
Pt–NiO/C anode electrocatalysts for direct methanol fuel cells
Upload
independentCategory
view
6download
0
ARTICLE IN PRESS
Available at www.sciencedirect.com
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1
0043-1354/$ - see frodoi:10.1016/j.watres
�Corresponding auE-mail address:
journal homepage: www.elsevier.com/locate/watres
Mineralization of salicylic acid in acidic aqueous mediumby electrochemical advanced oxidation processes usingplatinum and boron-doped diamond as anode andcathodically generated hydrogen peroxide
Elena Guinea, Conchita Arias, Pere Lluıs Cabot, Jose Antonio Garrido,Rosa Marıa Rodrıguez, Francesc Centellas, Enric Brillas�
Laboratori d’Electroquımica dels Materials i del Medi Ambient, Departament de Quımica Fısica, Facultat de Quımica,
Universitat de Barcelona, Martı i Franques 1-11, 08028 Barcelona, Spain
a r t i c l e i n f o
Article history:
Received 31 May 2007
Received in revised form
19 July 2007
Accepted 24 July 2007
Available online 1 August 2007
Keywords:
Salicylic acid
Anodic oxidation
Electro-Fenton
Photoelectro-Fenton
Solar photoelectro-Fenton
Oxidation products
nt matter & 2007 Elsevie.2007.07.046
thor. Tel.: +34 93 4021223;[email protected] (E. Brillas)
a b s t r a c t
Solutions containing 164 mg L�1 salicylic acid of pH 3.0 have been degraded by
electrochemical advanced oxidation processes such as anodic oxidation, anodic oxidation
with electrogenerated H2O2, electro-Fenton, photoelectro-Fenton and solar photoelectro-
Fenton at constant current density. Their oxidation power has been comparatively studied
in a one-compartment cell with a Pt or boron-doped diamond (BDD) anode and a graphite
or O2-diffusion cathode. In the three latter procedures, 0.5 mM Fe2+ is added to the solution
to form hydroxyl radical (dOH) from Fenton’s reaction between Fe2+ and H2O2 generated at
the O2-diffusion cathode. Total mineralization is attained for all methods with BDD and for
photoelectro-Fenton and solar photoelectro-Fenton with Pt. The poor decontamination
achieved in anodic oxidation and electro-Fenton with Pt is explained by the slow removal
of most pollutants by dOH formed from water oxidation at the Pt anode in comparison to
their quick destruction with dOH produced at BDD. dOH generated from Fenton’s reaction
oxidizes rapidly all aromatic pollutants, but it cannot destroy final Fe(III)–oxalate
complexes. Solar photoelectro-Fenton treatments always yield quicker degradation rate
due to the very fast photodecarboxylation of these complexes by UVA irradiation supplied
by solar light. The effect of current density on the degradation rate, efficiency and energy
cost of all methods is examined. The salicylic acid decay always follows a pseudo-
first-order kinetics. 2,3-Dihydroxybenzoic, 2,5-dihydroxybenzoic, 2,6-dihydroxybenzoic,
a-ketoglutaric, glycolic, glyoxylic, maleic, fumaric, malic, tartronic and oxalic acids are
detected as oxidation products. A general reaction sequence for salicylic acid mineraliza-
tion considering all these intermediates is proposed.
& 2007 Elsevier Ltd. All rights reserved.
1. Introduction
Advanced oxidation processes (AOPs) are chemical, photo-
chemical, photocatalytic and electrochemical methods char-
r Ltd. All rights reserved.
fax: +34 93 4021231..
acterized by the in situ generation of hydroxyl radical (dOH).
They are promising environmentally friendly technologies for
the treatment of wastewaters containing low contents of
toxic and biorefractory organics (Tarr, 2003). This is feasible

ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1500
because dOH is the second most strong oxidant known after
fluorine and has a very high standard potential (E1(dOH/
H2O) ¼ 2.80 V vs. NHE) that makes it able to non-selectively
react with organic pollutants giving dehydrogenated or
hydroxylated derivatives up to overall mineralization, i.e.,
total conversion into CO2 and inorganic ions. Recently, there
is increasing interest in the use of electrochemical methods
such as anodic oxidation (AO) and indirect electro-oxidation
methods with H2O2 electrogeneration, so-called electroche-
mical advanced oxidation processes (EAOPs), which can
produce dOH as the main oxidizing agent by different ways
(Brillas et al., 2000; Kraft et al., 2003).
The most popular EAOP is AO consisting in the destruction
of organics in an electrolytic cell under the action of hydroxyl
radical formed as intermediate from water oxidation to O2 at
the surface of a high O2-overvoltage anode (Marselli et al.,
2003; Panizza and Cerisola, 2005):
MþH2O!MðdOHÞ þHþ þ e�; (1)
where M(dOH) denotes the hydroxyl radical adsorbed on the
anode M or remaining near its surface. In the past years, AO
has attracted great attention for wastewater remediation due
to the use of a boron-doped diamond (BDD) thin-film anode
that has technologically important characteristics such as an
inert surface with low adsorption properties, remarkable
corrosion stability and a wide potential windows in aqueous
medium (Panizza and Cerisola, 2005). These properties confer
to the BDD electrode a much greater O2-overvoltage than a
conventional anode such as Pt, allowing the production of
higher amount of reactive BDD(dOH) than Pt(dOH) from
reaction (1) and enhancing the oxidation rate of most
organics. Different authors have shown that in aqueous
medium several organic pollutants can be completely miner-
alized by AO with BDD, whereas the use of a Pt anode under
comparable conditions leads to weak decontamination be-
cause of the formation of carboxylic acids that are hardly
oxidized with Pt(dOH) (Brillas et al., 2000, 2004, 2005; Montilla
et al., 2002; Kraft et al., 2003; Marselli et al., 2003; Canizares
et al., 2005; Panizza and Cerisola, 2005; Boulbaba et al., 2006;
Flox et al., 2006; Sires et al., 2007).
More potent indirect electro-oxidation methods with hy-
drogen peroxide electrogeneration are also being developed
for wastewater remediation. In these techniques, H2O2 is
continuously supplied to the contaminated solution from the
two-electron reduction of O2 usually at carbon-felt (Drogui
et al., 2001; Oturan et al., 2001; Gozmen et al., 2003; Hanna
et al., 2005; Irmak et al., 2006; Diagne et al., 2007) and carbon-
polytetrafluoroethylene (PTFE) O2-diffusion (Brillas et al.,
2000, 2004; Flox et al., 2006, 2007; Sires et al., 2006, 2007)
cathodes:
O2ðgÞ þ 2Hþ þ 2e� ! H2O2: (2)
When an undivided electrolytic cell is used, H2O2 is
oxidized to O2 at the anode with formation of hydroperoxyl
radical ðHOd2 Þ as intermediate, a much weaker oxidant than
dOH (Brillas et al., 2000):
H2O2 ! HO2d þHþ þ e�: (3)
Hydrogen peroxide is then accumulated in the medium up
to reaching a steady concentration directly proportional to
the applied current, just when the rates of reactions (2) and (3)
become equal. The direct use of this procedure, so-called
anodic oxidation with electrogenerated H2O2 (AO-H2O2),
involves the oxidation of organics mainly by M(dOH) formed
from reaction (1), although they can also be destroyed by
weaker oxidizing agents like H2O2 and HOd2 .
In acidic medium, the oxidizing power of H2O2 can be
strongly enhanced using the electro-Fenton method (EF),
where a small quantity of Fe2+ is added as catalyst to the
contaminated solution to generate dOH and Fe3+ from
Fenton’s reaction (Sun and Pignatello, 1993):
Fe2þ þH2O2 ! Fe3þ þ dOHþOH�: (4)
An advantage of EF is that reaction (4) is propagated from
Fe2+ regeneration that mainly takes place by reduction of Fe3+
at the cathode (Oturan et al., 2001). Our group has tested the
behavior of this method using an undivided cell with a Pt or
BDD anode and found that pollutants are destroyed by
Pt(dOH) or BDD(dOH) produced from reaction (1) and by dOH
formed in the medium from Fenton’s reaction (4) (Brillas et al.,
2000, 2004; Flox et al., 2006, 2007; Sires et al., 2006, 2007).
Another EAOP is the photoelectro-Fenton (PEF) process in
which the solution treated under EF conditions is simulta-
neously irradiated with UVA light. The action of this irradia-
tion is complex and can be accounted for by: (i) the
production of greater amount of dOH from photoreduction
of Fe(OH)2+, the predominant Fe3+ species in acid medium
(Sun and Pignatello, 1993), by reaction (5) and (ii) the
photolysis of complexes of Fe(III) with generated carboxylic
acids, as shown, for example, in reaction (6) for oxalic acid
(Zuo and Hoigne, 1992):
FeðOHÞ2þ þ hn! Fe2þ þ dOH; (5)
2FeðC2O4Þnð3�2nÞ þ hn! 2Fe2þ þ ð2n� 1Þ C2O4
2� þ 2CO2: (6)
Oxalic acid is produced during the oxidation of most
organics, and the fast photodecarboxylation of Fe(III)–oxalate
complexes ðFeðC2O4Þþ; FeðC2O4Þ
�2 ; FeðC2O4Þ
3�3 Þ favors the de-
contamination process (Brillas et al., 2000; Irmak et al., 2006;
Sires et al., 2006, 2007). Under these conditions, it is also
feasible to use sunlight as an alternative inexpensive source
of UVA light using the solar photoelectro-Fenton (SPEF)
method (Flox et al., 2007).
Thousands of tons of pharmaceutical drugs are consumed
yearly worldwide in human and veterinary medicine and
agricultural products. Because of the inefficient destruction of
their wastewaters in sewage treatment plants (STPs), a fairly
large number of these compounds have been recently
detected in surface, ground and even drinking waters at low
contents of up to micrograms per liter (Daughton and Jones-
Lepp, 2001; Kummerer, 2001; Heberer, 2002a, b; Andreozzi
et al., 2003; Nakada, et al., 2006). The possible interactions of
these pollutants with living beings in the environment are not
well documented, although available data indicate that some
drugs can affect the endocrine system of fishes, can exert
toxic effects on algae and invertebrates and can favor the
development of multi-resistant strains of microorganisms
(Balcioglu and Otker, 2003). This makes necessary the
development of powerful oxidation methods to efficiently
remove drugs and their metabolites from wastewaters for

ARTICLE IN PRESS
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 501
avoiding their potential adverse health effects on human
beings and animals.
In previous work, we have reported the degradation of
acidic solutions of the drug paracetamol (Sires et al., 2005,
2006) and the metabolite clofibric acid (Sires et al., 2007) by
AO, EF and PEF, showing that mineralization is more efficient
using a BDD anode than a Pt one. To gain a better knowledge
of the characteristics of EAOPs to decontaminate wastewaters
containing drugs and their by-products, we have undertaken
a comparative study on the destruction of salicylic acid
(2-hydroxybenzoic acid) by such procedures using both Pt and
BDD anodes, with special attention to the possible application
of SPEF that has not been previously tested for these
compounds. Salicylic acid is used in many pharmaceutical
and cosmetic formulations, being easily produced from
hydrolytic deacetylation of the common drug acetylsalicylic
acid (aspirin), which is the main source of its presence in STP
effluents and natural waters (Heberer, 2002a, b; Andreozzi et
al., 2003; Nakada et al., 2006). Several authors have treated
aqueous solutions of this product by AO. Thus, partial
decontamination with either a Pt or a C fiber anode
(Weichgrebe et al., 2004) and complete mineralization with a
BDD anode (Montilla et al., 2002; Marselli et al., 2003; Boulbaba
et al., 2006) have been found. In addition, partial degradation
of salicylic acid by AO-H2O2 with a DSA anode and a carbon-
felt cathode (Drogui et al., 2001) and generation of polyhy-
droxylated derivatives under the action of electrochemically
generated dOH (Oturan et al., 1992) have also been described.
This paper reports a study on the treatment of acidic
synthetic wastewaters of salicylic acid by AO, AO-H2O2, EF,
PEF and SPEF using a Pt or BDD anode under comparable
conditions. The influence of current density on the degrada-
tion rate, mineralization current efficiency and energy cost
for total mineralization of all methods was explored. The
effect of Fe2+ concentration and pH in the EF and PEF
processes was also examined. Oxidation products were
detected by gas chromatography–mass spectrometry
(GC–MS). The salicylic acid decay and the evolution of
intermediates were followed by chromatographic techniques.
2. Materials and methods
2.1. Chemicals
Salicylic acid was of analytical grade (499% purity), supplied
by Fluka, and used in the electrolytic experiments without
further purification. 2,3-Dihydroxybenzoic, 2,5-dihydroxyben-
zoic, 2,6-dihydroxybenzoic, a-ketoglutaric, glycolic, glyoxylic,
maleic, malic, fumaric, tartronic and oxalic acids were either
reagent or analytical grade, purchased from Sigma, Aldrich,
Merck, Panreac and Avocado. Analytical grade sulfuric acid
from Merck was used to adjust the initial solution pH.
Anhydrous sodium sulfate and heptahydrated ferrous sulfate
were of analytical grade from Fluka. All solutions were
prepared with pure water obtained from a Millipore Milli-Q
system, with resistivity 418 MO cm at 25 1C. Organic solvents
and other chemicals used were either HPLC or analytical
grade from Panreac and Aldrich.
2.2. Electrolytic systems
All electrolytic treatments were conducted in an open,
undivided and thermostated conic glass cell containing
100 mL of solution vigorously stirred with a magnetic bar.
Experiments were made at constant current density (j)
supplied with an Amel 2049 potentiostat-galvanostat. The
applied cell voltage was directly measured with a Demestres
605 BR digital multimeter. AO degradations (without H2O2 in
solution) were performed using either a 3 cm2 Pt sheet of
99.99% purity from SEMPSA (AO-Pt method) or a 3 cm2 BDD
thin film deposited on a conductive Si sheet from CSEM (AO-
BDD method) as anode and a 3 cm2 graphite bar from Sofacel
as cathode. A cathode of 3 cm2 carbon-PTFE cloth from E-TEK,
fed with pure O2 at 12 mL min�1, was used for continuous
H2O2 electrogeneration from reaction (2). The preparation and
characteristics of this O2-diffusion cathode are described
elsewhere (Brillas et al., 2000). AO-H2O2, EF, PEF and SPEF
treatments were then carried out using this cathode and
either the above Pt (AO-H2O2-Pt, EF-Pt, PEF-Pt and SPEF-Pt
methods) or BDD (AO-H2O2-BDD, EF-BDD, PEF-BDD and
SPEF-BDD methods) anode. PEF trials became operative
when the solution was irradiated with UVA light supplied
by a Philips 6 W fluorescent black light blue tube emitting
between 300 and 420 nm, with lmax ¼ 360 nm. The tube
was placed at the top of the open cell, at 7 cm above the
solution, giving a photoionization energy input to the
solution of 140mW cm�2, as detected with an NRC 820 laser
power meter working at 514 nm. In the SPEF treatments the
cell was directly exposed to solar irradiation, with a mirror at
its bottom to better collect the sun rays. These trials were
made in sunny and clear days during July 2006 in our
laboratory of Barcelona (longitude: 411210 N, latitude: 21100
E). The average solar irradiation intensity varied between 850
and 960 W m�2, with direct UV irradiation intensity between
20 and 23 W m�2, as measured by the weather station of our
center.
Solutions with 164 mg L�1 salicylic acid (corresponding to
100 mg L�1 total organic carbon (TOC)) and 0.05 M Na2SO4 as
background electrolyte of pH 3.0 were comparatively de-
graded by all methods at 33, 100 and 150 mA cm�2. The pH
value of 3.0 was chosen since it is close to the optimum pH of
2.8 for Fenton’s reaction (4) (Sun and Pignatello, 1993). The
effect of pH in the range 2.0–6.0 and Fe2+ concentration
between 0.2 and 2.0 mM in the EF and PEF processes was also
studied. All trials were made at 35 1C, which is the maximum
temperature to operate with the open cell without significant
water evaporation from solution (Sires et al., 2006).
2.3. Apparatus and analytical procedures
The solution pH was measured with a Crison 2000 pH-meter.
All samples withdrawn from treated solutions were filtered
with 0.45mm PTFE filters from Whatman before analysis.
The mineralization of salicylic acid solutions was monitored
from the decay of their TOC, determined on a Shimadzu 5050
TOC analyzer. Reproducible TOC values were obtained in all
cases, with an accuracy of 72%. These data allowed
calculating the mineralization current efficiency (MCE, in %)
for each treated solution at a given electrolysis time t (h) from

ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1502
the equation:
MCE ¼nFVsDðTOCÞexp
4:32� 107 mIt� 100, (7)
where n is the number of electrons consumed in the
mineralization process, F is the Faraday constant
( ¼ 96,487 C mol�1), Vs is the solution volume (L), D(TOC)exp is
the experimental TOC decay (mg L�1), 4.32�107 is a conver-
sion factor ( ¼ 3600 s h�1�12,000 mg of C mol�1), m is the
number of carbon atoms in a salicylic acid molecule ( ¼ 7) and
I is the applied current (A). The n value was taken as 28,
considering that salicylic acid is completely mineralized to
CO2 as follows:
C7H6O3 þ 11H2O! 7CO2 þ 28Hþ þ 28e�: (8)
The energy cost for total mineralization (ETM, in kWh m�3)
of a given solution at time tTM (h) was determined from the
expression
ETM ¼VItTM
Vs, (9)
where V is the average cell voltage (V).
To identify the intermediates formed during salicylic acid
degradation, several solutions with 164 mg L�1 of this com-
pound at pH 3.0 were electrolyzed by AO-Pt at 33 and
150 mA cm�2 for 3 h. Direct extraction of resulting organics
with CH2Cl2 only allowed identifying the residual starting
compound by GC–MS. However, stable aromatics and some
carboxylic acids were detected by this technique after their
silylation. To do this, about 15 mL of each degraded solution
was lyophilized and the remaining solid was treated with
100mL of N,O-bis-(trimethylsilyl)acetamide under stirring and
heating at 60–80 1C for 10 min. The resulting trimethylsilyl
derivatives were diluted with 1 mL of ethyl acetate and further
separated and detected by GC–MS using a Hewlett-Packard
5890 Series II gas chromatograph fitted with a HP-5 0.25mm,
30 m�0.25 mm (i.d.), column, and a Hewlett-Packard 5989 A
mass spectrophotometer operating in EI mode at 70 eV. The
temperature ramp for this column was 50 1C for 3 min,
10 1C min�1 up to 300 1C and hold time 5 min, and the
temperature of the inlet, transfer line and detector was 250,
250 and 290 1C, respectively.
The evolution of salicylic acid and its aromatic products
was followed by reversed-phase HPLC chromatography using
a Waters 600 high-performance liquid chromatograph fitted
with a Spherisorb ODS2 5mm, 150 mm�4.6 mm (i. d.), column
at room temperature, and coupled with a Waters 996
photodiode array detector, which was selected for each
compound at the maximum wavelength of its UV absorption
band. Generated carboxylic acids were detected and quanti-
fied by ion-exclusion chromatography using the above HPLC
chromatograph fitted with a Bio-rad Aminex HPX 87H,
300 mm�7.8 mm (i.d.), column at 35 1C and selecting the
photodiode detector at l ¼ 210 nm. For these analyses, a
50:45:5 (v/v/v) methanol/phosphate buffer (pH ¼ 2.5)/penta-
nol mixture at 0.5 mL min�1 for reversed-phase HPLC chro-
matography and 4 mM H2SO4 at 0.6 mL min�1 for ion-
exclusion HPLC chromatography were used as mobile phases.
3. Results and discussion
3.1. Comparative degradation by EAOPs
A first series of trials was carried out by electrolyzing
164 mg L�1 of salicylic acid at pH 3.0, 33 mA cm�2 and 35 1C
for 6 h to clarify the comparative oxidation power of EAOPs
tested. The EF, PEF and SPEF treatments were operative using
0.5 mM Fe2+ as catalyst. In these experiments, the solution pH
dropped slightly with prolonging electrolysis time due to the
generation of strong acid by-products, and then it was
continuously regulated to pH 3.0 by adding small volumes
of 1 M NaOH. Treated solutions by AO and AO-H2O2 were
always colorless. In contrast, they acquired an intense violet
color after a few minutes of degradation by all EAOPs with
Fe2+, probably due to the formation of complexes of the
remaining salicylic acid with Fe(III) largely generated from
Fenton’s reaction (4) (Ogawa and Tobe, 1966). Violet solutions
were further decolorized, progressively changing from brown
to pale yellow color and becoming colorless again at
60–90 min of electrolysis when all aromatics were already
removed.
The TOC abatement with specific charge (Q, in Ah L�1)
found for the above trials using a Pt and BDD anode is
presented in Fig. 1a and b, respectively. Table 1 summarizes
the corresponding percentage of TOC removal determined
after 60 and 180 min of electrolysis. Comparison of these
results allows concluding that AO, AO-H2O2 and EF treat-
ments yield lower decontamination with Pt than with BDD,
but the catalytic action of UVA and solar light is so effective
that the PEF and SPEF methods show a quite similar
degradation rate for both anodes. The oxidation power of
EAOPs for a given anode increases in the order: AOpAO-
H2O2oEFoPEFoSPEF.
As can be seen in Fig. 1a, the AO-Pt method has the lowest
oxidation power because it only leads to 13% mineralization
at 6 h, indicating that reaction (1) produces a small quantity of
Pt(dOH) with ability to oxidize the organic pollutants to CO2.
The use of AO-H2O2-Pt yields a quicker degradation from
2 Ah L�1 (120 min), yielding 29% of final TOC decay, as
expected if some intermediates can be slowly mineralized
with H2O2 electrogenerated from reaction (2) and/or HOd2
formed from reaction (3). In contrast, Fig. 1b shows that the
analogous AO-BDD and AO-H2O2-BDD methods give a much
faster and similar TOC removal, attaining 85% of the final
decontamination in both cases. These results evidence that
contaminants are mainly destroyed by BDD(dOH), which is
produced in greater amount than Pt(dOH) by reaction (1), with
small participation of weak oxidants like H2O2 and HOd2 .
A different behavior can also be observed in Fig. 1a and b for
the EF process depending on the anode employed. In EF-Pt,
TOC is rapidly reduced by 57% for 180 min (see Table 1), that
is, up to 3 Ah L�1, but at longer time it is slowly removed to
67% of the decontamination at 6 h. The faster degradation
found at the early stages of EF-Pt in comparison to AO-H2O2-
Pt (see Fig. 1a) can then be explained by the quicker reaction
of pollutants with additional dOH produced in the medium
from Fenton’s reaction (4), whereas the strong inhibition of
the EF-Pt process at long times can be ascribed to the
formation of stable complexes of Fe(III) with final short-chain

ARTICLE IN PRESS
TO
C /
mg
L-1
0
20
40
60
80
100
120
0
20
40
60
80
100
0 1 2 3 4 5 6 7
Q / Ah L-1
a
b
Fig. 1 – TOC decay with specific charge for 100 mL of
164 mg L�1 salicylic acid solutions in 0.05 M Na2SO4 of pH
3.0 at 33 mA cm�2 and at 35 1C treated with: (a) Pt and (b) BDD
anode, both of 3 cm2 area. Method: (J,K) anodic oxidation
using a 3 cm2 graphite cathode (AO); (&,’) anodic oxidation
with electrogenerated H2O2 (AO-H2O2) using a 3 cm2 O2-
diffusion cathode; (W,m) electro-Fenton (EF) with 0.5 mM
Fe2+ in solution as catalyst; (X,.) photoelectro-Fenton (PEF)
with 0.5 mM Fe2+ and 6 W UVA light of kmax ¼ 360 nm as
catalysts; and (B,E) solar photoelectro-Fenton (SPEF) with
0.5 mM Fe2+ under direct solar irradiation.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 503
carboxylic acids that cannot be destroyed by dOH and Pt(dOH)
(Brillas et al., 2000, 2004; Sires et al., 2006). Results of Fig. 1b
also confirm the greater oxidation power of dOH than
BDD(dOH), since TOC decays much more rapidly in EF-BDD
than in AO-H2O2-BDD. However, the degradation rate of the
EF-BDD treatment gradually decreases up to reach 91% of
TOC abatement at 6 h, suggesting that some products
including complexes of Fe(III) with final carboxylic acids are
slowly oxidized with BDD(dOH) because they cannot be
removed by dOH. The parallel quicker photodecomposition
of these complexes to CO2 under an UVA irradiation (Zuo and
Hoigne, 1992) can be accounted for by the total mineralization
(498% TOC decay) achieved after 3 Ah L�1 (180 min) of PEF-Pt
and PEF-BDD, as well as after 2 Ah L�1 (120 min) of SPEF-Pt
and SPEF-BDD (see Fig. 1a and b). These findings allow
concluding that the oxidation power of both methods is
independent of the anode employed, the SPEF procedure
always being more potent than the PEF one. Solar light can
then be used as source of UVA irradiation for an effective and
complete mineralization of acidic wastewaters containing
salicylic acid.
3.2. Effect of current density
The influence of current density on the oxidation power
of all EAOPs was further examined by treating the above
salicylic acid solutions of pH 3.0 at 33, 100 and 150 mA cm�2.
Selected results listed in Table 1 show a gradual rise in TOC
removal with increasing j from 33 to 150 mA cm�2 in all
cases. This trend evidences an enhancement of the oxi-
dation power of all methods under these conditions, which
can be related to a quicker destruction of organic pollutants
due to the increase in rate of reaction (1) to form more
amount of Pt(dOH) or BDD(dOH), as well as the greater H2O2
electrogeneration at the O2-diffusion cathode via reac-
tion (2) producing more quantity of dOH from Fenton’s
reaction (4). The fact that final carboxylic acids are more
rapidly formed under the action of these oxidants
suggests the concomitant acceleration of the photodecompo-
sition of their Fe(III) complexes in PEF-Pt, PEF-BDD, SPEF-Pt
and SPEF-BDD, in agreement with the higher oxidation
power found for these procedures when j rises (see Table 1).
Overall mineralization is then achieved for all EAOPs
with BDD and for PEF-Pt and SPEF-Pt at decreasing times
with increasing current density, as can be deduced from
data given in Table 2. Note that a similar tTM-value is found
either for the PEF or for the SPEF treatment with both anodes
at each j, thus confirming the quite effective action of
UVA irradiation to photolyze all complexes between Fe(III)
and final carboxylic acids. At 150 mA cm�2, for example,
the degradation of 164 mg L�1 salicylic acid solutions in
SPEF-Pt and SPEF-BDD is so rapid by the combined action
of oxidants and UVA irradiation from solar light that they
can be completely mineralized in a time as short as 60 min
(see Table 2).
Note that all methods need the consumption of more
specific charge to achieve a given TOC decay when j rises, as
can be observed in Fig. 2a for the SPEF-BDD treatment.
In this case, total mineralization is achieved by consum-
ing greater Q-values of 2.0, 3.6 and 4.5 Ah L�1 at increasing
current densities of 33, 100 and 150 mA cm�2. This behavior
seems opposite to the faster degradation found for all
processes as more j is applied. This apparent contradiction
can be explained if higher current density causes the
production of larger amounts of Pt(dOH), BDD(dOH) and/ordOH favoring the removal of organics with time, but consum-
ing greater specific charge because these oxidants are
generated to a less relative extent due to the faster accelera-
tion of their non-oxidizing reactions. So, in the AO and AO-
H2O2 processes, Pt(dOH) or BDD(dOH) is expected to be more
rapidly oxidized to O2 from reaction (10) and recombined into
H2O2 from reaction (11) with increasing j, whereas when BDD
is used less relative production of BDD(dOH) takes place
due to the enhancement of the anodic generation of weaker
oxidants such as S2O2�8 and O3 via reactions (12) and (13),
respectively (Panizza and Cerisola, 2005). In EF, PEF and
SPEF, it is expected that a larger proportion of dOH formed
from Fenton’s reaction (4) is wasted by Fe2+ from reaction (14)
(Sun and Pignatello, 1993), along with the relative decay of

ARTICLE IN PRESS
Table 1 – Percentage of TOC removal and mineralization current efficiency determined from Eq. (7) for 100 mL of 164 mg L�1
salicylic acid solutions in 0.05 M Na2SO4 of pH 3.0 at 35 1C treated by different electrochemical advanced oxidationprocesses under selected conditions
Methoda Current density (mA cm�2) After 60 min of treatment After 180 min of treatment
% TOC removal MCE % TOC removal MCE
AO-Pt 33 5.3 4.7 8.5 2.5
100 6.5 1.9 12 1.2
150 7.6 1.5 14 0.9
AO-H2O2-Pt 33 4.5 4.0 14 4.2
100 6.0 1.8 17 1.7
150 7.4 1.5 23 1.5
AO-BDD 33 22 20 64 19
100 24 7.1 69 6.8
150 27 5.4 81 5.4
AO-H2O2-BDD 33 22 20 61 18
100 25 7.4 73 7.2
150 31 6.1 84 5.6
EF-Ptb 33 30 27 57 17
100 47 14 62 6.1
150 54 11 64 4.2
EF-BDDb 33 41 37 73 22
100 53 16 85 8.4
150 67 13 90 6.0
PEF-Ptb 33 44 39 97 29
100 75 22 –c –c
150 77 15 –c –c
PEF-BDDb 33 54 48 96 28
100 67 20 –c –c
150 73 14 –c –c
SPEF-Ptb 33 85 76 –c –c
100 95 28 –c –c
150 97 19 –c –c
SPEF-BDDb 33 82 73 –c –c
100 96 28 –c –c
150 98 19 –c –c
a AO-Pt: anodic oxidation with a Pt anode and a graphite cathode; AO-H2O2-Pt: anodic oxidation with electrogenerated H2O2 using a Pt anode;
AO-BDD: anodic oxidation with a BDD anode and a graphite cathode; AO-H2O2-BDD: anodic oxidation with electrogenerated H2O2 using a BDD
anode; EF-Pt: electro-Fenton with Pt; EF-BDD: electro-Fenton with BDD; PEF-Pt: photoelectro-Fenton with Pt; PEF-BDD: photoelectro-Fenton
with BDD; SPEF-Pt: solar photoelectro-Fenton with Pt; SPEF-BDD: solar photoelectro-Fenton with BDD.b The initial solution contained 0.5 mM Fe2+ as catalyst.c Overall mineralization (498% TOC decay) attained at smaller time.
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1504
Pt(dOH) or BDD(dOH):
2dOH! O2ðgÞ þ 2Hþ þ 2e�; (10)
2dOH! H2O2; (11)
2SO42� ! S2O8
2� þ 2e�; (12)
3H2O! O3ðgÞ þ 6Hþ þ 6e�; (13)
Fe2þ þ dOH! Fe3þ þOH�: (14)
The influence of current density on specific charge affects
notably the mineralization current efficiency and energy cost
of each degradation process, as will be discussed below.
3.3. Effect of Fe2+ content and pH
The degradation rate of the EF, PEF and SPEF treatments of
salicylic acid is expected to depend on the initial concentra-
tion of catalyst Fe2+ and solution pH, because they limit, at
least, the quantity of oxidant dOH produced in the medium

ARTICLE IN PRESS
Table 2 – Energy cost for total mineralization calculatedfrom Eq. (9) for 164 mg L�1 salicylic acid solutionsdegraded by electrochemical advanced oxidation pro-cesses
Method Currentdensity
(mA cm�2)
Averagecell
voltage(V)
tTMa
(min)ETM
(kWh m�3)
AO-BDD 100 14.2 360 256
150 18.1 300 407
AO-
H2O2-
BDD
100 17.3 330 285
150 20.9 270 423
EF-BDDb 100 17.1 360 308
150 21.0 300 472
PEF-Ptb 33 5.3 180 34c
100 13.2 150 114c
150 17.5 120 170c
PEF-
BDDb
33 10.2 180 49c
100 17.0 150 142c
150 20.8 130 219c
SPEF-Ptb 33 5.0 120 10
100 12.8 85 54
150 17.1 60 77
SPEF-
BDDb
33 9.9 120 20
100 16.8 75 61
150 20.7 60 93
a Time for total mineralization.b The initial solution contained 0.5 mM Fe2+ as catalyst.c Value including the energy consumption of the 6 W UVA lamp
used to irradiate the solution.
0
20
40
60
80
100
120
0
20
40
60
80
100
TO
C /
mg
L-1
0
20
40
60
80
100
0 1 2 3 4 5 6 7
Q / Ah L-1
a
b
c
Fig. 2 – Effect of experimental parameters on TOC removal of
100 mL of 164 mg L�1 salicylic acid solutions in 0.05 M
Na2SO4 at 35 1C. In plot (a), SPEF-BDD process with 0.5 mM
Fe2+ at pH 3.0 applying: (K) 33 mA cm�2; (’) 100 mA cm�2;
and (m) 150 mA cm�2. In plot (b), EF-BDD degradation with a
Fe2+ concentration of: (K) 0.2 mM; (’) 0.5 mM; (m) 1.0 mM;
and (.) 2.0 mM, at pH 3.0 and at 33 mA cm�2. In plot (c), EF-Pt
treatment with 0.5 mM Fe2+ at pH: (K) 2.0; (’) 3.0; (m) 4.0;
and (.) 6.0, and at 33 mA cm�2.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 505
from Fenton’s reaction (4) (Sun and Pignatello, 1993). The
effects of such parameters were studied for the EF and PEF
processes, being more clearly observed in the first method
due to their lower oxidation power. Fig. 2b presents the TOC–Q
plots obtained for the EF-BDD degradations of 164 mg L�1 of
salicylic acid in the presence of 0.2–2.0 mM Fe2+, at pH 3.0 and
33 mA cm�2. A similar TOC decay can be observed within the
Fe2+ range 0.2–1.0 mM, leading to 91–96% mineralization at
6 h. However, the use of 2.0 mM Fe2+ causes a strong inhibition
of the degradation process, mainly from 1.5 Ah L�1 (90 min), so
that only 82% of the initial TOC is removed at the end of this
trial. This tendency evidences the production of similar
amounts of oxidants BDD(dOH) and dOH up to 1.0 mM Fe2+,
whereas at higher contents of this ion both species are
significantly wasted by reaction (14) taking place in both the
vicinity of the anode, where BDD(dOH) formed from reaction
(1) is accumulated, and the solution bulk, where dOH is
generated from Fenton’s reaction (4). A similar influence of
Fe2+ concentration was found for the analogous EF-Pt
treatments of the same solution. According to these results,
a small amount of this catalyst, even as low as 0.2 mM, is
enough to obtain the fastest destruction of all contaminants
by Pt(dOH), BDD(dOH) and/or dOH in all indirect electro-
oxidation methods.
Fig. 2c shows a significant effect of pH on the EF-Pt process
of 164 mg L�1 salicylic acid solutions with 0.5 mM Fe2+ at
33 mA cm�2. These trials were made with continuous regula-
tion of the solution pH to its initial value with 1 M NaOH. As
can be seen, decontamination is very fast at pH 3.0 and 4.0,
where TOC is reduced by 67% and 74%, respectively, at 6 h. At
pH 2.0, however, the degradation rate drops slightly to yield
59% of final TOC removal, but at pH 6.0 it is so strongly

ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1506
inhibited that a poor mineralization of 26% is attained. This
trend confirms that organics are more rapidly destroyed withdOH formed from Fenton’s reaction (4) than with Pt(dOH)
generated from reaction (1), since the rate of the first reaction
depends on pH (Sun and Pignatello, 1993), whereas the second
one is pH-independent (Sires et al., 2006). Similar treatments
by EF-BDD showed similar results but with greater removal of
pollutants, even at pH 6.0, because they can also be oxidized
with BDD(dOH). One can then establish that pH values of
3.0–4.0 are optimal to decontaminate salicylic acid waste-
waters by the indirect electro-oxidation methods.
3.4. Mineralization current efficiency and energy cost
The mineralization current efficiency for salicylic acid
degradation by all EAOPs was calculated from Eq. (7) and
selected values thus obtained are collected in Table 1. Fig. 3
shows the comparative MCE–Q plots determined for the trials
using a BDD anode at 33 mA cm�2 reported in Fig. 1b. As
expected, the efficiency increases in the same sequence as
the oxidation power of the methods, that is: AO-BDDEAO-
H2O2-BDDoEF-BDDoPEF-BDDoSPEF-BDD. For both AO pro-
cesses, a constant MCE value of 20% is reached approximately
up to 3 Ah L�1 (180 min), whereupon it decays slightly to 13%
at 6 h. This behavior suggests a practically constant destruc-
tion rate of all pollutants mainly by reaction with BDD(dOH) in
both cases. In contrast, all indirect electro-oxidation methods
present a maximum efficiency at 1 Ah L�1 (60 min), with
increasing values of 37% for EF-BDD, 48% for PEF-BDD and
73% for SPEF-BDD, which further fall to 14%, 29% and 44% at
the end of the corresponding runs. The increase in MCE at the
early stages of these processes can be ascribed to the initial
formation of large amounts of products easily mineralized
with BDD(dOH) and mainly with dOH, whereas the drop of this
parameter at longer electrolysis time can be associated with
the production of complexes of Fe(III) with final carboxylic
acids that are difficult to oxidize by BDD(dOH) and/or slowly
photolyze by UVA or solar light. The highest photoefficiency
of UVA irradiation supplied by solar light then explains the
0
10
20
30
40
50
60
70
80
0 1 2 3 4 5 6 7
MC
E /
%
Q / Ah L-1
Fig. 3 – Mineralization current efficiency calculated from Eq. (7)
vs. specific charge for the experiments reported in Fig. 1b.
greatest oxidation power and MCE of the SPEF-BDD method.
Similar relative efficiencies were found for the experiments
given in Fig. 1a using a Pt anode, although lower MCE values
were obtained for the AO-Pt, AO-H2O2-Pt and EF-Pt processes
due to the smaller oxidation power of Pt(dOH). On the other
hand, an inspection of Table 1 reveals a gradual decay of MCE
with rising j for all EAOPs, which can be related to the higher
increase in rate of non-oxidizing reactions of hydroxyl radical
involving its oxidation to O2 and reactions (10), (11) and/or
(14), as well as the enhancement of the formation of weaker
oxidants like S2O2�8 by reaction (12) and O3 by reaction (13).
This gives rise to the production of lower relative quantities of
Pt(dOH), BDD(dOH) and/or dOH with the consequent con-
sumption of more specific charge and loss of efficiency. This
effect is very notable even for the two SPEF treatments for
which the MCE value at 60 min decays from 73–76% at
33 mA cm�2 to 19% at 150 mA cm�2, with an increase in
mineralization from 82–85% to ca. 98%.
Table 2 summarizes the energy cost for total mineralization
calculated from Eq. (9) for the above trials. An analysis of
these data reveals an increase of ETM in each method when j
increases due to the higher specific charge consumed,
although this parameter strongly decreases for both PEF and
SPEF treatments since much more TOC is removed by their
higher oxidation power. In addition, indirect electro-oxidation
processes become more economic using Pt than BDD because
of the lower average cell voltage applied. This means that
SPEF-Pt is the most viable EAOP for the treatment of salicylic
acid solutions under the present conditions, since it yields
overall mineralization with the lowest energy consumption of
10 kWh m�3 at 33 mA cm�2 (see Table 2).
3.5. Salicylic acid decay
The kinetics for salicylic acid decay by the different EAOPs
tested was followed by reverse-phase HPLC chromatography,
in which it exhibited a well-defined absorption peak with a
retention time (tr) of 5.4 min. No change in the concentration
of this compound was previously found when 20 mM H2O2,
0.5 mM Fe2+ or 0.5 mM Fe3+ were separately added to a
164 mg L�1 salicylic acid solution in 0.05 M Na2SO4 of pH 3.0
and further exposed either to UVA or to solar light for 60 min
without applying current. This brings us to consider that in all
EAOPs the electroactive species (salicylic acid and/or mainly
its complexes with Fe(III)) is degraded neither by electro-
generated H2O2 nor by incident light, and hence it can
react only with Pt(dOH) or BDD(dOH) formed from reaction
(1) and/or dOH generated in the medium from reactions (4)
and/or (5).
Fig. 4a shows the change of salicylic acid concentration
with time for the AO-Pt, AO-H2O2-Pt, AO-BDD and AO-H2O2-
BDD methods at 33 mA cm�2. This compound is so slowly
removed under these conditions that it still remains bet-
ween 7 and 17 mg L�1 after 6 h of these treatments. A similar
decay rate can be observed for both AO and AO-H2O2
processes at each anode, as expected if salicylic acid
does not react with electrogenerated H2O2, although its
removal is slightly enhanced using BDD instead of Pt.
This behavior evidences that it is destroyed by a similar
amount of Pt(dOH) or BDD(dOH) during the AO procedures.

ARTICLE IN PRESS
[sal
icyl
ic a
cid]
/ m
g L
-1
0
50
100
150
200
0 60 120 180 240 300 360 420
time / min
ln (
C0/C
)0.0
0.5
1.0
1.5
2.0
0 60 120 180 240 300
0
50
100
150
200
0 10 20 30 40
time / min
time / min
ln (
C0/C
)
0.0
1.0
2.0
3.0
0 5 10 15 20 25
a
b
Fig. 4 – Salicylic acid decay with electrolysis time under the
same conditions as given in Fig. 1. In plot (a), (J) AO-Pt; (K)
AO-BDD; (&) AO-H2O2-Pt; and (’) AO-H2O2-BDD. In plot (b),
(W) EF-Pt; (m) EF-BDD; (X) PEF-Pt; and (.) PEF-BDD. The
corresponding kinetic analysis assuming a pseudo-first-
order reaction for salicylic acid is given in the inset panels.
Table 3 – Aromatic intermediates and generated car-boxylic acids detected during the degradation of salicylicacid by electrochemical advanced oxidation methods
Compound Analyticaltechniquea
Retentiontime(min)
Molecularmassb
(g mol�1)
2,3-
Dihydroxybenzoic
acid
GC-MS 17.7 385
HPLC 3.5
2,5-
Dihydroxybenzoic
acid
GC-MS 18.0 385
HPLC 3.8
2,6-
Dihydroxybenzoic
acid
HPLC 3.0
a-Ketoglutaric
acid
GC-MS 16.1 362
Glycolic acid HPLC 12.3
Glyoxylic acid HPLC 9.2
Malic acid GC-MS 14.6 350
Maleic Acid GC-MS 12.1 260
HPLC 8.0
Fumaric acid GC-MS 12.6 260
HPLC 15.5
Tartronic acid GC-MS 13.3 336
HPLC 7.7
Oxalic acid HPLC 6.5
a GC–MS analysis was carried out after treating 164 mg L�1 salicylic
acid solution of pH 3.0 by anodic oxidation at 33 and 150 mA cm�2
for 180 min, followed by silylation.b Value corresponding to the trimethylsilyl derivative.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 507
However, Figs. 1 and 4a show that only the BDD anode is able
to remove salicylic acid and simultaneously mineralize the
treated solution at practically the same rate for 360 min
(Q ¼ 6 Ah L�1), suggesting that the major part of reactive
BDD(dOH) produced by this anode reacts rapidly with its
oxidation products to yield CO2.
As can be seen in Fig. 4b, the comparative EF-Pt, PEF-Pt, EF-
BDD and PEF-BDD treatments with 0.5 mM Fe2+ at 33 mA cm�2
yield a very fast and overall removal of salicylic acid in about
30 min in all cases. The quite similar decay rate observed for
electro-Fenton and photoelectro-Fenton agrees with the fact
that Fe(III)–salicylate complexes are not photolyzed by UVA
irradiation, as stated above, and also evidences the existence
of a slow production of dOH via reaction (5). The large
enhancement of the decay rate of salicylic acid in these
indirect electro-oxidation methods in relation to the AO ones
corroborates that the main oxidant is dOH generated from
Fenton’s reaction (4), at least at short electrolysis times.
Good linear correlations were obtained by fitting the above
concentration decays with a pseudo-first-order kinetics, as
presented in the inset of Fig. 4a and b. This analysis allows
determining an analogous apparent first-order rate constant
(k1) of 9.2�10�5 s�1 (square regression coefficient (R2¼ 0.997)
for AO-Pt and 8.7�10�5 s�1 (R2¼ 0.992) for AO-H2O2-Pt, which
slightly rises to 1.1�10�4 s�1 (R2¼ 0.994) and 1.2�10�4 s�1
(R2¼ 0.993) for the same methods with BDD). In contrast,
much greater and similar k1-values of 2.2�10�3 s�1 (R2¼
0.989), 2.1�10�3 s�1 (R2¼ 0.992), 1.8�10�3 s�1 (R2
¼ 0.990) and
2.0�10�3 s�1 (R2¼ 0.990) are found for EF-Pt, PEF-Pt, EF-BDD
and PEF-BDD, respectively. The increase in k1 for these
procedures is due to the much faster degradation with dOH
than with Pt(dOH) or BDD(dOH), as pointed out above. Since
the absolute second-order rate constant for the reaction
between salicylic acid and dOH in acidic medium is
k2 ¼ 2.7�1010 M�1 s�1 (Amphlett et al., 1968), one can esti-
mate, as the first approach, a constant dOH generation of
about 10�13 M ( ¼ k1/k2) from reaction (4) in the indirect
electro-oxidation methods at 33 mA cm�2.
ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1508
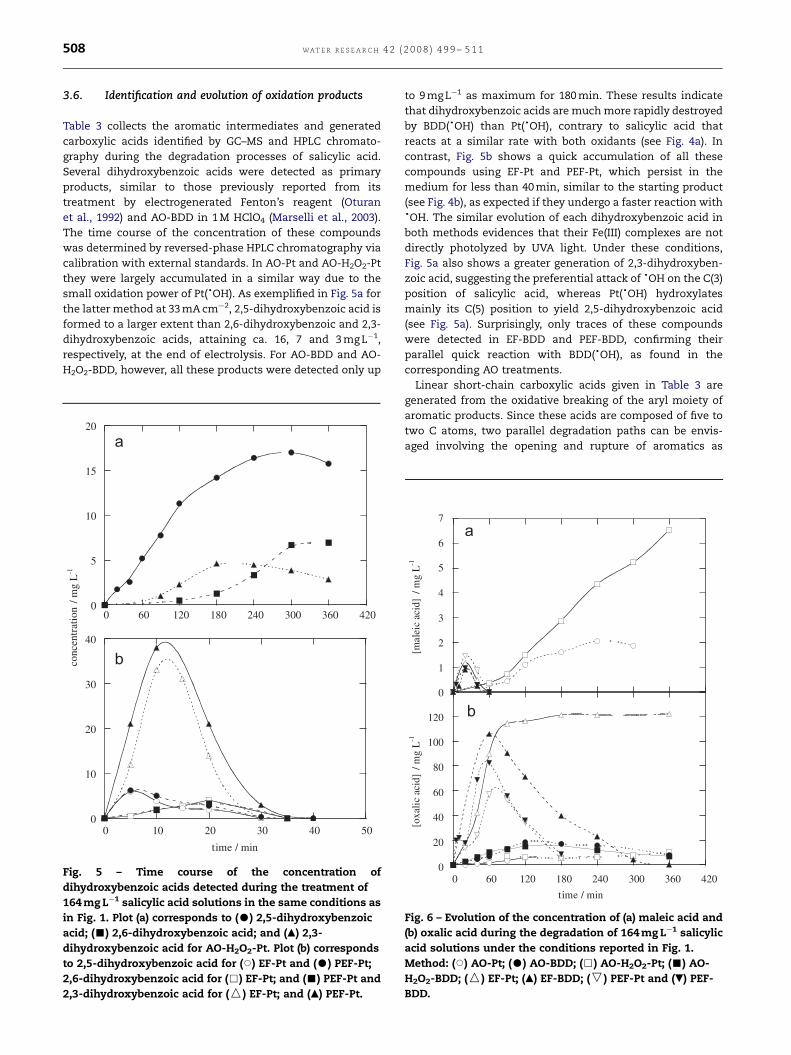
3.6. Identification and evolution of oxidation products
Table 3 collects the aromatic intermediates and generated
carboxylic acids identified by GC–MS and HPLC chromato-
graphy during the degradation processes of salicylic acid.
Several dihydroxybenzoic acids were detected as primary
products, similar to those previously reported from its
treatment by electrogenerated Fenton’s reagent (Oturan
et al., 1992) and AO-BDD in 1 M HClO4 (Marselli et al., 2003).
The time course of the concentration of these compounds
was determined by reversed-phase HPLC chromatography via
calibration with external standards. In AO-Pt and AO-H2O2-Pt
they were largely accumulated in a similar way due to the
small oxidation power of Pt(dOH). As exemplified in Fig. 5a for
the latter method at 33 mA cm�2, 2,5-dihydroxybenzoic acid is
formed to a larger extent than 2,6-dihydroxybenzoic and 2,3-
dihydroxybenzoic acids, attaining ca. 16, 7 and 3 mg L�1,
respectively, at the end of electrolysis. For AO-BDD and AO-
H2O2-BDD, however, all these products were detected only up
conc
entr
atio
n /
mg
L-1
0
10
20
30
40
0 10 20 30 40 50
time / min
0
5
10
15
20
0 60 120 180 240 300 360 420
a
b
Fig. 5 – Time course of the concentration of
dihydroxybenzoic acids detected during the treatment of
164 mg L�1 salicylic acid solutions in the same conditions as
in Fig. 1. Plot (a) corresponds to (K) 2,5-dihydroxybenzoic
acid; (’) 2,6-dihydroxybenzoic acid; and (m) 2,3-
dihydroxybenzoic acid for AO-H2O2-Pt. Plot (b) corresponds
to 2,5-dihydroxybenzoic acid for (J) EF-Pt and (K) PEF-Pt;
2,6-dihydroxybenzoic acid for (&) EF-Pt; and (’) PEF-Pt and
2,3-dihydroxybenzoic acid for (W) EF-Pt; and (m) PEF-Pt.
to 9 mg L�1 as maximum for 180 min. These results indicate
that dihydroxybenzoic acids are much more rapidly destroyed
by BDD(dOH) than Pt(dOH), contrary to salicylic acid that
reacts at a similar rate with both oxidants (see Fig. 4a). In
contrast, Fig. 5b shows a quick accumulation of all these
compounds using EF-Pt and PEF-Pt, which persist in the
medium for less than 40 min, similar to the starting product
(see Fig. 4b), as expected if they undergo a faster reaction withdOH. The similar evolution of each dihydroxybenzoic acid in
both methods evidences that their Fe(III) complexes are not
directly photolyzed by UVA light. Under these conditions,
Fig. 5a also shows a greater generation of 2,3-dihydroxyben-
zoic acid, suggesting the preferential attack of dOH on the C(3)
position of salicylic acid, whereas Pt(dOH) hydroxylates
mainly its C(5) position to yield 2,5-dihydroxybenzoic acid
(see Fig. 5a). Surprisingly, only traces of these compounds
were detected in EF-BDD and PEF-BDD, confirming their
parallel quick reaction with BDD(dOH), as found in the
corresponding AO treatments.
Linear short-chain carboxylic acids given in Table 3 are
generated from the oxidative breaking of the aryl moiety of
aromatic products. Since these acids are composed of five to
two C atoms, two parallel degradation paths can be envis-
aged involving the opening and rupture of aromatics as
0
1
2
3
4
5
6
7
[mal
eic
acid
] / m
g L
-1
0
20
40
60
80
100
120
0 60 120 180 240 300 360 420
[oxa
lic
acid
] / m
g L
-1
time / min
a
b
Fig. 6 – Evolution of the concentration of (a) maleic acid and
(b) oxalic acid during the degradation of 164 mg L�1 salicylic
acid solutions under the conditions reported in Fig. 1.
Method: (J) AO-Pt; (K) AO-BDD; (&) AO-H2O2-Pt; (’) AO-
H2O2-BDD; (W) EF-Pt; (m) EF-BDD; (X) PEF-Pt and (.) PEF-
BDD.

ARTICLE IN PRESS
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 509
dihydroxybenzoic acids containing seven C atoms to give two
carboxylic acids either with (i) five and two C atoms or with
(ii) four and three C atoms. It is then expected that
a-ketoglutaric and glycolic acids are formed in the first
pathway, whereas malic, maleic and fumaric acids along
with tartronic acid are produced in the second one. Glycolic
acid is further transformed into glyoxylic acid (Brillas et al.,
2000). All these acids are independently oxidized to oxalic
acid as the ultimate by-product (Brillas et al., 2005; Flox et al.,
2006, 2007), which is directly mineralized to CO2.
Ion-exclusion chromatograms of treated solutions revealed
the generation of small contents of maleic, fumaric and
tartronic acids, as well as traces of glycolic and glyoxylic
acids, for most processes. The time course of these com-
CO
HO
CO 2
COOH
COOH
O
2,3-dihydroxybenzoic acid
P
salicylic
OH
COOH
OH2,5-dihydroxyb
CO
HO
oxalic a
B
B
.
HOOC
α-ketoglutaric acid
HO
CH2OH
COOH
glycolic acid
COH
COOH
glyoxylic acid
HOOCOH+
OHPt( OH)BDD( OH).
..
+
OPB
.OHPt( OH)
BDD( OH)..
.
OH Pt( OH)
BDD( OH)...
OHPt( OH)
BDD( OH)..
.
Fig. 7 – Proposed reaction sequence for salicylic acid mineraliza
oxidation processes. Pt(dOH) and BDD(dOH) are the oxidant hydro
from water oxidation, whereas dOH is the hydroxyl radical form
pounds at 33 mA cm�2 was similar to that depicted in Fig. 6a
for maleic acid. A continuous accumulation of this acid takes
place in AO-Pt due to the low oxidation power of Pt(dOH),
although it is slightly removed in AO-H2O2-Pt probably by its
slow reaction with H2O2 and/or HOd2 . In contrast, maleic acid
was undetected for both AO treatments with BDD, indicating
its efficient destruction by BDD(dOH). For EF-Pt, PEF-Pt, EF-
BDD and PEF-BDD, this acid reaches a maximum accumula-
tion of 0.9–1.5 mg L�1 at 20 min and disappears at 60 min. This
evidences the most effective reaction of Fe(III)-maleate
complexes with dOH in the medium, without significant
photolysis under UVA irradiation.
A very different behavior can be seen in Fig. 6b for oxalic
acid detected in the trials at 33 mA cm�2. In both AO methods
OH
OH
COOH
OCHOOC
COOH
H
Fe3+
-Fe2+
t( OH)
acid
enzoic acid
OH
OH
2,6-dihydroxybenzoic acid
OH
COOH
HO
cid
Fe(III)-oxalate complexes
DD( OH)
hν
BDD( OH).
.
DD( OH)..
+
maleic acid fumaric acid
OC COOH
malic acidOH
HO
COOH
COOH
tartronic acid
+
Ht( OH)DD( OH).. OH
Pt( OH)BDD( OH).
..
OHPt( OH)BDD( OH).
..
tion in acidic aqueous medium by electrochemical advanced
xyl radicals produced at the Pt and BDD anode, respectively,
ed from Fenton’s reaction.

ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1510
with Pt this acid is progressively accumulated to 10 mg L�1 at
360 min, as expected if it does not react with Pt(dOH). The low
oxidation power of these methods can then be accounted for
by the inability of this oxidant to degrade most carboxylic
acids. In contrast, this acid shows a maximum accumulation
of 18 mg L�1 after 120 min of AO-BDD and AO-H2O2-BDD
treatments, decreasing at longer time because it is miner-
alized with BDD(dOH). The ability of this species to oxidize all
carboxylic acids explains the great oxidation power of the AO
methods using BDD, yielding the total mineralization of
salicylic acid at high current density. As can be seen in Fig.
6b, the use of EF-Pt leads to a steady concentration of oxalic
acid close to 120 mg L�1 from 180 min of electrolysis, as
expected if Fe(III)–oxalate complexes are not destroyed either
by dOH or by Pt(dOH). The remaining oxalic acid in the final
solution treated by this method corresponds to 32 mg L�1 of
TOC, in good agreement with the value of 33 mg L�1 experi-
mentally found (see Fig. 1a). This confirms the overall removal
of all generated products by this method, except Fe(III)–ox-
alate complexes that remain stable in the final solution.
Fig. 6b also shows that these complexes are slowly destroyed
by BDD(dOH) in EF-BDD and much more rapidly photodecom-
posed by UVA light in PEF-Pt and PEF-BDD. In the first case,
oxalic acid disappears in 360 min while the final solution still
has 9 mg L�1 of TOC (see Fig. 1b), suggesting that it contains
some undetected products that are hardly oxidized by
BDD(dOH) and dOH. These products are also efficiently
mineralized under the action of UVA light in PEF-Pt and
PEF-BDD, where solutions are completely decontaminated in
180 min (see Figs. 1a and b) when all Fe(III)–oxalate complexes
are already destroyed (see Fig. 6b). The faster photodecompo-
sition of all these final products by UVA irradiation supplied
by solar light then explains the highest oxidation power of the
SPEF-Pt and SPEF-BDD treatments.
3.7. Mineralization sequence
Taking into account the intermediates detected in the present
work, a plausible general reaction sequence for salicylic acid
mineralization is proposed in Fig. 7. This path only remarks
the formation of final Fe(III)–oxalate complexes in the indirect
electro-oxidation methods for sake of simplicity, although the
other carboxylic products also form large amounts of Fe(III)
complexes that are oxidized in parallel following the same
sequence. The main oxidants (dOH, Pt(dOH) and BDD(dOH))
are only specified, also being possible parallel and slower
reactions of some intermediates with weaker oxidizing
agents such as H2O2, HOd2 ; S2O2�
8 , O3, etc., as stated above.
2,3-Dihydroxybenzoic, 2,5-dihydroxybenzoic and 2,6-dihy-
droxybenzoic acids are initially formed from hydroxylation
on the C(3), C(5) and C(6) positions of salicylic acid by dOH,
Pt(dOH) and BDD(dOH), respectively. Further breaking of such
products by these oxidants leads to two mixtures of
carboxylic acids, one of them corresponding to a-ketoglutaric
and glycolic acids and the other to malic, maleic and fumaric
acids along with tartronic acid. Glycolic acid is subsequently
oxidized to glyoxylic acid. All these acids are then rapidly
transformed into oxalic acid by dOH and BDD(dOH), but are
much more slowly oxidized by Pt(dOH). In the AO treatments,
oxalic acid is converted into CO2 only by BDD(dOH) but not by
dOH and Pt(dOH), whereas in all indirect electro-oxidation
methods it forms complexes with Fe(III) that persist up to the
end of electrolysis. These Fe(III)–oxalate complexes remain
stable under EF-Pt conditions, being mineralized by BDD(dOH)
in EF-BDD and more quickly photodecarboxylated under UVA
irradiation in the PEF and SPEF processes, with loss of Fe2+
according to reaction (6).
4. Conclusions
Solutions containing 164 mg L�1 salicylic acid can be totally
mineralized by all EAOPs using a BDD anode, with increasing
oxidation power in the order AO-BDDEAO-H2O2-BDDoEF-
BDDoPEF-BDDoSPEF-BDD. The same sequence is followed
by the same procedures with a Pt anode, although the AO-Pt,
AO-H2O2-Pt and EF-Pt methods yield much lower decontami-
nation under comparable conditions. This behavior can be
accounted for by the slow removal of most pollutants by
Pt(dOH) in comparison to their quick destruction with
BDD(dOH). dOH oxidizes rapidly all aromatic pollutants, but
it cannot mineralize final Fe(III)–oxalate complexes. The very
fast photolysis of these complexes under UVA irradiation
explains the higher oxidation power found for PEF and SPEF.
Solar light is then an inexpensive and useful source of UVA
irradiation for an efficient treatment of acidic wastewaters
containing salicylic acid. The increase in current density
causes a gradual rise in the degradation rate of all methods
due to the higher production of Pt(dOH), BDD(dOH) and dOH.
Their efficiency, however, undergoes a progressive decay
because these oxidants are generated in less relative propor-
tion by the greater acceleration of non-oxidizing reactions
and the enhancement of parallel anodic reactions giving
weaker oxidants such as S2O2�8 and O3. Indirect electro-
oxidation processes operate in optimum conditions at pH
values 3.0–4.0 and with Fe2+ contents between 0.2 and 1.0 mM.
The lowest energy cost for total mineralization is attained for
the SPEF-Pt treatment, of 10 kWh m�3 at 33 mA cm�2. Salicylic
acid always follows a pseudo-first-order decay with a much
lower rate constant for the AO methods than for the indirect
electro-oxidation ones. 2,3-Dihydroxybenzoic, 2,5-dihydrox-
ybenzoic and 2,6-dihydroxybenzoic acids are detected as
primary aromatic products, which are further oxidized to a
mixture of carboxylic acids. The ultimate by-product is oxalic
acid, which can be mineralized only by BDD(dOH) under AO
conditions. In the indirect electro-oxidation methods, it forms
Fe(III) complexes that remain stable in EF-Pt, being miner-
alized by BDD(dOH) in EF-BDD and more quickly photodecar-
boxylated under UVA irradiation in the PEF and SPEF
processes.
Acknowledgments
The authors are grateful to MEC (Ministerio de Educacion y
Ciencia, Spain) for the financial support and the grant given to
E. Guinea to do this work under project CTQ2004-01954/BQU.

ARTICLE IN PRESS
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 4 9 9 – 5 1 1 511
R E F E R E N C E S
Amphlett, C.B., Adams, G.E., Michael, B.D., 1968. Pulse radiolysisstudies of deaerated aqueous salicylate solutions. In: Gould,R.F. (Ed.), Radiation Chemistry. I. Aqueous Media, Biology,Dosimetry. Proc. Advances in Chemistry Series 81. AmericanChemical Society, Washington, pp. 231–250.
Andreozzi, R., Marotta, R., Nicklas, P., 2003. Pharmaceuticals inSTP effluents and their solar photodegradation in aquaticenvironment. Chemosphere 50 (10), 1319–1330.
Balcioglu, A.I., Otker, M., 2003. Treatment of pharmaceuticalwastewater containing antibiotics by O3 and O3/H2O2 pro-cesses. Chemosphere 50 (1), 85–95.
Boulbaba, L., Nasr, B., Abdellatif, G., 2006. Electrochemicaloxidation of benzoic acid derivatives on boron doped dia-mond: voltammetric study and galvanostatic electrolyses.Chem. Eng. Technol. 29 (8), 944–950.
Brillas, E., Calpe, J.C., Casado, J., 2000. Mineralization of 2,4-D byadvanced electrochemical oxidation processes. Water Res. 34(8), 2253–2262.
Brillas, E., Boye, B., Sires, I., Garrido, J.A., Rodrıguez, R.M., Arias, C.,Cabot, P.L., Comninellis, Ch., 2004. Electrochemical destructionof chlorophenoxy herbicides by anodic oxidation and electro-Fenton using a boron-doped diamond electrode. Electrochim.Acta 49 (25), 4487–4496.
Brillas, E., Sires, I., Arias, C., Cabot, P.L., Centellas, F., Rodrıguez, R.M.,Garrido, J.A., 2005. Mineralization of paracetamol in aqueousmedium by anodic oxidation with a boron-doped diamondelectrode. Chemosphere 58 (4), 399–406.
Canizares, P., Lobato, J., Paz, R., Rodrigo, M.A., Saez, C., 2005.Electrochemical oxidation of phenolic wastes with boron-doped diamond anodes. Water Res. 39 (12), 2687–2703.
Daughton, C.G., Jones-Lepp, T.L., (Eds.), 2001. Pharmaceuticals andpersonal care products in the environment. Scientific andregulatory issues. ACS Symposium Series, Washington.
Diagne, M., Oturan, N., Oturan, M.A., 2007. Removal of methylparathion from water by electrochemically generated Fenton’sreagent. Chemosphere 66 (5), 841–848.
Drogui, P., Elmaleh, S., Rumeau, M., Bernard, C., Rambaud, A.,2001. Oxidising and disinfecting by hydrogen peroxide pro-duced in a two-electrode cell. Water Res. 35 (13), 3235–3241.
Flox, C., Ammar, S., Arias, C., Brillas, E., Vargas-Zavala, A.V.,Abdelhedi, R., 2006. Electro-Fenton and photoelectro-Fentondegradation of indigo carmine in acidic aqueous medium.Appl. Catal. B Environ. 67 (1–2), 93–104.
Flox, C., Cabot, P.L., Centellas, F., Garrido, J.A., Rodrıguez, R.M.,Arias, C., Brillas, E., 2007. Solar photoelectro-Fenton degrada-tion of cresols using a flow reactor with a boron-dopeddiamond anode. Appl. Catal. B Environ. 75 (1–2), 17–28.
Gozmen, B., Oturan, M.A., Oturan, N., Erbatur, O., 2003. Indirectelectrochemical treatment of bisphenol A in water viaelectrochemically generated Fenton’s reagent. Environ. Sci.Technol. 37 (16), 3716–3723.
Hanna, K., Chiron, S., Oturan, M.A., 2005. Coupling enhancedwater solubilization with cyclodextrin to indirect electroche-mical treatment for pentachlorophenol contaminated soilremediation. Water Res. 39 (12), 2763–2773.
Heberer, T., 2002a. Tracking persistent pharmaceutical residuesfrom municipal sewage to drinking water. J. Hydrol. 266 (3–4),175–189.
Heberer, T., 2002b. Occurrence, fate, and removal of pharmaceu-tical residues in the aquatic environment: a review of recentresearch data. Toxicol. Lett. 131 (1–2), 5–17.
Irmak, S., Yavuz, H.I., Erbatur, O., 2006. Degradation of 4-chloro-2-methylphenol in aqueous solution by electro-Fenton andphotoelectro-Fenton processes. Appl. Catal. B Environ. 63(3–4), 243–248.
Kraft, A., Stadelmann, M., Blaschke, M., 2003. Anodic oxidationwith doped diamond electrodes: a new advanced oxidationprocess. J. Hazard. Mater. 103 (3), 247–261.
Kummerer, K. (Ed.), 2001. Pharmaceuticals in the Environment.Sources, Fate and Risks. Springer, Berlin.
Marselli, B., Garcıa-Gomez, J., Michaud, P.A., Rodrigo, M.A.,Comninellis, Ch., 2003. Electrogeneration of hydroxyl radicalson boron-doped diamond electrodes. J. Electrochem. Soc. 150(3), D79–D83.
Montilla, F., Michaud, P.A., Morallon, E., Vazquez, J.L., Comninellis,Ch., 2002. Electrochemical oxidation of benzoic acid at boron-doped diamond electrodes. Electrochim. Acta 47 (21),3509–3513.
Nakada, N., Tanishima, T., Shinohara, H., Kiri, K., Takada, H., 2006.Pharmaceutical chemicals and endocrine disrupters in muni-cipal wastewater in Tokyo and their removal during activatedsludge treatment. Water Res. 40 (17), 3297–3303.
Ogawa, K., Tobe, N., 1966. A spectrophotometric study of thecomplex formation between iron(III) and salicylic acid. Bull.Soc. Chem. Jpn. 39 (2), 227–232.
Oturan, M.A., Pinson, J., Deprez, D., Terlain, B., 1992. Polyhydrox-ylation of salicyclic acid by electrochemically generated OHd
radicals. New J. Chem. 16 (6), 705–710.Oturan, M.A., Oturan, N., Lahitte, C., Trevin, S., 2001. Production of
hydroxyl radicals by electrochemically assisted Fenton’sreagent. Application to the mineralization of an organicmicropollutant, the pentachlorophenol. J. Electroanal. Chem.507 (1–2), 96–102.
Panizza, M., Cerisola, G., 2005. Application of diamond electrodes toelectrochemical processes. Electrochim. Acta 51 (2), 191–199.
Sires, I., Garrido, J.A., Rodrıguez, R.M., Cabot, P.L., Centellas, F.,Arias, C., Brillas, E., 2006. Electrochemical degradation ofparacetamol from water by catalytic action of Fe2+, Cu2+, andUVA light on electrogenerated hydrogen peroxide. J. Electro-chem. Soc. 153 (1), D1–D9.
Sires, I., Centellas, F., Garrido, J.A., Rodrıguez, R.M., Arias, C.,Cabot, P.L., Brillas, E., 2007. Mineralization of clofibric acid byelectrochemical advanced oxidation processes using a boron-doped diamond anode and Fe2+ and UVA light as catalysts.Appl. Catal. B Environ. 72 (3–4), 373–381.
Sun, Y., Pignatello, J.J., 1993. Photochemical reactions involved inthe total mineralization of 2,4-D by iron(3+)/hydrogen per-oxide/UV. Environ. Sci. Technol. 27 (2), 304–310.
Tarr, M. (Ed.), 2003. Chemical Degradation Methods for Wastesand Pollutants. Environmental and Industrial Applications.Marcel-Dekker, New York.
Weichgrebe, D., Danilova, E., Rosenwinkel, K.H., Vedenjapin, A.A.,Baturova, M., 2004. Electrochemical oxidation of drug residuesin water by the example of tetracycline, gentamicin andaspirin. Water Sci. Technol. 49 (4), 201–206.
Zuo, Y., Hoigne, J., 1992. Formation of hydrogen peroxide anddepletion of oxalic acid in atmospheric water by photolysis ofiron(III)–oxalate complexes. Environ. Sci. Technol. 26 (5),1014–1022.
Copyright © 2022 FDOKUMEN




















