
Performance of Palm-Based C16/18 Methyl Ester Sulphonate ...
Upload
independentCategory
view
4download
0
Hum Genet (1994) 93:639-648 human .. genet, cs
�9 Springer-Verlag 1994
Linkage analysis of the genetic determinants of high density lipoprotein concentrations and composition: evidence for involvement of the apolipoprotein A-II and cholesteryl ester transfer protein loci
Xiangdong Bu a, Craig H. Warden 2, Yu-Rong Xia 2, Cynthia De Meester 2, Donald L. Puppione 3, Scott Teruya 2, Beth Lokensgard 2, Siamac Daneshmand 2, Jane Brown 1, Richard J. Gray t, Jerome I. Rotter 1, Aldons J. Lusis 2
1 Divisions of Medical Genetics and Cardiology, Departments of Medicine and Pediatrics, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA 2Departments of Medicine and Microbiology and Molecular Genetics, and Molecular Biology Institute, University of California, Los Angeles, CA 90024, USA 3 Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90024, USA
Received: 3 September 1993 / Revised: 29 December 1993
Abstract. We have tested for evidence of linkage between the genetic loci determining concentrations and composi- tion of plasma high density lipoproteins (HDL) with the genes for the major apolipoproteins and enzymes partici- pating in lipoprotein metabolism. These genes include those encoding various apolipoproteins (apo), including apoA-I, apoA-II, apoA-IV, apoB, apoC-I, apoC-II, apoC- III, apoE, and apo(a), cholesteryl ester transfer protein (CETP), HDL-binding protein, lipoprotein lipase, and the low density lipoprotein (LDL) receptor. Polymorphisms of these genes, and nearby highly polymorphic simple se- quence repeat markers, were examined by quantitative sib-pair linkage analysis in 30 coronary artery disease families consisting of a total of 366 individuals. Evidence for linkage was observed between a marker locus D16S313 linked to the CETP locus and a locus determin- ing plasma HDL-cholesterol concentration (P = 0.002), and the genetic locus for apoA-II and a locus determining the levels of the major apolipoproteins of HDL, apoA-I and apoA-II (P = 0.009 and 0.02, respectively). HDL level was also influenced by the variation at the apo(a) lo- cus on chromosome 6 (P = 0.02). Thus, these data indi- cate the simultaneous involvement of at least two differ- ent genetic loci in the determination of the levels of HDL and its associated lipoproteins.
Introduction
The plasma concentrations of high density lipoprotein (HDL)-cholesterol and the most abundant HDL protein,
Correspondence to: J. I. Rotter, Division of Medical Genetics, SSB-3, Departments of Medicine and Pediatrics, 8700 W. Beverly Blvd., Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA
apolipoprotein A-I (apoA-I), are inversely correlated with the risk for coronary artery disease (Gordon et al. 1972; Schaefer 1984; Miller 1987; Tall 1990; Karathanasis 1992). The variations in levels of human HDL are due largely to genetic factors, although environmental influ- ences are also of considerable importance (Rao et al. 1983; Hamsten et al. 1986; Lusis 1988; Breslow 1989, 1991; Hunt et al. 1989; Vogler et al. 1989; Berg 1989; Tall 1992). Studies of rare human mutations have revealed that apoA-I is required for normal HDL assembly, as individ- uals who fail to express apoA-I have greatly reduced HDL-cholesterol levels (reviewed in Karathanasis 1992). In contrast, null mutations of the apoA-II gene, encoding the second most abundant protein of human HDL, do not exhibit dramatic effects on HDL-cholesterol levels (Deeb et al. 1990). Null mutations of cholesteryl ester transfer protein (CETP), on the other hand, are associated with markedly increased levels of HDL-cholesterol (Brown et al. 1989; Inazu et al. 1990). CETP mediates the transfer of cholesteryl esters from HDL to intermediate density lipoproteins (IDL) and low density lipoproteins (LDL). Deficiency states of lipoprotein lipase activity (LPL), in- volving mutations either of the LPL or apoC-II genes, are associated with decreased HDL levels (Doolittle et al. 1992). In one study, variation in HDL levels was shown to be associated with genetic variation at the apolipopro- tein(a) structural gene locus on chromosome 6 (Hegele et al. 1991). A variety of structural gene mutations that abol- ish the activity of lecithin-cholesterol ester transferase (LCAT), the enzyme responsible for esterification of HDL-cholesterol, also greatly decrease levels of HDL- cholesterol (Taramelli et al. 1990). Another potentially very interesting gene as regards its possible role in HDL metabolism is the HDL binding protein (HDLBP), whose structural locus has been mapped to chromosome 2 (Xia et al. 1993). Although this is a cellular HDL-binding pro- tein that recognizes apoA-I, its importance in the metabo-

640
l i sm or overal l regula t ion o f H D L levels is still poor ly un- ders tood (Graham and Oram 1987). Popula t ion associa- t ion and l inkage studies with po lymorph i sms o f the genes for the major apol ipopro te ins and enzymes involved in l ipoprote in me tabo l i sm have sugges ted that variat ions in the genes for apoA- I I (Scot t et al. 1985; Thorn et al. 1993; Warden et al. 1993c), C E T P (Kondo et al. 1989; Kess l ing et al. 1991; He iba et al. 1993), LPL (Heinzmann et al. 1991), and the a p o A I - C I I I - A I V gene complex (Wile et al. 1989; Kess l ing et al. 1991; Xu et al. 1993) inf luence ei- ther HDL-cho les t e ro l levels or H D L composi t ion .
The in v ivo regula t ion o f H D L and its major associ- ated apol ipopro te ins in man is complex . Many known or yet to be known genet ic loci are involved. Mouse models or t ransgenic mouse mode l s can, at least partly, decom- pose this complexi ty . Recent studies of natural ly occur- r ing var ia t ions in mice have revealed that the apoA-I I gene (Mehrabian et al. 1993) and ch romosomal loci not cor responding to any known candidate genes (Warden et al. 1993a) de te rmine HDL-cho les t e ro l levels. Our recent data f rom an apoA- I I t ransgenic mouse indica ted that mice overexpress ing the apoA- I I gene had e levated HDL- choles terol concentra t ions but, nevertheless , exhibi ted in- c reased a therosclerot ic lesion deve lopmen t as compared to normal mice (Warden et al. 1993b). Other studies with genet ica l ly mod i f i ed mice (either t ransgenic mice or gene- targe ted mice) have demons t ra ted that express ion o f apoA- I (Walsh et al. 1989; Rubin et al. 1991), CETP (Ag- el lon et al. 1991), and apoE (Plump et al. 1992; Zhang et al. 1992) each inf luence H D L levels.
We have recent ly repor ted observat ions , based on mouse /human synteny, that the apoA- I I locus is l inked to a locus de te rmin ing apoA- I I levels in man (Warden et al. 1993c). We now repor t a sys temat ic survey of candidate genes ( those genes shown to be b iochemica l ly involved in H D L me tabo l i sm or to encode for HDL-assoc ia t ed apol ipopro te ins ) by l inkage analysis in an enlarged panel o f 30 famil ies enr iched for coronary artery disease. Those genes examined inc lude those encoding the major apo- l ipoprote ins [apo A-I , A-II , A-IV, B, C-I, C-II , C-III , E, and apo(a)] , and prote ins involved in l ipoprote in metabo- l i sm (CETP, LPL, L D L receptor, HDL-b ind ing protein). Ana lys i s pe r fo rmed by the quanti ta t ive s ib-pai r l inkage method (Haseman and Els ton 1972; A m o s et al. 1989) y ie lded two signif icant f indings: a marker locus D 16S313 l inked to the CETP gene is l inked to a locus de termining HDL-cho les t e ro l levels, and the apoA- I I gene is l inked to a locus de te rmin ing the levels o f the major H D L apol ipo- prote ins (apoA-1 and apoA-I I ) .
Materia ls and methods
Pedigrees
At the time of analysis, the study sample consisted of 30 multiplex multigenerational Caucasian pedigrees ascertained through a proband with documented (surgically or angiographically) coro- nary artery disease (CAD) identified at Cedars-Sinai Medical Cen- ter in Los Angeles. Family inclusion criteria required that at least one other blood relative be affected with CAD. A total of 366 in- dividuals from these 30 families agreed to participate in this study
(6 to 33 members per family). The investigation was approved by the institutional Human Subjects Protection Committee.
Plasma biochemical analysis
Overnight fasting blood from participating family members was drawn into EDTA-containing tubes. Plasma concentrations of cho- lesterol and triglyceride (TG) were measured in duplicate using en- zymes and reagents described previously (Moll et al. 1986; War- den et al. 1993c). Surveillance samples measured quarterly using these protocols met the criteria of standardization required by the Centers for Disease Control, Atlanta, GA. Plasma apoA-II concen- trations were determined in triplicate using a turbidimetric assay with standards and antisera obtained from Boehringer Mannheim Biochemicals (Indianapolis, IN.).
Genetic markers
DNA was isolated from peripheral blood cells or transformed lym- phocytes and typed for polymorphisms at or near the following candidate genes: the apoA I-CII1-A IV gene cluster (chromosome 11), apoA-II (chromosome 1), apoB (chromosome 2), the apoE- CII-CI gene cluster (chromosome 19), LPL (chromosome 8), LDL receptor (chromosome 19), CETP (chromosome 16), HDL-binding protein (chromosome 2), and apo(a) (chromosome 6). These poly- morphisms are described in Table 1.
As regards genotyping at the apoC-III tetranucleotide repeat (Zuliani and Hobbs 1990a), novel primers were designed by the Oligo program to amplify the apoC-III tetranucleotide repeat in a single step. Sequences were hC3F (AGG CAG GAG AAT GGG TTG AA) and hC3R (CGG GAG AGA TGA CAG AGT TG). Polymerase chain reaction (PCR) was carried out in 96 well plates in an MJ research thermocycler. Reaction conditions were 95~ l min; 59~ 30 s; and 72~ 1 min; with 25 cycles.
Statistical analysis
The values of HDL and its two major apolipoprotein components, apoA-I and apoA-II, were adjusted by multiple regression analysis for age, sex, and body mass index (BMI). The methodology of ro- bust sib-pair test of linkage was used to test the hypotheses as to whether there was evidence for linkage between a genetic locus controlling a quantitative trait of interest and a specific polymor- phic marker locus (Haseman and Elston 1972; Amos et al. 1989). The underlying basis for this approach is to compare the quantita- tive variation in a trait between siblings as a function of the num- ber of marker alleles they share identical-by-descent (IBD). The actual sib-pair linkage analyses and ordering of marker loci are performed by utilizing the SIBPAL subroutine program of SAGE (1992), version 2.1. This program also calculates the relative prob- ability of the IBD status of each sib-pair. Additional linkage analy- ses were performed on those sib-pairs in which an exact determi- nation of the number of alleles IBD was possible from the pedigree structure. The intraclass correlations between those sib-pairs shar- ing exactly two and zero marker alleles IBD were also calculated. The ILINK subroutine program of LINKAGE 5.2 (Lathrop et al. 1984) was used to estimate the genetic distance between the re- striction fragment length polymorphism (RFLP) markers, which are usually at or only a few kilobases from the actual candidate gene locus, and additional more informative marker(s), such as (CA)n microsatellite repeats.
In addition to utilizing a robust linkage technology that mini- mizes assumptions regarding mode of inheritance, we have tried to avoid false positive results by the following analytic measures: (1) We have excluded from our analyses those sibs with extreme trait values, i.e. greater than the mean _+3 S.D. (2) We excluded those sib-pairs with very large squared trait differences, i.e., greater than the mean +3 S.D. (3) We used the more conservative unweighted least-squares option during sib-pair analysis. (4) Finally, to be
641
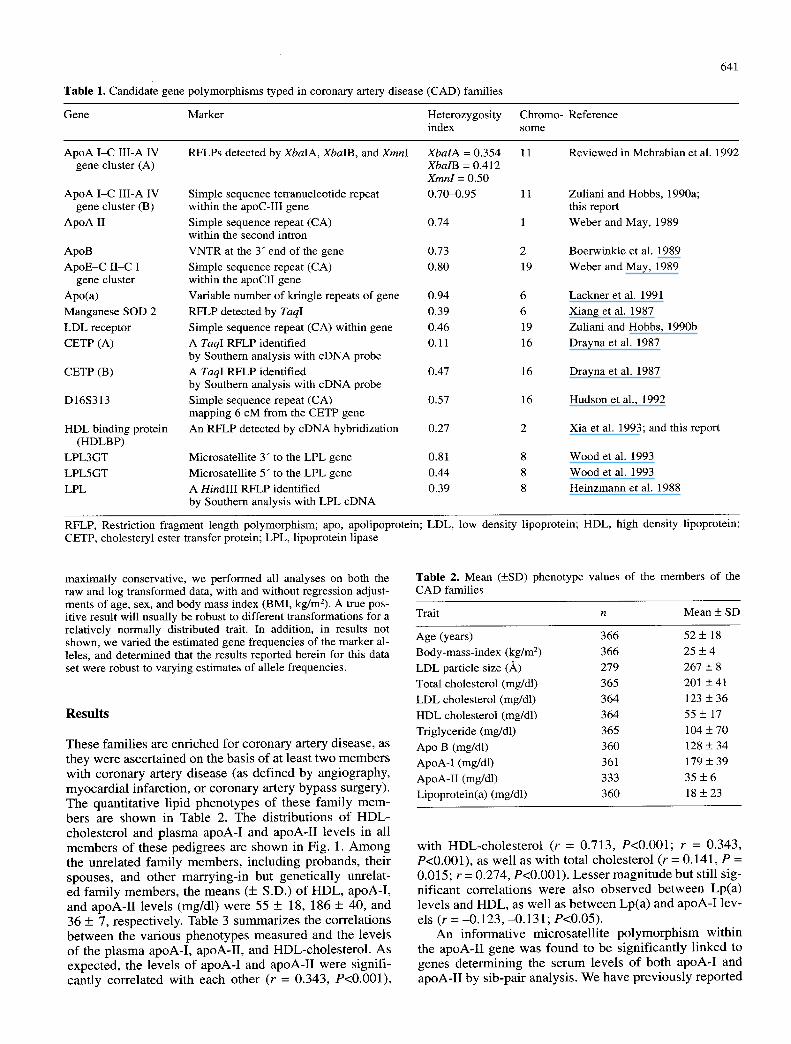
Table 1. Candidate gene polymorphisms typed in coronary artery disease (CAD) families
Gene Marker Heterozygosity Chromo- Reference index some
ApoA I-C III-A IV RFLPs detected by XbalA, XbalB, and XmnI XbalA = 0.354 11 Reviewed in Mehrabian et al. 1992 gene cluster (A) XbalB = 0.412
Xmnl = 0.50 0.70-0.95 11
0.74 1
0.73 2 0.80 19
0.94 6 0.39 6 O.46 19 0.11 16
0.47 16
0.57 16
0.27 2
0.81 8 0.44 8 0.39 8
ApoA I-C III-A IV gene cluster (B)
ApoA II
ApoB ApoE-C II-C I
gene cluster Apo(a) Manganese SOD 2 LDL receptor CETP (A)
CETP (B)
D16S313
HDL binding protein (HDLBP)
LPL3GT LPL5GT LPL
Simple sequence tetranucleotide repeat within the apoC-III gene Simple sequence repeat (CA) within the second intron VNTR at the 3" end of the gene Simple sequence repeat (CA) within the apoCII gene Variable number of kringle repeats of gene
RFLP detected by TaqI
Simple sequence repeat (CA) within gene A TaqI RFLP identified by Southern analysis with cDNA probe A TaqI RFLP identified by Southern analysis with cDNA probe Simple sequence repeat (CA) mapping 6 cM from the CETP gene An RFLP detected by cDNA hybridization
Microsatellite 3" to the LPL gene Microsatellite 5" to the LPL gene A HindIII RFLP identified by Southern analysis with LPL cDNA
Zuliani and Hobbs, 1990a; this report Weber and May, 1989
Boerwinkle et al. 1989 Weber and May, 1989
Lackner et al. 1991 Xiang et al. 1987 Zuliani and Hobbs, 1990b Drayna et al. 1987
Drayna et al. 1987
Hudson et al., 1992
Xia et al. 1993; and this report
Wood et al. 1993 Wood et al. 1993 Heinzmann et al. 1988
RFLP, Restriction fragment length polymorphism; apo, apolipoprotein; LDL, low density lipoprotein; HDL, high density lipoprotein; CETP, cholesteryl ester transfer protein; LPL, lipoprotein lipase
maximally conservative, we performed all analyses on both the raw and log transformed data, with and without regression adjust- ments of age, sex, and body mass index (BMI, kg/m2). A true pos- itive result will usually be robust to different transformations for a relatively normally distributed trait. In addition, in results not shown, we varied the estimated gene frequencies of the marker al- leles, and determined that the results reported herein for this data set were robust to varying estimates of allele frequencies.
Resu l t s
These families are enriched for coronary artery disease, as they were ascertained on the basis of at least two members with coronary artery disease (as defined by angiography, myocardial infarction, or coronary artery bypass surgery). The quantitative lipid phenotypes of these family mem- bers are shown in Table 2. The distr ibutions of HDL- cholesterol and plasma apoA-I and apoA-II levels in all members of these pedigrees are shown in Fig. 1. A mong the unrelated family members , including probands, their spouses, and other marrying- in but genetically unrelat- ed family members , the means (_+ S.D.) of HDL, apoA-I, and apoA-II levels (mg/dl) were 55 + 18, 186 _+ 40, and 36 _+ 7, respectively. Table 3 summarizes the correlations between the various phenotypes measured and the levels of the plasma apoA-I, apoA-II, and HDL-cholesterol . As expected, the levels of apoA-I and apoA-II were signifi- cantly correlated with each other (r = 0.343, P<0.001),
Table 2. Mean (+SD) phenotype values of the members of the CAD families
Trait n Mean + SD
Age (years) 366 52 + 18 Body-mass-index (kg/m 2) 366 25 + 4 LDL particle size (~) 279 267 + 8 Total cholesterol (mg/dl) 365 201 + 41 LDL cholesterol (mg/dl) 364 123 + 36 HDL cholesterol (mg/dl) 364 55 + 17 Triglyceride (mg/dl) 365 104 + 70 Apo B (mg/dl) 360 128 + 34 ApoA-I (mg/dl) 361 179 + 39 ApoA-II (mg/dl) 333 35 + 6 Lipoprotein(a) (mg/dl) 360 18 + 23
with HDL-cholesterol (r = 0.713, P<0.001; r = 0.343, P<0.001), as well as with total cholesterol (r = 0.141, P = 0.015; r = 0.274, P<0.001). Lesser magni tude but still sig- nif icant correlations were also observed between Lp(a) levels and HDL, as well as between Lp(a) and apoA-I lev- els (r = -0 .123, -0 .131 ; P<0.05).
A n informative microsatell i te po lymorphism within the apoA-II gene was found to be significantly l inked to genes determining the serum levels of both apoA-I and apoA-II by sib-pair analysis. We have previously reported

642
100 90 80 7o
b-, Z 60
50 O O 40
30 20 10 0
20 40 60 80 100 120 14050 100 150 200 250 300 350 15 20 25 30 35 40 45 50
HDL (mg/dl) ApoAI (mg/dl) ApoAlI (mg/dl)
Fig. 1. Distributions of HDL, apoA-I, and apoA-II levels (mg/dl) in the coronary artery disease families
Table 3. Correlations between plasma apo-A-I, apo-AII, and HDL-cholesterol concentration and other lipid variables a
Variable (mg/dl) ApoA I ApoA II HDL Cholesterol
n Correlation P value n Correlation P value n Correlation P value
Total cholesterol b 294 0.141 0.015" 274 0.274 0.001" HDL-cholesterol b 294 0.713 0.001" 274 0.343 0.001" LDL-cholesterol b 294 -0.042 0.470 274 0.042 0.493 Triglyceride b 294 -0.127 0.030* 274 0.170 0.005* ApoB b 294 -0.122 0.037* 269 0.092 0.131 ApoAI b - - - 269 0.343 0.001" ApoAII b 269 0.343 0.001" - - -
Lp(a) b 292 -0.131 0.025* 267 -0.036 0.557
300 0.117 0.043*
300 -0.136 0.018" 300 -0.374 0.001" 294 --0.240 0.001" 294 0.713 0.001" 274 0.343 0.001" 292 -0.123 0.036*
* P<0.05 a All variables adjusted for age, sex, and body mass index b Units: mg/dl
Table 4. P-values of sib-pair linkage analyses for ApoA-I levels (mg/dl) with apoA-II locus
Genetic Loci No. of P values sib- pairs Log ApoA-I
(ApoA-I)
ApoA-1 linkage (SIBPAL program)
(a) Without age, sex, bmi adjustment:
apoA-II 99 0.06 0.140
(b) With age, sex, bmi adjustment:
apoA-II 91 0.009 0.030
ApoA-1 linkage on those sib-pairs sharing exactly two, one, and zero apoAH alleles
(a) Without age, sex, bmi adjustment:
apoA-II 59 0.068 0.133
(b) With age, sex, bmi adjustment:
apoA-II 59 0.015 0.038
bmi, Body mass index
the l inkage of the apoA-II gene with a locus determining apoA-II levels after adjustment for sex, age, and body- mass index (Warden et al. 1993c). An additional 12 sib- pairs are included in the present study, yielding a similar
P-value of 0.02 for the total combined data. Fol lowing ad- jus tment for age, sex and body mass index, a P-value of 0.009 was obtained for the l inkage of log(apoA-I) levels with the apoA-II gene in 91 sib-pairs (Table 4). A P-value of 0.03 was obtained for the untransformed apoA-I levels. Similar significant P-values (P = 0.015 and 0.038 for log(apoA-I) and apoA-I levels, respectively) were ob- tained by limiting the analysis to the 59 sib-pairs sharing exactly two, one, and zero apoA-II alleles IBD (Fig. 2). Figure 3 shows the correlations of apoA-I levels among sib-pairs sharing two apoA-II alleles IBD (r = 0.71) and zero apoA-II alleles IBD (r = 0.27). The levels of HDL- cholesterol did not exhibi t s ignif icant ev idence for l ink- age with the apoA-II locus at the current sample size (P = 0.106, 99 sib-pairs), though this result is in the cor- rect direct ion for l inkage. We also examined the evi- dence for l inkage of a locus de te rmin ing apoA-I levels with two highly po lymorphic markers located 6 cM (D1S74) and 12 cM (D1S75) from the apoA-I I gene on h u m a n chromosome 1. Non-s ign i f ican t P-va lues (P = 0.19 and 0.67, respect ively) were obta ined with these two markers, indicating no evidence for l inkage at these distances.
Evidence for l inkage was also observed between a mi- crosatellite polymorphism (D 16S313) l inked to the gene for CETE and a locus determining HDL-cholesterol level and apoA-I level by the sib-pair l inkage method (Table 5). Analysis of a subset of the pedigrees (9 pedigrees) with a

S i b - p a i r s q u a r e d a p o A - I ( m g / d l ) d i f f e r e n c e
643
16,ooo
14,ooo
12,ooo
lO,OOO
8,ooo
6,ooo
4,000
2,000
-A
r = - 0 . 2 3 N=59
P = 0 . 0 3 8
A
-A
/x
&
/,,
1----- 1
N u m b e r o f a p o A - I I a l l e l e s s h a r e d I B D
A
2x
2
Fig. 2. Significant regression of squared trait differ- ences for unadjusted plasma apoA-I levels versus the number of apoA-II alleles that a sib-pair shares identi- cal-by-descent (IBD). This figure includes only those sib-pairs whose apoA-II marker IBD genotypes could be definitely assigned to sharing exactly two, one, and zero alleles at the apoA-II locus
250
200"
a. 150
..c, 100"
A0,71 "/f" g �9 �9
r=0,27 �9 �9 ~
e~
~5
Sib 2, ApoA-I (mg/dl)
Fig. 3 A, B. Intraclass correlations of apoA-I levels between sib- pairs. A Sib-pairs sharing exactly two (11) apoA-II marker alleles IBD; B Sib-pairs sharing zero (O) apoA-II marker alleles IBD
TaqI RFLP of the CETP gene [referred to in Table 1 as CETP(B)] yielded possible evidence for linkage (P = 0.07 and P = 0.02) of the CETP gene with log-transformed plasma HDL-cholesterol and apoA-I levels, following ad- justment for age, sex, and body mass index. To increase informativeness, a microsatellite polymorphism in the vicinity of the CETP gene (D 16S313) was typed among a larger set of pedigrees (104 sib-pairs). The estimated dis- tance between the microsatellite and the CETP gene was estimated at 6.2 cM by the ILINK program of LINKAGE
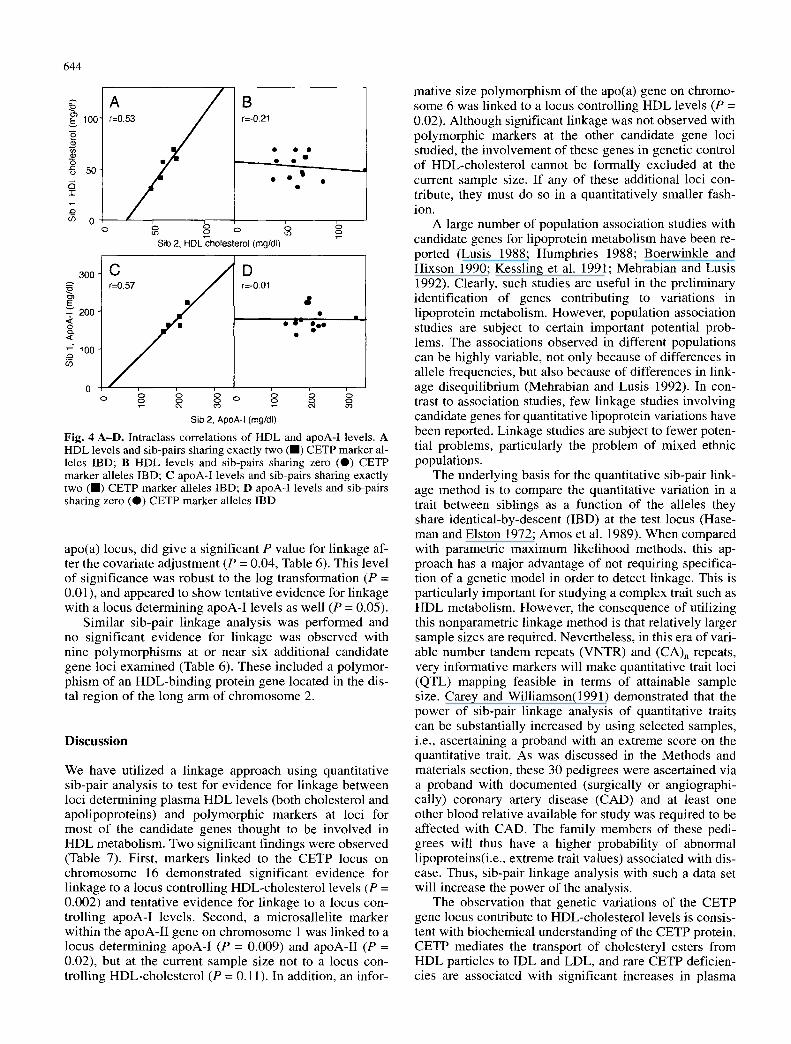
5.1 (Lathrop et al. 1984). Following adjustment for sex, age, and BMI, significant evidence for linkage was ob- served between D16S313 locus and log-transformed HDL-cholesterol levels by sib-pair analysis (P = 0.002; Table 5). The levels of significance for the apoA-I levels versus the CETP locus were more variable, and achieved statistical significance only if more extreme values (i.e. possible outliers) were included in the analysis (Table 5). No evidence for linkage was observed between D16S313 locus and the locus controlling apoA-II levels (P = 0.41). Figure 4 shows the intraclass correlations of HDL and apoA-I levels between sib-pairs sharing two or zero D16S313 alleles identical-by-descent. Consistent with the above sib-pair linkage analyses, sib-pairs sharing both D 16S313 alleles IBD have a much higher intraclass corre- lations for these quantitative traits than those of sib-pairs sharing no D16S313 alleles IBD.
Tentative evidence for linkage was also observed be- tween polymorphisms of the structural gene for apt(a), encoding a polypeptide of the Lp(a) particle, and a locus determining unadjusted HDL levels (P = 0.02, 104 sib- pairs; Table 6). However, after adjusting the HDL levels for age, sex, and BMI, the evidence of linkage did not quite reach statistical significance (P = 0.08). The results were not significant with log transformation. In contrast, a less informative RFLP marker for the manganese super- oxide dismutase 2 gene, which is about 10 cM from the
Table 5. P-values of sib-pair linkage analyses for HDL and apoA-I levels with the D16S313 locus, which is linked to CETP
Adjustment No. of P-values No. of P-values No. of P-values sib-pairs sib-pairs sib-pairs
Log HDL Log apoA-I (HDL) (apoA-I)
Log apoA-II (apoA-II)
Without age, sex, 104 0.030 0.174 99 0.054 0.080 97 0.40 0.40 bmi adjustment (0.015)*
With age, sex, 99 0.002 0.026 95 0.140 0.074 93 0.41 0.31 bmi adjustment (0.048)* (0.015)*
* P values in parentheses are obtained from the analyses using all sib-pairs (including possible outliers)
644
~ 100
o -~ 50-
"T
O3
B �9 r=-0.21
0o o ~ o ~ Sib 2, HDL cholesterol (mg/dl)
�9 �9 �9
300 C
200 �9
<
' - - 1 0 0
6'3
o o o o ~ ~ ~
Sib 2, ApoA-I (mg/dl)
Fig. 4 A-D. Intraclass correlations of HDL and apoA-I levels. A HDL levels and sib-pairs sharing exactly two (m) CETP marker al- leles IBD; B HDL levels and sib-pairs sharing zero (O) CETP marker alleles IBD; C apoA-I levels and sib-pairs sharing exactly two (1) CETP marker alleles IBD; D apoA-I levels and sib-pairs sharing zero (O) CETP marker alleles IBD
D r=-O.01
J
Q
apo(a) locus, did give a significant P value for linkage af- ter the covariate adjustment (P = 0.04, Table 6). This level of significance was robust to the log transformation (P = 0.01), and appeared to show tentative evidence for linkage with a locus determining apoA-I levels as well (P = 0.05).
Similar sib-pair linkage analysis was performed and no significant evidence for linkage was observed with nine polymorphisms at or near six additional candidate gene loci examined (Table 6). These included a polymor- phism of an HDL-binding protein gene located in the dis- tal region of the long arm of chromosome 2.
Discussion
We have utilized a linkage approach using quantitative sib-pair analysis to test for evidence for linkage between loci determining plasma HDL levels (both cholesterol and apolipoproteins) and polymorphic markers at loci for most of the candidate genes thought to be involved in HDL metabolism. Two significant findings were observed (Table 7). First, markers linked to the CETP locus on chromosome 16 demonstrated significant evidence for linkage to a locus controlling HDL-cholesterol levels (P = 0.002) and tentative evidence for linkage to a locus con- trolling apoA-I levels. Second, a microsallelite marker within the apoA-II gene on chromosome 1 was linked to a locus determining apoA-I (P = 0.009) and apoA-II (P = 0.02), but at the current sample size not to a locus con- trolling HDL-cholesterol (P = 0.11). In addition, an infor-
mative size polymorphism of the apo(a) gene on chromo- some 6 was linked to a locus controlling HDL levels (P = 0.02). Although significant linkage was not observed with polymorphic markers at the other candidate gene loci studied, the involvement of these genes in genetic control of HDL-cholesterol cannot be formally excluded at the current sample size. If any of these additional loci con- tribute, they must do so in a quantitatively smaller fash- ion.
A large number of population association studies with candidate genes for lipoprotein metabolism have been re- ported (Lusis 1988; Humphries 1988; Boerwinkle and Hixson 1990; Kessling et al. 1991; Mehrabian and Lusis 1992). Clearly, such studies are useful in the preliminary identification of genes contributing to variations in lipoprotein metabolism. However, population association studies are subject to certain important potential prob- lems. The associations observed in different populations can be highly variable, not only because of differences in allele frequencies, but also because of differences in link- age disequilibrium (Mehrabian and Lusis 1992). In con- trast to association studies, few linkage studies involving candidate genes for quantitative lipoprotein variations have been reported. Linkage studies are subject to fewer poten- tial problems, particularly the problem of mixed ethnic populations.
The underlying basis for the quantitative sib-pair link- age method is to compare the quantitative variation in a trait between siblings as a function of the alleles they share identical-by-descent (IBD) at the test locus (Hase- man and Elston 1972; Amos et al. 1989). When compared with parametric maximum likelihood methods, this ap- proach has a major advantage of not requiring specifica- tion of a genetic model in order to detect linkage. This is particularly important for studying a complex trait such as HDL metabolism. However, the consequence of utilizing this nonparametric linkage method is that relatively larger sample sizes are required. Nevertheless, in this era of vari- able number tandem repeats (VNTR) and (CA)n repeats, very informative markers will make quantitative trait loci (QTL) mapping feasible in terms of attainable sample size. Carey and Williamson(1991) demonstrated that the power of sib-pair linkage analysis of quantitative traits can be substantially increased by using selected samples, i.e., ascertaining a proband with an extreme score on the quantitative trait. As was discussed in the Methods and materials section, these 30 pedigrees were ascertained via a proband with documented (surgically or angiographi- cally) coronary artery disease (CAD) and at least one other blood relative available for study was required to be affected with CAD. The family members of these pedi- grees will thus have a higher probability of abnormal lipoproteins(i.e., extreme trait values) associated with dis- ease. Thus, sib-pair linkage analysis with such a data set will increase the power of the analysis.
The observation that genetic variations of the CETP gene locus contribute to HDL-cholesterol levels is consis- tent with biochemical understanding of the CETP protein. CETP mediates the transport of cholesteryl esters from HDL particles to IDL and LDL, and rare CETP deficien- cies are associated with significant increases in plasma

Table 6. P-values of sib-pair linkage analy- ses for other candidate loci
645
Candidate loci a No. of P-values No. of P-values sib-pairs sib-pairs
Log HDL Log apoA-I (HDL) (apoA-I)
Without age, sex, bmi adjustment
ApoAI-CIII-AIV(A) b 67 0.10 0.19 65 0.19 0.21 ApoAI-CIII-AIV(B) 95 0.26 0.47 88 0.10 0.09 apoB 101 0.88 0.86 96 0.39 0.31 apoE-CII-CI 35 0.99 1.00 30 0.90 0.96 apolipoprotein(a) 104 0.10 0.02 99 0.08 0.13 Manganese SOD 2 58 0.08 0.13 58 0.11 0.12 LDL-receptor 103 0.30 0.16 98 0.93 0.91 HDL binding protein 115 0.75 0.54 110 0.80 0.69 LPL3GT 86 0.66 0.41 84 0.70 0.74 LPL5GT 102 0.99 0.97 97 0.96 0.93 Lipoprotein lipase 82 0.92 0.85 81 0.96 0.97
With age, sex, bmi adjustment
ApoA I-C III-A IV(A) b 66 0.04 0.12 65 0.13 0.15 ApoA I-C III-A IV(B) 92 0.51 0.68 87 0.21 0.15 apoB 100 0.63 0.73 95 0.25 0.20 poCI-CII-E 35 0.95 0.98 30 0.81 0.95 apolipoprotein(a) 102 0.26 0.08 97 0.24 0.23 Manganese SOD 2 57 0.01 0.04 57 0.05 0.06 LDL-receptor 102 0.11 0.11 97 0.93 0.90 HDL binding protein 110 0.78 0.41 105 0.90 0.65 LPL3GT 81 0.64 0.51 79 0.67 0.73 LPL5GT 97 0.89 0.88 92 0.79 0.58 Lipoprotein lipase 77 0.98 0.97 76 0.95 0.95
a see Table 1 for definitions of candidate loci. b Based on the haplotypes generated by three RFLPs
Table 7. Summary of sib-pair linkage analyses for HDL and its as- sociated apolipoprotein levels with D16S313 (CETP linked) and with apoA-II
Quantitative trait Candidate gene locus
D16S313 (CETP) apoA-II
Without age, sex, bmi adjustment
HDL cholesterol Linked Unlinked a apoA-I levels Borderline Linked apoA-II levels Unlinked Linked
With age, sex, bmi adjustment
HDL cholesterol Linked Unlinked a apoA-I levels Borderline Linked apoA-II levels Unlinked Linked
a Unlinked in current sample, although the P-value is in the correct direction for indicating linkage
HDL-cho les t e ro l levels (Inazu et al. 1990). Moreover , previous associat ion (Kondo et al. 1989) and l inkage (Heiba et al. 1993) studies have sugges ted that the CETP locus contr ibutes to HDL-cho le s t e ro l levels. One cons idera t ion in the interpretat ion o f this f inding is that the gene for LCAT, invo lved in the es ter i f icat ion o f HDL-choles te ro l ,
res ides near the CETP gene on c h romosome 16. Unfor tu- nately, po lymorph i sms at the LCAT gene have not been ident i f ied in humans. Never theless , associa t ion studies showing that po lymorph i sms o f the CETP gene are asso- c ia ted with al terat ions in HDL-cho le s t e ro l levels suppor t the concept that the CETP locus d i rec t ly contr ibutes , at least in part, to the obse rved var ia t ions in HDL-cho le s - terol. This is so since l inkage d i sequi l ib r ium is un l ike ly to extend more than a few cent imorgans , and in fact usual ly only a few hundred k i lobases (a f ract ion o f a cen t imorgan in humans) , as has been demons t ra ted in the studies o f cyst ic f ibrosis and o f Hunt ington d isease (Kerem et al. 1989; Andrew et al. 1992). However , in our data set, the D16S313 locus was est imated to be 6.2 cM from the CETP locus and it may still be poss ib le that the var ia t ions at the D16S313 locus affecting H D L ref lect the var ia t ions at the LCAT locus rather than at the CETP locus, or even that both loci contribute.
The f inding that the apoA- I I gene locus is l inked to genes control l ing the levels of both a p o A - I and apoA- I I but not to HDL-cho les t e ro l is unexpected . We recent ly observed s ignif icant l inkage be tween the apoA- I I gene lo- cus and a locus control l ing the levels of p l a sma apoA- I I and p la sma free fatty acids in both mice and humans (Warden et al. 1993c). Genet ic studies and studies with t ransgenic animals have revea led that the level of apoA- I I

646
production has a profound effect on plasma HDL-choles- terol levels in mice, whereas apoA-I levels are only mini- mally influenced (Doolittle et al. 1990; Warden et al. 1993b). In contrast, the present findings indicate that apoA-II gene variations do not significantly affect levels of HDL-cholesterol but do significantly affect levels of apoA-I. (Alternatively, the effect on apoA-I is relatively greater than that on HDL). The explanation is unclear, but the difference in the findings in mice and humans may be related to certain important differences in HDL metabo- lism. For example, mice have little or no plasma CETP, and mouse apoA-II is a monomer rather than a dimer (dis- cussed in Hedrick et al. 1993). It should be noted that all our linkage results should still be considered somewhat tentative due to our studies involving several statistical comparisons. While the apoA-II locus appears to be linked to a locus (or loci) determining both apoA-I and apoA-II levels, this linkage methodology cannot distinguish whether the effects on apoA-I and apoA-II are independent effects of the locus, or indirectly linked, i.e., one quantitative trait mediating the other, and that the linkage of both is due to their consequent biochemical correlations.
The observation of tentative linkage between a locus controlling HDL levels and the apt(a) locus should be considered preliminary. As discussed above, this could simply be a type-I error. Further studies will be required to confirm this result. The biochemical basis of the ob- served linkage between the apt(a) gene and HDL levels is totally unknown. Interestingly, in one previous associa- tion study, HDL levels were shown to vary with genetic variation at the apt(a) locus (Hegele et al. 1991). As shown in Table 3, borderline significant correlations(neg- ative) between the Lp(a) level and the levels of apoA-I, apoA-II, or HDL-cholesterol were observed in our data. Nevertheless, the metabolism of the various lipoprotein particles is highly interrelated, and undoubtedly many in- teractions have remained undetected.
No evidence of linkage between HDL levels and the remaining candidate genes surveyed was observed. Some of these tested loci have revealed significant relationships with HDL levels or its associated apolipoprotein levels in previous association studies, including the apoAI-CIII- AIV gene cluster (reviewed in Humphries 1988; Lusis 1988) and the LPL gene (Heinzmann et al. 1991). There are several possible explanations: (1) the current sample size may have been inadequate to detect such a linkage if it exists; (2) the informativeness of the polymorphisms and closeness of the marker to the candidate genes may have been inadequate; (3) the associations previously ob- served may have been artifactual; (4) the current popula- tion studies may not exhibit variations that occur in other populations; and (5) the previously reported associations may indicate only quantitatively minor polygenic effects rather than major gene effects tested by the linkage meth- ods utilized herein. If the prior association results have been consistent over a sufficient number of studies and populations, the latter hypothesis is the most intriguing, and emphasizes the importance of a linkage approach to complement population association studies.
In conclusion, our results indicate that: (1) the CETP gene contributes to interindividual variation in HDL-cho-
lesterol levels; (2) variation of the apoA-II gene deter- mines, in part, HDL apolipoprotein composition, i.e., apoA-I and apoA-II levels; and (3) the apt(a) gene may also be involved in determining HDL levels. In consider- ing the limitations of robust sib-pair linkage analysis methodology and multiple candidate gene loci tested (i.e., relatively large number of statistical tests) in this paper, we feel that all these findings require further confirma- tion. Although we have examined many of the biochemi- cally defined genes likely to Contribute to HDL levels and the remaining few are to be examined in ongoing studies, this does not exhaust the potential of other, as yet unknown, genes. Indeed, it seems likely that many loci presently un- known (for example, those encoding regulators of gene transcription) are likely to participate in the control of HDL levels (Warden et al. 1993a).
Acknowledgements. The authors thank Judy Tyler (Cedars-Sinai Medical Center) for assistance with aspects of this work. This work was supported by NIH grants HL42481 (A.J.L. and J.I.R.) and HL28481 (A.J.L.). J.I.R. was also supported by a grant from the Stuart Foundations and the Cedars-Sinai Board of Governors' Chair in Medical Genetics. C.W. was supported, in part, by a grant-in aid from the AHA/GLAA. X.B. was supported, in part, by NIH endocrinology training grant DK07426. The results of sib- pair analysis were obtained by using the program package SAGE, which is supported by U.S. Public Health Service Resource Grant 1 P41 RR03655 from the Division of Research Resources.
References
Agellon LB, Walsh A, Hayek T, Moulin P, Jiang XC, Shelanski SA, Breslow JL, Tall AR (1991) Reduced high density lipo- protein cholesterol in human cholesterol ester transfer protein transgenic mice. J Biol Chem 266:101796-101801
Amos C1, Elston RC, Wilson AF, Bailey-Wilson JE (1989) A more powerful robust sib-pair test of linkage for quantitative traits. Genet Epidemiol 6:435-449
Andrew S, Theilmann J, Hedrick A, Mah D, Weber B, Hayden MR (1992) Nonrandom association between Huntington dis- ease and two loci separated about 3 Mb on 4pt6.3. Genomics 13:301-311
Berg K (1989) Impact of medical genetics on research and prac- tices in the area of cardiovascular disease. Clin Genet 36: 299-312
Boerwinkle E, Hixson JE (1990) Genes and normal lipid varia- tions. Curr Opin Lipids 1:151-159
Boerwinkle E, Xiong W, Fourest E, Chan L (1989) Rapid typing of tandemly repeated hypervariable loci by the polymerase chain reaction: application to the apolipoprotein B 3" hyper- variable region. Proc Natl Acad Sci USA 86:212-216
Breslow JL (1989) Familial disorders of high density lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. Mc- Graw-Hill, New York, pp 1251-1264
Breslow JL (1991) Lipoprotein transport gene abnormalities un- derlying coronary heart disease susceptibility. Annu Rev Med 42:357-371
Brown ML, Inazu A, Hesler CB, Agellon LB, Mann C, Whitlock ME, Marcel YL, Milne RW, Koizumi J, Mabuchi H, Takeda R, Tall AR (1989) Molecular basis of lipid transfer protein de- ficiency in a family with increased high density lipoproteins. Nature 342:448-451
Carey G, Williamson J (1991) Linkage analysis of quantitative traits: increased power by using selected samples. Am J Hum Genet 49:786-796

647
Deeb SS, Takata K, Peng R, Kajiyama G, Albers JJ (1990) A splice-junction mutation responsible for familial apolipopro- tein AI1 deficiency. Am J Hum Genet 46:822-827
Doolittle MH, LeBoeuf RC, Warden CH, Bee LM, Lusis AJ (1990) A polymorphism affecting apolipoprotein A-II transla- tional efficiency determines high density lipoprotein size and composition. J Biol Chem 265:161380-161388
Doolittle MH, Durstenfeld A, Garfinkel AS, Schotz MC (1992) Triglyceride lipases, hypertriglyceridemia and atherosclerosis. In: Lusis AJ, Rotter JI, Sparkes RS (eds) Molecular genetics of coronary artery disease: candidate genes and processes in ath- erosclerosis. Karger, Basel, pp 172-188
Drayna D, Lawn R (1987) Multiple RFLPs at the human choles- terol ester transfer protein (CETP) locus. Nucleic Acids Res 15:4698
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR (1972) High density lipoprotein as a protective factor against coronary heart disease: the Framingham study. Am J Med 63: 7 O0-714
Graham DL, t r a m JF (1987) Identification and characterization of a high density lipoprotein binding protein in cell membranes by ligand blotting. J Biol Chem 262:7435-7442
Hamsten A, Iselius L, Dahlen G, Faire U de (1986) Genetic and cultural inheritance of serum lipids, low and high density lipo- protein cholesterol and serum apolipoproteins A-I, A-II and B. Atherosclerosis 60:199-208
Haseman JK, Elston RC (1972) The investigation of linkage be- tween a quantitative trait and a marker locus. Behav Genet 2: 3-19
Hedrick CC, Castellani LW, Warden CH, Puppione DL, Lusis AJ (1993) Influence of mouse apolipoprotein A-II on plasma lipoproteins in transgenic mice. J Biol Chem 268:20676- 20 682
Hegele RA, Sutherland S, Robertson M, Wu L, Emi M, Hopkins PN, Williams RR, Lalouel JM (1991) The effect of genetic de- terminants of low density lipoprotein levels on lipoprotein(a). Clin Invest Med 14:146-152
Heiba IM, DeMeester CA, Xia YR, Diep A, George VT, Amos CI, Srinivasan SR, Berenson GA, Elston RC, Lusis AJ (1993) Ge- netic contributions to quantitative lipoprotein traits associated with coronary artery disease: analysis of a large pedigree from the Bogalusa Heart Study. Am J Med Genet 47:875-883
Heinzmann C, Ladias J, Antonarakis S, Diep A, Schotz M, Lusis AJ (1988) Two polymorphisms for the human hepatic lipase (HL) gene. Nucleic Acids Res 16:4739
Heinzmann C, Kirchgessner T, Kwiterovich PO, Ladias JA, Derby C, Antonarakis SE, Lusis AJ (1991) DNA polymorphism hap- lotypes of the human lipoprotein lipase gene: possible associa- tion with high density lipoprotein levels. Hum Genet 86:578- 584
Hudson T J, Engelstein M, Lee MK, Ho EC, Rubenfield M J, Adams CP, Houseman DE, Dracopoli NC (1992) Isolation and chromosomal assignment of 100 highly informative human simple sequence repeat polymorphisms. Genomics 13:622- 629
Humphries SE (1988) DNA polymorphisms of the apolipoprotein genes - their use in the investigation of the genetic component of hyperlipidemia and atherosclerosis. Atherosclerosis 72:89- 108
Hunt SC, Hasstedt S J, Kuida H, Stults BM, Hopkins PN, Williams RA (1989) Genetic heritability and common environmental components of resting and stressed blood pressures, lipids and body mass index in Utah pedigrees and twins. Am J Epidemiol 129:625-638
Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruhama Y, Mabuchi H, Tall AR (1990) Increased high density lipoprotein levels caused by a common cholesteryl-es- ter transfer protein gene mutation. N Engl J Med 323: 1234-1238
Karathanasis S (1992) Lipoprotein metabolism: high density lipoproteins. In: Lusis AJ, Rotter JI, Sparkes RS (eds) Molecu-
lar genetics of coronary artery disease: candidate genes and processes in atherosclerosis. Karger, Basel, pp 140-171
Kerem BS, Rommens JM, Buchnan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073-1080
Kessling A, Ovellette S, Bouffard O, Chamberland A, Bttard C, Selinger E, Xhignesse M, Lussier-Cacan S, Davignon J (1991) Patterns of association between genetic variability in apolipo- protein (apt) B, apt AI-CIII-AIV and cholesterol ester transfer protein gene regions and quantitative variation in lipid and lipoprotein traits: influence of gender and exogenous hor- mones. Am J Hum Genet 50:92-106
Kondo I, Berg K, Drayna D, Lawn R (1989) DNA polymorphism at the locus for human cholesterol ester transfer protein (CETP) is associated with high density lipoprotein cholesterol and apolipoprotein levels. Clin Genet 35:49-56
Lackner CE, Boerwinkle E, Leffert CC, Rahmig T, Hobbs HH (1991) Molecular basis of apolipoprotein(a) isoform size het- erogeneity as revealed by pulsed-field gel electrophoresis. J Clin Invest 87:2077-2086
Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for mul- tilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443-3446
Lusis AJ (1988) Genetic factors affecting blood lipoproteins: the candidate gene approach. J Lipid Res 29:397-429
Mehrabian M, Qiao J-H, Hyman R, Ruddle D, Laughton C, Lusis AJ (1993) Influence of the apoA-II gene locus on HDL levels and fatty streak development in mice. Arterioscl Thromb 13: 1-10
Mehrabian M, Lusis AJ (1992) Genetic markers for studies of ath- erosclerosis and related traits. In: Lusis AJ, Rotter JI, Sparkes RS (eds) Molecular genetics of coronary artery disease: candi- date genes and processes in atherosclerosis. Karger, Basel, pp 363-418
Miller NE (1987) Associations of high-density lipoprotein sub- classes and apolipoproteins with ischemic heart disease and coronary atherosclerosis. Am Heart J 113:589-597
Moll PP, Sing CF, Williams RR, Mat SJT, Kottke BA (1986) The genetic determination of plasma apolipoprotein A-I levels measured by radioimmunoassay: a study of high-risk pedi- grees. Am J Hum Genet 38:361-372
Plump AS, Smith JD, Hayek T, Aalto-Set~il~i K, Walsh A, Ver- stuyft JG, Rubin EM, Breslow JL (1992) Severe hypercholes- terolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71:343-353
Rat DC, Lalouel JM, Suarez BR, Schonfeld G, Glueck CJ, Sier- vogel RM (1983) A genetic study of hyper-alphalipoproteine- mia. Am J Med Genet 15:195-203
Rubin EM, Ishida BY, Clift SM, Krauss RM (1991) Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size particles. Proc Natl Acad Sci USA 88:434-438
SAGE, Statistical Analysis for Genetic Epidemiology, release 2.1 (1992) Computer program package available from the Depart- ment of Biometry and Genetics, Louisiana State University Medical Center, New Orleans
Schaefer EJ (1984) Clinical, biochemical and genetic features in familial disorders of high density lipoprotein deficiency. Arte- riosclerosis 4:303-322
Scott J, Knott TJ, Priestley LM, Robertson ME, Mann DV, Kost- ner G, Miller GJ Miller NE (1985) High-density lipoprotein composition is altered by a common DNA polymorphism adja- cent to apolipoprotein AII gene in man. Lancet I:771-773
Tall A (1990) Plasma high density iipoproteins: metabolism and relationship to atherogenesis. J Clin Invest 86:379-384
Tall A (1992) Metabolic and genetic control of HDL cholesterol levels~ J Intern Med 231:661-668
Taramelli R, Pontoglio M, Candiani G, Ottolenghi S, Dieplinger H, Catapano A, Albers J, Vergani C, McLean J (1990) Lecithin

648
cholesterol acyl transferase deficiency: molecular analysis of a mutated allele. Hum Genet 85:195-199
Thorn JA, Stocks J, Reichl D, Alcolado JC, Chamberlain JC, Gal- ton D (1993) Variability of plasma apolipoprotein (apo) A-II levels associated with an apoA-II gene polymorphism in monozygotic twins. Biochem Biophys Acta 1180:299-303
Vogler GP, Wette R, Laskarzewski PM, Perry TS, Rice T, Province MA, Rao DC (1989) Heterogeneity in the biological and cultural determinants of high-density lipoprotein choles- terol in five North American populations: the Lipid Research Clinics Family Study. Hum Hered 39:249-257
Walsh A, Ito Y, Breslow JL (1989) High levels of human apolipo- protein A-I transgenic mice result in increased plasma levels of small density lipoprotein (HDL) particles comparable to hu- man HDL3. J Biol Chem 264:6488-6494
Warden CH, Fisler JS, Pace MJ, Svenson KL, Lusis AJ (1993a) Coincidence of genetic loci for plasma cholesterol levels and obesity in a multifactorial mouse model. J Clin Invest 92:773- 779
Warden CH, Hedrick CC, Qiao JH, Castellani LW, Lusis AJ (1993b) Atherosclerosis in transgenic mice overexpressing apolipoprotein A-II. Science 261:469-472
Warden CH, Daluiski A, Bu X, Pnrcell-Huynh DA, DeMeester C, Shieh B-H, Puppione DL, Gray RJ, Reaven GM, Chen Y-DI, Rotter JI, Lusis AJ (1993c) The apolipoprotein AII gene deter- mines plasma apolipoprotein All levels and free fatty acid lev- els in both humans and mice. Proc Nail Acad Sci USA 90:101886-101890
Weber JL, May PE (1989) Abundant class of human DNA poly- morphisms which can be typed using the polymerase chain re- action. Am J Hum Genet 44:388-396
Wile DB, Barbir M, Gallagher J, Myant NB, Ritchie CD, Thomp- son GR, Humphries SE (1989) Apolipoprotein A-I gene poly- morphisms: frequency in patients with coronary artery disease and healthy controls and association with serum apoA-I and HDL-cholesterol concentration. Atherosclerosis 78:9-18
Wood S, Schertzer M, Hayden M, Ma Y (1993) Support for founder effect for two lipoprotein lipase (LPL) gene mutations in French Canadians by analysis of GT microsatellites flanking the LPL gene. Hum Genet 91:312-316
Xia YR, Klisak I, Sparkes RS, Oram J, Lusis AJ (1993) Localiza- tion of the gene for high-density lipoprotein binding protein (HDLBP) to human chromosome 2Q37. Genomics 16:524- 525
Xiang K, Cox NJ, Hallewell RA, Bell GI (1987) Multiple TaqI RFLPs at the human manganese superoxide dismutase (SOD2) locus on chromosome 6. Nucleic Acids Res 15:7654
Xu CF, Angelico F, Ben MD, Humphries S (1993) Role of genetic variation at the apo AI-CIII-A1V gene cluster in determining plasma apo A-I levels in boys and girls. Genet Epidemiol 10:113-122
Zhang SH, Reddick RL, Piedrahita JA, Maeda N (1992) Sponta- neous hypercholesterolemia and arterial lesions in mice lack- ing apolipoprotein E. Science 258:468-471
Zuliani G, Hobbs HH (1990a) Tetranucleotide repeat polymor- phism in the apolipoprotein C-Ill gene. Nucleic Acids Res 18: 4299
Zuliani G, Hobbs HH (1990b) Dinucleotide repeat polymorphism at the 3" end of the LDL receptor gene. Nucleic Acids Res 18:4300
Copyright © 2022 FDOKUMEN




















