
MICA polymorphism: biology and importance in immunity and disease
10
MICA polymorphism: biology and importance in immunity and disease Mun-Kit Choy 1 and Maude E. Phipps 2 1 Division of Cardiovascular Medicine, Department of Medicine, University of Cambridge, Box 110, Level 6, Addenbrooke’s Centre for Clinical Investigation (ACCI), Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0QQ, UK 2 School of Medicine and Health Sciences, Monash University (Sunway Campus), Jalan Lagoon Selatan, 46150 Sunway City, Selangor Darul Ehsan, Malaysia The human major histocompatibility complex class I chain-related gene A (MICA) is one of the genes in the HLA class I region of chromosome 6. Unlike HLA classical class I gene products, MICA does not present any antigen but acts as a ligand for several immune cells including natural killer (NK) cells bearing NKG2D receptors. MICA is the member of the non-classical class I family that dis- plays the greatest degree of polymorphism. MICA alleles can be divided into two large groups with the polymorph- isms found in a3 domains. This division could be explained by a possible polyphyletic origin that is in line with recent findings from evolutionary, population and functional studies of this gene. MICA polymorphisms are associated with a number of diseases related to NK activity, such as viral infection, cancer and allograft rejec- tion or graft-versus-host disease (GVHD). The mechan- isms underlying these associations include NK cell- mediated cytotoxicity and MICA shedding to produce immunosuppressive soluble MICA particles. The MICA- induced humoral response has attracted interest recently because of its possible role in graft rejection in solid organ transplantation. Here, we discuss the genetics and biology of the MICA gene and its products, and their importance in disease. Introduction Graft rejection after allotransplantation had led to the understanding that our bodies have the capability to dis- tinguish ‘‘self’’ and ‘‘non-self’’. From extensive studies in mice, a genomic region responsible for graft rejection was identified as the major histocompatibility complex (MHC). The MHC region contains numerous polymorphic and multicopy genes that play important roles not only in tissue histocompatibility but also other primary functions in the immune system that provide protection against pathogens. The first MHC products were discovered on the surface of leucocytes (white blood cells) and the human MHC was initially referred to as the human leucocyte antigen (HLA) complex. Human MHC genes are cate- gorised into three classes: MHC classes I, II and III. MHC class I (HLA-A, -B and -C) and class II (HLA-DR, - DQ and -DP) genes encode antigen-presenting molecules that are expressed on antigen-presenting cells and stimu- late CD8 + and CD4 + T cells, respectively. MHC class III or central MHC proteins are a diverse group of molecules that perform various immune functions in the body such as complement proteins involved in the antibody response, and inflammatory cytokines [1]. Although the antigen-presenting MHC molecules are known as classical MHC molecules, a number of MHC genes encode non-classical molecules that are structurally related to the classical molecules but do not present anti- gens. Non-classical MHC class II molecules (HLA-DM and -DO) regulate the editing and loading of peptides onto classical MHC class II molecules [2]. Non-classical MHC class I molecules are more diverse. They interact with NK cells and specialised classes of T cells [3]. In 1994, a new set of loci related to MHC class I genes called MHC class I chain-related genes (MIC) or Perth beta block transcript 11 (PERB11) were identified independently by Bahram et al. [4] and Leelayuwat et al. [5]. The nomenclature has now been standardised as MIC [6]. The MIC family was once considered to fulfil the five criteria of MHC non-classical I genes compared with the classical class I genes: i) sequence dissimilarity; ii) random tissue distribution; iii) presence of pseudogenes; iv) reduced polymorphism; and v) difference in functionality [7]. However, years later a member of the MIC family, MICA, was found to be the most polymorphic non-classical class I gene (IMGT/HLA database; http:// www.ebi.ac.uk/imgt/hla/). The polymorphism of MICA has been an enigma. Do the myriad allelic forms play major roles of physiological and pathological significance? Here, we discuss the genetics and biology of the MICA gene and its products, and their importance in disease. MICA gene and protein structures: from classical domains to non-classical flexibility The human MHC region encompasses 3.6 Mb on the short arm of chromosome 6 and contains 224 gene loci [8,9]. The two expressed members of the MIC family, MICA and MICB, and the five pseudogene members, MICC to MICG, are located in the MHC classical class I region [1,8–10]. MICA is at the centromeric end of the classical class I region approximately 46.4 Kb from HLA-B [11]. MICA has six exons separated by five introns with the first intron being the largest [10]. MICA (11.7 Kb) is transcribed into an mRNA of 1382 bp, giving rise to a 383-amino acid polypeptide of 43 kDa [12]. The domain structure of MICA, similar to those of classical class I molecules, consists of three extracellular domains, namely a1 (encoded by exon 2), a2 (encoded by exon 3) and a3 (encoded by exon 4), Opinion Corresponding author: Choy, M.-K. ([email protected]). 1471-4914/$ – see front matter ß 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.molmed.2010.01.002 Available online 12 February 2010 97
-
Upload
umonash-my -
Category
Documents
-
view
0 -
download
0
Transcript of MICA polymorphism: biology and importance in immunity and disease

MICA polymorphism: biology andimportance in immunity and diseaseMun-Kit Choy1 and Maude E. Phipps2
1 Division of Cardiovascular Medicine, Department of Medicine, University of Cambridge, Box 110, Level 6, Addenbrooke’s Centre
for Clinical Investigation (ACCI), Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0QQ, UK2 School of Medicine and Health Sciences, Monash University (Sunway Campus), Jalan Lagoon Selatan, 46150 Sunway City,
Selangor Darul Ehsan, Malaysia
Opinion
The human major histocompatibility complex class Ichain-related gene A (MICA) is one of the genes in theHLA class I region of chromosome 6. Unlike HLA classicalclass I gene products, MICA does not present any antigenbut acts as a ligand for several immune cells includingnatural killer (NK) cells bearing NKG2D receptors. MICA isthe member of the non-classical class I family that dis-plays the greatest degree of polymorphism. MICA allelescan be divided into two large groups with the polymorph-isms found in a3 domains. This division could beexplained by a possible polyphyletic origin that is in linewith recent findings from evolutionary, population andfunctional studies of this gene. MICA polymorphisms areassociated with a number of diseases related to NKactivity, such as viral infection, cancer and allograft rejec-tion or graft-versus-host disease (GVHD). The mechan-isms underlying these associations include NK cell-mediated cytotoxicity and MICA shedding to produceimmunosuppressive soluble MICA particles. The MICA-induced humoral response has attracted interest recentlybecause of its possible role in graft rejection in solid organtransplantation. Here, we discuss the genetics andbiology of the MICA gene and its products, and theirimportance in disease.
IntroductionGraft rejection after allotransplantation had led to theunderstanding that our bodies have the capability to dis-tinguish ‘‘self’’ and ‘‘non-self’’. From extensive studies inmice, a genomic region responsible for graft rejection wasidentified as the major histocompatibility complex (MHC).The MHC region contains numerous polymorphic andmulticopy genes that play important roles not only intissue histocompatibility but also other primary functionsin the immune system that provide protection againstpathogens. The first MHC products were discovered onthe surface of leucocytes (white blood cells) and the humanMHC was initially referred to as the human leucocyteantigen (HLA) complex. Human MHC genes are cate-gorised into three classes: MHC classes I, II and III.MHC class I (HLA-A, -B and -C) and class II (HLA-DR, -DQ and -DP) genes encode antigen-presenting moleculesthat are expressed on antigen-presenting cells and stimu-late CD8+ and CD4+ T cells, respectively. MHC class III orcentralMHC proteins are a diverse group of molecules that
Corresponding author: Choy, M.-K. ([email protected]).
1471-4914/$ – see front matter � 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.molmed.2
perform various immune functions in the body such ascomplement proteins involved in the antibody response,and inflammatory cytokines [1].
Although the antigen-presenting MHC molecules areknown as classical MHC molecules, a number of MHCgenes encode non-classical molecules that are structurallyrelated to the classical molecules but do not present anti-gens. Non-classical MHC class II molecules (HLA-DM and-DO) regulate the editing and loading of peptides ontoclassical MHC class II molecules [2]. Non-classical MHCclass I molecules are more diverse. They interact with NKcells and specialised classes of T cells [3]. In 1994, a new setof loci related to MHC class I genes called MHC class Ichain-related genes (MIC) or Perth beta block transcript 11(PERB11) were identified independently by Bahram et al.[4] and Leelayuwat et al. [5]. The nomenclature has nowbeen standardised as MIC [6]. The MIC family was onceconsidered to fulfil the five criteria of MHC non-classical Igenes compared with the classical class I genes: i) sequencedissimilarity; ii) random tissue distribution; iii) presence ofpseudogenes; iv) reduced polymorphism; and v) differencein functionality [7]. However, years later a member of theMIC family, MICA, was found to be the most polymorphicnon-classical class I gene (IMGT/HLA database; http://www.ebi.ac.uk/imgt/hla/). The polymorphism of MICAhas been an enigma. Do the myriad allelic forms playmajor roles of physiological and pathological significance?Here, we discuss the genetics and biology of theMICA geneand its products, and their importance in disease.
MICA gene and protein structures: from classicaldomains to non-classical flexibilityThe human MHC region encompasses 3.6 Mb on the shortarm of chromosome 6 and contains 224 gene loci [8,9]. Thetwo expressed members of the MIC family, MICA andMICB, and the five pseudogene members, MICC to MICG,are located in the MHC classical class I region [1,8–10].MICA is at the centromeric end of the classical class Iregion approximately 46.4 Kb from HLA-B [11]. MICA hassix exons separated by five introns with the first intronbeing the largest [10]. MICA (11.7 Kb) is transcribed intoan mRNA of 1382 bp, giving rise to a 383-amino acidpolypeptide of 43 kDa [12]. The domain structure ofMICA,similar to those of classical class I molecules, consists ofthree extracellular domains, namely a1 (encoded by exon2), a2 (encoded by exon 3) and a3 (encoded by exon 4),
010.01.002 Available online 12 February 2010 97

Glossary
Allotransplantation: a type of transplantation performed using cells, tissues or
organs sourced from a genetically non-identical member of the same species
as the recipient. The transplants involved are called allografts.
Chaperone: a type of protein that assists the noncovalent folding/unfolding or
the assembly/disassembly of other macromolecular structures, but ceases to
associate when the structures perform their biological functions.
Complement: a series of serum proteins triggered classically by the interaction
of antibodies with specific antigens to mediate immune reactions.
Cytokines: a number of regulatory proteins that are secreted by specific cells of
the immune system and act as intercellular mediators that affect other cells in
the generation of an immune response.
Dendritic cell: a special type of immune cell that processes and presents
antigenic material professionally to activate naıve T cells and stimulate the
growth and differentiation of B cells. At certain developmental stages they
grow branched projections, the dendrites, that give the cell its name.
Glycosylation: an enzymatic process that links saccharides to produce glycans
attached to proteins, lipids or other organic molecules.
Graft-versus-host disease (GVHD): one of the common complications of
allogeneic bone marrow transplantation. Functional immune cells such as T cells
in the transplanted bone marrow graft recognise the recipient as ‘‘foreign’’ and
attack host tissues. GVHD usually occurs in cases where the donor and the
recipient are not immunogenetically identical. GVHD takes two forms: an early
form called acute GVHD that occurs soon after transplantation and a late form
called chronic GVHD. The prevention of GVHD includes T cell depletion in the
graft and the administration of immunosuppressive drugs to the patient.
Haplotype: short for ‘‘haploid genotype’’. A combination of closely linked
genes/alleles at multiple loci that are transmitted together on the same
chromosome from one parent.
Humoral response: an immune response mediated by secreted antibodies (as
opposed to cell-mediated immunity, which involves T lymphocytes) produced
by cells of the B lymphocyte lineage (B cells). The response broadly refers to
antibody production, accessory processes that accompany it, and the effector
functions of the antibodies.
Isomerase: an enzyme that catalyses the conversion of one isomer into
another.
MicroRNAs (miRNAs): single-stranded, short, non-coding RNA molecules (�22
nucleotides) that regulate gene expression. miRNAs are transcribed from
endogenous genes but are not translated into proteins. A primary transcript
(pri-miRNA) is transcribed from an miRNA gene, processed into a short stem–
loop structure called a pre-miRNA, exported into the cytoplasm and finally
processed into a functional mature miRNA. Mature miRNA molecules are
either fully or partially complementary to one or more messenger RNA (mRNA)
molecules, and can downregulate gene expression at the transcriptional or
translational levels accordingly. It is now known that miRNAs regulate the
expression of more than half of all human protein-coding genes, and that the
proper functioning of miRNAs is important to prevent diseases.
Natural killer cell: a type of cytotoxic lymphocyte that kills cancer or virus-
infected cells by releasing small proteins called perforin and granzyme that
cause death of target cells by apoptosis.
Nuclear receptors: a large family of intracellular receptor proteins that binds to
hydrophobic signal molecules or intracellular metabolites and are then
activated to bind to specific DNA sequences, in concert with other proteins,
to regulate the transcription of specific genes.
Orthologues: genes in different species that are similar to each other because
they originated from a common ancestor.
Polyphyly: polyphyletic groups are taxa (or gene families) with members that
have descended through different ancestral lineages.
Toll-like receptors: a class of single membrane-spanning non-catalytic receptor
proteins that recognise structurally conserved molecules derived from
microorganisms, to activate the non-specific and innate immune system. They
share similarities with the Toll receptors in Drosophila, hence the name.
Ubiquitination: a post-translational modification to add small regulatory
proteins called ubiquitin onto a protein to commit the labelled protein to
degradation in the proteasome. The proteasome is a barrel-shaped protein
complex in which proteins are disassembled by proteases. Ubiquitination is
carried out sequentially by three enzymes: E1, E2 and E3. Ubiquitin is first
activated by the E1 ubiquitin-activating enzyme in a process requiring ATP as
an energy source before being transferred to the active site, the cysteine
residue, on E1. The ubiquitin molecule is then passed on to the cysteine active
site of the second enzyme, E2 ubiquitin-conjugating enzyme, before reaching
the final enzyme, E3, the ubiquitin protein ligase. The E3 ligase recognises and
binds the target substrate and labels it with the ubiquitin. The process can be
repeated until a short chain is formed with three or more ubiquitin molecules.
Opinion Trends in Molecular Medicine Vol.16 No.3
a transmembrane segment (encoded by exon 5) and acarboxy-terminal cytoplasmic tail (encoded by exon 6)[10]. The initially hypothesised role of MICA in peptidebinding and antigen presentation based on domain
98
similarity was dismissed because, unlike the classical classI molecules, MICA does not bind b2-microglobulin (b2-m)and is independent of any transporter-associated protein(TAP) [13]. In addition, attempts to identify peptides boundto MICA have been unsuccessful [14].
The discovery of MICA crystal structure, as a result ofthe collaboration between the Strong and Spies groups,further revealed the basis of the functional differencebetween MICA and its classical class I counterparts [15].Although there are many similarities between the struc-tures of MICA and classical class I molecules, MICA isevidently not a peptide/antigen-presenting molecule. Inthe putative antigen-presenting platform formed bydomains a1 and a2, the a2 helix of MICA, one of thegroove-defining helices, is flexible and not visible in thecrystal structure [15].Moreover, the putative platformflipsover by an angle of 113.58 pointing downwards to the cellmembrane and exposing the underside to the intercellularspace [15]. In the putative interaction interfaces with b2-m,some amino acid residues clash with the b2-m structureand occlude the binding between the molecules [15]. How-ever, when MICA is in the complex with its receptor(NKG2D, see later), the flexible a2 helix becomes ordered[16]. The a1-a2 structure flips back 968 in the ligand-re-ceptor complex, indicating that the linker between theplatform and the a3 stalk is also flexible [16].
MICA gene expression and protein functions: rolesas stress markers and NK ligandsBeing considered members of the non-classical class Ifamily, MIC proteins have limited tissue distribution incontrast to the ubiquitous expression of classical class Imolecules [6]. Although MIC products have been found invarious cells/tissues, the current consensus is that MICgenes aremainly expressed in gastrointestinal epithelium,endothelial cells and fibroblasts [6]. However, MIC tran-scripts have been observed in all the major organs exceptthe brain [17]. Because MIC expression is inducible byheat, viral infection, inflammation and DNA damage, themolecules have been thought to be stress markers for thecells [13,14,18–21]. MICA can be upregulated by Toll-likereceptors that are highly expressed in inflammatory andviral myopathies [19]. MIC proteins have been detected inrenal and pancreatic allografts with evidence of both acuteand chronic rejection [22]. MIC proteins might affect thesurvival of transplants through activating the complementsystem because anti-MICA antibodies can kill cells in thepresence of complement [23,24]. MIC products have beenobserved in transformed cell lines, and this has raised thepossibility of their role as tumour-associated antigens [25].These proteins were later detected in lesions and cell linesderived from epithelial tumours and in cutaneous mela-noma lesions [26], and this MICA expression in trans-formed or cancerous cells might be mediated bytranscription factors (such as specificity protein 1, SP1)[27] and nuclear receptors (such as the retinoic acid re-ceptor) [28]. However, no coherent pattern of MIC expres-sion emerges in several human tumours comparedwith therespective normal tissues [17].
Many genes in the human non-classical class I familyincludingMIC and those in themurine non-classical class I

Figure 1. Mechanism of MICA shedding. MICA, a ligand for NK cells bearing NKG2D receptors, forms a complex with a disulphide isomerase/chaperone, ERp5, to induce a
conformational change that enables proteolytic cleavage of MICA by ADAM proteases. The cleaved MICA then interacts with NKG2D, which, in turn, induces the
internalisation and degradation of the receptor and stimulates the population expansion of immunosuppressive T cells.
Opinion Trends in Molecular Medicine Vol.16 No.3
family are ‘‘functional’’ rather than genuine homologuesbecause of convergent evolution [29]. The fact that some ofthe murine non-classical class I molecules were found to beligands for gd T cells led to experiments designed toinvestigate lymphocyte lineages that recognise MICproteins [14,30]. Indeed, MIC proteins are recognised bya class of gd T cells with the Vd1 T cell receptor variableregion [14,30]. Further study by Spies and co-workers haseventually demonstrated that MICA acts as a ligand forNK cells, gd T cells and ab CD8+ T cells, which express acommon activating NK cell receptor NKG2D [31] . NKG2Drecognises the human MICA protein in conjunction with atransmembrane signalling adaptor protein, DNAX-acti-vation protein (DAP) [32]. Since then, investigators havefocused on the physiological/pathological perspective of theMIC–NKG2D interaction, especially in viral and tumoursurveillance mechanisms known to be mediated by NKcells [33].
Table 1. Non-synonymous amino acid substitutions in MICA exter
a1 domain encoded by exon 2
Codon: 6 14 24
MICA*001: Arg Trp Thr
Variant: Pro Gly Ala
a2 domain encoded by exon 3
Codon: 90 91 105
MICA*001: Leu Gln Arg
Variant: Phe Arg Lys
Codon: 142 151 156
MICA*001: Val Met His
Variant: Ile Val Leu/Arg
a3 domain encoded by exon 4
Codon: 206 208 210
MICA*001: Gly Tyr Trp
Variant: Ser Cys Arg
Codon: 256 268 271
MICA*001: Arg Ser Pro
Variant: Ser Gly Ala
Non-conservative amino acid substitutions are underlined. Amino acid substitutions th
Groh et al. [18] demonstrated that the MIC–NKG2Dinteraction enhances T cell cytotoxic responses againstvirus-infected cells when MHC class I antigen processingand expression are impaired after prolonged infection.However, viruses are able to downregulate MIC throughubiquitination [34] and microRNA [35,36]. In cancer, theMIC–NKG2D interaction should activate antitumour NKand T cell responses [31], but NKG2D is reduced in bothCD8+ tumour-infiltrating T cells and peripheral blood Tcells when associated with circulating tumour-derivedsoluble MICA, a phenomenon called ‘‘MICA shedding’’[37,38]. On the surface of tumour cells, the a3 domain ofMICA forms a complex with a disulphide isomerase/cha-peron, endoplasmic reticulum protein 5 (ERp5), to induce aconformational change enabling the proteolytic cleavage ofMICA by ADAM (a disintegrin and metalloproteinase)proteases [38,39]. The cleaved MICA then interacts withNKG2D, which, in turn, induces the internalisation and
nal domains
26 36
Val Cys
Gly Tyr
114 122 124 125 129
Gly Leu Thr Lys Met
Arg Val Ser Glu Val
173 175 176 181
Lys Gly Val Thr
Glu Ser Ile Arg
213 215 221 230 251
Thr Ser Val Trp Gln
Ile Thr Leu Ser Arg/Glu
at define LI and LII lineages of MICA are in bold. Table updated from [6].
99
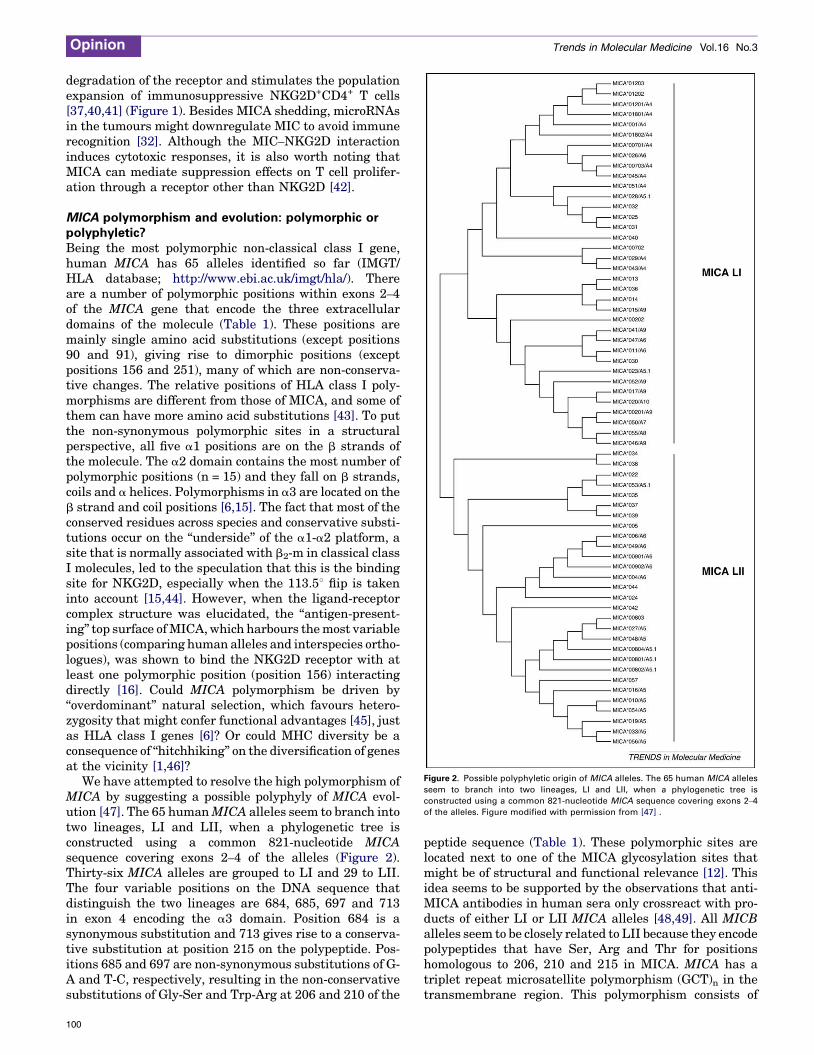
Figure 2. Possible polyphyletic origin of MICA alleles. The 65 human MICA alleles
seem to branch into two lineages, LI and LII, when a phylogenetic tree is
constructed using a common 821-nucleotide MICA sequence covering exons 2–4
of the alleles. Figure modified with permission from [47] .
Opinion Trends in Molecular Medicine Vol.16 No.3
degradation of the receptor and stimulates the populationexpansion of immunosuppressive NKG2D+CD4+ T cells[37,40,41] (Figure 1). Besides MICA shedding, microRNAsin the tumours might downregulate MIC to avoid immunerecognition [32]. Although the MIC–NKG2D interactioninduces cytotoxic responses, it is also worth noting thatMICA can mediate suppression effects on T cell prolifer-ation through a receptor other than NKG2D [42].
MICA polymorphism and evolution: polymorphic orpolyphyletic?Being the most polymorphic non-classical class I gene,human MICA has 65 alleles identified so far (IMGT/HLA database; http://www.ebi.ac.uk/imgt/hla/). Thereare a number of polymorphic positions within exons 2–4of the MICA gene that encode the three extracellulardomains of the molecule (Table 1). These positions aremainly single amino acid substitutions (except positions90 and 91), giving rise to dimorphic positions (exceptpositions 156 and 251), many of which are non-conserva-tive changes. The relative positions of HLA class I poly-morphisms are different from those of MICA, and some ofthem can have more amino acid substitutions [43]. To putthe non-synonymous polymorphic sites in a structuralperspective, all five a1 positions are on the b strands ofthe molecule. The a2 domain contains the most number ofpolymorphic positions (n = 15) and they fall on b strands,coils and a helices. Polymorphisms in a3 are located on theb strand and coil positions [6,15]. The fact that most of theconserved residues across species and conservative substi-tutions occur on the ‘‘underside’’ of the a1-a2 platform, asite that is normally associated with b2-m in classical classI molecules, led to the speculation that this is the bindingsite for NKG2D, especially when the 113.58 flip is takeninto account [15,44]. However, when the ligand-receptorcomplex structure was elucidated, the ‘‘antigen-present-ing’’ top surface ofMICA,which harbours themost variablepositions (comparing human alleles and interspecies ortho-logues), was shown to bind the NKG2D receptor with atleast one polymorphic position (position 156) interactingdirectly [16]. Could MICA polymorphism be driven by‘‘overdominant’’ natural selection, which favours hetero-zygosity that might confer functional advantages [45], justas HLA class I genes [6]? Or could MHC diversity be aconsequence of ‘‘hitchhiking’’ on the diversification of genesat the vicinity [1,46]?
We have attempted to resolve the high polymorphism ofMICA by suggesting a possible polyphyly of MICA evol-ution [47]. The 65 humanMICA alleles seem to branch intotwo lineages, LI and LII, when a phylogenetic tree isconstructed using a common 821-nucleotide MICAsequence covering exons 2–4 of the alleles (Figure 2).Thirty-six MICA alleles are grouped to LI and 29 to LII.The four variable positions on the DNA sequence thatdistinguish the two lineages are 684, 685, 697 and 713in exon 4 encoding the a3 domain. Position 684 is asynonymous substitution and 713 gives rise to a conserva-tive substitution at position 215 on the polypeptide. Pos-itions 685 and 697 are non-synonymous substitutions of G-A and T-C, respectively, resulting in the non-conservativesubstitutions of Gly-Ser and Trp-Arg at 206 and 210 of the
100
peptide sequence (Table 1). These polymorphic sites arelocated next to one of the MICA glycosylation sites thatmight be of structural and functional relevance [12]. Thisidea seems to be supported by the observations that anti-MICA antibodies in human sera only crossreact with pro-ducts of either LI or LII MICA alleles [48,49]. All MICBalleles seem to be closely related to LII because they encodepolypeptides that have Ser, Arg and Thr for positionshomologous to 206, 210 and 215 in MICA. MICA has atriplet repeat microsatellite polymorphism (GCT)n in thetransmembrane region. This polymorphism consists of

Opinion Trends in Molecular Medicine Vol.16 No.3
alleles with four, five, six, seven, eight, nine and tenrepetitions of GCT or five repetitions of GCT with anadditional nucleotide insertion (G), designated as A4,A5, A6, A7, A8, A9, A10 and A5.1, respectively [50]. Var-iants A5.1 and A6 are commonly found in both MICAlineages. However, A4, A7, A8, A9 and A10 are specificto LI and A5 is specific to LII [47] (Figure 2). The variationsof the triplet repeats in both lineages suggest that thispolymorphism might have gone through further diversifi-cation after the bifurcation of LI and LII.
MIC genes of non-human primates do not seem tobelong to any of these lineages except the MIC gene fromthe gorilla that shares a homology to LI [47]. However, bylooking at the positions homologous to 206, 210 and 215 inhuman MICA, the MICs of pygmy chimpanzee (Pan panis-cus) and chimpanzee (Pan troglodytes) are related to LII(data not shown). Phylogenetic studies on MIC genes ofnon-human primates have so far concluded thatmost of thespecies possess either MICA/B or MICD/E and very rarelyboth [51–53]. Taking together all the observations, theevolution of primate MIC genes seems to follow the ‘‘birthand death’’ model proposed for the evolution pattern ofmammalian MHC genes [54]. We propose that there werethree ancestral types of MIC genes: MICA-LI, MICA-LII/MICB and MICD/E (Figure 3). Different primate species,along the evolution process, could have acquired one ormore of the ancestral types that evolved into the existingorthologues/alleles:MICA-LI evolved into humanMICA LIalleles and gorilla MIC; MICA-LII/MICB evolved intohuman MICA LII and MICB alleles and non-humanprimate MICA LII, MICB and MICA/B alleles; andMICD/E evolved into human MICD and MICE pseudo-genes and non-human primate MICD and MICE genes.Within the human evolutionary history of MIC genes, the
Figure 3. Hypothetical model of the evolution of MIC genes. MIC genes are proposed t
Different primate species, along the evolutionary process, could have acquired one or
hypothesised that MICA-LI evolved into human MICA LI alleles and gorilla MIC; MICA-LII/
LII, MICB and MICA/B alleles; and MICD/E evolved into human MICD and MICE pseudo
bold. *Examples based on published work .
human ancestor could have got all three of the MICancestral types but some human offspring might havelost MICA-LI because the HLA-B15/4801-MICA-del-MICB*0107N haplotype with MICA deletion betweenHLA-B and MICB does occur in humans [55,56]. Someindividuals from this group of humans with MICA-LIdeletion could have re-acquired their MICA from MICA-LII/MICB, possibly because of re-emerged selection advan-tages, which evolved into MICA LII alleles (Figure 3).
This hypothesis for the evolution of MIC genes couldexplain the variable MICA allele frequencies in a numberof human populations.MICA LII alleles such asMICA*008and MICA*009 are the most frequent alleles in Japanese,Korean, Thai, Welsh, Brazilian, Euro-American and Mor-occan populations, whereas MICA*002, an LI allele, is themost frequent allele in several Amerindian populations[57]. This might suggest that MICA alleles evolved fromtwo different ancestral types and are, therefore, polyphy-letic.
MICA from a clinical perspective: disease associationsand transplantationBecause of MICA’s proximity to HLA-B, which is associ-ated to a number of autoimmune diseases, and the possiblerole of gd T cells, which recognise MICA through NKG2D,in autoimmune diseases, early efforts to study the role ofMICA in human disease mainly focused on its alleleassociations with various autoimmune diseases [6,50,58].It has also been proposed that the NKG2D–MIC inter-action triggers autoimmune responses evident in rheuma-toid arthritis, coeliac disease and insulin-dependentdiabetes where MIC molecules and NKG2D-bearing T/NK cells are aberrantly induced [59]. However, the proxi-mity ofMICA toHLA-B andHLA-C, two genes known to be
o have originated from three ancestral types: MICA-LI, MICA-LII/MICB and MICD/E.
more of the ancestral types that evolved into the existing orthologues/alleles. It is
MICB evolved into human MICA LII and MICB alleles and non-human primate MICA
genes and non-human primate MICD and MICE genes. Pseudogenes are shown in
101

Opinion Trends in Molecular Medicine Vol.16 No.3
associated with various disorders, necessitated furtherscrutiny about its specific role in autoimmune disease[6]. In addition, mostMICA and disease association studieswere performed in small populations, single-ethnic groupsand focused on polymorphisms of triplet repeats in exon 5,rather than exons 2–4 that encode the extracellulardomains recognisable by NKG2D [6]. For example,Behcet’s disease was among the first to be associated withMICA polymorphism [50]. Several later studies concludedthat the disease is instead associated with HLA-B*51 andthe perceived association with MICA alleles was due tolinkage disequilibrium [60–63]. Ankylosing spondylitis isknown to be primarily associated withHLA-B*27,whereasMICA association is secondary [64]. Therefore, morerefined disease studies on MICA polymorphism, indepen-dent of HLA association, need to be undertaken to confirmits pathological role.
Gonzalez et al. [65] showed aMICA*002 associationwithpsoriatic arthritis in Spanish patients independent of otherHLA class I alleles commonly associated with the disease,but this link was not seen in Korean patients [66]. Anothersetback in the endeavour to elucidate MICA’s role in auto-immune diseases is that autoreactive CD4+CD28�NKG2D+
T cells are not increased in patientswith rheumatoid arthri-tis and Sjogren syndrome [17] as expected [67]. In addition,MIC molecules were only found in fibroblastic/epithelialcells but not in inflammatory infiltrates in patients ofrheumatoid arthritis and Sjogren syndrome [17]. Morerecently, early onset ankylosing spondylitis was observedto be associated with a single nucleotide polymorphism(SNP), MICA-129met, in a cohort of Algerian patients inde-pendent ofHLA-B*27 [68], andKirsten et al. [69] discoveredthat a SNP, MICA-173lys, confers protection againstrheumatoid arthritis in French and German populationsindependent of known HLA-DRB1 risk alleles. Althoughthese findings need to be validated with larger and multi-ethnic populations, they have certainly provided an SNPapproach of studying autoimmunediseaseassociationswithMICA polymorphism (disease associations with MICA’sSNPs are summarised in Table 2).
Because MICA acts as a ligand for NK cells, the associ-ation betweenMICA polymorphism and tumour/virus sur-veillance has attracted attention. Cervical cancer wasamong the first malignant diseases investigated. Althoughno significant association was found betweenMICA allelesand cervical cancer [70–73], a number ofMICA polymorph-isms have been reported as risk factors for other cancertypes. For example, MICA*A9 in the Chinese Han popu-lation [74] and SNPMICA-129val in a Tunisian population[75] are associated with nasopharyngeal carcinoma.Nevertheless, MIC molecules are not specificallytumour-associated antigens because MIC transcripts arenot specifically present in tumours compared with respect-
Table 2. SNPs in MICA alleles associated with human diseases
SNP Disease
MICA-129met Early onset ankylosing spondylitis
MICA-213thr, MICA-251gln Cervical cancer
MICA-129val Chronic GVHD
MICA-129val Nasopharyngeal carcinoma
MICA-173lys Rheumatoid arthritis
102
ive normal tissues [17]. Thus, the associations of someMICA variants, such as MICA-129val, with cancers canbe better explained by their reduced affinity for NKG2D[76]. However, it also seems to be relevant to understandthe relationship betweenMICA variants and cancers in thecontext of ‘MICA shedding’ as described earlier.
The MICA*A5.1 allele carries a nucleotide insertionthat results in a premature stop codon in the transmem-brane region and might encode a soluble form of the MICAmolecule [50], although MICA*A5.1 molecules have beendescribed to be just truncated and ectopic [10]. Therefore.the shedding of MICA*5.1 without a transmembrane seg-ment by tumours is more likely to happen compared withother MICA variants. In Japan, MICA*A5.1 is associatedwith oral squamous cell carcinoma (OSCC) [77,78]. Notonly are OSCC patients more likely to have MICA*A5.1,patients with homozygousMICA*A5.1 had higher levels ofsoluble MICA and a lower survival rate [78]. In Spanishwomen, MICA*A5.1 also seems to confer susceptibility tobreast cancer [79]. The a3 domain ofMICA is important forMICA shedding [38], and polymorphic positions definingLI and LII MICA alleles are located in this domain [47].Therefore, it is likely that LI and LII are associated withcancer. Jumnainsong et al. found that two SNPs in exon 4encoding the a3 domain, MICA-213thr and MICA-251gln,are negatively associated with cervical cancer, although noassociation was established between conventional MICAalleles and the disease [73]. AllMICA LI alleles are MICA-213thr but only 66% of MICA LII alleles are MICA-213ile.Likewise, 94% of MICA alleles LI are MICA-251gln andonly 66% ofMICA LII alleles are MICA-251arg. It could bethatMICALImolecules are less prone to shedding becauseof their a3 structure, but further validation is required.
The surveillance of viral infection is one of the importantfunctions of NK cells. Viruses such as human cytomegalo-virus (HCMV) induce MIC expression that, in turn, aug-ments the virus-specific responses of NKG2D-bearing cells[18]. We now know that the glycoprotein UL142 of HCMVcan downregulate the cell surface expression of MICA,leading to a protection from NK cytotoxicity, but not thetruncated MICA*A5.1 [80–82]. It is possible that the cyto-plasmic tail of MIC is important for virus-mediated down-regulation because KSHV (Kaposi’s sarcoma-associatedherpes virus) encodes E3 ubiquitin ligases, K3 and K5, tointernalise MIC by ubiquitinating MIC’s cytoplasmic taillysine residues; however, this does not happen toMICA*A5.1 [34]. By contrast,MICA*A5.1hasbeen reportedas a risk factor for HCMV reactivation in HIV-1-infectedpatients [83]. No significant association has been reportedwith other viruses, althoughMICA*015might be associatedwith hepatitis B and C virus infections [84]. Cytokines suchas interleukin-15 (IL-15) and type I interferon (IFN) lead toMIC expression and the subsequent activation of NK cells,
Effect Population Reference
Susceptibility Algerian [68]
Protection Northeastern Thai [73]
Susceptibility French [111]
Susceptibility Tunisian [75]
Protection French, German [69]

Box 1. Outstanding questions
� What is the molecular structure of ERp5? It is important to
understand this to investigate compounds/drugs that target the
molecule either to prevent or enhance MICA shedding.
� Why and how are MICA molecules expressed in allografts? By
understanding this we can attempt to deplete MICA expression in
the grafts before transplantation.
� Are the MICA molecules produced from LI and LII alleles
structurally different? Human anti-MICA antibodies show specifi-
city for LI and LII MICA molecules. This might imply that MICA
molecules from the two groups are structurally and functionally
different.
Opinion Trends in Molecular Medicine Vol.16 No.3
but the production of IL-15 and, therefore, MIC expressionare impaired in hepatitis C virus-infected patients [85,86].The MICA polymorphism has also been reported to beassociated with the infection of Mycobacterium avium com-plex [87].
MICA is now widely recognised as an important non-classicalHLAantigen in the clinical transplantation of solidorgans [88]. Possible mechanisms for MIC-mediated organrejection include the following: the recognition of MIC onallografts and NKG2D-mediated cytotoxicity; the develop-ment of anti-MICA antibodies to increase rejection; theupregulation of NKG2D by interleukins and NK cell acti-vation (in case of inflammatory conditions); NK cell-induceddendritic cell maturation and the subsequent activation ofalloreactiveTcells; and theNKG2D-mediatedreductionofTregulatory cells that could contribute to the tolerance intransplantation [89]. However, an increasing amount ofevidence has led the focus to the link between anti-MICAantibodies and graft rejection [90]. Anti-MIC antibodieshave been identified in the sera of renal, pancreatic andcardiac transplant recipients who displayed signs of rejec-tion and associated with decreased graft survival [23,91–
99]. These anti-MIC antibodies seem to react with productsexpressed from a range ofMICA alleles but specifically fromeither LI or LII [48,49]. Therefore, the understanding of twolineages of MICA alleles might be useful to resolve thisclinical problem, especially because this might imply thatMICA molecules from the two groups are structurally andfunctionally different (see outstanding questions in Box 1).
Bone marrow or haematopoietic stem cell transplan-tation (HSCT) currently relies on the close HLA matchingof donors and recipients to prevent GVHD and increaseoverall survival [100–102]. Should donors’ and recipients’MICA alleles also be matched for better post-transplantoutcomes [10]? Instead of matching individual MHC loci,MHC block matching is a DNA-based MHC matchingtechnique that utilises non-HLA DNA polymorphisms inthe MHC as markers of blocks of ancestral haplotypes,such as beta block (the region including HLA-B, HLA-C,MICA and MICAB) and delta block (the region includingHLA-DR and HLA-DQ) [103,104]. MHC beta block match-ing was found to be the marker for MIC genes and corre-lated withMICmatching in addition toHLA-B andHLA-Cmatching [105,106]. The survival of HSCT patients whowere matched for HLA-B, HLA-C, HLA-DR and HLA-DQwas improved with additional matching for beta block,MIC or both [105]. Although these might just be betterhaplotype-matched pairs, there is a chance thatMIC genesplay a role in the survival after HSCT because there were
some HSCT cases with matched HLA but mismatchedMICA that showed an increased incidence of GVHD[107]. Recently, the polymorphism of NK receptors suchas killer cell immunoglobulin-like receptors (KIR) havealso been shown to play a role in HSCT, thereby suggestinga link between NK responses and GVHD [108].
Therapeutic implications and concluding remarksTo date, there has been no clear explanation of how andwhyMICA genes evolved such high allelic diversity. In theeffort to resolve this enigmatic issue, here we haverevisited the question of whether MICA alleles are poly-phyletic rather than highly polymorphic, which we pro-posed several years ago [47]. This theoretical model mightseem unconventional but it fits well with findings frommost evolutionary, population and functional studies onMICA to date. Our model can offer a better explanation asto why the gorilla has the MICA gene and some primatesonly have MICD/E genes. The alternative theory forprimate MIC gene evolution is that a 95 Kb deletionoccurred between MICA and MICB to produce some ofthe primateMICA/B genes evident today [109]. There hasbeen some consensus that the main driving factor ofMICApolymorphism is natural overdominant selection asexerted by infectious agents [6,10,57]. It has also beenproposed that immunodeficiency viruses might have wipedout the chimpanzee’s MIC gene diversity [109]; however,there is yet to be any evidence for this.
MICA polymorphism features significantly in diseaseassociations and donor–recipient matching for transplan-tations. The associations between MICA alleles and auto-immune diseases have been described [6]. However, asdiscussed above, most investigations could not excludethe linkage disequilibrium with other HLA genes nearbyand were performed with small and single-ethnic popu-lations. In addition, most of the cellular mechanisms pro-posed to explain MICA’s role in autoimmune diseases haveeither not been directly proven [59] or reproduced [17].Studies on MICA’s associations with cancers seem morepromising. A number of findings are concordant with thefunctional aspects of MICA shedding. As explained above,truncated MICA*A5.1 that might be soluble or more ‘‘shed-dable’’ is associated with OSCC and breast cancer [77–79].SNPs in exon4, encoding thea3 domain that is important inboth MICA shedding and defining LI/LII lineages, areweakly associated with cervical cancer [73]. The potentialtherapeutic targets forMICAshedding seemtobeERp5andsoluble MICA. Screening strategies need to be designed tolook for drugs or compounds that can inactivate ERp5 andsoluble MICA (see outstanding questions in Box 1). Anti-bodies against MICA could be used as therapeutics becausethese could reduce levels of circulating soluble MICA,antagonise immune suppression and stimulate antitumourcytotoxicity [110]. Antagomirs, cholesterol-linked single-stranded RNAs that can silence endogenous microRNAs,might also be useful if further studies show thatmicroRNAsdownregulate MIC in tumours. The functional aspect of theassociation ofMICA alleles with viral infection has yet to beelucidated. However, approaches that suppress ubiquitina-tion and microRNAs using chemical agents might helpretainMIC expression during viral infection, because these
103

Opinion Trends in Molecular Medicine Vol.16 No.3
are among the possible mechanisms deployed by viruses[34,35].
MHCmatching is important to ensure better post-trans-plant outcomes in allogeneic HSCT. In addition to classicalHLA class I and II matching, matches at MICA and MICBloci have been shown to increase patient survival [105].Therefore, techniques such as beta and delta block match-ing that can profile MIC genes and other HLA genes couldbe performed alongside other HLA matching techniquesbefore transplantation [103,104]. In fact, this is alreadybeing carried out routinely in several tissue typing labora-tories in Malaysia, Australia and Thailand. Similar tocancer, ERp5 and solubleMICA (see outstanding questionsin Box 1) could be considered for investigation as thera-peutic targets to prevent GVHD and allograft rejection intransplantation. For chronic GVHD in HSCT, Boukouaciet al. [111] reported recently thatMICA-129val and solubleMICA are risk factors for chronic GVHD, whereas thepresence of anti-MICA antibodies that can neutralisesoluble MICA confers protection. However, for solid organallograft rejection, drugs or compounds that can activateERp5 or induce soluble MICA should also be investigatedbecause there is a significant correlation between thepresence of soluble MICA and a lower incidence of rejec-tion, possibly caused by immune suppression arising fromthe interaction between soluble MICA and NKG2D, as hasbeen observed in cancer [112]. For solid organ transplan-tation, theMICA-induced humoral response could also be atherapeutic target for graft rejection because the presenceof anti-MICA antibodies is associated with decreased graftsurvival (see outstanding questions in Box 1).
The importance of MICA polymorphism and its func-tional aspects are gradually being resolved. Detailed un-derstanding of MICA, particularly its evolutionarydevelopment, pathobiological roles and mechanisms ofaction, should lead us to devise effective therapeutic strat-egies for diseases mediated by MICA, in particular forcancer, allograft rejection or GVHD.
AcknowledgementsWe are grateful to Alexander Steinle, Chanvit Leelayuwat and RogerFoo for their critical comments and Insiya Jafferji for an insightfuldiscussion. MKC is supported by British Heart Foundation (GrantNumber: FS/07/035).
References1 Horton, R. et al. (2004) Gene map of the extended human MHC. Nat.
Rev. Genet. 5, 889–8992 Brocke, P. et al. (2002) HLA-DM, HLA-DO and tapasin: functional
similarities and differences. Current Opinion in Immunology 14, 22–293 Arnaiz-Villena, A. (2007) 14th International HLA and
Immunogenetics Workshop: report on non-classical class I genes.Tissue Antigens 69, 130–131
4 Bahram, S. et al. (1994) A second lineage of mammalian majorhistocompatibility complex class I genes. Proc. Natl. Acad. Sci. U.S. A. 91, 6259–6263
5 Leelayuwat, C. et al. (1994) A new polymorphic and multicopy MHCgene family related to nonmammalian class I. Immunogenetics 40,339–351
6 Stephens, H.A.F. (2001) MICA and MICB genes: can the enigma oftheir polymorphism be resolved? Trends in Immunology 22, 378–385
7 Klein, J. andO’hUigin, C. (1994) The conundrum of nonclassical majorhistocompatibility complex genes. Proc. Natl. Acad. Sci. U. S. A. 91,6251–6252
104
8 The MHC sequencing consortium (1999) Complete sequence and genemap of a human major histocompatibility complex. The MHCsequencing consortium. Nature 401, 921–923
9 Horton, R. et al. (2004) Gene map of the extended human MHC. Nat.Rev. Genet. 5, 889–899
10 Bahram, S. (2000)MIC genes: from genetics to biology.Adv. Immunol.76, 1–60
11 Shiina, T. et al. (1999) Molecular dynamics of MHC genesis unraveledby sequence analysis of the 1,796,938-bp HLA class I region. Proc.Natl. Acad. Sci. U. S. A. 96, 13282–13287
12 Bahram, S. et al. (1994) A second lineage of mammalian majorhistocompatibility complex class I genes. Proc. Natl. Acad. Sci. U.S. A 91, 6259–6263
13 Groh, V. et al. (1996) Cell stress-regulated human majorhistocompatibility complex class I gene expressed ingastrointestinal epithelium. Proc. Natl. Acad. Sci. U. S. A. 93,12445–12450
14 Groh, V. et al. (1998) Recognition of stress-induced MHCmolecules byintestinal epithelial gammadelta T cells. Science 279, 1737–1740
15 Li, P. et al. (1999) Crystal structure of the MHC class I homolog MIC-A, a gammadelta T cell ligand. Immunity 10, 577–584
16 Li, P. et al. (2001) Complex structure of the activatingimmunoreceptor NKG2D and its MHC class I-like ligand MICA.Nat. Immunol. 2, 443–451
17 Schrambach, S. et al. (2007) In vivo expression pattern of MICA andMICB and its relevance to auto-immunity and cancer. PLoS ONE 2,e518
18 Groh, V. et al. (2001) Costimulation of CD8alphabeta T cells byNKG2D via engagement by MIC induced on virus-infected cells.Nat. Immunol. 2, 255–260
19 Schreiner, B. et al. (2006) Expression of toll-like receptors by humanmuscle cells in vitro and in vivo: TLR3 is highly expressed ininflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. Faseb. J. 20, 118–120
20 Gasser, S. et al. (2005) The DNA damage pathway regulates innateimmune system ligands of the NKG2D receptor. Nature 436, 1186–
119021 Tang, K.F. et al. (2008) Decreased Dicer expression elicits DNA
damage and up-regulation of MICA and MICB. J. Cell Biol. 182,233–239
22 Hankey, K.G. et al. (2002) MIC expression in renal and pancreaticallografts. Transplantation 73, 304–306
23 Mizutani, K. et al. (2005) Serial ten-year follow-up of HLA and MICAantibody production prior to kidney graft failure. Am. J. Transplant.5, 2265–2272
24 Zou, Y. et al. (2002) MICA is a target for complement-dependentcytotoxicity with mouse monoclonal antibodies and humanalloantibodies. Hum. Immunol. 63, 30–39
25 Zwirner, N.W. et al. (1998) MICA, a new polymorphic HLA-relatedantigen, is expressed mainly by keratinocytes, endothelial cells, andmonocytes. Immunogenetics 47, 139–148
26 Seliger, B. et al. (2003) HLA-G and MIC expression in tumors andtheir role in anti-tumor immunity. Trends Immunol. 24, 82–87
27 Andresen, L. et al. (2007) Molecular regulation of MHC class I chain-related protein A expression after HDAC-inhibitor treatment ofJurkat T cells. J. Immunol. 179, 8235–8242
28 Jinushi, M. et al. (2003) Expression and role of MICA and MICB inhuman hepatocellular carcinomas and their regulation by retinoicacid. Int. J. Cancer 104, 354–361
29 Kumanovics, A. et al. (2003) Genomic organization of the mammalianMHC. Annu. Rev. Immunol. 21, 629–657
30 Groh, V. et al. (1999) Broad tumor-associated expression andrecognition by tumor-derived gamma delta T cells of MICA andMICB. Proc. Natl. Acad. Sci. U. S. A. 96, 6879–6884
31 Bauer, S. et al. (1999) Activation of NK cells and T cells by NKG2D, areceptor for stress-inducible MICA. Science 285, 727–729
32 Wu, J. et al. (1999) An activating immunoreceptor complex formed byNKG2D and DAP10. Science 285, 730–732
33 Vivier, E. et al. (2008) Functions of natural killer cells.Nat. Immunol.9, 503–510
34 Thomas, M. et al. (2008) Natural killer cell evasion by an E3 ubiquitinligase from Kaposi’s sarcoma-associated herpesvirus. Biochem. Soc.Trans. 36, 459–463

Opinion Trends in Molecular Medicine Vol.16 No.3
35 Stern-Ginossar, N. et al. (2007) Host immune system gene targetingby a viral miRNA. Science 317, 376–381
36 Stern-Ginossar, N. et al. (2008) Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat.Immunol. 9, 1065–1073
37 Groh, V. et al. (2002) Tumour-derived soluble MIC ligands impairexpression of NKG2D and T-cell activation. Nature 419, 734–738
38 Kaiser, B.K. et al. (2007) Disulphide-isomerase-enabled shedding oftumour-associated NKG2D ligands. Nature 447, 482–486
39 Waldhauer, I. et al. (2008) Tumor-associated MICA is shed by ADAMproteases. Cancer Res. 68, 6368–6376
40 Groh,V. et al. (2006)Fas-ligand-mediatedparacrineT cell regulationbythe receptor NKG2D in tumor immunity. Nat. Immunol. 7, 755–762
41 Doubrovina, E.S. et al. (2003) Evasion fromNK cell immunity byMHCclass I chain-related molecules expressing colon adenocarcinoma. J.Immunol. 171, 6891–6899
42 Kriegeskorte, A.K. et al. (2005) NKG2D-independent suppression of Tcell proliferation byH60 andMICA.Proc. Natl. Acad. Sci. U. S. A. 102,11805–11810
43 Mason, P.M. and Parham, P. (1998) HLA class I region sequences,1998. Tissue Antigens 51, 417–466
44 Strong, R.K. (2000) Class (I) will come to order–not. Nat. Struct. Biol.7, 173–176
45 Penn, D.J. et al. (2002) MHC heterozygosity confers a selectiveadvantage against multiple-strain infections. Proc. Natl. Acad. Sci.U. S. A. 99, 11260–11264
46 Shiina, T. et al. (2006) Rapid evolution of major histocompatibilitycomplex class I genes in primates generates new disease alleles inhumans via hitchhiking diversity. Genetics 173, 1555–1570
47 Choy, M-K. and Phipps, M. (2003) Possible polyphyletic origin ofmajor histocompatibility complex class i chain-related gene A(MICA) alleles. Journal of Molecular Evolution 57, 38–43
48 Duquesnoy, R.J. et al. (2008) Structurally based epitope analysis ofmajor histocompatibility complex class I-related chain A (MICA)antibody specificity patterns. Hum. Immunol. 69, 826–832
49 Zou, Y. et al. (2009) Polymorphisms of MICA recognized by humanalloantibodies. Immunogenetics 61, 91–100
50 Mizuki, N. et al. (1997) Triplet repeat polymorphism in thetransmembrane region of the MICA gene: a strong association ofsix GCT repetitions with Behcet disease. Proc. Natl. Acad. Sci. U.S. A. 94, 1298–1303
51 Doxiadis, G.G. et al. (2007) MIC gene polymorphism and haplotypediversity in rhesus macaques. Tissue Antigens 69, 212–219
52 Cattley, S.K. et al. (1999) Phylogenetic analysis of primate MIC(PERB11) sequences suggests that the representation of the genefamily differs in different primates: comparison of MIC (PERB11) andC4. Eur. J. Immunogenet. 26, 233–238
53 Seo, J.W. et al. (1999) Major histocompatibility complex-linked MICgenes in rhesus macaques and other primates. Immunogenetics 50,358–362
54 Nei, M. and Rooney, A.P. (2005) Concerted and birth-and-deathevolution of multigene families*. Annu. Rev. Genet. 39, 121–152
55 Aida, K. et al. (2002) High frequency ofMIC null haplotype (HLA-B48-MICA-del-MICB*0107N) in the Angaite Amerindian community inParaguay. Immunogenetics 54, 439–441
56 Komatsu-Wakui, M. et al. (1999) MIC-A polymorphism in Japaneseand a MIC-A-MIC-B null haplotype. Immunogenetics 49, 620–628
57 Oliveira, L.A. et al. (2008) High frequencies of alleles MICA*020 andMICA*027 in Amerindians and evidence of positive selection on exon3. Genes Immun. 9, 697–705
58 Goto, K. et al. (1997) MICA gene and ankylosing spondylitis: linkageanalysis via a transmembrane-encoded triplet repeat polymorphism.Tissue Antigens 49, 503–507
59 Caillat-Zucman, S. (2006) How NKG2D ligands triggerautoimmunity? Hum Immunol. 67, 204–207
60 Mizuki, N. et al. (2007) Association of major histocompatibilitycomplex class i chain-related gene A and HLA-B alleles withBehcet’s disease in Turkey. Jpn. J. Ophthalmol. 51, 431–436
61 Nishiyama,M. et al. (2006)Microsatellite polymorphisms of theMICAgene among Japanese patients with Behcet’s disease. Can. J.Ophthalmol. 41, 210–215
62 Salvarani, C. et al. (2001) Association ofMICA alleles andHLA-B51 inItalian patients with Behcet’s disease. J. Rheumatol. 28, 1867–1870
63 Wallace, G.R. et al. (1999) MIC-A allele profiles and HLA class Iassociations in Behcet’s disease. Immunogenetics 49, 613–617
64 Martinez-Borra, J. et al. (2000) HLA-B27 alone rather than B27-related class I haplotypes contributes to ankylosing spondylitissusceptibility. Hum Immunol. 61, 131–139
65 Gonzalez, S. et al. (1999) The MICA-A9 triplet repeat polymorphismin the transmembrane region confers additional susceptibility to thedevelopment of psoriatic arthritis and is independent of theassociation of Cw*0602 in psoriasis. Arthritis Rheum. 42, 1010–1016
66 Choi, H.B. et al. (2000) MICA 5.1 allele is a susceptibility marker forpsoriasis in the Korean population. Tissue Antigens 56, 548–550
67 Groh, V. et al. (2003) Stimulation of T cell autoreactivity by anomalousexpression of NKG2D and its MIC ligands in rheumatoid arthritis.Proc. Natl. Acad. Sci. U. S. A. 100, 9452–9457
68 Amroun, H. et al. (2005) Early-onset ankylosing spondylitis isassociated with a functional MICA polymorphism. Hum. Immunol.66, 1057–1061
69 Kirsten, H. et al. (2009) Association of MICA with rheumatoidarthritis independent of known HLA-DRB1 risk alleles in a family-based and a case control study. Arthritis Res. Ther. 11, R60
70 Ghaderi, M. et al. (1999) MICA gene polymorphism and the risk todevelop cervical intraepithelial neoplasia.Hum. Immunol. 60, 970–973
71 Chen, J.R. et al. (2005) MHC class I chain-related gene A (MICA)polymorphism and the different histological types of cervical cancer.Neoplasma 52, 369–373
72 Ghaderi, M. et al. (2001) Tumor necrosis factor A and MHC class Ichain related gene A (MIC-A) polymorphisms in Swedish patientswith cervical cancer. Hum. Immunol. 62, 1153–1158
73 Jumnainsong, A. et al. (2007) Association of polymorphic extracellulardomains of MICA with cervical cancer in northeastern Thaipopulation. Tissue Antigens 69, 326–333
74 Tian,W. et al. (2006) Gender-specific associations betweenMICA-STRand nasopharyngeal carcinoma in a southern Chinese Hanpopulation. Immunogenetics 58, 113–121
75 Douik, H. et al. (2009) Association of MICA-129 polymorphism withnasopharyngeal cancer risk in a Tunisian population.Hum. Immunol.70, 45–48
76 Steinle, A. et al. (2001) Interactions of humanNKG2Dwith its ligandsMICA, MICB, and homologs of the mouse RAE-1 protein family.Immunogenetics 53, 279–287
77 Tamaki, S. et al. (2007) An association between the MICA-A5.1 alleleand an increased susceptibility to oral squamous cell carcinoma inJapanese patients. J. Oral. Pathol. Med. 36, 351–356
78 Tamaki, S. et al. (2009) Relationship between soluble MICA and theMICA A5.1 homozygous genotype in patients with oral squamous cellcarcinoma. Clin. Immunol. 130, 331–337
79 Lavado-Valenzuela, R. et al. (2009) MHC class I chain-related gene Atransmembrane polymorphism in Spanish women with breast cancer.Tissue Antigens 74, 46–49
80 Chalupny, N.J. et al. (2006) Down-regulation of the NKG2D ligandMICA by the human cytomegalovirus glycoprotein UL142. Biochem.Biophys. Res. Commun. 346, 175–181
81 Zou, Y. et al. (2005) Effect of human cytomegalovirus on expression ofMHC class I-related chains A. J. Immunol. 174, 3098–3104
82 Wills, M.R. et al. (2005) Human cytomegalovirus encodes an MHCclass I-like molecule (UL142) that functions to inhibit NK cell lysis. J.Immunol. 175, 7457–7465
83 Moenkemeyer, M. et al. (2009) Higher risk of cytomegalovirusreactivation in human immunodeficiency virus-1-infected patientshomozygous for MICA5.1. Hum. Immunol. 70, 175–178
84 Karacki, P.S. et al. (2004) MICA and recovery from hepatitis C virusand hepatitis B virus infections. Genes Immun. 5, 261–266
85 Jinushi, M. et al. (2003) Autocrine/paracrine IL-15 that is required fortype I IFN-mediated dendritic cell expression of MHC class I-relatedchain A and B is impaired in hepatitis C virus infection. J. Immunol.171, 5423–5429
86 Jinushi, M. et al. (2003) Critical role of MHC class I-related chain Aand B expression on IFN-alpha-stimulated dendritic cells in NK cellactivation: impairment in chronic hepatitis C virus infection. J.Immunol. 170, 1249–1256
87 Shojima, J. et al. (2009) Identification ofMICA as a susceptibility genefor pulmonary Mycobacterium avium complex infection. J. Infect. Dis.199, 1707–1715
105

Opinion Trends in Molecular Medicine Vol.16 No.3
88 Sumitran-Holgersson, S. (2008) Relevance of MICA and other non-HLA antibodies in clinical transplantation. Curr. Opin. Immunol. 20,607–613
89 Suarez-Alvarez, B. et al. (2009) Potential role of NKG2D and itsligands in organ transplantation: new target forimmunointervention. Am. J. Transplant. 9, 251–257
90 Zou, Y. and Stastny, P. (2009) The role of major histocompatibilitycomplex class I chain-related gene A antibodies in organtransplantation. Curr. Opin. Organ. Transplant. 14, 414–418
91 Suarez-Alvarez, B. et al. (2007) The relationship of anti-MICAantibodies and MICA expression with heart allograft rejection. Am.J. Transplant. 7, 1842–1848
92 Zou, Y. et al. (2006) Detection of anti-MICA antibodies in patientsawaiting kidney transplantation, during the post-transplant course,and in eluates from rejected kidney allografts by Luminex flowcytometry. Hum. Immunol. 67, 230–237
93 Mizutani, K. et al. (2006) Frequency of MIC antibody in rejectedrenal transplant patients without HLA antibody. Hum. Immunol.67, 223–229
94 Mizutani, K. et al. (2006) Detection of HLA and MICA antibodiesbefore kidney graft failure. Clin. Transpl. 255–264
95 Mizutani, K. et al. (2006) Association of kidney transplant failure andantibodies against MICA. Hum. Immunol. 67, 683–691
96 Panigrahi, A. et al. (2007) Post transplant development of MICA andanti-HLA antibodies is associated with acute rejection episodes andrenal allograft loss. Hum. Immunol. 68, 362–367
97 Zou, Y. et al. (2007) Antibodies against MICA antigens and kidney-transplant rejection. N. Engl. J. Med. 357, 1293–1300
98 Terasaki, P.I. et al. (2007) Four-year follow-up of a prospective trial ofHLA and MICA antibodies on kidney graft survival. Am. J.Transplant. 7, 408–415
99 Zwirner, N.W. et al. (2000) Identification of MICA as a newpolymorphic alloantigen recognized by antibodies in sera of organtransplant recipients. Hum. Immunol. 61, 917–924
100 Sasazuki, T. et al. (1998) Effect of matching of class I HLA alleles onclinical outcome after transplantation of hematopoietic stem cellsfrom an unrelated donor. Japan Marrow Donor Program. N. Engl. J.Med. 339, 1177–1185
106
101 Petersdorf, E.W. et al. (1998) Optimizing outcome after unrelatedmarrow transplantation by comprehensive matching of HLA class Iand II alleles in the donor and recipient. Blood 92, 3515–3520
102 Morishima, Y. et al. (2002) The clinical significance of humanleukocyte antigen (HLA) allele compatibility in patients receiving amarrow transplant from serologically HLA-A, HLA-B, and HLA-DRmatched unrelated donors. Blood 99, 4200–4206
103 Tay, G.K. et al. (1995) Matching for MHC haplotypes results inimproved survival following unrelated bone marrowtransplantation. Bone Marrow Transplant. 15, 381–385
104 Tay, G.K. et al. (1995) The identification of MHC identical siblingswithout HLA typing. Exp. Hematol. 23, 1655–1660
105 Kitcharoen, K. et al. (2006) MICA, MICB, and MHC beta blockmatching in bone marrow transplantation: relevance totransplantation outcome. Hum. Immunol. 67, 238–246
106 Gaudieri, S. et al. (2001) Sequence analysis of the MHC class I regionreveals the basis of the genomic matching technique.Hum. Immunol.62, 279–285
107 Parmar, S. et al. (2009) Donor-recipient mismatches in MHC class Ichain-related gene A in unrelated donor transplantation lead toincreased incidence of acute graft-versus-host disease. Blood 114,2884–2887
108 Moretta, A. et al. (2009) Activating and inhibitory killerimmunoglobulin-like receptors (KIR) in haploidenticalhaemopoietic stem cell transplantation to cure high-riskleukaemias. Clin. Exp. Immunol. 157, 325–331
109 de Groot, N.G. et al. (2005) Reduced MIC gene repertoire variation inWest African chimpanzees as compared to humans. Mol. Biol. Evol.22, 1375–1385
110 Jinushi, M. et al. (2006) Therapy-induced antibodies to MHC class Ichain-related proteinA antagonize immune suppression and stimulateantitumor cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 103, 9190–9195
111 Boukouaci, W. et al. (2009) MICA-129 genotype, soluble MICA, andanti-MICA antibodies as biomarkers of chronic graft-versus-hostdisease. Blood 114, 5216–5224
112 Suarez-Alvarez, B. et al. (2006) The predictive value of soluble majorhistocompatibility complex class I chain-related molecule A (MICA)levels on heart allograft rejection. Transplantation 82, 354–361




















