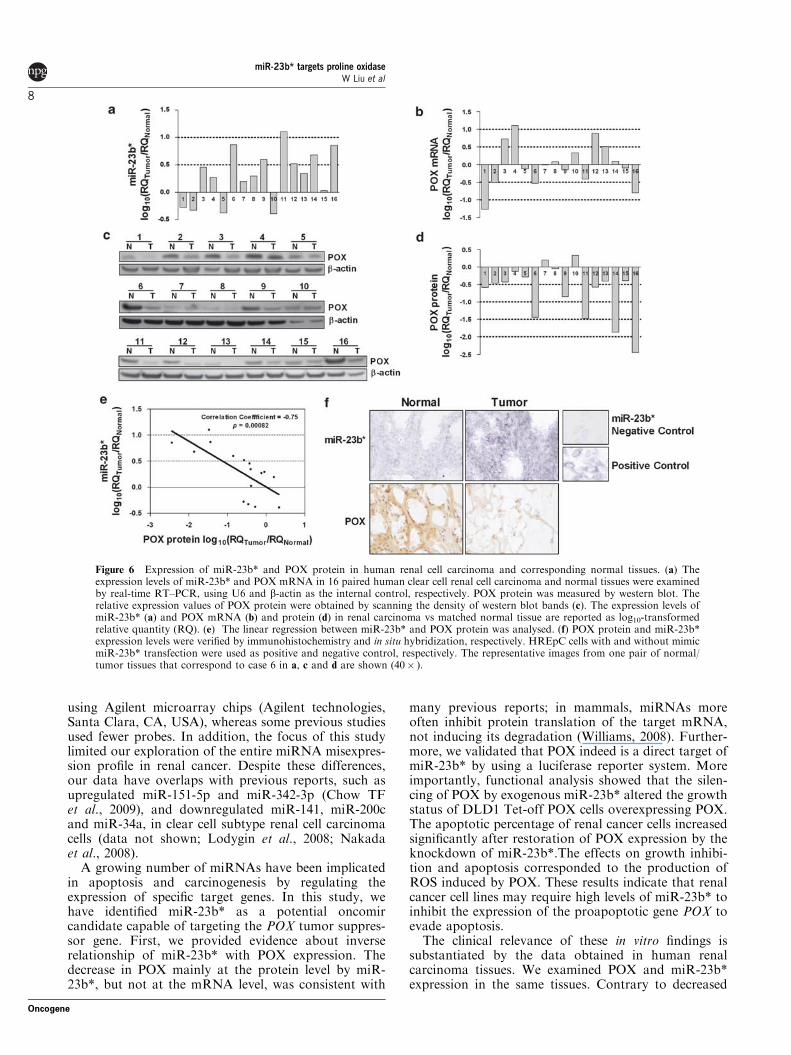
expression of miR-23b* and POX in the aforementioned16 paired clear cell renal cell carcinoma tissues andnormal counterparts by real-time RT–PCR and westernblot, respectively. Most tumor tissues (11 out of 16;68.8%) revealed 1.6- to 12.7-fold higher levels of miR-23b* expression relative to the corresponding normaltissues (Figure 6a). By contrast, only 7 out of 16 tumortissues showed a decrease in POX mRNA levels with
Figure 4 miR-23b* inhibits ROS production and promotes theproliferation of DLD1 Tet-off POX cells by decreasing POXexpression. (a) DLD1Tet-off POX cells were transfected with100 nM mimic miR-23b*, POX siRNA or negative siRNA. After6 h, the medium was replaced with new medium without DOX for48 h. Proliferation assay was performed by MTS method. (b) ROSproduction was performed by DCF assay after cells were treated asabove. All values represent means±s.e.m. (n¼ 3). P-value wasobtained in one-way analysis of variance. (c) HIF-1a expressionwas shown by western blot after cells were treated as above.
miR-23b* targets proline oxidaseW Liu et al
6
Oncogene
respect to normal counterparts (Figure 6b). There wasno significant negative correlation between miR-23b*and POX mRNA levels by Spearman’s correlationanalysis (correlation coefficient¼�0.11, P¼ 0.68).However, when we tested POX protein expression by
western blot, 13 out of 16 tumor tissues showed a 24.8–98.6% reduction with respect to normal counterparts(Figures 6c and d). By using log10-transformed data,paired t-tests indicated significant differences in bothmiR23b* (t¼�2.58, P¼ 0.02) and POX protein expres-sion (t¼ 3.56, P¼ 0.003) between the tumor andcorresponding normal tissues. Spearman’s correlationanalysis showed a significantly negative correlationbetween miR-23b* and POX protein (correlationcoefficient¼�0.75, P¼ 0.00082). Their linear regres-sion in Figure 6e showed this negative correlation.
Furthermore, we validated the miR-23b* expressionby in situ hybridization. The results showed a contentthat was consistent with those from real-time RT–PCR.Figure 6e shows the representative images of miR-23b*expression by in situ hybridization and POX proteinexpression by immunohistochemical staining frompaired cancer and normal tissues in the same patient.These results suggest that increased miR-23b* expres-sion is a frequent event in human renal carcinoma andmay be involved in renal carcinogenesis.
Discussion
In this study, we robustly confirmed the reducedexpression of POX in renal cancer in vitro and in vivo,strongly suggesting its tumor suppressor role. Thefunctional loss of POX may contribute to the resistanceof tumor cells to apoptosis, and consequently favorstumor progression. However, no genetic or epigeneticbasis for its reduced expression has been found inhuman tumors. POX was initially identified as a p53-induced gene (Polyak et al., 1997), and Maxwell andRivera et al. (2003). suggested that the reducedexpression of POX mRNA in renal cancer was due tomutated p53 . But p53 mutation is not a frequent eventin clear cell renal cell carcinoma (Suzuki et al., 1992;Girgin et al., 2001), which accounts for most cases ofkidney cancer. Furthermore, our results showed thatPOX expression was reduced in both p53-mutant (786-0and TK10) and p53 wild-type cell lines (UO31). There-fore, we considered other unknown factors that mayresult in the reduction of POX. Noticeably, we foundthat renal tumor cells had a much lower POX proteinexpression than normal renal cells in the face of a similarmRNA level; this prompted us to seek its possibleregulation through miRNAs, which has a prominentrole in inhibition of translation in mammals.
We examined miRNA effects on POX downregula-tion in renal carcinoma cells. By comparing clear cellrenal cell carcinoma cell lines with primary normal renalcells, we generated a miRNA overexpression profile.Several human miRNAs (miR-23b*, miR-151-5p, miR-30b*, miR-595 and miR-766) putatively related to POXwere shown to be significantly increased in those cancercells. Some of these have not been reported in previousstudies (Gottardo et al., 2007; Nakada et al., 2008),which may be because of biological and technicaldifferences, including different analytical platforms ornumber of miRNA probes used. We tested 723 probes
Figure 5 Knockdown of miR-23b* sensitizes renal cancer cells toapoptosis. (a) TK10 cells were transfected with 100 nM antagomirsof miR-23b* (anti-23b*) with or without 100 nM POX siRNA(POX si). Scrambled RNAs served as negative controls. Apoptosisassays were performed by flow cytometry analysis. Transfectionswere carried out in triplicates and repeated twice in independentexperiments. (b) TK10 cells were treated as described above. ROSproduction was measured by DCF assay. Values represent means±s.e.m. of the mean (n¼ 3). P-value was obtained in one-wayanalysis of variance. (c) HIF-1a expression was shown by westernblot after TK10 cells were treated as above.
miR-23b* targets proline oxidaseW Liu et al
7
Oncogene
using Agilent microarray chips (Agilent technologies,Santa Clara, CA, USA), whereas some previous studiesused fewer probes. In addition, the focus of this studylimited our exploration of the entire miRNA misexpres-sion profile in renal cancer. Despite these differences,our data have overlaps with previous reports, such asupregulated miR-151-5p and miR-342-3p (Chow TFet al., 2009), and downregulated miR-141, miR-200cand miR-34a, in clear cell subtype renal cell carcinomacells (data not shown; Lodygin et al., 2008; Nakadaet al., 2008).
A growing number of miRNAs have been implicatedin apoptosis and carcinogenesis by regulating theexpression of specific target genes. In this study, wehave identified miR-23b* as a potential oncomircandidate capable of targeting the POX tumor suppres-sor gene. First, we provided evidence about inverserelationship of miR-23b* with POX expression. Thedecrease in POX mainly at the protein level by miR-23b*, but not at the mRNA level, was consistent with
many previous reports; in mammals, miRNAs moreoften inhibit protein translation of the target mRNA,not inducing its degradation (Williams, 2008). Further-more, we validated that POX indeed is a direct target ofmiR-23b* by using a luciferase reporter system. Moreimportantly, functional analysis showed that the silen-cing of POX by exogenous miR-23b* altered the growthstatus of DLD1 Tet-off POX cells overexpressing POX.The apoptotic percentage of renal cancer cells increasedsignificantly after restoration of POX expression by theknockdown of miR-23b*.The effects on growth inhibi-tion and apoptosis corresponded to the production ofROS induced by POX. These results indicate that renalcancer cell lines may require high levels of miR-23b* toinhibit the expression of the proapoptotic gene POX toevade apoptosis.
The clinical relevance of these in vitro findings issubstantiated by the data obtained in human renalcarcinoma tissues. We examined POX and miR-23b*expression in the same tissues. Contrary to decreased
Figure 6 Expression of miR-23b* and POX protein in human renal cell carcinoma and corresponding normal tissues. (a) Theexpression levels of miR-23b* and POX mRNA in 16 paired human clear cell renal cell carcinoma and normal tissues were examinedby real-time RT–PCR, using U6 and b-actin as the internal control, respectively. POX protein was measured by western blot. Therelative expression values of POX protein were obtained by scanning the density of western blot bands (c). The expression levels ofmiR-23b* (a) and POX mRNA (b) and protein (d) in renal carcinoma vs matched normal tissue are reported as log10-transformedrelative quantity (RQ). (e) The linear regression between miR-23b* and POX protein was analysed. (f) POX protein and miR-23b*expression levels were verified by immunohistochemistry and in situ hybridization, respectively. HREpC cells with and without mimicmiR-23b* transfection were used as positive and negative control, respectively. The representative images from one pair of normal/tumor tissues that correspond to case 6 in a, c and d are shown (40� ).
miR-23b* targets proline oxidaseW Liu et al
8
Oncogene

POX protein, but not mRNA, miR-23b* was frequentlyincreased in renal carcinoma, strongly suggesting thatthe increased miR-23b* might contribute to renaloncogenesis and progression by downregulating POX.This provides a possible strategic opening to inhibittumor growth by decreasing the levels of miR-23b* orby blocking its function. However, the potential of miR-23b* and POX as diagnostic indicators or therapeutictargets in renal carcinoma remains to be investigated.
In fact, because of the identification of VHL tumorsuppressor as a negative regulator of HIF-a in renalcancer, the treatment of renal cell carcinoma bytargeting HIF-a or its downstream genes has showngreat promise (Kaelin, 2009; Smaldone and Maranchie,2009). However, the development of more effectivemanagement strategies remains necessary. As POX hasbeen found to downregulate HIF-1 signaling byincreasing a-ketoglutarate, a substrate of PHD (Liuet al., 2009), we examined the possible effects on HIF-1aby miR-23b* inhibition of POX expression. In DLD1Tet-off POX cells, miR-23b* did induce the expressionof HIF-1a by inhibiting POX. Although in the 786-0 cellline with a VHL-negative background, HIF-2a, but notHIF-1a, exhibits tumorigenic potential (Kondo et al.,2002; Maranchie et al., 2002), HIF-1a was inhibitedafter miR-23b* antagomirs increased POX expression inTK10 renal cancer cells without VHL mutation. Thisfinding provides a broader prospect for miR-23b*application in renal cancer therapeutics on the basis ofPOX-dependent HIF-1a signaling.
As mentioned above, POX is encoded by a p53-induced gene (Polyak et al., 1997). Maxwell SA et al.(2003) reported that reduced expression of POX mRNAin renal cancer was due to a p53 mutation (Maxwell andRivera, 2003). However, as p53 mutation is not afrequent event in clear cell renal cell carcinoma, wecould not exclude the possibility that wild-type p53regulates the final expression of POX by a miR-23b*-dependent mechanism in renal cancer. To verify thisview, we expressed exogenous p53 in p53-mutant renalcancer cell line TK10 to investigate the effect of p53 onmiR-23b*. The results showed that p53 significantlyincreased the expression of miR-23b* (SupplementaryFigure S3), suggesting that the upregulation of miR-23b* by p53 may counteract the direct role of p53 onPOX gene expression in clear cell renal cell carcinoma.This interaction might also account for discrepanciesbetween POX mRNA and protein expression.
In addition, current evidence suggests that miR-23b*could be regulated by factors other than p53. Forexample, several reports have shown the link betweenupregulation of miR-23b and hypoxia (Kulshreshthaet al., 2007, 2008; Guimbellot et al., 2009). As miR-23band miR-23b* share the same precursor, miR-23b* alsocould be regulated by HIF. In renal cell carcinoma, theconstitutive expression of HIF due to VHL deficiencymay link this regulation of miR-23b* with VHL. Inlymphoma cells, the oncogenic transcription factorc-Myc transcriptionally suppresses miR-23b to enhancemitochondrial glutaminase expression and glutaminemetabolism (Gao et al., 2009). Thus, c-Myc effects on
miR-23b* are a likely possibility. The fact that HIF-1negatively regulates mitochondrial biogenesis by inhibit-ing c-Myc activity in VHL-deficient renal carcinomacells (Zhang et al., 2007) further increases the possibilitythat miR-23b* could be regulated by VHL, HIF andc-Myc, thereby affecting the expression of POX. Theseregulatory interactions are of great interest and arebeing pursued.
An additional point worth mentioning is the defini-tion of miRNA*. As previously documented, pre-miRNAs are processed by the endonuclease Dicer intoa miRNA duplex: one strand becomes mature miRNA,whereas the other strand, commonly named as miR-NA*, was usually thought to be easily degraded andthus short lived (Denli et al., 2004; Gregory et al., 2004).Recent evidence indicates that some of the miRNAs*are also functional, such as miR-9* and miR-199* (Kimet al., 2008; Migliore et al., 2008; Yang et al., 2008; Nasset al., 2009). Our identification of miR-23b* providesanother example.
In summary, we show for the first time that the loss ofPOX expression in cancer is mediated by miRNAs, andprovide evidence supporting the oncogenic role ofmiRNAs by regulating a tumor suppressor gene.Knockdown of miR-23b* not only increased apoptosisby upregulating POX through ROS generation but alsodecreased HIF-1a signaling in renal cancer cells. Thesefindings suggest that miR-23b* might be a noveltherapeutic target for renal cell carcinoma. Thispotential role is being intensively investigated.
Materials and methods
Cell lines and tissue specimensThe human renal cancer cell lines (786-0, TK10 and UO31)were provided by the NCI cell line repository and werecultured in RPMI-1640 supplemented with 10% fetal bovineserum. Two normal human renal/renal cortical epithelial cells(HREpC and HRCEpC) were obtained from PromoCell(Heidelberg, Germany), and were cultured in renal epithelialcell medium provided by PromoCell. In addition, DLD1 Tet-off POX cells were cultured as described (Donald et al., 2001).Sixteen pairs of human clear cell renal cell carcinomas and
corresponding normal renal tissues were obtained withappropriate informed consent from the Cooperative HumanTissue Network. Their histological characteristics were con-firmed by the pathologist.
miRNA microarray expression profilingTotal RNA from each cell line was collected using an RNeasyMini Kit (Qiagen, Germantown, MD, USA). miRNA profilingwas performed using human miRNA microarray chips (V2)from Agilent Technologies (Santa Clara, CA, USA), whichcontains 723 human miRNA probes. Briefly, 100 ng of mRNAfrom each sample was labeled with Cy3 fluorescent dye,followed by dephosphorylation and denaturation. Hybridiza-tion and washing of the microarray slides were performed asrecommended. Scanning was performed using a G2505BMicroarray Scanner (Agilent Technologies). After scanningthe microarrays, we used Agilent Feature Extraction Softwareto generate raw intensity data. Average log2 ratios werecalculated on the basis of the two measurements of eachmiRNA.
miR-23b* targets proline oxidaseW Liu et al
9
Oncogene

Real-time RT–PCR analysis of miRNAs and POX mRNAexpressionTotal RNA was collected using an RNeasy Mini Kit (Qiagen).For POX mRNA analysis, complementary DNA was synthe-sized using the SuperScript II Reverse Transcriptase Kit(Invitrogen, Carlsbad, CA, USA). SYBR Green Supermix(Bio-Rad, Hercules, CA, USA) was used for real-time PCRapplications. The primer set for POX was 50-GCCATTAAGCTCACAGCACTGGG–30 (forward) and 50-CTGATGGCCGGCTGGAAGTAG-30 (reverse). b-actin served as aninternal control using primers 50-ATCCACGAAACTACCTTCAACTC-30 (forward) and 50-GAGGAGCAATGATCTTGATCTTC-30 (reverse). Stem-loop real-time RT–PCR formature miRNAs was carried out using the Taqman Micro-RNA assay (Applied Biosystems, Foster City, CA, USA) asdescribed (Ma et al., 2007), using U6 as an internal control.
RNA oligoribonucleotides and cell transfectionsMimic miRNAs were designed as described previously (Limet al., 2005). Antagomir against miR-23b* (anti-23b*) was theantisense sequence of mature miR-23b*. The siRNA targetingPOX mRNA was designated as POX siRNA. The controlRNA duplex for both mimic miRNAs and POX siRNA(designated as negative siRNA) and single-strand RNAcontrol for anti-miR-23b* (designated as negative RNA) werescrambled and nonhomologous to any human genomesequence. All the RNA oligoribonucleotides were synthesizedby Invitrogen. Renal cancer cells, normal renal cells or DLD1Tet-off POX cells were transfected with the above-mentionedRNA molecules by using Lipofectamine 2000 (Invitrogen)after 24 h of seeding. Briefly, Lipofectamine 2000 was mixedwith Opti MEM medium (Gibco, Carlsbad, CA, USA) andincubated for 5min. Meanwhile, RNA molecules were dilutedin Opti MEM reduced-serum medium (Gibco). Thereafter, thediluted miRNA and Lipofectamine 2000 were mixed andincubated for 20min at room temperature, and the resultantmixture was added to each well/flask.
Plasmid vector constructionThe 30UTR of POX (393 bp) containing the putative miR-23b*binding sequence was amplified using PCR from complemen-tary DNA synthesized from total RNA. The used primerscontained the following restriction sites (lowercase): forward,50-AGCactagtCAGCACACCCTTAGCC-30 (SpeI); reverse,
50-GCAGaagcttTGGTTTATTGACCAG-30 (HindIII). ThePCR product was cloned into the SpeI and HindIII restrictionsites downstream of the open reading frame of luciferase inpMIR-REPORT Vector (Ambion, Austin, TX, USA) togenerate the pPOX 30UTR reporter.Mutation of the miR-23b* binding site in the 30UTR of
POX mRNA was created using site-directed mutagenesis bythe megaprimer PCR method (Figure 3a). Primers include theforward and reverse primers shown above, and the mutatedprimer 50-ACACAGCCCGAtaaaCTTGGGGAG-30 (muta-tion sites are shown in lowercase). The mutated PCR productwas cloned into the cloning site of pMIR-REPORT togenerate the pPOX 30UTR-MUT reporter. All cloned productswere verified by sequencing before use.The p53 expression construct was generated by amplifying
p53 complementary DNA and was subsequently cloned intopTarget as described before (Pandhare et al., 2006). The pCicontrol vector was obtained from Promega (Fitchburg, WI,USA).The following procedures are included in Supplementary
Materials: western blot analysis, luciferase reporter assay,apoptosis assay, proliferation assay, measurement of intracel-lular ROS, POX immunohistochemical staining and miR-23b*in situ hybridization.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank Dr Anna E Maciag for insightful comments, DrMatthew J Fivash for statistical analysis, Dr Miriam R Anverfor evaluating POX immunohistochemical staining and miR-23b* in situ hybridization, Dr Chang H Kim for his help withthe miRNA microarray. This research was supported (in part)by the Intramural Research Program of the NIH, the NationalCancer Institute, Center for Cancer Research. The content ofthis publication does not necessarily reflect the views orpolicies of the Department of Health and Human Services, nordoes mention of trade names, commercial products ororganizations imply endorsement by the US Government.
References
Bartel DP. (2004). MicroRNAs: genomics, biogenesis, mechanism, andfunction. Cell 116: 281–297.
Chen K, Rajewsky N. (2007). The evolution of gene regulation bytranscription factors and microRNAs. Nat Rev Genet 8: 93–103.
Chow TF YY, Lianidou E, Romaschin AD, Honey RJ, Stewart R,Pace KT et al. (2009). Differential expression profiling of micro-RNAs and their potential involvement in renal cell carcinomapathogenesis. Clin Biochem 43: 150–158.
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. (2004).Processing of primary microRNAs by the microprocessor complex.Nature 432: 231–235.
Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D et al. (2001).Proline oxidase, encoded by p53-induced gene-6, catalyzes thegeneration of proline-dependent reactive oxygen species. Cancer Res
61: 1810–1815.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M
et al. (2009). miR-205 Exerts tumor-suppressive functions in humanprostate through down-regulation of protein kinase Cepsilon.Cancer Res 69: 2287–2295.
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T et al. (2009).c-Myc suppression of miR-23a/b enhances mitochondrial glutaminaseexpression and glutamine metabolism. Nature 458: 762–765.
Girgin C, Tarhan H, Hekimgil M, Sezer A, Gurel G. (2001). P53mutations and other prognostic factors of renal cell carcinoma. Urol
Int 66: 78–83.Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P et al.
(2007). Micro-RNA profiling in kidney and bladder cancers. Urol
Oncol 25: 387–392.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B,
Cooch N et al. (2004). The microprocessor complex mediates thegenesis of microRNAs. Nature 432: 235–240.
Guimbellot JS, Erickson SW, Mehta T, Wen H, Page GP, Sorscher EJet al. (2009). Correlation of microRNA levels during hypoxia withpredicted target mRNAs through genome-wide microarray analysis.BMC Med Genomics 2: 15.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni Set al. (2005). MicroRNA gene expression deregulation in humanbreast cancer. Cancer Res 65: 7065–7070.
miR-23b* targets proline oxidaseW Liu et al
10
Oncogene

Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng Aet al. (2005). RAS is regulated by the let-7 microRNA family. Cell
120: 635–647.Kaelin Jr WG. (2009). Treatment of kidney cancer: insights provided
by the VHL tumor-suppressor protein. Cancer 115: 2262–2272.Kim S, Lee UJ, Kim MN, Lee EJ, Kim JY, Lee MY et al. (2008).
MicroRNA miR-199a*regulates the MET proto-oncogene and thedownstream extracellular signal-regulated kinase 2 (ERK2). J Biol
Chem 283: 18158–18166.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin Jr WG.
(2002). Inhibition of HIF is necessary for tumor suppression by thevon Hippel-Lindau protein. Cancer Cell 1: 237–246.
Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. (2008). AmicroRNA component of the hypoxic response. Cell Death Differ
15: 667–671.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-
Perez FJ et al. (2007). A microRNA signature of hypoxia. Mol Cell
Biol 27: 1859–1867.Lewis BP, Burge CB, Bartel DP. (2005). Conserved seed pairing, often
flanked by adenosines, indicates that thousands of human genes aremicroRNA targets. Cell 120: 15–20.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM,Castle J et al. (2005). Microarray analysis shows that somemicroRNAs downregulate large numbers of target mRNAs. Nature
433: 769–773.Liu Y, Borchert GL, Donald SP, Diwan BA, Anver M, Phang JM.
(2009). Proline oxidase functions as a mitochondrial tumorsuppressor in human cancers. Cancer Res 69: 6414–6422.
Liu Y, Borchert GL, Donald SP, Surazynski A, Hu CA, Weydert CJet al. (2005). MnSOD inhibits proline oxidase-induced apoptosis incolorectal cancer cells. Carcinogenesis 26: 1335–1342.
Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. (2006). Prolineoxidase activates both intrinsic and extrinsic pathways forapoptosis: the role of ROS/superoxides, NFAT and MEK/ERKsignaling. Oncogene 25: 5640–5647.
Liu Y, Borchert GL, Surazynski A, Phang JM. (2008). Proline oxidase,a p53-induced gene, targets COX-2/PGE2 signaling to induceapoptosis and inhibit tumor growth in colorectal cancers. Oncogene
27: 6729–6737.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T,
Korner H et al. (2008). Inactivation of miR-34a by aberrant CpGmethylation in multiple types of cancer. Cell Cycle 7: 2591–2600.
Ma L, Teruya-Feldstein J, Weinberg RA. (2007). Tumour invasionand metastasis initiated by microRNA-10b in breast cancer. Nature
449: 682–688.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM,
Klausner RD. (2002). The contribution of VHL substrate bindingand HIF1-alpha to the phenotype of VHL loss in renal cellcarcinoma. Cancer Cell 1: 247–255.
Maxwell SA, Rivera A. (2003). Proline oxidase induces apoptosis intumor cells, and its expression is frequently absent or reduced inrenal carcinomas. J Biol Chem 278: 9784–9789.
Migliore C, Petrelli A, Ghiso E, Corso S, Capparuccia L, Eramo Aet al. (2008). MicroRNAs impair MET-mediated invasive growth.Cancer Res 68: 10128–10136.
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AMet al. (2006). A pattern-based method for the identification ofmicroRNA binding sites and their corresponding heteroduplexes.Cell 126: 1203–1217.
Nakada C, Matsuura K, Tsukamoto Y, Tanigawa M, Yoshimoto T,Narimatsu T et al. (2008). Genome-wide microRNA expressionprofiling in renal cell carcinoma: significant down-regulation ofmiR-141 and miR-200c. J Pathol 216: 418–427.
Nass D, Rosenwald S, Meiri E, Gilad S, Tabibian-Keissar H,Schlosberg A et al. (2009). MiR-92b and miR-9/9*arespecifically expressed in brain primary tumors and can be used todifferentiate primary from metastatic brain tumors. Brain Pathol 19:375–383.
Northcott PA, Fernandez LA, Hagan JP, Ellison DW, Grajkowska W,Gillespie Y et al. (2009). The miR-17/92 polycistron is up-regulatedin sonic hedgehog-driven medulloblastomas and induced by N-mycin sonic hedgehog-treated cerebellar neural precursors. Cancer Res
69: 3249–3255.Pandhare J, Cooper SK, Phang JM. (2006). Proline oxidase, a
proapoptotic gene, is induced by troglitazone: evidence for bothperoxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem 281: 2044–2052.
Phang JM. (1985). The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr Top Cell Regul 25: 91–132.
Phang JM, Pandhare J, Liu Y. (2008). The metabolism ofproline as microenvironmental stress substrate. J Nutr 138:2008S–2015S.
Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. (1997). Amodel for p53-induced apoptosis. Nature 389: 300–305.
Smaldone MC, Maranchie JK. (2009). Clinical implications of hypoxiainducible factor in renal cell carcinoma. Urol Oncol 27: 238–245.
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. (2003).Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptionalresponse to hypoxia. Cancer Res 63: 6130–6134.
Suzuki Y, Tamura G, Satodate R, Fujioka T. (1992). Infrequentmutation of p53 gene in human renal cell carcinoma detected bypolymerase chain reaction single-strand conformation polymorph-ism analysis. Jpn J Cancer Res 83: 233–235.
Williams AE. (2008). Functional aspects of animal microRNAs. Cell
Mol Life Sci 65: 545–562.Yang H, Kong W, He L, Zhao JJ, O’Donnell JD, Wang J et al. (2008).
MicroRNA expression profiling in human ovarian cancer: miR-214induces cell survival and cisplatin resistance by targeting PTEN.Cancer Res 68: 425–433.
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KIet al. (2007). HIF-1 inhibits mitochondrial biogenesis and cellularrespiration in VHL-deficient renal cell carcinoma by repression ofC-MYC activity. Cancer Cell 11: 407–420.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
miR-23b* targets proline oxidaseW Liu et al
11
Oncogene




















