
Lipoprotein Lipase Gene Polymorphisms and the Risk of Target Vessel Revascularization After...
8
Polymorphisms and Coronary Disease Lipoprotein Lipase Gene Polymorphisms and the Risk of Target Vessel Revascularization After Percutaneous Coronary Intervention Pascalle S. Monraats, MSC,*† Jamal S. Rana, MD,‡ Melchior C. Nierman, MD,‡ Nuno M. M. Pires, MSC,*§ Aeilko H. Zwinderman, PHD, John J. P. Kastelein, MD, PHD,‡ Jan Albert Kuivenhoven, PHD,‡ Moniek P. M. de Maat, PHD,¶ Saskia Z. H. Rittersma, MD# Abbey Schepers, MD,§** Pieter A. F. Doevendans, MD, PHD,†† Robbert J. de Winter, MD, PHD,# René A. Tio, MD, PHD,‡‡ Rune R. Frants, PHD,§§ Paul H. A. Quax, PHD,§** Arnoud van Der Laarse, PHD,* Ernst E. van Der Wall, MD, PHD,* J. Wouter Jukema, MD, PHD*† Leiden, Utrecht, Amsterdam, Rotterdam, and Groningen, the Netherlands OBJECTIVES We sought to identify polymorphisms in genes that predispose to restenosis. BACKGROUND Variations in the lipoprotein lipase (LPL) gene have been implicated in a number of pathophysiologic conditions associated with coronary heart disease. The present study examines the impact of polymorphisms in the LPL gene on restenosis (defined by target vessel revascularization [TVR]) in a large patient population undergoing percutaneous coronary intervention (PCI). A mouse model for restenosis was used to further investigate LPL’s role in restenosis. METHODS The GENetic DEterminants of Restenosis (GENDER) project is a multicenter, prospective study design that enrolled 3,104 consecutive patients after successful PCI. These patients were genotyped for four different LPL gene polymorphisms. In apolipoprotein E (ApoE)*3- Leiden transgenic mice, arterial messenger ribonucleic acid (mRNA) was used to assess LPL expression during a cuff-induced restenotic process. RESULTS Using multivariable analysis, carriers of the 447Ter allele of the LPL enzyme showed a lower risk of TVR compared with 447Ser homozygotes (p 0.005). In the mouse model, LPL mRNA levels were increased 40-fold compared with control arteries at 6 h after cuff placement. CONCLUSIONS The LPL C/G polymorphism (Ser447Ter), resulting in a truncation of the two C-terminal amino acids of the mature LPL protein, appears to be an important protective factor for TVR in humans. The role of LPL in this process was further established in a mouse model, where LPL expression was very strongly up-regulated in the target arterial wall, suggesting a contribution of this lipolytic enzyme to restenosis. Possibly, LPL Ser447Ter genotyping may lead to better risk stratification and tailored therapy in the prevention of restenosis after PCI. (J Am Coll Cardiol 2005;46:1093–100) © 2005 by the American College of Cardiology Foundation Lipoprotein lipase (LPL) is the rate-limiting enzyme in the lipolysis of plasma triglyceride-rich lipoproteins in the circula- tion. In adulthood, it is synthesized in parenchymal cells of adipose tissue, as well as in skeletal and cardiac muscle, followed by transfer to heparin sulfate-binding sites at the vascular side of the endothelium (1). The hydrolytic function of LPL is essential for the processing of triglyceride-rich chylo- microns and very-low-density lipoproteins (VLDLs) to rem- nant particles and also for the transfer of phospholipids and apolipoproteins to high-density lipoprotein (HDL). Further- more, LPL plays a key role in the receptor-mediated removal of lipoproteins from the circulation (2). The gene coding for LPL, located on chromosome 8p22, encompasses 10 exons and is rather polymorphic (3). Abnormal LPL function has been reported to be associated with a number of pathophysi- ologic conditions that underlie coronary heart disease (4,5). In From the *Department of Cardiology, Leiden University Medical Center, Leiden; †Interuniversity Cardiology Institute of the Netherlands (ICIN), Utrecht; ‡Depart- ment of Vascular Medicine, Academic Medical Center, Amsterdam; §Gaubius Laboratory, TNO-Quality of Life, Leiden; Department of Medical Statistics, Academic Medical Center, Amsterdam; ¶Department of Hematology, Erasmus University Medical Center, Rotterdam; #Department of Cardiology, Academic Medical Center, Amsterdam; **Department of Surgery, Leiden University Medical Center, Leiden; ††Department of Cardiology, University Medical Center, Utrecht; ‡‡Department of Cardiology, Academic Hospital, Groningen; and the §§Depart- ment of Human Genetics, Center for Human and Clinical Genetics, Leiden University Medical Center, Leiden, the Netherlands. The study sponsor had no role in the design or conduct of the study, the writing of this report, or the decision to submit it for publication. Dr. Monraats is supported by grant 99.210 from the Netherlands Heart Foundation and a grant from the Interuniversity Cardiology Institute of the Netherlands (ICIN). Dr. Quax (Established Investigator) and Dr. Schepers are supported by the Molecular Cardiology Program of the Netherlands Heart Foundation (M 93.001). Dr. Jukema is an Established Clinical Investigator of the Netherlands Heart Foundation (2001 D 032). Manuscript received April 25, 2005; revised manuscript received May 20, 2005, accepted May 24, 2005. Journal of the American College of Cardiology Vol. 46, No. 6, 2005 © 2005 by the American College of Cardiology Foundation ISSN 0735-1097/05/$30.00 Published by Elsevier Inc. doi:10.1016/j.jacc.2005.05.071
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Lipoprotein Lipase Gene Polymorphisms and the Risk of Target Vessel Revascularization After...

LaAPNJARAL
LltafvLm
†mLAUMC‡m
Journal of the American College of Cardiology Vol. 46, No. 6, 2005© 2005 by the American College of Cardiology Foundation ISSN 0735-1097/05/$30.00P
Polymorphisms and Coronary Disease
ipoprotein Lipase Gene Polymorphismsnd the Risk of Target Vessel Revascularizationfter Percutaneous Coronary Intervention
ascalle S. Monraats, MSC,*† Jamal S. Rana, MD,‡ Melchior C. Nierman, MD,‡uno M. M. Pires, MSC,*§ Aeilko H. Zwinderman, PHD,� John J. P. Kastelein, MD, PHD,‡
an Albert Kuivenhoven, PHD,‡ Moniek P. M. de Maat, PHD,¶ Saskia Z. H. Rittersma, MD#bbey Schepers, MD,§** Pieter A. F. Doevendans, MD, PHD,†† Robbert J. de Winter, MD, PHD,#ené A. Tio, MD, PHD,‡‡ Rune R. Frants, PHD,§§ Paul H. A. Quax, PHD,§**rnoud van Der Laarse, PHD,* Ernst E. van Der Wall, MD, PHD,* J. Wouter Jukema, MD, PHD*†eiden, Utrecht, Amsterdam, Rotterdam, and Groningen, the Netherlands
OBJECTIVES We sought to identify polymorphisms in genes that predispose to restenosis.BACKGROUND Variations in the lipoprotein lipase (LPL) gene have been implicated in a number of
pathophysiologic conditions associated with coronary heart disease. The present studyexamines the impact of polymorphisms in the LPL gene on restenosis (defined by targetvessel revascularization [TVR]) in a large patient population undergoing percutaneouscoronary intervention (PCI). A mouse model for restenosis was used to further investigateLPL’s role in restenosis.
METHODS The GENetic DEterminants of Restenosis (GENDER) project is a multicenter, prospectivestudy design that enrolled 3,104 consecutive patients after successful PCI. These patientswere genotyped for four different LPL gene polymorphisms. In apolipoprotein E (ApoE)*3-Leiden transgenic mice, arterial messenger ribonucleic acid (mRNA) was used to assess LPLexpression during a cuff-induced restenotic process.
RESULTS Using multivariable analysis, carriers of the 447Ter allele of the LPL enzyme showed a lowerrisk of TVR compared with 447Ser homozygotes (p � 0.005). In the mouse model, LPLmRNA levels were increased 40-fold compared with control arteries at 6 h after cuffplacement.
CONCLUSIONS The LPL C/G polymorphism (Ser447Ter), resulting in a truncation of the two C-terminalamino acids of the mature LPL protein, appears to be an important protective factor for TVRin humans. The role of LPL in this process was further established in a mouse model, whereLPL expression was very strongly up-regulated in the target arterial wall, suggesting acontribution of this lipolytic enzyme to restenosis. Possibly, LPL Ser447Ter genotyping maylead to better risk stratification and tailored therapy in the prevention of restenosis afterPCI. (J Am Coll Cardiol 2005;46:1093–100) © 2005 by the American College of
ublished by Elsevier Inc. doi:10.1016/j.jacc.2005.05.071
Cardiology Foundation
namoLabo
UisNISHt
ipoprotein lipase (LPL) is the rate-limiting enzyme in theipolysis of plasma triglyceride-rich lipoproteins in the circula-ion. In adulthood, it is synthesized in parenchymal cells ofdipose tissue, as well as in skeletal and cardiac muscle,ollowed by transfer to heparin sulfate-binding sites at theascular side of the endothelium (1). The hydrolytic function ofPL is essential for the processing of triglyceride-rich chylo-icrons and very-low-density lipoproteins (VLDLs) to rem-
From the *Department of Cardiology, Leiden University Medical Center, Leiden;Interuniversity Cardiology Institute of the Netherlands (ICIN), Utrecht; ‡Depart-ent of Vascular Medicine, Academic Medical Center, Amsterdam; §Gaubiusaboratory, TNO-Quality of Life, Leiden; �Department of Medical Statistics,cademic Medical Center, Amsterdam; ¶Department of Hematology, Erasmusniversity Medical Center, Rotterdam; #Department of Cardiology, Academicedical Center, Amsterdam; **Department of Surgery, Leiden University Medicalenter, Leiden; ††Department of Cardiology, University Medical Center, Utrecht;
‡Department of Cardiology, Academic Hospital, Groningen; and the §§Depart-ent of Human Genetics, Center for Human and Clinical Genetics, Leiden aant particles and also for the transfer of phospholipids andpolipoproteins to high-density lipoprotein (HDL). Further-ore, LPL plays a key role in the receptor-mediated removal
f lipoproteins from the circulation (2). The gene coding forPL, located on chromosome 8p22, encompasses 10 exonsnd is rather polymorphic (3). Abnormal LPL function haseen reported to be associated with a number of pathophysi-logic conditions that underlie coronary heart disease (4,5). In
niversity Medical Center, Leiden, the Netherlands. The study sponsor had no rolen the design or conduct of the study, the writing of this report, or the decision toubmit it for publication. Dr. Monraats is supported by grant 99.210 from theetherlands Heart Foundation and a grant from the Interuniversity Cardiology
nstitute of the Netherlands (ICIN). Dr. Quax (Established Investigator) and Dr.chepers are supported by the Molecular Cardiology Program of the Netherlandseart Foundation (M 93.001). Dr. Jukema is an Established Clinical Investigator of
he Netherlands Heart Foundation (2001 D 032).
Manuscript received April 25, 2005; revised manuscript received May 20, 2005,ccepted May 24, 2005.

ltti
tdefcst
rpckAtrrri
M
SdnscwePc4tM
HtcpTm
rrigoetsFnPtbcic
fslcctcGlL9tw(spbdecpsltptb
wi(nLvtos(
1094 Monraats et al. JACC Vol. 46, No. 6, 2005LPL Ser447Ter Polymorphism and TVR September 20, 2005:1093–100
ine, changes in LPL gene expression, or amino acid substitu-ions as a result of point mutation in the LPL gene, affectriglyceride and HDL cholesterol levels, which in turn aremplicated in atherosclerotic risk (6,7).
Percutaneous coronary intervention (PCI), an importantreatment for patients with atherosclerosis, is limited by theevelopment of restenosis, despite the advent of drug-luting stents. There is increasing evidence that inheritedactors may partly explain the excessive risk of restenosis inertain patients. Identifying such patients may improvetratification of patients to a more individually tailoredreatment (8,9).
To our knowledge, the role of LPL polymorphisms inestenosis has thus far not been investigated. Therefore, theurpose of the current study was to evaluate in a largeonsecutive study population whether four different well-nown variants in the LPL gene, denoted as �93T/G,sp9Asn, Asn291Ser, and Ser447Ter, have predictive value
oward the risk of restenosis (defined by target vesselevascularization [TVR]) after PCI. To further establish theole of LPL in restenosis, we quantified LPL messengeribonucleic acid (mRNA) expression in the restenotic vesseln an established mouse model for restenosis (10–12).
ETHODS
tudy design. The present study population has beenescribed previously (13). In brief, the GENetic DEtermi-ants of Restenosis (GENDER) project was designed totudy the association between genetic polymorphisms andlinical restenosis. Patients were eligible for inclusion if theyere successfully treated for stable angina, non–ST-segment
levation acute coronary syndromes, or silent ischemia byCI. Patients treated for acute ST-segment elevation myo-ardial infarction were excluded. All patients were treated inof the 13 referral centers for interventional cardiology in
he Netherlands. The overall inclusion period lasted fromarch 1999 until June 2001.The study protocol conforms to the Declaration ofelsinki and was approved by the Medical Ethics Commit-
ee of each participating institution. Written, informedonsent was obtained from each participant before the PCIrocedure.he PCI procedure. Standard angioplasty and stent place-
Abbreviations and AcronymsCABG � coronary artery bypass graft surgeryHDL � high-density lipoproteinLDL � low-density lipoproteinLPL � lipoprotein lipaseNO � nitric oxidePCI � percutaneous coronary interventionPCR � polymerase chain reactionTVR � target vessel revascularizationVLDL � very-low-density lipoprotein
ent were performed by experienced operators using a d
adial or femoral approach. Before the procedure, patientseceived 300 mg aspirin and 7,500 IU heparin. The use ofntracoronary stents and additional medication, such aslycoprotein IIb/IIIa inhibitors, was at the discretion of theperator. In case of stent implantation, patients receivedither ticlopidine or clopidogrel for at least one month afterhe procedure, depending on local practice. During thetudy, no drug-eluting stents were used.ollow-up and study end points. Follow-up lasted at leastine months, except when a coronary event occurred.atients were either seen in the outpatient clinic or con-
acted by telephone. Target vessel revascularization, eithery PCI or coronary artery bypass graft surgery (CABG), wasonsidered as restenosis and was our primary end point. Anndependent Clinical Events Committee adjudicated thelinical events.
Events occurring within the first month were excludedrom the analysis, as these events were attributable mainly toubacute stent thrombosis or occluding dissections, and lessikely to restenosis. Data were collected with standardizedase-report forms that were completed by the researchoordinator at each site, who was blinded to the genotype ofhe patients. Representatives from the data-coordinatingenter monitored all sites.
enotyping. Blood was collected in EDTA tubes at base-ine, and DNA was extracted after standard procedures. ThePL G/A, A/G, and C/G polymorphisms in exons 2, 6, and, respectively, resulting in the following amino acid substitu-ions—Asp9Asn, Asn291Ser, and Ser447Ter, respectively—ere determined by validated multilocus genotyping assay
Roche Molecular Systems, Alameda, Califormia) (14). Aimilar method was used to detect the LPL �93T/G promoterolymorphism. All four polymorphisms were selected on theasis of their previously described relation to coronary arteryisease and/or their influence on LPL activity (14,15). In short,ach DNA sample was amplified in a multiple polymerasehain reaction (PCR) using biotinylated primers. The PCRroduct pool was then hybridized to a matching panel ofequence-specific oligonucleotide probes immobilized in ainear array on nylon membrane strips. A colorimetric detec-ion method based on incubation with streptavidin-horseradisheroxidase conjugate, using hydrogen peroxide and 3,3=,5,5=-etramethylbenzidine as substrates, was used. Operatorslinded to restenosis status performed genotyping.
To confirm genotype assignments, the PCR procedureas performed in replicate on 10% of the samples. Two
ndependent observers carried out scoring. Disagreements�1%) were resolved by further joint reading, and whenecessary, genotyping was repeated.ipid analysis. To study the effect of the different LPLariants on lipid levels, we measured plasma triglycerides,otal cholesterol, and HDL cholesterol in a subpopulationf patients. Cholesterol and triglyceride concentrations inerum were measured with a fully automated Hitachi 747Hitachi, Tokyo, Japan). The HDL cholesterol level was
etermined by a turbidimetric assay on a Hitachi 911.
LidpAtmMeeSwamtNsTFwpTsa(tW
AACuBgctdsbawStStWwmaqnnf
pt
Nacpo
aiahwT
fsaewtl
Dvt
R
Pp(rettAD3pga
Wpe
t9acgagFem
1095JACC Vol. 46, No. 6, 2005 Monraats et al.September 20, 2005:1093–100 LPL Ser447Ter Polymorphism and TVR
ow-density lipoprotein cholesterol was calculated accord-ng to the equation of Friedewald et al. (16). Blood wasrawn before the PCI procedure. Two of the four partici-ating centers (Leiden University Medical Center andcademic Hospital Maastricht) systematically collected ex-
ra blood samples to perform additional laboratory measure-ents to examine other predictors of restenosis.ouse model of restenosis. We further studied LPL gene
xpression during the development of restenosis in anstablished mouse model for diet-induced atherosclerosis.pecifically, we analyzed LPL mRNA levels in the vesselall of apolipoprotein E (ApoE)*3-Leiden transgenic mice
fter cuff placement (10–12). Before cuff placement, theice were fed a Western-type diet containing 1% choles-
erol and 0.05% cholate (Hope Farms, Woerden, theetherlands) three weeks before surgery and continued after
urgery in order to obtain stable plasma cholesterol levels.his diet results in a human-like lipoprotein profile (10).emoral arteries, either cuffed or non-cuffed sham-operated,ere pooled (two arteries per sample, two samples per timeoint), and total RNA was isolated per time point using therizol protocol (Invitrogen, Breda, the Netherlands). Sub-
equently, cDNA synthesis of all RNA samples waschieved using Ready-To-Go real-time (RT)-PCR beadsAmersham Biosciences, Uppsala, Sweden). All experimen-al procedures in mice were approved by the Animal
elfare Committee of TNO-PG, Leiden, the Netherlands.Intron-spanning primers (forward: 5=GTGGCCGAG-
GCGAGAAC-3=, reverse:5=TCCACCTCCGTGTAA-TCAAGA-3=) and probe (5=TTCCCTTCACCCTGC-CGAGGTT-3=) for mouse LPL gene were designedsing Primer Express 1.5 software (Perkin-Elmer Appliediosystems, Foster City, California). The housekeepingenes—hypoxanthine-guanine phosphoribosyl transferase,yclophilin, and GAPDH—were used as controls. Real-ime PCR was performed on an ABI Prism 7700 sequenceetection system (Perkin Elmer Biosystems, Boston, Mas-achusetts). Cycle conditions were: 50°C for 2 min, followedy 10 min at 95°C, amplification phase of 45 cycles of 15 st 95°C, followed by 1 min at 60°C. The RT-PCR analysisas performed using RT-PCR mastermix (Eurogentec,eraing, Belgium). Aqua-dest was incorporated as a nega-ive control.tatistical methodology. Deviations of the genotype dis-
ribution from that expected for a population in Hardy-einberg equilibrium was tested using the chi-square test
ith one degree of freedom. Allele frequencies were deter-ined by counting; the 95% confidence intervals of the
llele frequencies were calculated from sample allele fre-uencies, based on the approximation of the binomial andormal distributions in large sample sizes. Polymorphismsot in Hardy-Weinberg equilibrium were excluded fromurther analysis.
In the first stage, the association between each LPLolymorphism and TVR was assessed using a Cox propor-
ional regression model under a co-dominant genetic model. Lo adjustment for co-variates was performed at this stage tollow for the assessment of their possible involvement in theausal pathway. If �10 patients were homozygous for aarticular allele, two groups were formed with the absencer presence of that allele as group variable.All polymorphisms were also assessed using dominant
nd recessive models, and the model with the lowest Akaikenformation criterion was used in multivariable regressionnalysis (17). The LPL polymorphisms were combined intoaplotypes, and the effect of haplotypes on restenosis riskas estimated according to the methods developed byanck et al. (18).Multivariable regression analysis of TVR risk was per-
ormed on all polymorphisms using a stepwise backwardelection algorithm. In the final step, clinical variablesssociated with TVR or associated with genotype werentered into the regression model. The Kruskal-Wallis testas used to examine the association of the different geno-
ypes with concentrations of HDL cholesterol, low-densityipoprotein (LDL) cholesterol, and triglyceride levels.
Animal data are presented as the mean value � SEM.ata were analyzed using the Mann-Whitney U test. A p
alue �0.05 was considered statistically significant. Statis-ical analysis was carried out using SPSS version 11.5.
ESULTS
atient characteristics. A total of 3,146 patients had com-lete follow-up (99.3%), with a median duration of 9.6 monthsinterquartile range 3.9). A total number of 42 patients expe-ienced an event in the first 30 days and were thereforexcluded from further analysis, according to the protocol. Inhe remaining 3,104 patients, we assessed the frequencies ofhe following LPL polymorphisms; �93T/G, Asp9Asn (G/), Asn291Ser (A/G), and Ser447Ter (C/G). SuccessfulNA genotyping was possible in 3,028, 3,031, 3,021, and
,054 patients, respectively. The results of the remainingatients were missing due to a lack of DNA or inconclusiveenotyping. The frequencies of the rare �93G, 9Asn, 291Ser,nd 447Ter alleles were 0.02, 0.02, 0.03, and 0.10, respectively.
The genotype distributions were consistent with Hardy-einberg equilibrium (p � 0.05), except for the �93T/G
olymorphism. Therefore, this promoter polymorphism wasxcluded from further analysis.
Comparisons of baseline characteristics among the geno-ypes are shown in Table 1. The low frequency of carriers ofAsn and 291Ser has prompted us to pool heterozygotesnd homozygotes for these two LPL variants. No statisti-ally significant differences were observed between theroups, with the following two exceptions: heterozygotesnd homozygotes for the allele encoding for a 291Serenotype had a higher rate of previous CABG (p � 0.05).urthermore, heterozygotes and homozygotes for the allelencoding for the 9Asn LPL variant used less lipid-loweringedication but had a higher use of beta-blocker medication.
esion-related and procedural parameters of the genotype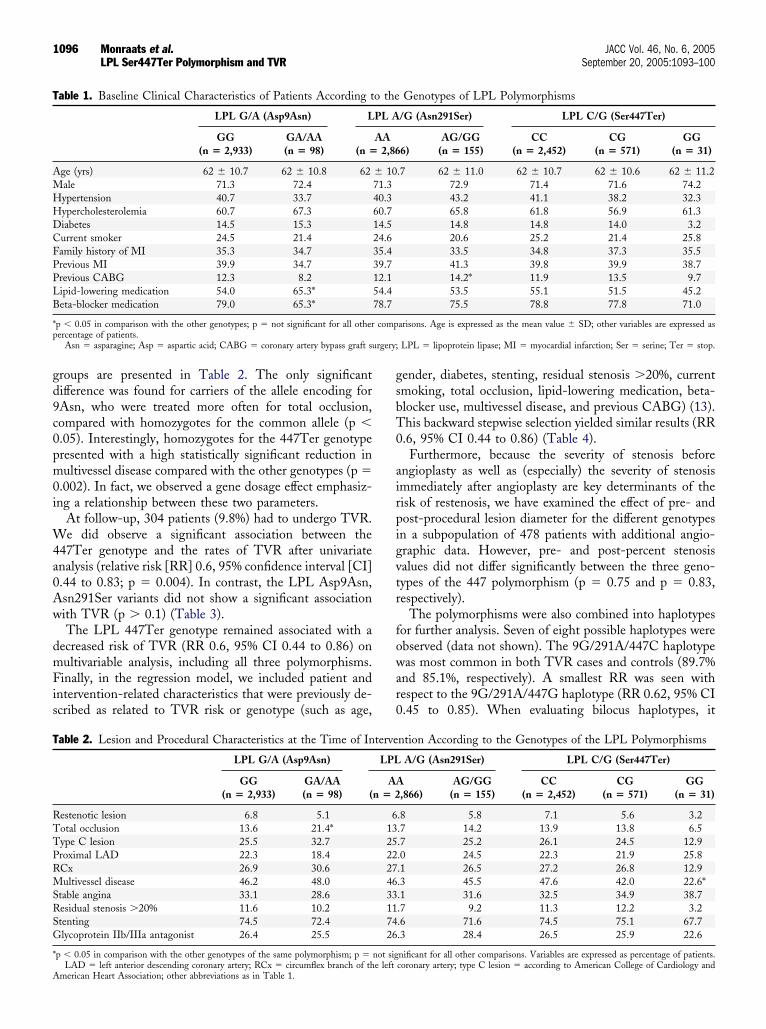
gd9c0pm0i
W4a0Aw
dmFis
gsbT0
airpigvtr
fowar0
T
AMHHDCFPPLB
*p
rgery;
T
RTTPRMSRSG
*
A
1096 Monraats et al. JACC Vol. 46, No. 6, 2005LPL Ser447Ter Polymorphism and TVR September 20, 2005:1093–100
roups are presented in Table 2. The only significantifference was found for carriers of the allele encoding forAsn, who were treated more often for total occlusion,ompared with homozygotes for the common allele (p �.05). Interestingly, homozygotes for the 447Ter genotyperesented with a high statistically significant reduction inultivessel disease compared with the other genotypes (p �
.002). In fact, we observed a gene dosage effect emphasiz-ng a relationship between these two parameters.
At follow-up, 304 patients (9.8%) had to undergo TVR.e did observe a significant association between the
47Ter genotype and the rates of TVR after univariatenalysis (relative risk [RR] 0.6, 95% confidence interval [CI].44 to 0.83; p � 0.004). In contrast, the LPL Asp9Asn,sn291Ser variants did not show a significant associationith TVR (p � 0.1) (Table 3).The LPL 447Ter genotype remained associated with a
ecreased risk of TVR (RR 0.6, 95% CI 0.44 to 0.86) onultivariable analysis, including all three polymorphisms.inally, in the regression model, we included patient and
ntervention-related characteristics that were previously de-cribed as related to TVR risk or genotype (such as age,
able 1. Baseline Clinical Characteristics of Patients According t
LPL G/A (Asp9Asn) L
GG(n � 2,933)
GA/AA(n � 98) (n �
ge (yrs) 62 � 10.7 62 � 10.8 62ale 71.3 72.4ypertension 40.7 33.7ypercholesterolemia 60.7 67.3iabetes 14.5 15.3urrent smoker 24.5 21.4amily history of MI 35.3 34.7revious MI 39.9 34.7revious CABG 12.3 8.2ipid-lowering medication 54.0 65.3*eta-blocker medication 79.0 65.3*
p � 0.05 in comparison with the other genotypes; p � not significant for all otherercentage of patients.
Asn � asparagine; Asp � aspartic acid; CABG � coronary artery bypass graft su
able 2. Lesion and Procedural Characteristics at the Time of In
LPL G/A (Asp9Asn)
GG(n � 2,933)
GA/AA(n � 98) (
estenotic lesion 6.8 5.1otal occlusion 13.6 21.4*ype C lesion 25.5 32.7roximal LAD 22.3 18.4Cx 26.9 30.6ultivessel disease 46.2 48.0
table angina 33.1 28.6esidual stenosis �20% 11.6 10.2tenting 74.5 72.4lycoprotein IIb/IIIa antagonist 26.4 25.5
p � 0.05 in comparison with the other genotypes of the same polymorphism; p �
LAD � left anterior descending coronary artery; RCx � circumflex branch of the leftmerican Heart Association; other abbreviations as in Table 1.
ender, diabetes, stenting, residual stenosis �20%, currentmoking, total occlusion, lipid-lowering medication, beta-locker use, multivessel disease, and previous CABG) (13).his backward stepwise selection yielded similar results (RR.6, 95% CI 0.44 to 0.86) (Table 4).Furthermore, because the severity of stenosis before
ngioplasty as well as (especially) the severity of stenosismmediately after angioplasty are key determinants of theisk of restenosis, we have examined the effect of pre- andost-procedural lesion diameter for the different genotypesn a subpopulation of 478 patients with additional angio-raphic data. However, pre- and post-percent stenosisalues did not differ significantly between the three geno-ypes of the 447 polymorphism (p � 0.75 and p � 0.83,espectively).
The polymorphisms were also combined into haplotypesor further analysis. Seven of eight possible haplotypes werebserved (data not shown). The 9G/291A/447C haplotypeas most common in both TVR cases and controls (89.7%
nd 85.1%, respectively). A smallest RR was seen withespect to the 9G/291A/447G haplotype (RR 0.62, 95% CI.45 to 0.85). When evaluating bilocus haplotypes, it
Genotypes of LPL Polymorphisms
/G (Asn291Ser) LPL C/G (Ser447Ter)
6)AG/GG
(n � 155)CC
(n � 2,452)CG
(n � 571)GG
(n � 31)
.7 62 � 11.0 62 � 10.7 62 � 10.6 62 � 11.272.9 71.4 71.6 74.243.2 41.1 38.2 32.365.8 61.8 56.9 61.314.8 14.8 14.0 3.220.6 25.2 21.4 25.833.5 34.8 37.3 35.541.3 39.8 39.9 38.714.2* 11.9 13.5 9.753.5 55.1 51.5 45.275.5 78.8 77.8 71.0
arisons. Age is expressed as the mean value � SD; other variables are expressed as
LPL � lipoprotein lipase; MI � myocardial infarction; Ser � serine; Ter � stop.
ntion According to the Genotypes of the LPL Polymorphisms
A/G (Asn291Ser) LPL C/G (Ser447Ter)
,866)AG/GG
(n � 155)CC
(n � 2,452)CG
(n � 571)GG
(n � 31)
.8 5.8 7.1 5.6 3.2
.7 14.2 13.9 13.8 6.5
.7 25.2 26.1 24.5 12.9
.0 24.5 22.3 21.9 25.8
.1 26.5 27.2 26.8 12.9
.3 45.5 47.6 42.0 22.6*
.1 31.6 32.5 34.9 38.7
.7 9.2 11.3 12.2 3.2
.6 71.6 74.5 75.1 67.7
.3 28.4 26.5 25.9 22.6
nificant for all other comparisons. Variables are expressed as percentage of patients.
o the
PL A
AA2,86
� 1071.340.360.714.524.635.439.712.154.478.7
comp
terve
LPL
AAn � 2
6132522274633117426
not sig
coronary artery; type C lesion � according to American College of Cardiology and
b4
(stlCia
pahp
ftTacwrpt
D
Ic
Sl(sIwaFSdlpttic(
p
sotaala
acedst
bccm
TA
P
A
A
S
T
Ta
DCSTRL
C
Flp
1097JACC Vol. 46, No. 6, 2005 Monraats et al.September 20, 2005:1093–100 LPL Ser447Ter Polymorphism and TVR
ecame evident that this effect was caused only by the LPL47 variant.Lipid profiles were investigated in a subgroup of patients
n � 942, data not shown). We were not able to find aignificant correlation between carriers and non-carriers ofhe polymorphisms investigated with regard to HDL cho-esterol, LDL cholesterol, and triglycerides (p � 0.20).orrection of lipid levels in this subgroup analysis had no
nfluence on the association of the different polymorphismsnd TVR.
Subsequently, we evaluated if LPL gene expressionlayed a role in the development of restenosis by analyzingrterial mRNA in an established model of restenosis inypercholesterolemic mice. At the time of surgery, the totallasma cholesterol level was 13.9 � 3.6 mmol/l.The mRNA encoding LPL, isolated from the cuffed right
emoral artery and untreated left femoral artery, was quan-ified at various time points after cuff placement (Fig. 1).he mRNA encoding LPL showed peak expression 6 h
fter cuff placement, where it showed a 40-fold increaseompared with the normal artery. The LPL mRNA levelsere back to baseline after 24 h of induction of the
estenotic process. Sham-operated vessels (femoral arteryrepared free, but without cuff placement) showed essen-ially the same results as non-operated vessels.
ISCUSSION
n a large, prospective, multicenter, follow-up study ofonsecutive patients, we demonstrated that the LPL
able 3. Univariate Analysis of LPL Polymorphisms inssociation With TVR and the Distributions of Polymorphisms
olymorphismsNo. of
Patients TVR (%)ModelUsed
pValue
sp9Asn 3,031 Dominant 0.77GG 9.7GA/AA 10.4
sn291Ser 3,021 Dominant 0.62AA 9.9AG/GG 8.5
er447Ter 3,054 Dominant 0.004CC 10.5CG 7.0GG 0
VR � target vessel revascularization; other abbreviations as in Table 1.
able 4. Multivariable Cox Regression of Clinical Variablesnd LPL Ser447Ter Polymorphism Associated With TVR
RR
95% CI
p ValueLow High
iabetes 1.52 1.14 2.01 0.004urrent smoker 0.73 0.54 0.97 0.03tenting 0.76 0.58 1.00 0.05otal occlusion 1.51 1.12 2.02 0.006esidual stenosis �20% 1.35 0.97 1.89 0.08PL 447Ter 0.62 0.44 0.86 0.005
I � confidence interval; RR � relative risk; other abbreviations as in Table 1.
er447Ter variant, present in �20% of the general popu-ation, is associated with a decreased risk of TVR after PCI3). This polymorphism has been shown in most but not alltudies to modulate lipid levels, as well as LPL (2,4,19–25).n fact, it was shown that this polymorphism is associatedith decreased triglycerides, increased HDL cholesterol,
nd a decreased risk of coronary artery disease (4,26,27).urthermore, a recent meta-analysis confirmed that theer447Ter variant has an effect on the lipoprotein profile, byecreasing plasma triglycerides and increasing HDL cho-
esterol (4). The mechanism responsible for cardiovascularrotection and beneficial lipid profile changes observed inhis LPL variant is not entirely clear. The data suggest thathis variant may be catalytically normal with normal stabil-ty, but may be present at higher concentrations in theirculation associated with a higher level of LPL activity2,4,21,28,29).
We did not find any associations for the other twoolymorphisms and TVR.The Asp9Asn substitution at the N-terminal end is
ituated near a glycosylation site, which may influenceverall catalytic activity, whereas the Asn291Ser substitu-ion is located in a heparin-binding cluster and may thusffect the interaction between LPL and the cell wall glycos-minoglycans. Both these two amino acid substitutions areocated in the N-domain and likely reduce enzyme activitynd consequently increase triglyceride levels.
The Ser447Ter substitution is located in the C-domainnd thus may cause increased binding affinity of the trun-ated LPL to receptors or may affect its subunit interaction,ither facilitating or otherwise affecting the formation ofimers, which would explain the opposite effect of thisubstitution compared with the other two, possibly forminghe basis of the observed association (4,7).
In our study, we did not find a significant associationetween the LPL447 polymorphism and HDL and LDLholesterol and triglyceride levels (data not shown). Thisould be due to the use of lipid-lowering medications inany of our patients.
igure 1. Messenger ribonucleic acid (mRNA) expression of lipoproteinipase in a cuffed femoral artery (as compared with hypoxathine-guaninehosphoribosyl transferase mRNA expression, cuff vs. normal artery).
Haplotype analyses showed that the difference in TVR

rp
hVbiaheSesSbTtr
idoasiNflac(pcsl(ttn
L(maao(
iwsclecm
vf
Lioelppfitrnnp
ddnwisScbolmttsanld
mmfcL
siitti
dmHd
1098 Monraats et al. JACC Vol. 46, No. 6, 2005LPL Ser447Ter Polymorphism and TVR September 20, 2005:1093–100
ate between the various haplotypes was completely ex-lained by the LPL 447 variant.In addition to the well-known role of LPL in the
ydrolysis of the triglycerides packaged in chylomicrons andLDL, several other functions of the enzyme have recentlyeen identified. In particular, it has been shown that LPLncreases monocyte adherence via a mechanism that requiresn interaction between the C-terminal domain of the LPL,eparin sulfate proteoglycans, and integrins (30–32). How-ver, Zhang et al. (29) showed that the affinity of LPLer447Ter for heparin sulfate proteoglycans was not differ-nt from wild-type LPL. Because LPL is also expressed inmooth muscle cells in the arterial media (33), the LPLer447Ter polymorphism may affect the level of interactionetween smooth muscle cells and the extracellular matrix.he former could result in less arterial stiffness (34), leading
o more distensible arteries and thus lesser propensity forestenosis.
On a completely different note, the LPL protein may alsonfluence vascular tone by affecting the synthesis or degra-ation of endothelium-derived relaxing factors such as nitricxide (NO). Endothelium-dependent vascular relaxation isbnormal in the setting of atherosclerosis, associated withubnormal endothelial nitric oxide synthase (eNOS) activ-ty, the key enzyme in basal endothelial cell NO production.
itric oxide dilates coronary arteries and promotes bloodow by inhibiting smooth muscle contraction, plateletggregation, and platelet adhesion to endothelial cells by ayclic guanosine monophosphate-mediated mechanism35,36) In fact, LPL has been reported to increase NOSroduction and consequential increased NO production inulture macrophages. Lipoprotein lipase may well have aimilar function in vivo in both macrophages and endothe-ial cells and may therefore have an effect on vascular tone1,37). Therefore, mutated levels of LPL, as observed withhe Ser447Ter polymorphism, may be beneficial for endo-helial function, an important contributor involved in reste-osis (38).In addition, Ziouzenkova et al. (39) found a link between
PL and peroxisome proliferator-activated receptorPPAR) activation, suggesting that impaired LPL enzy-atic activity might decrease endogenous PPAR-alpha
ctivation and its subsequent downstream effects, includingnti-inflammatory responses. Inflammation has been previ-usly reported as a very important component of restenosis40–42).
Clee et al. (43) found a decrease in blood pressurendependent of the lower level of triglycerides in patientsith the LPL Ser447Ter polymorphism. Another study also
howed that the LPL Ser447Ter polymorphism was asso-iated with lower systolic blood pressure and pulse pressureevels in women (34). Because inflammation (40–42) andlevated blood pressure (44) have been implicated in in-reased risk of restenosis, any positive effect of this poly-
orphism on arterial tone, inflammatory status, and ele- gated blood pressure may ultimately translate into lesser riskor restenosis.
Because we hypothesize a potential interaction betweenPL activity by genotype and inflammatory activity, we also
nvestigated several inflammatory markers, which could bef influence on the development of restenosis and theirffect on TVR. These markers examined are the fibrinogenevel, the erythrocyte sedimentation rate, and C-reactiverotein. These markers were determined in a subgroup ofatients from the GENDER project (in 753 patients forbrinogen, in 1,000 patients for the erythrocyte sedimenta-ion rate, and in 888 patients for C-reactive protein,espectively). All three factors, determined before PCI, didot have a statistically significant effect on TVR (p � 0.20),or did they influence the general results for the LPLolymorphisms.We further studied LPL gene expression during the
evelopment of restenosis in an established mouse model foriet-induced atherosclerosis. In the mouse model of reste-osis, LPL mRNA levels were increased 40-fold comparedith control arteries at 6 h after cuff placement. This
ndicates that LPL may play an important role in the earlytages of the restenotic process development.tudy limitations. In our study, we lack data on LPLoncentration and LPL activity in plasma. However, weelieve that plasma determinations have no added value,wing to a number of reasons. Circulating LPL proteinevels were not assessed here, as basal (before PCI) plasma
easurements of the gene product are not likely to reflecthe genetically determined differences in reaction to arauma such as PCI. Moreover, local differences in LPLensitive reactions, such as those occurring in the vessel wallt the site of PCI, cannot be measured systemically, as it isot yet possible to measure gene products in the vessel wall
ocally in the early phase of treatment or in the followingays.Furthermore, we made use of an atherosclerotic mouseodel to study the effect of LPL on restenosis; in thisodel, we were not able to test the LPL polymorphisms
ound in humans. However, we believe that this modelontributes to a better understanding of the involvement ofPL in the process of restenosis.Although the mice studies can be used for analysis, it
hould be realized that perivascular cuff placement resultsnitially in adventitial injury, whereas in patients with PCI,t results in intimal injury. It is not certain to what extenthese apparently different ways of vascular injury differ inheir reaction regarding vascular activation and the resultingntimal hyperplasia.
In addition, genotyping of some patients was missingue to a lack of DNA or inconclusive genotyping, andistakes could have been made in the genotyping.owever, patients who could not be genotyped did not
iffer in any characteristic from those who could be
enotyped. Furthermore, the PCR procedure was per-
fo
Wwo1epcaifc�alsgp
sdstasnp
spaCcwbFfirti
AWRoufgCJV
RLCJ
R
1
1
1
1
1
1
1
1
1
1099JACC Vol. 46, No. 6, 2005 Monraats et al.September 20, 2005:1093–100 LPL Ser447Ter Polymorphism and TVR
ormed in replicate on 10%, and there was a differencebserved in �1% of the samples.The �93T/G polymorphism was not in Hardy-einberg equilibrium; the observed numbers of patients
ere 2,914 for T/T, 109 for T/G, and 5 for G/G, aspposed to the expected number of patients: 2,910 for T/T,17 for T/G, and 1 for G/G. This discrepancy could bexplained by the fact that it is possible that this polymor-hism is associated with one or more inclusion/exclusionriteria; however, we cannot confirm this with our data, asll participants fulfilled those criteria. The most importantnclusion criterion was that all participants were scheduledor PCI, and obviously the classic risk factors of cardiovas-ular disease will be enriched in such a population. The93T/G polymorphism is sometimes found to be especially
ssociated with HDL2 cholesterol, and ApoA-I levels withower levels in �93G/G carriers (45). Based on this as-umption, one might expect more heterozygote or homozy-ote carriers, and the latter were indeed slightly moreresent than expected.Drug-eluting stents are now more widely used. In our
tudy, we do not have any data on these stents, as norug-eluting stents were used for our study, which can beeen as a limitation of our study. However, genes involved inhe process of restenosis after drug-eluting stents are prob-bly different from the process of restenosis after bare-metaltent placement or plain balloon angioplasty. Therefore,ew studies are needed to investigate genes involved in therocess of restenosis after drug-eluting stents.Finally, the polymorphism associated with TVR in our
tudy may be in linkage disequilibrium with other polymor-hisms in the gene or with other nearby genes that arectually responsible for the development of this condition.onclusions. We have demonstrated that LPL is signifi-
antly associated with TVR. The LPL C/G polymorphism,hich results in a 447 Ser¡Stop (X) mutation, appears toe an important independent protective factor in this.urthermore, LPL mRNA was highly up-regulated in therst 6 h after vascular damage in a mouse model ofestenosis. Determination of this genotype could contributeo better risk stratification and more tailored therapy for thendividual patient to prevent TVR after PCI.
cknowledgmentse thank S. Cheng, M. Grow, and their colleagues at
oche Molecular Systems (Alameda, California) for devel-ping and providing their multilocus genotyping assaysnder a research collaboration. We also thank P. Schiffersrom the University of Maastricht for assistance with theenotyping assay. The contribution of the members of thelinical Event Committee—J. J. Schipperheyn, MD, PhD,
. W. Viersma, MD, PhD, D. Düren, MD, PhD, and J.
ainer, MD—is greatly acknowledged.eprint requests and correspondence: Dr. J. Wouter Jukema,eiden University Medical Center, Department of Cardiology,5-P, P.O. Box 9600, 2300 RC Leiden, the Netherlands. E-mail:
EFERENCES
1. Kastelein JJ, Jukema JW, Zwinderman AH, et al., the REGRESSStudy Group. Lipoprotein lipase activity is associated with severity ofangina pectoris. Circulation 2000;102:1629–33.
2. Groenemeijer BE, Hallman MD, Reymer PW, et al., the REGRESSStudy Group. Genetic variant showing a positive interaction withbeta-blocking agents with a beneficial influence on lipoprotein lipaseactivity, HDL cholesterol, and triglyceride levels in coronary arterydisease patients. The Ser447-stop substitution in the lipoprotein lipasegene. Circulation 1997;95:2628–35.
3. Sing K, Ballantyne CM, Ferlic L, et al. Lipoprotein lipase genemutations, plasma lipid levels, progression/regression of coronaryatherosclerosis, response to therapy, and future clinical events: theLipoproteins and Coronary Atherosclerosis Study. Atherosclerosis1999;144:435–42.
4. Wittrup HH, Tybjaerg-Hansen A, Nordestgaard BG. Lipoproteinlipase mutations, plasma lipids and lipoproteins, and risk of ischemicheart disease: a meta-analysis. Circulation 1999;99:2901–7.
5. Jukema JW, van Boven AJ, Groenemeijer B, et al., the REressionGRowth Evaluation Statin Study (REGRESS) Study Group. TheAsp9 Asn mutation in the lipoprotein lipase gene is associated withincreased progression of coronary atherosclerosis. Circulation 1996;94:1913–8.
6. Murthy V, Julien P, Gagne C. Molecular pathobiology of the humanlipoprotein lipase gene. Pharmacol Ther 1996;70:101–35.
7. Ukkola O, Garenc C, Perusse L, et al. Genetic variation at thelipoprotein lipase locus and plasma lipoprotein and insulin levels in theQuebec Family Study. Atherosclerosis 2001;158:199–206.
8. Kastrati A, Schomig A, Elezi S, Schuhlen H, Wilhelm M, Dirsch-inger J. Interlesion dependence of the risk for restenosis in patientswith coronary stent placement in multiple lesions. Circulation 1998;97:2396–401.
9. Weintraub WS, Kosinski AS, Brown CL III, King SB III. Canrestenosis after coronary angioplasty be predicted from clinical vari-ables? J Am Coll Cardiol 1993;21:6–14.
0. Lardenoye JH, Delsing DJ, de Vries MR, et al. Accelerated athero-sclerosis by placement of a perivascular cuff and a cholesterol-rich dietin ApoE*3Leiden transgenic mice. Circ Res 2000;87:248–53.
1. Moroi M, Zhang L, Yasuda T, et al. Interaction of genetic deficiencyof endothelial nitric oxide, gender, and pregnancy in vascular responseto injury in mice. J Clin Invest 1998;101:1225–32.
2. Quax PH, Lamfers ML, Lardenoye JH, et al. Adenoviral expression ofa urokinase receptor-targeted protease inhibitor inhibits neointimaformation in murine and human blood vessels. Circulation 2001;103:562–9.
3. Agema WRP, Monraats PS, Zwinderman AH, et al. Current PTCApractice and clinical outcomes in the Netherlands: the real world in thepre-drug-eluting stent era. Eur Heart J 2004;25:1163–70.
4. Cheng S, Grow MA, Pallaud C, et al. A multilocus genotyping assayfor candidate markers of cardiovascular disease risk. Genome Res1999;9:936–49.
5. Barcellos LF, Begovich AB, Reynolds RL, et al. Linkage and associ-ation with the NOS2A locus on chromosome 17q11 in multiplesclerosis. Ann Neurol 2004;55:793–800.
6. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concen-tration of low-density lipoprotein cholesterol in plasma, without use ofthe preparative ultracentrifuge. Clin Chem 1972;18:499–502.
7. Li W, Nyholt DR. Marker selection by Akaike information criterionand Bayesian information criterion. Genet Epidemiol 2001;21 Suppl1:S272–7.
8. Tanck MW, Klerkx AH, Jukema JW, De Knijff P, Kastelein JJ,Zwinderman AH. Estimation of multilocus haplotype effects usingweighted penalised log-likelihood: analysis of five sequence variations
at the cholesteryl ester transfer protein gene locus. Ann Hum Genet2003;67:175–84.
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
4
1100 Monraats et al. JACC Vol. 46, No. 6, 2005LPL Ser447Ter Polymorphism and TVR September 20, 2005:1093–100
9. Chen W, Srinivasan SR, Elkasabany A, Ellsworth DL, Boerwinkle E,Berenson GS. Influence of lipoprotein lipase serine 447 stop polymor-phism on tracking of triglycerides and HDL cholesterol from child-hood to adulthood and familial risk of coronary artery disease: theBogalusa Heart Study. Atherosclerosis 2001;159:367–73.
0. Gagne SE, Larson MG, Pimstone SN, et al. A common truncationvariant of lipoprotein lipase (Ser447X) confers protection againstcoronary heart disease: the Framingham Offspring Study. Clin Genet1999;55:450–4.
1. Humphries SE, Nicaud V, Margalef J, Tiret L, Talmud PJ. Lipopro-tein lipase gene variation is associated with a paternal history ofpremature coronary artery disease and fasting and postprandial plasmatriglycerides: the European Atherosclerosis Research Study (EARS).Arterioscler Thromb Vasc Biol 1998;18:526–34.
2. Jemaa R, Fumeron F, Poirier O, et al. Lipoprotein lipase genepolymorphisms: associations with myocardial infarction and lipopro-tein levels, the Etude Cas Temoin sur l’Infarctus du Myocarde(ECTIM) study. J Lipid Res 1995;36:2141–6.
3. Kozaki K, Gotoda T, Kawamura M, et al. Mutational analysis ofhuman lipoprotein lipase by carboxy-terminal truncation. J Lipid Res1993;34:1765–72.
4. Mattu RK, Needham EW, Morgan R, et al. DNA variants at the LPLgene locus associate with angiographically defined severity of athero-sclerosis and serum lipoprotein levels in a Welsh population. Arterio-scler Thromb Vasc Biol 1994;14:1090–7.
5. van Bockxmeer FM, Liu Q, Mamotte C, Burke V, Taylor R.Lipoprotein lipase D9N, N291S and S447X polymorphisms: theirinfluence on premature coronary heart disease and plasma lipids.Atherosclerosis 2001;157:123–9.
6. Kuivenhoven JA, Groenemeyer BE, Boer JM, et al. Ser447stopmutation in lipoprotein lipase is associated with elevated HDLcholesterol levels in normolipidemic males. Arterioscler Thromb VascBiol 1997;17:595–9.
7. Sawano M, Watanabe Y, Ohmura H, et al. Potentially protectiveeffects of the Ser447-Ter mutation of the lipoprotein lipase geneagainst the development of coronary artery disease in Japanese subjectsvia a beneficial lipid profile. Jpn Circ J 2001;65:310–4.
8. Henderson HE, Kastelein JJ, Zwinderman AH, et al. Lipoproteinlipase activity is decreased in a large cohort of patients with coronaryartery disease and is associated with changes in lipids and lipoproteins.J Lipid Res 1999;40:735–43.
9. Zhang H, Henderson H, Gagne SE, et al. Common sequence variantsof lipoprotein lipase: standardized studies of in vitro expression andcatalytic function. Biochim Biophys Acta 1996;1302:159–66.
0. Nielsen MS, Brejning J, Garcia R, et al. Segments in the C-terminalfolding domain of lipoprotein lipase important for binding to the lowdensity lipoprotein receptor-related protein and to heparin sulfate
proteoglycans. J Biol Chem 1997;272:5821–7.1. Mamputu JC, Desfaits AC, Renier G. Lipoprotein lipase enhanceshuman monocyte adhesion to aortic endothelial cells. J Lipid Res1997;38:1722–9.
2. Obunike JC, Paka S, Pillarisetti S, Goldberg IJ. Lipoprotein lipase canfunction as a monocyte adhesion protein. Arterioscler Thromb VascBiol 1997;17:1414–20.
3. Camps L, Reina M, Llobera M, Vilaro S, Olivecrona T. Lipoproteinlipase: cellular origin and functional distribution. Am J Physiol1990;258:C673–81.
4. Sass C, Herbeth B, Siest G, Visvikis S. Lipoprotein lipase (C/G)447polymorphism and blood pressure in the Stanislas Cohort. J Hypertens2000;18:1775–81.
5. Moncada S, Higgs A. The L-arginine–nitric oxide pathway. N EnglJ Med 1993;329:2002–12.
6. Radomski MW, Salas E. Nitric oxide—biological mediator, modula-tor and factor of injury: its role in the pathogenesis of atherosclerosis.Atherosclerosis 1995;118 Suppl:S69–80.
7. Renier G, Lambert A. Lipoprotein lipase synergizes with interferongamma to induce macrophage nitric oxide synthetase mRNA expres-sion and nitric oxide production. Arterioscler Thromb Vasc Biol1995;15:392–9.
8. Horlitz M, Sigwart U, Niebauer J. Fighting restenosis after coronaryangioplasty: contemporary and future treatment options. Int J Cardiol2002;83:199–205.
9. Ziouzenkova O, Perrey S, Asatryan L, et al. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: evidence for an antiinflam-matory role for lipoprotein lipase. Proc Natl Acad Sci U S A2003;100:2730–5.
0. Hojo Y, Ikeda U, Katsuki T, et al. Chemokine expression in coronarycirculation after coronary angioplasty as a prognostic factor for reste-nosis. Atherosclerosis 2001;156:165–70.
1. Serrano CV Jr., Ramires JA, Venturinelli M, et al. Coronary angio-plasty results in leukocyte and platelet activation with adhesionmolecule expression: evidence of inflammatory responses in coronaryangioplasty. J Am Coll Cardiol 1997;29:1276–83.
2. Welt FG, Rogers C. Inflammation and restenosis in the stent era.Arterioscler Thromb Vasc Biol 2002;22:1769–76.
3. Clee SM, Loubser O, Collins J, Kastelein JJ, Hayden MR. The LPLS447X cSNP is associated with decreased blood pressure and plasmatriglycerides, and reduced risk of coronary artery disease. Clin Genet2001;60:293–300.
4. Cutlip DE, Chauhan MS, Baim DS, et al. Clinical restenosis aftercoronary stenting: perspectives from multicenter clinical trials. J AmColl Cardiol 2002;40:2082–9.
5. Ferencak G, Pasalic D, Grskovic B, et al. Lipoprotein lipase genepolymorphisms in Croatian patients with coronary artery disease. Clin
Chem Lab Med 2003;414:541–6.



















