
LETTERS TO THE EDITOR - NCBI
20
LETTERS TO THE EDITOR Volitional and stimulation induced neuromyotonic discharges: unusual electrophysiological pattern in acquired neuromyotonia Neuromyotonic discharges are electrophysi- ologically characterised as bursts of motor unit potentials firing at more than 150 Hz for 0.5 to 2 seconds. The amplitude of the response typically wanes. Discharges may occur spontaneously or be initiated by needle movement. 1 Walsh described a case of a mediastinal tumour and neuromyotonia with very high frequency discharges that outlasted voluntary eVort. 2 We report a case of an acquired paraneo- plastic neuromyotonia associated with thy- moma, clinically manifested myotonia-like muscle stiVness, and an unusual electrophysi- ological pattern of neuromyotonic discharges that were evoked voluntarily or with electrical stimulation but were absent spontaneously and were not elicited by needle displacement. A 71 year old women presented with a 6 month history of muscle stiVness, paraesthe- sias provoked mostly by movement, disturbed speech, and diYcult walking. At the time of examination she could not walk independ- ently. Clinical examination disclosed pro- nounced dysarthria and ataxic-like limb movement interrupted by superimposed tonic involuntary contractions. The muscle decontraction was prolonged and percussion myotonia was absent. Fasciculations and myokymia-like movements were seen in her arms, but occurred only sparsely and inter- mittently. The distal foot and hand muscles were slightly paretic and atrophic. Tendon reflexes were weak in the arms and absent in the legs. A decreased perception of vibration was present distally in all limbs. Computed tomography disclosed a tumour in the mediastinum, which was totally removed after initial therapy and clinical improvement; the thymoma was confirmed histologically. An examination of voltage gated K + channel (VGKC) antibody titres was per- formed using immunoprecipitation of 125 I-Æ dendrotoxin labelled VGKCs extracted from human frontal cortex 2 (Institute of Molecular Medicine, John RadcliVe Hospital, Oxford, UK). The first titre was positive (241 pM (positive titres >200 pM). During a course of intravenous immu- noglobulin (IVIg) infusions at a normal dose (0.4 g/kg on 5 subsequent days, total dose of 2 g/kg) both pseudomyotonic and sensory signs and symptoms started to improve and at the end of the IVIg treatment the patient was able to walk independently. After the initial IVIg therapy (administered 1 month before surgical removal of the thymoma), clinical signs and symptoms stabilised with the ability to walk independently for 20 metres. After a year of stabilisation, the stiVness, dysarthria, and walking ability worsened in the course of 3 months to the point at which the patient was once more unable to walk independently. The patient then received a second course of IVIg therapy (2 g/kg) and improved to the same degree as after the first treatment. An EMG at the beginning of clinical follow up disclosed sparse fasciculations and my- okymic discharges (with a short interburst interval of about 5–10 ms) and motor unit potentials with slightly higher amplitude, longer duration, mild waveform instability, and polyphasic pattern from distal muscles in the lower limbs. Voluntary contraction evoked repetitive bursts of high frequency discharges resembling motor unit potentials with amplitude decrement and a characteris- tic “pinging” sound (figure); the discharges lasted several hundred milliseconds and were present uniformly in all examined muscles. Nerve conduction studies disclosed bor- derline or slightly reduced amplitudes of compound muscle action potentials (CMAPs) with no conduction blocks, tempo- ral dispersion of CMAPs, or conduction slowing. The amplitudes of the sensory nerve action potentials were either unrecordable or decreased; sensory conduction velocities were borderline. The supramaximal stimulation of upper and lower limb motor nerves (median, ulnar, peroneal, and tibial nerves bilaterally) evoked CMAPs followed immediately or after a short period—up to 30 ms—by repetitive “neuro- myotonic” discharges of high frequency (about 230 Hz), waning amplitude, and duration of hundreds of milliseconds that could be recorded with the surface recording electrode. The complete blockade of ulnar and median nerves at the elbow by lidocaine did not interrupt the ability of shocks delivered distally to the site of the block to evoke neu- romyotonic discharges. The repetitive motor nerve stimulation study of ulnar and axillary nerves performed at a stimulation frequency of 2 Hz showed no decrement. The stimulation single fibre EMG from the extensor digitorum communis muscle on the right side showed a slightly abnormal jitter (19 recordings, mean jitter 34 μs, five record- ings above 40 μs), which together with a slight incerease in fibre density (2.3) indicated the reinnervation process. Second EMG and conduction studies per- formed 7 days after the end of the second IVIg treatment showed less frequent neuro- myotonic discharges evoked by electrical stimulation of the motor nerves and the voluntary contraction and the ability to evoke them waned; after several contractions they disappeared. Torbergsen et al 3 stated that, in addition to spontaneous occurrence, neuromyotonic dis- charges could also be registered during voluntary activation or after nerve stimula- tion; it was assumed that such a type of elec- trophysiological abnormality is caused by the slightest degree of hyperexcitability of axons when neuromyotonic discharges are triggered after a preceding impulse, simply voluntary or electrical, has passed, whereas spontaneous neuromyotonic discharges without an obvi- ous trigger are generated in the case of more increased hyperexcitability of axons. Clinically, as well as muscle stiVness, ataxia-like voluntary movement was present in our patient; this movement was inter- rupted repeatedly, probably due to repeated bursts of neuromyotonic discharges. Moreo- ver, the movement provoked corresponding sensory phenomena of dysaesthesias and par- aesthesias. It seems likely that these sensory phenomena of dysaesthesias and paraesthe- sias were evoked by similar types of sensory Needle EMG from abductor pollicis brevis muscle showing high frequency (about 200 Hz) neuromyotonic discharge with waning amplitude and duration of 250 ms, provoked by voluntary contraction (arrow). Spa record Abduc. Pol. Br.L 15:08:25 1 mV Foot switch status: Hold / Run Trig: –100 μV↑ 50 ms J Neurol Neurosurg Psychiatry 2001;70:406–425 406 www.jnnp.com
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of LETTERS TO THE EDITOR - NCBI

LETTERS TOTHE EDITOR
Volitional and stimulation inducedneuromyotonic discharges: unusualelectrophysiological pattern in acquiredneuromyotonia
Neuromyotonic discharges are electrophysi-ologically characterised as bursts of motorunit potentials firing at more than 150 Hz for0.5 to 2 seconds. The amplitude of theresponse typically wanes. Discharges mayoccur spontaneously or be initiated by needlemovement.1 Walsh described a case of amediastinal tumour and neuromyotonia withvery high frequency discharges that outlastedvoluntary eVort.2
We report a case of an acquired paraneo-plastic neuromyotonia associated with thy-moma, clinically manifested myotonia-likemuscle stiVness, and an unusual electrophysi-ological pattern of neuromyotonic dischargesthat were evoked voluntarily or with electricalstimulation but were absent spontaneouslyand were not elicited by needle displacement.
A 71 year old women presented with a 6month history of muscle stiVness, paraesthe-sias provoked mostly by movement, disturbedspeech, and diYcult walking. At the time ofexamination she could not walk independ-ently.
Clinical examination disclosed pro-nounced dysarthria and ataxic-like limbmovement interrupted by superimposedtonic involuntary contractions. The muscledecontraction was prolonged and percussionmyotonia was absent. Fasciculations andmyokymia-like movements were seen in herarms, but occurred only sparsely and inter-mittently. The distal foot and hand muscleswere slightly paretic and atrophic. Tendonreflexes were weak in the arms and absent inthe legs. A decreased perception of vibrationwas present distally in all limbs.
Computed tomography disclosed a tumourin the mediastinum, which was totallyremoved after initial therapy and clinical
improvement; the thymoma was confirmedhistologically.
An examination of voltage gated K+
channel (VGKC) antibody titres was per-formed using immunoprecipitation of 125I-ádendrotoxin labelled VGKCs extracted fromhuman frontal cortex2 (Institute of MolecularMedicine, John RadcliVe Hospital, Oxford,UK). The first titre was positive (241 pM(positive titres >200 pM).
During a course of intravenous immu-noglobulin (IVIg) infusions at a normal dose(0.4 g/kg on 5 subsequent days, total dose of2 g/kg) both pseudomyotonic and sensorysigns and symptoms started to improve and atthe end of the IVIg treatment the patient wasable to walk independently. After the initialIVIg therapy (administered 1 month beforesurgical removal of the thymoma), clinicalsigns and symptoms stabilised with the abilityto walk independently for 20 metres. After ayear of stabilisation, the stiVness, dysarthria,and walking ability worsened in the course of3 months to the point at which the patientwas once more unable to walk independently.The patient then received a second course ofIVIg therapy (2 g/kg) and improved to thesame degree as after the first treatment.
An EMG at the beginning of clinical followup disclosed sparse fasciculations and my-okymic discharges (with a short interburstinterval of about 5–10 ms) and motor unitpotentials with slightly higher amplitude,longer duration, mild waveform instability,and polyphasic pattern from distal muscles inthe lower limbs. Voluntary contractionevoked repetitive bursts of high frequencydischarges resembling motor unit potentialswith amplitude decrement and a characteris-tic “pinging” sound (figure); the dischargeslasted several hundred milliseconds and werepresent uniformly in all examined muscles.
Nerve conduction studies disclosed bor-derline or slightly reduced amplitudes ofcompound muscle action potentials(CMAPs) with no conduction blocks, tempo-ral dispersion of CMAPs, or conductionslowing. The amplitudes of the sensory nerveaction potentials were either unrecordable ordecreased; sensory conduction velocitieswere borderline.
The supramaximal stimulation of upperand lower limb motor nerves (median, ulnar,peroneal, and tibial nerves bilaterally) evoked
CMAPs followed immediately or after a shortperiod—up to 30 ms—by repetitive “neuro-myotonic” discharges of high frequency(about 230 Hz), waning amplitude, andduration of hundreds of milliseconds thatcould be recorded with the surface recordingelectrode.
The complete blockade of ulnar andmedian nerves at the elbow by lidocaine didnot interrupt the ability of shocks delivereddistally to the site of the block to evoke neu-romyotonic discharges.
The repetitive motor nerve stimulationstudy of ulnar and axillary nerves performedat a stimulation frequency of 2 Hz showed nodecrement.
The stimulation single fibre EMG from theextensor digitorum communis muscle on theright side showed a slightly abnormal jitter(19 recordings, mean jitter 34 µs, five record-ings above 40 µs), which together with a slightincerease in fibre density (2.3) indicated thereinnervation process.
Second EMG and conduction studies per-formed 7 days after the end of the secondIVIg treatment showed less frequent neuro-myotonic discharges evoked by electricalstimulation of the motor nerves and thevoluntary contraction and the ability to evokethem waned; after several contractions theydisappeared.
Torbergsen et al3 stated that, in addition tospontaneous occurrence, neuromyotonic dis-charges could also be registered duringvoluntary activation or after nerve stimula-tion; it was assumed that such a type of elec-trophysiological abnormality is caused by theslightest degree of hyperexcitability of axonswhen neuromyotonic discharges are triggeredafter a preceding impulse, simply voluntary orelectrical, has passed, whereas spontaneousneuromyotonic discharges without an obvi-ous trigger are generated in the case of moreincreased hyperexcitability of axons.
Clinically, as well as muscle stiVness,ataxia-like voluntary movement was presentin our patient; this movement was inter-rupted repeatedly, probably due to repeatedbursts of neuromyotonic discharges. Moreo-ver, the movement provoked correspondingsensory phenomena of dysaesthesias and par-aesthesias. It seems likely that these sensoryphenomena of dysaesthesias and paraesthe-sias were evoked by similar types of sensory
Needle EMG from abductor pollicis brevis muscle showing high frequency (about 200 Hz) neuromyotonic discharge with waning amplitude and durationof 250 ms, provoked by voluntary contraction (arrow).
Spa record Abduc. Pol. Br.L 15:08:25
1 mV Foot switch status: Hold / Run Trig: –100 µV↑ 50 ms
J Neurol Neurosurg Psychiatry 2001;70:406–425406
www.jnnp.com

neural hyperactivity. The high frequency dis-charges in our patient with neuromyotoniaconsisted of motor unit potential-like wave-forms, which did not arise spontaneously, butthe high frequency of about 200 Hz clearlyshowed their ectopic origin.
We think that actual definitions of neuro-myotonic discharges emphasising their spon-taneous occurrence or initiation by needlemovement should be reconsidered and modi-fied.
The presence of VGKC antibodies andclinical, immunological, and electrophysi-ological response to IVIg treatment are infavour of the role of VGKC blockade in thegeneration of “evoked” neuromuscular dis-charges. Antibodies to VGKC found in ourpatient may have accessed the paranodalregion (due to myelin disturbance inpolyneuropathy) and blocked fast K+ chan-nels similarly to 4-amidopyridine.4 5 Thiswould enhance supernormality and couldthereby allow a single action potential totrigger another discharge or a train todischarge.
J BEDNARÍKZ KADANKA
Department of Neurology, Faculty Hospital andMasaryk University Brno, Jihlavská 20, 639 00 Brno,
The Czech Republic
Correspondence to: J Bednarí[email protected]
1 Kimura J. Electrodiagnosis in diseases of nerve andmuscle: principles and practice. Philadelphia: FADavis, 1983:634.
2 Walsh JC. Neuromyotonia: an unusual presen-tation of intrathoracic malignancy. J NeurolNeurosurg Psychiatry 1976;39:1086–91.
3 Torbergsen T, Stålberg E, Brautaset NJ. Gen-erator sites for spontaneous activity in neuro-myotonia. An EMG study. ElectroencephalogrClin Neurophysiol 1996;101:69–78.
4 Vincent A. Understanding neuromyotonia.Muscle Nerve 2000;23:655–7.
5 Shilito P, Molenaar PC, Vincent A, et al.Acquired neuromyotonia: evidence for autoan-tibodies directed against K+ channels ofperipheral nerves. Ann Neurol 1995;38:714–22.
Zeta class glutathione transferasepolymorphisms and Parkinson’s disease
Glutathione transferase genes (GST) arecandidate genes for Parkinson’s disease be-cause they are involved with the metabolismof pesticides, dopamine, and glutathione.Recent reports have suggested an associationbetween Parkinson’s disease and polymor-phisms of GSTP11 or GSTM1 and GSTT1.2
Recently we discovered a new polymorphicsite in the zeta class G→T (GSTZ1) gene.3
This consists of a C6T transition at nucle-otide 245 in exon 5 that results in an aminoacid change at position 82 from methionineto threonine. The T substitution occurs in14% of white people. We have previouslyreported two other polymorphic sites atnucleotides 94 and 124 in exon 3.4 There arenow thought to be four alleles of GSTZ1:
Z1*A (A94A124C245), Z1*B (A94G124C245), Z1*C(G94G124C245,) and Z1*D (G94G124T245). Herewe investigated the association of Parkinson’sdisease, pesticide exposure, and these GSTZ1polymorphisms.
DNA was extracted from blood samplescollected from patients with Parkinson’sdisease and matched controls as describedpreviously.1 This study was approved by thePrincess Alexandra Hospital ethics com-mittee. Polymorphisms at nucleotide 94 and124 were detected by polymerase chainreaction/RFLP analysis as describedpreviously.4 To detect the nucleotide 245polymorphism, PCR was performed with thefollowing primers: 5'AAGAGGTGTAGTGATGGTGC3' and 5'GGTGCAAGTGTACAAGTGCC3'. The PCR was carried out in a20 µl reaction volume containing reactionbuVer IV (Advanced Biotechnologies, EpsomUK; 20 mM (NH4)2SO4, 75 mM Tris/HClpH 9.0, 0.1% Tween 20), dNTPs (0.2 mM),MgCl2 (1.5 mM), primers (0.3 µM each),thermostable DNA polymerase (AdvancedBiotechnologies, 0.5 U), and DNA (25 ng).No DNA was added to control reactions.Thermal cycling was carried out using a Cor-bett capillary thermal cycler under thefollowing conditions: initial denaturation at94°C for 2 minutes; subsequently 35 cycles of94°C for 20 seconds, 60°C for 20 seconds,72°C for 30 seconds; and a final extension of72°C for 2 minutes. Products of PCR weredigested overnight with restriction enzymeBsh 1236I (MBI fermentas) at 37°C andfragments were separated by 8% polyacryla-mide gel electrophoresis and stained withethidium bromide. The restriction enzymeBsh 1236I cleaves the C245 fragment gener-ating 12, 108, and 142 bp fragments and theT245 fragment generating 108 and 154 bpfragments.
We tested 307 Parkinson’s disease and 105control samples. The population sampleswere in Hardy-Weinberg equilibrium. Therewere no associations between the nucleotide245, 94, or 124 polymorphisms and Parkin-son’s disease (table). A total of 87 patientsand 53 controls reported a history of regularpesticide exposure. In this group there was aweak association between the nucleotide 245genotype and Parkinson’s disease (p=0.05)(table). Furthermore, in this group, the Z1*Cgenotype (G94G124C245) was less common inthe patients with Parkinson’s disease than inthe controls (39% v 52%, OR=0.58, 95%confidence interval (95%CI) 0.36–0.95,p=0.03, not corrected for multiple compari-sons).
There was no overall association betweenthe GSTZ polymorphisms and Parkinson’sdisease. However, we found a diVerencewhen only those who reported pesticideexposure were analysed. We also combinedthe data for the three polymorphic sites todetermine the frequency of the four GSTZ1alleles. The Z1*C allele is the most commonvariant in white control populations.
We found that this allele was less commonin patients with Parkinson’s disease thancontrols when stratified for pesticide expo-sure.
Studies of this nature have limitationsrelated to selection bias, case ascertainment,recall bias, diYculty in assessing extent ofexposure, and multiple comparisons. Ac-cordingly, our conclusion that there is apotential association between GSTZ, pesti-cide exposure, and Parkinson’s disease mustbe considered preliminary. Nevertheless, it isinteresting that there have now been severalreports suggesting an association betweenthe risk of Parkinson’s disease, polymorphicvariability in detoxification genes, and expo-sure to environmental toxins. These includeCYP2D6 and solvent exposure,5 GSTP andpesticide exposure1 and CYP2D6, pesticideexposure, and Parkinson’s disease withdementia.6 Thus, it has been recognised thatstudies examining the association ofpolymorphic variation in xenobiotic metabo-lism genes and Parkinson’s disease shouldtake into account the eVect of exposure totoxins.7
This study was funded by the National Health andMedical Research Council of Australia and theGeriatric Medical Foundation of Queensland.
M C TAYLORP G BOARD
A C BLACKBURNJohn Curtin School of Medical Research, Australian
National University, ACT 2601, Australia
G D MELLICKDepartment of Medicine, University of Queensland,
Australia
D G LE COUTEURCanberra Clinical School, University of Sydney,
Australia
Correspondence to:Professor P G Board
1 Menegon A, Board PG, Blackburn AC, et al.Parkinson’s disease, pesticides and glutathionetransferase polymorphisms. Lancet1998;352:1344–6.
2 Stroombergen MC, Waring R. Determinationof glutathione S-transferase ì and è polymor-phisms in neurological disease. Hum Exp Toxi-col 1999;18:141–5.
3 Blackburn AC, Tzeng H-F, Anders MW,et al. Activity of four allelic forms of humanglutathione transferase zeta: GST1a-1a possesshigher activity towards dichloroacetic acid. Pro-ceedings of the 9th North American ISSX confer-ence 1999;209.
4 Blackburn AC, Tzeng HF, Anders MW, et al.Discovery of a functional polymorphism inhuman glutathione transferase zeta by ex-pressed sequence tag database analysis. Phar-macogenetics 2000;10:49–57.
5 De Palma G, Mozzoni P, Mutti A, et al.Case-control study of interactions betweengenetic and environmental factors in Parkin-son’s disease. Lancet 1998;352:1986–7.
6 Hubble JP, Kurth JH, Glatt SL, et al. Gene-toxininteraction as a putative risk factor for Parkin-son’s disease with dementia. Neuroepidemiology1998;17:96–104.
7 Golbe LI. Parkinson’s disease: nature meetsnuture. Lancet 1998;352:1328–9.
A case of stiV limb syndrome responsiveto plasma exchange
StiV limb syndrome is a recently described,rare condition that is characterised by rigiditywithin the limbs that progresses in a relapsingand remitting fashion, often with involvementof the sphincters and brain stem.1 2 The axialmuscles are spared in the early stages of theillness, which helps distinguish it from stiVman syndrome, although it may still representa similar pathogenic mechanism to that
Association between the frequency of GSTZ1 polymorphisms and Parkinson’s disease
Polymorphism
Controls (n=105) Patients (n=307)
TT CT CC TT CT CC
245 C→T 0.04 0.33 0.63 0.02 0.35 0.64*245 C→T 0.04 0.25 0.72 0.01 0.44 0.55
AA AG GG AA AG GG94G→A 0.08 0.40 0.52 0.11 0.45 0.44124G→A 0 0.12 0.88 0.01 0.15 0.84
*Analysis restricted to subjects with a history of regular pesticide exposure (p=0.05).
J Neurol Neurosurg Psychiatry 2001;70:406–425 407
www.jnnp.com

proposed in stiV man syndrome, in whichanti-GAD antibodies are typically seen inabout 60% of patients.3 However, patientswith stiV limb syndrome seem to have diVer-ent neurophysiological abnormalities fromstiV man syndrome2 and fewer of thesepatients have anti-GAD antibodies; they alsotypically show a poorly sustained response tobaclofen and diazepam. The response toimmunotherapy in stiV limb syndrome is notknown, whereas patients with stiV mansyndrome may respond to intravenous immu-noglobulin4 5 as well as possibly plasmaexchange.6–8 We now report on a patient withstiV limb syndrome who responded dramati-cally to plasma exchange and in whom anantiaxonal antibody was isolated, suggestingthat this condition may have an immunologi-cal basis.
A 50 year old retired auxiliary nursepresented with a 10 year history of progres-sive pain, stiVness, and flexion contractures inher hands, followed by increasing immobility.Her neurological problems began at 24 yearsof age when she developed viral meningitisbased on a headache, fever, and a CSFlymphocytosis, that resolved after a week. Atthe age of 28 she complained of back and legpain with urinary retention but displayed noabnormal neurological signs and had amyelogram that was normal. Her leg symp-toms resolved, but she continued to complainof urinary retention and frequency, for whichno cause was found. She went on to have aurethrotomy which did not relieve her symp-toms. At the age of 40 she started to developstiVness and pain in the hands, which slowlyclawed, after which her arms and neckbecame progressively stiVer and her trunkbecame increasingly stooped on walking, withadditional diYculty raising her arms aboveher head. Five years after the onset of hersymptoms she was incapacitated, requiredassistance with all activities of daily living,and was permanently catheterised. At thisstage a seronegative polyarthritis was diag-nosed and she was treated with hydroxychlo-roquine, prothiaden, and corticosteroids, allwithout eVect. She subsequently had somespontaneous remission but at the time of herreferral to us she could only walk 10 yardswith one stick and continued to complain ofheaviness, pain, and stiVness in the limbsespecially the left arm. In addition she haddeveloped an intermittent tremor of the rightarm and leg, which sometimes aVected herjaw and she had diYculty swallowing largeboluses of food.
Examination at this time showed her tohave flexion contractures of all fingers. Shehad irregular jerking movements of her rightarm and leg that were accentuated by movingthe limb or walking a few steps. She had dif-ficulty standing up from a chair withoutassistance and although she was not weak onformal examination, all limb movements wereaccompanied by pain. No reflex or sensoryabnormalities were found and her plantarresponses were flexor.
Investigations showed normal nerve con-duction studies as well a normal CSF exam-ination including negative oligoclonal bands;brain and spine MRI, and routine hemato-logical and biochemical tests were alsonormal. Her autoantibody screen was nega-tive as were her anti-GAD and anti-neuronalantibodies although an antiaxonal antibodywas detected in her serum which is currentlybeing further characterised. More extensiveneurophysiology showed her to have anabnormal hypersegmented EMG pattern
during muscle spasms although no continu-ous motor unit activity was recorded at rest.The latency of responses to magnetic stimu-lation of the motor cortex was normal.
The diagnosis of stiV limb syndrome wasmade and she was initially treated withdiazepam and baclofen, but continued todeteriorate and gradually lost all useful func-tion of her arms. There was a similar lack ofresponse to intravenous methylprednisolone,so she was given intravenous immunoglobu-lin, to which she developed an anaphylacticreaction. Her disorder progressed and shedeveloped prominent rigidity and spasms ofthe face, trunk and limbs. Her speechdeveloped a strangled quality and she hadepisodes of involuntary tachypnoea appar-ently due to spasm of respiratory muscles.She was bed bound and totally dependent forall activities of daily living, needing constantnursing. As a result it was decided to give herempirical treatment with two courses ofplasma exchange in November and Decem-ber 1996. After the first exchange, the spasmsof her facial and respiratory muscles ceasedand after the second there was a slowsustained improvement in limb power, so thatafter 6 months she had regained independ-ence. For the next 18 months she walked andlived normally, even travelling on holiday.This improvement was punctuated by twoadmissions with chest infections and im-paired respiratory function, which respondedwell to antibiotics.
Unfortunately the stiVness and tremor ofher limbs returned in June 1998 and byNovember of that year, she was once againimmobile and dependent such that she couldbarely wash herself and was unable to walk atall. Any attempt to move her limbs causeddisabling tremor and stiVness. Again, reflexesand plantars were normal. She had a furthercourse of plasma exchange and again re-sponded slowly, such that 3 months later shewas able to walk on two sticks. This improve-ment was initially sustained although she hasrequired a further course of plasma exchangein February 2000 and has now been startedon oral cyclophosphamide with notable ben-efit. The patient has not had further neuro-physiological investigations.
This case has all the features of stiV limbsyndrome with the novel finding of antiaxonalantibodies detected in the serum. The patientfailed to respond to baclofen and diazepamand could not tolerate intravenous immu-noglobulin but did have a dramatic andsustained response to plasma exchange,although the need for repeated courses of thistreatment has led to her being started oncyclophosphamide. This has not been re-ported before for this condition and whereasthis case illustrates the possible therapeuticeVect of this treatment, it also raises thepossibility that stiV limb syndrome may havean immunological basis.
We thank John Pilling for permission to present hiscase, Peter Brown for performing the detailed neu-rophysiology on this patient, and Angela Vincent forthe serological testing and detection of the antiax-onal antibody.
A COLESDepartment of Neurology, Norfolk and Norwich
Health Care NHS Trust, Brunswick Road,Norwich Norfolk NR1 3SR, UK
A COLESR BARKER
Department of Neurology, Addenbrooke’s NHS Trust,Hills Road, Cambridge CB2 2QQ, UK
Correspondence to: Dr R Barker, Cambridge Cen-tre for Brain Repair, Forvie Site, Robinson Way,Cambridge CB2 2PY, UK
1 Brown P, Rothwell JC, Marsden CD. ThestiV-leg syndrome. J Neurol Neurosurg Psychia-try 1997;62:31–7.
2 Barker RA, Revesz T, Thom M, et al. Review of23 patients aVected by the stiV man syndrome:clinical subdivision into stiV trunk (man)syndrome, stiV limb syndrome, and progressiveencephalomyelitis with rigidity. J Neurol Neuro-surg Psychiatry 1998;65:633–40.
3 Solimena M, Folli F, Aparisi R, et al. Autoanti-bodies to GABA-ergic neurons and pancreaticbeta cells in stiV-man syndrome. N Engl J Med1990;322:1555–60.
4 Barker RA, Marsden CD. Successful treatmentof stiV man syndrome with intravenous immu-noglobulin. J Neurol Neurosurg Psychiatry 1997;62:426.
5 Armato AA, Cornman EW, Kissel JT. Treat-ment of stiV-man syndrome with intravenousimmunoglobulin. Neurology 1994;44:1652–4.
6 Thompson PD, Kocen RS, et al. Plasmaexchange and immunosuppression in the stiVman syndrome [letter]. Lancet 1989;ii:915.
7 Brashear HR, Phillips LH. Autoantibodies toGABAergic neurons and response to plas-mapheresis in stiV-man syndrome. Neurology1991;4:1588–92.
8 Vicari AM, Folli F, Pozza G, et al. Plasmapher-esis in the treatment of stiV-man syndrome. NEngl J Med 1989;320:1499.
Acute autonomic and sensoryneuropathy after interferon á-2btherapy for chronic hepatitis C
Acute autonomic and sensory neuropathy(AASN) is a disorder characterised by acuteautonomic and sensory nerve dysfunctions,and well preserved motor nerve function.1
Although the pathomechanism of AASN isnot clear, autonomic and sensory ganglionneuron cell bodies may be the main target ofthe immune mediated process underlyingAASN.2 On the other hand, patients treatedwith interferon may develop neurologicalcomplications including neuropathy.3 Wereport the first case of AASN which can beassociated with interferon á-2b therapy forchronic hepatitis C.
A 57 year old Japanese man with chronichepatitis C had been treated with interferoná-2b since June 1998. On 3 September, a skineruption abruptly emerged on his chest andrapidly spread over his whole body. There wasno history of exposure to toxins and drugsother than the interferon. The interferontherapy was stopped on 7 September; after atotal dose of 390 000 000 units. The skineruption gradually resolved, but 1 week later,numbness appeared in his limbs. Subse-quently he became unable to walk and stand.Further, he developed urinary overflow in-continence and bowel distension. He wasthen transfered to our neurology departmenton 2 October.
Physical examination disclosed orthostatichypotension without secondary tachycardia(120/60 mm Hg lying, 85/52 mm Hg sitting,fixed pulse rate 60 bpm) and paralytic ileus.He was catheterised for incontinence. He wasdrowsy. The pupils were anisocoric althoughthey reacted promptly to light. Other cranialnerves were unremarkable. Muscle strengthand bulk were normal. Deep tendon reflexeswere generally absent. There were no patho-logical reflexes. Light touch, pain, andtemperature sensations were impaired mod-erately over the trunk, more so in his limbs.Vibration and joint sensations were impairedseverely in the same distribution, and lost in
408 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

his fingers, knees, ankles, and toes. Sensoryataxia and pseudoathetosis in his fingers werenoted.
Routine laboratory examinations werenormal except for hyponatraemia due to thesyndrome of inappropriate secretion of anti-diuretic hormone (SIADH) (plasma sodium124 mEq/l, urinary sodium 182 mEq/l,plasma osmolarity 262 mOsmol/l, urineosmolarity 775 mOsmol/l, vasopressin 1.86pg/ml; and normal renal, thyroid, andadrenal function). Liver function wasnormal, and blood hepatitis C virus RNAwas negative. Immunoglobulins andcomplements were normal. Cryoglobulin,M-protein, antinuclear antibody, and anti-SS-A/-B antibodies were negative. We exam-ined various antiviral antibodies (coxsackieviruses, herpes simplex virus, varicella zostervirus, cytomegalovirus, Epstein-Barr virus,measles, rubella, mumps, adenovirus, andinfluenza A and B) in the serum or CSF,but they showed no remarkable change.Several tumour markers in the serum alsoshowed no particular change. Serum IgG
class anti-GQ1b antibody was presentwith low titre as demonstrated by enzymelinked immunosorbent assay (ELISA).Immunohistochemistry using frozensections of rat cerebral cortex, cerebellum,spinal cord, and dorsal root ganglion showedno antineuronal antibody in the serum fromthe patient, although the serum from apatient with anti-Hu antibody positive para-neoplastic syndrome showed positive reac-tions with these neurons (data not shown).ELISA for anti-Hu antibody was negative inthe serum and CSF. His CSF showed anincreased protein concentration (159 mg/dl)without pleocytosis but no oligoclonalbands.
Brain and spinal MRI were normal. Wholebody CT examination; colon fibroscopy anda 67Ga-citrate scintigram showed no malig-nancy.
On neurophysiological studies, an EEGshowed delta bursts in all leads. Motorconduction velocity and amplitude of com-pound muscle action potentials in the rightmedian, ulnar, and posterior tibial nerves
were within the normal range. By contrast,sensory nerve action potentials (SNAPs)could not be elicited in right median andsural nerves. In the right ulnar nerve,amplitude of SNAPs was markedly decreased(5 µV) with preservation of sensory conduc-tion velocity (53.3 m/s). A needle EMG gavenormal results. Sympathetic skin responsecould not be elicited in the upper and lowerlimbs. The coeYcient of variation of R-Rintervals on ECG was decreased (1.17% atrest; mean value and lower limit in the 50s agegroup 2.80, 1.41).
The sural nerve biopsy disclosed markedaxonal degeneration with a significant de-crease of both myelinated (1359/mm2) andunmyelinated fibres (13 791/mm2) (figure).There was no inflammatory cell infiltration orvasculitis.
The patient was treated with plasmapher-esis (3000 ml×3) beginning on 6 October.Soon after the plasmapheresis, joint sensationin his fingers was slightly improved andanisocoria disappeared. Plasma sodium con-centration, the patient’s level of conscious-ness, and the EEG were subsequently nor-malised. After the plasmapheresis, he wastreated with steroids (methylprednisolone(1000 mg intravenously), for the first 3 days,and then prednisone (60 mg orally), followedby a gradual taper). This did not furtherimprove his symptoms; severe sensory im-pairment, orthostatic hypotension, and con-stipation persisted 3 months after the onset ofthe disorder.
Our patient presented with acute onset ofsensory impairment, autonomic dysfunc-tions, selective impairment of sensory andautonomic nerves in electrophysiologicalstudies, and a raised CSF protein concentra-tion. These clinical features are compatiblewith a diagnosis of AASN. In addition, ourpatient showed SIADH and consciousnessdisturbance suggestive of involvement of theCNS.4
In AASN, episodes of infection beforethe onset are often seen, suggesting thatpreceding infection may induce the immunemediated process leading to AASN. Pavesiet al described a patient with Coxsackie Bvirus infection complicated by an acuteautonomic and sensory neuropathy.5 Intheir patient, diVuse mucosal and cutaneous erythema preceded neurologicalcomplications. Our patient also presented acutaneous lesion followed by an auto-nomic and sensory neuropathy. However,serum and CSF studies for antiviral antibod-ies showed no evidence for any viralinfection.
Peripheral neuropathy is a rare neurologi-cal side eVect of interferon. There have beenreports of multiple mononeuropathy, acutemotor or sensorimotor axonal polyneuropa-thy, and cranial nerve palsies. Although thepathomechanism underlying peripheral neu-ropathy associated with interferon is un-known, immunomodulatory eVects of inter-feron may cause disorders of the peripheralnervous system.3
In our patient, AASN developed after theinterferon therapy with an increased proteinconcentration in the CSF, and plasmapher-esis seemed to result in slight improvementand prevention of the disease progression.This is the first report suggesting associationof interferon and AASN. We suggest thatinterferon may induce an immune mediated
Pathological findings in the sural nerve. (A) Histologically, most of the myelinated fibres showformation of myelin ovoids indicating active axonal degeneration, which is also present in the teasedfibre preparations (B). (C) The unmyelinated fibres are also aVected showing swelling of the axons(arrows). ((A) epon embedded section stained with toluidine blue; bar=20 µm; (B) teased fibre,bar=100 µm; (C) electron micrograph; bar=2 µm).
J Neurol Neurosurg Psychiatry 2001;70:406–425 409
www.jnnp.com
damage to the autonomic and sensoryganglion neurons leading to clinical manifes-tation of AASN.
T IRIOKAM YAMADA
M YAMAWAKIY SAITO
H MIZUSAWADepartment of Neurology and Neurological Science,
Graduate School of Medicine,Tokyo Medical and Dental University,
1–5–45 Yushima Bunkyo-ku,Tokyo 113–8519, Japan
M YAMADADepartment of Neurology,
Kanazawa University School of Medicine, Japan
H MIURADepartment of Internal Medicine, Social Insurance
Chuo General Hospital, Japan
Correspondence to: Dr T [email protected]
1 Colan RV, Snead OC, Oh SJ, et al. Acute auto-nomic and sensory neuropathy. Ann Neurol1980;8:441–4.
2 Yasuda T, Sobue G, Mokuno K, et al. Clinico-pathophysiological features of acute autonomicand sensory neuropathy: a long-term follow-upstudy. J Neurol 1995;242:623–8.
3 Quattrini A, Comi G, Nemni R, et al. Axonalneuropathy associated with interferon-á treat-ment for hepatitis C: HLA-DR immunoreac-tivity in Schwann cells. Acta Neuropathol 1997;94:504–8.
4 Adachi H, Mukai E, Okuda S, et al. A severecase of acute autonomic and sensory neu-ropathy. Clinical Neurology (Tokyo) 1998;38:663–8. (In Japanese.)
5 Pavesi G, Gemignani F, Macaluso GM, et al.Acute sensory and autonomic neuropathy: pos-sible association with Coxsackie B virusinfection. J Neurol Neurosurg Psychiatry 1992;55:613–15.
Neuropathic pain with vesical and rectalhyperreflexia and cocontraction afterpelvic surgery
Pelvic and pudendal nerve injury can occurduring extirpative visceral surgery such asradical hysterectomy.1 2 Many of thesepatients develop severe chronic pelvic pain
and bladder symptoms, and are oftenreferred to neurologists with suspicion oflumbosacral plexus lesions or disc disease.There are few or no signs on examination,and patients are often considered to be “hys-terical”, despite having severe symptoms.Here, we describe two patients in whomsevere pelvic pain and bladder dysfunctiondeveloped after hysterectomy, and who dem-onstrated detrusor and rectal hyperreflexiawith cocontractions, features usually associ-ated with lesions of the CNS. Whereas spinalcord sensitisation is well recognised aftersomatic nerve injury, our studies provide thefirst clear evidence for its development aftervisceral nerve injury in humans, and amethod for its detection using ambulatoryurorectodynamics.
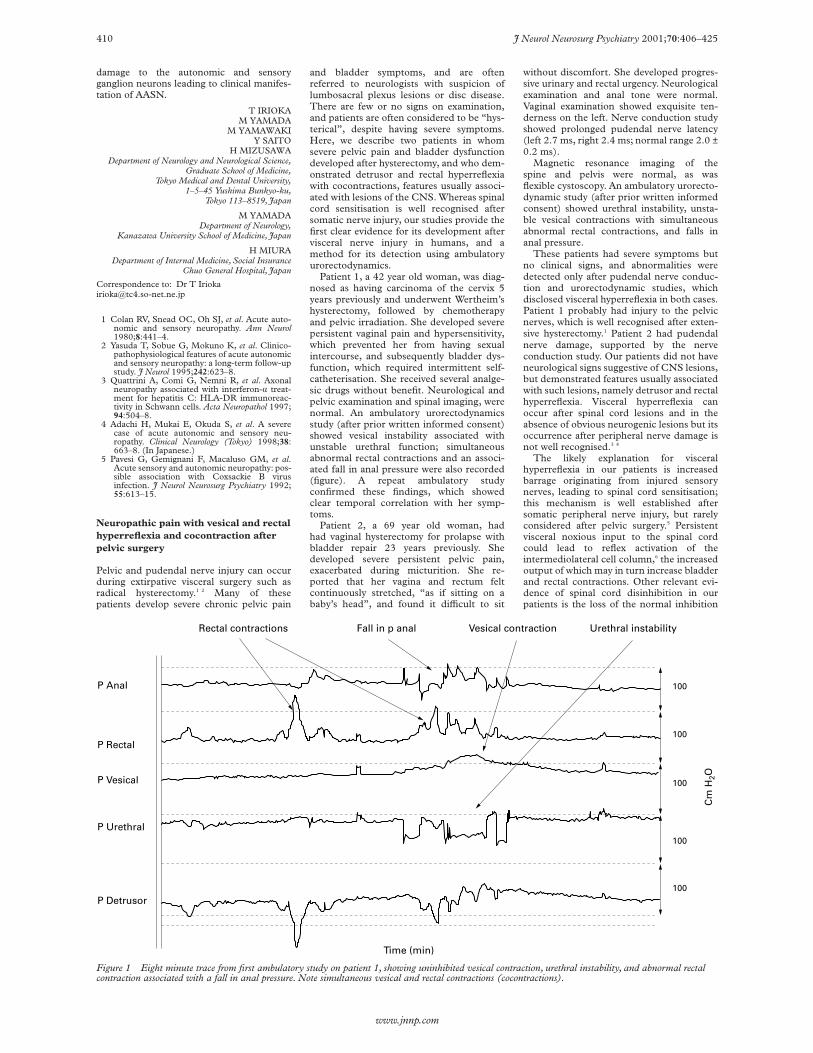
Patient 1, a 42 year old woman, was diag-nosed as having carcinoma of the cervix 5years previously and underwent Wertheim’shysterectomy, followed by chemotherapyand pelvic irradiation. She developed severepersistent vaginal pain and hypersensitivity,which prevented her from having sexualintercourse, and subsequently bladder dys-function, which required intermittent self-catheterisation. She received several analge-sic drugs without benefit. Neurological andpelvic examination and spinal imaging, werenormal. An ambulatory urorectodynamicsstudy (after prior written informed consent)showed vesical instability associated withunstable urethral function; simultaneousabnormal rectal contractions and an associ-ated fall in anal pressure were also recorded(figure). A repeat ambulatory studyconfirmed these findings, which showedclear temporal correlation with her symp-toms.
Patient 2, a 69 year old woman, hadhad vaginal hysterectomy for prolapse withbladder repair 23 years previously. Shedeveloped severe persistent pelvic pain,exacerbated during micturition. She re-ported that her vagina and rectum feltcontinuously stretched, “as if sitting on ababy’s head”, and found it diYcult to sit
without discomfort. She developed progres-sive urinary and rectal urgency. Neurologicalexamination and anal tone were normal.Vaginal examination showed exquisite ten-derness on the left. Nerve conduction studyshowed prolonged pudendal nerve latency(left 2.7 ms, right 2.4 ms; normal range 2.0 ±0.2 ms).
Magnetic resonance imaging of thespine and pelvis were normal, as wasflexible cystoscopy. An ambulatory urorecto-dynamic study (after prior written informedconsent) showed urethral instability, unsta-ble vesical contractions with simultaneousabnormal rectal contractions, and falls inanal pressure.
These patients had severe symptoms butno clinical signs, and abnormalities weredetected only after pudendal nerve conduc-tion and urorectodynamic studies, whichdisclosed visceral hyperreflexia in both cases.Patient 1 probably had injury to the pelvicnerves, which is well recognised after exten-sive hysterectomy.1 Patient 2 had pudendalnerve damage, supported by the nerveconduction study. Our patients did not haveneurological signs suggestive of CNS lesions,but demonstrated features usually associatedwith such lesions, namely detrusor and rectalhyperreflexia. Visceral hyperreflexia canoccur after spinal cord lesions and in theabsence of obvious neurogenic lesions but itsoccurrence after peripheral nerve damage isnot well recognised.3 4
The likely explanation for visceralhyperreflexia in our patients is increasedbarrage originating from injured sensorynerves, leading to spinal cord sensitisation;this mechanism is well established aftersomatic peripheral nerve injury, but rarelyconsidered after pelvic surgery.5 Persistentvisceral noxious input to the spinal cordcould lead to reflex activation of theintermediolateral cell column,6 the increasedoutput of which may in turn increase bladderand rectal contractions. Other relevant evi-dence of spinal cord disinhibition in ourpatients is the loss of the normal inhibition
Figure 1 Eight minute trace from first ambulatory study on patient 1, showing uninhibited vesical contraction, urethral instability, and abnormal rectalcontraction associated with a fall in anal pressure. Note simultaneous vesical and rectal contractions (cocontractions).
P Anal
P Rectal
P Vesical
Vesical contraction Urethral instabilityFall in p analRectal contractions
P Urethral
P Detrusor
100
100
100
100
100
Time (min)
Cm
H2O
410 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

of urinary bladder contraction induced byrectal and vaginal stimulation7 and thedevelopment of bladder and rectum cocon-tractions, which have not been reported pre-viously.
Our cases show how pelvic surgery couldbe complicated by persistent neuropathicpain and bladder and bowel hypersensitivity,and further studies of spinal cord excitabilityare needed to clarify underlying mecha-nisms. Early recognition and initiation ofanalgesic treatment for neuropathic painisessential to prevent the condition becomingintractable.
P SHEMBALKARP ANAND
Peripheral Neuropathy Unit, Department ofNeurology, Imperial College School of Medicine, Area
A, Ground Floor, Hammersmith Hospital, Du CaneRoad, London, W12 0NN, UK
I JUNAIDC FOWLER
Department of Urology, The Royal London Hospital,Whitechapel, London
E1 1BB, UK
N S WILLIAMSAcademic Department of Surgery, The Royal London
Hospital, Whitechapel, London E1 1BB, UK
Correspondence to: Professor P [email protected]
1 Shingleton HM, Thompson JD. Cancer of thecervix. In: Rock JA, Thompson JD, eds. Linde’soperative gynaecology. Philadelphia: LippincottRaven, 1997:1413–99.
2 Benson JT, McClellan E. The eVect of vaginaldissection on the pudendal nerve. Obstet Gyne-col 1993;82:387–9.
3 Fowler CJ. Neurological disorders of micturi-tion and their treatment. Brain 1999;122:1213–31.
4 Lowe E, Anand P, Terenghi G, et al. IncreasedNGF levels in the urinary bladder of womenwith idiopathic sensory urgency and interstitialcystitis. Br J Urol 1997;79:572–7.
5 Zermann DH, Ishigooka M, Doggweiler R,et al. Postoperative chronic pain andbladder dysfunction: windup and neuronalplasticity: do we need a more neurourologicalapproach in pelvic surgery ? J Urol 1998;160:102–5.
6 Anand P, Gibson SJ, McGregor GP, et al. A VIPcontaining system concentrated in the lum-bosacral region of human spinal cord. Nature1983;305:143–5.
7 Morrison JFB. The neural control of thebladder. In: Bloom SR, Polak JM, LindenlaubE, eds. Systemic role of regulatory peptides: sympo-sium, Oosterbeek, Netherlands, 2–6 May 1982.Stuttgart: FK Schattauer Verlag, 1982:381–96.
Peripheral nerve ischaemia afterinternal iliac artery ligation
Ligation of the internal iliac (hypogastric)arteries has been used to control seriousobstetric and pelvic bleeding. It is generallywell tolerated in the young obstetric orgynaecological patient, presumably becauseof an extensive collateral blood supply.1–3
Acute lumbosacral plexopathies have beendescribed, however, in older patients withvascular disease when the internal iliac arter-ies are interrupted.4–8 We report on a teenagepatient with similar peripheral nerve ischae-mia after bilateral internal iliac artery ligationfor postpartum haemorrhage.
An 18 year old woman presented at 40weeks gestation with mildly raised bloodpressures, trace proteinuria, oliguria, andgeneralised oedema. She was diagnosed withpre-eclampsia and admitted for induction.When induction was unsuccessful, she under-went a caesarean section, which was compli-cated by uterine atony and a postpartumhaemorrhage with an estimated blood loss of
2500 ml. After bilateral uterine artery ligationfailed to control the bleeding, bilateral inter-nal iliac artery ligation was performed withresultant haemostasis.
On the first postoperative day, she com-plained of left buttock pain and diYcultymoving her left leg. Superficial skin break-down over the sacrum and buttocks wasnoted on the second postoperative day. Shedeveloped a fever and fundal tenderness onday 4. Helical CT of the abdomen and pelvisdisclosed residual gas and fluid within theendometrial canal consistent with endometri-tis, which was treated with intravenousantibiotics. No retroperitoneal haematomawas present. Neurological evaluation on thefifth postoperative day was limited by painbut disclosed normal strength, sensation, andreflexes in the arms and the right leg.Strength in the left leg was 2 to 3/5 on hipflexion and knee extension and 3 to 4/5 onankle plantarflexion, ankle dorsiflexion, andtoe extension. Sensation was diminished to allmodalities in the entire left leg below the hip.The left patellar and ankle stretch reflexeswere absent.
Magnetic resonance imaging of the thora-columbar spinal cord was unremarkable. Aninitial magnetic resonance angiogram (MRA)of the pelvis showed segmental occlusions ofboth internal iliac arteries with distal recon-stitution greater on the right than on the left.The left superior gluteal artery was not visu-alised. Revascularisation was considered butdeferred due to the concomitant active pelvicinfection. Peripheral pulses remained strong,and Doppler ultrasound of both legs showedno evidence of distal thrombus.
Nerve conduction studies 1 week afterligation were extremely limited and diYcultto interpret due to generalised oedema. Suraland peroneal sensory responses were absentbilaterally. Right peroneal and left tibialmotor responses were normal. A small leftperoneal motor response was present in theanterior tibilais muscle. Electromyographywas not performed at that time.
The fevers and endometritis graduallycleared, and over the next month left legstrength improved slowly, but incompletely,with greater proximal (4–5 in hip flexion andknee extension) than distal (2–3 in ankleplantarflexion, 0 in ankle dorsiflexion) recov-ery. The left leg continued to have dimin-ished sensation to all modalities and re-mained areflexic. The superficial skinnecrosis progressed to an open non-healingulcer 7 cm×5 cm over the sacrum and leftgluteal musculature. Magnetic resonanceimaging of the region disclosed additionaltissue necrosis subcutaneously along the leftposterolateral buttock and inflammation inthe surrounding subcutaneous tissues andunderlying gluteal musculature with exten-sion into the left sacroiliac joint. There wasno evidence of rectal, uterine, or bladderischaemia.
A follow up MRA of the pelvis 6 weeksafter ligation demonstrated persistent seg-mental occlusion of both internal iliacarteries and less collateral flow on the leftcompared with the right. Again, the superiorgluteal artery was not visualised on the leftbut appeared to fill on the right.
Electromyography of selected muscles ofthe left leg at that time (6 weeks after ligation)showed 2+ to 4+ fibrillations and positivesharp waves in the vastus lateralis, tibialisanterior, and lateral gastrocnemius muscles,consistent with acute denervation. Therewere no voluntary units in the tibialis anterior
and low firing rates in the gastrocnemius.Low amplitude polyphasic motor units invastus lateralis suggested early proximalrecovery. Nerve conduction studies showeddiminished left sural sensory amplitudes andslowed velocities (2.8 µV, 36.0 m/s). The leftperoneal motor responses were markedlyattenuated, and the left posterior tibial motorvelocities were slowed (32.0 m/s). The rightsural sensory (13.2 µV, 42.0 m/s) andperoneal motor (2.3 mV, 46.0 m/s) responseswere normal.
In general, the internal iliac artery dividesinto an anterior and a posterior division. Theanterior division is formed by the inferiorgluteal artery and its branches, which supplythe pelvic viscera, the lower buttocks, and theback of the thigh. The posterior division isformed by the superior gluteal artery and itsbranches, which supply the gluteal muscula-ture, the femoral nerve, and the sciatic nerveroots.9
Ligation of the internal iliac arteries hasbeen accepted as a safe and eVective means ofcontrolling serious haemorrhaging from theuterus or lower pelvis after delivery or aftergynaecological surgery.1–3 The lack of ischae-mic complications from ligation of the inter-nal iliac artery is thought to be due to themultiple sources of collateral blood flowpresent in the pelvis.
There are, however, reports of buttockischaemia or lumbosacral plexopathies as acomplication of interruption of the internaliliac arteries during aortic bypass proceduresor aortoiliac aneurysm resection.4–5 In astudy of 11 patients (mean age 67, range 53to 87) with aortoiliac occlusive disease oraortoiliac aneurysmal disease, seven devel-oped ischaemic injury to the lumbosacralplexus after bilateral internal iliac arteryligation.6 In four of those patients, buttocknecrosis with extension to the bony pelviswas also seen. In another report, four women(mean age 37, range 33 to 47) with insulindependent diabetes and end stage renal dis-ease developed ipsilateral lumbosacral plex-opathies when the internal iliac artery wasligated during kidney transplantation.7 Elec-tromyography showed denervation of thetibialis anterior, gastrocnemius, and vastusmedialis in one patient and of the tibialisanterior and gastrocnemius in another.Ischaemia of the sciatic and femoral nervesand buttocks also occurred after internal iliacartery embolisation in patients with terminalpelvic malignancies who received radio-therapy.8
Our 18 year old patient developed a com-bination of leg weakness, leg numbness, andbuttock necrosis after internal iliac arteryligation, as described in older vasculopathicpatients. In the patients described in the lit-erature, as in our patient, the clinical andelectromyographic findings do not distin-guish between combined femoral and sciaticnerve lesions, a lumbosacral plexopathy, or acombination of the two. Our patient’spresentation, however, can be most suc-cinctly explained by an infarction in the ter-ritory of the left superior gluteal artery andits branches, resulting in ischaemia to thegluteal musculature, the femoral nerveproper, and the sciatic nerve roots. Thislocalisation is supported by serial MRangiograms of the pelvis in which the leftsuperior gluteal artery and its branches werenot visualised.
It has been shown that, in experimentalligation of the internal iliac artery of rats,moderate ischaemia is associated with demy-
J Neurol Neurosurg Psychiatry 2001;70:406–425 411
www.jnnp.com
elination, whereas severe ischaemia produceswallerian degeneration.10 In our patient, thereduction in the left sural sensory amplitudeand slowing of the left sural sensory and pos-terior tibial motor conduction velocities weremore consistent with axonal degenerationthan demyelination, implying a significantdegree of injury.
Our patient had no pre-existing vascularrisk factors that would have predisposed herto ischaemic complications, but possiblecontributing factors in her case includepre-eclampsia, prior bilateral uterine arteryligation, and postpartum endometritis.Pre-eclampsia is associated with changes inthe renin-angiotensin-aldosterone axis, anincreased thromboxane to prostacyclin ratio,and an increase in plasma endothelin.11 Thesefactors result in vasoconstriction and plateletaggregation, which could interfere with pelviccollateralisation. The ligation of both uterinearteries before the internal iliac arteryligations may have reduced pelvic collateralflow. Postpartum pelvic infections can alsoalter vascular tone or extend to involve ovar-ian and pelvic vessels, potentially interferingwith collateralisation.12
In summary, although internal iliac arteryligation is generally well tolerated because ofmultiple collateral sources of blood supply,complications such as peripheral nerveinjury and necrosis of the gluteal muscula-ture can occur. Although this occurs mostcommonly in patients with severe aortoiliacvascular disease or vascular risk factors suchas insulin-dependent diabetes mellitus orradiotherapy treatment, it can occur even intheir absence.
R K SHINM M STECKER
Department of Neurology, Hospital of the University ofPennsylvania, 3400 Spruce Street, Philadelphia,
PA 19104–4283, USA
S G IMBESIDepartment of Radiology
Correspondence to: Dr R K [email protected]
1 Likeman RK. The boldest procedure possiblefor checking the bleeding. Aust NZ J ObstetGynaecol 1992;32:256–62.
2 Das BN, Biswas AK. Ligation of internal iliacarteries in pelvic haemorrhage. J Obstet Gynae-col Res 1998;24:251–4.
3 Paraskevaides E, Noelke L, Afrasiabi M. Inter-nal iliac artery ligation (IIAL) in obstetrics andgynaecology. Eur J Obstet Gynecol Reprod Biol1993;52:73–5.
4 Picone AL, Green RM, Ricotta JR, et al. Spinalcord ischemia following operations onthe abdominal aorta. J Vasc Surg 1986;3:95–103.
5 Usubiaga JE, Kolodny J, Usubiaga LE. Neuro-logical complications of prevertebral surgeryunder regional anesthesia. Surgery 1970;68:304–9.
6 Iliopoulos JI, Howanitz PE, Pierce GE, et al.The critical hypogastric circulation. Am J Surg1987;154:671–5.
7 Hefty TR, Nelson KA, Hatch TR, et al. Acutelumbosacral plexopathy in diabetic womenafter renal transplantation. J Urol 1990;143:107–9.
8 Hare WSC, Holland CJ. Paresis following inter-nal iliac artery embolization. Radiology 1983;146:47–51.
9 Braithwaite JL. Variations in origin of theparietal branches of the internal iliac artery. JAnat 1952;86:423–30.
10 Nukada H, Powell HC, Myers RR. Spatialdistribution of nerve injury after occlusion ofindividual major vessels in rat sciatic nerves. JNeuropathol Exp Neurol 1993;52:452–9.
11 Brown MA. The physiology of pre-eclampsia.Clin Exp Pharmacol Physiol 1995;22:781–91.
12 Cox SM, Gilstrap LC. Postpartum endometri-tis. Obstet Gynecol Clin North Am 1989;16:363–71.
DiVusion weighted magnetic resonanceimaging in Neuro-Behçet’s disease
Neurological involvement is one of the mostdevastating manifestations of Behçet’s dis-ease.1 However, the pathogenic mechanismfor CNS lesions in patients with neuro-Behçet’s disease is unclear. Although vasculi-tis is usually considered to be the centralpathological feature in Behçet’s disease, avasculitic process was not usually demon-strated in the CNS.2
DiVusion weighted imaging can detectchanges in water diVusion associated withcellular dysfunction. It has been well docu-mented that acute infarction related tocytotoxic oedema is characterised by amarked decrease in diVusion, and also thatincreased interstitial water related to va-sogenic oedema shows increased diVusion.3
Conventional MRI cannot distinguish be-tween these diVerent types of oedema. Wereport on a patient with neuro-Behçet’sdisease with a significantly reversible T2 sig-nal and diVusion abnormalities in CNSlesions.
A 54 year old Asian man was admitted withdysarthria and left hemiparesis, whichevolved over a period of 2 days and was asso-ciated with gradual mental deterioration. Thepatient had a history of frequent orogenitalulcers and acneiform nodules on his face.Physical examination showed active genitalulceration. Neurological examination dis-
closed drowsy consciousness and disorienta-tion. Moderate degrees of hemiparesis andhemihypaesthesia involving the face, arm,and leg were found on the left side. Deep ten-don reflexes were increased and Babinski’ssign was extensor on the left side. Erythrocytesedimentation rate (54 mm/h) and C-reactiveprotein concentration (3.4 mg/100 ml) wereincreased. Examination of CSF showed mildpleocytosis (18 white blood cells/mm3) withnormal concentrations of protein and glu-cose. Fundus examination showed retinalvein occlusion and retinal haemorrhage onthe right side. The diagnosis of Behçet’s dis-ease was made based on the recurrentorogenital ulcerations, skin lesions, and eyeinvolvement.
The patient was examined on a 1.5T MRunit (Signa Horizon, Echospeed; GeneralElectric Medical Systems) with echoplanarimaging (EPI) capability. Fast spin echo, T2weighted images (T2 weighted images;TR/TE 4200/112 ms; field of view 21×21cm; matrix 256×192; and slice thickness 5mm) were obtained. DiVusion weightedimaging was obtained in the transverseplane using a single shot EPI (TR/TE 6500/125 ms; field of view 24×24 cm; matrix128×128; slice thickness 5 mm; and two bvalues 0 and 1000 s/mm2). The diVusiongradients were applied along the three axes(x, y, z) simultaneously. The apparentdiVusion coeYcient (ADC) was calculatedbased on the negative slope of the linear
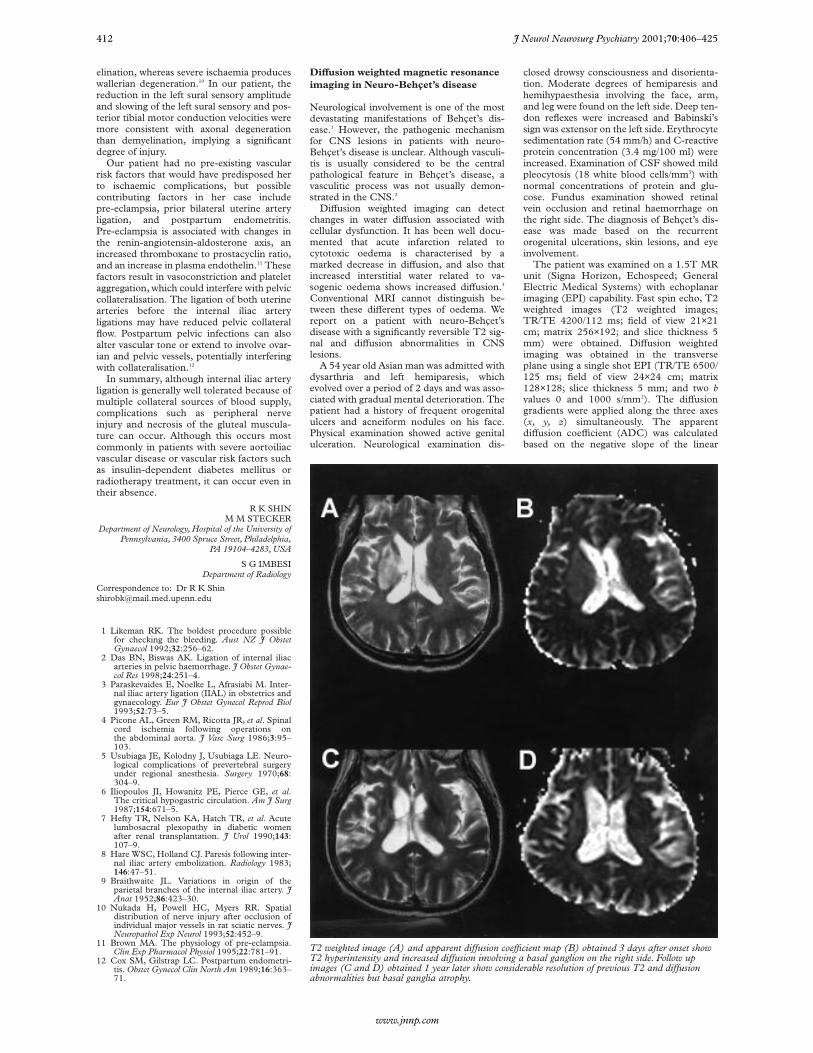
T2 weighted image (A) and apparent diVusion coeYcient map (B) obtained 3 days after onset showT2 hyperintensity and increased diVusion involving a basal ganglion on the right side. Follow upimages (C and D) obtained 1 year later show considerable resolution of previous T2 and diVusionabnormalities but basal ganglia atrophy.
412 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

regression line best fitting the points for bversus ln (SI); where SI is the signal intensityfrom a region of interest within the imagesacquired at each b value. Performing this cal-culation on a pixel by pixel basis created theADC maps.
Brain MRI performed 3 days aftersymptom onset showed extensive T2 hyper-intensities involving the corona radiata, inter-nal capsule, basal ganglion, thalamus, andmidbrain on the right side. Brain diVusionweighted imaging showed slight hyperinten-sities which were limited to the coronaradiata, the medial portion of the basal gan-glion, and the thalamus. Four sampledADCs in the corresponding regions of T2hyperintensity demonstrated increased diVu-sion (ranging from 1.17 to 1.26×10-5 cm2/s),compared with a matching location in theuninvolved contralateral hemisphere (rang-ing from 0.77 to 0.80×10-5 cm2/s, figure Aand B). Magnetic resonance angiographyshowed no abnormalities. The patient im-proved rapidly after treatment with a highdose of corticosteroid. Within 2 weeks allpreviously noted neurological abnormalitieshad resolved except for a slight left hemi-paresis. An MRI repeated at this timeshowed a partial decrease in the extent of theT2 hyperintensity. One year later he wasreadmitted with a slowly progressive bulbarweakness, frontal lobe dysfunction, urinaryincontinence, and depressive mood changes.Follow up MRI performed at this time,showed that the previous T2 abnormalitieshad improved, but the atrophy of thebrain stem and basal ganglia became evidentwith periventricular high signal intensities.Four ADCs sampled in locations corre-sponding to those of the initially increasedADCs decreased to values which rangedfrom 0.98 to 1.07×10-5 cm2/s (figure C andD).
In our patient the ADC maps and ADCvalues showed high proton mobility, whichsuggests vasogenic oedema in acute lesions ofneuro-Behçet’s disease. Vasogenic oedemadevelops when the blood-brain barrier is dis-rupted and is not primarily associated withcellular damage. Discrimination betweencytotoxic and vasogenic oedema has impor-tant clinical implications because vasogenicoedema can be reversed by proper manage-ment.
According to the MRI findings for neuro-Behçet’s disease, the most prevalent abnor-malities are located in the brain stem or thebasal ganglia extending to the diencephalicstructures during an acute attack, and brain-stem atrophy in chronic cases.4 The revers-ibility of CT or MRI abnormalities of acutelesions in neuro-Behçet’s disease has alsobeen documented and correlated with clini-cal improvement.4 5 The serial MRI findingsin our patient were consistent with thosedescribed in previous reports. The precisepathomechanism of CNS lesions in Behçet’sdisease has not been established. Studies ofpathology showed that lymphocytic or neu-trophilic meningoencephalitis with perivas-cular inflammatory cell cuYng aroundvenules and capillaries were predominant inthe brain stem and basal ganglia in neuro-Behçet’s disease.1 However, most studiesshowed histopathological changes at achronic stage of the disease and histopatho-logical findings may show various types oflesions according to the age of lesion at thetime of examination. A recent pathologicalreport in a fulminant form of neuro-Behçet’sdisease found no evidence of vasculitis but
an acute destructive inflammatory process.2
It has been postulated that at an early stageof the disease, the reversibility of lesionsmay reflect a reversible breakdown in theblood-brain barrier rather than gliosis orinfarction.5 The pattern of diVusion changesin the acute lesions in our patient stronglysupports the idea that there is increasedpermeability in the blood-brain barrieras a result of the primary inflammatoryprocess.
We thank Byung Kee Yoo for his assistancewith diVusion weighted MR data acquisition.
D-W KANGK CHU
J-Y CHOJ-S KOO
B-W YOONJ-K ROH
Department of Neurology, Seoul National UniversityCollege of Medicine, 28 Yongon-dong, Chongno-gu,
Seoul 110–744, Korea
I C SONGK H CHANG
Department of Radiology
Correspondence to: Dr J-K Roh,[email protected]
1 Kidd D, Steuer A, Denman AM, et al.Neurological complications in Behçet’s syn-drome. Brain 1999;122:2183–94.
2 Hadfield MG, Aydin F, Lippman HR, et al.Neuro-Behçet’s disease. Clin Neuropathol 1997;16:55–60.
3 Schäbitz WR, Fisher M. DiVusion weightedimaging for acute cerebral infarction. NeurolRes 1995;17:270–4.
4 Zuheir Al Kawi M, Bohlega S, Banna M. MRIfindings in neuro-Behçet’s disease. Neurology1991;41:405–8.
5 Patel DV, Neuman MJ, Hier DB. Reversibility ofCT and MR findings in neuro-Behçet disease.J Comput Assist Tomogr 1989;13:669–73.
Azathioprine and interferon â-1btreatment in relapsing-remittingmultiple sclerosis
Both interferon â-1b (IFNâ-1b) and azathio-prine (AZA) are eVective in reducing relapsefrequency in relapsing-remitting multiplesclerosis (RRMS).1 2 However, no prospective
study has compared the eYcacy of the twodrugs. To assess their clinical eYcacy andimpact on the patients’ quality of life, we per-formed a pilot study on a small group ofpatients with RRMS. Patients with at leasttwo relapses during the previous 2 years andEDSS lower than or equal to 3.5 were oVeredtreatment with IFNâ-1b or AZA afterinformation about the eYcacy, tolerability,and mode of administration of both drugs,and allocated to one of the two treatmentsaccording to the patient’s choice. Somepatients refused to be treated with eitherdrug, mainly because of the fear of sideeVects and the negative impact of chronictreatment on their lifestyle; they were fol-lowed up according to the same protocol (nottreated (NT) group). All patients gaveinformed consent. Serial neurological evalua-tions were performed every 3 months for 1year. At the same times a self administereddisease specific questionnaire (MSQOL-54),recently validated in the Italian MS popula-tion,3 was filled in and the Hamilton depres-sion rating scale (HD) was administered.Scores for the MSQOL-54 were analysed aspreviously described3; briefly, the raw scoreswere linearly transformed into 0–100 scales;the higher the transformed score, the betterthe patient’s quality of life. The two compos-ite scores mental health and physical healthwere also evaluated. A t test for unpairedsamples was used to compare the scoresbetween groups, with adjustment for multiplecomparisons. The Kruskal-Wallis test and thetwo sample Wilcoxon rank sum test wereused to compare the change in scoresbetween groups. The main clinical variableswere compared using a t test for unpaired andpaired data.
Thirty two patients were included inthe study (11 IFNâ-1b, 10 AZA, 11 NT).The clinical characteristics at entry weresimilar in the two actively treated groups,whereas in the NT group age was signifi-cantly higher than in the AZA group (but notthe IFN group) and pretreatment relapsefrequency (RF) was lower than in the IFNgroup (but not the AZA group). After 1 year,RF significantly decreased in both the
Data of patients before and after treatment
IFN AZA NT
No of patients 11 10 11*Sex (M/F) 3/8 2/8 3/8Age at entry (y) 33 (6.2) 31.2 (4.9)† 38 (6.3)†Disease duration (y) 8.3 (5) 6.95 (6.7) 8.4 (6.8*)EDSS at entry 2.32 (0.9) 2.35 (0.9) 1.83 (1.15)
At 12 months 2.2 (1.05) 2.1 (0.9) 1.9 (1.3)No of worsened patients at 12 months 1/11 0/10 2/10RF 2 year pretreatment 2.2 (0.8)† 2 (1) 1.4 (0.3)†
At 12 months 0.8 (0.7)‡ 0.9 (0.4)‡ 1.0 (0.9)No of relapse free patients at 12 months 4/11 7/10 4/10PHC at entry 68.02 (9.1) 61.7 (10.8) 61.7 (11.7)
Change at 12 months −2.64 (9.26) +7.9 (9.9) +3.66 (13.7)MHC at entry 74.7 (15.7) 61.25 (14.15) 58.7 (16.9)
Change at 12 months −6.04 (13.9)† +21.25 (11.9)† +6.37 (21.8)RLE at entry 83.34 (32.4) 37.5 (33.05) 55.55 (40.8)
Change at 12 months −10 (35.3)† +49.9 (35.6)† +16.6 (53.4)
IFN=Interferonâ-1b treated patients; AZA=azathioprine treated patients; NT=no actively treated patients;RF=relapse frequency (No of relapses/patient/y); worsened=increase of>1 EDSS point. PHC=physicalcomposite score; MHC=mental composite score; RLE=role limitation for emotional reasons.*One patient in NT group dropped out at 6 months.†Significant diVerences between groups :Age at entry : NT v AZA p=0.01.RF at entry : NT v IFN p=0.006.MHC change : IFN v AZA p=0.006.RLE change : IFN v AZA p=0.001.‡Significant diVerences within each group (12 months v entry):RF: IFN p<0.001.AZA p=0.005.
J Neurol Neurosurg Psychiatry 2001;70:406–425 413
www.jnnp.com
IFN and AZA treated groups withoutdiVerences between the two treatments,whereas it was unchanged in the NT group.The EDSS remained stable in the threegroups (table). Five of 11 patients treatedwith IFN had flu-like symptoms on one ormore occasions, whereas no side eVectsoccurred in the other two groups.
No significant diVerences in the HDscores and quality of life profile were foundbetween the three groups at entry. At 6 (datanot shown) and 12 months the mental healthcomposite score significantly increased inpatients treated with AZA compared with thepatients treated with IFN, mainly due to theincrease in role limitation for emotionalreasons item; no significant diVerences be-tween the NT group and actively treatedgroups were seen. No significant changes inHD scores in the three groups were found at12 months. These results suggest that bothAZA and IFNâ-1b are eVective in reducingrelapse frequency in patients with RRMS. Thetreatment eVect on quality of life has beenrarely investigated, with conflicting results: nosignificant change after 1 year of IFNâ-1btreatment was found by Schwartz et al,4
whereas an improvement on physicalitems after 5 years was reported by Rice et al.5
In our study, the impact on quality of life wasbetter in patients treated with AZA than inthose treated with IFN, mainly due to theimprovement in mental score. A direct eVect ofthe drugs on the CNS seems unlikely: nosymptoms of neurotoxicity were found ineither treatment group and no patients devel-oped depression according to the HD scale.Most likely the improvement of quality of lifein patients treated with AZA might be relatedto diVerent tolerability or to diVerences intreatment schedules, resulting in a morepronounced and persistent perception of thedisease in patients treated with IFN. Due tothe few patients, the results of this study needto be verified by a larger randomised compara-tive trial.
We are indebted to Dr Alessandra Solari, Labora-tory of Epidemiology, C Besta National Neurologi-cal Institute, Milan, Italy, for performing the statis-tical analysis of the data.
C MILANESEL LA MANTIAA SALMAGGI
Istituto Nazionale Neurologico C Besta, Via Celoria11, 20133 Milan, Italy
D CAPUTOIRCCS Fondazione Don Gnocchi, Milan, Italy
Correspondence to: Dr C [email protected]
1 The IFNB Multiple Sclerosis Study Group andthe University of British Columbia MS/MRIAnalysis Group. Interferon beta-1b in thetreatment of multiple sclerosis: final outcomeof the randomized controlled trial. Neurology1995;45:1277–85.
2 Yudkin PL, Ellison GW, Ghezzi A, et al.Overview of azathioprine treatment in multiplesclerosis. Lancet 1991;338:1051–5.
3 Solari A, Filippini G, Mendozzi L, et al. Valida-tion of Italian multiple sclerosis quality of life54 questionnaire. J Neurol Neurosurg Psychiatry1999;66:0–6.
4 Schwartz CE, Coulthard-Morris L, Cole B, et al.The quality-of-life eVects of interferon beta-1bin multiple sclerosis. An extended Q-TwiSTanalysis. Arch Neurol 1997;54:1475–80.
5 Rice GP, Oger J, Duquette P, et al. Treatmentwith interferon beta-1b improves quality of lifein multiple sclerosis. Can J Neurol Sci 1999;26:276–82.
Unilateral caudate head lesionsimulating brain tumour in X-linkedadult onset adrenoleukodystrophy
The appearance of X-linked adrenomyelo-neuropathy (AMN)/adrenoleukodystrophy(ALD) on MRI is usually specific, withbilateral symmetric areas of white matterabnormality surrounding the posterior hornsof the lateral ventricles with various degreesof atrophy of the spinal cord.1 Our patientwith AMN, however, showed a lesion in theright caudate head simulating a braintumour, which has not been a feature in thisdisease.
At the age of 25 the patient started to haveprogressive spastic paraparesis and mildataxia with genitourinary dysfunction (urgeurinary incontinence and erectile dysfunc-tion).2 On admission to our hospital at theage of 34, T2 weighted MR images showedsmall lesions in the bilateral internal capsulealthough no abnormality was seen in the spi-nal cord. Nerve conduction studies and thesural nerve biopsy showed evidence of
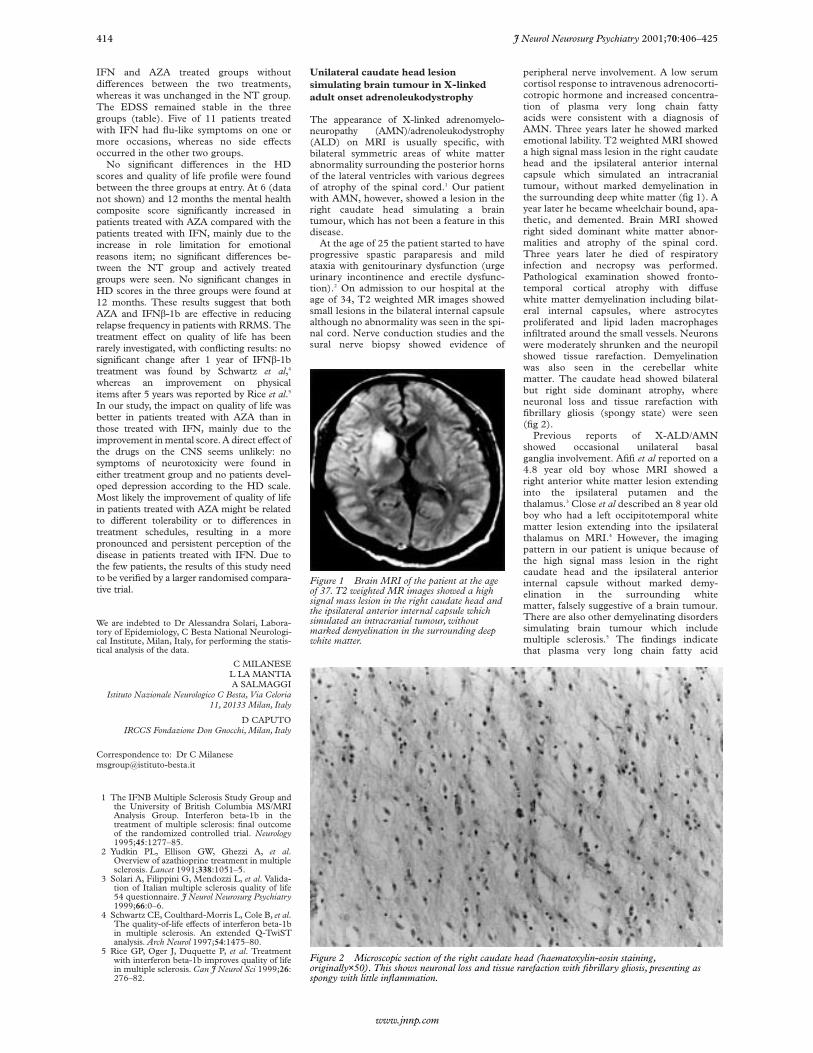
peripheral nerve involvement. A low serumcortisol response to intravenous adrenocorti-cotropic hormone and increased concentra-tion of plasma very long chain fattyacids were consistent with a diagnosis ofAMN. Three years later he showed markedemotional lability. T2 weighted MRI showeda high signal mass lesion in the right caudatehead and the ipsilateral anterior internalcapsule which simulated an intracranialtumour, without marked demyelination inthe surrounding deep white matter (fig 1). Ayear later he became wheelchair bound, apa-thetic, and demented. Brain MRI showedright sided dominant white matter abnor-malities and atrophy of the spinal cord.Three years later he died of respiratoryinfection and necropsy was performed.Pathological examination showed fronto-temporal cortical atrophy with diVusewhite matter demyelination including bilat-eral internal capsules, where astrocytesproliferated and lipid laden macrophagesinfiltrated around the small vessels. Neuronswere moderately shrunken and the neuropilshowed tissue rarefaction. Demyelinationwas also seen in the cerebellar whitematter. The caudate head showed bilateralbut right side dominant atrophy, whereneuronal loss and tissue rarefaction withfibrillary gliosis (spongy state) were seen(fig 2).
Previous reports of X-ALD/AMNshowed occasional unilateral basalganglia involvement. Afifi et al reported on a4.8 year old boy whose MRI showed aright anterior white matter lesion extendinginto the ipsilateral putamen and thethalamus.3 Close et al described an 8 year oldboy who had a left occipitotemporal whitematter lesion extending into the ipsilateralthalamus on MRI.4 However, the imagingpattern in our patient is unique because ofthe high signal mass lesion in the rightcaudate head and the ipsilateral anteriorinternal capsule without marked demy-elination in the surrounding whitematter, falsely suggestive of a brain tumour.There are also other demyelinating disorderssimulating brain tumour which includemultiple sclerosis.5 The findings indicatethat plasma very long chain fatty acid
Figure 1 Brain MRI of the patient at the ageof 37. T2 weighted MR images showed a highsignal mass lesion in the right caudate head andthe ipsilateral anterior internal capsule whichsimulated an intracranial tumour, withoutmarked demyelination in the surrounding deepwhite matter.
Figure 2 Microscopic section of the right caudate head (haematoxylin-eosin staining,originally×50). This shows neuronal loss and tissue rarefaction with fibrillary gliosis, presenting asspongy with little inflammation.
414 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

concentrations should be measured in pa-tients with unexplained basal ganglia abnor-malities on MRI.
R SAKAKIBARAT FUKUTAKE
K ARAIK KATAYAMA
M MORIT HATTORI
Neurology Department Chiba University, 1–8–1Inohana Chuo-Ku, Chiba 260–8670 Japan
R SAKAKIBARAT FUKUTAKEK
K KATAYAMAM MORI
Neurology Department Kashima Rosai Hospital,1–9108–2 Doai-Honmachi Hasaki,
Kashima 314–03 Japan
Correspondence to: Dr R [email protected]
1 Kumar AJ, Köhler W, Kruse B, et al. MRfindings in adult-onset adrenoleukodystrophy.AJNR Am J Neuroradiol 1995;16:1227–37.
2 Sakakibara R, Hattori T, Fukutake T, et al. Mic-turitional disturbance in a patient with adreno-myeloneuropathy (AMN). Neurourol Urodyn1998;17:207–12.
3 Afifi AK, Menezes AH, Reed LA, et al. Atypicalpresentation of X linked childhood adrenoleu-kodystrophy with an unusual magnetic reso-nance imaging pattern. J Child Neurol 1996;11:497–9.
4 Close PJ, Sinnott SJ, Nolan KT.Adrenoleukodystrophy; a case report demon-strating unilateral abnormalities. Pediatr Radiol1993;23:400–1.
5 Ernst T, Chang L, Walot I, et al. PhysiologicMRI of a tumefactive multiple sclerosis.Neurology 1998;51:1486–8.
Lymphadenopathy in patients withmultiple sclerosis undergoing treatmentwith glatiramer acetate
Glatiramer acetate (GA)—formerly known ascopolymer 1 or COP-1—has been shown toreduce the frequency of relapses and diseaseactivity and burden as measured by MRI inpatients with relapsing-remitting multiplesclerosis (RR-MS).1 The mechanism ofaction is thought to involve MHC-II block-ade2 and the induction of a Th2/Th3 cytokineresponse.3 Peripheral blood mononuclearcells from patients with multiple sclerosis andhealthy controls proliferate in reponse to GAin vitro.4 Therefore GA seems to have bothimmunostimulatory and immunomodulatorypotential.
In our centre 27 patients with relapsing-remitting or relapsing-progressive multiplesclerosis were treated with 20 mg subcutan-eous GA daily for 3 years as part of anopen label multicentre study. Safety evalua-tion and expanded disability status scale(EDSS) rating were performed every 3months and in the 3rd year every 6 monthsand when clinical relapses occurred. Re-lapses were defined according to Poser crite-ria and annual relapse rates were calculatedfor the 3 year study duration and a 2 yearprestudy period. As two patients reportedgeneralised tender swelling of lymph nodes
spontaneously in temporal relation to thebeginning of GA injections special attentionwas paid to the symptom and regular assess-ment of regional lymph nodes was per-formed in all patients. Only if patientsreported symptoms such as tenderness orpain, was the diagnosis of lymphadenopathymade. All patients completed the full 3 yearsof the study. In one patient with generalisedlymphadenopathy a lymph node biopsy wastaken to rule out malignancy. As controlspatients who were routinely treated withIFN-â injections at our multiple sclerosisoutpatient clinic were also examined forlymphadenopathy.
In nine out of 27 patients lymphadeno-pathy occurred 1 to 15 months afterinitiating GA treatment and persisted for thestudy (treatment) duration. There were nosignificant diVerences between the groupswith and without lymphadenopathy in theirmean age, disease duration, EDSS scores,and annual relapse rates at the beginning ofthe study. The size of the lymph nodesranged from 2 to 5 cm and lymphadenopathywas considered mild to moderate in eightpatients and severe in one patient. In sevenout of the nine patients lymphadenopathywas restricted to inguinal lymph nodes andin two patients it was generalised. Serologicaland haematological routine diagnostics ofperipheral blood were normal. The lymphnode biopsy in one patient with severegeneralised lymphadenopathy showed strongimmune stimulation with lymphofollicularhyperplasia but no atypical cells (thus rulingout malignancy). Lymphadenopathy did notnecessitate the discontinuation of GA treat-ment. The examiners were reassured that allpatients used a good (sterile) injection tech-nique. In the control patients no lymphaden-opathy was detected.
When analysing annual relapse rates, asignificant reduction of the mean annualrelapse rate was found under GA treatment.The annual relapse rate decreased from 1.8/year to 0.33/year and from 1.5/year to0.54/year in the group of patients with andwithout lymphadenopathy respectively.When comparing annual relapse rate forboth patient groups the diVerence did notreach significance (Mann-Whitney U test,p=0.076) with a trend to a slightly favourableresponse in the group with lymphadenopa-thy. Although in the group with lymphaden-opathy no patient showed an increase inrelapse rate, three patients in the group with-out lymphadenopathy did. In both groups ofpatients no significant change in medianEDSS over the 3 years of the study was noted(table 1).
The frequency of lymphadenopathy foundin this study (nine out of 27 patients) issignificantly higher than that reported in thepostmarketing surveillance of GA (55 reportsof lymphadenopathy out of about 30 000reports of other adverse events). The lym-phadenopathy in our study was mild to mod-erate, not accompanied by changes in routinelaboratory indices, and persisted as long asthe GA treatment was continued. In seven
out of nine patients lymphadenopathy re-mained localised to the draining lymphnodes. Lymph node swelling receded andreappeared depending on injection site. Lym-phadenopathy did not necessitate discontinu-ation of GA treatment. In one patient withgeneralised lymphadenopathy a biopsy wasperformed to rule out malignancy. Thepatient was then continued on GA withoutfurther problems and remained relapse free;GA injections were stopped 1 year after theend of the study, due to pregnancy, and lym-phadenopathy resolved completely within 4weeks.
Lymphadenopathy, if not due to malig-nancy, is a clinical sign of immune activationand has not yet been reported as an adverseevent of GA treatment in the literature. TheeVect might be due to direct stimulation of Tcells in vivo as GA has been shown to inducemRNA expression of IL-2 and T cell prolif-eration in vitro.4 In previous studies immu-nostimulatory cytokines such as IFN-ã orviral infections worsened the clinical courseof multiple sclerosis whereas immunosup-pression (for example, mitoxantrone, cyclo-phosphamide) was beneficial.5 It will beinteresting to study further whether lym-phadenopathy related to GA is associatedwith alteration of the clinical outcome meas-ures of multiple sclerosis. The cohort ofpatients with lymphadenopathy did not showa significant diVerence in annual relapse rateor EDSS compared with patients withoutlymphadenopathy, but the small sample sizeper group should be taken into account.Glatiramer acetate induces clinical signs ofimmune stimulation (lymphadenopathy) in asubgroup of patients with multiple sclerosisthat is not associated with clinical worsening.The finding is therefore interestingwith regard to the potential mechanism ofaction of GA in vivo. Larger numbers ofpatients need to be examined to determinewhether lymphadenopathy in patientsunder GA treatment is associated withdistinct immunological markers—for exam-ple, MHC-II type or cytokine secretion pat-tern.
Patients examined in this study were enrolled in ourcentre as part of the German open label phase IIIbtreatment study (protocol COP 1600) supported byTEVA and Aventis
A WINDHAGENS MANIAK
S MARCKMANNA WILKENINGR-B LINDERT
F HEIDENREICHNeurologische Klinik, Medizinische Hochschule
Hannover, Carl-Neuberg-Strasse 1, 30623 Hannover,Germany
R BLASCZYKAbteilung für Transfusionsmedizin
Correspondence to: Dr A [email protected]
1 Johnson KP, Brooks B, Cohen J, et al.Copolymer 1 reduces relapse rate and improvesdisability in relapsing-remitting multiple scle-rosis. Neurology 1995;45:1268–76.
2 Fridkis-Hareli M, Strominger J. Promiscuousbinding of synthetic copolymer 1 to purifiedHLA-DR molecules. J Immunol 1998;160:4386–97.
3 Duda P, Krieger J, Schmied M, et al. Glatirameracetate (Copaxone®) induces degenerate,Th2-polarized immune responses in patientswith multiple sclerosis J Clin Invest 2000;105:967–76.
Clinical data of patients with multiple sclerosis treated with glatiramer acetate
Annual relapse rateat start
Annual relapse ratein study
EDSS at start ofstudy
EDSS at end ofstudy
Lymph node swelling 1.8 (1–3.5) 0.33 (0–1) 2.5 (0–3.5) 2.5 (0–3.5)No lymph node swelling 1.5 (1–3.5) 0.54 (0–1.8) 2.5 (1–5) 3 (0–6.5)
Values are mean (relapse rate) or median (EDSS) (range).
J Neurol Neurosurg Psychiatry 2001;70:406–425 415
www.jnnp.com

4 Brosnan C, Litwak M, Neighbour P, et al.Immunogenic potential of copolymer 1 in nor-mal human lymphocytes. Neurology 1985;35:1754–9.
5 Arnason B. Immunologic therapy of multiplesclerosis. Annu Rev Med 1999;50:291–302.
CORRESPONDENCE
Neurovisual rehabilitation in Balint’ssyndrome
Further to the excellent review of neuro-visual rehabilitation by KerkhoV1, we thinkthat it is prudent to communicate ourexperience in the management of a patientwith Balint’s syndrome after traumatic braininjury. This was seen in a 41 year old righthanded manual worker whose initial cranialCT showed right extradural haematoma.Subsequent scans demonstrated left poste-rior occipital infarct. Brain MRI 3 monthsafter the injury showed high signal in theright occipitoparietal and left occipitotempo-ral regions. His physical recovery wassatisfactory in that he was fully mobileunaided. However, he presented with simul-tunagnosia, optic ataxia, and psychic paraly-sis of gaze.2 This had an adverse impact onhis functional independence; he had diY-culty in route finding—crashing intofurniture and walls—and other activitiesof daily living including dressing and toilet-ing. He failed most subtests in the River-mead perceptual assessment battery(RPAB).
We agree with the author that eVectivetreatment strategies are poorly developed andevaluated. We have identified three ap-proaches for the rehabilitation of the percep-tual deficits including those seen in Balint’ssyndrome.x The adaptive (functional) approach,3
which involves functional tasks utilisingthe person’s strengths and abilities, help-ing them to compensate for problems oraltering the environment to lessen theirdisabilities
x The remedial approach,4 which involvesrestoration of the damaged CNS by train-ing in the perceptual skills, which may begeneralised across all activities of daily liv-ing. This could be achieved by tabletopactivities or sensorimotor exercises
x The multicontext approach,5 which isbased on the fact that learning is not auto-matically transferred from one situation toanother. This involves practising of atargeted strategy in a multiple environ-ment with varied tasks and movementdemands, and it incorporates self aware-ness tasks.In this patient, we used the adaptive
approach, practising functional tasks repeat-edly with increasing complexity of the tasks asthe sessions continued. This approach as-sumes that treatment has little eVect onimpairment and that generalisation to othertasks is unlikely. It also assumes that the brainhas limited ability to improve and restoreitself after injury.3 The remaining abilities areused to oVset the deficits. This patient wasable to develop his own compensatorystrategies, learned to use his hands to acquiretactile feedback, and managed to direct his
gaze to visually locate objects when required.His performance on the RPAB was improvedand he was successfully discharged homewith little support.
As Balint’s syndrome is likely to be seen inclinical conditions such as Alzheimer’s dis-ease, multiple strokes, intracranial tumours,brain injury, and CNS complications of HIVinfection, we thought it important to outlinethe possible options for the management ofthis condition. Further work is required on alarger series of patients.
I AL-KHAWAJASussex Rehabilitation Centre,
Brighton General Hospital,Brighton, East Sussex BN2 3EX, UK
N H J HABOUBINorth StaVordshire Rehabilitation Centre,
Stoke-on-Trent ST6 7AG, UK
Correspondence to: Dr I [email protected]
1 KerkhoV G. Neurovisual rehabilitation: recentdevelopments and future directions. J NeurolNeurosurg Psychiatry 2000;68:691–706.
2 Rizzo M. Balint’s syndrome and associatedvisuo-spatial disorders. Bailliere’s Clinical Neu-rology 1993;2:415–37.
3 Neistadt ME. A critical analysis of occupationaltherapy approaches for perceptual deficits inadults with brain injuries. Am J Occup Ther1990;44:299–304.
4 Perez M, Trunkel RS, Lackman EA, et al.Balint’s syndrome arising from bilateral poste-rior cortical atrophy or infarction: rehabilita-tion strategy and their limitation. Disabil Reha-bil 1996;18:300–4.
5 Toglia JP. Generalization of treatment. A multi-context approach to cognitive perceptual im-pairment in adults with brain injury. Am JOccup Ther 1990;43:587–95.
KerkhoV replies:Al-Khawaja and Haboubi have reported suc-cessful neurovisual rehabilitation in a patientwith Balint’s syndrome due to a rightoccipitoparietal and a left occipitotemporallesion, using adaptive practising of functionaltasks with increasing complexity. This caseshows, together with some of the otherpublished cases,1 that individually tailoredrehabilitation strategies can be adapted suc-cessfully for some severely disabled patients.Due to the various aetiologies and subse-quent lesion localisations it is likely that mostpatients with Balint’s syndrome will havesome outstanding deficits, but also somespared abilities, which can be used forcompensatory purposes (for example, intacttactile feedback1).
In two patients with Balint’s syndrometreated for several months in our depart-ment, significant improvements could beachieved by systematic treatment so thatboth patients could live independently athome with only minimal assistance. Onepatient, who had received traumatic, urae-mic, and hypoxic brain damage at the age of27 years initially was nearly blind. Two yearslater, when treatment started in our unit, hecouldn’t perceive more than two visualstimuli simultaneously (simultanagnosia),was almost unable to read, showed opticataxia, and had severely impaired spatial-perceptual functions in the visual and tactilemodality. However, his memory, intelligence,and executive functions were largely pre-served, so that he relearned reading partially,learnt to dress himself partially, and wasfinally able to travel by train. He managed tolive alone in his flat, with only minorassistance from others.
The second patient, a 60 year old physi-cian, had bilateral vascular parieto-occipitallesions. She was initially (falsely) consideredas blind, although she could well see anddescribe faces and correctly identify thecolour of one’s eyes. She presented withseverely disturbed depth and horizontaldistance perception, simultanagnosia, andoptic ataxia as well as a peculiar deficit inidentifying spatial directions and locatingsound sources. For instance, it proveddiYcult for her to identify the direction inwhich someone pointed when describing aparticular route, or to decide in which direc-tion a train would move when looking at therailway track. However, as in the first patient,she had some spared abilities—that is, excel-lent introspection and awareness of herdisorder, preserved cognitive abilities, andshe was highly motivated to relearn routefinding in her town district. After intensivetraining for reading and route finding shecould be discharged, living independently athome. She continues to use public transportto go shopping, visit friends, see her neurolo-gist, the pharmacist, or going to a concerthall.
I think that systematic treatment in bothcases helped to improve basic visual abilitiesand activities of daily living so that bothpatients could live independently at home—which was hardly expected when seeing themat the onset of treatment. To conclude, I amconvinced that many patients with Balint’ssyndrome can learn to compensate for atleast some of their visual deficits by system-atic and individualised treatment. Thesearch for spared functions will undoudtedlydisclose multiple ways for compensation andwill increase our understanding of someunresolved aspects of this fascinatingsyndrome—for example, the tactile orauditory-spatial abilities of patients withBalint’s syndrome.
G KERKHOFFEKN-Clinical Neuropsychology Research Group,
Department of Neuropsychology, City, HospitalBogenhausen, Dachauerstr.164,D-80992
Munich, Germany
1 Perez FM, Tunkel RS, Lachmann EA, et al. Bal-int’s syndrome arising from bilateral posteriorcortical atrophy or infarction: rehabilitationstrategies and their limitation. Disabil Rehabil1996;18:300–4.
Arnold Chiari malformation andnystagmus of skew
I enjoyed Pieh and Gottlob’s article1 pointingout the association of a Chiari malformationwith a “unique” form of nystagmus that theycall “the nystagmus of skew.” The distinctivefeature of this nystagmus is a disjunctive ver-tical oscillation in which the fast phase of oneeye moves upward while, at the same time,the other eye moves downward.
The authors1 state that their secondpatient had a “rotatory component” bywhich I assume they mean torsional; thispattern of nystagmus is already established inthe literature and is known as “jerk-waveform see-saw nystagmus”.2 3 In theirfirst patient they point out that the amplitudeof the vertical nystagmus was so small thatthey were unable to confidently exclude atorsional component. It would have beenmost interesting to obtain recordings lookingfor a torsional component using the modified
416 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

scleral search coil technique4; I suspect that itwould have shown a torsional componentand that this patient also had jerk-waveformsee-saw nystagmus.
Jerk-waveform see-saw nystagmus occurswith unilateral mesodiencephalic lesions,presumed due to selective unilateral inactiva-tion of the torsional eye velocity integrator inthe interstitial nucleus of Cajal2; during thefast (jerk) phases the upper poles of both eyesrotate toward the side of the lesion. With lat-eral medullary injury the fast phases of thetorsional component jerk away from the sideof the lesion.5 In both situations the torsionalcomponent is always conjugate. With meso-diencephalic lesions the vertical componentis always disjunctive, but with medullarylesions it may be either conjugate (usuallyupward) or disjunctive.2
PATRICK LAVINDepartments of Neurology and Ophthalmology,
Vanderbilt University Medical Center, 2100 PierceAve. Nashville, TN 37212, USA
1 Pieh C, Gottlob I. Arnold Chiari malformationand nystagmus of skew. J Neurol Neurosurg Psy-chiatry 2000;69:124–6.
2 Halmagyi GM, Aw ST, Dahaene I, et al.Jerk-waveform see-saw nystagmus due to uni-lateral meso-diencephalic lesion. Brain 1994;117:789–803.
3 Lavin PJM. Diplopia, nystagmus and other ocu-lar oscillations. In: DaroV RB, Fenichel GM,Marsden CD, et al, eds. Neurology in clinicalpractice. Boston: Butterworth, 2000;16:203–27.
4 Collwejin H, Vaan der Steen J, Ferman L, et al.Human ocular counterroll: assessment of staticand dynamic properties from electromagneticsearch coil readings. Exp Brain Res 1985;59:185–1196.
5 Morrow MJ, Sharpe JA. Torsional nystagmus inthe lateral medullary syndrome. Ann Neurol1988;24:390–8.
The authors reply:We thank Lavin for his interesting comments.
We stated in our article that the possibilityof a fine see-saw nystagmus could not beexcluded. We did re-evaluate our patientswith a torsional coil and did not record a tor-sional component. However, because of thefast improvement in both patients, all the eyemovement abnormalities on re-evaluationwere minimal. Clinically, even in the stage ofmaximal abnormalities, in one patient we didnot detect any torsional component, whichsuggests that if there was an element ofsee-saw nystagmus, it was subclinical.
We did not state that the type of nystagmusassociated with the Arnold-Chiari malforma-tion was unique, precisely because we couldnot rule out with total certainty a see-sawnystagmus, which has been reported in onepatient with the malformation. We did, how-ever, point out that this association isunusual.
Because of the lack of strong evidence of atorsional component to the dissociated verti-cal nystagmus, we preferred the term, kindlysuggested by a reviewer, “nystagmus ofskew”. This would represent a more inclu-sive, descriptive term, of which both the pen-dular and the jerk see-saw nystagmus formsand the dissociated vertical nystagmus with-out demonstrable torsional componentwould represent subvariants.
C PIEHKantonsspital St Gallen,
Switzerland
I GOTTLOBDepartment of Ophthalmology,
Leicester Royal Infirmary,Leicester LE1 5WW, UK
Botulinum toxin for the treatment ofsialorrhoea in ALS: serious side eVectsof a transductal approach
We have read with interest the article by Giesset al,1 which showed that botulinum toxin A(BoNT/A) might be a new treatment optionfor sialorrhoea in patients with bulbar palsy.We have recently conducted a similar studywhich was interrupted due to serious sideeVects.
In September 1998 we injected 25 MUBotox into the parotid glands of a 59 year oldwomen who had ALS with pronouncedbulbar palsy. We noticed a reduction of thesialorrhoea but facial weakness on the leftside worsened significantly.
After this experience we developed aprotocol for the treatment of sialorrhoea inpatients with ALS with bulbar palsy by retro-grade injection of BoNT/A through thesalivary duct into the salivary glands. Wechose the retrograde way of administration ofBoNT/A for this pilot study because wethought that this technique would avoid facialweakness.
After informed consent the patients re-ceived 12.5 mouse units (MU) BoNT/A(BotoxR) retrogradly into each parotid andsublingual gland from a small catheterinserted into the salivary duct. Neurologicalexamination and quantification of saliva pro-duction were performed before the BoNT/Ainjection and on days 1, 3, 7, 14, and 28, aswell as after 2 and 3 months. Technetium99m scintigraphy was performed before and7 days after the injection. Quantification ofsaliva production was performed with a sim-ple method: the patients were asked to expec-torate as much saliva as possible into a paperhandkerchief for 10 minutes. This procedurewas repeated twice. The mean of thediVerence in weight of the handkerchiefbefore and after these procedures was takenas the maximum expectorated saliva produc-tion (MESP). Quality of life and the clinicaleVect of the treatment were evaluated by aquestionnaire.
We treated a 60 year old woman (patient1) and a 69 year old women (patient 2) withthis new technique. Both had certain ALSaccording to the El Escorial criteria, withsevere bulbar palsy with durations of 23 and28 months respectively. Both patients had asignificant reduction of MESP seven days
after the injection (76% and 58%; from 5420mg to 1301 mg, and 4365 to 1829 mg) whichlasted for 4 to 8 weeks. Technecium 99mscintigraphy showed a significant reductionof radiotracer uptake into the injectedsalivary glands in both patients (figure). Bothpatients estimated the injection procedure aspainful. Patient 1 developed a severe swellingof the right sublingual salivary gland andbase of the tongue 3 days after the injectionwhich was treated with antibiotics andcorticosteroids. Patient 2, who was able toswallow with diYculty before the injection,mentioned impairment of swallowing be-tween days 4 and 21. Both patients had a“moderate” improvement of sialorrhoea butdid not want the injections to be repeated.After these experiences we decided to stopthe pilot study.
The injection of BoNT/A through the sali-vary duct reduces the activity of the salivaryglands significantly for several weeks but hasserious side eVects. Local and systemiceVects of BoNT/A are probably pronouncedin ALS.2 Subclinical EMG abnormalitiesdistant to the injection sites have beendescribed in therapeutic doses, but also sys-temic weakness has been found.3 As there aresome reports that BoNT/A injections, evenin low doses, may exaggerate pre-existingneuromuscular diseases,2 4 careful monitor-ing of neurological symptoms, which is diY-cult in a progressive disease, is needed toexclude side eVects of BoNT/A. The drug iseVective in reducing drooling5 but we needmore data about the safety of BoNT/Abefore it can be used safely for the treatmentof sialorrhoea in ALS. The transductalapproach in particular seems to have unac-ceptable side eVects.
M G M WINTERHOLLERF J ERBGUTH
Department of Neurology,Friedrich-Alexander-Universität Erlangen,
Schwabachanlage 6, D-91054 Erlangen, Germany
S WOLFDepartment of Otorhinolaryngology
S KATDepartment of Nuclear Medicine
Correspondence to: Dr M GM Winterholler, [email protected]
Figure 1 Scintigraphy of the head of patient 1 showing technetium 99m uptake one day before (left)and 7 days after (right) retrograde transductal BoNT/A injection into the salivary glands, showingsignificant reduction of tracer uptake after the BoNT/A injection (quantification: parotid gland left–59%, right –48%; sublingual gland left –10%; right –56%).
J Neurol Neurosurg Psychiatry 2001;70:406–425 417
www.jnnp.com

1 Giess R, Naumann M, Werner E, et al.Injections of botulinum toxin A into thesalivary glands improve sialorrhoea in amyo-trophic lateral sclerosis. J Neurol Neurochir Psy-chiatry 2000;69:121–123.
2 Mezaki T, Kaji R, Kohara N, et al. Develop-ment of general weakness in a patient withamyotrophic lateral sclerosis after focal botuli-num toxin injection. Neurology 1996;46:845–6.
3 Bakheit AM, Ward CD, McLellanDL. General-ized botulism-like syndrome after intramuscu-lar injections of botulinum toxin type A. Areport of two cases. J Neurol Neurochir Psychia-try 1997;62:196–200.
4 Erbguth F, Claus D, Engelhardt A, et al.Systemic eVect of local botulinum toxininjections unmasks subclinical Lambert-Eatonmyasthenic syndrome. J Neurol Neurochir Psy-chiatry 1993;56:1235–36.
5 Jost WH. Treatment of drooling in Parkinson´sdisease with botulinum toxin. Mov Disord1999;14:1057.
The authors reply:We appreciate the comments by Winterhol-ler et al on our article1 on botulinum toxin(BTX/A) treatment of sialorrhoea in patientswith amyotrophic lateral sclerosis (ALS).Although we did not find any serious sideeVects after transcutaneous injections ofBTX/A into the parotid and submandibularglands Winterholler et al report on sublin-gual salivary gland infection in one patientand deterioration of dysphagia in anotherpatient after a transductal approach. Thesecomplications support our notion that theindividually tolerated dose of BTX/A inpatients with ALS may be low and also indi-cate that the transcutaneous approach asperformed in several studies1–3 may be saferthan the retrograde transductal injection.This is not unexpected as the transductalapproach has possibly a higher risk of infec-tion because of the reduced salivary glandsecretion rate found in patients with ALS.1 Inaddition, the total dose of 25 MU Botox forthe sublingual glands may be rather high inview of the close anatomical relation of theseglands to the pharyngeal muscles. We there-fore underscore our previous suggestion tostart with injections of the parotid glandsalone and to cautiously escalate the dose andnumber of injected glands.1 In view ofthe potential risk of BTX/A in deterioratingALS symptoms injections should be re-stricted to otherwise intractable and ex-tremely disabled patients with ALS who havesialorrhoea.
M NAUMANNR GIESS
K SCHWAGERK V TOYKA
Department of Neurology and Department ofOtorhinolaryngology,
Bayerische-Julius-Maximilians-Universität,Josef-Schneider-Strasse 11, 97080 Würzburg,
Germany
Correspondence to: Dr M [email protected]
1 Giess R, Naumann M, Werner E, et al.Injections of botulinum toxin A into thesalivary glands improve sialorrhoea in amyo-trophic lateral sclerosis. J Neurol Neurosurg Psy-chiatry 2000;69:121–3.
2 Jost WH. Treatment of drooling in Parkinson’sdisease with botulinum toxin. Mov Disord1999;14:1057.
3 Bhatia KP, Münchau A, Brown P. Botulinumtoxin is a useful treatment in excessive droolingof saliva [letter]. J Neurol Neurosurg Psychiatry1999;67:679.
Treatment of early onset Parkinson’sdisease with ropinirole
The recent editorial1 supporting initial treat-ment of early onset Parkinson’s disease with adopamine agonist hinged in part on the dem-onstration2 in 268 patients that treatment ofearly onset Parkinson’s disease with rop-inirole alone or with supplementarylevodopa/dopa decarboxylase inhibitor(benserazide) (LD/DDI) resulted in substan-tially less dyskinesia than with LD/DDIalone, with only slightly less motor benefit.Five per cent of patients on ropinirole alonedeveloped dyskinesia after 5 years, comparedwith 25% with ropinirole plus LD/DDI, and45% of those on LD/DDI alone). The trialdesign allowed LD/DDI supplementation ifresponse was inadequate and additional trialdrug could not be tolerated. Up to 24 mgropinirole and 1200 mg LD/DDI daily wereallowed. Sixty six per cent of patientscompleting the ropinirole arm required sup-plementation, the average mean daily dose ofropinirole at 5 years being 16.5 mg, com-pared with 753 mg of LD/DDI when the sec-ond was used alone.
It is unfortunate that the study required athree times daily dosage regime. It seemspossible that this accounts for the surprising33% of patients on LD/DDI alone who with-drew as a result of early adverse events, andfor the occurrence of nausea in 49.4% ofpatients on LD/DDI alone. Whether smaller,more frequent, dosage would have allowedbetter tolerance of and motor response toropinirole, it is not surprising that frequentdyskinesia was seen at 5 years on three timesdaily dosage of LD/DDI. A substantialproportion of patients on LD/DDI (43.8%)were also on selegiline, amplifying the eVectsubstantially. By comparison, the 5 year studyof immediate release (IR) and controlledrelease (CR) LD/DDI (carbidopa) in 681patients, ironically reported earlier in 1997,and later in 1999,3 in whom dosage could beadjusted up to five or more times a dayresulted not only in a lower frequency of dys-kinesia (20.6% IR, 21.7% CR) at 5 years butalso a lower mean total daily dose (426 mgIR; 510 mg CR (bioequivalent)).
Whereas it may be argued that diVerentdrug preparations and methods of assessmentinvalidate comparison, it may be simply thatless frequent higher pulsatile dosage provokesnot only greater peak dose dyskinesia butalso, as a mirroring eVect, greater oV time aspostsynaptic mechanisms adapt to cope withsurges of dopamine and perhaps lose sensitiv-ity to troughs. Patients seen during troughswould be liable to have their dose increased.If the interdose interval were fixed this wouldlead to a vicious circle.
Given reports of long term resolution ofdyskinesia and on/oV eVects in response tovarious methods of continuous stimulation,at an appropriate strength, including continu-ous daytime jejunal infusion of LD/DDI(with little or no change in LD/DDI dosagerequirement over 57 months),4 and of a neu-roprotective eVect of levodopa,5 the results ofRascol et al2 should not dissuade others frompursuing oral treatment with LD/DDI in amore frequent, lower dose regime. Withgradual (allowing for the long duration actionof levodopa) titration of slow release LD/DDIdosage and interdose interval (if necessaryusing a timer), against response and compli-ance of patient (or carer), it may in theoryand, with suYcient observation and titration,in practice be possible to approximate to such
a steady state stimulation and response. Thiswould have potentially less risk for developinghallucinations, and would cost less.
J R PONSFORDDepartment of Neurology, Walsgrave Hospital NHS
Trust, CliVord Bridge Road, Coventry CV2 2DX, UK
1 Brooks DJ. Dopamine agonists: Their role in thetreatment of Parkinson’s disease [editorial]. JNeurol Neurosurg Psychiatry 2000;68:685–90.
2 Rascol O, Brooks DJ, Korczyn AD, et al. A fiveyear study of the incidence of dyskinesia inpatients with early Parkinson’s disease whowere treated with ropinirole or levodopa. NEngl J Med 2000;342:1484–91.
3 Koller WC, Hulton JT, Tolosa E, et al. Immedi-ate release and controlled release carbidopa/levodopa in Parkinson’s disease. Neurology1999;53:1012–19.
4 Syed N, Murphy J, Zimmermann T Jr, et al. Tenyears experience with enteral levodopa infu-sions for motor fluctuations in Parkinson’s dis-ease. Mov Disord 1998;13:336–8.
5 Murer G, Dziewczapolski G, Menalled LB, et al.Chronic levodopa is not toxic for remainingdopamine neurons, but instead promotes theirrecovery in rats with moderate nigrostriatallesions. Ann Neurol 1998;43:561–75.
Brooks replies:Ponsford seems to focus primarily on thedesign and findings of the 056 trial ofropinirole versus levodopa in early Parkinson’sdisease recently reported in the N Engl J Med1
rather than the editorial as a whole; however, totake up his points:
Firstly, he suggests that it is unfortunatethat the 056 trial required a three times dailylevodopa dosage regime as use of morefrequent smaller doses could have reducedthe incidence of dyskinesias. We chose a threetimes daily regime in part to match the threetimes daily regime of ropinirole and alsobecause it was thought that this regimereflected common clinical practice in patientswith early Parkinson’s disease. A trial for-mally comparing use of multiple low doses oflevodopa versus a three times daily mediumdose regime in early Parkinson’s diseasewould, however, be of great interest. It mightwell be that the multiple low dose approach inearly disease would spare complications butthis has yet to be shown. Addition of acatechol-O-methyltransferase inhibitor tosmooth out the plasma levodopa profile inearly Parkinson’s disease might also provebeneficial.
Secondly, he suggests that the allowedpresence of selegeline may have magnified thetendency of levodopa to cause complications.This could indeed be the case althoughstratifying for selegeline usage in the levodopaarm did not highlight any such eVect.
Thirdly, he suggests that allowing the useof slow release levodopa preparations in earlyParkinson’s disease could have been benefi-cial. There is currently no trial data tosupport this viewpoint; on the contrary, earlyuse of either slow release madopar or sinemethas been reported to be associated with asimilar prevalence of complications as the useof standard preparations.
My current feeling is that early use ofdopamine agonists in suitable patients withParkinson’s disease remains a reasonablestrategy to delay complications. It may well bethat in the future, however, a more optimalway of delivering levodopa is devised whichachieves continuous and uniform dopaminer-gic stimulation and so reduces the prevalenceof fluctuations and dyskinesias.
D J BROOKSNeurology Department,
Imperial College School of Medicine,Hammersnith Hospital, London W12 0NN, UK
418 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

1 Rascol O, Brooks DJ, Korczyn AD, et al. A fiveyear study of the incidence of dyskinesia inpatients with early Parkinson’s disease whowere treated with ropinirole or levodopa.N Engl J Med 2000;342:1484–91.
Does disturbed homocysteine and folatemetabolism in depression result fromenhanced oxidative stress?
In their recent article, Bottiglieri et aldescribe increased homocysteine concomi-tant with decreased folate concentrationsin a subgroup of patients with depression.1
In addition, some relation betweenreduced folate availability and disturbedmonoamine metabolism was found. Theclose relation between increased homo-cysteine and reduced folate concentrations,which was described previously in otherclinical conditions such as cardiovascularand cerebrovascular diseases is usually as-cribed to a reduced dietary intake of folate,and dietary supplementation with folate iscapable of reducing hyperhomocysteinae-mia.
The coincidence described of disturbedhomocysteine and monoamine metabolismmay shed some additional light to the possi-ble mechanism underlying this metabolicabnormality. Both metabolic pathways de-pend on the presence of reduced pteridinespecies: (1) the biosynthesis of methioninerequires supply of methyl groups frommethyl-5,6,7,8-tetrahydrofolic acid, defi-ciency of which results in hyperhomocystei-naemia; (2) biosynthesis of serotonin,dopamine, and noradrenaline (norepine-phrine) depends on the presence of 5,6,7,8-tetrahydrobiopterin, deficiency of which thusresults in monoamine deficiency. Both tet-rahydropteridines are recycled by dihydrop-teridine reductases2; both compounds havestrong reducing capacities and are thus rap-idly oxidised by oxidising chemicals. Inter-estingly, recent studies show that depressionis associated with activation of the immunesystem,3 and it is even speculated that aninfectious agent might be involved. Immunesystem activation is accompanied by anincreased production of reactive species bycytotoxic cells such as activated monocytesand macrophages to achieve antimicrobialand cytocidal activities. Activated humanmonocytes/macrophages also release in-creased amounts of neopterin—another pte-ridine derivative—which is a sensitive indexfor the mediation of cell mediated immunereactions in patients.4 Recent data also pointto a new functional aspect of neopterin—namely, to enhance oxidative processes.4
Increased concentrations of neopterin havebeen described in patients with depression.3
Therefore the question arises whetheroxidative stress rather than insuYcient di-etary intake of folate is the basis of5,6,7,8-tetrahydrofolate and also 5,6,7,8-tetrahydrobiopterin deficiency. Interestingly,in patients with Alzheimer’s dementiaa similar relation has already beendemonstrated: hyperhomocysteinaemiawas associated with reduced folateconcentrations, but also an increaseddegree of immune activation could bedetected in the same patients.5 There isgood reason to think that the scenario mightbe similar in patients with depression,and enhanced oxidative stress due tochronic immune system activation is a majorcause of the loss of reductants such as
5,6,7,8-tetrahydrofolic acid and 5,6,7,8-tetrahydrobiopterin.
B WIDNERD FUCHS
Institute of Medical Chemistry and Biochemistry,Fritz Pregl Strasse 3, A-6020 Innsbruck, Austria;
University of Innsbruck, Ludwig Boltzmann Instituteof AIDS-Research, Innsbruck, Austria
F LEBLHUBERDepartment of Gerontology, Landesnervenklinik
Wagner-Jauregg, Linz, Austria
B SPERNER-UNTERWEGERDepartment of Psychiatry,
University of Innsbruck, Austria
Correspondence to: Dr D FuchsDietmar. [email protected]
1 Bottiglieri T, Laundy M, Crellin R, et al. Homo-cysteine, folate, methylation, and monoaminemetabolism in depression. J Neurol NeurosurgPsychiatry 2000;69:228–32.
2 Armarego WLF. A 20 year association withpteridines: chemistry, biochemistry and mo-lecular biology. Pteridines 1998;9:55–68.
3 Maes M, Bosmans E, Scharpe S, et al. Plasmasoluble interleukin-2-receptors in depression:relationships to plasma neopterin and serumIL-2 concentrations and HPA-axis activity. EurPsychiatry 1995;10:397–403.
4 Widner B, Baier-Bitterlich G, Wede I, et al.Neopterin: indicator of oxidative stress andpart of the cytotoxic armature of activatedmacrophages in humans. Pteridines 1998;9:91–102.
5 Leblhuber F, Walli J, Artner-Dworzak E, et al.Hyperhiomocysteinemia in dementia. J NeuralTransm 2000:107;1069–74.
Reynolds and Botiglieri reply:We thank Widner et al for suggesting anexplanation of our findings of impaired folateand monoamine metabolism in some patientswith depression.
The relation between homocysteine andfolate is well established, which is why weincluded it in our study. We agree that simplydietary deficiency is an inadequate explana-tion for folate deficiency in many patientswith depression as several studies have failedto confirm this.1 We have recently reported afall in CSF folate with aging2 and this may bea factor contributing to the high incidence offolate deficiency in psychogeriatric patients,including depression and dementia.1 We havealso reported a fall in CSF tetrahydrobiopt-erin (BH4) in depression which is correlatedwith folate deficiency, as reflected in a fall inred cell folate, and with impaired monoaminemetabolism—that is, a fall in CSF monaminemetabolites.3
The mechanisms of these relations be-tween impaired folate and monoamine me-tabolism remain uncertain but the suggestionthat oxidative stress plays a part is specula-tive. We are unaware of any clinical or experi-mental evidence that oxidative stress leads tofolate deficiency. It has been suggested thatfolates play a part in maintaining BH4synthesis.3 4 The relation of folate and BH4 toaging requires clarification. It is also relevantthat S-adenosyl-methionine, the major me-thyl donor in the brain which derives itsmethyl group from methyl folate can, likefolates, increase the turnover of monoaminesin the brain.5 This and other evidence suggestthat methylation mechanisms are involved inthese relations and in mood and cognitivefunction.6
E H REYNOLDSInstitute of Epileptology, Weston Education Centre,
King’s College, Denmark Hill Campus,Cutcombe Road, London SE5 9PJ, UK
T BOTTIGLIERIMetabolic Disease Center, Baylor Research Institute,
3812 Elm Street, Dallas, Texas 75226, USA
Correspondence to: Dr E H [email protected]
1 Bottiglieri T, Crellin RF, Reynolds EH. Folateand neuropsychiatry. In: Bailey LB, ed. Folate inhealth and disease. New York: Marcel Dekker,1995:435–62.
2 Bottiglieri T, Reynolds EH, Laundy MC Folatein CSF and age. J Neurol Neurosurg Psychiatry2000;69:562.
3 Bottiglieri T, Hyland K, Laundy M, et al. Folatedeficiency, biopterin and monoamine metabo-lism in depression. Psychol Med 1992;22:871–6.
4 Kaufman S. Some metabolic relationshipsbetween biopterin and folate:implications forthe “methyl folate trap hypothesis”. NeurochemRes 1991;16:1031–6.
5 Losada ME, Rubio MC. Acute eVects ofS-adenosyl-methionine on catacholaminergiccentral function. Eur J Pharmacol 1989;163:353–6.
6 Reynolds EH, Carney MWP, Toone BK. Meth-ylation and mood. Lancet 1984;ii:196–8.
Long term follow up afterperimesencephalic subarachnoidhaemorrhage
Marquardt et al describe the clinical courseand long term outcome of 21 patients theydiagnosed as having a perimesencephalichaemorrhage.1 The paper raises two ques-tions.
The first is an impression of an “emperor’snew clothes syndrome” given by the first fig-ure of the publication. This figure shows aslice of the CT made shortly after the initialepisode of headache in the patient reported tohave a recurrent episode of perimesen-cephalic haemorrhage. The legend of the fig-ure states that the CT shows extravasatedblood in the perimesencephalic subarachnoidspace, but we fail to see any blood at all.Thanks to the electronic availability of theJournal we were able to review not only thepaper version of the figure, but also anenlarged version on screen. Even afterenlargement no blood was seen; the slicenicely shows the tentorium adjacent to theambient cisterns, the proximal parts of theposterior cerebral arteries, and perfectly clearCSF in the perimesencephalic (chiasmaticand partly the ambient and quadrigeminal)cisterns and in the frontal interhemisphericand sylvian fissures.
There are several explanations for thisdiagnostic mystery. Firstly, the authors mayhave submitted an inappropriate slice of theCT. In some patients with perimesencephalichaemorrhage, the prepontine cistern is theonly site where CT shows blood.2 If bloodwas visible in the prepontine cistern in thisparticular patient, the authors have indeedfound a patient with a perimesencephalic,non-aneurysmal haemorrhage with a recur-rent haemorrhage. Given the unique charac-ter of this sequence of events, it would be fairto provide the appropriate slice to convincereaders of the Journal.
Secondly, if no evidence of blood is foundeven in the prepontine cistern, the patientmay have had a CT negative subarachnoidhaemorrhage. In patients with rupturedaneurysms CT can be negative, even ifperformed within 12 hours after onset of thehaemorrhage.3 But a negative CT plus anegative angiography does not add up to adiagnosis of perimesencephalic haemor-rhage.
Thirdly, the patient may not have had asubarachnoid haemorrhage at all. The casereport tells us that lumbar puncture waspositive, but does not give details. Because
J Neurol Neurosurg Psychiatry 2001;70:406–425 419
www.jnnp.com

lumbar puncture was performed before theCT, and CT was performed on the day of theheadache, lumbar puncture may have beenperformed too early to detect blood degrada-tion products in the CSF and therefore tooearly to distinguish a traumatic tap from agenuine haemorrhage. In the absence of deg-radation products the patient may have had anon-haemorrhagic cause of the headache.
With regard to figure 2, a slice of the CTmade after the second episode of headache, itwould be interesting to know the intervalbetween the onset of symptoms and the CT.Perimesencephalic haemorrhage can be diag-nosed reliably only if the initial CT isperformed within 3 days after onset of thesymptoms. After this interval distinctionbetween aneurysmal and perimesencephalicpatterns becomes unreliable.
The second issue is that of long term out-come. On long term follow up the authorsfound a high rate of persisting symptoms suchas headaches, irritability, depression, andfatigability. This contrasts with the goodquality of life (as measured by the sicknessimpact profile, a validated questionnaire onquality of life) found in a follow up study per-formed in The Netherlands.4 If the methodsused by Marquardt et al are valid, the diVer-ence in outcome between these two studypopulations requires an explanation. This iswhere the question on management strategyfor patients with perimesencephalic haemor-rhage comes in. Do the authors include anyrestriction in counselling their patients whohave had a perimesencephalic haemorrhage,or are such restrictions imposed on theseformer patients by physicians who assesspeople before employment or by medicaladvisors of insurance companies? We reassurepatients on discharge and again a couple ofweeks later, at an outpatients, consultation,that a perimesencephalic haemorrhage is nota warning for a major neurological event, wedo not impose any restrictions, and westimulate patients to take up all activities theyundertook before the haemorrhage. Wehypothesise that imposing restrictions orsharing uncertainties or worries with patientscan lead to subjective symptoms as describedabove. Given this possibility we are reluctantto start informing patients that perimesen-cephalic haemorrhages can reoccur, as longas recurrences have not been reportedconvincingly. We do agree that the patientreported on forms a diagnostic challenge, butwe have too little evidence to agree that thispatient has had two episodes of perimesen-cephalic haemorrhage.
G J E RINKELDepartment of Neurology, University Medical Centre,
PO Box 85500, 508 GA Utrecht, The Netherlands
B K VELTHUISDepartment of Radiology
Correspondence to: Dr G J E Rinkel
1 Marquardt G, Niebauer T, Schick U, et al. Longterm follow up after perimesencephalic sub-arachnoid haemorrhage. J Neurol NeurosurgPsychiatry. 2000;69:127–30.
2 Rinkel GJE, Wijdicks EFM, Vermeulen M, et al.Non-aneurysmal perimesencephalic subarach-noid hemorrhage: CT and MR patterns thatdiVer from aneurysmal rupture. AJNR Am JNeuroradiol 1991;12:829–34.
3 van der Wee N, Rinkel GJE, Hasan D, et al.Detection of subarachnoid haemorrhage onearly CT: is lumbar puncture still needed aftera negative scan? J Neurol Neurosurg Psychiatry1995;58:357–9.
4 Brilstra EH, Hop JW, Rinkel GJE. Quality of lifeafter perimesencephalic haemorrhage. J NeurolNeurosurg Psychiatry 1997;63:382–4.
The authors reply:We respond to some of the questions raisedby Rinkel and Velthuis on our recentpublication in this Journal.1 The patient ofinterest presented with typical clinical signsof subarachnoid haemorrhage. He com-plained of sudden onset of severe headaches,irradiation into the nuchal region, andnausea. Lumbar puncture was performedand blood stained CSF was found. Centrifu-gation of the CSF disclosed xanthochromiaof the supernatant fluid and cytology demon-strated siderophages indicating the presenceof intracranial haemorrhage as no lumbarpuncture was carried out earlier.2
Non-contrast enhanced CT showed bloodin the ambient cisterns and these findingswere interpreted as perimesencephalic sub-arachnoid haemorrhage in two diVerent hos-pitals.
Four vessel digital subtraction cerebralangiography with multiple views was negativeas was a repeated angiography 10 weeks later.A third angiography performed in the courseof the second episode of haemorrhage againdid not disclose any source of the bleeding,and thus the causes of bleeding remain unre-solved. A recent publication by Canhao et alstudied the prevalence of vascular risk factorsin patients who had perimesencephalic sub-arachnoid haemorrhage.3 They found thathypertension was more frequent amongpatients with perimesencephalic haemor-rhage than among two control groups andthat among women, smoking was more com-mon in perimesencephalic haemorrhage.However, the medical history of our patientwas not relevant, and there was no history ofprevious arterial hypertension.
Rinkel and Velthuis express their concernabout a high rate of persisting symptoms suchas headaches, irritability, depression, andfatiguability in long term follow up of ourpatients. They state that these findingscontrast with the good quality of life found ina follow up study performed by Brilstra et al4
and that these diVerences require explana-tion.
In this study, which was cited by us as well,quality of life was measured by means of thesickness impact profile and outcome ofpatients with perimesencephalic subarach-noid haemorrhage was compared with thatof a reference population. Analysing the sub-mitted data, however, significant diVerencestowards less dysfunction in patients wereproved only for the categories body care,movement, and household management. Sixof the 25 patients (24%) had more dysfunc-tion in the category work than the referencepopulation, and 11 patients (44%) reporteda change in their headache pattern asnon-specific headaches occurred more oftenthan before the haemorrhage in 10 patientsand less often than before in one patient.Two patients reported fear of rebleeding.Brilstra et al concluded that patients with aperimesencephalic haemorrhage have noreduction in quality of life but had to admit“that most consequences of the perimesen-cephalic subarachnoid haemorrhage arefound in the psychosocial domains.” Theyrelate the problems with short term memory,sleeping, fears, irritability, and nervousnesswith the haemorrhage itself and with theexperience of sudden illness leading toadmission to an intensive care unit.4 Theseresults imply that in the Dutch study as wellpersisting symptoms are frequent and thisdoes not contrast with our findings at all.However, the focus of our follow up study
was directly on these psychosocial implica-tions of perimesencephalic subarachnoidhaemorrhage. Only 38% of our patientsthought that they were fully recovered andcompletely well whereas 62% of the patientshad residual complaints. Moreover, only41% of the patients returned to theirprevious occupation whereas 53% of thepatients retired from work and one manbecame unemployed. Thus quality of lifeafter the haemorrhage is not as excellent assuggested and it becomes obvious thatperimesencephalic subarachnoid haemor-rhage has an enormous impact on individualpatients and social life.
We do agree with Rinkel and Velthuis onthe further management strategy for patientswith former PMSAH. We inform the patientsof the benign nature of the disease and do notimpose any restrictions at all. We alsoreassure the patients that they can return tothe same regular daily activities they under-took before the haemorrhage.
It is supposed that in 15% to 20% of thepatients with subarachnoid haemorrhage theangiogram is negative and that patients withPMSAH account for about half of thesepatients with angiogram negative subarach-noid haemorrhage.3 5 On these premises theremust be thousands of patients every year whoare treated for PMSAH throughout theworld. However, reviewing the literature in1996, Schwartz and Solomon could only find169 reported patients who had PMSAH.5 Itseems, therefore, reasonable to compile moredata to gain more information about thenatural course of PMSAH in significantlylarger cohorts of patients.
G MARQUARDTT NIEBAUER
U SCHICKR LORENZ
Neurosurgical Clinic, Johann WolfgangGoethe-University, Schleusenweg 2–16, 60528
Frankfurt am Main, Germany
Correspondence to: Dr G Marquardt
1 Marquardt G, Niebauer T, Schick U, et al. Longterm follow up after perimesencephalic sub-arachnoid haemorrhage. J Neurol NeurosurgPsychiatry 2000;69:127–30.
2 Vermeulen M, van Gijn J. The diagnosis of sub-arachnoid haemorrhage. J Neurol NeurosurgPsychiatry 1990;53:365–72.
3 Canhao P, Falcao F, Pinho E, et al. Vascular riskfactors for perimesencephalic non-aneurysmalsubarachnoid hemorrhage. J Neurol 1999;246:492–6.
4 Brilstra EH, Hop JW, Rinkel GJE. Quality of lifeafter perimesencephalic haemorrhage. J NeurolNeurosurg Psychiatry 1997;63:382–4.
5 Schwartz TH, Solomon RA. Perimesencephalicnon-aneurysmal subarachnoid hemorrhage: re-view of the literature. Neurosurgery 1996;39:433–40.
Idiopathic intracranial hypertensionand anticardolipin antibodies
The study by Kesler et al1 concludes with theassumption that the presence of anticardioli-pin antibodies (aCL-Abs) indicates a uniquesubgroup of patients with idiopathic intracra-nial hypertension. Their study does not sup-port this view. They regard as important inthis respect the fact that the three patientswith aCL-Abs (p<0.035) were significantlyolder than those without. This is hardlysurprising when it is known that the incidenceof these antibodies increases with age andmay be identifiable in up to 12% of healthypeople.2 3 Their control group therefore needsto be age matched. Further speculation for
420 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

this conclusion is in their statement that theremay have been an occult thrombosis of thecerebral venous sinuses, a fact that I agreewith as CT and MRI cannot exclude athrombosis for certain–hence intracranialhypertension would not be the diagnosis. It isnot stated how soon cerebral sinus imagingwas performed after the onset of symptoms.Thirdly, the concentration of raised aCL-Abin these patients is not very high apart fromthe initial measurement in patient 1. Titresless than 40 units are generally not thoughtto be pathological but this is quite anarbitrary figure as test systems are variableand not standardised.4 Fourthly, with aprevalence of aCL-Ab at 5% in Israel, thepresence of these antibodies in three of 37patients is not a significant finding (÷2;p>0.45). Finally, it is an accepted view thatthe presence of aCL-Ab may represent anepiphenomenon due to a non-specifiedinjury. This is supported by the incidentalfindings of aCL-Ab in symptom-free pa-tients.5 Hence the findings cannot supportthe authors’ proposal that the patients withaCL-Ab form a subgroup of patients withintracranial hypertension.
O C BACKHOUSEDepartment of Ophthalmology, Leeds General
Infirmary, The Leeds Teaching Hospitals NHS Trust,Leeds LS1 3EX, UK
Correspondence to: Dr O [email protected]
1 Kesler A, Ellis M, Reshef T, et al. Idiopathicintracranial hypertension and anticardolipinantibodies. J Neurol Neurosurg Psychiatry2000;68:379–80.
2 Vila P, Hernandez M, Lopez-Fernandez M, etal. Prevalence, follow up, and clinical signifi-cance of the anticardolipin antibodies innormal subjects. Thromb Haemost 1994;72:209–13.
3 Fields R, Toubbeh H, Searles R, et al. Theprevalence of anticardolipin antibodies in ahealthy elderly population and its associationwith antinuclear antibodies. J Rheumatol 1989;16:623–5.
4 Biousse V. Coagulation abnormalities and theirneuro-ophthalmic manifestations. Curr OpinOphthalmol 1999;10:382–93.
5 Greaves M. Antiphospholipid antibodies andthrombosis. Lancet 1999;353:1348–53.
BOOK REVIEWS
The Neuropathology of Schizophrenia.Progress and Interpretation. Edited byPAUL J HARRISON and GARETH W ROBERTS
(Pp374, £65.00). Published by OxfordUniversity Press, Oxford, 2000. ISBN0-19-262907-7.
The neuropathology of schizophrenia hasbeen for a long time perhaps one of the mostcontroversial fields of biomedical research. Inthe middle decades of the last century therewas an increased interest in the neuropathol-ogy of psychosis based on the assumptionthat structural alterations in the brain wouldprovide insight into the understanding of thiscomplex and devastating disease. However,the results of these investigations have beencontradictory and it has become a cliché tosay that schizophrenia is the graveyard ofneuropathologists. Indeed, the results of neu-ropathological investigations were confusing,
and resulted from both clinical and patho-logical problems. The clinical definition ofschizophrenia has been controversial and fora long time internationally accepted diagnos-tic criteria did not exist. Patients’ cohortswere extremely variable and clinical historiesfar from complete. Most patients had treat-ment which again varied from centre to cen-tre. The neuropathological methodology wasalso somewhat primitive and inappropriate todetect subtle changes. Moreover, the materialexamined varied considerably from centre tocentre and sometimes the pathologicalchanges described were the result of anotherdisease process, including epilepsy or minortraumas. It was thus not surprising that thesestudies were contradictory.
With the advent of neuroimaging a new erahas started. It was Johnstone and hercolleagues who showed structural alterations(enlarged ventricles) in the brain of psychoticpatients using at that time, the novel method-ology of CT scanning. Not much later areport of Stevens observing astrocytosis inthe brains of patients with schizophrenia,rekindled interest in the neuropathology ofschizophrenia.
This book is a comprehensive review ofcerebral changes associated with psychosis.The 15 chapters cover a wide range of struc-tural, functional, macroscopical, histological,neurochemical, and pharmacologicalchanges associated with the disease. In addi-tion there are chapters on animal models andmethodological issues, as well as on the con-sequences of treatment. The results of struc-tural and functional imaging are reviewed,the second in relation to neural circuitries.There is an excellent chapter on cerebralasymmetry, a feature important in the under-standing of the disease. Two chapters dealwith development, one more specifically withcortical development, giving a concise reviewof the molecular basis for the organisation ofthe forebrain and pattern formation inrelation to pathogenesis. Synaptic pathologyand the organisation of cortical circuitries, fora long time inaccessible to conventionalmethodology, have become the subject ofintense research, and recent developmentshave been suitably summarised in twoseparate chapters. The chapter on corticalpathology reviews a new generation of quan-titative microscopical studies in relation tothe GABA, glutamate, and dopamine sys-tems. The problems of gliosis are revisited ina separate chapter with the conclusion that itis unlikely to be a core feature of theneuropathology of schizophrenia. A chapterexamines schizophrenia from the perspectiveof other neurodegenerative diseases andlesions, including those which may causeschizophrenia-like symptoms—for example,metabolic diseases, epilepsy, and psychosis inneurodegenerative disorders. These provideuseful information in the diVerential diagno-sis of schizophrenia and other diseases of thenervous system with similar symptomatology.
This is a timely book, reviewing recentdevelopments in our understanding of thedisease mechanism in schizophrenia. Theeditors have brought together internationalexperts in the field to produce a book with atrue multidisciplinary approach. Theirachievement should be congratulated. How-ever, less praise should be lavished on thepublishing house, which has failed to invest inhigh quality reproductions. There is a singlecoloured plate of neuroimaging of MRI andPET and the black and white reproductionsof the same images have not been removed.
None the less, this is a book which should bepurchased by those who are interested inschizophrenia, both neuropathologists andpsychiatrists.
P L LANTOS
Philosophical and Ethical Problems inMental Handicap. By PETER BYRNE (Pp175,£40.00). Published by Macmillan Press,Basingstoke, 2000. ISBN 0 312 23460 0.
I cannot recommend this slim monographhighly enough to anyone working with peoplewho have the disorders of neurological devel-opment that give rise to what we term mentalhandicap, mental retardation, or learning dis-ability. It is tightly argued, well written, andthought provoking and bears reading muchmore than once.
Psychiatrists are still the dominant medicalprofession working with people with learningdisability, a fact of history that has been slowto change. Lothian has recently closed itslarge institution, one of several in Scotlandthat had around 1000 beds at its peak, withward sizes to match. It is easy to condemnthis partitioning of a whole section of society;it is much more diYcult to reconcile andaccept the fact that they do diVer in a host ofways from most people and yet at the sametime they have identical moral worth to anyother human being. Professor Byrne’s centraltenet is that these are not mutually exclusiveconcepts, but can be—indeed must be—integrated to make philosophical sense. Theother paths lead on the one hand to the con-cept that the labelling process has created afictional disability, that its use is a method ofsocial control, of maintaining power by thecreation of an underclass, and this denial ofintrinsic or primary disability is inherent insome of the concepts of full inclusion andnormalisation. The other position, perhapsmore worrying, is held by some philosophersof bioethics who would define humanity andmoral worth on the ability to reason, placingpeople with cognitive disability in a separatedomain in which it is permissible to useeugenic policies to select against them, to jus-tify infanticide.
Byrne argues with great care against boththese paths, his arguments grounded on thecommonality of humanity and what it is to behuman, carefully dissecting and dismissingthe accusation that this opens the way to thecharge of speciesism. He progresses from thedefinitions of mental handicap, the moral sta-tus of the disabled, through the diYcultterrain of euthanasia, abortion, genocide, andoppression, finally ending with theologicalinterpretations that oVer alternatives to hu-manism.
An appreciation of a common humanity isthe best protection for such people. I haveusually found that the persons are not soconcerned as to whether we describe theirintrinsic cognitive condition as a mentalhandicap or a learning disability but as towhether we remember their first and secondnames.
Psychiatrists may think that of course weknow all this, and that of course we would notfollow any of those extreme paths that aredescribed by Professor Byrne. The 86 menwho used to be herded together in one of theold wards that I inherited might not agree.
WALTER MUIR
J Neurol Neurosurg Psychiatry 2001;70:406–425 421
www.jnnp.com

Multiple sclerosis. Diagnosis, MedicalManagement and Rehabilitation. Editedby JACK S BURKS and KENNETH P JOHNSON. (Pp624, US$210.00). Published by DemosMedical Publishing, New York, 2000. ISBN1 888799 35 8.
The past few years have seen a plethora ofbooks on multiple sclerosis. Many of thesebooks simply decorate my bookshelf and arerarely consulted by any member of the team.
Some cynics have correlated a high numberof recent publications with pharmaceuticalinvestment in new drugs; there is no doubtthat many of these books are purchased bythe pharmaceutical companies and are oftenused as marketing tools.
Each new volume, therefore, needs tojustify its existence in some way. In most casesthe battle is between McAlpine’s MultipleSclerosis and each newcomer. Most youngpretenders are unable to match McAlpine inmost areas. This is certainly true for thepresent volume in the context of the history,epidemiology, pathology, diagnosis, and aeti-ology sections. Where this book is particularlystrong and justly deserves its place on themultiple sclerosis bookshelf is in the clinicalsections, most of which are very helpful.
The book is divided into eight sections,including most of the ones mentioned.Section 6 deals with symptom managementand rehabilitation, including specific chapterson weakness, mobility, fatigue, spastic pare-sis, vertigo and incoordination, neuro-ophthalmology, paroxysmal disorders, cogni-tive and emotional problems, pain anddysaesthesia, bladder dysfunction, bowel dis-turbance, sexual dysfunction, autonomic dis-orders, dysarthria, and dysphagia. Thesechapters are generally informative and wellwritten by specialists in their field. I wouldparticularly pick out excellent chapters onneuro-ophthalmic signs and symptoms, byElliot Frohman and colleagues, a very usefulchapter on dysarthria, and an excellentsection on clinical and rehabilitation outcomemeasures by Linda Coulthard-Morris, whichcontains analysis of most of the commonlyused measures together with examples of theforms.
The later chapters are involved with “soci-etal issues” and are more specific to theAmerican care system than European healthcare models.
This book is much weaker in the earliersections and it is here that I would reach formy trusty McAlpine.
A volume such as this is an opportunity notonly to review evidence, but also to interpretin a particular fashion. During this interpret-ation, authors may be tempted into contro-versial areas, occasionally using logic whichcan at best be described as woolly. Oftenthese ideas are promulgated at commerciallysponsored satellite symposia where the prom-ise of a free lunch and more books draws alarge audience. It is much more diYcult toget some of these theories into scientific peerreview publication than it is to express themat satellite symposia and in book form. Forexample, the concept of “asymptomaticdisease” and “symptomatic disease” has beenaround for a while and is again exposed byPat Coyle in this book. The logic goes some-thing like: early inflammation causes earlyaxonal damage, therefore we must treat earlyand aggressively. Jack Simon in his MRIchapter provides a good deal of commonsense information, including the fact thatcompletely unsuspected multiple sclerosis
may be detected at necropsy. At present, wehave no hard information to inform us thatany of the immunotherapies make a diVer-ence to secondary progression in the longterm, and the implication that we should betreating MRI rather than a patient may causedysphagia to some neurologists on this side ofthe Atlantic.
It is important to maintain a balancedviewpoint in life. Overall, the many goodchapters in this book outweigh the mediocreones, and the quality of the paper is wonder-ful!
JOHN ZAJICEK
Texture of the Nervous System of Manand the Vertebrates by Santiago Ramony Cajal. Volume 1. Translated and editedby PEDRO PASIK and TAUBA PASIK (Pp v1, 631.DM289, $US189.). Published by:Springer-Verlag, New York, 1999.84-07-00200-3 (complete series3-211-83056-1).
Everything we know about structure, func-tion, and physiology in the nervous system atthe cellular level, in health and disease,evolves from the concept that organisation isthrough the connectivity of functionally inde-pendent neurons and their processes. San-tiago Ramon y Cajal distinguished neuronsfrom glia; showed the variability of dendriticarborisations and axon terminations; estab-lished that axon cylinders end freely but formcontacts; conceived that the nerve impulse isconducted between axons, dendrites, and thecell body of neighbouring neurons; had theconcepts of trophism and tropism; andfollowing Rudolph Virchow, regarded the cellas the unit of all biological systems. His mostdetailed studies were of the cerebellum but, intime, no part of the brain and spinal cordwent unexplored. His great synthesis was tosettle debate on the neuron theory. Hisdescriptions were supplemented by beautifuldrawings based on Golgi stains. He and Golgiwere jointly awarded the Nobel prize formedicine in 1906. They disagreed publiclyduring the lectures in Stockholm.
Cajal is the most significant neuroscientistof the 20th century—Sir Charles Sherringtonbeing his only serious competitor. They metonly once when Sherrington hosted Cajal’sstay in London to deliver the 1894 RoyalSociety Croonian lecture. During the visit,Cajal was arrested as a vagrant at Cambridgerailway station when visiting the provinces toreceive an honorary doctorate. Cajal andSherrington fell over themselves to outpraiseeach other. Sherrington on Cajal: “He is thegreatest anatomist the nervous system hasever known . . .he solved at a stroke the direc-tion of nerve currents in their travel throughthe brain and spinal cord . . .it was a step ofgenius to study the embryonic nervoussystem.“
Between 1880 and 1933, Cajal wrote 288scientific publications including 22 mono-graphs. Much of his work remains untrans-lated from the original Spanish and henceunread. But the sustained admiration forCajal’s writings and their contemporaryrelevance for neuroscience is now matched bya welcome revival in publishing his works.Textura del sistema nervioso del hombre y de losvertebrados was published from Madrid inthree volumes (1897, 1898, and 1904). It wasupdated by Cajal with new text and illustra-tions for the translation into French asHistologie du systeme nerveux de l’homme et des
vertebres by Dr Leon Azoulay (2 volumes:1909-11). The complete French edition wasfirst translated into English by Neely Swan-son and Larry Swanson as Histology of theNervous System of Man and Vertebrates (Ox-ford University Press, 1995). Now theoriginal Spanish text is undergoing transla-tion by Pedro Pasik and Tauba Pasik asTexture of the Nervous System of Man and theVertebrates. The first of three volumes ap-peared in 1999; the other two are promisedfor 2000.
The advertising flysheet champions Cajal’sdiscovery of growth cones, chemoattractantsubstances, dendritic spines, and corticalinterneurons and claims absolute authorityover both the French and first Englisheditions. It boasts illustrations based onoriginal reproductions of drawings archivedin the Cajal Institute in Madrid (the evidenceis in the Museo-Cajal-Madrid stamp on manyfigures) with very little copied from previ-ously published editions. Facts and citationsare corrected from Cajal’s original text andauthenticated against contemporary sources.In which edition should the discerning Cajalreader invest? When complete, the Springerset will cost DM850/£330/$550 comparedwith £150 for the two Oxford volumes. ThediVerence is worth paying. The English-Spanish text is authentic: compare “the nerv-ous system represents the ultimate boundaryin the evolution of living matter, and the mostcomplicated machinery of noblest activitiesthat Nature has to oVer“ (English-Spanish)with “countless modifications during evolu-tion have provided living matter with aninstrument of unparalleled complexity andremarkable functions: the nervous system,the most highly organised structure in theanimal kingdom“ (English-French); or “itappears that with this [chemotactic] hypoth-esis we have shed light into a dark cave, whenin reality we have explored only the entrance,from which its imposing abyss appears evenmore distant and black“ (English-Spanish)versus “the theory of chemotaxis we ad-vanced . . .initially appeared to be pureconjecture with no hope of verification,although recently it has gained experimentalsupport“(English-French).
The text is authoritative and the produc-tion lavish. Pedro and Tauba Pasik include,and readily identify, translation of materialadded by Cajal for the French editionbetween 1904 and 1909. Original footnotesare retained but the citations are modernisedand gathered in a single section completingthe English-Spanish text. The lack of anindex will be put right when volume three ispublished. The illustrations are incomparablybetter in the Springer than the Oxfordvolume[s]. The line drawings are much morecrisp; the original figures of methylene bluestaining reproduce poorly as black and white(Oxford) but some of their polychromaticfigures are more subtle. Volume one dealswith the general principles of organisation inthe nervous system and Cajal’s methods, thedetails of neuronal structure and the spinalcord. Volumes two and three will completethe medulla and pons, cerebellum, midbrain,diencephalon including the retina, cortex,and autonomic nervous system. The originalSpanish and French editions are very expen-sive and virtually unobtainable. For thehistorian, physician, or scientist who studiesneuroscience, whether or not to invest in theSpringer set is simply not an issue—even ifyou already have the Oxford edition. Both aremagnificent publishing achievements . . .but
422 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

the Pasiks are on course to produce thedefinitive English language edition of thedefinitive Cajal.
ALASTAIR COMPSTON
Issues in preventive psychiatry. Editedby GEORGE N CHRISTODOULOU, DUSCIA
LECIC-TOSEVSKI, AND VASSILIS P KONTAXAKIS.(Pp148, US$97.50) Published by Karger,Basel, 1999. ISBN 3 8055 6912-2
This book comprises a selection of paperstaken from a world psychiatry associationsymposium on preventive psychiatry. Twocontributors are from the United States, onefrom Egypt, the remainder from Europe, par-ticularly south eastern Europe. The prefaceopens with a reference to an earlier WorldHealth Organisation (WHO) report whichestimated that as many as one third to onehalf of all mental and neurological disorderscould be averted by primary preventivemeasures. But it went on to note that in mostspheres primary prevention had been ne-glected due principally to a lack of awarenessof available eVective methods, a deficiencythat the book aimed to redress. Encouragedby this introduction the reader may then hopeto become acquainted with some of thestrategies and methodologies of preventivepsychiatry and even to read of a few of itssuccesses. If so, disappointment lies in store.Most of the writers approach their subjectthrough a protective smoke screen of broadgeneralisations; few emerge from it to oVer adetailed account of how any aspect ofpreventive psychiatry works on the ground.Some avoid the topic altogether: two of themore succinct chapters describe a process ofdeinstitutionalisation in Greece—a subjectnot without interest, but one that is onlyloosely connected with the book’s principalpurpose. Whether, and if so, how preventivepsychiatry succeeds receives little attention.Surprisingly little relevant outcome data arepresented. Much of the writing is stilted andlacks fluency. As with so many postsymposiaoVerings, thematic coherence is wanting.
It is diYcult to know who would benefitfrom reading this book. Preventive psychiatrymay not be the easiest subject to write about,but if it is to reach the audience it deserves, itwill need a more coherent and persuasiveplatform than this collection of contributionsprovides.
BRIAN TOONE
Microanatomical aspects forNeurosurgeons and Neuroradiologists.Edited by WOLFGANG SEEGER. (Pp 423,US$298.00). Published by Springer-Verlag,Wien, 2000. ISBN 3 211 83376 5.
The libraries of Cambridge Colleges containmany treasures. Among the particular treas-ures to be found in Peterhouse College is afirst edition of the Anatomical Engravings ofVesalius. This exquisite volume combinespractical instruction with wonderful aestheticpleasure in a manner that has long gone fromour more pedestrian age. Although aestheticpleasure is largely absent from our modernmanuals of practical instruction, neverthelesssuch manuals still have a place in the trainingand instruction of the young surgeon. Thisvolume will undoubtedly prove useful in thatrespect and indeed to some degree it followsdirectly in the tradition of Vesalius. Itrepresents the distillation of a lifetime’s work
of Dr Segar from Freiburg and is truly amonumental volume. It is beautifully illus-trated and a pleasure to handle. The labellingis clear and the diagrams are for the most partelegantly coloured, easy to follow, andstylishly executed. For a modern instructionmanual it comes surprisingly close to the aes-thetic tradition exemplified by the Vesalius.
However, all manuals of surgical anatomyhave the common characteristics that theyportray the normal anatomy, but as everysurgeon soon learns, in the vast majority oftheir cases the normal appearances have beendistorted or obliterated by the pathologywhich has caused the need for the surgery inthe first place. However, surgeons must alsoknow what things ought to look like evenwhen they are not visible when he first startsout upon his surgery. This volume illustratesthe diVering anatomical appearances to beencountered in various neurosurgical ap-proaches and this methodology is particularlylikely to be helpful for the junior trainee. It istherefore an ideal book for the departmentallibrary, but I fear may prove too costly for theaverage surgical trainee. The CambridgeColleges have traditionally chained theirmore valuable volumes to prevent theirdisappearance and I suspect this volume mayrequire an equivalent degree of supervision toprevent it disappearing into the eager regis-trar’s personal bookcase.
DAVID G HARDY
Hormones, Gender and the AgingBrain. The Endocrine Basis of GeriatricPsychiatry. Edited by MARY J MORRISON. (Pp359, £60.00). Published by CambridgeUniversity Press, Cambridge, 2000. ISBN0-521-65304-5.
Hormonal changes clearly influence brainfunction and certain mental disorders, suchas depression, are associated with, and mayeven result from, disorders of the endocrinesystem. As normal aging is associated withvarying degrees of dysregulation in the endo-crine system, this book addresses the hormo-nal basis of mental disorders in older people,which oVers the possibility of new therapeuticapproaches in an ever growing aged popula-tion.
The first section of the book provides aconcise and comprehensive overview of thediverse sites and cellular mechanisms ofaction of steroid and thyroid hormones in thebrain as well as their synthesis in theendocrine organs or in the brain itself. Thesecond section identifies age related changesin the prevailing levels of cortisol, thyroidhormones, and sex steroids (estrogen, proges-terone, testosterone, and dehydroepiandros-terone) and assesses the evidence for ascrib-ing a role for these changes in the emergenceof common mental disorders. For example,animal and human studies suggest that highcorticosteroid concentrations in elderly sub-groups are associated with a higher risk ofdeveloping cognitive defects; reduced respon-siveness of the hypothalamo-pituitary-thyroidaxis in aging seems to be related to mood dis-orders; therapy with estrogen (women) ortestosterone (men) may be protective againstdeveloping depressive symptoms and estro-gen may have beneficial eVects on cognitionand dementia. Sex diVerences and, by impli-cation, a role for sex steroid hormones, arealso noted in schizophrenia, anxiety disor-ders, pain perception, immune function, andpsychotropic drug metabolism. However,
many contributors emphasise the inconsist-encies in the scientific literature and the gen-eral lack of properly controlled hormonereplacement studies in elderly people. There-fore, the view that youthful hormonal profileswill promote healthier aging must remainspeculative until more conclusive evidence isavailable. In its critical approach, this bookshould be an impetus to such potentiallyimportant research and it provides valuableinformation for clinicians and basic research-ers alike in this complex and growing area.
GLENDA GILLIES
Essential Psychopharmacology:Neuroscientific Basis and PracticalApplications. By STEPHEN M STAHL (Pp 601,£39.95). Published by CambridgeUniversity Press, Cambridge, 2000. ISBN0-521-64615-4.
Psychiatry is a strange clinical subject. It hasby far the smallest knowledge base of any ofthe major subspecialties yet the argumentsover what should enter curricula and whatshould be in our postgraduate examinationssare as fierce as in any other Royal College.There are major requirements—for example,to understand psychotherapy and psycho-logical treatments. Yet as a treatment toolthey still play a subordinate role to biologicaltreatments in most settings for the manage-ment of the severely mentally ill. By contrast,time and time again surveys of trainee’s needspersistently cry out for good information onpsychopharmacology. In addition any teach-ing courses in psychopharmacology arealways voraciously snapped up. Our ownflawed Maudsley prescribing guidelines,which started life as a simple internaldocument, is thirstily sought after. EssentialPsychopharmacology is the book I alwayswanted to write but have been soundly beatento it by Stahl. The first edition was a finelycrafted book with logically distinct sectionson basic science, disease mechanisms, drugaction, and drug classes. This third edition isnow much improved again with copious col-our illustrations and bang up to date scientificinformation about both aetiological theoriesand new products and their associated modesof action. As the introduction states much haschanged since the publication of the first edi-tion 4 years ago. In one sense this is a slightpitfall of the book. The second edition hasbeen prepared in the middle of a majorresearch boom in psychopharmacology andin its attempt to be up to date it is in dangerof becoming rapidly out of date. A text bookformat with a fair publication lag may not bethe best vehicle for an attempt to cover abso-lutely up to date information. Nevertheless, Ithink the book is near perfect as a textbook ofbasic pharmacology. If I were to try andimprove it further, I wonder whether theauthor and publisher might think again aboutthe illustrations. It is superbly illustrated butsometimes the cartoon imagery is so meta-phorical that it serves occasionally to becumbersome and lacking clarity occasionallyobscuring rather than clarifying the issue.
The book strangely lacks an internationalfeel. There is a lot of Americanese (drugcombos) and the contents are largely basedaround around a United States formularywith some unfamiliar drugs as well as missingsome familiar European entities. All in allthough a benchmark book for modernpsychopharmacology teaching.
ROBERT KERWIN
J Neurol Neurosurg Psychiatry 2001;70:406–425 423
www.jnnp.com

Introduction to Clinical Neurology. ByDOUGLAS J GELB (Pp86, £22.50). Publishedby Butterworth Heinemann, Oxford, 2000.ISBN 0-7506-7202-1.
This is the second edition of Gelb’s system-atic approach to the neurological problemslikely to be encountered in general medicalpractice. The book seems to be aimed at gen-eral physicians in training, and medicalstudents. Although it also contains much toengage the interest of specialist trainees inneurology, I suspect that most of them willuse a more didactic text. Its appeal to medicalstudents may be diminished by the relativelack of illustrations; pictorial material ismainly limited to anatomical line drawings inthe early sections of the book. I did notencounter a single MR or CT brain scan; anomission giving the book questionable rel-evance to the starting point which is funda-mental to contemporary diagnosis and un-derstanding of neurological diseaseprocesses. At 365 text pages, it is a little onthe long side for medical students, who areattached to neurology for only a few weeks.On the positive side, many students will beintrigued by the frequent use of case historiesaround which the authors build discussion ofdiVerential diagnosis.
This second edition particularly introducesdetails of neurological examination which aresaid to be unavailable elsewhere. The au-thors’ programme for neurological examina-tion is highly detailed and comprehensive. Itis my impression that such exhaustive roteneurological examination is falling out offavour in specialist neurological practice inBritain. Yet it seems popular still with NorthAmerican neurologists who may benefit fromthe luxury of more time for patient consulta-tion, or may suVer more from the fear of legalretribution if they miss something. At the endof the chapter on neurological examinationthere is a brief section on “how to modify it”.This includes a screening neurological exam-ination. One could take issue with thesummary range of tests which have been cho-sen, particularly the value of looking for pro-nator drift, testing finger tapping, and testingboth heel-shin coordination and tandem gait.It always seems to me that a screening neuro-logical examination must avoid redundancy,and be rich in tests which provide unequivo-cal evidence of pathology. So why is fundo-scopic examination of the optic disc omittedfrom this recommended screening examina-tion, given that it can reveal the crucial physi-cal sign of papilloedema in a patient whomight otherwise be thought to have non-specific headache?
Each chapter starts with a set of thoughtprovoking case histories. Some of us are of themental disposition that enjoys learning fromsuch games, although others find it moreprofitable to digest more formally presentedtextbook information. I admit that I tend tofall into the second group, finding it less easyto evaluate “paper patients” than real patientsin a consulting room. My own thought proc-esses were impeded by finding that the casehistories, and the discussion of them, are dis-located to separate parts of the chapter,necessitating constant thumbing backwardsand forwards.
I suspect that this relatively wordy basictextbook will appeal more to students andjunior doctors in North America than in theUnited Kingdom. In the United Kingdommedical students seem to want short pithybooks. Junior staV in general medical train-
ing often seem to use the neurology entries ingeneral medical textbooks. Neurology train-ees and consultants seem to refer to special-ist textbooks. But if you like problemorientated learning, you should flick througha copy of this book to see whether it suitsyou.
MICHAEL DONAGHY
Parkinson’s Disease and Parkinsonismin the Elderly. Edited by JOLYON MEARA andWILLIAM C KOLLER (Pp251, £29.95).Published by Cambridge University Press,Cambridge, 2000. ISBN 0-521-62884-9.
This is a multiauthor book which aims to dis-cuss Parkinson’s disease and parkinsonism inthe elderly. The chapters are on the wholewell written and the overall style is very muchone of a practical handbook which isrelatively easy to dip in and out of. There islittle overlap in information across chapters.Chapters which stand out for their usefulcontent, particularly in relation to the olderage group, are those on diagnosis (Rodnit-sky), the overview chapter on Parkinson’sdisease and parkinsonism in the elderly(Meara and Bhowmick), and treatment(Zesiewicz and Hauser). However, the treat-ment presentation of the current use ofCOMT inhibitors could be improved: thesection meticulously describes the clinicaleVects of tolcapone, its half life, the appropri-ate dose, and the side eVects, and it is notuntil the last paragraph that the reader isalerted to the fact that the drug is no longerused in Europe and its usage in the UnitedStates is limited.
The main failing of the book is in theselection of chapters. I have three criticisms.The chapters on ancillary areas such asnursing, physiotherapy, speech, and occupa-tional therapy (of Parkinson’s disease) andspecific approaches to rehabilitation (in Par-kinson’s disease and parkinsonism), arebrought nicely into the context of medicalmanagement. However, depending on theintended audience, too large a proportion ofthe book is dedicated to these areas (no fewerthan five of the 14 chapters). As it stands thebook will satisfy neither medical nor para-medical readers as each group will find alarge portion of the book of limited practicalrelevance.
Secondly, in view of the many changesthat occur to the nervous system as part ofthe aging process (especially in relation togait), and the controversy which still existsabout the relation of Parkinson’s disease toage related attrition of dopaminergic neu-rons, it would have been useful to include achapter aimed at assisting the clinician todistinguish age related changes in motorfunction from Parkinsonian diseases.Thirdly, the book is far from comprehensivein the coverage of causes of parkinsonismother than Parkinson’s disease. The variouscauses are mentioned briefly in the summarychapters at the beginning of the book.Subsequently, however, only three chaptersare actually dedicated to other parkinsoniandisorders—and the choice of these seemsarbitrary—namely, gait apraxia and multi-infarct states; drug induced parkinsonism;and a chapter on essential tremor. The chap-ter on treatment deals only with Parkinson’sdisease. In view of the avowed subjectmatter of the book I would have liked tohave seen at least one chapter dealingspecifically with the diagnosis and manage-
ment of other parkinsonism plus disorders,such as multiple system atrophy, progressivesupranuclear palsy, and corticobasal degen-eration.
STAVIA BLUNT
Experimental and ClinicalNeurotoxicology, 2nd edition. Edited byPETER S SPENCER, HERBERT H SCHAUMBURG andALBERT C LUDOLPH. (Pp 1310, £145.00).Published by Oxford University Press, NewYork, 2000. ISBN 0-19-508477-2.
The first edition of this text was published 20years ago. The growing recognition of theimportance of neurotoxicology and the devel-opment of molecular and cellular pathologyand toxicology have resulted in a hugeincrease in knowledge since the publication.As a result the second edition of this veryinfluential publication is much increased incontent and mass. It is not a volume to becasually carried between library and home: itis now the definitive reference text on neuro-toxicology.
The book is in two sections. The first,comprising three chapters, provides informa-tive and well written introductions to the bio-logical principles of clinical neurotoxicology,and to the major aspects of human andveterinary neurotoxicology. The chapter onhuman neurotoxic disease describes diseaseprocesses by system rather than by class ofcompound; that on veterinary neurotoxicdisease describes disease by class of toxicagent. I found the first a much more valuableapproach. Despite that personal preference,these first three chapters should be requiredreading by any aspiring neurotoxicologist,neuropathologist, or neurologist whether sci-entist, clinician, or veterinarian.
The second section, about 1000 pages inlength, is a comprehensive listing of severalhundred substances with neurotoxic poten-tial. At its best, this section oVers mini-reviews of major neurotoxic industrialchemicals—for example, acrylamide,n-hexane—and therapeutic agents—for ex-ample, chlorpromazine. At times, however,the choice of compounds for inclusion seemsconfused, and cross referencing is not par-ticularly successful. For example, organo-phosphates and sarin and related organo-phosphate “nerve agents” are given separatesections, both dealt with in great detail butwith no cross reference at all. More problem-atic is the handling of natural poisons andvenoms and their respective toxic compo-nents (toxins). Venoms inoculated by bitingand stinging animals and the poisons respon-sible for events such as paralytic shellfish poi-soning and ciguatera are invariably complex.The clinical syndrome reflects the combinedactivities of numerous toxins. There is, there-fore, the possibility of defining the neurotoxicpotential of either the entire venom or poisonor only that of the neurotoxic toxins. Eachapproach has its strengths and weaknessesbut in this publication the editors have usedboth approaches without any clear strategy.This has resulted in some curious choicesbeing made. The venom of the sea snakes isgiven an entry, although sea snake bites arenow rather rare; the venom of the taipan isnot given an entry although eVective bites bythis snake constitute a neurological emer-gency. The postsynaptically active toxins ofcobra venoms are discussed but not thepresynaptically active toxins of krait venoms.Saxitoxin, the toxin primarily responsible for
424 J Neurol Neurosurg Psychiatry 2001;70:406–425
www.jnnp.com

paralytic shellfish poisoning, is a member ofmany related gonyautoxins, and interconver-sion is common. This is not mentioned in theentry on saxitoxins. Neither is the fact thatgonyautoxins are often found in blue-greenalgae. These confusions could relatively easilybe resolved in subsequent editions of thisbook by describing the neurotoxic potentialof major groups of toxin, “postsynapticallyactive toxins of snake venoms” for example.This may seem a complaint based on the per-sonal interests of the reviewer, but the editorsclearly feel that “natural” neurotoxic agentsare important.
Experimental and Clinical Neurotoxicology isan unusual book in structure, organisation,and content. But it is not easily put down. Ifound myself constantly moving to newsections exploring its contents much as onehandles a new dictionary. It is, quite simply, agood read. This new edition will become thedefinitive reference for the neurotoxicologist.It is an essential component of the library ofany respectable toxicology or pathology labo-ratory and of every neuropathologist or neu-rotoxicologist. I doubt we shall wait 20 yearsfor the third edition.
J B HARRIS
CORRECTION
Schrag A, Jahanshahi M, Quinn N. Whatcontributes to the quality of life in patientswith Parkinson’s disease? J Neurol NerosurgPsychiatry 2000;69;308-12. The numbersgiven for the PDQ-39 in depressed patients(BDI>17) and non-depressed patients(BDI<18) given in table 2 (top row of data)on page 309 should read 49.8 (21.4) and 23.6(14.3 instead of 39 (18.3) and 16.7 (11.2).
J Neurol Neurosurg Psychiatry 2001;70:406–425 425
www.jnnp.com




















