
Low-cost biogenic waste for phosphate capture from aqueous system
11
(This is a sample cover image for this issue. The actual cover is not yet available at this time.) This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
-
Upload
independent -
Category
Documents
-
view
6 -
download
0
Transcript of Low-cost biogenic waste for phosphate capture from aqueous system

(This is a sample cover image for this issue. The actual cover is not yet available at this time.)
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Low-cost biogenic waste for phosphate capture from aqueous system
N.A. Oladoja a,⇑, A.L. Ahmad b, O.A. Adesina a, R.O.A. Adelagun c
a Department of Chemistry, Adekunle Ajasin University, Akungba Akoko, Nigeriab School of Chemical Engineering, Universiti Sains Malaysia, Penang, Malaysiac Department of Chemistry, Wesley University of Science and Technology, Ondo, Nigeria
h i g h l i g h t s
" SS is a low cost biogenic sorbent for PO�34 sorption from aqua system.
" Anionic interference and organics negatively impacted the SS sorption capacity." SS-PO�3
4 interaction was not dominated by electrostatic attraction." Equilibrium is achieved faster at higher initial sorbate concentration." PO�3
4 laden sorbent could be used as fertilizer cum soil conditioner.
a r t i c l e i n f o
Article history:Received 17 June 2012Received in revised form 26 July 2012Accepted 26 July 2012Available online 3 August 2012
Keywords:Phosphate removalEutrophicationGastropod shellAfrican land snailAdsorptionWastewater treatment
a b s t r a c t
Premised on the principle of waste minimization and affordable water and wastewater treatment system,the ability of waste African land snail shell (SS), as a sorbent, in the capture of phosphate from aqueoussystem was evaluated. Batch sorption process assessment of the effects of process variables showed thatpH had no influence on phosphate sorption while anionic interference and organic load impacted nega-tively on the magnitude of phosphate uptake. Analysis of the sorption process using different kineticmodels showed that the reversible first order and pseudo first order kinetic models gave poor descrip-tions of the process (r2 = 0.6977–0.8819) while the pseudo second order kinetic model gave the bestdescription of the process (r2 > 0.99). Equilibrium isotherm analysis of the sorption process data showedthat the Freundlich isotherm gave the best description of the process than the Langmuir and Temkin iso-therm models. Estimation of the energy of sorption gave a value of 0.3318 kJ/mol, which is a pointer tothe fact that electrostatic interaction did not dominate the mechanism of interaction between phosphateand SS. A simple economic evaluation of the process showed that the monolayer sorption capacity of theSS for phosphate (222.22 mg/g) was higher than that of other sorbents that have been screened for phos-phate sorption from aqueous system and the phosphate laden SS could be used as soil conditioner cumfertilizer in agricultural practices.
� 2012 Elsevier B.V. All rights reserved.
1. Introduction
Consequent upon the position of phosphorus (P) as the limitingnutrient in eutrophication potentials of water bodies, removal of Pis one of the major constituents of the toolbox of environmentalmanagers. Chemical precipitation and enhanced biological phos-phorus removal (EBPR) approach are two conventional methodsof P removal from municipal and industrial wastewaters. Centeredon the recently published costs for some of the cationic salts, re-quired for chemical precipitation [1], the high cost of these chem-icals generally limit the use of this approach for wastewaterremediation. Furthermore, the use of chemical precipitation entails
additional costs for the purchase and installation of dosing equip-ment and operational costs for power, disposal of additional sludgeand manpower. Enhanced biological treatment can remove up to97% of the total phosphorous, but this process can be highly vari-able due to operational difficulties [2].
Premised on the challenges of the aforementioned conventionalP removal technologies, Investigators have opted for the use ofadsorption as a tertiary unitary process and novel sorbents for Pattenuation are being developed. Kioussis et al. [3] have developedgel-based sorbents for use in aquaculture applications, and werefound to be reusable for several cycles of sorption/regeneration.A P sorption media consisting of iron oxide nanoparticles embed-ded in an anion exchange resin that showed favorable P sorptioncapacities and rates have been developed [4]. Genz et al. [5] inves-tigated the use of granulated ferric hydroxide and activated alu-
1385-8947/$ - see front matter � 2012 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.cej.2012.07.125
⇑ Corresponding author. Tel.: +234 8055438642.E-mail address: [email protected] (N.A. Oladoja).
Chemical Engineering Journal 209 (2012) 170–179
Contents lists available at SciVerse ScienceDirect
Chemical Engineering Journal
journal homepage: www.elsevier .com/locate /ce j

Author's personal copy
mina for P removal from membrane bioreactor effluents. Raw min-erals and industrial waste products have also been screened for Pabstraction potential from aqua system. These include acid minedrainage sludge [6], calcium and iron oxides from steelmakingslags [7], calcium and aluminum silicate minerals [8], iron and alu-minum oxides from water treatment residuals [9], and coal com-bustion fly ash [10].
On the perspectives of waste minimization and cost reductionin the treatment of phosphate contaminated water, the shell ofAfrican land snail (SS) (Achatina achatina) is being proposed asthe Gastropod shell to be used as a low cost sorbent because ofthe ubiquity and the chemical and mineralogical assemblage. Gas-tropods have worldwide distribution from the near arctic and Ant-arctic zones to the tropics and very striking in its extraordinarydiversification of habitats. Large tons of Gastropod shell are dis-charged annually, as waste from food processing industries, whichmade it a low cost and abundant material that could be harnessedfor phosphate immobilization from aqueous system. SS has got thesame basic construction as other Mollusca shells and consists threelayers whose basic building block is aragonite [11,12].
In the present study, the SS was characterized and tested inphosphate removal from aqueous system in a batch adsorptionprocess. Process variables were optimized by method of continu-ous variations and the sorption process data required for processscale up were obtained via equilibrium isotherm analysis and ki-netic studies. Simple economic evaluation of the phosphate ladenSS derived from the process was also attempted.
2. Materials and methods
2.1. Gastropod shell preparation and characterization
The shell of the African land snail (A. achatina) was used as asource of the Gastropod shell. The snail shell (SS), whose some ofthe physicochemical parameters have been determined and re-ported in our earlier treatise was prepared as previously described[11,12]. Textural characteristics were determined using nitrogenabsorption procedure, to determine the specific surface area andthe pore diameter using an ASAP Micromeritics instrument by Bru-nauer–Emmett–Teller (BET) method (1938), and the Barrett–Joy-ner–Hanlenda (BJH) method (1951) was used to calculate themesopore distribution. The surface architecture and surficial ele-mental composition of the SS were determined by a scanning elec-tron microscope (SEM) equipped with energy dispersive analysis ofX-ray (EDAX). Thermogravimetric analyses of the SS were carriedout in Perkin Elmer model TG analyzer and the surface functionalgroups were determined using an FTIR spectrophotometer (Ther-mo Scientific, USA).
2.2. Sorption experiment
Potassium dihydrogen phosphate (KH2PO4) was used as thesource of orthophosphate in the present studies. A stock ortho-phosphate solution of 2000 mg/L was prepared with laboratory-grade water and working solutions of different orthophosphateconcentrations (25–300 mg/L) were prepared, as required, fromthe stock by serial dilution.
The kinetic parameters of phosphate sorption onto SS were de-rived by the addition of 2.0 g of SS into a litter of phosphate solu-tion of concentrations that ranged between 25 and 300 mg/L.Samples are withdrawn at intervals between 0 and 4 h, of sorption,centrifuged and the supernatant phosphate concentration wasdetermined in each case. The equilibrium isotherm analysis ofthe sorption process was evaluated by contacting 50 mL solutionof known phosphate concentration that ranged between 25 and
300 mg/L with 0.1 g of sorbent. The mixture was stirred at200 rpm in thermostatic shaker for 2 h, samples were removed,centrifuged and the supernatant was analyzed for residual phos-phate (expressed as phosphorous) by the molybdenum-blue ascor-bic acid method with a UV–VIS spectrophotometer. The amount ofphosphate sorbed per unit mass of the adsorbent (in mg/g) wascalculated using the mass balance equation.
The influence of some process variables on the sorption processwere evaluated thus: the effects of pH on the sorption process wasinvestigated by varying the pH of the initial phosphate solution be-tween pH 7 and 12; the effects of anionic interference was studiedby the addition of different potassium salts with different anion(NO2�
3 ; SO2�3 , and CO2�
3 ) concentrations (0.01, 0.1, and 0.25 M), or-ganic interference was simulated by the addition of humic acid(HA) of concentrations that range between 100 and 500 mg/L.
3. Results and discussion
3.1. SS characterization
Aragonite has been reported as the sole mineral constituent ofthe SS and the physicochemical profile revealed the very high inor-ganic fraction and the predominance of Ca2+ amongst the selectedmetal ions determined [11,12].
The summary of the textural characteristics and the nitrogenadsorption–desorption curve of the SS are presented in Table 1and Fig. 1, respectively. The BET surface area is 1.5312 m2/g, whileBJH adsorption/desorption surface area of pores is 0.6864 m2/g.The single point total pore volume of pores (d < 1098.594 Å) is0.003624 cm3/g, whereas cumulative adsorption/desorption porevolume of the pores (17 Å < d < 3000 Å) is 0.005939 cm3/g cm3/g,respectively. The average pore diameter by BET method is94.6641 Å, while the BJH adsorption/desorption average porediameter is 346.059 Å. Pore sizes are classified in accordance withthe classification adopted by the International Union of Pure andApplied Chemistry [13], that is, micropores (diameter (d) < 20 Å),mesopores (20 Å < d < 500 Å) and macropores (d > 500 Å). Microp-ores can be divided into ultra-micropores (d < 7 Å) and super-micropores (7 Å < d < 20 Å). Since the average pore diameter ofthe SS is greater than 20 Å and less than 500 Å the material is clas-sified as a mesoporous material.
The surface microstructure of the SS, studied using SEM, is pre-sented in Fig 2a. The different constituents of the SS appeared tohave been homogeneously processed into closely knitted fibrousstrands of varying lengths. The surficial elemental composition ofthe SS, determined via EDX analysis, which revealed the presenceof calcium, carbon and oxygen, is presented in Fig. 2b.
Table 1SS textural characteristics.
Textural properties Values
Single point surface area 1.4514 m2/gSBET 1.5312 m2/gSLangmuir 2.2866 m2/gt-Plot micropore area 0.0957 m2/gt-Plot external surface area 1.4355 m2/gSBJH 0.6864 m2/gVtotal 0.003624 cm3/gVmicro 0.000030 cm3/gVBJH 0.005939 cm3/gVBET average pore width 94.6641 ÅDBJH 346.059 Å
Vtotal = total pore volume; Vmicro = micropore volume; SBET = BET surface area;SLangmuir = surface area; SBJH = desorption cumulative surface area of pores;VBJH = BJH desorption cumulative volume of pores; VBET = BET average pore width;DBJH = desorption average pore diameter.
N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179 171

Author's personal copy
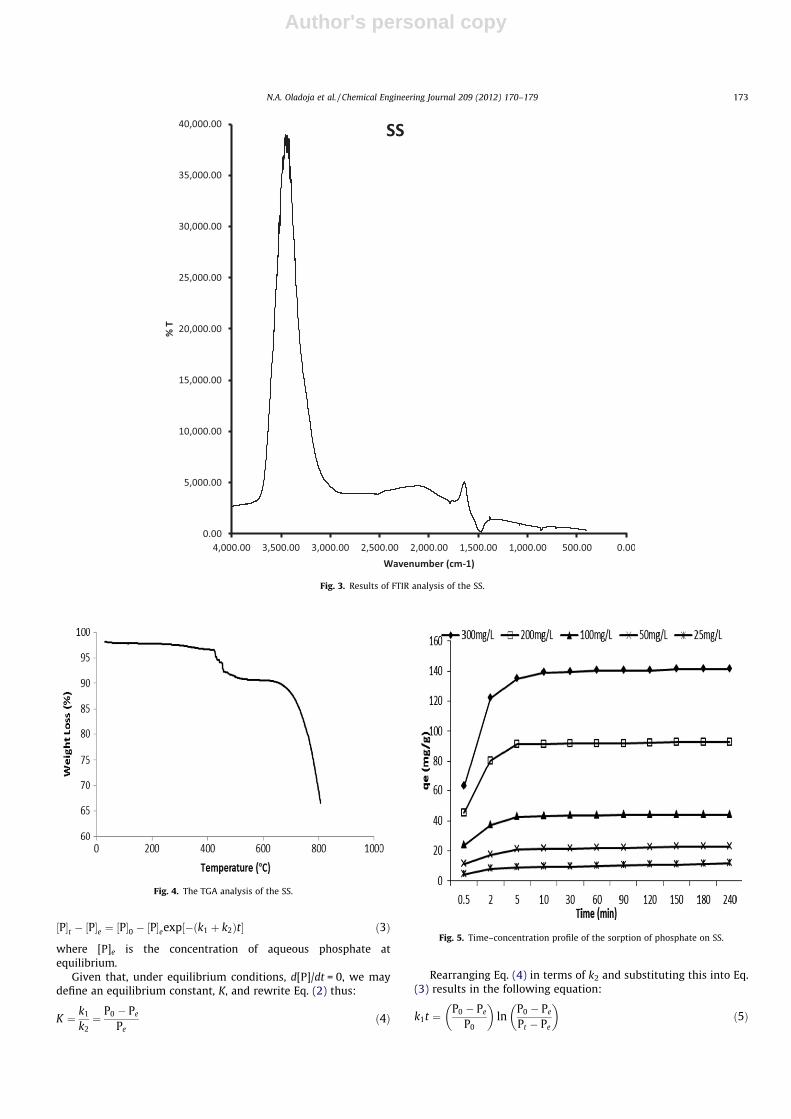
The determination of the functional group present in the SS viaFTIR (Fig. 3) showed multitudes of peaks (cm�1) between 1646 and2814 and prominent peaks at 3595.75, 3574.14, 3291.32, 1471.32and 1646.1. The multitude of peaks observed was not unexpectedbecause of the biogenic nature of this material and the presence ofconchin, a mixture of organic compounds, mostly of proteids in theSS. Thus, these ranges of peaks were ascribed to the presence of dif-ferent organic functional groups viz: AC@CA stretch; NAH bend;CAH rock; CAN stretch; CAC stretch (in-ring) and CAO stretch.The prominent peaks at 3595.75, 3574.14 are characteristic peaksrelated to the H-bonding stretching vibration of the OH while thepeak at 3291.32 was assigned to NAH stretch of primary, second-ary amines or amides. The peaks at 1471.32 and 1646.1 were as-cribed to the CO2�
3 band of CaCO3 and NAH bend of primaryamines respectively.
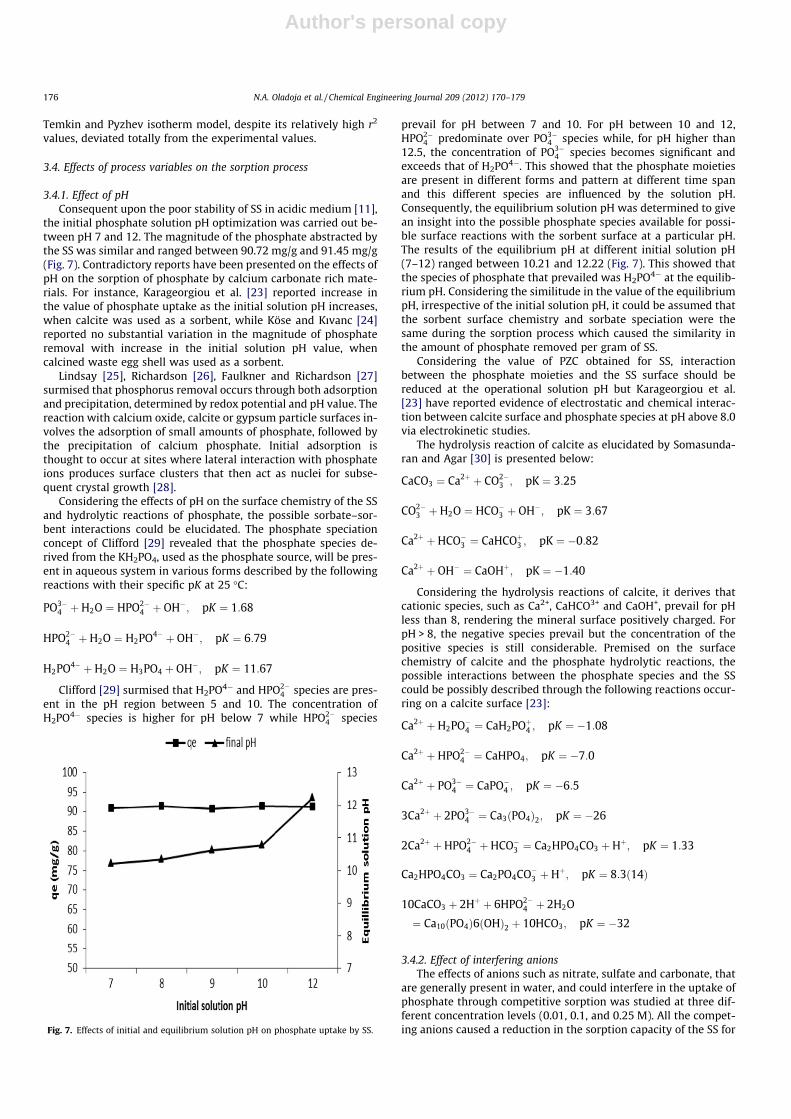
The thermogravimetric profiles of the SS, presented in Fig. 4,shows two major weight losses which represent the moisture(30–110 �C) and volatile matter (110–800 �C) present in the sam-ple. The first weight loss (2.16%) recorded was low as comparedto the second one (46.3384%). The former was assigned to the lossof superficial water while the latter was associated with the loss ofinterstitial water and CO2 in the SS.
3.2. Sorption process kinetic modeling
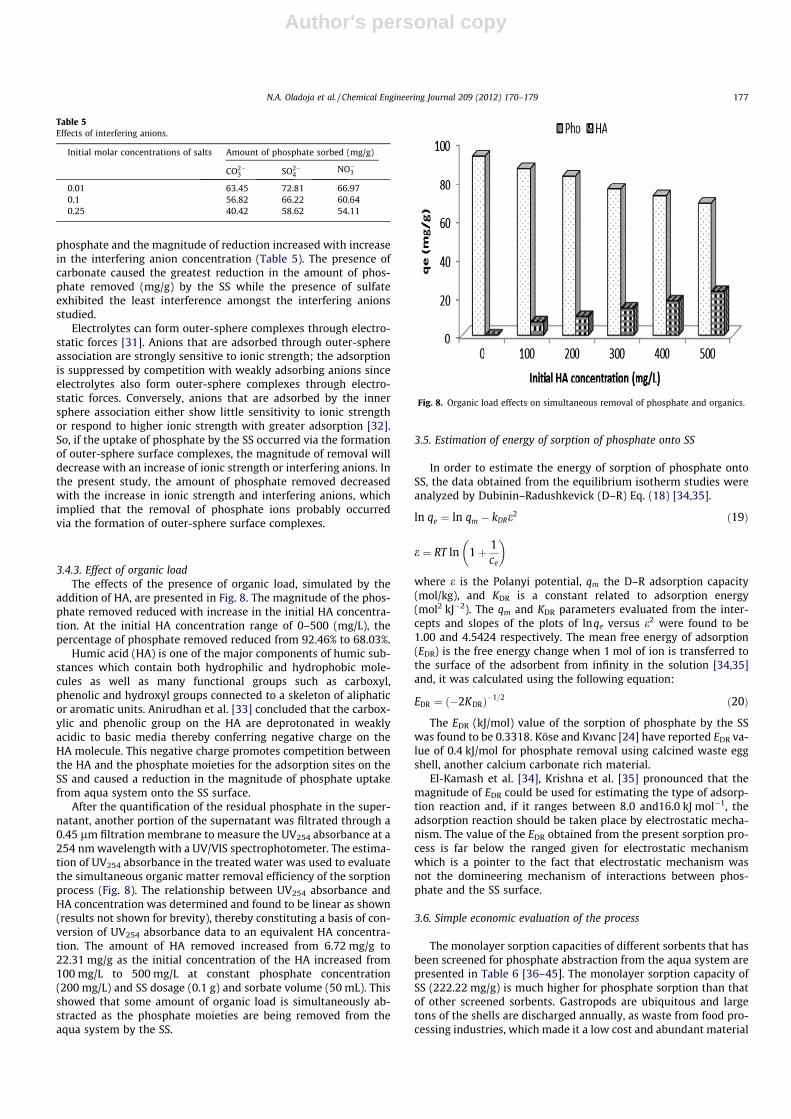
Results of the time–concentration profile of the sorption ofphosphate by the SS, at different initial phosphate concentrations,are presented in Fig. 5. The phosphate removal was fast at theinception of the studies up to the first 5 min and slowly attainedequilibrium within the first 120 min of the sorption process(Fig. 5) for all the concentration (25–300 mg/L) studied. The phos-phate uptake by the SS varied with the initial phosphate concen-tration. Values that ranged between 76% and 92% phosphateremoval were obtained for initial concentration that ranged be-tween 25 and 300 mg/L of phosphate at the first 30 min of thesorption process, whereas only 10–25% of additional removal oc-curred within 90 min of contact time, prior to the state of equilib-rium. The equilibrium adsorption time of 120 min can beconsidered economically favorable operational parameters for theremoval of phosphate via SS sorption. The initial rapid phosphateuptake was presumably due to the availability of bare active siteson the SS, at the inception of the sorption process, whereas the ob-served plateau corresponded to a slow step of adsorption duringwhich there was a saturation of active sites or a gradual diffusionof phosphate inside SS.
In order to understand the behavior of the static sorption pro-cess, phosphate sorption data were analyzed using different kineticmodels.
The uptake of phosphate by the SS is represented thus:
SSþ Pk1
k2SS � P ð1Þ
where k1 is the forward reaction rate constant, k2 the backwardreaction rate constant.
The experimental time–concentration data were analyzed usinga reversible-first-order kinetic model [14,15]. The rate equation forthis process is as follow:
d½P�dt¼ k1½P�t þ k2½SSP�t ð2Þ
The [P] and [SSP] represent the aqueous phosphate concentrationand solid phase (i.e., the SS adsorbed) phosphate concentrationsrespectively at time t. The migration of the phosphate moieties fromthe liquid phase to the SS surface, which caused reduction in theaqueous phosphate concentration, can be represented thus:[SSP]t = [P]0 � [P]t. Consequently, integration of Eq. (2) yields:
Fig. 1. The nitrogen adsorption–desorption curve of the SS.
Fig. 2a. The SEM image of the SS.
Fig. 2b. The EDAX image of the SS.
172 N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179
Author's personal copy
½P�t � ½P�e ¼ ½P�0 � ½P�eexp½�ðk1 þ k2Þt� ð3Þ
where [P]e is the concentration of aqueous phosphate atequilibrium.
Given that, under equilibrium conditions, d[P]/dt = 0, we maydefine an equilibrium constant, K, and rewrite Eq. (2) thus:
K ¼ k1
k2¼ P0 � Pe
Peð4Þ
Rearranging Eq. (4) in terms of k2 and substituting this into Eq.(3) results in the following equation:
k1t ¼ P0 � Pe
P0
� �ln
P0 � Pe
Pt � Pe
� �ð5Þ
Fig. 3. Results of FTIR analysis of the SS.
Fig. 4. The TGA analysis of the SS.
Fig. 5. Time–concentration profile of the sorption of phosphate on SS.
N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179 173

Author's personal copy
The gradient of a plot of the term on the right hand side of Eq. (5)versus time affords an estimate of the first order forward rate con-stant for the reaction, and thence from Eq. (4) an estimate of the re-verse reaction constant may be obtained. The linearity of thereversible first order kinetic model was poor (r2 range: 0.6977–0.8819) which is a pointer to the poor description of the experimen-tal data by this kinetic model (Table 2). The overall reaction rate, K,(min�1) increased from 8.42 to 16.10 with increase in the initialphosphate concentration from 25 mg/L to 300 mg/L while the rateof forward reaction, k1, was faster than that of the backward reac-tion, k2 (min�1), at all the concentrations studied (Table 2).
The system response time (�Iresp), defined as the reciprocal of thesummed rate constants, was used to evaluate concentrationdependence to attain equilibrium.
�Iresp ¼ ðk1 þ k2Þ�1 ð6Þ�Iresp, represents the time required for the system to reach the newequilibrium with varying concentration [15,16]. When the initialphosphate concentration increased from 25 to 300 mg/L, �Iresp wasfound to decrease from 81.30 to 38.68 min (Tables 3a and b). Thisrevealed that the new equilibrium was attained faster at higher ini-tial phosphate concentrations. The tendency of the system for rapidattainment of equilibrium at higher initial phosphate concentrationcould be ascribed to the increase in the driving force of overcomingall mass transfer resistance between the phosphate moieties andthe SS with the increase in the initial phosphate concentration. Con-sequently, an increase in the initial phosphate concentration in-creases the amount of phosphate uptake per gram of the SS.
In order to further elucidate the mechanism of the sorption pro-cess, the inclusion of instantaneous uptake of sorbate as a precursorto the first-order reaction was suggested by Martino et al. [17] viz:
SS0 þ PKD0 SS0 � P; SSþ Pk1
k2SS � P
In this case, sorption sites, SS0, on the SS are immediately occu-pied as SS particles are introduced into the system, an effect that isquantified by an instantaneous distribution coefficient, (KD)0 (mL/g). This constant is calculated from the difference in sorbate aque-ous concentration at the start of the experiment, [P]0, and at the‘‘effective’’ start of the experiment, ½P�00, defined as the first mea-surement in the adsorption time course.
KD0 ¼½P�0 � ½P
0�0½P0�0½m�
ð7Þ
where [m] represents the amount of SS particles in the reactor in g/mL. The value of the instantaneous distribution coefficient, (KD)0
(mL/g), increased with increase in the initial phosphate concentra-tion (Table 2).
The Lagergren pseudo first-order model [18] represented by Eq.(7) and pseudo second order model [19] represented by Eq. (8)were also employed to analyze the time–concentration data.
log½qe � qt� ¼ log½qe� �k1
2:303
� �t ð8Þ
tqt¼ 1
kqeþ 1
qet ð9Þ
where the initial sorption rate h, for the pseudo second order kineticmodel is represented by
h ¼ k2q2e ð10Þ
The results from the fitting of the data obtained from the sorp-tion process into the aforementioned kinetic models are presentedin Table 3a. The values of the pseudo first order equilibrium capac-ity (qe) and the pseudo first order sorption rate increased with in-crease in initial phosphate concentration (Table 4). The results ofthe pseudo second order parameters (Table 3a) also showed thatthe amount of phosphate uptake, (qe), the pseudo second order ratekinetics (k2) and the initial sorption rate (h) increased with in-crease in the initial phosphate concentrations. The qe value in-creased from 11.47 to 140.85 (mg/g), k2 value increased from0.226 to 2.290 (g/(mg min)) while h increased from 2.59 to322.59 (mg/(g min)). The experimental data showed better fittingto the pseudo second order kinetic equations (r2 > 0.99) than thepseudo first order (r2 < 0.80). This is a pointer to the fact that thesorption of the phosphate on the SS occurred via chemisorption.According to Ho and McKay [20] if the sorbate uptake is chemicallyrate controlled the pseudo second-order constants will be indepen-dent of particle diameter and flow rate and will depend on concen-tration of the ions in solution.
Premised on the opinion of Wu et al. [21], the parameter, k2qe
(min�1) was used to define kinetic performance. This parameterwas obtained by rearranging Eq. (10) to give Eq. (11) viz:
dqt
dt¼ k2ðqe � qtÞ ð11Þ
dq tqe
dt¼ k2qe 1� qt
qe
� �2
ð12Þ
Eq. (11) reveals that the time changes in dimensionless solid-phaseconcentration, d(qt/qe)/dt, which is another form of sorption rate, isproportional to the square of the residual amount of sorbate, 1 � (qt
/qe). Consequently, the proportionality constant k2qe can be definedas the second order rate index. Eq. (8) can be rewritten as:
1qt� 1
qe
� �t ¼ 1
k2q2e
ð13Þ
and
Table 2Kinetic parameters of the reversible first order kinetic model analysis of sorption ofphosphate on SS.
Initial conc.(mg/L)
K(min�1)
K1
(min�1)K2
(min�1)�Iresp KD0
(mL/g)R2
25 8.42 0.011 0.0013 81.30 5.08 0.881950 10.74 0.0228 0.0021 40.16 8.01 0.7615
100 7.32 0.0237 0.0032 37.17 8.83 0.6977200 13.12 0.0238 0.0018 39.06 8.27 0.7309300 16.10 0.0257 0.00015 38.68 7.19 0.7181
Table 3aResults of the kinetic parameters of the pseudo first and second order kinetic models analysis of sorption of phosphate on SS.
Initial conc. (mg/L) Pseudo 1st order parameters Pseudo 2nd order parameters
qe1 (mg/g) K1 (g/(mg min)) R2 qe2 (mg/g) K2 (g/(mg min)) h (mg/(g min)) R2
25 3.68 0.012 0.8819 11.47 0.226 2.59 0.997350 4.88 0.025 0.7615 22.88 0.613 14.03 0.9997
100 3.06 0.027 0.6977 44.05 2.671 117.66 1.0000200 6.07 0.026 0.7309 92.59 2.298 212.77 1.0000300 11.10 0.027 0.6977 140.85 2.290 322.55 1.0000
174 N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179

Author's personal copy
t ¼qt
ðqe�qtÞ
k2qeð14Þ
At the half-life of the sorption process (i.e., t = t0.5), we haveqt = 0.5qe and
t0:5 ¼1
k2qeð15Þ
The values for pseudo second order index k2qe, determined forSS–phosphate system are presented in Table 4 and found to in-crease with increase in initial concentration of phosphate. The val-ues of the half-life of the sorption process, presented in Table 3b,reduced with increasing initial concentrations of phosphate insolution. This implied that, as the initial concentration of phos-phate increased in solution, less time is needed to reduce the initialconcentration by half its original value.
3.3. Equilibrium isotherm studies of sorption process
At constant temperature, phosphate held onto the SS will be inequilibrium with phosphate in the aqueous medium. The saturatedmonolayer isotherm can be represented as:
qe ¼qmkace
1þ kaceð16Þ
where Ce is the equilibrium concentration (mg/dm3); qe is theamount of phosphate ion sorbed (mg/g); qm is qe for a completemonolayer (mg/g); Ka is sorption equilibrium constant (dm3/mg).The magnitude of Langmuir constant, Ka, is largely determined bythe heat of sorption. The higher magnitude of Langmuir constant,Ka, the higher heat of sorption and the stronger the bond formed.The monolayer coverage is obtained from a plot of Ce/qe versus Ce.The slope and the intercept of the linear graph obtained from thisplot give the value of qm and Ka. The Langmuir equilibrium adsorp-tion curves relating SS and aqua phase concentration of phosphateat equilibrium is given as:
qe ¼4:644ce
1þ 0:0209ce
The Freundlich isotherm (Eq. (16)) is an empirical equation thatencompasses the heterogeneity of sites and the exponential distri-bution of sites and their energies.
qe ¼ kfc1ne ð17Þ
The Freundlich coefficients n and Kf are obtained from the plots oflnqe versus InCe. The Freundlich equilibrium adsorption curvesrelating the SS and aqua phase concentration of phosphate at equi-librium are given as follows:
qe ¼ 3:697c0:8100e
Temkin and Pyzhev [22] studied the heat of sorption and thesorbate–sorbate interaction. The isotherm assumes that the heatof adsorption of all the molecules in layer decreases linearly withcoverage due to sorbent–sorbate interactions, and that the adsorp-tion is characterized by a uniform distribution of the bonding
energies, up to some maximum binding energy. The Temkin andPyzhev equilibrium adsorption curves relating solid and liquidphase concentration of phosphate at equilibrium is given as:
qe ¼ B lnðktCeÞ ð18Þ
where A (l/g) is the equilibrium binding constant, corresponding tothe maximum binding energy, and constant B is related to the heatof adsorption. A plot of qe versus lnCe enables the determination ofthe isotherm constants B and A from the slope and intercept of thestraight line plot. The Temkin and Pyzhev equilibrium adsorptioncurves relating the solid and liquid phase concentration of phos-phate at equilibrium are given as follows:
qe ¼ 61:712 lnð2:662CeÞ
The parameters obtained from the fitting of the equilibrium iso-therm data to the three isotherm equations are presented in pre-sented in Table 4. The Freundlich isotherm showed the bestcorrelation (r2) amongst the equilibrium isotherm models testedwith the experimental data.
In order to further assess the different isotherms and their cor-relation with experimental results, the theoretical plots obtainedfrom each isotherm analysis is shown with the experimental datafor the sorption of phosphate onto SS in Fig. 6. The graph is plottedin the form of phosphate sorbed per unit mass of SS, qe (mg/g),against the concentration of phosphate remaining in solution, Ce
(mg/L). The Freundlich isotherm models showed the best descrip-tion of the sorption process than the other two isotherm models(Fig. 6). The conformance of the Langmuir isotherm to the experi-mental data was very high at lower concentration (<50 mg/L) butstarted to deviate as the initial phosphate concentration was in-creased (>50 mg/L). The theoretical qe values obtained from the
Table 3bPseudo second order model parameters to determine sorption performance of SS forphosphate removal from aqua system.
2nd orderparameters
25 m/L
50 mg/L 100 mg/L 200 mg/L 300 mg/L
k2qe (min�1) 2.592 14.025 117.658 212.772 322.547t0.5 0.3858 0.0713 0.0085 0.0047 0.0031
Table 4Equilibrium isotherm parameters for the removal of phosphate by SS.
Langmuir isotherm Freundlich isotherm Temkin isotherm
qm = 222.22 n = 0.8100 B = 61.712Ka = 0.0209 Kf = 3.697 A = 2.662r2 = 0.6477 r2 = 0.9907 r2 = 0.9059
Fig. 6. Comparison of isotherm model with experimental values.
N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179 175
Author's personal copy
Temkin and Pyzhev isotherm model, despite its relatively high r2
values, deviated totally from the experimental values.
3.4. Effects of process variables on the sorption process
3.4.1. Effect of pHConsequent upon the poor stability of SS in acidic medium [11],
the initial phosphate solution pH optimization was carried out be-tween pH 7 and 12. The magnitude of the phosphate abstracted bythe SS was similar and ranged between 90.72 mg/g and 91.45 mg/g(Fig. 7). Contradictory reports have been presented on the effects ofpH on the sorption of phosphate by calcium carbonate rich mate-rials. For instance, Karageorgiou et al. [23] reported increase inthe value of phosphate uptake as the initial solution pH increases,when calcite was used as a sorbent, while Köse and Kıvanc [24]reported no substantial variation in the magnitude of phosphateremoval with increase in the initial solution pH value, whencalcined waste egg shell was used as a sorbent.
Lindsay [25], Richardson [26], Faulkner and Richardson [27]surmised that phosphorus removal occurs through both adsorptionand precipitation, determined by redox potential and pH value. Thereaction with calcium oxide, calcite or gypsum particle surfaces in-volves the adsorption of small amounts of phosphate, followed bythe precipitation of calcium phosphate. Initial adsorption isthought to occur at sites where lateral interaction with phosphateions produces surface clusters that then act as nuclei for subse-quent crystal growth [28].
Considering the effects of pH on the surface chemistry of the SSand hydrolytic reactions of phosphate, the possible sorbate–sor-bent interactions could be elucidated. The phosphate speciationconcept of Clifford [29] revealed that the phosphate species de-rived from the KH2PO4, used as the phosphate source, will be pres-ent in aqueous system in various forms described by the followingreactions with their specific pK at 25 �C:
PO3�4 þH2O ¼ HPO2�
4 þ OH�; pK ¼ 1:68
HPO2�4 þH2O ¼ H2PO4� þ OH�; pK ¼ 6:79
H2PO4� þH2O ¼ H3PO4 þ OH�; pK ¼ 11:67
Clifford [29] surmised that H2PO4� and HPO2�4 species are pres-
ent in the pH region between 5 and 10. The concentration ofH2PO4� species is higher for pH below 7 while HPO2�
4 species
prevail for pH between 7 and 10. For pH between 10 and 12,HPO2�
4 predominate over PO3�4 species while, for pH higher than
12.5, the concentration of PO3�4 species becomes significant and
exceeds that of H2PO4�. This showed that the phosphate moietiesare present in different forms and pattern at different time spanand this different species are influenced by the solution pH.Consequently, the equilibrium solution pH was determined to givean insight into the possible phosphate species available for possi-ble surface reactions with the sorbent surface at a particular pH.The results of the equilibrium pH at different initial solution pH(7–12) ranged between 10.21 and 12.22 (Fig. 7). This showed thatthe species of phosphate that prevailed was H2PO4� at the equilib-rium pH. Considering the similitude in the value of the equilibriumpH, irrespective of the initial solution pH, it could be assumed thatthe sorbent surface chemistry and sorbate speciation were thesame during the sorption process which caused the similarity inthe amount of phosphate removed per gram of SS.
Considering the value of PZC obtained for SS, interactionbetween the phosphate moieties and the SS surface should bereduced at the operational solution pH but Karageorgiou et al.[23] have reported evidence of electrostatic and chemical interac-tion between calcite surface and phosphate species at pH above 8.0via electrokinetic studies.
The hydrolysis reaction of calcite as elucidated by Somasunda-ran and Agar [30] is presented below:
CaCO3 ¼ Ca2þ þ CO2�3 ; pK ¼ 3:25
CO2�3 þH2O ¼ HCO�3 þ OH�; pK ¼ 3:67
Ca2þ þHCO�3 ¼ CaHCOþ3 ; pK ¼ �0:82
Ca2þ þ OH� ¼ CaOHþ; pK ¼ �1:40
Considering the hydrolysis reactions of calcite, it derives thatcationic species, such as Ca2+, CaHCO3+ and CaOH+, prevail for pHless than 8, rendering the mineral surface positively charged. ForpH > 8, the negative species prevail but the concentration of thepositive species is still considerable. Premised on the surfacechemistry of calcite and the phosphate hydrolytic reactions, thepossible interactions between the phosphate species and the SScould be possibly described through the following reactions occur-ring on a calcite surface [23]:
Ca2þ þH2PO�4 ¼ CaH2POþ4 ; pK ¼ �1:08
Ca2þ þHPO2�4 ¼ CaHPO4; pK ¼ �7:0
Ca2þ þ PO3�4 ¼ CaPO�4 ; pK ¼ �6:5
3Ca2þ þ 2PO3�4 ¼ Ca3ðPO4Þ2; pK ¼ �26
2Ca2þ þHPO2�4 þHCO�3 ¼ Ca2HPO4CO3 þHþ; pK ¼ 1:33
Ca2HPO4CO3 ¼ Ca2PO4CO�3 þHþ; pK ¼ 8:3ð14Þ
10CaCO3 þ 2Hþ þ 6HPO2�4 þ 2H2O
¼ Ca10ðPO4Þ6ðOHÞ2 þ 10HCO3; pK ¼ �32
3.4.2. Effect of interfering anionsThe effects of anions such as nitrate, sulfate and carbonate, that
are generally present in water, and could interfere in the uptake ofphosphate through competitive sorption was studied at three dif-ferent concentration levels (0.01, 0.1, and 0.25 M). All the compet-ing anions caused a reduction in the sorption capacity of the SS forFig. 7. Effects of initial and equilibrium solution pH on phosphate uptake by SS.
176 N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179
Author's personal copy
phosphate and the magnitude of reduction increased with increasein the interfering anion concentration (Table 5). The presence ofcarbonate caused the greatest reduction in the amount of phos-phate removed (mg/g) by the SS while the presence of sulfateexhibited the least interference amongst the interfering anionsstudied.
Electrolytes can form outer-sphere complexes through electro-static forces [31]. Anions that are adsorbed through outer-sphereassociation are strongly sensitive to ionic strength; the adsorptionis suppressed by competition with weakly adsorbing anions sinceelectrolytes also form outer-sphere complexes through electro-static forces. Conversely, anions that are adsorbed by the innersphere association either show little sensitivity to ionic strengthor respond to higher ionic strength with greater adsorption [32].So, if the uptake of phosphate by the SS occurred via the formationof outer-sphere surface complexes, the magnitude of removal willdecrease with an increase of ionic strength or interfering anions. Inthe present study, the amount of phosphate removed decreasedwith the increase in ionic strength and interfering anions, whichimplied that the removal of phosphate ions probably occurredvia the formation of outer-sphere surface complexes.
3.4.3. Effect of organic loadThe effects of the presence of organic load, simulated by the
addition of HA, are presented in Fig. 8. The magnitude of the phos-phate removed reduced with increase in the initial HA concentra-tion. At the initial HA concentration range of 0–500 (mg/L), thepercentage of phosphate removed reduced from 92.46% to 68.03%.
Humic acid (HA) is one of the major components of humic sub-stances which contain both hydrophilic and hydrophobic mole-cules as well as many functional groups such as carboxyl,phenolic and hydroxyl groups connected to a skeleton of aliphaticor aromatic units. Anirudhan et al. [33] concluded that the carbox-ylic and phenolic group on the HA are deprotonated in weaklyacidic to basic media thereby conferring negative charge on theHA molecule. This negative charge promotes competition betweenthe HA and the phosphate moieties for the adsorption sites on theSS and caused a reduction in the magnitude of phosphate uptakefrom aqua system onto the SS surface.
After the quantification of the residual phosphate in the super-natant, another portion of the supernatant was filtrated through a0.45 lm filtration membrane to measure the UV254 absorbance at a254 nm wavelength with a UV/VIS spectrophotometer. The estima-tion of UV254 absorbance in the treated water was used to evaluatethe simultaneous organic matter removal efficiency of the sorptionprocess (Fig. 8). The relationship between UV254 absorbance andHA concentration was determined and found to be linear as shown(results not shown for brevity), thereby constituting a basis of con-version of UV254 absorbance data to an equivalent HA concentra-tion. The amount of HA removed increased from 6.72 mg/g to22.31 mg/g as the initial concentration of the HA increased from100 mg/L to 500 mg/L at constant phosphate concentration(200 mg/L) and SS dosage (0.1 g) and sorbate volume (50 mL). Thisshowed that some amount of organic load is simultaneously ab-stracted as the phosphate moieties are being removed from theaqua system by the SS.
3.5. Estimation of energy of sorption of phosphate onto SS
In order to estimate the energy of sorption of phosphate ontoSS, the data obtained from the equilibrium isotherm studies wereanalyzed by Dubinin–Radushkevick (D–R) Eq. (18) [34,35].
ln qe ¼ ln qm � kDRe2 ð19Þ
e ¼ RT ln 1þ 1ce
� �
where e is the Polanyi potential, qm the D–R adsorption capacity(mol/kg), and KDR is a constant related to adsorption energy(mol2 kJ�2). The qm and KDR parameters evaluated from the inter-cepts and slopes of the plots of lnqe versus e2 were found to be1.00 and 4.5424 respectively. The mean free energy of adsorption(EDR) is the free energy change when 1 mol of ion is transferred tothe surface of the adsorbent from infinity in the solution [34,35]and, it was calculated using the following equation:
EDR ¼ ð�2KDRÞ�1=2 ð20Þ
The EDR (kJ/mol) value of the sorption of phosphate by the SSwas found to be 0.3318. Köse and Kıvanc [24] have reported EDR va-lue of 0.4 kJ/mol for phosphate removal using calcined waste eggshell, another calcium carbonate rich material.
El-Kamash et al. [34], Krishna et al. [35] pronounced that themagnitude of EDR could be used for estimating the type of adsorp-tion reaction and, if it ranges between 8.0 and16.0 kJ mol�1, theadsorption reaction should be taken place by electrostatic mecha-nism. The value of the EDR obtained from the present sorption pro-cess is far below the ranged given for electrostatic mechanismwhich is a pointer to the fact that electrostatic mechanism wasnot the domineering mechanism of interactions between phos-phate and the SS surface.
3.6. Simple economic evaluation of the process
The monolayer sorption capacities of different sorbents that hasbeen screened for phosphate abstraction from the aqua system arepresented in Table 6 [36–45]. The monolayer sorption capacity ofSS (222.22 mg/g) is much higher for phosphate sorption than thatof other screened sorbents. Gastropods are ubiquitous and largetons of the shells are discharged annually, as waste from food pro-cessing industries, which made it a low cost and abundant material
Table 5Effects of interfering anions.
Initial molar concentrations of salts Amount of phosphate sorbed (mg/g)
CO2�3 SO2�
4NO�3
0.01 63.45 72.81 66.970.1 56.82 66.22 60.640.25 40.42 58.62 54.11
Fig. 8. Organic load effects on simultaneous removal of phosphate and organics.
N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179 177

Author's personal copy
that could be harnessed for phosphate immobilization from aque-ous streams. The only associated cost is possibly the handlingcharges for the collection and transportation, therefore, the totalcost of SS will be extremely low in comparison to those for conven-tional adsorbent. The high cost of conventional adsorbent alsomakes phosphate recovery and sorbent regeneration a must,whereas, the exhausted SS with the adsorbed phosphate could bedried and used as soil conditioner cum fertilizer in agriculturalpractices.
The use of P fertilizers is becoming more expensive and less sus-tainable because phosphate ores are limited non-renewable re-sources, a fact that tends to increase the production costs of Pderivates [46]. Utilizing phosphate laden SS as soil amendmentsmay supply nutrients that could complement and reduce the useof phosphate fertilizers. A number of phosphates laden sorbentshave already shown great fertilizer potential when used as soilamendments in pot cultivation experiments. Studies on iron-richsands and light expanded clay aggregates have shown that phos-phate sorbed to these substrates is available to ryegrass plants[47]. Ochre, a by-product from iron mining, when saturated withphosphate and used as a slow-release fertilizer, has proven to beas effective as conventional phosphate fertilizer for grass and bar-ley crops [48]. The industrial by-product, blast furnace slag effi-ciently improved the yield of barley in another study [49]. Soilamended with Polonite also improved the yield of barley in thatstudy, and induced an increase in available soil phosphate in an-other pot experiment [50]. Cucarella et al. [51] have also proventhat natural calcium-rich materials, such as Polonite, may helpimproving soil condition and fertility. Industrial by-products withhigh concentrations of calcium have shown promising results asliming agents while supplying nutrients for plant uptake [52]. Con-sequent upon the high phosphate sorption capacity of SS and theinherent high calcium content, the phosphate laden SS could beharnessed both as a phosphate fertilizer source and a soilconditioner.
4. Conclusion
SS is a promising mesoporous sorbent for phosphate sequestra-tion from aqua system. Initial solution pH has no influence on thephosphate abstraction but anionic species and organic load reducedthe phosphate uptake. The pseudo second order kinetic model gavethe best description of the sorption process and the systemresponse time (Tresp) showed that equilibrium could be attainedfaster at higher initial phosphate concentration. The instantaneous
distribution coefficient (KD0) increased with increase in initial phos-phate concentration. Freundlich isotherm gave the best descriptionof the sorption process while the Langmuir isotherm models onlyfitted well to the experimental data only at low initial phosphateconcentration. Estimation of the energy of sorption showed thatthe phosphate – SS interaction is not dominated by electrostaticinteractions. The simple economic evaluation of the use of SSas a sorbent showed that the monolayer sorption capacity(222.22 mg/L) was higher than most of the others screened sorbentfor phosphate and that the phosphate laden sorbent could be usedas soil conditioner cum fertilizer in agricultural practices.
Acknowledgment
The Authors wish to acknowledge the efforts of the technicalstaff of the School of Chemical Engineering, Universiti Sains Malay-sia, Malaysia for the characterization of the SS.
References
[1] Chemical Market Reporter, 2006. Chemical Prices for Week Ending, ChemicalMarket Reporter, <http://www.icis.com/StaticPages/a-e.htm>, August 28,2006.
[2] T. Clark, T. Stephenson, P.A. Pearce, Water Res. 31 (10) (1997) 2557.[3] D.R. Kioussis, F.W. Wheaton, P. Kofinas, Phosphate polymeric hydrogels for
aquaculture wastewater remediation, Aquacul. Eng. 19 (1999) 163–178.[4] L.M. Blaney, S. Cinar, A.K. SenGupta, Hybrid anion exchanger for trace
phosphate removal from water and wastewater, Water Res. 41 (2007) 1603–1613.
[5] A. Genz, A. Kornmüller, M. Jekel, Advanced phosphorus removal frommembrane filtrates by adsorption on activated aluminium oxide andgranulated ferric hydroxide, Water Res. 38 (2004) 3523–3530.
[6] P. L Sibrella, G.A. Montgomery, K.L. Ritenoura, T.W. Tucker, Removal ofphosphorus from agricultural wastewaters using adsorption media preparedfrom acid mine drainage sludge, Water Res. 43 (2009) 2240–2250.
[7] A. Drizo, C. Forget, R.P. Chapuis, Y. Comeau, Phosphorus removal by electric arcfurnace steel slag and serpentine, Water Res. 40 (2006) 1547–1554.
[8] J.P. Gustafsson, A. Renman, G. Renman, K. Poll, Phosphate removal by mineral-based sorbents used in filters for smallscale wastewater treatment, Water Res.42 (2008) 189–197.
[9] K.C. Makris, H. El-Shall, W.G. Harris, G.A. O’Connor, T.A. Obreza, Intraparticlephosphorus diffusion in a drinking water treatment residual at roomtemperature, J. Colloid Interface Sci. 277 (2004) 417–423.
[10] S.E. O’Reilly, J.T. Sims, Phosphorus adsorption and desorption in a sandy soilamended with high rates of coal fly ash, Commun. Soil Sci. Plant Anal. 26 (17–18) (1995) 2983–2993.
[11] N.A. Oladoja, Y.D. Aliu, Snail shell as coagulant aid in the alum precipitation ofmalachite green from aqua system, J. Hazard. Mater. 164 (2009) 1494–1502.
[12] N.A. Oladoja, Y.D. Aliu, A.E. Ofomaja, Evaluation of snail shell as coagulant aidin the alum precipitation of aniline blue from aqua system, Environ. Technol.32 (6) (2011) 639–652.
[13] IUPAC Manual of Symbols and Terminology of Colloid Surface, Butterworths,London, 1982, pp. 1.
[14] J.M. Garnier, M.K. Pham, P. Ciffroy, J.M. Martin, Kinetics of trace metalcomplexation with suspended matter and with filterable ligands in freshwater,Environ. Sci. Technol. 31 (1997) 1597–1606.
[15] A. Turner, M. Crussel, G. Millward, A.C. Garcia, A. Fischer, Adsorption kinetics ofplatinum group elements in river water, Environ. Sci. Technol. 40 (2006)1524–1531.
[16] T.S. Anirudhan, S. Rijith, Glutaraldehyde cross-linked epoxyaminated chitosanas an adsorbent for the removal and recovery of copper (II) from aqueousmedia, Colloids Surf. A: Physicochem. Eng. Aspects 351 (2009) 52–59.
[17] M. Martino, A. Turner, G.E. Millward, Influence of organic complexation on theadsorption kinetics of nickel in river waters, Environ. Sci. Technol. 37 (2003)2383–2388.
[18] S. Lagergren, About the theory of so-called adsorption of soluble substances, K.Sven. Vetenskapsakad. Handl. Band. 24 (1898) 1–39.
[19] G. McKay, The adsorption of basic dye onto silica from aqueous solution–soliddiffusion model, Chem. Eng. Sci. 39 (1) (1984) 129–138.
[20] Y.S. Ho, G. Mckay, Sorption of dye from aqueous solution by peat, Chem. Eng. J.70 (1998) 115–124.
[21] F.C. Wu, R.L. Tseng, R.S. Juang, Initial behavior of intraparticle diffusion modelused in the description of adsorption kinetics, Chem. Eng. J. 153 (2009) 1–8.
[22] M.I. Temkin, V. Pyzhev, Acta Physiochim. URSS 12 (1940) 327.[23] K. Karageorgiou, M. Paschalis, G.N. Anastassakis, Removal of phosphate species
from solution by adsorption onto calcite used as natural adsorbent, J. Hazard.Mater. A139 (2007) 447–452.
[24] T.E. Köse, B. Kıvanc, Adsorption of phosphate from aqueous solutions usingcalcined waste eggshell, Chem. Eng. J. 178 (2011) 34–39.
Table 6Comparison of sorption capacity of phosphate on various adsorbents.
Sorbent Sorption capacity (qmax mg/g)
Source
SS 222.22 Presentstudies
Natural palygorskite 4 [36]Layered double hydroxides 42.4 [37]Akaganéite 59.6 [38]Fe-oxides-coated quartz sand 0.62 [39]Al-oxides-coated quartz sand 0.54 [40]Calcite 1.9 [23]Calcined metal hydroxide
sludge16.48 [41]
Date palm fibers 4.35 [42]Steel-making slag 215.7 [43]Coal fly ash treated with
NaOH57.1 [44]
Zeolite synthesized from flyash
40.7 [45]
Aluminum oxide hydroxide 45 [46]
178 N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179

Author's personal copy
[25] W.L. Lindsay, Phosphate. Chemical Equilibria in Soils, JohnWiley & Sons, Inc.,1979. pp. 162–209.
[26] C.J. Richardson, Mechanisms controlling P retention capacity in fresh waterwetlands, Science 228 (1985) 1424.
[27] S.P. Faulkner, B.C. Richardson, Physical and chemical characteristic offreshwater wetlands, in: D.A. Hammer (Ed.), Constructed Wetlands forWastewater Treatment: Municipal, Industrial and Agricultural, LewisPublishers, Chelsea, 1989, pp. 41–72.
[28] Y. H Moon, J.G. Kim, J.S. Ahn, G.H. Lee, Hi-Soo Moon, Phosphate removal usingsludge from fuller’s earth production, J. Hazard. Mater. 143 (2007) 41–48.
[29] A.F. Clifford, Inorganic Chemistry of Quantitative Analysis, Prentice Hall, NewJersey, 1961.
[30] P. Somasundaran, G.E. Agar, The zero point of charge of calcite, J. ColloidInterface Sci. 24 (1967) 433–440.
[31] S. Fendorf, M.J. Eick, P. Grossl, D.L. Sparks, Arsenate and chromate retentionmechanisms on goethite. 1. Surface structure, Environ. Sci. Technol. 31 (1997)315–319.
[32] T.H. Hsia, S.L. Lo, C.F. Lin, D.Y. Lee, Characterization of arsenate adsorption onhydrous iron-oxide using chemical and physical methods, Colloid Surf. A 85(1994) 1–7.
[33] T.S. Anirudhan, P.S. Suchithra, S. Rijith, Amine-modified polyacrylamidebentonite composite for the adsorption of humic acid in aqueous solutions,Colloids Surf. A Physicochem. Eng. Asp. 326 (2008) 147–156.
[34] A.M. El-Kamash, A.A. Zaki, M.A. El Geleel, Modeling batch kinetics andthermodynamics of zinc and cadmium ions removal from waste solutionsusing synthetic zeolite A, J. Hazard. Mater. 127 (2005) 211–220.
[35] B.S. Krishna, D.S.R. Murty, B.S. Jai Prakash, Thermodynamics of chromium(VI)anionic species sorption onto surfactant-modified montmorillonite clay, J.Colloid Interface Sci. 229 (2000) 230–236.
[36] H. Ye, F. Chen, Y. Sheng, G. Sheng, J. Fu, Adsorption of phosphate from aqueoussolution onto modified palygorskites, Sep. Purif. Technol. 50 (2006) 283–290.
[37] J. Das, B.S. Patra, N. Baliarsingh, K.M. Parida, Adsorption of phosphate bylayered double hydroxides in aqueous solutions, Appl. Clay Sci. 32 (2006) 252–260.
[38] E.A. Deliyanni, E.N. Peleka, N.K. Lazaridis, Comparative study of phosphatesremoval from aqueous solutions by nanocrystalline akaganéite and hybridsurfactant-akaganéite, Sep. Purif. Technol. 52 (2007) 478–486.
[39] L. Zeng, X. Li, J. Liu, Adsorptive removal of phosphate from aqueous solutionsusing iron oxide tailings, Water Res. 38 (2004) 1318–1326.
[40] A.K. Golder, A.N. Samanta, S. Ray, Removal of phosphate from aqueoussolutions using calcined metal hydroxides sludge waste generated fromelectrocoagulation, Sep. Purif. Technol. 52 (2006) 102–109.
[41] K. Riahia, B. Ben Thayer, A. Ben Mammoub, A. Ben Ammarc, M.H. Jaafoura,Biosorption characteristics of phosphates from aqueous solution onto Phoenixdactylifera L. date palm fibers, J. Hazard. Mater. A 170 (2009) 511–519.
[42] V.K. Jha, Y. Kameshima, A. Nakajima, K. Okada, Utilization of steel-making slagfor the uptake of ammonium and phosphate ions from aqueous solution, J.Hazard. Mater. 156 (2008) 156–162.
[43] P. Pengthamkeerati, T. Satapanajaru, P. Chularuengoaksorn, Chemicalmodification of coal fly ash for the removal of phosphate from aqueoussolution, Fuel 87 (2008) 2469–2476.
[44] J.G. Chen, H.N. Kong, D.Y. Wu, Z.B. Hu, Z.S. Wang, Y.H. Wang, Removal ofphosphate from aqueous solution by zeolite synthesized from fly ash, J. ColloidInterface Sci. 300 (2006) 491–497.
[45] S. Tanada, M. Kabayama, N. Kawasaki, T. Sakiyama, T. Nakamura, M. Araki, T.Tamura, Removal of phosphate by aluminum oxide hydroxide, J. ColloidInterface Sci. 257 (2003) 135–140.
[46] J. Driver, D. Lijmbach, I. Steen, Why recover phosphorus for recycling, andhow?, Environ Technol. 20 (7) (1999) 651–662.
[47] M.E. Kvarnstrom, A.L. Morel, T. Krogstad, Plant availability of phosphorus infilter substrates derived from small-scale wastewater treatment systems, Ecol.Eng. 22 (2004) 1–15.
[48] K.E. Dobbie, K.V. Heal, K.A. Smith, Assessing the performance of phosphorus –saturated ochre as a fertilizer and its environmental acceptability, Soil UseManage. 21 (2) (2005) 231–239.
[49] L. Hylander, A. Kietlinska, G. Renman, G. Sima, Phosphorus retention in filtermaterials for wastewater treatment and its subsequent suitability for plantproduction, Bioresour. Technol. 97 (2006) 914–921.
[50] V. Cucarella, T. Zaleski, R. Mazurek, G. Renman, Fertilizer potential of calcium-rich substrates used for phosphorus removal from wastewater, Polish J.Environ. Stud. 16 (6) (2007) 817–822.
[51] V. Cucarella, T. Zaleski, R. Mazurek, G. Renman, Effect of reactive substratesused for the removal of phosphorus from wastewater on the fertility of acidsoils, Bioresour. Technol. 99 (2008) 4308–4314.
[52] V. Cucarella, R. Mazurek, T. Zaleski, M. Kopec, G. Renman, Effect of poloniteused for phosphorus removal from wastewater on soil properties and fertilityof a mountain meadow, Environ. Pollut. 157 (2009) 2147–2152.
N.A. Oladoja et al. / Chemical Engineering Journal 209 (2012) 170–179 179




















