
Mapping the positions of beads on a string: dethreading rotaxanes by molecular force spectroscopy
8
IOP PUBLISHING NANOTECHNOLOGY Nanotechnology 19 (2008) 345706 (8pp) doi:10.1088/0957-4484/19/34/345706 Mapping the positions of beads on a string: dethreading rotaxanes by molecular force spectroscopy Alex Dunlop 1 , Jirut Wattoom 2 , Erol A Hasan 2 , Terence Cosgrove 2 and Andrew N Round 1,3 1 H H Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol BS8 1TL, UK 2 School of Chemistry, University of Bristol, Cantocks Close, Bristol BS8 1TS, UK E-mail: [email protected] Received 4 April 2008, in final form 23 June 2008 Published 16 July 2008 Online at stacks.iop.org/Nano/19/345706 Abstract The direct manipulation by atomic force microscopy (AFM) of individual macrocycles within a rotaxane offers a potential route to a new sequencing tool for complex macromolecules such as polysaccharides, glycoproteins and nucleic acids. In this paper we demonstrate for the first time that a sliding contact made between a macrocycle, α-cyclodextrin, and its polymer axle by an AFM tip can be used to map the positions of specific groups along the polymer as if they were beads along a string, thereby generating sequence information. We find very good agreement (linear fit with slope = 1.03, R 2 = 0.968) between the calculated and measured positions of phenylenediamine and benzenetricarboxylic acid groups within polymers of polyethylene oxide (PEO). The rupture force profiles attributable to the dethreading interactions of phenylenediamine and benzenetricarboxylic acid differ observably from each other and, in the latter case, from the rupture of the corresponding host–guest complex. As well as opening the way to a macromolecular sequencing technique, the ability demonstrated by this method to manipulate the dethreading of a rotaxane offers a new tool for investigating the process energetics in a wide array of spontaneously forming and forced rotaxane systems. (Some figures in this article are in colour only in the electronic version) 1. Introduction Since their discovery, rotaxanes (molecular complexes consisting of a macrocycle through whose inner pore is threaded an ‘axle’ typically comprising a linear polymer) have found a wide array of applications [1, 2], both as functional materials in their own right and as models for non-covalent recognition interactions in nature. The rather intuitive concept underlying their structure and formation would appear to make them ideal candidates for exploration by the tools now available for single molecule manipulation such as scanning probe microscopes, yet few attempts have been made to interact directly with individual rotaxane complexes [3, 4]. 3 Author to whom any correspondence should be addressed. Present address: School of Chemical Sciences and Pharmacy, University of East Anglia, Earlham Road, Norwich NR4 7TJ, UK. Recently the use of dynamic force spectroscopy to make a sliding contact with a macrocycle threaded over DNA has been proposed as a route to single molecule DNA sequencing [5], and the nature of the inclusion phenomenon offers considerable potential for application to any system for which a threading interaction with a suitable macrocycle can be induced. This fact raises the possibility that a system similar to that described here may be developed into a sequencing tool for macromolecules whose structures are at present difficult to characterize in molecular detail by other methods, such as polysaccharides and glycoproteins. Here we demonstrate for the first time that a sliding contact made between a macrocycle and its polymer axle by an atomic force microscope (AFM) tip can be used to map the positions of specific groups along the polymer as if they were beads along a string, thereby generating sequence information. 0957-4484/08/345706+08$30.00 © 2008 IOP Publishing Ltd Printed in the UK 1
Transcript of Mapping the positions of beads on a string: dethreading rotaxanes by molecular force spectroscopy

IOP PUBLISHING NANOTECHNOLOGY
Nanotechnology 19 (2008) 345706 (8pp) doi:10.1088/0957-4484/19/34/345706
Mapping the positions of beads on astring: dethreading rotaxanes bymolecular force spectroscopyAlex Dunlop1, Jirut Wattoom2, Erol A Hasan2, Terence Cosgrove2
and Andrew N Round1,3
1 H H Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol BS8 1TL, UK2 School of Chemistry, University of Bristol, Cantocks Close, Bristol BS8 1TS, UK
E-mail: [email protected]
Received 4 April 2008, in final form 23 June 2008Published 16 July 2008Online at stacks.iop.org/Nano/19/345706
AbstractThe direct manipulation by atomic force microscopy (AFM) of individual macrocycles within arotaxane offers a potential route to a new sequencing tool for complex macromolecules such aspolysaccharides, glycoproteins and nucleic acids. In this paper we demonstrate for the first timethat a sliding contact made between a macrocycle, α-cyclodextrin, and its polymer axle by anAFM tip can be used to map the positions of specific groups along the polymer as if they werebeads along a string, thereby generating sequence information. We find very good agreement(linear fit with slope = 1.03, R2 = 0.968) between the calculated and measured positions ofphenylenediamine and benzenetricarboxylic acid groups within polymers of polyethyleneoxide (PEO). The rupture force profiles attributable to the dethreading interactions ofphenylenediamine and benzenetricarboxylic acid differ observably from each other and, in thelatter case, from the rupture of the corresponding host–guest complex. As well as opening theway to a macromolecular sequencing technique, the ability demonstrated by this methodto manipulate the dethreading of a rotaxane offers a new tool for investigating theprocess energetics in a wide array of spontaneously forming and forced rotaxanesystems.
(Some figures in this article are in colour only in the electronic version)
1. Introduction
Since their discovery, rotaxanes (molecular complexesconsisting of a macrocycle through whose inner pore isthreaded an ‘axle’ typically comprising a linear polymer) havefound a wide array of applications [1, 2], both as functionalmaterials in their own right and as models for non-covalentrecognition interactions in nature. The rather intuitive conceptunderlying their structure and formation would appear tomake them ideal candidates for exploration by the tools nowavailable for single molecule manipulation such as scanningprobe microscopes, yet few attempts have been made tointeract directly with individual rotaxane complexes [3, 4].
3 Author to whom any correspondence should be addressed. Present address:School of Chemical Sciences and Pharmacy, University of East Anglia,Earlham Road, Norwich NR4 7TJ, UK.
Recently the use of dynamic force spectroscopy to make asliding contact with a macrocycle threaded over DNA has beenproposed as a route to single molecule DNA sequencing [5],and the nature of the inclusion phenomenon offers considerablepotential for application to any system for which a threadinginteraction with a suitable macrocycle can be induced. Thisfact raises the possibility that a system similar to thatdescribed here may be developed into a sequencing tool formacromolecules whose structures are at present difficult tocharacterize in molecular detail by other methods, such aspolysaccharides and glycoproteins. Here we demonstrate forthe first time that a sliding contact made between a macrocycleand its polymer axle by an atomic force microscope (AFM)tip can be used to map the positions of specific groups alongthe polymer as if they were beads along a string, therebygenerating sequence information.
0957-4484/08/345706+08$30.00 © 2008 IOP Publishing Ltd Printed in the UK1

Nanotechnology 19 (2008) 345706 A Dunlop et al
Scheme 1. Structures of polymers 1–3 which form rotaxanes 4–6 when mixed with the tethered α-CD. Circles represent thiomethylbenzoylgroups, squares phenylenediamine and triangles benzenetricarboxylic acid. Polymer 1 has MW = 17 000, corresponding to an expectedmaximum contour length of 107 nm.
2. Results and discussion
Our demonstration is made with a series of polymers basedupon polyethylene oxide (PEO) and the rotaxanes theyform with α-cyclodextrin (α-CD) (scheme 1). The firstpoly(pseudo)rotaxane to be described was that between PEOand α-CD [6] and the factors determining the formation of thiscomplex have been extensively studied since [1]. Aromaticderivatives were chosen to function as ‘beads’ along the PEO‘string’ as they are known to act either as stoppers or toform specific host–guest complexes with α and β-CDs [7, 8].The α-CD is functionalized with an amino-terminated tether,designed to bind specifically on the timescale of a forcespectroscopy experiment to a succinimidyl group attached toan AFM probe following the procedure of Wagner et al [9].The formation of an inclusion complex between α-CD andbenzene is a necessary step in the process of threading of theα-CD over the benzoyl end-groups on to the PEO and can befollowed by NMR titration [10]. Figure 1(a) shows that thedoublet at δ = 8.04 ppm arising from the benzoyl end-group inthe model polymer α,ω-4-bromomethylbenzoyl PEO24 shiftswith increasing amounts of α-CD until, at a CD:PEO ratioof 6:1, it occurs at δ = 8.13 ppm, indicating that thereis an interaction between the benzene and the cyclodextrin.Figure 1(b) then shows that the signal attributable to theethylene units of PEO (at δ = 3.65 ppm) is shifted anddecreased in magnitude as the ratio of CD:polymer is increasedfrom 1:1 to 6:1, demonstrating that α-CD threads over thebromomethylbenzoyl end-group and onto PEO, and that in thisway polyrotaxanes are formed.
Figure 2 depicts the force spectroscopy experimentscarried out, and figure 3 presents the results of three keyexperiments. Firstly, the formation of a strong bond onthe timescale of a dynamic force spectroscopy experimentbetween the amine group of the tether and the succinimidylgroup on the AFM probe was tested by grafting the tether
Figure 1. 1HNMR spectrum showing chemical shifts (ppm) for(a) the protons of the benzoyl end-group and (b) the ethylene protonsof the PEO. The concentration of PEO is 3 mM and the ratios arePEO: α-CD. Y -axes are arbitrary units.
to a gold surface and interrogating it with the succinimidyl-functionalized probe. As shown by Gaussian fits to thehistogram data, the peak rupture length for the tether is 12.4 ±5.5 nm (centre ± half-width) from the surface with a series offour peak rupture forces at 357.1±43.6, 593.4±113.1, 834.4±66.8 and 1030.6 ± 133.9 pN. Similarly, a second experimentutilized the same polymer-probe chemistry to determinethe rupture length of an amine-terminated PEO400 polymer(formed by the reaction of polymer 1 with phenylenediamine).
2

Nanotechnology 19 (2008) 345706 A Dunlop et al
Figure 2. (a) A simulated force spectrum, indicating the processoccurring at the AFM probe at each stage as follows: (1) thefunctionalized AFM probe binds to the amine group on the CDtether; (2) the CD slides along the polymer axle until it reaches astopper. Stopped from sliding, the polymer behaves as an entropicspring until (3) the CD slides over the stopper. The process repeats(4–6) for each subsequent stopper until it slides over the terminalstopper, dethreading itself from the polymer (7). (b) A forcespectrum obtained experimentally for the sliding and dethreading ofrotaxane 6.
The generated rupture force profile again produces four peakrupture forces at 276.0 ± 65.5, 590.7 ± 92.3, 818.7 ± 69.3and 1057.9 ± 60.6 pN together with a peak rupture lengthof 72.6 ± 6.5 nm, consistent with the length of the polymeronce geometric considerations such as probe tip radius andthe angle between the polymer anchoring point and its tetherto the AFM probe have been accounted for. The series ofpeak rupture forces for these two polymers can be fitted toa model representing multiple specific unbinding events andgives rise to a rupture force quantum of 270.8 pN. As thisis similar to the magnitude of the first rupture events in eachsample we interpret these events as the rupture of single bondsbetween the amine-terminated polymers and the succinimidyl-functionalized probe, with the subsequent events at ∼590 pN,∼830 pN and ∼1050 pN representing 2, 3 and 4 parallelunbinding events respectively. From the fact that the forcequantum is significantly lower than that found for the ruptureof the covalent bonds formed within the polymer or with thesubstrate [11] we are able to infer that this specific unbindingevent represents the disruption of a strong but non-covalentinteraction between the amine and succinimide groups. Thuswe have established that a strong and specific bond betweenthe tethered CD and our AFM probe can be made.
With this knowledge, the rotaxanes 4–6 formed betweenthe mixed population of polymers 1–3 and the tethered CDwere interrogated with the succinimidyl probe. The ruptureforce and length data (red datapoints in figure 3) are clusteredaround three loci, which we interpret as three distinct events.Two of these events occur at identical rupture forces (114.1 ±44.0 pN), indicating that they represent identical events, and atlengths (93.0 ± 9.2 nm and at 220.3 ± 6.7 nm) commensuratewith the positions of aromatic rings within rotaxanes 5 and 6while the third locus, that of events occurring over a wide rangeof lengths below 75 nm, shows a broad range of forces with afirst peak rupture force of 205.9 ± 73.8 pN, and a significantproportion of events occurring at much higher forces. Figure 4shows an analysis of the force profiles for each of the threeevents. Rotaxanes 5 and 6 have the same benzenetricarboxylicacid (BtcA) end-groups and thus we expect to see similarpatterns of rupture forces for the terminal unthreading eventsas CD passes over them. In both cases the absence of higher-order maxima suggests that we are observing predominantlysingle rupture events. The events occurring below 75 nmare shown here (figure 4(b)) to have a force distribution witha higher peak (above 200 pN) and a greater proportion ofhigher force events. The force distribution of rupture eventson a sample of the mixed polymers 1–3 in the absence of CD(figure 4(e)) shows a similar pattern. In both of these cases wedo not see the force quantization seen in the amine-terminatedpolymer samples (as free amine termini are not present in thesample) and attribute these force distributions to non-specificadhesions between the PEO or BtcA and the probe. A furtherexperiment in which rotaxane 5 alone was investigated showedsimilar results to those presented for the mixed system above,save for the absence of the locus at 220 nm (data not shown).In interpreting this set of experiments, the following threeobservations are pertinent: (i) the event characterized by thepeak rupture force of 114 pN only occurs in experiments whererotaxanes formed between the tethered CD and polymerscontaining the aromatic rings are present, (ii) the peak rupturelength in the case of the first such event (93.0 ± 9.2 nm)exceeds the peak rupture length of polymer 1 (72.6 ± 6.5 nm)by a distance consistent with the additional length of the CDtether (12.4 ± 5.5 nm) (the 220 nm event likewise fits a modelof twice the polymer 1 length plus the tether length whengeometric considerations are accounted for) and (iii) the clusterof rupture events at 220 nm, attributable to the sliding of theCD over the terminal aromatic ring in rotaxane 6, is isolatedand discrete. On the basis of the patterns of rupture lengthand force distributions described here, we identify the eventsat 93 and 220 nm as the threading of the CD over BtcAend-groups, with the events occurring at shorter distances anda broader range of higher forces consisting of non-specificrupture events.
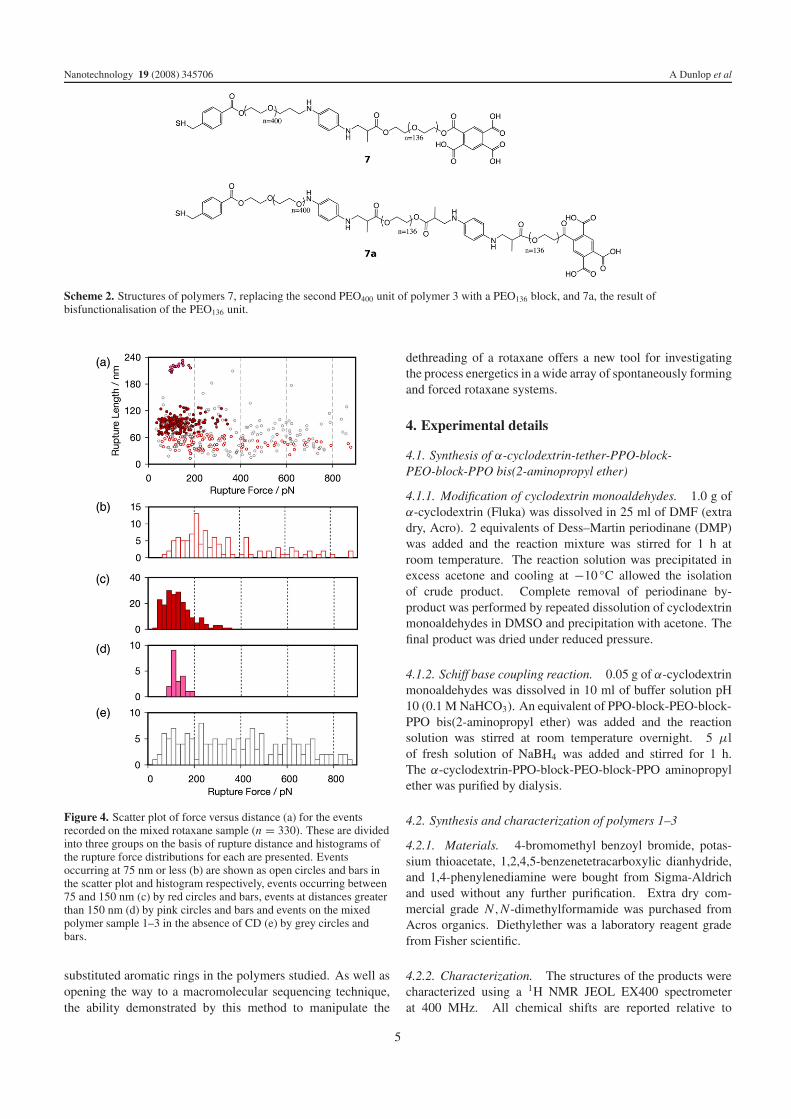
We next synthesized polymers 7 and 7a (scheme 2), beinganalogs of polymer 3 but with one or two PEO136 blocksreplacing one of the PEO400 blocks, and performed forcespectroscopy experiments on their corresponding rotaxanesas above. Figure 5 shows that the distribution of rupturelengths (maxima at 94.1 ± 8.8, 127.3 ± 6.0 and 150.9 ±6.2 nm) reflects the expected positions of aromatic rings in
3

Nanotechnology 19 (2008) 345706 A Dunlop et al
Figure 3. Scatter plot (a) and histograms of force (b) and distance (c) values for rupture events recorded for three systems; the tether (trianglesin (a), histogram (i) in (b) and (c), blue online), amine-terminated polymer 1 (open circles in (a), histogram (ii) in (b) and (c)) and the mixedrotaxane system 4–6 (filled circles in (a), histogram (iii) in (b) and (c), red online). n = 620, 208 and 330 for the tether, amine-terminatedpolymer and rotaxane systems respectively. Continuous lines represent multiple Gaussian fits to the data. (d) shows representations of thepolymers and rotaxanes in each sample with the same key as in scheme 1.
polymers 7 and 7a. Furthermore, in cases where multiplepeaks were observed in the same force curve, a histogramof distances between consecutive events in the same spectrareveals a clear maximum at 27.0 ± 3.3 nm, corresponding tothe distance between the aromatic rings in polymers 7 and7a. As can be seen from the structures in scheme 2, theevents occurring at around 90 nm will derive from dethreadingover a different ‘bead’ (phenylenediamine) to those eventsoccurring at around 150 nm (terminal BtcA), while thoseoccurring at around 120 nm may result from the dethreadingof either group (phenylenediamine in the case of polymer 7a,and terminal BtcA in the case of polymer 7). Gaussian fitsto the force distributions at each length indicate that the firstmaxima shift from 92.0 ± 24.1 pN for events at 90 nm to103.7 ± 39.8 and 116.8 ± 36.9 pN for events at 120 and150 nm respectively (the latter being similar to the force valuesrecorded for the BtcA groups in the mixed rotaxane systemat 93 and 220 nm), suggesting that the difference betweenthe dethreading forces for phenylenediamine and BtcA isobservable.
Figure 6 shows the excellent agreement between theexpected ring positions calculated from the molecular weightdistributions and the peak rupture lengths at which thesignature peak rupture forces identified above occur, giving riseto a linear fit with a slope of 1.03 (R2 = 0.968).
Finally, in order to test the hypothesis that we areobserving not dethreading events but instead host–guestcomplexation between terminal BtcA and α-CD, we probedthe rupture of such complexes. Instead of adding CD to the
polymer and forming a polyrotaxane in solution, the tetheredCD is bound to a succinimidyl-functionalized AFM probeprior to its introduction to polymer 2 immobilized on a goldsubstrate. As presented in figure 7, the interaction between theBtcA and the α-CD occurs with a peak rupture force of 55.7 ±12.1 pN. The difference between the rupture force magnitudesof the host–guest interaction and the threading interactiondescribed above (with its peak at 114 pN) emphasizestheir differing origins, reflecting the fact that the threadinginteraction involves the steric distortion of the CD and/oraromatic ring as one passes entirely over the other. Thevalue of 56 pN is consistent with previous force spectroscopymeasurements of the rupturing of host–guest interactionsbetween CDs and phenyl derivatives [7, 8].
3. Conclusions
This work represents the first example of the dethreading ofa rotaxane under the control of an AFM probe being used tomap the positions of specific groups along a polymer. Ouridentification of the features described here as dethreadingevents is based on the key observations that the events occurat positions equal to the sum of the lengths of the polymer andof the CD tether, show the expected intrapolymer distances andthat the peak rupture forces are signatures of an interaction notpresent in the absence of formation of the rotaxane. Employingthese criteria, we see very good correspondence between theobserved distribution of rupture lengths and the positions of
4
Nanotechnology 19 (2008) 345706 A Dunlop et al
Scheme 2. Structures of polymers 7, replacing the second PEO400 unit of polymer 3 with a PEO136 block, and 7a, the result ofbisfunctionalisation of the PEO136 unit.
Figure 4. Scatter plot of force versus distance (a) for the eventsrecorded on the mixed rotaxane sample (n = 330). These are dividedinto three groups on the basis of rupture distance and histograms ofthe rupture force distributions for each are presented. Eventsoccurring at 75 nm or less (b) are shown as open circles and bars inthe scatter plot and histogram respectively, events occurring between75 and 150 nm (c) by red circles and bars, events at distances greaterthan 150 nm (d) by pink circles and bars and events on the mixedpolymer sample 1–3 in the absence of CD (e) by grey circles andbars.
substituted aromatic rings in the polymers studied. As well asopening the way to a macromolecular sequencing technique,the ability demonstrated by this method to manipulate the
dethreading of a rotaxane offers a new tool for investigatingthe process energetics in a wide array of spontaneously formingand forced rotaxane systems.
4. Experimental details
4.1. Synthesis of α-cyclodextrin-tether-PPO-block-PEO-block-PPO bis(2-aminopropyl ether)
4.1.1. Modification of cyclodextrin monoaldehydes. 1.0 g ofα-cyclodextrin (Fluka) was dissolved in 25 ml of DMF (extradry, Acro). 2 equivalents of Dess–Martin periodinane (DMP)was added and the reaction mixture was stirred for 1 h atroom temperature. The reaction solution was precipitated inexcess acetone and cooling at −10 ◦C allowed the isolationof crude product. Complete removal of periodinane by-product was performed by repeated dissolution of cyclodextrinmonoaldehydes in DMSO and precipitation with acetone. Thefinal product was dried under reduced pressure.
4.1.2. Schiff base coupling reaction. 0.05 g of α-cyclodextrinmonoaldehydes was dissolved in 10 ml of buffer solution pH10 (0.1 M NaHCO3). An equivalent of PPO-block-PEO-block-PPO bis(2-aminopropyl ether) was added and the reactionsolution was stirred at room temperature overnight. 5 μlof fresh solution of NaBH4 was added and stirred for 1 h.The α-cyclodextrin-PPO-block-PEO-block-PPO aminopropylether was purified by dialysis.
4.2. Synthesis and characterization of polymers 1–3
4.2.1. Materials. 4-bromomethyl benzoyl bromide, potas-sium thioacetate, 1,2,4,5-benzenetetracarboxylic dianhydride,and 1,4-phenylenediamine were bought from Sigma-Aldrichand used without any further purification. Extra dry com-mercial grade N,N-dimethylformamide was purchased fromAcros organics. Diethylether was a laboratory reagent gradefrom Fisher scientific.
4.2.2. Characterization. The structures of the products werecharacterized using a 1H NMR JEOL EX400 spectrometerat 400 MHz. All chemical shifts are reported relative to
5

Nanotechnology 19 (2008) 345706 A Dunlop et al
Figure 5. Scatter plot of force versus distance (a) and histograms ofdistance (b), inter-event distance (c) and force (d) values for ruptureevents recorded for the rotaxane formed between polymers 7 and 7aand the tethered CD (n = 89). Dark circles in (a) are rupture eventsin force spectra showing multiple such events and distances betweenthese events in the same spectra are shown in histogram (c). (e) is aforce curve obtained on this sample.
DMSO (1H: δ = 2.49). Molecular weights of the polymerswere estimated by gel permeation chromatography in water(GPC, PL aquagel-OH guard plus 2× PL aquagel-OH mixedcolumn) relative to a poly(ethylene glycol)/poly(ethyleneoxide) standard.
4.2.3. Preparation of α-4-bromomethylbenzoyl-ω-formylPEO400. 1 g of α-hydroxy-ω-formyl PEO was dissolved inDMF (10 ml) at 40 ◦C until a clear solution of PEO wasobtained, upon which it was cooled to room temperature. Then,0.15 g of 4-bromomethyl benzoyl bromide (10 equivalentmole) and 1 mol equivalent (hydroxyl group) of triethylaminewas added to the reaction. The reaction was stirred and kept
Figure 6. Scatter plot of the expected position of aromatic rings(= polymer length + CD tether length) versus measured position forthe rotaxanes examined in figures 3 and 5. X-axis error bars are thewidth of the Gaussian peak fitting the GPC data, Y -axis error bars arethe widths of the Gaussian peaks fitting each set of rotaxane data.The continuous line represents the linear fit to the data points, with aslope of 1.03.
Figure 7. Histogram of rupture forces for the rupture of theinteraction between btCA and a-CD tethered to the AFM probe, asdepicted in the inset.
at room temperature for 5 days. Then, salt was filtered off thereaction solution. The solution was directly poured into diethylether, the white precipitate formed was washed with excessof diethyl ether and the product dried. 1H NMR chemicalshifts (δ): 3.51 (–O–CH2CH2–O–), 4.39 (–COO–CH2–), 4.77(–CH2–Br), 7.94 (–CH), 7.61(–CH).
4.2.4. Preparation of α-4-thiomethylbenzoyl-ω-formyl PEO400
(1). The product (1 g) obtained in step 4.2.3 was dissolved inbenzene. Then, 0.06 g of potassium thioacetate was added andstirred at 40 ◦C. After 5 days the product was precipitated indiethyl ether and washed using diethyl ether (3×150 ml) beforedrying under vacuum. The thioacetate functional group wasconverted using hydrolysis to give α-4-thiomethyl benzoyl-ω-formyl PEO. The product was purified by dialysis in water(MW cut off = 12 000). 1H NMR chemical shifts (δ): 3.51(–O–CH2CH2–O–), 4.18 (–COO–CH2–), 4.36 (–CH2–SH),7.46 (–CH), 7.89 (–CH).
4.2.5. Preparation of α-2,4,5-benzenetricarboxylic acid-ω-formyl PEO400. 1 g of α-hydroxy-ω-formyl PEO in DMF(10 ml) was stirred with triethylamine and left to equilibrate.Then, 0.12 g of 1,2,4,5-benzenetetracarboxylic dianhydridewas added to the reaction. The reaction was stirred and
6

Nanotechnology 19 (2008) 345706 A Dunlop et al
kept at room temperature for 5 days. The solution was laterprecipitated in excess of diethyl ether and the white powderproduct obtained. The product was washed with diethyl ether(3×150 ml). 1H NMR chemical shift (δ): 3.51 (–O–CH2CH2–O–), 4.22 (–COO–CH2–), 8.03 (–CH), 8.45 (–CH).
4.2.6. Preparation of α-2,4,5-benzenetricarboxylic acid-ω-4-amino-phenylamino PEO400 (2). The product (4.2.5) (1 g)was reacted with 1,4-phenylenediamine (10 equivalent molesto formyl group of PEO) via a Schiff base reaction. Thereaction was performed in carbonate buffer pH 10. Thereaction was vigorously stirred at room temperature for 2 days.Then, the reducing agent (NaBH4) was added. The solutionwas purified by dialysis and freeze-dried. 1H NMR chemicalshift (δ): 1.74 (–NH), 2.67 (–NH–CH2), 3.51 (–O–CH2CH2–O–), 4.28 (–COO–CH2–).
4.2.7. Preparation of α-4-thiomethylbenzoyl PEO400-α′-2,4,5-benzenetricarboxylic acid PEO400-diaminophenylene(3). Polymers 1 and 2 were connected together via a Schiffbase reaction. Molar equivalents of both modified polymerswere dissolved in carbonate buffer pH 10. The reaction wasvigorously stirred at room temperature for 2 days. Then, thereducing agent (NaBH4) was added. The solution was purifiedby dialysis. Then, the solution was freeze-dried and kept at4 ◦C until use. Finally, the product was characterized by GPC(data not shown). The molecular weight is 28 100 and polydis-persity is 2.00. The percentage of peak area of the additionalmolecular weight peak is 6.30%, which thus corresponds to theyield of polymer 3.
4.3. Synthesis and characterization of polymers 7 and 7a
4.3.1. Materials. Poly(ethylene oxide) MW = 6000, sodiumhydride (60%) in mineral oil, 1,2,4,5-Benzenetetracarboxylicdianhydride and pyruvic acid were bought from Aldrich andused without further purification. N,N-dimethylformamide(extra dry) was bought from Acros.
4.3.2. Characterization. The structures of the products werecharacterized using a 1H NMR JEOL EX400 spectrometerat 400 MHz. All chemical shifts are reported relative toDMSO (1H: δ = 2.49). Molecular weights of the polymerswere estimated by gel permeation chromatography in water(GPC, PL aquagel-OH guard plus 2× PL aquagel-OH mixedcolumn) relative to a poly(ethylene glycol)/poly(ethyleneoxide) standard.
4.3.3. Preparation of α-hydroxy-ω-pyruvic PEO136. Poly-(ethylene oxide) 1.50 g was dissolved in 5 ml of DMF at40 ◦C and the solution cooled to troom temperature. Then,0.0073 g (1 equivalent mole per a hydroxyl unit) of NaH wasadded and the solution stirred at room temperature for 2 h.Then, 25 μl of pyruvic acid was added dropwise and themixture was kept stirring overnight. The solution was laterprecipitated in diethyl ether and washed with excess amountof fresh diethyl ether three times. The white solid productwas obtained and dried under reduced pressure. 1H NMR
chemical shift (δ): 2.01(–OC–CH3), 3.51 (–O–CH2CH2–O–),4.60 (–COO–CH2–).
4.3.4. Preparation of α-pyruvic-ω-2,4,5-benzenetricarboxylicacid PEO136. 1 g of product (4.3.3) was dissolved in 5 ml ofDMF at 40 ◦C. Then, a drop of ethylene diamine was addedand kept stirring at room temperature. 0.0691 g (2 equivalentmoles per a hydroxyl unit) of 1,2,4,5-benzenetetracarboxylicdianhydride was added. The reaction was stirred at roomtemperature overnight. The final product was precipitatedby adding excess diethyl ether and was dried under reducedpressure. 1H NMR chemical shift (δ): 2.04 (–OC–CH3), 3.51(–O–CH2CH2–O–), 4.31 (–COO–CH2–), 8.52 (Ar–H).
4.3.5. Preparation of α-4-thiomethylbenzoyl PEO400-α′-2,4,5-benzenetricarboxylic acid PEO136-diaminophenylene-(7). Product (4.3.4) and polymer 1 were coupled viaa Schiff base reaction. The equivalent mole of bothpolymers were dissolved in carbonate buffer pH 10 with 1,4-phenylenediamine (10 equivalent moles to formyl group ofpolymer 1). The reaction was vigorously stirred at roomtemperature for 2 days. Then, the reducing agent (NaBH4)
was added. The solution was purified by dialysis. Then, thesolution was freeze-dried and kept at 4 ◦C until use.
4.3.6. Formation of α-4-thiomethylbenzoyl PEO400-α′-2,4,5-benzenetricarboxylic acid (PEO136-diaminophenyl)-PEO136-diaminophenylene (7a). In reaction (4.3.3) a minority ofPEO is bisfunctionalized with pyruvic acid so that it doesnot react with 1,2,4,5-benzenetetracarboxylic dianhydride butinstead is capable of reacting with two phenylenediamines instep 4.3.5. The Schiff base coupling of polymer 1, α,ω-dipyruvic PEO136 and α-pyruvic-ω-2,4,5-benzenetricarboxylicacid PEO136 (product (4.3.4)) with phenylenediamine givesrise to polymer 7a as a by-product of reaction (4.3.5).
4.4. AFM probe and sample preparation
Samples for investigation by AFM were prepared by depositingaqueous solutions of the polymers and rotaxanes on template-stripped gold as follows: 0.4% w/w of polymers 1–3 wasmixed with a 1:1 mol equivalent of α-CD for 24 h, anddeposited onto template-stripped gold from water for 24 h.AFM probes (MLCT silicon nitride from Veeco Instruments,Santa Barbara, CA, USA) with nominal spring constants of10 and 20 pN nm−1 were coated under vacuum with 1 nmCr and 10 nm Au (both from Goodfellow Corp., Berwyn,PA, USA) before incubation with 1 mM 11-11′-dithio-bis(succinimidylundecanoate) (DSU) in 1,4-dioxane for 10 min.DSU-functionalized probes were used immediately or storedin an inert atmosphere. Force spectroscopy experiments werecarried out at a pulling rate of 500 nm s−1 using a MultimodeAFM with Nanoscope IIIa or V controllers (Veeco Instruments,Santa Barbara, CA, USA) in water with cantilevers calibratedafter Cr/Au coating using the thermal tune method in theNanoscope V controller. Calibrated spring constants rangedfrom 14.3 to 25.1 pN nm−1.
7

Nanotechnology 19 (2008) 345706 A Dunlop et al
Acknowledgments
We are grateful to the Interdisciplinary Research Collaborationin Nanotechnology and the Royal Society of ChemistryAnalytical Chemistry Trust Fund (ACTF) for financial support.AD and JW are in receipt of ACTF Analytical ScienceStudentships.
References
[1] Wenz G, Han B H and Muller A 2006 Chem. Rev. 106 782[2] Frampton M J and Anderson H L 2007 Angew. Chem. Int. Edn
46 1028
[3] Shigekawa H, Miyake K, Sumaoka J, Harada A andKomiyama M 2000 J. Am. Chem. Soc. 122 5411
[4] Brough B, Northrop B H, Schmidt J J, Tseng H R, Houk K N,Stoddart J F and Ho C M 2006 Proc. Natl Acad. Sci. USA103 8583
[5] Voulgarakis N K, Redondo A, Bishop A R and Rasmussen K O2006 Nano Lett. 6 1483
[6] Harada A and Kamachi M 1990 Macromolecules 23 2821[7] Auletta T et al 2004 J. Am. Chem. Soc. 126 1577[8] Chen W, Chang C E and Gilson M K 2004 Biophys. J. 87 3035[9] Wagner P, Hegner M, Kernen P, Zaugg F and Semenza G 1996
Biophys. J. 70 2052[10] Hasan E A, Cosgrove T and Round A N 2008 Macromolecules
41 1393[11] Grandbois M, Beyer M, Rief M, Clausen-Schaumann H and
Gaub H E 1999 Science 283 1727
8




















