
Lehne's Pharmacology for Nursing Care - Digital Library ARS ...
210
404 UNIT V Central Nervous System Drugs PTSD develops in 4% of men at some time in their lives and in 10% to 14% of women. Traumatic events that involve interpersonal violence (e.g., assault, rape, torture) are more likely to cause PTSD than are traumatic events that do not (e.g., car accidents, natural disasters). For example, among rape victims, the incidence of PTSD is 45.9% for women and 65% for men. In contrast, among natural disaster survivors, the incidence is 5.4% for women and 3.7% for men. Combat carries a high risk of PTSD; the disorder develops in up to 40% of soldiers who go to war. Treatment PTSD can be treated with psychotherapy and with drugs, as described in an evidence-based guideline—VA/DoD Clinical Practice Guideline for the Management of Post-Traumatic Stress—released by the Department of Veterans Affairs and Department of Defense in 2010. Two basic types of psycho- therapy are recommended: trauma-focused therapy and stress inoculation training. Trauma-focused therapy uses a variety of cognitive behavioral techniques, including a very effective one known as exposure therapy, in which patients repeatedly reimagine traumatic events as a way to make those events lose their power. Stress inoculation training helps patients identify cues that can trigger fear and anxiety, and then teaches them techniques to cope with those disturbing reactions. Regarding drugs, evidence of efficacy is strongest for three SSRIs (fluoxetine, paroxetine, and sertraline) and one SNRI (venlafaxine). Of these four drugs, only two—paroxetine [Paxil] and sertraline [Zoloft]—are FDA approved for PTSD. If none of the first-line drugs is effective, the guidelines suggest several alternatives: mirtazapine, a TCA (amitriptyline or imipramine), or an MAOI (phenelzine). Current evidence does not support the use of monotherapy with bupropion, buspirone, trazodone, or a benzodiazepine. Benzodiazepines (e.g., clonazepam [Klonopin, Rivotril ], alprazolam [Xanax]) are an option for some patients. These drugs are well tolerated and their benefits are immediate, unlike those of the SSRIs. As a result, benzodiazepines can provide rapid relief and can be used PRN. Accordingly, these drugs are well suited for people whose fear is limited to performance situations and who must face those situations only occasionally. The usual dosage is 1 to 3 mg/day for clonazepam, and 1 to 6 mg/day for alprazolam. Propranolol [Inderal] and other beta blockers can benefit patients with performance anxiety. When taken 1 to 2 hours before a scheduled performance, beta blockers can reduce symptoms caused by autonomic hyperactivity (e.g., tremors, sweating, tachycardia, palpitations). Doses are relatively small—only 10 to 80 mg for propranolol. POST-TRAUMATIC STRESS DISORDER Characteristics Post-traumatic stress disorder (PTSD) develops following a traumatic event that elicited an immediate reaction of fear, helplessness, or horror. PTSD has three core symptoms: re- experiencing the event, avoiding reminders of the event (coupled with generalized emotional numbing), and a persistent state of hyperarousal. A traumatic event is one that involves a threat of injury or death, or a threat to one’s physical integrity. Many events meet this criterion. Among these are physical or sexual assault, rape, torture, combat, industrial explosions, serious accidents, natural disasters, being taken hostage, displacement as a refugee, and terrorist attacks. It should be noted that PTSD can affect persons who were only witnesses to a traumatic event—not just those who were directly involved. The epidemiology of PTSD is revealing. In the United States, more than 8 million Americans have PTSD in any given year, making PTSD the fourth most common psychiatric disorder. KEY POINTS ■ Anxiety is an uncomfortable state that has psychologic manifestations (fear, apprehension, dread, uneasiness) and physical manifestations (tachycardia, palpitations, trembling, dry mouth, sweating, weakness, fatigue, shortness of breath). ■ When anxiety is persistent and disabling, intervention is indicated. ■ As a rule, optimal therapy of anxiety disorders consists of psychotherapy combined with drug therapy. ■ The drugs used most often for anxiety disorders are serotonergic reuptake inhibitors and benzodiazepines. ■ Benzodiazepines are used primarily for panic disorder (PD) and generalized anxiety disorder (GAD), whereas SRIs are used for all anxiety disorders. ■ GAD is a chronic condition characterized by uncontrollable worrying. ■ First-line drugs for GAD are SRIs, buspirone, and benzodiazepines. ■ SRIs (venlafaxine, paroxetine, escitalopram, and duloxetine) are especially well suited for treating patients who have depression in addition to GAD. However, they are also effective even when depression is absent. ■ With buspirone, venlafaxine, paroxetine, escitalopram, and duloxetine, anxiolytic effects are delayed. Accordingly, these drugs are best suited for long-term management—not rapid relief. ■ Buspirone has three advantages over benzodiazepines: It does not cause CNS depression, has no abuse potential, and does not intensify the effects of CNS depressants. ■ Buspirone levels can be increased by erythromycin, ketoconazole, and grapefruit juice. ■ Benzodiazepines suppress symptoms of GAD immediately. Accordingly, these drugs are preferred agents for rapid stabilization, especially when anxiety is severe. ■ Benzodiazepines are CNS depressants and hence can cause sedation and psychomotor slowing. In addition, they can intensify CNS depression caused by other drugs. ■ Benzodiazepines have some potential for abuse, and hence should be used with caution in patients known to abuse alcohol or other psychoactive drugs. ■ When taken long term, benzodiazepines can cause physical dependence. To minimize withdrawal symptoms, dosage
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Lehne's Pharmacology for Nursing Care - Digital Library ARS ...

404
UNIT V Central Nervous System Drugs
PTSD develops in 4% of men at some time in their lives and in 10% to 14% of women. Traumatic events that involve interpersonal violence (e.g., assault, rape, torture) are more likely to cause PTSD than are traumatic events that do not (e.g., car accidents, natural disasters). For example, among rape victims, the incidence of PTSD is 45.9% for women and 65% for men. In contrast, among natural disaster survivors, the incidence is 5.4% for women and 3.7% for men. Combat carries a high risk of PTSD; the disorder develops in up to 40% of soldiers who go to war.
TreatmentPTSD can be treated with psychotherapy and with drugs, as described in an evidence-based guideline—VA/DoD Clinical Practice Guideline for the Management of Post-Traumatic Stress—released by the Department of Veterans Affairs and Department of Defense in 2010. Two basic types of psycho-therapy are recommended: trauma-focused therapy and stress inoculation training. Trauma-focused therapy uses a variety of cognitive behavioral techniques, including a very effective one known as exposure therapy, in which patients repeatedly reimagine traumatic events as a way to make those events lose their power. Stress inoculation training helps patients identify cues that can trigger fear and anxiety, and then teaches them techniques to cope with those disturbing reactions.
Regarding drugs, evidence of efficacy is strongest for three SSRIs (fluoxetine, paroxetine, and sertraline) and one SNRI (venlafaxine). Of these four drugs, only two—paroxetine [Paxil] and sertraline [Zoloft]—are FDA approved for PTSD. If none of the first-line drugs is effective, the guidelines suggest several alternatives: mirtazapine, a TCA (amitriptyline or imipramine), or an MAOI (phenelzine). Current evidence does not support the use of monotherapy with bupropion, buspirone, trazodone, or a benzodiazepine.
Benzodiazepines (e.g., clonazepam [Klonopin, Rivotril ], alprazolam [Xanax]) are an option for some patients. These drugs are well tolerated and their benefits are immediate, unlike those of the SSRIs. As a result, benzodiazepines can provide rapid relief and can be used PRN. Accordingly, these drugs are well suited for people whose fear is limited to performance situations and who must face those situations only occasionally. The usual dosage is 1 to 3 mg/day for clonazepam, and 1 to 6 mg/day for alprazolam.
Propranolol [Inderal] and other beta blockers can benefit patients with performance anxiety. When taken 1 to 2 hours before a scheduled performance, beta blockers can reduce symptoms caused by autonomic hyperactivity (e.g., tremors, sweating, tachycardia, palpitations). Doses are relatively small—only 10 to 80 mg for propranolol.
POST-TRAUMATIC STRESS DISORDERCharacteristicsPost-traumatic stress disorder (PTSD) develops following a traumatic event that elicited an immediate reaction of fear, helplessness, or horror. PTSD has three core symptoms: re-experiencing the event, avoiding reminders of the event (coupled with generalized emotional numbing), and a persistent state of hyperarousal. A traumatic event is one that involves a threat of injury or death, or a threat to one’s physical integrity. Many events meet this criterion. Among these are physical or sexual assault, rape, torture, combat, industrial explosions, serious accidents, natural disasters, being taken hostage, displacement as a refugee, and terrorist attacks. It should be noted that PTSD can affect persons who were only witnesses to a traumatic event—not just those who were directly involved.
The epidemiology of PTSD is revealing. In the United States, more than 8 million Americans have PTSD in any given year, making PTSD the fourth most common psychiatric disorder.
KEY POINTS
■ Anxiety is an uncomfortable state that has psychologic manifestations (fear, apprehension, dread, uneasiness) and physical manifestations (tachycardia, palpitations, trembling, dry mouth, sweating, weakness, fatigue, shortness of breath).
■ When anxiety is persistent and disabling, intervention is indicated.
■ As a rule, optimal therapy of anxiety disorders consists of psychotherapy combined with drug therapy.
■ The drugs used most often for anxiety disorders are serotonergic reuptake inhibitors and benzodiazepines.
■ Benzodiazepines are used primarily for panic disorder (PD) and generalized anxiety disorder (GAD), whereas SRIs are used for all anxiety disorders.
■ GAD is a chronic condition characterized by uncontrollable worrying.
■ First-line drugs for GAD are SRIs, buspirone, and benzodiazepines.
■ SRIs (venlafaxine, paroxetine, escitalopram, and duloxetine) are especially well suited for treating patients who have depression in addition to GAD. However, they are also effective even when depression is absent.
■ With buspirone, venlafaxine, paroxetine, escitalopram, and duloxetine, anxiolytic effects are delayed. Accordingly, these drugs are best suited for long-term management—not rapid relief.
■ Buspirone has three advantages over benzodiazepines: It does not cause CNS depression, has no abuse potential, and does not intensify the effects of CNS depressants.
■ Buspirone levels can be increased by erythromycin, ketoconazole, and grapefruit juice.
■ Benzodiazepines suppress symptoms of GAD immediately. Accordingly, these drugs are preferred agents for rapid stabilization, especially when anxiety is severe.
■ Benzodiazepines are CNS depressants and hence can cause sedation and psychomotor slowing. In addition, they can intensify CNS depression caused by other drugs.
■ Benzodiazepines have some potential for abuse, and hence should be used with caution in patients known to abuse alcohol or other psychoactive drugs.
■ When taken long term, benzodiazepines can cause physical dependence. To minimize withdrawal symptoms, dosage

405
CHAPTER 35 Management of Anxiety Disorders
should be tapered gradually—over a period of several months.
■ Patients with panic disorder experience recurrent panic attacks, characterized by palpitations, pounding heart, chest pain, derealization or depersonalization, and fear of dying or going crazy.
■ Many patients with panic disorder also experience agora-phobia, a condition characterized by anxiety about being in places or situations from which escape might be difficult or embarrassing, or in which help might be unavailable if a panic attack should occur.
■ SSRIs are first-line drugs for panic disorder.■ SSRIs decrease the frequency and intensity of panic attacks,
anticipatory anxiety, and avoidance behavior, and they work regardless of whether the patient has depression.
■ Obsessive-compulsive disorder (OCD) is characterized by persistent obsessions and compulsions that cause marked distress, consume at least 1 hour a day, and significantly interfere with daily living.
■ SSRIs are first-line drugs for OCD.■ Social anxiety disorder, formerly known as social phobia,
is characterized by an intense, irrational fear of being scrutinized by others or of doing something that could be embarrassing or humiliating.
■ The SSRIs are first-line drugs for most patients with social anxiety disorder.
■ When social anxiety disorder is limited to fear of speaking or performing in public and when these situations arise infrequently, PRN treatment with a benzodiazepine may be preferred to long-term treatment with an SSRI.
■ Post-traumatic stress disorder (PTSD) develops following a traumatic event that elicited an immediate reaction of fear, helplessness, or horror.
■ PTSD has three core symptoms: re-experiencing, avoidance/emotional numbing, and hyperarousal.
■ Events that can lead to PTSD include physical or sexual assault, rape, torture, combat, industrial explosions, serious accidents, natural disasters, being taken hostage, displace-ment as a refugee, and terrorist attacks.
■ According to a VA/DoD guideline, PTSD can be treated with psychotherapy and with drugs.
■ Two SSRIs—paroxetine and sertraline—are approved by the FDA for first-line drug treatment of PTSD. Additional drugs used for treatment of PTSD include venlafaxine (an SNRI), TCAs, and MAOIs.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

406
C H A P T E R
36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
CENTRAL NERVOUS SYSTEM STIMULANTS, p. 406Amphetamines, p. 406Methylphenidate and Dexmethylphenidate,
p. 408Methylphenidate, p. 408Dexmethylphenidate, p. 409
Methylxanthines, p. 409Caffeine, p. 409
Miscellaneous CNS Stimulants, p. 411Modafinil, p. 411Armodafinil, p. 411Doxapram, p. 411
ATTENTION-DEFICIT/HYPERACTIVITY DISORDER, p. 411
Basic Considerations, p. 411ADHD in Children, p. 411ADHD in Adults, p. 412
Drugs Used for ADHD, p. 412CNS Stimulants, p. 412Nonstimulants, p. 412
Key Points, p. 415Summary of Major Nursing Implications,
p. 415
AMPHETAMINES
The amphetamine family consists of amphetamine, dextroam-phetamine, methamphetamine, and lisdexamfetamine. All are powerful CNS stimulants. In addition to their CNS actions, amphetamines have significant peripheral actions—actions that can cause cardiac stimulation and vasoconstriction. The amphetamines have a high potential for abuse.
ChemistryDextroamphetamine and Levamphetamine. Amphet-
amines are molecules with an asymmetric carbon atom. As a result, amphetamines can exist as mirror images of each other. Such compounds are termed optical isomers or enantiomers. Dextroamphetamine and levamphetamine both contain the same atomic components, but those components are arranged differently around the asymmetric carbon. Because of this structural difference, these compounds have somewhat different properties. For example, dextroamphetamine is more selective than levamphetamine for causing stimulation of the CNS, and hence produces fewer peripheral side effects.
CENTRAL NERVOUS SYSTEM STIMULANTS
Central nervous system (CNS) stimulants increase the activity of CNS neurons. Most stimulants act by enhancing neuronal excitation. A few act by suppressing neuronal inhibition. In sufficient doses, all stimulants can cause convulsions.
Clinical applications of the CNS stimulants are limited. Currently these drugs have two principal indications: attention-deficit/hyperactivity disorder (ADHD) and narcolepsy.
Please note that CNS stimulants are not the same as anti-depressants. The antidepressants act selectively to elevate mood, and hence can relieve depression without affecting other CNS functions. In contrast, CNS stimulants cannot elevate mood without producing generalized excitation. Accordingly, the role of stimulants in treating depression is minor.
Our principal focus is on amphetamines, methylphenidate [Ritalin, others], and methylxanthines (e.g., caffeine). These are by far the most widely used stimulant drugs.
Prototype DrugsCentral Nervous System StimulantsAmphetamines
Amphetamine sulfate
Amphetamine-like Drugs
Methylphenidate
Methylxanthines
Caffeine
Drugs for Attention-Deficit/Hyperactivity DisorderCNS Stimulants
Methylphenidate
Nonstimulants
Atomoxetine
Amphetamine. The term amphetamine refers not to a single compound but rather to a 50 : 50 mixture of dextroam-phetamine and levamphetamine. (In chemistry, we refer to such equimolar mixtures of enantiomers as racemic.)

407
CHAPTER 36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
to those of cocaine.) Because of their abuse potential, all amphetamines, including lisdexamfetamine, are classified under Schedule II of the Controlled Substances Act and must be dispensed accordingly. Whenever amphetamines are used therapeutically, their potential for abuse must be weighed against their potential benefits.
Adverse EffectsCNS Stimulation. Stimulation of the CNS can cause
insomnia, restlessness, and extreme loquaciousness. These effects can occur at therapeutic doses.
Weight Loss. By suppressing appetite, amphetamines can cause weight loss.
Cardiovascular Effects. At recommended doses, stimulants produce a small increase in heart rate and blood pressure. For most patients, these increases lack clinical significance. However, for patients with pre-existing cardiovascular disease, stimulants may cause dysrhythmias, anginal pain, or hyper-tension. Accordingly, amphetamines must be employed with extreme caution in these people. Any patient who develops cardiovascular symptoms (e.g., chest pain, shortness of breath, fainting) while using a stimulant should be evaluated immediately.
Do amphetamines increase the risk of sudden death? Prob-ably not. Sudden death in children on these medications is very rare, and evidence is conflicting regarding the risk of sudden death. Should children routinely receive an electrocardiogram (ECG) before using these drugs? Probably not—despite a 2008 statement from the American Heart Association (AHA) saying it would be reasonable to consider obtaining an ECG in children being evaluated for stimulant therapy of ADHD. Why is the AHA concerned? Because 14 children, 5 with heart defects, died suddenly while using Adderall, a mixture of amphetamine and dextroamphetamine. However, given that millions of children have used the drug, the death rate is no greater than would be expected for a group this size, whether or not Adderall was being used. The bottom line? First, there are conflicting data showing that stimulants increase the risk of sudden death, even in children with heart disease. Second, there are no data showing that limiting the use of stimulants in children with heart defects will protect them from sudden death. And third, there are no data showing that screening for heart disease with an ECG before start-ing stimulants will be of benefit. Therefore, it would seem that routine ECGs are unnecessary before starting a child on stimulant therapy, especially if there is no evidence of heart disease. However, if there is evidence of heart disease, or evidence of hereditary cardiovascular defects, an ECG might be appropriate.
Psychosis. Excessive amphetamine use produces a state of paranoid psychosis, characterized by hallucinations and paranoid delusions (suspiciousness, feelings of being watched). Amphetamine-induced psychosis looks very much like schizophrenia. Symptoms are thought to result from the release of DA. Consistent with this hypothesis is the obser-vation that symptoms can be alleviated with a DA receptor blocking agent (e.g., haloperidol). Following amphetamine withdrawal, psychosis usually resolves spontaneously within a week.
In some individuals, amphetamines can unmask latent schizophrenia. For these people, symptoms of psychosis do not clear spontaneously, and hence psychiatric care is indicated.
Lisdexamfetamine. Lisdexamfetamine [Vyvanse] is a prodrug composed of dextroamphetamine covalently linked to L-lysine. Following oral dosing, the drug undergoes rapid hydrolysis by enzymes in the intestine and liver to yield lysine and free dextroamphetamine, the active form of the drug. If lisdexamfetamine is inhaled or injected, hydrolysis will not take place, and hence the drug is not effective by these routes. Accordingly, it may have a lower abuse potential than other forms of amphetamine.
Methamphetamine. Methamphetamine is simply dextro-amphetamine with an additional methyl group.
Mechanism of ActionThe amphetamines act primarily by causing release of norepi-nephrine (NE) and dopamine (DA), and partly by inhibiting reuptake of both transmitters. These actions take place in the CNS and in peripheral nerves. Most pharmacologic effects result from release of NE.
Pharmacologic EffectsCentral Nervous System. The amphetamines have
prominent effects on mood and arousal. At usual doses, they increase wakefulness and alertness, reduce fatigue, elevate mood, and augment self-confidence and initiative. Euphoria, talkativeness, and increased motor activity are likely. Task performance that had been reduced by fatigue or boredom improves.
Amphetamines can stimulate respiration and suppress appetite and the perception of pain. Stimulation of the medullary respiratory center increases respiration. Effects on the hypo-thalamic feeding center depress appetite. By a mechanism that is not understood, amphetamines can enhance the analgesic effects of morphine and other opioids.
Cardiovascular System. Cardiovascular effects occur secondary to release of NE from sympathetic neurons. Nor-epinephrine acts in the heart to increase heart rate, atrioven-tricular conduction, and force of contraction. Excessive cardiac stimulation can cause dysrhythmias. In blood vessels, NE promotes constriction. Excessive vasoconstriction can cause hypertension.
ToleranceWith regular amphetamine use, tolerance develops to elevation of mood, suppression of appetite, and stimulation of the heart and blood vessels. In highly tolerant users, doses up to 1000 mg (IV) every few hours may be required to maintain euphoric effects. This compares with daily doses of 5 to 30 mg for nontolerant individuals.
Physical DependenceChronic amphetamine use produces physical dependence. If amphetamines are abruptly withdrawn from a dependent person, an abstinence syndrome will ensue. Symptoms include exhaus-tion, depression, prolonged sleep, excessive eating, and a craving for more amphetamine. Sleep patterns may take months to normalize.
AbuseBecause amphetamines can produce euphoria (extreme mood elevation), they have a high potential for abuse. Psychologic dependence can occur. (Users familiar with CNS stimulants find the psychologic effects of amphetamines nearly identical

408
UNIT V Central Nervous System Drugs
METHYLPHENIDATE AND DEXMETHYLPHENIDATE
Methylphenidate and dexmethylphenidate are nearly identical in structure and pharmacologic actions. Furthermore, the pharmacology of both drugs is nearly identical to that of the amphetamines.
MethylphenidateAlthough methylphenidate [Ritalin, Metadate, Methylin, Concerta, Daytrana, Biphentin ] is structurally dissimilar from the amphetamines, the pharmacologic actions of these drugs are essentially the same. Consequently, methylphenidate can be considered an amphetamine in all but structure and name. Methylphenidate and amphetamine share the same mechanism of action (promotion of NE and DA release, and inhibition of NE and DA reuptake), adverse effects (insomnia, reduced appetite, emotional lability), and abuse liability (Schedule II). Like amphetamine, methylphenidate is not a single compound, but rather a 50 : 50 mixture of dextro and levo isomers. The dextro isomer is highly active; the levo isomer is not. Methylphenidate has two indications: ADHD and narcolepsy.
Preparations, Dosage, and AdministrationMethylphenidate is available in three types of formulations: IR, sustained-release (SR), and once-daily doses. All three are indicated for ADHD. As a rule, the SR and IR formulations must be taken 2 or 3 times a day.
Immediate Release. Ritalin and Methylin are available in standard tablets (5, 10, and 20 mg), chewable tablets (2.5, 5, and 10 mg), and an oral solution (5 and 10 mg/5 mL). Effects begin rapidly and last 3 to 5 hours. Because effects are brief, dosing must be done 2 or 3 times a day. The usual maintenance dosage for ADHD is 5 mg twice daily, with a maximum dose of 60 mg daily in two or three divided doses.
Sustained Release. Ritalin-SR and Metadate ER are available in 20-mg tablets, and Quillivant XR is available in a 25-mg/5 mL oral suspension or as Quillichew ER in 20-, 30-, and 40-mg chewable tablets. Effects are delayed and last 6 to 8 hours. Dosing is done once or twice daily. For children with ADHD, the usual maintenance dosage is 20 to 40 mg in the morning, supple-mented with 20 mg in the early afternoon if needed.
Once-Daily Dosing. Five products are available. Their brand names are Concerta, Metadate CD, Aptensio XR, Ritalin LA, and Daytrana. With all five, dosing is done once daily in the morning; no afternoon dose is needed.
Concerta. Concerta tablets—formulated as an osmotic-release oral system (OROS)—consist of an outer coating of IR methylphenidate and a special inner core that releases the remainder of each dose gradually. As a result, effects begin rapidly and last 10 to 12 hours. Because of their special archi-tecture, Concerta tablets must be swallowed whole, not crushed or chewed. The tablet shell may not dissolve fully in the GI tract. Accordingly, patients should be informed that they may see tablet “ghosts” in the stool. Concerta tablets are available in four strengths: 18, 27, 36, and 54 mg.
Dosage depends on whether the patient is already taking methylphenidate (IR or SR). For children not already taking methylphenidate, the initial dosage is 18 mg once daily in the morning. Dosage can be increased to a maximum of 72 mg once daily. For children who are taking methylphenidate (IR or SR), the initial dosage of Concerta is as follows:
• For those taking 5 mg (2 or 3 times a day) of IR methylphenidate or 20 mg once daily of SR methylphenidate, start with 18 mg of Concerta.
• For those taking 10 mg (2 or 3 times a day) of IR or 40 mg once daily of SR, start with 36 mg of Concerta.
• For those taking 15 mg (2 or 3 times a day) of IR or 60 mg once daily of SR, start with 54 mg of Concerta.
Metadate CD. Metadate CD is available in 10-, 20-, 30-, 40-, 50-, and 60-mg capsules that contain IR and delayed-release beads. The beads release
Acute ToxicitySymptoms. Overdose produces dizziness, confusion, hallucinations,
paranoid delusions, palpitations, dysrhythmias, and hypertension. Death is rare. Fatal overdose is associated with convulsions, coma, and cerebral hemorrhage.
Treatment. Hallucinations can be controlled with atypical antipsychotic drugs (e.g., olanzapine). An alpha-adrenergic blocker (e.g., phentolamine) can reduce hypertension (by promoting vasodilation). Owing to its ability to block alpha receptors, chlorpromazine helps lower blood pressure. Seizures can be managed with diazepam.
Therapeutic UsesAttention-Deficit/Hyperactivity Disorder. The role of
amphetamines in ADHD is discussed later in this chapter.Narcolepsy. Narcolepsy is a disorder characterized by
daytime somnolence and uncontrollable attacks of sleep. By stimulating the CNS, amphetamines can promote arousal and thereby alleviate symptoms.
Safety Alert
AMPHETAMINESAmphetamines have a high potential for abuse and dependence. In patients who use amphetamines chronically, withdrawal may occur if use of these medications is suddenly stopped.
Preparations, Dosage, and AdministrationFour members of the amphetamine family are used clinically: dextroamphet-amine sulfate, an amphetamine/dextroamphetamine mixture, lisdexamfetamine, and methamphetamine. In clinical practice, amphetamines are given orally. (These drugs are not approved for IV administration. Amphetamines for IV use are available only through illegal sources.) All amphetamines are regulated under Schedule II of the Controlled Substances Act and must be dispensed accordingly.
Dextroamphetamine Sulfate. Dextroamphetamine is available in immediate-release (IR) and extended-release (ER) formulations. Both are indicated for ADHD.
Immediate Release. IR dextroamphetamine is available in 2.5-, 5-, 7.5-, 10-, 15-, 20-, and 30-mg tablets sold as Zenzedi and as a 5-mg/mL solution sold as Procentra. Effects begin rapidly and last 4 to 6 hours. The usual maintenance dosage for ADHD is 5 to 10 mg once or twice daily, up to 40 mg/day.
Extended Release. ER dextroamphetamine [Dexedrine] is available in 5-, 10-, and 15-mg capsules. Effects begin rapidly and last 6 to 10 hours. The usual maintenance dosage for ADHD is 5 mg once or twice daily.
Amphetamine/Dextroamphetamine Mixture. Amphetamine/dextroamphetamine mixture is available in IR [Adderall] and ER [Adderall XR] formulations. Both are used for ADHD.
Immediate Release. Adderall is available in IR tablets (5, 7.5, 10, 12.5, 15, 20, and 30 mg). Effects begin rapidly and last 4 to 6 hours. The usual maintenance dosage for ADHD is 5 mg twice daily, taken in the morning and 4 to 6 hours later.
Extended Release. Adderall XR is available in 5-, 10-, 15-, 20-, 25-, and 30-mg ER capsules. Half the dose is released immediately, and the remainder is released 4 hours later. As a result, effects begin rapidly and last 10 to 12 hours. The usual maintenance dosage for ADHD is 20 mg once daily in the morning. This is equivalent to taking 10 mg of IR Adderall at 8:00 AM and again around noon.
Lisdexamfetamine. Lisdexamfetamine [Vyvanse] is available in capsules (10, 20, 30, 40, 50, 60, and 70 mg) and chewable tablets (10, 20, 30, 40, 50, and 60 mg). Effects begin rapidly and persist about 13 hours. Dosing is done once daily in the morning without regard to meals. The capsules may be swallowed intact, or their contents may be dissolved in water and swallowed immediately. The usual daily maintenance dosage for ADHD is 30 mg.
Methamphetamine. Methamphetamine [Desoxyn] is indicated for ADHD, although it is not a preferred treatment for this condition. The drug is available in 5-mg IR tablets. The usual regimen for ADHD is 20 to 25 mg/day, administered in two divided doses.

409
CHAPTER 36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
METHYLXANTHINES
The methylxanthines are methylated derivatives of xanthine, hence the family name. These compounds consist of a xanthine nucleus with one or more methyl groups attached. Caffeine, the most familiar member of the family, will serve as our prototype.
30% of the dose rapidly and the remaining 70% 4 hours later. As a result, plasma levels peak twice—at 1.5 and 4.5 hours. This is the same pattern produced by taking IR methylphenidate twice daily. For ADHD patients not already taking methylphenidate, the initial dosage is 20 mg once daily in the morning. This can be gradually increased to a maximum of 60 mg once daily. For patients who are already taking methylphenidate, start with 20 mg of Metadate CD once daily (for those taking 10 mg of IR methylphenidate twice daily) or with 40 mg of Metadate CD once daily (for those taking 20 mg of IR methylphenidate twice daily). If needed, Metadate CD capsules can be opened and sprinkled on a small amount of soft food (e.g., applesauce) right before ingestion.
Aptensio XR. Aptensio XR is formulated in capsules (10, 15, 20, 30, 40, 50, and 60 mg) that contain delayed-release beads. Therapeutic effects begin rapidly and persist for 12 hours. The capsules may be swallowed intact or opened to permit sprinkling the beads onto applesauce or some other soft food. Aptensio XR capsules are approved for treating ADHD in children, adolescents, and adults.
Ritalin LA. Ritalin LA is formulated as ER capsules (10, 20, 30, 40, and 60 mg). The product is much like Metadate CD in that some of the dose is released immediately and the rest 4 hours later. Dosing is done once daily in the morning. As with Metadate CD and Concerta, dosage depends on whether the patient is already taking methylphenidate (IR or SR). For children not already taking methylphenidate, the initial dosage is 20 mg. Dosage can be gradually increased to a maximum of 60 mg. For children who are taking methylphenidate (IR or SR), the initial dosage is as follows:
• For those taking 10 mg twice daily of IR methylphenidate or 20 mg once daily of SR methylphenidate, start with 20 mg of Ritalin LA.
• For those taking 15 mg twice daily of IR, start with 30 mg of Ritalin LA.
• For those taking 20 mg twice daily of IR or 40 mg once daily of SR, start with 40 mg of Ritalin LA.
• For those taking 30 mg twice daily of IR or 60 mg once daily of SR, start with 60 mg of Ritalin LA.
Daytrana. Daytrana—a transdermal methylphenidate patch—is the first nonoral treatment for ADHD. Following patch application, blood levels of methylphenidate rise slowly and peak in about 9 hours, after which the patch should be removed. Because of the slow rise, effects are delayed about 2 hours. Furthermore, effects will persist for about 3 hours after patch removal. Daytrana patches are available in four sizes—12.5, 18.75, 25, and 37.5 cm2—that deliver 10, 15, 20, and 30 mg/9 hr, respectively. Treatment should begin with the smallest patch, even in patients already taking meth-ylphenidate PO. If needed, larger patches can be tried at weekly intervals. Patients should apply the patch to the hip in the morning—alternating hips each day—and remove it no more than 9 hours later. (They can remove it sooner to terminate effects early.) Application to inflamed skin or application of heat will accelerate drug absorption, and hence should be avoided. Patients should be informed that bathing, showering, and swimming will not dislodge the patch.
Side effects of the patch are like those of oral methylphenidate, with two exceptions. First, users may experience erythema and pruritus at the application site. Second, exposing the skin to methylphenidate can cause a hypersensitivity reaction. If hypersensitivity develops, the patient may be unable to use any methylphenidate formulation—transdermal or oral—ever again.
DexmethylphenidateDexmethylphenidate [Focalin, Focalin XR], a drug for ADHD, is simply the dextro isomer of methylphenidate. As noted, the dextro isomer accounts for most of the pharmacologic activity of methylphenidate, a 50 : 50 mixture of dextro and levo isomers. Accordingly, the pharmacology of dexmethylphenidate is nearly identical to that of methylphenidate. The only difference is that the dosage of dexmethylphenidate is one-half the dosage of methylphenidate. Dexmethylphenidate is available in IR tablets (2.5, 5, and 10 mg) marketed as Focalin, and in ER capsules (5, 10, 15, 20, 25, 30, 35, and 40 mg) marketed as Focalin XR. Both formulations may be administered with or without food. For children currently treated with methylphenidate, the initial dosage of dexmethylphenidate is one-half the methylphenidate dosage. For children who are not currently being treated, the initial dosage is 2.5 mg twice daily using Focalin or 10 mg once daily using Focalin XR. The maximum dosage is 10 mg twice daily for Focalin and 40 mg once daily for Focalin XR. Dexmethylphenidate is a Schedule II drug and must be dispensed accordingly.
Stimulants
Life Stage Patient Care Concerns
Infants Caffeine citrate [Cafcit] is used for neonatal apnea. Other CNS stimulants should be avoided in this population.
Children The stimulant class of drugs for treatment of ADHD has been proven safe and effective for this population. Atomoxetine, a nonstimulant for ADHD, may cause suicidal thinking in children and adolescents.
Pregnant women
Caffeine may pose a small risk of birth defects, although human data are lacking. Methylphenidate and atomoxetine are classified as FDA Pregnancy Risk Category C,a as adverse fetal effects have been demonstrated in animal studies.
Breast-feeding women
Stimulants, such as methylphenidate, do not have any reported side effects in the breast-feeding infant. There are limited to no data on nonstimulants and the effects on breast-feeding infants.
Older adults Most studies focus on patients older than 65 years, since stimulants are often used for the treatment of apathy, depression, and fatigue in the older adult population. Stimulants should be avoided in patients with cardiac disease or glaucoma. Consider a lower starting dose, and monitor heart rate, blood pressure, and weight.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
CaffeineCaffeine is consumed worldwide for its stimulant effects. In the United States, per capita consumption is about 200 mg/day, mostly in the form of coffee. Although clinical applications of caffeine are few, caffeine remains of interest because of its widespread ingestion for nonmedical purposes.
Dietary SourcesCaffeine can be found in chocolates, desserts, soft drinks, and beverages prepared from various natural products. Common dietary sources are coffee, tea, and cola drinks. The caffeine in cola drinks derives partly from the cola nut and partly from caffeine added by the manufacturer. Caffeine is also present in many noncola soft drinks. The caffeine content of some common foods and beverages is shown in Table 36.1.

410
UNIT V Central Nervous System Drugs
vessels is thought to underlie the drug’s ability to relieve headache.
Bronchi. Caffeine and other methylxanthines cause relaxation of bronchial smooth muscle and thereby promote bronchodilation. Theophylline is an especially effective bronchodilator, and hence can be used to treat asthma (see Chapter 76).
Kidney. Caffeine is a diuretic. The mechanism underlying increased urine formation is likely related to suppression of antidiuretic hormone in the posterior pituitary.
Reproduction. Caffeine readily crosses the placenta and may pose a risk of birth defects, although that risk appears low. When applied to cells in culture, caffeine can cause chro-mosomal damage and mutations. However, the concentrations required are much greater than can be achieved by drinking caffeinated beverages. Also, although there is clear proof that caffeine can cause birth defects in animals, studies have failed to document birth defects in humans. Although caffeine-induced birth defects seem unlikely, caffeine has been associated with low birth weight.
According to a meta-analysis reported in 2010, consuming less than 300 mg of caffeine daily does not increase the risk of preterm birth. An additional review in 2013 revealed that restricting caffeine consumption during the second and third trimesters of pregnancy did not affect birth weight or length of gestation. Whether higher doses might increase risk is unclear.
PharmacokineticsCaffeine is readily absorbed from the GI tract and achieves peak plasma levels within 1 hour. Plasma half-life ranges from 3 to 7 hours. Elimination is by hepatic metabolism.
Therapeutic UsesNeonatal Apnea. Premature infants may experience
prolonged apnea (lasting 15 seconds or more) along with bradycardia. Hypoxemia and neurologic damage may result. Caffeine and other methylxanthines can reduce the number and duration of apnea episodes and can promote a more regular pattern of breathing.
Promoting Wakefulness. Caffeine is used commonly to aid in staying awake. The drug is marketed in various over-the-counter preparations [Maximum Strength NoDoz, Vivarin, others] for this purpose. Of course, individuals desiring increased alertness can get just as much caffeine by drinking coffee or some other caffeine-containing beverage.
Other Applications. Intravenous caffeine can help relieve headache induced by spinal puncture. The drug is used orally to enhance analgesia induced by opioids and by nonopioid analgesics (e.g., aspirin).
Acute ToxicityCaffeine toxicity is characterized by intensification of the responses seen at low doses. Stimulation of the CNS results in excitement, restlessness, and insomnia; if the dosage is very high, convulsions may occur. Tachycardia and respiratory stimulation are likely. Sensory phenomena (ringing in the ears, flashing lights) are common. Death from caffeine overdose is rare. When fatalities have occurred, between 5 and 10 gm have been ingested.
Preparations, Dosage, and AdministrationFor Promoting Wakefulness. Caffeine is available in three formulations
for promoting wakefulness: 200-mg tablets, 200-mg capsules, and 75-mg lozenges. The usual dosage is 100 to 200 mg every 3 to 4 hours as needed.
For Neonatal Apnea. Caffeine citrate [Cafcit] is used for neonatal apnea. The drug is available in oral and IV solutions. Both have the same concentration: 20 mg/mL. Treatment consists of an IV loading dose (20 mg/
Mechanism of ActionSeveral mechanisms of action have been proposed. These include (1) reversible blockade of adenosine receptors, (2) enhancement of calcium permeability in the sarcoplasmic reticulum, and (3) inhibition of cyclic nucleotide phosphodi-esterase, resulting in accumulation of cyclic adenosine monophosphate (cyclic AMP). Blockade of adenosine receptors appears responsible for most effects.
Pharmacologic EffectsCentral Nervous System. In low doses, caffeine decreases
drowsiness and fatigue and increases the capacity for prolonged intellectual exertion. With increasing dosage, caffeine produces nervousness, insomnia, and tremors. When administered in very large doses, caffeine can cause convulsions. Despite popular belief, there is little evidence that caffeine can restore mental function during intoxication with alcohol, although it might delay passing out.
Heart. High doses of caffeine stimulate the heart. When caffeinated beverages are consumed in excessive amounts, dysrhythmias may result.
Blood Vessels. Caffeine affects blood vessels in the periphery differently from those in the CNS. In the periphery, caffeine promotes vasodilation, whereas in the CNS, caffeine promotes vasoconstriction. Constriction of cerebral blood
Product Amount Caffeine (mg)
COFFEE
Brewed (typical) 8 oz 60–180
Instant 8 oz 30–120
Espresso 1.5 oz 77
Decaffeinated 8 oz 1–5
TEA
Brewed 8 oz 35–40
Herbal tea 8 oz 0
Iced tea mix, decaffeinated 8 oz <5
SODA AND ENERGY DRINKS
Energy drink 12 oz 71
Diet cola 12 oz 47
Cola 12 oz 45
Orange soda 12 oz 40
Concentrated energy drink 2 oz 200
Caffeinated water 16 oz 100
ICE CREAM AND YOGURT
Coffee ice cream 12 cup 20–30
Coffee yogurt 4 oz 22
MISCELLANEOUS
Cocoa 8 oz 2–50
Chocolate milk 1.5 oz 3–11
Milk chocolate bar 1.5 oz 10
Dark chocolate bar 1.5 oz 31
TABLE 36.1 ■ Dietary Caffeine

411
CHAPTER 36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
of modafinil. Otherwise, the two drugs are essentially identical, although armodafinil costs more. Armodafinil has the same indications as modafinil—improving wakefulness in people with narcolepsy, SWSD, and OSAHS—and has similar adverse effects, including the potential for rare but severe skin reactions. Like modafinil, armodafinil is classified as a Schedule IV substance. Armodafinil is available in 50-, 150-, and 250-mg tablets. The recommended dosage for narcolepsy and OSAHS is 150 or 250 mg, taken in the morning. The recommended dosage for SWSD is 150 mg, taken 1 hour before the work shift.
DoxapramDoxapram [Dopram] stimulates the CNS at all levels. The drug is employed clinically to stimulate respiration. However, since the doses required are close to those that can produce generalized CNS stimulation and convulsions, doxapram must be used with great care. Furthermore, although doxapram is labeled for the treatment of general CNS depressant overdose, its use for this purpose should be discontinued: Experience has shown that respiratory depression from CNS depressant overdose can be managed more safely and effectively with mechanical support of ventilation than with pharmacologic stimulation of respiration.
ATTENTION-DEFICIT/HYPERACTIVITY DISORDER
Our discussion of ADHD has two parts. We begin by addressing basic concepts in ADHD—specifically, signs and symptoms, etiology, and treatment strategy. After that, we discuss the pharmacology of the drugs used for treatment.
BASIC CONSIDERATIONSADHD in ChildrenADHD is the most common neuropsychiatric disorder of child-hood, affecting 5% to 11% of school-age children. The incidence in boys is two to three times the incidence in girls. Symptoms begin between ages 3 and 7, usually persist into the teens, and often persist into adulthood. The majority (60% to 70%) of children respond well to stimulant drugs. Methylphenidate [Ritalin, Concerta, others] is the agent employed most.
Signs and SymptomsADHD is characterized by inattention, hyperactivity, and impulsivity. Affected children are fidgety, unable to concentrate on schoolwork, and unable to wait their turn; switch excessively from one activity to another; call out excessively in class; and never complete tasks. To make a diagnosis, symptoms must appear before age 12 years and be present for at least 6 months. Since other disorders—especially anxiety and depression—may cause similar symptoms, diagnosis must be done carefully.
ADHD can be subclassified as predominantly inattentive type, predominantly hyperactive-impulsive type, or combined type, depending on the symptom profile. Former names for ADHD—hyperkinetic syndrome and minimal brain dysfunction— are misleading and have been abandoned.
EtiologyAlthough various theories have been proposed, the underlying pathophysiology of ADHD is only partly understood. Neuro-imaging studies indicate structural and functional abnormalities in multiple brain areas, including the frontal cortex, basal ganglia, brainstem, and cerebellum—regions involved with regulating attention, impulsive behavior, and motor activity. Several theories implicate dysregulation in neuronal pathways that employ NE, DA, and serotonin as transmitters. These
kg) followed every 24 hours by an oral or IV maintenance dose (5 mg/kg). Note: The amount of caffeine base in a 20-mg dose of caffeine citrate is only 10 mg (i.e., one-half of the total dose on a milligram basis).
MISCELLANEOUS CNS STIMULANTS
ModafinilTherapeutic UseModafinil [Provigil, Alertec ], a unique nonamphetamine stimulant, is approved for promoting wakefulness in patients with excessive sleepi-ness associated with three disorders: narcolepsy, shift-work sleep disorder (SWSD), and obstructive sleep apnea/hypopnea syndrome (OSAHS). However, although the drug has only three approved uses, most prescriptions (95%) are written for off-label uses, including fatigue, depression, ADHD, jet lag, and sleepiness caused by medications. Investigational uses include ADHD and fatigue associated with multiple sclerosis. The military studied the drug for use in sustaining alertness in helicopter pilots and found it superior to placebo.
In clinical trials, modafinil has been moderately effective. In patients with narcolepsy, modafinil increased wakefulness, but only to about 50% of the level seen in normal people. In contrast, methylphenidate and dextroamphet-amine increase wakefulness to about 70% of normal. In patients with SWSD and OSAHS, benefits are about the same as those seen in narcolepsy.
Mechanism of ActionHow modafinil works remains unclear. The drug does seem to influence hypothalamic areas involved in maintaining the normal sleep-wakefulness cycle. Also, there is evidence that modafinil inhibits the activity of sleep-promoting neurons (in the ventrolateral preoptic nucleus) by blocking reuptake of norepinephrine.
PharmacokineticsModafinil is rapidly absorbed from the GI tract. Plasma levels peak in 2 to 4 hours. Food decreases the rate of absorption but not the extent. Elimination is by hepatic metabolism followed by renal excretion. The half-life is about 15 hours.
Adverse EffectsModafinil is generally well tolerated. The most common adverse effects are headache, nausea, nervousness, diarrhea, and rhinitis. Modafinil does not disrupt nighttime sleep. In clinical trials, only 5% of patients dropped out because of undesired effects. Initially, the drug was believed devoid of car-diovascular effects. However, we now know it can increase heart rate and blood pressure, apparently by altering autonomic function. Subjective effects—euphoria; altered perception, thinking, and feeling—are like those of other CNS stimulants. However, modafinil has less abuse potential, and hence is regulated as a Schedule IV substance. Physical dependence and withdrawal have not been reported. Modafinil is embryotoxic in laboratory animals, and hence should be avoided during pregnancy.
Postmarketing reports link modafinil to rare cases of serious skin reactions, including Stevens-Johnson syndrome, erythema multiforme, and toxic epidermal necrolysis. Patients should be informed about signs of these reactions—swelling or rash, especially in the presence of fever or changes in the oral mucosa—and instructed to discontinue the drug if they develop.
Drug InteractionsModafinil inhibits some forms of cytochrome P450 (CYP) and induces others. Induction of CYP3A4 (the 3A4 isoenzyme of P450) may accelerate the metabolism of oral contraceptives, cyclosporine, and certain other drugs, thereby causing their levels to decline. Caution is advised.
Preparations, Dosage, and AdministrationModafinil is available in 100- and 200-mg tablets. For patients with narcolepsy or OSAHS, the usual dosage is 200 mg/day, taken as a single dose in the morning. For patients with SWSD, the usual dosage is 200 mg/day, taken as a single dose 1 hour before the shift starts. For patients with severe hepatic impairment, doses should be decreased by 50%. Dosage reduction may also be needed in older adults.
ArmodafinilArmodafinil [Nuvigil] is simply the R-enantiomer of modafinil, a mixture of R- and S-enantiomers. Armodafinil differs from modafinil in that the R-enantiomer (armodafinil) has a somewhat longer half-life than the S-enantiomer component

412
UNIT V Central Nervous System Drugs
Although reduction of impulsiveness and hyperactivity with a stimulant may seem paradoxical, it isn’t. Stimulants don’t suppress rowdy behavior directly. Rather, they improve attention and focus. Impulsiveness and hyperactivity decline because the child is now able to concentrate on the task at hand. It should be noted that stimulants do not create positive behavior; they only reduce negative behavior. Accordingly, stimulants cannot give a child good study skills and other appropriate behaviors. Rather, these must be learned once the disruptive behavior is no longer an impediment.
The dosing schedule employed is important and is determined by the time course of the formulation selected. As discussed previously and shown in Table 36.2, CNS stimulants are available in IR, SR, and 24-hour formulations. With the IR and SR formulations, the child usually takes two or three doses a day. In contrast, the 24-hour formulations are taken just once a day (in the morning). Not only is once-daily dosing more convenient, it spares the child any embarrassment or stigma associated with taking medicine at school. Accordingly, 24-hour formulations (e.g., Adderall XR, Concerta, Daytrana) are generally preferred. With all formulations, dosage should be low initially and then gradually increased. Maintenance dosage is determined by monitoring for improvement in symptoms and the appearance of side effects.
Principal adverse effects of the stimulants are insomnia and growth suppression. Insomnia results from CNS stimulation and can be minimized by reducing the size of the afternoon dose and taking it no later than 4:00 PM. Growth suppression occurs secondary to appetite suppression. Growth reduction can be minimized by administering stimulants during or after meals (which reduces the impact of appetite suppression). In addition, some clinicians recommend taking “drug holidays” on weekends and in the summer (which creates an opportunity for growth to catch up). However, other clinicians argue against this strategy because depriving children of medication during these unstructured times can be hard on them. When stimulants are discontinued, a rebound increase in growth will take place; as a result, adult height may not be affected. Other adverse effects include headache and abdominal pain, which have an incidence of 10%, and lethargy and listlessness, which can occur when dosage is excessive.
NonstimulantsSeveral nonstimulants are used for ADHD, although only three of them—atomoxetine, guanfacine, and clonidine—are approved by the U.S. Food and Drug Administration (FDA) for this use. The nonstimulants are less effective than the stimulants, and hence are considered second-choice drugs. For treatment of ADHD, the nonstimulants may be employed as monotherapy or as add-on therapy with a stimulant. Unlike the stimulants, the nonstimulants are not regulated as controlled substances.
Atomoxetine, a Norepinephrine Uptake InhibitorDescription and Therapeutic Effects. Atomoxetine
[Strattera] is a unique drug approved for ADHD in children and adults. It was the first nonstimulant approved for ADHD,a and one of only three drugs approved for ADHD in adults (the
theories would be consistent with the effects of atomoxetine (which blocks NE reuptake), imipramine (which blocks NE and serotonin uptake), and stimulant drugs (which promote the release of NE and DA and to some degree block their uptake). Genetic factors play a significant role.
Management OverviewMultiple strategies may be employed to manage ADHD. In addition to drugs, which are considered first-line treatment, the management program can include family therapy, parent training, and cognitive therapy for the child. Guidelines issued by the American Academy of Pediatrics emphasize the impor-tance of a comprehensive treatment program involving col-laboration among clinicians, families, and educators. For long-term gains, a combination of cognitive therapy and stimulant drugs appears most effective. Of the drugs employed for ADHD, stimulants are most effective, and so are considered agents of choice. The nonstimulants (e.g., atomoxetine, guanfacine, clonidine) are less effective than stimulants, and hence are considered second-choice drugs.
ADHD in AdultsIn about 30% to 60% of cases, childhood ADHD persists into adulthood. In the United States, about 8 million adults are afflicted, although an estimated 90% are undiagnosed and untreated. Symptoms include poor concentration, stress intoler-ance, antisocial behavior, outbursts of anger, and inability to maintain a routine. Also, adults with ADHD experience more job loss, divorce, and driving accidents. As in childhood ADHD, therapy with a stimulant drug is the foundation of treatment. Methylphenidate is prescribed most often. About 33% of adults fail to respond to stimulants or cannot tolerate their side effects. For these patients, a trial with a nonstimulant may help. Combining behavioral therapy with drug therapy may be more effective than drug therapy alone.
DRUGS USED FOR ADHDCNS StimulantsStimulant drugs are the mainstay of ADHD therapy. Drugs with proven efficacy include methylphenidate [Ritalin, Concerta, others], dexmethylphenidate [Focalin], dextroamphetamine/amphetamine mixture [Adderall], and lisdexamfetamine [Vyvanse]. There are no data to support the use of one stimulant over another. If one stimulant is ineffective, another should be tried before considering a second-line agent.
The response to stimulants can be dramatic. These drugs can increase attention span and goal-oriented behavior while decreasing impulsiveness, distractibility, hyperactivity, and restlessness. Tests of cognitive function (memory, reading, arithmetic) often improve significantly. Unfortunately, although benefits can be dramatic initially, in children they diminish after 2 to 3 years. This finding was initially reported in a 2009 paper: MTA at 8 Years: Prospective Follow-up of Children Treated for Combined-Type ADHD in a Multisite Study. The findings were corroborated in a systematic review of long-term outcomes in ADHD in 2012. Nonetheless, stimulant therapy can still buy time to teach children behavioral strategies to help them combat inattention and hyperactivity over the long term.
aSome older nonstimulants, such as imipramine and bupropion, although used for ADHD, are not actually approved for ADHD.

413
CHAPTER 36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
which a stimulant—methylphenidate [Concerta]—was again clearly superior to atomoxetine.
Mechanism of Action. Atomoxetine is a selective inhibitor of NE reuptake, and hence causes NE to accumulate at synapses. Although the precise relationship between this neurochemical action and symptom relief is unknown, it would appear that adaptive changes that occur following uptake blockade underlie benefits. Uptake blockade occurs immediately, whereas full therapeutic effects are not seen for at least a week—suggesting that, after uptake blockade occurs, additional processes must take place before benefits can be seen.
Pharmacokinetics. Atomoxetine is rapidly and completely absorbed following oral administration. Plasma levels peak in 1 to 3 hours, depending on whether the drug was taken without or with food. Atomoxetine is metabolized in the liver, primarily by CYP2D6 (the 2D6 isoenzyme of cytochrome P450). For most patients, the half-life is 5 hours. However, for 5% to 10% of patients, the half-life is much longer: 24 hours. These patients have an atypical form of CYP2D6, which metabolizes atomoxetine slowly. Dosage should be reduced in these people.
Adverse Effects. Like the CNS stimulants, atomoxetine is generally well tolerated. In clinical trials, the most common
others are amphetamine/dextroamphetamine mixture [Adderall XR] and lisdexamfetamine [Vyvanse]). In contrast to the CNS stimulants, atomoxetine has no potential for abuse, and hence is not regulated as a controlled substance. As a result, prescrip-tions can be refilled over the phone, making atomoxetine more convenient than the stimulants. Like the long-acting stimulants, atomoxetine can be administered just once a day.
In clinical trials comparing atomoxetine with placebo in children or adults with ADHD, atomoxetine was clearly superior at reducing symptoms. Benefits were similar whether the drug was given once a day or in two divided doses. It should be noted that responses develop slowly: The initial response takes a few days to develop, and the maximal response is seen in 1 to 3 weeks. This contrasts with the CNS stimulants, whose effects are near maximal with the first dose.
How does atomoxetine compare with stimulants for treating children with ADHD? In two older 3-week randomized trials, comparing atomoxetine with either methylphenidate [Concerta] or an amphetamine [Adderall XR], the stimulants were superior: More children responded to the stimulants, symptom reduction was greater, and benefits developed more quickly. These results were reinforced in a large 6-week placebo-controlled trial, in
Drug Brand Name Duration (hr) Dosing Schedule Usual Pediatric Maintenance Dosage
STIMULANTS
Methylphenidate
Immediate Release Ritalin, Methylin 3–5 2 or 3 times daily 10 mg at 8:00 AM and noon, and 5 mg at 4:00 PM
Sustained Release Ritalin-SR, Metadate ER, Quillivant XR,
Quillichew ER
6–8 Once or twice daily 20 or 40 mg in AM plus 20 mg in the early PM if needed
24-Hour Concerta 10–12 Once daily 36 mg in the AMAptensio XR 12 Once daily 10 mg in the AMMetadate CD 8–12 Once daily 30 mg in the AMRitalin LA 8–12 Once daily 30 mg in the AMDaytrana 10–12 Once daily One 15- or 20-mg patch, applied in the
AM and removed 9 hr later
Dexmethylphenidate
Immediate Release Focalin 4–5 Twice daily 10 mg in the AM plus 10 mg in the early PM
Sustained Release Focalin XR 12 Once daily 10 mg in the AM
Dextroamphetamine
Immediate Release Zenzedi, Procentra 4–6 Once or twice daily 5 mg in the AM
Sustained Release Dexedrine 6–10 Once or twice daily 10 mg at 8:00 AM
Amphetamine Mixture
Immediate Release Adderall 4–6 Twice daily 5 mg in the AM and 4–6 hr laterSustained Release Adderall XR 10–12 Once daily 20 mg in the AM
Lisdexamfetamine
Sustained Release Vyvanse 13 Once daily 30 mg in the AM
NONSTIMULANTS
Atomoxetine Strattera 24 Once or twice daily 80 mg in the AM or 40 mg in the AM and early PM
Guanfacine Intuniv 24 Once daily 1–4 mg in the AM
Clonidine Kapvay 24 Twice daily 0.1–0.2 mg in the AM and PM
TABLE 36.2 ■ Major Drugs for Attention-Deficit/Hyperactivity Disorder

414
UNIT V Central Nervous System Drugs
Two dosing schedules may be used: Patients may either (1) take the total daily dose all at once in the morning or (2) divide the dosage up, taking half in the morning and half in the late afternoon or early evening. Note that, with either schedule, dosing during school hours is unnecessary.
Dosage should be reduced in patients who are slow metabolizers, either because of hepatic insufficiency or atypical CYP2D6.
Alpha2-Adrenergic AgonistsTwo alpha2-adrenergic agonists—guanfacine and clonidine—are approved for ADHD. Both drugs appear less effective than CNS stimulants. Principal side effects are sedation, hypotension, and fatigue. Unlike the CNS stimulants, guanfacine and clonidine are not controlled substances and do not cause anorexia or insomnia. The basic pharmacology of these drugs is discussed in Chapter 19.
Guanfacine. Available in an ER formulation, sold as Intuniv, guanfacine is used for treating children and adolescents with ADHD. In clinical trials, ER guanfacine improved hyperactivity and inattention. Benefits were greater than with placebo, but less than reported with stimulants. The exact mechanism behind guanfacine is unknown. We do know that guanfacine activates presynaptic alpha2-adrenergic receptors in the brain. However, we don’t know how this action relates to clinical benefits. Principal side effects are somnolence, fatigue, and reduced blood pressure. Effects on blood pressure are most pronounced during initial therapy and whenever dosage is increased. Abrupt discontinuation can cause rebound hypertension. In contrast to the stimulants, guanfacine causes weight gain rather than weight loss, causes somnolence rather than insomnia, and is not regulated under the Controlled Substances Act. Who should receive guanfacine? Because the drug does not cause anorexia or insomnia, it might be especially good for children who cannot tolerate these effects of stimulants. Guanfacine can also be combined with a stimulant to treat severe ADHD.
For treatment of ADHD, guanfacine [Intuniv] is available in ER tablets (1, 2, 3, and 4 mg), which should be swallowed intact, without chewing, cutting, or crushing. Dosing with a high-fat meal increases absorption and should be avoided. Dosage starts at 1 mg/day for at least 1 week, and can be increased at intervals of 1 week (or longer) by 1 mg/day. Children switching from IR guanfacine should use the same titration schedule, regardless of the dosage they had been taking. For all children, the maximum dosage is 4 mg/day. When guanfacine is discontinued, dosage should be tapered by 1 mg/day every 3 to 7 days. Abrupt discontinuation should be avoided, owing to a risk of rebound hypertension. The IR formulation used for hypertension, sold as Tenex, is discussed in Chapter 19.
Clonidine. ER clonidine [Kapvay] is much like ER guanfacine [Intuniv]. Both drugs are alpha2 agonists, both were developed for hypertension, and both had been used off-label in ADHD for years. In clinical trials of ADHD, ER clonidine was superior to placebo when used alone and provided additional symptom relief when combined with a stimulant. As with guanfacine, principal side effects are somnolence, fatigue, and hypotension. Somnolence can be made worse by alcohol and other CNS depressants. Hypotension can be made worse by antihypertensive agents. Because clonidine can lower blood pressure (and slow heart rate too), blood pressure and heart rate should be measured at baseline, following each dose increase, and periodically thereafter. Like guanfacine, clonidine does not cause anorexia or insomnia and is not a controlled substance. For treatment of ADHD, clonidine [Kapvay] is supplied in 0.1- and 0.2-mg ER tablets, which should be swallowed whole without crushing, cutting, or chewing. Dosing may be done with or without food. The initial dosage is 0.1 mg in the evening, and the maximum dosage is 0.2 mg twice a day. Dosage is titrated, at intervals of 1 week or longer, as follows:
• 0.1 mg in PM• 0.1 mg in AM and 0.1 mg in PM
effects were GI reactions (dyspepsia, nausea, and vomiting), reduced appetite, dizziness, somnolence, mood swings, and trouble sleeping. Sexual dysfunction and urinary retention were seen in adults. Severe allergic reactions, including angioneurotic edema, occurred rarely. If allergy develops, patients should discontinue the drug and contact their prescriber immediately.
Atomoxetine may cause suicidal thinking in children and adolescents, but not in adults. Fortunately, the incidence is relatively low: about 4 cases per 1000 patients. Risk is greatest during the first few months of treatment. Young patients should be monitored closely for suicidal thinking and behavior, and for signs of clinical worsening (e.g., agitation, irritability).
Appetite suppression may result in weight loss and growth delay. Among children who took atomoxetine for 18 months or longer, mean height and weight percentiles declined. Because experience with the drug is limited, we don’t know whether expected adult height will be affected, nor do we know whether “drug holidays” would have an impact on growth.
Atomoxetine poses a small risk of severe liver injury that may progress to outright liver failure, resulting in death or the need for a liver transplant. Patients should be informed about signs of liver injury—jaundice, dark urine, abdominal tenderness, unexplained flu-like symptoms—and instructed to report these immediately. In the event of jaundice or laboratory evidence of liver injury, atomoxetine should be discontinued.
Atomoxetine may raise or lower blood pressure. During clinical trials, some patients experienced a small increase in blood pressure and heart rate. Accordingly, atomoxetine should be used with caution by patients with hypertension or tachy-cardia. During postmarketing surveillance, some patients experienced hypotension and syncope (fainting). Patients should be informed of this possibility and advised to sit or lie down if they feel faint.
Drug Interactions. Combining atomoxetine with a mono-amine oxidase inhibitor (e.g., isocarboxazid [Marplan], phenelzine [Nardil]) can cause hypertensive crisis, owing to accumulation of NE at synapses in the periphery. Accordingly, these drugs must not be used together or within 3 weeks of each other.
Inhibitors of CYP2D6 can increase levels of atomoxetine, and hence must be used with caution. Common examples include paroxetine [Paxil], fluoxetine [Prozac], and quinidine.
Role in ADHD Therapy. Atomoxetine is recommended for treatment of ADHD in cases in which there may be concern for stimulant abuse or there exists a strong aversion to treatment with stimulant medications. Because CNS stimulants are more effective and have a long record of safety and efficacy, it would seem prudent to reserve atomoxetine for patients who are unresponsive to or intolerant of the stimulants. In the absence of a compelling reason, patients doing well on the stimulants shouldn’t switch.
Preparations, Dosage, and Administration. Atomoxetine [Strattera] is available in capsules (10, 18, 25, 40, 60, 80, and 100 mg) that should be swallowed whole, with or without food. Dosage is based on body weight as follows:
• Children who weigh less than 70 kg—Start with 0.5 mg/kg/day and then, after at least 3 days, increase to the recommended target of 1.2 mg/kg/day. The maximum dosage is 1.4 mg/kg/day or 100 mg, whichever is smaller.
• Children who weigh more than 70 kg and all adults—Start with 40 mg/day and then, after at least 3 days, increase to the recommended target of 80 mg/day. Do not exceed 100 mg/day.

415
CHAPTER 36 Central Nervous System Stimulants and Attention-Deficit/Hyperactivity Disorder
be taken continuously. Adverse effects include sedation and anticholinergic effects (e.g., dry mouth, blurred vision, urinary retention, constipation). More importantly, sudden death (from cardiotoxicity) has occurred in at least three children. Compared with stimulants, antidepressants have their benefits (no insomnia, abuse potential, or suppression of appetite and growth) as well as their drawbacks (anticholinergic effects, delayed onset, tolerance, less efficacy, risk of sudden death). Because these antidepressants are less effective and more dangerous than the stimulants, they are considered second-line drugs. Dosages for ADHD range from 2 to 5 mg/kg/day, administered in two or three divided doses. The basic pharmacology of the antidepressants is presented in Chapter 32.
Bupropion. Bupropion [Wellbutrin] can reduce behavioral symptoms of ADHD, but is less effective than stimulants. The drug lacks the adverse effects associated with tricyclic antidepressants (e.g., cardiotoxicity, anticho-linergic effects) but does pose a risk of seizures. Like the tricyclic antidepres-sants, bupropion is considered a second-line drug for ADHD. Dosage is 100 to 150 mg twice a day. The basic pharmacology of bupropion is presented in Chapter 32.
• 0.1 mg in AM and 0.2 mg in PM• 0.2 mg in AM and 0.2 mg in PM
When treatment stops, dosage should be reduced by 0.1 mg every 3 to 7 days to avoid rebound hypertension. Clonidine formulations used for hypertension, marketed as Catapres and Catapres-TTS, are discussed in Chapter 19.
AntidepressantsThree antidepressants—desipramine, imipramine, and bupropion—can reduce behavioral symptoms in children with ADHD. However, these antidepressants are less effective than CNS stimulants and are not approved for ADHD. Accordingly, they are generally reserved for children who have not responded to trials with at least two different stimulants.
Tricyclic Antidepressants. Desipramine [Norpramin] and imipramine [Tofranil] can reduce symptoms in children with ADHD. These drugs decrease hyperactivity but have little effect on impulsivity and inattention. Responses develop slowly. Beneficial effects begin in 2 to 3 weeks and reach a maximum at around 6 weeks. Tolerance frequently develops within a few months. In contrast to the stimulants, which can be discontinued on weekends, antidepressants must
KEY POINTS
■ The amphetamine family consists of dextroamphetamine, amphetamine (a racemic mixture of dextroamphetamine and levamphetamine), methamphetamine, and lisdexamfetamine.
■ The amphetamines work primarily by promoting neuronal release of NE and DA, and partly by blocking NE and DA reuptake.
■ Through actions in the CNS, the amphetamines can increase wakefulness and alertness, reduce fatigue, elevate mood, stimulate respiration, and suppress appetite.
■ By promoting release of NE from peripheral neurons, amphetamines can cause vasoconstriction and cardiac effects (increased heart rate, increased atrioventricular conduction, and increased force of contraction).
■ The most common adverse effects of amphetamines are insomnia and weight loss. Amphetamines may also cause psychosis and cardiovascular effects (dysrhythmias, angina, hypertension).
■ Amphetamines have a high abuse potential (owing to their ability to elevate mood), and hence are classified as Schedule II drugs.
■ The principal indication for amphetamines is ADHD.■ The pharmacology of methylphenidate is nearly identical
to that of the amphetamines.
■ Methylphenidate and other CNS stimulants are the most effective drugs for ADHD, and hence are considered agents of first choice.
■ Methylphenidate and other CNS stimulants reduce symp-toms of ADHD by enhancing the patient’s ability to focus.
■ Only three nonstimulants—atomoxetine, guanfacine, and clonidine—are approved for ADHD.
■ In treatment of ADHD, the nonstimulants may be used alone or as add-on therapy with a stimulant.
■ Compared with the CNS stimulants, the nonstimulants are less effective in ADHD, but also are safer and have a lower potential for abuse.
■ Caffeine and other methylxanthines act primarily by block-ing adenosine receptors.
■ Responses to caffeine are dose dependent: low doses decrease drowsiness and fatigue; higher doses cause nervousness, insomnia, and tremors; and huge doses cause convulsions.
■ Caffeine has two principal uses: treatment of apnea in premature infants and reversal of drowsiness.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
AMPHETAMINES, METHYLPHENIDATE, AND DEXMETHYLPHENIDATEPreadministration AssessmentTherapeutic GoalReduction of symptoms in children and adults with ADHD. Reduction of sleep attacks in patients with narcolepsy.
Baseline DataChildren With ADHD. Document the degree of inatten-
tion, impulsivity, hyperactivity, and other symptoms of ADHD.
Symptoms must be present for at least 6 months to allow a diagnosis of ADHD. Obtain baseline values of height and weight.
Narcolepsy. Document the degree of daytime sleepiness and the frequency and circumstances of sleep attacks.
Identifying High-Risk PatientsAll amphetamines are contraindicated for patients with symptomatic cardiovascular disease, advanced atherosclerosis, hypertension, hyperthyroidism, agitated states, and a history of drug abuse, and in those who have taken monoamine
Continued

416
UNIT V Central Nervous System Drugs
aPatient education information is highlighted as blue text.
oxidase inhibitors within the previous 2 weeks. Amphetamine/dextroamphetamine mixture [Adderall XR] is generally contraindicated for patients with structural cardiac defects.
Implementation: AdministrationRoutes
Oral. Amphetamines, methylphenidate, and dexmethyl-phenidate.
Transdermal. Methylphenidate only.
AdministrationOral. Instruct patients to swallow long-acting formulations
intact, without crushing or chewing.Advise parents that children with ADHD should take the
morning dose after breakfast and the last daily dose by 4:00 PM.
Transdermal. Instruct patients using transdermal meth-ylphenidate [Daytrana] to apply one patch to alternating hips each morning and to remove each patch not more than 9 hours after applying it. Instruct patients to avoid application to skin that is inflamed.
Ongoing Evaluation and InterventionsEvaluating Therapeutic Effects
Children With ADHD. Monitor for reductions in symp-toms (impulsiveness, hyperactivity, inattention) and for improvement in cognitive function.
Minimizing Adverse EffectsExcessive CNS Stimulation. CNS stimulants can cause
restlessness and insomnia. Advise patients to use the smallest dose required and to avoid dosing late in the day. Advise patients to minimize or eliminate dietary caffeine (e.g., coffee, tea, caffeinated soft drinks).
Weight Loss. Appetite suppression can cause weight loss. Advise patients to take the morning dose after breakfast and the last daily dose early in the afternoon to minimize interference with eating.
Cardiovascular Effects. Warn patients about cardiovas-cular responses (palpitations, hypertension, angina, dysrhyth-mias), and instruct them to notify the prescriber if these develop.
Very rarely, children using stimulants for ADHD have experienced sudden cardiac death. In response, the AHA says it is reasonable to consider giving a child an ECG before
starting stimulant therapy. However, there is conflicting evidence that stimulants actually cause sudden death, or that withholding stimulants will protect from sudden death, or that screening for cardiac defects with an ECG will be of any benefit. Therefore, it would seem that routine ECG screening is unnecessary, especially in children with no signs or symptoms of heart defects. However, if there is evidence of existing heart disease, or evidence of hereditary cardio-vascular defects, an ECG might be appropriate.
Psychosis. If amphetamine-induced psychosis develops, therapy should be discontinued. For most individuals, symptoms resolve within a week. For some patients, drug-induced psychosis may represent unmasking of latent schizophrenia, indicating a need for psychiatric care.
Withdrawal Reactions. Abrupt discontinuation can produce extreme fatigue and depression. Minimize by withdrawing amphetamines and methylphenidate gradually.
Hypersensitivity Reactions. Transdermal methylpheni-date [Daytrana] can cause hypersensitivity reactions, which may necessitate discontinuing all methylphenidate products, oral as well as transdermal. Inform patients about signs of hypersensitivity—erythema, edema, papules, vesicles—and instruct them to inform the prescriber if these develop.
Minimizing AbuseIf the medical history reveals that the patient is prone to drug abuse, monitor use of these drugs closely.
CAFFEINEGeneral ConsiderationsCaffeine is usually administered to promote wakefulness. Warn patients against habitual caffeine use to compensate for chronic lack of sleep. Advise patients to consult the prescriber if fatigue is persistent or recurrent.
Minimizing Adverse EffectsCardiovascular EffectsInform patients about cardiovascular responses to caffeine (palpitations, rapid pulse, dizziness), and instruct them to discontinue caffeine if these occur.
Excessive CNS StimulationWarn patients that overdose can cause convulsions. Advise them to ingest no more caffeine than needed.
Summary of Major Nursing Implicationsa—cont’d

417
C H A P T E R
37 Substance Use Disorders I: Basic Considerations
Definitions, p. 417Drug Abuse, p. 417Substance Use Disorder, p. 418Other Definitions, p. 418
Diagnostic Criteria Regarding Substance Use Disorder, p. 418
Factors That Contribute to Substance Use Disorder, p. 418Reinforcing Properties of Drugs, p. 418Physical Dependence, p. 419Psychologic Dependence, p. 419Social Factors, p. 419Drug Availability, p. 419Vulnerability of the Individual, p. 419
Neurobiology of Substance Use Disorder, p. 419Principles of Substance Use Disorder Treatment,
p. 420The Controlled Substances Act, p. 421
Record Keeping, p. 421DEA Schedules, p. 421Prescriptions, p. 421Labeling, p. 422State Laws, p. 422
Key Points, p. 423
secondary medical complications of drug abuse, (3) the facilita-tion of drug withdrawal, and (4) education and counseling to maintain long-term abstinence.
Our discussion of drug abuse occurs in two stages. In this chapter, we discuss basic concepts in drug abuse. In Chapters 38, 39, and 40, we focus on the pharmacology of specific abused agents and methods of treatment.
DEFINITIONSDrug AbuseDrug abuse can be defined as using a drug in a fashion inconsistent with medical or social norms. Traditionally, the term also implies drug usage that is harmful to the individual or society. As we shall see, although we can give abuse a general definition, deciding whether a particular instance of drug use constitutes “abuse” is often difficult.
Whether drug use is considered abuse depends, in part, on the purpose for which a drug is taken. Not everyone who takes large doses of psychoactive agents has a disorder. For example, we do not consider it abuse to take large doses of opioids long term to relieve pain caused by cancer. However, we do consider it abusive for an otherwise healthy individual to take those same opioids in the same doses to produce euphoria.
Abuse can have different degrees of severity. Some people, for example, use heroin only occasionally, whereas others use it habitually and compulsively. Although both patterns of drug use are socially condemned, and therefore constitute abuse, there is an obvious quantitative difference between taking heroin once or twice and taking it routinely and compulsively.
Note that, by the definition earlier in this section, drug abuse is culturally defined. Because abuse is culturally defined and because societies differ from one another and are change-able, there can be wide variations in what is labeled abuse. What is defined as abuse can vary from one culture to another. For example, in the United States, moderate consumption of alcohol is not usually considered abuse. In contrast, any inges-tion of alcohol may be considered abuse in some Muslim societies. Furthermore, what is defined as abuse can vary from one time to another within the same culture. For example, when a few Americans first experimented with lysergic acid diethylamide (LSD) and other psychedelic drugs, these agents were legal and their use was not generally disapproved. However, when use of psychedelics became widespread, our societal posture changed and legislation was passed to make the manufacture, sale, and use of these drugs illegal.
Within the United States, there is divergence of opinion about what constitutes drug abuse. For example, some people would consider any use of marijuana to be abuse, whereas others would call smoking marijuana abusive only if it were
Mind-altering drugs have intrigued human beings since the dawn of civilization. Throughout history, people have taken drugs to elevate mood, release inhibitions, distort perceptions, induce hallucinations, and modify thinking. Many of those who take mind-altering drugs restrict usage to socially approved patterns. However, many others self-administer drugs to excess. Excessive drug use is our focus in this chapter and the three that follow.
Drug abuse extracts a huge toll on the individual and on society. Tobacco alone kills about 480,000 Americans each year. Alcohol and illicit drugs kill an additional 100,000. In addition to putting people at risk of death, drug abuse puts them at risk of long-term illness and impairs their ability to fulfill role obligations at home, school, and work. The economic burden of drug abuse is staggering: The combined direct and indirect costs from abusing nicotine, alcohol, and illicit sub-stances are estimated at over $700 billion each year.
Drug abuse confronts clinicians in a variety of ways, making knowledge of abuse a necessity. Important areas in which expertise on drug abuse may be applied include (1) diagnosis and treatment of acute toxicity, (2) diagnosis and treatment of

418
UNIT V Central Nervous System Drugs
DIAGNOSTIC CRITERIA REGARDING SUBSTANCE USE DISORDER
Substance use disorder is best defined as continued use of a substance despite significant substance-related problems. There exists a change in brain circuitry that persists despite detoxifica-tion. Diagnosis of substance abuse disorder is based on behaviors related to continued use of a substance.
Tolerance and withdrawal are among the criteria established by the APA for having a substance use disorder. Please note, however, that tolerance and withdrawal, by themselves, are neither necessary nor sufficient for a substance use disorder to exist. Put another way, the pattern of drug use that constitutes a substance use disorder can exist in persons who are not physically dependent on drugs and who have not developed tolerance. Because this distinction is extremely important, we will express it another way: Being physically dependent on a drug is not the same as having a disorder! Many people are physically dependent but do not meet the criteria for a substance use disorder. These people are not considered to have SUD because they do not demonstrate the behavior pattern that constitutes substance dependence. Patients with terminal cancer, for example, are often physically dependent on opioids. However, since their lives are not disrupted by their medication (quite the contrary), their drug use does not meet the criteria for a substance use disorder. Similarly, some degree of physical dependence occurs in all patients who take phenobarbital to control seizure disorders. However, despite their physical dependence, patients with seizure do not carry out stereotypic addictive behavior and therefore do not have a substance use disorder.
Having stressed that physical dependence and SUD are different from each other, we must note that the two states are not entirely unrelated. As discussed in the sections that follow, although physical dependence is not the same as SUD, physical dependence often contributes to addictive behavior.
FACTORS THAT CONTRIBUTE TO SUBSTANCE USE DISORDERSubstance use disorder is the end result of a progressive involvement with drugs. Taking psychoactive drugs is usually initiated out of curiosity. From this initial involvement, the user can progress to occasional use. Occasional use can then evolve into compulsive use. Factors that play a role in the progression from experimental use to compulsive use are discussed in the sections that follow.
Reinforcing Properties of DrugsAlthough there are several reasons for initiating drug use (e.g., curiosity, peer pressure), individuals would not continue drug use unless drugs produced desirable feelings or experiences. By making people feel “good,” drugs reinforce the reasons for their use. Conversely, if drugs did not give people experi-ences that they found desirable, the reasons for initiating drug use would not be reinforced, and drug use would stop.
Reinforcement by drugs can occur in two ways. First, drugs can give the individual an experience that is pleasurable. Cocaine, for example, produces a state of euphoria. Second,
done habitually. Similarly, although many Americans do not consider cigarette smoking abuse (even though the practice is compulsive and clearly harmful to the individual and society), others believe very firmly that cigarette smoking is a blatant form of abuse.
As we can see, distinguishing between culturally acceptable drug use and drug use that is to be called abuse is more in the realm of social science than pharmacology. Accordingly, since this is a pharmacology text and not a sociology text, we will not attempt to define just what patterns of drug use do or do not constitute abuse. Instead, we will focus on the pharmacologic properties of abused drugs—leaving distinctions about what is and is not abuse to sociologists and legislators. Fortunately, we can identify the drugs that tend to be abused and discuss their pharmacology without having to resolve all arguments about what patterns of use should or should not be considered abusive.
Substance Use DisorderAccording to the American Psychiatric Association (APA), substance use disorder (SUD) is defined as a cluster of cognitive, behavioral, and physiological symptoms indicating that the individual continues using the substance despite significant substance-related problems. Please note that nowhere in this definition is SUD equated with physical dependence. As discussed elsewhere in this chapter, although physical depen-dence can contribute to addictive behavior, it is neither necessary nor sufficient for addiction to occur.
Other DefinitionsTolerance results from regular drug use and can be defined as a state in which a particular dose elicits a smaller response than it did with initial use. As tolerance increases, higher and higher doses are needed to elicit desired effects.
Cross-tolerance is a state in which tolerance to one drug confers tolerance to another. Cross-tolerance generally develops among drugs within a particular class, and not between drugs in different classes. For example, tolerance to one opioid (e.g., heroin) confers cross-tolerance to other opioids (e.g., morphine), but not to central nervous system (CNS) depressants, psycho-stimulants, psychedelics, or nicotine.
Psychologic dependence can be defined as an intense subjec-tive need for a particular psychoactive drug.
Physical dependence can be defined as a state in which an abstinence syndrome will occur if drug use is discontinued. Physical dependence is the result of neuroadaptive processes that take place in response to prolonged drug exposure.
Cross-dependence refers to the ability of one drug to support physical dependence on another drug. When cross-dependence exists between drug A and drug B, taking drug A will prevent withdrawal in a patient physically dependent on drug B, and vice versa. As with cross-tolerance, cross-dependence generally exists among drugs in the same pharmacologic family, but not between drugs in different families.
A withdrawal syndrome is a constellation of signs and symptoms that occurs in physically dependent individuals when they discontinue drug use. Quite often, the symptoms seen during withdrawal are opposite to effects the drug produced before it was withdrawn. For example, discontinuation of a CNS depressant can cause CNS excitation.

419
CHAPTER 37 Substance Use Disorders I: Basic Considerations
after the initial exposure. For example, most people do not especially enjoy their first cigarette; were it not for peer pressure, many would quit before they smoked enough for it to become pleasurable. Similarly, initial use of heroin, with its associated nausea and vomiting, is often deemed unpleasant; peer pressure is a common reason for continuing heroin use long enough to develop tolerance to these undesirable effects.
Drug AvailabilityDrug availability is clearly a factor in the development and maintenance of abuse. Abuse can flourish only in environ-ments where drugs can be readily obtained. In contrast, where procurement is difficult, abuse is minimal. The ready availability of drugs in hospitals and clinics is a major reason for the unusually high rate of addiction among pharmacists, nurses, and physicians.
Vulnerability of the IndividualSome individuals are more prone to becoming drug abusers than others. By way of illustration, let’s consider three individu-als from the same social setting who have equal access to the same psychoactive drug. The first person experiments with the drug briefly and never uses it again. The second person progresses from experimentation to occasional use. The third goes on to take the drug compulsively. Since social factors, drug availability, and the properties of the drug itself are the same for all three people, these factors cannot explain the three different patterns of drug use. We must conclude, therefore, that the differences must lie in the people: one individual was not prone to drug abuse, one had only moderate tendencies toward abuse, and the third was highly vulnerable to becoming an abuser.
Several psychologic factors have been associated with tendencies toward drug abuse. Drug abusers are frequently individuals who are impulsive, have a low tolerance for frustra-tion, and are rebellious against social norms. Other psychologic factors that seem to predispose individuals to abusing drugs include depressive disorders, anxiety disorders, and antisocial personality. It is also clear that individuals who abuse one type of drug are likely to abuse other drugs.
There is speculation that some instances of drug abuse may actually represent self-medication to relieve emotional dis-comfort. For example, some people may use alcohol and other depressants to control severe anxiety. Although their drug use may appear excessive, it may be no more than they need to neutralize intolerable feelings.
Genetics also contributes to drug abuse. Vulnerability to alcoholism, for example, may result from an inherited predisposition.
NEUROBIOLOGY OF SUBSTANCE USE DISORDERRepeated use of a drug contributes to the transition from voluntary drug use to compulsive use by causing molecular changes in the brain. Each time the drug is taken, it causes changes that promote further drug use. With repeated drug exposure, these changes are reinforced, making drug use increasingly more difficult to control.
drugs can reduce the intensity of unpleasant experiences. For example, drugs can reduce anxiety and stress.
The reinforcing properties of drugs can be clearly demon-strated in experiments with animals. In the laboratory, animals will self-administer most of the drugs that are abused by humans (e.g., opioids, barbiturates, alcohol, cocaine, amphetamines, phencyclidine, nicotine, caffeine). When these drugs are made freely available, animals develop patterns of drug use that are similar to those of humans. Animals will self-administer these drugs (except for nicotine and caffeine) in preference to eating, drinking, and sex. When permitted, they often die from lack of food and fluid. These observations strongly suggest that pre-existing psychopathology is not necessary for drug abuse to develop. Rather, these studies suggest that drug abuse results, in large part, from the reinforcing properties of drugs themselves.
Physical DependenceAs defined earlier in this chapter, physical dependence is a state in which an abstinence syndrome will occur if drug use is discontinued. The degree of physical dependence is deter-mined largely by dosage and duration of drug use. Physical dependence is greatest in people who take large doses for a long time. The more physically dependent a person is, the more intense the withdrawal syndrome. Substantial physical dependence develops to the opioids (e.g., morphine, heroin) and CNS depressants (e.g., barbiturates, alcohol). Physical dependence tends to be less prominent with other abused drugs (e.g., psychostimulants, psychedelics, marijuana).
Physical dependence can contribute to compulsive drug use. Once dependence has developed, the desire to avoid withdrawal becomes a motivator for continued dosing. Fur-thermore, if the drug is administered after the onset of with-drawal, its ability to alleviate the discomfort of withdrawal can reinforce its desirability. Please note, however, that although physical dependence plays a role in the abuse of drugs, physical dependence should not be viewed as the primary cause of addictive behavior. Rather, physical dependence is just one of several factors that can contribute to the development and continuation of compulsive use.
Psychologic DependencePsychologic dependence is defined as an intense subjective need for a drug. Individuals who are psychologically dependent feel very strongly that their sense of well-being is dependent upon continued drug use; a sense of “craving” is felt when the drug is unavailable. There is no question that psychologic dependence can be a major factor in addictive behavior. For example, it is psychologic dependence—and not physical dependence—that plays the principal role in causing renewed use of opioids by addicts who had previously gone through withdrawal.
Social FactorsSocial factors can play an important role in the development of SUD. The desire for social status and approval is a common reason for initiating drug use. Also, since initial drug experiences are frequently unpleasant, the desire for social approval can be one of the most compelling reasons for repeating drug use

420
UNIT V Central Nervous System Drugs
amounts of dopamine, the reward circuit is put in a state of excessive activation. In response, the brain (1) produces less dopamine and (2) reduces the number of dopamine receptors. As a result, responses to drugs are reduced. Unfortunately, the ability of natural stimuli to activate the circuit is reduced as well. In the absence of pleasurable feelings from natural stimuli, the abuser is left feeling flat, lifeless, and depressed. The good news is that, when drug use stops, neural remodeling tends to gradually reverse.
PRINCIPLES OF SUBSTANCE USE DISORDER TREATMENTSubstance use disorder is a treatable disease of the brain. With therapy, between 40% and 60% of addicts can reduce drug use. The first science-based guide on addiction therapy—Principles of Drug Addiction Treatment—was published by the National Institute on Drug Abuse in 1999, and later revised in 2009 and 2012. The guide centers on 13 principles of effective treatment, shown in Table 37.1.
Molecular changes occur in the so-called reward circuit—a system that normally serves to reinforce behaviors essential for survival, such as eating and reproductive activities. Neurons of the reward circuit originate in the ventral tegmental area of the midbrain and project to the nucleus accumbens. Their major transmitter is dopamine. Under normal circumstances, biologi-cally critical behavior, such as sexual intercourse, activates the circuit. The resultant release of dopamine rewards and reinforces the behavior. Like natural positive stimuli, addictive drugs can also activate the circuit and thereby cause dopamine release. In fact, drugs are so effective at activating the circuit that the amount of dopamine released may be 2 to 10 times the amount released by natural stimuli. Ultimately, whether the system is activated by use of drugs or by behavior essential for survival, the outcome is the same: a tendency to repeat the behavior that turned the system on. With repeated activation over time, the system undergoes synaptic remodeling, thereby consolidating changes in brain function. This neural remodeling persists after drug use has ceased.
An important aspect of drug-induced remodeling is a phenomenon known as down-regulation, which serves to reduce the response to drugs. Because drugs release abnormally large
Adapted from National Institute on Drug Abuse: Principles of Drug Addiction Treatment: A Research-Based Guide, 3rd ed. (Publication No. 12-4180). Bethesda, MD: National Institutes of Health, 2012.
1. Substance use disorder is a complex but treatable disease that affects brain function and behavior. Drugs of abuse alter brain structure and function, resulting in changes that persist long after drug use has stopped. These persistent changes may explain why former abusers are at the risk of relapse after prolonged abstinence.
2. No single treatment is appropriate for everyone. It is critical to match treatment settings, interventions, and services to each patient’s problems and needs.
3. Treatment must be readily available. Treatment applicants can be lost if treatment is not immediately available or readily accessible. As with other chronic diseases, the earlier treatment is offered in the disease process, the greater the likelihood of positive outcomes.
4. Effective treatment must attend to multiple needs of the individual, not solely drug use. In addition to addressing drug use, treatment must address the individual’s medical, psychologic, social, vocational, and legal problems.
5. Remaining in treatment for an adequate time is critical. Treatment duration is based on individual need. Most patients require at least 3 months of treatment to significantly reduce or stop drug use. Additional treatment can produce further progress. As with other chronic illnesses, relapses can occur, signaling a need for treatment to be reinstated or adjusted. Programs should include strategies to prevent patients from leaving prematurely.
6. Individual and/or group counseling and other behavioral therapies are the most common forms of drug abuse treatment. In therapy, patients address motivation, build skills to resist drug use, replace drug-using activities with constructive and rewarding activities, and improve problem-solving abilities. Behavioral therapy also addresses incentives for abstinence and facilitates interpersonal relationships. Ongoing group therapy and other peer support programs can help maintain abstinence.
7. Medication can be an important element of treatment, especially when combined with counseling and other behavioral therapies. Methadone, buprenorphine, and naltrexone can help persons addicted to opioids. Nicotine replacement therapy (e.g., patches, gum), bupropion, and varenicline can help persons addicted to nicotine. Disulfiram, naltrexone, topiramate, and acamprosate can help persons addicted to alcohol.
8. Because needs of the individual can change, the plan for treatment and services must be reassessed continually and modified as indicated. At different times during treatment, a patient may develop a need for medications, medical services, family therapy, parenting instruction, vocational rehabilitation, and social and legal services.
9. Many individuals with substance use disorder also have other mental disorders, which must be addressed. Because drug addiction often co-occurs with other mental illnesses, patients presenting with one condition should be assessed for other conditions and treated as indicated.
10. Medically assisted detoxification is only the first stage of treatment and, by itself, does little to change long-term drug use. Medical detoxification manages the acute physical symptoms of withdrawal—and can serve as a precursor to effective long-term treatment.
11. Treatment needn’t be voluntary to be effective. Sanctions or enticements coming from the family, employer, or criminal justice system can significantly increase treatment entry, retention, and success.
12. Drug use during treatment must be monitored continuously, as relapses during treatment do occur. Knowing that drug use is being monitored (e.g., through urinalysis) can help the patient withstand urges to use drugs. Monitoring also can provide early evidence of drug use, thereby allowing timely adjustment of the treatment program.
13. Treatment programs should provide assessment for HIV/AIDS, hepatitis B and C, tuberculosis, and other infectious diseases, along with counseling, to help patients modify behaviors that place them or others at risk.
TABLE 37.1 ■ Principles of Substance Use Treatment
HIV/AIDS, Human immunodeficiency virus/acquired immunodeficiency syndrome.

421
CHAPTER 37 Substance Use Disorders I: Basic Considerations
dispensing machines that count the controlled substances dispensed during each shift.
DEA SchedulesEach drug preparation regulated under the CSA has been assigned to one of five categories: Schedule I, II, III, IV, or V. Drugs in Schedule I have a high potential for abuse and no approved medical use in the United States. In contrast, drugs in Schedules II through V all have approved applications. Assignment to Schedules II through V is based on abuse potential and potential for causing physical or psychologic dependence. Of the drugs that have medical applications, those in Schedule II have the highest potential for abuse and depen-dence. Drugs in the remaining schedules have decreasing abuse and dependence liabilities. Table 37.2 lists the primary drugs that come under the five DEA Schedules.
Scheduling of drugs under the CSA undergoes periodic re-evaluation. With increased understanding of the abuse and dependence liabilities of a drug, the DEA may choose to reassign it to a different Schedule. For example, hydrocodone (a commonly used opioid) was switched from Schedule III to Schedule II.
PrescriptionsThe CSA places restrictions on prescribing drugs in Schedules II through V. (Drugs in Schedule I have no approved uses, and hence are not prescribed.) Only prescribers registered with the DEA are authorized to prescribe controlled drugs. Regula-tions on prescribing controlled substances are summarized in the sections that follow.
Schedule IIAll prescriptions for Schedule II drugs must be typed or filled out in ink or indelible pencil and signed by the prescriber. Alternatively, prescribers may submit prescriptions using an electronic prescribing procedure. Oral prescriptions may be called in, but only in emergencies, and a written prescription must follow within 72 hours. Prescriptions for Schedule II drugs cannot be refilled. However, a DEA rule allows a pre-scriber to write multiple prescriptions on the same day—for the same patient and same drug—to be filled sequentially for up to a 90-day supply.
Schedules III and IVPrescriptions for drugs in Schedules III and IV may be oral, written, or electronic. If authorized by the prescriber, these prescriptions may be refilled up to 5 times. Refills must be made within 6 months of the original order. If additional medication is needed beyond the amount provided for in the original prescription, a new prescription must be written.
Schedule VThe same regulations for prescribing drugs in Schedules III and IV apply to drugs in Schedule V. In addition, Schedule V drugs may be dispensed without a prescription provided the following conditions are met: (1) the drug is dispensed by a pharmacist; (2) the amount dispensed is very limited; (3) the recipient is at least 18 years old and can prove it; (4) the pharmacist writes and initials a record indicating the date, the name and amount of the drug, and the name and address
Ideally, the goal of treatment is complete cessation of drug use. However, total abstinence is not the only outcome that can be considered successful. Treatment that changes drug use from compulsive to moderate will permit increased productivity, better health, and a decrease in socially unacceptable behavior. Clearly, this outcome is beneficial both to the individual and to society—even though some degree of drug use continues. It must be noted, however, that in the treatment of some forms of abuse, nothing short of total abstinence can be considered a true success. Experience has shown that abusers of cigarettes, alcohol, and opioids are rarely capable of sustained moderation. Hence, for many of these individuals, abstinence must be complete if there is to be any hope of avoiding a return to compulsive use.
Recovery from addiction is a prolonged process that typically requires multiple treatment episodes because addiction is a chronic, relapsing illness. As such, periods of treatment-induced abstinence will very likely be followed by relapse. This does not mean that treatment has failed. Rather, it simply means that at least one more treatment episode is needed. Eventually, many patients achieve stable, long-term abstinence, along with a more productive and rewarding life.
Because addiction is a complex illness that affects all aspects of life, the treatment program must be comprehensive and multifaceted. In addition to addressing drug use itself, the program should address any related medical, psychologic, social, vocational, and legal problems. Obviously, treatment must be tailored to the individual; no single approach works for all people. Multiple techniques are employed. Techniques with proven success include (1) group and individual therapy directed at resolving emotional problems that underlie drug use, (2) substituting alternative rewards for the rewards of drug use, (3) threats and external pressure to discourage drug use, and (4) the use of pharmacologic agents to modify the effects of abused drugs. The most effective treatment programs incorporate two or more of these methods.
THE CONTROLLED SUBSTANCES ACTThe Comprehensive Drug Abuse Prevention and Control Act of 1970, known informally as the Controlled Substances Act (CSA), is the principal federal legislation addressing drug abuse. One objective of the CSA is to reduce the chances that drugs originating from legitimate sources will be diverted to abusers. To accomplish this goal, the CSA sets forth regulations for the handling of controlled substances by manufacturers, distribu-tors, pharmacists, nurses, and physicians. Enforcement of the CSA is the responsibility of the Drug Enforcement Agency (DEA), an arm of the U.S. Department of Justice.
Record KeepingTo keep track of controlled substances that originate from legitimate sources, a written record must be made of all transac-tions involving these agents. Every time a controlled substance is purchased or dispensed, the transfer must be recorded. Physicians, pharmacists, and hospitals must keep an inventory of all controlled substances in stock. This inventory must be reported to the DEA every 2 years. Although not specifically obliged to do so by the CSA, many hospitals use medication

422
UNIT V Central Nervous System Drugs
State LawsAll states have their own laws regulating drugs of abuse. In many cases, state laws are more stringent than federal laws. As a rule, whenever there is a difference between state and federal laws, the more restrictive of the two takes precedence.
of the recipient; and (5) state and local laws do not prohibit dispensing Schedule V drugs without a prescription.
LabelingWhen drugs in Schedules II, III, and IV are dispensed, their containers must bear this label: Caution—Federal law prohibits the transfer of this drug to any person other than the patient for whom it was prescribed. The label must also indicate whether the drug belongs to Schedule II, III, or IV. The symbols C-II, C-III, and C-IV are used to indicate the Schedule.
Schedule I Drugs Schedule II Drugs Schedule III Drugs Schedule IV Drugs Schedule V Drugs
OpioidsAcetylmethadolHeroinNormethadoneMany othersPsychedelicsBufoteninDiethyltryptamineDimethyltryptamineIbogained-Lysergic acid diethylamide
(LSD)Mescaline3,4-Methylenedioxy-
methamphetamine (MDMA)
PsilocinPsilocybinCannabis DerivativesMarijuanaOthersGamma-hydroxybutyrateMethaqualone
OpioidsAlfentanilCodeineFentanylHydrocodoneHydromorphoneLevorphanolMeperidineMethadoneMorphineOpium tinctureOxycodoneOxymorphoneRemifentanilSufentanilPsychostimulantsAmphetamineCocaineDextroamphetamineMethamphetamineMethylphenidatePhenmetrazineBarbituratesAmobarbitalPentobarbitalSecobarbitalMiscellaneous DepressantsGlutethimide
OpioidsBuprenorphineParegoricCannabinoidsDronabinol (THC)StimulantsBenzphetaminePhendimetrazineBarbituratesAprobarbitalButabarbitalTalbutalThiamylalThiopentalMiscellaneous DepressantsMethyprylonAnabolic SteroidsFluoxymesteroneMethyltestosteroneNandroloneOxandroloneStanozololTestosteroneMany othersOthersKetamine
OpioidsButorphanolPentazocineStimulantsDiethylpropionFenfluramineMazindolPemolinePhentermineBarbituratesMethohexitalPhenobarbitalBenzodiazepinesAlprazolamChlordiazepoxideClonazepamClorazepateDiazepamEstazolamFlurazepamLorazepamMidazolamOxazepamPrazepamQuazepamTemazepamTriazolamBenzodiazepine-like DrugsZaleplonZolpidemMiscellaneous DepressantsChloral hydrateDichloralphenazoneEthchlorvynolEthinamateMeprobamateParaldehydeTramadol
OpioidsDiphenoxylate plus
atropineMiscellaneousPregabalin
TABLE 37.2 ■ Drug Enforcement Agency Classification of Controlled Substances

423
CHAPTER 37 Substance Use Disorders I: Basic Considerations
KEY POINTS
■ Drug abuse can be defined as drug use that is inconsistent with medical or social norms.
■ Drug abuse is a culturally defined term. What is considered abuse can vary from one culture to another and from one time to another within the same culture.
■ Substance use disorder can be defined as a chronic, relapsing brain disease characterized by compulsive drug seeking and use, despite harmful consequences. Note that physical dependence is not required for SUD to exist.
■ Tolerance is a state in which a particular drug dose elicits a smaller response than it formerly did.
■ Cross-tolerance is a state in which tolerance to one drug confers tolerance to another drug.
■ Psychologic dependence is defined as an intense subjective need for a particular psychoactive drug.
■ Physical dependence is a state in which an abstinence syndrome will occur if drug use is discontinued. Physical dependence is not the same as addiction.
■ Cross-dependence refers to the ability of one drug to support physical dependence on another drug.
■ A withdrawal syndrome is a group of signs and symptoms that occur in physically dependent individuals when they discontinue drug use.
■ Although tolerance and withdrawal are among the diagnostic criteria for substance dependence, they are neither necessary nor sufficient for a diagnosis.
■ Although physical dependence is not the same as SUD, physical dependence can certainly contribute to addictive behavior.
■ Drugs can reinforce their own use by providing pleasurable experiences, reducing the intensity of unpleasant experi-ences, and warding off a withdrawal syndrome.
■ All addictive drugs activate the brain’s dopamine reward circuit. Over time, they cause adaptive changes in the circuit that make it more and more difficult to control use.
■ Some individuals, because of psychologic or genetic factors, are more prone to SUD than others.
■ Because SUD is a chronic, relapsing illness, recovery is a prolonged process that typically requires multiple episodes of treatment.
■ The ideal goal of treatment is complete abstinence. However, treatment that substantially reduces drug use can still be considered a success.
■ Under the Controlled Substances Act, drugs in Schedule I have a high potential for abuse and no medically approved use in the United States. Drugs in Schedules II through V have progressively less abuse potential and are all medically approved.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

424
C H A P T E R
38 Substance Use Disorders II: Alcohol
Basic Pharmacology of Alcohol, p. 424Central Nervous System Effects, p. 424Other Pharmacologic Effects, p. 425Impact on Longevity, p. 426Pharmacokinetics, p. 427Tolerance, p. 428Physical Dependence, p. 428Drug Interactions, p. 428Acute Overdose, p. 428Precautions and Contraindications, p. 428Therapeutic Uses, p. 429
Alcohol Use Disorder, p. 429Drugs for Alcohol Use Disorder, p. 431
Drugs Used to Facilitate Withdrawal, p. 431Drugs Used to Maintain Abstinence, p. 431Nutritional Support, Fluid Replacement, and
Antibiotics, p. 433Key Points, p. 433Summary of Major Nursing Implications, p. 434
for serotonin (5-hydroxytryptamine [5-HT]). The depressant effects of alcohol result from binding with receptors for GABA (the principal inhibitory transmitter in the CNS) and receptors for glutamate (a major excitatory transmitter in the CNS). When alcohol binds with GABA receptors, it enhances GABA-mediated inhibition, causing widespread depression of CNS activity. When alcohol binds with glutamate receptors, it blocks glutamate-mediated excitation and thereby reduces overall CNS activity. The rewarding effects of alcohol result from binding with 5-HT3 receptors in the brain’s reward circuit. When these receptors are activated (by serotonin), they promote release of dopamine, the major transmitter of the reward system. When alcohol binds with these receptors, it enhances serotonin-mediated release of dopamine and intensifies the reward process.
The depressant effects of alcohol are dose dependent. When dosage is low, higher brain centers (cortical areas) are primarily affected. As dosage increases, more primitive brain areas (e.g., medulla) become depressed. With depression of cortical func-tion, thought processes and learned behaviors are altered, inhibitions are released, and self-restraint is replaced by increased sociability and expansiveness. Cortical depression also impairs motor function. As CNS depression deepens, reflexes diminish greatly and consciousness becomes impaired. At very high doses, alcohol produces a state of general anes-thesia. (Alcohol can’t be used for anesthesia because the doses required are close to lethal.) Table 38.1 shows the effects of alcohol as a function of blood alcohol level and indicates the brain areas involved.
Chronic EffectsWhen consumed chronically and in excess, alcohol can produce severe neurologic and psychiatric disorders. Injury to the CNS is caused by the direct actions of alcohol and by the nutritional deficiencies often seen in chronic heavy drinkers.
Two neuropsychiatric syndromes are common in patients with AUD: Wernicke’s encephalopathy and Korsakoff’s psychosis. Both disorders are caused by thiamine deficiency, which results from poor diet and alcohol-induced suppression of thiamine absorption. Wernicke’s encephalopathy is characterized by confusion, nystagmus, and abnormal ocular movements. This syndrome is readily reversible with thiamine. Korsakoff’s psy-chosis is characterized by polyneuropathy, inability to convert short-term memory into long-term memory, and confabulation (unconscious filling of gaps in memory with fabricated facts and experiences). Korsakoff’s psychosis is not reversible.
Perhaps the most dramatic effect of long-term excessive alcohol consumption is enlargement of the cerebral ventricles, presumably in response to atrophy of the cerebrum itself. These gross anatomic changes are associated with impairment of memory and intellectual function. With cessation of drinking, ventricular enlargement and cognitive deficits partially reverse, but only in some individuals.
Alcohol (ethyl alcohol, ethanol) is the most commonly used and abused psychoactive agent in the United States. Although alcohol does have some therapeutic applications, the drug is of interest primarily for its nonmedical use. When consumed in moderation, alcohol prolongs life and reduces the risk of dementia and cardiovascular disorders. Conversely, when consumed in excess, alcohol does nothing but diminish life in both quality and quantity.
In approaching our study of alcohol, we begin by discussing the basic pharmacology of alcohol, and then we discuss alcohol use disorder and the drugs employed for treatment.
BASIC PHARMACOLOGY OF ALCOHOLCentral Nervous System EffectsAcute EffectsAlcohol has two acute effects on the brain: (1) general depres-sion of central nervous system (CNS) function and (2) activation of the reward circuit.
For many years, we believed that alcohol simply dissolved into the neuronal membrane, thereby disrupting the ordered arrangement of membrane phospholipids. However, we now know that alcohol interacts with specific proteins—certain receptors, ion channels, and enzymes—that regulate neuronal excitability. Three target proteins are of particular importance, namely (1) receptors for gamma-aminobutyric acid (GABA), (2) receptors for glutamate, and (3) the 5-HT3 subset of receptors

425
CHAPTER 38 Substance Use Disorders II: Alcohol
Not all of the cardiovascular effects of alcohol are deleteri-ous: There is clear evidence that people who drink moderately (2 drinks a day or less for men, 1 drink a day or less for women) experience less ischemic stroke, coronary artery disease (CAD), myocardial infarction (MI), and heart failure than do abstainers. It is important to note, however, that heavy drinking (5 or more drinks/day) increases the risk of heart disease and stroke. Moderate drinking protects against heart disease primar-ily by raising levels of high-density lipoprotein (HDL) cho-lesterol. As discussed in Chapter 50, HDL cholesterol protects against CAD, whereas low-density lipoprotein (LDL) cholesterol promotes CAD. Of all the agents that can raise HDL cholesterol, alcohol is one of the most effective known. In addition to raising HDL cholesterol, alcohol may confer protection through four other mechanisms: decreasing platelet aggregation, decreas-ing levels of fibrinogen (the precursor of fibrin, which reinforces clots), increasing levels of tissue plasminogen activator (a clot-dissolving enzyme), and suppressing the inflammatory component of atherosclerosis. The degree of cardiovascular protection is nearly equal for beer, wine, and distilled spirits. That is, protection is determined primarily by the amount of alcohol consumed—not by the particular beverage the alcohol is in. Also, the pattern of drinking matters: protection is greater for people who drink moderately 3 or 4 days a week than for people who drink just 1 or 2 days a week. Finally, cardioprotec-tion is greatest for those with an unhealthy lifestyle: Among people who exercise, eat fruits and vegetables, and do not smoke, alcohol has little or no effect on the incidence of coro-nary events; conversely, among people who lack these behaviors, moderate alcohol intake is associated with a 50% reduction in coronary risk.
Glucose MetabolismAlcohol has several effects on glucose metabolism that may decrease the risk for type 2 diabetes. For example, alcohol raises levels of adiponectin, a compound that enhances insulin sensitivity. In addition, alcohol suppresses gluconeogenesis, blunts the postprandial rise in blood glucose, and lowers fasting levels of both glucose and insulin.
Bone HealthAlcohol increases bone mineral density, probably by increasing levels of sex hormones.
RespirationLike all other CNS depressants, alcohol depresses respiration. Respiratory depression from moderate drinking is negligible. However, when consumed in excess, alcohol can cause death by respiratory arrest. The respiratory depressant effects of alcohol are potentiated by other CNS depressants (e.g., ben-zodiazepines, opioids, barbiturates).
LiverAlcohol-induced liver damage can progress from fatty liver to hepatitis to cirrhosis, depending on the amount consumed. Acute drinking causes reversible accumulation of fat and protein in the liver. With more chronic drinking, nonviral hepatitis develops in about 90% of heavy users. In 8% to 20% of patients with chronic alcohol use disorder (AUD), hepatitis evolves into cirrhosis—a condition characterized by proliferation of fibrous tissue and destruction of liver parenchymal cells. Although various factors other than alcohol can cause cirrhosis,
Impact on Cognitive FunctionLow to moderate drinking helps preserve cognitive function in older people and may protect against development of dementia.
Effect on SleepAlthough alcohol is commonly used as a sleep aid, it actually disrupts sleep. Drinking can alter sleep cycles, decrease total sleeping time, and reduce the quality of sleep. In addition, alcohol can intensify snoring and exacerbate obstructive sleep apnea. Having a drink with dinner won’t affect sleep, but drinking late in the evening will.
Other Pharmacologic EffectsCardiovascular SystemWhen alcohol is consumed acutely and in moderate doses, cardiovascular effects are minor. The most prominent effect is dilation of cutaneous blood vessels, causing increased blood flow to the skin. By doing so, alcohol imparts a sensation of warmth—but at the same time promotes loss of heat.
Although the cardiovascular effects of moderate alcohol consumption are unremarkable, chronic and excessive consump-tion is clearly harmful. Abuse of alcohol results in direct damage to the myocardium, thereby increasing the risk of heart failure. Some investigators believe that alcohol may be the major cause of cardiomyopathy in the Western world.
In addition to damaging the heart, alcohol produces a dose-dependent elevation of blood pressure. The cause is vasocon-striction in vascular beds of skeletal muscle brought on by increased activity of the sympathetic nervous system. Estimates suggest that heavy drinking may be responsible for 10% of all cases of hypertension.
TABLE 38.1 ■ Central Nervous System Responses at Various Blood Alcohol Levelsa
aAccording to the National Institute on Alcohol Abuse and Alcoholism.
Blood Alcohol Level (%) Pharmacologic Response
Brain Area Affected
–0.50 Peripheral collapseMedulla
–0.45 Respiratory depression–0.40 Stupor, coma
Diencephalon–0.35 Apathy, inertia–0.30 Altered equilibrium Cerebellum
Double visionOccipital lobe
–0.25 Altered perception–0.20 Reduced motor skills
Slurred speechParietal lobe
–0.15 TremorsAtaxiaReduced attention
Frontal lobe
–0.10 LoquaciousnessAltered judgment
–0.05Euphoria, decreased
inhibitions

426
UNIT V Central Nervous System Drugs
alcohol spectrum disorder (FASD) is used in reference to the full range of outcomes—from mild to severe—that drinking during pregnancy can cause. In contrast, the term fetal alcohol syndrome (FAS) is reserved for the most severe cases of FASD, characterized by craniofacial malformations, growth restriction (including microcephaly), and neurodevelopmental abnormali-ties, manifesting during childhood as cognitive and social dysfunction. In addition to causing FASD and FAS, heavy drinking during pregnancy can result in stillbirth, spontaneous abortion, and giving birth to an alcohol-dependent infant.
Is light drinking safe during pregnancy? The data are unclear. Two studies published in 2010 suggest that light drinking may carry little risk. One study, conducted in the United Kingdom, found no clinically relevant behavioral or cognitive problems in 5-year-olds whose mothers consumed 1 to 2 drinks a week during pregnancy. The other study, conducted in Australia, found no link between low to moderate alcohol consumption during pregnancy and alcohol-related birth defects (ARBDs), although the same study did show that heavy drinking was associated with a fourfold increased risk of an ARBD. These results are consistent with other recent studies, which have failed to show a relationship between occasional or light drinking during pregnancy and abnormalities in newborns or older children. However, since all of these studies were observational, rather than randomized controlled trials, the negative results might be explained by confounding factors, especially educational level, income, or access to prenatal care. Furthermore, since the follow-up time for these studies was relatively short (only 5 years), the long-term effects of light drinking remain unknown. What’s the bottom line? If there is some amount of alcohol that is safe during pregnancy, that amount is very low. Accordingly, despite the studies noted, the American College of Obstetricians and Gynecologists (ACOG) continues to maintain its long-held position that no amount of alcohol can be considered safe during pregnancy. Therefore, in the interests of fetal health, all women should be advised to avoid alcohol entirely while pregnant or trying to conceive. Having said that, it is important to appreciate that a few drinks early in pregnancy are not likely to harm the fetus. Consequently, if a woman consumed a little alcohol before realizing she was pregnant, she should be reassured that the risk to the fetus—if any—is extremely low.
LactationThe concentration of alcohol in breast milk parallels the concentration in blood. Recent data indicate that drinking while breast-feeding can adversely affect the infant’s feeding and behavior.
Impact on LongevityThe effects of alcohol on life span depend on the amount consumed. Heavy drinkers have a higher mortality rate than the population at large. Causes of death include cirrhosis, respiratory disease, cancer, and fatal accidents. The risk of mortality associated with alcohol abuse increases markedly in individuals who consume 6 or more drinks a day.
Interestingly, people who consume moderate amounts of alcohol live longer than those who abstain—and combining regular exercise with moderate drinking prolongs life even more. Compared with nondrinkers, moderate drinkers have a 30% lower mortality rate, a 50% lower incidence of MI, and
alcohol abuse is unquestionably the major cause of fatal cirrhosis.
StomachExcessive use of alcohol can cause erosive gastritis. About one-third of patients with AUD have this disorder. Two mechanisms are involved. First, alcohol stimulates secretion of gastric acid. Second, when present in high concentrations, alcohol can injure the gastric mucosa directly.
KidneyAlcohol is a diuretic. It promotes urine formation by inhibiting the release of antidiuretic hormone (ADH) from the pituitary. Because ADH acts on the kidney to promote water reabsorption, thereby decreasing urine formation, a reduction in circulating ADH will increase urine production.
PancreasApproximately 35% of cases of acute pancreatitis can be attributed to alcohol, making alcohol the second most common cause of the disorder. Flare-ups typically occur after a bout of heavy drinking. Only 10% of patients with AUD develop pancreatitis, and then only after years of overindulgence.
Sexual FunctionAlcohol has both psychologic and physiologic effects related to human sexual behavior. Although alcohol is not exactly an aphrodisiac, its ability to release inhibitions has been known to motivate sexual activity. Ironically, the physiologic effects of alcohol may frustrate attempts at consummating the activity that alcohol inspired: Objective measurements in males and females show that alcohol significantly decreases our physi-ologic capacity for sexual responsiveness. In males, long-term use of alcohol may induce feminization. Symptoms include testicular atrophy, impotence, sterility, and breast enlargement.
CancerAlcohol—even in moderate amounts—is associated with an increased risk of several common cancers. Among these are cancers of the breast, liver, rectum, and aerodigestive tract, which includes the lips, tongue, mouth, nose, throat, vocal cords, and portions of the esophagus and trachea. According to a 2016 study, alcohol consumption is linked to seven cancers. The fraction attributable to alcohol is highest for aerodigestive tract cancers. People who drink more than 50 gm of alcohol daily (12 oz beer, 5 oz wine, or 1.25 oz of hard alcohol are all about 14 gm) are at 4 to 7 times increased risk of developing these types of cancers. The risk is somewhat lower for liver, breast, and colorectal cancer (1.5 times increased risk). The bottom line? Data suggest that, regarding cancer risk, no amount of alcohol can be considered safe—although risk is lowest with moderate drinking (2 drinks or less a day for men and 1 drink or less a day for women).
PregnancyEffects of alcohol on the developing fetus are dose-dependent. The risk of fetal injury is greatest with heavy drinking and much lower with light drinking. Is there some low level of drinking that is completely safe? We don’t know.
Fetal alcohol exposure can cause structural and functional abnormalities, ranging from mild neurobehavioral deficits to facial malformation and developmental delay. The term fetal

427
CHAPTER 38 Substance Use Disorders II: Alcohol
Because alcohol is metabolized at a slow and constant rate, there is a limit to how much alcohol one can consume without having the drug accumulate. For practical purposes, that limit is about 1 drink per hour. Consumption of more than 1 drink per hour—be that drink beer, wine, straight whiskey, or a cocktail—will result in alcohol accumulation.
The information in Table 38.2 helps explain why we can’t metabolize more than 1 drink’s worth of alcohol per hour. Beer, wine, and whiskey differ from one another with respect to alcohol concentration and usual serving size. However, despite these differences, it turns out that the average can of beer, the average glass of wine, and the average shot of whiskey all contain the same amount of alcohol—namely, 18 mL (0.6 oz). Since the liver can metabolize about 15 mL of alcohol per hour and since the average alcoholic drink contains 18 mL of alcohol, 1 drink contains just about the amount of alcohol that the liver can comfortably process each hour. Consumption of more than 1 drink per hour will overwhelm the capacity of the liver for alcohol metabolism, and therefore alcohol will accumulate.
When used on a regular basis, alcohol induces hepatic drug-metabolizing enzymes, thereby increasing the rate of its own metabolism and that of other drugs. As a result, individuals who consume alcohol routinely in high amounts can metabolize the drug faster than people who drink occasionally and moderately.
Males and females differ with respect to activity of alcohol dehydrogenase in the stomach. Specifically, women have lower activity than men. As a result, gastric metabolism of alcohol is significantly less in women. This difference partly explains why women achieve higher blood alcohol levels than men after consuming the same number of drinks.
a 59% lower incidence of heart failure. According to a study by the American Medical Association, if all Americans were to give up drinking, deaths from heart disease would increase by 81,000 a year. Hence, for people who already are moderate drinkers, continued moderate drinking would seem beneficial. Conversely, despite the apparent benefits of drinking—and the apparent health disadvantage of abstinence—no one is recom-mending that abstainers take up drinking. Furthermore, when the risks of alcohol outweigh any possible benefits—such as in pregnancy—then alcohol consumption should be avoided entirely.
How does alcohol prolong life? In large part by reducing cardiovascular disease. For people who drink red wine, a small benefit may come from resveratrol, although the amount present appears too small to have a significant effect (see Chapter 108).
PharmacokineticsAbsorptionAlcohol is absorbed from the stomach and small intestine. About 20% of ingested alcohol is absorbed from the stomach. Gastric absorption is relatively slow and is delayed even further by the presence of food. Milk is especially effective at delaying absorption. Absorption from the small intestine is rapid and largely independent of food; about 80% of ingested alcohol is absorbed from this site. Because most alcohol is absorbed from the small intestine, gastric emptying time is a major determinant of individual variation in alcohol absorption.
DistributionBecause alcohol is both nonionic and water soluble, it distributes well to all tissues and body fluids. The drug crosses the blood-brain barrier with ease, allowing alcohol in the brain to equilibrate rapidly with alcohol in the blood. Alcohol also crosses the placenta and hence can affect the developing fetus.
Distribution in body water partly explains why women are more sensitive to alcohol than men. As a rule, women have a lower percentage of body water than men. Hence, when a woman drinks, the alcohol is diluted in a smaller volume of water, causing the concentration of alcohol in tissues and fluids to be relatively high, which causes the effects of alcohol to be more intense.
MetabolismAlcohol is metabolized in both the liver and stomach. The liver is the primary site. The process begins with conversion of alcohol to acetaldehyde, a reaction catalyzed by alcohol dehydrogenase. This reaction is slow and puts a limit on the rate at which alcohol can be inactivated. Once formed, acet-aldehyde undergoes rapid conversion by aldehyde dehydro-genase to acetic acid. Through a series of reactions, acetic acid is then used to synthesize cholesterol, fatty acids, and other compounds.
The kinetics of alcohol metabolism differ from those of most other drugs. With most drugs, as plasma drug levels rise, the amount of drug metabolized per unit of time increases too. This is not true for alcohol: As the alcohol content of blood increases, there is almost no change in the speed of alcohol breakdown. That is, alcohol is metabolized at a relatively constant rate—regardless of how much alcohol is present. The average rate at which individuals can metabolize alcohol is about 15 mL (0.5 oz) per hour.
Wine Beer Whiskey
Usual serving 1 glass 1 can or bottle 1 shotServing size 150 mL
(5 oz)360 mL(12 oz)
45 mL(1.5 oz)
Alcohol concentration 12%a 5%b 40%c
Alcohol per serving 18 mLd
(0.6 oz)18 mLe
(0.6 oz)18 mLf
(0.6 oz)
TABLE 38.2 ■ Alcohol Content of Beer, Wine, and Whiskey
aThe alcohol content of wine varies from 8% to 20%; typical table wines contain 12%.bThe alcohol content of beer varies: 5% alcohol is typical of American premium beers; cheaper American beers and light beers have less alcohol (2.4% to 5%); and imported or craft beers may have more alcohol (6% to 10%). Beer sold in Europe may have 7% to 8% alcohol.cWhiskeys and other distilled spirits (e.g., rum, vodka, gin) are usually 80 proof (40% alcohol) but may also be 100 proof (50% alcohol).dThe alcohol in a 5-ounce glass of wine varies from 12 to 30 mL, depending on the alcohol concentration in the wine. Wine with 12% alcohol has 18 mL of alcohol per 5-ounce glass.eThe alcohol in a 12-ounce can of beer varies from 9 to 29 mL, depending on the alcohol concentration in the beer. Beer with 5% alcohol has 18 mL per 12-ounce can.fThe alcohol in a 1.5-ounce shot of whiskey can be either 18 or 22.5 mL, depending on the proof of the whiskey. Eighty-proof whiskey has 18 mL of alcohol per 1.5-ounce serving.

428
UNIT V Central Nervous System Drugs
interaction between alcohol and acetaminophen is discussed further in Chapter 71.Blood Levels of Alcohol
Since alcohol in the brain rapidly equilibrates with alcohol in the blood, blood levels of alcohol are predictive of CNS effects. The behavioral effects associated with specific blood levels are shown in Table 38.1. The earliest effects (euphoria, reduced inhibitions, increased confidence) are seen when blood alcohol content is about 0.05%. As blood alcohol rises, intoxication becomes more intense. When blood alcohol exceeds 0.4%, there is a substantial risk of respiratory depression, peripheral collapse, and death. In the United States, a level of 0.08% defines intoxication.
ToleranceChronic consumption of alcohol produces tolerance. As a result, to alter consciousness, people who drink on a regular basis require larger amounts of alcohol than people who drink occa-sionally. Tolerance to alcohol confers cross-tolerance to general anesthetics, barbiturates, and other general CNS depressants. However, no cross-tolerance develops to opioids. Tolerance subsides within a few weeks following drinking cessation.
Although tolerance develops to many of the effects of alcohol, very little tolerance develops to respiratory depression. Consequently, the lethal dose of alcohol for chronic heavy drinkers is not much bigger than the lethal dose for nondrinkers. Individuals with AUD may tolerate blood alcohol levels as high as 0.4% (5 times the amount defined by law as intoxicating) with no marked reduction in consciousness. However, if blood levels rise only slightly above this level, death may ensue.
Physical DependenceChronic use of alcohol produces physical dependence. If alcohol is withdrawn abruptly, an abstinence syndrome will result. The intensity of the abstinence syndrome is proportional to the degree of physical dependence. Individuals who are physi-cally dependent on alcohol show cross-dependence with other general CNS depressants (e.g., barbiturates, chloral hydrate, benzodiazepines) but not with opioids. The alcohol withdrawal syndrome and its management are discussed in detail later in this chapter.
Drug InteractionsCNS DepressantsThe CNS effects of alcohol are additive with those of other CNS depressants (e.g., barbiturates, benzodiazepines, opioids). Consumption of alcohol with other CNS depressants intensifies the psychologic and physiologic manifestations of CNS depres-sion and greatly increases the risk of death from respiratory depression.
Nonsteroidal Anti-Inflammatory DrugsLike alcohol, aspirin, ibuprofen, and other nonsteroidal anti-inflammatory drugs (NSAIDs) can injure the GI mucosa. The combined effects of alcohol and NSAIDs can result in significant gastric bleeding.
AcetaminophenThe combination of acetaminophen [Tylenol, others] with alcohol poses a risk of potentially fatal liver injury. The
Safety Alert
ACETAMINOPHEN AND ALCOHOLThere is evidence that relatively modest alcohol consumption (2 to 4 drinks a day) can cause fatal liver damage when combined with acetaminophen taken in normal therapeutic doses. Accordingly, some authorities recommend that people who drink take no more than 2 gm of acetaminophen a day (i.e., half the normal dosage).
DisulfiramThe combination of alcohol with disulfiram [Antabuse] can cause a variety of adverse effects, some of which are dangerous. These effects, and the use of disulfiram to maintain abstinence, are discussed later.
Antihypertensive DrugsSince alcohol raises blood pressure, it tends to counteract the effects of antihypertensive medications. However, elevation of blood pressure is significant only when alcohol dosage is high. Conversely, when the dosage is low, alcohol may actually help: Among hypertensive men, light to moderate alcohol consumption is associated with a reduced risk for both car-diovascular mortality and all-cause mortality.
Acute OverdoseAcute overdose produces vomiting, coma, pronounced hypoten-sion, and respiratory depression. The combination of vomiting and unconsciousness can result in aspiration, which in turn can result in pulmonary obstruction and pneumonia. Alcohol-induced hypotension results from a direct effect on peripheral blood vessels and cannot be corrected with vasoconstrictors (e.g., epinephrine). Hypotension can lead to renal failure (secondary to compromised renal blood flow) and cardiovascular shock, a common cause of alcohol-related death. Although death can also result from respiratory depression, this is not the usual cause.
Because symptoms of acute alcohol poisoning can mimic symptoms of other pathologies (e.g., diabetic coma, skull fracture), a definitive diagnosis may not be possible without measuring alcohol in the blood, urine, or expired air. The smell of “alcohol” on the breath is not a reliable means of diagnosis, since the breath odors we associate with alcohol are due to impurities in alcoholic beverages—and not to alcohol itself. Hence, these odors may or may not be present.
Alcohol poisoning is treated like poisoning with all other general CNS depressants. Details of management are discussed in Chapter 34. Alcohol can be removed from the body by gastric lavage and dialysis. Stimulants (e.g., caffeine) should not be given.
Precautions and ContraindicationsAlcohol can injure the GI mucosa and should not be consumed by persons with peptic ulcer disease. Alcohol is harmful to

429
CHAPTER 38 Substance Use Disorders II: Alcohol
AUD. Alcohol abuse during pregnancy can result in FASD (including FAS), stillbirth, and spontaneous abortion. Lastly, chronic alcohol abuse is harmful to the body; consequences include liver disease, cardiomyopathy, and brain damage—not to mention injury and death from accidents.
Chronic alcohol consumption produces substantial tolerance. Tolerance is both pharmacokinetic (accelerated alcohol metabolism) and pharmacodynamic. Pharmacodynamic toler-ance is evidenced by an increase in the blood alcohol level required to produce intoxication. Individuals with AUD may tolerate blood alcohol levels of 200 to 400 mg/dL—2.5 to 5 times the level that defines legal intoxication—with no marked reduction in consciousness. It should be noted, however, that very little tolerance develops to respiratory depression. Hence, as the person with AUD consumes increasing amounts in an effort to feel good, the risk of death from respiratory arrest gets increasingly high. Cross-tolerance exists with general anesthetics and other CNS depressants, but not with opioids.
Chronic use of alcohol produces physical dependence, and abrupt withdrawal produces an abstinence syndrome. When the degree of physical dependence is low, withdrawal symptoms are mild (disturbed sleep, weakness, nausea, anxiety, mild tremors) and last less than a day. In contrast, when the degree of dependence is high, withdrawal symptoms can be severe. Initial symptoms appear 12 to 72 hours after the last drink and continue 5 to 7 days. Early manifestations include cramps, vomiting, hallucinations, and intense tremors; heart rate, blood pressure, and temperature may rise, and tonic-clonic seizures may develop. As the syndrome progresses, disorientation and loss of insight occur. A few individuals with AUD (fewer than 5%) experience delirium tremens (severe persecutory hallucina-tions). Hallucinations can be so vivid and lifelike that people often can’t distinguish them from reality. In extreme cases,
the liver and should not be used by individuals with liver disease. Alcohol should be avoided during pregnancy, owing to the risk of FASD (including FAS), stillbirth, spontaneous abortion, and neurodevelopmental abnormalities.
Alcohol must be used with caution by patients with epilepsy. During alcohol use, the CNS is depressed. When alcohol consumption ceases, the CNS undergoes rebound excitation; seizures can result.
Alcohol causes a dose-related increase in the risk of breast cancer. All women—and especially those at high risk—should minimize alcohol consumption. Alcohol also increases the risk of cancer of the liver, rectum, and aerodigestive tract.
Alcohol can cause serious adverse effects if combined with CNS depressants, NSAIDs, acetaminophen, vasodilators, and disulfiram. These combinations should be avoided.
Therapeutic UsesAlthough our emphasis has been on the nonmedical use of alcohol, it should be remembered that alcohol does have therapeutic applications.
TopicalTopical alcohol can be an effective skin disinfectant.
OralOral alcohol is frequently used as self-medication for insomnia—although it can actually disrupt sleep.
Local InjectionInjection of alcohol in the vicinity of nerves produces nerve block. This technique can relieve pain from trigeminal neuralgia, inoperable carcinoma, and other causes.
ALCOHOL USE DISORDER (AUD)Alcohol use disorder is a chronic, relapsing disorder character-ized by impaired control over drinking, preoccupation with alcohol consumption, use of alcohol despite awareness of adverse consequences, and distortions in thinking, especially as evidenced by denial of a drinking problem. The development and manifestations of AUD are influenced by genetic, psycho-social, and environmental factors. The disease is progressive and often fatal. In the United States, about 6% of the adult population are diagnosed with AUD.
Alcohol use disorder is defined as a problematic pattern of alcohol use leading to clinically significant impairment or distress occurring within a 12-month period. Manifestations of alcohol use disorder can include recurrent alcohol use in situations in which it is physically hazardous; recurrent use resulting in a failure to fulfill major role obligations at work, school, or home; and spending a great deal of time in activities necessary to obtain alcohol, use alcohol, or recover from alcohol.
In the United States, misuse of alcohol is responsible for 6 million nonfatal injuries and 85,000 deaths each year. Causes of death range from liver disease to automobile wrecks. Fully 50% of all fatal highway crashes are alcohol related. Among teens, alcohol-related crashes are the leading cause of death. Alcohol also causes industrial accidents and is responsible for 40% of industrial fatalities.
Alcohol abuse is a major public health problem, and its con-sequences are numerous. AUD produces psychologic derange-ments, including anxiety, depression, and suicidal ideation. Malnutrition, secondary to inadequate diet and malabsorption, is common. Poor work performance and disruption of family life reflect the social deterioration suffered by individuals with
Alcohol
Life Stage Patient Care Concerns
Infants See “Breast-feeding women,” below.Children/
adolescentsAdolescents are more sensitive to alcohol-
induced memory impairment than adults, but less sensitive to the motor effects of alcohol. Alcohol exposure during adolescence affects brain functioning during adulthood.
Pregnant women
The use of alcohol while pregnant can cause structural and functional abnormalities in the fetus. These abnormalities are termed fetal alcohol spectrum disorder (FASD). It is recommended that pregnant women abstain from alcohol.
Breast-feeding women
The concentration of alcohol in breast milk parallels the concentration in blood. Breast-feeding while consuming alcohol can affect infant behavior.
Older adults Aging lowers the body’s tolerance for alcohol. Many older adults may not metabolize alcohol as efficiently as younger adults. Many older adults take medications that interact with alcohol.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN

430
UNIT V Central Nervous System Drugs
• Advise and assist (brief intervention).• At follow-up: continue support.
This process is founded in part on two lines of evidence. First, we can identify people who misuse alcohol with an easily administered questionnaire, such as the AUDIT (Table 38.3).a Second, for many people, alcohol consumption can be reduced through brief interventions, such as offering feedback and advice about drinking and about setting goals. Long-term follow-up studies have shown that these simple interventions can decrease hospitalization and lower mortality rates. The guide is available at www.niaaa.nih.gov/guide.
alcohol withdrawal can result in cardiovascular collapse and death. Drugs used to ease withdrawal are discussed later in this chapter.
In 2008, the National Institute on Alcohol Abuse and Alcoholism (NIAAA) updated its document titled Helping Patients Who Drink Too Much: A Clinician’s Guide, which contains clear, concise information on screening, counseling, and treatment of alcohol use disorders. By following this guide, clinicians can help reduce morbidity and mortality among people who drink more than is safe, defined as more than 4 drinks in a day (or 14/week) for men, or more than 3 drinks in a day (or 7/week) for women. Helping patients involves four simple steps:
• Ask about alcohol use.• Assess for alcohol use disorders using the Alcohol Use
Disorders Identification Test (AUDIT).
aRapid screening can be accomplished with a single question: How many times in the past year have you had x or more drinks in a day? (x = 5 for men and 4 for women). A positive response is defined as 1 or more. If this simple screen is positive, a more detailed diagnostic interview is indicated.
0 1 2 3 4 Score
1. How often do you have a drink containing alcohol?
Never Monthly or less 2 to 4 times a month
2 to 3 times a week
4 or more times a week
2. How many drinks containing alcohol do you have on a typical day when you are drinking?
1 or 2 3 or 4 5 or 6 7 to 9 10 or more
3. How often do you have 5 or more drinks on one occasion?
Never Less than monthly
Monthly Weekly Daily or almost daily
4. How often during the last year have you found that you were not able to stop drinking once you had started?
Never Less than monthly
Monthly Weekly Daily or almost daily
5. How often during the last year have you failed to do what was normally expected of you because of drinking?
Never Less than monthly
Monthly Weekly Daily or almost daily
6. How often during the last year have you needed a first drink in the morning to get yourself going after a heavy drinking session?
Never Less than monthly
Monthly Weekly Daily or almost daily
7. How often during the last year have you had a feeling of guilt or remorse after drinking?
Never Less than monthly
Monthly Weekly Daily or almost daily
8. How often during the last year have you been unable to remember what happened the night before because of your drinking?
Never Less than monthly
Monthly Weekly Daily or almost daily
9. Have you or someone else been injured because of your drinking?
No Yes, but not in the last year
Yes, during the last year
10. Has a relative, friend, doctor, or other healthcare worker been concerned about your drinking or suggested you cut down?
No Yes, but not in the last year
Yes, during the last year
Total Score
TABLE 38.3 ■ Screening Instrument: The Alcohol Use Disorders Identification Test (AUDIT)
Instructions to patient: Circle the option that best describes your answer to each question.Scoring: Record the score (0, 1, 2, 3, or 4) for each response in the blank box at the end of each line, and then add up the total score. The maximum possible is 40. A total score of 8 or more for men up to age 60 (or 4 or more for women, adolescents, and men over 60) is considered a positive screen. For patients with totals near the cut-points, clinicians may wish to examine individual responses to questions and clarify them during the clinical examination.Reprinted with permission from the World Health Organization. To reflect standard drink sizes in the United States, the number of drinks in question 3 was changed from 6 to 5.

431
CHAPTER 38 Substance Use Disorders II: Alcohol
stabilize vital signs, reduce symptom intensity, and decrease the risk of seizures and delirium tremens. Although all ben-zodiazepines are effective, agents with longer half-lives are generally preferred because they provide the greatest protection against seizures and breakthrough symptoms. The benzodiaz-epines employed most often are chlordiazepoxide [Librium, others], clorazepate [Tranxene], oxazepam (generic only), and lorazepam [Ativan]. Traditionally, benzodiazepines have been administered around-the-clock on a fixed schedule. However, PRN administration (in response to symptoms) is just as effective and permits speedier withdrawal.
Adjuncts to BenzodiazepinesCombining a benzodiazepine with another drug may improve withdrawal outcome. Agents that have been tried include carbamazepine (an antiepileptic drug), clonidine (an alpha2-adrenergic agonist), and atenolol and propranolol (beta-adrenergic blockers). Carbamazepine may reduce withdrawal symptoms and the risk of seizures. Clonidine and the beta blockers reduce the autonomic component of withdrawal symptoms. In addition, the beta blockers may improve vital signs and decrease craving. It should be stressed, however, that these drugs are not very effective as monotherapy. Hence, they should be viewed only as adjuncts to benzodiazepines—not as substitutes.
Drugs Used to Maintain AbstinenceOnce detoxification has been accomplished, the goal is to prevent—or at least minimize—future drinking. The ideal goal is complete abstinence. However, if drinking must resume, keeping it to a minimum is still beneficial, since doing so will reduce alcohol-related morbidity.
In trials of drugs used to maintain abstinence, several parameters are used to measure efficacy. These include:
• Proportion of patients who maintain complete abstinence• Days to relapse• Number of drinking days• Number of drinks per drinking day
In the United States, only three drugs—disulfiram, naltrexone, and acamprosate—are approved for maintaining abstinence. Disulfiram works by causing an unpleasant reaction if alcohol is consumed. Naltrexone blocks the pleasurable effects of alcohol and decreases craving. Acamprosate reduces some of the unpleasant feelings (e.g., tension, dysphoria, anxiety) brought on by alcohol abstinence. Of the three drugs, naltrexone appears most effective. However, even with this agent, benefits are modest.
Owing to the risk of relapse, prolonged treatment is needed. The minimum duration is 3 months. However, continuing for a year or more is not unreasonable. If the first drug fails, clini-cians often try a different one.
Disulfiram Aversion TherapyTherapeutic Effects. Disulfiram [Antabuse] helps individu-
als with AUD avoid drinking by causing unpleasant effects if alcohol is ingested. Disulfiram has no applications outside the treatment of AUD.
Although disulfiram has been employed for decades, its efficacy is only moderate. In clinical trials, there is emerging evidence that the drug may be only slightly better than placebo
To help individuals who drink too much, the NIAAA created an interactive web site, located at rethinkingdrinking.niaaa .nih.gov. Content includes tools to identify and manage problem drinking, plus a calculator for determining the alcohol content of various beverages.
DRUGS FOR ALCOHOL USE DISORDERIn the United States, about 1 million people with AUD seek treatment every year. Although the success rate is discouraging—nearly 50% relapse during the first few months—treatment should nonetheless be tried. The objective is to modify drinking patterns (i.e., to reduce or completely eliminate alcohol con-sumption). Drugs can help in two ways. First, they can facilitate withdrawal. Second, they can help maintain abstinence once withdrawal has been accomplished.
Drugs Used to Facilitate WithdrawalManagement of withdrawal depends on the degree of depen-dence. When dependence is mild, withdrawal can be accom-plished on an outpatient basis without drugs. However, when dependence is great, withdrawal carries a risk of death. Accordingly, hospitalization and drug therapy are indicated. The goals of management are to minimize symptoms of withdrawal, prevent seizures and delirium tremens, and facilitate transition to a program for maintaining abstinence. In theory, any drug that has cross-dependence with alcohol (i.e., any of the general CNS depressants) should be effective. However, in actual practice, benzodiazepines are the drugs of choice. The benefits of benzodiazepines and other drugs used during withdrawal are shown in Table 38.4.
BenzodiazepinesOf the drugs used to facilitate alcohol withdrawal, benzodi-azepines are the most effective. Furthermore, they are safe. In patients with severe alcohol dependence, benzodiazepines can
Drug Benefit During Withdrawal
BENZODIAZEPINES
ChlordiazepoxideClorazepateDiazepamLorazepamOxazepam
Decrease withdrawal symptoms; stabilize vital signs; prevent seizures and delirium tremens
BETA-ADRENERGIC BLOCKERS
AtenololPropranolol
Improve vital signs; decrease craving; decrease autonomic component of withdrawal symptoms
CENTRAL ALPHA2-ADRENERGIC AGONIST
Clonidine Decreases autonomic component of withdrawal symptoms
ANTIEPILEPTIC DRUG
Carbamazepine Decreases withdrawal symptoms; prevents seizures
TABLE 38.4 ■ Drugs Used to Facilitate Alcohol Withdrawal

432
UNIT V Central Nervous System Drugs
NaltrexoneNaltrexone [ReVia, Vivitrol] is a pure opioid antagonist that decreases craving for alcohol and blocks alcohol’s reinforcing (pleasurable) effects. Patients with AUD report that naltrexone decreases their “high.” Although the mechanism underlying these effects is uncertain, one possibility is blockade of dopamine release secondary to blockade of opioid receptors. Naltrexone is generally well tolerated. Nausea is the most common adverse effect, followed by headache, anxiety, and sedation. Because naltrexone is an opioid antagonist, the drug will precipitate withdrawal if given to a patient who is opioid dependent. Conversely, if a patient taking naltrexone needs emergency treatment with an opioid analgesic, high doses of the opioid will be required.
Naltrexone was approved for AUD on the basis of random-ized clinical trials that combined extensive counseling along with the drug. In these trials, naltrexone cut the relapse rate by 50%. Compared with patients taking placebo, those taking naltrexone reported less craving for alcohol, fewer days drinking, fewer drinks per occasion, and reduced severity of alcohol-related problems. In contrast to the original trials, a more recent trial, conducted by the U.S. Department of Veterans Affairs, failed to show any benefit of naltrexone in maintaining absti-nence. Why did naltrexone work in the original trials but not in the more recent one? The most likely reason is that the subjects in the two trials were very different: The veterans with AUD suffered from chronic AUD, had little or no social support, and received minimal counseling during the trial, whereas subjects in the earlier studies were younger, had good support systems, and received extensive counseling along with naltrexone. Hence, the new study does not prove that naltrexone doesn’t work. Rather, it proves only that naltrexone doesn’t work for all individuals and that it doesn’t work in the absence of adequate counseling.
Naltrexone is available in two formulations: 50-mg tablets [ReVia] for oral use and a 380-mg depot formulation [Vivitrol] for IM injection. The oral dosage is 50 mg once a day. The IM dosage is 380 mg once a month. Depot naltrexone is especially good when patient adherence is a concern. As with disulfiram, patients must stop drinking before starting naltrexone.
The basic pharmacology of naltrexone is discussed in Chapter 28.
AcamprosateTherapeutic Use. Acamprosate is approved for maintaining
abstinence in patients with alcohol dependence following detoxification. Benefits derive from reducing unpleasant feelings (e.g., tension, dysphoria, anxiety) brought on by abstinence. This effect contrasts with the effects of disulfiram (which makes drinking unpleasant) and naltrexone (which blocks the pleasant feelings that alcohol can cause). Acamprosate should be used only as part of a comprehensive management program that includes psychosocial support.
In clinical trials, acamprosate was moderately effective. Compared with patients taking placebo, those taking acam-prosate abstained from their first drink longer, had greater rates of complete abstinence, and were abstinent for more total days. Benefits may be related to the degree of alcohol dependence: The greater the dependence, the more likely that acamprosate will help. Among patients who lack psychosocial support, little or no benefit is seen.
at maintaining long-term abstinence; however, long-term studies have not been completed. Disulfiram does decrease the fre-quency of drinking after relapse has occurred—presumably because of the unpleasant reaction that the patient is now familiar with. Supervised administration of disulfiram may be more effective than when patients self-administer the drug.
Mechanism of Action. Disulfiram disrupts alcohol metabolism by causing irreversible inhibition of aldehyde dehydrogenase, the enzyme that converts acetaldehyde to acetic acid. As a result, if alcohol is ingested, acetaldehyde will accumulate to toxic levels, producing unpleasant and potentially harmful effects.
Pharmacologic Effects. The constellation of effects caused by alcohol plus disulfiram is referred to as acetaldehyde syndrome, a potentially dangerous event. In its “mild” form, the syndrome manifests as nausea, copious vomiting, flushing, palpitations, headache, sweating, thirst, chest pain, weakness, blurred vision, and hypotension; blood pressure may ultimately decline to shock levels. This reaction, which may last from 30 minutes to several hours, can be brought on by consuming as little as 7 mL of alcohol.
In its most severe manifestation, the acetaldehyde syndrome is life threatening. Possible reactions include marked respiratory depression, cardiovascular collapse, cardiac dysrhythmias, MI, acute congestive heart failure, convulsions, and death. Clearly, the acetaldehyde syndrome is not simply unpleasant; this syndrome can be extremely hazardous and must be avoided.
In the absence of alcohol, disulfiram rarely causes significant effects. Drowsiness and skin eruptions may occur during initial use, but they diminish with time.
Patient Selection. Owing to the severity of the acetalde-hyde syndrome, candidates must be carefully chosen. People with AUD who lack the determination to stop drinking should not receive disulfiram. In other words, disulfiram must not be administered to individuals who are likely to attempt drinking while undergoing treatment.
Patient Education. Patient education is an extremely important component of therapy. Patients must be thoroughly informed about the potential hazards of treatment. Patients should be made aware that the effects of disulfiram will persist about 2 weeks after the last dose, and hence continued absti-nence is necessary. Individuals using disulfiram should be encouraged to carry identification indicating their status.
Safety Alert
DISULFIRAMPatients must be made aware that consuming any alcohol while taking disulfiram may produce a severe, potentially fatal, reaction. Patients must be warned to avoid all forms of alcohol, including alcohol found in sauces and cough syrups, and alcohol applied to the skin in aftershave lotions, colognes, and liniments.
Preparations, Dosage, and Administration. Disulfiram [Antabuse] is supplied in 250- and 500-mg tablets. At least 12 hours must elapse between the patient’s last drink and starting treatment. The initial dosage is 500 mg once daily for 1 to 2 weeks. Maintenance dosages range from 125 to 500 mg/day, usually taken as a single dose in the morning. Therapy may last months or even years.

433
CHAPTER 38 Substance Use Disorders II: Alcohol
since alternatives are available. Acamprosate has no potential for dependence or abuse, and appears devoid of significant drug interactions.
Preparations, Dosage, and Administration. Acamprosate is available in 333-mg delayed-release tablets. The recommended dosage is 2 tablets (666 mg) 3 times a day, taken with meals. (The reason for administration with meals is to promote compliance—not to influence absorption or GI effects.) Dosing should start immediately after detoxification is over and should continue even if relapse occurs. For patients with mild renal impairment, the recommended initial dosage is 333 mg 3 times a day. If the patient has severe renal impairment, acamprosate should not be used.
Nutritional Support, Fluid Replacement, and AntibioticsMalnutrition is a common problem in the patient with chronic AUD. The underlying causes are poor diet and malabsorption of nutrients and vitamins. Malabsorption results from alcohol-induced damage to the GI mucosa. Poor diet occurs in part because individuals with AUD meet up to 50% of their caloric needs with alcohol and therefore consume suboptimal amounts of foods with high nutritional value. Because of their poor nutritional state, these patients are in need of fat, protein, and vitamins. The B vitamins (thiamine, folic acid, cyanocobalamin) are especially needed. To correct nutritional deficiencies, a program of dietary modification and vitamin supplements should be implemented.
Individuals with AUD frequently require fluid replacement therapy. Fluids are needed to replace fluids lost because of gastritis or because of vomiting associated with withdrawal.
Mechanism of Action. Just how acamprosate works is unknown. One theory suggests that acamprosate enhances inhibitory neurotransmission (mediated by GABA) and sup-presses excitatory neurotransmission (mediated by glutamate), and thereby restores a balance between these transmitter systems. When given to alcohol-dependent animals, the drug reduces voluntary alcohol intake. Acamprosate is devoid of direct anxiolytic, anticonvulsant, and antidepressant activity, and does not cause alcohol aversion.
Pharmacokinetics. Acamprosate is administered orally, and bioavailability is low (11%). Food reduces absorption even further. The drug has a long half-life (20 to 33 hours), and hence about 5 days are required for plasma levels to reach a plateau. Acamprosate does not undergo metabolism, and is excreted unchanged in the urine.
Adverse Effects and Drug Interactions. Acamprosate is generally well tolerated. With most adverse effects, the incidence is no greater than with placebo. The principal excep-tion is diarrhea, which occurs in 17% of acamprosate users compared with 10% of those taking placebo. Reports of suicide-related events (suicidal ideation, suicide attempts, completed suicide) are rare, but more common than with placebo. Acamprosate can cause fetal malformations in animals (at doses close to those used by humans). Accordingly, it would seem prudent to avoid this drug during pregnancy, especially
KEY POINTS
■ Alcohol is generally beneficial when consumed in modera-tion and always detrimental when consumed in excess.
■ Alcohol causes CNS depression by enhancing the depressant effects of GABA and reducing the excitatory effects of glutamate.
■ As blood levels of alcohol rise, CNS depression progresses from cortical areas to more primitive brain areas (e.g., medulla).
■ Long-term, excessive drinking reduces the size of the cerebrum.
■ Alcohol produces a dose-dependent increase in blood pressure.
■ Moderate drinking is defined as 2 drinks per day or less for men and 1 drink per day or less for women.
■ Moderate drinking significantly reduces the risk of CAD, MI, ischemic stroke, and heart failure—primarily by raising HDL cholesterol and partly by suppressing platelet aggregation, reducing fibrin formation, enhancing fibri-nolysis, and suppressing the inflammatory component of atherosclerosis.
■ Excessive drinking causes direct damage to the myocardium.■ Like all other CNS depressants, alcohol depresses
respiration.■ Chronic, heavy drinking can cause hepatitis and cirrhosis.
People with liver disease should avoid alcohol.■ Heavy drinking can cause erosive gastritis.■ Alcohol is a diuretic.■ Alcohol, even in low doses, increases the risk of breast
cancer, as well as cancers of the liver, rectum, and aerodiges-tive tract.
■ Excessive drinkers die younger than the population at large.■ Because of the cardioprotective effects of alcohol, moderate
drinkers live longer than those who abstain.■ Alcohol dehydrogenase is the rate-limiting enzyme in
alcohol metabolism.■ Alcohol is metabolized at a constant rate, regardless of
how high blood levels rise. In contrast, the rate of metabo-lism of most drugs increases as their blood levels rise.
■ Most people can metabolize about 1 drink per hour— be it beer, wine, straight whiskey, or a cocktail. Consuming more than 1 drink per hour causes alcohol to accumulate.
■ Chronic drinking produces tolerance to many of alcohol’s effects—but not to respiratory depression.
■ Tolerance to alcohol confers cross-tolerance to general anes-thetics, barbiturates, and other general CNS depressants— but not to opioids.
■ The CNS-depressant effects of alcohol are additive with those of other CNS depressants.
■ The combined effects of alcohol and NSAIDs can cause significant gastric bleeding. People with peptic ulcer disease should avoid alcohol.
■ The combination of alcohol and acetaminophen can cause fatal hepatic failure.
■ Alcohol use during pregnancy can result in FASD (including FAS), stillbirth, and spontaneous abortion. Women who are pregnant or trying to conceive should not drink.
■ Benzodiazepines (e.g., chlordiazepoxide, diazepam, lorazepam) are drugs of choice for facilitating withdrawal in alcohol-dependent individuals. Benzodiazepines suppress symptoms because of cross-dependence with alcohol.
Continued

434
UNIT V Central Nervous System Drugs
■ Three drugs are approved for maintaining alcohol absti-nence: disulfiram, naltrexone, and acamprosate.
■ Disulfiram blocks aldehyde dehydrogenase. As a result, if alcohol is consumed, acetaldehyde will accumulate, thereby causing a host of unpleasant and potentially dangerous symptoms.
■ Naltrexone blocks opioid receptors and thereby decreases craving for alcohol and blocks alcohol’s reinforcing effects.
■ Acamprosate decreases tension, anxiety, and other unpleas-ant feelings caused by the absence of alcohol. The underly-ing mechanism is unclear.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
aPatient education information is highlighted as blue text.
Summary of Major Nursing Implicationsa
DISULFIRAMPreadministration AssessmentTherapeutic GoalMaintaining alcohol abstinence.
Patient SelectionCandidates for therapy must be chosen carefully. Disulfiram must not be given to patients who are likely to attempt drinking while taking this drug.
Identifying High-Risk PatientsDisulfiram is contraindicated for patients suspected of being incapable of abstinence from alcohol; for patients with myocardial disease, coronary occlusion, or psychosis; and for patients who have recently received alcohol, metronidazole, or alcohol-containing medications (e.g., cough syrups, tonics).
Implementation: AdministrationRouteOral.
AdministrationInstruct the patient not to administer the first dose until at least 12 hours after his or her last drink.
Dosing is done once daily and may continue for months or even years.
Inform patients that tablets may be crushed or mixed with liquid.
Implementation: Measures to Enhance Therapeutic EffectsPatient education is essential for safety. Inform patients about the potential hazards of treatment, and warn them to avoid all forms of alcohol, including alcohol in vinegar, sauces, and cough syrups, and alcohol applied to the skin in aftershave lotions, colognes, and liniments. Inform patients that the effects of disulfiram will persist about 2 weeks after the last dose and that alcohol must not be consumed during this time. Encourage patients to carry identification to alert emergency healthcare personnel to their condition.

435
C H A P T E R
39 Substance Use Disorders III: Nicotine and Smoking
Basic Pharmacology of Nicotine, p. 435Mechanism of Action, p. 435Pharmacokinetics, p. 435Pharmacologic Effects, p. 435Tolerance and Dependence, p. 436Acute Poisoning, p. 436Chronic Toxicity From Smoking, p. 436
Pharmacologic Aids to Smoking Cessation, p. 437Nicotine Replacement Therapy, p. 438Bupropion SR, p. 440Varenicline, p. 440Products That Are Not Recommended, p. 441
Key Points, p. 442
Box 39.1. Smoking Cessation During Pregnancy, p. 437
• Strengthen advertising restrictions, including the prohibi-tion on marketing to youth.
• Require revised and more prominent warning labels.• Require disclosure of all ingredients in tobacco products
and restrict harmful additives.• Monitor nicotine yields, and mandate gradual nicotine
reduction to nonaddictive levels.
BASIC PHARMACOLOGY OF NICOTINEMechanism of ActionThe effects of nicotine result from actions at nicotinic receptors. Whether these receptors are activated or inhibited depends on nicotine dosage. Low doses activate nicotinic receptors; high doses block them. The amount of nicotine received from ciga-rettes is relatively low. Accordingly, cigarette smoking causes receptor activation.
Nicotine can activate nicotinic receptors at several locations. Most effects result from activating nicotinic receptors in autonomic ganglia and the adrenal medulla. In addition, nicotine can activate nicotinic receptors in the carotid body, aortic arch, and central nervous system (CNS). As discussed later, actions in the CNS mimic those of cocaine and other highly addictive substances. When present at the levels produced by smoking, nicotine has no significant effect on nicotinic receptors of the neuromuscular junction.
PharmacokineticsAbsorption of nicotine depends on whether the delivery system is a cigarette or e-cigarette, a cigar, or smokeless tobacco. Nicotine in cigarette smoke is absorbed primarily from the lungs. When cigarette smoke is inhaled, between 90% and 98% of nicotine in the lungs enters the blood. Unlike nicotine in cigarette smoke, nicotine in cigar smoke is absorbed primarily from the mouth, as is nicotine in smokeless tobacco.
Nicotine can cross membranes easily and is widely distrib-uted throughout the body. The drug readily enters breast milk, reaching levels that can be toxic to the nursing infant. Nicotine also crosses the placental barrier and can cause fetal harm. When inhaled in cigarette smoke, nicotine reaches the brain in just 10 seconds.
Nicotine is rapidly metabolized to inactive products. Nicotine and its metabolites are excreted by the kidney. The drug’s half-life is 1 to 2 hours.
Pharmacologic EffectsThe pharmacologic effects discussed in this section are associ-ated with low doses of nicotine. These are the effects caused
Cigarette smoking remains the greatest single cause of pre-ventable illness and premature death. In the United States, smoking kills more than 480,000 adults each year—about 1 of every 5 deaths. Around the world, tobacco kills more than 6 million people each year. On average, male smokers die 13.2 years prematurely, and female smokers die 14.5 years prematurely. According to the Centers for Disease Control and Prevention, most deaths result from lung cancer (127,700), coronary heart disease (99,300), and chronic airway obstruction (100,600). Not only do cigarettes kill people who smoke, but every year, through secondhand smoke, cigarettes kill about 41,000 nonsmoking Americans, and about 600,000 nonsmokers worldwide. The direct medical costs of smoking exceed $170 billion a year. Indirect costs, including lost time from work and disability, add up to an additional $156 billion. In the United States, the prevalence of smoking among adults fell steadily from 1965 (42%) through the 1980s and 1990s, but decreased only slightly between 2004 (20.9%) and 2015 (15.1%).
Although tobacco smoke contains many dangerous com-pounds, nicotine is of greatest concern. Other hazardous com-ponents in tobacco smoke include carbon monoxide, hydrogen cyanide, ammonia, nitrosamines, and tar. Tar is composed of various polycyclic hydrocarbons, some of which are proven carcinogens.
What is the regulatory status of cigarettes? Good question, given that cigarettes are the single most dangerous product available to U.S. consumers. Until recently, cigarettes had avoided virtually all federal regulation. However, strong regula-tions are now in place. Under the Family Smoking Prevention and Tobacco Control Act, the U.S. Food and Drug Administra-tion (FDA) now has the authority to:

436
UNIT V Central Nervous System Drugs
report more discomfort than men. Experience has shown that abrupt discontinuation may be preferable to gradual reduction. (All that gradual reduction seems to do is prolong the suffering.)
Acute PoisoningNicotine is highly toxic. Doses as low as 40 mg can be fatal. Toxicity is underscored by the use of nicotine as an insecticide. Common causes of nicotine poisoning include ingestion of tobacco by children and exposure to nicotine-containing insecticides.
SymptomsThe most prominent symptoms involve the cardiovascular, GI, and central nervous systems. Specific symptoms include nausea, salivation, vomiting, diarrhea, cold sweats, disturbed hearing and vision, confusion, and faintness; pulses may be rapid, weak, and irregular. Death results from respiratory paralysis, which is caused by direct effects of nicotine on the muscles of respiration, as well as by effects in the CNS.
TreatmentManagement centers on reducing nicotine absorption and supporting respiration; there is no specific antidote to nicotine poisoning. Absorption of ingested nicotine can be reduced by giving activated charcoal. If respiration is depressed, ventilatory assistance is indicated. Since nicotine undergoes rapid metabolic inactivation, recovery from the acute phase of poisoning can occur within hours.
Chronic Toxicity From SmokingAccording to a 2004 report from the U.S. Surgeon General, the adverse consequences of smoking are more extensive than previously understood. It is now clear that chronic smoking can injure nearly every organ of the body. We already knew that smoking could cause cardiovascular disease, chronic lung disease, and cancers of the larynx, lung, esophagus, oral cavity, and bladder. New additions to the list include leukemia, cata-racts, pneumonia, periodontal disease, type 2 diabetes, abdominal aortic aneurysm, and cancers of the cervix, kidney, pancreas, and stomach. Smoking during pregnancy increases the risk of low birth weight, preterm labor, stillbirth, miscarriage, spontane-ous abortion, perinatal mortality, and sudden infant death. The leading causes of smoking-related death are lung cancer, ischemic heart disease, and chronic airway obstruction.
by smoking cigarettes. Responses to high doses are discussed under Acute Poisoning.
Cardiovascular EffectsThe cardiovascular effects of nicotine result primarily from activating nicotinic receptors in sympathetic ganglia and the adrenal medulla. Activation of these receptors promotes the release of norepinephrine from sympathetic nerves and the release of epinephrine (and some norepinephrine) from the adrenals. Norepinephrine and epinephrine act on the cardio-vascular system to constrict blood vessels, accelerate the heart, and increase the force of ventricular contraction. The net result is elevation of blood pressure and increased cardiac work. These effects underlie cardiovascular deaths.
GI EffectsNicotine influences GI function primarily by activating nicotinic receptors in parasympathetic ganglia, thereby increasing secre-tion of gastric acid and augmenting tone and motility of GI smooth muscle. In addition, nicotine can promote vomiting. Nicotine-induced vomiting results from a complex process that involves nicotinic receptors in the aortic arch, carotid sinus, and CNS.
CNS EffectsNicotine is a CNS stimulant. The drug stimulates respiration and produces an arousal pattern on an electroencephalograph. Moderate doses can cause tremors, and high doses can cause convulsions.
Nicotine has multiple psychologic effects. The drug increases alertness, facilitates memory, improves cognition, reduces aggression, and suppresses appetite. In addition, by promoting the release of dopamine, nicotine activates the brain’s “pleasure system” located in the mesolimbic area. The effects of nicotine on the pleasure system are identical to those of other highly addictive drugs, including cocaine, amphetamines, and opioids.
Effects During Pregnancy and LactationNicotine exposure during gestation can harm the fetus, and nicotine in breast milk can harm the nursing infant. Nonetheless, as discussed in Box 39.1, since pharmaceutical nicotine is safer than tobacco smoke, it is reasonable to consider using nicotine therapy during pregnancy to help a patient quit smoking.
Tolerance and DependenceToleranceTolerance develops to some effects of nicotine but not to others. Tolerance does develop to nausea and dizziness, which are common in the unseasoned smoker. In contrast, very little tolerance develops to the cardiovascular effects: Veteran smokers continue to experience increased blood pressure and increased cardiac work whenever they smoke.
DependenceChronic cigarette smoking results in dependence. By defini-tion, this means that individuals who discontinue smoking will experience an abstinence syndrome. Prominent symptoms are craving, nervousness, restlessness, irritability, impatience, increased hostility, insomnia, impaired concentration, increased appetite, and weight gain. Symptoms begin about 24 hours after smoking has ceased and can last for weeks to months. Women
Prototype Drugs
DRUGS TO AID SMOKING CESSATIONNicotine-Based ProductsNicotine patch [Nicoderm CQ]Nicotine gum [Nicorette]Nicotine lozenge [Nicorette Lozenge]Nicotine nasal spray [Nicotrol NS]Nicotine inhaler [Nicotrol Inhaler]
Nicotine-Free ProductsVareniclineBupropion

437
CHAPTER 39 Substance Use Disorders III: Nicotine and Smoking
apply not only to people who quit while they are young and healthy, but also to people who quit after age 65 years and to those with established tobacco-related disease. Data from the Nurses’ Health Study indicate that former smokers eventually achieve the same disease-risk status as never smokers, even with respect to lung cancer. The risk of chronic obstructive pulmonary disease or death from a heart attack declines to that of never smokers in 20 years, and the risk of lung cancer reaches that of never smokers in 30 years.
Seven drug products have been shown to aid smoking cessation (Table 39.1). Of these seven products, five contain nicotine and two don’t. The nicotine-based products—nicotine gum, nicotine lozenge, nicotine patch, nicotine inhaler, and nicotine nasal spray—are employed as nicotine replacement therapy (NRT). The nicotine-free products—sustained-release bupropion (bupropion SR) [Zyban, Buproban] and varenicline [Chantix, Champix ]—are taken to decrease nicotine craving and to suppress symptoms of withdrawal. The most effective drug therapies for smoking cessation are varenicline alone and the nicotine patch combined with a short-acting nicotine product (i.e., nasal spray or gum). At this time, we cannot predict who
PHARMACOLOGIC AIDS TO SMOKING CESSATION
Cigarettes are highly addictive, which makes giving them up very hard. Nonetheless, abstinence can be achieved. Every year, about 41% of Americans who smoke make one or more attempts to quit. Of those who try to quit without formal help, only 4% to 7% achieve long-term success. In contrast, when a combination of counseling and drugs is employed, the 6-month abstinence rate approaches 25%. However, even with the aid of counseling and drugs, the first attempt usually fails. In fact, most people try to quit 5 to 7 times before they ultimately succeed. As time without a cigarette increases, the chances of relapse get progressively smaller: Of those who quit for a year, only 15% smoke again; and of those who quit for 5 years, only 3% smoke again.
Long-term smokers should be assured that quitting offers important health benefits. Regardless of how long you have smoked, quitting can reduce the risk of developing a tobacco-related disease, slow the progression of an established tobacco-related disease, and increase life expectancy. These benefits
BOX 39.1 ■ SPECIAL INTEREST TOPIC
SMOKING CESSATION DURING PREGNANCYSmoking is the largest modifiable risk factor for pregnancy-related morbidity and mortality. Smoking increases the risk of ectopic pregnancy, placenta previa, placental abruption, chorioamnionitis, stillbirth, preterm birth, and spontaneous abortion. In addition, fetal exposure increases the risk of low birth weight, perinatal mortality, sudden infant death syndrome (SIDS), and cognitive, behavioral, and emotional deficits in childhood.
Of the many harmful chemicals in tobacco smoke, reproductive toxicity is due in large part to just three: nicotine, carbon monoxide, and oxidizing agents. Nicotine reduces placental blood flow (by promoting vasoconstriction), delays or impairs fetal brain development (by direct neurotoxic effects), inhibits matura-tion of fetal pulmonary cells, and increases the risk of SIDS. Carbon monoxide reduces the oxygen-carrying capacity of blood and, in high levels, is neuroteratogenic. Oxidizing agents increase the risk of thrombotic events, and by decreasing the availability of nitrous oxide (a smooth muscle relaxant), they contribute to placental vasoconstriction and preterm labor.
Clearly, smoking during pregnancy is dangerous. Ideally, female smokers should quit before conception or early in pregnancy. However, quitting later is still beneficial. To aid in smoking cessation, the authors of Treating Tobacco Use and Dependence: 2008 Update recommend that clinicians offer effective interventions at the first prenatal visit and throughout the course of pregnancy as needed. Intensive person-to-person psychosocial intervention should be offered to all pregnant smokers. Pharmacologic intervention—mainly nicotine replace-ment therapy (NRT)—may also be offered, but only if psycho-social intervention alone has failed. Of note, quit rates with a combination of psychosocial intervention plus NRT are higher than with psychosocial intervention alone.
What do we know about NRT during pregnancy? Not as much as we would like. Studies on the efficacy of NRT in pregnant smokers have been inconclusive—probably because the nicotine dosage was too low. (During the later stages of pregnancy, nicotine is metabolized at a high rate. Hence, if conventional NRT doses are used, nicotine blood levels may be too low to be effective.) Nonetheless, even if we are uncertain about NRT efficacy, it seems likely that NRT is much safer than smoking. After all, cigarette smoke contains thousands of harmful chemicals (in addition to nicotine), whereas NRT contains nicotine only. In fact, among pregnant patients who switched from smoking to NRT, there was no evidence of serious adverse effects, and there was an important benefit: birth weight was increased.
Who should use NRT? According to a 2009 review,a the use of NRT should be based on the number of cigarettes smoked. Pregnant patients who smoke no more than five cigarettes a day should be offered psychosocial support, but not NRT. Conversely, pregnant patients who smoke more should be offered NRT along with psychosocial support.
What about bupropion and varenicline? Compared with NRT, bupropion lacks the potential adverse effects of nicotine; however, its side effect of appetite suppression is not desirable for the pregnant patient and developing fetus. One prominent voice—the American College of Obstetricians and Gynecologists (ACOG)—says that bupropion may be considered when behavioral interven-tions have failed. However, another prominent voice—the Motherisk Program—says that bupropion should be avoided until we know more about its safety and efficacy. As for vareni-cline, we have no human data on safety in pregnancy, but we do have animal data showing fetal harm. Accordingly, varenicline should not be used.
aOsadchy A, Kazmin A, Koren G: Nicotine replacement therapy during pregnancy: recommended or not recommended? J Obstet Gynaecol Can 31:744–747, 2009.

438
UNIT V Central Nervous System Drugs
For additional information on smoking cessation, visit the Internet sites for Canada and the United States listed in Table 39.2.
Nicotine Replacement TherapyNRT allows smokers to substitute a pharmaceutical source of nicotine for the nicotine in cigarettes—and then gradually to withdraw the replacement nicotine. This is analogous to using methadone to wean addicts from heroin.
Five FDA-approved formulations are available: chewing gum, lozenges, transdermal patches, a nasal spray, and an
will respond best to a particular product. Accordingly, selection should be based on patient preference, success with a particular product in the past, and side effects.
Interventions for smoking cessation can be found in Treating Tobacco Use and Dependence: 2008 Update, a clinical practice guideline issued by the U.S. Public Health Service. As stated in the guideline, tobacco dependence is a chronic condition that warrants repeated intervention until long-term abstinence is achieved. This is the same philosophy that guides the treat-ment of dependence on other highly addictive substances, including cocaine and heroin. Tobacco dependence can be treated with two methods: drugs and counseling. Both methods are effective, but a combination of both is more effective than either one alone. Accordingly, the guidelines recommend that all patients who want to quit be offered (1) at least one smoking cessation drug (bupropion, varenicline, or a nicotine-based product) along with (2) counseling, be it one-on-one, in a group, or over the phone (dial 1-800-QUITNOW in the United States or 1-877-513-5333 in Canada). The overall intervention strategy is summarized in the “5 A’s” model for treating tobacco use and dependence:
Ask (screen all patients for tobacco use).Advise tobacco users to quit.Assess willingness to make a quit attempt.Assist with quitting (offer medication and provide or refer to
counseling).Arrange follow-up contacts, beginning within the first week
after the quit date.
Product Common Side Effects Advantages Disadvantages
NICOTINE-BASED PRODUCTS
Nicotine patch [Nicorette Invisipatch , NicoDerm CQ]
Transient itching, burning, and redness under the patch; insomnia
Nonprescription; provides a steady level of nicotine; easy to use; unobtrusive
User cannot adjust dose if craving occurs; nicotine released more slowly than in other products
Nicotine gum [Nicorette, others]
Mouth and throat irritation, aching jaw muscles, dyspepsia
Nonprescription; user controls dose Unpleasant taste; requires proper chewing technique; cannot eat or drink while chewing the gum; can damage dental work and is difficult for denture wearers to use
Nicotine lozenge [Nicorette Lozenge]
Hiccups, dyspepsia, mouth irritation, nausea
Nonprescription; user controls dose; easier to use than nicotine gum
Cannot eat or drink while the lozenge is in the mouth
Nicotine nasal spray [Nicotrol NS]
During first week: mouth and throat irritation, rhinitis, sneezing, coughing, teary eyes
User controls dose; fastest nicotine delivery and highest nicotine levels of all nicotine-based products
Prescription required; most irritating nicotine-based product; device visible when used
Nicotine inhaler [Nicotrol Inhaler]
Mouth and throat irritation, cough
User controls dose; mimics hand-to-mouth motion of smoking
Prescription required; slow onset and low nicotine levels; frequent puffing needed; device visible when used
NICOTINE-FREE PRODUCTS
Varenicline [Chantix, Champix ]
Nausea, sleep disturbances, headaches, abnormal dreams
Easy to use (pill); no nicotine; most effective pharmacologic aid to smoking cessation
Prescription required; may cause neuropsychiatric disturbances, including suicidal thoughts and actions
Bupropion [Zyban, Buproban]
Insomnia, dry mouth, agitation
Easy to use (pill); no nicotine; promotes weight loss, which may limit cessation-related weight gain; first-choice drug for smokers with depression
Prescription required; carries a small risk of seizures
TABLE 39.1 ■ Pharmacologic Aids for Smoking Cessation
UNITED STATES
• U.S. Department of Health and Human Services: https://www.surgeongeneral.gov/library/reports/index.html
• Centers for Disease Control and Prevention: http://www.cdc.gov/tobacco/quit_smoking/
• American Lung Association: http://www.lungusa.org/stop-smoking/
CANADA
• Health Canada: https://www.canada.ca/en/health-canada/services/health-concerns/tobacco.html
• The Lung Association: https://www.lung.ca/lung-health/smoking-and-tobacco/how-quit-smoking
TABLE 39.2 ■ Internet-Based Resources for Smoking Cessation

439
CHAPTER 39 Substance Use Disorders III: Nicotine and Smoking
4-mg strength. The average adult dosage is 9 to 12 pieces a day. The maximum daily dosage is 24 pieces. Experience indicates that dosing on a fixed schedule (one piece every 2 to 3 hours) is more effective than PRN dosing for achieving abstinence.
After 3 months without cigarettes, patients should discontinue nicotine use. Withdrawal should be done gradually. Use of nicotine gum beyond 6 months is not recommended.
Nicotine Lozenges (Nicotine Polacrilex)The pharmacology of nicotine lozenges [Nicorette Lozenge] is very similar to that of nicotine gum. Both products contain nicotine bound to polacrilex. Sucking on the lozenge releases nicotine, which is then absorbed across the oral mucosa into the systemic circulation. Like nicotine gum and other forms of NRT, nicotine lozenges double the cessation success rate.
The most common adverse effects are mouth irritation, dyspepsia, nausea, and hiccups—all of which can be made worse by taking two lozenges at once or by taking several lozenges in immediate succession.
Administration consists of placing the lozenge in the mouth and allowing it to dissolve, which takes 20 to 30 minutes. Users should not eat or drink for 15 minutes before dosing and while the lozenge is in the mouth. Also, they should not chew or swallow the lozenge.
Like nicotine gum, nicotine lozenges are available in two strengths: 2 mg and 4 mg. Dosing with the lozenges is the same as with the gum. Users should consume no more than 5 lozenges every 6 hours and no more than 20 lozenges per day. The recommended dosing schedule is 1 lozenge every 1 to 2 hours for the first 6 weeks; 1 every 2 to 4 hours for the next 3 weeks; and 1 every 4 to 8 hours for the next 3 weeks, after which dosing should stop.
Nicotine Transdermal Systems (Patches)Nicotine transdermal systems are nicotine-containing adhesive patches that, after application to the skin, slowly release their nicotine content. The nicotine is absorbed into the skin and then into the blood, producing steady blood levels. Use of the patch about doubles the cessation success rate.
Two systems are available: NicoDerm CQ and Nicorette Invisipatch . Both can be purchased without a prescription. As indicated in Table 39.3, the patches come in different sizes. The larger patches release more nicotine.
Nicotine patches are applied once a day to clean, dry, non-hairy skin of the upper body or upper arm. The site should be changed daily and not reused for at least 1 week. NicoDerm
inhaler (see Table 39.1). With the gum, lozenges, patches, and inhaler, blood levels of nicotine rise slowly and remain relatively steady. Because nicotine levels rise slowly, these delivery systems produce less pleasure than cigarettes, but nonetheless do relieve symptoms of withdrawal. With the nasal spray, blood levels of nicotine rise rapidly, much as they do with smoking. Hence the nasal spray provides some of the subjective pleasure that smoking does.
Long-term quit rates are significantly greater with NRT than with placebo—although absolute success rates remain low. For example, the 1-year success with nicotine patches is about 25%, compared with 9% for placebo. Success rates are highest when replacement therapy is combined with counseling.
Nicotine products are classified in FDA Pregnancy Risk Category Ca (chewing gum, lozenge) or Category D (patch, inhaler, nasal spray). Accordingly, they should generally be avoided during pregnancy. However, since smoking is probably more harmful than NRT, use of NRT during pregnancy is worth consideration (see Box 39.1).
Nicotine Chewing Gum (Nicotine Polacrilex)Nicotine chewing gum [Nicorette, others] is composed of a gum base plus nicotine polacrilex, an ion exchange resin to which nicotine is bound. The gum must be chewed to release the nicotine. Following release, nicotine is absorbed across the oral mucosa into the systemic circulation. Like other forms of NRT, nicotine gum doubles the cessation success rate.
The most common adverse effects are mouth and throat soreness, jaw muscle ache, eructation (belching), and hiccups. Using optimal chewing technique minimizes these problems.
Patients should be advised to chew the gum slowly and intermittently for about 30 minutes. Rapid chewing can release too much nicotine at one time, resulting in effects similar to those of excessive smoking (e.g., nausea, throat irritation, hiccups). Because foods and beverages can reduce nicotine absorption, patients should not eat or drink while chewing or for 15 minutes before chewing.
Nicotine gum is available in two strengths: 2 mg/piece and 4 mg/piece. Dosing is individualized and based on the degree of nicotine dependence. For initial therapy, patients with low to moderate nicotine dependence (those who smoke their first cigarette after 30 minutes of waking) should use the 2-mg strength; highly dependent patients (those who smoke their first cigarette within 30 minutes of waking) should use the
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
Brand NameSurface Area (cm2)
Hours/Day in Place Dose Absorbed
Duration of Use
Per Patch Size Total
Nicoderm CQ Step 1 30 24 21 mg over 24 hr First 4–6 wk 8–10 wkNicoderm CQ Step 2 20 24 14 mg over 24 hr Next 2 wkNicoderm CQ Step 3 10 24 7 mg over 24 hr Next 2 wk
Nicorette Invisipatch Step 1 22.5 16 25 mg over 16 hr First 8 wk 12 wk
Nicorette Invisipatch Step 2 13.5 16 15 mg over 16 hr Next 2 wk
Nicorette Invisipatch Step 3 9 16 10 mg over 16 hr Next 2 wk
TABLE 39.3 ■ Nicotine Transdermal Systems (Patches)

440
UNIT V Central Nervous System Drugs
27% of users avoided smoking for 1 year—about twice the abstinence rate achieved with placebo. The bad news is that many patients continued to use the spray, being unwilling or unable to give it up. Nonetheless, since the spray delivers nicotine without the additional hazards in cigarettes, using the spray is clearly preferable to smoking.
Adverse effects are mild and temporary. At first, most users experience rhinitis, sneezing, coughing, watering eyes, and nasal and throat irritation. Fortunately, these effects abate in a few days. Nicotine nasal spray should be avoided by patients with sinus problems, allergies, or asthma.
Bupropion SRBupropion SR [Zyban, Buproban], an atypical antidepressant, was the first non-nicotine drug approved as an aid to smoking cessation. The drug is structurally similar to amphetamine and, like amphetamine, causes CNS stimulation and suppresses appetite. In people trying to quit cigarettes, bupropion reduces the urge to smoke and reduces some symptoms of nicotine withdrawal (e.g., irritability, anxiety). The drug is effective in the presence and absence of depression. Although the mecha-nism of action is uncertain, benefits may derive from blocking uptake of norepinephrine and dopamine. For use in depression, bupropion is sold under the brand name Wellbutrin.
Like the NRT products, bupropion SR doubles the cessation success rate. In one trial, patients were given bupropion SR (100, 150, or 300 mg/day) or placebo. At 7 weeks, abstinence rates were 19% with placebo, and 29%, 39%, and 44% with increasing dosages of bupropion SR. At 12 weeks, abstinence rates were lower: 12% with placebo and 20%, 23%, and 23% with increasing dosages of bupropion SR. Combining a nicotine patch with bupropion SR is somewhat more effective than either treatment alone.
Adverse effects are generally mild. The most common are dry mouth and insomnia. High doses (above 450 mg/day) are associated with a 0.4% risk of seizures. However, at the doses employed for smoking cessation (300 mg/day), seizures have not been reported. Nonetheless, bupropion SR should be avoided in patients with seizure risk factors, such as head trauma, history of seizures, anorexia nervosa, cocaine use, and alcohol with-drawal. Because it suppresses appetite, bupropion SR can cause weight loss. Bupropion SR should not be combined with a monoamine oxidase inhibitor. Nor should it be given to patients taking Wellbutrin, which is just another name for bupropion itself.
The usual regimen is 150 mg in the morning for 3 days, followed by 150 mg twice a day for 7 to 12 weeks. To minimize interference with sleep, the second dose should be taken as early in the day as possible—but at least 8 hours after the morning dose. Because onset of effects is delayed, dosing should begin 1 to 2 weeks before attempting to give up cigarettes.
The basic pharmacology of bupropion is discussed in Chapter 32.
VareniclineVarenicline [Chantix, Champix ], a partial agonist at nicotinic receptors, is our most effective aid to smoking cessation. In clinical trials, more patients achieved abstinence with varenicline than with bupropion SR or the nicotine patch. Estimated abstinence rates after 6 months were 33.2% with varenicline,
CQ patches are left in place for 24 hours and then immediately replaced with a fresh one. In contrast, Nicorette patches are applied in the morning and removed 16 hours later at bedtime. This pattern is intended to simulate nicotine dosing produced by smoking.
Most patients begin with a large patch and then use progres-sively smaller patches over several weeks. Certain patients (those with cardiovascular disease, those who weigh less than 100 pounds, or those who smoke less than one-half pack of cigarettes a day) should begin with a smaller patch.
Adverse effects are generally mild. Short-lived erythema, itching, and burning occur under the patch in 35% to 50% of users. In 14% to 17% of users, persistent erythema occurs, lasting up to 24 hours after patch removal. Patients who experience severe, persistent local reactions (e.g., severe erythema, itching, edema) should discontinue the patch and contact a physician or nurse practitioner.
Nicotine InhalerThe nicotine inhaler [Nicotrol Inhaler] differs from other NRT products in that it looks much like a cigarette. Puffing on it delivers the nicotine. Because of this delivery method, using the inhaler can substitute for the hand-to-mouth behavior of smoking. In addition to nicotine, the inhaler contains menthol, which is intended to create a sensation in the back of the throat reminiscent of that caused by smoke. Like other forms of NRT, the inhaler doubles cessation success rates.
The nicotine inhaler consists of a mouthpiece and a sealed tubular cartridge. Inside the cartridge is a porous plug containing 10 mg of nicotine. Inserting the cartridge into the mouthpiece breaks the seal. Puffing on the mouthpiece draws air over the plug and thereby draws nicotine vapor into the mouth. Most of the nicotine is absorbed through the oral mucosa—not in the lungs. As a result, blood levels rise slowly and peak 10 to 15 minutes after puffing stops. Blood levels are less than half those achieved with cigarettes. Each cartridge can deliver 300 to 400 puffs. Benefits are greatest with frequent puffing over 20 minutes, after which the cartridge is discarded. Patients generally use 6 to 16 cartridges a day for 3 months, and then taper off over 2 to 3 months.
Adverse effects are mild. The most frequent are dyspepsia, coughing, throat irritation, oral burning, and rhinitis. The inhaler should not be used by patients with asthma. Because the cartridges contain dangerous amounts of nicotine, they should be kept away from children and pets.
Nicotine Nasal SprayNicotine nasal spray [Nicotrol NS] differs from other NRT formulations in that blood levels of nicotine rise rapidly after each administration, thereby closely simulating smoking. Because nicotine levels rise rapidly, the spray provides some of the subjective pleasure associated with cigarettes. As with other forms of NRT, the spray doubles smoking cessation rates.
The spray device delivers 0.5 mg of nicotine per activation. Two sprays (one in each nostril) constitute one dose and are equivalent to the amount of nicotine absorbed from one cigarette. Treatment should be started with 1 or 2 doses per hour—and never more than 5 doses per hour, or 40 doses a day. After 4 to 6 weeks, dosing should be gradually reduced and then stopped.
Quitting success with the spray has been good news and bad news. The good news, as reported in one study, is that

441
CHAPTER 39 Substance Use Disorders III: Nicotine and Smoking
revealed a similar risk in patients without cardiovascular disease. Fortunately, cardiovascular risk appears to be small—much smaller than the risk posed by smoking. Nonetheless, patients should be warned about cardiovascular risk and instructed to notify the prescriber if they experience new or worsening cardiovascular symptoms and to seek immediate medical attention if symptoms of myocardial infarction appear.
Owing to concerns about unpredictable physical and psy-chiatric adverse effects, authorities in the United States have banned the use of varenicline by truck drivers, bus drivers, airplane pilots, and air traffic controllers.
Drug InteractionsVarenicline does not affect the major components of the cytochrome P450 system. Studies with bupropion, transdermal nicotine, digoxin, warfarin, cimetidine, and metformin have shown no significant interactions. To date, no clinically sig-nificant interactions with other drugs have been reported.
Preparations, Dosage, and AdministrationVarenicline is formulated in 0.5- and 1-mg tablets. To reduce nausea, each dose should be taken after eating and with a full glass of water. Dosing should begin 8 to 35 days before smoking is stopped. Titrate dosage as follows: on days 1 through 3, take 0.5 mg once daily; on days 4 through 7, take 0.5 mg twice daily; then take 1 mg twice daily for 12 weeks. If abstinence has been achieved, an additional 12 weeks of treatment is recommended. Patients who fail to stop smoking after the initial 12 weeks or who relapse after a full course of treatment should be encouraged to try again when conditions are deemed favorable. Patients with severe renal impairment should begin therapy at 0.5 mg once daily and increase to 0.5 mg twice daily if tolerated. Patients with end-stage renal disease undergo-ing dialysis should take a maximum of 0.5 mg once daily. Dosage adjustment is unnecessary in patients with mild to moderate renal impairment.
Products That Are Not RecommendedAccording to Treating Tobacco Use and Dependence: 2008 Update, there is insufficient proof to recommend the following drugs as aids to smoking cessation: naltrexone, silver acetate, beta blockers, benzodiazepines, and antidepressants other than bupropion SR, including the selective serotonin reuptake inhibitors.
Although not mentioned in the 2008 Update, electronic cigarettes, or e-cigarettes, should be avoided. E-cigarettes are battery-powered, cigarette-shaped devices that release a puff of vaporized nicotine, sometimes together with flavoring and other chemicals. According to analyses conducted by the FDA, the amount of nicotine per puff can vary widely, and the vapor may contain trace amounts of diethylene glycol and/or other contaminants. E-cigarettes are promoted on the Internet as aids to quit smoking, but are not FDA-approved for this use (or any other use, for that matter). At this writing, the FDA is attempting to regulate e-cigarettes as drug-delivery devices (which seems reasonable), but is meeting resistance from the courts. The bottom line: Because the dose of nicotine with e-cigarettes is unpredictable and because data on safety and efficacy are lacking, the use of e-cigarettes should be discouraged—especially since products of known safety and efficacy are available.
24.2% with bupropion SR, and 23.4% with a nicotine patch. The most common side effect is nausea. The most troubling side effects are psychologic changes. Unlike bupropion SR and NRT, varenicline does not cause weight loss.
Mechanism of ActionVarenicline acts as a partial agonist at a subset of nicotinic receptors—known as alpha4beta2 nicotinic receptors—whose activation promotes release of dopamine, the compound that mediates the pleasurable effects of nicotine. Compared with nicotine, varenicline binds alpha4beta2 receptors with greater affinity. Hence, when varenicline is present, access of nicotine to these receptors is blocked. Because varenicline is a partial agonist, receptor binding results in mild activation, which promotes some dopamine release and thereby helps reduce both nicotine craving and the intensity of withdrawal symptoms. At the same time, the presence of varenicline prevents intense receptor activation by nicotine itself and thereby blocks the reward that nicotine can provide.
PharmacokineticsVarenicline is readily absorbed from the GI tract, both in the presence and absence of food. Plasma levels peak about 4 hours after dosing. Binding to plasma proteins is low (20%). Metabolism is minimal, and hence most of each dose (92%) is excreted unchanged in the urine. The plasma half-life is 17 to 24 hours. Moderate to severe renal impairment delays excretion and increases varenicline blood levels.
Adverse EffectsIn clinical trials, dose-dependent nausea was the most common adverse effect, occurring in 30% to 40% of users. Nausea is mild to moderate initially and becomes less severe over time. Other common reactions include sleep disturbances, headaches, abnormal dreams, constipation, dry mouth, flatulence, vomiting, and altered sense of taste. Mild physical dependence develops, but there have been no reports of abuse or addictive behavior. Rarely, varenicline has been associated with seizures, diabetes, dizziness, disturbed vision, and moderate and severe skin reac-tions, although a causal relationship has not been established.
Early postmarketing reports indicated that varenicline can cause serious neuropsychiatric effects, including mood changes, erratic behavior, and suicidality. At that time, the FDA placed a black box warning on varenicline for this reason. In 2016, the FDA removed the warning, as these cases were deemed more rare than initially expected. Nevertheless, all patients should be advised to contact their prescriber if they experience a significant change in behavior or mental status. Varenicline should be used with caution in patients with a history of psychiatric disease.
In 2011, the FDA warned that varenicline can increase the risk of cardiovascular events (e.g., angina pectoris, peripheral edema, hypertension, nonfatal myocardial infarction) in patients with stable cardiovascular disease. After that, a Canadian study

442
UNIT V Central Nervous System Drugs
KEY POINTS
■ Cigarette smoking kills about 480,000 American adults each year, making smoking the largest preventable cause of premature death.
■ The principal cause of death among smokers is lung cancer, followed closely by heart disease.
■ Nicotine in cigarette smoke is absorbed from the lungs, whereas nicotine in cigar smoke and smokeless tobacco is absorbed from the mouth.
■ By activating nicotinic receptors in sympathetic ganglia and the adrenal medulla, nicotine causes vasoconstriction, increases heart rate, and increases force of ventricular contraction, thereby elevating blood pressure and increasing cardiac work. These effects underlie cardiovascular deaths.
■ Through actions in the CNS, nicotine increases alertness, facilitates memory, improves cognitive function, reduces aggression, and suppresses appetite. In addition, by promot-ing the release of dopamine, nicotine activates the same pleasure circuit involved in addiction to cocaine, amphet-amines, and opioids.
■ Although tolerance develops to some effects of nicotine, very little tolerance develops to cardiovascular effects: Veteran smokers continue to experience an increase in blood pressure and cardiac work whenever they smoke.
■ Nicotine causes physical dependence. Withdrawal is characterized by craving, nervousness, restlessness, irritabil-ity, impatience, increased hostility, insomnia, impaired concentration, increased appetite, and weight gain.
■ Nicotine for replacement therapy is available in five FDA-approved delivery systems: chewing gum, lozenges, transdermal patches, nasal spray, and an inhaler.
■ Although nicotine is harmful during pregnancy, NRT is probably safer than smoking, and hence use of NRT during pregnancy is worth consideration.
■ Bupropion SR [Zyban, Buproban], which blocks reuptake of norepinephrine and dopamine, helps smokers quit by reducing nicotine craving and withdrawal symptoms.
■ Varenicline [Chantix, Champix ] acts as a partial agonist at a specific subset of nicotinic receptors and thereby reduces nicotine craving and withdrawal symptoms. In addition, the drug blocks access of nicotine itself to those receptors and thereby prevents nicotine from producing pleasurable effects.
■ The most effective drug/therapies for smoking cessation are varenicline alone and the nicotine patch combined with PRN nicotine nasal spray or nicotine gum.
■ With the aid of counseling and pharmacotherapy, about 30% of smokers who attempt to quit can expect to achieve long-term abstinence.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

443
C H A P T E R
40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
Heroin, Oxycodone, and Other Opioids, p. 443Patterns of Use, p. 443Subjective and Behavioral Effects, p. 444Preferred Drugs and Routes of Administration,
p. 444Tolerance and Physical Dependence, p. 445Treatment of Acute Toxicity, p. 445Detoxification, p. 445Drugs for Long-Term Management of Opioid Use
Disorder, p. 445Sequelae of Compulsive Opioid Use, p. 447
General CNS Depressants, p. 447Barbiturates, p. 447Benzodiazepines, p. 447
Psychostimulants, p. 448Cocaine, p. 448Methamphetamine, p. 449Synthetic Cathinones, p. 450
Marijuana and Related Preparations, p. 450Cannabis sativa: The Source of Marijuana, p. 450Psychoactive Component, p. 450Mechanism of Action, p. 450Pharmacokinetics, p. 450Behavioral and Subjective Effects, p. 450Physiologic Effects, p. 451
Tolerance and Dependence, p. 451Therapeutic Use, p. 451Comparison of Marijuana with Alcohol, p. 452Synthetic Marijuana, p. 452
Psychedelics, p. 453d-Lysergic Acid Diethylamide (LSD), p. 453Salvia, p. 454Mescaline, Psilocin, Psilocybin, and
Dimethyltryptamine, p. 454Dissociative Drugs, p. 454
Phencyclidine, p. 454Ketamine, p. 455
Dextromethorphan, p. 4553,4-Methylenedioxymethamphetamine (MDMA,
Ecstasy), p. 456Time Course and Dosage, p. 456Who Uses MDMA and Why?, p. 456Adverse Effects, p. 456Potential Medical Use, p. 456
Inhalants, p. 456Anesthetics, p. 456Volatile Nitrites, p. 456Organic Solvents, p. 457
Anabolic Steroids, p. 457Key Points, p. 457
Schedule II substances. The basic pharmacology of the opioids is discussed in Chapter 28.
Patterns of UseOpioid use disorder (OUD) is encountered in all segments of society. Formerly, OUD was limited almost exclusively to lower socioeconomic groups residing in cities. Today, however, opioid abuse is more widespread, occurring in small towns as well as big cities, and among the rich and middle class as well as the poor.
For most people with OUD, initial exposure to opioids occurs either recreationally (i.e., illicitly) or in the context of pain management in a medical setting. The overwhelming majority of individuals who go on to abuse opioids begin their drug use illicitly. Only an exceedingly small percentage of those exposed to opioids therapeutically develop a pattern of compulsive drug use.
In this chapter, we discuss all of the major drugs of abuse except alcohol (see Chapter 38) and nicotine (see Chapter 39). As indicated in Table 40.1, abused drugs fall into seven major categories: (1) opioids, (2) psychostimulants, (3) depressants, (4) psychedelics, (5) dissociative drugs, (6) anabolic steroids, and (7) miscellaneous drugs of abuse. The basic pharmacology of many of these drugs is presented in previous chapters, so their discussion here is brief. Agents that have not been addressed previously (e.g., marijuana, d-lysergic acid dieth-ylamide [LSD]) are discussed in depth.
HEROIN, OXYCODONE, AND OTHER OPIOIDSThe opioids (e.g., heroin, oxycodone, meperidine) are major drugs of abuse. As a result, most opioids are classified as

444
UNIT V Central Nervous System Drugs
Preferred Drugs and Routes of AdministrationWhich opioids do people abuse? Practically all of them. In the past, heroin was the most commonly abused opioid drug, but no longer, although its use is increasing. Prescription opioid analgesics were abused much more commonly than heroin, but with the opioid epidemic and increases in deaths from overdose, heroin is becoming more available and is cheaper than opioids. In 2015, more than 3.4 million Americans reported past-month abuse of opioids and 325,000 people reported that they had used heroin within the past month (www.samhsa.gov/data/sites/default/files/NSDUH).
HeroinAmong street users, heroin is the traditional opioid of choice. Because of its high lipid solubility, heroin crosses the blood-brain barrier with ease, causing effects that are both immediate and intense. This combination of speed and intensity sets heroin apart from other opioids.
Heroin can be administered in several ways. The order of preference is IV injection, smoking, and nasal inhalation (known as sniffing or snorting). Intravenous injection produces effects with the greatest intensity and fastest onset (7 to 8 seconds). When heroin is smoked or snorted, effects develop more slowly, peaking in 10 to 15 minutes. Among users who seek treatment, injection is the predominant method of administration. However, because sniffing and smoking are safer and easier than injection, these routes have become increasingly popular.
It should be noted that, when heroin is administered orally or subcutaneously, as opposed to intravenously, its effects cannot be distinguished from those of morphine and other opioids. This observation is not surprising given that, once in the brain, heroin is rapidly converted into morphine, its active form.
OxycodoneIn many parts of the United States, people are abusing the controlled-release formulation of oxycodone [OxyContin], an opioid similar to morphine. The controlled-release tablets were designed to provide steady levels of oxycodone over an extended time, and are safe and effective when swallowed intact. However, abusers do not ingest the tablets whole. Rather, they crush the tablets; then they either snort the powder or dissolve it in water and then inject it intravenously. As a result, the entire dose is absorbed immediately, producing blood levels that are dangerously high. Thousands of deaths have been reported. The risk of respiratory depression and death is greatest in people who have not developed tolerance to opioids.
In an effort to reduce OxyContin abuse, the controlled-release tablets were reformulated in 2010. The new formulation bears the imprint OP, rather than OC, which appeared on the old formulation. Compared with the old tablets, OxyContin OP tablets are much harder to crush into a powder. And if exposed to water or alcohol, the tablets just form a gel, rather than a solution that can be drawn into a syringe and injected. However, there is no evidence that OxyContin OP tablets are less subject to abuse, diversion, overdose, or development of OUD than the old tablets.
MeperidineNurses and physicians who abuse opioids may select meperidine [Demerol], a drug with distinct advantages for these users.
Opioid abuse by healthcare providers deserves special consideration. It is well established that physicians, nurses, and pharmacists, as a group, abuse opioids to a greater extent than all other groups with similar educational backgrounds. The vulnerability of healthcare professionals to opioid abuse is due primarily to drug access.
Subjective and Behavioral EffectsMoments after IV injection, heroin produces sensations of pleasure, relaxation, warmth, and thirst. This initial reaction, known as a “rush” or “kick,” persists for about 45 seconds. After this, the user experiences a prolonged sense of euphoria (well-being); there is a feeling that “all is well with the world.” These extended effects, rather than the initial rush, are the primary reason for opioid abuse.
Interestingly, when individuals first use opioids, nausea and vomiting are prominent, and an overall sense of dysphoria may be felt. In many cases, were it not for peer pressure, individuals would not continue opioid use long enough to allow these unpleasant reactions to be replaced by a more agreeable experience.
Category Examples
OPIOIDS HeroinHydromorphoneMeperidineMorphineOxycodone
PSYCHOSTIMULANTS CocaineDextroamphetamineMethamphetamineMethylphenidate
DEPRESSANTS
Barbiturates AmobarbitalPentobarbitalPhenobarbitalSecobarbital
Benzodiazepines DiazepamLorazepam
Miscellaneous AlcoholGamma-hydroxybutyrateMeprobamateMethaqualone
PSYCHEDELICS DimethyltryptamineLSDMescalinePsilocybin
DISSOCIATIVE DRUGS KetaminePhencyclidine
ANABOLIC STEROIDS NandroloneOxandroloneTestosterone
MISCELLANEOUS Amyl nitriteDextromethorphanMarijuanaNicotineNitrous oxide
TABLE 40.1 ■ Pharmacologic Categorization of Abused Drugs

445
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
methadone-aided withdrawal is to substitute methadone for the opioid upon which the individual is dependent. Because opioids display cross-dependence with one another, methadone will prevent an abstinence syndrome. Once the subject has been stabilized on methadone, withdrawal is accomplished by administering methadone in gradually smaller doses. The resultant abstinence syndrome is mild, with symptoms resembling those of moderate influenza. The entire process of methadone substitution and withdrawal takes about 10 days.
When substituting methadone for another opioid, suppression of the abstinence syndrome requires that methadone dosage be closely matched to the existing degree of physical depen-dence. Hence, to ensure that methadone dosing is adequate, the extent of physical dependence must be assessed. This can be accomplished by taking a history on the extent of drug use and by observing the patient for symptoms of withdrawal. Of the two approaches, observation is the more reliable. Estimates of drug use based on patient histories may be unreliable because (1) street users don’t know the purity of the drugs they have taken, (2) claims of drug use may be inflated in hopes of receiving larger doses of methadone, and (3) healthcare profes-sionals with OUD may report minimal consumption to downplay the extent of use. Because information from patients with OUD is not likely to permit accurate assessment of dependence, it is essential to observe the patient to make certain methadone dosage is sufficient to suppress withdrawal.
The use of methadone for maintenance therapy and sup-pressive therapy is discussed later in this chapter.BuprenorphineBuprenorphine is an agonist-antagonist opioid. Like methadone, buprenorphine can be substituted for the opioid on which an individual with OUD is physically dependent and can thereby prevent symptoms of withdrawal. After the individual is stabilized on buprenorphine, the dosage is gradually reduced, thereby keeping symptoms of withdrawal to a minimum. Use of buprenorphine for maintenance therapy is discussed later.
Clonidine-Assisted WithdrawalClonidine is a centrally acting alpha2-adrenergic agonist. When administered to an individual physically dependent on opioids, clonidine can suppress some symptoms of abstinence. Clonidine is most effective against symptoms related to autonomic hyperactivity (nausea, vomiting, diarrhea). Modest relief is provided from muscle aches, restlessness, anxiety, and insomnia. Opioid craving is not diminished. The basic pharmacology of clonidine is discussed in Chapter 19.
Rapid and Ultrarapid WithdrawalIn both procedures, the patient is given an opioid antagonist (naloxone or naltrexone) to precipitate immediate withdrawal and thereby accelerate the withdrawal process. The ultrarapid procedure is carried out under general anesthesia or heavy sedation with IV midazolam [Versed]. In both procedures, clonidine may be added to ease symptoms. These procedures permit a rapid switch to maintenance therapy with an opioid antagonist. However, they are no more effective than standard withdrawal techniques, and they are considerably more expensive.
Drugs for Long-Term Management of Opioid Use DisorderThree kinds of drugs are employed for long-term management: opioid agonists, opioid agonist-antagonists, and opioid antago-nists. Opioid agonists (methadone) and agonist-antagonists (buprenorphine) substitute for the abused opioid and are given to patients who are not yet ready for detoxification. In contrast, opioid antagonists (naltrexone) are used to discourage renewed opioid use after detoxification has been accomplished. Drugs
First, unlike heroin, meperidine is highly effective when administered orally, and hence abuse need not be associated with telltale signs of repeated injections. Second, meperidine produces less pupillary constriction than other opioids, thereby minimizing awkward questions about miosis. Lastly, meperidine has minimal effects on smooth muscle function, making constipation and urinary retention less problematic than with other opioids.
Tolerance and Physical DependenceToleranceWith prolonged opioid use, tolerance develops to some phar-macologic effects, but not others. Effects to which tolerance does develop include euphoria, respiratory depression, and nausea. In contrast, little or no tolerance develops to constipation and miosis. Because tolerance to respiratory depression develops in parallel with tolerance to euphoria, respiratory depression does not increase as higher doses are taken to produce desired subjective effects. Persons tolerant to one opioid are cross-tolerant to other opioids. However, there is no cross-tolerance between opioids and general central nervous system (CNS) depressants (e.g., barbiturates, benzodiazepines, alcohol).
Physical DependenceLong-term use produces substantial physical dependence. The abstinence syndrome resulting from opioid withdrawal is described in Chapter 28. It is important to note that, although the opioid withdrawal syndrome can be extremely unpleasant, it is rarely dangerous.
Following the acute abstinence syndrome, which fades in 10 days, patients with opioid use disorder may experience a milder but protracted phase of withdrawal. This second phase, which may persist for months, is characterized by insomnia, irritability, and fatigue. Gastrointestinal hyperactivity and premature ejaculation may also occur.
Treatment of Acute ToxicityTreatment of acute opioid toxicity is discussed in Chapter 28 and summarized here. Overdose produces a classic triad of symptoms: respiratory depression, coma, and pinpoint pupils. Naloxone [Narcan], an opioid antagonist, is the treatment of choice. This agent rapidly reverses all signs of opioid poisoning. However, dosage must be titrated carefully because if too much is given, the patient will swing from a state of intoxication to one of withdrawal. Owing to its short half-life, naloxone must be readministered every few hours until opioid concentrations have dropped to nontoxic levels, which may take days. Failure to repeat naloxone dosing may result in the death of patients who had earlier been rendered symptom free.
DetoxificationPersons who are physically dependent on opioids experience unpleasant symptoms if drug use is abruptly discontinued. Techniques for minimizing discomfort are presented later in this chapter.
Methadone SubstitutionMethadone, a long-acting oral opioid, is the agent most commonly employed for easing withdrawal. The first step in

446
UNIT V Central Nervous System Drugs
BuprenorphineBuprenorphine [Suboxone] is an agonist-antagonist opioid. The drug is a partial agonist at mu receptors and a full antagonist at kappa receptors. Buprenorphine can be used for maintenance therapy and to facilitate detoxification (see earlier section in this chapter). When used for maintenance, buprenorphine alleviates craving, reduces the use of illicit opioids, and increases retention in therapeutic programs.
Unlike methadone, which is available only through certified Opioid Treatment Programs, buprenorphine can be prescribed and dispensed in general medical settings, such as primary care offices. Prescribers must receive at least 8 hours of authorized training and must register with the Substance Abuse and Mental Health Services Administration.
Buprenorphine has several properties that make it attractive for treating OUD. Because it is a partial agonist at mu recep-tors, it has a low potential for abuse—but can still suppress craving for opioids. If the dosage is sufficiently high, buprenor-phine can completely block access of strong opioids to mu receptors and can thereby prevent opioid-induced euphoria. With buprenorphine, there is a ceiling to respiratory depres-sion, which makes it safer than methadone. Development of physical dependence is low, and hence withdrawal is relatively mild.
Buprenorphine is currently available in three formulations that are dosed once a day. One formulation—sublingual tablets— contains buprenorphine alone. The other two formulations—sublingual tablets and sublingual films, both marketed as Suboxone—contain buprenorphine combined with naloxone. Generic buprenorphine is used for the first few days of treat-ment, and then Suboxone is used for long-term maintenance. The naloxone in Suboxone is there to discourage IV abuse. If taken intravaneously, the naloxone in Suboxone will precipitate withdrawal. However, with sublingual administration, very little
used for long-term management of opioid use disorder are shown in Table 40.2.
MethadoneIn addition to its role in facilitating opioid withdrawal, methadone [Methadose] can be used for maintenance therapy and suppressive therapy. These strategies are employed to modify drug-using behavior in patients who are not ready to try withdrawal.
Methadone maintenance consists of transferring the patient from the abused opioid to oral methadone. By taking methadone, the individual avoids both withdrawal and the need to procure illegal drugs. Maintenance dosing is done once a day. Main-tenance is most effective when done in conjunction with nondrug measures directed at altering patterns of drug use.
Suppressive therapy is done to prevent the reinforcing effects of opioid-induced euphoria. Suppression is achieved by giving the patient progressively larger doses of methadone until a very high dose (120 mg/day) is reached. Building up to this dose creates a high degree of tolerance, and hence no subjective effects are experienced from the methadone itself. Because cross-tolerance exists among opioids, once the patient is tolerant to methadone, taking street drugs, even in high doses, cannot produce significant desirable effects. As a result, individuals made tolerant with methadone will be less likely to seek out illicit opioids.
The use of methadone to treat opioid use disorder is restricted to Opioid Treatment Programs approved by the designated state authority and certified by the federal Substance Abuse and Mental Health Services Administration. These restrictions on the nonanalgesic use of methadone are needed to control abuse of methadone, a Schedule II drug with the same abuse liability as morphine and other strong opioids.
The basic pharmacology of methadone is presented in Chapter 28.
DrugBrand Name Formulation
Dosing Schedule
CSA Schedule Comments
OPIOID AGONIST
Methadone Methadose Concentrated oral liquid
Once daily II Methadone maintenance may be provided only by Opioid Treatment Programs certified by the federal Substance Abuse and Mental Health Services Administration and approved by the designated state authority.
OPIOID AGONIST-ANTAGONIST
Buprenorphine Suboxonea Sublingual tablet Once daily III Suboxone may be prescribed in a primary care setting by any physician or nurse practitioner who has received authorized training and has registered with the Substance Abuse and Mental Health Services Administration.
Suboxonea Sublingual film Once daily
OPIOID ANTAGONIST
Naltrexone ReVia Oral tablet Once daily NR Naltrexone is not a controlled substance, and hence prescribers do not require special training or certification. Intramuscular naltrexone [Vivitrol] is the only drug approved for opioid use disorder that is given monthly, rather than daily. Before receiving naltrexone, patients must undergo opioid detoxification.
Vivitrol Extended-release suspension for IM injection
Once a month
TABLE 40.2 ■ Drugs for Long-Term Management of Opioid Use Disorder
aIn addition to buprenorphine, Suboxone contains naloxone, an opioid antagonist, to discourage IV dosing.CSA, Controlled Substances Act; NR, not regulated under the CSA.

447
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
BarbituratesThe barbiturates embody all of the properties that typify general CNS depressants, and hence can be considered prototypes of the group. Depressant effects are dose dependent and range from mild sedation to sleep to coma to death. With prolonged use, barbiturates produce tolerance and physical dependence.
The abuse liability of the barbiturates stems from their ability to produce subjective effects similar to those of alcohol. The barbiturates with the highest potential for abuse have a short to intermediate duration of action. These agents—amobarbital, pentobarbital, and secobarbital—are classified under Schedule II of the Controlled Substances Act. Other barbiturates appear under Schedules III and IV. Despite legal restrictions, barbi-turates are available cheaply and in abundance.
ToleranceRegular use of barbiturates produces tolerance to some effects, but not to others. Tolerance to subjective effects is significant. As a result, progressively larger doses are needed to produce desired psychologic responses. Unfortunately, very little toler-ance develops to respiratory depression. Consequently, as barbiturate use continues, the dose needed to produce subjective effects moves closer and closer to the dose that can cause respiratory arrest. (Note that this differs from the pattern seen with opioids, in which tolerance to subjective effects and to respiratory depression develop in parallel.) Individuals tolerant to barbiturates show cross-tolerance with other CNS depressants (e.g., alcohol, benzodiazepines, general anesthetics). However, little or no cross-tolerance develops to opioids.
Physical Dependence and Withdrawal TechniquesChronic barbiturate use can produce substantial physical dependence. Cross-dependence exists between barbiturates and other CNS depressants, but not with opioids. When physical dependence is great, the associated abstinence syndrome can be severe—sometimes fatal. In contrast, the opioid abstinence syndrome, although unpleasant, is rarely life threatening.
One technique for easing barbiturate withdrawal employs phenobarbital, a barbiturate with a long half-life. Because of cross-dependence, substitution of phenobarbital for the abused barbiturate suppresses symptoms of abstinence. Once the patient has been stabilized, the dosage of phenobarbital is gradually tapered off, thereby minimizing symptoms of abstinence.
Acute ToxicityOverdose with barbiturates produces a triad of symptoms: respiratory depression, coma, and pinpoint pupils—the same symptoms that accompany opioid poisoning. Treatment is directed at maintaining respiration and removing the drug; endotracheal intubation and ventilatory assistance may be required. Details of management are presented in Chapter 34. Barbiturate overdose has no specific antidote. Naloxone, which reverses poisoning by opioids, is not effective against poisoning by barbiturates.
BenzodiazepinesBenzodiazepines differ significantly from barbiturates. Ben-zodiazepines are much safer than the barbiturates, and overdose with oral benzodiazepines alone is rarely lethal. However, the
naloxone is absorbed, and hence, when the drug is administered as intended, the risk of withdrawal is low. Nonetheless, because there is a small risk with sublingual Suboxone, treatment is initi-ated with generic buprenorphine, thereby allowing substitution of buprenorphine for the abused opioid. Thereafter, Suboxone is taken for maintenance.
The basic pharmacology of buprenorphine is presented in Chapter 28.
NaltrexoneAfter a patient has undergone opioid detoxification, naltrexone [ReVia, Vivitrol], a pure opioid antagonist, can be used to discourage renewed opioid abuse. Benefits derive from blocking euphoria and all other opioid-induced effects. By preventing pleasurable effects, naltrexone eliminates the reinforcing properties of opioid use. When the recovering patient learns that taking an opioid cannot produce the desired response, drug-using behavior will cease. Naltrexone is not a controlled substance, and hence prescribers require no special training or certification.
Naltrexone is available in oral and IM formulations. The oral formulation, sold as ReVia, is dosed once a day. The IM formulation, sold as Vivitrol, is dosed once a month. At this time, Vivitrol is the only long-acting drug for managing opioid use disorder. All other drugs must be taken every day.
The basic pharmacology of naltrexone is presented in Chapter 28.
Sequelae of Compulsive Opioid UseSurprisingly, chronic opioid use has very few direct detrimental effects. Patients in treatment programs have been maintained on high doses of methadone for a decade with no significant impairment of health. Furthermore, individuals on metha-done maintenance can be successful socially and at work. It appears, then, that opioid use is not necessarily associated with poor health, lack of productivity, or inadequate social interaction.
Although opioids have few direct ill effects, there are many indirect hazards. These risks stem largely from the lifestyle of the opioid user and from impurities common to street drugs. Infections secondary to sharing nonsterile needles occur fre-quently. The infections that opioid abusers acquire include septicemia, cellulitis, abscesses, endocarditis, tuberculosis, hepatitis C, and HIV. Foreign-body emboli have resulted from impurities in opioid preparations. Opioid users suffer an unusu-ally high death rate. Some deaths reflect the violent nature of the subculture in which opioid use often takes place. Many others result from accidental overdose.
GENERAL CNS DEPRESSANTSThe family of CNS depressants consists of barbiturates, benzodiazepines, alcohol, and other agents. With the exception of the benzodiazepines, all of these drugs are more alike than different. The benzodiazepines have properties that set them apart. The basic pharmacology of the benzodiazepines, barbi-turates, and most other CNS depressants is presented in Chapter 34; the pharmacology of alcohol is presented in Chapter 38. Discussion here is limited to abuse of these drugs.

448
UNIT V Central Nervous System Drugs
into the bloodstream. A few users (about 5%) administer cocaine hydrochloride IV. Cocaine hydrochloride cannot be smoked because it is unstable at high temperature.
Cocaine base is administered by smoking, a process referred to as “freebasing.” Smoking delivers large amounts of cocaine to the lungs, where absorption is very rapid. Subjective and physiologic effects are equivalent to those elicited by IV injection.
Subjective Effects and Cocaine Use DisorderAt usual doses, cocaine produces euphoria similar to that produced by amphetamines. In a laboratory setting, individuals familiar with the effects of cocaine are unable to distinguish between cocaine and amphetamine. Cocaine causes euphoria through inhibition of neuronal reuptake of dopamine and thereby increases activation of dopamine receptors in the brain’s reward circuit.
As with many other psychoactive drugs, the intensity of subjective responses depends on the rate at which plasma drug levels rise. Because cocaine levels rise relatively slowly with intranasal administration and almost instantaneously with IV injection or smoking, responses produced by intranasal cocaine are much less intense than those produced by the other two routes.
When crack cocaine is smoked, desirable subjective effects begin to fade within minutes and are often replaced by dys-phoria. In an attempt to avoid dysphoria and regain euphoria, the user may administer repeated doses at short intervals. This usage pattern—termed bingeing—can rapidly lead to cocaine use disorder.
Acute Toxicity: Symptoms and TreatmentOverdose is frequent, and deaths have occurred. Mild overdose produces agitation, dizziness, tremor, and blurred vision. Severe overdose can produce hyperpyrexia, convulsions, ventricular dysrhythmias, and hemorrhagic stroke. Angina pectoris and myocardial infarction may develop secondary to coronary artery spasm. Psychologic manifestations of overdose include severe anxiety, paranoid ideation, and hallucinations (visual, auditory, and/or tactile). Because cocaine has a short half-life, symptoms subside in 1 to 2 hours.
Although there is no specific antidote to cocaine toxicity, most symptoms can be controlled with drugs. Intravenous diaz-epam or lorazepam can reduce anxiety and suppress seizures. Diazepam may also alleviate hypertension and dysrhythmias, since these result from increased central sympathetic activity. If hypertension is severe, it can be corrected with IV nitroprusside. Dysrhythmias associated with prolonging the QT interval may respond to hypertonic sodium bicarbonate. Although beta block-ers can suppress dysrhythmias, they might further compromise coronary perfusion (by preventing beta2-mediated coronary vasodilation). Reduction of thrombus formation with aspirin can lower the risk of myocardial ischemia. Hyperthermia should be reduced with external cooling.
Chronic ToxicityWhen administered intranasally on a long-term basis, cocaine can cause atrophy of the nasal mucosa and loss of sense of smell. In extreme cases, necrosis and perforation of the nasal septum occur. Nasal pathology results from local ischemia secondary to chronic vasoconstriction. Injury to the lungs can occur from smoking cocaine base.
risk of death is greatly increased when oral benzodiazepines are combined with other CNS depressants (e.g., alcohol, barbiturates) or when benzodiazepines are administered IV. If severe overdose occurs, signs and symptoms can be reversed with flumazenil [Romazicon, Anexate ], a benzodiazepine antagonist. As a rule, tolerance and physical dependence are only moderate when benzodiazepines are taken for legitimate indications, but can be substantial when these drugs are abused. In patients who develop physical dependence, the abstinence syndrome can be minimized by withdrawing benzodiazepines very slowly—over a period of months. The abuse liability of the benzodiazepines is much lower than that of the barbiturates. As a result, all benzodiazepines are classified under Schedule IV of the Controlled Substances Act. Benzodiazepines are discussed in Chapter 34.
PSYCHOSTIMULANTSDiscussion here focuses on two CNS stimulants that have a high potential for abuse: cocaine and methamphetamine. Because of their considerable abuse liability, these drugs are classified as Schedule II agents. (If they lacked approved medical uses, they would be classified in Schedule I.) In addition to stimulating the CNS, methamphetamine and cocaine can stimulate the heart, blood vessels, and other structures under sympathetic control. Because of these peripheral actions, these stimulants are also referred to as sympathomimetics.
Stimulants with little or no abuse potential are not addressed in this chapter. Included in this group are Schedule III stimulants (e.g., benzphetamine), Schedule IV stimulants (e.g., diethyl-propion), and stimulants that are not regulated at all (e.g., caffeine, ephedrine).
CocaineCocaine is a stimulant extracted from the leaves of the coca plant. The drug has CNS effects similar to those of the amphetamines. In addition, cocaine can produce local anesthesia as well as vasoconstriction and cardiac stimulation. Among abusers, a form of cocaine known as “crack” is used widely. Crack is extremely addictive, and the risk of lethal overdose is high.
According to the National Survey on Drug Use and Health, cocaine use has declined. In 2015, 2.4 million Americans age 18 and older reported using cocaine in any form, compared with 5.5 million in 2005.
FormsCocaine is available in two forms: cocaine hydrochloride and cocaine base (alkaloidal cocaine, freebase cocaine, “crack”). Cocaine base is heat stabile, whereas cocaine hydrochloride is not. Cocaine hydrochloride is available as a white powder that is frequently diluted (“cut”) before sale. Cocaine base is sold in the form of crystals (“rocks”) that consist of nearly pure cocaine. Cocaine base is widely known by the street name “crack,” a term inspired by the sound the crystals make when heated.
Routes of AdministrationCocaine hydrochloride is usually administered intranasally. The drug is “snorted” and absorbed across the nasal mucosa

449
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
to the 2015 National Survey on Drug Use and Health, use of methamphetamine within the past month by Americans age 12 years and older increased from 569,000 in 2014 to 898,000 in 2015.
Subjective and Behavioral EffectsAs discussed in Chapter 36, amphetamines act primarily by increasing the release of norepinephrine and dopamine, and partly by reducing the reuptake of both transmitters. By doing so, methamphetamine produces arousal and elevation of mood. Euphoria is likely and talkativeness is prominent. A sense of increased physical strength and mental capacity occurs. Self-confidence rises. Users feel little or no need for food and sleep.
Adverse Psychologic EffectsAll amphetamines can produce a psychotic state characterized by delusions, paranoia, and auditory and visual hallucinations, making patients look as if they suffer from schizophrenia. Although psychosis can be triggered by a single dose, it occurs more commonly with long-term abuse. Methamphetamine-induced psychosis usually resolves spontaneously following drug withdrawal. If needed, an antipsychotic agent (e.g., olanzapine) can be given to suppress symptoms.
Adverse Cardiovascular EffectsBecause of its sympathomimetic actions, methamphetamine can cause vasoconstriction and excessive stimulation of the heart, leading to hypertension, angina pectoris, and dysrhyth-mias. Overdose may also cause cerebral and systemic vasculitis and renal failure. Changes in cerebral blood vessels can lead to stroke. Vasoconstriction can be relieved with an alpha-adrenergic blocker (e.g., phentolamine). Cardiac stimulation can be reduced with a mixed alpha and beta blocker (e.g., labetalol).
Other Adverse EffectsBy suppressing appetite, methamphetamine can cause significant weight loss. Use during pregnancy increases the risk of preterm birth, hypertension, placental abruption, intrauterine growth restriction, and neonatal death. Heavy use can promote severe tooth decay, known informally as “meth mouth.” Causes include reduced salivation, grinding and clenching of the teeth, increased consumption of sugary drinks, and neglect of oral hygiene. Lastly, methamphetamine can cause direct injury to dopami-nergic nerve terminals in the brain, leading to prolonged deficits in cognition and memory.
Tolerance, Dependence, and WithdrawalLong-term use results in tolerance to mood elevation, appetite suppression, and cardiovascular effects. Although physical dependence is only moderate, psychologic dependence can be intense. Methamphetamine withdrawal can produce dysphoria and a strong sense of craving. Other symptoms include fatigue, prolonged sleep, excessive eating, and depression. Depression can persist for months and is a common reason for resuming drug use.
TreatmentAmphetamine-type substance use disorder responds well to cognitive behavioral therapy. One such approach, known as the Matrix Model, combines group therapy, individual therapy, family education, drug testing, and encouragement to participate
Use During PregnancyCocaine is highly lipid soluble and readily crosses the placenta, allowing it to accumulate in the fetal circulation. Data from 2013 revealed that babies born to mothers who used cocaine during pregnancy were more likely to be born early, to have smaller heads and decreased length, and to have lower birth weights than babies born to mothers who did not use cocaine. In addition, the effects on babies born to prenatal users of cocaine can last well into childhood, causing deficits in attention, memory, and language development.
Tolerance, Dependence, and WithdrawalIn animal models, regular administration of cocaine results in increased sensitivity to the drug, not tolerance. Whether this holds true for humans is not clear.
The degree of physical dependence produced by cocaine is in dispute. Some observers report little or no evidence of withdrawal following cocaine discontinuation. In contrast, others report symptoms similar to those associated with amphetamine withdrawal: dysphoria, craving, fatigue, depres-sion, and prolonged sleep.
Treatment of Cocaine Use DisorderAlthough achieving complete abstinence from cocaine is extremely difficult, treatment can greatly reduce cocaine use. For the individual with cocaine use disorder, psychosocial therapy is the cornerstone of treatment. This therapy is directed at motivating users to commit to a drug-free life, and then helping them work toward that goal. A combination of individual therapy and group drug counseling is most effective, producing a 70% reduction in cocaine use at 12-month follow-up.
Can medication help with cocaine use disorder? To date, no drug has been proved broadly effective in treating cocaine abuse. However, ongoing work with two agents is encouraging:
• Anticocaine vaccine—Subjects receiving the vaccine develop antibodies that bind with cocaine and thereby render the cocaine inactive. The higher the antibody titer, the greater the reduction in cocaine use. Phase I human testing recruitment began in 2016.
• Disulfiram [Antabuse]—Subjects receiving a combination of disulfiram plus cognitive behavioral therapy reduced their cocaine use from 2 or 3 times daily to 0.5 times daily. Disulfiram is the same drug we discussed in Chapter 38 for treating alcohol abuse.
MethamphetamineThe basic pharmacology of the amphetamine family is dis-cussed in Chapter 36. Discussion here is limited to abuse of methamphetamine.
Description and RoutesMethamphetamine is a white crystalline powder that readily dissolves in water or alcohol. The drug may be swallowed, “snorted,” smoked, or injected IV. Owing to its potential for abuse, methamphetamine is classified as a Schedule II drug.
Patterns of UseUse of methamphetamine declined between the years of 2006 and 2012. Yet, we are now seeing an increase in use. According

450
UNIT V Central Nervous System Drugs
preparation of the resinous exudate from female flowers. Hashish is considerably more potent than marijuana.
Psychoactive ComponentThe major psychoactive substance in Cannabis sativa is delta-9-tetrahydrocannabinol (THC), an oily chemical with high lipid solubility.
The THC content of Cannabis preparations is variable. The highest concentrations are found in the flowers of the female plant. The lowest concentrations are in the seeds. Depending on growing conditions and the strain of the plant, THC in marijuana preparations may range from 1% to 11%.
Mechanism of ActionPsychologic effects of THC result from activating specific cannabinoid receptors in the brain. The endogenous ligand for these receptors appears to be anandamide, a derivative of arachidonic acid unique to the brain. The concentration of cannabinoid receptors is highest in brain regions associated with pleasure, memory, thinking, concentration, appetite, sensory perception, time perception, and coordination of movement.
There is evidence that marijuana may act in part through the same reward system as do opioids and cocaine. Both heroin and cocaine produce pleasurable sensations by promoting a release of dopamine in the brain’s reward circuit. In rats, intravenous THC also causes dopamine release. Interestingly, the release of dopamine by THC is blocked by naloxone, a drug that blocks the effects of opioids. This suggests that THC causes the release of dopamine by first causing a release of endogenous opioids.
PharmacokineticsAdministration by SmokingWhen marijuana or hashish is smoked, about 60% of the THC content is absorbed. Absorption from the lungs is rapid. Subjec-tive effects begin in minutes and peak 10 to 20 minutes later. Effects from a single marijuana cigarette may persist for 2 to 3 hours. Termination results from metabolism of THC to inactive products.
Oral AdministrationWhen marijuana or hashish is ingested, practically all of the THC undergoes absorption. However, the majority is inactivated on its first pass through the liver. Hence, only 6% to 20% of absorbed drug actually reaches the systemic circulation. Because of this extensive first-pass metabolism, oral doses must be 3 to 10 times greater than smoked doses to produce equivalent effects. With oral dosing, effects are delayed and prolonged: Responses begin in 30 to 50 minutes and persist up to 12 hours.
Behavioral and Subjective EffectsMarijuana produces three principal subjective effects: euphoria, sedation, and hallucinations. This set of responses is unique to marijuana; no other psychoactive drug causes all three. Because of this singular pattern of effects, marijuana is in a class by itself.
in non–drug-related activities. At this time, no medications are approved for treatment. However, encouraging results have been achieved with two drugs: bupropion [Wellbutrin, Zyban], currently approved for major depression and smoking cessation, and modafinil [Provigil, Alertec ], a nonamphetamine stimulant currently approved for narcolepsy, shift-work sleep disorder, and obstructive sleep apnea/hypopnea syndrome. An additional drug, ibudilast, is in clinical trials. Preliminary results indicate that ibudilast may dampen cravings for methamphetamine and improve cognitive functioning.
Synthetic CathinonesSynthetic cathinones are drugs better known by their street name, “bath salts.” These drugs are closely related to the natu-rally occurring khat plant, found in East Africa and southern Arabia. Bath salts have appeared on the market within the past few years, and are touted as substitutes to other psychostimu-lants, such as cocaine or methamphetamine.
Bath salts are consumed by injecting, swallowing, snorting, or smoking. Often, bath salts are sold in packets of white power or tablets labeled “not for human consumption” or as plant food or cleaning substances. They are structurally related to amphetamines and are thought to increase dopamine, serotonin, and norepinephrine levels. This leads to feelings of euphoria and alertness. Onset of duration is fairly quick with overall duration of 2 to 4 hours.
Multiple negative psychologic effects have been noted with the use of bath salts, including paranoia, aggression, hallucina-tions, panic attacks, agitation, and delirium. Physical effects range from nose bleeds and sweating to renal failure and death. Bath salts were formerly available over the counter in gas stations and convenience stores, but due to their negative effects and many emergency room visits, bath salts are banned for sale and use in all 50 states.
MARIJUANA AND RELATED PREPARATIONSMarijuana is the most commonly used illicit drug in the United States. Over 128 million Americans have tried it at least once. In 2015, over 22 million Americans age 12 and older used marijuana. Among youth, rates of daily marijuana use are the highest since the 1970s. Results of the 2016 Monitoring the Future survey show that 5.4% of 8th-graders and 14% of 10th-graders reported using marijuana in the past month. Monthly marijuana use among 12th-graders has risen to 22.5%.
Cannabis sativa: The Source of MarijuanaMarijuana is prepared from Cannabis sativa, the Indian hemp plant—an unusual plant in that it has separate male and female forms. Psychoactive compounds are present in all parts of the male and female plants. However, the greatest concentration of psychoactive substances is found in the flowering tops of the female plants.
The two most common Cannabis derivatives are marijuana and hashish. Marijuana is a preparation consisting of leaves and flowers of male and female plants. Alternative names for marijuana include grass, weed, pot, and dope. The terms joint and reefer refer to marijuana cigarettes. Hashish is a dried

451
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
up to 140 beats/min are not uncommon. Pretreatment with propranolol prevents marijuana-induced tachycardia but does not block the drug’s subjective effects. Marijuana causes orthostatic hypotension and pronounced reddening of the conjunctivae. These responses apparently result from vasodilation.
Respiratory EffectsWhen used acutely, marijuana produces bronchodilation. However, when smoked chronically, the drug causes airway constriction. In addition, chronic use is closely associated with the development of bronchitis, sinusitis, and asthma. Lung cancer is another possible outcome. Animal studies have shown that tar from marijuana smoke is a more potent carcinogen than tar from cigarette smoke.
Effects on ReproductionResearch in animals has shown multiple effects on reproduction. In males, marijuana decreases spermatogenesis and testosterone levels. In females, the drug reduces levels of follicle-stimulating hormone, luteinizing hormone, and prolactin.
Multiple effects may be seen in babies and children who were exposed to marijuana in utero. Some babies present with trembling, altered responses to visual stimuli, and a high-pitched cry. Preschoolers may have a decreased ability to perform tasks that involve memory and sustained attention. School-children may exhibit deficits in memory, attentiveness, and problem solving.
Altered Brain StructureLong-term marijuana use is associated with structural changes in the brain. Specifically, the volume of the hippocampus and amygdala is reduced, by an average of 12% and 7.1%, respec-tively. We don’t know whether volume reduction is due to reduced cell size, reduced synaptic density, or loss of glial cells and/or neurons. Interestingly, hippocampal volume loss occurs primarily in the left hemisphere.
Tolerance and DependenceWhen taken in extremely high doses, marijuana can produce tolerance and physical dependence. Neither effect, however, is remarkable. Some tolerance develops to the cardiovascular, perceptual, and motor effects of marijuana. Little or no tolerance develops to subjective effects.
To demonstrate physical dependence on marijuana, the drug must be given in very high doses—and even then the degree of dependence is only moderate. Symptoms brought on by abrupt discontinuation of high-dose marijuana include irritabil-ity, restlessness, nervousness, insomnia, reduced appetite, and weight loss. Tremor, hyperthermia, and chills may occur too. Symptoms subside in 3 to 5 days. With moderate marijuana use, no withdrawal symptoms occur.
Therapeutic UseIn the United States, there are no approved medical uses for marijuana itself. However, there are U.S. Food and Drug Administration (FDA)–approved uses for two purified cannabinoids: THC and dronabinol, an analog of THC. A third cannabinoid preparation—nabiximols—is approved in Canada.
Effects of Low to Moderate DosesResponses to low doses of THC are variable and depend on several factors, including dosage size and route, setting of drug use, and expectations and previous experience of the user. The following effects are common: euphoria and relaxation; gaiety and a heightened sense of the humorous; increased sensitivity to visual and auditory stimuli; enhanced sense of touch, taste, and smell; increased appetite and ability to appreci-ate the flavor of food; and distortion of time perception such that short spans seem much longer than they really are. In addition to these effects, which might be considered pleasurable (or at least innocuous), moderate doses can produce undesirable responses. Among these are impairment of short-term memory; decreased capacity to perform multistep tasks; slowed reaction time and impairment of motor coordination (which can make driving dangerous); altered judgment and decision making (which can lead to high-risk sexual behavior); temporal dis-integration (inability to distinguish between past, present, and future); depersonalization (a sense of strangeness about the self); decreased ability to perceive the emotions of others; and reduced interpersonal interaction.
High-Dose EffectsIn high doses, marijuana can have serious adverse psychologic effects. The user may experience hallucinations, delusions, and paranoia. Euphoria may be displaced by intense anxiety, and a dissociative state may occur in which the user feels “outside of himself or herself.” In extremely high doses, marijuana can produce a state resembling toxic psychosis, which may persist for weeks. Because of the widespread use of marijuana, psychiatric emergencies caused by the drug are relatively common.
Not all users are equally vulnerable to the adverse psycho-logic effects of marijuana. Some individuals experience ill effects only at extremely high doses. In contrast, others routinely experience adverse effects at moderate doses.
Effects of Chronic UseChronic, excessive use of marijuana is associated with a behavioral phenomenon known as an amotivational syndrome, characterized by apathy, dullness, poor grooming, reduced interest in achievement, and disinterest in the pursuit of con-ventional goals. The precise relationship between marijuana and development of the syndrome is not known, nor is it certain what other factors may contribute. Available data do not suggest that the amotivational syndrome is due to organic brain damage.
Role in SchizophreniaMarijuana use is associated with an increased risk of schizo-phrenia. In young people with no history of psychotic symptoms, marijuana increases the risk of symptom occurrence. In people who already have symptoms, marijuana may prolong symptom persistence. In the stabilized schizophrenic, marijuana may precipitate an acute psychotic episode.
Physiologic EffectsCardiovascular EffectsMarijuana produces a dose-related increase in heart rate. Increases of 20 to 50 beats/min are typical. However, rates

452
UNIT V Central Nervous System Drugs
increase in the legalization of marijuana, medical studies have increased. In 2016 multiple studies were conducted regarding marijuana’s role in the treatment of epilepsy and migraine headaches, as well as its effects on the general health of frequent users.
Legal Status of Medical MarijuanaUnited States. Twenty-eight statesa and the District of
Columbia have enacted laws that eliminate criminal penalties for medical use of marijuana, and more states are considering doing the same. Because of the new state laws, patients can now possess and use small amounts of marijuana for medical purposes. In most of these states, qualified patients must have a debilitating medical condition plus documentation from their physicians that medical use of marijuana “may be of benefit.”
What about federal marijuana regulations? Because marijuana is classified by the DEA as a Schedule I substance, physicians still cannot prescribe the drug—all they can do is suggest it may be of benefit. Furthermore, in 2005, the U.S. Supreme Court ruled that DEA legislation trumps the new state laws, and hence people who use or provide medical marijuana can still be prosecuted under federal law, even in states where medical marijuana has been legalized. In a (delayed) response to this ruling, the Department of Justice, in 2009, instructed U.S. attorneys not to use federal resources to prosecute people whose actions comply with state laws that allow medical marijuana use. Hence, although patients may be breaking federal law, the Department of Justice will not prosecute them.
Canada. Medical use of marijuana has been legal in Canada since 2001, when the Marijuana Medical Access Regulations took effect. Patients with documentation from a physician can get their marijuana through Health Canada, or they can get a license to grow their own. Marijuana supplies for Health Canada are grown and distributed by Prairie Plant Systems.
Comparison of Marijuana With AlcoholIn several important ways, responses to marijuana and alcohol are quite different. Whereas increased hostility and aggression are common sequelae of alcohol consumption, aggressive behavior is rare among marijuana users. Although loss of judgment and control can occur with either drug, these losses are greater with alcohol. For the marijuana user, increased appetite and food intake are typical. In contrast, heavy drinkers often suffer nutritional deficiencies. Lastly, whereas marijuana can cause toxic psychosis, dissociative phenomena, and paranoia, these severe acute psychologic reactions rarely occur with alcohol.
Synthetic MarijuanaSynthetic cannabis blends showed up on the market in the early 2000s. They became popular because of their availability and their lack of traces in drug tests. They were initially legal, as they were thought to contain blends of natural herbs sprayed
Approved Uses for CannabinoidsSuppression of Emesis. Intense nausea and vomiting are
common side effects of cancer chemotherapy. In certain patients, these responses can be suppressed more effectively with can-nabinoids than with traditional antiemetics (e.g., prochlorpera-zine, metoclopramide). At this time, two cannabinoids—dronabinol [Marinol] and nabilone [Cesamet]—are available for antiemetic use. Dronabinol, a synthetic form of THC, is a Schedule III drug. Nabilone, a THC derivative, is a Schedule II drug. Dosage forms and dosages are presented in Chapter 80.
Appetite Stimulation. Dronabinol is FDA approved for stimulating appetite in patients with AIDS. By relieving anorexia, treatment may prevent or reverse loss of weight.
Relief of Neuropathic Pain. In 2005, Canadian regulators approved nabiximols [Sativex ], administered by oral spray, for treating neuropathic pain caused by multiple sclerosis (MS). Nabiximols is a mixture of two cannabinoids: THC and can-nabidiol. Because the cannabinoids in Sativex are absorbed through the oral mucosa, the product has a rapid onset (like smoked marijuana), while being devoid of the dangerous tars in marijuana smoke. In the United States, nabiximols is under study for treating intractable cancer pain. However, the drug is not yet approved in the United States, and cannot be legally imported, owing to its current classification as a Schedule I substance.
Unapproved Uses for CannabinoidsGlaucoma. In patients with glaucoma, smoking marijuana
may reduce intraocular pressure. Unfortunately, marijuana may also reduce blood flow to the optic nerve. We don’t know whether the drug improves vision.
Multiple Sclerosis. Whether smoked or ingested, marijuana appears to reduce spasticity and tremor of MS. Oral cannabi-noids may also reduce urge incontinence. As previously mentioned, cannabinoids can reduce neuropathic pain of MS.
Medical Research on MarijuanaProponents of making marijuana available by prescription argue that smoked marijuana can reduce chronic pain, suppress nausea caused by chemotherapy, improve appetite in patients with AIDS, lower intraocular pressure in patients with glaucoma, and suppress spasticity associated with MS and spinal cord injury. However, the evidence supporting most of these claims is weak—largely because federal regulations had effectively barred marijuana research.
In 1999, two developments opened the doors to marijuana research. First, an expert panel, convened by the National Academy of Sciences’ Institute of Medicine, recommended that clinical trials on marijuana proceed. Because smoking marijuana poses a risk of lung cancer and other respiratory disorders, the panel also recommended development of a rapid-onset nonsmoked delivery system. In response to this report and to pressure from scientists and voters, the gov-ernment created new guidelines that loosened restraints on marijuana research. Under the guidelines, researchers will be allowed to purchase marijuana directly from the federal government. (On behalf of the government, the University of Mississippi maintains a plot of marijuana on 1.8 closely guarded acres.) The only catch is that all proposed research must be approved by the FDA, the National Institute on Drug Abuse, and the Drug Enforcement Agency (DEA). Due to the
aAlaska, Arizona, Arkansas, California, Colorado, Connecticut, Delaware, Florida, Hawaii, Illinois, Louisiana, Maine, Maryland, Michigan, Minnesota, Montana, Nevada, New Hampshire, New Jersey, New Mexico, New York, North Dakota, Ohio, Oregon, Pennsylvania, Rhode Island, Vermont, and Washington.

453
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
Mechanism of ActionLSD acts at multiple sites in the brain and spinal cord. However, effects are most prominent in the cerebral cortex and the locus ceruleus. Effects are thought to result from activation of serotonin2 receptors. This concept has been reinforced by the observation that ritanserin, a selective blocker of serotonin2 receptors, can prevent the effects of LSD in animals.
Time CourseLSD is usually administered orally but can also be injected or smoked. With oral dosing, initial effects can be felt in minutes. Over the next few hours, responses become progressively more intense, and then subside 8 to 12 hours later.
Subjective and Behavioral EffectsResponses to LSD can be diverse, complex, and changeable. The drug can alter thinking, feeling, perception, sense of self, and sense of relationship with the environment and other people. LSD-induced experiences may be sublime or terrifying. Just what will be experienced during any particular “trip” cannot be predicted.
Perceptual alterations can be dramatic. Colors may appear iridescent or glowing, kaleidoscopic images may appear, and vivid hallucinations may occur. Sensory experiences may merge so that colors seem to be heard and sounds seem to be visible. Afterimages may occur, causing current perceptions to overlap with preceding perceptions. The LSD user may feel a sense of wonderment and awe at the beauty of commonplace things.
LSD can have a profound impact on affect. Emotions may range from elation, good humor, and euphoria to sadness, dysphoria, and fear. The intensity of emotion may be overwhelming.
Thoughts may turn inward. Attitudes may be re-evaluated, and old values assigned new priorities. A sense of new and important insight may be felt. However, despite the intensity of these experiences, enduring changes in beliefs, behavior, and personality are rare.
Physiologic EffectsLSD has few physiologic effects. Activation of the sympathetic nervous system can produce tachycardia, elevation of blood pressure, mydriasis, piloerection, and hyperthermia. Neuro-muscular effects (tremor, incoordination, hyperreflexia, and muscular weakness) may also occur.
Tolerance and DependenceTolerance to LSD develops rapidly. Substantial tolerance can be seen after just three or four daily doses. Tolerance to subjec-tive and behavioral effects develops to a greater extent than to cardiovascular effects. Cross-tolerance exists with mescaline and psilocybin, but not with DMT. Since DMT is similar to LSD, the absence of cross-tolerance is surprising. There is no cross-tolerance with amphetamines or THC. Upon cessation of LSD use, tolerance rapidly fades. Abrupt withdrawal of LSD is not associated with an abstinence syndrome. Hence there is no evidence for physical dependence.
ToxicityToxic reactions are primarily psychologic. LSD has never been a direct cause of death, although fatalities have occurred from accidents and suicides.
with chemicals that mimic the effects of THC. These chemicals come in two classes: THC analogs and other compounds.
Although synthetic marijuana was once thought harmless, the American Association of Poison Control Centers reported more than 7000 calls regarding synthetic marijuana in a 2-year period. In addition, several deaths and many episodes of florid psychosis and toxicity are possibly related to synthetic marijuana use. Side effects include hypertension, nausea, vomiting, anxiety, agitation, paranoid behavior, hallucinations, and catatonic state. By 2011, the FDA had placed many of these chemicals on the controlled substances list as Schedule I drugs, making them illegal to possess. However, more than 500 compounds exist; therefore, synthetic marijuana is still available. Many organiza-tions, including the U.S. military, have banned all similar compounds.
PSYCHEDELICSThe psychedelics are a fascinating drug family for which LSD can be considered the prototype. Other family members include mescaline, dimethyltryptamine (DMT), psilocin, and salvia. The psychedelics are so named because of their ability to produce what has been termed a psychedelic state. Individuals in this state show an increased awareness of sensory stimuli and are likely to perceive the world around them as beautiful and harmonious; the normally insignificant may assume exceptional meaning, the “self” may seem split into an “observer” and a “doer,” and boundaries between “self” and “nonself” may fade, producing a sense of unity with the cosmos.
Psychedelic drugs are often referred to as hallucinogens or psychotomimetics. These names reflect their ability to produce hallucinations as well as mental states that resemble psychoses.
Although psychedelics can cause hallucinations and psychotic-like states, these are not their most characteristic effects. The characteristic that truly distinguishes the psychedel-ics from other agents is their ability to bring on the same types of alterations in thought, perception, and feeling that otherwise occur only in dreams. In essence, the psychedelics seem able to activate mechanisms for dreaming without causing unconsciousness.
d-Lysergic Acid Diethylamide (LSD)HistoryThe first person to experience LSD was a Swiss chemist named Albert Hofmann. In 1943, 5 years after LSD was first synthe-sized, Hofmann accidentally ingested a minute amount of the drug. The result was a dream-like state accompanied by per-ceptual distortions and vivid hallucinations. The high potency and unusual actions of LSD led to speculation that it might provide a model for studying psychosis. Unfortunately, that speculation did not prove correct: Extensive research has shown that the effects of LSD cannot be equated with idiopathic psychosis. With the realization that LSD did not produce a “model psychosis,” medical interest in the drug declined. Not everyone, however, lost interest; during the 1960s, nonmedical experimentation flourished. This widespread use caused substantial societal concern, and by 1970 LSD had been clas-sified as a Schedule I substance. Nonetheless, street use of LSD continues.

454
UNIT V Central Nervous System Drugs
Like other psychedelic drugs, salvia induces a dream-like state of unreality. Users may lose awareness of their own bodies and of the room they are in. They may feel they are floating, traveling through time and space, or merging with or transform-ing into objects. Some feel they are being twisted or pulled. There may be a sense of overlapping realities and of being in several places at once. Speech may become slurred, and sentences may lack fluent structure. Uncontrollable laughter may break out. Possible physical effects include chills, dizziness, nausea, incoordination, and bradycardia. Whether saliva poses long-term health risks has not been studied. However, we do know that Mexican Indians have used the drug for generations, with no apparent ill effects.
Mescaline, Psilocin, Psilocybin, and DimethyltryptamineIn addition to LSD and salvia, the family of psychedelic drugs includes mescaline, psilocin, psilocybin, DMT, and several related compounds. Some psychedelics are synthetic, and some occur naturally. DMT and LSD represent the synthetic com-pounds. Mescaline, a constituent of the peyote cactus, and psilocin and psilocybin, constituents of “magic mushrooms,” represent compounds found in nature.
The subjective and behavioral effects of the miscellaneous psychedelic drugs are similar to those of LSD. Like LSD, these drugs can elicit modes of thought, perception, and feeling that are normally restricted to dreams. In addition, they can cause hallucinations and induce mental states that resemble psychosis.
The miscellaneous psychedelics differ from LSD with respect to potency and time course. LSD is the most potent of the psychedelics, producing its full spectrum of effects at doses as low as 0.5 mcg/kg. Psilocin and psilocybin are 100 times less potent than LSD, and mescaline is 4000 times less potent than LSD. Whereas the effects of LSD are prolonged (responses may last 12 or more hours), the effects of mescaline and DMT are shorter: Responses to mescaline usually fade within 8 to 12 hours, and responses to DMT fade within 1 to 2 hours.
None of these psychedelics is approved for medical use. The use of psilocybin in patients with terminal cancer and obsessive-compulsive disorder is under investigation.
DISSOCIATIVE DRUGSThe dissociative drugs—phencyclidine and ketamine—were originally developed as surgical anesthetics. When taken recreationally, these drugs distort perception of sight and sound, and produce feelings of dissociation (detachment) from the environment. High doses can produce sedation, immobility, analgesia, and amnesia.
PhencyclidinePhencyclidine (“PCP,” “angel dust,” “peace pill”) was originally developed as an anesthetic for animals. The drug was tried briefly as a general anesthetic for humans, but was withdrawn, owing to severe emergence delirium. Although rejected for therapeutic use, phencyclidine has become widely used as a drug of abuse, largely because it can be synthesized easily by amateur chemists, making it cheap and abundant. The popularity
Acute panic reactions are relatively common and may be associated with a fear of disintegration of the self. Such “bad trips” can usually be managed by a process of “talking down” (providing emotional support and reassurance in a nonthreaten-ing environment). Panic episodes can also be managed with an antianxiety agent, such as diazepam. Neuroleptics (e.g., haloperidol, chlorpromazine) may actually intensify the experi-ence, and hence their use is questionable.
A small percentage of former LSD users experience episodic visual disturbances, referred to as flashbacks by users and hallucinogen persisting perception disorder (HPPD) by clini-cians. These disturbances may manifest as geometric pseudohal-lucinations, flashes of color, or positive afterimages. Visual disturbances may be precipitated by several factors, including marijuana use, fatigue, stress, and anxiety. Phenothiazines exacerbate these experiences rather than providing relief. HPPD appears to be caused by permanent changes in the visual system.
In addition to panic reactions and visual disturbances, LSD can cause other adverse psychologic effects. Depressive epi-sodes, dissociative reactions, and distortions of body image may occur. When an LSD experience has been intensely ter-rifying, the user may be left with persistent residual fear. The drug may also cause prolonged psychotic reactions. In contrast to acute effects, which differ substantially from symptoms of schizophrenia, prolonged psychotic reactions mimic schizo-phrenia faithfully.
Potential Therapeutic UsesLSD has no recognized therapeutic applications. The drug has been evaluated in subjects with alcohol use disorder, opioid use disorder, and psychiatric disorders, including depression, anxiety, and obsessive-compulsive disorder. In addition, LSD has been studied as a possible means of promoting psychologic well-being in patients with terminal cancer. With the possible exception of some psychiatric disorders (e.g., depression, anxiety), LSD has proved either ineffective or impractical.
SalviaSalvia divinorum is a hallucinogenic herb native to southern Mexico and to Central and South America. Its primary psychoac-tive component is salvinorin A, a potent activator of kappa opioid receptors. The genus Salvia, a part of the mint family, is commonly known as sage—hence the colorful street names for S. divinorum: Magic Mint, Diviner’s Sage, and Sage of the Seers. Salvia is legal in some states, illegal in others, and not yet regulated under the Controlled Substances Act.
Salvia is used extensively in the United States, primarily by teens and young adults. In 2015, 1.8% of high school seniors reported using the drug in the past year. About 15% of Americans older than 12 years report having used a hal-lucinogen such as salvia at least once in their lives (https://www.drugabuse.gov/drugs-abuse/hallucinogens).
How is salvia administered? Among Mexican Indians, the traditional method is to chew the leaves or drink a liquid extract. In contrast, recreational users usually smoke the dried leaves, either in a pipe or rolled in a joint. When the smoke is inhaled, salvinorin A undergoes rapid absorption from the lungs. As a result, psychologic effects begin quickly (in less than 1 minute) and then quickly fade (typically in 5 to 10 minutes).

455
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
antipsychotic drugs, such as haloperidol, are limited. Physical restraint may be needed to prevent self-inflicted harm and to protect others from assault. If respiration is depressed, mechani-cal support of ventilation may be needed. Severe hypertension can be managed with nitroprusside, a vasodilator. Seizures can be controlled with IV diazepam. If fever is high, external cooling can lower temperature. By promoting muscle relaxation, dantrolene [Dantrium] can reduce heat generation and the risk of rhabdomyolysis.
Elimination of phencyclidine can be accelerated by continu-ous gastric lavage and by acidification of the urine with ammonium chloride. Continuous lavage is effective because gastroenteric recirculation keeps delivering drug to the stomach. Acidification of urine may promote phencyclidine excretion by reducing tubular reabsorption of this weak base.
KetamineKetamine is a dissociative anesthetic used in animals and humans (see Chapter 27). The drug can also produce rapid relief of depression. Medicinal ketamine is formulated as an injectable liquid. For recreational use, the liquid is evaporated off, leaving a powder that can be snorted or ingested. Doses typically range from 50 to 200 mg. Much of the ketamine sold on the street was diverted from veterinary offices. Ketamine is regulated as a Schedule III substance.
Ketamine is similar to phencyclidine in structure, mechanism, and effects, although its duration of action is shorter. When the drug is snorted, effects begin in 5 to 15 minutes and persist about an hour. Moderate doses produce effects ranging from a dream-like state to a pleasant sensation of floating to a sense of being separated from one’s body. Some users experience a state known as the K-hole, likened to a near-death experience in which there is a sense of rising above one’s body. For some, the K-hole experience is accompanied by a sense of inner peace and radiant light. For others, the K-hole experience is characterized by a terrifying sense of nearly complete sensory detachment. At high doses, ketamine can cause hallucinations, delirium, amnesia, elevation of blood pressure, and potentially fatal disruption of respiration. Even higher doses can produce unconsciousness and fatal cardiovascular collapse.
Because of its depressant and amnesic effects, ketamine has been used to facilitate sexual assault. The drug is colorless, tasteless, and odorless, and hence can be added to a beverage without detection.
DEXTROMETHORPHANDextromethorphan (DXM) is a cough suppressant widely available in over-the-counter cough and cold remedies (see Chapter 77). At the low doses needed for cough suppression, DXM is devoid of psychologic effects. However, at doses 5 to 10 times higher, DXM can cause euphoria, disorientation, paranoia, and altered sense of time, as well as visual, auditory, and tactile hallucinations. These effects are produced by dextror-phan, a metabolite of DXM that blocks receptors for NMDA. Note that this is the same mechanism used by phencyclidine and ketamine. Many of the products that contain DXM also contain other drugs, including acetaminophen, antihistamines, phenylephrine, and pseudoephedrine. Therefore, when exces-sive doses are taken, users are subject to toxicity from these
of phencyclidine is disturbing in that a high incidence of adverse effects makes phencyclidine one of the most dangerous drugs of abuse.
Chemistry and PharmacokineticsChemistry. Phencyclidine is a weak organic base with high
lipid solubility. The drug is chemically related to ketamine, an unusual general anesthetic (see Chapter 27 and later in this chapter).
Pharmacokinetics. Phencyclidine can be administered orally, intranasally, intravenously, and by smoking. For admin-istration by smoking, the drug is usually sprinkled on plant matter (e.g., oregano, parsley, tobacco, marijuana). Because of its high lipid solubility, phencyclidine is readily absorbed from all sites.
Once absorbed, phencyclidine undergoes substantial gas-troenteric recirculation. Because it is a base, phencyclidine in the blood can be drawn into the acidic environment of the stomach (by the pH partitioning effect); from the stomach, the drug reenters the intestine, from which it is reabsorbed into the blood. This cycling from blood to GI tract and back prolongs the drug’s sojourn in the body. Elimination occurs eventually through a combination of hepatic metabolism and renal excretion.
Mechanism of ActionPhencyclidine acts in the cerebral cortex and limbic system, where it blocks a subset of receptors for glutamate, known as N-methyl-D-aspartate (NMDA) receptor complexes. These glutamate receptors are involved in multiple processes, including learning, memory, emotionality, and perception of pain.
Subjective and Behavioral EffectsPhencyclidine produces a unique set of effects. Hallucinations are prominent. In addition, the drug can produce CNS depres-sion, CNS excitation, and analgesia.
Effects of Low to Moderate Doses. At low doses, phencyclidine produces effects like those of alcohol. Low-dose intoxication is characterized by euphoria, release of inhibitions, and emotional lability. Nystagmus, slurred speech, and motor incoordination may occur too.
As dosage increases, the clinical picture becomes more variable and complex. Symptoms include excitation, disorienta-tion, anxiety, disorganized thoughts, altered body image, and reduced perception of tactile and painful stimuli. Mood may be volatile and hostile. Bizarre behavior may develop. Heart rate and blood pressure are elevated.
High-Dose (Toxic) Effects. High doses can cause severe adverse physiologic and psychologic effects. Death may result from several causes.
Psychologic effects include hallucinations, confusional states, combativeness, and psychosis. The psychosis closely resembles schizophrenia and may persist for weeks. Individuals with pre-existing psychoses are especially vulnerable to psychoto-genic effects and may attempt suicide.
Physiologic effects of high-dose phencyclidine are varied. Extreme overdose can produce hypertension, coma, seizures, and muscular rigidity associated with severe hyperthermia and rhabdomyolysis (disintegration of muscle tissue).
Managing Toxicity. Treatment is primarily supportive. Psychotic reactions are best managed by isolation from external stimuli. “Talking down” is not effective, and the benefits of

456
UNIT V Central Nervous System Drugs
of memory, a brain function mediated in part by serotonin. Memory impairment persists long after MDMA was last taken. Second, the cerebrospinal fluid of long-term MDMA users contains abnormally low concentrations of serotonin metabolites, suggesting a loss of serotonergic neurons. And third, using positron emission tomography to study former MDMA users, researchers demonstrated decreased binding of a ligand selective for the serotonin transporter, indicating damage to serotonergic neurons. In this study, reductions in ligand binding correlated with the extent of MDMA use and not with the duration of abstinence.
MDMA can cause hyperthermia in association with dehydra-tion, hyponatremia, and rhabdomyolysis. Treatment consists of rapid cooling, rehydration, and the administration of dan-trolene [Dantrium], a drug that relaxes skeletal muscle, thereby reducing heat generation and the risk of rhabdomyolysis. The risk of hyperthermia and dehydration could be greatly reduced by providing ample fluids at dance parties known as “raves” and other events where MDMA is likely to be used.
Because of its amphetamine-like actions, MDMA can increase heart rate, blood pressure, and myocardial oxygen consumption. Remarkably, the increases in heart rate and blood pressure equal those produced by maximal doses of dobutamine, a powerful adrenergic agonist (see Chapter 17). Cardiovascular stimulation poses a special risk to users with heart disease.
Potential Medical UseDespite its potential for adverse effects, MDMA also has the potential for therapeutic good, owing largely to its ability to decrease feelings of fear and defensiveness and promote feelings of love, trust, and compassion. Small clinical trials of MDMA for the treatment of post-traumatic stress disorder and in patients with severe anxiety related to terminal cancer were promising, revealing reduced fear of death and decreased anxiety in patients taking MDMA.
INHALANTSThe inhalants are a diverse group of drugs that have one characteristic in common: administration by inhalation. These drugs can be divided into three classes: anesthetics, volatile nitrites, and organic solvents.
AnestheticsProvided that dosage is modest, anesthetics produce subjective effects similar to those of alcohol (euphoria, exhilaration, loss of inhibitions). The anesthetics that have been abused most are nitrous oxide (“laughing gas”) and ether. One reason for the popularity of these drugs is ease of administration: Both agents can be used without exotic equipment. For nitrous oxide, ready availability also promotes use: Small cylinders of the drug, marketed for aerating whipping cream, can be purchased without restriction.
Volatile NitritesFour volatile nitrites—amyl nitrite, butyl nitrite, isobutyl nitrite, and cyclohexyl nitrite—are subject to abuse. These drugs are abused by homosexual males because of an ability to relax
compounds as well as from DXM itself. The principal users of DXM are adolescents and teenagers. Between 1999 and 2004, reports of DXM abuse in these groups rose 10-fold. Many products contain DXM, including Coricidin HBP Cough & Cold, Robitussin DM, and Vicks NyQuil Cough Syrup.
3,4-METHYLENEDIOXYMETHAMPHETAMINE (MDMA, ECSTASY)MDMA, also known as “ecstasy,” is a complex drug with stimulant and psychedelic properties. The drug is structurally related to methamphetamine (a stimulant) and mescaline (a hallucinogen). Low doses produce mild LSD-like psychedelic effects; higher doses produce amphetamine-like stimulant effects. These effects result from (1) blocking reuptake of serotonin and (2) promoting the release of serotonin, dopamine, and norepinephrine. Although MDMA can produce effects that are clearly pleasurable, it can also be dangerous; the biggest concerns are neurotoxicity, seizures, excessive cardiovascular stimulation, and hyperthermia and its sequelae. MDMA is classified as a Schedule I drug.
Time Course and DosageMDMA is usually dosed orally, but may also be snorted, injected, or inserted as a rectal suppository. With oral administra-tion, effects begin in 20 minutes, peak in 2 to 3 hours, and persist 4 to 5 hours. The usual dose is 100 mg or less.
Who Uses MDMA and Why?MDMA is used primarily by adolescents and young adults in cities, in the suburbs, and in rural areas. According to a survey conducted in 2015, the use of ecstasy in school-age children has continued to steadily decline.
Why do people take MDMA? Because it makes them feel very good. The drug can elevate mood, increase sensory awareness, and heighten sensitivity to music. It can also facilitate interpersonal relationships: Users report a sense of closeness with others, a lowering of defenses, reduced anxiety, enhanced communication, and increased sociability.
Adverse EffectsMDMA is not free of risks. The drug can injure serotonergic neurons, stimulate the heart, and raise body temperature to a dangerous level. In addition, it can cause neurologic effects (e.g., seizures, spasmodic jerking, jaw clenching, teeth grinding) and a host of adverse psychologic effects (e.g., confusion, anxiety, paranoia, panic attacks, visual hallucinations, and suicidal thoughts and behavior). Every year, MDMA is associ-ated with several thousand admissions to emergency depart-ments, mainly because of seizures.
MDMA can damage serotonergic neurons, perhaps irrevers-ibly. When administered to rats and primates in doses only 2 to 4 times greater than those that produce hallucinations in humans, MDMA causes irreversible destruction of serotonergic neurons, resulting in passivity and insomnia. At least three lines of evidence suggest that MDMA is also neurotoxic in humans. First, MDMA causes dose-related impairment

457
CHAPTER 40 Substance Use Disorders IV: Major Drugs of Abuse Other Than Alcohol and Nicotine
Acute Pharmacologic EffectsThe acute effects of organic solvents are somewhat like those of alcohol (euphoria, impaired judgment, slurred speech, flushing, CNS depression). In addition, these compounds can cause visual hallucinations and disorientation with respect to time and place. High doses can result in sudden death. Possible causes include anoxia, respiratory depression, vagal stimulation (which slows heart rate), and dysrhythmias.
Chronic ToxicityProlonged use is associated with multiple toxicities. For example, chloroform is toxic to the heart, liver, and kidneys, and toluene can cause severe brain damage and bone marrow depression. Many solvents can damage the heart; fatal dys-rhythmias have occurred secondary to drug-induced heart block.
ManagementManagement of acute toxicity is strictly supportive. The objec-tive is to stabilize vital signs. We have no antidotes for volatile solvents.
ANABOLIC STEROIDSMany athletes take anabolic steroids (androgens) to enhance athletic performance. The principal benefit is increased muscle mass and strength. Because of the massive doses that are employed, the risk of adverse effects is substantial. With long-term steroid use, a disorder develops. Because of their abuse potential, most androgens are now classified as Schedule III drugs. The basic pharmacology of androgens and their abuse by athletes are discussed in Chapter 65.
the anal sphincter, and by males in general because of a reputed ability to prolong and intensify sexual orgasm.
The most pronounced pharmacologic effect of volatile nitrites is venodilation, which causes a pooling of blood in veins, which in turn causes a profound drop in systolic blood pressure. The result is dizziness, light-headedness, palpitations, and possibly pulsatile headache. Effects begin seconds after inhala-tion and fade rapidly. The primary toxicity is methemoglobin-emia, which can be treated with methylene blue and supplemental oxygen.
Nitrites are available from medical and nonmedical sources. Amyl nitrite is used for angina pectoris. Cyclohexyl nitrite is present in room odorizers. Butyl nitrite and isobutyl nitrite are present in products made solely for recreational use. On the street, preparations of amyl nitrite are known as “poppers” or “snappers.” These terms reflect the popping sound made by amyl nitrite ampules when snapped open to allow inhalation.
Organic SolventsA wide assortment of solvents have been inhaled to induce intoxication. These compounds include toluene, gasoline, lighter fluid, paint thinner, nail-polish remover, benzene, acetone, chloroform, and model-airplane glue. These agents are used primarily by children and the very poor—people who, because of age or insufficient funds, lack access to more conventional drugs of abuse. In recent years, the use of inhalants by young children and teens has been rising.
AdministrationSolvents are administered by three processes, referred to as “bagging,” “huffing,” and “sniffing.” Bagging is performed by pouring solvent in a bag and inhaling the vapor. Huffing is performed by pouring the solvent on a rag and inhaling the vapor. Sniffing is performed by inhaling the solvent directly from its container.
KEY POINTS
■ Abuse of OxyContin and other opioid analgesics remains more common than abuse of heroin, although the use of heroin is increasing.
■ Because heroin is very lipid soluble, initial effects are more intense and occur faster than with other opioids.
■ With opioids, tolerance to respiratory depression develops in parallel with tolerance to euphoria. As a result, respiratory depression does not increase as higher doses are taken to produce desired subjective effects.
■ Persons tolerant to one opioid are cross-tolerant to all other opioids.
■ Although the opioid withdrawal syndrome can be extremely unpleasant, it is rarely dangerous.
■ Opioid overdose produces a classic triad of symptoms: respiratory depression, coma, and pinpoint pupils. Death can result.
■ Naloxone, an opioid antagonist, is the treatment of choice for opioid overdose.
■ Naloxone dosage must be titrated carefully because too much naloxone will transport the patient from a state of intoxication to one of withdrawal. Also, since the half-life of naloxone is shorter than the half-lives of the opioids, naloxone must be administered repeatedly until the crisis is over.
■ Because of cross-dependence, methadone can ease with-drawal symptoms in opioid-dependent individuals. To ease withdrawal, methadone is substituted for the abused opioid and then gradually tapered.
■ For patients with opioid use disorder who are not ready for withdrawal, methadone can be used for maintenance therapy or for suppressive therapy. In maintenance therapy, the methadone dosage is equivalent to the dosage of the abused opioid, thereby preventing withdrawal. In suppres-sive therapy, the abuser is rendered opioid tolerant with very high doses of methadone; as a result, the use of street opioids can no longer produce subjective effects.
Continued

458
UNIT V Central Nervous System Drugs
■ Buprenorphine is an alternative to methadone for detoxifica-tion and maintenance of patients with opioid use disorder.
■ In contrast to methadone, which is available only through approved Opioid Treatment Programs, buprenorphine can be prescribed in a primary care setting by any physician or nurse practitioner who has (1) received at least 8 hours of approved training and (2) registered with the Substance Abuse and Mental Health Services Administration.
■ After a patient has undergone opioid detoxification, nal-trexone, an opioid antagonist, can be used to discourage renewed opioid abuse.
■ A sustained-release IM formulation of naltrexone can be dosed just once a month, unlike all other drugs for managing opioid use disorder, which must be dosed once a day.
■ With barbiturates, tolerance develops to subjective effects but not to respiratory depression. As a result, as increasingly large doses are taken to produce subjective effects, the risk of serious respiratory depression increases. (Note that this differs from the situation with opioids.)
■ Individuals who are tolerant to barbiturates show cross-tolerance with other CNS depressants (e.g., alcohol, benzodiazepines, general anesthetics) but not with opioids.
■ Individuals who are physically dependent on barbiturates show cross-dependence with other CNS depressants, but not with opioids.
■ When physical dependence on barbiturates (and other CNS depressants) is great, the associated abstinence syndrome can be severe—sometimes fatal. (Note that this differs from the situation with opioids.)
■ Overdose with barbiturates produces the same triad of symptoms seen with opioids: respiratory depression, coma, and pinpoint pupils. Death can result.
■ In contrast to opioid overdose, barbiturate overdose has no antidote, and hence treatment is only supportive.
■ In contrast to overdose with opioids or barbiturates, overdose with benzodiazepines alone is rarely fatal.
■ If necessary, benzodiazepine overdose can be treated with flumazenil, a benzodiazepine antagonist.
■ The psychologic effects of cocaine result from activation of dopamine receptors secondary to cocaine-induced blockade of dopamine reuptake.
■ Severe overdose with cocaine can produce hyperpyrexia, convulsions, ventricular dysrhythmias, and hemorrhagic stroke; death has occurred. Psychologic effects of overdose include severe anxiety, paranoid ideation, and hallucinations.
■ There is no specific antidote to cocaine overdose. Intravenous diazepam can suppress anxiety, seizures, hypertension, and dysrhythmias. Intravenous nitroprusside or phentolamine can treat severe hypertension.
■ In animals, regular use of cocaine produces sensitization—not tolerance. Whether this is true for humans is unclear.
■ Whether cocaine causes significant physical dependence is in dispute.
■ Psychosocial therapy is considered the cornerstone of treatment for cocaine use disorder. Adding disulfiram may also help.
■ In addition to CNS stimulation, methamphetamine causes vasoconstriction and stimulates the heart. Cardiovascular stimulation may result in hypertension, angina, dysrhyth-mias, and stroke.
■ Regular use of methamphetamine can produce a state that closely resembles paranoid schizophrenia.
■ Although physical dependence on methamphetamine is only moderate, psychologic dependence can be intense. Withdrawal can produce dysphoria and a strong sense of craving.
■ The major psychoactive substance in marijuana is delta-9-tetrahydrocannabinol (THC).
■ THC produces its psychologic effects by activating can-nabinoid receptors in the brain.
■ Marijuana has three principal subjective effects: euphoria, sedation, and hallucinations.
■ Physiologic effects of marijuana, as well as tolerance and physical dependence, are minimal.
■ Marijuana has no approved medical uses, although THC and other purified cannabinoids do.
■ Psychedelic drugs produce alterations in thought, perception, and feeling that otherwise occur only in dreams.
■ Psychedelic drugs are also known as hallucinogens or psychotomimetics—names that reflect their ability to produce hallucinations and mental states that resemble psychosis.
■ Lysergic acid diethylamide (LSD) can be considered the prototype of the psychedelic drugs.
■ LSD produces its effects by activating serotonin2 receptors in the brain.
■ Although tolerance develops to LSD, physiologic effects and physical dependence are minimal.
■ Acute panic reactions to LSD can be managed by “talking down” and by treatment with benzodiazepines. Neuroleptic drugs (e.g., haloperidol) may intensify the reaction.
■ LSD users may experience episodic visual disturbances after discontinuing the drug. In many cases, the underlying cause is a permanent change in the visual system.
■ Some LSD users experience prolonged psychotic reactions that closely resemble schizophrenia.
■ Phencyclidine (PCP) is a dissociative anesthetic that produces alcohol-like effects at low doses and hallucinations and psychotic reactions at high doses.
■ Extreme overdose with phencyclidine can produce hyper-tension, coma, and seizures, as well as muscular rigidity associated with severe hyperthermia and rhabdomyolysis.
■ There is no specific antidote to phencyclidine overdose. “Talking down” is not effective, and antipsychotic drugs are of limited help.
■ Ecstasy (MDMA) produces psychedelic effects at low doses and amphetamine-like stimulation at higher doses.
■ Ecstasy can cause irreversible destruction of serotonergic neurons.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

459
U N I T
VIDRUGS THAT AFFECT FLUID AND ELECTROLYTE BALANCE
C H A P T E R
41 Diuretics
Review of Renal Anatomy and Physiology, p. 459Anatomy, p. 459Physiology, p. 459
Introduction to Diuretics, p. 462How Diuretics Work, p. 462Adverse Impact on Extracellular Fluid, p. 462Classification of Diuretics, p. 462
Loop Diuretics, p. 462Furosemide, p. 462Other Loop Diuretics, p. 464
Thiazides and Related Diuretics, p. 464Hydrochlorothiazide, p. 464Other Thiazide-Type Diuretics, p. 465
Potassium-Sparing Diuretics, p. 465Spironolactone, p. 466Triamterene, p. 466Amiloride, p. 467
Mannitol: An Osmotic Diuretic, p. 467Key Points, p. 468Summary of Major Nursing Implications, p. 468
portion of Henle’s loop is located in the renal cortex and the lower end of the loop descends toward the renal medulla. Without this orientation, the kidney could not produce con-centrated urine.
In addition to the nephrons, the collecting ducts (the tubules into which the nephrons pour their contents) play a critical role in kidney function. The final segment of the distal convoluted tubule (4b) plus the collecting duct into which it empties (5) can be considered a single functional unit: the distal nephron.
PhysiologyOverview of Kidney FunctionsThe kidney serves three basic functions: (1) cleansing of extracellular fluid (ECF) and maintenance of ECF volume and composition; (2) maintenance of acid-base balance; and (3) excretion of metabolic wastes and foreign substances (e.g., drugs, toxins). Of the three, maintenance of ECF volume and composition is the one that diuretics affect most.
The Three Basic Renal ProcessesEffects of the kidney on ECF are the net result of three basic processes: (1) filtration, (2) reabsorption, and (3) active secre-tion. To cleanse the entire ECF, a huge volume of plasma must be filtered. Furthermore, to maintain homeostasis, practically everything that has been filtered must be reabsorbed—leaving behind only a small volume of urine for excretion.
Filtration. Filtration occurs at the glomerulus and is the first step in urine formation. Virtually all small molecules (electrolytes, amino acids, glucose, drugs, metabolic wastes) that are present in plasma undergo filtration. In contrast, cells and large molecules (lipids, proteins) remain behind in the blood. The most prevalent constituents of the filtrate are sodium ions and chloride ions. Bicarbonate ions and potassium ions are also present, but in smaller amounts.
The filtration capacity of the kidney is very large. Each minute the kidney produces 125 mL of filtrate, which adds up to 180 L/day. Since the total volume of ECF is only 12.5 L, the kidneys can process the equivalent of all the ECF in the body every 100 minutes. Hence, the ECF undergoes complete cleansing about 14 times each day.
Be aware that filtration is a nonselective process and there-fore cannot regulate the composition of urine. Reabsorption and secretion—processes that display a significant degree of selectivity—are the primary determinants of what the urine ultimately contains. Of the two, reabsorption is by far the more important.
Diuretics are drugs that increase the output of urine. These agents have two major applications: (1) treatment of hyperten-sion and (2) mobilization of edematous fluid associated with heart failure, cirrhosis, or kidney disease. In addition, because of their ability to maintain urine flow, diuretics are used to prevent renal failure.
REVIEW OF RENAL ANATOMY AND PHYSIOLOGYUnderstanding the diuretic drugs requires a basic knowledge of the anatomy and physiology of the kidney. Therefore, let’s review these topics before discussing the diuretics themselves.
AnatomyThe basic functional unit of the kidney is the nephron. As indicated in Fig. 41.1, the nephron has four functionally distinct regions: (1) the glomerulus, (2) the proximal convoluted tubule, (3) the loop of Henle, and (4a, 4b) the distal convoluted tubule. All nephrons are oriented within the kidney such that the upper

460
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
filtrate, reabsorption of these ions is of greatest interest. As we discuss reabsorption, numeric values are given for the percentage of solute reabsorbed at specific sites along the nephron. Bear in mind that these values are only approximate. Fig. 41.2 depicts the sites of sodium and chloride reabsorption, indicating the amount of reabsorption that occurs at each site.
Proximal Convoluted Tubule. The proximal convoluted tubule (PCT) has a high reabsorptive capacity. A large fraction (about 65%) of filtered sodium and chloride is reabsorbed at the PCT. In addition, essentially all of the bicarbonate and potassium in the filtrate is reabsorbed here. As sodium, chloride, and other solutes are actively reabsorbed, water follows pas-sively. Since solutes and water are reabsorbed to an equal extent, the tubular urine remains isotonic (300 mOsm/L). By the time the filtrate leaves the PCT, sodium and chloride are the only solutes that remain in significant amounts.
Loop of Henle. The descending limb of the loop of Henle is freely permeable to water. Hence, as tubular urine moves down the loop and passes through the hypertonic environment of the renal medulla, water is drawn from the loop into the interstitial space. This process decreases the volume of the tubular urine and causes the urine to become concentrated (tonicity increases to about 1200 mOsm/L).
Within the thick segment of the ascending limb of the loop of Henle, about 20% of filtered sodium and chloride is
Reabsorption. More than 99% of the water, electrolytes, and nutrients that are filtered at the glomerulus undergo reabsorption. This conserves valuable constituents of the filtrate while allowing wastes to undergo excretion. Reabsorption of solutes (e.g., electrolytes, amino acids, glucose) takes place by way of active transport. Water then follows passively along the osmotic gradient created by solute reuptake. Specific sites along the nephron at which reabsorption takes place are dis-cussed later in this chapter. Diuretics work primarily by interfering with reabsorption.
Active Tubular Secretion. The kidney has two major kinds of “pumps” for active secretion. These pumps transport compounds from the plasma into the lumen of the nephron. One pump transports organic acids and the other transports organic bases. Together, these pumps can promote the excretion of a wide assortment of molecules, including metabolic wastes, drugs, and toxins. The pumps for active secretion are located in the proximal convoluted tubule.
Processes of Reabsorption That Occur at Specific Sites Along the NephronBecause most diuretics act by disrupting solute reabsorption, to understand the diuretics, we must first understand the major processes by which nephrons reabsorb filtered solutes. Because sodium and chloride ions are the predominant solutes in the
21
4a
4b
53 Collectingduct
Fig. 41.1 ■ Schematic representation of a nephron and collecting duct.

461
CHAPTER 41 Diuretics
SPIRONOLACTONE,TRIAMTERENE
Thick segmentascendinglimb ofHenle’s loop
Late distalconvolutedtubule andcollectingduct (distalnephron)
Early distalconvoluted tubule
Proximalconvoluted tubule
Fig. 41.2 ■ Schematic diagram of a nephron showing sites of sodium absorption and diuretic action. The percentages indicate how much of the filtered sodium and chloride are reabsorbed at each site.
potassium excretion—can be viewed as an exchange mechanism. This exchange is shown in Fig. 41.2. Aldosterone promotes sodium-potassium exchange by stimulating cells of the distal nephron to synthesize more of the pumps responsible for sodium and potassium transport.
reabsorbed (see Fig. 41.2). Since, unlike the descending limb, the ascending limb is not permeable to water, water must remain in the loop as reabsorption of sodium and chloride takes place. This process causes the tonicity of the tubular urine to return to that of the original filtrate (300 mOsm/L).
Distal Convoluted Tubule (Early Segment). About 10% of filtered sodium and chloride is reabsorbed in the early segment of the distal convoluted tubule. Water follows passively.
Distal Nephron: Late Distal Convoluted Tubule and Collecting Duct. The distal nephron is the site of two important processes. The first involves exchange of sodium for potas-sium and is under the influence of aldosterone. The second determines the final concentration of the urine and is regulated by antidiuretic hormone (ADH). Although sodium-potassium exchange is discussed in more detail, we will not continue discussion of ADH, as it has little to do with the actions of diuretics.
Sodium-Potassium Exchange. Aldosterone, the principal mineralocorticoid of the adrenal cortex, stimulates reabsorp-tion of sodium from the distal nephron. At the same time, aldosterone causes potassium to be secreted. Although not directly coupled, these two processes—sodium retention and
Prototype Drugs
DIURETICSLoop DiureticsFurosemide
Thiazide DiureticsHydrochlorothiazide
Potassium-Sparing DiureticsSpironolactoneTriamterene

462
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
these drugs are employed primarily to lower intraocular pressure (IOP) and not to increase urine production. Conse-quently, the carbonic anhydrase inhibitors are discussed in Chapter 104.
LOOP DIURETICSThe loop agents are the most effective diuretics available. These drugs produce more loss of fluid and electrolytes than any other diuretics. They are known as loop diuretics because their site of action is in the loop of Henle.
FurosemideFurosemide [Lasix] is the most frequently prescribed loop diuretic and will serve as our prototype for the group.
Mechanism of ActionFurosemide acts in the thick segment of the ascending limb of Henle’s loop to block reabsorption of sodium and chloride (see Fig. 41.2). By blocking solute reabsorption, furosemide prevents passive reabsorption of water. Since a substantial amount (20%) of filtered NaCl is normally reabsorbed in the loop of Henle, interference with reabsorption here can produce profound diuresis.
PharmacokineticsFurosemide can be administered orally, IV, and IM. With oral administration, diuresis begins in 60 minutes and persists for 8 hours. Oral therapy is used when rapid onset is not required. Effects of IV furosemide begin within 5 minutes and last for 2 hours. Intravenous therapy is used in critical situations (e.g., pulmonary edema) that demand immediate mobilization of fluid. Furosemide undergoes hepatic metabolism followed by renal excretion.
Therapeutic UsesFurosemide is a powerful drug that is generally reserved for situations that require rapid or massive mobilization of fluid. This drug should be avoided when less efficacious diuretics (thiazides) will suffice. Conditions that justify use of furosemide include (1) pulmonary edema associated with congestive heart failure (CHF); (2) edema of hepatic, cardiac, or renal origin that has been unresponsive to less efficacious diuretics; and (3) hypertension that cannot be controlled with other diuretics. Furosemide is especially useful in patients with severe renal impairment, since, unlike the thiazides (see later in the chapter), the drug can promote diuresis even when renal blood flow and glomerular filtration rate (GFR) are low. If treatment with furosemide alone is insufficient, a thiazide diuretic may be added to the regimen. There is no benefit to combining furo-semide with another loop diuretic.
Adverse EffectsHyponatremia, Hypochloremia, and Dehydration. Furo-
semide can produce excessive loss of sodium, chloride, and water. Severe dehydration can result. Signs of evolving dehydra-tion include dry mouth, unusual thirst, and oliguria (scanty urine output). Impending dehydration can also be anticipated from excessive loss of weight. If dehydration occurs, furosemide should be withheld.
INTRODUCTION TO DIURETICSHow Diuretics WorkMost diuretics share the same basic mechanism of action: blockade of sodium and chloride reabsorption. By blocking the reabsorption of these prominent solutes, diuretics create osmotic pressure within the nephron that prevents the passive reabsorption of water. Hence, diuretics cause water and solutes to be retained within the nephron and thereby promote the excretion of both.
The increase in urine flow that a diuretic produces is directly related to the amount of sodium and chloride reabsorption that it blocks. Accordingly, drugs that block solute reabsorption to the greatest degree produce the most profound diuresis. Since the amount of solute in the nephron becomes progressively smaller as filtrate flows from the proximal tubule to the col-lecting duct, drugs that act early in the nephron have the opportunity to block the greatest amount of solute reabsorption. As a result, these agents produce the greatest diuresis. Con-versely, since most of the filtered solute has already been reabsorbed by the time the filtrate reaches the distal parts of the nephron, diuretics that act at distal sites have very little reabsorption available to block. Consequently, such agents produce relatively scant diuresis.
It is instructive to look at the quantitative relationship between blockade of solute reabsorption and production of diuresis. Recall that the kidneys produce 180 L of filtrate a day, practically all of which is normally reabsorbed. With filtrate production at this volume, a diuretic will increase daily urine output by 1.8 L for each 1% of solute reabsorption that is blocked. A 3% blockade of solute reabsorption will produce 5.4 L of urine a day—a rate of fluid loss that would reduce body weight by 12 pounds in 24 hours. Clearly, with only a small blockade of reabsorption, diuretics can produce a profound effect on the fluid and electrolyte composition of the body.
Adverse Impact on Extracellular FluidTo promote excretion of water, diuretics must interfere with the normal operation of the kidney. By doing so, diuretics can cause hypovolemia (from excessive fluid loss), acid-base imbalance, and altered electrolyte levels. These adverse effects can be minimized by using short-acting diuretics and by timing drug administration such that the kidney is allowed to operate in a drug-free manner between periods of diuresis. Both measures will give the kidney periodic opportunities to readjust the ECF so as to compensate for any undesired alterations produced under the influence of diuretics.
Classification of DiureticsThere are four major categories of diuretic drugs: (1) loop diuretics (e.g., furosemide); (2) thiazide diuretics (e.g., hydro-chlorothiazide); (3) osmotic diuretics (e.g., mannitol); and (4) potassium-sparing diuretics. The last group, the potassium-sparing agents, can be subdivided into aldosterone antagonists (e.g., spironolactone) and nonaldosterone antagonists (e.g., triamterene).
In addition to the four major categories of diuretics, there is a fifth group: the carbonic anhydrase inhibitors. Although the carbonic anhydrase inhibitors are classified as diuretics,

463
CHAPTER 41 Diuretics
(LDL) cholesterol and triglycerides. Although these undesirable effects by themselves can increase the risk of coronary heart disease, they are more than balanced by the beneficial effects of the diuretic therapy on the heart. That is, despite adverse effects on lipids, loop diuretics reduce the risk of coronary mortality by 25%.
Furosemide increases urinary excretion of magnesium, posing a risk of magnesium deficiency. Symptoms include muscle weakness, tremor, twitching, and dysrhythmias.
Furosemide increases urinary excretion of calcium. This action has been exploited to treat hypercalcemia.
Drug InteractionsDigoxin. Digoxin is used for heart failure (see Chapter 48)
and cardiac dysrhythmias (see Chapter 49). In the presence of low potassium, the risk of serious digoxin-induced toxicity (ventricular dysrhythmias) is greatly increased. Because loop diuretics promote potassium loss, the use of these drugs in combination with digoxin can increase the risk of dysrhythmia. This interaction is unfortunate in that most patients who take digoxin for heart failure must also take a diuretic as well. To reduce the risk of toxicity, potassium levels should be monitored routinely; when indicated, potassium supplements or a potassium-sparing diuretic should be given.
Ototoxic Drugs. The risk of furosemide-induced hearing loss is increased by concurrent use of other ototoxic drugs—especially aminoglycoside antibiotics (e.g., gentamicin). Accordingly, combined use of these drugs should be avoided.
Potassium-Sparing Diuretics. The potassium-sparing diuretics (e.g., spironolactone, triamterene) can help counterbal-ance the potassium-wasting effects of furosemide, thereby reducing the risk of hypokalemia.
Lithium. Lithium is used to treat bipolar disorder (see Chapter 33). In patients with low sodium, excretion of lithium is reduced. Hence, by lowering sodium levels, furosemide can cause lithium to accumulate to toxic levels. Accordingly, lithium levels should be monitored; if they climb too high, lithium dosage should be reduced.
Antihypertensive Agents. The hypotensive effects of furosemide add to those of other hypotensive drugs. To avoid excessive reduction of blood pressure, patients may need to reduce or eliminate the use of other hypotensive medications.
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). Aspirin and other NSAIDs can attenuate the diuretic effects of furosemide. The mechanism
Dehydration can promote thrombosis and embolism. Symptoms include headache and pain in the chest, calves, or pelvis. The prescriber should be notified if these develop.
The risk of dehydration and its sequelae can be minimized by initiating therapy with low doses, adjusting the dosage carefully, monitoring weight loss every day, and administering furosemide on an intermittent schedule.
Safety Alert
FUROSEMIDEUse of furosemide can produce excessive loss of sodium, chloride, and water. Severe dehydration can result. Dehydration can promote hypotension, thrombosis, and embolism.
Diuretics
Life Stage Patient Care Concerns
Infants See “Breast-feeding women,” below.Children/
adolescentsDiuretics can be used safely in children, just in
smaller doses. Side effect profiles are similar to those for adults.
Pregnant women
Animal studies revealed that furosemide can cause maternal death, abortion, and fetal resorption. Risks and benefits must be considered for administration during pregnancy.
Breast-feeding women
Furosemide may decrease breast milk production through excessive diuresis. Data are lacking regarding transmission of drug from mother to infant via breast milk.
Older adults Diuretics are the most common cause of adverse medication reactions and interactions in older adults. Monitor closely for dehydration and cardiac dysrhythmias.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Hypotension. Furosemide can cause a substantial drop in blood pressure. At least two mechanisms are involved: (1) loss of volume and (2) relaxation of venous smooth muscle, which reduces venous return to the heart. Signs of hypotension include dizziness, light-headedness, and fainting. If blood pressure falls precipitously, furosemide should be discontinued. Because of the risk of hypotension, blood pressure should be monitored routinely.
Outpatients should be taught to monitor their blood pressure and instructed to notify the prescriber if it drops substantially. Also, patients should be informed about symptoms of postural hypotension (dizziness, light-headedness) and advised to sit or lie down if these occur. Patients should be taught that postural hypotension can be minimized by rising slowly.
Hypokalemia. Potassium is lost through increased secretion in the distal nephron. If serum potassium falls below 3.5 mEq/L, fatal dysrhythmias may result. As discussed later under Drug Interactions, loss of potassium is of special concern for patients taking digoxin, a drug for heart failure. Hypokalemia can be minimized by consuming potassium-rich foods (e.g., dried fruits, nuts, spinach, potatoes, bananas), taking potassium supplements, or using a potassium-sparing diuretic.
Ototoxicity. Rarely, loop diuretics cause hearing impair-ment. With furosemide, deafness is transient. With ethacrynic acid (another loop diuretic), irreversible hearing loss may occur. The ability to impair hearing is unique to the loop diuretics. Diuretics in other classes are not ototoxic. Because of the risk of hearing loss, caution is needed when loop diuretics are used in combination with other ototoxic drugs (e.g., aminoglycoside antibiotics).
Hyperglycemia. Elevation of plasma glucose is a potential, albeit uncommon, complication of furosemide therapy. Hyperglycemia appears to result from inhibition of insulin release. Increased glycogenolysis and decreased glycogen synthesis may also contribute. When furosemide is taken by a diabetic patient, he or she should be especially diligent about monitoring blood glucose content.
Hyperuricemia. Elevation of plasma uric acid is a frequent side effect of treatment. For most patients, furosemide-induced hyperuricemia is asymp-tomatic. However, for patients predisposed to gout, elevation of uric acid may precipitate a gouty attack. Patients should be informed about symptoms of gout (tenderness or swelling in joints) and instructed to notify the prescriber if these develop.
Use in Pregnancy. When administered to pregnant laboratory animals, loop diuretics have caused maternal death, abortion, fetal resorption, and other adverse effects. There are no definitive studies on loop diuretics during human pregnancy. However, given the toxicity displayed in animals, prudence dictates that pregnant patients use these drugs only if absolutely required.
Impact on Lipids, Magnesium, and Calcium. Furosemide reduces high-density lipoprotein (HDL) cholesterol and raises low-density lipoprotein

464
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
use in hypertension, a very common disorder, hydrochloro-thiazide is one of our most widely used drugs.
Mechanism of ActionHydrochlorothiazide promotes urine production by blocking the reabsorption of sodium and chloride in the early segment of the distal convoluted tubule (see Fig. 41.2). Retention of sodium and chloride in the nephron causes water to be retained as well, thereby producing an increased flow of urine. Because only 10% of filtered sodium and chloride is normally reabsorbed at the site where thiazides act, the maximum urine flow these drugs can produce is lower than with the loop diuretics.
The ability of thiazides to promote diuresis is dependent on adequate kidney function. These drugs are ineffective when GFR is low (less than 15 to 20 mL/min). Hence, in contrast to the loop diuretics, thiazides cannot be used to promote fluid loss in patients with severe renal impairment.
PharmacokineticsDiuresis begins about 2 hours after oral administration. Effects peak within 4 to 6 hours and may persist up to 12 hours. Most of the drug is excreted unchanged in the urine.
Therapeutic UsesEssential Hypertension. The primary indication for
hydrochlorothiazide is hypertension, a condition for which thiazides are often drugs of first choice. For many hypertensive patients, blood pressure can be controlled with a thiazide alone, although many other patients require multiple-drug therapy. The role of thiazides in hypertension is discussed in Chapter 47.
Edema. Thiazides are preferred drugs for mobilizing edema associated with mild to moderate heart failure. They are also given to mobilize edema associated with hepatic or renal disease.
Diabetes Insipidus. Diabetes insipidus is a rare condition characterized by excessive production of urine. In patients with this disorder, thiazides reduce urine production by 30% to 50%. The mechanism of this paradoxical effect is unclear.
Protection Against Postmenopausal Osteoporosis. Thiazides promote tubular reabsorption of calcium and may thereby decrease the risk of osteoporosis in postmenopausal women. Here’s how. Before menopause, estrogen from the ovaries acts on renal tubules to promote calcium reabsorption. When menopause occurs, estrogen levels drop, allowing renal excretion of calcium to increase. The resultant decrease in circulating calcium promotes mobilization of calcium from bone and thereby increases the risk of osteoporosis. Because thiazides promote renal calcium retention, they may counteract the calcium loss associated with menopause and may thereby help preserve bone integrity.
appears to be inhibition of prostaglandin synthesis in the kidney. Part of the diuretic effect of furosemide results from increasing renal blood flow, which is thought to occur through a prostaglandin-mediated process. By inhibiting prostaglandin synthesis, NSAIDs prevent the increase in renal blood flow and thereby partially blunt diuretic effects.
Preparations, Dosage, and AdministrationOral. Furosemide [Lasix] is available in tablets (20, 40, and 80 mg) and
in solution (10 mg/mL, 40 mg/5 mL) for oral use. The initial dosage for adults is 20 to 80 mg/day as a single dose. The maximum daily dosage is 600 mg. Twice-daily dosing (8:00 AM and 2:00 PM) is common. Dosing late in the day produces nocturia and should be avoided.
Parenteral. Furosemide is available in solution (10 mg/mL) for IV and IM administration. The usual dosage for adults is 20 to 40 mg, repeated in 1 or 2 hours if needed. Intravenous administration should be done slowly (over 1 to 2 minutes). For high-dose therapy, furosemide can be administered by continuous infusion at a rate of 4 mg/min or slower.
Other Loop DiureticsIn addition to furosemide, three other loop diuretics are available: ethacrynic acid [Edecrin], torsemide [Demadex], and bumetanide [Burinex , generic only in United States]. All three are much like furosemide. They all promote diuresis by inhibiting sodium and chloride reabsorption in the thick ascending limb of the loop of Henle. All are approved for edema caused by heart failure, chronic renal disease, and cirrhosis, but only torsemide, like furosemide, is also approved for hypertension. All can cause ototoxicity, hypovolemia, hypotension, hypokalemia, hyperuricemia, hyperglycemia, and disruption of lipid metabolism, specifically, reduction of HDL cholesterol and elevation of LDL cholesterol and triglycerides. Lastly, they all share the same drug interac-tions: Their effects can be blunted by NSAIDs, they can intensify ototoxicity caused by aminoglycosides, they can increase cardiotoxicity caused by digoxin, and they can cause lithium to accumulate to toxic levels. Routes, dosages, and time courses are shown in Table 41.1.
THIAZIDES AND RELATED DIURETICSThe thiazide diuretics (also known as benzothiadiazides) have effects similar to those of the loop diuretics. Like the loop diuretics, thiazides increase renal excretion of sodium, chloride, potassium, and water. In addition, thiazides elevate plasma levels of uric acid and glucose. The principal difference between the thiazides and loop diuretics is that the maximum diuresis produced by the thiazides is considerably lower than the maximum diuresis produced by the loop diuretics. In addition, whereas loop diuretics can be effective even when urine flow is decreased, thiazides cannot.
HydrochlorothiazideHydrochlorothiazide is the most widely used thiazide diuretic and will serve as our prototype for the group. Because of its
Drug Route
Time Course
Dosage (mg) Doses/DayOnset (min) Duration (hr)
Furosemide [Lasix] Oral Within 60 6–8 20–80 1–2IV or IM Within 5 2 20–40 1–2
Ethacrynic acid [Edecrin] Oral Within 30 6–8 50–100 1–2IV Within 5 2 50 1–2
Bumetanide [Burinex , generic only in United States] Oral 30–60 4–6 0.5–2 1IV Within a few 0.5–1 0.5–1 1–3
Torsemide [Demadex] Oral Within 60 6–8 5–20 1IV Within 10 6–8 5–20 1
TABLE 41.1 ■ Loop Diuretics: Routes, Time Course, and Dosage

465
CHAPTER 41 Diuretics
In contrast to the loop diuretics, the thiazides can be combined with ototoxic agents without an increased risk of hearing loss.Preparations, Dosage, and AdministrationHydrochlorothiazide is supplied in capsules (12.5 mg) and tablets (12.5, 25, and 50 mg). Like most other thiazides, hydrochlorothiazide is administered only by mouth. The usual adult dosage is 25 to 50 mg once or twice daily. To minimize nocturia, the drug should not be administered late in the day. To minimize electrolyte imbalance, the drug should be administered on an intermittent basis (e.g., every other day). In addition to being marketed alone, hydrochlorothiazide is available in fixed-dose combinations with potassium-sparing diuretics and a long list of other drugs: beta blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers, hydralazine, clonidine, and methyldopa (see Chapter 47).
Other Thiazide-Type DiureticsIn addition to hydrochlorothiazide, five other thiazides (and related drugs) are approved for use in the United States (Table 41.2). All have pharmacologic properties similar to those of hydrochlorothiazide. With the exception of chlorothiazide, these drugs are administered only by mouth. Chlorothiazide can be administered IV as well as PO. Although the thiazides differ from one another in milligram potency (see Table 41.2), at therapeutically equivalent doses, all elicit the same degree of diuresis. Although most have the same onset time (1 to 2 hours), these drugs differ significantly with respect to duration of action. As with hydrochlorothiazide, disturbance of electrolyte balance can be minimized through alternate-day dosing. Nocturia can be minimized by avoiding dosing in the late afternoon. Table 41.2 lists three drugs—chlorthalidone, indapamide, and metolazone—that are not true thiazides. However, these agents are very similar to thiazides both in structure and function, and hence are included in the group.
POTASSIUM-SPARING DIURETICSThe potassium-sparing diuretics can elicit two potentially useful responses. First, they produce a modest increase in urine production. Second, they produce a substantial decrease in potassium excretion. Because their diuretic effects are limited, the potassium-sparing drugs are rarely employed alone to promote diuresis. However, because of their marked ability to decrease potassium excretion, these drugs are often used to counteract potassium loss caused by thiazide and loop diuretics.
There are two subcategories of potassium-sparing diuret-ics: aldosterone antagonists and nonaldosterone antagonists. In the United States, only one aldosterone antagonist—spironolactone—is used for diuresis.a Two nonaldosterone
Adverse EffectsThe adverse effects of thiazide diuretics are similar to those of the loop diuretics. In fact, with the exception that thiazides are not ototoxic, the adverse effects of the thiazides and loop diuretics are nearly identical.
Hyponatremia, Hypochloremia, and Dehydration. Loss of sodium, chloride, and water can lead to hyponatremia, hypochloremia, and dehydration. However, since the diuresis produced by thiazides is moderate, these drugs have a smaller impact on sodium, chloride, and water than do the loop diuretics. To evaluate fluid and electrolyte status, electrolyte levels should be determined periodically, and the patient should be weighed on a regular basis.
Hypokalemia. Like the loop diuretics, the thiazides can cause hypokalemia from excessive potassium excretion. As noted, potassium loss is of particular concern for patients taking digoxin. Potassium levels should be measured periodically; if serum potassium falls below 3.5 mEq/L, treatment with potas-sium supplements or a potassium-sparing diuretic should be instituted. Hypokalemia can be minimized by eating potassium-rich foods.
Hyperglycemia. Like the loop diuretics, the thiazides can elevate plasma levels of glucose. Significant hyperglycemia develops only in diabetic patients, who should be especially diligent about monitoring blood glucose. To maintain normal glucose levels, the diabetic patient may require larger doses of insulin or an oral hypoglycemic drug.
Hyperuricemia. The thiazides, like the loop diuretics, can cause retention of uric acid, thereby elevating plasma uric acid. Although hyperuricemia is usually asymptomatic, it may precipitate gouty arthritis in patients with a history of the disorder. Plasma levels of uric acid should be measured periodically.
Impact on Lipids and Magnesium. Thiazides can increase levels of LDL cholesterol, total cholesterol, and triglycerides. Thiazides increase excretion of magnesium, sometimes causing magnesium deficiency. Symptoms include muscle weakness, tremor, twitching, and dysrhythmias.
Drug InteractionsThe important drug interactions of the thiazides are nearly identical to those of the loop diuretics. By promoting potassium loss, thiazides can increase the risk of toxicity from digoxin. By lowering blood pressure, thiazides can augment the effects of other antihypertensive drugs. By promoting sodium loss, thiazides can reduce renal excretion of lithium, thereby causing the drug to accumulate, possibly to toxic levels. NSAIDs may blunt the diuretic effects of thiazides. By counterbalancing the potassium-wasting effects of the thiazides, the potassium-sparing diuretics can help prevent excessive potassium loss.
Generic Name Brand Name
Time Course Optimal Oral Adult Dosage (mg/day)Onset (hr) Duration (hr)
THIAZIDES
Chlorothiazide Diuril 1–2 6–12 500–1000Hydrochlorothiazide Microzide 2 6–12 12.5–25Methyclothiazide Enduron 2 24 2.5–5
RELATED DRUGS
Chlorthalidone Thalitone 2 24–72 50–100Indapamide Lozide , generic only in the United States 1–2 Up to 36 2.5–5
Metolazone Generic only 1 12–24 2.5–20
TABLE 41.2 ■ Thiazides and Related Diuretics: Dosages and Time Course of Effects
aAnother aldosterone antagonist—eplerenone [Inspra]—is available, but the drug is not considered a diuretic. The basic pharmacology of eplerenone and its main use, heart failure, are discussed in Chapters 44 and 47, respectively.

466
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
abnormal heart rhythm), spironolactone should be discontin-ued and potassium intake restricted. Injection of insulin can help lower potassium levels by promoting potassium uptake into cells.
Endocrine Effects. Spironolactone is a steroid derivative with a structure similar to that of steroid hormones (e.g., progesterone, estradiol, testosterone). As a result, spironolactone can cause a variety of endocrine effects, including gynecomastia, menstrual irregularities, impotence, hirsutism, and deepening of the voice.
Benign and Malignant Tumors. When given long term to rats in doses 25 to 250 times those used in humans, spironolactone has caused benign adenomas of the thyroid and testes, malignant mammary tumors, and prolifera-tive changes in the liver. The risk of tumors in humans from use of normal doses is unknown.
Drug InteractionsThiazide and Loop Diuretics. Spironolactone is frequently
combined with thiazide and loop diuretics. The goal is to counteract the potassium-wasting effects of the more powerful diuretic.
Agents That Raise Potassium Levels. Because of the risk of hyperkalemia, caution must be employed when combining spironolactone with potassium supplements, salt substitutes (which contain potassium chloride), or another potassium-sparing diuretic. In addition, three groups of drugs—angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, and direct renin inhibitors—can elevate potassium levels (by suppressing aldosterone secretion), and hence should be combined with spironolactone only when clearly necessary.Preparations, Dosage, and AdministrationSpironolactone [Aldactone] is dispensed in tablets (25, 50, and 100 mg) for oral dosing. The usual adult dosage is 25 to 100 mg/day. Spironolactone is also marketed in a fixed-dose combination with hydrochlorothiazide under the brand name Aldactazide.
Safe Handling and Administration. In 2016, the National Institute for Occupational Safety and Health (NIOSH), a division of the Centers for Disease Control and Prevention, published a list of drugs considered to be potentially hazardous in healthcare settings. Spironolactone was included in this list secondary to its potential to cause fetal harm. Exposure to these drugs can pose a reproductive risk to healthcare workers who administer these drugs. To promote safe administration, NIOSH suggests donning a protective gown and two sets of gloves when cutting or crushing tablets. For further information on this report, visit https://www.cdc.gov/niosh/topics/antineoplastic/pdf/hazardous-drugs-list_2016-161.pdf.
TriamtereneMechanism of ActionLike spironolactone, triamterene [Dyrenium] disrupts sodium-potassium exchange in the distal nephron. However, in contrast to spironolactone, which reduces ion transport indirectly through blockade of aldosterone, triamterene is a direct inhibitor of the exchange mechanism itself. The net effect of inhibition is
antagonists—triamterene and amiloride—are currently employed.
SpironolactoneMechanism of ActionSpironolactone [Aldactone] blocks the actions of aldosterone in the distal nephron. Since aldosterone acts to promote sodium uptake in exchange for potassium secretion (see Fig. 41.2), inhibition of aldosterone has the opposite effect: retention of potassium and increased excretion of sodium. The diuresis caused by spironolactone is scanty because most of the filtered sodium load has already been reabsorbed by the time the filtrate reaches the distal nephron. (Recall that the degree of diuresis a drug produces is directly proportional to the amount of sodium reuptake that it blocks.)
The effects of spironolactone are delayed, taking up to 48 hours to develop (Table 41.3). Recall that aldosterone acts by stimulating cells of the distal nephron to synthesize the proteins required for sodium and potassium transport. By preventing aldosterone’s action, spironolactone blocks the synthesis of new proteins, but does not stop existing transport proteins from doing their job. Therefore, effects are not visible until the existing proteins complete their normal life cycle—a process that takes 1 or 2 days.
Therapeutic UsesHypertension and Edema. Spironolactone is used primar-
ily for hypertension and edema. Although it can be employed alone, the drug is used most commonly in combination with a thiazide or loop diuretic. The purpose of spironolactone in these combinations is to counteract the potassium-wasting effects of the more powerful diuretics. Spironolactone also makes a small contribution to diuresis.
Heart Failure. In patients with severe heart failure, spi-ronolactone reduces mortality and hospital admissions. Benefits derive from protective effects of aldosterone blockade in the heart and blood vessels (see Chapter 48).
Other Uses. In addition to the applications already discussed, spirono-lactone can be used for primary hyperaldosteronism (see Chapter 60), pre-menstrual syndrome (see Chapter 61), polycystic ovary syndrome (see Chapter 63), and acne in young women (see Chapter 105).
Adverse EffectsHyperkalemia. The potassium-sparing effects of spi-
ronolactone can result in hyperkalemia, a condition that can produce fatal dysrhythmias. Although hyperkalemia is most likely when spironolactone is used alone, it can also develop when spironolactone is used in conjunction with potassium-wasting agents (thiazides and loop diuretics). If serum potassium rises above 5 mEq/L or if signs of hyperkalemia develop (e.g.,
Generic Name Brand Name
Time Course
Usual Adult Dosage (mg/day)Onset (hr) Duration (hr)
Spironolactone Aldactone 24–48 48–72 25–200Triamterene Dyrenium 2–4 12–16 50–300Amiloride Midamor 2 24 5–20
TABLE 41.3 ■ Potassium-Sparing Diuretics: Names, Dosages, and Time Course of Effects

467
CHAPTER 41 Diuretics
• Undergoes minimal metabolism.• Is pharmacologically inert (i.e., it has no direct effects on the bio-
chemistry or physiology of cells).Following IV administration, mannitol is filtered by the glomerulus. However, unlike other solutes, the drug undergoes minimal reabsorption. As a result, most of the filtered drug remains in the nephron, creating an osmotic force that inhibits passive reabsorption of water. Hence, urine flow increases. The degree of diuresis produced is directly related to the concentration of mannitol in the filtrate: The more mannitol present, the greater the diuresis. Mannitol has no significant effect on the excretion of potassium and other electrolytes.
PharmacokineticsMannitol does not diffuse across the GI epithelium and cannot be transported by the uptake systems that absorb dietary sugars. Accordingly, to reach the circulation, the drug must be given parenterally. Following IV injection, mannitol distributes freely to extracellular water. Diuresis begins in 30 to 60 minutes and persists 6 to 8 hours. Most of the drug is excreted intact in the urine.
Therapeutic UsesProphylaxis of Renal Failure. Under certain conditions (e.g., dehydra-
tion, severe hypotension, hypovolemic shock), blood flow to the kidney is decreased, causing a great reduction in filtrate volume. When the volume of filtrate is this low, transport mechanisms of the nephron are able to reabsorb virtually all of the sodium and chloride present, causing complete reabsorption of water as well. As a result, urine production ceases, and kidney failure ensues. The risk of renal failure can be reduced with mannitol. Here’s how. Because filtered mannitol is not reabsorbed—even when filtrate volume is small—filtered mannitol will remain in the nephron, drawing water with it. Hence, mannitol can preserve urine flow and may thereby prevent renal failure. Thiazides and loop diuretics are not as effective for this applica-tion because, under conditions of low filtrate production, there is such an excess of reabsorptive capacity (relative to the amount of filtrate) that these drugs are unable to produce sufficient blockade of reabsorption to promote diuresis.
Reduction of Intracranial Pressure. Intracranial pressure (ICP) that has been elevated by cerebral edema can be reduced with mannitol. The drug lowers ICP because its presence in the blood vessels of the brain creates an osmotic force that draws edematous fluid from the brain into the blood. There is no risk of increasing cerebral edema because mannitol cannot exit the capillary beds of the brain.
Reduction of Intraocular Pressure. Mannitol and other osmotic agents can lower IOP by rendering the plasma hyperosmotic with respect to intraocular fluids. The hyperosmotic plasma creates an osmotic force that draws ocular fluid into the blood. Use of mannitol to lower IOP is reserved for patients who have not responded to more conventional treatment.
Adverse EffectsEdema. Mannitol can leave the vascular system at all capillary beds
except those of the brain. When the drug exits capillaries, it draws water along, causing edema. Mannitol must be used with extreme caution in patients with heart disease, since it may precipitate CHF and pulmonary edema. If signs of pulmonary congestion or CHF develop, use of the drug must cease immediately. Mannitol must also be discontinued if patients with heart failure or pulmonary edema develop renal failure, because the resultant accumulation of mannitol would increase the risk of cardiac or pulmonary injury.
Other Adverse Effects. Common responses include headache, nausea, and vomiting. Fluid and electrolyte imbalance may also occur.
Preparations, Dosage, and AdministrationMannitol [Osmitrol] is administered by IV infusion. Solutions for IV use range in concentration from 5% to 25%. Dosing is complex and varies with the objective of therapy (prevention of renal failure, lowering of ICP, lowering of IOP). The usual adult dosage for preventing renal failure is 50 to 100 gm over 24 hours. The infusion rate should be set to elicit a urine flow of at least 30 to 50 mL/hr. It should be noted that mannitol may crystallize out of solution if exposed to low temperature. Accordingly, preparations should be observed for crystals before use. Preparations that contain crystals should be warmed (to redissolve the mannitol) and then cooled to body temperature for administra-tion. A filter needle is employed to withdraw mannitol from the vial, and an in-line filter is used to prevent crystals from entering the circulation. If urine flow declines to a very low rate or ceases entirely, the infusion should be stopped.
a decrease in sodium reabsorption and a reduction in potassium secretion. Hence, sodium excretion is increased, while potassium is conserved. Because it inhibits ion transport directly, triam-terene acts much more quickly than spironolactone. Initial responses develop in hours, compared with days for spirono-lactone. As with spironolactone, diuresis with triamterene is minimal.
Therapeutic UsesTriamterene can be used alone or in combination with other diuretics to treat hypertension and edema. When used alone, triamterene produces mild diuresis. When combined with other diuretics (e.g., furosemide, hydrochlorothiazide), triamterene augments diuresis and helps counteract the potassium-wasting effects of the more powerful diuretic. It is the latter effect for which triamterene is principally employed.
Adverse EffectsHyperkalemia. Excessive potassium accumulation is the
most significant adverse effect. Hyperkalemia is most likely when triamterene is used alone, but can also occur when the drug is combined with thiazides or loop diuretics. Caution should be employed when triamterene is used in conjunction with another potassium-sparing diuretic or with potassium supplements or salt substitutes. In addition, caution is needed if the drug is combined with an ACE inhibitor, angiotensin receptor blocker, or direct renin inhibitor.
Other Adverse Effects. Relatively common side effects include nausea, vomiting, leg cramps, and dizziness. Blood dyscrasias occur rarely.Preparations, Dosage, and AdministrationTriamterene [Dyrenium] is available in 50- and 100-mg capsules for oral use. The usual initial dosage is 100 mg twice a day. The maximum dosage is 300 mg/day. Triamterene is also marketed in fixed-dose combinations with hydrochlorothiazide under the brand names Dyazide and Maxzide.
AmiloridePharmacologic PropertiesAmiloride has actions similar to those of triamterene. Both drugs inhibit potassium loss by direct blockade of sodium-potassium exchange in the distal nephron. Also, both drugs produce only modest diuresis. Although it can be employed alone as a diuretic, amiloride is used primarily to counteract potassium loss caused by more powerful diuretics (thiazides, loop diuretics). The major adverse effect is hyperkalemia. Accordingly, concurrent use of other potassium-sparing diuretics or potassium supplements must be monitored closely. Caution is needed if the drug is combined with an ACE inhibitor, angiotensin receptor blocker, or direct renin inhibitor.
Preparations, Dosage, and AdministrationAmiloride is supplied in 5-mg tablets for oral use. Dosing is begun at 5 mg/day and may be increased to a maximum of 20 mg/day. Amiloride is available in a fixed-dose combination with hydrochlorothiazide.
MANNITOL: AN OSMOTIC DIURETIC
Osmotic diuretics differ from other diuretics with regard to mechanism and uses. At this time, mannitol is the only osmotic diuretic available in the United States. Three related drugs—urea, glycerin, and isosorbide—have been withdrawn.
Mechanism of Diuretic ActionMannitol [Osmitrol] is a simple six-carbon sugar that embodies the four properties of an ideal osmotic diuretic. Specifically, the drug:
• Is freely filtered at the glomerulus.• Undergoes minimal tubular reabsorption.

468
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
KEY POINTS
■ More than 99% of the water, electrolytes, and nutrients that are filtered at the glomerulus undergo reabsorption.
■ Most diuretics block active reabsorption of sodium and chloride, and thereby prevent passive reabsorption of water.
■ The amount of diuresis produced is directly related to the amount of sodium and chloride reabsorption blocked.
■ Drugs that act early in the nephron are in a position to block the greatest amount of solute reabsorption, and hence produce the greatest diuresis.
■ Loop diuretics block sodium and chloride reabsorption in the loop of Henle.
■ Loop diuretics produce the greatest diuresis.■ In contrast to thiazide diuretics, loop diuretics are effective
even when the glomerular filtration rate is low.■ Loop diuretics can cause dehydration through excessive
fluid loss.■ Loop diuretics can cause hypotension by decreasing blood
volume and relaxing venous smooth muscle.■ Loop diuretics can cause hearing loss which, fortunately,
is usually reversible.■ Hypokalemia caused by loop diuretics is a special problem
for patients taking digoxin.■ Thiazide diuretics block sodium and water reabsorption
in the early distal convoluted tubule.■ Thiazide diuretics produce less diuresis than loop diuretics.■ Thiazide diuretics are ineffective when glomerular filtration
rate is low.
■ Like the loop diuretics, thiazide diuretics can cause dehydra-tion and hypokalemia. However, thiazides do not cause hearing loss.
■ Thiazide-induced hypokalemia is a special problem for patients taking digoxin.
■ Potassium-sparing diuretics act by directly or indirectly blocking sodium-potassium “exchange” in the distal convoluted tubule.
■ Potassium-sparing diuretics cause only modest diuresis.■ Potassium-sparing diuretics are used primarily to counteract
potassium loss in patients taking loop diuretics or thiazides.■ The principal adverse effect of potassium-sparing diuretics
is hyperkalemia.■ Because of the risk of hyperkalemia, use caution when
combining potassium-sparing diuretics with one another or with potassium supplements, and in patients taking ACE inhibitors, angiotensin receptor blockers, or direct renin inhibitors.
■ Loop diuretics and thiazides are used to treat hypertension and edema associated with heart failure, cirrhosis, and kidney disease.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
LOOP DIURETICSBumetanideEthacrynic acidFurosemideTorsemide
Preadministration AssessmentTherapeutic GoalLoop diuretics are indicated for patients with (1) pulmonary edema associated with congestive heart failure; (2) edema of hepatic, cardiac, or renal origin that has been unresponsive to less effective diuretics; (3) hypertension that cannot be controlled with thiazide and potassium-sparing diuretics; and (4) all patients who need diuretic therapy but have low renal blood flow.
Baseline DataFor all patients, obtain baseline values for weight, blood pressure (sitting and supine), pulse, respiration, and electrolytes (sodium, potassium, chloride). For patients with edema, record sites and extent of edema. For patients with ascites, measure abdominal girth. For acutely ill patients (e.g., severe CHF), assess lung sounds.
Identifying High-Risk PatientsUse with caution in patients with cardiovascular disease, renal impairment, diabetes mellitus, or a history of gout, and in patients who are pregnant or taking digoxin, lithium, ototoxic drugs, NSAIDs, or antihypertensive drugs.
Implementation: AdministrationRoutes
Furosemide and Bumetanide. Oral, IV, IM.Ethacrynic Acid and Torsemide. Oral, IV.
AdministrationOral. Dosing may be done once daily, twice daily, or on
alternate days. Instruct patients who are using once-a-day or alternate-day dosing to take their medication in the morning. Instruct patients using twice-a-day dosing to take their medication at 8:00 AM and 2:00 PM (to minimize nocturia).
Advise patients to administer furosemide with food if GI upset occurs.
Parenteral. Administer IV injections slowly (over 1 to 2 minutes). For high-dose therapy, administer by continuous infusion. Discard discolored solutions.

469
CHAPTER 41 Diuretics
Promoting AdherenceIncreased frequency of urination is inconvenient and can discourage adherence. To promote adherence, inform patients that treatment will increase urine volume and frequency of voiding, and that these effects will subside 6 to 8 hours after dosing. Inform patients that nighttime diuresis can be mini-mized by avoiding dosing late in the day.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsMonitor blood pressure and pulse rate, weigh the patient daily, and evaluate for decreased edema.
Monitor intake and output. Notify the prescriber if oliguria (urine output less than 25 mL/hr) or anuria (no urine output) develops.
Instruct outpatients to weigh themselves daily (using the same scale), preferably in the morning before eating. Also, instruct them to maintain a weight record and to report excessive weight gain or loss.
In acute conditions requiring rapid diuresis and careful monitoring, a Foley catheter may be used. The catheter should be emptied before drug injection, and output should be monitored hourly and recorded.
Minimizing Adverse EffectsHyponatremia, Hypochloremia, and Dehydration.
Loss of sodium, chloride, and water can cause hyponatremia, hypochloremia, and severe dehydration. Signs of dehydration include dry mouth, unusual thirst, and oliguria. Withhold the drug if these appear.
Dehydration can promote thromboembolism. Monitor the patient for symptoms (headache; pain in the chest, calves, or pelvis), and notify the prescriber if these develop.
The risk of dehydration and its sequelae can be minimized by (1) initiating therapy with low doses, (2) adjusting the dosage carefully, (3) monitoring weight loss daily, and (4) using an intermittent dosing schedule.
Hypotension. Monitor blood pressure. If it falls precipi-tously, withhold medication and notify the prescriber.
Teach patients to monitor their blood pressure, and instruct them to notify the prescriber if it drops substantially.
Inform patients about signs of postural hypotension (diz-ziness, light-headedness), and advise them to sit or lie down if these occur. Inform patients that postural hypotension can be minimized by rising slowly and by dangling legs off the bed before standing.
Hypokalemia. If serum potassium falls below 3.5 mEq/L, fatal dysrhythmias may result. Hypokalemia can be minimized by consuming potassium-rich foods (e.g., nuts, dried fruits, spinach, citrus fruits, potatoes, bananas), taking potassium supplements, or using a potassium-sparing diuretic. Teach patients the signs and symptoms of hypokalemia (e.g., irregular heartbeat, muscle weakness, cramping, flaccid paralysis, leg discomfort, extreme thirst, confusion), and stress the importance of showing up for regular blood tests.
Ototoxicity. Inform patients about possible hearing loss and instruct them to notify the prescriber if a hearing
deficit develops. Exercise caution when loop diuretics are used concurrently with other ototoxic drugs, especially aminoglycosides.
Hyperglycemia. Loop diuretics may elevate blood glucose levels in diabetic patients. Advise these patients to be especially diligent about monitoring blood glucose.
Hyperuricemia. Loop diuretics frequently cause asymp-tomatic hyperuricemia, although gout-prone patients may experience a gouty attack. Inform patients about signs of gout (tenderness or swelling in joints), and instruct them to notify the prescriber if these occur.
Minimizing Adverse InteractionsDigoxin. By lowering potassium levels, loop diuretics
increase the risk of fatal dysrhythmias from digoxin. Serum potassium levels must be monitored and maintained above 3.5 mEq/L.
Lithium. Loop diuretics can suppress lithium excretion, thereby causing the drug to accumulate, possibly to toxic levels. Plasma lithium should be monitored routinely. If drug levels become elevated, lithium dosage should be reduced.
Ototoxic Drugs. The risk of hearing loss from loop diuretics is increased in the presence of other ototoxic drugs, especially aminoglycosides. Exercise caution when such combinations are employed.
THIAZIDES AND RELATED DIURETICSChlorothiazideChlorthalidoneHydrochlorothiazideIndapamideMethyclothiazideMetolazone
Thiazide diuretics have actions much like those of the loop diuretics. Hence, nursing implications for the thiazides are nearly identical to those of the loop diuretics.
Preadministration AssessmentTherapeutic GoalThiazide diuretics are indicated for hypertension and edema.
Baseline DataFor all patients, obtain baseline values for weight, blood pressure (sitting and supine), pulse, respiration, and electrolytes (sodium, chloride, potassium). For patients with edema, record sites and extent of edema.
Identifying High-Risk PatientsUse with caution in patients with cardiovascular disease, renal impairment, diabetes mellitus, or a history of gout and in patients taking digoxin, lithium, or antihypertensive drugs.
Implementation: AdministrationRoutes
Oral. All thiazide-type diuretics.Intravenous. Chlorothiazide.
Summary of Major Nursing Implicationsa—cont’d
Continued

470
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
AdministrationDosing may be done once daily, twice daily, or on alternate days. When once-a-day dosing is employed, instruct patients to take their medicine early in the day to minimize nocturia. When twice-a-day dosing is employed, instruct patients to take their medicine at 8:00 AM and 2:00 PM.
Advise patients to administer thiazides with or after meals if GI upset occurs.
Promoting AdherenceSee nursing implications for Loop Diuretics.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsSee nursing implications for Loop Diuretics.
Minimizing Adverse EffectsLike the loop diuretics, thiazides can cause hyponatremia, hypochloremia, dehydration, hypokalemia, hypotension, hyperglycemia, and hyperuricemia. For implications regarding these effects, see nursing implications for Loop Diuretics.
Minimizing Adverse InteractionsLike loop diuretics, thiazides can interact adversely with digoxin and lithium. For implications regarding these interac-tions, see nursing implications for Loop Diuretics.
POTASSIUM-SPARING DIURETICSAmilorideSpironolactoneTriamterene
Preadministration AssessmentTherapeutic GoalPotassium-sparing diuretics are given primarily to counterbal-ance the potassium-losing effects of thiazide diuretics and loop diuretics.
Baseline DataObtain baseline values for serum potassium, along with baseline values for weight, blood pressure (sitting and supine), pulse, respiration, sodium, and chloride. For patients with edema, record sites and extent of edema.
Identifying High-Risk PatientsPotassium-sparing diuretics are contraindicated for patients with hyperkalemia and should be used with caution in patients taking potassium supplements or another potassium-sparing diuretic. Use with caution in patients taking ACE inhibitors, angiotensin receptor blockers, and direct renin inhibitors.
Implementation: AdministrationRouteOral. Spironolactone has the potential to cause reproductive harm to healthcare workers exposed during administration. NIOSH suggests donning a protective gown and two sets of gloves when crushing or splitting tablets.
AdministrationAdvise patients to take these drugs with or after meals if GI upset occurs.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsMonitor serum potassium levels on a regular basis. The objective is to maintain serum potassium levels between 3.5 and 5 mEq/L.
Minimizing Adverse EffectsHyperkalemia. Hyperkalemia is the principal adverse
effect. Instruct patients to restrict intake of potassium-rich foods. If serum potassium levels rise above 5 mEq/L, or if signs of hyperkalemia develop (e.g., abnormal cardiac rhythm), withhold medication and notify the prescriber. Insulin can be given to (temporarily) decrease potassium levels.
Endocrine Effects. Spironolactone may cause menstrual irregularities and impotence. Inform patients about these effects, and instruct them to notify the prescriber if they occur.
Minimizing Adverse InteractionsDrugs That Raise Potassium Levels. Owing to a risk
of hyperkalemia, use caution when combining a potassium-sparing diuretic with potassium supplements, salt substitutes, or with another potassium-sparing diuretic. Generally avoid combined use with ACE inhibitors, angiotensin receptor blockers, and direct renin inhibitors.
Summary of Major Nursing Implicationsa—cont’d
aPatient education information is highlighted as blue text.

471
C H A P T E R
42 Agents Affecting the Volume and Ion Content of Body Fluids
Disorders of Fluid Volume and Osmolality, p. 471Volume Contraction, p. 471Volume Expansion, p. 472
Acid-Base Disturbances, p. 472Respiratory Alkalosis, p. 472Respiratory Acidosis, p. 472Metabolic Alkalosis, p. 472Metabolic Acidosis, p. 473
Potassium Imbalances, p. 473Regulation of Potassium Levels, p. 473Hypokalemia, p. 473Hyperkalemia, p. 474
Magnesium Imbalances, p. 474Hypomagnesemia, p. 474Hypermagnesemia, p. 475
Key Points, p. 475
isotonic contraction, hypertonic contraction, and hypotonic contraction. Volume expansion may also be subclassified as isotonic, hypertonic, or hypotonic. Descriptions and causes of these abnormal states are discussed in the sections that follow.
In the clinical setting, changes in osmolality are described in terms of the sodium content of plasma. Sodium is used as the reference for classification because this ion is the principal extracellular solute. (Recall that plasma sodium content ranges from 135 to 145 mEq/L.) In most cases, the total osmolality of plasma is about 2 times the osmolality of sodium. That is, total plasma osmolality usually ranges from 280 to 300 mOsm/kg water.
Volume ContractionIsotonic Contraction
Definition and Causes. Isotonic contraction is defined as volume contraction in which sodium and water are lost in isotonic proportions. Hence, although there is a decrease in the total volume of extracellular fluid, there is no change in osmolality. Causes of isotonic contraction include vomiting, diarrhea, kidney disease, and misuse of diuretics. Isotonic contraction is characteristic of cholera, an infection that produces vomiting and severe diarrhea.
Treatment. Lost volume should be replaced with fluids that are isotonic to plasma. This can be accomplished by infusing isotonic (0.9%) sodium chloride in sterile water, a solution in which both sodium and chloride are present at a concentration of 145 mEq/L. Volume should be replenished slowly to avoid pulmonary edema.
Hypertonic ContractionDefinition and Causes. Hypertonic contraction is defined
as volume contraction in which loss of water exceeds loss of sodium. Hence, there is a reduction in extracellular fluid volume coupled with an increase in osmolality. Because of extracellular hypertonicity, water is drawn out of cells, thereby producing intracellular dehydration and partial compensation for lost extracellular volume.
Causes of hypertonic contraction include excessive sweating, osmotic diuresis, and feeding excessively concentrated foods to infants. Hypertonic contraction may also develop secondary to extensive burns or disorders of the central nervous system (CNS) that render the patient unable to experience or report thirst.
Treatment. Volume replacement in hypertonic contraction should be accomplished with hypotonic fluids (e.g., 0.45% sodium chloride) or with fluids that contain no solutes at all. Initial therapy may consist simply of drinking water.
The drugs discussed in this chapter are used to correct distur-bances in the volume and ionic composition of body fluids. Three groups of agents are considered: (1) drugs used to correct disorders of fluid volume and osmolality, (2) drugs used to correct disturbances of hydrogen ion concentration (acid-base status), and (3) drugs used to correct electrolyte imbalances.
DISORDERS OF FLUID VOLUME AND OSMOLALITYGood health requires that both the volume and osmolality of extracellular and intracellular fluids remain within a normal range. If a substantial alteration in either the volume or osmolal-ity of these fluids develops, significant harm can result.
Maintenance of fluid volume and osmolality is primarily the job of the kidneys, and, even under adverse conditions, renal mechanisms usually succeed in keeping the volume and composition of body fluids within acceptable limits. However, circumstances can arise in which the regulatory capacity of the kidneys is exceeded. When this occurs, disruption of fluid volume, osmolality, or both can result.
Abnormal states of hydration can be divided into two major categories: volume contraction and volume expansion. Volume contraction is defined as a decrease in total body water; conversely, volume expansion is defined as an increase in total body water. States of volume contraction and volume expansion have three subclassifications based on alterations in extracel-lular osmolality. For volume contraction, the subcategories are

472
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
of CO2 (secondary to respiratory slowing) tends to lower pH. The kidneys influence pH by regulating bicarbonate excretion. By retaining bicarbonate, the kidneys can raise pH. Conversely, by increasing bicarbonate excretion, the kidneys can lower pH and thereby compensate for alkalosis.
There are four principal types of acid-base imbalance: (1) respiratory alkalosis, (2) respiratory acidosis, (3) metabolic alkalosis, and (4) metabolic acidosis. Causes and treatments are discussed in the sections that follow.
Respiratory AlkalosisCausesRespiratory alkalosis is produced by hyperventilation. Deep and rapid breathing increases CO2 loss, which in turn lowers the pCO2 (partial pressure of carbon dioxide) of blood and increases pH. Mild hyperventilation may result from a number of causes, including hypoxia, pulmonary disease, and drugs (especially aspirin and other salicylates). Severe hyperventilation can be caused by CNS injury and hysteria.
TreatmentManagement of respiratory alkalosis is dictated by the severity of pH elevation. When alkalosis is mild, no specific treatment is indicated. Severe respiratory alkalosis resulting from hysteria can be controlled by having the patient rebreathe his or her CO2-laden expired breath. This can be accomplished by holding a paper bag over the nose and mouth. A sedative (e.g., diazepam [Valium]) can help suppress the hysteria.
Respiratory AcidosisCausesRespiratory acidosis results from retention of CO2 secondary to hypoventilation. Reduced CO2 exhalation raises plasma pCO2, which in turn causes plasma pH to fall. Primary causes of impaired ventilation are (1) depression of the medullary respiratory center and (2) pathologic changes in the lungs (e.g., status asthmaticus, airway obstruction). Over time, the kidneys compensate for respiratory acidosis by excreting less bicarbonate.
TreatmentPrimary treatment of respiratory acidosis is directed at correcting respiratory impairment. The patient may also need oxygen and ventilatory assistance. Infusion of sodium bicarbonate may be indicated if acidosis is severe.
Metabolic AlkalosisCausesMetabolic alkalosis is characterized by increases in both the pH and bicarbonate content of plasma. Causes include excessive loss of gastric acid (through vomiting or suctioning) and administration of alkalinizing salts (e.g., sodium bicarbonate). The body compensates for metabolic alkalosis by (1) hypoven-tilation (which causes retention of CO2), (2) increased renal excretion of bicarbonate, and (3) accumulation of organic acids.
TreatmentIn most cases, metabolic alkalosis can be corrected by infusing a solution of sodium chloride plus potassium chloride. This
Alternatively, 5% dextrose can be infused intravenously. (Since dextrose is rapidly metabolized to carbon dioxide and water, dextrose solutions can be viewed as the osmotic equivalent of water alone.) Volume replenishment should be done in stages. About 50% of the estimated loss should be replaced during the first few hours of treatment. The remainder should be replenished over 1 to 2 days.
Hypotonic ContractionDefinition and Causes. Hypotonic contraction is defined
as volume contraction in which loss of sodium exceeds loss of water. Hence, both the volume and osmolality of extracellular fluid are reduced. Because intracellular osmolality now exceeds extracellular osmolality, extracellular volume becomes dimin-ished further by movement of water into cells.
The principal cause of hypotonic contraction is excessive loss of sodium through the kidneys. This may occur because of diuretic therapy, chronic renal insufficiency, or lack of aldosterone (the adrenocortical hormone that promotes renal retention of sodium).
Treatment. If hyponatremia is mild, and if renal function is adequate, hypotonic contraction can be corrected by infusing isotonic sodium chloride solution for injection. When this is done, plasma tonicity will be adjusted by the kidneys. However, if the sodium loss is severe, a hypertonic (e.g., 3%) solution of sodium chloride should be infused. Administration should continue until plasma sodium concentration has been raised to about 130 mEq/L. Patients should be monitored for signs of fluid overload (distention of neck veins, peripheral or pulmonary edema). When hypotonic contraction is due to aldosterone insufficiency, patients should receive hormone replacement therapy along with intravenous infusion of isotonic sodium chloride.
Volume ExpansionVolume expansion is defined as an increase in the total volume of body fluid. As with volume contraction, volume expansion may be isotonic, hypertonic, or hypotonic. Volume expansion may result from an overdose with therapeutic fluids (e.g., sodium chloride infusion) or may be associated with disease states, such as heart failure, nephrotic syndrome, or cirrhosis of the liver with ascites. The principal drugs employed to correct volume expansion are diuretics and the agents used for heart failure. These drugs are discussed in Chapters 41 and 48, respectively. A specific form of volume expansion known as hypervolemic hyponatremia can be treated with a vasopressin antagonist, such as conivaptan or tolvaptan (see Chapter 59).
ACID-BASE DISTURBANCESMaintenance of acid-base balance is a complex process, the full discussion of which is beyond the scope of this text. Hence, discussion here is condensed.
Acid-base status is regulated by multiple systems. The most important are (1) the bicarbonate–carbonic acid buffer system, (2) the respiratory system, and (3) the kidneys. The respiratory system influences pH through control of CO2 exhalation. Because CO2 represents volatile carbonic acid, exhalation of CO2 tends to elevate pH (reduce acidity), whereas retention

473
CHAPTER 42 Agents Affecting the Volume and Ion Content of Body Fluids
HypokalemiaCauses and ConsequencesHypokalemia is defined as a deficiency of potassium in the blood. By definition, hypokalemia exists when serum potassium levels fall below 3.5 mEq/L. The most common cause is treatment with a thiazide or loop diuretic (see Chapter 41). Other causes include insufficient potassium intake; alkalosis and excessive insulin (both of which decrease extracellular potassium levels by driving potassium into cells); increased renal excretion of potassium (e.g., as caused by aldosterone); and potassium loss associated with vomiting, diarrhea, and abuse of laxatives. Hypokalemia may also occur because of excessive potassium loss in sweat. As a rule, potassium depletion is accompanied by loss of chloride. Insufficiency of both ions produces hypokalemic alkalosis.
Hypokalemia has adverse effects on skeletal muscle, smooth muscle, blood pressure, and the heart. Symptoms include weakness or paralysis of skeletal muscle, a risk of fatal dys-rhythmias, and intestinal dilation and ileus. In patients taking digoxin (a cardiac drug), hypokalemia is the principal cause of digoxin toxicity. For all people, hypokalemia increases the risk of hypertension and stroke.
Prevention and TreatmentPotassium depletion can be treated with three potassium salts: potassium chloride, potassium phosphate, and potassium bicarbonate. These may also be used for prophylaxis against potassium insufficiency. For either treatment or prophylaxis, the preferred salt is potassium chloride because chloride deficiency frequently coexists with potassium deficiency.
Potassium chloride may be administered PO or IV. Oral therapy is preferred for prophylaxis and for treating mild deficiency. Intravenous therapy is reserved for severe deficiency and for patients who cannot take potassium by mouth.
Oral Potassium ChlorideUses, Dosage, and Preparations. Oral potassium chloride
may be used for both prevention and treatment of potassium deficiency. Dosages for prevention range from 16 to 24 mEq/day. Dosages for deficiency range from 40 to 100 mEq/day.
Oral potassium chloride is available in solution and in solid formulations: immediate-release tablets, sustained-release tablets, effervescent tablets, and powders. The sustained-release tablets (e.g., Klor-Con, Micro-K) are preferred because they are more convenient and better tolerated than the other formulations, and hence offer the best chance of patient adherence.
Adverse Effects. Potassium chloride irritates the GI tract, frequently causing abdominal discomfort, nausea, vomiting, and diarrhea. With the exception of the sustained-release tablets, solid formulations can produce high local concentrations of potassium, resulting in severe intestinal injury (ulcerative lesions, bleeding, perforation); death has occurred. To minimize GI effects, oral potassium chloride should be taken with meals or a full glass of water. If symptoms of irritation occur, dosing should be discontinued. Rarely, oral potassium chloride produces hyperkalemia. This dangerous development is much more likely with IV therapy.
Intravenous Potassium Chloride. Intravenous potassium chloride is indicated for prevention and treatment of hypoka-lemia. Intravenous solutions must be diluted (preferably to 40 mEq/L or less) as they are extremely irritating to the veins.
facilitates renal excretion of bicarbonate and thereby promotes normalization of plasma pH. When alkalosis is severe, direct correction of pH is indicated. This can be accomplished by infusing dilute (0.1 N) hydrochloric acid through a central venous catheter or by administering an acid-forming salt, such as ammonium chloride. However, ammonium chloride must not be given to patients with liver failure because the drug is likely to cause hepatic encephalopathy.
Metabolic AcidosisCausesPrincipal causes of metabolic acidosis are chronic renal failure, loss of bicarbonate during severe diarrhea, and metabolic disorders that result in overproduction of lactic acid (lactic acidosis) or ketoacids (ketoacidosis). Metabolic acidosis may also result from poisoning by methanol and certain medications (e.g., aspirin and other salicylates).
TreatmentTreatment consists of correcting the underlying cause of acidosis and administering an alkalinizing salt (e.g., sodium bicarbonate, sodium carbonate) if the acidosis is severe.
When an alkalinizing salt is indicated, sodium bicarbonate is generally preferred. Administration may be oral or intrave-nous. If acidosis is mild, oral administration is preferred. Intravenous infusion is usually reserved for severe reductions of pH. When sodium bicarbonate is given IV to treat acute severe acidosis, caution must be exercised to avoid excessive elevation of plasma pH because rapid conversion from acidosis to alkalosis can be hazardous. Also, because of the sodium content of sodium bicarbonate, care should be taken to avoid hypernatremia.
POTASSIUM IMBALANCESPotassium is the most abundant intracellular cation, having a concentration within cells of about 150 mEq/L. In contrast, extracellular concentrations are low (4 to 5 mEq/L). Potassium plays a major role in conducting nerve impulses and maintaining the electrical excitability of muscle. Potassium also helps regulate acid-base balance.
Regulation of Potassium LevelsSerum levels of potassium are regulated primarily by the kidneys. Under steady-state conditions, urinary output of potas-sium equals intake. Renal excretion of potassium is increased by aldosterone, an adrenal steroid that promotes conservation of sodium while increasing potassium loss. Potassium excretion is also increased by most diuretics. Potassium-sparing diuretics (e.g., spironolactone) are the exception.
Potassium levels are influenced by extracellular pH. In the presence of extracellular alkalosis, potassium uptake by cells is enhanced, causing a reduction in extracellular potas-sium levels. Conversely, extracellular acidosis promotes the exit of potassium from cells, thereby causing extracellular hyperkalemia.
Insulin has a profound effect on potassium: In high doses, insulin stimulates potassium uptake by cells. This ability has been used to treat hyperkalemia.

474
UNIT VI Drugs That Affect Fluid and Electrolyte Balance
extracellular potassium levels; and (3) if acidosis is present (which is likely), infusion of sodium bicarbonate to move pH toward alkalinity and thereby increase cellular uptake of potassium. If these measures prove inadequate, steps can be taken to remove potassium. These include (1) oral or rectal administration of sodium polystyrene sulfonate [Kayexalate, Kionex], an exchange resin that absorbs potassium; and (2) peritoneal or extracorporeal dialysis.
MAGNESIUM IMBALANCESMagnesium is required for the activity of many enzymes and for binding of messenger RNA to ribosomes. In addition, magnesium helps regulate neurochemical transmission and the excitability of muscle. The concentration of magnesium within cells is about 40 mEq/L, much higher than its concentration outside cells (about 2 mEq/L).
HypomagnesemiaCauses and ConsequencesLow levels of magnesium may result from a variety of causes, including diarrhea, hemodialysis, kidney disease, and prolonged IV feeding with magnesium-free solutions. Hypomagnesemia may also be seen in chronic alcoholics and in people with diabetes or pancreatitis. Frequently, patients with magnesium deficiency also present with hypocalcemia and hypokalemia.
Prominent symptoms of hypomagnesemia involve cardiac and skeletal muscle. In the presence of low levels of magnesium, release of acetylcholine at the neuromuscular junction is enhanced. This can increase muscle excitability to the point of tetany. Hypomagnesemia also increases excitability of neurons in the CNS, causing disorientation, psychoses, and seizures.
In the kidneys, hypomagnesemia may lead to nephrocalci-nosis (formation of minuscule calcium stones within nephrons). Renal injury occurs when the stones become large enough to block the flow of tubular urine.
Prevention and TreatmentFrank hypomagnesemia is treated with parenteral magnesium sulfate. For prophylaxis against magnesium deficiency, an oral preparation (magnesium oxide) may be used.
Magnesium Oxide. Tablets of magnesium oxide may be taken as supplements to dietary magnesium to help prevent hypomagnesemia. With any oral magnesium preparation, excessive doses may cause diarrhea. The adult dosage for preventing deficiency is 400 to 800 mg daily.
Magnesium SulfateUses, Administration, and Dosage. Magnesium sulfate
(IM or IV) is the preferred treatment for severe hypomagne-semia. The IM dosage is 0.5 to 1 gm 4 times a day. For IV therapy, a 10% solution can be used, infused at a rate of 1.5 mL/min or less.
Adverse Effects. Excessive levels of magnesium cause neuromuscular blockade. Paralysis of the respiratory muscles is of particular concern. By suppressing neuromuscular transmis-sion, magnesium excess can intensify the effects of neuromus-cular blocking agents (e.g., succinylcholine, atracurium). Hence, caution must be exercised in patients receiving these drugs. The neuromuscular blocking actions of magnesium can be
The principal complication is hyperkalemia, which can prove fatal. To reduce the risk of hyperkalemia, serum potassium levels should be measured before the infusion and periodically throughout the treatment interval. Also, renal function should be assessed before and during treatment to ensure adequate output of urine. If renal failure develops, the infusion should be stopped immediately. Changes in the electrocardiogram (ECG) can be an early indication that potassium toxicity is developing.
Contraindications to Potassium Use. Potassium should be avoided under conditions that predispose to hyperkalemia (e.g., severe renal impairment, use of potassium-sparing diuret-ics, hypoaldosteronism). Potassium must also be avoided when hyperkalemia already exists.
HyperkalemiaCausesHyperkalemia (excessive elevation of serum potassium) can result from a number of causes. These include severe tissue trauma, untreated Addison’s disease, acute acidosis (which draws potassium out of cells), acute renal failure, misuse of potassium-sparing diuretics, and overdose with IV potassium.
ConsequencesThe most serious consequence of hyperkalemia is disruption of the electrical activity of the heart. Because hyperkalemia alters the generation and conduction of cardiac impulses, alterations in the ECG and cardiac rhythm are usually the earliest signs that potassium levels are growing dangerously high. With mild elevation of serum potassium (5 to 7 mEq/L), the T wave heightens and the PR interval becomes prolonged. When serum potassium reaches 8 to 9 mEq/L, cardiac arrest can occur, possibly preceded by ventricular tachycardia or fibrillation.
Effects of hyperkalemia are not limited to the heart. Non-cardiac effects include confusion, anxiety, dyspnea, weakness or heaviness of the legs, and numbness or tingling of the hands, feet, and lips.
TreatmentTreatment is begun by withholding any foods that contain potassium and any medicines that promote potassium accumula-tion (e.g., potassium-sparing diuretics, potassium supplements). After this, management consists of measures that (1) counteract potassium-induced cardiotoxicity and (2) lower extracellular levels of potassium. Specific steps include (1) infusion of a calcium salt (e.g., calcium gluconate) to offset effects of hyperkalemia on the heart; (2) infusion of glucose and insulin to promote uptake of potassium by cells and thereby decrease
Safety Alert
POTASSIUMIntravenous potassium must be infused slowly (generally no faster than 10 mEq/hr in adults). Potassium chloride must never be administered by IV push. In fact, potassium chloride is one of the agents used in lethal injections, as rapid infusion results in cardiac arrest.

475
CHAPTER 42 Agents Affecting the Volume and Ion Content of Body Fluids
HypermagnesemiaToxic elevation of magnesium levels is most common in patients with renal insufficiency, especially when magnesium-containing antacids or cathartics are being used. Symptoms of mild intoxication include muscle weakness (resulting from inhibition of acetylcholine release), hypotension, sedation, and ECG changes. As noted, respiratory paralysis is likely when plasma levels reach 12 to 15 mEq/L. At higher magnesium concentra-tions, there is a risk of cardiac arrest. Muscle weakness and paralysis can be counteracted with IV calcium.
counteracted with calcium. Accordingly, when parenteral magnesium is being employed, an injectable form of calcium (e.g., calcium gluconate) should be immediately available.
In the heart, excessive magnesium can suppress impulse conduction through the atrioventricular (AV) node. Accordingly, magnesium sulfate is contraindicated for patients with AV heart block.
To minimize the risk of toxicity, serum magnesium levels should be monitored. Respiratory paralysis occurs at 12 to 15 mEq/L. When magnesium levels exceed 25 mEq/L, cardiac arrest may set in.
KEY POINTS
■ Treat isotonic volume contraction with isotonic (0.9%) sodium chloride.
■ Treat hypertonic volume contraction with hypotonic (e.g., 0.45%) sodium chloride.
■ Treat hypotonic volume contraction with hypertonic (e.g., 3%) sodium chloride.
■ Treat volume expansion with diuretics.■ Treat respiratory or metabolic acidosis with sodium
bicarbonate.■ Treat respiratory alkalosis by having patients inhale 5%
CO2 or rebreathe their expired air.■ Treat metabolic alkalosis with an infusion of sodium
chloride plus potassium chloride. For severe cases, infuse 0.1% hydrochloric acid or ammonium chloride.
■ Treat moderate hypokalemia with potassium chloride in sustained-release tablets.
■ Treat severe hypokalemia with IV potassium chloride.■ To treat hyperkalemia, begin by withdrawing potassium-
containing foods and drugs that promote potassium accumulation (e.g., potassium supplements, potassium-sparing diuretics). Subsequent measures include (1) infusing a calcium salt to offset the cardiac effects of potassium, (2) infusing glucose and insulin to promote potassium uptake by cells, and (3) infusing sodium bicarbonate if acidosis is present.
■ Treat hypomagnesemia with IM or IV magnesium sulfate. For prophylaxis, give oral magnesium (e.g., magnesium oxide).
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

476
U N I T
VIIDRUGS THAT AFFECT THE HEART, BLOOD VESSELS, AND BLOOD
C H A P T E R
43 Review of Hemodynamics
Overview of the Circulatory System, p. 476Components of the Circulatory System, p. 476Distribution of Blood, p. 476What Makes Blood Flow?, p. 476How Does Blood Get Back to the Heart?, p. 477
Regulation of Cardiac Output, p. 477Determinants of Cardiac Output, p. 477Starling’s Law of the Heart, p. 478Factors That Determine Venous Return,
p. 479Starling’s Law and Maintenance of
Systemic-Pulmonary Balance, p. 479Regulation of Arterial Pressure, p. 479
Overview of Control Systems, p. 479Steady-State Control by the ANS, p. 480Rapid Control by the ANS: The Baroreceptor
Reflex, p. 480The Renin-Angiotensin-Aldosterone System,
p. 480Renal Retention of Water, p. 480Postural Hypotension, p. 480Natriuretic Peptides, p. 480
Key Points, p. 481
circulation delivers blood to the lungs. The systemic circulation delivers blood to all other organs and tissues. The systemic circulation is also known as the greater circulation or peripheral circulation.
Components of the Circulatory SystemThe circulatory system is composed of the heart and blood vessels. The heart is the pump that moves blood through the arterial tree. The blood vessels have several functions:
• Arteries transport blood under high pressure to tissues.• Arterioles are control valves that regulate local blood
flow.• Capillaries are the sites for exchange of fluid, oxygen,
carbon dioxide, nutrients, hormones, and wastes.• Venules collect blood from the capillaries.• Veins transport blood back to the heart. In addition, veins
serve as a major reservoir for blood.
Arteries and veins differ with respect to distensibility (elasticity). Arteries are very muscular, and hence do not readily stretch. As a result, large increases in arterial pressure (AP) cause only small increases in arterial diameter. Veins are much less muscular, and hence are 6 to 10 times more distensible. As a result, small increases in venous pressure cause large increases in vessel diameter; this produces a large increase in venous volume.
Distribution of BloodThe adult circulatory system contains about 5 L of blood, which is distributed throughout the system. As indicated in Fig. 43.1, 9% is in the pulmonary circulation, 7% is in the heart, and 84% is in the systemic circulation. Within the systemic circulation, however, distribution is uneven: most (64%) of the blood is in veins, venules, and venous sinuses; the remaining 20% is in arteries (13%) and arterioles or capillaries (7%). The large volume of blood in the venous system serves as a reservoir.
What Makes Blood Flow?Blood moves within vessels because the force that drives flow is greater than the resistance to flow. As shown in Fig. 43.2, the force that drives blood flow is the pressure gradi-ent between two points in a vessel. Blood will flow from the point where pressure is higher toward the point where pressure is lower. Resistance to flow is determined by the
Hemodynamics is the study of the movement of blood through-out the circulatory system, along with the regulatory mechanisms and driving forces involved. Concepts introduced here reappear throughout the chapters on cardiovascular drugs, so we urge you to review these now. Because this is a pharmacology text and not a physiology text, discussion is limited to hemo-dynamic factors that have particular relevance to the actions of drugs.
OVERVIEW OF THE CIRCULATORY SYSTEMThe circulatory system has two primary functions: (1) delivery of oxygen, nutrients, hormones, electrolytes, and other essentials to cells and (2) removal of carbon dioxide and metabolic wastes from cells. In addition, the system helps fight infection.
The circulatory system has two major divisions: the pul-monary circulation and the systemic circulation. The pulmonary

477
CHAPTER 43 Review of Hemodynamics
How Does Blood Get Back to the Heart?As indicated in Fig. 43.3, pressure falls progressively as blood moves through the systemic circulation. Pressure is 120 mm Hg when blood enters the aorta, 30 mm Hg when blood enters capillaries, and only 18 mm Hg when blood leaves capillaries; pressure then drops to negative values (0 to −5 mm Hg) in the right atrium. (Negative atrial pressure is generated by expansion of the chest during inspiration.)
Given that pressure is only 18 mm Hg when blood leaves capillaries, we must ask, “How does blood get back to the heart?” In addition to the small pressure head in venules, three mechanisms help ensure venous return. First, negative pressure in the right atrium helps “suck” blood toward the heart. Second, constriction of smooth muscle in the venous wall increases venous pressure, which helps drive blood toward the heart. Third, and most important, the combination of venous valves and skeletal muscle contraction constitutes an auxiliary “venous pump.” As shown in Fig. 43.4A, the veins are equipped with a system of one-way valves. When skeletal muscles contract (Fig. 43.4B), venous blood is squeezed toward the heart—the only direction the valves will permit.
REGULATION OF CARDIAC OUTPUTIn the average adult, cardiac output is about 5 L/min. Hence, every minute the heart pumps the equivalent of all the blood in the body. In this section, we consider the major factors that determine how much blood the heart pumps.
Determinants of Cardiac OutputThe basic equation for cardiac output is:
CO HR SV= ×
diameter and length of the vessel and by blood viscosity. From a pharmacologic viewpoint, the most important determinant of resistance is vessel diameter: The larger the vessel, the smaller the resistance. Accordingly, when vessels dilate, resistance declines, causing blood flow to increase. When vessels constrict, resistance rises, causing blood flow to decline. To maintain adequate flow when resistance rises, blood pressure must rise as well.
Pulmonary circulation: 9%
Heart: 7%
Arterioles and capillaries: 7%
Arteries: 13%
Veins,venules,
venous sinuses: 64%
Fig. 43.1 ■ Distribution of blood in the circulatory system. A large percentage of the blood resides in the venous system.
Resistance: vessel diameter vessel length blood viscosity
Higherpressure
Pressure gradient
Flow
Lowerpressure
Fig. 43.2 ■ Forces that promote and impede flow of blood. Blood flows from the point of higher pressure toward the point of lower pressure. Resistance to flow is determined by vessel diameter, vessel length, and blood viscosity.
120
110
40
Blood pressure(in mm Hg)
Aorta
Largearteries
Arterioles
Capillaries
Venules
Muscularveins
Central veins
Right atrium
3018
16
12
7
0 to –5
Fig. 43.3 ■ Distribution of pressure within the systemic circulation. Pressure is highest when blood leaves the left ventricle, falls to only 18 mm Hg as blood exits capillaries, and reaches negative values within the right atrium.
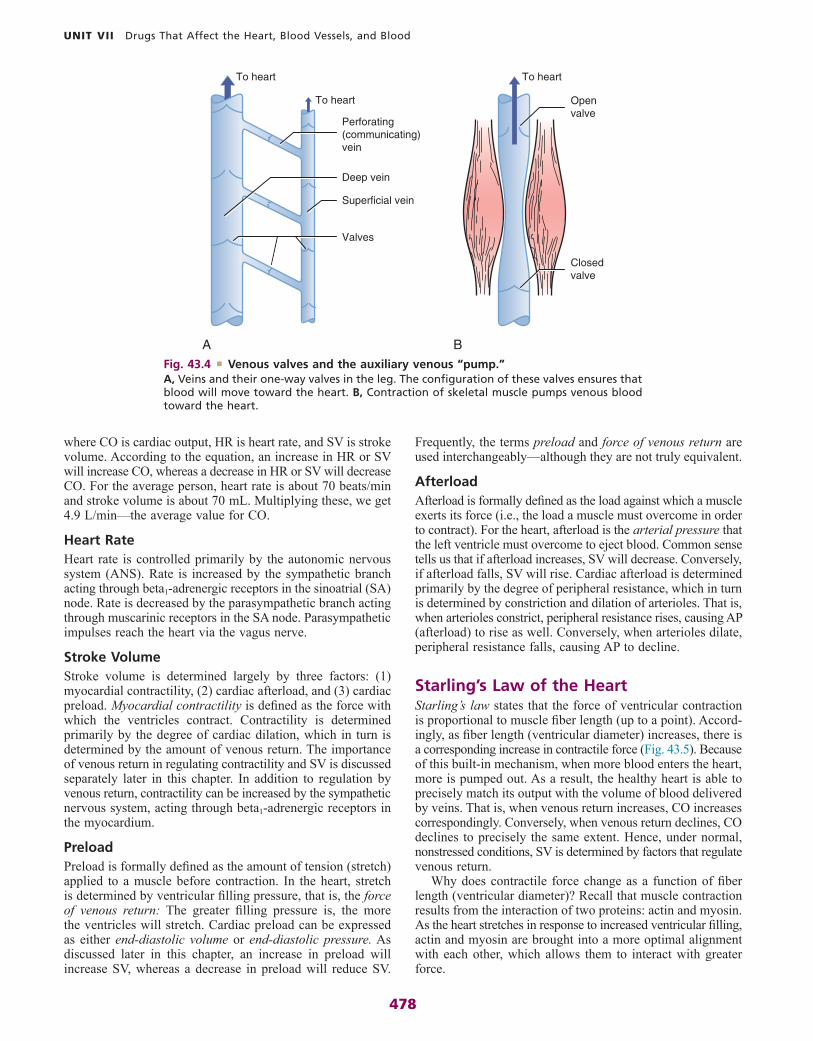
478
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Frequently, the terms preload and force of venous return are used interchangeably—although they are not truly equivalent.
AfterloadAfterload is formally defined as the load against which a muscle exerts its force (i.e., the load a muscle must overcome in order to contract). For the heart, afterload is the arterial pressure that the left ventricle must overcome to eject blood. Common sense tells us that if afterload increases, SV will decrease. Conversely, if afterload falls, SV will rise. Cardiac afterload is determined primarily by the degree of peripheral resistance, which in turn is determined by constriction and dilation of arterioles. That is, when arterioles constrict, peripheral resistance rises, causing AP (afterload) to rise as well. Conversely, when arterioles dilate, peripheral resistance falls, causing AP to decline.
Starling’s Law of the HeartStarling’s law states that the force of ventricular contraction is proportional to muscle fiber length (up to a point). Accord-ingly, as fiber length (ventricular diameter) increases, there is a corresponding increase in contractile force (Fig. 43.5). Because of this built-in mechanism, when more blood enters the heart, more is pumped out. As a result, the healthy heart is able to precisely match its output with the volume of blood delivered by veins. That is, when venous return increases, CO increases correspondingly. Conversely, when venous return declines, CO declines to precisely the same extent. Hence, under normal, nonstressed conditions, SV is determined by factors that regulate venous return.
Why does contractile force change as a function of fiber length (ventricular diameter)? Recall that muscle contraction results from the interaction of two proteins: actin and myosin. As the heart stretches in response to increased ventricular filling, actin and myosin are brought into a more optimal alignment with each other, which allows them to interact with greater force.
where CO is cardiac output, HR is heart rate, and SV is stroke volume. According to the equation, an increase in HR or SV will increase CO, whereas a decrease in HR or SV will decrease CO. For the average person, heart rate is about 70 beats/min and stroke volume is about 70 mL. Multiplying these, we get 4.9 L/min—the average value for CO.
Heart RateHeart rate is controlled primarily by the autonomic nervous system (ANS). Rate is increased by the sympathetic branch acting through beta1-adrenergic receptors in the sinoatrial (SA) node. Rate is decreased by the parasympathetic branch acting through muscarinic receptors in the SA node. Parasympathetic impulses reach the heart via the vagus nerve.
Stroke VolumeStroke volume is determined largely by three factors: (1) myocardial contractility, (2) cardiac afterload, and (3) cardiac preload. Myocardial contractility is defined as the force with which the ventricles contract. Contractility is determined primarily by the degree of cardiac dilation, which in turn is determined by the amount of venous return. The importance of venous return in regulating contractility and SV is discussed separately later in this chapter. In addition to regulation by venous return, contractility can be increased by the sympathetic nervous system, acting through beta1-adrenergic receptors in the myocardium.
PreloadPreload is formally defined as the amount of tension (stretch) applied to a muscle before contraction. In the heart, stretch is determined by ventricular filling pressure, that is, the force of venous return: The greater filling pressure is, the more the ventricles will stretch. Cardiac preload can be expressed as either end-diastolic volume or end-diastolic pressure. As discussed later in this chapter, an increase in preload will increase SV, whereas a decrease in preload will reduce SV.
Deep vein
Closedvalve
Superficial vein
Valves
Perforating(communicating)vein
To heart
To heart
To heart
A B
Openvalve
Fig. 43.4 ■ Venous valves and the auxiliary venous “pump.” A, Veins and their one-way valves in the leg. The configuration of these valves ensures that blood will move toward the heart. B, Contraction of skeletal muscle pumps venous blood toward the heart.

479
CHAPTER 43 Review of Hemodynamics
Fiber length (ventricular diameter)
Con
trac
tile
forc
e (s
trok
e vo
lum
e)
Fig. 43.5 ■ The Starling relationship between myocardial fiber length and contractile force. An increase in fiber length produces a corresponding increase in contractile force. Fiber length increases as the ventricles enlarge during filling. Increased contractile force is reflected by increased stroke volume.
the pulmonary circulation. In less than 40 minutes, death from pulmonary congestion would ensue. This example underscores the importance of systemic-pulmonary balance and the critical role of Starling’s mechanism in maintaining it.
REGULATION OF ARTERIAL PRESSUREArterial pressure is the driving force that moves blood through the arterial side of the systemic circulation. The general formula for AP is:
AP PR CO= ×
where AP is arterial pressure, PR is peripheral resistance, and CO is cardiac output. Accordingly, an increase in PR or CO will increase AP, whereas a decrease in PR or CO will decrease AP. Peripheral resistance is regulated primarily through constric-tion and dilation of arterioles. Cardiac output is regulated by the mechanisms discussed previously. Regulation of AP through processes that alter PR and CO is discussed in the sections that follow.
Overview of Control SystemsUnder normal circumstances, AP is regulated primarily by three systems: the ANS, the renin-angiotensin-aldosterone system (RAAS), and the kidneys. These systems differ greatly with regard to time frame of response. The ANS acts in two ways: (1) it responds rapidly (in seconds or minutes) to acute changes in blood pressure and (2) it provides steady-state control. The RAAS responds more slowly, taking hours or days to influence AP. The kidneys are responsible for long-term control, and hence may take days or weeks to adjust AP.
Factors That Determine Venous ReturnHaving established that venous return is the primary determinant of SV (and hence CO), we need to understand the factors that determine venous return. With regard to pharmacology, the most important factor is systemic filling pressure (i.e., the force that returns blood to the heart). The normal value for filling pressure is 7 mm Hg. This value can be raised to 17 mm Hg by constriction of veins. Filling pressure can also be raised by an increase in blood volume. Conversely, filling pressure, and hence venous return, can be lowered by venodilation or by reducing blood volume. Blood volume and venous tone can both be altered with drugs.
In addition to systemic filling pressure, three other factors influence venous return: (1) the auxiliary muscle pumps dis-cussed earlier, (2) resistance to flow between peripheral vessels and the right atrium, and (3) right atrial pressure, elevation of which will impede venous return. None of these factors can be directly influenced with drugs.
Starling’s Law and Maintenance of Systemic-Pulmonary BalanceBecause the myocardium operates in accord with Starling’s law, the right and left ventricles always pump exactly the same amount of blood. When venous return increases, SV of the right ventricle increases, thereby increasing delivery of blood to the pulmonary circulation, which in turn delivers more blood to the left ventricle; this increases filling of the left ventricle, which causes its SV to increase. Because an increase in venous return causes the output of both ventricles to increase, blood flow through the systemic and pulmonary circulations is always in balance, as long as the heart is healthy.
In the failing heart, Starling’s law breaks down. That is, force of contraction no longer increases in proportion to increased ventricular filling. As a result, blood backs up behind the failing ventricle (Fig. 43.6). In this example, output of the left ventricle is 1% less than the output of the right ventricle, which causes blood to back up in the pulmonary circulation. In only 20 minutes, this small imbalance between left and right ventricular output shifts a liter of blood from the systemic circulation to
5.00L/min
4.95L/minRV LV
0.5 L
4.5 L
RV LV
1.5 L
3.5 L
Systemic
Pulmonary
BEGIN 20 MINUTES LATER
Pulmonary
Systemic
Fig. 43.6 ■ Systemic-pulmonary imbalance that develops when the output of the left and right ventricles is not identical. In this example, the output of the left ventricle (LV) is 1% less than the output of the right ventricle (RV). Hence, while the right ventricle pumps 5000 mL/min, the left pumps only 4950 mL/min—50 mL/min less than the right side. This causes blood to back up in the pulmonary circulation. After 20 minutes, 1000 mL of blood has shifted from the systemic circulation to the pul-monary circulation. Death would ensue in less than 40 minutes. Numbers in the pulmonary and systemic circulations indicate volume of blood in liters.

480
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Renal Retention of WaterWhen AP remains low for a long time, the kidneys respond by retaining water, which in turn causes AP to rise. Pressure rises because fluid retention increases blood volume, which increases venous pressure, which increases venous return, which increases CO, which increases AP. Water retention is a mecha-nism for maintaining AP over long periods (weeks, months, years).
Reduction in AP causes the kidneys to retain water because low AP reduces renal blood flow (RBF), which in turn reduces glomerular filtration rate (GFR). Because less fluid is filtered, less urine is produced, and therefore more water is retained. Low AP activates the RAAS, causing levels of angiotensin II and aldosterone to rise. Angiotensin II causes constriction of renal blood vessels and thereby further decreases RBF and GFR. Aldosterone promotes renal retention of sodium, which causes water to be retained along with it.
Postural HypotensionPostural hypotension, also known as orthostatic hypotension, is a reduction in AP that can occur when we move from a supine or seated position to an upright position. The cause of hypotension is pooling of blood in veins, which decreases venous return, which in turn decreases CO. Between 300 and 800 mL of blood can pool in veins when we stand, causing CO to drop by as much as 2 L/min. Blood collects in veins when we stand, as gravity increases the pressure that blood exerts on veins. Because veins are not very muscular, they are unable to retain their shape when pressure increases, and hence they stretch. The resultant increase in venous volume allows blood to pool.
Two mechanisms help overcome postural hypotension. One is the system of auxiliary venous pumps, which promote venous return. In fact, in healthy individuals, these auxiliary pumps usually prevent postural hypotension from occurring in the first place. When postural hypotension does occur, the barorecep-tor reflex can restore AP by (1) constricting veins and arterioles and (2) increasing HR.
In patients taking drugs that interfere with venoconstriction, postural hypotension is more intense and more prolonged. Hypotension is more intense because venous pooling is greater. Hypotension is more prolonged because there is no venoconstric-tion to help reverse venous pooling. As with drugs that reduce AP by dilating arterioles, drugs that reduce AP by relaxing veins can trigger the baroreceptor reflex and can thereby cause reflex tachycardia.
Natriuretic PeptidesNatriuretic peptides serve to protect the cardiovascular system in the event of volume overload, a condition that increases preload and thereby increases CO and AP. Volume overload is caused by excessive retention of sodium and water. Natriuretic peptides work primarily by (1) reducing blood volume and (2) promoting dilation of arterioles and veins. Both actions lower AP.
The family of natriuretic peptides has three principal members: atrial natriuretic peptide (ANP), B- or brain natri-uretic peptide (BNP), and C-natriuretic peptide (CNP). ANP
Arterial pressure is also regulated by a fourth system: a family of natriuretic peptides. These peptides come into play primarily under conditions of volume overload.
Steady-State Control by the ANSThe ANS regulates AP by adjusting CO and PR. Sympathetic tone to the heart increases HR and contractility, thereby increas-ing CO. In contrast, parasympathetic tone slows the heart and thereby reduces CO. As discussed in Chapter 13, constriction of blood vessels is regulated exclusively by the sympathetic branch of the ANS; blood vessels have no parasympathetic innervation. Steady-state sympathetic tone provides a moderate level of vasoconstriction. The resultant resistance to blood flow maintains AP. Complete elimination of sympathetic tone would cause AP to fall by 50%.
Rapid Control by the ANS: The Baroreceptor ReflexThe baroreceptor reflex serves to maintain AP at a predetermined level. When AP changes, the reflex immediately attempts to restore AP to the preset value.
The reflex works as follows. Baroreceptors (pressure sensors) in the aortic arch and carotid sinus sense AP and relay this information to the vasoconstrictor center of the medulla. When AP changes, the vasoconstrictor center compensates by sending appropriate instructions to arterioles, veins, and the heart. For example, when AP drops, the vasoconstrictor center causes (1) constriction of nearly all arterioles, thereby increasing PR; (2) constriction of veins, thereby increasing venous return; and (3) acceleration of HR (by increasing sympathetic impulses to the heart and decreasing parasympathetic impulses). The combined effect of these responses is to restore AP to the preset level. When AP rises too high, opposite responses occur: The reflex dilates arterioles and veins and slows the heart.
The baroreceptor reflex is poised for rapid action—but not for sustained action. When AP falls or rises, the reflex acts within seconds to restore the preset pressure. However, when AP remains elevated or lowered, the system resets to the new pressure within 1 to 2 days. After this, the system perceives the new (elevated or reduced) pressure as “normal” and ceases to respond.
Drugs that lower AP will trigger the baroreceptor reflex. For example, if we administer a drug that dilates arterioles, the resultant drop in PR will reduce AP, causing the baroreceptor reflex to activate. The most noticeable response is reflex tachycardia. The baroreceptor reflex can temporarily negate efforts to lower AP with drugs.
The Renin-Angiotensin- Aldosterone SystemThe RAAS supports AP by causing (1) constriction of arterioles and veins and (2) retention of water by the kidneys. Vasocon-striction is mediated by a hormone named angiotensin II. Water retention is mediated in part by aldosterone through retention of sodium. Responses develop in hours (vasoconstriction) to days (water retention). The RAAS and its role in controlling blood pressure are discussed in Chapter 44.

481
CHAPTER 43 Review of Hemodynamics
extravascular compartment; the underlying mechanism is increased vascular permeability. Second, these peptides act on the kidney to cause diuresis (loss of water) and natriuresis (loss of sodium). Third, they promote dilation of arterioles and veins, in part by suppressing sympathetic outflow from the central nervous system. In addition to these actions, ANP and BNP help protect the heart during the early phase of heart failure by suppressing both the RAAS and sympathetic outflow, and by inhibiting proliferation of myocytes. Although CNP shares some actions of ANP and BNP, its primary action is to promote vasodilation.
is produced by myocytes of the atria; BNP is produced by myocytes of the ventricles (and to a lesser extent by cells in the brain, where BNP was discovered); and CNP is produced by cells of the vascular endothelium. When blood volume is excessive, all three peptides are released. (Release of ANP and BNP is triggered by stretching of the atria and ventricles, which occurs because of increased preload.)
ANP and BNP have similar actions. Both peptides reduce blood volume and increase venous capacitance, and thereby reduce cardiac preload. Three processes are involved. First, ANP and BNP shift fluid from the vascular system to the
KEY POINTS
■ Arterioles serve as control valves to regulate local blood flow.
■ Veins are a reservoir for blood.■ Arteries are not very distensible. As a result, large increases
in arterial pressure (AP) cause only small increases in arterial diameter.
■ Veins are highly distensible. As a result, small increases in venous pressure cause large increases in venous diameter.
■ The adult circulatory system contains 5 L of blood, 64% of which is in systemic veins.
■ Vasodilation reduces resistance to blood flow, whereas vasoconstriction increases resistance to flow.
■ In addition to the small pressure head in venules, three mechanisms help ensure venous return to the heart: (1) negative pressure in the right atrium sucks blood toward the heart; (2) constriction of veins increases venous pressure and thereby drives blood toward the heart; and (3) contrac-tion of skeletal muscles, in conjunction with one-way venous valves, pumps blood toward the heart.
■ Heart rate is increased by sympathetic nerve impulses and decreased by parasympathetic impulses.
■ Stroke volume is determined by myocardial contractility, cardiac preload, and cardiac afterload.
■ Preload is defined as the amount of tension (stretch) applied to a muscle before contraction. In the heart, preload is determined by the force of venous return.
■ Afterload is defined as the load against which a muscle exerts its force. For the heart, afterload is the AP that the left ventricle must overcome to eject blood.
■ Cardiac afterload is determined primarily by peripheral resistance, which in turn is determined by the degree of constriction in arterioles.
■ Starling’s law states that the force of ventricular contraction is proportional to myocardial fiber length. Because of this relationship, when more blood enters the heart, more is pumped out. As a result, the healthy heart is able to precisely match output with venous return.
■ The most important determinant of venous return is systemic filling pressure, which can be raised by constricting veins and increasing blood volume.
■ Because cardiac muscle operates under Starling’s law, the right and left ventricles always pump exactly the same amount of blood (assuming the heart is healthy). Hence, balance between the pulmonary and systemic circulations is maintained.
■ Arterial pressure is regulated by the ANS, the RAAS, the kidneys, and natriuretic peptides.
■ The ANS regulates AP (1) through tonic control of heart rate and peripheral resistance and (2) through the barorecep-tor reflex.
■ The baroreceptor reflex is useful only for short-term control of AP. When pressure remains elevated or lowered, the system resets to the new pressure within 1 to 2 days, and hence ceases to respond.
■ Drugs that lower AP trigger the baroreceptor reflex and thereby cause reflex tachycardia. Hence, the baroreceptor reflex can temporarily negate efforts to lower AP with drugs.
■ The RAAS supports AP by causing (1) constriction of arterioles and veins and (2) retention of water by the kidneys. Vasoconstriction is mediated by angiotensin II; water retention is mediated in part by aldosterone.
■ The kidneys provide long-term control of blood pressure by regulating blood volume.
■ Postural (orthostatic) hypotension is caused by decreased venous return secondary to pooling of blood in veins, which can occur when we assume an erect posture.
■ Drugs that dilate veins intensify and prolong postural hypo-tension. As with other drugs that reduce AP, venodilators can trigger the baroreceptor reflex and can thereby cause reflex tachycardia.
■ Natriuretic peptides defend the cardiovascular system from volume overload—primarily by reducing blood volume and promoting vasodilation.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

482
C H A P T E R
44 Drugs Acting on the Renin- Angiotensin-Aldosterone System
Physiology of the Renin-Angiotensin-Aldosterone System, p. 482Types of Angiotensin, p. 482Actions of Angiotensin II, p. 482Actions of Aldosterone, p. 484Formation of Angiotensin II by Renin and
Angiotensin-Converting Enzyme, p. 484Regulation of Blood Pressure by the
Renin-Angiotensin-Aldosterone System, p. 485
Tissue (Local) Angiotensin II Production, p. 485Angiotensin-Converting Enzyme Inhibitors, p. 485Angiotensin II Receptor Blockers, p. 489Aliskiren, a Direct Renin Inhibitor, p. 491Aldosterone Antagonists, p. 492
Eplerenone, p. 492Spironolactone, p. 493
Key Points, p. 493Summary of Major Nursing Implications, p. 494
associated with hypertension, heart failure, and MI. The RAAS exerts its effects through angiotensin II and aldosterone.
Types of AngiotensinBefore considering the physiology of the RAAS, we need to introduce the angiotensin family, which consists of angiotensin I, angiotensin II, and angiotensin III. All three compounds are small polypeptides. Angiotensin I is the precursor of angiotensin II (Fig. 44.1) and has only weak biologic activity. In contrast, angiotensin II has strong biologic activity. Angiotensin III, which is formed by degradation of angiotensin II, has moderate biologic activity.
Actions of Angiotensin IIAngiotensin II participates in all processes regulated by the RAAS. The most prominent actions of angiotensin II are vasoconstriction and stimulation of aldosterone release. Both actions raise blood pressure. In addition, angiotensin II (as well as aldosterone) can act on the heart and blood vessels to cause pathologic changes in their structure and function.
VasoconstrictionAngiotensin II is a powerful vasoconstrictor. The compound acts directly on vascular smooth muscle (VSM) to cause contrac-tion. Vasoconstriction is prominent in arterioles and less so in veins. As a result of angiotensin-induced vasoconstriction, blood pressure rises. In addition to its direct action on blood vessels, angiotensin II can cause vasoconstriction indirectly by acting on (1) sympathetic neurons to promote norepinephrine release, (2) the adrenal medulla to promote epinephrine release, and (3) the central nervous system to increase sympathetic outflow to blood vessels.
Release of AldosteroneAngiotensin II acts on the adrenal cortex to promote synthesis and secretion of aldosterone, whose actions are discussed in an upcoming section. The adrenal cortex is highly sensitive to angiotensin II, and hence angiotensin II can stimulate aldo-sterone release even when angiotensin II levels are too low to induce vasoconstriction. Aldosterone secretion is enhanced when sodium levels are low and when potassium levels are high.
Alteration of Cardiac and Vascular StructureAngiotensin II may cause pathologic structural changes in the heart and blood vessels. In the heart, it may cause hypertrophy (increased cardiac mass) and remodeling (redistribution of mass within the heart). In hypertension, angiotensin II may be responsible for increasing the thickness of blood vessel walls. In atherosclerosis, it may be responsible for thickening the
In this chapter we consider four families of drugs: angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), direct renin inhibitors (DRIs), and aldosterone antagonists. With all four groups, effects result from interfering with the renin-angiotensin-aldosterone system (RAAS). The ACE inhibitors, available for more than three decades, have established roles in the treatment of hypertension, heart failure, and diabetic nephropathy; in addition, these drugs are indicated for myocardial infarction (MI) and prevention of cardiovascular events in patients at risk. Indications for ARBs are limited to hypertension, heart failure, diabetic nephropathy, and prevention of cardiovascular events in patients at risk. The aldosterone antagonist eplerenone has only two indications: hypertension and heart failure; spironolactone is also used to prevent diuretic-induced hypokalemia and to treat hyperaldosteronism. Current indications for DRIs are limited to hypertension. We begin by reviewing the physiology of the RAAS, and then we discuss the drugs that affect it.
PHYSIOLOGY OF THE RENIN-ANGIOTENSIN- ALDOSTERONE SYSTEMThe RAAS plays an important role in regulating blood pressure, blood volume, and fluid and electrolyte balance. In addition, the system appears to mediate certain pathophysiologic changes

483
Retention of sodium and water(and loss of potassium)
ALDOSTERONE
RENIN
Sodium /potassiumexchange
Renalvasoconstriction
Renalbloodflow
Glomerularfiltration
Blood volume
Blood pressure
Blood pressure
Blood volume
Renal perfusion
Sodium depletion
Beta1 stimulation
Systemicvasoconstriction
(arteriolesand veins)
ANGIOTENSIN I I(active)
Angiotensin- converting
enzyme (ACE)
Angiotensin I(inactive)
Angiotensinogen
Blocked byACE inhibitors
Blocked byaldosteroneantagonists
Blockedby ARBs
Blockedby ARBs
Blockedby ARBs
Blocked by directrenin inhibitors
Fig. 44.1 ■ Regulation of blood pressure by the renin-angiotensin-aldosterone system. In addition to the mechanisms depicted, angiotensin II can raise blood pressure by (1) acting on the distal nephron to promote reabsorption of sodium and (2) increasing vasoconstriction by three mechanisms: promoting release of norepinephrine from sympathetic nerves; promoting release of epinephrine from the adrenal medulla; and acting in the central nervous system to increase sympathetic outflow to blood vessels. (ARBs, Angiotensin receptor blockers.)

484
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Formation of Angiotensin II by Renin and Angiotensin- Converting EnzymeAngiotensin II is formed through two sequential reactions. The first is catalyzed by renin, the second by ACE.
ReninRenin catalyzes the formation of angiotensin I from angio-tensinogen. This reaction is the rate-limiting step in angio-tensin II formation. Renin is produced by juxtaglomerular cells of the kidney and undergoes controlled release into the bloodstream, where it cleaves angiotensinogen into angiotensin I.
Regulation of Renin Release. Since renin catalyzes the rate-limiting step in angiotensin II formation and since renin must be released into the blood in order to act, the factors that regulate renin release regulate the rate of angiotensin II formation.
Release of renin can be triggered by multiple factors (see Fig. 44.1). Release increases in response to a decline in blood pressure, blood volume, plasma sodium content, or renal perfusion pressure. Reduced renal perfusion pressure is an especially important stimulus for renin release and can occur in response to (1) stenosis of the renal arteries, (2) reduced systemic blood pressure, and (3) reduced plasma volume (brought on by dehydration, hemorrhage, or chronic sodium depletion). For the most part, these factors increase renin release through effects exerted locally in the kidney. However, some of these factors may also promote renin release through activation of the sympathetic nervous system. (Sympathetic nerves increase secretion of renin by causing stimulation of beta1-adrenergic receptors on juxtaglomerular cells.)
Release of renin is suppressed by factors opposite to those that cause release. That is, renin secretion is inhibited by elevation of blood pressure, blood volume, and plasma sodium content. Hence, as blood pressure, blood volume, and plasma sodium content increase in response to renin release, further release of renin is suppressed. In this regard, we can view release of renin as being regulated by a classic negative feedback loop.
Angiotensin-Converting Enzyme (Kinase II)ACE catalyzes the conversion of angiotensin I (inactive) into angiotensin II (highly active). ACE is located on the luminal surface of all blood vessels. The vasculature of the lungs is especially rich in the enzyme. Because ACE is abundant, conversion of angiotensin I into angiotensin II occurs almost instantaneously after angiotensin I has been formed. ACE is a relatively nonspecific enzyme that can act on a variety of substrates in addition to angiotensin I.
Nomenclature regarding ACE can be confusing and requires comment. As just noted, ACE can act on several substrates. When the substrate is angiotensin I, we refer to the enzyme as ACE. However, when the enzyme is acting on other substrates, we refer to it by different names. Of importance to us, when the substrate is a hormone known as bradykinin, we refer to the enzyme as kinase II. So, please remember, whether we call it ACE or kinase II, we’re talking about the same enzyme.
intimal surface of blood vessels. And in heart failure and MI, it may be responsible for causing cardiac hypertrophy and fibrosis. Known effects of angiotensin II that could underlie these pathologic changes include:
• Increased migration, proliferation, and hypertrophy of VSM cells
• Increased production of extracellular matrix by VSM cells
• Hypertrophy of cardiac myocytes• Increased production of extracellular matrix by cardiac
fibroblasts
Actions of AldosteroneRegulation of Blood Volume and Blood PressureAfter being released from the adrenal cortex, aldosterone acts on distal tubules of the kidney to cause retention of sodium and excretion of potassium and hydrogen. Because retention of sodium causes water to be retained as well, aldosterone increases blood volume, which causes blood pressure to rise.
Pathologic Cardiovascular EffectsUntil recently, knowledge of aldosterone’s actions was limited to effects on the kidney. Now, however, we know that aldo-sterone can cause more harmful effects. Like angiotensin II, aldosterone can promote cardiac remodeling and fibrosis. In addition, aldosterone can activate the sympathetic nervous system and suppress uptake of norepinephrine in the heart, thereby predisposing the heart to dysrhythmias. Also, aldoste-rone can promote vascular fibrosis (which decreases arterial compliance), and it can disrupt the baroreceptor reflex. These adverse effects appear to be limited to states such as heart failure, in which levels of aldosterone can be extremely high.
RAAS Inhibitors
Life Stage Patient Care Concerns
Infants Captopril and enalapril have been used in infants safely for management of hypertension (HTN).
Children/adolescents
Some ACE inhibitors and ARBs are approved for use in children over the age of 6 for treatment of HTN.
Pregnant women
Animal studies revealed that drugs that block the RAAS should be avoided in pregnancy, especially in the second and third trimesters. ACE inhibitors, ARBs, and DRIs are classified in FDA Pregnancy Risk Category D.a
Breast-feeding women
Data are lacking regarding effects on the infant when breast-feeding. Caution is advised.
Older adults The SCOPE and LIFE trials revealed a 25% decrease in stroke in patients ages 55–80 years using losartan when compared with atenolol. A 20% decreased risk of new-onset diabetes was seen with candesartan when compared with placebo.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.

485
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
ANGIOTENSIN-CONVERTING ENZYME INHIBITORS
The ACE inhibitors are important drugs for treating hyperten-sion, heart failure, diabetic nephropathy, and MI. In addition, they are used to prevent adverse cardiovascular events in patients at risk. Their most prominent adverse effects are cough, angioedema, first-dose hypotension, and hyperkalemia. For all of these agents, beneficial effects result largely from suppressing formation of angiotensin II. Because the similarities among ACE inhibitors are much more striking than their differences, we will discuss these drugs as a group, rather than selecting a prototype to represent them.
Mechanism of Action and Overview of Pharmacologic EffectsAs shown in Fig. 44.2, ACE inhibitors produce their beneficial effects and adverse effects by (1) reducing levels of angiotensin II (through inhibition of ACE) and (2) increasing levels of bradykinin (through inhibition of kinase II). By reducing levels of angiotensin II, ACE inhibitors can dilate blood vessels (primarily arterioles and to a lesser extent veins), reduce blood volume (through effects on the kidney), and, importantly, prevent or reverse pathologic changes in the heart and blood vessels mediated by angiotensin II and aldosterone. Inhibition of ACE can also cause hyperkalemia and fetal injury. Elevation of
Regulation of Blood Pressure by the Renin-Angiotensin- Aldosterone SystemThe RAAS is poised to help regulate blood pressure. Factors that lower blood pressure turn the RAAS on; factors that raise blood pressure turn it off. However, although the RAAS does indeed contribute to blood pressure control, its role in normovolemic, sodium-replete individuals is only modest. In contrast, the system can be a major factor in maintaining blood pressure in the presence of hemorrhage, dehydration, or sodium depletion.
The RAAS, acting through angiotensin II, raises blood pressure through two basic processes: vasoconstriction and renal retention of water and sodium. Vasoconstriction raises blood pressure by increasing total peripheral resistance; retention of water and sodium raises blood pressure by increasing blood volume. Vasoconstriction occurs within minutes to hours of activating the system, and hence can raise blood pressure quickly. In contrast, days, weeks, or even months are required for the kidney to raise blood pressure by increasing blood volume.
Angiotensin II acts in two ways to promote renal retention of water. First, by constricting renal blood vessels, angiotensin II reduces renal blood flow and thereby reduces glomerular filtration. Second, angiotensin II stimulates release of aldoste-rone from the adrenal cortex. Aldosterone then acts on renal tubules to promote retention of sodium and water and excretion of potassium.
Tissue (Local) Angiotensin II ProductionIn addition to the traditional RAAS that we’ve been discussing, in which angiotensin II is produced in the blood and then carried to target tissues, angiotensin II is produced in individual tissues. This permits discrete, local effects of angiotensin II independent of the main system. Interference with local produc-tion of angiotensin II may underlie some effects of the ACE inhibitors.
It is important to note that some angiotensin II is produced by pathways that do not involve ACE. As a result, drugs that inhibit ACE cannot completely block angiotensin II production.
Prototype Drugs
DRUGS ACTING ON THE RAAS SYSTEMAngiotensin-Converting Enzyme (ACE) InhibitorCaptopril
Angiotensin II Receptor BlockerLosartan
Direct Renin InhibitorAliskiren
Aldosterone AntagonistEplerenone
Angiotensin I Inactive product
Blockedby ACE
inhibitors
BradykininAngiotensin II
ACE KinaseII
Vasodilation
Potassiumretention
Fetal injury
Angiotensin IIresults in
•
Blood volume
Cardiac andvascularremodeling
Bradykininresults in
Vasodilation
Cough
Angioedema(rarely)
•
•
•
•
•
•
•
Fig. 44.2 ■ Overview of ACE inhibitor actions and pharma-cologic effects. Angiotensin-converting enzyme (ACE) and kinase II are two names for the same enzyme. When angiotensin II is the substrate, we call the enzyme ACE; when bradykinin is the substrate, we call it kinase II. Inhibition of this enzyme decreases production of angiotensin II (thereby reducing angiotensin II levels) and decreases breakdown of bradykinin (thereby increasing brady-kinin levels).

486
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Therapeutic UsesWhen the ACE inhibitors were introduced, their only indication was hypertension. Today, they are also used for heart failure, acute MI, left ventricular (LV) dysfunction, and diabetic and nondiabetic nephropathy. In addition, they can help prevent MI, stroke, and death in patients at high risk for cardiovascular events. It should be noted that no single ACE inhibitor is approved for all of these conditions (Table 44.1). However, given that all ACE inhibitors are very similar, it seems likely that all may produce similar benefits.
Hypertension. All ACE inhibitors are approved for hypertension. These drugs are especially effective against malignant hypertension and hypertension secondary to renal arterial stenosis. They are also useful against essential hyperten-sion of mild to moderate intensity—although maximal benefits may take several weeks to develop.
In patients with essential hypertension, the mechanism underlying blood pressure reduction is not fully understood. Initial responses are proportional to circulating angiotensin II levels and are clearly related to reduced formation of that compound. (By lowering angiotensin II levels, ACE inhibitors dilate blood vessels and reduce blood volume; both actions help lower blood pressure.) However, with prolonged therapy, blood pressure often undergoes additional decline. During this phase, there is no relationship between reductions in blood
bradykinin causes vasodilation (secondary to increased produc-tion of prostaglandins and nitric oxide) and can also promote cough and angioedema.
PharmacokineticsRegarding pharmacokinetics, the following generalizations apply:
• Nearly all ACE inhibitors are administered orally. The only exception is enalaprilat (the active form of enalapril), which is given IV.
• Except for captopril and moexipril, all oral ACE inhibitors can be administered with food.
• With the exception of captopril, all ACE inhibitors have prolonged half-lives, and hence can be administered just once or twice a day. Captopril is administered 2 or 3 times a day.
• With the exception of lisinopril, all ACE inhibitors are prodrugs that must undergo conversion to their active form in the small intestine and liver. Lisinopril is active as given.
• All ACE inhibitors are excreted by the kidneys. As a result, nearly all can accumulate to dangerous levels in patients with kidney disease, and hence dosages must be reduced in these patients. Only one agent—fosinopril—does not require a dosage reduction.
Generic Name Brand Name Approved Indications Starting Dosagea Usual Maintenance Dosagea
Benazepril Lotensin Hypertension 10 mg once/day 20–80 mg/day in 1 or 2 dosesCaptopril Capoten Hypertension 25 mg 2 or 3 times/day 25–50 mg 2 or 3 times/day
Heart failure 6.25–12.5 mg 3 times/day 50 mg 3 times/dayLVD after MI 12.5 mg 3 times/day 50 mg 3 times/dayDiabetic nephropathy 25 mg 3 times/day 25 mg 3 times/day
Enalapril Vasotec, Epaned
Hypertension 2.5–5 mg once/day 10–40 mg/day in 1 or 2 dosesHeart failure 2.5 mg twice/day 10–20 mg twice/dayAsymptomatic LVD 2.5 mg twice/day 10 mg twice/day
Enalaprilat Generic only Hypertension 1.25 mg every 6 hr Not used for maintenanceFosinopril Generic only Hypertension 10 mg once/day 20–40 mg/day in 1 or 2 doses
Heart failure 5–10 mg once/day 20–40 mg once/dayLisinopril Prinivil, Zestril,
QbrelisHypertension 10 mg once/day 10–40 mg once/dayHeart failure 2.5–5 mg once/day 20–40 mg once/dayAcute MI 5 mg once/day 10 mg once/day
Moexipril Generic only Hypertension 7.5 mg once/day 7.5–30 mg/day in 1 or 2 dosesPerindopril Aceon,
Coversyl Hypertension 4 mg once/day 4–8 mg/day in 1 or 2 dosesStable CAD 4 mg once/day 8 mg once/day
Quinapril Accupril Hypertension 1020 mg/day 20–80 mg/day in 1 or 2 dosesHeart failure 5 mg twice/day 20–40 mg twice/day
Ramipril Altace Hypertension 2.5 mg once/day 2.5–20 mg/day in 1 or 2 dosesHeart failure after MI 1.25–2.5 mg twice/day 5 mg twice/dayPrevention of MI, stroke, and death
in people at high risk for CVD2.5 mg/day for 1 wk 10 mg once/day
Trandolapril Mavik Hypertension 1 mg once/day 2–4 mg once/dayHeart failure after MI 1 mg once/day 4 mg once/dayLVD after MI 1 mg once/day 4 mg once/day
TABLE 44.1 ■ ACE Inhibitors: Approved Indications and Adult Dosages
aFor all ACE inhibitors except fosinopril, dosage must be reduced in patients with significant renal impairment.CAD, Coronary artery disease; CVD, cardiovascular disease; LVD, left ventricular dysfunction; MI, myocardial infarction.

487
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
pressure by reducing levels of angiotensin II, a compound that can raise filtration pressure by two mechanisms. First, angio-tensin II raises systemic blood pressure, which raises pressure in the afferent arteriole of the glomerulus (Fig. 44.3). Second, it constricts the efferent arteriole, thereby generating back-pressure in the glomerulus. The resultant increase in filtration pressure promotes injury. By reducing levels of angiotensin II, ACE inhibitors lower glomerular filtration pressure and thereby slow development of renal injury.
At this time, the only ACE inhibitor approved for nephropa-thy is captopril. However, the American Diabetes Association considers benefits in diabetic nephropathy to be a class effect, and hence recommends choosing an ACE inhibitor based on its cost and likelihood of patient adherence.
Can ACE inhibitors be used for primary prevention of diabetic nephropathy? No. Although use of these agents can slow progression of kidney disease, they ultimately do not prevent it. This conclusion is based on multiple small studies, as well as the Renin-Angiotensin System Study (RASS), which evaluated the effects of an ACE inhibitor (enalapril [Vasotec]) and an ARB (losartan [Cozaar]) in patients with type 1 diabetes who did not have hypertension or any signs of early kidney disease. Both drugs failed to protect the kidney: Compared with patients receiving placebo, those receiving enalapril or losartan developed the same degree of microalbuminuria (an early sign of kidney damage), the same decline in kidney function, and the same changes in glomerular structure (as shown by microscopic analysis of kidney biopsy samples). Hence, although ACE inhibitors may slow progression of established nephropathy, they do not protect against kidney damage.
Prevention of MI, Stroke, and Death in Patients at High Cardiovascular Risk. One ACE inhibitor (ramipril [Altace]) is approved for reducing the risk of MI, stroke, and death from cardiovascular causes in patients at high risk for a major cardiovascular event—high risk being defined by (1) a history of stroke, coronary artery disease, peripheral vascular disease, or diabetes, combined with (2) at least one other risk factor, such as hypertension, high LDL cholesterol, low HDL cholesterol, or cigarette smoking. Ramipril was approved for this use based on results of the Heart Outcomes Prevention Evaluation (HOPE) trial, a large study in which patients at high cardiovascular risk took either ramipril (10 mg/day) or
pressure and reductions in circulating angiotensin II. It may be that the delayed response is due to reductions in local angiotensin II levels—reductions that would not be revealed by measuring angiotensin II in the blood.
ACE inhibitors offer several advantages over most other antihypertensive drugs. In contrast to the sympatholytic agents, ACE inhibitors do not interfere with cardiovascular reflexes. Hence, exercise capacity is not impaired and orthostatic hypotension is minimal. In addition, these drugs can be used safely in patients with bronchial asthma, a condition that precludes the use of beta2-adrenergic antagonists. ACE inhibitors do not promote hypokalemia, hyperuricemia, or hyperglycemia—side effects seen with thiazide diuretics. Furthermore, they do not induce lethargy, weakness, or sexual dysfunction—responses that are common with other antihypertensive agents. Most importantly, ACE inhibitors reduce the risk of cardiovascular mortality caused by hypertension. The only other drugs proved to reduce hypertension-associated mortality are beta blockers and diuretics (see Chapter 47).
Heart Failure. ACE inhibitors produce multiple benefits in heart failure. By lowering arteriolar tone, these drugs improve regional blood flow, and, by reducing cardiac afterload, they increase cardiac output. By causing venous dilation, they reduce pulmonary congestion and peripheral edema. By dilating blood vessels in the kidney, they increase renal blood flow and thereby promote excretion of sodium and water. This loss of fluid has two beneficial effects: (1) it helps reduce edema, and (2) by lowering blood volume, it decreases venous return to the heart and thereby reduces right-heart workload. Lastly, by suppressing aldosterone and reducing local production of angiotensin II in the heart, ACE inhibitors may prevent or reverse pathologic changes in cardiac structure. Although only seven ACE inhibi-tors are approved for heart failure (see Table 44.1), both the American Heart Association and the American College of Cardiology have concluded that the ability to improve symptoms and prolong survival is a class effect. The use of ACE inhibitors in heart failure is discussed further in Chapter 48.
Myocardial Infarction. ACE inhibitors can reduce mortality following acute MI (heart attack). In addition, they decrease the chance of developing overt heart failure. Treatment should begin as soon as possible after infarction and should continue for at least 6 weeks. In patients who develop overt heart failure, treatment should continue long term. As for patients who do not develop heart failure, there are no data to indicate whether or not continued treatment would be beneficial. At this time, only three ACE inhibitors—captopril, lisinopril, and trandolapril—are approved for patients with MI.
Diabetic and Nondiabetic Nephropathy. ACE inhibitors can benefit patients with diabetic nephropathy, the leading cause of end-stage renal disease in the United States. In patients with overt nephropathy, as indicated by proteinuria of more than 500 mg/day, ACE inhibitors can slow progression of renal disease. In patients with less advanced nephropathy (30 to 300 mg proteinuria/day), ACE inhibitors can delay onset of overt nephropathy. These benefits were first demonstrated in patients with type 1 diabetes (insulin-dependent diabetes mellitus) and were later demonstrated in patients with type 2 diabetes (non–insulin-dependent diabetes mellitus). More recently, ACE inhibitors have been shown to provide similar benefits in patients with nephropathy unrelated to diabetes.
The principal protective mechanism appears to be reduction of glomerular filtration pressure. ACE inhibitors lower filtration
Afferent arteriole
Efferent arteriole
Glomerulus
Bowman’s capsule
Fig. 44.3 ■ Elevation of glomerular filtration pressure by angiotensin II. Angiotensin II increases filtration pressure by (1) increasing pres-sure in the afferent arteriole (secondary to increasing systemic arterial pressure) and (2) constricting the efferent arteriole, thereby generating back-pressure in the glomerulus.

488
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
should be instructed to avoid potassium supplements and potassium-containing salt substitutes unless they are prescribed.
Renal Failure. ACE inhibitors can cause severe renal insufficiency in patients with bilateral renal artery stenosis or stenosis in the artery to a single remaining kidney. In patients with renal artery stenosis, the kidneys release large amounts of renin. The resulting high levels of angiotensin II serve to maintain glomerular filtration by two mechanisms: elevation of blood pressure and constriction of efferent glomerular arterioles (see Fig. 44.3). When ACE is inhibited, causing angiotensin II levels to fall, the mechanisms that had been supporting glomerular filtration fail, causing urine production to drop precipitously. Not surprisingly, ACE inhibitors are contraindicated for patients with bilateral renal artery stenosis (or stenosis in the artery to a single remaining kidney).
Fetal Injury. For a long time, we have known that use of ACE inhibitors during the second and third trimesters of pregnancy can injure the developing fetus. Specific effects include hypotension, hyperkalemia, skull hypoplasia, pulmonary hypoplasia, anuria, renal failure (reversible and irreversible), and death. Women who become pregnant while using ACE inhibitors should discontinue treatment as soon as possible. Infants who have been exposed to ACE inhibitors during the second or third trimester should be closely monitored for hypotension, oliguria, and hyperkalemia.
Are ACE inhibitors safe early in pregnancy? Possibly. Even though an article in the New England Journal of Medicine reported that among 209 children exposed to ACE inhibitors during the first trimester, 18 (8.7%) had major congenital malformations, compared with 3.2% of controls, these data contrast with animal studies, which suggest that such malforma-tions are not likely. Furthermore, no mechanism by which ACE inhibitors might disrupt early embryogenesis is known. It is now thought that the malformations attributed to ACE inhibitors are more related to hypertension in pregnancy, not the drug itself. As there is lack of complete data, however, the U.S. Food and Drug Administration (FDA) still classifies ACE inhibitors in Pregnancy Risk Category D,a which indicates they should be avoided in pregnancy.
Angioedema. Angioedema is a potentially fatal reaction that develops in up to 1% of patients. Symptoms, which result from increased capillary permeability, include giant wheals and edema of the tongue, glottis, lips, eyes, and pharynx. Severe reactions should be treated with subcutaneous epinephrine. If angioedema develops, ACE inhibitors should be discontinued and never used again. Angioedema is caused by accumulation of bradykinin secondary to inhibition of kinase II.
placebo. Follow-up time was 5 years. The combined endpoint of MI, stroke, or death from cardiovascular causes was sig-nificantly lower in the ramipril group (14% vs. 18%)—a 22% reduction in risk. Possible mechanisms underlying benefits include reduced vascular resistance and protection of the heart, blood vessels, and kidneys from the damage that angiotensin II and aldosterone can cause over time.
Like ramipril, perindopril [Aceon, Coversyl ] can reduce morbidity and mortality in patients at risk for major cardio-vascular events. However, the drug is not yet approved for this use. Benefits were demonstrated in the EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease (EUROPA). Patients in EUROPA were at lower risk than those in HOPE.
Can ACE inhibitors other than ramipril and perindopril also reduce cardiovascular risk? Possibly. However, at this time there is insufficient evidence to say for sure.
Diabetic Retinopathy. The RASS trial showed that at least one ACE inhibitor—enalapril—can reduce the risk of diabetic retinopathy in some patients. Specifically, in patients with type 1 diabetes who do not have hypertension, nephropathy, or established retinopathy, enalapril prevented or slowed development of retinal change. However, in patients with type 1 diabetes and established retinopathy, enalapril had no benefit. In patients with type 2 diabetes, enalapril had no benefit, regardless of retinopathy status.
Adverse EffectsACE inhibitors are generally well tolerated. Some adverse effects (e.g., first-dose hypotension, hyperkalemia) are due to a reduction in angiotensin II, whereas others (cough, angio-edema) are due to elevation of bradykinin.
First-Dose Hypotension. A precipitous drop in blood pressure may occur following the first dose of an ACE inhibitor. This reaction is caused by widespread vasodilation secondary to abrupt lowering of angiotensin II levels. First-dose hypotension is most likely in patients with severe hypertension, in patients taking diuretics, and in patients who are sodium depleted or volume depleted. To minimize the first-dose effect, initial doses should be low. Also, diuretics should be temporarily discontinued, starting 2 to 3 days before beginning an ACE inhibitor. Blood pressure should be monitored for several hours following the first dose of an ACE inhibitor. If hypotension develops, the patient should assume a supine position. If neces-sary, blood pressure can be raised with an infusion of normal saline.
Cough. All ACE inhibitors can cause persistent dry, irritat-ing, nonproductive cough. Severity can range from a scratchy throat to severe hacking cough. The underlying cause is accumulation of bradykinin secondary to inhibition of kinase II (another name for ACE). Cough occurs in about 10% of patients and is the most common reason for discontinuing therapy. Factors that increase the risk of cough include advanced age, female sex, and Asian ancestry. Cough begins to subside 3 days after discontinuing an ACE inhibitor and is gone within 10 days.
Hyperkalemia. Inhibition of aldosterone release (secondary to inhibition of angiotensin II production) can cause potassium retention by the kidney. As a rule, significant potassium accumu-lation is limited to patients taking potassium supplements, salt substitutes (which contain potassium), or a potassium-sparing diuretic. For most other patients, hyperkalemia is rare. Patients
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
Safety Alert
ACE INHIBITORSACE inhibitors can cause angioedema, a potentially life-threatening reaction. If patients report edema of the tongue, lips, or eyes, emergency care should be sought immediately, and the patient must never take ACE inhibitors again.

489
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
• Enalaprilat, the active form of enalapril, is available in solution (1.25 mg/mL) for IV therapy of severe hyper-tension. Enalaprilat is the only ACE inhibitor that is given IV.
• Fosinopril is available alone (10-, 20-, and 40-mg tablets) as Monopril, and combined with hydrochlorothiazide.
• Lisinopril is available alone (2.5-, 5-, 10-, 20-, 30-, and 40-mg tablets) as Prinivil and Zestril, as a 1 mg/mL oral solution sold as Qbrelis, and combined with hydrochlorothiazide.
• Moexipril is available alone (7.5- and 15-mg tablets) and combined with hydrochlorothiazide.
• Perindopril is available alone (2-, 4-, and 8-mg tablets) as Aceon and Coversyl , in combination with amlodipine (3.5/2.5-, 7/5-, and 14/10-mg tablets) sold as Prestalia, and combined with indapamide (2/0.625, 4/1.25, and 8/2.5 mg) as Coversyl Plus .
• Quinapril is available alone (5-, 10-, 20-, and 40-mg tablets) as Accupril, and combined with hydrochloro-thiazide as Accuretic.
• Ramipril is available alone (1.25-, 2.5-, 5-, and 10-mg capsules) as Altace, and combined with hydrochloro-thiazide (2.5/12.5, 5/12.5, 10/12.5, 5/25, 10/25 mg) as Altace HCT .
• Trandolapril is available alone (1-, 2-, and 4-mg tablets) as Mavik, and combined with verapamil as Tarka. The drug is not available in combination with hydrochlorothiazide.
ANGIOTENSIN II RECEPTOR BLOCKERSThe ARBs are relatively new, and their indications are evolving. Initially, ARBs were approved only for hypertension. Today, they also are approved for treating heart failure, diabetic nephropathy, and MI, and for prevention of MI, stroke, and death in people at high risk for cardiovascular events.
Like the ACE inhibitors, ARBs decrease the influence of angiotensin II. However, the mechanisms involved differ: Whereas ACE inhibitors block production of angiotensin II, ARBs block the actions of angiotensin II. Because both groups interfere with angiotensin II, they both have similar effects. They differ primarily in that ARBs pose a much lower risk of cough or hyperkalemia.
Given that ACE inhibitors and ARBs have very similar effects, are these drugs clinically interchangeable? No. We have clear and extensive evidence that ACE inhibitors decrease cardiovascular morbidity and mortality. The evidence for ARBs is less convincing. Accordingly, until more is known, ACE inhibitors are preferred. For patients who cannot tolerate ACE inhibitors, ARBs are an appropriate second choice.
Eight ARBs are available. All eight are very similar; hence, we will discuss them as a group, rather than choosing one as a prototype.
Mechanism of Action and Overview of Pharmacologic EffectsARBs block access of angiotensin II to its receptors in blood vessels, the adrenals, and all other tissues. As a result, ARBs have effects much like those of the ACE inhibitors. By blocking angiotensin II receptors on blood vessels, ARBs cause dilation of arterioles and veins. By blocking angiotensin II receptors
Neutropenia. Neutropenia, with its associated risk of infection, is a rare but serious complication. Neutropenia is most likely in patients with renal impairment and in those with collagen vascular diseases (e.g., systemic lupus erythematosus, scleroderma). These patients should be followed closely. Fortunately, neutropenia is reversible when detected early. If neutropenia develops, ACE inhibitors should be withdrawn immediately. Neutrophil counts should normalize in approximately 2 weeks. In the absence of early detection, neutropenia may progress to fatal agranulocytosis. Patients should be informed about early signs of infection (e.g., fever, sore throat) and instructed to report them immediately. Neutropenia is more common with captopril than with other ACE inhibitors.
Drug InteractionsDiuretics. Diuretics may intensify first-dose hypotension.
To prevent this interaction, diuretics should be withdrawn 2 to 3 days before giving an ACE inhibitor. Diuretic therapy can be resumed later if needed.
Antihypertensive Agents. The hypotensive effects of ACE inhibitors are often additive with those of other antihy-pertensive drugs (e.g., diuretics, sympatholytics, vasodilators, calcium channel blockers). When an ACE inhibitor is added to an antihypertensive regimen, dosages of other drugs may require reduction.
Drugs That Raise Potassium Levels. ACE inhibitors increase the risk of hyperkalemia caused by potassium supplements and potassium-sparing diuretics. The risk of hyperkalemia is increased because, by suppressing aldosterone secretion, ACE inhibitors can reduce excretion of potassium. To minimize the risk of hyperkalemia, potassium supplements and potassium-sparing diuretics should be employed only when clearly indicated.
Lithium. ACE inhibitors can cause lithium to accumulate to toxic levels. Lithium levels should be monitored frequently.
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). Aspirin, ibuprofen, and other NSAIDs may reduce the anti-hypertensive effects of ACE inhibitors.
Preparations, Dosage, and AdministrationExcept for enalaprilat, all ACE inhibitors are administered orally. Of the oral products, all are available in single-drug formulations. Most are also available in fixed-dose combinations with hydrochlorothiazide, a thiazide diuretic, and one agent—perindopril—is available combined with indapamide. Three ACE inhibitors—benazepril, perindopril, and trandolapril—are available combined with calcium channel blockers. Except for captopril and moexipril, all oral formulations may be admin-istered without regard to meals; captopril and moexipril should be administered 1 hour before meals. Dosages for all ACE inhibitors (except fosinopril) should be reduced in patients with renal impairment. Dosages for specific indications are shown in Table 44.1. Formulations are described in the list that follows:
• Benazepril is available alone (5-, 10-, 20-, and 40-mg tablets) as Lotensin, combined with hydrochlorothiazide as Lotensin HCT, and combined with amlodipine as Lotrel.
• Captopril is available alone (12.5-, 25-, 50-, and 100-mg tablets) as Capoten, and combined with hydrochlorothiazide.
• Enalapril is available alone (2.5-, 5-, 10-, and 20-mg tablets) as Vasotec, as a 1 mg/mL oral solution sold as Epaned, and combined with hydrochlorothiazide as Vaseretic.

490
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
cardiovascular morbidity and mortality. Of note, combining telmisartan with ramipril was no more effective than either agent alone, but did increase the risk of adverse events. Can other ARBs reduce cardiovascular risk? Possibly, but proof is lacking.
Diabetic Retinopathy. In the RASS study mentioned earlier, benefits of the ARB losartan [Cozaar] were like those of enalapril: In patients with type 1 diabetes without established retinopathy, losartan slowed the development and progression of retinopathy, but it had no benefit in patients with established retinopathy. In patients with type 2 diabetes, the drug offered no benefit at all, regardless of retinopathy status.
Adverse EffectsAll of the ARBs are well tolerated. In contrast to ACE inhibitors, ARBs do not cause clinically significant hyperkalemia. Fur-thermore, because ARBs do not promote accumulation of bradykinin in the lung, they have a lower incidence of cough.
Angioedema. Like the ACE inhibitors, ARBs can cause angioedema, although the incidence may be lower with ARBs. If angioedema occurs, ARBs should be withdrawn immediately and never used again. Severe reactions are treated with sub-cutaneous epinephrine.
ARBs cause angioedema possibly by increasing bradykinin availability. Unlike ACE inhibitors, ARBs do not inhibit bra-dykinin breakdown. However, through an indirect mechanism, ARBs may be able to increase local bradykinin synthesis.
Is it reasonable to give an ARB to a patient who developed angioedema with an ACE inhibitor? Sometimes. About 8% of patients who experience angioedema with an ACE inhibitor will also develop angioedema if given an ARB. Nonetheless, switching to an ARB may be worth the risk for specific patients, namely, those with a disorder for which ARBs are known to improve outcomes (i.e., heart failure, diabetes, and MI).
Fetal Harm. Like the ACE inhibitors, ARBs can injure the developing fetus if taken during the second or third trimester of pregnancy, and hence are contraindicated during this period. Also, there is concern that ARBs and ACE inhibitors may harm the fetus earlier in pregnancy, and hence should be discontinued as soon as pregnancy is discovered.
Renal Failure. Like the ACE inhibitors, ARBs can cause renal failure in patients with bilateral renal artery stenosis or stenosis in the artery to a single remaining kidney. Accordingly, ARBs must be used with extreme caution in patients with these conditions.
Drug InteractionsThe hypotensive effects of ARBs are additive with those of other antihypertensive drugs. When an ARB is added to an antihypertensive regimen, dosages of the other drugs may require reduction.
Preparations, Dosage, and AdministrationAll ARBs are administered PO, and all may be taken with or without food. All are available alone, and all but azilsartan are also available in fixed-dose combinations with hydrochlo-rothiazide, a thiazide diuretic. Dosages for specific indications are shown in Table 44.2. Formulations are described in the list that follows:
• Azilsartan is available alone (40- and 80-mg tablets) as Edarbi. Unlike other ARBs, the drug is not available
in the heart, ARBs can prevent angiotensin II from inducing pathologic changes in cardiac structure. By blocking angiotensin II receptors in the adrenals, ARBs decrease release of aldo-sterone and can thereby increase renal excretion of sodium and water. Sodium and water excretion is further increased through dilation of renal blood vessels.
In contrast to the ACE inhibitors, ARBs do not inhibit kinase II, and hence do not increase levels of bradykinin in the lung. As a result, ARBs have a lower risk of cough, the most common reason for stopping ACE inhibitors.
Therapeutic UsesHypertension. All ARBs are approved for hypertension.
Reductions in blood pressure equal those seen with ACE inhibitors. Whether ARBs share the ability of ACE inhibitors to reduce mortality has not been established.
Heart Failure. Currently, only two ARBs—valsartan [Diovan] and candesartan [Atacand]—are approved for heart failure. In clinical trials, these drugs reduced symptoms, decreased hospitalizations, improved functional capacity, and increased LV ejection fraction. More importantly, they prolonged survival. Because experience with these drugs is limited, they should be reserved for patients who cannot tolerate ACE inhibitors (because of cough). Although the other ARBs are not yet approved for heart failure, most authorities believe they are effective.
Diabetic Nephropathy. Two ARBs—irbesartan [Avapro] and losartan [Cozaar]—are approved for managing nephropathy in hypertensive patients with type 2 diabetes. In clinical trials, these drugs delayed development of overt nephropathy and slowed progression of established nephropathy. Benefits are due in part to reductions in blood pressure and in part to mechanisms that have not been determined. How do ARBs compare with ACE inhibitors? Although both groups of drugs can delay progression of nephropathy, only the ACE inhibitors have been shown to reduce mortality. As noted previously, neither ARBs nor ACE inhibitors are effective for primary prevention of diabetic nephropathy.
Myocardial Infarction. One ARB—valsartan [Diovan]—is approved for reducing cardiovascular mortality in post-MI patients with heart failure or LV dysfunction. Approval was based on the results of a major trial—the Valsartan in Acute Myocardial Infarction Trial (VALIANT)—that showed that valsartan was as effective as captopril at reducing short-term and long-term mortality in these patients.
Stroke Prevention. One ARB—losartan [Cozaar]—is approved for reducing the risk of stroke in patients with hypertension and LV hypertrophy. In clinical studies, stroke prevention with losartan was better than with atenolol (a beta blocker), even though both drugs produced an equivalent decrease in blood pressure. This observation indicates that the benefits of losartan cannot be explained on the basis of reduced blood pressure alone.
Prevention of MI, Stroke, and Death in Patients at High Cardiovascular Risk. One ARB—telmisartan [Micar-dis]—is approved for reducing the risk of MI, stroke, and death from cardiovascular causes in patients age 55 years and older, but only if they are intolerant of ACE inhibitors. (Recall that ramipril, an ACE inhibitor, is also approved for preventing MI, stroke, and death in high-risk patients.) Approval of telmisartan was based on the ONTARGET study, which showed that telmisartan was similar to ramipril with regard to reducing

491
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
ALISKIREN, A DIRECT RENIN INHIBITOR
DRIs are drugs that act on renin to inhibit the conversion of angiotensinogen into angiotensin I. By decreasing production of angiotensin I, DRIs can suppress the entire RAAS. Currently, only one DRI—aliskiren [Tekturna, Rasilez ]—is available.
Blood pressure reduction with aliskiren equals that seen with ACE inhibitors. Aliskiren causes less cough and angio-edema than the ACE inhibitors, but poses similar risks to the developing fetus.
Mechanism of ActionAliskiren binds tightly with renin and thereby inhibits the cleavage of angiotensinogen into angiotensin I. Since this reaction is the first and rate-limiting step in the production of angiotensin II and aldosterone, aliskiren can reduce the influence of the entire RAAS. In clinical trials, the drug decreased plasma renin activity by 50% to 80%. Although aliskiren works at an earlier step than either the ACE inhibitors or ARBs, there is no proof that doing so results in superior clinical outcomes.
Therapeutic UseAliskiren is approved only for hypertension. It may be used alone or in combination with other antihypertensives. In clinical trials, aliskiren reduced blood pressure to the same extent as did ACE inhibitors, ARBs, or calcium channel blockers. Maximal effects developed within 2 weeks. Although aliskiren can reduce blood pressure in hypertensive patients, we don’t know whether the drug also reduces negative outcomes (i.e., blindness, stroke, kidney disease, death). In contrast, the ability of ACE inhibitors and ARBs to improve outcomes is well established. Until the long-term benefits and safety of aliskiren are known, older antihypertensives should be considered first. In addition to its
combined with hydrochlorothiazide but is paired with chlorthalidone and marketed as Edarbyclor.
• Candesartan is available alone (4-, 8-, 16-, and 32-mg tablets) as Atacand, and combined with hydrochloro-thiazide as Atacand HCT.
• Eprosartan is available alone (400- and 600-mg tablets) as Teveten, and combined with hydrochlorothiazide as Teveten HCT.
• Irbesartan is available alone (75-, 150-, and 300-mg tablets) as Avapro, and combined with hydrochlorothiazide as Avalide.
• Losartan is available alone (25-, 50-, and 100-mg tablets) as Cozaar, and combined with hydrochlorothiazide as Hyzaar.
• Olmesartan is available alone (5-, 20-, and 40-mg tablets) as Benicar and Olmetec , combined with hydrochlo-rothiazide as Benicar HCT and Olmetec Plus , combined with amlodipine as Azor, and combined with amlodipine plus hydrochlorothiazide as Tribenzor.
• Telmisartan is available alone (20-, 40-, and 80-mg tablets) as Micardis, combined with hydrochlorothiazide as Micardis HCT, and combined with amlodipine as Twynsta.
• Valsartan is available alone (40-, 80-, 160-, and 320-mg tablets) as Diovan, combined with hydrochlorothiazide as Diovan HCT, combined with amlodipine as Exforge, combined with nebivolol as Byvalson, and combined with amlodipine plus hydrochlorothiazide as Exforge HCT.
A new combination of valsartan and sacubitril, sold as Entresto, is approved only for heart failure and is discussed in Chapter 48.
Generic Name Brand Name Approved Indications Initial Dosage Maintenance Dosage
Azilsartan Edarbi Hypertension 40–80 mg once/day 80 mg once/dayCandesartan Atacand Hypertension 16 mg once/day 8–32 mg/day in 1 or 2 doses
Heart failure 4 mg once/day 32 mg once/dayEprosartan Teveten Hypertension 600 mg once/day 400–800 mg/day in 1 or 2 dosesIrbesartan Avapro Hypertension 150 mg once/day 150–300 mg once/day
Diabetic nephropathya 300 mg once/day 300 mg once/dayLosartan Cozaar Hypertension 25–50 mg once/day 25–100 mg/day in 1 or 2 doses
Stroke preventionb 50 mg once/day 50–100 mg once/dayDiabetic nephropathya 50 mg once/day 100 mg once/day
Olmesartan Benicar, Olmetec Hypertension 20 mg once/day 20–40 mg once/day
Telmisartan Micardis Hypertension 40 mg once/day 20–80 mg once/dayPrevention of MI, stroke, and death
in people at high risk for CVD but who can’t take an ACE inhibitor
80 mg once/day 80 mg once/day
Valsartan Diovan Hypertension 80–160 mg once/day 80–320 mg once/dayHeart failure 40 mg twice/day 40–160 mg twice/dayMI 20 mg twice/day 160 mg twice/day
TABLE 44.2 ■ Angiotensin II Receptor Blockers: Approved Indications and Adult Dosages
aIn patients with type 2 diabetes.bIn patients with hypertension and left ventricular hypertrophy.ACE, Angiotensin-converting enzyme; CVD, cardiovascular disease; MI, myocardial infarction.

492
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Aliskiren/hydrochlorothiazide [Tekturna HCT, Rasilez HCT ] tablets are available in four strengths—150 mg/12.5 mg, 150 mg/25 mg, 300 mg/12.5 mg, and 300 mg/25 mg—for once-daily dosing. As with Tekturna, each daily dose should be taken at the same time with respect to meals.
Aliskiren/amlodipine [Tekamlo] tablets are available in four strengths— 150 mg/5 mg, 150 mg/10 mg, 300 mg/5 mg, and 300 mg/10 mg—for once-daily dosing. As with Tekturna and Tekturna HCT, each daily dose should be taken at the same time with respect to meals.
ALDOSTERONE ANTAGONISTS
Aldosterone antagonists are drugs that block receptors for aldosterone. Two such agents are available: eplerenone and spironolactone. Both drugs have similar structures and actions, and both are used for the same disorders: hypertension and heart failure. They differ, however, in that spironolactone is less selective than eplerenone. As a result, spironolactone causes more side effects.
EplerenoneEplerenone [Inspra] is a first-in-class selective aldosterone receptor blocker. The drug is used for hypertension and heart failure and has one significant side effect: hyperkalemia.
Mechanism of ActionEplerenone produces selective blockade of aldosterone receptors, having little or no effect on receptors for other steroid hormones (e.g., glucocorticoids, progesterone, androgens). In the kidney, activation of aldosterone receptors promotes excretion of potassium and retention of sodium and water. Receptor blockade has the opposite effect: retention of potassium and increased excretion of sodium and water. Loss of sodium and water reduces blood volume, and hence blood pressure. Blockade of aldosterone receptors at nonrenal sites may prevent or reverse pathologic effects of aldosterone on cardiovascular structure and function.
Therapeutic UsesHypertension. For treatment of hypertension, eplerenone
may be used alone or in combination with other antihypertensive agents. Maximal reductions in blood pressure take about 4 weeks to develop. In clinical trials, reductions in blood pressure were equivalent to those produced by spironolactone and superior to those produced by losartan (an ARB). In patients already using an ACE inhibitor or an ARB, adding eplerenone produced a further reduction in blood pressure.
Although it is clear that eplerenone can lower blood pressure, we have no information on what really matters: the drug’s ability to reduce morbidity and mortality in patients with isolated hypertension and lack of LV hypertrophy. Until more is known, eplerenone should be reserved for patients who have not responded to traditional antihypertensive drugs.
Heart Failure. In patients with heart failure, eplerenone can improve symptoms, reduce hospitalizations, and prolong life. Benefits appear to derive from blocking the adverse effects of aldosterone on cardiovascular structure and function. Use of eplerenone in heart failure is discussed in Chapter 48.
PharmacokineticsEplerenone is administered orally, and absorption is not affected by food. Plasma levels peak about 1.5 hours after dosing. Absolute bioavailability is
use in hypertension, aliskiren was evaluated for treating heart failure and renal failure associated with diabetes. Unfortunately, in the ATMOSPHERE and ALTITUDE trials, aliskiren was no more effective than other current treatments.
PharmacokineticsAliskiren is administered orally, and bioavailability is low (only 2.5%). Dosing with a high-fat meal makes availability much lower (about 0.8%). Aliskiren undergoes some metabolism by CYP3A4 (the 3A4 isoenzyme of cytochrome P450), but the extent of metabolism is not known. About 25% of the drug is eliminated unchanged in the urine. The half-life is about 24 hours.
Adverse EffectsAliskiren is generally well tolerated. At usual doses, the risk of angioedema, cough, or hyperkalemia is low. At high thera-peutic doses, some patients experience diarrhea. Like other drugs that affect the RAAS, aliskiren should be avoided during pregnancy.
Angioedema and Cough. With ACE inhibitors, angio-edema and cough result from inhibition of kinase II. Because aliskiren does not inhibit kinase II, the risk of these effects is low. In clinical trials, the incidence of cough was 1.1% with aliskiren versus about 10% with an ACE inhibitor. Similarly, the incidence of angioedema was 0.06% with aliskiren versus 1% with an ACE inhibitor. If angioedema does occur, aliskiren should be discontinued immediately.
Gastrointestinal Effects. Aliskiren causes dose-dependent diarrhea, seen in 2.3% of patients taking 300 mg/day. Women and older adults are most susceptible. Excessive doses (600 mg/day) are associated with abdominal pain and dyspepsia.
Hyperkalemia. Like the ACE inhibitors, aliskiren rarely causes hyperkalemia when used alone. However, hyperkale-mia might be expected if aliskiren were combined with an ACE inhibitor, a potassium-sparing diuretic, or potassium supplements.
Fetal Injury and Death. Although aliskiren has not been studied in pregnant women, the drug is likely to pose a risk of major congenital malformations and fetal death because the risk of these events is well established with other drugs that suppress the RAAS. Therefore, like the ACE inhibitors and ARBs, aliskiren is contraindicated during the second and third trimesters, and should be discontinued as soon as possible when pregnancy occurs.Drug InteractionsAliskiren undergoes some metabolism by CYP3A4, but it neither induces nor inhibits the P450 system. In clinical trials, aliskiren had no significant interac-tions with atenolol, digoxin, amlodipine, or hydrochlorothiazide. However, levels of aliskiren were significantly raised by atorvastatin and ketoconazole (a P450 inhibitor), and significantly lowered by irbesartan. Levels of furosemide were lowered by aliskiren.
Preparations, Dosage, and AdministrationAliskiren is available alone as Tekturna and Rasilez , in combination with hydrochlorothiazide as Tekturna HCT and Rasilez HCT , and in combination with amlodipine as Tekamlo. All formulations are indicated for hypertension.
Aliskiren alone [Tekturna, Rasilez ] is available in 150- and 300-mg tablets. The initial dosage is 150 mg once a day. If control of blood pressure is inadequate, dosage may be increased to 300 mg once a day. Daily doses above 300 mg will not increase benefits, but will increase the risk of diarrhea. Since high-fat meals decrease absorption substantially, each daily dose should be taken at the same time with respect to meals (e.g., 1 hour before dinner), so as to achieve a consistent response.

493
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
Drugs that raise potassium levels can increase the risk of hyperkalemia. Eplerenone should not be combined with potas-sium supplements, salt substitutes, or potassium-sparing diuretics. Combining eplerenone with ACE inhibitors or ARBs should be done with caution.
Drugs similar to eplerenone (e.g., ACE inhibitors and diuretics) are known to increase levels of lithium. Although the combination of eplerenone and lithium has not been studied, caution is nonetheless advised. Lithium levels should be measured frequently.Preparations, Dosage, and AdministrationEplerenone [Inspra] is available in 25- and 50-mg tablets. The usual starting dosage is 50 mg once a day, taken with or without food. After 4 weeks, dosage can be increased to 50 mg twice daily (if the hypotensive response has been inadequate). Raising the dosage above 100 mg/day is not recommended because doing so is unlikely to increase the therapeutic response, but will increase the risk of hyperkalemia. In patients taking weak inhibitors of CYP3A4, the initial dosage should be reduced by 50% (to 25 mg once a day).
SpironolactoneSpironolactone [Aldactone], a much older drug than eplerenone, blocks receptors for aldosterone, but also binds with receptors for other steroid hormones (e.g., glucocorticoids, progesterone, androgens). Blockade of aldosterone receptors underlies beneficial effects in hypertension and heart failure, as well as the drug’s major adverse effect: hyperkalemia. Binding with receptors for other steroid hormones underlies additional adverse effects: gynecomastia, menstrual irregularities, impotence, hirsutism, and deepening of the voice. The basic pharmacology of spironolactone and its use in heart failure are discussed in Chapters 41 and 48, respectively.
unknown. Eplerenone undergoes metabolism by CYP3A4, followed by excretion in the urine (67%) and feces (32%). The elimination half-life is 4 to 6 hours.
Adverse EffectsEplerenone is generally well tolerated. The incidence of adverse effects is nearly identical to that of placebo. A few adverse effects—diarrhea, abdominal pain, cough, fatigue, gynecomastia, flu-like syndrome—occur slightly (1% to 2%) more often with eplerenone than with placebo.
Hyperkalemia. The greatest concern is hyperkalemia, which can occur secondary to potassium retention. Because of this risk, combined use with potassium supplements, salt substitutes, or potassium-sparing diuretics (e.g., spironolactone, triamterene) is contraindicated. Combined use with ACE inhibitors or ARBs is permissible, but should be done with caution. Eplerenone is contraindicated for patients with high serum potassium (above 5.5 mEq/L) and for patients with impaired renal function or type 2 diabetes with microalbumin-uria, both of which can promote hyperkalemia. Monitoring potassium levels is recommended for patients at risk (e.g., those taking ACE inhibitors or ARBs).
Drug InteractionsInhibitors of CYP3A4 can increase levels of eplerenone, thereby posing a risk of toxicity. Weak inhibitors (e.g., erythromycin, saquinavir, verapamil, fluconazole) can double eplerenone levels. Strong inhibitors (e.g., ketoconazole, itraconazole) can increase levels fivefold. If eplerenone is combined with a weak inhibitor, eplerenone dosage should be reduced. Eplerenone should not be combined with a strong inhibitor.
KEY POINTS
■ The RAAS helps regulate blood pressure, blood volume, and fluid and electrolyte balance. The system can promote cardiovascular pathology.
■ The RAAS acts through production of angiotensin II and aldosterone.
■ Angiotensin II has much greater biologic activity than angiotensin I or angiotensin III.
■ Angiotensin II is formed by the actions of two enzymes: renin and ACE.
■ Angiotensin II causes vasoconstriction (primarily in arterioles) and release of aldosterone. In addition, angio-tensin II can promote pathologic changes in the heart and blood vessels.
■ Aldosterone acts on the kidneys to promote retention of sodium and water. In addition, aldosterone can also mediate pathologic changes in cardiovascular function.
■ The RAAS raises blood pressure by causing vasoconstriction and by increasing blood volume (secondary to aldosterone-mediated retention of sodium and water).
■ In addition to the traditional RAAS, in which angiotensin II is produced in the blood and then carried to target tissues, angiotensin II can be produced locally by individual tissues.
■ Beneficial effects of ACE inhibitors result largely from inhibition of ACE and partly from inhibition of kinase II (the name for ACE when the substrate is bradykinin).
■ By inhibiting ACE, ACE inhibitors decrease production of angiotensin II. The result is vasodilation, decreased blood volume, and prevention or reversal of pathologic changes in the heart and blood vessels mediated by angiotensin II and aldosterone.
■ ACE inhibitors (and ARBs) are used to treat patients with hypertension, heart failure, MI, and established diabetic nephropathy. In addition, they are used to prevent MI, stroke, and death from cardiovascular causes in patients at high risk for a cardiovascular event. Of note, ACE inhibitors (and ARBs) are not effective for primary preven-tion of diabetic nephropathy.
■ Preliminary data indicate that ACE inhibitors (and ARBs) can reduce the risk of developing diabetic retinopathy, although they can’t slow the progression of established retinopathy.
■ ACE inhibitors can produce significant first-dose hypoten-sion by causing a sharp drop in circulating angiotensin II.
■ Cough, secondary to accumulation of bradykinin, is the most common reason for discontinuing ACE inhibitors.
■ By suppressing aldosterone release, ACE inhibitors can cause hyperkalemia. Exercise caution in patients taking potassium supplements, salt substitutes, or potassium-sparing diuretics.
■ ACE inhibitors can cause major fetal malformations and should be avoided during pregnancy. Until recently,
Continued

494
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
we thought that risk was limited to exposure during the second and third trimesters. However, new data indicate that exposure during the first trimester may be dangerous as well.
■ ACE inhibitors can cause a precipitous drop in blood pressure in patients with bilateral renal artery stenosis (or stenosis in the artery to a single remaining kidney).
■ ARBs block the actions of angiotensin II in blood vessels, the adrenals, and all other tissues.
■ ARBs are similar to ACE inhibitors in that they cause vasodilation, suppress aldosterone release, promote excre-tion of sodium and water, reduce blood pressure, and cause birth defects and angioedema.
■ ARBs differ from ACE inhibitors in that they have a much lower incidence of hyperkalemia or cough.
■ Aliskiren, a DRI, binds tightly with renin and thereby inhibits cleavage of angiotensinogen into angiotensin I. As a result, the drug suppresses the entire RAAS.
■ Like the ACE inhibitors and ARBs, aliskiren causes vasodilation, suppresses aldosterone release, promotes excretion of sodium and water, reduces blood pressure, and causes birth defects and angioedema.
■ Despite their similarities, aliskiren, ARBs, and ACE inhibi-tors are not clinically interchangeable.
■ Aldosterone antagonists (spironolactone, eplerenone) block receptors for aldosterone.
■ By blocking aldosterone receptors, aldosterone antagonists can (1) promote renal excretion of sodium and water (and can thereby reduce blood volume and blood pressure) and (2) prevent or reverse pathologic effects of aldosterone on cardiovascular structure and function.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
ANGIOTENSIN-CONVERTING ENZYME INHIBITORSBenazeprilCaptoprilEnalaprilEnalaprilatFosinoprilLisinoprilMoexiprilPerindoprilQuinaprilRamiprilTrandolapril
Unless indicated otherwise, the implications summarized in the sections that follow pertain to all of the ACE inhibitors.
Preadministration AssessmentTherapeutic GoalsACE inhibitors are used to:
• Reduce blood pressure in patients with hypertension (all ACE inhibitors).
• Improve hemodynamics in patients with heart failure (captopril, enalapril, fosinopril, lisinopril, quinapril).
• Slow progression of established diabetic nephropathy (captopril).
• Reduce mortality following acute MI (lisinopril).• Treat heart failure after MI (ramipril, trandolapril).• Reduce the risk of MI, stroke, or death from cardiovascular
causes in patients at high risk (ramipril).• Reduce cardiovascular mortality or nonfatal MI in patients
with stable coronary artery disease (perindopril).
Baseline DataDetermine blood pressure, and obtain a white blood cell count and differential.
Identifying High-Risk PatientsACE inhibitors are contraindicated during the second and third trimesters of pregnancy and for patients with (1) bilateral renal artery stenosis (or stenosis in the artery to a single remaining kidney) or (2) a history of hypersensitivity reactions (especially angioedema) to ACE inhibitors.
Exercise caution in patients with salt or volume depletion, renal impairment, or collagen vascular disease, and in those taking potassium supplements, salt substitutes, potassium-sparing diuretics, ARBs, aliskiren, or lithium.
Implementation: AdministrationRoutes
Oral. All ACE inhibitors (except enalaprilat).Intravenous. Enalaprilat.
Dosage and AdministrationDosage is low initially and then gradually increased.
Instruct patients to administer captopril and moexipril at least 1 hour before meals. All other oral ACE inhibitors can be administered with food.
Ongoing Evaluation and InterventionsMonitoring SummaryMonitor blood pressure closely for 2 hours after the first dose and periodically thereafter. Obtain a white blood cell count and differential every 2 weeks for the first 3 months of therapy and periodically thereafter.
Evaluating Therapeutic EffectsHypertension. Monitor for reduced blood pressure. The
usual target pressure is systolic/diastolic of 140/90 mm Hg or 130/80 in patients with diabetes.
Heart Failure. Monitor for a lessening of signs and symptoms (e.g., dyspnea, cyanosis, jugular vein distention, edema).

495
CHAPTER 44 Drugs Acting on the Renin-Angiotensin-Aldosterone System
Continued
Diabetic Nephropathy. Monitor for proteinuria and altered glomerular filtration rate.
Minimizing Adverse EffectsFirst-Dose Hypotension. Severe hypotension can occur
with the first dose. Minimize hypotension by (1) withdrawing diuretics 2 to 3 days before initiating ACE inhibitors and (2) using low initial doses. Monitor blood pressure for 2 hours following the first dose. Instruct patients to lie down if hypotension develops. If necessary, infuse normal saline to restore pressure.
Cough. Warn patients about the possibility of persistent dry, irritating, nonproductive cough. Instruct them to consult the prescriber if cough is bothersome. Stopping the ACE inhibitor may be indicated.
Hyperkalemia. ACE inhibitors may increase potassium levels. Instruct patients to avoid potassium supplements and potassium-containing salt substitutes unless they are pre-scribed by the provider. Potassium-sparing diuretics must also be avoided.
Fetal Injury. Warn women of childbearing age that taking ACE inhibitors during the second and third trimesters of pregnancy can cause major fetal injury (hypotension, hyper-kalemia, skull hypoplasia, anuria, reversible and irreversible renal failure, death) and that taking these drugs earlier in pregnancy may pose a risk as well. If the patient becomes pregnant, withdraw ACE inhibitors as soon as possible. Closely monitor infants who have been exposed to ACE inhibitors during the second or third trimester for hypotension, oliguria, and hyperkalemia.
Angioedema. This rare and potentially fatal reaction is characterized by giant wheals and edema of the tongue, glottis, and pharynx. Instruct patients to seek immediate medical attention if these symptoms develop. If angioedema is diagnosed, ACE inhibitors should be discontinued and never used again. Treat severe reactions with subcutaneous epinephrine.
Renal Failure. Renal failure is a risk for patients with bilateral renal artery stenosis or stenosis in the artery to a single remaining kidney. ACE inhibitors must be used with extreme caution in these people.
Neutropenia (Mainly With Captopril). Neutropenia poses a high risk of infection. Inform patients about early signs of infection (fever, sore throat, mouth sores), and instruct them to notify the prescriber if these occur. If neutropenia develops, withdraw the drug immediately; neutrophil counts should normalize in approximately 2 weeks. Neutropenia is most likely in patients with renal impairment and collagen vascular diseases (e.g., systemic lupus erythematosus, scleroderma); monitor these patients closely.
Minimizing Adverse InteractionsDiuretics. Diuretics may intensify first-dose hypotension.
Withdraw diuretics 2 to 3 days before beginning an ACE inhibitor. Diuretics may be resumed later if needed.
Antihypertensive Agents. The antihypertensive effects of ACE inhibitors are additive with those of other antihyperten-sive drugs (e.g., ARBs, diuretics, sympatholytics, vasodilators,
calcium channel blockers). When an ACE inhibitor is added to an antihypertensive regimen, dosages of the other drugs may require reduction.
Drugs That Elevate Potassium Levels. ACE inhibitors increase the risk of hyperkalemia associated with potassium supplements, potassium-sparing diuretics, and possibly aliskiren. Risk can be minimized by avoiding potassium supplements and potassium-sparing diuretics except when they are clearly indicated.
Lithium. ACE inhibitors can increase serum levels of lithium, causing toxicity. Monitor lithium levels frequently.
Nonsteroidal Anti-Inflammatory Drugs. NSAIDs (e.g., aspirin, ibuprofen) can interfere with the antihypertensive effects of ACE inhibitors. Advise patients to minimize NSAID use.
ANGIOTENSIN II RECEPTOR BLOCKERSAzilsartanCandesartanEprosartanIrbesartanLosartanOlmesartanTelmisartanValsartan
Unless indicated otherwise, the implications summarized here pertain to all of the ARBs.
Preadministration AssessmentTherapeutic GoalsARBs are used to:
• Reduce blood pressure in patients with hypertension (all ARBs).
• Treat heart failure (candesartan, valsartan).• Slow the progression of established diabetic nephropathy
(irbesartan, losartan).• Prevent stroke in patients with hypertension and LV
hypertrophy (losartan).• Protect against MI, stroke, and death from cardiovascular
causes in high-risk patients, but only if they can’t tolerate ACE inhibitors (telmisartan).
• Treat heart failure after MI (valsartan).
Baseline DataDetermine blood pressure.
Identifying High-Risk PatientsARBs are contraindicated during the second and third tri-mesters of pregnancy and for patients with either (1) bilateral renal artery stenosis (or stenosis in the artery to a single remaining kidney) or (2) a history of hypersensitivity reactions (especially angioedema) to ARBs.
Implementation: AdministrationRouteOral.
Summary of Major Nursing Implicationsa—cont’d

496
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Dosage and AdministrationInform patients that ARBs may be taken with or without food.
Ongoing Evaluation and InterventionsEvaluating Therapeutic Effects
Hypertension. Monitor for reduced blood pressure. The usual target pressure is systolic/diastolic of 140/90 mm Hg or 130/80 in patients with diabetes.
Heart Failure. Monitor for a lessening of signs and symptoms (e.g., dyspnea, cyanosis, jugular vein distention, edema).
Diabetic Nephropathy. Monitor for proteinuria and altered glomerular filtration rate.
Minimizing Adverse EffectsAngioedema. This rare and potentially fatal reaction
is characterized by giant wheals and edema of the tongue, glottis, and pharynx. Instruct patients to seek immediate medical attention if these symptoms develop. If angioedema is diagnosed, ARBs should be discontinued and never used again. Treat severe reactions with subcutaneous epinephrine.
Fetal Injury. Warn women of childbearing age that ARBs can cause fetal injury during the second and third trimesters of pregnancy, and may pose a risk earlier in pregnancy as well. If the patient becomes pregnant, withdraw ARBs as soon as possible. Closely monitor infants who have been exposed to ARBs during the second or third trimester for hypotension, oliguria, and hyperkalemia.
Renal Failure. Renal failure is a risk for patients with bilateral renal artery stenosis or stenosis in the artery to a single remaining kidney. ARBs are contraindicated for these people.
Minimizing Adverse InteractionsAntihypertensive Agents. The antihypertensive effects
of ARBs are additive with those of other antihypertensive drugs (e.g., diuretics, sympatholytics, vasodilators, ACE inhibitors, calcium channel blockers). When an ARB is added to an antihypertensive regimen, dosages of the other drugs may require reduction.
ALISKIREN, A DIRECT RENIN INHIBITORPreadministration AssessmentTherapeutic GoalReduction of blood pressure in patients with hypertension.
Baseline DataDetermine blood pressure.
Identifying High-Risk PatientsAliskiren is contraindicated during the second and third trimesters of pregnancy.
Exercise caution in patients taking potassium supplements, salt substitutes, potassium-sparing diuretics, or ACE inhibitors.
Implementation: AdministrationRouteOral.
Dosage and AdministrationAdvise patients to take each daily dose at the same time with respect to meals (e.g., 1 hour before dinner). Dosage should be low (150 mg/day) initially, and increased to a maximum of 300 mg/day if needed.
Ongoing Evaluation and InterventionsMinimizing Adverse Effects
Hyperkalemia. Aliskiren may increase potassium levels. Instruct patients to avoid potassium supplements and potassium-containing salt substitutes unless they are pre-scribed by the provider. Potassium-sparing diuretics must also be avoided. Exercise caution in patients taking an ACE inhibitor.
Fetal Injury. Warn women of childbearing age that aliskiren taken during the second and third trimesters of pregnancy can cause fetal injury (hypotension, hyperkalemia, skull hypoplasia, anuria, reversible and irreversible renal failure, death). If the patient becomes pregnant, withdraw aliskiren as soon as possible. Closely monitor infants who have been exposed to aliskiren during the second or third trimester for hypotension, oliguria, and hyperkalemia.
Angioedema. This rare and potentially fatal reaction is characterized by giant wheals and edema of the tongue, glottis, and pharynx. Instruct patients to seek immediate medical attention if these symptoms develop. If angioedema is diagnosed, aliskiren should be discontinued and never used again. Treat severe reactions with subcutaneous epinephrine.
Minimizing Adverse InteractionsDrugs That Elevate Potassium Levels. Aliskiren
increases the risk of hyperkalemia associated with ACE inhibitors, potassium supplements, and potassium-sparing diuretics. Risk can be minimized by avoiding ACE inhibitors, potassium supplements, and potassium-sparing diuretics except when they are clearly indicated.
Antihypertensive Agents. The antihypertensive effects of aliskiren are additive with those of other antihypertensive drugs (e.g., ACE inhibitors, ARBs, diuretics, sympatholytics, vasodilators, calcium channel blockers). When aliskiren is added to an antihypertensive regimen, dosages of the other drugs may require reduction.
Summary of Major Nursing Implicationsa—cont’d
aPatient education information is highlighted as blue text.

497
C H A P T E R
45 Calcium Channel Blockers
Calcium Channels: Physiologic Functions and Consequences of Blockade, p. 497Vascular Smooth Muscle, p. 497Heart, p. 497
Calcium Channel Blockers: Classification and Sites of Action, p. 497Classification, p. 497Sites of Action, p. 498
Verapamil and Diltiazem: Agents That Act on Vascular Smooth Muscle and the Heart, p. 498Verapamil, p. 498Diltiazem, p. 500
Dihydropyridines: Agents That Act Mainly on Vascular Smooth Muscle, p. 501Nifedipine, p. 501Other Dihydropyridines, p. 502
Key Points, p. 503Summary of Major Nursing Implications,
p. 503
HeartIn the heart, calcium channels help regulate the myocardium, the sinoatrial (SA) node, and the atrioventricular (AV) node. Calcium channels at all three sites are coupled to beta1-adrenergic receptors.
MyocardiumIn cardiac muscle, calcium entry has a positive inotropic effect. That is, calcium increases force of contraction. If calcium channels in atrial and ventricular muscle are blocked, contractile force will diminish.
SA NodePacemaker activity of the SA node is regulated by calcium influx. When calcium channels are open, spontaneous discharge of the SA node increases. Conversely, when calcium channels close, pacemaker activity declines. Hence, the effect of calcium channel blockade is to reduce heart rate.
AV NodeImpulses that originate in the SA node must pass through the AV node on their way to the ventricles. Because of this arrange-ment, regulation of AV conduction plays a critical role in coordinating contraction of the ventricles with contraction of the atria.
The excitability of AV nodal cells is regulated by calcium entry. When calcium channels are open, calcium entry increases and cells of the AV node discharge more readily. Conversely, when calcium channels are closed, discharge of AV nodal cells is suppressed. Hence, the effect of calcium channel blockade is to decrease velocity of conduction through the AV node.
Coupling of Cardiac Calcium Channels to Beta1-Adrenergic ReceptorsIn the heart, calcium channels are coupled to beta1-adrenergic receptors (Fig. 45.1). As a result, when cardiac beta1 receptors are activated, calcium influx is enhanced. Conversely, when beta1 receptors are blocked, calcium influx is suppressed. Because of this relationship, CCBs and beta blockers have identical effects on the heart. That is, they both reduce force of contraction, slow heart rate, and suppress conduction through the AV node.
CALCIUM CHANNEL BLOCKERS: CLASSIFICATION AND SITES OF ACTIONClassificationThe CCBs used in the United States belong to three chemical families (Table 45.1). The largest family is the dihydropyridines, for which nifedipine is the prototype. This family name is encountered frequently, and hence the name is worth remember-ing. The other two families consist of orphans: verapamil is the only phenylalkylamine, and diltiazem is the only benzothiaz-epine. The drug names are important; the family names are not.
Calcium channel blockers (CCBs) are drugs that prevent calcium ions from entering cells. These agents have their greatest effects on the heart and blood vessels. CCBs are used widely to treat hypertension, angina pectoris, and cardiac dysrhythmias. There remains controversy about the safety of CCBs in patients with heart failure. Alternative names for CCBs are calcium antago-nists and slow channel blockers.
CALCIUM CHANNELS: PHYSIOLOGIC FUNCTIONS AND CONSEQUENCES OF BLOCKADECalcium channels are gated pores in the cytoplasmic membrane that regulate entry of calcium ions into cells. Calcium entry plays a critical role in the function of vascular smooth muscle (VSM) and the heart.
Vascular Smooth MuscleIn VSM, calcium channels regulate contraction. When an action potential travels down the surface of a smooth muscle cell, calcium channels open and calcium ions flow inward, thereby initiating the contractile process. If calcium channels are blocked, contraction will be prevented and vasodilation will result.
At therapeutic doses, CCBs act selectively on peripheral arterioles and arteries and arterioles of the heart. CCBs have no significant effect on veins.

498
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
and diltiazem can). The differences in selectivity among CCBs are based on structural differences among the drugs themselves and structural differences among calcium channels.
VERAPAMIL AND DILTIAZEM: AGENTS THAT ACT ON VASCULAR SMOOTH MUSCLE AND THE HEARTVerapamilVerapamil [Calan, Verelan, Covera-HS ] blocks calcium channels in blood vessels and in the heart. Major indications are angina pectoris, essential hypertension, and cardiac dys-rhythmias. Verapamil was the first CCB available and will serve as our prototype for the group.
Hemodynamic EffectsThe overall hemodynamic response to verapamil is the net result of (1) direct effects on the heart and blood vessels and (2) reflex responses.
SA node AV node Myocardium
Ca++
Heart rateConductionvelocity
Force ofcontraction
GTP PO4
ATP
cAMPProteinkinase
(inactive)
Proteinkinase(active)
Ca++
αGDP
GDP
GTP Adenylyl cyclase
Cell membrane
Outside
Inside
β agonist
αG protein
β receptor
Cal
cium
Cha
nnel
Fig. 45.1 ■ Coupling of cardiac calcium channels with beta1-adrenergic receptors. In the heart, beta1 receptors are coupled to calcium channels. As a result, when cardiac beta1 receptors are activated, calcium influx is enhanced. The process works as follows: Binding of an agonist (e.g., norepinephrine) causes a conformational change in the beta receptor, which in turn causes a change in G protein, converting it from an inactive state (in which GDP is bound to the alpha subunit) to an active state (in which GTP is bound to the alpha subunit). (G protein is so named because it binds guanine nucleotides: GDP and GTP.) Following activation, the alpha subunit dissociates from the rest of G protein and activates adenylyl cyclase, an enzyme that converts ATP to cyclic AMP (cAMP). cAMP then activates protein kinase, an enzyme that phosphorylates proteins—in this case, the calcium channel. Phosphorylation changes the channel such that calcium entry is enhanced when the channel opens. (Opening of the channel is triggered by a change in membrane voltage [i.e., by passage of an action potential].) The effect of calcium entry on cardiac function is determined by the type of cell involved. If the cell is in the SA node, heart rate increases; if the cell is in the AV node, impulse conduction through the node accelerates; and if the cell is part of the myocardium, force of contraction is increased. Because binding of a single agonist molecule to a single beta receptor stimulates the synthesis of many cAMP molecules, with the subsequent activation of many protein kinase molecules, causing the phosphorylation of many calcium channels, this system can greatly amplify the signal initiated by the agonist. (GDP, Guanosine diphosphate; GTP, guanosine-5’-triphosphate; cAMP, cyclic AMP phospho-diesterase; ATP, adenosine triphosphate.)
Prototype Drugs
CALCIUM CHANNEL BLOCKERSAgents That Affect the Heart and Blood VesselsVerapamil
Dihydropyridines: Agents That Act Mainly on Blood VesselsNifedipine
Sites of ActionAt therapeutic doses, the dihydropyridines act primarily on arterioles; in contrast, verapamil and diltiazem act on arterioles and the heart (see Table 45.1). However, although dihydropyri-dines don’t affect the heart at therapeutic doses, toxic doses can produce dangerous cardiac suppression (just like verapamil

499
CHAPTER 45 Calcium Channel Blockers
Elimination is primarily by hepatic metabolism. Because the drug is eliminated by the liver, doses must be reduced sub-stantially in patients with hepatic impairment.
Therapeutic UsesAngina Pectoris. Verapamil is used widely to treat angina
pectoris. The drug is approved for vasospastic angina and angina of effort. Benefits in both disorders derive from vasodilation. The role of verapamil in angina is discussed in Chapter 51.
Essential Hypertension. Verapamil is a second-line agent for chronic hypertension, used after thiazide diuretics. The drug lowers blood pressure by dilating arterioles. The role of verapamil and other CCBs in hypertension is discussed in Chapter 47.
Cardiac Dysrhythmias. Verapamil, administered IV, is used to slow ventricular rate in patients with atrial flutter, atrial fibrillation, and paroxysmal supraventricular tachycardia. Benefits derive from suppressing impulse conduction through the AV node, thereby preventing the atria from driving the ventricles at an excessive rate. Antidysrhythmic applications are discussed in Chapter 49.
Adverse EffectsCommon Effects. Verapamil is generally well tolerated.
Constipation occurs frequently and is the most common complaint. This problem, which can be especially severe in older adults, can be minimized by increasing dietary fluids and fiber. Constipation results from blockade of calcium channels in smooth muscle of the intestine. Other common effects—dizziness, facial flushing, headache, and edema of the ankles and feet—occur secondary to vasodilation.
Cardiac Effects. Blockade of calcium channels in the heart can compromise cardiac function. In the SA node, calcium channel blockade can cause bradycardia; in the AV node, blockade can cause partial or complete AV block; and in the
Direct Effects. By blocking calcium channels in the heart and blood vessels, verapamil has five direct effects:
• Blockade at peripheral arterioles causes dilation, and thereby reduces arterial pressure.
• Blockade at arteries and arterioles of the heart increases coronary perfusion.
• Blockade at the SA node reduces heart rate.• Blockade at the AV node decreases AV nodal conduction.• Blockade in the myocardium decreases the force of
contraction.
Of the direct effects on the heart, reduced AV conduction is the most important.
Indirect (Reflex) Effects. Verapamil-induced lowering of blood pressure activates the baroreceptor reflex, causing increased firing of sympathetic nerves to the heart. Norepi-nephrine released from these nerves acts to increase heart rate, AV conduction, and force of contraction. However, since these same three parameters are suppressed by the direct actions of verapamil, the direct and indirect effects tend to neutralize each other.
Net Effect. Because the direct effects of verapamil on the heart are counterbalanced by indirect effects, the drug has little or no net effect on cardiac performance: For most patients, heart rate, AV conduction, and contractility are not noticeably altered. Consequently, the overall cardiovascular effect of verapamil is simply vasodilation accompanied by reduced arterial pressure and increased coronary perfusion.
PharmacokineticsVerapamil may be administered orally or IV. The drug is well absorbed following oral administration, but undergoes extensive metabolism on its first pass through the liver. Consequently, only about 20% of an oral dose reaches the systemic circulation. Effects begin 30 minutes after dosing and peak within 5 hours.
Sites of Action
Indications
Classification Hypertension Angina Dysrhythmias Others
DIHYDROPYRIDINES
Nifedipine [Adalat CC, Nifediac, Nifedical, Procardia] Arterioles ✔ ✔a
Amlodipine [Norvasc] Arterioles ✔ ✔
Clevidipine [Cleviprex] Arterioles ✔b
Felodipine [Plendil, Renedil ] Arterioles ✔
Isradipine [DynaCirc CR] Arterioles ✔
Nicardipine [Cardene SR] Arterioles ✔ ✔
Nimodipine [Nymalize, Nimotop ] Arterioles c
Nisoldipine [Sular] Arterioles ✔
PHENYLALKYLAMINE
Verapamil [Calan, Covera-HS , Verelan] Arterioles/heart ✔ ✔ ✔
BENZOTHIAZEPINE
Diltiazem [Cardizem, Dilacor XR, Tiazac, others] Arterioles/heart ✔ ✔ ✔
TABLE 45.1 ■ Calcium Channel Blockers: Classification, Sites of Action, and Indications
aSuppression of preterm labor (off-label use).bOnly for IV treatment of severe hypertension.cProphylaxis of neurologic injury after rupture of an intracranial aneurysm.

500
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
day (using IR tablets), 180 mg of an ER formulation (administered once a day in the morning with food), or 200 mg of Verelan PM (administered once a day at bedtime). Dosages should be reduced for older adult patients and for patients with advanced renal or liver disease. Dosages for dysrhythmias are presented in Chapter 49.
Intravenous. Intravenous verapamil is used for dysrhythmias. Because IV verapamil can cause severe adverse cardiovascular effects, blood pressure and the electrocardiogram (ECG) should be monitored and equipment for resuscitation should be immediately available. Intravenous dosages for dys-rhythmias are presented in Chapter 49.
DiltiazemActions and UsesLike verapamil, diltiazem [Cardizem, Dilacor XR, Tiazac, others] blocks calcium channels in the heart and blood vessels. As a result, the actions and applications of verapamil and diltiazem are very similar. Diltiazem has the same effects on cardiovascular function as verapamil. Both drugs lower blood pressure through arteriolar dilation, and because their direct suppressant actions are balanced by reflex cardiac stimulation, both have little net effect on the heart. Like verapamil, diltiazem is used for angina pectoris, essential hyperten-sion, and cardiac dysrhythmias (atrial flutter, atrial fibrillation, and paroxysmal supraventricular tachycardia).
PharmacokineticsOral diltiazem is well absorbed and then extensively metabolized on its first pass through the liver. As a result, bioavailability is only about 50%. Effects begin rapidly (within a few minutes) and peak within half an hour. The drug undergoes nearly complete metabolism before elimination in the urine and feces.
Adverse EffectsAdverse effects are like those of verapamil, except that diltiazem causes less constipation. The most common effects are dizziness, flushing, headache, and edema of the ankles and feet. Like verapamil, diltiazem can exacerbate cardiac dysfunction in patients with bradycardia, sick sinus syndrome, heart failure, or second-degree or third-degree AV block. Like other CCBs, diltiazem may cause chronic eczematous rash in older adults.
Drug and Food InteractionsLike verapamil, diltiazem can exacerbate digoxin-induced suppression of AV conduction and can intensify the cardiosuppressant effects of beta blockers.
myocardium, blockade can decrease contractility. When the heart is healthy, these effects rarely have clinical significance. However, in patients with certain cardiac diseases, verapamil can seriously exacerbate dysfunction. Accordingly, the drug must be used with special caution in patients with cardiac failure, and it must not be used at all in patients with sick sinus syndrome or second-degree or third-degree AV block.
Other Effects. In older patients, CCBs have been associated with chronic eczematous eruptions, typically starting 3 to 6 months after treatment onset. If the reaction is mild, switching to a different CCB may help. If the condition is severe, use of verapamil and other CCBs should stop.
Gingival hyperplasia (overgrowth of gum tissue) has been reported.
Drug and Food InteractionsDigoxin. Like verapamil, digoxin suppresses impulse
conduction through the AV node. Accordingly, when these drugs are used concurrently, the risk of AV block is increased. Patients receiving the combination should be monitored closely.
Verapamil increases plasma levels of digoxin by about 60%, thereby increasing the risk of digoxin toxicity. If signs of toxicity appear, digoxin dosage should be reduced.
Beta-Adrenergic Blocking Agents. Beta blockers and verapamil have the same effects on the heart: They decrease heart rate, AV conduction, and contractility. Hence, when a beta blocker and verapamil are used together, there is a risk of excessive cardiosuppression. To minimize risk, beta blockers and IV verapamil should be administered several hours apart.
Grapefruit Juice. Grapefruit juice can inhibit the intestinal and hepatic metabolism of many drugs and thus raise their levels. In a case report on verapamil toxicity, consumption of grapefruit juice and verapamil (360 mg over 24 hours) led to a verapamil blood level of 2772 ng/mL—approximately 8 to 24 times higher than would have been achieved without grapefruit juice.
ToxicityClinical Manifestations. Overdose can produce severe hypotension
and cardiotoxicity (bradycardia, AV block).TreatmentGeneral Measures. Verapamil can be removed from the GI tract with
gastric lavage followed by activated charcoal. Intravenous calcium gluconate can counteract both vasodilation and negative inotropic effects, but will not reverse AV block.
Hypotension. Hypotension can be treated with IV norepinephrine, which promotes vasoconstriction (by activating alpha1 receptors on blood vessels) and increases cardiac output (by activating beta1 receptors in the heart). Placing the patient in modified Trendelenburg’s position (legs elevated) and administer-ing IV fluids may also help.
Bradycardia and AV Block. Bradycardia and AV block can be treated with atropine (an anticholinergic drug that blocks parasympathetic influences on the heart). If pharmacologic measures are inadequate, electronic pacing may be required. Use of glucagon in animal models has improved heart rate through increasing amounts of intracellular cyclic AMP. It has been used successfully in treating human cases of CCB toxicity.
Preparations, Dosage, and AdministrationOral. Verapamil is available in immediate-release (IR) tablets (40, 80,
and 120 mg) as Calan; extended-release (ER) tablets (120, 180, and 240 mg) as Calan SR and Covera-HS ; and ER capsules (120, 180, 240, and 360 mg) as Verelan. In addition, verapamil is available as Verelan PM (100-, 200-, and 300-mg capsules), a timed-release formulation that, when administered at bedtime, produces maximum verapamil levels in the morning. The sustained- and timed-release formulations are approved only for hypertension. Instruct patients to swallow these products intact, without crushing or chewing. A fixed-dose combination with trandolapril (an angiotensin-converting enzyme [ACE] inhibitor) is available under the brand name Tarka.
The usual initial dosage for angina pectoris is 80 to 120 mg 3 times a day. The usual initial dosage for essential hypertension is 80 mg 3 times a
Calcium Channel Blockers
Life Stage Patient Care Concerns
Infants Verapamil can be used in infants for conversion of certain heart dysrhythmias.
Children/adolescents
Calcium channel blockers are used in children for hypertension, hypertensive emergencies, and hypertrophic cardiomyopathy.
Pregnant women
Calcium channel blockers are classified in U.S. Food and Drug Administration Pregnancy Risk Category C.a There is a risk of fetal death based on animal data, but inadequate human data exist. Therefore, caution is advised during pregnancy.
Breast-feeding women
Certain drugs such as verapamil may pose harm to the infant. For other drugs such as nifedipine, data are lacking regarding transmission of drug from mother to infant via breast milk.
Older adults In older patients, calcium channel blockers have been associated with chronic eczematous eruptions.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.

501
CHAPTER 45 Calcium Channel Blockers
drops quickly and the reflex is turned on. Conversely, with the SR formulation, blood levels of nifedipine rise slowly, so blood pressure falls slowly and the reflex is blunted.
Net Effect. The overall hemodynamic response to nifedipine is simply the sum of its direct effect (vasodilation) and indirect effect (reflex cardiac stimulation). Accordingly, nifedipine (1) lowers blood pressure, (2) increases heart rate, and (3) increases contractile force. Please note, however, that the reflex increases in heart rate and contractile force are transient and occur primar-ily with the IR formulation.PharmacokineticsNifedipine is well absorbed following oral administration, but undergoes extensive first-pass metabolism. As a result, only about 50% of an oral dose reaches the systemic circulation. With the IR formulation, effects begin rapidly and peak in 30 minutes; with the SR formulation, effects begin in 20 minutes and peak in 6 hours. Nifedipine is fully metabolized before excretion in the urine.
Therapeutic UsesAngina Pectoris. Nifedipine is indicated for vasospastic
angina and angina of effort. The drug is usually combined
Patients receiving diltiazem concurrently with digoxin or a beta blocker should be monitored closely for cardiac status. As with verapamil, grapefruit juice can significantly increase levels of diltiazem.
Preparations, Dosage, and AdministrationOral diltiazem is available in IR tablets (30, 60, 90, and 120 mg) as Cardizem, extended-release (ER) tablets (120, 180, 240, 300, 360, and 420 mg) as Cardizem LA, and SR capsules (120, 180, 240, 300, 360, and 420 mg) as Cardizem CD, Cartia XT, Dilacor XR, Dilt-CD, Dilt-XR, Diltia XT, Taztia XT, and Tiazac. The drug is also available in solution (5 mg/mL) for IV administration. The usual initial dosage for hypertension is 180 mg once a day with Cardizem CD, 60 to 120 mg twice a day with Cardizem, and 180 to 240 mg once a day with Cardizem LA or Dilacor XR. Angina pectoris can be treated with IR tablets (30 mg 4 times a day initially and 60 mg 4 times a day for maintenance). Intravenous diltiazem is reserved for dysrhythmias.
DIHYDROPYRIDINES: AGENTS THAT ACT MAINLY ON VASCULAR SMOOTH MUSCLE
All of the drugs discussed in this section belong to the dihy-dropyridine family. At therapeutic doses, these drugs produce significant blockade of calcium channels in blood vessels and minimal blockade of calcium channels in the heart. The dihydropyridines are similar to verapamil in some respects, but quite different in others.
NifedipineNifedipine [Adalat CC, Nifedical XL, Nifediac CC, Procardia, Procardia XL] was the first dihydropyridine available and will serve as our prototype for the group. Like verapamil, nifedipine blocks calcium channels in VSM and thereby promotes vasodilation. However, in contrast to verapamil, nifedipine produces very little blockade of calcium channels in the heart. As a result, nifedipine cannot be used to treat dysrhythmias, does not cause cardiac suppression, and is less likely than verapamil to exacerbate pre-existing cardiac disorders. Nife-dipine also differs from verapamil in that nifedipine is more likely to cause reflex tachycardia. Contrasts between nifedipine and verapamil are shown in Table 45.2.
Hemodynamic EffectsDirect Effects. The direct effects of nifedipine on the
cardiovascular system are limited to blockade of calcium channels in VSM. Blockade of calcium channels in peripheral arterioles causes vasodilation and thus lowers arterial pressure. Calcium channel blockade in arteries and arterioles of the heart increases coronary perfusion. Because nifedipine does not block cardiac calcium channels at usual therapeutic doses, the drug has no direct suppressant effects on automaticity, AV conduction, or contractile force.
Indirect (Reflex) Effects. By lowering blood pressure, nifedipine activates the baroreceptor reflex, thereby causing sympathetic stimulation of the heart. Because nifedipine lacks direct cardiosuppressant actions, cardiac stimulation is unop-posed, and hence heart rate and contractile force increase.
It is important to note that reflex effects occur primarily with the immediate-release formulation of nifedipine, not with the SR formulation. This is because the baroreceptor reflex is turned on only by a rapid fall in blood pressure; a gradual decline will not activate the reflex. With the IR formulation, blood levels of nifedipine rise quickly, and hence blood pressure
Property
Drug
Nifedipine Verapamil
DIRECT EFFECTS ON THE HEART AND ARTERIOLES
Arteriolar dilation Yes YesEffects on the heart Reduced automaticity No Yes Reduced AV conduction No Yes Reduced contractile force No Yes
MAJOR INDICATIONS
Hypertension Yes YesAngina pectoris (classic and variant) Yes YesDysrhythmias No Yes
ADVERSE EFFECTS
Exacerbation of AV block No Yes Sick sinus syndrome No Yes Heart failure No YesEffects secondary to vasodilation Edema (ankles and feet) Yes Yes Flushing Yes Yes Headaches Yes Yes Dizziness Yes Yes Reflex tachycardia Yes NoConstipation No Yes
DRUG INTERACTIONS
Intensifies digoxin-induced AV block No YesIntensifies cardiosuppressant effects
of beta blockersNo Yes
Often combined with a beta blocker to suppress reflex tachycardia
Yes No
TABLE 45.2 ■ Comparisons and Contrasts Between Nifedipine and Verapamil
AV, Atrioventricular.

502
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Preparations, Dosage, and AdministrationNifedipine is available in IR capsules (10 mg) as Procardia and in SR tablets (30, 60, and 90 mg) as Adalat CC, Nifedical XL, Nifediac CC, and Procardia XL. Instruct patients to swallow SR tablets whole, without crushing or chewing.
For treatment of angina pectoris, the usual initial dosage is 10 mg 3 times a day. The usual maintenance dosage is 10 to 20 mg 3 times a day. The maximum recommended dosage is 180 mg/day.
For essential hypertension, only the SR tablets are approved. The usual initial dosage is 30 mg once a day.
Other DihydropyridinesIn addition to nifedipine, seven other dihydropyridines are available. All are similar to nifedipine. Like nifedipine, these drugs produce greater blockade of calcium channels in VSM than in the heart.
NicardipineAt therapeutic doses, nicardipine [Cardene, Cardene SR] produces selective blockade of calcium channels in blood vessels and has minimal direct effects on the heart. The drug has two indications: essential hypertension and effort-induced angina pectoris. The most common adverse effects are flushing, headache, asthenia (weakness), dizziness, palpitations, and edema of the ankles and feet. As with other CCBs, eczematous rash may develop in older patients. Gingival hyperplasia (overgrowth of gum tissue) has been reported. Like nifedipine, nicardipine can be combined with a beta blocker to promote therapeutic effects and suppress reflex tachycardia. Nicardipine is available in 20- and 30-mg IR capsules (generic only) and in ER capsules (30, 45, and 60 mg) sold as Cardene SR. The usual initial dosage for angina pectoris is 20 mg 3 times a day using the IR capsules. The usual initial dosage for essential hypertension is 20 mg 3 times a day (using IR capsules) or 30 mg twice a day (using ER capsules).
AmlodipineAt therapeutic doses, amlodipine [Norvasc] produces selective blockade of calcium channels in blood vessels, having minimal direct effects on the heart. Approved indications are essential hypertension and angina pectoris (effort induced and vasospastic). Amlodipine is administered orally and absorbed slowly; peak levels develop in 6 to 12 hours. The drug has a long half-life (30 to 50 hours) and therefore is effective with once-a-day dosing. Principal adverse effects are peripheral and facial edema. Flushing, dizziness, and headache may also occur, as may eczematous rash in older patients. In contrast to other dihydropyridines, amlodipine causes little reflex tachycardia. Amlodipine is available in 2.5-, 5-, and 10-mg tablets. The usual initial dosage for hypertension or angina pectoris is 5 mg once a day. Fixed-dose combinations are also available: amlodipine/benazepril [Lotrel], amlodipine/telmisartan [Twynsta], amlodipine/atorvastatin [Caduet], amlodipine/aliskiren [Tekamlo], amlodipine/olmesartin [Azor], amlodipine/valsartan [Exforge], aliskiren/amlodipine/hydrochlorothiazide [Amturnide], amlodipine/valsartan/hydrochlorothiazide [Exforge HCT], and amlodipine/hydrochlorothiazide/olmesartan [Tribenzor].
IsradipineLike nifedipine, isradipine (generic only) produces relatively selective blockade of calcium channels in blood vessels. In the United States, the drug is approved only for hypertension. Isradipine is rapidly absorbed following oral administra-tion, but undergoes extensive first-pass metabolism. Parent drug and metabolites are excreted in the urine. The most common side effects are facial flushing, headache, dizziness, and ankle edema. Eczematous rash may develop in older patients. In contrast to nifedipine, isradipine causes minimal reflex tachycardia. The drug is available in capsules (2.5 and 5 mg). The usual antihypertensive dosage is 2.5 to 5 mg twice a day.
FelodipineFelodipine [Plendil, Renedil ] produces selective blockade of calcium channels in blood vessels. In the United States, the drug is approved only for hypertension. Felodipine is well absorbed following oral administration but undergoes extensive first-pass metabolism. As a result, bioavailability is low—only 20%. Plasma levels peak in 2.5 to 5 hours and then decay with a half-life of 24 hours. Because of its prolonged half-life, felodipine is effective with once-a-day dosing. Characteristic adverse effects are reflex tachycardia, peripheral edema, headache, facial flushing, and dizziness. Eczematous rash may develop in older patients. Gingival hyperplasia has been reported. Felodipine is available in ER tablets (2.5, 5, and 10 mg). The usual dosage for hypertension is 5 to 10 mg once a day.
NimodipineNimodipine [Nymalize, Nimotop ] produces selective blockade of calcium channels in cerebral blood vessels. The only approved application is prophylaxis
with a beta blocker to prevent reflex stimulation of the heart, which could intensify anginal pain. Long-term use reduces the rates of overt heart failure, coronary angiography, and coronary bypass surgery—but not rates of stroke, myocardial infarction, or death. The role of nifedipine in angina is discussed in Chapter 51.
Hypertension. Nifedipine is used widely to treat essential hypertension. Only the ER formulation should be used. In the past, nifedipine was used for hypertensive emergencies, but it has largely been replaced by drugs that are safer. The use of CCBs in essential hypertension is discussed in Chapter 47.
Investigational Uses. Nifedipine has been used off-label to suppress preterm labor (see Chapter 64).
Adverse Effects
Some adverse effects are like those of verapamil; others are quite different. Like verapamil, nifedipine can cause flushing, dizziness, headache, peripheral edema, and gingival hyperplasia, and may pose a risk of chronic eczematous rash in older patients. In contrast to verapamil, nifedipine causes very little constipa-tion. Also, since nifedipine causes minimal blockade of calcium channels in the heart, the drug is not likely to exacerbate AV block, heart failure, bradycardia, or sick sinus syndrome. Accordingly, nifedipine is preferred to verapamil for patients with these disorders.
A response that occurs with nifedipine that does not occur with verapamil is reflex tachycardia. This response is prob-lematic in that it increases cardiac oxygen demand and can thereby increase pain in patients with angina. To prevent reflex tachycardia, nifedipine can be combined with a beta blocker (e.g., metoprolol).
Safety Alert
IMMEDIATE-RELEASE NIFEDIPINEImmediate-release nifedipine has been associated with increased mortality in patients with myocardial infarction and unstable angina. Other IR CCBs have been associated with an increased risk of myocardial infarction in patients with hypertension. However, in both cases, a causal relationship has not been established. Nonetheless, the National Heart, Lung, and Blood Institute has recommended that immediate-release nifedipine, especially in higher doses, be used with great caution, if at all. It is important to note that these adverse effects have not been associated with sustained-release nifedipine or with any other long-acting CCB.
Drug InteractionsBeta-Adrenergic Blockers. Beta blockers are combined
with nifedipine to prevent reflex tachycardia. It is important to note that, whereas beta blockers can decrease the adverse cardiac effects of nifedipine, they can intensify the adverse cardiac effects of verapamil and diltiazem.
ToxicityWhen taken in excessive dosage, nifedipine loses selectivity. Hence, toxic doses affect the heart in addition to blood vessels. Consequently, the manifestations and treatment of nifedipine overdose are the same as described previously for verapamil.

503
CHAPTER 45 Calcium Channel Blockers
ClevidipineClevidipine [Cleviprex] is indicated only for intravenous therapy of severe hypertension, defined as systolic blood pressure above 180 mm Hg or diastolic pressure above 110 mm Hg. The drug has an ultrashort half-life (about 1 minute), owing to rapid inactivation by plasma esterases. Effects are not altered by impairment of liver or kidney function. Because of IV dosing and rapid inactivation, blood pressure falls quickly and then rises quickly when the infusion is slowed or stopped. As a result, responses can be easily titrated. Clevidipine is formulated in a lipid emulsion made from soybean oil and egg yolk phospholipids, and hence is contraindicated for patients allergic to soybeans or eggs. Clevidipine is supplied in single-dose vials (50, 100, or 250 mL) at a concentration of 0.5 mg/mL. For patients with severe hypertension, the infusion rate is 1 to 2 mg/hr initially, and can be doubled every 90 seconds up to a maximum of 32 mg/hr. In clinical trials, the average time to reach the target blood pressure was 10.9 minutes. The most common side effects are headache, nausea, and vomiting. Like other dihydropyridines, clevidipine can cause hypotension and reflex tachycardia.
of neurologic injury following rupture of an intracranial aneurysm. Benefits derive from preventing cerebral arterial spasm that follows subarachnoid hemorrhage (SAH) and can result in ischemic neurologic injury. Dosing (60 mg every 4 hours) should begin within 96 hours of SAH and continue for 21 days. Nimotop is available in 30-mg liquid-filled capsules for oral administration and as a 60 mg/20 mL oral solution [Nymalize]. Nimodipine must never be given intravenously, owing to a risk of potentially fatal cardiovascular events.
NisoldipineLike nifedipine, nisoldipine [Sular] produces selective blockade of calcium channels in blood vessels; the drug has minimal direct effects on the heart. The only approved indication is hypertension. Nisoldipine is well absorbed following oral administration, but the first-pass effect limits bioavailability to 5%. Plasma levels peak 6 hours after administration. The most common side effects are dizziness, headache, and peripheral edema. Reflex tachycardia may also occur. As with other CCBs, eczematous rash may develop in older patients. Nisoldipine is available in ER tablets (8.5, 17, 20, 25.5, 30, 34, and 40 mg). The dosage for hypertension is 17 to 60 mg once a day.
KEY POINTS
■ Calcium channels are gated pores in the cytoplasmic membrane that regulate calcium entry into cells.
■ In blood vessels, calcium entry causes vasoconstriction, and hence calcium channel blockade causes vasodilation.
■ In the heart, calcium entry increases heart rate, AV conduc-tion, and myocardial contractility, so calcium channel blockade has the opposite effects.
■ In the heart, calcium channels are coupled to beta1 receptors, activation of which enhances calcium entry. As a result, calcium channel blockade and beta blockade have identical effects on cardiac function.
■ At therapeutic doses, nifedipine and the other dihydro-pyridines act primarily on VSM; in contrast, verapamil and diltiazem act on VSM and on the heart.
■ All CCBs promote vasodilation, and hence are useful in hypertension and angina pectoris.
■ Because they suppress AV conduction, verapamil and diltiazem are useful for treating cardiac dysrhythmias (in addition to hypertension and angina pectoris).
■ Because of their cardiosuppressant effects, verapamil and diltiazem can cause bradycardia, partial or complete AV block, and exacerbation of heart failure.
■ Beta blockers intensify cardiosuppression caused by verapamil and diltiazem.
■ Nifedipine and other dihydropyridines can cause reflex tachycardia. Tachycardia is most intense with immediate-release formulations, and much less intense with sustained-release formulations.
■ Beta blockers can be used to suppress reflex tachycardia caused by nifedipine and other dihydropyridines.
■ Because they cause vasodilation, all CCBs can cause dizziness, headache, and peripheral edema.
■ In toxic doses, nifedipine and other dihydropyridines can cause cardiosuppression, just like verapamil and diltiazem.
■ Immediate-release nifedipine has been associated with increased mortality in patients with myocardial infarction and unstable angina, although a causal relationship has not been established. The National Heart, Lung, and Blood Institute recommends that immediate-release nifedipine, especially in higher doses, be used with great caution, if at all.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
VERAPAMIL AND DILTIAZEMPreadministration AssessmentTherapeutic GoalVerapamil and diltiazem are indicated for hypertension, angina pectoris, and cardiac dysrhythmias (atrial fibrillation, atrial flutter, and paroxysmal supraventricular tachycardia).
Baseline DataFor all patients, determine blood pressure and pulse rate, and obtain laboratory evaluations of liver and kidney function. For patients with angina pectoris, obtain baseline data on the frequency and severity of anginal attacks. For baseline data relevant to hypertension, see Chapter 47.
Identifying High-Risk PatientsVerapamil and diltiazem are contraindicated for patients with severe hypotension, sick sinus syndrome (in the absence of electronic pacing), and second-degree or third-degree AV block. Use with caution in patients with heart failure or liver impairment and in patients taking digoxin or beta blockers.
Implementation: AdministrationRoutesOral, IV.
AdministrationOral. Verapamil and diltiazem may be used for angina
pectoris and essential hypertension. Verapamil may be used
Continued

504
Continued
with digoxin to control ventricular rate in patients with atrial fibrillation and atrial flutter.
Extended-release formulations are reserved for essential hypertension. Instruct patients to swallow extended-release formulations whole, without crushing or chewing.
Before dosing, measure blood pressure and pulse rate. If hypotension or bradycardia is detected, withhold medication and notify the prescriber.
Intravenous. Intravenous therapy with verapamil or diltiazem is reserved for cardiac dysrhythmias. Perform injections slowly (over 2 to 3 minutes). Monitor the ECG for AV block, sudden reduction in heart rate, and prolongation of the PR or QT interval. Have facilities for cardioversion and cardiac pacing immediately available.
Ongoing Evaluation and InterventionsEvaluating Therapeutic Effects
Angina Pectoris. Keep an ongoing record of anginal attacks, noting the time and intensity of each attack and the likely precipitating event. Teach outpatients to chart the time, intensity, and circumstances of their attacks, and to notify the prescriber if attacks increase.
Essential Hypertension. Monitor blood pressure periodically. For most patients, the goal is to reduce systolic/diastolic pressure to a value below 140/90 mm Hg. Teach patients to self-monitor their blood pressure and to maintain a blood pressure record.
Minimizing Adverse EffectsCardiosuppression. Verapamil and diltiazem can cause
bradycardia, AV block, and heart failure. Inform patients about manifestations of cardiac effects (e.g., slow heartbeat, shortness of breath, weight gain) and instruct them to notify the prescriber if these occur. If cardiac impairment is severe, drug use should stop.
Peripheral Edema. Inform patients about signs of edema (swelling in ankles or feet), and instruct them to notify the prescriber if these occur. If necessary, edema can be reduced with a diuretic.
Constipation. Constipation occurs primarily with vera-pamil. Advise patients that constipation can be minimized by increasing dietary fluid and fiber.
Minimizing Adverse InteractionsDigoxin. The combination of digoxin with verapamil or
diltiazem increases the risk of partial or complete AV block. Monitor for indications of impaired AV conduction (missed beats, slowed ventricular rate).
Verapamil (and possibly diltiazem) can increase plasma levels of digoxin. Digoxin dosage should be reduced.
Beta Blockers. Concurrent use of a beta blocker with vera-pamil or diltiazem can cause bradycardia, AV block, or heart failure. Monitor closely for cardiac suppression. Administer intravenous verapamil and beta blockers several hours apart.
Grapefruit Juice. Grapefruit juice can raise levels of vera-pamil and diltiazem. Toxicity may result. Advise patients that it may be prudent to minimize grapefruit juice consumption.
Managing Acute ToxicityRemove unabsorbed drug with gastric lavage followed by activated charcoal. Give IV calcium to help counteract exces-sive vasodilation and reduced myocardial contractility.
To raise blood pressure, give IV norepinephrine. Intrave-nous fluids and placing the patient in modified Trendelenburg’s position can also help.
Bradycardia and AV block can be reversed with atropine and glucagon. If these are inadequate, electronic pacing may be required.
DIHYDROPYRIDINESAmlodipineClevidipineFelodipineIsradipineNicardipineNifedipineNimodipineNisoldipine
Preadministration AssessmentTherapeutic GoalAmlodipine, nifedipine, and nicardipine are approved for essential hypertension and angina pectoris.
Isradipine, felodipine, and nisoldipine are approved for hypertension only.
Nimodipine is used only for subarachnoid hemorrhage.Clevidipine is used only for IV therapy of severe
hypertension.
Baseline DataSee nursing implications for Verapamil and Diltiazem.
Identifying High-Risk PatientsUse dihydropyridines with caution in patients with hypoten-sion, sick sinus syndrome (in the absence of electronic pacing), angina pectoris (because of reflex tachycardia), heart failure, and second-degree or third-degree AV block.
Implementation: AdministrationRoute
Oral. All dihydropyridines except clevidipine.Intravenous. Nicardipine, clevidipine.
AdministrationInstruct patients to swallow sustained-release formulations whole, without crushing or chewing.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsSee nursing implications for Verapamil and Diltiazem.
Minimizing Adverse EffectsReflex Tachycardia. Reflex tachycardia can be suppressed
with a beta blocker.Peripheral Edema. Inform patients about signs of edema
and instruct them to notify the prescriber if these occur. If necessary, edema can be reduced with a diuretic.
Managing Acute ToxicitySee nursing implications for Verapamil and Diltiazem.
Summary of Major Nursing Implicationsa—cont’d
aPatient education information is highlighted as blue text.

505
C H A P T E R
46 Vasodilators
Basic Concepts in Vasodilator Pharmacology, p. 505Selectivity of Vasodilatory Effects, p. 505Overview of Therapeutic Uses, p. 505Adverse Effects Related to Vasodilation, p. 505
Pharmacology of Individual Vasodilators, p. 506Hydralazine, p. 506Minoxidil, p. 507Sodium Nitroprusside, p. 508
Key Points, p. 509
Because hemodynamic responses to dilation of arterioles and veins differ, the selectivity of a vasodilator is a major determinant of its effects, both therapeutic and undesired. Undesired effects related to selective dilation of arterioles and veins are discussed later in this chapter. Therapeutic implications of selective dilation are discussed in Chapters 47, 48, 51, 53, and 107.
Overview of Therapeutic UsesThe vasodilators, as a group, have a broad spectrum of uses. Principal indications are essential hypertension, hypertensive crisis, angina pectoris, heart failure, and myocardial infarction. Additional indications include pheochromocytoma, peripheral vascular disease, pulmonary arterial hypertension, and produc-tion of controlled hypotension during surgery. The specific applications of any particular agent are determined by its pharmacologic profile. Important facets of that profile are route of administration, site of vasodilation (arterioles, veins, or both), and intensity and duration of effects.
Adverse Effects Related to VasodilationPostural HypotensionPostural (orthostatic) hypotension is defined as a fall in blood pressure brought on by moving from a supine or seated position to an upright position. The underlying cause is relaxation of smooth muscle in veins. Because of venous relaxation, gravity causes blood to “pool” in veins, thereby decreasing venous return to the heart. Reduced venous return causes a decrease in cardiac output and a corresponding decrease in blood pressure. Hypotension from venous dilation is minimal in recumbent subjects because when we are lying down, the impact of gravity on venous return is small.
Vasodilation can be produced with a variety of drugs. The major classes of vasodilators, along with representative agents and the chapters in which they are discussed, are shown in Table 46.1. Some of these drugs act primarily on arterioles, some act primarily on veins, and some act on both types of vessels. The vasodilators are widely used, with indications ranging from hypertension to angina pectoris to heart failure. Many of the vasodilators have been discussed in previous chapters. Three agents—hydralazine, minoxidil, and nitroprusside—are introduced here.
In approaching the vasodilators, we begin by considering concepts that apply to the vasodilators as a group. After that we discuss the pharmacology of individual agents.
BASIC CONCEPTS IN VASODILATOR PHARMACOLOGYSelectivity of Vasodilatory EffectsVasodilators differ from one another with respect to the types of blood vessels they affect. Some agents (e.g., hydralazine) produce selective dilation of arterioles. Others (e.g., nitroglyc-erin) produce selective dilation of veins. Still others (e.g., prazosin) dilate arterioles and veins. The selectivity of some important vasodilators is shown in Table 46.2.
The selectivity of a vasodilator determines its hemodynamic effects. For example, drugs that dilate resistance vessels (arterioles) cause a decrease in cardiac afterload (the force the heart works against to pump blood). By decreasing afterload, arteriolar dilators reduce cardiac work while causing cardiac output and tissue perfusion to increase. In contrast, drugs that dilate capacitance vessels (veins) reduce the force with which blood is returned to the heart, which reduces ventricular filling. This reduction in filling decreases cardiac preload (the degree of stretch of the ventricular muscle before contraction), which in turn decreases the force of ventricular contraction. Hence, by decreasing preload, venous dilators cause a decrease in cardiac work, along with a decrease in cardiac output and tissue perfusion.
Safety Alert
FALLSVasodilators place patients at increased risk of falls. Patients receiving vasodilators should be informed about symptoms of hypotension (light-headedness, dizziness) and advised to sit or lie down if these occur. Failure to follow this advice may result in fainting. Patients should also be taught that they can minimize hypotension by avoiding abrupt transitions from a supine or seated position to an upright position.
Reflex TachycardiaReflex tachycardia can be produced by dilation of arterioles or veins. The mechanism is this: (1a) arteriolar dilation causes a direct decrease in arterial pressure or (1b) venous dilation

506
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
reduces cardiac output, which in turn reduces arterial pressure; (2) baroreceptors in the aortic arch and carotid sinus sense the drop in pressure and relay this information to the vasomotor center of the medulla; and (3) in an attempt to bring blood pressure back up, the medulla sends impulses along sympathetic nerves instructing the heart to beat faster.
Reflex tachycardia is undesirable for two reasons. First, tachycardia can put an unacceptable burden on the heart. Second, if the vasodilator was given to reduce blood pressure, tachy-cardia would raise pressure and thereby negate the desired effect.
To help prevent vasodilator-induced reflex tachycardia, patients can be pretreated with a beta blocker (e.g., metoprolol), which will block sympathetic stimulation of the heart.
Expansion of Blood VolumeProlonged use of arteriolar or venous dilators can cause an increase in blood volume (secondary to prolonged reduction of blood pressure). The increase in volume represents an attempt by the body to restore blood pressure to pretreatment levels.
First, reduced blood pressure triggers secretion of aldosterone by the adrenal glands. Aldosterone then acts on the kidney to promote retention of sodium and water, thereby increasing blood volume. Second, by reducing arterial pressure, vasodila-tors decrease both renal blood flow and glomerular filtration rate; because filtrate volume is decreased, the kidney is able to reabsorb an increased fraction of filtered sodium and water, which causes blood volume to expand.
Increased blood volume can negate the beneficial effects of the vasodilator. For example, if volume increases during the treatment of hypertension, blood pressure will rise and the benefits of therapy will be canceled. To prevent the kidney from neutralizing the beneficial effects of vasodilation, patients often receive concurrent therapy with a diuretic, which prevents fluid retention and volume expansion.
PHARMACOLOGY OF INDIVIDUAL VASODILATORSIn this section we focus on three drugs: hydralazine, minoxidil, and sodium nitroprusside. All of the other vasodilators are discussed in other chapters (see Table 46.1).
HydralazineCardiovascular EffectsHydralazine causes selective dilation of arterioles. The drug has little or no effect on veins. Arteriolar dilation results from a direct action on vascular smooth muscle (VSM). The exact mechanism is unknown. In response to arteriolar dilation, peripheral resistance and arterial blood pressure fall. In addition, heart rate and myocardial contractility increase, largely by reflex mechanisms. Because hydralazine acts selectively on arterioles, postural hypotension is minimal.
PharmacokineticsAbsorption and Time Course of Action. Hydralazine
is readily absorbed following oral administration. Effects begin within 45 minutes and persist for 6 hours or longer. With parenteral administration, effects begin faster (within 10 minutes) and last 2 to 4 hours.
Category Examples Chapter
DRUGS ACTING ON THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
Angiotensin-converting enzyme inhibitors
CaptoprilEnalapril
44
Angiotensin II receptor blockers
LosartanValsartan
44
Direct renin inhibitors Aliskiren 44Organic nitrates Isosorbide dinitrate
Nitroglycerin51
Calcium channel blockers DiltiazemNifedipineVerapamil
45
SYMPATHOLYTIC DRUGS
Alpha-adrenergic blockers PhenoxybenzaminePhentolaminePrazosinTerazosin
18
Adrenergic neuron blockers Reserpine 19Centrally acting agents Clonidine
Methyldopa19
Drugs for pulmonary arterial hypertension
BosentanEpoprostenol
107
Other important vasodilators
HydralazineMinoxidilNitroprusside
46
TABLE 46.1 ■ Types of Vasodilators
Vasodilator
Site of Vasodilation
Arterioles Veins
Hydralazine +
Minoxidil +
Diltiazem +
Nifedipine +
Verapamil +
Prazosin + +
Terazosin + +
Phentolamine + +
Nitroprusside + +
Captopril + +
Enalapril + +
Losartan + +
Aliskiren + +
Nitroglycerin +
Isosorbide dinitrate +
TABLE 46.2 ■ Vasodilator Selectivity

507
CHAPTER 46 Vasodilators
Preparations, Dosage, and AdministrationPreparations. Hydralazine is available in tablets (10, 25, 50, and 100 mg)
for oral use and in solution (20 mg/mL in 1-mL ampules) for parenteral administration. Hydralazine is also available in a fixed-dose combination with isosorbide dinitrate (a vasodilator), sold as BiDil. As discussed in Chapters 8 and 48, BiDil is the first drug product approved for treating a specific ethnic group—namely, African Americans.
Oral Therapy. Dosage should be low initially (10 mg 4 times a day) and then gradually increased. Rapid increases may produce excessive hypoten-sion. Usual maintenance dosages for adults range from 25 to 100 mg twice a day. Daily doses greater than 200 mg are associated with an increased incidence of adverse effects and should be avoided.
Parenteral Therapy. Parenteral administration (IV and IM) is reserved for hypertensive crises. The usual dose is 20 to 40 mg, repeated as needed. Blood pressure should be monitored frequently to minimize excessive hypoten-sion. In most cases, patients can be switched from parenteral hydralazine to oral therapy within 48 hours.
MinoxidilMinoxidil produces more intense vasodilation than hydralazine but also causes more severe adverse reactions. Because it is both very effective and very dangerous, minoxidil is reserved for patients with severe hypertension unresponsive to safer drugs.
Cardiovascular EffectsLike hydralazine, minoxidil produces selective dilation of arterioles. Little or no venous dilation occurs. Arteriolar dilation decreases peripheral resistance and arterial blood pressure. In response, reflex mechanisms increase heart rate and myocardial contractility. Both responses can increase cardiac oxygen demand and can thereby exacerbate angina pectoris.
Vasodilation results from a direct action on VSM. To relax VSM, minoxidil must first be metabolized to minoxidil sulfate. This metabolite then causes potassium channels in VSM to open. The resultant efflux of potassium hyperpolarizes VSM cells, thereby reducing their ability to contract.PharmacokineticsMinoxidil is rapidly and completely absorbed following oral administration. Vasodilation is maximal within 2 to 3 hours and then gradually declines. Residual effects may persist for 2 days or more. Minoxidil is extensively metabolized. Metabolites and parent drug are eliminated in the urine. The drug’s half-life is 4.2 hours.
Therapeutic UsesThe only cardiovascular indication for minoxidil is severe hypertension. Because of its serious adverse effects, minoxidil is reserved for patients who have not responded to safer drugs. To minimize adverse responses (reflex tachycardia, expansion of blood volume, pericardial effusion), minoxidil should be used with a beta blocker plus intensive diuretic therapy.
Topical minoxidil [Rogaine, others] is used to promote hair growth in balding men and women (see Chapter 105).
Adverse EffectsReflex Tachycardia. Blood pressure reduction triggers
reflex tachycardia, a serious effect that can be minimized by co-treatment with a beta blocker.
Sodium and Water Retention. Fluid retention is both common and serious. Volume expansion may be so severe as to cause cardiac decompensation. Management of fluid retention requires a loop diuretic (e.g., furosemide) used alone or in combination with a thiazide diuretic. If diuretics are inadequate, dialysis must be employed or minoxidil must be withdrawn.
Metabolism. Hydralazine is inactivated by a metabolic process known as acetylation. The ability to acetylate drugs is genetically determined. Some people are rapid acetylators; some are slow acetylators. The distinction between rapid and slow acetylators can be clinically significant because individuals who acetylate hydralazine slowly are likely to have higher blood levels of the drug, which can result in excessive vasodilation and other undesired effects. To avoid hydralazine accumulation, dosage should be reduced in slow acetylators.
Therapeutic UsesEssential Hypertension. Oral hydralazine can be used to
lower blood pressure in patients with essential hypertension. The regimen almost always includes a beta blocker, and may include a diuretic as well. Although commonly employed in the past, hydralazine has been largely replaced by newer antihypertensive agents (see Chapter 47).
Hypertensive Crisis. Parenteral hydralazine is used to lower blood pressure rapidly in severe hypertensive episodes. The drug should be administered in small incremental doses. If dosage is excessive, severe hypotension may replace the hypertension.
Heart Failure. As discussed in Chapter 48, hydralazine (usually in combination with isosorbide dinitrate) can be used short term to reduce afterload in patients with heart failure. With prolonged therapy, tolerance to hydralazine develops.
Adverse EffectsReflex Tachycardia. By lowering arterial blood pressure,
hydralazine can trigger reflex stimulation of the heart, thereby causing cardiac work and myocardial oxygen demand to increase. Because hydralazine-induced reflex tachycardia is frequently severe, the drug is usually combined with a beta blocker.
Increased Blood Volume. Hydralazine-induced hypoten-sion can cause sodium and water retention and a corresponding increase in blood volume. A diuretic can prevent volume expansion.
Systemic Lupus Erythematosus–like Syndrome. Hydrala-zine can cause an acute rheumatoid syndrome that closely resembles systemic lupus erythematosus (SLE). Symptoms include muscle pain, joint pain, fever, nephritis, pericarditis, and the presence of antinuclear antibodies. The syndrome occurs most frequently in slow acetylators and is rare when dosage is kept below 200 mg/day. If an SLE-like reaction occurs, hydralazine should be discontinued. Symptoms are usually reversible but may take 6 or more months to resolve. In some cases, rheumatoid symptoms persist for years.
Other Adverse Effects. Common responses include headache, dizziness, weakness, and fatigue. These reactions are related to hydralazine-induced hypotension.
Drug InteractionsHydralazine can be combined with a beta blocker to protect against reflex tachycardia and with a diuretic to prevent sodium and water retention and expansion of blood volume. Drugs that lower blood pressure will intensify hypotensive responses to hydralazine. Accordingly, if hydralazine is used with other antihypertensive agents, care is needed to avoid excessive hypotension. In the treatment of heart failure, hydralazine is usually combined with isosorbide dinitrate, a drug that dilates veins.

508
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Because of these qualities, nitroprusside is a drug of choice for hypertensive emergencies.
Cardiovascular EffectsIn contrast to hydralazine and minoxidil, nitroprusside causes venous dilation in addition to arteriolar dilation. Curiously, although nitroprusside is an effective arteriolar dilator, reflex tachycardia is minimal. Administration is by IV infusion, and effects begin at once. By adjusting the infusion rate, blood pressure can be decreased to almost any level desired. When the infusion is stopped, blood pressure returns to pretreat-ment levels in minutes. Nitroprusside can trigger retention of sodium and water; furosemide can help counteract this effect.Mechanism of ActionOnce in the body, nitroprusside breaks down to release nitric oxide, which then activates guanylate cyclase, an enzyme present in VSM. Guanylate cyclase catalyzes the production of cyclic GMP, which, through a series of reactions, causes vasodilation. This mechanism is similar to that of nitroglycerin.
MetabolismNitroprusside contains five cyanide groups, which are split free in the first step of nitroprusside metabolism. Nitric oxide, the active component, is released next. Both reactions take place in smooth muscle. Once freed, the cyanide groups are converted to thiocyanate by the liver, using thiosulfate as a cofactor. Thiocyanate is eliminated by the kidneys over several days.
Therapeutic UsesHypertensive Emergencies. Nitroprusside is used to lower
blood pressure rapidly in hypertensive emergencies. Oral antihypertensive medication should be initiated simultaneously. During nitroprusside treatment, furosemide may be needed to prevent excessive retention of fluid.
Other Uses. Nitroprusside is approved for producing controlled hypoten-sion during surgery (to reduce bleeding in the surgical field) and for acute decompensated heart failure. In addition, the drug has been employed investigationally to treat myocardial infarction.
Adverse EffectsExcessive Hypotension. If administered too rapidly,
nitroprusside can cause a precipitous drop in blood pressure, resulting in headache, palpitations, nausea, vomiting, and sweating. Blood pressure should be monitored continuously.
Cyanide Poisoning. Rarely, lethal amounts of cyanide have accumulated. Cyanide buildup is most likely in patients with liver disease and in those with low stores of thiosulfate, the cofactor needed for cyanide detoxification. The chances of cyanide poisoning can be minimized by avoiding rapid infusion (faster than 5 mcg/kg/min) and by coadministering thiosulfate. If cyanide toxicity occurs, nitroprusside should be withdrawn.
Thiocyanate Toxicity. When nitroprusside is given for several days, thiocyanate may accumulate. Although much less hazardous than cyanide, thiocyanate can also cause adverse effects. These effects, which involve the central nervous system, include disorientation, psychotic behavior, and delirium. To minimize toxicity, patients receiving nitroprusside for more than 3 days should undergo monitoring of plasma thiocyanate, which must be kept below 0.1 mg/mL.
Hypertrichosis. About 80% of patients taking minoxidil for 4 weeks or more develop hypertrichosis (excessive growth of hair). Hair growth begins on the face and later develops on the arms, legs, and back. Hypertrichosis appears to result from proliferation of epithelial cells at the base of the hair follicle; vasodilation may also be involved. Hairiness is a cosmetic problem that can be controlled by shaving or using a depilatory. However, many patients find hypertrichosis both unmanageable and intolerable and refuse to continue treatment.
Pericardial Effusion. Rarely, minoxidil-induced fluid retention results in pericardial effusion (fluid accumulation beneath the pericardium). In most cases, pericardial effusion is asymptomatic. However, in some cases, fluid accumulation becomes so great as to cause cardiac tamponade (compression of the heart with a resultant decrease in cardiac performance). If tamponade occurs, it must be treated by pericardiocentesis or surgical drainage.
Other Adverse Effects. Minoxidil may cause nausea, headache, fatigue, breast tenderness, glucose intolerance, thrombocytopenia, and skin reactions (rashes, Stevens-Johnson syndrome). In addition, the drug has caused hemor-rhagic cardiac lesions in experimental animals.
Preparations, Dosage, and AdministrationMinoxidil is supplied in 2.5- and 10-mg tablets. The initial dosage is 5 mg once a day. The maximum dosage is 100 mg/day. The usual adult dosage is 10 to 40 mg/day administered in single or divided doses. When a rapid response is needed, a loading dose of 5 to 20 mg is given, followed by doses of 2.5 to 10 mg every 4 hours.
Vasodilators
Life Stage Patient Care Concerns
Infants Hydralazine is used in infants as young as 1 month for management of hypertensive crisis and chronic hypertension. Sodium nitroprusside is used in heart failure and for management of hypertensive emergency.
Children/adolescents
Hydralazine and sodium nitroprusside can be used safely in children, just in smaller doses. Side effect profiles are similar to those of adults.
Pregnant women
Hydralazine, sodium nitroprusside, and minoxidil are classified in U.S. Food and Drug Administration Pregnancy Risk Category C.a Benefits should outweigh the risks.
Breast-feeding women
Data are lacking regarding transmission of drug from mother to infant via breast milk. Sodium nitroprusside causes potential adverse effects in the infant.
Older adults Monitor for falls, as there is increased risk with polypharmacy and associated orthostatic hypotension.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
Sodium NitroprussideSodium nitroprusside [Nitropress, Nipride ] is potent and efficacious and acts faster than any other vasodilator available.

509
CHAPTER 46 Vasodilators
The maximal rate is 10 mcg/kg/min. If infusion at the maximal rate for 10 minutes fails to produce an adequate drop in blood pressure, administration should stop. During the infusion, blood pressure should be monitored continu-ously, with either an arterial line or an electronic monitoring device. No other drugs should be mixed with the nitroprusside solution.
Preparations, Dosage, and AdministrationSodium nitroprusside [Nitropress] is available in powdered form (50 mg) to be dissolved and then diluted for IV infusion. Fresh solutions may have a faint brown color. Solutions that are deeply colored (blue, green, dark red) should be discarded. Nitroprusside in solution can be degraded by light, and hence should be protected with an opaque material.
Blood pressure can be adjusted to practically any level by increasing or decreasing the rate of infusion. The initial infusion rate is 0.3 mcg/kg/min.
KEY POINTS
■ Some vasodilators are selective for arterioles, some are selective for veins, and some dilate both types of vessel.
■ Drugs that dilate arterioles reduce cardiac afterload and can thereby reduce cardiac work while increasing cardiac output and tissue perfusion.
■ Drugs that dilate veins reduce cardiac preload and can thereby reduce cardiac work, cardiac output, and tissue perfusion.
■ Principal indications for vasodilators are essential hyperten-sion, hypertensive crisis, angina pectoris, heart failure, and myocardial infarction.
■ Drugs that dilate veins can cause orthostatic hypotension.■ Drugs that dilate arterioles or veins can cause reflex
tachycardia, which increases cardiac work and elevates blood pressure. Reflex tachycardia can be blunted with a beta blocker.
■ Drugs that dilate arterioles or veins can cause fluid retention, which can be blunted with a diuretic.
■ Hydralazine causes selective dilation of arterioles.■ Hydralazine can cause a syndrome that resembles SLE.■ Minoxidil causes selective and profound dilation of
arterioles.■ Minoxidil can cause hypertrichosis.■ Sodium nitroprusside dilates arterioles and veins.■ Prolonged infusion of nitroprusside can result in toxic
accumulation of cyanide and thiocyanate.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

510
C H A P T E R
47 Drugs for Hypertension
BASIC CONSIDERATIONS IN HYPERTENSION, p. 510Classification of Blood Pressure, p. 510Types of Hypertension, p. 511
Primary (Essential) Hypertension, p. 511Secondary Hypertension, p. 511
Consequences of Hypertension, p. 511MANAGEMENT OF CHRONIC HYPERTENSION, p. 511Basic Considerations, p. 511
Diagnosis, p. 511Benefits of Lowering Blood Pressure, p. 511Patient Evaluation, p. 512Treatment Goals, p. 512Therapeutic Interventions, p. 512
Lifestyle Modifications, p. 512Sodium Restriction, p. 512The DASH Eating Plan, p. 512Alcohol Restriction, p. 513Aerobic Exercise, p. 513Smoking Cessation, p. 513Weight Loss, p. 513Maintenance of Potassium and Calcium Intake,
p. 513Drug Therapy, p. 513
Review of Blood Pressure Control, p. 513Antihypertensive Mechanisms: Sites of Drug
Action and Effects Produced, p. 514Classes of Antihypertensive Drugs, p. 516Fundamentals of Hypertension Drug Therapy,
p. 520Individualizing Therapy, p. 521Minimizing Adverse Effects, p. 523Promoting Adherence, p. 523
DRUGS FOR HYPERTENSIVE EMERGENCIES, p. 524Sodium Nitroprusside, p. 524Fenoldopam, p. 524Labetalol, p. 525Clevidipine, p. 525
DRUGS FOR HYPERTENSIVE DISORDERS OF PREGNANCY, p. 525
Chronic Hypertension, p. 525Preeclampsia and Eclampsia, p. 525Key Points, p. 526Summary of Major Nursing Implications, p. 527
Hypertension (elevated blood pressure [BP]) is a common chronic disorder that affects about 2 million American chil-dren, 85 million American adults, and over 1 billion people worldwide. According to the World Health Organization, hypertension is the leading global risk for mortality, and has increased nearly 20% from 25.1 million in 1990 to 30.8 million in 2013. Left untreated, hypertension can lead to heart disease, kidney disease, and stroke. Conversely, a treatment program of lifestyle modifications and drug therapy can reduce BP and the risk of long-term complications. However, although we can reduce symptoms and long-term consequences, we can’t cure hypertension. As a result, treatment must continue lifelong, making nonadherence a significant problem. Despite advances in management, hypertension remains undertreated: Among Americans with the disease, only 74% undergo treatment, and only 48% take sufficient medicine to bring their BP under control.
We can treat hypertension with 14 classes of drugs. Fortu-nately, all 14 were introduced in previous chapters. In this chapter, rather than struggling with a huge array of new drugs, all you have to do is learn the antihypertensive applications of drugs you already know about.
In 2017 the American College of Cardiology, the American Heart Association, and The Task Force on Clinical Practice Guidelines issued a new guideline for hypertension entitled 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation and Management of High Blood Pressure in Adults. Clinical practice recommendations throughout this chapter reflect those in the 2017 hypertension guidelines except where noted otherwise.a
BASIC CONSIDERATIONS IN HYPERTENSION
In this section, we consider three issues: (1) classification of BP based on values for systolic and diastolic pressure, (2) types of hypertension, and (3) the damaging effects of chronic hypertension.
CLASSIFICATION OF BLOOD PRESSUREThe 2017 hypertension guidelines define four BP categories: normal, elevated, stage 1 hypertension, and stage 2 hypertension.
aAlthough the 2017 hypertension guideline is the most influential guideline in the United States, it is not the only authoritative guideline available. Treatment guidelines have been released by several organizations, including the American Society of Hypertension, the Canadian Hypertension Education Program, the European Society of Hypertension in conjunction with the European Society of Cardiology, and the World Health Organization in conjunction with the International Society of Hypertension.

511
CHAPTER 47 Drugs for Hypertension
lead to heart disease (myocardial infarction [MI], heart failure, angina pectoris), kidney disease, and stroke. The degree of injury is directly related to the degree of pressure elevation: The higher the pressure, the greater the risk. Among people 40 to 70 years old, the risk of cardiovascular disease is doubled for each 20 mm Hg increase in SBP or each 10 mm Hg increase in DBP—beginning at 115/75 mm Hg and continuing through 185/155 mm Hg. For people older than 50 years, elevated systolic BP poses a greater risk than elevated diastolic BP. For patients of all ages, hypertension-related deaths result largely from cerebral hemorrhage, renal failure, heart failure, and MI.
Unfortunately, despite its potential for serious harm, hypertension usually remains asymptomatic until long after injury has begun to develop. As a result, the disease can exist for years before overt pathology is evident. Because injury develops slowly and progressively, and because hypertension rarely causes discomfort, many people who have the disease don’t know it. Furthermore, many who do know it forgo treatment anyway, largely because hypertension doesn’t make them feel bad—that is, until it’s too late.
MANAGEMENT OF CHRONIC HYPERTENSION
In this section we consider treatments for chronic hypertension. We begin by addressing patient evaluation and other basic issues, after which we discuss the two modes of management: lifestyle modifications and drug therapy.
BASIC CONSIDERATIONSDiagnosisDiagnosis should be based on several BP readings, not just one. If an initial screen shows that BP is elevated (but does not represent an immediate danger), measurement should be repeated on two subsequent office visits. At each visit, two measurements should be made, at least 5 minutes apart. The patient should be seated in a chair—not on an examination table—with his or her feet on the floor. High readings should be confirmed in the contralateral arm. If the mean of all readings shows that SBP is indeed greater than 130 mm Hg or that DBP is greater than 80 mm Hg, a diagnosis of hypertension can be made.
Ideally, diagnosis would be based on ambulatory blood pressure monitoring (ABPM) because office-based measure-ments are often abnormally high, causing individuals to be diagnosed with hypertension when they don’t really have it. By contrast, when BP is measured with ABPM, false-positive diagnoses can be avoided. Accordingly, some experts recom-mend that office-based measurements be used only for screening and that treatment be postponed until the diagnosis is confirmed using ABPM. In this way, the risks and expense of unnecessary treatment will be avoided.
Benefits of Lowering Blood PressureMultiple clinical trials have demonstrated unequivocally that when the BP of hypertensive individuals is lowered, morbidity is decreased and life is prolonged. Treatment reduces the
These categories are defined solely by systolic and diastolic blood pressures. These new guidelines do not differentiate between patient age or comorbidities.
TYPES OF HYPERTENSIONThere are two broad categories of hypertension: primary hypertension and secondary hypertension. Primary hypertension is by far the most common form of hypertensive disease. Less than 10% of people with hypertension have a secondary form.
Primary (Essential) HypertensionPrimary hypertension is defined as hypertension that has no identifiable cause. A diagnosis of primary hypertension is made by ruling out probable specific causes of BP elevation. Primary hypertension is a chronic, progressive disorder. In the absence of treatment, patients will experience a continuous, gradual rise in BP over the rest of their lives.
In the United States, primary hypertension affects about 30% of adults. However, not all groups are at equal risk: Older people are at higher risk than younger people; African Americans are at higher risk than Caucasian Americans; and postmeno-pausal women are at higher risk than premenopausal women.
Although the cause of primary hypertension is unknown, the condition can be successfully treated. Please understand, however, that treatment is not curative: Drugs can lower BP, but they can’t eliminate the underlying pathology. Consequently, treatment must continue lifelong.
Primary hypertension is also referred to as essential hypertension. This alternative name preceded the term primary hypertension and reflects our ignorance about the cause of the problem. Historically, it had been noted that as people grew older, their BP rose. Why older people had elevated BP was (and remains) unknown. One hypothesis noted that as people aged, their vascular systems offered greater resistance to blood flow. To move blood against this increased resistance, a compensatory increase in BP was required. Therefore, the hypertension that occurred with age was seen as being “essential” for providing adequate tissue perfusion—hence, the term essential hyperten-sion. Over time, the term came to be applied to all cases of hypertension for which an underlying cause could not be found.
Secondary HypertensionSecondary hypertension is defined as an elevation of BP brought on by an identifiable primary cause. Because secondary hyper-tension results from an identifiable cause, it may be possible to treat that cause directly, rather than relying on antihypertensive drugs for symptomatic relief. As a result, some individuals can actually be cured. For example, if hypertension occurs secondary to pheochromocytoma (a catecholamine-secreting tumor), surgical removal of the tumor may produce permanent cure. When cure is not possible, secondary hypertension can be managed with the same drugs used for primary hypertension.
CONSEQUENCES OF HYPERTENSIONChronic hypertension is associated with increased morbidity and mortality. Left untreated, prolonged elevation of BP can

512
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Treatment GoalsThe ultimate goal in treating hypertension is to reduce cardiovascular and renal morbidity and mortality. Hopefully this can be accomplished without decreasing quality of life with the drugs used. For all patients regardless of age, the goal is to maintain SBP below 130 mm Hg and DBP below 80 mm Hg. These numbers have decreased from the prior guidelines.
Therapeutic InterventionsWe can reduce BP in two ways: We can implement healthy lifestyle changes, and we can treat with antihypertensive drugs. For all patients, a combination of lifestyle changes and drugs is indicated. Lifestyle changes and drug therapy are discussed in detail in the sections that follow.
LIFESTYLE MODIFICATIONSLifestyle changes offer multiple cardiovascular benefits with little cost and minimal risk. When implemented before hyperten-sion develops, they may actually prevent hypertension. When implemented after hypertension has developed, they can lower BP, possibly decreasing or eliminating the need for drugs. Lastly, lifestyle modifications can decrease other cardiovascular risk factors. Accordingly, all patients should be strongly encour-aged to adopt a healthy lifestyle. Key components are discussed in the sections that follow.
Sodium RestrictionReducing sodium chloride (salt) intake can lower BP in people with hypertension and can help prevent overt hypertension in those with prehypertension. In addition, salt restriction can enhance the hypotensive effects of drugs. However, the benefits of sodium restriction are both small and short lasting: Over time, BP returns to its original level, despite continued salt restriction. Nonetheless, the Department of Health and Human Services Dietary Guidelines for Americans 2015-2020 recom-mends that people consume no more than 2300 mg of sodium a day. In patients at higher risk for cardiovascular disease (African Americans, patients older than 51 years, and those who have hypertension, diabetes, or chronic kidney disease), the recommendation is even lower, at 1500 mg of sodium daily. This recommendation is undergoing evaluation, because this decrease has not yet shown any benefit in patient outcomes. To facilitate salt restriction, patients should be given information on the salt content of foods.
Experts disagree about the relationship between salt intake and BP in normotensive patients. In particular, they disagree as to whether a high-salt diet causes hypertension. For people with normal BP, a low-salt diet may be consid-ered healthy or unnecessary, depending on the expert you consult.
The DASH Eating PlanTwo studies have shown that we can reduce BP by adopting a healthy diet, known as the Dietary Approaches to Stop Hypertension (DASH) eating plan. This diet is rich in fruits,
incidence of stroke by 35% to 40%, MI by 20% to 25%, and heart failure by more than 50%. Although reductions in mortality are less dramatic, they are nonetheless significant: Among patients with stage 1 hypertension plus additional cardiovascular risk factors, one death would be prevented for every 11 patients who reduced SBP by 12 mm Hg for a period of 10 years. Among those with hypertension plus cardiovascular disease or target-organ damage, one death would be prevented for every 9 patients who achieved a sustained 12 mm Hg reduction in pressure.
Patient EvaluationEvaluation of patients with hypertension has two major objec-tives. Specifically, we must assess for (1) identifiable causes of hypertension and (2) factors that increase cardiovascular risk. To aid evaluation, diagnostic tests are required.
Hypertension With a Treatable CauseAs discussed earlier, some forms of hypertension result from a treatable cause, such as Cushing’s syndrome, pheochromo-cytoma, and the use of oral contraceptives. Patients should be evaluated for these causes and managed appropriately. In many cases, direct treatment of the underlying cause can control BP, eliminating the need for further antihypertensive therapy.
Factors That Increase Cardiovascular RiskTwo types of factors—existing target-organ damage and major cardiovascular risk factors—increase the risk of cardiovascular events in people with hypertension. When these factors are present, aggressive therapy is indicated. Accordingly, to select appropriate interventions, we must identify patients with the following types of target-organ damage:
• Heart disease:• Left ventricular hypertrophy• Angina pectoris• Prior MI• Prior coronary revascularization• Heart failure
• Stroke or transient ischemic attack• Chronic kidney disease• Peripheral arterial disease• Retinopathy
We must also identify patients with the following major cardiovascular risk factors (other than hypertension):
• Cigarette smoking• Physical inactivity• Dyslipidemia• Diabetes• Microalbuminuria• Advancing age (older than 55 years for men, older than
65 years for women)• Family history of premature cardiovascular disease
Diagnostic TestsThe following tests should be done in all patients: electrocar-diogram; complete urinalysis; hemoglobin and hematocrit; and blood levels of sodium, potassium, calcium, creatinine, glucose, uric acid, triglycerides, and cholesterol (total, LDL, and HDL cholesterol).

513
CHAPTER 47 Drugs for Hypertension
Although adequate calcium is needed for overall good health, the impact of calcium on BP is only modest. In epidemiologic studies, high calcium intake is associated with a reduced incidence of hypertension. Among patients with hypertension, a few may be helped by increasing calcium intake. To maintain good health, calcium intake should be 1000 mg/day for men ages 25 to 65 years and women ages 19 to 50 years, and 1500 mg/day for men and women older than 65.
DRUG THERAPYDrug therapy, together with lifestyle modifications, can control BP in all patients with chronic hypertension. The decision to use drugs should be the result of collaboration between pre-scriber and patient. We have a wide assortment of antihyper-tensive drugs. Consequently, for the majority of patients, it should be possible to establish a program that is effective and yet devoid of objectionable side effects.
vegetables, and low-fat dairy products, and low in total fat, saturated fats, and cholesterol. In addition, the plan encourages intake of whole-grain products, fish, poultry, and nuts, and recommends minimal intake of red meat and sweets. Details are available online at https://www.nhlbi.nih.gov/health/health-topics/topics/dash/.
Alcohol RestrictionExcessive alcohol consumption can raise BP and create resistance to antihypertensive drugs. Accordingly, patients should limit alcohol intake: Most men should consume no more than 1 ounce/day; women and lighter weight men should consume no more than 0.5 ounce/day. (One ounce of ethanol is equivalent to about two mixed drinks, two glasses of wine, or two cans of beer.)
Aerobic ExerciseRegular aerobic exercise (e.g., walking, swimming, bicycling) can reduce BP by about 10 mm Hg. In addition, exercise reduces the risk of cardiovascular disease and reduces all-cause mortal-ity. In normotensive people, exercise decreases the risk of developing hypertension. Accordingly, patients should be encouraged to develop an exercise program if they have not already done so. An activity as simple as brisk walking 30 to 45 minutes most days of the week is beneficial.
Smoking CessationSmoking is a major risk factor for cardiovascular disease. Each time a cigarette is smoked, BP rises. In patients with hyperten-sion, smoking can reduce the effects of antihypertensive drugs. Clearly, all patients who smoke should be strongly encouraged to quit. (Pharmacologic aids to smoking cessation are discussed in Chapter 39.) As a rule, the use of nicotine replacement products (e.g., nicotine gum, nicotine patch) does not elevate BP. The cardiovascular benefits of quitting become evident within 1 year.
Weight LossWeight loss can reduce BP in 60% to 80% of overweight hypertensive individuals and can enhance responses to antihypertensive drugs. Consequently, a program of weight management and exercise is recommended for patients who are overweight.
Maintenance of Potassium and Calcium IntakePotassium has a beneficial effect on BP. In patients with hypertension, potassium can lower BP. In normotensive people, high potassium intake helps protect against hypertension, whereas low intake elevates BP. For optimal cardiovascular effects, all people should take in 4700 mg of potassium a day. Preferred sources are fresh fruits and vegetables. If hypokalemia develops secondary to diuretic therapy, dietary intake may be insufficient to correct the problem. In this case, the patient may need to use a potassium supplement, a potassium-sparing diuretic, or a potassium-containing salt substitute.
Prototype Drugs
DRUGS FOR HYPERTENSIONDiureticsHydrochlorothiazideSpironolactone
Beta-Adrenergic BlockersPropranololMetoprolol
Inhibitors of the Renin-Angiotensin- Aldosterone SystemCaptopril (ACE inhibitor)Losartan (angiotensin II receptor blocker)Aliskiren (direct renin inhibitor)Eplerenone (aldosterone antagonist)
Calcium Channel BlockersVerapamilNifedipine
Review of Blood Pressure ControlBefore discussing the antihypertensive drugs, we need to review the major mechanisms by which BP is controlled. This information will help you understand the mechanisms by which drugs lower BP.
Principal Determinants of Blood PressureThe principal determinants of BP are shown in Fig. 47.1. As indicated, arterial pressure is the product of cardiac output and peripheral resistance. An increase in either will increase BP.
Cardiac Output. Cardiac output is influenced by four factors: (1) heart rate, (2) myocardial contractility (force of contraction), (3) blood volume, and (4) venous return of blood to the heart. An increase in any of these will increase cardiac output, thereby causing BP to rise. Conversely, a decrease in

514
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
level. Consequently, as therapy proceeds, sympathetic reflexes offer progressively less resistance to the hypotensive effects of medication.
Renin-Angiotensin-Aldosterone System. The RAAS can elevate BP, negating the hypotensive effects of drugs. The RAAS is discussed in Chapter 44 and reviewed briefly here.
The RAAS elevates BP beginning with the release of renin from juxtaglomerular cells of the kidney. These cells release renin in response to reduced renal blood flow, reduced blood volume, reduced BP, and activation of beta1-adrenergic recep-tors on the cell surface. Following its release, renin catalyzes the conversion of angiotensinogen into angiotensin I, a weak vasoconstrictor. After this, angiotensin-converting enzyme (ACE) acts on angiotensin I to form angiotensin II, a compound that constricts systemic and renal blood vessels. Constriction of systemic blood vessels elevates BP by increasing peripheral resistance. Constriction of renal blood vessels elevates BP by reducing glomerular filtration, which causes retention of salt and water, which in turn increases blood volume and BP. In addition to causing vasoconstriction, angiotensin II causes release of aldosterone from the adrenal cortex. Aldosterone acts on the kidney to further increase retention of sodium and water.
Because drug-induced reductions in BP can activate the RAAS, this system can counteract the effect we are trying to achieve. We have five ways to cope with this problem. First, we can suppress renin release with beta blockers. Second, we can prevent conversion of angiotensinogen to angiotensin I with a direct renin inhibitor. Third, we can prevent the conver-sion of angiotensin I into angiotensin II with an ACE inhibitor. Fourth, we can block receptors for angiotensin II with an angiotensin II receptor blocker. And fifth, we can block receptors for aldosterone with an aldosterone antagonist.
Renal Regulation of Blood Pressure. As discussed in Chapter 43, the kidney plays a central role in long-term regula-tion of BP. When BP falls, glomerular filtration rate (GFR) falls too, thereby promoting retention of sodium, chloride, and water. The resultant increase in blood volume increases venous return to the heart, causing an increase in cardiac output, which in turn increases arterial pressure. We can neutralize renal effects on BP with diuretics.
Antihypertensive Mechanisms: Sites of Drug Action and Effects ProducedAs discussed earlier, drugs can lower BP by reducing heart rate, myocardial contractility, blood volume, venous return, and the tone of arteriolar smooth muscle. In this section we survey the principal mechanisms by which drugs produce these effects.
The major mechanisms for lowering BP are shown in Fig. 47.2 and Table 47.1. The figure depicts the principal sites at which antihypertensive drugs act. The table shows the effects elicited when drugs act at these sites. The numbering system used in the sections that follow corresponds with the system used in Fig. 47.2 and Table 47.1.
1—BrainstemAntihypertensive drugs acting in the brainstem suppress sympathetic outflow to the heart and blood vessels, resulting in decreased heart rate, decreased myocardial contractility, and vasodilation. Vasodilation contributes the most to reducing
these factors will make BP fall. Hence, to reduce BP, we might give a beta blocker to reduce cardiac output, or a diuretic to reduce blood volume, or a venodilator to reduce venous return.
Peripheral Vascular Resistance. Vascular resistance is increased by arteriolar constriction. Accordingly, we can reduce BP with drugs that promote arteriolar dilation.
Systems That Help Regulate Blood PressureHaving established that BP is determined by heart rate, myo-cardial contractility, blood volume, venous return, and arteriolar constriction, we can now examine how these factors are regu-lated. Three regulatory systems are of particular significance: (1) the sympathetic nervous system, (2) the renin-angiotensin-aldosterone system (RAAS), and (3) the kidney.
Sympathetic Baroreceptor Reflex. The sympathetic nervous system employs a reflex circuit—the baroreceptor reflex—to keep BP at a preset level. This circuit operates as follows:
1. Baroreceptors in the aortic arch and carotid sinus sense BP and relay this information to the brainstem.
2. When BP is perceived as too low, the brainstem sends impulses along sympathetic nerves to stimulate the heart and blood vessels.
3. BP is then elevated by (a) activation of beta1 receptors in the heart, resulting in increased cardiac output; and (b) activation of vascular alpha1 receptors, resulting in vasoconstriction.
4. When BP has been restored to an acceptable level, sympathetic stimulation of the heart and vascular smooth muscle subsides.
The baroreceptor reflex frequently opposes our attempts to reduce BP with drugs. Opposition occurs because the “set point” of the baroreceptors is high in people with hypertension. That is, the baroreceptors are set to perceive excessively high BP as “normal” (i.e., appropriate). As a result, the system oper-ates to maintain BP at pathologic levels. Consequently, when we attempt to lower BP using drugs, the reduced (healthier) pressure is interpreted by the baroreceptors as below what it should be, and, in response, signals are sent along sympathetic nerves to “correct” the reduction. These signals produce reflex tachycardia and vasoconstriction—responses that can counteract the hypotensive effects of drugs. Clearly, if treatment is to succeed, the regimen must compensate for the resistance offered by this reflex. Taking a beta blocker, which will block reflex tachycardia, can be an effective method of compensation. Fortunately, when BP has been suppressed with drugs for an extended time, the baroreceptors become reset at a lower
Peripheral resistanceis determined by:Arteriolar constriction
Cardiac output isdetermined by:
ARTERIALPRESSURE
CARDIACOUTPUT
PERIPHERALRESISTANCEX=
Heart rateContractilityBlood volumeVenous return
Fig. 47.1 ■ Primary determinants of arterial blood pressure.

515
CHAPTER 47 Drugs for Hypertension
rarely, and then only for hypertensive emergencies. Because use is so limited, the last one available—mecamylamine—was voluntarily withdrawn from the U.S. market.
3—Terminals of Adrenergic NervesAntihypertensive agents that act at adrenergic nerve terminals decrease the release of norepinephrine, resulting in decreased sympathetic stimulation of the heart and blood vessels. These drugs, known as adrenergic neuron blocking agents, are used
BP. Dilation of arterioles reduces BP by decreasing vascular resistance. Dilation of veins reduces BP by decreasing venous return to the heart.
2—Sympathetic GangliaGanglionic blockade reduces sympathetic stimulation of the heart and blood vessels. Antihypertensive effects result primarily from dilation of arterioles and veins. Ganglionic blocking agents produce such a profound reduction in BP that they are used
BRAINSTEM
SYMPATHETIC GANGLIA
ADRENERGICTERMINALS
VASCULAR SMOOTH MUSCLE
CARDIAC β1 RECEPTORS
VASCULAR α1 RECEPTORS
RENAL TUBULES
COMPONENTS OF THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
Mecamylamine*
ANGIOTENSIN IIRECEPTORS
LosartanValsartanOther ARBs
ALDOSTERONERECEPTORS
EplerenoneSpironolactone
β1 RECEPTORS ONJUXTAGLOMERULARCELLS
8a 8b
PropranololMetoprololOther � blockers
HydralazineMinoxidilNitroprussideCa channel blockersThiazides
PrazosinTerazosinLaetalol
Thiazide diureticsFurosemideK+-sparing diuretics
Reserpine
PropranololMetoprololOther β blockers
ClonidineMethyldopa
RENIN
8
Aliskiren
8c 8d 8eANGIOTENSIN-CONVERTINGENZYME
CaptoprilEnalaprilOther ACE inhibitors
Fig. 47.2 ■ Sites of action of antihypertensive drugs. Note that some antihypertensive agents act at more than one site: beta (β) blockers act at sites 4 and 8a, and thiazides act at sites 6 and 7. The hemodynamic consequences of drug actions at the sites depicted are shown in Table 47–1. (ACE, Angiotensin-converting enzyme; ARB, angiotensin II receptor blocker.) *No longer available in the United States.

516
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
8b—Renin. Inhibition of renin decreases conversion of angiotensinogen into angiotensin I and thereby suppresses the entire RAAS. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8c—Angiotensin-Converting Enzyme. Inhibitors of ACE suppress formation of angiotensin II. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8d—Angiotensin II Receptors. Blockade of angiotensin II receptors prevents the actions of angiotensin II. Hence blockade results in peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8e—Aldosterone Receptors. Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water, and thereby reduces blood volume.
Classes of Antihypertensive DrugsIn this section we consider the principal drugs employed to treat chronic hypertension. Drugs for hypertensive emergen-cies and hypertensive disorders of pregnancy are considered separately.
Individual antihypertensive drugs and their classes are shown in Table 47.2. Combination products are shown in Table 47.3.
DiureticsDiuretics are a mainstay of antihypertensive therapy. These drugs reduce BP when used alone, and they can enhance the
only rarely. In the United States, reserpine is the only drug in this class still on the market.
4—Beta1-Adrenergic Receptors on the HeartBlockade of cardiac beta1 receptors prevents sympathetic stimulation of the heart. As a result, heart rate and myocardial contractility decline.
5—Alpha1-Adrenergic Receptors on Blood VesselsBlockade of vascular alpha1 receptors promotes dilation of arterioles and veins. Arteriolar dilation reduces peripheral resistance. Venous dilation reduces venous return to the heart.
6—Vascular Smooth MuscleSeveral antihypertensive drugs (see Fig. 47.2) act directly on vascular smooth muscle to cause relaxation. One of these agents—sodium nitroprusside—is used only for hypertensive emergencies. The rest are used for chronic hypertension.
7—Renal TubulesDiuretics act on renal tubules to promote salt and water excre-tion. As a result, blood volume declines, causing BP to fall.
8—Components of the RAAS (8a to 8e)8a—Beta1 Receptors on Juxtaglomerular Cells. Blockade
of beta1 receptors on juxtaglomerular cells suppresses release of renin. The resultant decrease in angiotensin II levels has three effects: peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
Site of Drug Actiona Representative Drug Drug Effects
1. Brainstem Clonidine Suppression of sympathetic outflow decreases sympathetic stimulation of the heart and blood vessels.
2. Sympathetic ganglia Mecamylamineb Ganglionic blockade reduces sympathetic stimulation of the heart and blood vessels.3. Adrenergic nerve
terminalsReserpine Reduced norepinephrine release decreases sympathetic stimulation of the heart and
blood vessels.4. Cardiac beta1 receptors Metoprolol Beta1 blockade decreases heart rate and myocardial contractility.5. Vascular alpha1 receptors Prazosin Alpha1 blockade causes vasodilation.6. Vascular smooth muscle Hydralazine Relaxation of vascular smooth muscle causes vasodilation.7. Renal tubules Hydrochlorothiazide Promotion of diuresis decreases blood volume.Components of the renin-angiotensin-aldosterone system (8a to 8e)8a. Beta1 receptors on
juxtaglomerular cellsMetoprolol Beta1 blockade suppresses renin release, resulting in (1) vasodilation secondary to
reduced production of angiotensin II and (2) prevention of aldosterone-mediated volume expansion.
8b. Renin Aliskiren Inhibition of renin suppresses formation of angiotensin I, which in turn decreases formation of angiotensin II and thereby reduces (1) vasoconstriction and (2) aldosterone-mediated volume expansion.
8c. Angiotensin-converting enzyme (ACE)
Captopril Inhibition of ACE decreases formation of angiotensin II and thereby prevents (1) vasoconstriction and (2) aldosterone-mediated volume expansion.
8d. Angiotensin II receptors Losartan Blockade of angiotensin II receptors prevents angiotensin-mediated vasoconstriction and aldosterone-mediated volume expansion.
8e. Aldosterone receptors Eplerenone Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water and thereby reduces blood volume.
TABLE 47.1 ■ Antihypertensive Effects Elicited by Drug Actions at Specific Sites
aSite numbers in this table correspond with site numbers in Fig. 47.2.bNo longer available in the United States.

517
CHAPTER 47 Drugs for Hypertension
hypertension. Rather, they are reserved for (1) patients who need greater diuresis than can be achieved with thiazides and (2) patients with a low GFR (because thiazides won’t work when GFR is low). Like the thiazides, the loop diuretics lower BP by reducing blood volume and promoting vasodilation.
Most adverse effects are like those of the thiazides: hypo-kalemia, dehydration, hyperglycemia, and hyperuricemia. In addition, loop diuretics can cause hearing loss.
Potassium-Sparing Diuretics. The degree of diuresis induced by the potassium-sparing agents (e.g., spironolactone) is small. Consequently, these drugs have only modest hypo-tensive effects. However, because of their ability to conserve potassium, these drugs can play an important role in an antihypertensive regimen. Specifically, they can balance potas-sium loss caused by thiazides or loop diuretics. The most significant adverse effect of the potassium-sparing agents is hyperkalemia. Because of the risk of hyperkalemia, potassium-sparing diuretics must not be used in combination with one another or with potassium supplements. Also, they should not be used routinely with ACE inhibitors, angiotensin II receptor blockers, or aldosterone antagonists, all of which promote significant hyperkalemia.
Sympatholytics (Antiadrenergic Drugs)Sympatholytic drugs suppress the influence of the sympathetic nervous system on the heart, blood vessels, and other structures. These drugs are used widely for hypertension.
effects of other hypotensive drugs. The basic pharmacology of the diuretics is discussed in Chapter 41.
Thiazide Diuretics. Thiazide diuretics (e.g., hydrochloro-thiazide, chlorthalidone) are first-line drugs for hypertension. They reduce BP by two mechanisms: reduction of blood volume and reduction of arterial resistance. Reduced blood volume is responsible for initial antihypertensive effects. Reduced vascular resistance develops over time and is responsible for long-term antihypertensive effects. The mechanism by which thiazides reduce vascular resistance has not been determined.
Of the thiazides available, hydrochlorothiazide is used most widely. In fact, hydrochlorothiazide is used more widely than any other antihypertensive drug. Nonetheless, other thiazides, especially chlorthalidone, may be more effective.
The principal adverse effect of thiazides is hypokalemia. This can be minimized by consuming potassium-rich foods (e.g., bananas, citrus fruits) and using potassium supplements or a potassium-sparing diuretic. Other side effects include dehydration, hyperglycemia, and hyperuricemia.
Thiazides are superior to calcium channel blockers and ACE inhibitors as monotherapy, and therefore are preferred.
Loop Diuretics. Loop diuretics (e.g., furosemide) produce much greater diuresis than the thiazides. For most individuals with chronic hypertension, the amount of fluid loss that loop diuretics can produce is greater than needed or desirable. Consequently, loop diuretics are not used routinely for
Diuretics Sympatholytics RAAS Suppressants Others
Thiazides and Related Diuretics
ChlorothiazideChlorthalidoneHydrochlorothiazideIndapamideMethyclothiazideMetolazone
Loop Diuretics
BumetanideEthacrynic acidFurosemideTorsemide
Potassium-Sparing Diuretics
AmilorideSpironolactoneTriamterene
Beta Blockers
Acebutolol (has ISA)AtenololBetaxololBisoprololMetoprololNadololNebivololPenbutolol (has ISA)Pindolol (has ISA)PropranololTimolol
Alpha1 Blockers
DoxazosinPrazosinTerazosin
Alpha/Beta Blockers
CarvedilolLabetalol
Centrally Acting Alpha2 Agonists
ClonidineGuanabenzGuanfacineMethyldopa
Adrenergic Neuron Blockers
Reserpine
ACE Inhibitors
BenazeprilCaptoprilEnalaprilFosinoprilLisinoprilMoexiprilPerindoprilQuinaprilRamiprilTrandolapril
Angiotensin II Receptor Blockers
AzilsartanCandesartanEprosartanIrbesartanLosartanOlmesartanTelmisartanValsartan
Direct Renin Inhibitor
Aliskiren
Aldosterone Antagonists
EplerenoneSpironolactone
Direct-Acting Vasodilators
HydralazineMinoxidil
Calcium Channel Blockers
AmlodipineDiltiazem (non-DHP)FelodipineIsradipineNicardipineNifedipineNimodipineNisoldipineVerapamil (non-DHP)
TABLE 47.2 ■ Drugs for Chronic Hypertension
DHP, Dihydropyridine; ISA, intrinsic sympathomimetic activity; RAAS, renin-angiotensin-aldosterone system.

518
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
beta receptors while blocking receptor activation by strong agonists (e.g., norepinephrine). As a result, heart rate at rest is slowed less than with other beta blockers. Accordingly, if a patient develops symptomatic bradycardia with another beta blocker, switching to one of these may help.
Beta blockers can produce several adverse effects. Blockade of cardiac beta1 receptors can produce bradycardia, decreased atrioventricular (AV) conduction, and reduced contractility. Consequently, beta blockers should not be used by patients with sick sinus syndrome or second- or third-degree AV block, and they must be used with care in patients with heart failure. Blockade of beta2 receptors in the lung can promote broncho-constriction. Accordingly, beta blockers should be avoided by patients with asthma. If an asthmatic individual absolutely must use a beta blocker, a beta1-selective agent (e.g., metoprolol) should be employed. Beta blockers can mask signs of hypo-glycemia; therefore, they must be used with caution in patients with diabetes. Potential side effects of beta blockers include depression, insomnia, bizarre dreams, and sexual dysfunction; however, a review of older clinical trials has shown that the risk is small or nonexistent.
The basic pharmacology of the beta blockers is discussed in Chapter 18.
As indicated in Table 47.2, there are five subcategories of sympatholytic drugs: (1) beta blockers, (2) alpha1 blockers, (3) alpha/beta blockers, (4) centrally acting alpha2 agonists, and (5) adrenergic neuron blockers.
Beta-Adrenergic Blockers. Like the thiazides, beta block-ers (e.g., propranolol, metoprolol) are widely used antihyper-tensive drugs. However, despite their efficacy and frequent use, the exact mechanism by which they reduce BP is somewhat uncertain. Beta blockers are less effective in African Americans than in whites.
The beta blockers have at least four useful actions in hypertension. First, blockade of cardiac beta1 receptors decreases heart rate and contractility, thereby causing cardiac output to decline. Second, beta blockers can suppress reflex tachycardia caused by vasodilators. Third, blockade of beta1 receptors on juxtaglomerular cells of the kidney reduces release of renin and thereby reduces angiotensin II–mediated vasoconstriction and aldosterone-mediated volume expansion. Fourth, long-term use of beta blockers reduces peripheral vascular resistance by a mechanism that is unknown. This action could readily account for most of their antihypertensive effects.
Three beta blockers have intrinsic sympathomimetic activity (see Table 47.2). That is, they can produce mild activation of
Generic Name Brand Name
TWO-DRUG COMBINATIONS
Thiazide Plus a Beta Blocker
Hydrochlorothiazide + metoprolol Lopressor HCT
Hydrochlorothiazide + bisoprolol Ziac
Hydrochlorothiazide + pindolol Viskazide
Bendroflumethiazide + nadolol Corzide
Chlorthalidone + atenolol Tenoretic
Thiazide Plus an ACE Inhibitor
Hydrochlorothiazide + captopril Generic only
Hydrochlorothiazide + benazepril Lotensin HCT
Hydrochlorothiazide + enalapril Vaseretic
Hydrochlorothiazide + fosinopril Generic only
Hydrochlorothiazide + lisinopril Zestoretic
Hydrochlorothiazide + moexipril Generic only
Hydrochlorothiazide + quinapril Accuretic
Indapamide + perindopril Coversyl Plus
Thiazide Plus an ARB
Hydrochlorothiazide + losartan Hyzaar
Hydrochlorothiazide + valsartan Diovan HCT
Hydrochlorothiazide + candesartan Atacand HCT
Hydrochlorothiazide + eprosartan Teveten HCT
Hydrochlorothiazide + irbesartan Avalide
Hydrochlorothiazide + telmisartan Micardis HCT
Hydrochlorothiazide + olmesartan Benicar HCT, Olmetec Plus
Generic Name Brand Name
Thiazide Plus a Potassium-Sparing Diuretic
Hydrochlorothiazide + spironolactone Aldactazide
Hydrochlorothiazide + triamterene Dyazide, Maxzide
Hydrochlorothiazide + amiloride Moduretic
Thiazide Plus an Alpha2 Agonist
Chlorthalidone + clonidine Clorpres
Hydrochlorothiazide + methyldopa Generic only
Thiazide Plus a Direct-Acting Vasodilator
Hydrochlorothiazide + hydralazine Generic only
CCB Plus an ACE Inhibitor
Amlodipine + benazepril Lotrel
Felodipine + enalapril Generic only
Verapamil + trandolapril Tarka
CCB Plus an ARB
Amlodipine + olmesartan Azor
Amlodipine + valsartan Exforge
Amlodipine + telmisartan Twynsta
Aliskiren (a DRI) Plus Another Drug
Aliskiren + amlodipine Tekamlo
Aliskiren + hydrochlorothiazide Tekturna HCT, Rasilez HCT
THREE-DRUG COMBINATIONS
Hydrochlorothiazide + amlodipine + valsartan Exforge HCT
Hydrochlorothiazide + amlodipine + olmesartan Tribenzor
Hydrochlorothiazide + amlodipine + aliskiren Amturnide
TABLE 47.3 ■ Combination Products for Chronic Hypertension
ACE, Angiotensin-converting enzyme; ARB, angiotensin II receptor blocker; CCB, calcium channel blocker; DRI, direct renin inhibitor.

519
CHAPTER 47 Drugs for Hypertension
hypotension is low. With both drugs, the lowering of BP may be followed by reflex tachycardia, renin release, and fluid retention. Reflex tachycardia and release of renin can be prevented with a beta blocker. Fluid retention can be prevented with a diuretic.
The most disturbing adverse effect of hydralazine is a syndrome resembling systemic lupus erythematosus (SLE). Fortunately, this reaction is rare at recommended doses. If an SLE-like reaction occurs, hydralazine should be withdrawn. Hydralazine is considered a third-line drug for chronic hypertension.
Minoxidil is potentially more harmful than hydralazine. By causing fluid retention, minoxidil can promote pericardial effusion (accumulation of fluid beneath the myocardium) that in some cases progresses to cardiac tamponade (compression of the heart). A less serious effect is hypertrichosis (excessive hair growth). Because of its capacity for significant side effects, minoxidil is not used routinely for chronic hypertension. Instead, the drug is reserved for patients with severe hypertension that has not responded to safer drugs.
The basic pharmacology of hydralazine and minoxidil is discussed in Chapter 46.
Calcium Channel BlockersThe calcium channel blockers (CCBs) fall into two groups: dihydropyridines (e.g., nifedipine) and nondihydropyridines (e.g., verapamil and diltiazem). Drugs in both groups promote dilation of arterioles. In addition, verapamil and diltiazem have direct suppressant effects on the heart.
Like other vasodilators, CCBs can cause reflex tachycardia. This reaction is greatest with the dihydropyridines and minimal with verapamil and diltiazem. Reflex tachycardia is low with verapamil and diltiazem because of cardiosuppression. Since dihydropyridines do not block cardiac calcium channels, reflex tachycardia with these drugs can be substantial.
Because of their ability to compromise cardiac performance, verapamil and diltiazem must be used cautiously in patients with bradycardia, heart failure, or AV heart block. These precau-tions do not apply to dihydropyridines.
The immediate-release formulation of nifedipine has been associated with increased mortality in patients with MI and unstable angina. Thus the National Heart, Lung, and Blood Institute has recommended that the use of immediate-release nifedipine be discontinued for treatment of hypertensive emergency.
The basic pharmacology of the CCBs is discussed in Chapter 45.
Drugs That Suppress the RAASBecause the RAAS plays an important role in controlling BP, drugs that suppress the system—especially the ACE inhibitors—have a significant role in controlling hypertension. The basic pharmacology of these drugs is discussed in Chapter 44.
ACE Inhibitors. The ACE inhibitors (e.g., captopril, enalapril) lower BP by preventing the formation of angiotensin II and thereby preventing angiotensin II–mediated vasoconstric-tion and aldosterone-mediated volume expansion. In hyper-tensive diabetic patients with renal damage, these actions slow the progression of kidney injury. Like the beta blockers, ACE inhibitors are less effective in African Americans than in whites. Principal adverse effects are persistent cough, first-dose hypotension, angioedema, and hyperkalemia (secondary to suppression of aldosterone release). Because of the risk of
Alpha1 Blockers. The alpha1 blockers (e.g., doxazosin, terazosin) prevent stimulation of alpha1 receptors on arterioles and veins, thereby preventing sympathetically mediated vaso-constriction. The resultant vasodilation reduces both peripheral resistance and venous return to the heart.
The most disturbing side effect of alpha blockers is ortho-static hypotension. Hypotension can be especially severe with the initial dose. Significant hypotension continues with sub-sequent doses but is less profound.
The American College of Cardiology recommends that alpha blockers not be used as first-line therapy for hypertension. In a huge clinical trial known as the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), in which doxazosin was compared with chlorthalidone (a thia-zide diuretic), patients taking doxazosin experienced 25% more cardiovascular events and were twice as likely to be hospitalized for heart failure. It is not clear whether doxazosin increased cardiovascular risk or chlorthalidone decreased risk. Either way, the diuretic is clearly preferred to the alpha blocker.
The basic pharmacology of the alpha blockers is discussed in Chapter 18.
Alpha/Beta Blockers: Carvedilol and Labetalol. Carvedilol and labetalol are unusual in that they can block alpha1 receptors as well as beta receptors. Blood pressure reduction results from a combination of actions: (1) alpha1 blockade promotes dilation of arterioles and veins, (2) blockade of cardiac beta1 receptors reduces heart rate and contractility, and (3) blockade of beta1 receptors on juxtaglomerular cells suppresses release of renin. Presumably, these drugs also share the ability of other beta blockers to reduce peripheral vascular resistance. Like other nonselective beta blockers, labetalol and carvedilol can exacerbate bradycardia, AV heart block, and asthma. Blockade of venous alpha1 receptors can produce postural hypotension.
Centrally Acting Alpha2 Agonists. As discussed in Chapter 19, these drugs (e.g., clonidine, methyldopa) act within the brainstem to suppress sympathetic outflow to the heart and blood vessels. The result is vasodilation and reduced cardiac output, both of which help lower BP. All central alpha2 agonists can cause dry mouth and sedation. In addition, clonidine can cause severe rebound hypertension if treatment is abruptly discontinued. Additional adverse effects of methyldopa are hemolytic anemia (accompanied by a positive direct Coombs’ test) and liver disorders.
Adrenergic Neuron Blockers. Reserpine—the only adrenergic neuron blocker still available—depletes norepineph-rine from postganglionic sympathetic nerve terminals, reducing sympathetic stimulation of the heart and blood vessels. The result is a drop in cardiac output and blood pressure. In addition to its peripheral effects, reserpine depletes serotonin and catecholamines from neurons in the central nervous system, causing deep emotional depression. Accordingly, reserpine is absolutely contraindicated for patients with a history of depres-sive illness. Because reserpine can cause depression and because more desirable antihypertensive drugs are available, reserpine is not a preferred agent for treating hypertension. The basic pharmacology of reserpine is discussed in Chapter 19.
Direct-Acting Vasodilators: Hydralazine and MinoxidilHydralazine and minoxidil reduce BP by promoting dilation of arterioles. Neither drug causes significant dilation of veins. Because venous dilation is minimal, the risk of orthostatic

520
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
As shown in the algorithm at this link, lifestyle changes should be instituted first. If these fail to lower BP enough, drug therapy should be started, and the lifestyle changes should continue. Treatment often begins with a single drug. If needed, another drug may be added (if the initial drug was well tolerated but inadequate) or substituted (if the initial drug was poorly toler-ated). However, before another drug is considered, possible reasons for failure of the initial drug should be assessed. Among these are insufficient dosage, poor adherence, excessive salt intake, and the presence of secondary hypertension. If treatment with two drugs is unsuccessful, a third and even fourth may be added.
Initial Drug SelectionInitial drug selection is determined by the presence or absence of a compelling indication, defined as a comorbid condition for which a specific class of antihypertensive drugs has been shown to improve outcomes. Initial drugs for patients with and without compelling indications are discussed in the sections that follow.
Patients Without Compelling Indications. For initial therapy in the absence of a compelling indication, a thiazide diuretic is currently recommended for most patients. This preference is based on long-term controlled trials showing conclusively that thiazides can reduce morbidity and mortality in hypertensive patients, and are well tolerated and inexpen-sive too. Other options for initial therapy—ACE inhibitors, ARBs, and CCBs—equal diuretics in their ability to lower BP. However, they may not be as effective at reducing morbidity and mortality. Accordingly, these drugs should be reserved for special indications and for patients who have not responded to thiazides. Certain other alternatives—centrally acting sympatholytics, adrenergic neuron blockers, and direct-acting vasodilators—are associated with a high incidence of adverse effects, and hence are not well suited for initial monotherapy. One last alternative—alpha1 blockers—is no longer recom-mended as first-line therapy. As noted, when the alpha blocker doxazosin was compared with the diuretic chlorthalidone, doxazosin was associated with a much higher incidence of adverse cardiovascular events.
Patients With Compelling Indications. For patients with hypertension plus certain comorbid conditions (e.g., heart failure, diabetes), there is strong evidence that specific antihypertensive drugs can reduce morbidity and mortality. Drugs shown to improve outcomes for six comorbid conditions are indicated in Table 47.4. Clearly, these drugs should be used for initial therapy. If needed, other antihypertensive agents can be added to the regimen. Management of hypertension in patients with diabetes and renal disease—two specific comorbid conditions—is discussed further under Individualizing Therapy.
Adding Drugs to the RegimenRationale for Drug Selection. When using two or
more drugs to treat hypertension, each drug should come from a different class. That is, each drug should have a different mechanism of action. In accord with this guide-line, it would be appropriate to combine a beta blocker, a diuretic, and a vasodilator, since each lowers BP by a dif-ferent mechanism. In contrast, it would be inappropriate to combine two thiazide diuretics or two beta blockers or two vasodilators.
hyperkalemia, combined use with potassium supplements or potassium-sparing diuretics is generally avoided. ACE inhibitors can cause serious fetal harm, especially during the second and third trimesters of pregnancy, and hence must not be given to pregnant women. ACE inhibitors—along with angiotensin receptor blockers (ARBs) and direct renin inhibitors (DRIs)—are the only antihypertensive drugs specifically contraindicated during pregnancy.
Angiotensin II Receptor Blockers. ARBs lower BP in much the same way as do ACE inhibitors. Like the ACE inhibitors, ARBs prevent angiotensin II–mediated vasoconstric-tion and release of aldosterone. The only difference is that ARBs do so by blocking the actions of angiotensin II, whereas ACE inhibitors block the formation of angiotensin II. Both groups lower BP to the same extent. Like the ACE inhibitors, ARBs can cause fetal harm and must not be used during pregnancy. In contrast to ACE inhibitors, ARBs have a low incidence of inducing cough or significant hyperkalemia, but they do cause angioedema.
Direct Renin Inhibitors. DRIs act directly on renin to inhibit conversion of angiotensinogen into angiotensin I. As a result, DRIs can suppress the entire RAAS. At this time, only one DRI—aliskiren [Tekturna, Rasilez ]—is available. Antihypertensive effects equal those of ACE inhibitors, ARBs, and CCBs. Compared with ACE inhibitors, aliskiren causes less hyperkalemia, cough, or angioedema—but poses a similar risk of fetal harm. In addition, aliskiren causes diarrhea in 2.3% of patients. Also, in patients with type 2 diabetes mellitus, the use of aliskiren has demonstrated an increased incidence of renal impairment, hypotension, and hyperkalemia. Because of these findings, the use of aliskiren is contraindicated in patients with diabetes mellitus who are also taking an ACE inhibitor or ARB. Although we know that aliskiren can lower BP, we don’t yet know if it reduces adverse outcomes (e.g., stroke, kidney failure, MI). Accordingly, until experience with the drug is more extensive, other antihypertensives should be considered first.
Aldosterone Antagonists. Aldosterone antagonists lower BP by promoting renal excretion of sodium and water. Only two agents are available: eplerenone and spironolactone. (In case you’re confused about spironolactone, yes, it’s the same drug we discussed earlier under potassium-sparing diuretics. We’re discussing it here because it produces diuresis through aldosterone receptor blockade.) Both spironolactone and eplerenone promote renal retention of potassium, and hence pose a risk of hyperkalemia. Accordingly, they should not be given to patients with existing hyperkalemia and should not be combined with potassium-sparing diuretics or potassium supplements. Combined use with ACE inhibitors, ARBs, and DRIs is permissible, but must be done with caution. Spirono-lactone is discussed in Chapter 41, and eplerenone is discussed in Chapter 44.
Fundamentals of Hypertension Drug TherapyTreatment AlgorithmThe basic approach to treating hypertension was published with the 2017 guidelines in the Journal of the American College of Cardiology and can be found online at http://www .onlinejacc.org/content/early/2017/11/04/j.jacc.2017.11.006?_ga =2.176790566.2093167245.1516654950-136371117.1515608281.

521
CHAPTER 47 Drugs for Hypertension
discussed here. Preferred drugs for patients with these and other comorbid conditions are shown in Table 47.4. Drugs to avoid in patients with specific comorbid conditions are sum-marized in Table 47.5.
Renal Disease. Nephrosclerosis (hardening of the kidney) secondary to hypertension is among the most common causes of progressive renal disease. Pathophysiologic changes include degeneration of renal tubules and fibrotic thickening of the glomeruli, both of which contribute to renal insufficiency. Nephrosclerosis sets the stage for a downward spiral: Renal insufficiency causes water retention, which in turn causes BP to rise higher, which in turn promotes even more renal injury, and so forth. Accordingly, early detection and treat-ment are essential. To slow progression of renal damage, the most important action is to lower BP. The target BP in all patients is now below 120/80 mm Hg. Although all classes of antihypertensive agents are effective in nephrosclerosis, ACE inhibitors and ARBs work best. Hence, in the absence of contraindications, all patients should get one of these drugs. In most cases, a diuretic is used too. In patients with advanced renal insufficiency, thiazide diuretics are ineffective, hence a loop diuretic should be employed. Potassium-sparing diuretics should be avoided.
Diabetes. In patients with diabetes, the target BP is the same as with all other populations. Preferred antihypertensive drugs are ACE inhibitors, ARBs, CCBs, and diuretics (in low doses). In patients with diabetic nephropathy, ACE inhibitors and ARBs can slow the progression of renal damage and reduce albuminuria. In diabetic patients, as in nondiabet-ics, beta blockers and diuretics can decrease morbidity and mortality. Keep in mind, however, that beta blockers can sup-press glycogenolysis and mask early signs of hypoglycemia; therefore, they must be used with caution. Thiazides and loop diuretics promote hyperglycemia, and hence should be used with care.
How do ACE inhibitors compare with CCBs in patients with hypertension and diabetes? In one large study, patients taking nisoldipine (a CCB) had a higher incidence of MI than did patients taking enalapril (an ACE inhibitor). Because the study was not placebo controlled, it was impossible to distinguish between two possible interpretations: (1) the CCB increased the risk of MI or (2) the ACE inhibitor protected against MI.
Benefits of Multidrug Therapy. Treatment with multiple drugs offers significant benefits. First, by employing drugs that have different mechanisms, we can increase the chance of success: Targeting BP control at several sites is likely to be more effective than targeting at one site. Second, when drugs are used in combination, each can be administered in a lower dosage than would be possible if it were used alone. As a result, both the frequency and the intensity of side effects are reduced. Third, when proper combinations are selected, one agent can offset the adverse effects of another. For example, if a vasodilator is used alone, reflex tachycardia is likely. However, if a vasodilator is combined with a beta blocker, reflex tachycardia will be minimal.
DosingFor each drug in the regimen, dosage should be low initially and then gradually increased. For most people with chronic hypertension, the disease poses no immediate threat. Hence, there is no need to lower BP rapidly using large doses. Also, when BP is reduced slowly, baroreceptors gradually reset to the new lower pressure. As a result, sympathetic reflexes offer less resistance to the hypotensive effects of therapy. Finally, since there is no need to drop BP rapidly and since higher doses carry a higher risk of adverse effects, the use of high initial doses would needlessly increase the risk of adverse effects.
Step-Down TherapyAfter BP has been controlled for at least 1 year, an attempt should be made to reduce dosages and the number of drugs in the regimen. Of course, lifestyle modifications should continue. When reductions are made slowly and progressively, many patients are able to maintain BP control with less medication—and some can be maintained with no medication at all. If drugs are discontinued, regular follow-up is essential, because BP usually returns to hypertensive levels—although it may take years to do so.
Individualizing TherapyPatients With Comorbid ConditionsComorbid conditions complicate treatment. Two conditions that are especially problematic—renal disease and diabetes—are
Data from The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA 289:2560–2572, 2003.
Heart Failure
Recurrent Stroke Prevention
High Risk of CAD
Post–Myocardial Infarction Diabetes
Chronic Kidney Disease
Recommended Drug Classes for Treatment of HTN
• ACEinhibitor
• Aldosteroneantagonist
• ARB• Betablocker• CCB• Diuretic
• ACEinhibitor
• Diuretic
• ACEinhibitor• Betablocker• CCB• Diuretic
• ACEinhibitor• Aldosterone
antagonist• Betablocker
• ACEinhibitor
• ARB• Betablocker• CCB• Diuretic
• ACEinhibitor• ARB
TABLE 47.4 ■ Classes of Antihypertensive Drugs Recommended for Initial Therapy of Hypertension in Patients With Certain High-Risk Comorbid Conditions
ACE, Angiotensin-converting enzyme; ARB, angiotensin II receptor blocker; CAD, coronary artery disease; CCB, calcium channel blocker.

522
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
The primary problem is that hypertension often goes untreated among African Americans until after significant organ damage has developed. If hypertension were diagnosed and treated earlier, the prognosis would be greatly improved. Accordingly, it is important that African Americans undergo routine monitor-ing of BP. If hypertension is diagnosed, treatment should begin at once. Because African Americans have a high incidence of salt sensitivity and cigarette use, lifestyle modifications are an important component of treatment.
African Americans respond better to some antihypertensive drugs than to others. Controlled trials have shown that diuretics can decrease morbidity and mortality in blacks. Accordingly, diuretics are drugs of first choice. CCBs and alpha/beta blockers are also effective. In contrast, monotherapy with beta blockers or ACE inhibitors is less effective in blacks than in whites. Nonetheless, beta blockers and ACE inhibitors should be used
Either way, it seems clear that ACE inhibitors are better than CCBs for patients with hypertension and diabetes.
Patients in Special PopulationsAfrican Americans. Hypertension is a major health problem
for African American adults. Hypertension develops earlier, has a much higher incidence, and is likely to be more severe. As a result, African Americans face a greater risk of heart disease, end-stage renal disease, and stroke. Compared with the general population, African Americans experience a 50% higher rate of death from heart disease, are twice as likely to die from stroke, and are six times more likely to experience hypertension-related end-stage renal disease.
With timely treatment, this disparity can be greatly reduced, if not eliminated. We know that blacks and whites respond equally to treatment (although not always to the same drugs).
Comorbid ConditionDrugs to Be Avoided or Used With Caution Reason for Concern
CARDIOVASCULAR DISORDERS
Heart failure VerapamilDiltiazem
These drugs act on the heart to decrease myocardial contractility and can thereby further reduce cardiac output.
AV heart block Beta blockersLabetalolVerapamilDiltiazem
These drugs act on the heart to suppress AV conduction and can thereby intensify AV block.
Coronary artery disease Hydralazine Reflex tachycardia induced by hydralazine can precipitate an anginal attack.Post–myocardial infarction Hydralazine Reflex tachycardia induced by hydralazine can increase cardiac work and
oxygen demand.
OTHER DISORDERS
Dyslipidemia Beta blockersDiuretics
These drugs may exacerbate dyslipidemia.
Renal insufficiency K+-sparing diureticsK+ supplements
Use of these agents can lead to dangerous accumulations of potassium.
Asthma Beta blockersLabetalol
Beta2 blockade promotes bronchoconstriction.
Depression Reserpine Reserpine can cause depression.Diabetes mellitus Thiazides
FurosemideBeta blockers
Thiazides and furosemide promote hyperglycemia, and beta blockers suppress glycogenolysis and can mask signs of hypoglycemia.
Gout ThiazidesFurosemide
These diuretics promote hyperuricemia.
Hyperkalemia K+-sparing diureticsACE inhibitorsDirect renin inhibitorsAldosterone antagonists
These drugs cause potassium accumulation.
Hypokalemia ThiazidesFurosemide
These drugs cause potassium loss.
Collagen diseases Hydralazine Hydralazine can precipitate a lupus erythematosus–like syndrome.Liver disease Methyldopa Methyldopa is hepatotoxic.Preeclampsia ACE inhibitors
ARBsDirect renin inhibitors
These drugs can injure the fetus.
TABLE 47.5 ■ Comorbid Conditions That Require Cautious Use or Complete Avoidance of Certain Antihypertensive Drugs
ACE, Angiotensin-converting enzyme; ARBs, angiotensin II receptor blockers; AV, atrioventricular.

523
CHAPTER 47 Drugs for Hypertension
High initial doses and rapid dosage escalation can increase the incidence and severity of adverse effects. Accordingly, doses should be low at first and then gradually increased. Remember, there is usually no need to reduce BP rapidly. Hence, large initial doses that can produce a rapid fall in BP but also produce adverse effects should be avoided.
Promoting AdherenceThe major cause of treatment failure in patients with chronic hypertension is lack of adherence to the prescribed regimen. In this section we consider the causes of nonadherence and discuss some solutions.
Why Adherence Is Often Hard to AchieveMuch of the difficulty in promoting adherence stems from the nature of hypertension itself. Hypertension is a chronic, slowly progressing disease that is devoid of overt symptoms through much of its course. Because symptoms are absent, it can be difficult to convince patients that they are ill and need treatment. In addition, since there are no symptoms to relieve, drugs cannot produce an obvious therapeutic response. In the absence of such a response, it can be difficult for patients to believe that their medication is doing anything useful.
Because hypertension progresses very slowly, the disease tends to encourage procrastination. For most people, the adverse
if they are strongly indicated for a comorbid condition. For example, ACE inhibitors should be used in black patients who have type 1 diabetes with proteinuria. Also, ACE inhibitors should be used in patients with hypertensive nephrosclerosis, a condition for which ACE inhibitors are superior to CCBs. When BP cannot be adequately controlled with a single drug, several two-drug combinations are recommended: an ACE inhibitor plus a thiazide diuretic, an ACE inhibitor plus a CCB, and a beta blocker plus a thiazide.
In 2010, the International Society on Hypertension in Blacks (ISHIB) issued updated guidelines on managing hypertension in African Americans. Because hypertension takes a high toll on the black community, these guidelines call for strict new BP goals: under 135/85 mm Hg for most patients, and under 130/80 mm Hg for those at high risk of a cardiovascular event. Some experts, however, have criticized these goals, arguing there is insufficient evidence to support them.
Children and Adolescents. The incidence of secondary hypertension in children is much higher than in adults. Accord-ingly, efforts to diagnose and treat an underlying cause should be especially diligent. For children with primary hypertension, treatment is the same as for adults—although doses are lower and should be adjusted with care. Because ACE inhibitors and ARBs can cause fetal harm, they should be avoided in girls who are sexually active or pregnant.
Older Adults. By age 65 years, most Americans have hypertension. Furthermore, high BP in this group almost always presents as isolated systolic hypertension; DBP is usually normal or low. The good news, as shown in the Hypertension in the Very Elderly Trial (HYVET), is that treatment can reduce the incidence of heart failure, fatal stroke, and all-cause mortality. The bad news is that most older people are not treated.
Because cardiovascular reflexes are blunted in older adults, treatment carries a significant risk of orthostatic hypotension. Accordingly, initial doses should be low—about one-half those used for younger adults—and dosage escalation should be done slowly. Drugs that are especially likely to cause orthostatic hypotension (e.g., reserpine, alpha1 blockers, alpha/beta block-ers) should be used with caution.
Minimizing Adverse EffectsAntihypertensive drugs can produce many unwanted effects, including hypotension, sedation, and sexual dysfunction. (Although not stressed previously, practically all antihyper-tensive drugs can interfere with sexual function.)
The fundamental strategy for decreasing side effects is to tailor the regimen to the sensitivities of the patient. Simply put, if one drug causes effects that are objectionable, a more acceptable drug should be substituted. The best way to identify unacceptable responses is to encourage patients to report them.
Adverse effects caused by the exacerbation of comorbid diseases are both predictable and avoidable. We know, for example, that beta blockers can intensify AV block, and hence should not be taken by people with these disorders. Other conditions that can be aggravated by antihypertensive drugs are listed in Table 47.5. To help avoid drug-disease mismatches, the medical history should identify all comorbid conditions. With this information, the prescriber can choose drugs that are least likely to make the comorbid condition worse.
Hypertension
Life Stage Patient Care Concerns
Infants See “Breast-feeding women,” below.Children/
adolescentsNo data are available on the long-term effects
of antihypertensive drugs on growth and development of children. Drugs recommended for treatment of hypertension in children 1 to 18 years old include ACE inhibitors, diuretics, beta blockers, and calcium channel blockers.
Pregnant women Drugs of choice in treating pregnant women with mild preeclampsia include labetalol and methyldopa. Magnesium sulfate is used in the prevention of seizures in severe preeclampsia or for the treatment of seizures in eclampsia.
Breast-feeding women
Effects of RAAS-blocking drugs have not been studied in breast-feeding. Beta blockers, such as metoprolol, appear safe for the breast-feeding infant. Diuretics appear safe, but may suppress lactation.
Older adults Older adults benefit from SBPs <145 mm Hg. Treatment with ACE inhibitors, diuretics, and/or beta blockers is reasonable. Caution must be taken to avoid overdiuresis when using diuretics in the older adult population.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN

524
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
can be promoted by scheduling office visits at convenient times and by following up when appointments are missed. For many patients, antihypertensive therapy represents a significant economic burden; devising a regimen that is effective but inexpensive will help.
DRUGS FOR HYPERTENSIVE EMERGENCIES
A hypertensive emergency exists when diastolic BP exceeds 120 mm Hg. The severity of the emergency is determined by the likelihood of organ damage. When excessive BP is associated with papilledema (edema of the retina), intracranial hemorrhage, MI, or acute congestive heart failure, a severe emergency exists—and BP must be lowered rapidly (within 1 hour). If severe hypertension is present but does not yet pose an immedi-ate threat of organ damage, reducing BP more slowly (over 24 to 48 hours) is preferable. Because rapid reductions can cause cerebral ischemia, MI, and renal failure, pressure should be reduced gradually whenever possible.
The major drugs used for hypertensive emergencies are discussed here. All reduce BP by causing vasodilation, and all are given IV.
Sodium NitroprussideWhen acute severe hypertension demands a rapid but controlled reduction in BP, IV nitroprusside [Nitropress] is usually the drug of first choice. Nitroprusside is a direct-acting vasodilator that relaxes smooth muscle of arterioles and veins. Effects begin in seconds and then fade rapidly when administration ceases. Nitroprusside is administered by continuous IV infusion using an infusion pump to control the rate. The usual rate is 0.3 to 3 mcg/kg/min. To avoid hypotension, continuous BP monitoring is required. Because nitroprusside has an extremely short duration, hypotension can be corrected quickly by reducing the rate of infusion. Prolonged infusion (longer than 72 hours) can produce toxic accumulation of thiocyanate and should be avoided. The basic pharmacology of nitroprusside is discussed in Chapter 46.
FenoldopamFenoldopam [Corlopam] is an IV drug indicated for short-term management of hypertensive emergencies. Benefits equal those of nitroprusside. Fenoldopam lowers BP by activating dopamine1 receptors on arterioles to cause vasodilation. In animal models, the drug dilates renal, coronary, mesenteric, and peripheral vessels.
Fenoldopam differs from other antihypertensives in that it helps maintain (or even improve) renal function. Two mechanisms are involved. First, the drug dilates renal blood vessels, increasing renal blood flow (despite reducing arterial pressure). Second, fenoldopam promotes sodium and water excretion through direct effects on renal tubules.
Fenoldopam has a rapid onset and short duration. Effects begin in less than 5 minutes. The drug undergoes rapid hepatic metabolism followed by renal excretion. Its plasma half-life is only 5 minutes.
Fenoldopam is generally well tolerated. The most common side effects are hypotension, headache, flushing, dizziness, and reflex tachycardia—all of which occur secondary to vasodilation. Tachycardia may cause ischemia in patients with angina. Combined use with a beta blocker can minimize tachycardia, but may also result in excessive lowering of BP. Fenoldopam can elevate intraocular pressure, and hence should be used with caution in patients with glaucoma.
Fenoldopam is administered by continuous IV infusion. To minimize tachycardia, the initial dosage should be low. The typical infusion rate is 0.25
effects of hypertension will not become manifest for many years. Realizing this, patients may reason (incorrectly) that they can postpone therapy without significantly increasing risk.
The negative aspects of treatment also contribute to non-adherence. Antihypertensive regimens can be complex and expensive. In addition, treatment must continue lifelong. Lastly, antihypertensive drugs can cause a number of adverse effects, ranging from sedation to hypotension to impaired sexual function. It is difficult to convince people who are feeling good to take drugs that may make them feel worse. Some people may decide that exposing themselves to the negative effects of therapy today is paying too high a price to avoid the adverse consequences of hypertension at some indefinite time in the future.
Ways to Promote AdherencePatient Education. Adherence requires motivation, and
patient education can help provide it. Patients should be taught about the consequences of hypertension and the benefits of treatment. Because hypertension does not cause discomfort, it may not be clear to patients that their condition is indeed serious. Patients must be helped to understand that, left untreated, hypertension can cause heart disease, kidney disease, and stroke. In addition, patients should appreciate that, with proper therapy, the risks of these long-term complications can be minimized, resulting in a longer and healthier life. Lastly, patients must understand that drugs do not cure hypertension—they only control symptoms. Hence, for treatment to be effective, medication must be taken lifelong.
Teach Self-Monitoring. Patients should be taught the goal of treatment (usually maintenance of BP below 120/80 mm Hg), and they should be taught to monitor and record their BP daily. This increases patient involvement and provides positive feedback that can help promote adherence.
Minimize Side Effects. If we expect patients to comply with long-term treatment, we must keep adverse effects to a minimum. As discussed previously, adverse effects can be minimized by (1) encouraging patients to report side effects, (2) discontinuing objectionable drugs and substituting more acceptable ones, (3) avoiding drugs that can exacerbate comorbid conditions, and (4) using doses that are initially low and gradually increased.
Establish a Collaborative Relationship. The patient who feels like a collaborative partner in the treatment program is more likely to comply than is the patient who feels that treatment is being imposed. Collaboration allows the patient to help set treatment goals, create the treatment program, and evaluate progress. In addition, a collaborative relationship facilitates communication about side effects.
Simplify the Regimen. Antihypertensive regimens may consist of several drugs taken multiple times a day. Such complex regimens deter adherence. To promote adherence, the dosing schedule should be as simple as possible. Once an effective regimen has been established, dosing just once or twice daily should be tried. If an appropriate combination product is available (e.g., a fixed-dose combination of a thiazide diuretic plus an ACE inhibitor), the combination product may be substituted for its components.
Other Measures. Adherence can be promoted by giving positive reinforcement when therapeutic goals are achieved. Involvement of family members can be helpful. Also, adherence

525
CHAPTER 47 Drugs for Hypertension
does not. (The ACOG defines severe hypertension as SBP above 160 mm Hg or DBP above 110 mm Hg, and mild hypertension as SBP 140 to 159 mm Hg or DBP 90 to 109 mm Hg.) There is good evidence that treating severe hypertension reduces risk. In contrast, there is little evidence that treating mild hypertension offers significant benefit.
Patients who have chronic hypertension during preg-nancy are at increased risk of developing preeclampsia (see next section). Unfortunately, reducing BP does not lower this risk.
PREECLAMPSIA AND ECLAMPSIAPreeclampsia is a multisystem disorder characterized by the combination of elevated BP (above 130/90 mm Hg) and proteinuria (300 mg or more in 24 hours) that develops after the 20th week of gestation. The disorder occurs in about 5% of pregnancies. Rarely, women with preeclampsia develop seizures. If seizures do develop, the condition is then termed eclampsia. Risk factors for preeclampsia include black race, chronic hypertension, diabetes, collagen vascular disorders, and previous preeclampsia. The etiology of preeclampsia is complex and incompletely understood.
Preeclampsia poses serious risks for the fetus and mother. Risks for the fetus include intrauterine growth restriction, premature birth, and even death. The mother is at risk for seizures (eclampsia), renal failure, pulmonary edema, stroke, and death.
Management of preeclampsia is based on the severity of the disease, the status of mother and fetus, and the length of gestation. The objective is to preserve the health of the mother and deliver an infant who will not require intensive and prolonged neonatal care. Success requires close maternal and fetal monitoring. Although drugs can help reduce BP, delivery is the only cure.
Management of mild preeclampsia is controversial and depends on the duration of gestation. If preeclampsia develops near term, and if fetal maturity is certain, induction of labor is advised. However, if mild preeclampsia develops earlier in gestation, experts disagree about what to do. Suggested measures include bed rest, prolonged hospitalization, treatment with antihypertensive drugs, and prophylaxis with an anticonvulsant. Studies to evaluate these strategies have generally failed to demonstrate benefits from any of them, including treatment with antihypertensive drugs.
The definitive intervention for severe preeclampsia is delivery. However, making the choice to induce labor presents a dilemma. Since preeclampsia can deteriorate rapidly, with grave consequences for the patient and fetus, immediate delivery is recommended. However, if the fetus is not sufficiently mature, immediate delivery could threaten its life. Do we deliver the fetus immediately, which would eliminate risk for the patient but present a serious risk for the fetus—or do we postpone delivery, which would reduce risk for the fetus but greatly increase risk for the patient? If the patient elects to postpone delivery, then BP can be lowered with drugs. Because severe preeclampsia can be life threatening, treatment must be done in a tertiary care center to permit close monitoring. The major objective is to prevent maternal cerebral complications (e.g., hemorrhage, encephalopathy). The drug of choice for lowering
to 0.5 mcg/kg/min. With continuous 24-hour infusion, no tolerance develops to antihypertensive effects, and there is no rebound increase in BP when the infusion is stopped. With a 48-hour infusion, some tolerance may develop. Oral antihypertensive therapy can be added as soon as BP has stabilized.
LabetalolLabetalol blocks alpha- and beta-adrenergic receptors. Blood pressure is reduced by arteriolar dilation secondary to alpha blockade. Beta blockade prevents reflex tachycardia in response to reduced arterial pressure, and hence the drug is probably safe for patients with angina or MI. Beta blockade can aggravate bronchial asthma, heart failure, AV block, cardiogenic shock, and bradycardia. Accordingly, labetalol should not be given to patients with these disorders. Administration is by slow IV injection.
ClevidipineClevidipine [Cleviprex] is a dihydropyridine CCB with an ultrashort half-life (about 1 minute). Administration is by IV infusion. As with nitroprusside, effects begin rapidly and then fade rapidly when the infusion is slowed or stopped. As a result, BP can be easily titrated. For patients with severe hypertension, the infusion rate is 1 to 2 mg/hr initially, and can be doubled every 3 minutes up to a maximum of 32 mg/hr. In clinical trials, the average time to reach the target BP was 10.9 minutes. The most common side effects are headache, nausea, and vomiting. The basic pharmacology of clevidipine is discussed in Chapter 45.
DRUGS FOR HYPERTENSIVE DISORDERS OF PREGNANCY
Hypertension is the most common complication of pregnancy, with an incidence of about 10%. When hypertension develops, it is essential to distinguish between chronic hypertension and preeclampsia. Chronic hypertension is relatively benign, whereas preeclampsia can lead to life-threatening complications for the patient and the fetus.
CHRONIC HYPERTENSIONChronic hypertension, seen in 5% of pregnancies, is defined as hypertension that was present before pregnancy or that developed before the 20th week of gestation. Persistent severe hypertension carries a risk to both the patient and the fetus. Potential adverse outcomes include placental abruption, maternal cardiac decompensation, premature birth, fetal growth delay, central nervous system hemorrhage, and renal failure. The goal of treatment is to minimize the risk of hypertension to the patient and fetus while avoiding drug-induced harm to the fetus. With the exception of ACE inhibitors, ARBs, and DRIs, antihypertensive drugs that were being taken before pregnancy can be continued. ACE inhibitors, ARBs, and DRIs are con-traindicated owing to their potential for harm (fetal growth delay, congenital malformations, neonatal renal failure, neonatal death). When drug therapy is initiated during pregnancy, methyldopa or labetalol are the traditional agents of choice. These drugs have limited effects on uteroplacental and fetal hemodynamics, and do not adversely affect the fetus or neonate. Regardless of the drug selected, treatment should not be too aggressive because an excessive drop in BP could compromise uteroplacental blood flow.
According to guidelines issued by the American College of Obstetricians and Gynecologists (ACOG), “severe” hyperten-sion requires treatment, whereas “mild” hypertension generally

526
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
or 5 gm IM injected into alternating buttocks every 4 hours. To ensure therapeutic effects and prevent toxicity, blood levels of magnesium, as well as presence of patellar reflex, should be monitored. The target range for serum magnesium is 4 to 7 mEq/L (the normal range for magnesium is 1.5 to 2 mEq/L).
Can drugs help prevent preeclampsia in those at risk? Yes. When started before 16 weeks of gestation, low-dose aspirin reduces risk by about 50%. Similarly, L-arginine (com-bined with antioxidant vitamins) can also help. By contrast, several other preparations—magnesium, zinc, vitamin C, vitamin E, fish oil, and diuretics—appear to offer no protection at all.
BP is labetalol (20 mg by IV bolus over 2 minutes); dosing may be repeated at 10-minute intervals up to a total of 300 mg.
Because severe preeclampsia can evolve into eclampsia, an antiseizure drug may be given for prophylaxis. Magnesium sulfate is the drug of choice. In one study, prophylaxis with magnesium sulfate reduced the risk of eclampsia by 58% and the risk of death by 45%. Dosing is the same as for treating eclampsia.
If eclampsia develops, magnesium sulfate is the preferred drug for seizure control. Initial dosing consists of a 4- to 6-gm IV loading dose followed by 5 gm IM injected into each buttock. Maintenance consists of continuous IV infusion of 1 to 2 gm/hr
KEY POINTS
■ Hypertension is defined as SBP greater than 130 mm Hg or DBP greater than 80 mm Hg.
■ Primary hypertension (essential hypertension), defined as hypertension with no identifiable cause, is the most common form of hypertension.
■ Untreated hypertension can lead to heart disease, kidney disease, and stroke.
■ In patients older than 50, elevated systolic BP represents a greater cardiovascular risk than elevated diastolic BP.
■ The goal of antihypertensive therapy is to decrease morbid-ity and mortality without decreasing quality of life. For most patients, this goal is achieved by maintaining BP between 120/80 and 130/80 mm Hg.
■ To reduce BP, two types of treatment may be used: drug therapy and lifestyle modification (smoking cessation, reduction of salt and alcohol intake, following the DASH diet, and increasing aerobic exercise).
■ The baroreceptor reflex, the kidneys, and the RAAS can oppose our attempts to lower BP with drugs. We can counteract the baroreceptor reflex with a beta blocker, the kidneys with a diuretic, and the RAAS with an ACE inhibitor, ARB, DRI, or aldosterone antagonist.
■ Thiazide diuretics (e.g., hydrochlorothiazide, chlorthalidone) and loop diuretics (e.g., furosemide) reduce BP in two ways: they reduce blood volume (by promoting diuresis) and they reduce arterial resistance (by an unknown mechanism).
■ Loop diuretics should be reserved for (1) patients who need greater diuresis than can be achieved with thiazides and (2) patients with a low GFR (because thiazides don’t work when GFR is low).
■ Beta blockers (e.g., metoprolol) appear to lower BP primar-ily by reducing peripheral vascular resistance; the mecha-nism is unknown. They may also lower BP by decreasing myocardial contractility and suppressing reflex tachycardia (through beta1 blockade in the heart), and by decreasing renin release (through beta1 blockade in the kidney).
■ Calcium channel blockers (e.g., diltiazem, nifedipine) reduce BP by promoting dilation of arterioles.
■ ACE inhibitors, ARBs, and DRIs lower BP by preventing angiotensin II–mediated vasoconstriction and aldosterone-mediated volume expansion. ACE inhibitors work by blocking the formation of angiotensin II, whereas ARBs block the actions of angiotensin II. DRIs prevent formation of angiotensin I and thereby shut down the entire RAAS.
■ Aldosterone antagonists lower BP by preventing aldosterone-mediated retention of sodium and water in the kidney.
■ Thiazide diuretics are preferred drugs for initial therapy of uncomplicated hypertension.
■ When a combination of drugs is used for hypertension, each drug should have a different mechanism of action.
■ Dosages of antihypertensive drugs should be low initially and increased gradually. This approach minimizes adverse effects and permits baroreceptors to reset to a lower pressure.
■ Lack of patient adherence is the major cause of treatment failure in antihypertensive therapy.
■ Adherence is difficult to achieve because (1) hypertension has no symptoms (so drug benefits aren’t obvious); (2) hypertension progresses slowly (so patients think they can postpone treatment); and (3) treatment is complex and expensive, continues lifelong, and can cause adverse effects.
■ A severe hypertensive emergency exists when diastolic BP exceeds 120 mm Hg and there is ongoing end-organ damage.
■ Nitroprusside (IV) is a drug of choice for hypertensive emergencies.
■ Hypertension is the most common complication of pregnancy.
■ Methyldopa and labetalol are drugs of choice for treating chronic hypertension of pregnancy.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

527
CHAPTER 47 Drugs for Hypertension
Summary of Major Nursing Implicationsa
ANTIHYPERTENSIVE DRUGSPreadministration AssessmentTherapeutic GoalThe goal of antihypertensive therapy is to prevent the long-term sequelae of hypertension (heart disease, kidney disease, stroke) while minimizing drug effects that can reduce quality of life. For most patients, BP should be reduced to less than 130/80 mm Hg.
Baseline DataThe following tests should be done in all patients: BP; electrocardiogram; complete urinalysis; hemoglobin and hematocrit; and blood levels of sodium, potassium, calcium, creatinine, glucose, uric acid, triglycerides, and cholesterol (total, LDL, and HDL cholesterol).
Identifying High-Risk PatientsWhen taking the patient’s drug history, attempt to identify drugs that can raise BP or that can interfere with the effects of antihypertensive drugs. Some drugs of concern are listed later under Minimizing Adverse Interactions.
The patient history should identify comorbid conditions that either contraindicate the use of specific agents (e.g., AV block contraindicates use of beta blockers) or require that drugs be used with special caution (e.g., thiazide diuretics must be used with caution in patients with gout or diabetes). For risk factors that pertain to specific antihypertensive drugs, see the chapters in which those drugs are discussed.
Implementation: AdministrationRoutesAll drugs for chronic hypertension are administered orally.
DosageTo minimize adverse effects, dosages should be low initially and increased gradually. It is counterproductive to employ high initial dosages that produce a rapid fall in pressure while also producing intense adverse effects that can discourage adherence. After 12 months of successful treatment, dosage reductions should be tried.
Implementation: Measures to Enhance Therapeutic EffectsLifestyle ModificationsIn hypertensive patients, lifestyle changes can reduce BP and increase responsiveness to antihypertensive drugs. These changes should be tried for 6 to 12 months before implement-ing drug therapy and should continue even if drugs are required.
Weight Reduction. Help patients develop an exercise and weight management program if needed.
Sodium Restriction. Encourage patients to consume no more than 2300 mg of salt daily and provide them with information on the salt content of foods.
DASH Diet. Encourage patients to adopt a diet rich in fruits, vegetables, and low-fat dairy products, and low in total fat, unsaturated fat, and cholesterol.
Alcohol Restriction. Encourage patients to limit alcohol consumption to 1 ounce/day (for most men) and 0.5 ounce/day (for women and lighter weight men). One ounce of ethanol is equivalent to about two mixed drinks, two glasses of wine, or two cans of beer.
Exercise. Encourage patients with a sedentary lifestyle to perform 30 to 45 minutes of aerobic exercise (e.g., walking, swimming, bicycling) most days of the week.
Smoking Cessation. Strongly encourage patients to quit smoking. Teach patients about aids for smoking cessation (e.g., nicotine patch, bupropion, varenicline).
Promoting AdherenceNonadherence is the major cause of treatment failure. Achiev-ing adherence is difficult for several reasons: hypertension is devoid of overt symptoms; drugs don’t make people feel better (but can make them feel worse); regimens can be complex and expensive; complications of hypertension take years to develop, thereby providing a misguided ratio-nale for postponing treatment; and treatment usually lasts lifelong.
Provide Patient Education. Educate patients about the long-term consequences of hypertension and the ability of lifestyle changes and drug therapy to decrease morbidity and prolong life. Inform patients that drugs do not cure hypertension; therefore, the medication prescribed must usually be taken lifelong.
Encourage Self-Monitoring. Make certain that patients know the treatment goal (usually reduction of BP to less than 130/80 mm Hg), and teach them to monitor and chart their own BP. This will increase their involvement and help them see the benefits of treatment.
Minimize Adverse Effects. Adverse drug effects are an obvious deterrent to adherence. Measures to reduce undesired effects are discussed under Minimizing Adverse Effects.
Establish a Collaborative Relationship. Encourage patients to be active partners in setting treatment goals, creating a treatment program, and evaluating progress.
Simplify the Regimen. An antihypertensive regimen can consist of several drugs taken multiple times a day. Once an effective regimen has been established, attempt to switch to once-a-day or twice-a-day dosing. If an appropriate combina-tion product is available (e.g., a fixed-dose combination of a thiazide diuretic plus an ACE inhibitor), substitute the combination product for its components.
Other Measures. Additional measures to promote adherence include providing positive reinforcement when treatment goals are achieved, involving family members in the treatment program, scheduling office visits at convenient times, following up on patients who miss an appointment, and devising a program that is effective but keeps costs low.
Continued

528
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
aPatient education information is highlighted as blue text.
Ongoing Evaluation and InterventionsEvaluating TreatmentMonitor BP periodically. The usual goal is to reduce it to less than 130/80 mm Hg. Teach patients to self-monitor their BP and to maintain a BP record.
Minimizing Adverse EffectsGeneral Considerations. The fundamental strategy for
decreasing adverse effects is to tailor the regimen to the sensitivities of the patient. If a drug causes objectionable effects, a more acceptable drug should be substituted.
Inform patients about the potential side effects of treat-ment, and encourage them to report objectionable responses.
Avoid drugs that can exacerbate comorbid conditions. For example, don’t give beta blockers to patients who have bradycardia, AV block, or asthma. Table 47.5 lists drugs to avoid in patients with specific disorders.
Initiate therapy with low doses and increase them gradually.
Adverse Effects of Specific Drugs. For measures to minimize adverse effects of specific antihypertensive drugs (e.g., beta blockers, diuretics, ACE inhibitors), see the chapters in which those drugs are discussed.
Minimizing Adverse InteractionsWhen taking the patient history, identify drugs that can raise BP or interfere with the effects of antihypertensive drugs. Drugs of concern include oral contraceptives, nonsteroidal anti-inflammatory drugs, glucocorticoids, appetite suppres-sants, tricyclic antidepressants, monoamine oxidase inhibitors, cyclosporine, erythropoietin, alcohol (in large quantities), and nasal decongestants and other cold remedies.
Antihypertensive regimens frequently contain two or more drugs, posing a potential risk of adverse interactions (e.g., ACE inhibitors can increase the risk of hyperkalemia caused by potassium-sparing diuretics). For interactions that pertain to specific antihypertensive drugs, see the chapters in which those drugs are discussed.
Summary of Major Nursing Implicationsa—cont’d

529
C H A P T E R
48 Drugs for Heart Failure
Pathophysiology of Heart Failure, p. 529Cardiac Remodeling, p. 530Physiologic Adaptations to Reduced Cardiac
Output, p. 530The Vicious Cycle of “Compensatory” Physiologic
Responses, p. 531Signs and Symptoms of Heart Failure, p. 531Classification of Heart Failure Severity, p. 531
Overview of Drugs Used to Treat Heart Failure, p. 532Diuretics, p. 532Drugs That Inhibit the RAAS, p. 532Beta Blockers, p. 534Ivabradine (Corlanor), p. 535Inotropic Agents, p. 535Vasodilators (Other Than ACE Inhibitors and
ARBs), p. 535Digoxin, a Cardiac Glycoside, p. 536
Chemistry, p. 536Mechanical Effects on the Heart, p. 536Hemodynamic Benefits in Heart Failure, p. 538Neurohormonal Benefits in Heart Failure,
p. 538Electrical Effects on the Heart, p. 538Adverse Effects I: Cardiac Dysrhythmias, p. 539Adverse Effects II: Noncardiac Adverse Effects,
p. 539Adverse Effects III: Measures to Reduce Adverse
Effects, p. 540Drug Interactions, p. 540Pharmacokinetics, p. 540Preparations, Dosage, and Administration,
p. 541Management of Heart Failure, p. 541
Stage A, p. 541Stage B, p. 542Stage C, p. 542Stage D, p. 543
Key Points, p. 543Summary of Major Nursing Implications, p. 545
Box 48.1. Attention: Digoxin May Be Hazardous to Women’s Health, p. 537
Heart failure is a disease with two major forms: (1) heart failure with left ventricular (LV) systolic dysfunction, also known as heart failure with reduced LV ejection fraction (HFrEF) and (2) diastolic heart failure, also known as heart failure with preserved LV ejection fraction (HFpEF). In this chapter, discussion is limited to the first form. Accordingly, for the rest of this chapter, the term heart failure (HF) will be used to denote the first form only.
Heart failure is a progressive, often fatal disorder character-ized by ventricular dysfunction, reduced cardiac output, insufficient tissue perfusion, and signs of fluid retention (e.g., peripheral edema, shortness of breath). The disease affects nearly 6 million Americans and is responsible for about 285,000 deaths every year. Of those who have HF, 20% are likely to die within 1 year, and 50% within 5 years. Heart failure is primarily a disease of older adults, affecting 4% to 8% of those at age 65 years and more than 9% to 12% of those older than 80 years. Direct and indirect healthcare costs of HF are estimated at more than $30 billion yearly. With improved evaluation and care, many hospitalizations could be prevented, quality of life could be improved, and life expectancy could be extended.
In the past, HF was commonly referred to as congestive heart failure. This term was used because HF frequently causes fluid accumulation (congestion) in the lungs and peripheral tissues. However, because many patients do not have signs of pulmonary or systemic congestion, the term heart failure is now preferred.
Drugs recommended for treatment include diuretics, inhibi-tors of the renin-angiotensin-aldosterone system (RAAS), beta blockers, and digoxin. In this chapter, only digoxin is discussed at length. The other drugs are presented at length in previous chapters, so discussion here is limited to their use in heart failure.
To understand HF and its treatment, you need a basic understanding of hemodynamics. In particular, you need to understand the role of venous pressure, afterload, and Starling’s mechanism in determining cardiac output. You also need to understand the roles of the baroreceptor reflex, the RAAS, and the kidneys in regulating arterial pressure. You can refresh your memory of these concepts by reading Chapter 43.
PATHOPHYSIOLOGY OF HEART FAILUREHeart failure is a syndrome in which the heart is unable to pump sufficient blood to meet the metabolic needs of tissues. The syndrome is characterized by signs of inadequate tissue perfusion (fatigue, shortness of breath, exercise intolerance) and/or signs of volume overload (venous distention, peripheral and pulmonary edema). The major underlying causes of HF are chronic hypertension and myocardial infarction. Other causes include valvular heart disease, coronary artery disease, congenital heart disease, dysrhythmias, and aging of the

530
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
insufficient time for complete ventricular filling, and cardiac output will fall.
• Increased contractility. Increased myocardial contractility has the obvious benefit of increasing cardiac output. The only detriment is an increase in cardiac oxygen demand.
• Increased venous tone. Elevation of venous tone increases venous pressure and thereby increases ventricular filling. Because of Starling’s mechanism, increased filling increases stroke volume. Unfortunately, if venous pressure is excessive, blood will back up behind the failing ventricles, thereby aggravating pulmonary and peripheral edema. Furthermore, excessive filling pressure can dilate the heart so much that stroke volume will begin to decline.
• Increased arteriolar tone. Elevation of arteriolar tone increases arterial pressure, thereby increasing perfusion of vital organs. Unfortunately, increased arterial pressure also means the heart must pump against greater resistance. Since cardiac reserve is minimal in HF, the heart may be unable to meet this challenge, and output may fall.
Water Retention and Increased Blood VolumeMechanisms. Water retention results from two mechanisms.
First, reduced cardiac output causes a reduction in renal blood flow, which in turn decreases glomerular filtration rate (GFR). As a result, urine production is decreased and water is retained. Retention of water increases blood volume.
Second, HF activates the RAAS. Activation occurs in response to reduced blood pressure and reduced renal blood flow. Once activated, the RAAS promotes water retention by increasing circulating levels of aldosterone and angiotensin II. Aldosterone acts directly on the kidneys to promote retention of sodium and water. Angiotensin II causes constriction of renal blood vessels, which decreases renal blood flow and thereby further decreases urine production. In addition, angio-tensin II causes constriction of systemic arterioles and veins, and thereby increases venous and arterial pressure.
myocardium. In its earliest stage, HF is asymptomatic. As failure progresses, fatigue and shortness of breath develop. As cardiac performance declines further, blood backs up behind the failing ventricles, causing venous distention, peripheral edema, and pulmonary edema. Heart failure is a chronic disorder that requires continuous treatment with drugs.
Cardiac RemodelingIn the initial phase of failure, the heart undergoes remodeling, a process in which the ventricles dilate (grow larger), hyper-trophy (increase in wall thickness), and become more spherical (less cylindrical). These alterations in cardiac geometry increase wall stress and reduce LV ejection fraction. Remodeling occurs in response to cardiac injury brought on by infarction and other causes. The remodeling process is driven primarily by neurohormonal systems, including the sympathetic nervous system (SNS) and the RAAS. In addition to promoting remodel-ing, neurohormonal factors promote cardiac fibrosis and myocyte death. The net result of these pathologic changes—remodeling, fibrosis, and cell death—is progressive decline in cardiac output. As a rule, cardiac remodeling precedes development of symptoms and continues after they appear. As a result, cardiac performance continues to decline.
Physiologic Adaptations to Reduced Cardiac OutputIn response to reductions in cardiac pumping ability, the body undergoes several adaptive changes. Some of these help improve tissue perfusion; others compound existing problems.
Cardiac DilationDilation of the heart is characteristic of HF. Cardiac dilation results from a combination of increased venous pressure (see the following text) and reduced contractile force. Reduced contractility lowers the amount of blood ejected during systole, causing end-systolic volume to rise. The increase in venous pressure increases diastolic filling, which causes the heart to expand even further.
Because of Starling’s mechanism, the increase in heart size that occurs in HF helps improve cardiac output. That is, as the heart fails and its volume expands, contractile force increases, causing a corresponding increase in stroke volume. However, please note that the maximal contractile force that can be developed by the failing heart is considerably lower than the maximal force of the healthy heart. This limitation is reflected in the curve for the failing heart shown in Fig. 48.1.
If cardiac dilation is insufficient to maintain cardiac output, other factors come into play. As discussed further in the text, these are not always beneficial.
Increased Sympathetic ToneHeart failure causes arterial pressure to fall. In response, the baroreceptor reflex increases sympathetic output to the heart, veins, and arterioles. At the same time, parasympathetic effects on the heart are reduced. The consequences of increased sympathetic tone are summarized as follows:
• Increased heart rate. Acceleration of heart rate increases cardiac output, thereby helping improve tissue perfusion. However, if heart rate increases too much, there will be
Failure plus Digoxin
Normal
Failure
Fiber Length(Ventricular Diameter)
Con
trac
tile
For
ce(S
trok
e V
olum
e)
Fig. 48.1 ■ Relationship of ventricular diameter to contractile force. In the normal heart and the failing heart, increased fiber length produces increased contractile force. However, for any given fiber length, contractile force in the failing heart is much less than in the healthy heart. By increasing cardiac contractility, digoxin shifts the relationship between fiber length and stroke volume in the failing heart toward that in the normal heart.

531
CHAPTER 48 Drugs for Heart Failure
and myocardial hypertrophy result in cardiomegaly (increased heart size). The combination of increased venous tone plus increased blood volume helps cause pulmonary edema, peripheral edema, hepatomegaly (increased liver size), and distention of the jugular veins. Weight gain results from fluid retention.
Classification of Heart Failure SeverityThere are two major schemes for classifying HF severity. One scheme, established by the New York Heart Association (NYHA), classifies HF based on the functional limitations it causes. A newer scheme, proposed jointly by the American College of Cardiology (ACC) and the American Heart Associa-tion (AHA), is based on the observation that HF is a progressive disease that moves through stages of increasing severity.
The NYHA scheme, which has four classes, can be sum-marized as follows:
• Class I—No limitation of ordinary physical activity• Class II—Slight limitation of physical activity: normal
activity produces fatigue, dyspnea, palpitations, or angina• Class III—Marked limitation of physical activity: even
mild activity produces symptoms• Class IV—Symptoms occur at rest
The ACC/AHA scheme, which also has four stages, can be summarized as follows:
• Stage A—At high risk for HF but without structural heart disease or symptoms of HF
• Stage B—Structural heart disease but without symptoms of HF
• Stage C—Structural heart disease with prior or current symptoms of HF
• Stage D—Advanced structural heart disease with marked symptoms of HF at rest, and requiring specialized
Consequences. As with other adaptive responses to HF, increased blood volume can be beneficial or harmful. Increased blood volume increases venous pressure and thereby increases venous return. As a result, ventricular filling and stroke volume are increased. The resultant increase in cardiac output can improve tissue perfusion. However, as noted, if venous pressure is too high, edema of the lungs and periphery may result. More importantly, if the increase in cardiac output is insufficient to maintain adequate kidney function, renal retention of water will progress unabated. The resultant accumulation of fluid will cause severe cardiac, pulmonary, and peripheral edema—and, ultimately, death.
Natriuretic PeptidesIn response to stretching of the atria and dilation of the ventricles, the heart releases two natriuretic peptides: atrial natriuretic peptide (ANP) and B-natriuretic peptide (BNP). As discussed in Chapter 43, these hormones promote dilation of arterioles and veins, and also promote a loss of sodium and water through the kidneys. Hence, they tend to counterbalance vasoconstriction caused by the SNS and angiotensin II, as well as retention of sodium and water caused by the RAAS. However, as HF progresses, the effects of ANP and BNP eventually become overwhelmed by the effects of the SNS and RAAS.
Levels of circulating BNP are an important index of cardiac status in HF patients and so can be a predictor of long-term survival. High levels of BNP indicate poor cardiac health and can predict a lower chance of survival. Conversely, low levels of BNP indicate better cardiac health and can predict a higher chance of survival. This information can be helpful when assessing the hospitalized patient at discharge: The lower the BNP level, the greater the chances of long-term survival.
The Vicious Cycle of “Compensatory” Physiologic ResponsesAs discussed earlier, reduced cardiac output leads to compensa-tory responses: (1) cardiac dilation, (2) activation of the SNS, (3) activation of the RAAS, and (4) retention of water and expansion of blood volume. Although these responses represent the body’s attempt to compensate for reduced cardiac output, they can actually make matters worse: Excessive heart rate can reduce ventricular filling; excessive arterial pressure can lower cardiac output; and excessive venous pressure can cause pulmonary and peripheral edema. Thus, as depicted in Fig. 48.2, the “compensatory” responses can create a self-sustaining cycle of maladaptation that further impairs cardiac output and tissue perfusion. If cardiac output becomes too low to maintain sufficient production of urine, the resultant accumulation of water will eventually be fatal. The actual cause of death is complete cardiac failure secondary to excessive cardiac dilation and cardiac edema.
Signs and Symptoms of Heart FailureThe prominent signs and symptoms of HF are a direct conse-quence of the pathophysiology just described. Decreased tissue perfusion results in reduced exercise tolerance, fatigue, and shortness of breath; shortness of breath may also reflect pul-monary edema. Increased sympathetic tone produces tachy-cardia. Increased ventricular filling, reduced systolic ejection,
Cardiac remodeling
Reduced cardiac output
“Compensatory”responses
Cardiac dilationActivation of the sympatheticnervous systemActivation of the renin-angiotensin-aldosterone systemRetention of water and increased blood volume
1.2.
3.
4.
Heart rateVenous pressureArterial pressure
Fig. 48.2 ■ The vicious cycle of maladaptive compensatory responses to a failing heart.

532
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Loop DiureticsThe loop diuretics (e.g., furosemide) produce profound diuresis. In contrast to the thiazides, these drugs can promote fluid loss even when GFR is low. Therefore, loop diuretics are preferred to thiazides when cardiac output is greatly reduced. Administra-tion may be oral or IV. Because they can mobilize large volumes of water and because they work when GFR is low, loop diuretics are drugs of choice for patients with severe HF. Like the thia-zides, these drugs can cause hypokalemia, thereby increasing the risk of digoxin toxicity. In addition, loop diuretics can cause severe hypotension secondary to excessive volume reduction.
Potassium-Sparing DiureticsIn contrast to the thiazides and loop diuretics, the potassium-sparing diuretics (e.g., spironolactone, triamterene) promote only scant diuresis. In patients with HF, these drugs are employed to counteract potassium loss caused by thiazide and loop diuretics, thereby lowering the risk of digoxin-induced dysrhythmias. Not surprisingly, the principal adverse effect of the potassium-sparing drugs is hyperkalemia. Because angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) also carry a risk of hyperkalemia, caution is needed if these drugs are combined with a potassium-sparing diuretic. Accordingly, when therapy with an ACE inhibitor or ARB is initiated, the potassium-sparing diuretic should be discontinued. It can be resumed later if needed.
One potassium-sparing diuretic—spironolactone—prolongs survival in patients with HF primarily by blocking receptors for aldosterone, not by causing diuresis. This drug and a related agent—eplerenone—are discussed later under Aldosterone Antagonists.
Drugs That Inhibit the RAASThe RAAS plays an important role both in cardiac remodeling and in the hemodynamic changes that occur in response to reduced cardiac output. Accordingly, agents that inhibit the
interventions (e.g., heart transplant, mechanical assist device)
Please note that the ACC/AHA scheme is intended to comple-ment the NYHA scheme, not replace it. The relationship between the two is shown in Fig. 48.3.
OVERVIEW OF DRUGS USED TO TREAT HEART FAILUREFor routine therapy, heart failure is treated with three types of drugs: (1) diuretics, (2) agents that inhibit the RAAS, and (3) beta blockers. Other agents (e.g., digoxin, dopamine, hydrala-zine) may be used as well.
DiureticsDiuretics are first-line drugs for all patients with signs of volume overload or with a history of volume overload. By reducing blood volume, these drugs can decrease venous pressure, arterial pressure (afterload), pulmonary edema, peripheral edema, and cardiac dilation. However, excessive diuresis must be avoided: If blood volume drops too low, cardiac output and blood pressure may fall precipitously, thereby further compromising tissue perfusion. For the most part, the benefits of diuretics are limited to symptom reduction. As a rule, these drugs do not prolong survival. The basic pharmacology of the diuretics is discussed in Chapter 41.
Thiazide DiureticsThe thiazide diuretics (e.g., hydrochlorothiazide) produce moderate diuresis. These oral agents are used for long-term therapy of HF when edema is not too great. Since thiazides are ineffective when GFR is low, these drugs cannot be used if cardiac output is greatly reduced. The principal adverse effect of the thiazides is hypokalemia, which increases the risk of digoxin-induced dysrhythmias (see later in this chapter).
A At high risk for HF but without structuralheart disease or symptoms of HF
B Structural heart disease but withoutsymptoms of HF
I Asymptomatic
C Structural heart disease with prior orcurrent symptoms of HF
D Advanced structural heart disease withmarked symptoms of HF at rest despitemaximal medical therapy. Specializedinterventions (e.g., heart transplant,mechanical assist device) required
II Symptomatic with moderate exertion
III Symptomatic with minimal exertion
IV Symptomatic at rest
ACC/AHA Stage NYHA Functional Classification
Fig. 48.3 ■ American College of Cardiology/American Heart Association (ACC/AHA) stages and New York Heart Association (NYHA) functional classification of heart failure.

533
CHAPTER 48 Drugs for Heart Failure
Interestingly, suppression of angiotension II production diminishes over time, suggesting that long-term benefits are the result of some other action.
Impact on Cardiac Remodeling. With continued use, ACE inhibitors have a favorable impact on cardiac remodeling. Elevation of kinins is largely responsible. This statement is based in part on the observation that, in experimental models, giving a kinin receptor blocker decreases beneficial effects on remodeling. Also, we know that suppression of angiotensin II production diminishes over time, and so reduced angiotensin II cannot fully explain long-term benefits.
Adverse Effects. The principal adverse effects of the ACE inhibitors are hypotension (secondary to arteriolar dilation), hyperkalemia (secondary to decreased aldosterone release), intractable cough, and angioedema. In addition, these drugs can cause renal failure in patients with bilateral renal artery stenosis. If taken during pregnancy—especially the second and third trimesters—ACE inhibitors can cause fetal injury. Accordingly, if pregnancy occurs, these drugs should be discontinued. Because of their ability to elevate potassium levels, ACE inhibitors should be used with caution in patients taking potassium supplements or a potassium-sparing diuretic (e.g., spironolactone, triamterene).
Dosage. Adequate dosage is still debated in the literature: Higher dosages may be associated with increased survival, but conflicting evidence remains. Results of the Assessment of Treatment with Lisinopril and Survival (ATLAS) trial indicate that the doses needed to increase survival are higher than those needed to produce hemodynamic changes. In contrast to the ATLAS trial, the NETWORK trial and the High Dose Enalapril Study Group did not find a difference in mortality between patients on low-dose or high-dose ACE inhibitors. Treatment with ACE inhibitors should be initiated at low doses and slowly titrated upward if the patient tolerates treatment. Target dosages associated with disease modification are shown in Table 48.1. These dosages should be used unless side effects make them intolerable.
Angiotensin II Receptor BlockersIn patients with HF, the effects of ARBs are similar—but not identical—to those of ACE inhibitors. Hemodynamic effects of both groups are much the same. Clinical trials have shown that ARBs improve LV ejection fraction, reduce HF symptoms, increase exercise tolerance, decrease hospitalization, enhance quality of life, and, most importantly, reduce mortality. However, because ARBs do not increase levels of kinins, their effects on cardiac remodeling are less favorable than those of ACE inhibitors. For this reason, and because clinical experience with ACE inhibitors is much greater than with ARBs, ACE inhibitors are generally preferred. For now, ARBs should be reserved for HF patients who cannot tolerate ACE inhibitors, usually owing to intractable cough. (Because ARBs do not increase bradykinin levels, they do not cause cough.)
Angiotensin Receptor Neprilysin InhibitorSacubitril/Valsartan [Entresto]. Sacubitril/valsartan [En-
tresto] is a newly approved drug that functions in two different manners. Sacubitril is a new class of drug, called an ARNI (angiotensin receptor neprilysin inhibitor). In simple terms, Entresto increases natriuretic peptides while suppressing the negative effects of the RAAS. As discussed earlier in the chapter, ANP and BNP are important indices of cardiac status in HF
RAAS can be highly beneficial. Five groups of drugs are available: ACE inhibitors, ARBs, angiotensin receptor neprilysin inhibitors (ARNIs), direct renin inhibitors (DRIs), and aldo-sterone antagonists. Of the five, the ACE inhibitors have been studied most thoroughly in HF. The basic pharmacology of the RAAS inhibitors is presented in Chapter 44.
Prototype Drugs
DRUGS FOR HEART FAILUREDiureticsHydrochlorothiazideFurosemide
Inhibitors of the Renin-Angiotensin- Aldosterone SystemCaptopril (ACE inhibitor)Losartan (angiotensin II receptor blocker)Entresto (angiotensin receptor neprilysin inhibitor)Eplerenone (aldosterone antagonist)
Beta BlockersMetoprolol
Inotropic AgentsDigoxin (a cardiac glycoside)Dopamine (a sympathomimetic)
VasodilatorsIsosorbide dinitrate plus hydralazine
ACE InhibitorsACE inhibitors (e.g., captopril, enalapril) are a cornerstone of HF therapy. These drugs can improve functional status and prolong life. In one trial, the 2-year mortality rate for patients taking enalapril was 47% lower than the rate for patients taking placebo. Other large, controlled trials have shown similar benefits. Accordingly, in the absence of specific contraindica-tions, all patients with HF should receive one of these drugs. Although ACE inhibitors can be used alone, they are usually combined with a beta blocker and a diuretic.
ACE inhibitors help by blocking the production of angio-tensin II, decreasing the release of aldosterone, and suppressing the degradation of kinins. As a result, they improve hemodynam-ics and favorably alter cardiac remodeling.
Hemodynamic Benefits. By suppressing production of angiotensin II, ACE inhibitors cause dilation of arterioles and veins, and they decrease release of aldosterone. Resulting benefits in HF are as follows:
• Arteriolar dilation improves regional blood flow in the kidneys and other tissues. By reducing afterload, it increases stroke volume and cardiac output. Increased renal blood flow promotes excretion of sodium and water.
• Venous dilation reduces venous pressure and thereby reduces pulmonary congestion, peripheral edema, preload, and cardiac dilation.
• Suppression of aldosterone release enhances excretion of sodium and water, while causing retention of potassium.

534
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
In the past, researchers believed that aldosterone’s only action was to promote renal retention of sodium (and water) in exchange for excretion of potassium. However, we now know that aldosterone has additional—and more harmful—effects. Among these are:
• Promotion of myocardial remodeling (which impairs pumping)
• Promotion of myocardial fibrosis (which increases the risk of dysrhythmias)
• Activation of the SNS and suppression of norepinephrine uptake in the heart (both of which can promote dys-rhythmias and ischemia)
• Promotion of vascular fibrosis (which decreases arterial compliance)
• Promotion of baroreceptor dysfunction
During HF, activation of the RAAS causes levels of aldosterone to rise. In some patients, levels reach 20 times normal. As aldosterone levels grow higher, harmful effects increase, and prognosis becomes progressively worse.
Drugs can reduce the impact of aldosterone by either decreasing aldosterone production or blocking aldosterone receptors. ACE inhibitors, ARBs, and DRIs decrease aldo-sterone production; spironolactone and eplerenone block aldosterone receptors. Although ACE inhibitors and ARBs can reduce aldosterone production, they do not block it entirely. Furthermore, production is suppressed for only a relatively short time. Hence, when ACE inhibitors or ARBs are used alone, detrimental effects of aldosterone can persist. However, when an aldosterone antagonist is added to the regimen, any residual effects are eliminated. As a result, symptoms of HF are improved and life is prolonged.
Aldosterone antagonists have one major adverse effect: hyperkalemia. The underlying cause is renal retention of potas-sium. Risk is increased by renal impairment and by using an ACE inhibitor or ARB. To minimize risk, potassium levels and renal function should be measured at baseline and periodically thereafter. Potassium supplements should be discontinued.
Spironolactone—but not eplerenone—poses a significant risk of gynecomastia (breast enlargement) in men, a condition that can be both cosmetically troublesome and painful. In the RALES trial, 10% of males experienced painful breast enlargement.
Direct Renin InhibitorsAs discussed in Chapter 44, DRIs can shut down the entire RAAS. In theory, their benefits in HF should equal those of the ACE inhibitors and ARBs. At this time, only one DRI—aliskiren [Tekturna]—is available. In recent trials, aliskiren did not improve outcomes in hospitalized patients with heart failure. Because of these findings, aliskiren is approved for hypertension, but is not approved for HF.
Beta BlockersThe role of beta blockers in HF continues to evolve. Previously, HF was considered an absolute contraindication to these drugs. After all, blockade of cardiac beta1-adrenergic receptors reduces contractility—an effect that is clearly detrimental, given that contractility is already compromised in the failing heart. However, it is now clear that with careful control of dosage, beta blockers can improve patient status. Controlled trials have
patients. Entresto is approved for patients with Class II–IV HF to be used in place of an ACE inhibitor or ARB.
In the PARADIGM-HF study, Entresto was superior to enalapril alone when looking at the overall endpoints of reduc-tion in hospitalizations, risk of all-cause mortality, and risk of death from cardiovascular causes. In fact, the study was ter-minated early secondary to the overwhelmingly positive results.
As Entresto contains an ARB, the contraindications and side effects are similar. Entresto can cause angioedema, hyperkalemia, and hypotension. Administration should be avoided in pregnancy, as its use can cause fetal harm.
Aldosterone AntagonistsIn patients with HF, aldosterone antagonists—spironolactone [Aldactone] and eplerenone [Inspra]—can reduce symptoms, decrease hospitalizations, and prolong life. These benefits were first demonstrated with spironolactone in the Randomized Aldactone Evaluation Study (RALES). Similar results were later obtained with eplerenone. Current guidelines recommend adding an aldosterone antagonist to standard HF therapy, but only in patients with persistent symptoms despite adequate treatment with an ACE inhibitor and a beta blocker.
Aldosterone antagonists work primarily by blocking aldo-sterone receptors in the heart and blood vessels. To understand these effects, we need to review the role of aldosterone in HF.
Drug Initial Daily DoseMaximum Daily Dose
ACE INHIBITORS
Captopril [Capoten] 6.25 mg 3 times 150 mg 3 timesEnalapril [Vasotec] 2.5 mg twice 10–20 mg twiceFosinopril [generic
only]10 mg once 80 mg once
Lisinopril [Zestril, Prinivil]
2.5–5 mg once 20–40 mg once
Quinapril [Accupril]
5 mg once 20 mg twice
Ramipril [Altace] 2.5 mg twice 5 mg twiceTrandolapril
[Mavik]1 mg once 4 mg once
ANGIOTENSIN II RECEPTOR BLOCKERS
Candesartan [Atacand]
4 mg once 32 mg once
Losartan [Cozaar] 25–50 mg once 50–100 mg onceValsartan [Diovan] 40 mg twice 160 mg twice
ANGIOTENSIN RECEPTOR NEPRILYSIN INHIBITOR
Sacubitril/Valsartan [Entresto]
24/26 mg twice 97/103 mg twice
ALDOSTERONE ANTAGONISTS
Eplerenone [Inspra] 25 mg once 50 mg onceSpironolactone
[Aldactone]25 mg once 25 mg once or
twice
TABLE 48.1 ■ Inhibitors of the Renin-Angiotensin-Aldosterone System Used in Heart Failure

535
CHAPTER 48 Drugs for Heart Failure
Sympathomimetic Drugs: Dopamine and Dobutamine. The basic pharmacology of dopamine and dobutamine is presented in Chapter 17. Discus-sion here is limited to their use in HF. Both drugs are administered by IV infusion.
Dopamine. Dopamine is a catecholamine that can activate (1) beta1-adrenergic receptors in the heart, (2) dopamine receptors in the kidney, and (3) at high doses, alpha1-adrenergic receptors in blood vessels. Activation of beta1 receptors increases myocardial contractility, thereby improving cardiac performance. Beta1 activation also increases heart rate, creating a risk of tachycardia. Activation of dopamine receptors dilates renal blood vessels, thereby increasing renal blood flow and urine output. Activation of alpha1 receptors increases vascular resistance (afterload) and can thereby reduce cardiac output. Dopamine is administered by continuous infusion. Constant monitoring of blood pressure, the electrocardiogram (ECG), and urine output is required. Dopamine is employed as a short-term rescue measure for patients with severe acute cardiac failure.
Dobutamine. Dobutamine is a synthetic catecholamine that causes selective activation of beta1-adrenergic receptors. By doing so, the drug can increase myocardial contractility and can thereby improve cardiac performance. Like dopamine, dobutamine can cause tachycardia and induce myocardial ischemia. In contrast to dopamine, dobutamine does not activate alpha1 receptors and therefore does not increase vascular resistance. As a result, the drug is generally preferred to dopamine for short-term treatment of acute HF. Administration is by continuous infusion.
Phosphodiesterase InhibitorsMilrinone. Milrinone has been called an inodilator because it increases
myocardial contractility and promotes vasodilation. Increased contractility results from accumulation of cyclic AMP (cAMP) secondary to inhibition of phosphodiesterase type 3 (PDE3), an enzyme that degrades cAMP. Milrinone is administered by IV infusion and is indicated only for short-term therapy of severe HF. The initial dose is 25 to 75 mcg/kg over 10 to 20 minutes. The maintenance infusion is 0.375 to 0.75 mcg/kg/min. Use should be reserved for patients with severe reduction in cardiac output resulting in decreased organ perfusion, as inotropes can induce dysrhythmias and cause myocardial ischemia from increased metabolic demand.
Vasodilators (Other Than ACE Inhibitors and ARBs)Isosorbide Dinitrate Plus HydralazineFor treatment of HF, isosorbide dinitrate (ISDN) and hydralazine are usually combined. The combination represents an alternative to ACE inhibitors or ARBs. However, ACE inhibitors and ARBs are generally preferred.
Isosorbide dinitrate [Isordil Titradose] belongs to the same family as nitroglycerin. Like nitroglycerin, ISDN causes selective dilation of veins. In patients with severe refractory HF, the drug can reduce congestive symptoms and improve exercise capacity. In addition to its hemodynamic actions, ISDN may inhibit abnormal myocyte growth and thus may delay cardiac remodeling. Principal adverse effects are orthostatic hypotension and reflex tachycardia. The basic pharmacology of ISDN and other organic nitrates is discussed in Chapter 51.
Hydralazine causes selective dilation of arterioles. By doing so, the drug can improve cardiac output and renal blood flow. For treatment of HF, hydralazine is always used in combination with ISDN, since hydralazine by itself is not very effective. Principal adverse effects are hypotension, tachycardia, and a syndrome that resembles systemic lupus erythematosus. The basic pharmacology of hydralazine is discussed in Chapter 46.
BiDil, a fixed-dose combination of hydralazine and isosorbide dinitrate, is indicated for treating HF—but only in African Americans, making BiDil the first medication approved for a specific ethnic group. Can BiDil help people in other ethnic groups? Probably, but data are lacking: The manufacturer tested the product only in African Americans. As discussed in Chapter 8, testing was limited primarily because of regulatory and market incentives, not because there were data suggesting it wouldn’t work for others. Of course, now that BiDil is approved, clinicians may prescribe it for anyone they see fit. Each BiDil tablet contains 37.5 mg hydralazine and 20 mg isosorbide dinitrate. The recommended dosage is 1 or 2 tablets 3 times a day.
Intravenous Vasodilators for Acute CareNitroglycerin. Intravenous nitroglycerin is a powerful venodilator that
produces a dramatic reduction in venous pressure. Effects have been described as being equivalent to “pharmacologic phlebotomy.” In HF, nitroglycerin is used to relieve acute severe pulmonary edema. Principal adverse effects are hypotension and resultant reflex tachycardia. The basic pharmacology of nitroglycerin is discussed in Chapter 51.
shown that three beta blockers—carvedilol [Coreg], bisoprolol [Zebeta], and sustained-release metoprolol [Toprol XL]—when added to conventional therapy, can improve LV ejection fraction, increase exercise tolerance, slow progression of HF, reduce the need for hospitalization, and, most importantly, prolong survival. Accordingly, beta blockers are now recommended as first-line therapy for most patients. These drugs can even be used in patients with severe disease (NYHA Class IV), provided the patient is euvolemic and hemodynamically stable. Although the mechanism underlying benefits is uncertain, likely possibilities include protecting the heart from excessive sympathetic stimulation and protecting against dysrhythmias. Because excessive beta blockade can reduce contractility, doses must be very low initially and then gradually increased. Full benefits may not be seen for 1 to 3 months. Among patients with HF, the principal adverse effects are (1) fluid retention and worsening of HF, (2) fatigue, (3) hypotension, and (4) bradycardia or heart block. The basic pharmacology of the beta blockers is discussed in Chapter 18.
Ivabradine (Corlanor)In 2015, the FDA approved ivabradine [Corlanor] for use in patients with stable, symptomatic heart failure and LVEF <35% in sinus rhythm with heart rates >70 BPM on maximally toler-ated doses of beta blockers. It may also be used in patients who have a contraindication to beta blocker use.
Ivabradine causes a dose-dependent reduction in heart rate by blocking channels responsible for cardiac pacemaker current. Although the drug slows heart rate, it does not possess negative inotropic effects or cause QTc prolongation. When used in recommended doses, heart rate reduction is approximately 10 beats/min.
The SHIFT (Systolic Heart failure treatment with the If inhibitor ivabradine Trial) study was a randomized, double-blind trial comparing ivabradine to placebo in 6558 adults. End results revealed that the use of ivabradine reduced the risk of hospitalization for worsening heart failure or cardiovascular death. Given these findings, ivabradine may be a useful alterna-tive for patients with heart failure who need additional beta blockade above what is currently available.
Inotropic AgentsDigoxinDigoxin belongs to a class of drugs known as cardiac glycosides, agents best known for their positive inotropic actions, that is, their ability to increase myocardial contractile force. By increasing contractile force, digoxin can increase cardiac output. In addition, it can alter the electrical activity of the heart, and it can favorably affect neurohormonal systems. Unfortunately, although digoxin can reduce symptoms of HF, it does not prolong life. Used widely in the past, digoxin is considered a second-line agent today. The pharmacology of digoxin is discussed later.
Inotropic Agents (Other Than Digoxin)In addition to digoxin, we have two other types of inotropic drugs: sympatho-mimetics and phosphodiesterase (PDE) inhibitors. Unlike digoxin, which can be taken orally, these other inotropics must be given by IV infusion. Accordingly, their use is restricted to acute care of hospitalized patients. Because digoxin can be given PO, it is the only inotropic agent suited for long-term therapy.

536
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Digitalis is a dangerous drug because it can cause severe dysrhythmias at doses close to the therapeutic range. Owing to its prodysrhythmic actions, digoxin must be used with respect, caution, and skill.
Digoxin is indicated for HF and for control of dysrhythmias (see Chapter 49). When used for HF, digoxin can reduce symptoms, increase exercise tolerance, and decrease hospitaliza-tions. However, the drug does not prolong life. Furthermore, when used by women, it may actually shorten life (Box 48.1). Because benefits are limited to symptomatic relief and because the risk of toxicity is substantial, digoxin is now considered a second-line drug for treating HF.
ChemistryDigoxin consists of three components: a steroid nucleus, a lactone ring, and three molecules of digitoxose (a sugar). It is because of the sugars that digoxin is known as a glycoside. The region of the molecule composed of the steroid nucleus plus the lactone ring (i.e., the region without the sugar molecules) is responsible for the pharmacologic effects of digoxin. The sugars only increase solubility.
Mechanical Effects on the HeartDigoxin exerts a positive inotropic action on the heart. That is, the drug increases the force of ventricular contraction and can thereby increase cardiac output.
Mechanism of Inotropic ActionDigoxin increases myocardial contractility by inhibiting an enzyme known as sodium, potassium-ATPase (Na+/K+-ATPase). By way of an indirect process described in the paragraphs that follow, inhibition of Na+/K+-ATPase promotes calcium accu-mulation within myocytes. The calcium then augments con-tractile force by facilitating the interaction of myocardial contractile proteins: actin and myosin.
To understand how inhibition of Na+/K+-ATPase causes intracellular calcium to rise, we must first understand the normal role of Na+/K+-ATPase in myocytes. That role is shown in Fig. 48.4. As indicated, when an action potential passes along the myocyte membrane (sarcolemma), Na+ ions and Ca++ ions enter the cell, and K+ ions exit. Once the action potential has passed, these ion fluxes must be reversed so that the original ionic balance of the cell can be restored. Na+/K+-ATPase is critical to this process. Na+/K+-ATPase acts as a “pump” to draw extracellular K+ ions into the cell, while simultaneously extruding intracellular Na+. The energy required for pumping Na+ and K+ is provided by the breakdown of ATP—hence the name Na+/K+-ATPase. To complete the normalization of cellular ionic composition, Ca++ ions must leave the cell. Extrusion of Ca++ is accomplished through an exchange process in which extracellular Na+ ions are taken into the cell while Ca++ ions exit. This exchange of Na+ for Ca++ is a passive (energy-independent) process.
We can now answer the question, how does inhibition of Na+/K+-ATPase increase intracellular Ca++? By inhibiting Na+/K+-ATPase, digoxin prevents the myocyte from restoring its proper ionic composition following the passage of an action potential. Inhibition of Na+/K+-ATPase blocks uptake of K+ and extrusion of Na+. Therefore, with each successive action potential, intracellular K+ levels decline and intracellular Na+ levels rise. It is this rise in Na+ that leads to the rise in intracellular Ca++. In the presence of excess intracellular Na+, further Na+ entry is suppressed. Since Na+ entry is suppressed, the passive exchange of Ca++ for Na+ cannot take place, and so Ca++ accumulates within the cell.
Relationship of Potassium to Inotropic ActionPotassium ions compete with digoxin for binding to Na+/K+-ATPase. This competition is of great clinical significance. Because potassium competes with digoxin, when potassium levels are low, binding of digoxin to Na+/K+-ATPase increases. This increase can produce excessive inhibition of Na+/K+-ATPase
Sodium Nitroprusside. Sodium nitroprusside [Nitropress] acts rapidly to dilate arterioles and veins. Arteriolar dilation reduces afterload and thereby increases cardiac output. Venodilation reduces venous pressure and thereby decreases pulmonary and peripheral congestion. The drug is indicated for short-term therapy of severe refractory HF. The principal adverse effect is profound hypotension. Blood pressure must be monitored continuously. The basic pharmacology of nitroprusside is discussed in Chapter 46.
Nesiritide. Nesiritide [Natrecor] is a synthetic form of human BNP indicated only for short-term IV therapy of hospitalized patients with acutely decompensated HF, characterized by increased pulmonary capillary wedge pressure (PCWP) and dyspnea at rest. Nesiritide is produced by recombinant DNA technology and has the same amino acid sequence as naturally occurring BNP. The drug was approved after a relatively small trial—Vasodilation in the Management of Acute Congestive Heart Failure (VMAC)—and showed a modest decrease in dyspnea and PCWP. However, a much larger trial—Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF)—failed to show any benefit: The incidence of dyspnea, rehospitalization, and 30-day mortality was the same for patients receiving nesiritide as it was for patients receiving placebo. Worse yet, although nesiritide offered no benefit, it nearly doubled the incidence of hypotension. These results led the authors to conclude that “Nesiritide cannot be recommended for routine use in the broad population of patients with heart failure.”
Mechanism of Action. Nesiritide affects hemodynamics by three mechanisms: suppression of the RAAS, suppression of sympathetic outflow from the central nervous system (CNS), and direct dilation of arterioles and veins. In patients with HF, benefits derive primarily from direct vasodilation. To promote vasodilation, nesiritide binds with receptors on vascular smooth muscle (VSM), and thereby stimulates production of cyclic GMP (cGMP), a second messenger that causes VSM to relax. This mechanism is similar to that of nitroglycerin, which also stimulates cGMP production. However, whereas nitroglycerin acts primarily on veins, nesiritide dilates arterioles as well. By dilating arterioles and veins, nesiritide reduces both preload and afterload. The net result is a decrease in PCWP and increased cardiac output. Also, by dilating afferent renal arterioles, nesiritide increases GFR, and thereby increases excretion of sodium and water. The result is a reduction in blood volume, which further reduces cardiac preload.
Pharmacokinetics. With continuous infusion, nesiritide achieves steady-state levels that are 3 to 6 times greater than the level of endogenous BNP present at baseline. Nesiritide is eliminated by three mechanisms: (1) proteolytic cleavage by endopeptidases present on the luminal surface of blood vessels; (2) binding to clearance receptors on the surface of cells, followed by cellular uptake and proteolytic cleavage; and (3) renal filtration. The drug’s half-life is short, about 18 minutes.
Adverse Effects. The principal adverse effect is symptomatic hypotension. In the ASCEND-HF trial, hypotension developed in 26.6% of patients receiving nesiritide, compared with 15.3% of those receiving placebo. The risk of hypotension is increased by high doses of nesiritide and by concurrent use of ACE inhibitors and other vasodilators. In addition to causing hypotension, nesiritide can cause ventricular tachycardia, headache, back pain, dizziness, and nausea. An analysis of several clinical trials suggested that nesiritide may cause renal damage. However, ASCEND-HF revealed no evidence of renal harm.
Preparations, Dosage, and Administration. Nesiritide [Natrecor] is available in 1.5-mg, single-use vials. The powder must be dissolved and then diluted to a final concentration of 6 mcg/mL. Dosing consists of an initial IV bolus (2 mcg/kg) followed by continuous infusion (0.01 mcg/kg/min), typically lasting 48 hours or less. If symptomatic hypotension develops, the infusion should be slowed or stopped.
DIGOXIN, A CARDIAC GLYCOSIDE
Digoxin [Lanoxin] belongs to a family of drugs known as cardiac glycosides. These drugs are prepared by extraction from Digitalis purpurea (purple foxglove) and Digitalis lanata (Grecian foxglove), and so are also known as digitalis glyco-sides. In the United States, digoxin is the only cardiac glycoside available.
Digoxin has profound effects on the mechanical and electrical properties of the heart. In addition, it has important neurohor-monal effects. In patients with HF, benefits derive from increased myocardial contractility and from effects on neurohormonal systems as well.

537
CHAPTER 48 Drugs for Heart Failure
�� �
�� � �
� �
Na�, K�-ATPase
Fig. 48.4 ■ Ion fluxes across the cardiac cell membrane. During the action potential, Na+ and Ca++ enter the cardiac cell and K+ exits. Following the action potential, Na+/K+-ATPase pumps Na+ out of the cell and takes up K+. Ca++ leaves the cell in exchange for the uptake of Na+. By inhibiting Na+/K+-ATPase, digoxin prevents the extrusion of Na+, causing Na+ to accumulate inside the cell. The resulting buildup of intracellular Na+ suppresses the Na+-Ca++ exchange process, thereby causing intracellular levels of Ca++ to rise.
BOX 48.1 ■ SPECIAL INTEREST TOPIC
ATTENTION: DIGOXIN MAY BE HAZARDOUS TO WOMEN’S HEALTH
Researchers who conducted a reanalysis of older data discovered that for women with heart failure digoxin may do more harm than good. The Digitalis Investigation Groupa (DIG) reported the results of a large randomized, placebo-controlled trial designed to assess the impact of digoxin on morbidity and mortality in patients with heart failure. The study enrolled 6801 patients (men and women) with heart failure and followed them for an average of 37 months. They all took an ACE inhibitor and a diuretic; half also received digoxin, and the other half received a placebo. The result? Digoxin improved symptoms and decreased hospitalizations, but did not reduce mortality. The overall death rate was 35%, regardless of whether patients took digoxin or placebo. However, the data were not analyzed for possible gender-related effects. Accordingly, Rathore et al.b performed a retrospective analysis of the DIG data to determine whether digoxin had different effects in men and women. Among men, digoxin had no significant impact on mortality, mirroring the overall mortality seen previously. However, among women, digoxin produced a small but significant increase in mortality: After 37 months, the death rate was 28.9% for women taking placebo compared with 33.1% for those taking digoxin—an increase of 4.2%.
Although the Rathore et al. study suggests that for female patients the benefits of digoxin therapy (primarily a small [4%]
decrease in the risk of hospitalization) may not justify the risk of possible drug-induced death, a further study including over 35,000 patients demonstrated no difference in mortality between men and women taking digoxin for the treatment of heart failure.c
Why did digoxin possibly increase the mortality rate in women in the Rathore et al. study? We don’t know. Possibilities include sex-based differences in autonomic function, muscle metabolism, signal transduction, or myocardial cell growth and function. However, there may be a simpler answer: Among the women who died, digoxin plasma levels may have been excessive. It is well established that digoxin can be lethal at high levels. In the DIG trial, digoxin levels were measured only in randomly selected patients, and so Rathore et al. lacked the data needed to determine whether deaths were related to high drug levels. If high digoxin levels were indeed responsible for the observed mortality increase, then the take-home message is obvious: We must keep digoxin doses low.
As a rule, the drug should be reserved for patients who have not responded adequately to first-line medicines: ACE inhibitors or ARBs, diuretics, and beta blockers. Furthermore, digoxin levels should be kept as low as possible (0.5 to 0.8 ng/mL is a reasonable initial target).
aThe Digitalis Investigation Group: The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 336:525–533, 1997.bRathore SS, Wang Y, Krumholz H: Sex-based differences in the effect of digoxin for the treatment of heart failure. N Engl J Med 347:1403–1411, 2002.cFlory JH, Ky B, Haynes K, et al: Observational cohort study of the safety of digoxin use in women with heart failure. BMJ Open 2:e000888:1–7, 2012.

538
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
can produce substantial improvement in HF symptoms, it does not prolong life.
Neurohormonal Benefits in Heart FailureAt dosages below those needed for positive inotropic effects, digoxin can modulate the activity of neurohormonal systems. The underlying mechanism is inhibition of Na+/K+-ATPase.
In the kidney, digoxin can suppress renin release. By inhibit-ing Na+/K+-ATPase in renal tubules, digoxin decreases tubular absorption of sodium. As a result, less sodium is presented to the distal tubule, and so renin release is suppressed.
Through effects on the vagus nerve, digoxin can decrease sympathetic outflow from the CNS. Specifically, by inhibiting Na+/K+-ATPase in vagal afferent fibers, digoxin increases the sensitivity of cardiac baroreceptors. As a result, these receptors discharge more readily, thereby signaling the CNS to reduce sympathetic traffic to the periphery.
How important are these effects on renin and sympathetic tone? No one knows for sure. However, they are probably just as important as inotropic effects, and perhaps even more important.
Electrical Effects on the HeartThe effects of digoxin on the electrical activity of the heart are of therapeutic and toxicologic importance. It is because of its electrical effects that digoxin is useful for treating dys-rhythmias (see Chapter 49). Ironically, these same electrical effects are responsible for causing dysrhythmias, the most serious adverse effect of digoxin.
The electrical effects of digoxin can be bewildering in their complexity. Through a combination of actions, digoxin can alter the electrical activity in noncontractile tissue (sinoatrial [SA] node, atrioventricular [AV] node, Purkinje fibers) as well as in ventricular muscle. In these various regions, digoxin can alter automaticity, refractoriness, and impulse conduction. Whether these parameters are increased or decreased depends on cardiac status, digoxin dosage, and the region involved.
Although the electrical effects of digoxin are many and varied, only a few are clinically significant. These are discussed in the sections that follow.
Mechanisms for Altering Electrical Activity of the HeartDigoxin alters the electrical properties of the heart by inhibiting Na+/K+-ATPase and by enhancing vagal influences on the heart. By inhibiting Na+/K+-ATPase, digoxin alters the distribution of ions (Na+, K+, Ca++) across the cardiac cell membrane. This change in ion distribution can alter the electrical responsiveness of the cells involved. Since hypokalemia intensifies inhibition of Na+/K+-ATPase, hypokalemia intensifies alterations in cardiac electrical properties.
Digoxin acts in two ways to enhance vagal effects on the heart. First, the drug acts in the CNS to increase the firing rate of vagal fibers that innervate the heart. Second, digoxin increases the responsiveness of the SA node to acetylcholine (the neurotransmitter released by the vagus). The net result of these vagotonic effects is (1) decreased automaticity of the SA node and (2) decreased conduction through the AV node.
Effects on Specific Regions of the HeartIn the SA node, digoxin decreases automaticity (by the vagotonic mechanisms just mentioned). In the AV node, digoxin decreases conduction velocity and prolongs the effective refractory period. These effects, which can promote varying degrees of AV block, result primarily from the drug’s vagotonic actions. In Purkinje fibers, digoxin-induced inhibition of Na+/K+-ATPase results in increased automaticity; this increase can generate ectopic foci that, in turn, can cause ventricular dysrhythmias. In the ventricular myocardium, digoxin acts to shorten the effective refractory period and (possibly) increase automaticity.
with resultant toxicity. Conversely, when levels of potassium are high, inhibition of Na+/K+-ATPase by digoxin is reduced, causing a reduction in the therapeutic response. Because an increase in potassium can impair therapeutic responses, whereas a decrease in potassium can cause toxicity, it is imperative that potassium levels be kept within the normal physiologic range: 3.5 to 5 mEq/L.
Hemodynamic Benefits in Heart FailureIncreased Cardiac OutputIn patients with HF, increased myocardial contractility increases cardiac output. By increasing contractility, digoxin shifts the relationship of fiber length to stroke volume in the failing heart toward that in the healthy heart. Consequently, at any given heart size, the stroke volume of the failing heart increases, causing cardiac output to rise.
Consequences of Increased Cardiac OutputAs a result of increased cardiac output, three major second-ary responses occur: (1) sympathetic tone declines, (2) urine production increases, and (3) renin release declines. These responses can reverse virtually all signs and symptoms of HF. However, they do not correct the underlying problem of cardiac remodeling.
Decreased Sympathetic Tone. By increasing contractile force and cardiac output, digoxin increases arterial pressure. In response, sympathetic nerve traffic to the heart and blood vessels is reduced via the baroreceptor reflex. (Recall that a compensatory increase in sympathetic tone had taken place because of HF.)
The decrease in sympathetic tone has several beneficial effects. First, heart rate is reduced, thereby allowing more complete ventricular filling. Second, afterload is reduced (because of reduced arteriolar constriction), thereby allowing more complete ventricular emptying. Third, venous pressure is reduced (because of reduced venous constriction), thereby reducing cardiac distention, pulmonary congestion, and peripheral edema.
Increased Urine Production. The increase in cardiac output increases renal blood flow and thereby increases produc-tion of urine. The resultant loss of water reduces blood volume, which in turn reduces cardiac distention, pulmonary congestion, and peripheral edema.
Decreased Renin Release. In response to increased arterial pressure, renin release declines, causing levels of aldosterone and angiotensin II to decline as well. The decrease in angiotensin II decreases vasoconstriction, thereby further reducing afterload and venous pressure. The decrease in aldosterone reduces retention of sodium and water, which reduces blood volume, which in turn further reduces venous pressure.
Summary of Hemodynamic EffectsWe can see that, through direct and indirect mechanisms, digoxin has the potential to reverse all of the overt manifestations of HF: cardiac output improves, heart rate decreases, heart size declines, constriction of arterioles and veins decreases, water retention reverses, blood volume declines, peripheral and pulmonary edema decrease, and water weight is lost. In addition, exercise tolerance improves and fatigue is reduced. There is, however, one important caveat: Although digoxin

539
CHAPTER 48 Drugs for Heart Failure
can precipitate serious dysrhythmias in patients with HF. The probability and severity of a dysrhythmia are directly related to the severity of the underlying disease. Since heart disease is the reason for taking digoxin, it should be no surprise that people taking the drug are at risk of dysrhythmias.
Diagnosing Digoxin-Induced DysrhythmiasDiagnosis is not easy, largely because the failing heart is prone to spontaneous dysrhythmias. Hence, when a dysrhythmia occurs, we cannot simply assume that digoxin is the cause: The possibility that the dysrhythmia is the direct result of heart disease must be considered. Compounding diagnostic difficulties is the poor correlation between plasma digoxin levels and dysrhythmia onset. Because of this loose association, the presence of an apparently excessive digoxin level does not necessarily indicate that digoxin is responsible for the problem. Laboratory data required for diagnosis include digoxin level, serum electrolytes, and an ECG. Ultimately, diagnosis is based on experience and clinical judgment. Resolution of the dysrhythmia following digoxin withdrawal confirms the diagnosis.
Managing Digoxin-Induced DysrhythmiasWith proper treatment, digoxin-induced dysrhythmias can almost always be controlled. Basic management measures are as follows:
• Withdraw digoxin and potassium-wasting diuretics. For many patients, no additional treatment is needed. To help ensure that medication is stopped, a written order to withhold digoxin should be made.
• Monitor serum potassium. If the potassium level is low or nearly normal, potassium (IV or PO) should be administered. Potassium displaces digoxin from Na+/K+- ATPase and thereby helps reverse toxicity. However, if potassium levels are high or if AV block is present, no more potassium should be given. Under these conditions, more potassium may cause complete AV block.
• Some patients may require an antidysrhythmic drug. Phenytoin and lidocaine are most effective. Quinidine, another antidysrhythmic drug, can cause plasma levels of digoxin to rise, and so should not be used.
• Patients who develop bradycardia or AV block can be treated with atropine. (Atropine blocks the vagal influ-ences that underlie bradycardia and AV block.) Alterna-tively, electronic pacing may be employed.
• When overdose is especially severe, digoxin levels can be lowered using Fab antibody fragments [Digifab]. Following IV administration, these fragments bind digoxin and thereby prevent it from acting. Treatment is expen-sive: A full neutralizing dose costs $3000 to $4000. Cholestyramine and activated charcoal, agents that also bind digoxin, can be administered orally to suppress absorption of digoxin from the GI tract.
Adverse Effects II: Noncardiac Adverse EffectsThe principal noncardiac toxicities of digoxin concern the GI system and the CNS. Since adverse effects on these systems frequently precede development of dysrhythmias, symptoms involving the GI tract and CNS can provide advance warning
Adverse Effects I: Cardiac DysrhythmiasDysrhythmias are the most serious adverse effect of digoxin. They result from altering the electrical properties of the heart. Fortunately, when used in the dosages recommended today, dysrhythmias are uncommon.
What kinds of dysrhythmias can occur? Digoxin can mimic practically all types of dysrhythmias. Atrioventricular block with escape beats is among the most common. Ventricular flutter and ventricular fibrillation are the most dangerous.
Because serious dysrhythmias are a potential consequence of therapy, all patients should be evaluated frequently for changes in heart rate and rhythm. If significant changes occur, digoxin should be withheld and the prescriber consulted. Outpatients should be taught to monitor their pulses and instructed to report any significant changes in rate or regularity.Mechanism of Ventricular Dysrhythmia GenerationDigoxin-induced ventricular dysrhythmias result from a combination of four factors:
• Decreased automaticity of the SA node• Decreased impulse conduction through the AV node• Spontaneous discharge of Purkinje fibers (caused in part by increased
automaticity)• Shortening of the effective refractory period in ventricular muscle
Increased Purkinje fiber discharge and shortening of the ventricular effective refractory period predispose the ventricles to developing ectopic beats. Potential ectopic beats become manifest because the effects of digoxin on the SA and AV nodes decrease the ability of the normal pacemaker to drive the ventricles, allowing ventricular ectopic beats to take over.
Predisposing FactorsHypokalemia. The most common cause of dysrhythmias
in patients receiving digoxin is hypokalemia secondary to the use of diuretics. Less common causes include vomiting and diarrhea. Hypokalemia promotes dysrhythmias by increasing digoxin-induced inhibition of Na+/K+-ATPase, which in turn leads to increased automaticity of Purkinje fibers. Because low potassium can precipitate dysrhythmias, it is imperative that serum potassium levels be kept within the normal range. If diuretic therapy causes potassium levels to fall, a potassium-sparing diuretic (e.g., spironolactone) can be prescribed to correct the problem. Potassium supplements may be used too. Patients should be taught to recognize symptoms of hypokalemia (e.g., muscle weakness) and instructed to notify the prescriber if these develop.
Elevated Digoxin Levels. Digoxin has a narrow thera-peutic range: Drug levels only slightly higher than the thera-peutic range greatly increase the risk of toxicity. Possible causes of excessive digoxin levels include (1) intentional or accidental overdose, (2) increased digoxin absorption, and (3) decreased digoxin elimination.
If digoxin levels are kept within the optimal therapeutic range—now considered to be 0.5 to 0.8 ng/mL—the chances of a dysrhythmia will be reduced. However, it is important to note that careful control over drug levels does not eliminate the risk. As already discussed, there is only a loose relationship between digoxin levels and clinical effects. As a result, some patients may experience dysrhythmias even when drug levels are within what is normally considered a safe range.
Heart Disease. The ability of digoxin to cause dysrhythmias is greatly increased by the presence of heart disease. Doses of digoxin that have no adverse effects on healthy volunteers

540
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
VerapamilVerapamil, a calcium channel blocker, can significantly increase plasma levels of digoxin. If the combination is employed, digoxin dosage must be reduced. In addition, verapamil can suppress myocardial contractility and can thereby counteract the benefits of digoxin.
PharmacokineticsAbsorptionAbsorption with digoxin tablets is variable, ranging between 60% and 80%, and can be decreased by certain foods and drugs. Meals high in bran can decrease absorption significantly, as can cholestyramine, kaolin-pectin, and certain other drugs (see Table 48.2). Of note, taking digoxin with meals decreases the rate of absorption but not the extent.
In the past, there was considerable variability in the absorp-tion of digoxin from tablets prepared by different manufacturers. This variability resulted from differences in the rate and extent of tablet dissolution. Because of this variable bioavailability, it had been recommended that patients not switch between different digoxin brands. Today, bioavailability of digoxin in tablets produced by different companies is fairly uniform, making brands of digoxin more interchangeable than in the
of more serious toxicity. Accordingly, patients should be taught to recognize these effects and instructed to notify the prescriber if they occur.
Anorexia, nausea, and vomiting are the most common GI side effects. These responses result primarily from stimulation of the chemoreceptor trigger zone of the medulla. Digoxin rarely causes diarrhea.
Fatigue is the most frequent CNS effect. Visual disturbances (e.g., blurred vision, yellow tinge to vision, appearance of halos around dark objects) are also relatively common.
Adverse Effects III: Measures to Reduce Adverse EffectsPatient education can help reduce the incidence of toxicity. Patients should be warned about digoxin-induced dysrhythmias and instructed to take their medication exactly as prescribed. In addition, they should be informed about symptoms of developing toxicity (altered heart rate or rhythm, visual or GI disturbances) and instructed to notify the prescriber if these develop. If a potassium supplement or potassium-sparing diuretic is part of the regimen, it should be taken exactly as ordered.
Drug InteractionsDigoxin is subject to a large number of significant drug interac-tions. Some are pharmacodynamic and some are pharmaco-kinetic. Several important interactions are discussed in the sections that follow. Interactions are shown in Table 48.2.
DiureticsThiazide diuretics and loop diuretics promote loss of potassium and thereby increase the risk of digoxin-induced dysrhythmias. Accordingly, when digoxin and these diuretics are used concur-rently, serum potassium levels must be monitored and maintained within the normal range (3.5 to 5 mEq/L). If hypokalemia develops, potassium levels can be restored with potassium supplements, a potassium-sparing diuretic, or both.
ACE Inhibitors and ARBsThese drugs can increase potassium levels and can thereby decrease therapeutic responses to digoxin. Exercise caution if an ACE inhibitor or ARB is combined with potassium supple-ments or a potassium-sparing diuretic.
SympathomimeticsSympathomimetic drugs (e.g., dopamine, dobutamine) act on the heart to increase the rate and force of contraction. The increase in contractile force can add to the positive inotropic effects of digoxin. These complementary actions can be beneficial. In contrast, the ability of sympathomimetics to increase heart rate may be detrimental in that the risk of a tachydysrhythmia is increased.
QuinidineQuinidine is an antidysrhythmic drug that can cause plasma levels of digoxin to rise. Quinidine increases digoxin levels by (1) displacing digoxin from tissue binding sites and (2) reducing renal excretion of digoxin. By elevating levels of free digoxin, quinidine can promote digoxin toxicity. Accordingly, concurrent use of quinidine and digoxin should be avoided.
Drug Effect
PHARMACODYNAMIC INTERACTIONS
Thiazide diureticsLoop diureticsSuccinylcholine
Promote potassium loss and thereby increase the risk of digoxin-induced dysrhythmias
Beta blockersVerapamilDiltiazem
Decrease contractility and heart rate
Sympathomimetics Increase contractility and heart rate
PHARMACOKINETIC INTERACTIONS
CholestyramineKaolin-pectinMetoclopramideNeomycinSulfasalazine
Decrease digoxin levels by decreasing digoxin absorption or bioavailability
AminoglycosidesAntacidsColestipolAzithromycinClarithromycinErythromycinOmeprazoleTetracycline
Increase digoxin levels by increasing digoxin absorption or bioavailability
AlprazolamAmiodaroneAtorvastatinCaptoprilDiltiazemNifedipineNitrendipinePropafenoneQuinidineVerapamil
Increase digoxin levels by decreasing excretion of digoxin, altering distribution of digoxin, or both
TABLE 48.2 ■ Drug Interactions With Digoxin

541
CHAPTER 48 Drugs for Heart Failure
on too heavily. Rather, this information should be seen as but one factor among several to be considered when evaluating clinical responses.
Preparations, Dosage, and AdministrationPreparationsDigoxin is available in three formulations:
• Tablets—0.125 and 0.25 mg• Pediatric elixir—0.05 mg/mL• Solution for injection—0.1 and 0.25 mg/mL
AdministrationDigoxin can be administered orally and intravenously. Intramuscular administra-tion should be avoided, owing to a risk of tissue damage and severe pain. Before dosing, the rate and regularity of the heartbeat should be determined. If heart rate is less than 60 beats/min or if a change in rhythm is detected, digoxin should be withheld and the prescriber notified. When digoxin is given IV, cardiac status should be monitored continuously for 1 to 2 hours.
Dosage in Heart FailureMost patients can be treated with initial and maintenance dosages of 0.125 mg/day. Doses above 0.25 mg/day are rarely used or needed. The target plasma drug level is 0.5 to 0.8 ng/mL.
DigitalizationThe term digitalization refers to the use of a loading dose to achieve high plasma levels of digoxin quickly. (As noted, 6 days are needed for drug levels to reach plateau if no loading dose is employed.) Although digitalization was common in the past, the practice is now considered both unnecessary and inappropriate in the treatment of chronic heart failure.
MANAGEMENT OF HEART FAILUREOur discussion of HF management is based on recommendations in the practice guideline: 2013 ACCF/AHA Guideline for the Management of Heart Failure, and on an update—2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure. These guidelines approach HF as a progres-sive disease that advances through four stages of increasing severity. Management for each stage is discussed next.
Stage ABy definition, patients in ACC/AHA Stage A have no symptoms of HF and no structural or functional cardiac abnormalities—but they do have behaviors or conditions strongly associated with developing HF. Important among these are hypertension, coronary artery disease, diabetes, family history of cardiomy-opathy, and a personal history of alcohol abuse, rheumatic fever, or treatment with a cardiotoxic drug (e.g., doxorubicin, trastuzumab).
Management is directed at reducing risk. Hypertension, hyperlipidemia, and diabetes should be controlled, as should ventricular rate in patients with supraventricular tachycardias. An ACE inhibitor or ARB can be useful for patients with diabetes, atherosclerosis, or hypertension. Patients should cease behaviors that increase HF risk, especially smoking and alcohol abuse. (Excessive, chronic consumption of alcohol is a leading cause of cardiomyopathy. In patients with HF, acute alcohol consumption can suppress contractility.) There is no evidence that getting regular exercise can prevent development of HF, although exercise does have other health benefits. Routine use of dietary supplements to prevent structural heart disease is not recommended.
past. However, given the narrow therapeutic range of digoxin, some authorities still recommend that patients not switch between brands of digoxin tablets—even when prescriptions are written generically—except with the approval and supervi-sion of the prescriber.
DistributionDigoxin is distributed widely and crosses the placenta. High levels are achieved in cardiac and skeletal muscle, owing largely to binding to Na+/K+-ATPase. About 23% of digoxin in plasma is bound to proteins, mainly albumin.
EliminationDigoxin is eliminated primarily by renal excretion. Hepatic metabolism is minimal. Because digoxin is eliminated by the kidneys, renal impairment can lead to toxic accumulation. Accordingly, dosage must be reduced if kidney function declines. Because digoxin is not metabolized to a significant extent, changes in liver function do not affect digoxin levels.
Half-Life and Time to PlateauThe half-life of digoxin is about 1.5 days. Therefore, in the absence of a loading dose, about 6 days (four half-lives) are required to reach plateau. When use of the drug is discon-tinued, another 6 days are required for digoxin stores to be eliminated.
Single-Dose Time CourseEffects of a single oral dose begin 30 minutes to 2 hours after administration and peak within 4 to 6 hours. Effects of IV digoxin begin rapidly (within 5 to 30 minutes) and peak in 1 to 4 hours.
A Note on Plasma Digoxin LevelsMost hospitals are equipped to measure plasma levels of digoxin. The optimal range is 0.5 to 0.8 ng/mL. Levels above 1 ng/mL offer no additional benefits but do increase the risk of toxicity. Knowledge of plasma levels can be useful for
• Establishing dosage• Monitoring compliance• Diagnosing toxicity• Determining the cause of therapeutic failure
Once a stable blood level has been achieved, routine measure-ment of digoxin levels can be replaced with an annual measure-ment. Additional measurements may be useful when
• Digoxin dosage is changed• Symptoms of HF intensify• Kidney function deteriorates• Signs of toxicity appear• Drugs that can affect digoxin levels are added to or
deleted from the regimen
Although knowledge of digoxin plasma levels can aid the clinician, it must be understood that the extent of this aid is limited. The correlation between plasma levels of digoxin and clinical effects—both therapeutic and adverse—is not very tight: Drug levels that are safe and effective for patient A may be subtherapeutic for patient B and toxic for patient C. Because of interpatient variability, knowledge of digoxin levels does not permit precise predictions of therapeutic effects or toxicity. Hence, information regarding drug levels must not be relied

542
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
may take weeks or even months to develop. However, even in the absence of symptomatic improvement, ACE inhibitors may prolong life. Dosage should be sufficient to reduce mortality (see Table 48.1). For patients who cannot tolerate ACE inhibitors (owing to intractable cough or angioedema), ARBs remain the recommended alternative.
Beta Blockers. In the absence of specific contraindications, all patients with Stage C HF should receive an approved beta blocker (e.g., carvedilol). As with ACE inhibitors, symptomatic improvement may not be evident for months. Nonethe-less, life may be prolonged even in the absence of clinical improvement.
Aldosterone Antagonists. Adding an aldosterone antago-nist (spironolactone or eplerenone) to standard therapy (i.e., diuretic, ACE inhibitor or ARB, and a beta blocker) is reasonable in patients with moderately severe or severe symptoms of HF following a heart attack. However, aldosterone antagonists must not be used if kidney function is impaired or serum potassium is elevated. Monitoring renal function and potassium levels is imperative.
Digoxin. Digoxin may be used in combination with ACE inhibitors (or ARBs), diuretics, and beta blockers to improve clinical status. However, although digoxin can reduce symptoms, it does not prolong life. The usual dosage is 0.125 mg/day. Adjustments are based on clinical response. Digoxin may be started early to help improve symptoms, or it may be reserved for patients who have not responded adequately to a diuretic, ACE inhibitor or ARB, and beta blocker.
Isosorbide Dinitrate/Hydralazine. Adding ISDN/hydralazine is recommended to improve outcomes in self-described African Americans who have moderate to severe symptoms despite optimal therapy with ACE inhibitors, beta blockers, and diuretics. For all other patients who continue to have symptoms despite treatment with standard therapy, adding ISDN/hydralazine to the regimen is considered reasonable. For patients who cannot tolerate ACE inhibitors or ARBs, substitution of ISDN/hydralazine is considered reasonable.
Drugs to AvoidPatients in Stage C should avoid three classes of drugs: antidysrhythmics, calcium channel blockers (CCBs), and NSAIDs (e.g., aspirin). Reasons for not using these drugs are as follows:
• Antidysrhythmic agents—These drugs have cardiosup-pressant and prodysrhythmic actions that can make HF worse. Only two agents—amiodarone [Cordarone] and dofetilide [Tikosyn]—have been proven not to reduce survival.
• Calcium channel blockers—These drugs can make HF worse and may increase the risk of adverse cardiovascular events. Only the long-acting dihydropyridine CCBs, such as amlodipine [Norvasc], have been shown not to reduce survival.
• NSAIDs—These drugs promote sodium retention and peripheral vasoconstriction. Both actions can make HF worse. In addition, NSAIDs can reduce the effi-cacy and intensify the toxicity of diuretics and ACE inhibitors. Hence, even though aspirin has beneficial effects on coagulation, it should still be avoided unless clinically indicated for conditions such as myocardial infarction.
Stage BLike patients in Stage A, those in Stage B have no signs or symptoms of HF, but they do have structural heart disease that is strongly associated with the development of HF. Among these structural changes are LV hypertrophy or fibrosis, LV dilation or hypocontractility, valvular heart disease, and previous myocardial infarction.
The goal of management is to prevent development of symptomatic HF. The approach is to implement measures that can prevent further cardiac injury, delaying the progression of remodeling and LV dysfunction. Specific measures include all those discussed for Stage A. In addition, treatment with an ACE inhibitor plus a beta blocker is recommended for all patients with a reduced ejection fraction, history of myocardial infarction, or both. For patients who cannot tolerate ACE inhibitors, an ARB may be used instead. As in Stage A, there is no evidence that using dietary supplements or getting regular exercise can help prevent progression to symptomatic HF.
Stage CPatients in Stage C have symptoms of HF and also have structural heart disease. As discussed earlier, symptoms include dyspnea, fatigue, peripheral edema, and distention of the jugular veins. Treatment has four major goals: (1) relief of pulmonary and peripheral congestive symptoms, (2) improvement of functional capacity and quality of life, (3) slowing of cardiac remodeling and progression of LV dysfunction, and (4) pro-longation of life. Treatment measures include those recom-mended for Stages A and B, plus those discussed in the sections that follow.
Drug TherapyDrug therapy for HF has changed dramatically over the past 15 to 20 years. Formerly, digoxin was a mainstay of treatment. Today, its role is secondary. First-line therapy now consists of three drugs: a diuretic, an ACE inhibitor or ARB, and a beta blocker. As a rule, digoxin is added only when symptoms cannot be managed with the preferred agents.
Diuretics. All patients with evidence of fluid retention should restrict salt intake and use a diuretic. Diuretics are the only reliable means of correcting fluid overload. Furthermore, these drugs produce symptomatic improvement faster than any other drugs. If renal function is good, a thiazide diuretic will work. However, if renal function is significantly impaired, as it is in most patients, a loop diuretic will be needed. Efficacy of diuresis is best assessed by daily measurement of body weight. Once fluid overload has been corrected, diuretic therapy should continue to prevent recurrence. Diuretics should not be used alone. Rather, for most patients, they should be combined with an ACE inhibitor (or ARB) plus a beta blocker. Since aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) can decrease the effects of diuretics and increase the incidence of acute kidney injury when used in combination with an ACE inhibitor and a diuretic, these agents should be avoided. As noted, although diuretics reduce symptoms, they do not prolong survival.
ACE Inhibitors and ARBs. In the absence of specific contraindications (e.g., pregnancy), all patients with Stage C HF should receive an ACE inhibitor. If fluid retention is evident, a diuretic should be used as well. Symptomatic improvement

543
CHAPTER 48 Drugs for Heart Failure
memory, concentration), and ability to participate in usual social, recreational, and work activities.
Routine measurement of ejection fraction or maximal exercise capacity is not recommended. Although the degree of reduction in ejection fraction measured at the beginning of therapy is predictive of outcome, improvement in the ejection fraction does not necessarily indicate the prognosis has changed.
As noted earlier, a reduction in circulating BNP indicates improvement. The lower BNP is, the better the odds of long-term survival.
Stage DPatients in Stage D have advanced structural heart disease and marked symptoms of HF at rest, despite treatment with maximal dosages of medications used in Stage C. Repeated and prolonged hospitalization is common. For eligible candidates, the best long-term solution is a heart transplant. An implantable LV mechanical assist device can be used as a “bridge” in patients awaiting a transplant and to prolong life in those who are not transplant eligible.
Management focuses largely on the control of fluid retention, which underlies most signs and symptoms. Intake and output should be monitored closely, and the patient should be weighed daily. Fluid retention can usually be treated with a loop diuretic, perhaps combined with a thiazide. If volume overload becomes severe, the patient should be hospitalized and given an IV diuretic. If needed, IV dopamine or IV dobutamine can be added to increase renal blood flow, thereby enhancing diuresis. Patients should not be discharged until a stable and effective oral diuretic regimen has been established.
What about beta blockers and ACE inhibitors? These agents may be tried, but doses should be low and responses monitored with care. In Stage D, beta blockers pose a significant risk of making HF worse, and ACE inhibitors may induce profound hypotension or renal failure.
When severe symptoms persist despite application of all recommended therapies, options for end-of-life care should be discussed with the patient and family.
Device TherapyImplanted Cardioverter-Defibrillators. Cardiac arrest
and fatal ventricular dysrhythmias are relatively common complications of HF. Accordingly, implantable cardioverter-defibrillators are now recommended for primary or secondary prevention to reduce mortality in selected patients.
Cardiac Resynchronization. When the left and right ventricles fail to contract at the same time, cardiac output is further compromised. Synchronized contractions can be restored with a biventricular pacemaker. In clinical trials, cardiac resynchronization improved exercise tolerance and quality of life and reduced all-cause mortality.
Exercise TrainingIn the past, bed rest was recommended owing to concern that physical activity might accelerate progression of LV dysfunction. However, we now know that inactivity is actually detrimental: It reduces conditioning, worsens exercise intolerance, and contributes to HF symptoms. Conversely, studies have shown that exercise training can improve clinical status, increase exercise capacity, and improve quality of life. Accordingly, exercise training should be considered for all stable patients.
Evaluating TreatmentEvaluation is based on symptoms and physical findings. Reduc-tions in dyspnea on exertion, paroxysmal nocturnal dyspnea, and orthopnea (difficulty breathing, except in the upright position) indicate success. The physical examination should assess for reductions in jugular distention, edema, and crackles. Success is also indicated by increased capacity for physical activity. Accordingly, patients should be interviewed to deter-mine improvements in the maximal activity they can perform without symptoms, the type of activity that regularly produces symptoms, and the maximal activity they can tolerate. (Activity is defined as walking, stair climbing, activities of daily living, or any other activity that is appropriate.) Successful treatment should also improve health-related quality of life in general. Thus the interview should look for improvements in sleep, sexual function, outlook on life, cognitive function (alertness,
KEY POINTS
■ Heart failure with LV systolic dysfunction, referred to simply as heart failure (HF) in this chapter, is characterized by ventricular dysfunction, reduced cardiac output, signs of inadequate tissue perfusion (fatigue, shortness of breath, exercise intolerance), and signs of fluid overload (venous distention, peripheral edema, pulmonary edema).
■ The initial phase of HF consists of cardiac remodeling—a process in which the ventricles dilate (grow larger), hypertrophy (increase in wall thickness), and become more spherical—coupled with cardiac fibrosis and myocyte death. As a result of these changes, cardiac output is reduced.
■ Reduced cardiac output leads to compensatory responses: (1) activation of the SNS, (2) activation of the RAAS, and (3) retention of water and expansion of blood volume. As a result of volume expansion, cardiac dilation increases.
■ If the compensatory responses are not sufficient to maintain adequate production of urine, body water will continue to accumulate, eventually causing death (from complete cardiac failure secondary to excessive cardiac dilation and cardiac edema).
■ There are three major groups of drugs for heart failure: diuretics, ACE inhibitors or ARBs, and beta blockers. Digoxin, which had been used widely in the past, may be added as indicated.
■ Diuretics are first-line drugs for all patients with fluid overload. By reducing blood volume, these drugs can decrease venous pressure, arterial pressure, pulmonary edema, peripheral edema, and cardiac dilation.
■ Although diuretics can reduce symptoms of HF, they do not prolong survival.
Continued

544
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
■ Thiazide diuretics are ineffective when GFR is low, and cannot be used if cardiac output is greatly reduced.
■ Loop diuretics are effective even when GFR is low, and are preferred to thiazides for most patients.
■ Thiazide diuretics and loop diuretics can cause hypokalemia and can increase the risk of digoxin-induced dysrhythmias.
■ Potassium-sparing diuretics are used to counteract potassium loss caused by thiazide diuretics and loop diuretics.
■ Potassium-sparing diuretics can cause hyperkalemia. By doing so, they can increase the risk of hyperkalemia in patients taking ACE inhibitors or ARBs.
■ In patients with HF, ACE inhibitors improve functional status and reduce mortality. In the absence of specific contraindications, all patients should be prescribed one.
■ ACE inhibitors block formation of angiotensin II, promote accumulation of kinins, and reduce aldosterone release. As a result, these drugs cause dilation of veins and arterioles, promote renal excretion of water, and favorably alter cardiac remodeling.
■ By dilating arterioles, ACE inhibitors (1) improve regional blood flow in the kidneys and other tissues and (2) reduce cardiac afterload, which causes stroke volume and cardiac output to rise.
■ By dilating veins, ACE inhibitors reduce venous pressure, which in turn reduces pulmonary congestion, peripheral edema, preload, and cardiac dilation.
■ By suppressing aldosterone release, ACE inhibitors increase excretion of sodium and water, and decrease excretion of potassium.
■ By increasing levels of kinins (and partly by decreasing levels of angiotensin II), ACE inhibitors can favorably alter cardiac remodeling.
■ Major side effects of ACE inhibitors are hypotension, hyperkalemia, cough, angioedema, and birth defects.
■ ARBs share the beneficial hemodynamic effects of ACE inhibitors, but not the beneficial effects on cardiac remodeling.
■ In patients with HF, ARBs should be reserved for patients intolerant of ACE inhibitors (usually owing to cough).
■ In patients with HF, aldosterone antagonists (e.g., spirono-lactone, eplerenone) reduce symptoms and prolong life. Benefits derive from blocking aldosterone receptors in the heart and blood vessels.
■ Beta blockers can prolong survival in patients with HF, and are considered first-line therapy.
■ To avoid excessive cardiosuppression, beta-blocker dosage must be very low initially and then gradually increased.
■ Isosorbide dinitrate (which dilates veins) plus hydralazine (which dilates arterioles) can be used in place of an ACE inhibitor (or ARB) if an ACE inhibitor (or ARB) cannot be used.
■ BiDil, a fixed-dose combination of hydralazine and iso-sorbide dinitrate, is approved specifically for treating HF in African Americans.
■ Digoxin and other inotropic agents increase the force of myocardial contraction and thereby increase cardiac output.
■ Of the available inotropic agents, digoxin is the only one that is both effective and safe when used orally and the only one suitable for long-term use.
■ Digoxin increases contractility by inhibiting myocardial Na+/K+-ATPase, thereby (indirectly) increasing intracellular calcium, which in turn facilitates the interaction of actin and myosin.
■ Potassium competes with digoxin for binding to Na+/K+-ATPase. Therefore, if potassium levels are low, excessive inhibition of Na+/K+-ATPase can occur, resulting in toxicity. Conversely, if potassium levels are high, insufficient inhibition can occur, resulting in therapeutic failure. Accordingly, it is imperative to keep potassium levels in the normal physiologic range: 3.5 to 5 mEq/L.
■ By increasing cardiac output, digoxin can reverse all of the overt manifestations of HF: cardiac output improves, heart rate decreases, heart size declines, constriction of arterioles and veins decreases, water retention reverses, blood volume declines, peripheral and pulmonary edema decrease, water weight is lost, and exercise tolerance improves. Unfortunately, although digoxin can improve symptoms, it does not prolong life.
■ In patients with HF, benefits of digoxin are not due solely to improved cardiac output; neurohormonal effects are important too.
■ Digoxin causes dysrhythmias by altering the electrical properties of the heart (secondary to inhibition of Na+/K+- ATPase).
■ The most common reason for digoxin-related dysrhythmias is diuretic-induced hypokalemia.
■ If a severe digoxin overdose is responsible for dysrhythmias, digoxin levels can be lowered using Fab antibody fragments [Digifab].
■ In addition to dysrhythmias, digoxin can cause GI effects (anorexia, nausea, vomiting) and CNS effects (fatigue, visual disturbances). Gastrointestinal and CNS effects often precede dysrhythmias and therefore can provide advance warning of serious toxicity.
■ Digoxin has a narrow therapeutic range.■ Digoxin is eliminated by renal excretion.■ Although routine monitoring of digoxin levels is generally
unnecessary, monitoring can be helpful when dosage is changed, symptoms of HF intensify, kidney function declines, signs of toxicity appear, or drugs that affect digoxin levels are added to or deleted from the regimen.
■ Maintenance doses of digoxin are based primarily on observation of the patient: Doses should be large enough to minimize symptoms of HF but not so large as to cause adverse effects.
■ Maintenance doses of digoxin must be reduced if renal function declines.
■ Therapy of Stage C HF has four major goals: (1) relief of pulmonary and peripheral congestion, (2) improvement of functional status and quality of life, (3) delay of progression of cardiac remodeling and LV dysfunction, and (4) prolonga-tion of life.
■ For routine therapy, Stage C HF is treated with a diuretic, an ACE inhibitor or an ARB, and a beta blocker.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

Summary of Major Nursing Implicationsa
DIGOXINPreadministration AssessmentTherapeutic GoalDigoxin is used to treat HF and dysrhythmias. Be sure to confirm which disorder the drug has been ordered for.
Baseline DataAssess for signs and symptoms of HF, including fatigue, weakness, cough, breathing difficulty (orthopnea, dyspnea on exertion, paroxysmal nocturnal dyspnea), jugular distention, and edema.
Determine baseline values for maximal activity without symptoms, activity that regularly causes symptoms, and maximal tolerated activity.
Laboratory tests should include an ECG, serum electrolytes, measurement of ejection fraction, and evaluation of kidney function.
Identifying High-Risk PatientsDigoxin is contraindicated for patients experiencing ventricu-lar fibrillation, ventricular tachycardia, or digoxin toxicity.
Exercise caution in the presence of conditions that can predispose the patient to serious adverse responses to digoxin, such as hypokalemia, partial AV block, advanced HF, or renal impairment.
Implementation: AdministrationRoutesOral, slow IV injection.
AdministrationOral. Determine heart rate and rhythm before administra-
tion. If heart rate is less than 60 beats/min or if a change in rhythm is detected, withhold digoxin and notify the prescriber.
Warn patients not to “double up” on doses in attempts to compensate for missed doses.
Intravenous. Monitor cardiac status closely for 1 to 2 hours following IV injection.
Promoting AdherenceBecause digoxin has a narrow therapeutic range, rigid adher-ence to the prescribed dosage is essential. Inform patients that failure to take digoxin exactly as prescribed may lead to toxicity or therapeutic failure. If poor adherence is sus-pected, serum drug levels may help in assessing the extent of nonadherence.
Implementation: Measures to Enhance Therapeutic EffectsAdvise patients to limit salt intake to 1500 mg/day and to avoid excessive fluids. Advise patients who drink alcohol to consume no more than one drink each day. Help patients establish an appropriate program of regular mild exercise (e.g., walking, cycling). Precipitating factors for HF (e.g., hypertension, valvular heart disease) should be corrected.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsEvaluation is based on symptoms and physical findings. Assess for reductions in orthopnea, dyspnea on exertion, paroxysmal nocturnal dyspnea, neck vein distention, edema, and crackles, and for increased capacity for physical activity. In addition,
assess for improvements in sleep, sexual function, outlook on life, cognitive function, and ability to participate in social, recreational, and work activities.
Plasma BNP levels reflect cardiac status: The lower the level, the better the odds for long-term survival.
Measurement of plasma drug levels can help determine the cause of therapeutic failure. The optimal range for digoxin is 0.5 to 0.8 ng/mL.
Minimizing Adverse EffectsCardiotoxicity. Dysrhythmias are the most serious adverse
effect of digoxin. Monitor hospitalized patients for alterations in heart rate or rhythm, and withhold digoxin if significant changes develop.
Inform outpatients about the danger of dysrhythmias. Teach them to monitor their pulses for rate and rhythm, and instruct them to notify the prescriber if significant changes occur. Provide the patient with an ECG rhythm strip; this can be used by providers unfamiliar with the patient (e.g., when the patient is traveling) to verify suspected changes in rhythm.
Hypokalemia, usually diuretic induced, is the most frequent underlying cause of dysrhythmias. Monitor serum potassium concentrations. If hypokalemia develops, potassium levels can be raised with potassium supplements, a potassium-sparing diuretic, or both. Teach patients to recognize early signs of hypokalemia (e.g., muscle weakness), and instruct them to notify the prescriber if these develop. Severe vomiting and diarrhea can increase potassium loss; exercise caution if these events occur.
To treat digoxin-induced dysrhythmias: (1) withdraw digoxin and diuretics (make sure that a written order for digoxin withdrawal is made); (2) administer potassium (unless potassium levels are above normal or AV block is present); (3) administer an antidysrhythmic drug (phenytoin or lidocaine, but not quinidine) if indicated; (4) manage bradycardia with atropine or electrical pacing; and (5) treat with Fab fragments if toxicity is life threatening.
Noncardiac Effects. Nausea, vomiting, anorexia, fatigue, and visual disturbances (blurred or yellow vision) frequently foreshadow more serious toxicity (dysrhythmias) and should be reported immediately. Inform patients about these early indications of toxicity, and instruct them to notify the pre-scriber if they develop.
Minimizing Adverse InteractionsDiuretics. Thiazide diuretics and loop diuretics increase
the risk of dysrhythmias by promoting potassium loss. Monitor potassium levels. If hypokalemia develops, it should be corrected with potassium supplements, a potassium-sparing diuretic, or both.
ACE Inhibitors and ARBs. These drugs can elevate potas-sium levels and decrease therapeutic responses to digoxin. Exercise caution if an ACE inhibitor or ARB is combined with potassium supplements or a potassium-sparing diuretic.
Sympathomimetic Agents. Sympathomimetic drugs (e.g., dopamine, dobutamine) stimulate the heart, thereby increasing the risk of tachydysrhythmias and ectopic pace-maker activity. When sympathomimetics are combined with digoxin, monitor closely for dysrhythmias.
Quinidine. Quinidine can elevate plasma levels of digoxin. If quinidine is employed concurrently with digoxin, digoxin dosage must be reduced. Do not use quinidine to treat digoxin-induced dysrhythmias.
aPatient education information is highlighted as blue text.

546
C H A P T E R
49 Antidysrhythmic Drugs
INTRODUCTION TO CARDIAC ELECTROPHYSIOLOGY, DYSRHYTHMIAS, AND THE ANTIDYSRHYTHMIC DRUGS, p. 546
Electrical Properties of the Heart, p. 547Impulse Conduction: Pathways and Timing, p. 547Cardiac Action Potentials, p. 547The Electrocardiogram, p. 549
Generation of Dysrhythmias, p. 549Disturbances of Automaticity, p. 549Disturbances of Conduction, p. 550
Classification of Antidysrhythmic Drugs, p. 550Class I: Sodium Channel Blockers, p. 550Class II: Beta Blockers, p. 550Class III: Potassium Channel Blockers (Drugs That
Delay Repolarization), p. 551Class IV: Calcium Channel Blockers, p. 551Other Antidysrhythmic Drugs, p. 551
Prodysrhythmic Effects of Antidysrhythmic Drugs, p. 551
Overview of Common Dysrhythmias and Their Treatment, p. 552Supraventricular Dysrhythmias, p. 552Ventricular Dysrhythmias, p. 553
Principles of Antidysrhythmic Drug Therapy, p. 553Balancing Risks and Benefits, p. 553Properties of the Dysrhythmia to Be Considered,
p. 554
Phases of Treatment, p. 554Long-Term Treatment: Drug Selection and
Evaluation, p. 554Minimizing Risks, p. 554
PHARMACOLOGY OF THE ANTIDYSRHYTHMIC DRUGS, p. 554
Class I: Sodium Channel Blockers, p. 554Class IA Agents, p. 554Class IB Agents, p. 557Class IC Agents, p. 558
Class II: Beta Blockers, p. 558Propranolol, p. 558Acebutolol, p. 559Esmolol, p. 559
Class III: Potassium Channel Blockers (Drugs That Delay Repolarization), p. 559Amiodarone, p. 559Dronedarone, p. 560Sotalol, p. 561Dofetilide, p. 562Ibutilide, p. 562
Class IV: Calcium Channel Blockers, p. 562Other Antidysrhythmic Drugs, p. 563
Adenosine, p. 563Digoxin, p. 563
Key Points, p. 564Summary of Major Nursing Implications, p. 565
drugs should be employed only when the benefits of treatment clearly outweigh the risks.
For two reasons, the use of antidysrhythmic drugs is declining. First, research has shown that some of these agents actually increase the risk of death. Second, nonpharmacologic therapies—especially implantable defibrillators and radiofre-quency ablation—have begun to replace drugs as the preferred treatment for many dysrhythmia types.
A note on terminology: Dysrhythmias are also known as arrhythmias. Since the term arrhythmia denotes an absence of cardiac rhythm, whereas dysrhythmia denotes an abnormal rhythm, dysrhythmia would seem the more appropriate term.
CARDIAC ELECTROPHYSIOLOGY, DYSRHYTHMIAS, AND THE ANTIDYSRHYTHMIC DRUGS
In this section we discuss background information that will help you understand the actions and uses of antidysrhythmic
A dysrhythmia is defined as an abnormality in the rhythm of the heartbeat. In their mildest forms, dysrhythmias have only modest effects on cardiac output. However, in their most severe forms, dysrhythmias can so disable the heart that no blood is pumped at all. Because of their ability to compromise cardiac function, dysrhythmias are associated with a high degree of morbidity and mortality.
There are two basic types of dysrhythmias: tachydysrhyth-mias (dysrhythmias in which heart rate is increased) and bradydysrhythmias (dysrhythmias in which heart rate is slowed). In this chapter, we consider only the tachydysrhythmias. This is by far the largest group of dysrhythmias and the group that responds best to drugs. We do not discuss the bradydysrhythmias because they are few in number and are commonly treated with electronic pacing. When drugs are indicated, atropine is usually the agent of choice.
It is important to appreciate that virtually all of the drugs used to treat dysrhythmias can also cause dysrhythmias. These drugs can create new dysrhythmias and worsen existing ones. Because of these prodysrhythmic actions, antidysrhythmic

547
CHAPTER 49 Antidysrhythmic Drugs
AV NodeImpulses originating in the atria must travel through the AV node to reach the ventricles. In the healthy heart, impulses arriving at the AV node are delayed before going on to excite the ventricles. This delay provides time for blood to fill the ventricles before ventricular contraction.
His-Purkinje SystemThe fibers of the His-Purkinje system consist of specialized conducting tissue. The function of these fibers is to conduct electrical excitation very rapidly to all parts of the ventricles. Stimulation of the His-Purkinje system is caused by impulses leaving the AV node. These impulses are conducted rapidly down the bundle of His, enter the right and left bundle branches, and then distribute to the many fine branches of the Purkinje fibers (see Fig. 49.1). Because impulses travel quickly through this system, all regions of the ventricles are stimulated almost simultaneously, producing synchronized ventricular contraction with resultant forceful ejection of blood.
Cardiac Action PotentialsCardiac cells can initiate and conduct action potentials, consist-ing of self-propagating waves of depolarization followed by repolarization. As in neurons, cardiac action potentials are generated by the movement of ions into and out of cells. These ion fluxes take place by way of specific channels in the cell membrane. In the resting cardiac cell, negatively charged ions cover the inner surface of the cell membrane while positively charged ions cover the external surface. Because of this separa-tion of charge, the cell membrane is said to be polarized. Under proper conditions, channels in the cell membrane open, allowing positively charged ions to rush in. This influx eliminates the charge difference across the cell membrane, and so the cell is said to depolarize. Following depolarization, positively charged ions are extruded from the cell, causing the cell to return to its original polarized state.
In the heart, two kinds of action potentials occur: fast potentials and slow potentials. These potentials differ with respect to the mechanisms by which they are generated, the kinds of cells in which they occur, and the drugs to which they respond.
Profiles of fast and slow potentials are depicted in Fig. 49.2. Please note that action potentials in this figure represent the electrical activity of single cardiac cells. Such single-cell recordings, which are made using experimental preparations, should not be confused with the ECG, which is made using surface electrodes, and thus reflects the electrical activity of the entire heart.
Fast PotentialsFast potentials occur in fibers of the His-Purkinje system and in atrial and ventricular muscle. These responses serve to conduct electrical impulses rapidly throughout the heart.
As shown in Fig. 49.2A, fast potentials have five distinct phases, labeled 0, 1, 2, 3, and 4. As we discuss each phase, we focus on its ionic basis and its relationship to the actions of antidysrhythmic drugs.
Phase 0. In phase 0, the cell undergoes rapid depolarization in response to influx of sodium ions. Phase 0 is important in that the speed of phase 0 depolarization determines the velocity
drugs. We begin by reviewing the electrical properties of the heart and the electrocardiogram (ECG). Next, we discuss how dysrhythmias are generated. After that, we discuss classification of the antidysrhythmic drugs, as well as the ability of these drugs to cause dysrhythmias. We conclude by discussing the major dysrhythmias and the basic principles that guide anti-dysrhythmic therapy.
ELECTRICAL PROPERTIES OF THE HEARTDysrhythmias result from alteration of the electrical impulses that regulate cardiac rhythm—and antidysrhythmic drugs control rhythm by correcting or compensating for these alterations. Accordingly, to understand both the generation and treatment of dysrhythmias, we must first understand the electrical proper-ties of the heart. Therefore, we begin by reviewing (1) pathways and timing of impulse conduction, (2) cardiac action potentials, and (3) basic elements of the ECG.
Impulse Conduction: Pathways and TimingFor the heart to pump effectively, contraction of the atria and ventricles must be coordinated. Coordination is achieved through precise timing and routing of impulse conduction. In the healthy heart, impulses originate in the sinoatrial (SA) node, spread rapidly through the atria, pass slowly through the atrioventricular (AV) node, and then spread rapidly through the ventricles via the His-Purkinje system (Fig. 49.1).
SA NodeUnder normal circumstances, the SA node serves as the pacemaker for the heart. Pacemaker activity results from spontaneous phase 4 depolarization (discussed later in this chapter). Because cells of the sinus node usually discharge faster than other cells that display automaticity, the SA node normally dominates all other potential pacemakers.
After the SA node discharges, impulses spread rapidly through the atria along the internodal pathways. This rapid conduction allows the atria to contract in unison.
Internodalpathways
Atrioventricular(AV) node
Purkinje fibers
Sinoatrial(SA) node
Bundle of His
Left bundle branch
Right bundle branch
Fig. 49.1 ■ Cardiac conduction pathways.

548
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
a cell is unable to respond to excitation and initiate a new action potential. Therefore, extending the ERP prolongs the minimum interval between two propagating responses.) Phase 3 repolarization can be delayed by drugs that block potassium channels.
Phase 4. During phase 4, two types of electrical activity are possible: (1) the membrane potential may remain stable (solid line in Fig. 49.2A) or (2) the membrane may undergo spontaneous depolarization (dashed line in Fig. 49.2A). In cells undergoing spontaneous depolarization, the membrane potential gradually rises until a threshold potential is reached. At this point, rapid phase 0 depolarization takes place, setting off a new action potential. Hence, it is phase 4 depolarization that gives cardiac cells automaticity (i.e., the ability to initiate an action potential through self-excitation). The capacity for self-excitation makes potential pacemakers of all cells that have it.
of impulse conduction. Drugs that decrease the rate of phase 0 depolarization (by blocking sodium channels) slow impulse conduction through the His-Purkinje system and myocardium.
Phase 1. During phase 1, rapid (but partial) repolarization takes place. Phase 1 has no relevance to antidysrhythmic drugs.
Phase 2. Phase 2 consists of a prolonged plateau in which the membrane potential remains relatively stable. During this phase, calcium enters the cell and promotes contraction of atrial and ventricular muscle. Drugs that reduce calcium entry during phase 2 do not influence cardiac rhythm. However, since calcium influx is required for contraction, these drugs can reduce myocardial contractility.
Phase 3. In phase 3, rapid repolarization takes place. This repolarization is caused by extrusion of potassium from the cell. Phase 3 is relevant in that delay of repolarization prolongs the action potential duration and thereby prolongs the effective refractory period (ERP). (The ERP is the time during which
Myocardium and His-Purkinje System
SA Node and AV Node
ContractionDepolarization
Depolarization
�90 mV
�60 mV
3
4
4
2
2
0
1
3
0
Repolarization
Class II: Beta BlockersClass IV: Calcium Channel Blockers
Class II: Beta BlockersClass IV: Calcium Channel BlockersAdenosine
Class IQuinidineProcainamideLidocaine
Class II: Beta BlockersClass IV: Calcium Channel Blockers
Class IIIBretyliumAmiodarone
K+
Ca
++
Na+
Ca ++
?
A
BFig. 49.2 ■ Ion fluxes during cardiac action potentials and effects of antidysrhythmic drugs. A, Fast potential of the His-Purkinje system and atrial and ventricular myocardium. Blockade of sodium influx by class I drugs slows conduction in the His-Purkinje system. Blockade of calcium influx by beta blockers and calcium channel blockers decreases contractility. Blockade of potassium efflux by class III drugs delays repolarization and thereby prolongs the effective refractory period. B, Slow potential of the sinoatrial (SA) node and atrioventricular (AV) node. Blockade of calcium influx by beta blockers, calcium channel blockers, and adenosine slows AV conduction. Beta blockers and calcium channel blockers decrease SA nodal auto-maticity (phase 4 depolarization); the ionic basis of this effect is not understood.

549
CHAPTER 49 Antidysrhythmic Drugs
In addition to the features just described, the ECG has three other components of interest: the PR interval, the QT interval, and the ST segment. The PR interval is defined as the time between the onset of the P wave and the onset of the QRS complex. Lengthening of this interval indicates a delay in conduction through the AV node. Several drugs increase the PR interval. The QT interval is defined as the time between the onset of the QRS complex and completion of the T wave. This interval is prolonged by drugs that delay ventricular repolarization. The ST segment is the portion of the ECG that lies between the end of the QRS complex and the beginning of the T wave. Digoxin depresses the ST segment.
GENERATION OF DYSRHYTHMIASDysrhythmias arise from two fundamental causes: disturbances of impulse formation (automaticity) and disturbances of impulse conduction. One or both of these disturbances underlie all dysrhythmias. Factors that may alter automaticity or conduction include hypoxia, electrolyte imbalance, cardiac surgery, reduced coronary blood flow, myocardial infarction, and antidysrhythmic drugs.
Disturbances of AutomaticityDisturbances of automaticity can occur in any part of the heart. Cells normally capable of automaticity (cells of the SA node, AV node, and His-Purkinje system) can produce dysrhythmias if their normal rate of discharge changes. In addition, dys-rhythmias may be produced if tissues that do not normally express automaticity (atrial and ventricular muscle) develop spontaneous phase 4 depolarization.
Altered automaticity in the SA node can produce tachycardia or bradycardia. Excessive discharge of sympathetic neurons that innervate the SA node can augment automaticity to such a degree that sinus tachycardia results. Excessive vagal (parasympathetic) discharge can suppress automaticity to such a degree that sinus bradycardia results.
Increased automaticity of Purkinje fibers is a common cause of dysrhythmias. The increase can be brought on by injury and by excessive stimulation of Purkinje fibers by the sym-pathetic nervous system. If Purkinje fibers begin to discharge
Under normal conditions, His-Purkinje cells undergo very slow spontaneous depolarization, and myocardial cells do not undergo any. However, under pathologic conditions, significant phase 4 depolarization may occur in all of these cells, espe-cially in Purkinje fibers. When this happens, a dysrhythmia can result.
Slow PotentialsSlow potentials occur in cells of the SA node and AV node. The profile of a slow potential is depicted in Fig. 49.2B. Like fast potentials, slow potentials are generated by ion fluxes. However, the specific ions involved are not the same for every phase.
From a physiologic and pharmacologic perspective, slow potentials have three features of special significance: (1) phase 0 depolarization is slow and mediated by calcium influx, (2) these potentials conduct slowly, and (3) spontaneous phase 4 depolarization in the SA node normally determines heart rate.
Phase 0. Phase 0 (depolarization phase) of slow potentials differs significantly from phase 0 of fast potentials. As we can see from Fig. 49.2, whereas phase 0 of fast potentials is caused by a rapid influx of sodium, phase 0 of slow potentials is caused by slow influx of calcium. Because calcium influx is slow, the rate of depolarization is slow; and because depolariza-tion is slow, these potentials conduct slowly. This explains why impulse conduction through the AV node is delayed. Phase 0 of the slow potential is of therapeutic significance in that drugs that suppress calcium influx during phase 0 can slow (or stop) AV conduction.
Phases 2 and 3. Slow potentials lack a phase 1 (see Fig. 49.2B). Phases 2 and 3 of the slow potential are not significant with respect to the actions of antidysrhythmic drugs.
Phase 4. Cells of the SA node and AV node undergo spontaneous phase 4 depolarization. The ionic basis of this phenomenon is complex and incompletely understood.
Under normal conditions, the rate of phase 4 depolarization in cells of the SA node is faster than in all other cells of the heart. As a result, the SA node discharges first and determines heart rate. Hence, the SA node is referred to as the cardiac pacemaker.
As shown in Fig. 49.2B, two classes of drugs (beta blockers and calcium channel blockers) can suppress phase 4 depolariza-tion. By doing so, these agents can decrease automaticity in the SA node.
The ElectrocardiogramThe ECG provides a graphic representation of cardiac electrical activity. The ECG can be used to identify dysrhythmias and monitor responses to therapy. (Note: In referring to the elec-trocardiogram, two abbreviations may be used: EKG and ECG.)
The major components of an ECG are shown in Fig. 49.3. As we can see, three features are especially prominent: the P wave, the QRS complex, and the T wave. The P wave is caused by depolarization in the atria. Therefore, the P wave corresponds to atrial contraction. The QRS complex is caused by depolariza-tion of the ventricles, so the QRS complex corresponds to ventricular contraction. If conduction through the ventricles is slowed, the QRS complex will widen. The T wave is caused by repolarization of the ventricles, so this wave is not associated with overt physical activity of the heart.
Fig. 49.3 ■ The electrocardiogram.

550
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
In normal impulse conduction, electrical impulses travel down both branches of the Purkinje fiber to cause excitation of the muscle at two locations (Fig. 49.4A). Impulses created within the muscle travel in both directions (to the right and to the left) away from their sites of origin. Those impulses that are moving toward each other meet midway between the two branches of the Purkinje fiber. Since in the wake of both impulses the muscle is in a refractory state, neither impulse can proceed further, so both impulses stop.
In a reentrant circuit (Fig. 49.4B), there is a region of one-way conduction block of one branch of the Purkinje fiber. This region prevents conduction of impulses downward (toward the muscle) but does not prevent impulses from traveling upward. (Impulses can travel back up the block because impulses in muscle are very strong, and hence are able to pass the block, whereas impulses in the Purkinje fiber are weaker, and so are unable to pass.) A region of one-way block is essential for reentrant activation.
How does one-way block lead to reentrant activation? As an impulse travels down the Purkinje fiber, it is blocked in one branch but continues unimpeded in the other branch. Upon reaching the tip of the second (unblocked) branch, the impulse stimulates the muscle. As previously described, the impulse in the muscle travels to the right and to the left away from its site of origin. However, in this new situation, as the impulse travels toward the impaired branch of the Purkinje fiber, it meets no impulse coming from the other direction and continues on, resulting in stimulation of the terminal end of the first (blocked) branch. This stimulation causes an impulse to travel backward up the blocked branch of the Purkinje fiber. Since blockade of conduction in that branch is one way (downward only), the impulse can pass upward through the region of block and then back down into the unblocked branch, causing reentrant activation of this branch. Under proper conditions, the impulse will continue to cycle indefinitely, resulting in repetitive ectopic beats.
There are two mechanisms by which drugs can abolish a reentrant dys-rhythmia. First, drugs can improve conduction in the sick branch of the Purkinje fiber and can thereby eliminate the one-way block (Fig. 49.4C). Alternatively, drugs can suppress conduction in the sick branch, thereby converting one-way block into two-way block (Fig. 49.4D).
CLASSIFICATION OF ANTIDYSRHYTHMIC DRUGS
According to the Vaughan Williams classification scheme, the antidysrhythmic drugs fall into five groups (Table 49.1). There are four major classes of antidysrhythmic drugs (classes I, II, III, and IV) and a fifth group that includes adenosine and digoxin. Membership in classes I through IV is determined by the effects on ion movements during slow and fast potentials (see Fig. 49.2).
Class I: Sodium Channel BlockersClass I drugs block cardiac sodium channels (see Fig. 49.2A). By doing so, these drugs slow impulse conduction in the atria, ventricles, and His-Purkinje system. Class I constitutes the largest group of antidysrhythmic drugs.
Class II: Beta BlockersClass II consists of beta-adrenergic blocking agents. As sug-gested by Fig. 49.2, these drugs reduce calcium entry (during fast and slow potentials), and they depress phase 4 depolarization (in slow potentials only). Beta blockers have three prominent effects on the heart:
• In the SA node, they reduce automaticity.• In the AV node, they slow conduction velocity.• In the atria and ventricles, they reduce contractility.
Cardiac effects of the beta blockers are nearly identical to those of the calcium channel blockers.
faster than the SA node, they will escape control by the SA node; potentially serious dysrhythmias can result.
Under special conditions, automaticity may develop in cells of atrial and ventricular muscle. If these cells fire faster than the SA node, dysrhythmias will result.
Antidysrhythmic Drugs
Life Stage Patient Care Concerns
Infants See “Breast-feeding women,” below.Children/
adolescentsSome antidysrhythmic drugs can be used safely
in children, just in smaller doses. These include disopyramide, flecainide, and sotalol. Side effect profiles are similar to those of adults.
Pregnant women
Many of the drugs discussed in this chapter are classified in FDA Pregnancy Risk Category C or D.a Animal studies show adverse fetal effects, and in Category D,a there is evidence of human fetal risk. Benefits should outweigh the risks. Dronedarone is classified in Pregnancy Risk Category X.a
Breast-feeding women
For most of the drugs discussed in this chapter, data are lacking regarding transmission of drug from mother to infant via breast milk. Breast-feeding is contraindicated in women taking dronedarone.
Older adults Aging alters the absorption, distribution, metabolism, and elimination of antidysrhythmic drugs. Liver and kidney function must be monitored, and antiarrhythmic dosing may need to be adjusted for age. Older adult patients are also more susceptible to the side effects of many antidysrhythmics, including bradycardia, orthostatic hypotension, urinary retention, and falls.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
Disturbances of ConductionAtrioventricular BlockImpaired conduction through the AV node produces varying degrees of AV block. If impulse conduction is delayed (but not prevented entirely), the block is termed first degree. If some impulses pass through the node but others do not, the block is termed second degree. If all traffic through the AV node stops, the block is termed third degree.
Reentry (Recirculating Activation)Reentry, also referred to as recirculating activation, is a general-ized mechanism by which dysrhythmias can be produced. Reentry causes dysrhythmias by establishing a localized, self-sustaining circuit capable of repetitive cardiac stimulation. Reentry results from a unique form of conduction disturbance. The mechanism of reentrant activation and the effects of drugs on this process are described here.

551
CHAPTER 49 Antidysrhythmic Drugs
by decreasing conduction through the AV node and reducing automaticity in the SA node.
PRODYSRHYTHMIC EFFECTS OF ANTIDYSRHYTHMIC DRUGSVirtually all of the drugs used to treat dysrhythmias have prodysrhythmic (proarrhythmic) effects. That is, all of these drugs can worsen existing dysrhythmias and generate new ones. This ability was documented dramatically in the Cardiac Arrhythmia Suppression Trial (CAST), in which use of class IC drugs (encainide and flecainide) to prevent dysrhythmias after myocardial infarction actually doubled the rate of mortality. Because of their prodysrhythmic actions, antidysrhythmic drugs should be used only when dysrhythmias are symptomatically significant and only when the potential benefits clearly outweigh the risks. Applying this guideline, it would be inappropriate to give antidysrhythmic drugs to a patient with nonsustained ventricular tachycardia, since this dysrhythmia does not sig-nificantly reduce cardiac output. Conversely, when a patient is facing death from ventricular fibrillation, any therapy that might work must be tried. In this case, the risk of prodysrhythmic
Class III: Potassium Channel Blockers (Drugs That Delay Repolarization)Class III drugs block potassium channels (see Fig. 49.2A) and thereby delay repolarization of fast potentials. By delaying repolarization, these drugs prolong both the action potential duration and the effective refractory period.
Class IV: Calcium Channel BlockersOnly two calcium channel blockers—verapamil and diltiazem—are employed as antidysrhythmics. As indicated in Fig. 49.2, calcium channel blockade has the same impact on cardiac action potentials as does beta blockade. Accordingly, verapamil, diltiazem, and beta blockers have nearly identical effects on cardiac function—namely, reduction of automaticity in the SA node, delay of conduction through the AV node, and reduction of myocardial contractility. Antidysrhythmic benefits derive from suppressing AV nodal conduction.
Other Antidysrhythmic DrugsAdenosine and digoxin do not fit into the four major classes of antidysrhythmic drugs. Both drugs suppress dysrhythmias
A
C
B
D
Normal Conduction Reentrant Activation
Drug Effect IIDrug Effect I
Purkinjefiber
Region ofone-way block
Ventricularmuscle
Branch 1 Branch 2
Conversion totwo-way block
Eliminationof block
Fig. 49.4 ■ Reentrant activation: mechanism and drug effects. A, In normal conduction, impulses from the branched Purkinje fiber stimulate the strip of ventricular muscle in two places. Within the muscle, waves of excitation spread from both points of excitation, meet between the Purkinje fibers, and cease further travel. B, In the presence of one-way block, the strip of muscle is excited at only one location. Impulses spreading from this area meet no impulses coming from the left and, therefore, can travel far enough to stimulate branch 1 of the Purkinje fiber. This stimulation passes back up the fiber, past the region of one-way block, and then stimulates branch 2, causing reentrant activation. C, Elimination of reentry by a drug that improves conduction in the sick branch of the Purkinje fiber. D, Elimination of reentry by a drug that further suppresses conduction in the sick branch, thereby converting one-way block into two-way block.

552
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
dangerous than supraventricular dysrhythmias. With either type, intervention is required only if the dysrhythmia interferes with effective ventricular pumping. Treatment often proceeds in two phases: (1) termination of the dysrhythmia (with electrical countershock, drugs, or both), followed by (2) long-term suppression with drugs. Dysrhythmias can also be treated with an implantable cardioverter-defibrillator (ICD) or by destroying small areas of cardiac tissue using radiofrequency (RF) catheter ablation.
It is important to appreciate that drug therapy of dysrhythmias is highly empiric (i.e., based largely on the response of the patient and not on scientific principles). In practice, this means that even after a dysrhythmia has been identified, we cannot predict with certainty just which drugs will be effective. Frequently, trials with several drugs are required before the control of rhythm is achieved. In the discussion that follows, only first-choice drugs are considered.
Supraventricular DysrhythmiasSupraventricular dysrhythmias are dysrhythmias that arise in areas of the heart above the ventricles (atria, SA node, AV node). Supraventricular dysrhythmias per se are not especially harmful because dysrhythmic activity within the atria does not significantly reduce cardiac output (except in patients with valvular disorders and heart failure [HF]). Supraventricular tachydysrhythmias can be dangerous, however, in that atrial impulses are likely to traverse the AV node, resulting in excita-tion of the ventricles. If the atria drive the ventricles at an excessive rate, diastolic filling will be incomplete and cardiac output will decline. Hence, when treating supraventricular tachydysrhythmias, the objective is frequently the slowing of ventricular rate (by blocking impulse conduction through the AV node) and not elimination of the dysrhythmia itself. Of course, if treatment did abolish the dysrhythmia, this outcome would not be unwelcome. Acute treatment of supraventricular dysrhythmias is accomplished with vagotonic maneuvers, direct-current (DC) cardioversion, and certain drugs: class II agents, class IV agents, adenosine, and digoxin.
Atrial FibrillationAtrial fibrillation is the most common sustained dysrhythmia, affecting about 4 million people in the United States. The disorder is caused by multiple atrial ectopic foci firing randomly; each focus stimulates a small area of atrial muscle. This chaotic excitation produces a highly irregular atrial rhythm. Depending upon the extent of impulse transmission through the AV node, ventricular rate may be very rapid or nearly normal.
In addition to compromising cardiac performance, atrial fibrillation carries a high risk of stroke because, in patients with atrial fibrillation, some blood can become trapped in the atria (rather than flowing straight through to the ventricles), thereby permitting formation of a clot. When normal sinus rhythm is restored, the clot may become dislodged; then it may travel to the brain to cause stroke.
Treatment of atrial fibrillation has two goals: improvement of ventricular pumping and prevention of stroke. Pumping can be improved by either (1) restoring normal sinus rhythm or (2) slowing ventricular rate. The preferred method is to slow ventricular rate by long-term therapy with a beta blocker (atenolol or metoprolol) or a cardioselective calcium channel blocker (diltiazem or verapamil), both of which impede
CLASS I: SODIUM CHANNEL BLOCKERS
Class IA
QuinidineProcainamide [Procan , generic in United States]Disopyramide [Norpace, Norpace CR]
Class IB
Lidocaine [Xylocaine]Phenytoin [Dilantin]Mexiletine
Class IC
FlecainidePropafenone [Rythmol, Rythmol SR]
CLASS II: BETA BLOCKERS
Propranolol [Inderal LA]Acebutolol [Sectral]Esmolol [Brevibloc]
CLASS III: POTASSIUM CHANNEL BLOCKERS (DRUGS THAT DELAY REPOLARIZATION)
Amiodarone [Cordarone, Pacerone]Dronedarone [Multaq]Sotalol [Betapace, Betapace AF]Dofetilide [Tikosyn]Ibutilide [Corvert]
CLASS IV: CALCIUM CHANNEL BLOCKERS
Verapamil [Calan, Covera-HS, Verelan]Diltiazem [Cardizem, Dilacor-XR, Tiazac, others]
OTHER ANTIDYSRHYTHMIC DRUGS
Adenosine [Adenocard]Digoxin [Lanoxin]
TABLE 49.1 ■ Vaughan Williams Classification of Antidysrhythmic Drugs
Safety Alert
ANTIDYSRHYTHMIC DRUGSAll of these drugs can worsen existing dysrhythmias and generate new ones. Regardless of the particular circumstances of drug use, all patients must be followed closely.
Of the mechanisms by which drugs can cause dysrhythmias, one deserves special mention: prolongation of the QT interval. As discussed in Chapter 7, drugs that prolong the QT interval increase the risk of torsades de pointes, a dysrhythmia that can progress to fatal ventricular fibrillation. All class IA and class III agents cause QT prolongation, and so must be used with special caution.
effects is clearly outweighed by the potential benefits of stopping the fibrillation.
OVERVIEW OF COMMON DYSRHYTHMIAS AND THEIR TREATMENTThe common dysrhythmias can be divided into two major groups: supraventricular dysrhythmias and ventricular dys-rhythmias. In general, ventricular dysrhythmias are more

553
CHAPTER 49 Antidysrhythmic Drugs
rhythm, IV procainamide should be administered; lidocaine and amiodarone are alternatives. For long-term management, drugs (e.g., sotalol, amiodarone) or an ICD may be employed.
Ventricular FibrillationVentricular fibrillation is a life-threatening emergency that requires immediate treatment. This dysrhythmia results from the asynchronous discharge of multiple ventricular ectopic foci. Because many different foci are firing and because each focus initiates contraction in its immediate vicinity, localized twitching takes place all over the ventricles, making coordinated ventricular contraction impossible. As a result, the pumping action of the heart stops. In the absence of blood flow, the patient becomes unconscious and cyanotic. If heartbeat is not restored rapidly, death soon follows. Electrical countershock (defibrillation) is applied to eliminate fibrillation and restore cardiac function. Amiodarone can be used for acute and long-term suppression.
Premature Ventricular Complexes (PVCs)PVCs are beats that occur before they should in the cardiac cycle. These beats are caused by ectopic ventricular foci. PVCs may arise from a single ectopic focus or from several foci. In the absence of additional signs of heart disease, PVCs are benign and not usually treated. However, in the presence of acute myocardial infarction, PVCs may predispose the patient to ventricular fibrillation. In this case, therapy is required. A beta blocker is the agent of choice.
Digoxin-Induced Ventricular DysrhythmiasDigoxin toxicity can mimic practically all types of dysrhythmias. Varying degrees of AV block are among the most common. Ventricular flutter and ventricular fibrillation are the most dangerous. Digoxin causes dysrhythmias by increasing auto-maticity in the atria, ventricles, and His-Purkinje system, and by decreasing conduction through the AV node.
With proper treatment, digoxin-induced dysrhythmias can almost always be controlled. Treatment is discussed in Chapter 48. If antidysrhythmic drugs are required, lidocaine and phenytoin are the agents of choice. In patients with digoxin toxicity, DC cardioversion may bring on ventricular fibrillation. Accordingly, this procedure should be used only when absolutely required.
Torsades de PointesTorsades de pointes is an atypical, rapid, undulating ventricular tachydysrhythmia that can evolve into potentially fatal ven-tricular fibrillation. The main factor associated with development of torsades de pointes is prolongation of the QT interval, which can be caused by a variety of drugs, including class IA and class III antidysrhythmic agents. Acute management consists of IV magnesium plus cardioversion for sustained ventricular tachycardia.
PRINCIPLES OF ANTIDYSRHYTHMIC DRUG THERAPYBalancing Risks and BenefitsTherapy with antidysrhythmic drugs is based on a simple but important concept: Treat only if there is a clear benefit—and
conduction through the AV node. For patients who elect to restore normal rhythm, options are DC cardioversion, short-term treatment with drugs (e.g., amiodarone, sotalol), or RF ablation of the dysrhythmia source.
To prevent stroke, patients are treated with warfarin or newer anticoagulants. For those undergoing treatment to restore normal sinus rhythm, warfarin should be taken for 3 weeks before the procedure and for 4 weeks after. For those taking an antidysrhythmic drug long term to control ventricular rate, warfarin must be taken long term too. Alternatives to warfarin include four oral anticoagulants (apixaban [Eliquis], dabigatran [Pradaxa], edoxaban [Savaysa], and rivaroxaban [Xarelto]) and antiplatelet drugs (aspirin alone or aspirin plus clopidogrel).
Atrial FlutterAtrial flutter is caused by an ectopic atrial focus discharging at a rate of 250 to 350 times a minute. Ventricular rate is considerably slower, however, because the AV node is unable to transmit impulses at this high rate. Typically, one atrial impulse out of two reaches the ventricles. The treatment of choice is DC cardioversion, which almost always converts atrial flutter to normal sinus rhythm. Cardioversion may also be achieved with IV ibutilide. To prevent the dysrhythmia from recurring, patients may need long-term therapy with drugs—either a class IC agent (flecainide or propafenone) or a class III agent (amiodarone, dronedarone, sotalol, dofetilide).
There are two alternatives to cardioversion: (1) RF ablation of the dysrhythmia focus and (2) control of ventricular rate with drugs. As with atrial fibrillation, ventricular rate is con-trolled with drugs that suppress AV conduction: verapamil, diltiazem, or a beta blocker.
Like atrial fibrillation, atrial flutter poses a risk of stroke, which can be reduced by treatment with anticoagulants.
Sustained Supraventricular Tachycardia (SVT)Sustained SVT is usually caused by an AV nodal reentrant circuit. Heart rate is increased to 150 to 250 beats/min. SVT often responds to interventions that increase vagal tone, such as carotid sinus massage or the Valsalva maneuver. If these are ineffective, an IV beta blocker or calcium channel blocker can be tried. With these drugs, ventricular rate will be slowed even if the dysrhythmia persists. Once the dysrhythmia has been controlled, beta blockers and/or calcium channel blockers can be taken orally to prevent recurrence. As a last resort, amiodarone can be used for prevention.
Ventricular DysrhythmiasIn contrast to atrial dysrhythmias, which are generally benign, ventricular dysrhythmias can cause significant disruption of cardiac pumping. Accordingly, the usual objective is to abolish the dysrhythmia. Cardioversion is often the treatment of choice. When antidysrhythmic drugs are indicated, agents in class I or class III are usually employed.
Sustained Ventricular TachycardiaVentricular tachycardia arises from a single, rapidly firing ventricular ectopic focus, typically located at the border of an old infarction. The focus drives the ventricles at a rate of 150 to 250 beats/min. Because the ventricles cannot pump effectively at these rates, immediate intervention is required. Cardioversion is the treatment of choice. If cardioversion fails to normalize

554
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Long-Term Treatment: Drug Selection and EvaluationSelecting a drug for long-term therapy is largely empiric. There are many drugs that might be employed, and we usually can’t predict which one is going to work. Therefore, finding an effective drug is done by trial and error.
Drug selection can be aided with electrophysiologic testing. In these tests, a dysrhythmia is generated artificially by pro-grammed electrical stimulation of the heart. If a candidate drug is able to suppress the electrophysiologically induced dysrhythmia, it may also work against the real thing.
Holter monitoring can be used to evaluate treatment. A Holter monitor is a portable ECG device that is worn by the patient around-the-clock. If Holter monitoring indicates that dysrhythmias are still occurring with the present drug, a different drug should be tried.
Minimizing RisksSeveral measures can help minimize risk. These include:
• Starting with low doses and increasing them gradually.• Using a Holter monitor during initial therapy to detect
danger signs—especially QT prolongation, which can precede torsades de pointes.
• Monitoring plasma drug levels. Unfortunately, although drug levels can be good predictors of noncardiac toxicity (e.g., quinidine-induced nausea), they are less helpful for predicting adverse cardiac effects.
PHARMACOLOGY OF THE ANTIDYSRHYTHMIC DRUGS
As discussed earlier in this chapter, the antidysrhythmic drugs fall into four main groups—classes I, II, III, and IV—plus a fifth group that includes adenosine and digoxin. The pharmacol-ogy of these drugs is presented here and in Table 49.2.
CLASS I: SODIUM CHANNEL BLOCKERSClass I antidysrhythmic drugs block cardiac sodium channels. By doing so, they decrease conduction velocity in the atria, ventricles, and His-Purkinje system.
There are three subgroups of class I agents. Drugs in all three groups block sodium channels. In addition, class IA agents delay repolarization, whereas class IB agents accelerate repolarization. Class IC agents have pronounced prodysrhythmic actions.
The class I drugs are similar in action and structure to the local anesthetics. In fact, one of these drugs—lidocaine—has both local anesthetic and antidysrhythmic applications.
Class IA AgentsQuinidineQuinidine is the oldest, best studied, and most widely used class IA drug. Accordingly, quinidine will serve as our prototype for the group. Like other antidysrhythmic agents, quinidine has prodysrhythmic actions.
then only if the benefit outweighs the risks. As a rule, this means that intervention is needed only when the dysrhythmia interferes with ventricular pumping.
Treatment offers two potential benefits: reduction of symptoms and reduction of mortality. Symptoms that can be reduced include palpitations, angina, dyspnea, and faintness. For most antidysrhythmic drugs, there is little or no evidence of reduced mortality. In fact, mortality may actually increase.
Antidysrhythmic therapy carries considerable risk. Because of their prodysrhythmic actions, antidysrhythmic drugs can exacerbate existing dysrhythmias and generate new ones. Examples abound: Toxic doses of digoxin can generate a wide variety of dysrhythmias; drugs that prolong the QT interval can cause torsades de pointes; many drugs can cause ventricular ectopic beats; several drugs (quinidine, flecainide, propafenone) can cause atrial flutter; and one drug—flecainide—can produce incessant ventricular tachycardia. Because of their prodys-rhythmic actions, antidysrhythmic drugs can increase mortality. Other adverse effects include HF and third-degree AV block (caused by calcium channel blockers and beta blockers), as well as many noncardiac effects, including severe diarrhea (quinidine), a lupus-like syndrome (procainamide), and pul-monary toxicity (amiodarone).
Properties of the Dysrhythmia to Be ConsideredSustained Versus Nonsustained DysrhythmiasAs a rule, nonsustained dysrhythmias require intervention only when they are symptomatic; in the absence of symptoms, treatment is usually unnecessary. In contrast, sustained dys-rhythmias can be dangerous; therefore, the benefits of treatment generally outweigh the risks.
Asymptomatic Versus Symptomatic DysrhythmiasNo study has demonstrated a benefit to treating dysrhythmias that are asymptomatic or minimally symptomatic. In contrast, therapy may be beneficial for dysrhythmias that produce symptoms (palpitations, angina, dyspnea, faintness).
Supraventricular Versus Ventricular DysrhythmiasSupraventricular dysrhythmias are generally benign. The primary harm comes from driving the ventricles too rapidly to allow adequate filling. The goal of treatment is to either (1) terminate the dysrhythmia or (2) prevent excessive atrial beats from reaching the ventricles (using a beta blocker, calcium channel blocker, or digoxin). In contrast to supraventricular dysrhythmias, ventricular dysrhythmias frequently interfere with pumping. Accordingly, the goal of treatment is to terminate the dysrhythmia and prevent its recurrence.
Phases of TreatmentTreatment has two phases: acute and long term. The goal of acute treatment is to terminate the dysrhythmia. For many dysrhythmias, termination is accomplished with DC cardiover-sion (electrical countershock) or vagotonic maneuvers (e.g., carotid sinus massage) rather than drugs. The goal of long-term therapy is to prevent dysrhythmias from recurring. Quite often, the risks of long-term prophylactic therapy outweigh the benefits.

555
CHAPTER 49 Antidysrhythmic Drugs
Chemistry and Source. Quinidine is similar to quinine in structure and actions. The natural source of both drugs is the bark of the South American cinchona tree. Accordingly, these agents are referred to as cinchona alkaloids. Like quinine, quinidine has antimalarial and antipyretic properties.
Effects on the Heart. By blocking sodium channels, quinidine slows impulse conduction in the atria, ventricles, and His-Purkinje system. In addition, the drug delays repolariza-tion at these sites, apparently by blocking potassium channels. Both actions contribute to suppression of dysrhythmias.
Quinidine is strongly anticholinergic (atropine-like) and blocks vagal input to the heart. The resultant increase in SA nodal automaticity and AV conduction can drive the ventricles at an excessive rate. To prevent excessive ventricular stimula-tion, patients are usually pretreated with digoxin, verapamil, or a beta blocker, all of which suppress AV conduction.
Drug Usual Route Effects on the ECG Major Antidysrhythmic Applications
CLASS IA
Quinidine PO Widens QRS, prolongs QT Broad spectrum: used for long-term suppression of ventricular and supraventricular dysrhythmias
Procainamide PO Widens QRS, prolongs QT Broad spectrum: similar to quinidine, but toxicity makes it less desirable for long-term use
Disopyramide PO Widens QRS, prolongs QT Ventricular dysrhythmias
CLASS IB
Lidocaine IV No significant change Ventricular dysrhythmiasPhenytoin PO No significant change Digoxin-induced ventricular dysrhythmiasMexiletine PO No significant change Ventricular dysrhythmias
CLASS IC
Flecainide PO Widens QRS, prolongs PR Maintenance therapy of supraventricular dysrhythmiasPropafenone PO Widens QRS, prolongs PR Maintenance therapy of supraventricular dysrhythmias
CLASS II
Propranolol PO Prolongs PR, bradycardia Dysrhythmias caused by excessive sympathetic activity; control of ventricular rate in patients with supraventricular tachydysrhythmias
Acebutolol PO Prolongs PR, bradycardia Premature ventricular beatsEsmolol IV Prolongs PR, bradycardia Control of ventricular rate in patients with supraventricular
tachydysrhythmias
CLASS III
Amiodarone PO, IV Prolongs QT and PR, widens QRS Life-threatening ventricular dysrhythmias, atrial fibrillationa
Dronedarone PO Prolongs QT and PR, widens QRS Atrial flutter, atrial fibrillationSotalol PO, IV Prolongs QT and PR, bradycardia Life-threatening ventricular dysrhythmias, atrial fibrillation/flutterDofetilide PO Prolongs QT Highly symptomatic atrial dysrhythmiasIbutilide IV Prolongs QT Atrial flutter, atrial fibrillation
CLASS IV
Verapamil PO Prolongs PR, bradycardia Control of ventricular rate in patients with supraventricular tachydysrhythmias
Diltiazem IV Prolongs PR, bradycardia Same as verapamil
OTHERS
Adenosine IV Prolongs PR Termination of paroxysmal supraventricular tachycardiaDigoxin PO, IV Prolongs PR, depresses ST Control of ventricular rate in patients with supraventricular
tachydysrhythmias
TABLE 49.2 ■ Properties of Antidysrhythmic Drugs
aAmiodarone is widely used for atrial fibrillation, but it is not approved for this use.
Prototype Drugs
ANTIDYSRHYTHMIC DRUGSClass I: Sodium Channel BlockersQuinidine (class IA)Lidocaine (class IB)
Class II: Beta BlockersPropranolol
Class III: Drugs That Delay RepolarizationAmiodarone
Class IV: Calcium Channel BlockersVerapamil
OthersAdenosineDigoxin

556
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
decreasing digoxin elimination. When these drugs are used concurrently, digoxin dosage must be reduced. Also, patients should be monitored closely for digoxin toxicity (dysrhythmias). Because of its interaction with digoxin, quinidine is a last-choice drug for treating digoxin-induced dysrhythmias.
Other Interactions. Because of its anticholinergic actions, quinidine can intensify the effects of other atropine-like drugs; one possible result is excessive tachycardia. Phenobarbital, phenytoin, and other drugs that induce hepatic drug metabolism can shorten the half-life of quinidine by as much as 50%. Quinidine can intensify the effects of warfarin by an unknown mechanism.
Preparations, Dosage, and AdministrationPreparations. Quinidine is available as two salts: quinidine sulfate and
quinidine gluconate. Because these salts have different molecular weights, equal doses (on a milligram basis) do not provide equal amounts of quinidine. A 200-mg dose of quinidine sulfate is equivalent to 275 mg of quinidine gluconate. Quinidine sulfate is available in immediate-release tablets (200 and 300 mg) and extended-release tablets (300 mg). Quinidine gluconate is available in sustained-release tablets (324 mg) and in solution (80 mg/mL) for parenteral use.
Dosage. The usual dosage of quinidine sulfate is 200 to 400 mg every 6 hours. The usual dosage of quinidine gluconate is 324 to 648 mg every 8 to 12 hours. Dosage is adjusted to produce plasma quinidine levels between 2 and 5 mcg/mL.
Administration. Quinidine is almost always administered by mouth. Intramuscular administration is painful and produces erratic absorption. Intravenous injection carries a high risk of adverse cardiovascular reactions; therefore, continuous cardiovascular monitoring is required.
ProcainamideProcainamide [Procan ] is similar to quinidine in actions and uses. Like quinidine, procainamide is active against a broad spectrum of dysrhythmias. Unfortunately, serious side effects frequently limit its use.
Effects on the Heart and ECG. Like quinidine, procainamide blocks cardiac sodium channels, thereby decreasing conduction velocity in the atria, ventricles, and His-Purkinje system. Also, the drug delays repolarization. In contrast to quinidine, procainamide is only weakly anticholinergic, and hence is not likely to increase ventricular rate. Effects on the ECG are the same as with quinidine: widening of the QRS complex and prolongation of the QT interval.
Therapeutic Uses. Procainamide is effective against a broad spectrum of atrial and ventricular dysrhythmias. Like quinidine, the drug can be used for long-term suppression. However, since prolonged therapy is often associated with serious adverse effects, procainamide is less desirable than quinidine for long-term use. In contrast to quinidine, procainamide can be used to terminate ventricular tachycardia and ventricular fibrillation.
Adverse EffectsSystemic Lupus Erythematosus–Like Syndrome. Prolonged treatment
with procainamide is associated with severe immunologic reactions. Within a year, about 70% of patients develop antinuclear antibodies (ANAs)—anti-bodies directed against the patient’s own nucleic acids. If procainamide is continued, between 20% and 30% of patients with ANAs go on to develop symptoms resembling those of systemic lupus erythematosus (SLE). These symptoms include pain and inflammation of the joints, pericarditis, fever, and hepatomegaly. When procainamide is withdrawn, symptoms usually slowly subside. If the patient has a life-threatening dysrhythmia for which no alternative drug is available, procainamide can be continued and the symptoms of SLE controlled with a nonsteroidal anti-inflammatory drug (e.g., aspirin)
Effects on the ECG. Quinidine has two pronounced effects on the ECG. The drug widens the QRS complex (by slowing depolarization of the ventricles) and prolongs the QT interval (by delaying ventricular repolarization).
Therapeutic Uses. Quinidine is a broad-spectrum agent active against supraventricular and ventricular dysrhythmias. The drug’s principal indication is long-term suppression of dysrhythmias, including SVT, atrial flutter, atrial fibrillation, and sustained ventricular tachycardia. An analysis of older studies indicates that quinidine may actually increase mortality in patients with atrial flutter and atrial fibrillation.
In addition to its antidysrhythmic applications, quinidine is a drug of choice for severe malaria (see Chapter 98). The pharmacokinetics of quinidine and other antidysrhythmics can be found in Table 49.3.
Adverse EffectsDiarrhea. Diarrhea and other GI symptoms develop in
about 33% of patients. These reactions can be immediate and intense, frequently forcing discontinuation of treatment. Gastric upset can be reduced by administering quinidine with food.
Cinchonism. Cinchonism is characterized by tinnitus (ringing in the ears), headache, nausea, vertigo, and disturbed vision. These can develop with just one dose.
Cardiotoxicity. At high concentrations, quinidine can cause severe cardiotoxicity (sinus arrest, AV block, ventricular tachydysrhythmias, asystole). These reactions occur secondary to increased automaticity of Purkinje fibers and reduced conduc-tion throughout all regions of the heart.
As cardiotoxicity develops, the ECG changes. Important danger signals are widening of the QRS complex (by 50% or more) and excessive prolongation of the QT interval. Notify the prescriber immediately if these changes occur.
Arterial Embolism. Embolism is a potential complication of treating atrial fibrillation. During atrial fibrillation, thrombi may form in the atria. When sinus rhythm is restored, these thrombi may be dislodged and cause embolism. To reduce the risk of embolism, anticoagulant therapy is given for 3 to 4 weeks before quinidine and is maintained for an additional 4 weeks. Signs of embolism (e.g., sudden chest pain, dyspnea) should be reported immediately.
Other Adverse Effects. Quinidine can cause alpha-adrenergic blockade, resulting in vasodilation and subsequent hypotension. This reaction is much more serious with IV therapy than with oral therapy. Rarely, quinidine has caused hypersen-sitivity reactions, including fever, anaphylactic reactions, and thrombocytopenia.
Drug InteractionsDigoxin. Quinidine can double digoxin levels. The increase
is caused by displacing digoxin from plasma albumin and by
Drug Usual Route Peak (hr) Half-Life (hr) Metabolism Excretion
Quinidine PO 0.5–1.5 6–8 Hepatic Renal
Procainamide [Procan ] IV — 3–4 Hepatic Renal
Lidocaine [Xylocaine] IV — 1.5–2 Hepatic RenalAmiodarone PO — 600–2640 Hepatic Gastrointestinal (bile)Dronedarone [Multaq] PO 3–6 13–19 Hepatic Gastrointestinal (feces)Dofetilide [Tikosyn] PO 2–3 10 Hepatic (minor) Renal
TABLE 49.3 ■ Pharmacokinetic Properties of Antidysrhythmic Drugs

557
CHAPTER 49 Antidysrhythmic Drugs
LidocaineLidocaine [Xylocaine], an intravenous agent, is used only for ventricular dysrhythmias. In addition to its antidysrhythmic applications, lidocaine is employed as a local anesthetic (see Chapter 26).
Effects on the Heart and ECG. Lidocaine has three significant effects on the heart: (1) like other class I drugs, lidocaine blocks cardiac sodium channels and thereby slows conduction in the atria, ventricles, and His-Purkinje system; (2) the drug reduces automaticity in the ventricles and His-Purkinje system by a mechanism that is poorly understood; and (3) lidocaine accelerates repolarization (shortens the action potential duration and ERP). In contrast to quinidine and procainamide, lidocaine is devoid of anticholinergic properties. Also, lidocaine has no significant impact on the ECG: A small reduction in the QT interval may occur, but there is no QRS widening.
Antidysrhythmic Use. Antidysrhythmic use of lidocaine is limited to short-term therapy of ventricular dysrhythmias. Lidocaine is not active against supraventricular dysrhythmias.
Adverse Effects. Lidocaine is generally well tolerated. However, adverse central nervous system (CNS) effects can occur. High therapeutic doses can cause drowsiness, confusion, and paresthesias. Toxic doses may produce seizures and respiratory arrest. Consequently, whenever lidocaine is used, equipment for resuscitation must be available. Seizures can be managed with diazepam.
Preparations, Dosage, and Administration. Administra-tion is parenteral only. The usual route is IV. Intramuscular injection can be used in emergencies. Blood pressure and the ECG should be monitored for signs of toxicity.
Intravenous. Lidocaine [Xylocaine] preparations intended for IV administration are clearly labeled as such. They contain no preservatives or catecholamines. (Lidocaine used for local anesthesia frequently contains epinephrine.) Preparations that contain epinephrine or another catecholamine must never be administered IV, since doing so can cause severe hypertension and life-threatening dysrhythmias.
Intravenous therapy is initiated with a loading dose followed by continuous infusion for maintenance. The usual loading dose is 50 to 100 mg (1 mg/kg) administered at a rate of 25 to 50 mg/min. An infusion rate of 1 to 4 mg/min is used for maintenance; the rate is adjusted on the basis of cardiac response. Intravenous lidocaine should be discontinued as soon as possible, usually within 24 hours. Lidocaine for IV administration is supplied in concentrated and dilute formulations. The concentrated formulations must be diluted with 5% dextrose in water.
To avoid toxicity, dosage should be reduced in patients with impaired hepatic function or impaired hepatic blood flow (e.g., older adult patients; patients with cirrhosis, shock, or HF).
PhenytoinPhenytoin is an antiseizure drug that is also used to treat digoxin-induced dysrhythmias. The basic pharmacology of phenytoin is shown in Chapter 24. Discussion here is limited to antidysrhythmic applications.
Effects on the Heart and ECG. Like lidocaine, phenytoin reduces automaticity (especially in the ventricles) and has little or no effect on the ECG. In contrast to lidocaine (and practically all other antidysrhythmic agents), phenytoin increases AV nodal conduction.
Pharmacokinetics. Phenytoin has two unfortunate kinetics properties. First, metabolism of the drug is subject to wide interpatient variation. Second, doses only slightly greater than therapeutic are likely to cause toxicity. Because of these characteristics, maintenance of therapeutic plasma levels (5 to 20 mcg/mL) is difficult.
Adverse Effects and Interactions. The most common adverse reactions are sedation, ataxia, and nystagmus. With too-rapid IV administration, phenytoin can cause hypotension, dysrhythmias, and cardiac arrest. Gingival hyperplasia is a frequent complication of long-term treatment. Phenytoin is subject to multiple undesirable drug interactions (see Chapter 24).
or a glucocorticoid. All patients taking procainamide chronically should be tested for ANAs. If the ANA titer rises, discontinuation of treatment should be considered.
Blood Dyscrasias. About 0.5% of patients develop blood dyscrasias, including neutropenia, thrombocytopenia, and agranulocytosis. Fatalities have occurred. These reactions usually develop during the first 12 weeks of treatment. Complete blood counts should be obtained weekly during this time and periodically thereafter. Also, complete blood counts should be obtained promptly at the first sign of infection, bruising, or bleeding. If blood counts indicate bone marrow suppression, procainamide should be withdrawn. Hematologic status usually returns to baseline within 1 month.
Cardiotoxicity. Procainamide has cardiotoxic actions like those of quinidine. Warning signs are QRS widening (more than 50%) and excessive QT prolongation. If these develop, the drug should be withheld and the prescriber informed.
Other Adverse Effects. Like quinidine, procainamide can cause GI symptoms and hypotension. However, these are much less prominent than with quinidine. Procainamide is a derivative of procaine (a local anesthetic); therefore, its use is contraindicated in patients with a history of procaine allergy. As with quinidine, arterial embolism may occur during treatment of atrial fibrillation.
Preparations, Dosage, and AdministrationOral. Procainamide is available in capsules (250, 375, and 500 mg) and
sustained-release tablets (250, 500, and 750 mg) in Canada. The usual maintenance dosage is 50 mg/kg/day in divided doses. The capsules are administered every 3 to 4 hours and the sustained-release tablets every 6 hours. Dosage is adjusted to maintain plasma drug levels between 4 and 8 mcg/mL.
Parenteral. Procainamide is available in solution (100 and 500 mg/mL) for IM and IV administration. Intramuscular injection is made deep into the gluteal muscle; dosage is 0.5 to 1 gm repeated every 4 to 8 hours.
Intravenous infusion may be performed at an initial rate of 20 to 50 mg/min (maximal loading dose is 1500 mg). After the loading period, an infusion rate of 2 to 6 mg/min should be employed. Once the dysrhythmia has been controlled, the patient should be switched to oral procainamide. Three hours should elapse between terminating the infusion and the first oral dose.
DisopyramideDisopyramide [Norpace, Norpace CR] is a class I drug with actions like those of quinidine. However, because of prominent side effects, indications for disopyramide are limited.
Effects on the Heart and ECG. Cardiac effects are similar to those of quinidine. By blocking sodium channels, disopyramide decreases conduction velocity in the atria, ventricles, and His-Purkinje system. In addition, the drug delays repolarization. Anticholinergic actions are greater than those of quinidine. In contrast to quinidine, disopyramide causes a pronounced reduction in contractility. Like quinidine, disopyramide widens the QRS complex and prolongs the QT interval.
Adverse Effects. Anticholinergic responses are most common. These include dry mouth, blurred vision, constipation, and urinary hesitancy or retention. Urinary retention frequently requires discontinuation of treatment.
Because of its negative inotropic effects, disopyramide can cause severe hypotension (secondary to reduced cardiac output) and can exacerbate heart failure. The drug should not be administered to patients with HF or to patients taking a beta blocker. Whenever disopyramide is used, pressor drugs should be immediately available.
Therapeutic Uses. Disopyramide is indicated only for ventricular dysrhythmias (PVCs, ventricular tachycardia, ventricular fibrillation). The drug is reserved for patients who cannot tolerate safer medications (e.g., quinidine, procainamide).
Preparations, Dosage, and Administration. Disopyramide [Norpace, Norpace CR] is available in immediate- and extended-release capsules (100 and 150 mg). An initial loading dose (200 to 300 mg) is followed by main-tenance doses (100 to 200 mg) every 6 hours.
Class IB AgentsAs a group, class IB agents differ from quinidine and the other class IA agents in two respects: (1) whereas class IA agents delay repolarization, class IB agents accelerate repo-larization; and (2) class IB agents have little or no effect on the ECG.

558
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
not known if propafenone, like flecainide, increases mortality in patients with asymptomatic ventricular dysrhythmias after myocardial infarction. Propafenone has beta-adrenergic blocking properties and can thereby decrease myocardial contractility and promote bronchospasm. Accordingly, the drug should be used with caution in patients with HF, AV block, or asthma. Noncardiac adverse effects are generally mild and include dizziness, altered taste, blurred vision, and GI symptoms (abdominal discomfort, anorexia, nausea, vomiting). Because of its prodysrhythmic actions, propafenone should be reserved for patients who have not responded to safer drugs. Propafenone is available in immediate-release tablets (150 and 225 mg) and extended-release capsules (225, 325, and 425 mg). For the immediate-release tablets, the dosage is 150 mg every 8 hours initially, and can be gradually increased to 300 mg every 8 hours. For the extended-release capsules, the dosage is 225 mg every 12 hours initially; it can be gradually increased to 425 mg every 12 hours.
CLASS II: BETA BLOCKERS
Class II consists of beta-adrenergic blocking agents. At this time only four beta blockers—propranolol, acebutolol, esmolol, and sotalol—are approved for treating dysrhythmias. One of these drugs—sotalol—also blocks potassium channels; it is discussed under class III. The basic pharmacology of the beta blockers is presented in Chapter 18. Discussion here is limited to their antidysrhythmic use.
PropranololPropranolol [Inderal LA] is considered a nonselective beta-adrenergic antagonist, in that it blocks both beta1- and beta2-adrenergic receptors. Beta1 blockade affects the heart, and beta2 blockade affects the bronchi.
Effects on the Heart and ECGBlockade of cardiac beta1 receptors attenuates sympathetic stimulation of the heart. The result is (1) decreased automaticity of the SA node, (2) decreased velocity of conduction through the AV node, and (3) decreased myocardial contractility. The reduction in AV conduction velocity translates to a prolonged PR interval on the ECG.
It is worth noting that cardiac beta1 receptors are functionally coupled to calcium channels and that beta1 blockade causes these channels to close. Therefore, the effects of beta blockers on heart rate, AV conduction, and contractility all result from decreased calcium influx. Because beta blockers and calcium channel blockers both decrease calcium entry, the cardiac effects of these drugs are very similar.
Therapeutic UsePropranolol is especially useful for treating dysrhythmias caused by excessive sympathetic stimulation of the heart. Among these are sinus tachycardia, severe recurrent ventricular tachycardia, exercise-induced tachydysrhythmias, and paroxysmal atrial tachycardia evoked by emotion or exercise. In patients with supraventricular tachydysrhythmias, propranolol has two beneficial effects: (1) suppression of excessive discharge of the SA node and (2) slowing of ventricular rate by decreasing transmission of atrial impulses through the AV node.
Adverse EffectsBeta blockers are generally well tolerated. Principal adverse effects concern the heart and bronchi. By blocking cardiac beta1 receptors, propranolol can cause heart failure, AV block,
Antidysrhythmic Applications. Phenytoin has been used for digoxin-induced dysrhythmias and for acute and chronic suppression of ventricular dysrhythmias. The ability of phenytoin to increase AV nodal conduction can help counteract the reduction in AV conduction caused by digoxin intoxication. Phenytoin should not be used to treat atrial fibrillation or atrial flutter because enhanced AV conduction could increase the number of atrial impulses reaching the ventricles, thereby driving the ventricles at an excessive rate.
Dosage and Administration. Phenytoin can be given PO or IV. For PO therapy, a loading dose (14 mg/kg) is followed by daily maintenance doses (200 to 400 mg).
Intravenous dosing is reserved for severe acute dysrhythmias. Blood pressure and the ECG must be monitored continuously. Phenytoin is not soluble in water and must be diluted in the medium supplied by the manufacturer. This medium is highly alkaline (pH 12) and will cause phlebitis if given by continuous infusion. Consequently, dosing is by intermittent injections. Intravenous injections must be performed slowly (50 mg/min or less), because rapid injection can cause cardiovascular collapse. Treatment is begun with a series of loading doses (100 mg every 5 minutes until the dysrhythmia has been controlled or until a maximum dose of 20 mg/kg is reached). Maintenance dosages range from 200 to 400 mg/day.
MexiletineMexiletine is an oral analog of lidocaine used for symptomatic ventricular dysrhythmias. Principal indications are PVCs and sustained ventricular tachycardia. Like lidocaine, mexiletine does not alter the ECG. The drug is eliminated by hepatic metabolism, so effects may be prolonged in patients with liver disease or reduced hepatic blood flow. The most common adverse effects are GI (nausea, vomiting, diarrhea, constipation) and neurologic (tremor, dizziness, sleep disturbances, psychosis, convulsions). About 40% of patients find these intolerable. Like other class I agents, mexiletine has prodysrhythmic properties. The initial dosage is 200 mg every 8 hours. The maintenance dosage is 200 to 300 mg every 8 hours. All doses should be taken with food.
Mexiletine is also used to alleviate persistent pain of diabetic neuropathy. Benefits derive from lidocaine-like anesthetic actions. Because mexiletine can cause dysrhythmias, it should not be used by diabetic patients with heart disease.
Class IC AgentsClass IC antidysrhythmics block cardiac sodium channels and thereby reduce conduction velocity in the atria, ventricles, and His-Purkinje system. In addition, these drugs delay ventricular repolarization, causing a small increase in the effective refractory period. All class IC agents can exacerbate existing dys-rhythmias and create new ones. Currently, only two class IC agents are available: flecainide and propafenone.
FlecainideFlecainide is active against a variety of ventricular and supraventricular dysrhythmias. However, use is restricted largely to maintenance therapy of supraventricular dysrhythmias. Like other class IC agents, flecainide decreases cardiac conduction and increases the effective refractory period. Prominent effects on the ECG are prolongation of the PR interval and widening of the QRS complex. Excessive QRS widening indicates a need for dosage reduction. Flecainide has prodysrhythmic effects. As a result, the drug can intensify existing dysrhythmias and provoke new ones. In patients with asymptomatic ventricular tachycardia associated with acute myocardial infarction, flecainide has caused a twofold increase in mortality. Flecainide decreases myocardial contractility and can thereby exacerbate or precipitate HF. Accordingly, the drug should not be combined with other agents that can decrease contractile force (e.g., beta blockers, verapamil, diltiazem). Elimination is by hepatic metabolism and renal excretion. Flecainide is available in tablets (50, 100, and 150 mg) for oral dosing. Dosage is low initially (50 to 100 mg every 12 hours) and then gradually increased to a maximum of 400 mg/day. Because of its potential for serious side effects, flecainide should be reserved for severe ventricular dysrhythmias that have not responded to safer drugs. Patients should be monitored closely.
PropafenonePropafenone [Rythmol, Rythmol SR] is similar to flecainide in actions and uses. By blocking cardiac sodium channels, the drug decreases conduction velocity in the atria, ventricles, and His-Purkinje system. In addition, it causes a small increase in the ventricular ERP. Prominent effects on the ECG are QRS widening and PR prolongation. Like flecainide, propafenone has prodysrhythmic actions that can exacerbate existing dysrhythmias and create new ones. It is

559
CHAPTER 49 Antidysrhythmic Drugs
Oral TherapyTherapeutic Use. Although amiodarone is very effective,
concerns about toxicity limit its indications. In the United States, oral amiodarone is approved only for long-term therapy of two life-threatening ventricular dysrhythmias: recurrent ventricular fibrillation and recurrent hemodynamically unstable ventricular tachycardia. Treatment should be reserved for patients who have not responded to safer drugs.
Amiodarone is our most effective drug for atrial fibrillation and is prescribed widely to treat this dysrhythmia—even though it is not approved for this use. The drug is given to convert atrial fibrillation to normal sinus rhythm and to maintain normal sinus rhythm following conversion.
Effects on the Heart and ECG. Amiodarone has complex effects on the heart. Like all other drugs in this class, amiodarone delays repolarization, and thereby prolongs the action potential duration and ERP. The underlying cause of these effects may be blockade of potassium channels. Additional cardiac effects include reduced automaticity in the SA node, reduced contractil-ity, and reduced conduction velocity in the AV node, ventricles, and His-Purkinje system. These occur secondary to blockade of sodium channels, calcium channels, and beta receptors. Prominent effects on the ECG are QRS widening and prolonga-tion of the PR and QT intervals. Amiodarone also acts on coronary and peripheral blood vessels to promote dilation.
Adverse Effects. Amiodarone produces many serious adverse effects. Furthermore, because the drug’s half-life is protracted, toxicity can continue for weeks or months after drug withdrawal. To reduce adverse events, the U.S. Food and Drug Administration (FDA) requires that all patients using amiodarone be given a Medication Guide describing potential toxicities.
Pulmonary Toxicity. Lung damage—hypersensitivity pneumonitis, interstitial/alveolar pneumonitis, pulmonary fibrosis—is the greatest concern. Symptoms (dyspnea, cough, chest pain) resemble those of HF and pneumonia. Pulmonary toxicity develops in 2% to 17% of patients and carries a 10% risk of mortality. Patients at highest risk are those receiving long-term, high-dose therapy. A baseline chest x-ray and pulmonary function test are recommended. Pulmonary function should be monitored throughout treatment. If lung injury develops, amiodarone should be withdrawn.
Cardiotoxicity. Amiodarone may cause a paradoxical increase in dysrhythmic activity. In addition, by suppressing the SA and AV nodes, the drug can cause sinus bradycardia and AV block. By reducing contractility, amiodarone can precipitate HF.
Thyroid Toxicity. Amiodarone may cause hypothyroidism or hyperthyroidism. Accordingly, thyroid function should be assessed at baseline and periodically during treatment. Hypo-thyroidism can be treated with thyroid hormone supplements. Hyperthyroidism can be treated with an antithyroid drug (e.g., methimazole) or thyroidectomy. Discontinuing amiodarone should be considered.
Liver Toxicity. Amiodarone can injure the liver. Accordingly, tests of liver function should be obtained at baseline and periodically throughout treatment. If circulating liver enzymes exceed 3 times the normal level, amiodarone should be dis-continued. Signs and symptoms of liver injury, which are seen only rarely, include anorexia, nausea, vomiting, malaise, fatigue, itching, jaundice, and dark urine.
and sinus arrest. Hypotension can occur secondary to reduced cardiac output. In patients with asthma, blocking beta2 receptors in the lung can cause bronchospasm. Because of its cardiac and pulmonary effects, propranolol should be used cautiously in patients with asthma, and it is contraindicated in patients with sinus bradycardia, high-degree heart block, and HF.Dosage and AdministrationPropranolol can be administered orally and, in life-threatening emergencies, by IV injection. Dosages with either route show wide individual variation. Oral dosages range from 10 to 30 mg every 6 to 8 hours. The usual IV dose is 1 to 3 mg injected at a rate of 1 mg/min.
AcebutololAcebutolol [Sectral] is a cardioselective beta blocker approved for oral therapy of PVCs. Adverse effects are like those of propranolol: bradycardia, HF, AV block, and—despite cardioselectivity—bronchospasm. Accordingly, acebutolol should be used cautiously in patients with asthma, and is contraindicated in patients with HF, severe bradycardia, and AV block. Acebutolol can also cause adverse immunologic reactions; titers of ANAs may rise, resulting in myalgia, arthralgia, and arthritis. For suppression of PVCs, the initial dosage is 200 mg twice daily. Usual maintenance dosages range from 600 to 1200 mg/day.
EsmololEsmolol [Brevibloc] is a cardioselective beta blocker with a very short half-life (9 minutes). Administration is by IV infusion. The drug is employed for immediate control of ventricular rate in patients with SVT, atrial flutter, and atrial fibrillation. Use is short term only (e.g., in patients with dysrhythmias associated with surgery). The most common adverse reaction is hypotension. However, like other beta blockers, esmolol can also cause bradycardia, heart block, HF, and bronchospasm (at higher doses). In addition, pain can occur at the infusion site. Esmolol is available in two concentrations: 10 and 20 mg/mL. Treatment begins with a loading dose (500 mcg/kg) infused over 1 minute. The usual maintenance infusion rate is 100 mcg/kg/min.
CLASS III: POTASSIUM CHANNEL BLOCKERS (DRUGS THAT DELAY REPOLARIZATION)Five class III antidysrhythmics are available: amiodarone, dronedarone, dofetilide, ibutilide, and sotalol (which is also a beta blocker). All five delay repolarization of fast potentials. Hence, all five prolong the action potential duration and ERP. By doing so, they prolong the QT interval. In addition, each drug can affect the heart in other ways, so they are not interchangeable.
AmiodaroneAmiodarone [Cordarone, Pacerone] is a class III antidysrhythmic agent that has complex effects on the heart. The drug is highly effective against both atrial and ventricular dysrhythmias. Unfortunately, serious toxicities (e.g., lung damage, visual impairment) are common and may persist for months after treatment has stopped. Because of toxicity, amiodarone is approved only for life-threatening ventricular dysrhythmias that have been refractory to safer agents. Nonetheless, because of its efficacy, amiodarone is one of our most frequently prescribed antidysrhythmic drugs, used for atrial and ventricular dysrhythmias alike.
Amiodarone is available for oral and IV use. Indications, electrophysiologic effects, time course of action, and adverse effects differ for each route. Accordingly, oral and IV therapy are discussed separately.

560
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
fibrillation, AV nodal reentrant tachycardia, and shock-resistant ventricular fibrillation.
Effects on the Heart and ECG. In contrast to oral amio-darone, which affects multiple aspects of cardiac function, IV amiodarone affects primarily the AV node. Specifically, the drug slows AV conduction and prolongs AV refractoriness. Both effects probably result from antiadrenergic actions. The mechanism underlying antidysrhythmic effects is unknown.
Adverse Effects. The most common adverse effects are hypotension and bradydysrhythmias. Hypotension develops in 15% to 20% of patients, and may require discontinuation of treatment. Bradycardia or AV block occurs in 5% of patients; discontinuation of treatment or insertion of a pacemaker may be needed. Infusions containing more than 2 mg/mL (in 5% dextrose in water) produce a high incidence of phlebitis, and hence they should be administered through a central venous line. Torsades de pointes in association with QT prolongation occurs rarely.
Dosage. Dosing is complex. During the first 24 hours, a total dose of 1050 mg is infused. After that, a maintenance infusion (0.5 mg/min) is given around-the-clock. The usual duration of treatment is 2 to 4 days. However, maintenance infusions may be continued for up to 3 weeks before switching to oral amiodarone.
DronedaroneDronedarone [Multaq], a derivative of amiodarone, is indicated for oral therapy of atrial flutter and paroxysmal or persistent atrial fibrillation, but not permanent atrial fibrillation. The manufacturer hoped to create a drug with the high efficacy of amiodarone, but with less toxicity. Unfortunately, although dronedarone is somewhat less toxic than amiodarone, it is also less effective. Furthermore, in patients with HF or permanent atrial fibrillation, dronedarone doubles the risk of death. Dronedarone has a much shorter half-life than amiodarone, so adverse effects resolve more quickly.
Effects on the Heart and ECGLike other class III agents, dronedarone blocks cardiac potas-sium channels and thereby delays repolarization. In addition, dronedarone can block sodium channels (like class I agents), beta-adrenergic receptors (like class II agents), and calcium channels (like class IV agents). Just how these actions contribute to antidysrhythmic benefits is unclear. Prominent effects on the ECG are PR and QT prolongation and widening of the QRS complex.
Adverse EffectsThe most common side effects are diarrhea, weakness, nausea, and skin reactions. In contrast to amiodarone, dronedarone does not cause significant thyroid toxicity, pulmonary toxicity (e.g., pulmonary fibrosis, pneumonitis), or ocular toxicity (e.g., corneal microdeposits, optic neuropathy)—although it can cause liver toxicity. Dronedarone can increase skin sensitivity to sunlight, but it does not cause the bluish-gray skin discoloration seen with amiodarone. Furthermore, because dronedarone has a much shorter half-life than amiodarone, adverse effects that do occur have a much shorter duration.
Cardiac Effects. In patients with severe heart failure, dronedarone doubles the risk of death, as shown in the ANDROMIDA trial. Accordingly, dronedarone is contraindi-cated in patients with New York Heart Association (NYHA)
Ophthalmic Effects. Rarely, amiodarone has been associ-ated with optic neuropathy and optic neuritis, sometimes progressing to blindness. However, the absolute risk is small. Patients who develop changes in visual acuity or peripheral vision should undergo ophthalmologic evaluation. If optic neuropathy or neuritis is diagnosed, discontinuation of amio-darone should be considered.
Virtually all patients taking amiodarone develop corneal microdeposits. Fortunately, these deposits have little or no effect on vision, and so rarely necessitate the cessation of amiodarone.
Toxicity in Pregnancy and Breast-Feeding. Amiodarone crosses the placental barrier and enters breast milk, and can thereby harm the developing fetus and breast-feeding infant. Accordingly, pregnancy and breast-feeding should be avoided while using the drug and for several months after stopping it.
Dermatologic Toxicity. Patients frequently experience photosensitivity reactions (skin reactions triggered by exposure to ultraviolet radiation). To reduce risk, patients should avoid sunlamps, and should wear sunblock and protective clothing when outdoors. With frequent and prolonged sun exposure, exposed skin may turn bluish-gray. Fortunately, this discolor-ation resolves within months after amiodarone is discontinued.
Other Adverse Effects. Possible CNS reactions include ataxia, dizziness, tremor, mood alteration, and hallucinations. Gastrointestinal reactions (anorexia, nausea, vomiting) are common.
Drug Interactions. Amiodarone is subject to significant interactions with many drugs. The result can be toxicity or reduced therapeutic effects. Accordingly, combined use with these drugs should be avoided. When it cannot, the patient should be monitored closely. Interactions of concern include the following:
• Amiodarone can increase levels of several drugs, includ-ing quinidine, procainamide, phenytoin, digoxin, diltia-zem, warfarin, cyclosporine, and three statins: lovastatin, simvastatin, and atorvastatin. Dosages of these agents often require reduction.
• Amiodarone levels can be increased by grapefruit juice and by inhibitors of CYP3A4. Toxicity can result.
• Amiodarone levels can be reduced by cholestyramine (which decreases amiodarone absorption) and by agents that induce CYP3A4 (e.g., St. John’s wort, rifampin).
• The risk of severe dysrhythmias is increased by diuretics (because they can reduce levels of potassium and magnesium) and by drugs that prolong the QT interval, of which there are many.
• Combining amiodarone with a beta blocker, verapamil, or diltiazem can lead to excessive slowing of heart rate.
Dosage. Oral amiodarone [Cordarone, Pacerone] is available in tablets (100, 200, and 400 mg). Treatment should be initiated in a hospital. The following schedule is used for loading: 800 to 1600 mg daily for 1 to 3 weeks, followed by 600 to 800 mg daily for 4 weeks. The daily maintenance dosage is 400 mg.
Intravenous TherapyTherapeutic Use. Intravenous amiodarone is approved
only for initial treatment and prophylaxis of recurrent ventricular fibrillation and hemodynamically unstable ventricular tachy-cardia in patients refractory to safer drugs. For these indications, amiodarone may be lifesaving.
In addition to its approved uses, IV amiodarone has been used with success against other dysrhythmias, including atrial

561
CHAPTER 49 Antidysrhythmic Drugs
• Dronedarone can inhibit CYP2D6 and can thereby increase levels of CYP2D6 substrates (e.g., beta blockers, tricyclic antidepressants). Concurrent use of these agents should be done with caution.
• Beta blockers, which suppress the SA node and AV conduction, can intensify dronedarone-induced brady-cardia and can also increase the risk of AV block.
• Like the beta blockers, two calcium channel blockers—verapamil and diltiazem—also suppress the SA node and AV conduction and pose the same risk as the beta blockers do. In addition, verapamil and diltiazem can inhibit CYP3A4 and can thereby raise dronedarone levels, making the risk of cardiosuppression even greater.
• Drugs and supplements that prolong the QT interval (e.g., phenothiazines, tricyclic antidepressants, class I and class III antidysrhythmics) can intensify dronedarone-induced QT prolongation, and can thereby increase the risk of torsades de pointes. Accordingly, these drugs are contraindicated for use with dronedarone.
ContraindicationsDronedarone has the following contraindications:
• NYHA Class IV HF or NYHA Class II or III HF with recent decompensation requiring hospitalization
• Liver or lung toxicity related to previous amiodarone use
• Permanent atrial fibrillation• Second- or third-degree AV block or sick sinus syndrome
(except in patients using a pacemaker)• Bradycardia below 50 beats/min• PR interval greater than 280 msec• QT interval greater than 500 msec• Use of drugs or supplements that prolong the QT
interval• Use of strong inhibitors of CYP3A4• Pregnancy• Breast-feeding• Severe liver impairment
Preparations, Dosage, and AdministrationDronedarone [Multaq] is supplied in 400-mg tablets for oral dosing. The recommended dosage is 400 mg twice daily, taken with the morning and evening meals. Note that, unlike amiodarone, dronedarone does not require a loading dose.
SotalolActions and UsesSotalol [Betapace, Betapace AF] is a beta blocker that also delays repolarization. Hence, the drug has combined class II and class III antidysrhythmic properties. Prodysrhythmic properties are pronounced. Sotalol was initially approved only for ventricular dysrhythmias, such as sustained ventricular tachycardia, that are considered life threatening. Later, it was approved for prophylaxis and treatment of atrial flutter and fibrillation, but only if symptoms are severe. The drug is not approved for hypertension or angina pectoris (the primary indications for other beta blockers).
PharmacokineticsSotalol is administered orally and undergoes nearly complete absorption. The drug is excreted unchanged in the urine. Its half-life is 12 hours.
Adverse EffectsThe major adverse effect is torsades de pointes, a serious dysrhythmia that develops in about 5% of patients. Risk is increased by hypokalemia and by other drugs that prolong the QT interval.
Class IV HF and in patients with NYHA Class II or III HF with recent decompensation that required hospitalization.
In patients with permanent atrial fibrillation (as opposed to paroxysmal or persistent atrial fibrillation), dronedarone doubles the risk of death, as shown in the PALLAS trial. Accordingly, dronedarone should not be used in these patients.
Dronedarone reduces SA nodal automaticity and AV nodal conduction, posing a risk of bradycardia and heart block. Accordingly, the drug is contraindicated in patients with sick sinus syndrome or second- or third-degree AV block (unless a pacemaker is in use), and in patients with bradycardia below 50 beats/min.
Dronedarone prolongs the QT interval (by about 10 msec). Accordingly, the drug should not be used in patients with a QT interval greater than 500 msec or in patients taking drugs or supplements that cause QT prolongation.
Liver Toxicity. Dronedarone has been associated with rare cases of severe liver injury, including two that required a liver transplant. Accordingly, patients should be warned about signs and symptoms of liver injury (e.g., anorexia, nausea, vomiting, malaise, fatigue, itching, jaundice, dark urine), and instructed to contact their provider immediately if these develop. Providers should consider monitoring for liver enzymes in blood, espe-cially during the first 6 months of treatment.
Toxicity in Pregnancy and Breast-Feeding. Dronedarone is a proven teratogen and must not be used during pregnancy. In animal studies, doses at or below the mean recommended human dose have produced visceral, skeletal, and external malformations. Accordingly, dronedarone is classified in FDA Pregnancy Risk Category Xa: risks to the develop-ing fetus clearly outweigh any possible benefit. Women of childbearing age should be counseled about using effective contraception.
We know that dronedarone is excreted in the milk of rats, but information in lactating women is lacking. Nonetheless, owing to the potential risk to nursing infants, dronedarone is contraindicated for use by breast-feeding mothers.
Drug InteractionsDronedarone is subject to multiple drug interactions, many involving CYP3A4.
• Strong inhibitors of hepatic CYP3A4 (e.g., ketoconazole, clarithromycin, ritonavir) can make dronedarone accu-mulate to dangerous levels. Accordingly, concurrent use of these inhibitors is contraindicated. Grapefruit juice, which strongly inhibits intestinal CYP3A4, can raise dronedarone levels threefold and so should also be avoided. Moderate inhibitors of hepatic CYP3A4 (e.g., verapamil, diltiazem) should be used with caution.
• Strong inducers of CYP3A4 (e.g., rifampin, carbamaze-pine, St. John’s wort) can reduce dronedarone levels by as much as 80% and can thereby greatly reduce dys-rhythmia control.
• In addition to being a substrate for CYP3A4, dronedarone can inhibit this enzyme. Accordingly, dronedarone can raise levels of other drugs that are CYP3A4 substrates. Substrates with a narrow therapeutic range (e.g., tacro-limus, sirolimus, warfarin) should be used with caution.
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.

562
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Conversion to sinus rhythm occurs during the infusion or within 90 minutes of its termination. Ibutilide is more effective against atrial flutter (49% to 70% success) than atrial fibrillation (22% to 43% success). Like other class III agents, ibutilide blocks potassium channels and thereby prolongs the action potential duration and QT interval. Up to 8% of patients develop torsades de pointes, frequently in association with QT prolongation. Oral doses are tera-togenic and embryocidal in rats. For patients who weigh over 60 kg, the dosage is 1 mg infused over 10 minutes. If the dysrhythmia does not convert within 10 minutes of terminating the infusion, a second 1-mg infusion may be tried.
CLASS IV: CALCIUM CHANNEL BLOCKERS
Only two calcium channel blockers—verapamil [Calan, Verelan] and diltiazem [Cardizem, Dilacor-XR, Tiazac, others]—are able to block calcium channels in the heart. Accordingly, they are the only calcium channel blockers used to treat dysrhythmias. Their basic pharmacology is discussed in Chapter 45. Con-sideration here is limited to their use against dysrhythmias.
Effects on the Heart and ECGBlockade of cardiac calcium channels has three effects:
• Slowing of SA nodal automaticity• Delay of AV nodal conduction• Reduction of myocardial contractility
Note that these are identical to the effects of beta blockers, which makes sense in that beta blockers promote calcium channel closure in the heart. The principal effect on the ECG is prolongation of the PR interval, reflecting delayed AV conduction.
Therapeutic UsesVerapamil and diltiazem have two antidysrhythmic uses. First, they can slow ventricular rate in patients with atrial fibrillation or atrial flutter. Second, they can terminate SVT caused by an AV nodal reentrant circuit. In both cases, benefits derive from suppressing AV nodal conduction. With IV administration, effects can be seen in 2 to 3 minutes. Verapamil and diltiazem are not active against ventricular dysrhythmias.
Adverse EffectsAlthough generally safe, these drugs can cause undesired effects. Blockade of cardiac calcium channels can cause bradycardia, AV block, and heart failure. Blockade of calcium channels in vascular smooth muscle can cause vasodilation, resulting in hypotension and peripheral edema. Blockade of calcium channels in intestinal smooth muscle can produce constipation.
Drug InteractionsBoth verapamil and diltiazem can elevate levels of digoxin, thereby increasing the risk of digoxin toxicity. Also, since digoxin shares with verapamil and diltiazem the ability to decrease AV conduction, combining digoxin with either drug increases the risk of AV block.
Because verapamil, diltiazem, and beta blockers have nearly identical suppressant effects on the heart, combining verapamil or diltiazem with a beta blocker increases the risk of bradycardia, AV block, and HF.
Preparations, Dosage, and AdministrationVerapamil. Dosing may be IV or oral. Intravenous therapy is preferred
for initial treatment. Oral therapy is used for maintenance.
At therapeutic doses, sotalol produces substantial beta blockade. It can cause bradycardia, AV block, HF, and bronchospasm. Accordingly, the usual contraindications to beta blockers apply.
Preparations, Dosage, and AdministrationSotalol is available under two brand names: Betapace and Betapace AF. Betapace (80-, 120-, 160-, and 240-mg tablets) is intended for treating ventricular dysrhythmias. Betapace AF (80-, 120-, and 160-mg tablets) is intended for treating atrial fibrillation and atrial flutter. Although tablets are the same under both brand names, packaging differs: Packaging for Betapace provides information specific for treating ventricular dysrhythmias, whereas packaging for Betapace AF provides information specific for treating atrial dysrhythmias. For both types of dysrhythmias, treatment should start in a hospital. Dosing for both is the same: The initial dosage is 160 mg/day (in two divided doses), and the usual maintenance dosage is 160 to 320 mg/day in two or three divided doses. The dosing interval should be increased in patients with renal impairment.
DofetilideTherapeutic UseDofetilide [Tikosyn] is an oral class III antidysrhythmic indicated for restoring and maintaining normal sinus rhythm in patients with atrial flutter or atrial fibrillation. The drug causes dose-related QT prolongation and thereby poses a serious risk of torsades de pointes. Accordingly, it should be reserved for patients with highly symptomatic atrial dysrhythmias. Initial treatment requires continuous ECG monitoring in a hospital. Dosage must be carefully titrated on the basis of renal function tests. Dofetilide is available only through authorized hospitals and prescribers.
Effects on the Heart and ECGLike other class III agents, dofetilide blocks cardiac potassium channels and delays repolarization, prolonging the QT interval. Dofetilide does not affect the PR interval or widen the QRS complex, and it has no effect on cardiac beta receptors or sodium channels.
Adverse EffectsBy increasing the QT interval, dofetilide predisposes to torsades de pointes, which can progress to fatal ventricular fibrillation. The risk is directly related to dofetilide blood levels and is increased by hypokalemia and by other drugs that cause QT prolongation. To assess risk, an ECG should be obtained at baseline, and ECG monitoring should be continuous during initial treatment. Dofetilide is contraindicated for patients with a baseline QT interval greater than 440 msec (or greater than 500 msec in patients with ventricular conduction abnormalities). Other side effects include headache, chest pain, and dizziness.
Drug InteractionsDrugs that are excreted by renal cation pumps can interfere with the excretion of dofetilide, thereby causing its levels to rise. Accordingly, concurrent use of these drugs (e.g., cimetidine, trimethoprim, ketoconazole, prochlorperazine, megestrol) is contraindicated.
Drugs that prolong the QT interval may increase the risk of dysrhythmias and so should be avoided. Among these are class I and class III antidysrhythmics, phenothiazines, tricyclic antidepressants, and some macrolide antibiotics.
Combining verapamil with dofetilide increases the risk of torsades de pointes and should be avoided.
Preparations, Dosage, and AdministrationDofetilide [Tikosyn] is available in capsules (125, 250, and 500 mcg) for oral dosing. Because of the risk of dysrhythmias, treatment must be initiated in a hospital with continuous ECG monitoring for at least 3 days. Because dys-rhythmia risk is directly related to plasma drug levels, which in turn are directly related to creatinine clearance (a measure of renal function), creatinine clearance must be monitored. Dosage should be reduced with decreasing creatinine clearance as follows: For patients with normal renal function (creatinine clearance greater than 60 mL/min), give 500 mcg twice a day; for creatinine clearance 40 to 60 mL/min, give 250 mcg twice a day; for creatinine clearance 20 to 39 mL/min, give 125 mcg twice a day; and for creatinine clearance below 20 mL/min, withhold dofetilide. If the QT interval becomes excessively prolonged (greater than 500 msec, or greater than 550 msec in patients with ventricular conduction abnormalities), dosage should be reduced.
IbutilideIbutilide [Corvert] is an IV agent used to terminate atrial flutter and atrial fibrillation of recent onset (i.e., that has been present no longer than 90 days).

563
CHAPTER 49 Antidysrhythmic Drugs
Drug InteractionsMethylxanthines (aminophylline, theophylline, caffeine) block receptors for adenosine. Hence, asthma patients taking ami-nophylline or theophylline need larger doses of adenosine, and even then adenosine may not work.
Dipyridamole, an antiplatelet drug, blocks cellular uptake of adenosine and can thereby intensify its effects.Preparations, Dosage, and AdministrationAdenosine [Adenocard] is supplied in solution (3 mg/mL) for bolus IV administration. The injection should be made as close to the heart as possible and should be followed by a saline flush. The initial dose is 6 mg. If there is no response in 1 or 2 minutes, 12 mg may be tried and repeated once. If a response is going to occur, it should happen as soon as the drug reaches the AV node.
DigoxinAlthough its primary indication is HF, digoxin [Lanoxin] is also used to treat supraventricular dysrhythmias. The basic pharmacology of digoxin is discussed in Chapter 48. Consideration here is limited to treatment of dysrhythmias.
Effects on the HeartDigoxin suppresses dysrhythmias by decreasing conduction through the AV node and by decreasing automaticity in the SA node. The drug decreases AV conduction by (1) a direct depressant effect on the AV node and by (2) acting in the CNS to increase vagal (parasympathetic) impulses to the AV node. Digoxin decreases automaticity of the SA node by increasing vagal traffic to the node and by decreasing sympathetic traffic. It should be noted that, although digoxin decreases automaticity in the SA node, it can increase automaticity in Purkinje fibers. The latter effect contributes to dysrhythmias caused by digoxin.
Effects on the ECGBy slowing AV conduction, digoxin prolongs the PR interval. The QT interval may be shortened, reflecting accelerated repolarization of the ventricles. Depression of the ST segment is common. The T wave may be depressed or even inverted. There is little or no change in the QRS complex.
Adverse Effects and InteractionsThe major adverse effect is cardiotoxicity (dysrhythmias). Risk is increased by hypokalemia, which can result from concurrent therapy with diuretics (thiazides and loop diuretics). Accordingly, it is essential that potassium levels be kept within the normal range (3.5 to 5 mEq/L). The most common adverse effects are GI disturbances (anorexia, nausea, vomiting, abdominal discomfort). CNS responses (fatigue, visual disturbances) are also relatively common.
Antidysrhythmic UsesDigoxin is used only for supraventricular dysrhythmias. The drug is inactive against ventricular dysrhythmias.
Atrial Fibrillation and Atrial Flutter. Digoxin can be used to slow ventricular rate in patients with atrial fibrillation and atrial flutter. Ventricular rate is decreased by reducing the number of atrial impulses that pass through the AV node.
Supraventricular Tachycardia. Digoxin may be employed acutely and chronically to treat SVT. Acute therapy is used to abolish the dysrhythmia. Chronic therapy is used to prevent its return. Digoxin suppresses SVT by increasing cardiac vagal tone and by decreasing sympathetic tone.
Dosage and AdministrationOral therapy is generally preferred. The initial dosage is 1 to 1.5 mg administered in three or four doses over 24 hours. The maintenance dosage is 0.125 to 0.5 mg/day. This dose should be decreased in patients with renal impairment.
Verapamil for intravenous use is supplied in solution (5 mg/2 mL). The initial dose is 5 to 10 mg injected slowly (over 2 to 3 minutes). If the dys-rhythmia persists, an additional 10 mg may be administered in 30 minutes. An IV infusion (0.375 mg/min) can be used for maintenance. Intravenous verapamil can cause serious cardiovascular effects. Accordingly, blood pressure and the ECG should be monitored, and equipment for resuscitation should be immediately available.
Verapamil for oral use is available in immediate- and sustained-release tablets. The maintenance dosage is 40 to 120 mg 3 or 4 times a day.
Diltiazem. Like verapamil, diltiazem may be given IV or PO. Intravenous therapy is preferred for initial treatment, and oral therapy is used for maintenance.
Intravenous therapy is initiated with an IV bolus (0.25 mg/kg). If the response is inadequate, a second bolus (0.35 mg/kg) may be administered in 15 minutes. If appropriate, initial therapy may be followed with a continuous IV infusion (up to 24 hours’ duration) at a rate of 5 to 15 mg/hr.
Diltiazem for oral use is available in immediate-release and extended-release tablets. Maintenance dosing is 360 to 480 mg daily taken in four divided doses.
OTHER ANTIDYSRHYTHMIC DRUGSAdenosineAdenosine [Adenocard], a naturally occurring nucleotide, is a drug of choice for terminating paroxysmal SVT. Adenosine has an extremely short half-life and thus must be administered IV. Adverse effects are minimal because adenosine is rapidly cleared from the blood.
Effects on the Heart and ECGAdenosine decreases automaticity in the SA node and greatly slows conduction through the AV node. The most prominent ECG change is prolongation of the PR interval, brought on by delayed AV conduction. Adenosine works in part by inhibit-ing cyclic AMP–induced calcium influx, thereby suppressing calcium-dependent action potentials in the SA and AV nodes.
Therapeutic UseAdenosine is approved only for termination of paroxysmal SVT, including Wolff-Parkinson-White syndrome. The drug is not active against atrial fibrillation, atrial flutter, or ventricular dysrhythmias.
PharmacokineticsAdenosine has an extremely short half-life (estimated at 1.5 to 10 seconds), owing primarily to rapid uptake by cells and partly to deactivation by circulating adenosine deaminase. Because of its rapid clearance, adenosine must be administered by IV bolus, as close to the heart as possible.
Adverse EffectsAdverse effects are short lived, lasting less than 1 minute. The most common are sinus bradycardia, dyspnea (from broncho-constriction), hypotension and facial flushing (from vasodilation), and chest discomfort (perhaps from stimulation of pain receptors in the heart).

564
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
KEY POINTS
■ Dysrhythmias result from alteration of the electrical impulses that regulate cardiac rhythm. Antidysrhythmic drugs control rhythm by correcting or compensating for these alterations.
■ In the healthy heart, the SA node is the pacemaker.■ Impulses originating in the SA node must travel through
the AV node to reach the ventricles. Impulses arriving at the AV node are delayed before going on to excite the ventricles.
■ The His-Purkinje system conducts impulses rapidly throughout the ventricles, thereby causing all parts of the ventricles to contract in near synchrony.
■ The heart employs two kinds of action potentials: fast potentials and slow potentials.
■ Fast potentials occur in the His-Purkinje system, atrial muscle, and ventricular muscle.
■ Slow potentials occur in the SA node and AV node.■ Phase 0 of fast potentials (depolarization) is generated by
rapid influx of sodium. Because depolarization is fast, these potentials conduct rapidly.
■ During phase 2 of fast potentials, calcium enters myocardial cells, thereby promoting contraction.
■ Phase 3 of fast potentials (repolarization) is generated by rapid extrusion of potassium.
■ Phase 0 of slow potentials (depolarization) is caused by slow influx of calcium. Because depolarization is slow, these potentials conduct slowly.
■ Spontaneous phase 4 depolarization—of fast or slow potentials—gives cells automaticity.
■ Spontaneous phase 4 depolarization of cells in the SA node normally determines heart rate.
■ The P wave of an ECG is caused by depolarization of the atria.
■ The QRS complex is caused by depolarization of the ventricles. Widening of the QRS complex indicates slowed conduction through the ventricles.
■ The T wave is caused by repolarization of the ventricles.■ The PR interval represents the time between onset of the
P wave and onset of the QRS complex. PR prolongation indicates delayed AV conduction.
■ The QT interval represents the time between onset of the QRS complex and completion of the T wave. QT prolonga-tion indicates delayed ventricular repolarization.
■ Dysrhythmias arise from disturbances of impulse formation (automaticity) or impulse conduction.
■ Reentrant dysrhythmias result from a localized, self-sustaining circuit capable of repetitive cardiac stimulation.
■ Tachydysrhythmias can be divided into two major groups: supraventricular tachydysrhythmias and ventricular tachy-dysrhythmias. In general, ventricular tachydysrhythmias disrupt cardiac pumping more than do supraventricular tachydysrhythmias.
■ Treatment of supraventricular tachydysrhythmias is often directed at blocking impulse conduction through the AV node, rather than at eliminating the dysrhythmia.
■ Treatment of ventricular dysrhythmias is usually directed at eliminating the dysrhythmia.
■ All antidysrhythmic drugs are also prodysrhythmic (proar-rhythmic). That is, they all can worsen existing dysrhythmias and generate new ones.
■ Class I antidysrhythmic drugs block cardiac sodium chan-nels and thereby slow impulse conduction through the atria, ventricles, and His-Purkinje system.
■ Slowing ventricular conduction widens the QRS complex.■ Quinidine (a class IA drug) blocks sodium channels and
delays ventricular repolarization. Delaying ventricular repolarization prolongs the QT interval.
■ Quinidine causes diarrhea and other GI symptoms in 33% of patients. These effects frequently force drug withdrawal.
■ Quinidine can cause dysrhythmias. Widening of the QRS complex (by 50% or more) and excessive prolongation of the QT interval are warning signs.
■ Quinidine can raise digoxin levels. If the drugs are used together, digoxin dosage must be reduced.
■ Class IB agents differ from class IA agents in two ways: they accelerate repolarization and have little or no effect on the ECG.
■ Lidocaine (a class IB agent) is used only for ventricular dysrhythmias. The drug is not active against supraventricular dysrhythmias.
■ Lidocaine undergoes rapid inactivation by the liver. As a result, it must be administered by continuous IV infusion.
■ Propranolol and other class II drugs block cardiac beta1 receptors.
■ By blocking cardiac beta1 receptors, propranolol attenuates sympathetic stimulation of the heart and thereby decreases SA nodal automaticity, AV conduction velocity, and myocardial contractility.
■ By decreasing AV conduction velocity, propranolol prolongs the PR interval.
■ The effects of propranolol on the heart result (ultimately) from suppressing calcium entry. Therefore, the cardiac effects of propranolol and the effects of calcium channel blockers are nearly identical.
■ Propranolol is especially useful for treating dysrhythmias caused by excessive sympathetic stimulation of the heart.
■ In patients with supraventricular tachydysrhythmias, propranolol helps by (1) slowing discharge of the SA node and (2) decreasing impulse conduction through the AV node, which prevents the atria from driving the ventricles at an excessive rate.
■ Class III antidysrhythmics block potassium channels and thereby delay repolarization of fast potentials. As a result, they prolong the action potential duration and the effective refractory period. By delaying ventricular repolarization, they prolong the QT interval.
■ Amiodarone (a class III agent) is highly effective against atrial and ventricular dysrhythmias, but can cause multiple serious adverse effects, including damage to the lungs, eyes, liver, and thyroid.
■ Dronedarone, a derivative of amiodarone, is somewhat less toxic than amiodarone, but also less effective. In patients with heart failure or permanent atrial fibrillation, dronedarone doubles the risk of death.

565
CHAPTER 49 Antidysrhythmic Drugs
■ Verapamil and diltiazem (class IV antidysrhythmics) block cardiac calcium channels and thereby reduce automaticity of the SA node, slow conduction through the AV node, and decrease myocardial contractility. These effects are identical to those of the beta blockers.
■ By suppressing AV conduction, verapamil and diltiazem prolong the PR interval.
■ Verapamil and diltiazem are used to slow ventricular rate in patients with atrial fibrillation or atrial flutter and to terminate SVT caused by an AV nodal reentrant circuit.
In both cases, benefits derive from suppressing AV nodal conduction.
■ Adenosine is a drug of choice for terminating paroxysmal SVT.
■ Adenosine has a very short half-life (less than 10 seconds) and must be given by IV bolus.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
Summaries are limited to the major antidysrhythmic drugs. Summaries for beta blockers (propranolol, acebutolol, and esmolol), phenytoin, calcium channel blockers (verapamil and diltiazem), and digoxin appear in Chapters 18, 24, 45, and 48, respectively.
QUINIDINEPreadministration AssessmentTherapeutic GoalThe usual goal is long-term suppression of atrial and ven-tricular dysrhythmias.
Baseline DataObtain a baseline ECG and laboratory evaluation of liver function. Determine blood pressure.
Identifying High-Risk PatientsQuinidine is contraindicated for patients with a history of hypersensitivity to quinidine or other cinchona alkaloids and for patients with complete heart block, digoxin toxicity, or conduction disturbances associated with marked QRS widening and QT prolongation.
Exercise caution in patients with partial AV block, HF, hypotensive states, and hepatic dysfunction.
Implementation: AdministrationRoutes
Usual Route. Oral.Rare Routes. IM and IV.
AdministrationAdvise patients to take quinidine with meals. Warn them not to crush or chew sustained-release formulations.
Dosage size depends on the particular quinidine salt being used: 200 mg of quinidine sulfate is equivalent to 275 mg of quinidine gluconate.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsMonitor for beneficial changes in the ECG. Plasma drug levels should be kept between 2 and 5 mcg/mL.
Minimizing Adverse EffectsDiarrhea. Diarrhea and other GI disturbances occur in
one-third of patients and frequently force drug withdrawal. Inform patients that they can reduce GI effects by taking quinidine with meals.
Cinchonism. Inform patients about symptoms of cincho-nism (tinnitus, headache, nausea, vertigo, disturbed vision), and instruct them to notify the prescriber if these develop.
Cardiotoxicity. Monitor the ECG for signs of cardiotoxic-ity, especially widening of the QRS complex (by 50% or more) and excessive prolongation of the QT interval. Monitor pulses for significant changes in rate or regularity. If signs of cardiotoxicity develop, withhold quinidine and notify the prescriber.
Arterial Embolism. Embolism may occur during therapy of atrial fibrillation. Risk is reduced by treatment with an anticoagulant (e.g., warfarin, dabigatran). Observe for signs of thromboembolism (e.g., sudden chest pain, dyspnea), and report these immediately.
Minimizing Adverse InteractionsDigoxin. Quinidine can double digoxin levels. When these
drugs are combined, digoxin dosage should be reduced. Monitor patients for digoxin toxicity (dysrhythmias).
PROCAINAMIDEPreadministration AssessmentTherapeutic GoalProcainamide is indicated for acute and long-term management of ventricular and supraventricular dysrhythmias. Because procainamide can be toxic with long-term use, quinidine is preferred to procainamide for chronic suppression.
Baseline DataObtain a baseline ECG, complete blood count, and laboratory evaluations of liver and kidney function. Determine blood pressure.
Identifying High-Risk PatientsProcainamide is contraindicated for patients with systemic lupus erythematosus (SLE), complete AV block, and second-
Continued

566
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
degree or third-degree AV block in the absence of an elec-tronic pacemaker, and patients with a history of procaine allergy.
Exercise caution in patients with hepatic or renal dysfunction.
Implementation: AdministrationRoutesOral, IM, IV.
AdministrationInstruct patients to administer procainamide at evenly spaced intervals around-the-clock. Warn patients not to crush or chew sustained-release formulations.
When switching from IV procainamide to oral procain-amide, allow 3 hours to elapse between stopping the infusion and giving the first oral dose.
Give IM injections deep into the gluteal muscle.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsMonitor the ECG for beneficial changes. Plasma drug levels should be kept between 3 and 10 mcg/mL.
Minimizing Adverse EffectsSLE-like Syndrome. Prolonged therapy can produce a
syndrome resembling SLE. Inform patients about manifesta-tions of SLE (joint pain and inflammation; hepatomegaly; unexplained fever; soreness of the mouth, throat, or gums), and instruct them to notify the prescriber if these develop. If SLE is diagnosed, procainamide should be discontinued. If discontinuation is impossible, signs and symptoms can be controlled with a nonsteroidal anti-inflammatory drug (e.g., aspirin) or a glucocorticoid. The ANA titer should be measured periodically, and if it rises, procainamide withdrawal should be considered.
Blood Dyscrasias. Procainamide can cause agranulocy-tosis, thrombocytopenia, and neutropenia. Deaths have occurred. Obtain complete blood counts weekly during the first 3 months of treatment and periodically thereafter. Instruct patients to inform the prescriber at the first sign of infection (fever, chills, sore throat), bruising, or bleeding. If subsequent blood counts indicate hematologic disturbance, discontinue procainamide immediately.
Cardiotoxicity. Procainamide can cause dysrhythmias. Monitor pulses for changes in rate or regularity. Monitor the ECG for excessive QRS widening (greater than 50%) and for PR prolongation. If these occur, withhold procainamide and notify the prescriber.
Arterial Embolism. Embolism may occur during therapy of atrial fibrillation. Risk is reduced by treatment with an anticoagulant (e.g., warfarin, dabigatran). Observe for signs of thromboembolism (e.g., sudden chest pain, dyspnea), and report these immediately.
LIDOCAINEPreadministration AssessmentTherapeutic GoalAcute management of ventricular dysrhythmias.
Baseline DataObtain a baseline ECG and determine blood pressure.
Identifying High-Risk PatientsLidocaine is contraindicated for patients with Stokes-Adams syndrome, Wolff-Parkinson-White syndrome, and severe degrees of SA, AV, or intraventricular block in the absence of electronic pacing.
Exercise caution in patients with hepatic dysfunction or impaired hepatic blood flow.
Implementation: AdministrationRoutes
Usual. IV.Emergencies. IM.
AdministrationIntravenous. Make certain the lidocaine preparation is
labeled for IV use (i.e., is devoid of preservatives and catechol-amines). Dilute concentrated preparations with 5% dextrose in water.
The initial dose is 50 to 100 mg (1 mg/kg) infused at a rate of 25 to 50 mg/min. For maintenance, monitor the ECG and adjust the infusion rate on the basis of cardiac response. The usual rate is 1 to 4 mg/min.
Intramuscular. Reserve for emergencies. The usual dose is 300 mg injected into the deltoid muscle. Switch to IV lidocaine as soon as possible.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsContinuous ECG monitoring is required. Plasma drug levels should be kept between 1.5 and 5 mcg/mL.
Minimizing Adverse EffectsExcessive doses can cause convulsions and respiratory arrest. Equipment for resuscitation should be available. Seizures can be managed with diazepam.
AMIODARONEPreadministration AssessmentTherapeutic Goal
Oral Therapy. Long-term treatment of (1) atrial fibrillation and (2) life-threatening recurrent ventricular fibrillation or recurrent hemodynamically unstable ventricular tachycardia in patients who have not responded to safer drugs.
Intravenous Therapy. Initial treatment of recurrent ventricular fibrillation, shock-resistant ventricular fibrillation,
Summary of Major Nursing Implicationsa—cont’d

567
CHAPTER 49 Antidysrhythmic Drugs
aPatient education information is highlighted as blue text.
recurrent hemodynamically unstable ventricular tachycardia, atrial fibrillation, and AV nodal reentrant tachycardia.
Baseline DataObtain a baseline ECG, eye examination, and chest x-ray, along with potassium and magnesium levels, and tests for thyroid, pulmonary, and liver function.
Identifying High-Risk PatientsAmiodarone is contraindicated for patients with severe sinus node dysfunction or second- or third-degree AV block, and for women who are pregnant or breast-feeding.
Exercise caution in patients with thyroid disorders, hypokalemia, or hypomagnesemia.
Implementation: AdministrationRoutes
Oral. Used for maintenance therapy of atrial and ven-tricular dysrhythmias.
Intravenous. Used for acute therapy of atrial and ven-tricular dysrhythmias.
Administration and DosageOral. Initiate treatment in a hospital. High doses are used
initially (800 to 1600 mg/day for 1 to 3 weeks). The usual maintenance dosage is 400 mg/day.
Intravenous. Administer by continuous IV infusion, starting with a rapid infusion rate and later reducing the rate for maintenance. Intravenous treatment may last from 2 days to 3 weeks.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsMonitor for beneficial changes in the ECG.
Minimizing Adverse EffectsPulmonary Toxicity. Amiodarone can cause potentially
fatal lung damage (hypersensitivity pneumonitis, interstitial/alveolar pneumonitis, and pulmonary fibrosis). Obtain a baseline chest x-ray and pulmonary function test, and monitor pulmonary function throughout treatment. Inform patients about signs of lung injury (dyspnea, cough, chest pain), and instruct them to report these immediately. Treatment consists of withdrawing amiodarone and providing supportive care, sometimes including glucocorticoids.
Cardiotoxicity. Amiodarone can cause HF and atrial and ventricular dysrhythmias. Patients with pre-existing heart failure must not use the drug. Warn patients about signs of HF (e.g., shortness of breath, reduced exercise tolerance, fatigue, tachycardia, weight gain), and instruct them to report these immediately.
Liver Toxicity. Amiodarone can injure the liver. Obtain tests of liver function at baseline and periodically during treatment. If circulating liver enzymes exceed 3 times the normal level, amiodarone should be withdrawn. Inform
patients about signs and symptoms of liver injury (e.g., anorexia, nausea, vomiting, malaise, fatigue, itching, jaundice, dark urine), and instruct them to report them immediately.
Thyroid Toxicity. Amiodarone can cause hypothyroidism and hyperthyroidism. Obtain tests of thyroid function at baseline and periodically during treatment. Treat hypothyroid-ism with thyroid hormone supplements. Treat hyperthyroidism with an antithyroid drug (e.g., methimazole) or thyroidectomy. Stopping amiodarone should be considered.
Toxicity in Pregnancy and Breast-Feeding. Amiodarone can harm the developing fetus and breast-feeding infant. Warn patients to avoid pregnancy and breast-feeding while using amiodarone and for several months after stopping.
Ophthalmic Effects. Amiodarone has been associated with optic neuropathy and optic neuritis, sometimes progressing to blindness. Obtain ophthalmic tests, including funduscopy and a slit-lamp examination, at baseline and periodically during treatment. Advise patients to report reductions in visual acuity or peripheral vision. If optic neuropathy or neuritis is diagnosed, discontinuing amiodarone should be considered.
Virtually all patients develop corneal microdeposits. In most cases, the deposits have no effect on vision and thus only rarely lead to the discontinuation of amiodarone.
Dermatologic Effects. Photosensitivity reactions are common. Advise patients to avoid sunlamps and to wear sunscreen and protective clothing when outdoors. With prolonged sun exposure, skin may develop a bluish-gray discoloration, which typically resolves a few months after amiodarone is stopped.
Minimizing Adverse InteractionsAmiodarone is subject to significant interactions with many drugs. Interactions of special concern are presented here.
Drugs Whose Levels Can Be Increased by Amioda-rone. Amiodarone can increase levels of several drugs, including quinidine, procainamide, phenytoin, digoxin, dil-tiazem, warfarin, cyclosporine, and three statins: lovastatin, simvastatin, and atorvastatin. Dosages of these agents often require reduction.
Drugs That Can Reduce Amiodarone Levels. Amio-darone levels can be reduced by cholestyramine (which decreases amiodarone absorption) and by agents that induce CYP3A4 (e.g., St. John’s wort, rifampin). Monitor to ensure that amiodarone is still effective.
Drugs That Can Increase the Risk of Dysrhyth-mias. The risk of severe dysrhythmias is increased by diuretics (because they can reduce levels of potassium and magnesium) and by drugs that prolong the QT interval.
Drugs That Can Cause Bradycardia. Combining amio-darone with a beta blocker, verapamil, or diltiazem can lead to excessive slowing of heart rate.
Grapefruit Juice. Grapefruit juice inhibits CYP3A4 and can raise levels of amiodarone. Toxicity can result. Advise patients to avoid grapefruit juice.
Summary of Major Nursing Implicationsa—cont’d

568
C H A P T E R
50 Prophylaxis of Atherosclerotic Cardiovascular Disease: Drugs That Help Normalize Cholesterol and Triglyceride Levels
Cholesterol, p. 568Plasma Lipoproteins, p. 569
Structure and Function of Lipoproteins, p. 569Classes of Lipoproteins, p. 569LDL Cholesterol Versus HDL Cholesterol, p. 570
Role of LDL Cholesterol in Atherosclerosis, p. 5702013 ACC/AHA Guidelines on the Treatment of
Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk, p. 571Cholesterol Screening, p. 571ASCVD Risk Assessment, p. 573Treatment of High LDL Cholesterol, p. 573Secondary Treatment Targets, p. 577
Drugs and Other Products Used to Improve Plasma Lipid Levels, p. 578HMG-CoA Reductase Inhibitors (Statins), p. 578A Word Regarding Niacin (Nicotinic Acid), p. 583Bile-Acid Sequestrants, p. 583Ezetimibe, p. 584Fibric Acid Derivatives (Fibrates), p. 585Monoclonal Antibodies (Proprotein Convertase
Subtilisin/Kexin Type 9 [PCSK9] Inhibitors), p. 586
Drug Combinations, p. 586Fish Oil, p. 587Plant Stanol and Sterol Esters, p. 587Cholestin, p. 587
Key Points, p. 588Summary of Major Nursing Implications, p. 588
killer of men and women, causing 801,000 deaths in 2013. According to the American Heart Association, about 92 million Americans have heart disease or a history of stroke. More than half of these people are women.
How does ASCVD develop? Very briefly, it begins as a fatty streak in the arterial wall. This is followed by deposition of fibrous plaque. As atherosclerotic plaque grows, it impedes coronary blood flow, causing anginal pain. Worse yet, athero-sclerosis encourages formation of thrombi, which can block flow to the brain and heart entirely, thereby causing MI and stroke.
It is important to appreciate that atherosclerosis is not limited to arteries of the heart or brain: Atherosclerotic plaque can develop in any artery and can thereby compromise circulation to any tissue. Furthermore, adverse effects can occur at sites distant from the original lesion: A ruptured lesion can produce a thrombus, which can travel downstream to block a new vessel. Blockage in the lungs is of particular concern.
The risk of developing ASCVD is directly related to increased levels of blood cholesterol in the form of low-density lipopro-teins (LDLs). By reducing levels of LDL cholesterol, we can slow progression of atherosclerosis, reduce the risk of serious ASCVD and its potential consequences, and prolong life. The preferred method for lowering LDL cholesterol is modification of diet combined with exercise. Drugs are employed only when diet modification and exercise are insufficient.
We approach our primary topic—cholesterol and its impact on ASCVD—in three stages. First, we discuss cholesterol itself, plasma lipoproteins (structures that transport cholesterol in blood), and the process of atherogenesis. Second, we discuss guidelines for cholesterol screening and the management of high cholesterol. Third, we discuss the pharmacology of the cholesterol-lowering drugs, as well as drugs used to lower TGs.
CHOLESTEROLCholesterol has several physiologic roles. Of greatest impor-tance, cholesterol is a component of all cell membranes and membranes of intracellular organelles. In addition, cholesterol is required for synthesis of certain hormones (e.g., estrogen, progesterone, testosterone) and for synthesis of bile salts, which are needed to absorb and digest dietary fats. Also, cholesterol is deposited in the stratum corneum of the skin, where it reduces
Our main topic for the chapter is drugs used to lower cholesterol. Drugs used to lower triglycerides (TGs) are considered as well. Cholesterol has a large impact on atherosclerosis (thicken-ing of the coronary arteries), also known as atherosclerotic cardiovascular disease (ASCVD). ASCVD includes the vessels of the heart as well as the brain. Damage to these vessels can result in myocardial infarction (MI) or stroke. Moderate cardiac ASCVD usually manifests first as anginal pain. Severe cardiac ASCVD sets the stage for acute coronary syndrome (ACS) and MI. In the United States, cardiac ASCVD is the leading

569
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
ApolipoproteinsAll lipoproteins have one or more apolipoprotein molecules embedded in their shell (see Fig. 50.1). Apolipoproteins, which constitute the protein component of lipoproteins, have three functions:
• They serve as recognition sites for cell-surface recep-tors and thereby allow cells to bind with and ingest lipoproteins.
• They activate enzymes that metabolize lipoproteins.• They increase the structural stability of lipoproteins.
The apolipoproteins of greatest clinical interest are labeled A-I, A-II, and B-100. All lipoproteins that deliver cholesterol and TGs to nonhepatic tissues contain apolipoprotein B-100. Conversely, all lipoproteins that transport lipids from nonhepatic tissues back to the liver (i.e., that remove lipids from tissues) contain apolipoprotein A-I.
Classes of LipoproteinsThere are six major classes of plasma lipoproteins. Distinctions among classes are based on size, density, apolipoprotein content, transport function, and primary core lipids (cholesterol or TG). From a pharmacologic perspective, the features of greatest interest are lipid content, apolipoprotein content, and transport function.
For two reasons, the topic of lipoprotein density deserves comment. First, naming of lipoprotein types is based on their density. Second, differences in density provide the basis for the physical isolation and subsequent measurement of plasma lipoproteins, as is done in research and clinical laboratories. The various classes of lipoproteins differ in density because they differ in their percentage of composition of lipid and protein. Because protein is denser than lipid, lipoproteins that have a high percentage of protein (and a low percentage of lipid) have a relatively high density. Conversely, lipoproteins with a lower percentage of protein have a lower density.
Of the six major classes of lipoproteins, three are especially important in coronary atherosclerosis. These classes are named (1) very-low-density lipoproteins (VLDLs), (2) low-density lipoproteins (LDLs), and (3) high-density lipoproteins (HDLs). Properties of these classes are shown in Table 50.1.
Very-Low-Density LipoproteinsVLDLs contain mainly triglycerides (and some cholesterol), and they account for nearly all of the TGs in blood. The main physiologic role of VLDLs is to deliver triglycerides from the liver to adipose tissue and muscle, which can use the TGs as fuel. Each VLDL particle contains one molecule of apolipo-protein B-100, which allows VLDLs to bind with cell-surface receptors and thereby transfer their lipid content to cells.
The role of VLDLs in atherosclerosis is unclear. Although several studies suggest a link between elevated levels of VLDLs and development of atherosclerosis, this link has not been firmly established. However, we do know that elevation of TG levels (above 500 mg/dL) increases the risk of pancreatitis.
Low-Density LipoproteinsLDLs contain cholesterol as their primary core lipid, and they account for the majority (60% to 70%) of all cholesterol in blood. The physiologic role of LDLs is delivery of cholesterol
evaporation of water and blocks transdermal absorption of water-soluble compounds.
Some of our cholesterol comes from dietary sources (exogenous cholesterol), and some is manufactured by cells (endogenous cholesterol), primarily in the liver. More cholesterol comes from endogenous production than from the diet. A critical step in hepatic cholesterol synthesis is catalyzed by an enzyme named 3-hydroxy-3-methylglutaryl coenzyme A reductase, or simply HMG-CoA reductase. As discussed later in the chapter, drugs that inhibit this enzyme—the statins—are our most effective and widely used cholesterol-lowering agents.
An increase in dietary cholesterol produces only a small increase in cholesterol in the blood, primarily because a rise in cholesterol intake inhibits endogenous cholesterol synthesis. Interestingly, an increase in dietary saturated fats produces a substantial (15% to 25%) increase in circulating cholesterol because the liver uses saturated fats to make cholesterol. Accordingly, when we want to reduce cholesterol levels, it is more important to reduce intake of saturated fats than to reduce intake of cholesterol itself, although cholesterol intake should definitely be lowered.
PLASMA LIPOPROTEINSStructure and Function of LipoproteinsFunctionLipoproteins serve as carriers for transporting lipids—cholesterol and TGs—in blood. Like all other nutrients and metabolites, lipids use the bloodstream to move throughout the body. However, being lipids, cholesterol and TGs are not water soluble, and hence cannot dissolve directly in plasma. Lipo-proteins provide a means of solubilizing these lipids, thereby permitting transport.
Basic StructureThe basic structure of lipoproteins is depicted in Fig. 50.1. As indicated, lipoproteins are tiny spherical structures that consist of a hydrophobic core, composed of cholesterol and TGs, surrounded by a hydrophilic shell, composed primarily of phospholipids. Because the hydrophilic shell completely covers the lipid core, the entire structure is soluble in the aqueous environment of the plasma.
Hydrophobic core: cholesterol and triglycerides
Apolipoprotein
Hydrophilic shell: phospholipids
Fig. 50.1 ■ Basic structure of plasma lipoproteins.

570
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
need to distinguish between cholesterol that is associated with HDLs and cholesterol that is associated with LDLs. To make this distinction, we use the terms HDL cholesterol and LDL cholesterol. Because LDL cholesterol promotes atherosclerosis, it has been dubbed bad cholesterol. Conversely, because HDL seems to protect against atherosclerosis, it is often called good cholesterol or healthy cholesterol.
ROLE OF LDL CHOLESTEROL IN ATHEROSCLEROSISLDLs initiate and fuel development of atherosclerosis. The process begins with the transport of LDLs from the arterial lumen into endothelial cells that line the lumens of blood vessels. From there, they move into the space that underlies the arterial epithelium. Once in the subendothelial space, components of LDLs undergo oxidation. This step is critical in that oxidized LDLs:
• Attract monocytes from the circulation into the suben-dothelial space, after which the monocytes are converted to macrophages (which are critical to atherogenesis)
• Inhibit macrophage mobility, thereby keeping macro-phages at the site of atherogenesis
• Undergo uptake by macrophages (macrophages do not take up LDLs that have not been oxidized)
• Are cytotoxic, and hence can damage the vascular endothelium directly
As macrophages engulf more and more cholesterol, they become large and develop large vacuoles. When macrophages assume this form, they are referred to as foam cells. Foam cell accumula-tion beneath the arterial epithelium produces a fatty streak, which makes the surface of the arterial wall lumpy, causing blood flow to become turbulent. Continued accumulation of foam cells can eventually cause rupture of the endothelium, thereby exposing the underlying tissue to the blood. This results in platelet adhesion and formation of microthrombi. As the process continues, smooth muscle cells migrate to the site, synthesis of collagen increases, and there can be repeated rupturing and healing of the endothelium. The end result is a mature atherosclerotic lesion, characterized by a large lipid core and a tough fibrous cap. In less mature lesions, the fibrous cap is not strong, and hence the lesions are unstable and more likely to rupture. As a result, arterial pressure and shear forces (from turbulent blood flow) can cause the cap to rupture. Accumulation of platelets at the site of rupture can rapidly
to nonhepatic tissues. Each LDL particle contains one molecule of apolipoprotein B-100, which is needed for the binding of LDL particles to LDL receptors on cells. LDLs can be viewed as by-products of VLDL metabolism, in that the lipids and apolipoproteins that compose LDLs are remnants of VLDL degradation.
Cells that require cholesterol meet their needs through endocytosis (engulfment) of LDLs from the blood. The process begins with the binding of LDL particles to LDL receptors on the cell surface. When cellular demand for cholesterol increases, cells synthesize more LDL receptors and thereby increase their capacity for LDL uptake. Accordingly, cells that are unable to make more LDL receptors cannot increase cholesterol absorption. Increasing the number of LDL receptors on cells is an important mechanism by which certain drugs increase LDL uptake and thereby reduce LDL levels in blood.
Of all lipoproteins, LDLs make the greatest contribution to coronary atherosclerosis. The probability of developing ASCVD is directly related to the level of LDLs in blood. Conversely, by reducing LDL levels, we decrease the risk of ASCVD. Accordingly, when cholesterol-lowering drugs are used, the main goal is to reduce elevated LDL levels. Multiple studies have shown that by reducing LDL levels we can arrest or perhaps even reverse atherosclerosis and can thereby reduce mortality from ASCVD. In fact, for each 1% reduction in the LDL level, there is about a 1% reduction in the risk of a major cardiovascular (CV) event.
High-Density LipoproteinsLike LDLs, HDLs contain cholesterol as their primary core lipid and account for 20% to 30% of all cholesterol in the blood. In contrast to LDLs, whose function is the delivery of cholesterol to peripheral tissues, HDLs carry cholesterol from peripheral tissues back to the liver. That is, HDLs promote cholesterol removal.
The influence of HDLs on ASCVD is dramatically different from that of LDLs. Whereas elevation of LDLs increases the risk of ASCVD, elevation of HDLs reduces the risk of ASCVD. That is, high HDL levels actively protect against ASCVD.
LDL Cholesterol Versus HDL CholesterolThe previous discussion shows that not all cholesterol in plasma has the same impact on ASCVD. As stated, a rise in cholesterol associated with LDLs increases the risk of ASCVD. In contrast, a rise in cholesterol associated with HDLs lowers the risk. Consequently, when speaking of plasma cholesterol levels, we
Lipoprotein Class
Major Core Lipids Apolipoproteins Transport Function Influence on Atherosclerosis
VLDL Triglycerides B-100, E, others Delivery of triglycerides to nonhepatic tissues
Probably contribute to atherosclerosis
LDL Cholesterol B-100 Delivery of cholesterol to nonhepatic tissues
Definitely contribute to atherosclerosis
HDL Cholesterol A-I, A-II, A-IV Transport of cholesterol from nonhepatic tissues back to the liver
Protect against atherosclerosis
TABLE 50.1 ■ Properties of the Plasma Lipoproteins That Affect Atherosclerosis
HDL, High-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.

571
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
cause thrombosis and can thereby cause infarction. Infarction is less likely at sites of mature atherosclerotic lesions. The atherosclerotic process is depicted in Fig. 50.2.
It is important to appreciate that atherogenesis involves more than just deposition of lipids. In fact, atherogenesis is now considered primarily a chronic inflammatory process. When LDLs penetrate the arterial wall, they cause mild injury. The injury, in turn, triggers an inflammatory response that includes infiltration of macrophages, T lymphocytes, and other potentially noxious chemicals (e.g., C-reactive protein [CRP]). In the late stage of the disease process, inflammation can weaken atherosclerotic plaque, leading to plaque rupture and subsequent thrombosis.
2013 ACC/AHA GUIDELINES ON THE TREATMENT OF BLOOD CHOLESTEROL TO REDUCE ATHEROSCLEROTIC CARDIOVASCULAR RISKIt is well established that high levels of cholesterol (primarily LDL cholesterol) cause substantial morbidity and mortality, and that aggressive treatment can save lives. Accordingly, periodic cholesterol screening and risk assessment are recom-mended. If the assessment indicates ASCVD risk, lifestyle changes—especially diet and exercise—should be implemented. If ASCVD risk is high, LDL-lowering drugs should be added to the regimen.
In 1988, the National Cholesterol Education Program (NCEP) began issuing guidelines on cholesterol detection and management. The most recent update was issued in 2001 and amended in 2004, and new guidelines were developed in 2013 in partnership with the American College of Cardiology (ACC) and the American Heart Association (AHA). A summary of the 2013 guidelines—2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines—was published in Circulation and is available at http://circ.ahajournals.org/content/early/ 2013/11/11/01.cir.0000437738.63853.7a.
Like earlier NCEP guidelines, the 2013 ACC/AHA choles-terol guideline focuses on the role of high cholesterol in ASCVD and stresses the importance of treatment. However, the new guideline focuses specifically on identifying patients who are most likely to benefit from cholesterol-lowering therapy instead of targeting specific cholesterol goals.
Cholesterol ScreeningAdultsManagement of high LDL cholesterol begins with screening, generally done every 5 years for adults older than 20 years. The ATP III guidelines recommended a more thorough screen than before, consisting of total cholesterol, LDL cholesterol, HDL cholesterol, and TGs. Blood for these tests should be drawn after fasting. Previous to the new guidelines, total cholesterol and LDL cholesterol levels were placed into clas-sifications. With the introduction of the new guidelines, patients should be considered for statin treatment if they fall into one of four different risk categories (Table 50.2).
Children and AdolescentsElevated cholesterol in pediatric patients is a growing concern and is not addressed in the 2013 ACC/AHA blood cholesterol guidelines. However, it is addressed in other guidelines, includ-ing one created in 2011 by an expert panel appointed by the National Heart, Lung, and Blood Institute, and endorsed by the American Academy of Pediatrics. This report—Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents—is available at www.nhlbi.nih.gov/guidelines/cvd_ped/summary.htm#chap9.
The guideline recommends lipid screening for all children between ages 9 and 11 years, followed by another screen between ages 18 and 21 years. For children with a family history of high cholesterol or heart disease, screening should start sooner: between ages 2 and 8 years. Cholesterol classifica-tion for children and adolescents is presented in Table 50.3.
If LDL cholesterol is high, all patients and their families should receive nutritional counseling. In addition, patients should focus on weight control and increased activity, as indicated. Should children use cholesterol-lowering drugs? For two reasons, the answer is “Probably not.” First, these children are in no immediate danger: Their risk of developing clinically significant ASCVD in the next 20 years is close to zero. And second, the only data from randomized, controlled trials were for children with familial hypercholesterolemia. No data exist to show that these drugs will improve outcomes when given to children with secondary lipid disorders.
Individuals with clinical atherosclerotic cardiovascular disease (ASCVD)
Individuals with primary elevations of LDL cholesterol (LDL-C) ≥190 mg/dL
Individuals 40 to 75 years of age with diabetes with LDL-C 70 to 189 mg/dL
Individuals without clinical ASCVD or diabetes who are 40 to 75 years of age with LDL-C 70 to 189 mg/dL and an estimated 10-year ASCVD risk of 7.5% or higher
TABLE 50.2 ■ Statin Benefit Groups as Defined by the 2013 ACC/AHA Blood Cholesterol Guidelines
Data from National Cholesterol Education Program: Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics 89(3 Pt 2):525–584, 1992.
CategoryTotal Cholesterol (mg/dL)
LDL Cholesterol (mg/dL)
Acceptable <170 <110Borderline 170–199 110–129Elevated ≥200 ≥130
TABLE 50.3 ■ NCEP Classification of Cholesterol Levels for Children and Adolescentsa
aHDL levels should be greater than or equal to 35 mg/dL and triglycerides should be less than or equal to 150 mg/dL.

Fatty streak
Response to injury
Damaged endothelium:Chronic endothelial injury
Fibrous plaque
Complicated lesion
HypertensionSmokingHyperlipidemiaHyperhomocystinemiaHemodynamic factorsToxinsVirusesImmune reactions
________
Endothelium
Tunica intima
Tunica media
Adventitia
Lipids
Foamy macrophageingesting lipids
Fibroblast
Lipid pool
Lipid accumulation
Fibroblast
Collagen cap(fibrous tissue)
Fissure in plaque
Thrombus
Thinning collagen cap
Lipid pool
Platelets attach toendothelium
Migration ofsmooth muscleinto the intima
Damagedendothelium
Platelets
Macrophage
Monocyte
A
B
C
D
Fig. 50.2 ■ Progression of atherosclerosis. A, Damaged endothelium. B, Diagram of fatty streak and lipid core formation. C, Diagram of fibrous plaque. Raised plaques are visible: some are yellow and some are white. D, Diagram of a complicated lesion, showing a thrombus (in red) and collagen (in blue). (From McCance KL, Huether SE: Pathophysiology: The Biologic Basis for Disease in Adults and Children, 7th ed. St. Louis: Elsevier, 2014.)

573
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
The instrument employed most often is the Framingham Risk Prediction Score, which takes five factors into account: age, total cholesterol, HDL cholesterol, smoking status, and systolic blood pressure. These are similar to risk factors noted earlier. Framingham scores can be determined using either (1) the tables for men and women shown in Fig. 50.3 or (2) a web-based risk calculator, such as the one provided by the ACC/AHA at http://tools.acc.org/ASCVD-Risk-Estimator-Plus/#!/calculate/estimate/.
Identifying ASCVD Risk Equivalents. An ASCVD risk equivalent is a condition that poses the same risk of a major coronary event as does established ASCVD (i.e., more than 20% risk of a major event within 10 years). There are two basic ASCVD risk equivalents:
• Diabetes• The presence of multiple risk factors that confer an
ASCVD 10-year risk score 7.5% or more
Identifying an Individual’s ASCVD Risk CategoryUnder the 2013 ACC/AHA cholesterol guideline, there are four categories of patients who would benefit from statin treatment of cholesterol (see Table 50.2). Category assignment is based on (1) the presence or absence of ASCVD (or an ASCVD risk equivalent, such as diabetes), (2) the number of risk factors the individual has (other than high LDL cholesterol), and (3) the individual’s 10-year ASCVD score. Although this assessment sounds complicated, it’s not. Let’s consider the hypothetical case of Ralph J. and follow along by looking at Fig. 50.3. Mr. J. is 62 years old, hypertensive, and he smokes—but, remarkably, his HDL cholesterol is high (above 60 mg/dL). He has no family history of premature ASCVD, does not have ASCVD himself, and does not have diabetes. His 10-year Framingham Risk Prediction Score is 11%. Because his estimated 10-year ASCVD score is greater than 7.5%, Mr. J. should be considered for moderate- to high-intensity drug therapy (Fig. 50.4). This is even easier if you use an online risk assessment tool, such as the ones available at https://www.cvdriskchecksecure.com/framinghamriskscore.aspx.
Final Note: Each Type of Dyslipidemia a Patient Has Contributes Independently to ASCVD RiskPatients are likely to have more than one type of dyslipidemia—for example, high LDL cholesterol combined with low HDL cholesterol and high TGs—and each of these disorders con-tributes independently to CV risk. This means that fixing just one of these problems will not eliminate the risk posed by the others. Accordingly, to get maximal risk reduction, we must correct all lipid abnormalities that are present.
Treatment of High LDL CholesterolTreatment of high LDL cholesterol is based on the individual’s ASCVD risk category or the presence of other comorbidities such as diabetes. Treatment may be started with a high-intensity statin or a moderate-intensity statin depending on the patient’s risk factors (see Fig. 50.4 and Table 50.4). To reduce LDL levels, the 2013 ACC/AHA guideline recommends two forms of intervention: (1) therapeutic lifestyle changes (TLCs) and (2) drug therapy. For some people, cholesterol can be reduced adequately with TLCs alone. Others require TLCs plus cholesterol-lowering drugs. Please note: Drugs should be used only as an adjunct to TLCs—not as a substitute.
Treating Dyslipidemia
Life Stage Patient Care Concerns
Infants See “Breast-feeding women,” below.Children/
adolescentsLovastatin, simvastatin, pravastatin, and
atorvastatin are approved for use in children. It is recommended to avoid statin use in children younger than 10 years.
Pregnant women
Statins are classified in FDA Pregnancy Risk Category X.a They are contraindicated in pregnancy. Ezetimibe and fibrates are classified in Pregnancy Risk Category C,a hence benefit should outweigh risk.
Breast-feeding women
Effects of statins, ezetimibe, and fibrates have not been studied in breast-feeding. Given the possibility of harm, benefit should outweigh risk.
Older adults In patients age 65 years and older, statins, compared to placebo, significantly reduced the risk of MI, as well as the risk of stroke, by 23.8%. However, the cost-benefit evaluation of treatment in older adults should be considered.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
ASCVD Risk AssessmentUnder the 2013 ACC/AHA guidelines, ASCVD risk assessment is directed at determining the patient’s absolute risk of develop-ing clinical coronary disease over the next 10 years. The mode of intervention is then determined by the individual’s degree of risk.
Factors in Risk AssessmentTo assess the ASCVD risk for an individual, we need three kinds of information. Specifically, we need to (1) identify ASCVD risk factors, (2) calculate 10-year ASCVD risk, and (3) identify ASCVD risk equivalents.
Identifying ASCVD Risk Factors. Major risk factors that modify LDL treatment goals include positive risk factors (advancing age, African American race, hypertension, cigarette smoking, and low HDL cholesterol) and one negative risk factor (high HDL cholesterol). (LDL itself is not listed because the reason for counting these risk factors is to modify treatment of high LDL.)
We know that diabetes is a very strong predictor of develop-ing ASCVD. Accordingly, we no longer consider diabetes to be a risk factor. Instead, for the purpose of risk assessment, diabetes is now considered an ASCVD risk equivalent. That is, having diabetes is considered equivalent to having ASCVD as a predictor of a major coronary event.
Calculating 10-Year ASCVD Risk. The 2013 ACC/AHA cholesterol guideline defines high ASCVD risk as 7.5% or greater. Some people are automatically in this risk group—specifically, those with existing ASCVD and those with diabetes. For all other people, 10-year risk must be calculated.
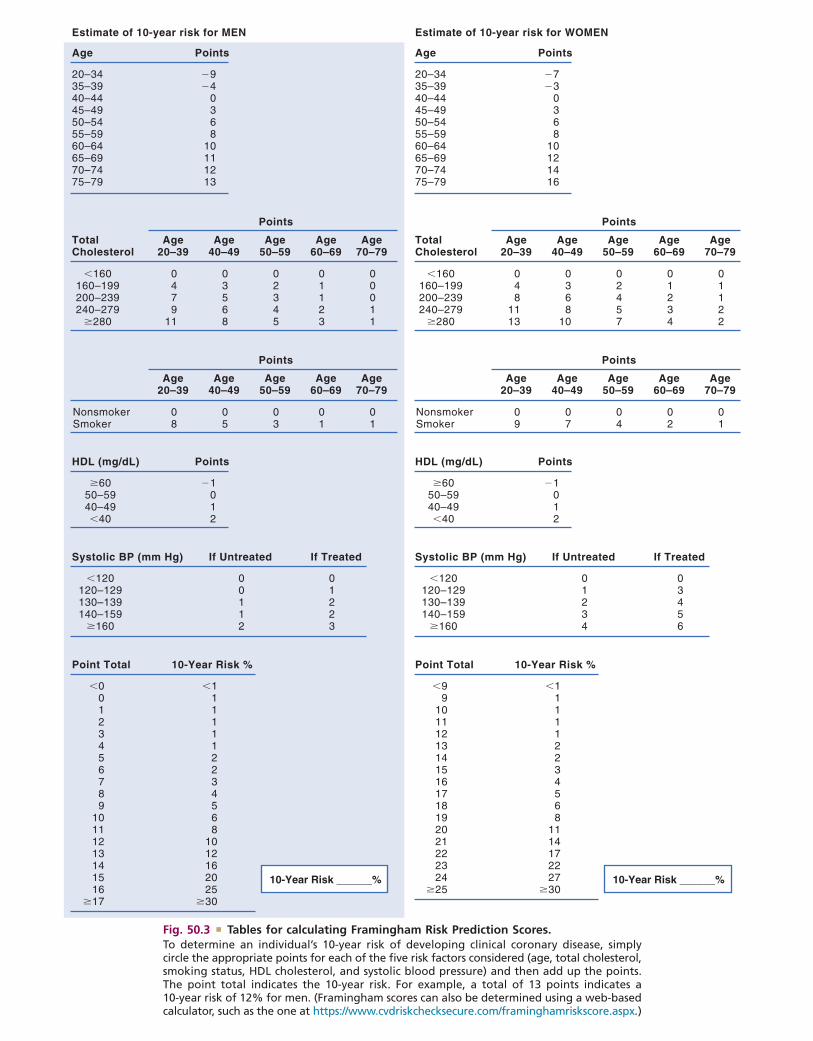
Estimate of 10-year risk for MEN
Age Points
20–3435–3940–4445–4950–5455–5960–6465–6970–7475–79
�9�4
0368
10111213
HDL (mg/dL) Points
�6050–5940–49�40
�1012
Point Total 10-Year Risk %
10-Year Risk ______%
�00123456789
10111213141516
�17
�1111112234568
1012162025
�30
Systolic BP (mm Hg) If Untreated
�120120–129130–139140–159
�160
00112
If Treated
01223
TotalCholesterol
Age20–39
Points
�160160–199200–239240–279
�280
0479
11
Age40–49
03568
Age50–59
02345
Age60–69
01123
Age70–79
00011
Age20–39
Points
NonsmokerSmoker
08
Age40–49
05
Age50–59
03
Age60–69
01
Age70–79
01
Estimate of 10-year risk for WOMEN
Age Points
20–3435–3940–4445–4950–5455–5960–6465–6970–7475–79
�7�3
0368
10121416
HDL (mg/dL) Points
�6050–5940–49�40
�1012
Point Total 10-Year Risk %
10-Year Risk ______%
�99
101112131415161718192021222324
�25
�111112234568
1114172227
�30
Systolic BP (mm Hg) If Untreated
�120120–129130–139140–159
�160
01234
If Treated
03456
TotalCholesterol
Age20–39
Points
�160160–199200–239240–279
�280
048
1113
Age40–49
0368
10
Age50–59
02457
Age60–69
01234
Age70–79
01122
Age20–39
Points
NonsmokerSmoker
09
Age40–49
07
Age50–59
04
Age60–69
02
Age70–79
01
Fig. 50.3 ■ Tables for calculating Framingham Risk Prediction Scores. To determine an individual’s 10-year risk of developing clinical coronary disease, simply circle the appropriate points for each of the five risk factors considered (age, total cholesterol, smoking status, HDL cholesterol, and systolic blood pressure) and then add up the points. The point total indicates the 10-year risk. For example, a total of 13 points indicates a 10-year risk of 12% for men. (Framingham scores can also be determined using a web-based calculator, such as the one at https://www.cvdriskchecksecure.com/framinghamriskscore.aspx.)

575
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
ModerateDaily dose lowers
LDL-C by approximately 30% to <50%
�7.5% estimated10-year ASCVD riskand age 40–75 years
DiabetesType 1 or 2
age 40–75 years
LDL-C �190 mg/dL
ClinicalASCVD
Age �75 yearsHigh-intensity statin
(Moderate-intensity statin if notcandidate for high-intensity statin)
ASCVD prevention benefit of statintherapy may be less clear in other groups
In selected individuals, consider additional factorsinfluencing ASCVD risk and potential ASCVD risk
benefits and adverse effects, drug-drug interactions,and patient preferences for statin treatment
ASCVD Statin Benefit GroupsHeart healthy lifestyle habits are the foundation of ASCVD prevention.
In individuals not receiving cholesterol-lowering drug therapy, recalculate estimated10-year ASCVD risk every 4–6 years in individuals aged 40–75 years without clinical ASCVD or
diabetes and with LDL-C 70–189 mg/dL.
Age >75 years OR if not candidate forhigh-intensity statin
Moderate-intensity statin
High-intensity statin(Moderate-intensity statin if not
candidate for high-intensity statin)
Moderate-intensity statin
Moderate- to high-intensity statin
Estimated 10-year ASCVD risk �7.5%High-intensity statin
Estimate 10-year ASCVD Riskwith pooled cohort equations
Adults age >21 years anda candidate for statin therapy
HighDaily dose lowers
LDL-C by approximately �50%
Yes
Yes
Yes
Yes
Yes
Yes
Yes
No
No
No
No
Definitions of High- andModerate-Intensity Statin therapy
Fig. 50.4 ■ Recommendations for statin therapy for ASCVD prevention.

576
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
total time spent exercising than on the intensity of exercise or improvements in fitness.
Smoking Cessation. Smoking cigarettes raises LDL cholesterol and lowers HDL cholesterol, thereby increasing the risk of ASCVD. Smokers should be strongly encouraged to quit—and nonsmokers should be urged not to start. Drugs to aid smoking cessation are discussed in Chapter 39.
Weight Control. Weight loss can reduce both LDL cholesterol and ASCVD risk. This is especially important for people with metabolic syndrome (discussed in an upcoming section).
Drug TherapyDrugs are not the first-line therapy for lowering LDL cholesterol. Rather, drugs should be employed only if TLCs fail to reduce LDL cholesterol to an acceptable level—and then only if the combination of elevated LDL cholesterol and the patient’s ASCVD risk category justify drug use. When drugs are used, it is essential that lifestyle modification continues because the beneficial effects of diet and drugs are additive; drugs alone may be unable to achieve the LDL goal. It is important to note that the principal benefit of drug therapy is primary prevention: Drugs are much better at preventing or slowing ASCVD than at promoting regression of established coronary atherosclerosis. Furthermore, because LDL cholesterol levels will return to pretreatment values if drugs are withdrawn, treatment must continue lifelong. Patients should be made aware of this requirement.
Table 50.5 shows properties of the drug families used to lower LDL cholesterol. The most effective agents are the HMG-CoA reductase inhibitors (e.g., atorvastatin [Lipitor]), usually referred to simply as statins. Lesser used alternatives are bile-acid sequestrants (e.g., cholestyramine) and niacin (nicotinic acid). Although fibrates are listed in Table 50.5, these drugs are used primarily to reduce levels of TGs—not LDLs. Treatment is initiated with a single drug, almost always a statin. If the statin is ineffective, a bile-acid sequestrant can be added to the regimen.
In addition to lowering LDL cholesterol, drugs may be used to raise HDL cholesterol. The most effective agents are niacin and the fibrates. However, virtually all of the drugs that we use to lower LDL cholesterol have the added benefit of
Therapeutic Lifestyle ChangesTherapeutic lifestyle changes are nondrug measures used to lower LDL cholesterol. TLCs focus on four main issues: diet, exercise, weight control, and smoking cessation. These measures are first-line treatment for LDL reduction and should be implemented before drug therapy. However, TLCs can be a challenge because some people do not eat healthier diets or exercise. Physical conditions such as arthritis can limit attempts at exercise, and economic and time limitations can be a barrier to healthier eating.
The TLC Diet. This diet has two objectives: (1) reducing LDL cholesterol and (2) establishing and maintaining a healthy weight. The central feature of the diet is reduced intake of cholesterol and saturated fats: Individuals should limit intake of cholesterol to 200 mg/day or less and intake of saturated fat to 7% or less of total calories. Intake of trans fats—found primarily in snack crackers, commercial baked goods, and fried foods—should be minimized. (Many food manufacturers are adding “no trans fat” labels to their product labels, making shopping somewhat easier.)
If the basic TLC diet fails to lower LDL cholesterol ade-quately, ATP III recommends two additional measures: increased intake of soluble fiber (10 to 25 gm/day; oatmeal is a good source) and increased intake of plant stanols and sterols (2 gm/day). Plant stanols and sterols are cholesterol-lowering chemicals found (albeit in very small amounts) in certain vegetable oils (e.g., canola), nuts (walnuts are a good source), certain fruits, and most beans and many other vegetables. They are also found in some of the cholesterol-lowering margarines, commonly advertised as “buttery spreads” (see later in this chapter under Plant Stanol and Sterol Esters).
Exercise. An inactive lifestyle carries an increased risk of ASCVD. Conversely, participating in regular exercise lowers ASCVD risk. Running and swimming, for example, can decrease LDL cholesterol and elevate HDL cholesterol, thereby reducing risk. In addition, exercise can reduce blood pressure, improve overall CV performance, and decrease insulin resistance (important because many people with high cholesterol also have diabetes). Accordingly, ATP III encourages regular physical activity (defined as 30 to 60 minutes of activity on most days). Improvements in the plasma lipid profile depend more on the
Adapted from 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Available at http://circ.ahajournals .org/content/early/2013/11/11/01.cir.0000437738.63853.7a.
High-Intensity Therapy Moderate-Intensity Therapy Low-Intensity Therapy
Daily dose lowers LCL-C on average by ≥ 50%
Daily dose lowers LDL-C on average by ~30% to <50%
Daily dose lowers LDL-C on average by <30%
Atorvastatin: 40–80 mgRosuvastatin: 20 mg
Atorvastatin: 10 mgRosuvastatin: 10 mgSimvastatin: 20–40 mgPravastatin: 40 mgLovastatin: 40 mg
Simvastatin: 10 mgPravastatin: 10–20 mgLovastatin: 20 mg
TABLE 50.4 ■ High-, Moderate-, and Low-Intensity Statin Therapy
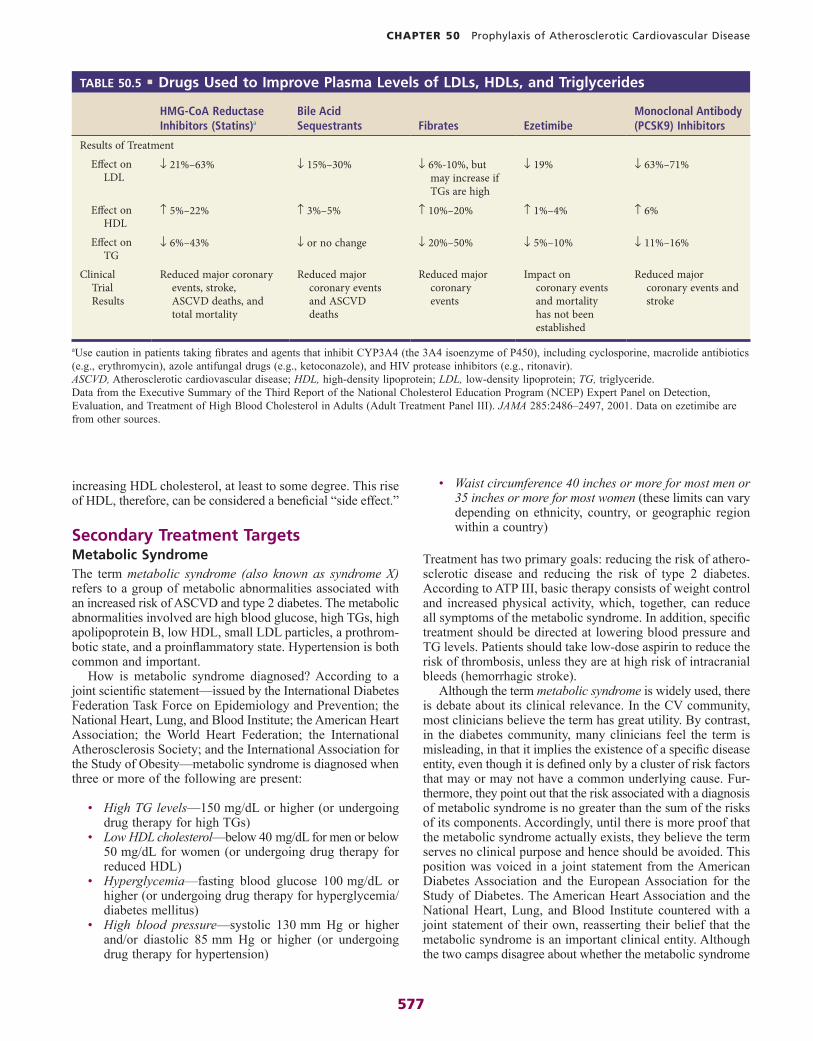
577
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
• Waist circumference 40 inches or more for most men or 35 inches or more for most women (these limits can vary depending on ethnicity, country, or geographic region within a country)
Treatment has two primary goals: reducing the risk of athero-sclerotic disease and reducing the risk of type 2 diabetes. According to ATP III, basic therapy consists of weight control and increased physical activity, which, together, can reduce all symptoms of the metabolic syndrome. In addition, specific treatment should be directed at lowering blood pressure and TG levels. Patients should take low-dose aspirin to reduce the risk of thrombosis, unless they are at high risk of intracranial bleeds (hemorrhagic stroke).
Although the term metabolic syndrome is widely used, there is debate about its clinical relevance. In the CV community, most clinicians believe the term has great utility. By contrast, in the diabetes community, many clinicians feel the term is misleading, in that it implies the existence of a specific disease entity, even though it is defined only by a cluster of risk factors that may or may not have a common underlying cause. Fur-thermore, they point out that the risk associated with a diagnosis of metabolic syndrome is no greater than the sum of the risks of its components. Accordingly, until there is more proof that the metabolic syndrome actually exists, they believe the term serves no clinical purpose and hence should be avoided. This position was voiced in a joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. The American Heart Association and the National Heart, Lung, and Blood Institute countered with a joint statement of their own, reasserting their belief that the metabolic syndrome is an important clinical entity. Although the two camps disagree about whether the metabolic syndrome
increasing HDL cholesterol, at least to some degree. This rise of HDL, therefore, can be considered a beneficial “side effect.”
Secondary Treatment TargetsMetabolic SyndromeThe term metabolic syndrome (also known as syndrome X) refers to a group of metabolic abnormalities associated with an increased risk of ASCVD and type 2 diabetes. The metabolic abnormalities involved are high blood glucose, high TGs, high apolipoprotein B, low HDL, small LDL particles, a prothrom-botic state, and a proinflammatory state. Hypertension is both common and important.
How is metabolic syndrome diagnosed? According to a joint scientific statement—issued by the International Diabetes Federation Task Force on Epidemiology and Prevention; the National Heart, Lung, and Blood Institute; the American Heart Association; the World Heart Federation; the International Atherosclerosis Society; and the International Association for the Study of Obesity—metabolic syndrome is diagnosed when three or more of the following are present:
• High TG levels—150 mg/dL or higher (or undergoing drug therapy for high TGs)
• Low HDL cholesterol—below 40 mg/dL for men or below 50 mg/dL for women (or undergoing drug therapy for reduced HDL)
• Hyperglycemia—fasting blood glucose 100 mg/dL or higher (or undergoing drug therapy for hyperglycemia/diabetes mellitus)
• High blood pressure—systolic 130 mm Hg or higher and/or diastolic 85 mm Hg or higher (or undergoing drug therapy for hypertension)
HMG-CoA Reductase Inhibitors (Statins)a
Bile Acid Sequestrants Fibrates Ezetimibe
Monoclonal Antibody (PCSK9) Inhibitors
Results of Treatment
Effect on LDL
↓ 21%–63% ↓ 15%–30% ↓ 6%-10%, but may increase if TGs are high
↓ 19% ↓ 63%–71%
Effect on HDL
↑ 5%–22% ↑ 3%–5% ↑ 10%–20% ↑ 1%–4% ↑ 6%
Effect on TG
↓ 6%–43% ↓ or no change ↓ 20%–50% ↓ 5%–10% ↓ 11%–16%
Clinical Trial Results
Reduced major coronary events, stroke, ASCVD deaths, and total mortality
Reduced major coronary events and ASCVD deaths
Reduced major coronary events
Impact on coronary events and mortality has not been established
Reduced major coronary events and stroke
TABLE 50.5 ■ Drugs Used to Improve Plasma Levels of LDLs, HDLs, and Triglycerides
ASCVD, Atherosclerotic cardiovascular disease; HDL, high-density lipoprotein; LDL, low-density lipoprotein; TG, triglyceride.Data from the Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 285:2486–2497, 2001. Data on ezetimibe are from other sources.
aUse caution in patients taking fibrates and agents that inhibit CYP3A4 (the 3A4 isoenzyme of P450), including cyclosporine, macrolide antibiotics (e.g., erythromycin), azole antifungal drugs (e.g., ketoconazole), and HIV protease inhibitors (e.g., ritonavir).

578
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
HMG-CoA Reductase Inhibitors (Statins)HMG-CoA reductase inhibitors, commonly called statins (because their generic names all end in statin), are the most effective drugs for lowering LDL and total cholesterol. In addition, they can raise HDL cholesterol and lower TGs in some patients. Most important, these drugs have been shown to improve clinical outcomes, including a lowering of the risk of heart failure, MI, and sudden death. Because of these benefits, and because so many people have ASCVD risks associated with dyslipidemias, statins are among our most widely prescribed drugs—and have earned tens of billions for their makers.
Beneficial ActionsThe statins have several actions that can benefit patients with (or at risk of) atherosclerosis. The most obvious and important are reductions of LDL cholesterol.
Reduction of LDL Cholesterol. Statins have a profound effect on LDL cholesterol. Low doses decrease LDL cholesterol by about 25%, and larger doses decrease levels by as much as 63% (Table 50.6). Reductions are significant within 2 weeks and maximal within 4 to 6 weeks. Because cholesterol synthesis normally increases during the night, statins are most effective when given in the evening. If statin therapy is stopped, serum cholesterol will return to pretreatment levels within weeks to months. Therefore, treatment should continue lifelong, unless serious adverse effects or specific contraindications (especially pregnancy or muscle damage) arise. The mechanism by which statins reduce cholesterol levels is discussed under Mechanism of Cholesterol Reduction.
Elevation of HDL Cholesterol. Statins can increase levels of HDL cholesterol. Recall that low levels of HDL cholesterol (below 40 mg/dL) are an independent risk factor for ASCVD. Hence, by raising HDL cholesterol, statins may help reduce the risk of CV events in yet another way. The objective is to raise levels to 50 mg/dL or more.
Reduction of Triglyceride Levels. Although statins mainly affect cholesterol synthesis and thereby lower LDL cholesterol levels, these drugs may also lower TGs. Just why these “anti-cholesterol” drugs lower TGs is unknown, but the response has been amply documented. Please note that although statins may reduce TG levels, they are not actually prescribed for this action. Hence TG reduction is usually a beneficial side effect in patients taking statins to lower their LDL cholesterol. Of note, the ability to lower TGs seems to be short lived, and hence a drug designed to lower TGs eventually may need to be added.
is an actual disease, both sides strongly agree that the risk factors that define the syndrome should be identified and treated.
One final point: Whether or not you think metabolic syn-drome is a useful term, you will still see lots of patients who meet the criteria. The fact is, patients with several of these risk factors are much more common than patients with just one.
High TriglyceridesHigh TG levels (above 200 mg/dL) may be an independent risk factor for ASCVD. In clinical practice, high TGs are seen most often in patients with metabolic syndrome. However, high levels may also be associated with inactive lifestyle, cigarette smoking, excessive alcohol intake, type 2 diabetes, certain genetic disorders, and high carbohydrate intake (when carbohydrates account for more than 60% of total caloric intake). In most patients with high TG levels, the first treatment goal is to achieve the original LDL goal. Dietary modification is always recommended. Statins being taken to lower cholesterol may help lower TGs as well, perhaps to a satisfactory level. However, if TG levels remain unacceptably high, medications specific to TGs—and fibrates—may be needed. Unfortunately, when these drugs are combined with cholesterol-lowering drugs (as they often are), the adverse effects of cholesterol-lowering agents may be intensified.
DRUGS AND OTHER PRODUCTS USED TO IMPROVE PLASMA LIPID LEVELSAs discussed earlier in this chapter, the lipid abnormality that contributes most to CV disease is high LDL cholesterol. Accordingly, we will focus primarily on drugs for this disorder. Nonetheless, we also need to consider other lipid abnormalities, especially (1) high total cholesterol,a (2) low HDL cholesterol, and (3) high TGs.
Some drugs for dyslipidemias are more selective than others. That is, whereas some drugs may improve just one dyslipidemia (e.g., high TGs), others may improve two or more dyslipidemias. The highly selective agents can be useful as add-ons, to “target” a particular lipid abnormality when other medications prove inadequate.
Drugs that lower LDL cholesterol levels include HMG-CoA reductase inhibitors (statins), bile-acid sequestrants, and ezetimibe. All are effective to varying degrees. The HMG-CoA reductase inhibitors—the statins—are more effective than the others, cause fewer adverse effects, are better tolerated, and are more likely to improve clinical outcomes.
As we consider the drugs for lipid disorders, you should be aware of the following: Although all of these drugs can improve lipid profiles, not all of them improve clinical outcomes (reduced morbidity and mortality). This leads us to question whether some of the lipid abnormalities are a true cause of pathophysiology and ultimate death, or whether they are simply “associated markers” of some other pathophysiology that we don’t yet understand.
Prototype Drugs
LDL CHOLESTEROL- AND TRIGLYCERIDE-LOWERING DRUGS
HMG-CoA Reductase Inhibitors (Statins)Lovastatin
Bile-Acid SequestrantsColesevelam
OthersEzetimibe
aNote that total cholesterol is slightly different from the simple sum of LDL cholesterol (LDL-C) plus HDL cholesterol (HDL-C); triglycerides (TG) also contribute to the value, as in the following equation: total cholesterol = HDL-C + LDL-C + (TG/5) (provided TG levels are below 400 mg/dL).

579
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
indicating that (1) inhibition of cholesterol synthesis, by itself, is not sufficient to explain cholesterol-lowering effects; and (2) for statins to be effective, synthesis of LDL receptors must increase.
In addition to inhibiting HMG-CoA reductase, statins decrease production of apolipoprotein B-100. As a result, hepatocytes decrease production of VLDLs. This lowers VLDL levels and TG levels too since they’re the main lipid in VLDLs. Also, statins raise HDL levels by 5% to 22%.
Clinical TrialsStatins slow the progression of ASCVD and decrease the risk of stroke, hospitalization, cardiac events, peripheral vascular disease, and death. Benefits are seen in men and women, and in apparently healthy people, as well as in those with a history of CV events. Hence, the statins are useful for both primary and secondary prevention. Furthermore, these drugs can help people with even normal LDL levels, in addition to those whose LDL level is high. Statins may also have some added protective effects in people with diabetes.
Secondary Prevention Studies. In patients with evidence of existing ASCVD (angina pectoris or previous MI), statins reduce the risk of death from cardiac causes. This was first demonstrated conclusively in the landmark Scandinavian Simvastatin Survival Study (4S). After 4.9 to 6.3 years of follow-up, the death rate was 12% among patients taking placebo and 8% among those taking simvastatin—a 30% decrease in overall mortality. Benefits were due to a decrease in cardiac-related mortality; deaths from noncardiac causes were the same in both groups.
The Cholesterol and Recurrent Events (CARE) trial demonstrated the ability of statins to reduce the risk of stroke in addition to coronary events. In this study, 4159 people with a history of MI were given pravastatin (40 mg daily) or placebo. After 5 years, the incidence of MI (fatal or nonfatal) was 13.2% in those taking placebo and 10.2% in those taking the drug. Pravastatin also produced a 26% decrease in the risk of stroke.
The Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) trial was the first to show that intensive reductions in LDL with statin
Nonlipid Beneficial CV Actions. There is increasing evidence that statins do more than just alter lipid levels. Specifi-cally, they can promote atherosclerotic plaque stability (by decreasing plaque cholesterol content), reduce inflammation at the plaque site, slow progression of coronary artery calcifica-tion, improve abnormal endothelial function, enhance the ability of blood vessels to dilate, reduce the risk of atrial fibrillation, and reduce the risk of thrombosis by (1) inhibiting platelet deposition and aggregation and (2) suppressing production of thrombin, a key factor in clot formation. All of these actions help reduce the risk of CV events.
Increased Bone Formation. There is evidence that statins may promote bone formation and may thereby reduce the risk of osteoporosis and related fractures. This has been shown in several case-control studies in humans. However, other case-control studies have failed to demonstrate a protective effect. In addition, six randomized controlled trials of statins in 3022 post-menopausal women failed to show a difference in bone density. Until randomized controlled trials are completed in other populations, osteoporosis should be managed with bisphosphonates and/or other drugs with proven efficacy (see Chapter 75).
Mechanism of Cholesterol ReductionThe mechanism by which statins decrease LDL cholesterol levels is complex and depends ultimately on increasing the number of LDL receptors on hepatocytes (liver cells). The process begins with inhibition of hepatic HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. In response to decreased cholesterol production, hepatocytes synthesize more HMG-CoA reductase. As a result, cholesterol synthesis is largely restored to pretreatment levels. However—and for reasons that are not fully understood—inhibition of cholesterol synthesis causes hepatocytes to synthesize more LDL receptors. As a result, hepatocytes are better able to remove more LDLs from the blood. In patients who are genetically unable to synthesize LDL receptors, statins fail to reduce LDL levels,
Drug
% Change in Serum LipidsaEffect of CYP3A4 Inhibitors on Statin Levelsb
Effect of Renal or Hepatic Impairment on Statin LevelsLDL-C HDL-C TGs
Atorvastatin [Lipitor]
↓ 25–60 ↑ 5–15 ↓ 15–50 Moderate ↑ No change with renal disease; significant ↑ with hepatic impairment
Fluvastatin [Lescol, Lescol XL]
↓ 20–40 ↑ 2–11 ↓ 10–25 None No change with renal disease; possible ↑ with hepatic impairment
Lovastatin [Altoprev, Mevacor]
↓ 20–40 ↑ 5–10 ↓ 5–25 Significant ↑ ↑ with significant renal impairment; no change with hepatic impairment
Pitavastatin [Livalo] ↓ 40–45 ↑ 6–8 ↓ 15–30 Little or none ↑ with significant renal impairment; little or no change with hepatic impairment
Pravastatin [Pravachol]
↓ 20–40 ↑ 1–15 ↓ 10–25 None Potential ↑ with either renal or hepatic impairment
Rosuvastatin [Crestor]
↓ 30–60 ↑ 3–20 ↓ 10–40 None ↑ levels with severe renal impairment or hepatic dysfunction
Simvastatin [Zocor] ↓ 25–50 ↑ 7–15 ↓ 8–40 Significant ↑ Potential ↑ with severe renal or hepatic impairment
TABLE 50.6 ■ HMG-CoA Reductase Inhibitors: Selected Aspects of Clinical Pharmacology
aThe values were obtained from a variety of studies and do not reflect dose dependency of drug responses.bInhibitors of CYP3A4 (the 3A4 isoenzyme of P450) include itraconazole, ketoconazole, erythromycin, clarithromycin, HIV protease inhibitors, cyclosporine, nefazodone, and substances in grapefruit juice.↑, Increase; ↓, decrease.HDL-C, High-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TGs, triglycerides.

580
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Obviously, doing so would greatly expand the number of patients receiving statin therapy.
A more recent trial—Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)—reinforced the results of the Heart Protection Study. The study demonstrated that rosuvastatin can reduce the risk of coronary events in people with normal LDL levels, but with high levels of CRP and other risk factors for ASCVD.
Prevention in Patients With Diabetes. Results of the Collaborative Atorvastatin Diabetes Study (CARDS) indicate that statin therapy can reduce the risk of CV events in diabetes patients, even if LDL levels are normal. This randomized trial, conducted in Britain and Ireland, enrolled 2838 patients with type 2 diabetes who had no history of CV disease. Half received 10 mg of atorvastatin [Lipitor] daily, and half received a placebo. After a mean of 4 years, the combined incidence of acute coronary events, coronary revascu-larization, and stroke was only 5.8% in the atorvastatin group, compared with 9% in the placebo group, representing a 36% reduction in risk. These results suggest that statin therapy could benefit most patients with diabetes, regardless of their LDL level.
Therapeutic UsesWhen the statins were introduced, they were approved only for hypercholesterolemia (elevated LDL cholesterol levels) in adults. As understanding of their benefits grew, so has the list of indications. Today the statins have nearly a dozen U.S. Food and Drug Administration (FDA)–approved indications, and these drugs can be prescribed for young patients, as well as for adults. Indications for individual statins are shown in Table 50.7. Major indications are discussed in the sections that follow.
Hypercholesterolemia. Statins are the most effective drugs we have for lowering LDL cholesterol. In sufficient dosage, statins can decrease LDL cholesterol by more than 60%. For many patients, the treatment goal is to drop LDL cholesterol to below 100 mg/dL. For patients at very high CV risk, a target of 70 mg/dL may be appropriate.
Primary and Secondary Prevention of CV Events. As discussed, statins can reduce the risk of CV events (e.g., MI,
therapy provide more CV protection than moderate reductions. In PROVE-IT, 4162 patients with ACS were randomized to either a moderate statin regimen (pravastatin, 40 mg daily) or an intensive statin regimen (atorvastatin, 80 mg daily). The result? LDL levels in the moderate group dropped to 95 mg/dL, compared with 62 mg/dL in the intensive group. Furthermore, not only did intensive therapy produce a greater decrease in LDL cholesterol, it also produced a greater reduction in adverse outcomes: After 24 months, the incidence of CV events (death, MI, unstable angina, or revascularization) was only 22.4% in the intensive group compared with 26.3% in the moderate group. These results led the ATP III panel to recommend lower target LDL levels in patients at very high CV risk.
Primary Prevention Studies. Two major studies have demonstrated the ability of statins to reduce mortality in people with no previous history of coronary events. In the first trial—the West of Scotland Coronary Prevention Study (WOSCOPS)—6595 men with high cholesterol were given either pravastatin (40 mg/day) or placebo. During an average follow-up of 4.9 years, 4.1% of those taking placebo died, compared with only 3.2% of those taking the statin. The second trial—the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS)—enrolled 6605 low-risk patients: men and women with average cholesterol levels (221 mg/dL) and no history of CV events. The subjects were randomly assigned to receive lovastatin (20 to 40 mg/day) or placebo. After an average follow-up of 5.5 years, the incidence of first major coronary events was 5.5% for those taking placebo and 3.5% for those taking the drug—representing a 36% decrease in risk.
Primary Prevention in Patients With Normal Cholesterol Levels. The landmark Heart Protection Study, published in 2002, was the first major trial to demonstrate that statins can reduce the risk of major coronary events in people who have normal levels of cholesterol. This double-blind, placebo-controlled trial enrolled 20,536 high-risk British patients: men and women with diabetes, prior MI, stroke, or prior angioplasty. Some had high levels of LDL and total cholesterol; others had normal levels. Subjects were randomly assigned to receive either simvastatin (40 mg/day) or placebo. After 5 years, the incidence of death was 12.9% in the treatment group, compared with 14.7% in the placebo group. Death from ASCVD was reduced by 18%. In addition, simvastatin reduced the risk of nonfatal MI by 38% and of stroke by 25%, and reduced the need for coronary revascularization (e.g., angioplasty) by 30%. Most strikingly, benefits were seen in patients whose LDL cholesterol was normal or low, as well as in those whose levels were high. These data suggest a radical shift in practice. Specifically, they suggest we should treat people at high ASCVD risk—not just those with high cholesterol levels.
IndicationAtorvastatin [Lipitor]
Fluvastatin [Lescol, Lescol XL]
Lovastatin [Altoprev, Mevacor]
Pitavastatin [Livalo]
Pravastatin [Pravachol]
Rosuvastatin [Crestor]
Simvastatin [Zocor]
Primary hypercholesterolemia ✓ ✓ ✓ ✓ ✓ ✓ ✓Homozygous familial
hyperlipidemia✓ ✓ ✓
Heterozygous familial hypercholesterolemia in adolescents
✓ ✓ ✓ ✓ ✓
Mixed dyslipidemia ✓ ✓ ✓ ✓ ✓ ✓ ✓Primary
dysbetalipoproteinemia✓ ✓ ✓ ✓
Primary prevention of coronary events
✓ ✓ ✓ ✓a ✓
Secondary prevention of CV events
✓ ✓ ✓ ✓ ✓
Increasing HDL cholesterol in primary hypercholesterolemia
✓ ✓ ✓ ✓ ✓
Slowing progression of coronary atherosclerosis
✓ ✓ ✓ ✓
TABLE 50.7 ■ HMG-CoA Reductase Inhibitors: FDA-Approved Indications
aRosuvastatin is approved for primary prevention in patients who have normal LDL cholesterol and no clinical evidence of ASCVD, but who do have high levels of C-reactive protein combined with other risk factors for CV disease.

581
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
and simvastatin—undergo clinically significant (10% to 20%) excretion in the urine.
Three statins—atorvastatin, lovastatin, and simvastatin—are metabolized by CYP3A4 (the 3A4 isoenzyme of cytochrome P450). As a result, levels of these drugs can be lowered by agents that induce CYP3A4 synthesis and speed up the meta-bolic inactivation of the statin. More importantly, statin levels can be increased—sometimes dramatically—by agents that inhibit CYP3A4 (see With Drugs That Inhibit CYP3A4 later in this chapter).
One agent—rosuvastatin—reaches abnormally high levels in people of Asian heritage. At usual therapeutic doses, rosu-vastatin levels in these patients are about twice those in Caucasians. Accordingly, if rosuvastatin is used by Asians, dosage should be reduced.
Adverse EffectsStatins are generally well tolerated. Side effects are uncommon. Some patients develop headache, rash, memory loss, or GI dis-turbances (dyspepsia, cramps, flatulence, constipation, abdominal pain). However, these effects are usually mild and transient. Serious adverse effects—hepatotoxicity and myopathy— are relatively rare. Some statins pose a greater risk than others, as noted here.
Myopathy/Rhabdomyolysis. Statins can injure muscle tissue. Mild injury occurs in 5% to 10% of patients. Charac-teristic symptoms are muscles aches, tenderness, or weakness that may be diffuse or localized to certain muscle groups. Rarely, mild injury progresses to myositis, defined as muscle inflammation associated with moderate elevation of creatine kinase (CK), an enzyme released from injured muscle. Release of potassium from muscle may cause blood potassium concentra-tions to rise. Rarely, myositis progresses to potentially fatal rhabdomyolysis, defined as muscle disintegration or dissolution. Release of muscle components leads to marked elevations of blood CK (greater than 10 times the upper limit of normal [ULN]) and elevations of free myoglobin. High levels of CK, in turn, may cause renal impairment, as excess CK can plug up the glomeruli, thereby preventing normal filtration.
Fortunately, fatal rhabdomyolysis is extremely rare: The overall incidence is less than 0.15 case per 1 million prescrip-tions. Nonetheless, patients should be informed about the risk of myopathy and instructed to notify the prescriber if unex-plained muscle pain or tenderness occurs. How statins cause myopathy is unknown.
Several factors increase the risk of myopathy. Among these are advanced age, small body frame, frailty, multisystem disease (e.g., chronic renal insufficiency, especially associated with diabetes), use of statins in high doses, low vitamin D and coenzyme Q levels, concurrent use of fibrates (which can cause myopathy too), and the use of drugs that can raise statin levels (see the following paragraphs). In addition, hypothyroidism increases risk. Accordingly, if muscle pain develops, thyroid function should be assessed. Measurement of CK levels can facilitate diagnosis. Levels should be determined at baseline and again if symptoms of myopathy appear. If the CK level is more than 10 times the ULN, the statin should be discon-tinued. If the level is less than 10 times the ULN, the statin can be continued, provided myopathy symptoms and the CK level are followed weekly. However, given that weekly blood tests are expensive and inconvenient, it may be best to stop the statin and re-evaluate therapy, even when CK levels are
angina, stroke) in patients who have never had one (primary prevention), and they can reduce the risk of a subsequent event after one has occurred (secondary prevention). Risk reduction is related to the reduction in LDL: the greater the LDL reduction, the greater the reduction in risk.
Primary Prevention in People With Normal LDL Levels. One agent—rosuvastatin [Crestor]—is now approved for reduc-ing the risk of CV events in people with normal levels of LDL and no clinically evident ASCVD, but who do have an increased risk based on advancing age, high levels of high-sensitivity C-reactive protein, and at least one other risk factor for CV disease (e.g., hypertension, low HDL, smoking). Approval for this use was based in large part on results of the JUPITER trial.
Post-MI Therapy. Patients who have survived an MI and who were not on statin therapy at the time of the event are routinely started on a statin, the rationale being “better late than never.” The current trend is to begin statins as soon as the patient is stabilized and able to take oral drugs. Other drugs for MI are discussed in Chapter 53.
Diabetes. Cardiovascular disease is the primary cause of death in people with diabetes. Hence, to reduce mortality, controlling CV risk factors—especially hypertension and high cholesterol—is as important as controlling high blood glucose. The American Diabetes Association recommends a statin for all patients older than 40 years whose LDL cholesterol is greater than 100 mg/dL. The American College of Physicians recom-mends a statin for (1) all patients with type 2 diabetes plus diagnosed ASCVD—even if they don’t have high cholesterol; and (2) all adults with type 2 diabetes plus one additional risk factor (e.g., hypertension, smoking, age older than 55 years)—even if they don’t have high cholesterol. Taken together, these guidelines suggest that most patients with diabetes should receive a statin.
Potential Uses. Potential uses of statins extend well beyond diabetes and CV disorders. Judging from preliminary evidence, these drugs may eventually be used to prevent and/or treat a variety of conditions, including Alzheimer’s disease, kidney disease, multiple sclerosis, macular degeneration, glaucoma, rheumatoid arthritis, weak or brittle bones, and even certain cancers.
Influenza is associated with an increase in the risk for ACS, stroke, and venous thromboembolism. This is related to the increase in proinflammatory cytokines released during influenza. It is thought that statins can decrease this cytokine release and improve influenza-associated morbidity and mortality. A 10-year retrospective cohort study of older adults revealed that statins can protect against influenza morbidity in people taking statins as an outpatient. It also noted that there was an increase in the mortality of hospitalized patients admitted with influenza when their statin was stopped. Although this information is promising, more research is needed.
PharmacokineticsStatins are administered orally. The amount absorbed ranges between 30% and 90%, depending on the drug. Regardless of how much is absorbed, most of an absorbed dose is extracted from the blood on its first pass through the liver, the princi-pal site at which statins act. Only a small fraction of each dose reaches the systemic circulation. Statins undergo rapid hepatic metabolism followed by excretion primarily in the bile. Only four agents—lovastatin, pitavastatin, pravastatin,

582
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
and similar results were found. The reason for this association is unknown. The greatest risk appears to be in patients taking simvastatin, lovastatin, and atorvastatin. Overall, the risk remains small, and statin therapy should be continued in patients when indicated.
Drug InteractionsWith Other Lipid-Lowering Drugs. Combining a statin
with most other lipid-lowering drugs (except probably the bile-acid sequestrants) can increase the incidence and severity of the most serious statin-related adverse events: muscle injury, liver injury, and kidney damage. The increase in risk occurs primarily with fibrates (gemfibrozil, fenofibrate), which are commonly combined with statins. The bottom line: When statins are combined with other lipid-lowering agents, use extra caution and monitor for adverse effects more frequently.
With Drugs That Inhibit CYP3A4. Drugs that inhibit CYP3A4 can raise levels of lovastatin and simvastatin sub-stantially, and can raise levels of atorvastatin moderately, by slowing their inactivation. Important inhibitors of CYP3A4 include macrolide antibiotics (e.g., erythromycin), azole antifungal drugs (e.g., ketoconazole, itraconazole), HIV protease inhibitors (e.g., ritonavir), amiodarone (an antidysrhythmic drug), and cyclosporine (an immunosuppressant). If these drugs are combined with a statin, increased caution is advised. Some authorities recommend an automatic reduction in statin dosage if these inhibitors are used.
As discussed in Chapter 6, chemicals in grapefruit and grapefruit juice can inhibit CYP3A4. Furthermore, the inhibition may persist for 3 days or more after eating the fruit or drinking its juice. Accordingly, statin users should avoid grapefruits and their juice.
Use in PregnancyStatins are classified in FDA Pregnancy Risk Category Xb: The risks to the fetus outweigh any potential benefits of treat-ment. Some statins have caused fetal malformation in animal models—but only at doses far higher than those used in humans. To date, teratogenic effects in humans have not been reported. Nonetheless, because statins inhibit synthesis of cholesterol and because cholesterol is required for the synthesis of cell membranes as well as several fetal hormones, concern regarding human fetal injury remains. Moreover, there is no compelling reason to continue lipid-lowering drugs during pregnancy: Stopping the statin for 9 months is not going to cause a sudden, dangerous rise in cholesterol levels or risk of ASCVD. Women of childbearing age should be informed about the potential for fetal harm and warned against becoming pregnant. If pregnancy occurs and the patient plans to continue the pregnancy, statins should be discontinued.
Preparations, Dosage, and AdministrationStatins are available alone and in fixed-dose combinations. The single-ingredient products are discussed here. The combina-tion products are discussed later in the chapter under the heading Drug Combinations.
Seven statins are available for use alone: atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin,
less than 10 times the ULN. Routine monitoring of CK in asymptomatic patients is unnecessary.
What is the rhabdomyolysis risk with individual statins? Of the seven statins in current use, rosuvastatin [Crestor] poses the highest risk of rhabdomyolysis. But even with this drug, the absolute number of cases is extremely low. With the other statins, the risk is even lower.
Should concerns about myopathy discourage statin use? Definitely not! Remember: The risk of serious myopathy is extremely low, whereas the risk of untreated LDL cholesterol is very high. Accordingly, when statins are used to lower cholesterol, the benefits of therapy (reduction of CV events) far outweigh the small risk of myopathy. Additional strategies for the management of myalgia include replacement of vitamin D and coenzyme Q and switching statins. Studies reveal that replacement of vitamin D and coenzyme Q can reduce myalgias in patients with low levels. Switching statins can be effective, as patients may not have myalgias when taking a different drug, even if it is within the same class.
Hepatotoxicity. Liver injury, as evidenced by elevations in serum transaminase levels, develops in 0.5% to 2% of patients treated 1 year or longer. However, jaundice and other clinical signs are rare. Progression to outright liver failure occurs very rarely. Because of the risk of liver injury, product labeling recommends that liver function tests (LFTs) be done before treatment and then if clinically indicated after starting the drug. If serum transaminase levels rise to 3 times the ULN and remain there, statins should be discontinued. Transaminase levels decline to pretreatment levels following drug withdrawal.
Should statins be used by patients with active liver disease? The answer depends on the disease. In patients with viral or alcoholic hepatitis, statins should be avoided. However, in patients with the most common cause of hepatitis—nonalcoholic fatty liver disease—statins are acceptable therapy. In fact, in these patients, not only can statins reduce cholesterol levels, they may also decrease liver inflammation, improve LFTs, and reduce steatosis (fatty infiltration in the liver). Should LFTs be monitored? Yes—at baseline and as clinically indicated thereafter. If LFTs climb to 3 times the ULN, statin use should stop.
New-Onset Diabetes. The risk of developing new-onset diabetes while taking a statin is 1 in 500 patients prescribed a statin. Yet many of the patients in these studies had prediabetes before taking a statin. It is unclear whether taking a statin accelerates the advancement from prediabetes to diabetes. Despite the possibility, the CV benefits of taking a statin far outweigh the risk, and management should not change.
Memory Loss. Some patients have reported reversible memory loss or confusion that improves after stopping statin therapy. Although memory loss may occur, there is an overall lack of evidence surrounding these reports. A large review of 41 different studies completed in 2013 found no connection between memory loss and statin use. What the evidence does reveal is a possible increase in memory loss in patients taking any type of cholesterol-reducing medication, not just statins. As this issue remains unclear, patients should still report confusion or memory loss to their provider.
Cataracts. An analysis of military healthcare records between 2003 and 2010 revealed a 27% increase of cataracts in patients who had taken a statin for at least 90 days when compared with patients not taking statins. More recently, two larger studies were completed in the older adult population,
bAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.

583
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
Safety in Asians. The same dose of rosuvastatin, when given to Asian and Caucasian subjects, may produce twofold higher blood levels in the Asians. Accordingly, when rosu-vastatin is used in Asians, start with the lowest available dosage and monitor diligently.
Price. Six statins—lovastatin, pravastatin, atorvastatin, fluvastatin, rosuvastatin, and simvastatin—are now available as generic products, and hence are cheaper than all other statins.
A Word Regarding Niacin (Nicotinic Acid)Niacin was once thought to be beneficial in the reduction of LDL and TG levels. However, despite these previously dem-onstrated favorable effects on lipid levels, niacin does little to improve outcomes and carries much potential harm. Therefore, in 2016, a panel of experts recommended that niacin be removed from the guidelines for use in hypertriglyceridemia and in lowering LDL cholesterol.
Bile-Acid SequestrantsBile-acid sequestrants reduce LDL cholesterol levels. In the past, these drugs were a mainstay of lipid-lowering therapy. Today, they are used primarily as adjuncts to statins. Three
and simvastatin. Information on preparations, dosage, and administration is shown in Table 50.8.
Dosing is done once daily, preferably in the evening either with the evening meal or at bedtime. Because endogenous cholesterol synthesis increases during the night, statins have the greatest impact when given in the evening.
Drug SelectionSeveral factors bear on statin selection, including the LDL goal, drug interactions, kidney function, safety in Asian patients, and price.
LDL Goal. If a 30% to 40% reduction in LDL is deemed sufficient, any statin will do. However, if LDL must be lowered by more than 40%, then atorvastatin or simvastatin may be preferred. Furthermore, not only are these two drugs highly effective, clinical experience with them is extensive.
Drug Interactions. Drugs that inhibit CYP3A4 can raise levels of atorvastatin, lovastatin, and simvastatin, thereby increasing the risk of toxicity, especially myopathy and liver injury. Accordingly, in patients taking a CYP3A4 inhibitor, other statins may be preferred.
Kidney Function. For patients with normal renal function, any statin is acceptable. However, for patients with significant renal impairment, atorvastatin and fluvastatin are preferred (because no dosage adjustment is needed).
Drug DosageAdministration With Regard to Meals
Dosage Changes in Special Populations Preparations
Atorvastatin [Lipitor] Initial: 10 mg at bedtimeMaximum: 80 mg at
bedtime
Take without regard to meals
No changes needed Lipitor (tablets): 10, 20, 40, 80 mg
Fluvastatin [Lescol, Lescol XL]
Initial: 20–40 mg at bedtime
Maximum, Lescol: 40 mg twice a day
Maximum, Lescol XL: 80 mg at bedtime
Take without regard to meals
Reduce dosage for severe renal impairment
Lescol (capsules): 20, 40 mg
Lescol XL (extended-release tablets): 80 mg
Lovastatin [Altoprev, Mevacor; generics]
Initial: 20 mgMaximum: 40 mg twice
daily or 80 mg at bedtime
Take immediate-release tablets with evening meal to increase absorption. Take extended-release tablets at bedtime.
Reduce dosage for severe renal impairment
Altoprev (extended-release tablets): 20, 40, 60 mg
Mevacor and generics (tablets): 10, 20, 40 mg
Pitavastatin [Livalo] Initial: 2 mg once daily at any time of day
Maximum: 4 mg once daily
Take without regard to meals
Reduce dosage for moderate to severe renal impairment
Livalo (tablets): 1, 2, 4 mg
Pravastatin [Pravachol; generics]
Initial: 40 mg at bedtimeMaximum: 80 mg at
bedtime
Take without regard to meals
Reduce dosage for moderate to severe renal impairment
Pravachol and generics (tablets): 10, 20, 40, 80 mg
Rosuvastatin [Crestor] Initial: 5–20 mg at bedtime
Maximum: 40 mg at bedtime
Take without regard to meals
Reduce dosage for severe renal impairment
Reduce dosage in Asian patients
Crestor (tablets): 5, 10, 20, 40 mg
Simvastatin [Zocor; generics]
Initial: 10–20 mg at bedtime
Maximum: 40 mg/day
Take without regard to meals
Reduce dosage for severe renal impairment
Zocor and generics (tablets): 5, 10, 20, 40, 80 mg
TABLE 50.8 ■ HMG-CoA Reductase Inhibitors: Preparations, Dosage, and Administration

584
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
colesevelam alone can lower LDL cholesterol by about 20% (the typical range is between 15% and 30%). In contrast, combined therapy with a statin can reduce LDL cholesterol by up to 50%.
Adverse Effects. The bile-acid sequestrants are not absorbed from the GI tract, and hence are devoid of systemic effects. Accordingly, they are safer than all other lipid-lowering drugs.
Adverse effects are limited to the GI tract. Constipation is the main complaint. This can be minimized by increasing dietary fiber and fluids. If necessary, a mild laxative may be used. Other GI effects include bloating, indigestion, and nausea. The older agents—cholestyramine and colestipol—can decrease fat absorption and may thereby decrease uptake of fat-soluble vitamins. However, this does not seem to be a problem with colesevelam.
Drug Interactions. The bile-acid sequestrants can form insoluble complexes with other drugs. Medications that undergo binding cannot be absorbed, and hence are not available for systemic effects. Drugs known to form complexes with the sequestrants include thiazide diuretics, digoxin, warfarin, and some antibiotics. To reduce formation of sequestrant-drug complexes, oral medications that are known to interact should be administered either 1 hour before the sequestrant or 4 hours after.
Preparations, Dosage, and Administration. Colesevelam [Welchol] is supplied in tablets (625 mg) and as a powder (1.875 and 3.75 gm) for making an oral suspension. With the tablets, the initial adult dosage is 3 tablets (1.9 gm) twice daily or 6 tablets (3.8 gm) once daily. With the oral suspension, the initial adult dosage is 1.875 gm twice daily or 3.75 gm once daily. All doses are taken with food and water. Of note, the dosage for colesevelam is much smaller than that of cholestyramine (4 to 24 gm/day) or colestipol (5 to 30 gm/day).
Older Agents: Cholestyramine and ColestipolCholestyramine and colestipol have been available for decades, but have been largely replaced by colesevelam because colesevelam is better tolerated, does not impede absorption of fat-soluble vitamins, and has minimal effects on other drugs. Although cholestyramine and colestipol are very safe, they fre-quently cause constipation, abdominal discomfort, and bloating.
Cholestyramine [Questran, Questran Light, Prevalite] is supplied in powdered form. Instruct patients to mix the powder with fluid, because swal-lowing it dry can cause esophageal irritation and impaction. Appropriate liquids for mixing include water, fruit juices, and soups. Pulpy fruits with a high fluid content (e.g., applesauce, crushed pineapple) may also be used. The dosage range is 4 to 24 gm/day.
Colestipol hydrochloride [Colestid] is supplied in granular form (5 gm) and in 1-gm tablets. The dosage for the granules is 5 to 30 gm/day administered in one or more doses. Instruct patients to mix the granules with fluids or pulpy fruits before ingestion. The dosage for the tablets is 2 to 16 gm/day administered in one or more doses. Tablets should be swallowed whole and taken with fluid.
EzetimibeEzetimibe [Zetia, Ezetrol ] is a unique drug for reducing plasma cholesterol. Benefits derive from blocking cholesterol absorption.
Mechanism of Action and Effect on Plasma LipoproteinsEzetimibe acts on cells of the brush border of the small intestine to inhibit dietary cholesterol absorption. The drug also inhibits reabsorption of cholesterol secreted in the bile. Treatment reduces plasma levels of total cholesterol, LDL cholesterol, TGs, and apolipoprotein B. In addition, ezetimibe can produce a small increase in HDL cholesterol.
agents are available: colesevelam, cholestyramine, and colesti-pol. Colesevelam is newer than the other two and is better tolerated.
ColesevelamColesevelam [Welchol] is the drug of choice when a bile-acid sequestrant is indicated. Like the older sequestrants, colesevelam is a nonabsorbable resin that binds (sequesters) bile acids and other substances in the GI tract and thereby prevents their absorption and promotes their excretion. Colesevelam is preferred to the older sequestrants for three reasons: (1) the drug is better tolerated (less constipation, flatulence, bloating, and cramping); (2) it does not reduce absorption of fat-soluble vitamins (A, D, E, and K); and (3) it does not significantly reduce the absorption of statins, digoxin, warfarin, and most other drugs studied.
In addition to its beneficial effects on plasma lipids, cole-sevelam is approved for adjunctive therapy of hyperglycemia in patients with type 2 diabetes. Diabetes and its management are the subject of Chapter 57.
Effect on Plasma Lipoproteins. The main response to bile-acid sequestrants is a reduction in LDL cholesterol. LDL decline begins during the first week of therapy and becomes maximal (about a 20% drop) within about a month. When these drugs are discontinued, LDL cholesterol returns to pretreat-ment levels in 3 to 4 weeks.
Bile-acid sequestrants may increase VLDL levels in some patients. In most cases, the elevation is transient and mild. However, if VLDL levels are elevated before treatment, the increase induced by the bile-acid sequestrants may be sustained and substantial. Accordingly, bile-acid sequestrants are not drugs of choice for lowering LDL cholesterol in patients with high VLDL levels.
Pharmacokinetics. Bile-acid sequestrants are biologically inert. Also, they are insoluble in water, cannot be absorbed from the GI tract, and are not attacked by digestive enzymes. Following oral administration, they simply pass through the intestine and are excreted in the feces.
Mechanism of Action. The bile-acid sequestrants lower LDL cholesterol through a mechanism that ultimately depends on increasing LDL receptors on hepatocytes. As background, you need to know that bile acids secreted into the intestine are normally reabsorbed and reused. Bile-acid sequestrants prevent this reabsorption. Following oral dosing, these drugs form an insoluble complex with bile acids in the intestine; this complex prevents the reabsorption of bile acids and thereby accelerates their excretion. Because bile acids are normally reabsorbed, the increase in excretion creates a demand for increased synthesis, which takes place in the liver. Since bile acids are made from cholesterol, liver cells require an increased cholesterol supply to increase bile acid production. The required cholesterol is provided by LDL. To avail themselves of more LDL cholesterol, liver cells increase their number of LDL receptors, thereby increasing their capacity for LDL uptake. The result is an increase in LDL uptake from plasma, which decreases circulating LDL levels. Individuals who are geneti-cally incapable of increasing LDL receptor synthesis are unable to benefit from these drugs.
Therapeutic Use. Colesevelam is indicated as adjunctive therapy to diet and exercise for reducing LDL cholesterol in patients with primary hypercholesterolemia. The drug may be used alone, but usually is combined with a statin. On average,

585
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
Fibric Acid Derivatives (Fibrates)The fibric acid derivatives, also known as fibrates, are the most effective drugs we have for lowering TG levels. In addition, these drugs can raise HDL cholesterol, but have little or no effect on LDL cholesterol. Furthermore, there is no proof that fibrates reduce mortality from ASCVD. Fibrates can increase the risk of bleeding in patients taking warfarin (an anticoagulant) and the risk of rhabdomyolysis in patients taking statins. Because of these and other undesired effects, and because mortality is not reduced, fibrates are considered third-line drugs for managing lipid disorders. In the United States, three preparations are available: gemfibrozil [Lopid], fenofibrate [Tricor, others], and fenofibric acid [TriLipix, Fibricor], a delayed-release preparation.
GemfibrozilGemfibrozil [Lopid] decreases TG (VLDL) levels and raises HDL cholesterol levels. The drug does not reduce LDL cho-lesterol to a significant degree. Its principal indication is hypertriglyceridemia.
Effect on Plasma Lipoproteins. Gemfibrozil decreases plasma TG content by lowering VLDL levels. Maximum reduc-tions in VLDLs range from 40% to 55%, and are achieved within 3 to 4 weeks of treatment. Gemfibrozil can raise HDL cholesterol by 6% to 10%. In patients with normal TG levels, the drug can produce a small reduction in LDL levels. However, if TG levels are high, gemfibrozil may actually increase LDL levels.
Mechanism of Action. Gemfibrozil and other fibrates appear to work by interacting with a specific receptor subtype—known as peroxisome proliferator-activated receptor alpha (PPAR alpha)—present in the liver and brown adipose tissue. Activation of PPAR alpha leads to (1) increased synthesis of lipoprotein lipase (LPL) and (2) reduced production of apolipoprotein C-III (an inhibitor of LPL). Both actions accelerate the clearance of VLDLs and thereby reduce levels of TGs. How do fibrates elevate HDL levels? By activating PPAR alpha, fibrates increase production of apolipoproteins A-I and A-II, which in turn facilitates HDL formation.
Therapeutic Use. Gemfibrozil is used primarily to reduce high levels of plasma triglycerides (VLDLs). Treatment is limited to patients who have not responded adequately to weight control and diet modification. Gemfibrozil can also reduce LDL cholesterol slightly. However, other drugs (statins, cholestyramine, colestipol) are much more effective.
Gemfibrozil can be used to raise HDL cholesterol, although it is not approved for this application. When tested in patients with normal LDL cholesterol and low HDL cholesterol, gemfibrozil reduced the risk of major CV events—but did not reduce mortality from ASCVD. Because LDL cholesterol was normal, it appears that benefits were due primarily to elevation of HDL cholesterol, along with reduction of plasma TGs.
Adverse Effects. Gemfibrozil is generally well tolerated. The most common reactions are rash and GI disturbances (nausea, abdominal pain, diarrhea).
Gallstones. Gemfibrozil increases biliary cholesterol satura-tion, thereby increasing the risk of gallstones. Patients should be informed about manifestations of gallbladder disease (e.g., upper abdominal discomfort, intolerance of fried foods, bloating) and instructed to notify the prescriber at once if these develop. Patients with pre-existing gallbladder disease should not take the drug.
Therapeutic UseEzetimibe is indicated as an adjunct to diet modification for reducing total cholesterol, LDL cholesterol, and apolipoprotein B in patients with primary hypercholesteremia. The drug is approved for monotherapy and for combined use with a statin. In clinical trials, ezetimibe alone reduced LDL cholesterol by about 19%, increased HDL cholesterol by 1% to 4%, and decreased TGs by 5% to 10%. When ezetimibe was combined with a statin, the reduction in LDL cholesterol was about 25% greater than with the statin alone. Despite these desirable effects on blood lipids, there is no evidence that ezetimibe reduces atherosclerosis or improves clinical outcomes.PharmacokineticsEzetimibe is administered orally, and absorption is not affected by food. In the intestinal wall and liver, ezetimibe undergoes extensive conversion to ezetimibe glucuronide, an active metabolite. Both compounds—ezetimibe itself and its main metabolite—are eliminated primarily in the bile. The elimination half-life is about 22 hours.
Adverse EffectsEzetimibe is generally well tolerated. During clinical trials, the incidence of significant side effects was nearly identical to that seen with placebo. However, during postmarketing surveillance, there have been reports of myopathy, rhabdomy-olysis, hepatitis, pancreatitis, and thrombocytopenia. In contrast to the bile-acid sequestrants, ezetimibe does not cause constipa-tion and other adverse GI effects.
Drug InteractionsStatins. In patients taking a statin, adding ezetimibe slightly
increases the risk of liver damage (as indicated by elevated transaminase levels). If the drugs are combined, transaminase levels should be monitored before starting therapy, as well as whenever clinically indicated thereafter. Combining ezetimibe with a statin may also increase the risk of myopathy.
Fibrates. Both ezetimibe and fibrates (gemfibrozil and fenofibrate) can increase the cholesterol content of bile and can thereby increase the risk of gallstones. Both also increase the risk of myopathy. Accordingly, combined use is not recommended.
Bile-Acid Sequestrants. Cholestyramine (and possibly colestipol) can significantly decrease the absorption of ezetimibe. To minimize effects on absorption, ezetimibe should be administered at least 2 hours before a sequestrant or more than 4 hours after.
Cyclosporine. Cyclosporine may greatly increase levels of ezetimibe. If the drugs are combined, careful monitoring is needed.
CautionIn patients with hepatic impairment, bioavailability of ezetimibe is significantly increased. At this time, we do not know if increased availability is harmful. Until more is known, patients with moderate or severe hepatic insufficiency should not be given the drug.
Preparations, Dosage, and AdministrationEzetimibe [Zetia, Ezetrol ] is available in 10-mg tablets. The recommended dosage is 10 mg once a day, taken with or without food. If ezetimibe is combined with a statin, both drugs can be taken at the same time. If ezetimibe is combined with a bile-acid sequestrant, ezetimibe should be taken 2 hours before the sequestrant or 4 hours after.

586
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
levels, specifically in patients with heterozygous familial hypercholesterolemia or with atherosclerotic heart problems who need additional lowering of LDL cholesterol. The PCSK9 inhibitors are indicated as an adjunct to diet modification and maximally tolerated statin therapy for reducing total LDL cholesterol.
Mechanism of Action and Effect on Plasma LipoproteinsProprotein convertase subtilisin kexin type 9 (PCSK9) is a protein that binds to low-density lipoprotein receptors (LDLR) within the liver. LDLR is the primary receptor that clears circulating LDL. When PCSK9 binds to LDLR, there is an increase in LDL cholesterol, because LDLR cannot clear LDL. By inhibiting PCSK9, we can free up LDLR and decrease circulating levels of LDL in the blood.
PharmacokineticsPCSK9 inhibitors are administered subcutaneously. Because monoclonal antibodies are composed of protein, no spe-cific metabolism studies were conducted. It is thought that the proteins degrade to small peptides and amino acids within the body. Both drugs have a long half-life of 11 to 20 days.
Adverse EffectsHypersensitivity. Hypersensitivity reactions, including
vasculitis, rash, and urticaria requiring hospitalization, have been noted with use of PCSK9 inhibitors.
Immunogenicity. As this class of drug is composed of protein, there is a risk for developing antibodies; 4.8% of patients treated with alirocumab and 0.1% treated with evo-locumab developed antibodies to the drug after initiating treatment. These patients also had a higher incidence of injection site reactions compared with patients who did not develop antibodies.
Drug InteractionsNeither drug is noted to have any significant drug interactions.Preparations, Dosage, and AdministrationAlirocumab [Praluent] is available in 75 and 150 mg/mL single-dose pre-filled pens and syringes. The recommended dosage is 75 mg subcutaneously every 2 weeks. If the LDL cholesterol response is inadequate, the dose may be increased to 150 mg every 2 weeks. Injection may be administered into the thigh, abdomen, or upper arm.
Evolocumab [Repatha], like alirocumab, is administered subcutaneously every 2 weeks. The recommended dose is 140 mg. Repatha is available in 140 mg/mL single-use pre-filled syringes or in a 140 mg/mL single-use SureClick auto-injector. Alternate dosing of 420 mg can also be administered every 4 weeks.
Drug CombinationsSimvastatin/Ezetimibe [Vytorin]
Actions and Uses. Simvastatin and ezetimibe are available in fixed-dose combination tablets sold as Vytorin. Vytorin has only one indication: hyper-cholesterolemia. Because ezetimibe has a different mechanism of action than simvastatin, the combination can lower cholesterol more effectively than simvastatin alone.
With this combination, the dose of simvastatin required to effectively lower cholesterol may be lower than the dose required when simvastatin is used alone. As a result, the risk of statin-related adverse effects can be reduced. Additional benefits of the combination are convenience (take just one pill instead of two) and reduced cost (the combination costs less than both drugs purchased separately).
Myopathy. Like the statins, gemfibrozil and other fibrates can cause myopathy. Warn patients to report any signs of muscle injury, such as tenderness, weakness, or unusual muscle pain.
Liver Injury. Gemfibrozil is hepatotoxic. The drug can disrupt liver function and may also pose a risk of liver cancer. Periodic tests of liver function are recommended.
Drug Interactions. Gemfibrozil displaces warfarin from plasma albumin, thereby increasing anticoagulant effects. Prothrombin time (international normalized ratio) should be measured frequently to assess coagulation status. Warfarin dosage may need to be reduced.
As noted, gemfibrozil increases the risk of statin-induced myopathy. Accordingly, the combination of a statin with gemfibrozil should be used with great caution, if at all.
Preparations, Dosage, and Administration. Gemfibrozil [Lopid] is available in 600-mg tablets. The adult dosage is 600 mg twice a day. Dosing is done 30 minutes before the morning and evening meals.
FenofibrateActions and Uses. Fenofibrate [Tricor, Antara, Lofibra, Triglide, Lipofen,
Lipidil ] is indicated for hypertriglyceridemia in patients who have not responded to dietary measures. The drug lowers TGs by decreasing levels of VLDLs.
Pharmacokinetics. Fenofibrate is well absorbed from the GI tract, especially in the presence of food. Once absorbed, the drug is rapidly converted to fenofibric acid, its active form. In the blood, the drug is 98% protein bound. Elimination is the result of hepatic metabolism followed by renal excretion. The plasma half-life is about 20 hours.
Adverse Effects and Drug Interactions. The most common adverse effects are rash and GI disturbances. Like gemfibrozil, fenofibrate can cause gallstones and liver injury. In animal models, doses 1 to 6 times the maximum human dose caused cancers of the pancreas and liver. Like gemfibrozil, fenofibrate can increase the risk of bleeding with warfarin and the risk of myopathy with statins.
Preparations, Dosage, and Administration. Fenofibrate is available in several formulations that differ with respect to dosage and the impact of food on absorption. Four products are discussed here.
Tricor tablets (48 and 145 mg) are made using NanoCrystal technology to enhance absorption. As a result, dosing can be done with or without food. The dosage range is 48 to 145 mg/day.
Triglide tablets (160 mg), like Tricor tablets, may be administered with or without food. The dosage is 160 mg/day.
Antara capsules (30 and 90 mg), which contain micronized particles, must be administered with food to maximize absorption. The dosage range is 30 to 90 mg/day.
Lofibra capsules (67, 134, and 200 mg) contain micronized particles. Like Antara, Lofibra must be administered with food to maximize absorption. The dosage range is 67 to 200 mg/day.
Fenofibric AcidFenofibric acid [TriLipix, Fibricor] is the active metabolite of fenofibrate. Accordingly, the pharmacology of the drug is much like that of the parent compound. Fenofibric acid stands out from other fibrates for being the only group member approved for use with a statin. However, there is no proof that combining the drug with a statin reduces the risk of a major CV event. Furthermore, just like other fibrates, fenofibric acid can cause myopathy, and hence combined use with a statin still poses significant myopathy risk. Therefore, the combination must be employed with great care. Fenofibric acid is available in delayed-release capsules (45 and 135 mg, sold as TriLipix; 35 and 105 mg, sold as Fibricor). Daily dosages for hypertriglyceridemia range from 35 to 135 mg. In patients with mild to moderate renal impairment, a low dosage (45 mg/day) should be used. When combined with a statin for patients with mixed dyslipidemias, fenofibric acid should be dosed at 135 mg/day.
Monoclonal Antibodies (Proprotein Convertase Subtilisin/Kexin Type 9 [PCSK9] Inhibitors)Alirocumab [Praluent] and evolocumab [Repatha] compose a new type of drug class used for patients with high LDL

587
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
carries some risk. Methylmercury can cause heart disease as well as neurologic damage, manifesting as tremor, numbness, tingling, altered vision, and impaired concentration. Exposure in utero or during early childhood can lead to developmental delay, blindness, and seizures. With dioxin and PCBs, carci-nogenesis is the major concern. Does this mean we should avoid eating fish? No. For postmenopausal women and for men who are middle-aged or older, the benefits of fish outweigh the risks. For women who are pregnant or breast-feeding, fish consumption should be limited to 12 ounces a week, and certain species—swordfish, king mackerel, shark, and golden snapper, all of which may have high levels of methylmercury—should be avoided entirely. Young children should limit fish consumption too. For people who like salmon, dioxin exposure can be reduced by eating wild salmon, which contains much less dioxin than farm-raised salmon. Exposure to all contaminants can be reduced by using fish-oil supplements, which have much less contamination than fish themselves.
LovazaLovaza is the brand name for the first preparation of omega-3-acid ethyl esters approved by the FDA. The product, available only by prescription, contains a combination of EPA and DHA. Lovaza is approved as an adjunct to dietary measures to reduce very high levels of TGs (500 mg/dL or greater). When used alone, Lovaza can reduce TG levels by 20% to 50%. Combining it with simvastatin produces a further decrease. Because large doses of omega-3 fatty acids can impair platelet function, leading to prolonged bleeding time, the product should be used with care in patients taking anticoagulants or antiplatelet drugs, including aspirin. Lovaza is supplied in 1-gm, liquid-filled, soft-gelatin capsules that contain approximately 465 mg of EPA and approximately 375 mg of DHA. The recommended dosage is 4 gm/day, taken either all at once (4 capsules) or in two doses (2 capsules twice a day).
Plant Stanol and Sterol EstersStanol esters and sterol esters, which are analogs of cholesterol, can reduce intestinal absorption of cholesterol (by 10%) and can thereby reduce levels of LDL cholesterol (by 14%). These compounds do not affect HDL levels or TG levels. ATP III recommends adding plant stanols or sterols to the diet if the basic TLC diet fails to reduce LDL cholesterol to the target level. Where can you get plant stanols and sterols? Two good sources are the Benecol brand of margarine and soft spreads sold under the brand name Promise.
CholestinCholestin is the brand name for a dietary supplement that can lower cholesterol levels. The product is made from rice fermented with red yeast. Its principal active ingredient—lovastatin—is identical to the active ingredient in Mevacor, a brand-name, cholesterol-lowering drug. In addition to lovastatin, Cholestin contains at least seven other HMG-CoA reductase inhibitors (statins).
Several clinical trials have demonstrated that Cholestin can lower cholesterol levels, although none has studied its effects on CV events. In a trial conducted at Tufts University School of Medicine, Cholestin reduced total cholesterol by 11.4% and LDL cholesterol by 21%, and increased HDL cholesterol by 14.6%. Similarly, in a study conducted at the University of California at Los Angeles Medical School, Cholestin reduced total cholesterol by 16% and LDL cholesterol by 22%. Whether Cholestin also reduces the incidence of ASCVD is unknown.
Information on Cholestin is lacking in four important areas: clinical benefits, adverse effects, drug interactions, and precise mechanism of action. As noted, there are no data on the ability of Cholestin to reduce the risk of MI, stroke, or any other CV event. In contrast, the clinical benefits of prescription statins (lovastatin and all the others) are fully documented. There is little or no information on the adverse effects or drug interactions of Cholestin. In contrast, the safety (and hazards) of prescription statins, as well as their drug interactions, have been studied extensively.
The mechanism by which Cholestin lowers cholesterol levels is only partly understood. The recommended daily dose of Cholestin contains only 5 mg of lovastatin and varying doses of other HMG-CoA reductase inhibitors, compared with 10 mg for the lowest recommended dose of Mevacor. Hence, it seems unlikely that the statins in Cholestin can fully account for the supple-ment’s ability to reduce cholesterol levels. This implies that Cholestin has one or more active ingredients that have not yet been identified. What they are and how they may work is a mystery.
What’s the bottom line? Until more is known about Cholestin, stick with statins—medications of proven safety and efficacy. Furthermore, for people with health insurance, using statins is cheaper: Most insurers will cover the cost of statins, but will not pay for Cholestin.
Despite the advantages of Vytorin, some authorities are concerned that the combination may be less beneficial than simvastatin alone. This concern is based on four facts:
• We have proof that simvastatin alone can decrease adverse outcomes (i.e., MI and other CV events).
• We have no proof that the combination can decrease adverse outcomes of elevated cholesterol (even though it can reduce levels of cholesterol).
• In addition to lowering cholesterol, statins have other beneficial actions (e.g., they often lower elevated TGs).
• When ezetimibe and simvastatin are combined, cholesterol goals can be met using simvastatin in reduced dosage (which is a problem for reasons discussed in the paragraph that follows).
Because the combination permits a reduction in simvastatin dosage, there is concern that although the target cholesterol goal may be reached, the reduction in adverse outcomes may be smaller than when cholesterol is lowered using simvastatin alone. Despite concerns, the FDA reviewed the evidence and determined that, although there was no significant change in carotid artery thickness after taking Vytorin, there was a significant decrease in LDL cho-lesterol in people taking Vytorin, which may decrease CV risk. For this reason, the FDA recommends that patients continue treatment on Vytorin.
Adverse Effects and Drug Interactions. Vytorin is generally well tolerated. However, myopathy is a concern (because both drugs can cause muscle injury). Concurrent use of a fibrate, which can also cause myopathy, increases risk. The risk of myopathy and other adverse effects is also increased by inhibitors of CYP3A4, the enzyme that inactivates simvastatin. Because Vytorin contains a statin, the product is contraindicated for women who are pregnant and for patients with liver disease.
Preparations, Dosage, and Administration. Vytorin tablets contain 10 mg of ezetimibe plus either 10, 20, 40, or 80 mg of simvastatin. The usual starting dosage is 10 mg ezetimibe/20 mg simvastatin each day. Dosing is done once daily, preferably in the evening. The simvastatin dosage can be increased as needed and tolerated.
Atorvastatin/Amlodipine [Caduet]Atorvastatin and amlodipine (a calcium channel blocker) are available in fixed-dose combination tablets under the brand name Caduet. This is the first single product indicated for dyslipidemia combined with hypertension and/or angina. The combination has one advantage over taking each drug separately: fewer pills to swallow. Eleven amlodipine/atorvastatin combinations are available: 2.5 mg amlodipine with either 10, 20, or 40 mg atorvastatin; 5 mg amlodipine with either 10, 20, 40, or 80 mg atorvastatin; and 10 mg amlodipine with either 10, 20, 40, or 80 mg atorvastatin. Dosage is individual-ized on the basis of therapeutic response and tolerance of adverse effects. The pharmacology of amlodipine and other calcium channel blockers is discussed in Chapter 45.
Fish OilConsuming fatty fish or fish-oil supplements was once associated with a decreased risk of ASCVD and ASCVD-related death. Unfortunately, recent studies have revealed that consuming fish oil provides no advantage in the prevention of heart disease in high-risk populations. Yet, it is still believed that taking fish oil can reduce the incidence of heart dysrhythmias after MI or heart failure.
Fish oil may be beneficial in the prevention of heart dysrhythmias because it contains two “heart healthy” compounds: eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Both compounds are long-chain, omega-3 polyunsaturated fatty acids, with a methyl group at one end and a carboxyl group at the other. They are called omega-3 fatty acids because they have a double bond located three carbons in from the methyl terminus.
How do omega-3 fatty acids help us? The answer is unclear. Benefits of lower doses (850 mg to 1 gm) may result from reducing platelet aggregation; reducing thrombosis (by effects on platelets and the vascular endothelium); reducing inflammation (which may help stabilize atherosclerotic plaques); and reducing blood pressure and cardiac dysrhythmias.
As the evidence is so new regarding lack of benefit in the primary prevention of heart disease, the American Heart Association still recommends eating at least two servings of fish a week. Fish with high concentrations of EPA and DHA are preferred. Among these are mackerel, halibut, herring, salmon, albacore tuna, and trout. The goal is to take in, on average, about 1 gm of fish oil a day.
Because fish concentrate certain environmental contaminants—especially methylmercury, dioxins, and polychlorinated biphenyls (PCBs)—eating fish

588
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
KEY POINTS
■ Lipoproteins are structures that transport lipids (cholesterol and triglycerides [TGs]) in blood.
■ Lipoproteins consist of a hydrophobic core, a hydrophilic shell, plus at least one apolipoprotein, which serves as a recognition site for receptors on cells.
■ Lipoproteins that contain apolipoprotein B-100 transport cholesterol and/or TGs from the liver to peripheral tissues.
■ Lipoproteins that contain apolipoproteins A-I or A-II transport cholesterol from peripheral tissues back to the liver.
■ There are three major types of lipoproteins: VLDLs (very-low-density lipoproteins), LDLs (low-density lipoproteins), and HDLs (high-density lipoproteins).
■ VLDLs transport TGs to peripheral tissues.■ The contribution of VLDLs to ASCVD is unclear.■ LDLs transport cholesterol to peripheral tissues.■ Elevation of LDL cholesterol greatly increases the risk of
ASCVD.■ By reducing LDL cholesterol levels, we can arrest or reverse
atherosclerosis, and can thereby reduce morbidity and mortality from ASCVD.
■ HDLs transport cholesterol back to the liver.■ HDLs protect against ASCVD.■ Atherogenesis is a chronic inflammatory process that begins
with accumulation of LDLs beneath the arterial endothe-lium, followed by oxidation of LDLs.
■ All adults older than 20 years should be screened every 5 years for total cholesterol, LDL cholesterol, HDL choles-terol, and TGs.
■ Treatment of high LDL cholesterol is based on the indi-vidual’s 10-year risk of having a major coronary event.
■ Individuals with established ASCVD or an ASCVD risk equivalent (e.g., diabetes) are in the highest 10-year risk group.
■ Diet modification along with exercise is the primary method for reducing LDL cholesterol. Drugs are employed only if diet modification and exercise fail to reduce LDL cholesterol to the target level.
■ Therapy with cholesterol-lowering drugs must continue lifelong. If these drugs are withdrawn, cholesterol levels will return to pretreatment values.
■ Statins (HMG-CoA reductase inhibitors) are the most effective drugs for lowering LDL cholesterol, and they cause few adverse effects.
■ Statins can slow progression of ASCVD, decrease the number of adverse cardiac events, and reduce mortality.
■ Statins reduce LDL cholesterol levels by increasing the number of LDL receptors on hepatocytes, thereby enabling hepatocytes to remove more LDLs from the blood. The process by which LDL receptor number is increased begins with inhibition of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis.
■ Four statins—atorvastatin, fluvastatin, lovastatin, and simvastatin—are metabolized by CYP3A4, and hence their levels can be increased by CYP3A4 inhibitors (e.g., cyclosporine, erythromycin, ketoconazole, ritonavir).
■ Statins can cause liver damage. Tests of liver function should be done at baseline and as clinically indicated thereafter.
■ Statins can cause myopathy. Patients who experience unusual muscle pain, soreness, tenderness, and/or weakness should inform their provider. A marker for muscle injury—creatine kinase (CK)—should be measured at baseline, before starting the drug, and whenever signs or symptoms that could be due to myositis or myopathy develop.
■ Statins should not be used during pregnancy.■ Bile-acid sequestrants (e.g., colesevelam) reduce LDL cho-
lesterol levels by increasing the number of LDL receptors on hepatocytes. The mechanism is complex and begins with preventing reabsorption of bile acids in the intestine.
■ Bile-acid sequestrants are not absorbed from the GI tract, and hence do not cause systemic adverse effects. However, they can cause constipation and other GI effects. (GI effects with one agent—colesevelam—are minimal.)
■ Older bile-acid sequestrants form complexes with other drugs and thereby prevent their absorption. Accordingly, oral medications should be administered 1 hour before the sequestrant or 4 hours after. With a newer sequestrant—colesevelam—these interactions are minimal.
■ Ezetimibe lowers LDL cholesterol by reducing cholesterol absorption in the small intestine.
■ Like the statins, ezetimibe can cause muscle injury.■ Gemfibrozil and other fibrates are the most effective drugs
for lowering TG levels.■ Like the statins, the fibrates can cause muscle injury.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.
Summary of Major Nursing Implicationsa
IMPLICATIONS THAT APPLY TO ALL DRUGS THAT LOWER LDL CHOLESTEROLPreadministration AssessmentBaseline DataObtain laboratory values for total cholesterol, LDL cholesterol, HDL cholesterol, and TGs (VLDLs).
Identifying ASCVD Risk FactorsThe patient history and physical examination should identify ASCVD risk factors. These include smoking, advancing age, a personal history of ASCVD, reduced levels of HDL cho-lesterol (below 40 mg/dL), and hypertension.
In the past, diabetes was considered an ASCVD risk factor. However, because the association between diabetes and

589
CHAPTER 50 Prophylaxis of Atherosclerotic Cardiovascular Disease
ASCVD is so strong, diabetes is now considered an ASCVD risk equivalent (i.e., it poses the same 10-year risk of a major coronary event as ASCVD itself).
Measures to Enhance Therapeutic EffectsDiet ModificationDiet modification should precede and accompany drug therapy for elevated LDL cholesterol. Inform patients about the importance of diet in controlling cholesterol levels, and arrange for dietary counseling. Advise patients to limit consumption of cholesterol (to below 200 mg/day) and saturated fat (to below 7% of caloric intake). If these measures fail to reduce LDL cholesterol to the target level, advise patients to add soluble fiber and plant stanols or sterols to the regimen.
ExerciseRegular exercise can reduce LDL cholesterol and elevate HDL cholesterol, reducing the risk of ASCVD. Help the patient establish an appropriate exercise program.
Reduction of ASCVD Risk FactorsCorrectable ASCVD risk factors should be addressed. Encour-age smokers to quit. Disease states that promote ASCVD—diabetes mellitus and hypertension—must be treated.
Promoting ComplianceDrug therapy for elevated LDL cholesterol must continue lifelong; if drugs are withdrawn, cholesterol levels will return to pretreatment values. Inform patients about the need for continuous therapy, and encourage them to adhere to the prescribed regimen.
HMG-COA REDUCTASE INHIBITORS (STATINS)AtorvastatinFluvastatinLovastatinPitavastatinPravastatinRosuvastatinSimvastatin
In addition to the implications discussed below, see earlier in this summary for implications that apply to all drugs that lower LDL cholesterol.
Preadministration AssessmentTherapeutic GoalStatins, in combination with diet modification and exercise, are used primarily to lower levels of LDL cholesterol. Additional indications are shown in Table 50.7.
Baseline DataObtain a baseline lipid profile, consisting of total cholesterol, LDL cholesterol, HDL cholesterol, and TGs (VLDLs). Also, obtain baseline LFTs and a CK level.
Identifying High-Risk PatientsStatins are contraindicated for patients with viral or alcoholic hepatitis and for women who are pregnant.
Exercise caution in patients with nonalcoholic fatty liver disease, in those who consume alcohol to excess, and in those taking fibrates or ezetimibe or agents that inhibit CYP3A4 (e.g., cyclosporine, erythromycin, ketoconazole, ritonavir). Use rosuvastatin with caution in Asian patients.
Implementation: AdministrationRouteOral.
AdministrationInstruct patients to take lovastatin with the evening meal; all other statins can be administered without regard to meals. Advise patients that dosing in the evening is preferred for all statins.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsCholesterol levels should be monitored monthly early in treatment and at longer intervals thereafter.
Minimizing Adverse EffectsStatins are very well tolerated. Side effects are uncommon, and serious adverse effects—hepatotoxicity and myopathy—are relatively rare.
Hepatotoxicity. Statins can injure the liver, but jaundice and other clinical signs are rare. Liver function should be assessed before treatment and as clinically indicated thereafter. If serum transaminase becomes persistently excessive (more than 3 times the ULN), statins should be discontinued. Statins should be avoided in patients with alcoholic or viral hepatitis, but may be used in patients with nonalcoholic fatty liver disease.
Myopathy. Statins can cause muscle injury. If statins are not withdrawn, injury may progress to severe myositis or potentially fatal rhabdomyolysis. Inform patients about the risk of myopathy, and instruct them to notify the prescriber if unexplained muscle pain or tenderness develops. If muscle pain does develop, the CK level should be measured, and if it is more than 10 times the ULN, the statin should be withdrawn or changed.
Minimizing Adverse InteractionsThe risk of myopathy is increased by (1) gemfibrozil, feno-fibrate, and ezetimibe, which promote myopathy themselves; and by (2) inhibitors of CYP3A4—such as cyclosporine, macrolide antibiotics (e.g., erythromycin), azole antifungal drugs (e.g., ketoconazole), and HIV protease inhibitors (e.g., ritonavir)—which can cause statin levels to rise. The combina-tion of a statin with any of these drugs should be used with caution.
Use in PregnancyStatins are contraindicated during pregnancy. Inform women of childbearing age about the potential for fetal harm and warn them against becoming pregnant. If pregnancy occurs and the patient intends to continue the pregnancy, statins should be withdrawn.
Summary of Major Nursing Implicationsa—cont’d
Continued

590
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
aPatient education information is highlighted as blue text.
BILE-ACID SEQUESTRANTSCholestyramineColesevelamColestipol
In addition to the implications discussed here, see earlier in this summary for implications that apply to all drugs that lower LDL cholesterol.
Preadministration AssessmentTherapeutic GoalBile-acid sequestrants, in conjunction with diet modification and exercise (and a statin if necessary), are used to reduce elevated levels of LDL cholesterol.
Baseline DataObtain laboratory values for total cholesterol, LDL cholesterol, HDL cholesterol, and TGs (VLDLs).
Implementation: AdministrationRouteOral.
AdministrationInstruct patients to mix cholestyramine powder and colestipol granules with water, fruit juice, soup, or pulpy fruit (e.g., applesauce, crushed pineapple) to reduce the risk of esopha-geal irritation and impaction. Inform patients that the sequestrants are not water soluble, so the mixtures will be cloudy suspensions, not clear solutions.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsCholesterol levels should be monitored monthly early in treatment and at longer intervals thereafter.
Minimizing Adverse EffectsConstipation. Cholestyramine and colestipol—but not
colesevelam—can cause constipation. Inform patients that constipation can be minimized by increasing dietary fiber and fluids. A mild laxative may be used if needed. Instruct patients taking cholestyramine or colestipol to notify the prescriber if constipation becomes bothersome, in which case a switch to colesevelam should be considered.
Vitamin Deficiency. Cholestyramine and colestipol—but not colesevelam—can impair absorption of fat-soluble vitamins (A, D, E, and K). Vitamin supplements may be required. Colesevelam does not reduce vitamin absorption.
Minimizing Adverse InteractionsCholestyramine and colestipol—but not colesevelam—can bind with other drugs and prevent their absorption. Advise patients to administer other medications 1 hour before these sequestrants or 4 hours after.
GEMFIBROZILPreadministration AssessmentTherapeutic GoalGemfibrozil, in conjunction with diet modification, is used to reduce elevated levels of TGs (VLDLs). The drug is not very effective at lowering LDL cholesterol. It may also be used to raise low levels of HDL cholesterol.
Baseline DataObtain laboratory values for total cholesterol, LDL cholesterol, HDL cholesterol, and TGs (VLDLs).
Identifying High-Risk PatientsGemfibrozil is contraindicated for patients with liver disease, severe renal dysfunction, and gallbladder disease.
Use with caution in patients taking statins or warfarin.
Implementation: AdministrationRouteOral.
AdministrationInstruct patients to administer gemfibrozil 30 minutes before the morning and evening meals.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsObtain periodic tests of blood lipids.
Minimizing Adverse EffectsGallstones. Gemfibrozil increases gallstone development.
Inform patients about symptoms of gallbladder disease (e.g., upper abdominal discomfort, intolerance of fried foods, bloating), and instruct them to notify the prescriber if these develop.
Myopathy. Gemfibrozil can cause muscle damage. Warn patients to report any signs of muscle injury, such as tender-ness, weakness, or unusual muscle pain.
Liver Disease. Gemfibrozil may disrupt liver function. Cancer of the liver may also be a risk. Obtain periodic tests of liver function.
Minimizing Adverse InteractionsWarfarin. Gemfibrozil enhances the effects of warfarin,
thereby increasing the risk of bleeding. Obtain more frequent measurements of prothrombin time and assess the patient for signs of bleeding. Reduction of warfarin dosage may be required, and reassessment and readjustment of the warfarin dosage may be needed if the fibrate is stopped.
Statins. Gemfibrozil and statins both cause muscle injury. Risks rise when both are used. Use the combination with caution.
Summary of Major Nursing Implicationsa—cont’d

591
C H A P T E R
51 Drugs for Angina Pectoris
Determinants of Cardiac Oxygen Demand and Oxygen Supply, p. 591Oxygen Demand, p. 591Oxygen Supply, p. 591
Angina Pectoris: Pathophysiology and Treatment Strategy, p. 591Chronic Stable Angina (Exertional Angina),
p. 591Variant Angina (Prinzmetal’s Angina, Vasospastic
Angina), p. 593Unstable Angina, p. 593
Organic Nitrates, p. 593Nitroglycerin, p. 593Isosorbide Mononitrate and Isosorbide Dinitrate,
p. 597Beta Blockers, p. 597Calcium Channel Blockers, p. 597Ranolazine, p. 598Treatment Measures, p. 598
Guidelines for Management of Chronic Stable Angina, p. 598
Management of Variant Angina, p. 601Key Points, p. 601Summary of Major Nursing Implications, p. 602
are discussed at length in previous chapters; consideration of these drugs here is limited to their use in angina.
DETERMINANTS OF CARDIAC OXYGEN DEMAND AND OXYGEN SUPPLYBefore discussing angina pectoris, we need to review the major factors that determine cardiac oxygen demand and supply.
Oxygen DemandThe principal determinants of cardiac oxygen demand are heart rate, myocardial contractility, and, most importantly, intra-myocardial wall tension. Wall tension is determined by two factors: cardiac preload and cardiac afterload. (Preload and afterload are defined in Chapter 43.) In summary, cardiac oxygen demand is determined by (1) heart rate, (2) contractility, (3) preload, and (4) afterload. Drugs that reduce these factors reduce oxygen demand.
Oxygen SupplyCardiac oxygen supply is determined by myocardial blood flow. Under resting conditions, the heart extracts nearly all of the oxygen delivered to it by the coronary vessels. There-fore, the only way to accommodate an increase in oxygen demand is to increase blood flow. When oxygen demand increases, coronary arterioles dilate; the resultant decrease in vascular resistance allows blood flow to increase. During exertion, coronary blood flow increases four- to fivefold. It is important to note that myocardial perfusion takes place only during diastole (i.e., when the heart relaxes). Perfusion does not take place during systole because the vessels that supply the myocardium are squeezed shut when the myocardium contracts.
ANGINA PECTORIS: PATHOPHYSIOLOGY AND TREATMENT STRATEGYAngina pectoris has three forms: (1) chronic stable angina (exertional angina), (2) variant angina (Prinzmetal’s or vaso-spastic angina), and (3) unstable angina. Our focus is on stable angina and variant angina. Consideration of unstable angina is brief.
Chronic Stable Angina (Exertional Angina)PathophysiologyStable angina is triggered most often by an increase in physical activity. Emotional excitement, large meals, and cold exposure
Angina pectoris is defined as sudden pain beneath the sternum, often radiating to the left shoulder, left arm, and jaw. Anginal pain is precipitated when the oxygen supply to the heart is insufficient to meet oxygen demand. Most often, angina occurs secondary to atherosclerosis of the coronary arteries, so angina should be seen as a symptom of a disease and not as a disease in its own right. In the United States, more than 10 million people have chronic stable angina; about 500,000 new cases develop annually.
Drug therapy of angina has two goals: (1) prevention of myocardial infarction (MI) and death and (2) prevention of myocardial ischemia and anginal pain. Two types of drugs are employed to decrease the risk of MI and death: cholesterol-lowering drugs and antiplatelet drugs. These agents are discussed in Chapters 50 and 52, respectively.
In this chapter, we focus on antianginal drugs (i.e., drugs that prevent myocardial ischemia and anginal pain). There are three main families of antianginal agents: organic nitrates (e.g., nitroglycerin), beta blockers (e.g., metoprolol), and calcium channel blockers (e.g., verapamil). In addition, a fourth agent—ranolazine—can be combined with these drugs to supplement their effects. Most of the chapter focuses on the organic nitrates. Beta blockers and calcium channel blockers

592
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
both the healthy heart and the heart with CAD, oxygen supply and oxygen demand are in balance during rest. In the presence of CAD, resting oxygen demand is met through dilation of arterioles distal to the partial occlusion. This dilation reduces resistance to blood flow, compensating for the increase in resistance created by plaque.
The picture is very different during exertion. In the healthy heart, as cardiac oxygen demand rises, coronary arterioles dilate, causing blood flow to increase. The increase keeps oxygen supply in balance with oxygen demand. By contrast, in people with CAD, arterioles in the affected region are already fully dilated during rest. Thus, when exertion occurs, there is no way to increase blood flow to compensate for the increase in oxygen demand. The resultant imbalance between oxygen supply and oxygen demand causes anginal pain.
Treatment StrategyThe goal of antianginal therapy is to reduce the intensity and frequency of anginal attacks. Because anginal pain results from an imbalance between oxygen supply and oxygen demand, logic dictates two possible remedies: (1) increase cardiac oxygen supply or (2) decrease oxygen demand. Since the underlying cause of stable angina is occlusion of the coronary arteries, there is little we can do to increase cardiac oxygen supply. Therefore, the first remedy is not a real option. Consequently, all we really can do is decrease cardiac oxygen demand. As discussed earlier, oxygen demand can be reduced with drugs that decrease heart rate, contractility, afterload, and preload.
Overview of Therapeutic AgentsStable angina can be treated with three main types of drugs: organic nitrates, beta blockers, and calcium channel blockers. As previously noted, ranolazine can be combined with these drugs for additional benefit. All four groups relieve the pain of stable angina primarily by decreasing cardiac oxygen demand (Table 51.1). Please note that drugs provide only symptomatic relief; they do not affect the underlying pathology. To reduce the risk of MI, all patients should receive an antiplatelet drug (e.g., aspirin) unless it is contraindicated. Other measures to reduce the risk of infarction are discussed later under Drugs Used to Prevent Myocardial Infarction and Death.
Nondrug TherapyPatients should attempt to avoid factors that can precipitate angina. These include overexertion, heavy meals, emotional stress, and exposure to cold.
DURING RESTHealthy Heart and Heart With CAD
Healthy Heart Heart With CAD
DURING EXERTION
O2
DEMAND
O2
DEMAND
O2
SUPPLY
O2SUPPLY
O2DEMAND
O2
SUPPLY
Fig. 51.1 ■ Effect of exertion on the balance between oxygen supply and oxygen demand in the healthy heart and the heart with CAD. In the healthy heart, O2 supply and O2 demand are always in balance; during exertion, coronary arteries dilate, producing an increase in blood flow to meet the increase in O2 demand. In the heart with CAD, O2 supply and O2 demand are in balance only during rest. During exertion, dilation of coronary arteries cannot compensate for the increase in O2 demand, and an imbalance results.
Drug Class
Mechanism of Pain Relief
Stable Angina Variant Angina
Nitrates Decrease oxygen demand by dilating veins, which decreases preload Increase oxygen supply by relaxing coronary vasospasm
Beta Blockers Decrease oxygen demand by decreasing heart rate and contractility Not usedCalcium Channel Blockers Decrease oxygen demand by dilating arterioles, which decreases
afterload (all calcium blockers), and by decreasing heart rate and contractility (verapamil and diltiazem)
Increase oxygen supply by relaxing coronary vasospasm
Ranolazine Appears to decrease oxygen demand, possibly by helping the myocardium generate energy more efficiently
Not used
TABLE 51.1 ■ Mechanisms of Antianginal Action
may also precipitate an attack. Because stable angina usually occurs in response to strain, this condition is also known as exertional angina or angina of effort.
The underlying cause of exertional angina is coronary artery disease (CAD), a condition characterized by deposition of fatty plaque in the arterial wall. If an artery is only partially blocked by plaque, blood flow will be reduced and angina pectoris will result. However, if complete vessel blockage occurs, blood flow will stop and MI (heart attack) will result.
The impact of CAD on the balance between myocardial oxygen demand and oxygen supply is shown in Fig. 51.1. In

593
CHAPTER 51 Drugs for Angina Pectoris
Anti-ischemic therapy consists of:• Nitroglycerin—give three doses sublingually every 5 minutes (tablet or
spray) and follow with IV therapy in the event of persistent ischemia or hypertension.
• A beta blocker—give the first dose IV if chest pain is ongoing. If beta blockers are contraindicated, substitute a nondihydropyridine calcium channel blocker (verapamil or diltiazem).
• Supplemental oxygen—for patients with cyanosis or respiratory distress.• Intravenous morphine sulfate—if pain is not relieved immediately by
nitroglycerin, or if pulmonary congestion or severe agitation is present.• An angiotensin-converting enzyme inhibitor—for patients with left
ventricular dysfunction or congestive heart failure. Angiotensin receptor blockers are a reasonable alternative in patients who have intolerance to angiotensin-converting enzyme inhibitors.
Antiplatelet therapy, which should be started promptly, consists of:• Aspirin—continue indefinitely.• Clopidogrel [Plavix], prasugrel [Effient], or ticagrelor [Brilinta]—con-
tinue for up to 2 months.• Abciximab [ReoPro], a glycoprotein IIb/IIIa inhibitor—but only if
angioplasty is planned.• Eptifibatide [Integrilin] or tirofiban [Aggrastat] (both are glycoprotein
IIb/IIIa inhibitors)—but only in high-risk patients with continuing ischemia, and only if angioplasty is not planned.
Anticoagulant therapy consists of subcutaneous low-molecular-weight heparin (e.g., enoxaparin [Lovenox]), direct thrombin inhibitors (bivalirudin [Angio-max]), factor Xa inhibitors (fondaparinux [Arixtra]), or IV unfractionated heparin.
ORGANIC NITRATES
The organic nitrates are the oldest and most frequently used antianginal drugs. These agents relieve angina by causing vasodilation. Nitroglycerin, the most familiar organic nitrate, will serve as our prototype.
Risk factors for stable angina should be corrected. Important among these are smoking, hypertension, hyperlipidemia, and a sedentary lifestyle. Patients should be strongly encouraged to quit smoking. Patients with a sedentary lifestyle should be encouraged to establish a regular program of aerobic exercise (e.g., walking, jogging, swimming, biking). Hypertension and hyperlipidemia are major risk factors and should be treated. These disorders are discussed in Chapters 47 and 50, respectively.
Variant Angina (Prinzmetal’s Angina, Vasospastic Angina)PathophysiologyVariant angina is caused by coronary artery spasm, which restricts blood flow to the myocardium. Hence, as in stable angina, pain is secondary to insufficient oxygenation of the heart. In contrast to stable angina, whose symptoms occur primarily at times of exertion, variant angina can produce pain at any time, even during rest and sleep. Frequently, variant angina occurs in conjunction with stable angina. Alternative names for variant angina are vasospastic angina and Prinzmetal’s angina.
Treatment StrategyThe goal is to reduce the incidence and severity of attacks. In contrast to stable angina, which is treated primarily by reducing oxygen demand, variant angina is treated by increasing cardiac oxygen supply. This makes sense in that the pain is caused by a reduction in oxygen supply, rather than by an increase in demand. Oxygen supply is increased with vasodilators, which prevent or relieve coronary artery spasm.
Overview of Therapeutic AgentsVasospastic angina is treated with two groups of drugs: calcium channel blockers and organic nitrates. Both relax coronary artery spasm. Beta blockers and ranolazine, which are effective in stable angina, are not effective in variant angina. As with stable angina, therapy is symptomatic only; drugs do not alter the underlying pathology.
Unstable AnginaPathophysiologyUnstable angina is a medical emergency. Symptoms result from severe CAD complicated by vasospasm, platelet aggrega-tion, and transient coronary thrombi or emboli. The patient may present with either symptoms of angina at rest, new-onset exertional angina, or intensification of existing angina. Unstable angina poses a much greater risk of death than stable angina, but a smaller risk of death than MI. The risk of dying is greatest initially and then declines to baseline in about 2 months.
TreatmentIn March 2012, the American College of Cardiology (ACC) and the American Heart Association (AHA) issued updated guidelines for the diagnosis and management of unstable angina. The document—2012 ACCF/AHA Focused Update Incorporated Into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina and Non–ST-Segment Elevation Myocardial Infarction—is available free at www.acc.org and www.americanheart.org. According to the guideline, the treatment strategy is to maintain oxygen supply and decrease oxygen demand. The goal is to reduce pain and prevent progression to MI or death. All patients should be hospitalized. Acute management consists of anti-ischemic therapy combined with antiplatelet and anticoagulation therapy.
Prototype Drugs
DRUGS FOR ANGINA PECTORISOrganic NitratesNitroglycerin
Beta BlockersMetoprololPropranolol
Calcium Channel BlockersNifedipineVerapamil
Drug That Increases Myocardial EfficiencyRanolazine
NitroglycerinNitroglycerin has been used to treat angina since 1879. The drug is effective, fast acting, and inexpensive. Despite avail-ability of newer antianginal agents, nitroglycerin remains the drug of choice for relieving an acute anginal attack.
Vasodilator ActionsNitroglycerin acts directly on vascular smooth muscle (VSM) to promote vasodilation. At usual therapeutic doses, the drug acts primarily on veins. Dilation of arterioles is only modest.

594
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
In patients with stable angina, nitroglycerin does not appear to increase blood flow to ischemic areas of the heart. This statement is based on two observations. First, nitroglycerin does not dilate atherosclerotic coronary arteries. Second, when nitroglycerin is injected directly into coronary arteries during an anginal attack, it does not relieve pain. Both observations suggest that pain relief results from effects of nitroglycerin on peripheral blood vessels—not from effects on coronary blood flow.
Variant Angina. In patients with variant angina, nitro-glycerin acts by relaxing or preventing spasm in coronary arteries. Hence, the drug increases oxygen supply. It does not reduce oxygen demand.
PharmacokineticsAbsorption. Nitroglycerin is highly lipid soluble and crosses
membranes with ease. Because of this property, nitroglycerin can be administered by uncommon routes (sublingual, buccal, transdermal), as well as by more conventional routes (oral, intravenous).
Metabolism. Nitroglycerin undergoes rapid inactivation by hepatic enzymes (organic nitrate reductases). As a result, the drug has a plasma half-life of only 5 to 7 minutes. When nitroglycerin is administered orally, most of each dose is destroyed on its first pass through the liver.
Adverse EffectsNitroglycerin is generally well tolerated. Principal adverse effects—headache, hypotension, and tachycardia—occur secondary to vasodilation.
Headache. Initial therapy can produce severe headache. This response diminishes over the first few weeks of treatment. In the meantime, headache can be reduced with aspirin, acetaminophen, or some other mild analgesic.
Orthostatic Hypotension. Relaxation of VSM causes blood to pool in veins when the patient assumes an erect posture. Pooling decreases venous return to the heart, which reduces cardiac output, causing blood pressure to fall. Symptoms of orthostatic hypotension include light-headedness and dizziness. Patients should be instructed to sit or lie down if these occur. Lying with the feet elevated promotes venous return and can help restore blood pressure.
Reflex Tachycardia. Nitroglycerin lowers blood pressure— primarily by decreasing venous return and partly by dilating arterioles. By lowering blood pressure, the drug can activate the baroreceptor reflex, causing sympathetic stimulation of the heart. The resultant increase in both heart rate and contractile force increases cardiac oxygen demand, which negates the benefits of therapy. Pretreatment with a beta blocker or verapamil (a calcium channel blocker that directly suppresses the heart) can prevent sympathetic cardiac stimulation.
Drug InteractionsHypotensive Drugs. Nitroglycerin can intensify the effects
of other hypotensive agents. Consequently, care should be exercised when nitroglycerin is used concurrently with beta blockers, calcium channel blockers, diuretics, and all other drugs that can lower blood pressure, including inhibitors of phosphodiesterase type 5 (PDE5). Also, patients should be advised to avoid alcohol.
Nitroglycerin and the PDE5 inhibitors both increase cGMP (nitrates increase cGMP formation, and PDE5 inhibitors decrease
The biochemical events that lead to vasodilation are outlined in Fig. 51.2. The process begins with uptake of nitrate by VSM, followed by conversion of nitrate to its active form: nitric oxide. As indicated, conversion requires the presence of sulfhydryl groups. Nitric oxide then activates guanylyl cyclase, an enzyme that catalyzes the formation of cyclic GMP (cGMP). Through a series of reactions, elevation of cGMP leads to dephosphorylation of light-chain myosin in VSM. (Recall that, in all muscles, phosphorylated myosin interacts with actin to produce contraction.) As a result of dephosphorylation, myosin is unable to interact with actin, and so VSM relaxes, causing vasodilation. For our purposes, the most important aspect of this sequence is the conversion of nitrate to its active form—nitric oxide—in the presence of a sulfhydryl source.
Mechanism of Antianginal EffectsStable Angina. Nitroglycerin decreases the pain of
exertional angina primarily by decreasing cardiac oxygen demand. Oxygen demand is decreased as follows: By dilating veins, nitroglycerin decreases venous return to the heart, and thereby decreases ventricular filling; the resultant decrease in wall tension (preload) decreases oxygen demand.
Dephosphorylation oflight-chain myosin
Activation ofguanylyl cyclase
Nitrate(within VSM)
Sulfhydrylgroups
Mitochondrialaldehyde
dehydrogenase
Nitrate(extracellular)
Relaxation of VSM
Cyclic GMP
Nitric oxide
Vasodilation
Fig. 51.2 ■ Biochemistry of nitrate-induced vasodilation. Note that sulfhydryl groups are needed to catalyze the conver-sion of nitrate to its active form, nitric oxide. If sulfhydryl groups are depleted from VSM, tolerance to nitrates will occur.

595
CHAPTER 51 Drugs for Angina Pectoris
as pain begins. Rapid-acting preparations can also be used for acute prophylaxis of angina. For this purpose, they are taken just before anticipated exertion. Long-acting preparations are used to provide sustained protection against anginal attacks. To provide protection, they are administered on a fixed schedule (but one that permits at least 8 drug-free hours each day).
Brand names and dosages for nitroglycerin preparations are shown in Table 51.3.
Sublingual Tablets and Powder. When administered sublingually (beneath the tongue), nitroglycerin is absorbed directly through the oral mucosa into the bloodstream. Hence, unlike orally administered drugs, which must pass through the liver on their way to the systemic circulation, sublingual nitroglycerin bypasses the liver and thereby temporarily avoids inactivation. Because the liver is bypassed, sublingual doses can be low (between 0.3 and 0.6 mg). These doses are about 10 times lower than those required when nitroglycerin is dosed orally.
Effects of sublingual nitroglycerin begin rapidly—in 1 to 3 minutes—and persist up to 1 hour. Because sublingual administration works fast, this route is ideal for (1) terminating an ongoing attack and (2) short-term prophylaxis when exertion is anticipated.
To terminate an acute anginal attack, sublingual nitroglycerin should be administered as soon as pain begins. Administration should not be delayed until the pain has become severe. Accord-ing to current guidelines, if pain is not relieved in 5 minutes, the
cGMP breakdown). Therefore, if these drugs are combined, levels of cGMP can rise dangerously high, causing excessive vasodilation and a precipitous drop in blood pressure.
Beta Blockers, Verapamil, and Diltiazem. These drugs can suppress nitroglycerin-induced tachycardia. Beta blockers do so by preventing sympathetic activation of beta1-adrenergic receptors on the heart. Verapamil and diltiazem prevent tachy-cardia through direct suppression of pacemaker activity in the sinoatrial node.
ToleranceTolerance to nitroglycerin-induced vasodilation can develop rapidly (over the course of a single day). One possible mecha-nism is depletion of sulfhydryl groups in VSM: In the absence of sulfhydryl groups, nitroglycerin cannot be converted to nitric oxide, its active form. Another possible mechanism is reversible oxidative injury to mitochondrial aldehyde dehydrogenase, an enzyme needed to convert nitroglycerin into nitric oxide. Patients who develop tolerance to nitroglycerin display cross-tolerance to all other nitrates and vice versa. Development of tolerance is most likely with high-dose therapy and uninterrupted therapy. To prevent tolerance, nitroglycerin and other nitrates should be used in the lowest effective dosages; long-acting formulations (e.g., patches, sustained-release preparations) should be used on an intermittent schedule that allows at least 8 drug-free hours every day, usually during the night. If pain occurs during the nitrate-free interval, it can be managed with sparing use of a short-acting nitrate (e.g., sublingual nitroglyc-erin) or by adding a beta blocker or calcium channel blocker to the regimen. Tolerance can be reversed by withholding nitrates for a short time.
Preparations and Routes of AdministrationNitroglycerin is available in several formulations for administra-tion by several routes. This proliferation of dosage forms reflects efforts to delay hepatic metabolism and prolong therapeutic effects.
All nitroglycerin preparations produce qualitatively similar responses; differences relate only to onset and duration of action (Table 51.2). With two preparations, effects begin rapidly (in 1 to 5 minutes) and then diminish in less than 1 hour. With three others, effects begin slowly but last several hours. Only one preparation—sublingual isosorbide dinitrate tablets—has both a rapid onset and a long duration.
Applications of specific preparations are based on their time course. Preparations with a rapid onset are employed to terminate an ongoing anginal attack. When used for this purpose, rapid-acting preparations are administered as soon
Safety Alert
PHOSPHODIESTERASE TYPE 5 INHIBITORSAs discussed in Chapter 66, PDE5 inhibitors—sildenafil [Viagra], tadalafil [Cialis], avanafil [Stendra], and vardenafil [Levitra]—are used for erectile dysfunction. All of these drugs can greatly intensify nitroglycerin-induced vasodilation. Life-threatening hypotension can result. Accordingly, concurrent use of PDE5 inhibitors with nitroglycerin is absolutely contraindicated.
Drug and Dosage Form Onseta Durationb
NITROGLYCERIN
Sublingual tablets Rapid (1–3 min) Brief (30–60 min)Sublingual powder Rapid (1–3 min) Brief (30–60 min)Translingual spray Rapid (2–3 min) Brief (30–60 min)Oral capsules, SR Slow (20–45 min) Long (3–8 hr)Transdermal patches Slow (30–60 min) Long (24 hr)c
Topical ointment Slow (20–60 min) Long (2–12 hr)
ISOSORBIDE MONONITRATE
Oral tablets, IR Slow (30–60 min) Long (6–10 hr)Oral tablets, SR Slow (30–60 min) Long (7–12 hr)
ISOSORBIDE DINITRATE
Sublingual tablets Rapid (2–5 min) Long (1–3 hr)Oral tablets, IR Slow (20–40 min) Long (4–6 hr)Oral tablets, SR Slow (30 min) Long (6–8 hr)Oral capsules, SR Slow (30 min) Long (6–8 hr)
TABLE 51.2 ■ Organic Nitrates: Time Course of Action
aNitrates with a rapid onset have two uses: (1) termination of an ongoing anginal attack and (2) short-term prophylaxis before anticipated exertion. Of the rapid-acting nitrates, nitroglycerin (sublingual tablet, sublingual powder, or translingual spray) is preferred to the others for terminating an ongoing attack.bLong-acting nitrates are used for sustained prophylaxis (prevention) of anginal attacks. All cause tolerance if used without interruption.cAlthough patches can release nitroglycerin for up to 24 hours, they should be removed after 12 to 14 hours to avoid tolerance.IR, Immediate release; SR, sustained release.

596
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Transdermal Delivery Systems. Nitroglycerin patches contain a reservoir from which nitroglycerin is slowly released. Following release, the drug is absorbed through the skin and then into the blood. The rate of release is constant and, depend-ing on the patch used, can range from 0.1 to 0.8 mg/hr. Effects begin within 30 to 60 minutes and persist as long as the patch remains in place (up to 14 hours). Patches are applied once daily to a hairless area of skin. The site should be rotated to avoid local irritation.
Tolerance develops if patches are used continuously (24 hours a day every day). Accordingly, a daily “patch-free” interval of 10 to 12 hours is recommended. This can be accomplished by applying a new patch each morning, leaving it in place for 12 to 14 hours, and then removing it in the evening.
Because of their long duration, patches are well suited for sustained prophylaxis. Since patches have a delayed onset, they cannot be used to abort an ongoing attack.
Translingual Spray. Nitroglycerin can be delivered to the oral mucosa using a metered-dose spray device. Each activation delivers a 0.4-mg dose. Indications for nitroglycerin spray are the same as for sublingual tablets: suppression of an acute anginal attack and prophylaxis of angina when exertion is anticipated. As with sublingual tablets, no more than three doses should be administered within a 15-minute interval. Instruct patients not to inhale the spray.
Topical Ointment. Topical nitroglycerin ointment is used for sustained protection against anginal attacks. The ointment is applied to the skin of the chest, back, abdomen, or anterior thigh. (Since nitroglycerin acts primarily by dilating peripheral veins, there is no mechanistic advantage to applying topical nitroglycerin directly over the heart.) Following topical application, nitroglycerin is absorbed through the skin and then into the blood. Effects begin in 20 to 60 minutes and may persist up to 12 hours.
patient should call 911 or report to an emergency department, since anginal pain that does not respond to nitroglycerin may indicate MI. While awaiting emergency care, the patient can take 1 more tablet, and then a third tablet 5 minutes later.
Sublingual administration is unfamiliar to most patients. Accordingly, education is needed. The patient should be instructed to place the tablet or empty the powder packet under the tongue and leave it there while it dissolves. Nitroglycerin tablets and powder formulated for sublingual use are ineffective if swallowed.
Nitroglycerin tablets available today have good chemical stability. When stored properly, they should remain effective until the expiration date on the container. To ensure good stability, the tablets should be stored moisture free at room temperature in their original container, which should be closed tightly after each use.
Sustained-Release Oral Capsules. Sustained-release oral capsules are intended for long-term prophylaxis only; these formulations cannot act fast enough to terminate an ongoing anginal attack. Sustained-release capsules contain a large dose of nitroglycerin that is slowly absorbed across the GI wall. In theory, doses are large enough so that amounts of nitroglycerin sufficient to produce a therapeutic response will survive passage through the liver. Because they produce sustained blood levels of nitroglycerin, these formulations can cause tolerance. To reduce the risk of tolerance, these products should be taken only once or twice daily. Patients should be instructed to swallow sustained-release capsules intact.
Drug and Formulation Brand Name Usual Dosage
NITROGLYCERIN
Sublingual tablets Nitrostat 0.3–0.6 mg as needed every 5 min for maximum of three dosesSublingual powder GONITRO 1–2 packets (400 mcg/packet, up to three packets in a 15-min period)Translingual spray Nitrolingual Pumpspray,
NitroMist1–2 sprays (up to 3 sprays in a 15-min period)
Oral capsules, SR Nitro-Time 2.5–6.5 mg 3 or 4 times daily; to avoid tolerance, administer only once or twice daily; do not crush or chew
Transdermal patches Nitro-Dur 1 patch a day; to avoid tolerance, remove after 12–14 hr, allowing 10–12 patch-free hours each day. Patches come in sizes that release 0.1–0.8 mg/hr.
Topical ointment Nitro-Bid 1–2 inches (7.5–40 mg) every 4–6 hrIntravenous Generic only 5 mcg/min initially, then increased gradually as needed (max 200 mcg/min); tolerance
develops with prolonged continuous infusion
ISOSORBIDE MONONITRATE
Oral tablets, IR Generic only 20 mg twice daily; to avoid tolerance, take the first dose upon awakening and the second dose 7 hr later
Oral tablets, SR Imdur 60–240 mg once daily; do not crush or chew
ISOSORBIDE DINITRATE
Sublingual tablets Generic only 2.5–5 mg before activities that may cause angina. For acute angina, 2.5–5 mg every 5 min for maximum of three doses. Do not crush or chew
Oral tablets, IR Isordil Oral Titradose 10–40 mg 2 or 3 times daily; to avoid tolerance, take the last dose no later than 7:00 PM
Oral tablets, SR Generic only 40 mg every 6–12 hr; to avoid tolerance, take only once or twice daily (at 8:00 AM and 2:00 PM)
Oral capsules, SR Dilatrate-SR 40 mg every 6–12 hr; to avoid tolerance, take only once or twice daily (at 8:00 AM and 2:00 PM)
TABLE 51.3 ■ Organic Nitrates: Brand Names and Dosages
IR, Immediate release; SR, sustained release.

597
CHAPTER 51 Drugs for Angina Pectoris
intensity of anginal attacks are lowered. All of the beta blockers appear equally effective. In addition to reducing anginal pain, beta blockers decrease the risk of death, especially in patients with a prior MI.
Beta blockers reduce anginal pain primarily by decreasing cardiac oxygen demand, principally through blockade of beta1 receptors in the heart, which decreases heart rate and contractil-ity. Beta blockers reduce oxygen demand further by causing a modest reduction in arterial pressure (afterload). In addition to decreasing oxygen demand, beta blockers help increase oxygen supply. By slowing heart rate, they increase time in diastole and thereby increase the time during which blood flows through myocardial vessels. (Recall that blood does not flow in these vessels during systole.) In patients taking vasodila-tors (e.g., nitroglycerin), beta blockers provide the additional benefit of blunting reflex tachycardia.
For treatment of stable angina, dosage should be low initially and then gradually increased. The dosing goal is to reduce resting heart rate to 50 to 60 beats/min and to limit exertional heart rate to about 100 beats/min. Beta blockers should not be withdrawn abruptly, since doing so can increase the incidence and intensity of anginal attacks, and may even precipitate MI.
Beta blockers can produce a variety of adverse effects. Blockade of cardiac beta1 receptors can produce bradycardia, decreased atrioventricular (AV) conduction, and reduction of contractility. Consequently, beta blockers should not be used by patients with sick sinus syndrome, heart failure, or second-degree or third-degree AV block. Blockade of beta2 receptors in the lung can promote bronchoconstriction. Accordingly, beta blockers should be used with caution by patients with asthma. If an asthmatic individual absolutely must use a beta blocker, a beta1-selective agent (e.g., metoprolol) should be chosen. Beta blockers can mask signs of hypoglycemia and therefore must be used with caution in patients with diabetes. Rarely, these drugs cause adverse central nervous system effects, including insomnia, depression, and bizarre dreams.
The basic pharmacology of the beta blockers is discussed in Chapter 18.
CALCIUM CHANNEL BLOCKERSThe calcium channel blockers (CCBs) used most frequently are verapamil, diltiazem, and nifedipine (a dihydropyridine-type calcium channel blocker). Accordingly, our discussion focuses on these three drugs. All three can block calcium channels in VSM, primarily in arterioles. The result is arteriolar dilation and reduction of peripheral resistance (afterload). In addition, all three can relax coronary vasospasm. Verapamil and diltiazem also block calcium channels in the heart and can thereby decrease heart rate, AV conduction, and contractility.
Calcium channel blockers are used to treat both stable and variant angina. In variant angina, these drugs promote relaxation of coronary artery spasm, increasing cardiac oxygen supply. In stable angina, they promote relaxation of peripheral arte-rioles; the resultant decrease in afterload reduces cardiac oxygen demand. Verapamil and diltiazem can produce modest additional reductions in oxygen demand by suppressing heart rate and contractility.
The major adverse effects of the CCBs are cardiovascular. Dilation of peripheral arterioles lowers blood pressure and can thereby induce reflex tachycardia. This reaction is greatest
Nitroglycerin ointment (2%) is dispensed from a tube, and the length of the ribbon squeezed from the tube determines dosage. (One inch contains about 15 mg of nitroglycerin.) The usual adult dosage is 1 to 2 inches applied every 4 to 6 hours. The ointment should be spread over an area at least 2.5 inches by 3.5 inches and then covered with plastic wrap. Sites of application should be rotated to minimize skin irritation. As with other long-acting formula-tions, uninterrupted use can cause tolerance.
Intravenous Infusion. Intravenous nitroglycerin is employed only rarely to treat angina pectoris. When used for angina, IV nitroglycerin is limited to patients who have failed to respond to other medications. Additional uses of IV nitroglycerin include treatment of heart failure associated with acute MI, treatment of perioperative hypertension, and production of controlled hypoten-sion for surgery.
Intravenous nitroglycerin has a very short duration, so continuous infusion is required. The infusion rate is 5 mcg/min initially; it is then increased gradually until an adequate response has been achieved. Heart rate and blood pressure must be monitored continuously.
Stock solutions of nitroglycerin must be diluted for IV use. Because ampules of nitroglycerin prepared by different manufacturers can differ in both volume and nitroglycerin concentration, the label must be read carefully when dilutions are made.
Administer using a glass IV bottle and the administration set provided by the manufacturer. Nitroglycerin absorbs into standard polyvinyl chloride tubing, so this tubing should be avoided.
Discontinuing NitroglycerinLong-acting preparations (transdermal patches, topical ointment, sustained-release oral tablets or capsules) should be discontinued slowly. If they are withdrawn abruptly, vasospasm may result.
Summary of Therapeutic UsesAcute Therapy of Angina. For acute treatment of angina
pectoris, nitroglycerin is administered in sublingual tablets and a translingual spray. Both formulations can be used to abort an ongoing anginal attack and to provide prophylaxis in anticipation of exertion.
Sustained Therapy of Angina. For sustained prophylaxis against angina, nitroglycerin is administered in the follow-ing formulations: transdermal patches, topical ointment, and sustained-release oral capsules.
Intravenous Therapy. Intravenous nitroglycerin is indicated for perioperative control of blood pressure, production of controlled hypotension during surgery, and treatment of heart failure associated with acute MI. In addition, IV nitroglycerin is used to treat unstable angina and chronic angina when symptoms cannot be controlled with preferred medications.
Isosorbide Mononitrate and Isosorbide DinitrateBoth of these drugs have pharmacologic actions identical to those of nitro-glycerin. Both drugs are used for angina, both are taken orally, and both produce headache, hypotension, and reflex tachycardia. Differences between them relate only to route of administration and time course of action. Time course determines whether a particular drug or dosage form will be used for acute therapy, sustained prophylaxis, or both. As with nitroglycerin, tolerance can develop to long-acting preparations. To avoid tolerance, the dosing schedule for long-acting preparations should allow at least 12 drug-free hours a day. Time courses are shown in Table 51.2. Brand names and dosages are shown in Table 51.3. A fixed-dose combination of isosorbide dinitrate plus hydralazine is discussed in Chapter 48.
BETA BLOCKERSBeta blockers (e.g., propranolol, metoprolol) are first-line drugs for angina of effort, but are not effective against vasospastic angina. When administered on a fixed schedule, beta blockers can provide sustained protection against effort-induced anginal pain. Exercise tolerance is increased, and the frequency and

598
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
juice, HIV protease inhibitors (e.g., ritonavir), macrolide antibiotics (e.g., erythromycin), azole antifungal drugs (e.g., itraconazole), and some calcium channel blockers.
QT Drugs. Drugs that prolong the QT interval (e.g., quinidine, sotalol) can increase the risk of torsades de pointes in patients taking ranolazine, and hence should be avoided. Chapter 7 presents a comprehensive list of QT drugs.
Calcium Channel Blockers. Most CCBs—but not amlodipine—can inhibit CYP3A4 and thus increase levels of ranolazine. Accordingly, when the use of ranolazine plus a CCB is indicated, amlodipine is the only CCB that should be used.Preparations, Dosage, and AdministrationRanolazine [Ranexa] is formulated in extended-release tablets (500 and 1000 mg) that should be swallowed intact, with or without food. Dosing begins at 500 mg twice daily, and it may be increased to a maximum of 1000 mg twice daily. Ranolazine may be used in combination with a nitrate, beta blocker, or amlodipine (a CCB), and other drugs for angina.
TREATMENT MEASURESGuidelines for Management of Chronic Stable AnginaIn 1999, three organizations—the American Heart Association, the American College of Cardiology, and the American College of Physicians–American Society of Internal Medicine—joined forces to produce the first national guidelines on the management of chronic stable angina. The 1999 guidelines were updated in 2002 and again in 2007. Both updates—ACC/AHA 2002 Guideline Update for the Management of Patients with Chronic Stable Angina, and 2007 Chronic Angina Focused Update of the ACC/AHA 2002 Guidelines for the Management of Patients with Chronic Stable Angina—are available free online at circ .ahajournals.org. The discussion that follows reflects recom-mendations in these guidelines.
Treatment of stable angina has two objectives: (1) prevention of MI and death, and (2) reduction of cardiac ischemia and associated anginal pain. Although both goals are desirable, prevention of MI and death is clearly more important. If two treatments are equally effective at decreasing anginal pain but one also decreases the risk of death, then the latter is preferred.
Drugs Used to Prevent Myocardial Infarction and DeathWe now have medical treatments that can decrease the risk of MI and death in patients with chronic stable angina. Therapy directed at preventing MI and death is a new paradigm in the management of stable angina, and all practitioners should become familiar with it.
Antiplatelet Drugs. These agents decrease platelet aggrega-tion and thereby decrease the risk of thrombus formation in coronary arteries. The most effective agents are aspirin and clopidogrel. In patients with stable angina, low-dose aspirin produces a 33% decrease in the risk of adverse cardiovascular events. Benefits of clopidogrel seem equal to those of aspirin, although they are not as well documented. The guidelines recommend that all patients with stable angina take 75 to 162 mg of aspirin daily, unless there is a specific reason not to. Aspirin, clopidogrel, and other antiplatelet drugs are discussed in Chapter 52.
Cholesterol-Lowering Drugs. Elevated cholesterol is a major risk factor for coronary atherosclerosis. Drugs that lower
with nifedipine and minimal with verapamil and diltiazem. Because of their suppressant effects on the heart, verapamil and diltiazem must be used cautiously in patients taking beta blockers and in patients with bradycardia, heart failure, or AV block. These precautions do not apply to nifedipine or other dihydropyridines.
The basic pharmacology of the CCBs is discussed in Chapter 45.
RANOLAZINEActions and Therapeutic UseRanolazine [Ranexa] represented the first new class of anti-anginal agents to be approved in more than 25 years. In clinical trials, the drug reduced the number of angina episodes per week and increased exercise tolerance. However, these benefits were modest, and were smaller in women than in men. Unlike most other antianginal drugs, ranolazine does not reduce heart rate, blood pressure, or vascular resistance. However, it can prolong the QT interval, and is subject to multiple drug interactions. Ranolazine works by reducing accumulation of sodium and calcium in myocardial cells, which might help the myocardium use energy more efficiently. However, the exact mechanism of action is unknown. Despite limited efficacy, many drug interactions, and a risk of dysrhythmias (see text that follows), ranolazine is now approved as a first-line drug for angina. It may be combined with nitrates, beta blockers, amlodipine (a CCB), and other drugs used for angina treatment.
PharmacokineticsAbsorption from the GI tract is highly variable, but not affected by food. Plasma levels peak 2 to 5 hours after dosing. In the liver, ranolazine undergoes rapid and extensive metabolism, mainly by CYP3A4 (the 3A4 isoenzyme of cytochrome P450). The drug has a plasma half-life of 7 hours and is excreted in the urine (75%) and feces (25%), almost entirely as metabolites.
Adverse EffectsQT Prolongation. Ranolazine can cause a dose-related
increase in the QT interval and may thereby increase the risk of torsades de pointes, a serious ventricular dysrhythmia. Accordingly, the drug is contraindicated for patients with pre-existing QT prolongation and for those taking other drugs that can increase the QT interval. In addition, ranolazine is contraindicated for patients at risk of developing high levels of the drug—namely, patients with hepatic impairment or those taking drugs that inhibit CYP3A4. The issue of drug-induced QT prolongation is discussed in Chapter 7.
Elevation of Blood Pressure. In patients with severe renal impairment, ranolazine can raise blood pressure by about 15 mm Hg. Accordingly, blood pressure should be monitored often in these people.
Other Adverse Effects. The most common adverse effects are constipation, dizziness, nausea, and headache.
Drug InteractionsCYP3A4 Inhibitors. Agents that inhibit CYP3A4 can
increase levels of ranolazine and can thereby increase the risk of torsades de pointes. Accordingly, moderate or strong CYP3A4 inhibitors should be avoided. Among these agents are grapefruit

599
CHAPTER 51 Drugs for Angina Pectoris
Note that, as we proceed along the drug-selection flow plan, drugs are added to the regimen, resulting in treatment with two or more agents. Combination therapy increases our chances of success because oxygen demand is decreased by multiple mechanisms: Beta blockers reduce heart rate and contractility; CCBs reduce afterload (by dilating arterioles); and nitrates reduce preload (by dilating veins).
If combined treatment with a beta blocker, CCB, and long-acting nitrate fails to provide relief, coronary artery bypass graft (CABG) surgery or percutaneous coronary intervention (PCI) may be indicated. Note that these invasive procedures
cholesterol can slow the progression of CAD, stabilize ath-erosclerotic plaques, and even cause plaque regression. Therapies that reduce cholesterol are associated with decreased mortality from coronary heart disease. For example, in patients with established CAD, taking simvastatin can decrease the risk of mortality by 35%. Because of the well-established benefits of cholesterol-lowering therapy, the guidelines recommend that all patients with stable angina receive a cholesterol-lowering drug. The pharmacology of the cholesterol-lowering drugs is discussed in Chapter 50.
Angiotensin-Converting Enzyme (ACE) Inhibitors. There is strong evidence that, in patients with CAD, ACE inhibitors greatly reduce the incidence of adverse outcomes. In the Heart Outcomes Prevention Evaluation (HOPE) trial, for example, ramipril reduced the incidence of stroke, MI, and cardiovascular death. Among one subset of patients—those with diabetes—benefits were particularly striking. Ramipril decreased the risk of stroke by 33%, MI by 22%, and cardiovascular death by 37%. In addition, ramipril reduced the risk of nephropathy, retinopathy, and other microvascular complications of diabetes. Because of these well-documented benefits, the guidelines recommend ACE inhibitors for most patients with established CAD, and especially for those with diabetes. The pharmacology of the ACE inhibitors is discussed in Chapter 44.
Antianginal Agents: Drugs Used to Reduce Anginal PainThe goal of antianginal therapy is to achieve complete (or nearly complete) elimination of anginal pain, along with a return to normal activities. This should be accomplished with a minimum of adverse drug effects.
The basic strategy of antianginal therapy is to provide baseline protection using one or more long-acting drugs (beta blocker, CCB, long-acting nitrate) supplemented with sublingual nitroglycerin when breakthrough pain occurs. A flow plan for drug selection is shown in Fig. 51.3. As indicated, treatment is approached sequentially. Progression from one step to the next is based on patient response. Some patients can be treated with a single long-acting drug, some require two or three, and some require revascularization.
Initial treatment consists of sublingual nitroglycerin plus a long-acting antianginal drug. Beta blockers are the preferred agents for baseline therapy because they can decrease mortality, especially in patients with a prior MI. In addition to providing prophylaxis, beta blockers suppress nitrate-induced reflex tachycardia.
If a beta blocker is inadequate, or if there are contraindica-tions to beta blockade, a long-acting CCB should be added or substituted. Dihydropyridine-type CCBs (e.g., nifedipine) lack cardiosuppressant actions and are safer than beta block-ers for patients with bradycardia, AV block, or heart failure. When a CCB is to be combined with a beta blocker, a dihy-dropyridine is preferred to verapamil or diltiazem because verapamil and diltiazem will intensify the cardiosuppressant actions of the beta blocker, whereas a dihydropyridine CCB will not.
If a CCB is inadequate, or if there are contraindications to calcium channel blockade, a long-acting nitrate (e.g., trans-dermal nitroglycerin) should be added or substituted. However, because tolerance can develop quickly, these nitrate preparations are less well suited than beta blockers or CCBs for continuous protection.
If seriouscontraindications
If seriouscontraindications
Successfultreatment?
No
Yes
Yes
Add a beta blocker(especially if prior MI)
Sublingual nitroglycerin
If seriouscontraindications
Successfultreatment?
No
Yes
Add or substitute acalcium channel blocker1
Add a long-acting nitrate Successfultreatment?
No
Consider revascularization(CABG or PCI)2
Fig. 51.3 ■ Flow plan for antianginal drug selection in patients with chronic stable angina. 1Avoid short-acting dihydropyridines. 2At any point in this process, based on coronary anatomy, severity of angina symptoms, and patient preference, it is reasonable to consider evaluation for coronary revascularization (PCI or CABG). Unless a patient is documented to have left main, three-vessel, or two-vessel CAD with significant stenosis of the proximal left anterior descending coronary artery, there is no demonstrated survival advantage associated with CABG or PCI in low-risk patients with chronic stable angina. Accordingly, medical therapy should be attempted in most patients before considering PCI or CABG. (Adapted from Gibbons RJ, Chatterjee K, Daley J, et al: ACC/AHA 2002 Guideline Update for the Management of Patients with Chronic Stable Angina: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines [Committee to Update the 1999 Guidelines for the Management of Patients with Chronic Stable Angina]. 2002. Available online at circ.ahajournals.org.)
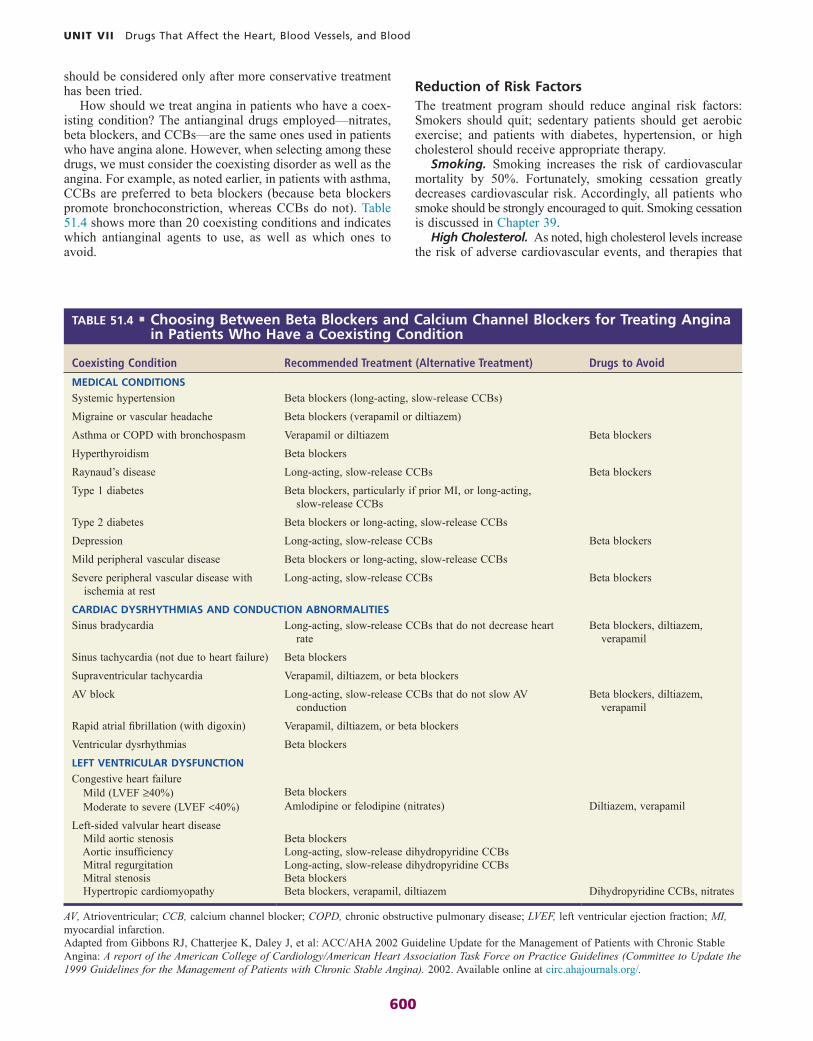
600
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Reduction of Risk FactorsThe treatment program should reduce anginal risk factors: Smokers should quit; sedentary patients should get aerobic exercise; and patients with diabetes, hypertension, or high cholesterol should receive appropriate therapy.
Smoking. Smoking increases the risk of cardiovascular mortality by 50%. Fortunately, smoking cessation greatly decreases cardiovascular risk. Accordingly, all patients who smoke should be strongly encouraged to quit. Smoking cessation is discussed in Chapter 39.
High Cholesterol. As noted, high cholesterol levels increase the risk of adverse cardiovascular events, and therapies that
should be considered only after more conservative treatment has been tried.
How should we treat angina in patients who have a coex-isting condition? The antianginal drugs employed—nitrates, beta blockers, and CCBs—are the same ones used in patients who have angina alone. However, when selecting among these drugs, we must consider the coexisting disorder as well as the angina. For example, as noted earlier, in patients with asthma, CCBs are preferred to beta blockers (because beta blockers promote bronchoconstriction, whereas CCBs do not). Table 51.4 shows more than 20 coexisting conditions and indicates which antianginal agents to use, as well as which ones to avoid.
Coexisting Condition Recommended Treatment (Alternative Treatment) Drugs to Avoid
MEDICAL CONDITIONS
Systemic hypertension Beta blockers (long-acting, slow-release CCBs)Migraine or vascular headache Beta blockers (verapamil or diltiazem)Asthma or COPD with bronchospasm Verapamil or diltiazem Beta blockersHyperthyroidism Beta blockersRaynaud’s disease Long-acting, slow-release CCBs Beta blockersType 1 diabetes Beta blockers, particularly if prior MI, or long-acting,
slow-release CCBsType 2 diabetes Beta blockers or long-acting, slow-release CCBsDepression Long-acting, slow-release CCBs Beta blockersMild peripheral vascular disease Beta blockers or long-acting, slow-release CCBsSevere peripheral vascular disease with
ischemia at restLong-acting, slow-release CCBs Beta blockers
CARDIAC DYSRHYTHMIAS AND CONDUCTION ABNORMALITIES
Sinus bradycardia Long-acting, slow-release CCBs that do not decrease heart rate
Beta blockers, diltiazem, verapamil
Sinus tachycardia (not due to heart failure) Beta blockersSupraventricular tachycardia Verapamil, diltiazem, or beta blockersAV block Long-acting, slow-release CCBs that do not slow AV
conductionBeta blockers, diltiazem,
verapamilRapid atrial fibrillation (with digoxin) Verapamil, diltiazem, or beta blockersVentricular dysrhythmias Beta blockers
LEFT VENTRICULAR DYSFUNCTION
Congestive heart failure Mild (LVEF ≥40%) Beta blockers Moderate to severe (LVEF <40%) Amlodipine or felodipine (nitrates) Diltiazem, verapamil
Left-sided valvular heart disease Mild aortic stenosis Beta blockers Aortic insufficiency Long-acting, slow-release dihydropyridine CCBs Mitral regurgitation Long-acting, slow-release dihydropyridine CCBs Mitral stenosis Beta blockers Hypertropic cardiomyopathy Beta blockers, verapamil, diltiazem Dihydropyridine CCBs, nitrates
TABLE 51.4 ■ Choosing Between Beta Blockers and Calcium Channel Blockers for Treating Angina in Patients Who Have a Coexisting Condition
Adapted from Gibbons RJ, Chatterjee K, Daley J, et al: ACC/AHA 2002 Guideline Update for the Management of Patients with Chronic Stable Angina: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1999 Guidelines for the Management of Patients with Chronic Stable Angina). 2002. Available online at circ.ahajournals.org/.
AV, Atrioventricular; CCB, calcium channel blocker; COPD, chronic obstructive pulmonary disease; LVEF, left ventricular ejection fraction; MI, myocardial infarction.

601
CHAPTER 51 Drugs for Angina Pectoris
Physical Inactivity. Increased physical activity has mul-tiple benefits. In patients with chronic stable angina, exercise increases exercise tolerance and the sense of well-being, and decreases anginal symptoms, cholesterol levels, and objective measures of ischemia. Accordingly, the guidelines recommend that patients perform 30 to 60 minutes of a moderate-intensity activity 3 to 4 times a week. Such activities include walking, jogging, cycling, and other aerobic exercises. Exercise by moderate- to high-risk patients should be medically supervised.
Management of Variant AnginaTreatment of vasospastic angina can proceed in three steps. For initial therapy, either a calcium channel blocker or a long-acting nitrate is selected. If either drug alone is inadequate, then combined therapy with a calcium channel blocker plus a nitrate should be tried. If the combination fails to control symptoms, CABG surgery may be indicated. Beta blockers are not effective in vasospastic angina.
reduce cholesterol reduce that risk. Accordingly, all patients with high cholesterol levels should receive cholesterol-lowering therapy.
Hypertension. High blood pressure increases the risk of cardiovascular mortality, and lowering blood pressure reduces the risk. Accordingly, all patients with hypertension should receive treatment. Blood pressure should be reduced to 140/90 mm Hg or less. In patients with additional risk factors (e.g., diabetes, heart failure, retinopathy), the target blood pressure is 130/80 mm Hg or less. Management of hypertension is discussed in Chapter 47.
Diabetes. Both type 1 (insulin-dependent) and type 2 (non–insulin-dependent) diabetes increase the risk of cardio-vascular mortality. Type 1 increases the risk 3- to 10-fold; type 2 increases the risk 2- to 4-fold. Although there is good evidence that tight glycemic control decreases the risk of microvascular complications of diabetes, there is little evidence to show that tight glycemic control decreases the risk of cardiovascular complications. Nonetheless, it is prudent to strive for optimal glycemic control.
KEY POINTS
■ Anginal pain occurs when cardiac oxygen supply is insuf-ficient to meet cardiac oxygen demand.
■ Cardiac oxygen demand is determined by heart rate, contractility, preload, and afterload. Drugs that reduce these factors can help relieve anginal pain.
■ Cardiac oxygen supply is determined by myocardial blood flow. Drugs that increase oxygen supply will reduce anginal pain.
■ Angina pectoris has three forms: chronic stable angina, variant (vasospastic) angina, and unstable angina.
■ The underlying cause of stable angina is coronary artery atherosclerosis.
■ The underlying cause of variant angina is coronary artery spasm.
■ Drugs relieve pain of stable angina by decreasing cardiac oxygen demand. They do not increase oxygen supply.
■ Drugs relieve pain of variant angina by increasing cardiac oxygen supply. They do not decrease oxygen demand.
■ Nitroglycerin and other organic nitrates are vasodilators.■ To cause vasodilation, nitroglycerin must first be converted
to nitric oxide, its active form. This reaction requires a sulfhydryl source.
■ Nitroglycerin relieves pain of stable angina by dilating veins, which decreases venous return, which decreases preload, which decreases oxygen demand.
■ Nitroglycerin relieves pain of variant angina by relaxing coronary vasospasm, which increases oxygen supply.
■ Nitroglycerin is highly lipid soluble, and therefore is readily absorbed through the skin and oral mucosa.
■ Nitroglycerin undergoes very rapid inactivation in the liver. Hence, when the drug is administered orally, most of each dose is destroyed before reaching the systemic circulation.
■ When nitroglycerin is administered sublingually, it is absorbed directly into the systemic circulation and therefore
temporarily bypasses the liver. Hence, to produce equivalent effects, sublingual doses can be much smaller than oral doses.
■ Nitroglycerin causes three characteristic side effects: headache, orthostatic hypotension, and reflex tachycardia. All three occur secondary to vasodilation.
■ Reflex tachycardia from nitroglycerin can be prevented with a beta blocker, verapamil, or diltiazem.
■ Continuous use of nitroglycerin can produce tolerance within 24 hours. The mechanism may be depletion of sulfhydryl groups.
■ To prevent tolerance, nitroglycerin should be used in the lowest effective dosage, and long-acting formulations should be used on an intermittent schedule that allows at least 8 drug-free hours every day, usually during the night.
■ Nitroglycerin preparations that have a rapid onset (e.g., sublingual nitroglycerin) are used to abort an ongoing anginal attack and to provide acute prophylaxis when exertion is expected. Administration is PRN.
■ Nitroglycerin preparations that have a long duration (e.g., patches, sustained-release oral capsules) are used for extended protection against anginal attacks. Administration is on a fixed schedule (but one that allows at least 8 drug-free hours a day).
■ Nitroglycerin should be used cautiously with most vasodila-tors and must not be used at all with sildenafil [Viagra] and other PDE5 inhibitors.
■ Beta blockers prevent pain of stable angina primarily by decreasing heart rate and contractility, which reduces cardiac oxygen demand.
■ Beta blockers are administered on a fixed schedule, not PRN.
■ Beta blockers are not used for variant angina.■ CCBs relieve the pain of stable angina by reducing cardiac
oxygen demand. Two mechanisms are involved. First, all
Continued

602
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Summary of Major Nursing Implicationsa
NITROGLYCERINPreadministration AssessmentTherapeutic GoalReduction of the frequency and intensity of anginal attacks.
Baseline DataObtain baseline data on the frequency and intensity of anginal attacks, the location of anginal pain, and the factors that precipitate attacks.
The patient interview and physical examination should identify risk factors for angina pectoris, including treatable contributing pathophysiologic conditions (e.g., hypertension, hyperlipidemia).
Identifying High-Risk PatientsUse with caution in hypotensive patients and in patients taking drugs that can lower blood pressure, including alcohol and antihypertensive medications. Use with sildenafil [Viagra] and other PDE5 inhibitors is contraindicated.
Implementation: AdministrationRoutes and Administration
Sublingual Tablets or PowderUse. Prophylaxis or termination of an acute anginal
attack.Technique of Administration. Instruct patients to place
the tablet or empty the powder under the tongue and leave it there until fully dissolved; these medications should not be swallowed.
Instruct patients to call 911 or go to an emergency depart-ment if pain is not relieved in 5 minutes. While awaiting emergency care, they can take 1 more dose, and then a third 5 minutes later.
Instruct patients to store tablets in a dry place at room temperature in their original container, which should be closed tightly after each use. Under these conditions, the tablets should remain effective until the expiration date on the container.
Sustained-Release Oral CapsulesUse. Sustained protection against anginal attacks. To avoid
tolerance, administer only once or twice daily.Technique of Administration. Instruct patients to
swallow these preparations intact, without chewing or crushing.
Transdermal Delivery SystemsUse. Sustained protection against anginal attacks.Technique of Administration. Instruct patients to apply
transdermal patches to a hairless area of skin, using a new patch and a different site each day.
Instruct patients to remove the patch after 12 to 14 hours, allowing 10 to 12 “patch-free” hours each day. This will prevent tolerance.
Translingual SprayUse. Prophylaxis or termination of an acute anginal attack.Technique of Administration. Instruct patients to direct
the spray against the oral mucosa. Warn patients not to inhale the spray.
Topical OintmentUse. Sustained protection against anginal attacks. Instruct
patients to remove any remaining ointment before applying a new dose.
Technique of Administration. (1) Squeeze a ribbon of ointment of prescribed length onto the applicator paper provided; (2) using the applicator paper, spread the ointment over an area at least 2.5 inches by 3.5 inches (application may be made to the chest, back, abdomen, upper arm, or anterior thigh); and (3) cover the ointment with plastic wrap. Avoid touching the ointment.
Instruct patients to rotate the application site to minimize local irritation.
CCBs relax peripheral arterioles and decrease afterload. Second, verapamil and diltiazem reduce heart rate and contractility (in addition to decreasing afterload).
■ CCBs relieve pain of variant angina by increasing cardiac oxygen supply. The mechanism is relaxation of coronary artery spasm.
■ When a CCB is combined with a beta blocker, a dihydro-pyridine (e.g., nifedipine) is preferred to verapamil or diltiazem. Verapamil and diltiazem will intensify cardiosup-pression caused by the beta blocker, whereas a dihydro-pyridine will not.
■ Ranolazine appears to reduce anginal pain by helping the heart generate energy more efficiently.
■ Ranolazine should not be used alone. Rather, it should be combined with a nitrate, a beta blocker, or the CCB amlodipine.
■ Ranolazine increases the QT interval and may pose a risk of torsades de pointes, a serious ventricular dysrhythmia.
■ In patients with chronic stable angina, treatment has two objectives: (1) prevention of MI and death and (2) preven-tion of anginal pain.
■ The risk of MI and death can be decreased with two types of drugs: (1) antiplatelet agents (e.g., aspirin, clopidogrel) and (2) cholesterol-lowering drugs.
■ Anginal pain is prevented with one or more long-acting antianginal drugs (beta blocker, CCB, long-acting nitrate) supplemented with sublingual nitroglycerin when break-through pain occurs.
■ As a rule, revascularization with CABG surgery or PCI is indicated only after treatment with two or three antianginal drugs has failed.
Please visit http://evolve.elsevier.com/Lehne for chapter-specific NCLEX® examination review questions.

603
CHAPTER 51 Drugs for Angina Pectoris
aPatient education information is highlighted as blue text.
IntravenousUses. (1) Angina pectoris refractory to more conventional
therapy, (2) perioperative control of blood pressure, (3) production of controlled hypotension during surgery, and (4) heart failure associated with acute MI.
Technique of Administration. Infuse IV using a glass IV bottle and the administration set provided by the manu-facturer; avoid standard IV tubing. Check the stock solution label to verify volume and concentration, which can differ among manufacturers. Dilute stock solutions before use.
Administer by continuous infusion. The rate is slow initially (5 mcg/min) and then gradually increased until an adequate response is achieved.
Monitor cardiovascular status constantly.
Terminating TherapyWarn patients against abrupt withdrawal of long-acting preparations (transdermal systems, topical ointment, sustained-release tablets and capsules).
Implementation: Measures to Enhance Therapeutic EffectsReducing Risk Factors
Precipitating Factors. Advise patients to avoid activities that are likely to elicit an anginal attack (e.g., overexertion, heavy meals, emotional stress, cold exposure).
Exercise. Encourage patients who have a sedentary lifestyle to establish a regular program of aerobic exercise (e.g., walking, jogging, swimming, biking).
Smoking Cessation. Strongly encourage patients to quit smoking.
Contributing Disease States. Ensure that patients with contributing pathology (especially hypertension or hyper-cholesterolemia) are receiving appropriate treatment.
Ongoing Evaluation and InterventionsEvaluating Therapeutic EffectsInstruct patients to keep a record of the frequency and intensity of anginal attacks, the location of anginal pain, and the factors that precipitate attacks.
Summary of Major Nursing Implicationsa—cont’d
Minimizing Adverse EffectsHeadache. Inform patients that headache will diminish
with continued drug use. Advise patients that headache can be relieved with aspirin, acetaminophen, or some other mild analgesic.
Orthostatic Hypotension. Inform patients about symp-toms of hypotension (e.g., dizziness, light-headedness), and advise them to sit or lie down if these occur. Inform patients that hypotension can be minimized by moving slowly when changing from a sitting or supine position to an upright posture.
Reflex Tachycardia. This reaction can be suppressed by concurrent treatment with a beta blocker, verapamil, or diltiazem.
Minimizing Adverse InteractionsHypotensive Agents, Including PDE5 Inhibitors. Nitro-
glycerin can interact with other hypotensive drugs to produce excessive lowering of blood pressure. Advise patients to avoid alcohol. Exercise caution when nitroglycerin is used in combination with beta blockers, calcium channel blockers, diuretics, and all other drugs that can lower blood pressure.
Warn patients not to combine nitroglycerin with a PDE5 inhibitor (e.g., sildenafil [Viagra]), because life-threatening hypotension can result.
ISOSORBIDE MONONITRATE AND ISOSORBIDE DINITRATEBoth drugs have pharmacologic actions identical to those of nitroglycerin. Differences relate only to dosage forms, routes of administration, and time course of action. Therefore, the implications presented for nitroglycerin apply to these drugs as well.

604
C H A P T E R
52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
COAGULATION: PHYSIOLOGY AND PATHOPHYSIOLOGY, p. 604
Hemostasis, p. 604Stage One: Formation of a Platelet Plug, p. 604Stage Two: Coagulation, p. 604Keeping Hemostasis Under Control, p. 605Physiologic Removal of Clots, p. 606
Thrombosis, p. 606Arterial Thrombosis, p. 606Venous Thrombosis, p. 606
OVERVIEW OF DRUGS FOR THROMBOEMBOLIC DISORDERS, p. 606
Anticoagulants, p. 606Heparin and Its Derivatives: Drugs That Activate
Antithrombin, p. 606Warfarin, a Vitamin K Antagonist, p. 612Direct Thrombin Inhibitors, p. 616Direct Factor Xa Inhibitors, p. 619Antithrombin, p. 620
Antiplatelet Drugs, p. 621Aspirin, p. 621P2Y12 Adenosine Diphosphate Receptor
Antagonists, p. 622Protease-Activated Receptor-1 (PAR-1)
Antagonists, p. 624Glycoprotein IIb/IIIa Receptor Antagonists,
p. 624Other Antiplatelet Drugs, p. 625
THROMBOLYTIC (FIBRINOLYTIC) DRUGS, p. 626Alteplase (tPA), p. 626Tenecteplase, p. 627Reteplase, p. 628
Key Points, p. 628Summary of Major Nursing Implications,
p. 629
COAGULATION: PHYSIOLOGY AND PATHOPHYSIOLOGY
HEMOSTASIS
Hemostasis is the physiologic process by which bleeding is stopped. Hemostasis occurs in two stages: (1) formation of a platelet plug, followed by (2) reinforcement of the platelet plug with fibrin. Both processes are set in motion by blood vessel injury.
Stage One: Formation of a Platelet PlugPlatelet aggregation is initiated when platelets come in contact with collagen on the exposed surface of a damaged blood vessel. In response to contact with collagen, platelets adhere to the site of vessel injury. Adhesion initiates platelet activation, which in turn leads to massive platelet aggregation.
Platelet aggregation is a complex process that ends with formation of fibrinogen bridges between glycoprotein IIb/IIIa (GP IIb/IIIa) receptors on adjacent platelets (Fig. 52.1). For these bridges to form, GP IIb/IIIa receptors must first undergo activation—that is, they must undergo a configurational change that allows them to bind with fibrinogen. As indicated in Fig. 52.1A, activation of GP IIb/IIIa can be stimulated by multiple factors, including thromboxane A2 (TXA2), thrombin, collagen, platelet activation factor, and ADP. Under the influence of these factors, GP IIb/IIIa changes its shape, binds with fibrinogen, and thereby causes aggregation (Fig. 52.1B). The aggregated platelets constitute a plug that stops bleeding. This plug is unstable, however, and must be reinforced with fibrin if protection is to last.
Stage Two: CoagulationCoagulation is defined as production of fibrin, a thread-like protein that reinforces the platelet plug. Fibrin is produced by two convergent pathways (Fig. 52.2), referred to as the contact activation pathway (also known as the intrinsic pathway) and the tissue factor pathway (also known as the extrinsic pathway). The two pathways converge at factor Xa, after which they employ the same final series of reactions. In both pathways, each reaction in the sequence amplifies the reaction that follows. Hence, once this sequence is initiated, it becomes self-sustaining and self-reinforcing.
The tissue factor pathway is turned on by trauma to the vascular wall, which triggers release of tissue factor,a also
The drugs discussed here are used to prevent formation of thrombi (intravascular blood clots) and to dissolve thrombi that have already formed. These drugs act in several ways: some suppress coagulation, some inhibit platelet aggregation, and some promote clot degradation. They all interfere with normal hemostasis. As a result, they all carry a significant risk of bleeding.
aFYI: The term tissue factor refers not to a single compound but rather to a complex of several compounds, including a proteolytic enzyme and phospho-lipids released from tissue membranes.
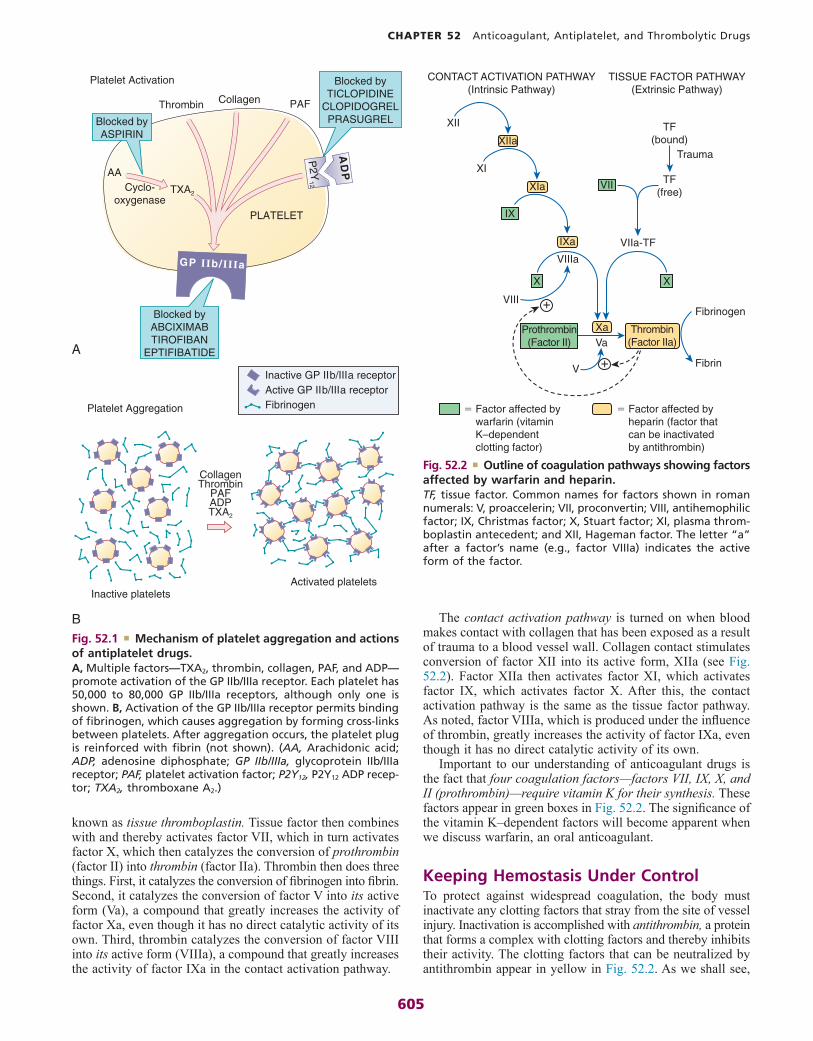
605
CHAPTER 52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
known as tissue thromboplastin. Tissue factor then combines with and thereby activates factor VII, which in turn activates factor X, which then catalyzes the conversion of prothrombin (factor II) into thrombin (factor IIa). Thrombin then does three things. First, it catalyzes the conversion of fibrinogen into fibrin. Second, it catalyzes the conversion of factor V into its active form (Va), a compound that greatly increases the activity of factor Xa, even though it has no direct catalytic activity of its own. Third, thrombin catalyzes the conversion of factor VIII into its active form (VIIIa), a compound that greatly increases the activity of factor IXa in the contact activation pathway.
Platelet Activation
Platelet Aggregation
AD
P
PAF
P2Y
12
CollagenThrombin
AA
TXA2
Inactive platelets
TXA2
Cyclo-oxygenase
Blocked byASPIRIN
Blocked byTICLOPIDINE
CLOPIDOGRELPRASUGREL
Blocked byABCIXIMABTIROFIBAN
EPTIFIBATIDE
Active GP IIb/IIIa receptorInactive GP IIb/IIIa receptor
Fibrinogen
PAFADP
Collagen
Activated platelets
Thrombin
PLATELET
A
B
GP IIb/IIIa
Fig. 52.1 ■ Mechanism of platelet aggregation and actions of antiplatelet drugs. A, Multiple factors—TXA2, thrombin, collagen, PAF, and ADP—promote activation of the GP IIb/IIIa receptor. Each platelet has 50,000 to 80,000 GP IIb/IIIa receptors, although only one is shown. B, Activation of the GP IIb/IIIa receptor permits binding of fibrinogen, which causes aggregation by forming cross-links between platelets. After aggregation occurs, the platelet plug is reinforced with fibrin (not shown). (AA, Arachidonic acid; ADP, adenosine diphosphate; GP IIb/IIIa, glycoprotein IIb/IIIa receptor; PAF, platelet activation factor; P2Y12, P2Y12 ADP recep-tor; TXA2, thromboxane A2.)
The contact activation pathway is turned on when blood makes contact with collagen that has been exposed as a result of trauma to a blood vessel wall. Collagen contact stimulates conversion of factor XII into its active form, XIIa (see Fig. 52.2). Factor XIIa then activates factor XI, which activates factor IX, which activates factor X. After this, the contact activation pathway is the same as the tissue factor pathway. As noted, factor VIIIa, which is produced under the influence of thrombin, greatly increases the activity of factor IXa, even though it has no direct catalytic activity of its own.
Important to our understanding of anticoagulant drugs is the fact that four coagulation factors—factors VII, IX, X, and II (prothrombin)—require vitamin K for their synthesis. These factors appear in green boxes in Fig. 52.2. The significance of the vitamin K–dependent factors will become apparent when we discuss warfarin, an oral anticoagulant.
Keeping Hemostasis Under ControlTo protect against widespread coagulation, the body must inactivate any clotting factors that stray from the site of vessel injury. Inactivation is accomplished with antithrombin, a protein that forms a complex with clotting factors and thereby inhibits their activity. The clotting factors that can be neutralized by antithrombin appear in yellow in Fig. 52.2. As we shall see,
CONTACT ACTIVATION PATHWAY(Intrinsic Pathway)
XIIa
Xa
Va
V
XII
XI
� Factor affected by heparin (factor that can be inactivated by antithrombin)
� Factor affected by warfarin (vitamin K–dependent clotting factor)
Trauma
TF(bound)
TF(free)
Prothrombin(Factor II)
Thrombin(Factor IIa)
VIIIa
VIII �
�
IX
VIIa-TF
VII
IXa
XIa
X X
TISSUE FACTOR PATHWAY(Extrinsic Pathway)
Fibrin
Fibrinogen
Fig. 52.2 ■ Outline of coagulation pathways showing factors affected by warfarin and heparin. TF, tissue factor. Common names for factors shown in roman numerals: V, proaccelerin; VII, proconvertin; VIII, antihemophilic factor; IX, Christmas factor; X, Stuart factor; XI, plasma throm-boplastin antecedent; and XII, Hageman factor. The letter “a” after a factor’s name (e.g., factor VIIIa) indicates the active form of the factor.

606
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
antithrombin is intimately involved in the action of heparin, an injectable anticoagulant drug.
Physiologic Removal of ClotsAs healing of an injured vessel proceeds, removal of the clot is eventually necessary. The body accomplishes this with plasmin, an enzyme that degrades the fibrin meshwork of the clot. Plasmin is produced through the activation of its precursor, plasminogen. The fibrinolytic drugs (e.g., alteplase) act by promoting conversion of plasminogen into plasmin.
THROMBOSISA thrombus is a blood clot formed within a blood vessel or within the heart. Thrombosis (thrombus formation) reflects pathologic functioning of hemostatic mechanisms.
Arterial ThrombosisFormation of an arterial thrombus begins with adhesion of platelets to the arterial wall. (Adhesion is stimulated by damage to the wall or rupture of an atherosclerotic plaque.) Following adhesion, platelets release ADP and TXA2, and thereby attract additional platelets to the evolving thrombus. With continued platelet aggregation, occlusion of the artery takes place. As blood flow comes to a stop, the coagulation cascade is initiated, causing the original plug to undergo reinforcement with fibrin.
The consequence of an arterial thrombus is localized tissue injury owing to lack of perfusion.
Venous ThrombosisVenous thrombi develop at sites where blood flow is slow. Stagnation of blood initiates the coagulation cascade, resulting in the production of fibrin, which enmeshes red blood cells and platelets to form the thrombus. The typical venous thrombus has a long tail that can break off to produce an embolus. Such emboli travel within the vascular system and become lodged at faraway sites, frequently the pulmonary arteries. Hence, unlike an arterial thrombus, whose harmful effects are localized, injury from a venous thrombus occurs secondary to embolization at a site distant from the original thrombus.
OVERVIEW OF DRUGS FOR THROMBOEMBOLIC DISORDERS
The drugs considered fall into three major groups: (1) anti-coagulants, (2) antiplatelet drugs, and (3) thrombolytic drugs, also known as fibrinolytic drugs. Anticoagulants (e.g., heparin, warfarin, dabigatran) disrupt the coagulation cascade and thereby suppress production of fibrin. Antiplatelet drugs (e.g., aspirin, clopidogrel) inhibit platelet aggregation. Thrombolytic drugs (e.g., alteplase) promote lysis of fibrin, causing dissolution of thrombi. Drugs that belong to these groups are shown in Table 52.1.
Although the anticoagulants and the antiplatelet drugs both suppress thrombosis, they do so by different mechanisms. As a result, they differ in their effects and applications. The antiplatelet drugs are most effective at preventing arterial thrombosis, whereas anticoagulants are most effective against venous thrombosis.
ANTICOAGULANTSBy definition, anticoagulants are drugs that reduce formation of fibrin. Two basic mechanisms are involved. One anticoagulant—warfarin—inhibits the synthesis of clotting factors, including factor X and thrombin. All other anticoagulants inhibit the activity of clotting factors: either factor Xa or thrombin, or both.
Traditionally, anticoagulants have been grouped into two major classes: oral anticoagulants and parenteral anticoagu-lants. This scheme was reasonable because, until recently, all oral anticoagulants belonged to just one pharmacologic class: the vitamin K antagonists, of which warfarin is the principal member. Today, however, anticoagulants in two other pharmacologic classes—direct factor Xa inhibitors and direct thrombin inhibitors—can also be administered by mouth (see Table 52.1). Hence, grouping the anticoagulants by route of administration makes less sense than in the past. Accordingly, these drugs are grouped only by pharmacologic class and not by whether they are given orally or by injection.
Heparin and Its Derivatives: Drugs That Activate AntithrombinAll drugs in this group share the same mechanism of action. Specifically, they greatly enhance the activity of antithrombin,
Prototype Drugs
ANTICOAGULANT, ANTIPLATELET, AND THROMBOLYTIC DRUGS
AnticoagulantsDrugs That Activate Antithrombin
Heparin (unfractionated)Enoxaparin (low-molecular-weight heparin)
Vitamin K Antagonist
Warfarin
Direct Thrombin Inhibitors
Dabigatran
Direct Factor Xa Inhibitors
RivaroxabanApixaban
Antiplatelet and Thrombolytic DrugsAntiplatelet Drugs
Aspirin (cyclooxygenase [COX] inhibitor)Clopidogrel (P2Y12 ADP receptor antagonist)Abciximab (glycoprotein IIb/IIIa receptor antagonist)
Thrombolytic Drugs
StreptokinaseAlteplase (tissue-type plasminogen activator)

607
CHAPTER 52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
more or less equally; the LMW heparins reduce the activity of factor Xa more than they reduce the activity of thrombin; and fondaparinux causes selective inhibition of factor Xa, having no effect on thrombin. Properties of the three preparations are shown in Table 52.2.
Heparin (Unfractionated)Heparin is a rapid-acting anticoagulant administered only by injection. Heparin differs from warfarin (an oral anticoagulant)
a protein that inactivates two major clotting factors: throm-bin and factor Xa. In the absence of thrombin and factor Xa, production of fibrin is reduced, and hence clotting is suppressed.
Our discussion focuses on three preparations: unfraction-ated heparin, the low-molecular-weight (LMW) heparins, and fondaparinux. Although all three activate antithrombin, they do not have equal effects on thrombin and factor Xa. Specifi-cally, heparin reduces the activity of thrombin and factor Xa
Generic Name Brand Name Route Action Therapeutic Use
ANTICOAGULANTS Anticoagulants decrease formation of fibrin
Used primarily to prevent thrombosis in veins and the atria of the heart
Vitamin K Antagonist
Warfarin Coumadin PO
Heparin and Its Derivatives: Drugs That Activate Antithrombin
Heparin (unfractionated) SubQ, IVLMW heparins Dalteparin Fragmin SubQ Enoxaparin Lovenox SubQFondaparinux Arixtra SubQ
Direct Thrombin Inhibitors
Hirudin Analogs
Bivalirudin Angiomax IVDesirudin Iprivask SubQ
Other Direct Thrombin Inhibitors
Argatroban Acova IVDabigatran Pradaxa, Pradax PO
Direct Factor Xa Inhibitors
Rivaroxaban Xarelto POApixaban Eliquis POEdoxaban Savaysa PO
Antithrombin (AT)
Recombinant human AT ATryn IVPlasma-derived AT Thrombate III IV
ANTIPLATELET DRUGS Antiplatelet drugs suppress platelet aggregation
Used primarily to prevent thrombosis in arteries
Cyclooxygenase Inhibitor
Aspirin PO
P2Y12 Adenosine Diphosphate Receptor Antagonists
Clopidogrel Plavix POPrasugrel Effient POTicagrelor Brilinta PO
Protease-Activated Receptor-1 (PAR-1) Antagonists
Vorapaxar Zontivity PO
Glycoprotein IIb/IIIa Receptor Antagonists
Abciximab ReoPro IVEptifibatide Integrilin IVTirofiban Aggrastat IV
Other Antiplatelet Drugs
Dipyridamole Persantine POCilostazol Pletal PO
THROMBOLYTIC (FIBRINOLYTIC) DRUGS Thrombolytic drugs promote breakdown of fibrin in thrombi
Used to dissolve newly formed thrombi
Alteplase Activase IVReteplase Retavase IVTenecteplase TNKase IV
TABLE 52.1 ■ Overview of Drugs for Thromboembolic Disorders
LMW, Low molecular weight.

608
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
in several respects, including mechanism, time course, indica-tions, and management of overdose.
Chemistry. Heparin is not a single molecule, but rather a mixture of long polysaccharide chains, with molecular weights that range from 3000 to 30,000. The active region is a unique pentasaccharide (five-sugar) sequence found randomly along the chain. An important feature of heparin’s structure is the presence of many negatively charged groups. Because of these negative charges, heparin is highly polar, and hence cannot readily cross membranes.
Mechanism of Anticoagulant Action. Heparin suppresses coagulation by helping antithrombin inactivate clotting factors, primarily thrombin and factor Xa. As shown in Fig. 52.3, binding of heparin to antithrombin produces a conformational change in antithrombin that greatly enhances its ability to inactivate both thrombin and factor Xa. However, the process of inactivating these two clotting factors is distinct. To inactivate thrombin, heparin must simultaneously bind with both thrombin and antithrombin, thereby forming a ternary complex. In contrast, to inactivate factor Xa, heparin binds only with antithrombin; heparin itself does not bind with factor Xa.
By activating antithrombin, and thereby promoting the inactivation of thrombin and factor Xa, heparin ultimately suppresses the formation of fibrin. Since fibrin forms the framework of thrombi in veins, heparin is especially useful for prophylaxis of venous thrombosis. Because thrombin and factor Xa are inhibited as soon as they bind with the heparin-antithrombin complex, the anticoagulant effects of heparin develop quickly (within minutes of IV administration). This contrasts with warfarin, whose full effects are not seen for days.
PharmacokineticsAbsorption and Distribution. Because of its polarity and
large size, heparin is unable to cross membranes, including those of the GI tract. Consequently, heparin cannot be absorbed if given orally and therefore must be given by injection (IV or subQ). Since it cannot cross membranes, heparin does not traverse the placenta and does not enter breast milk.
Protein and Tissue Binding. Heparin binds nonspecifically to plasma proteins, mononuclear cells, and endothelial cells. As a result, plasma levels of free heparin can be highly variable. Because of this variability, intensive monitoring is required (see later in this section).
Metabolism and Excretion. Heparin undergoes hepatic metabolism followed by renal excretion. Under normal condi-tions, the half-life is short (about 1.5 hours). However, in patients with hepatic or renal impairment, the half-life is increased.
Time Course. Therapy is sometimes initiated with a bolus IV injection, and effects begin immediately. Duration of action is brief (hours) and varies with dosage. Effects are prolonged in patients with hepatic or renal impairment.
Therapeutic Uses. Heparin is a preferred anticoagulant for use during pregnancy (because it doesn’t cross the placenta) and in situations that require rapid onset of anticoagulant effects, including pulmonary embolism (PE) and massive deep vein thrombosis (DVT). In addition, heparin is used for patients undergoing open heart surgery and renal dialysis; during these procedures, heparin serves to prevent coagulation in devices of extracorporeal circulation (heart-lung machines, dialyzers). Low-dose therapy is used to prevent postoperative venous thrombosis. Heparin may also be useful for treating disseminated intravascular coagulation, a complex disorder in which fibrin clots form throughout the vascular system and in which bleeding tendencies may be present; bleeding can occur because massive fibrin production consumes available supplies of clotting factors. Heparin is also used as an adjunct to thrombolytic therapy of acute myocardial infarction (MI).
Adverse EffectsHemorrhage. Bleeding develops in about 10% of patients
and is the principal complication of treatment. Hemorrhage can occur at any site and may be fatal. Patients should be monitored closely for signs of blood loss. These include reduced blood pressure, increased heart rate, bruises, petechiae, hema-tomas, red or black stools, cloudy or discolored urine, pelvic pain (suggesting ovarian hemorrhage), headache or faintness (suggesting cerebral hemorrhage), and lumbar pain (suggesting adrenal hemorrhage). If bleeding develops, heparin should be withdrawn. Severe overdose can be treated with protamine sulfate (see Protamine Sulfate for Heparin Overdose).
The risk of hemorrhage can be decreased in several ways. First, dosage should be carefully controlled so that the activated partial thromboplastin time or anti-factor Xa levels (see later in this chapter) do not exceed the recommended safe limits. In addition, candidates for heparin therapy should be screened
Property Unfractionated Heparin Low-Molecular-Weight Heparins Fondaparinux
Molecular weight range 3000–30,000 1000–9000 1728Mean molecular weight 12,000–15,000 4000–5000 1728Mechanism of action Activation of antithrombin,
resulting in the inactivation of factor Xa and thrombin
Activation of antithrombin, resulting in preferential inactivation of factor Xa, plus some inactivation of thrombin
Activation of antithrombin, resulting in selective inactivation of factor Xa
Routes IV, subQ SubQ only SubQ onlyNonspecific binding Widespread Minimal MinimalLaboratory monitoring aPTT monitoring is essential No aPTT monitoring required No aPTT monitoring requiredDosage Dosage must be adjusted on
the basis of aPTTDosage is fixed Dosage is fixed
Setting for use Hospital Hospital or home Hospital or home
TABLE 52.2 ■ Comparison of Drugs That Activate Antithrombin
aPTT, Activated partial thromboplastin time; LMW, low molecular weight.

609
CHAPTER 52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
for risk factors (see Warnings and Contraindications). Finally, antiplatelet drugs (e.g., aspirin, clopidogrel) should be avoided.
Spinal/Epidural Hematoma. Heparin and all other anti-coagulants pose a risk of spinal or epidural hematoma in patients undergoing spinal puncture or spinal/epidural anesthesia. Pressure on the spinal cord caused by the bleed can result in prolonged or permanent paralysis. Risk of hematoma is increased by:
• Use of an indwelling epidural catheter• Use of other anticoagulants (e.g., warfarin, dabigatran)• Use of antiplatelet drugs (e.g., aspirin, clopidogrel)• History of traumatic or repeated epidural or spinal
puncture• History of spinal deformity, spinal injury, or spinal surgery
Patients should be monitored for signs and symptoms of neurologic impairment. If impairment develops, immediate intervention is needed.
Heparin-Induced Thrombocytopenia. Heparin-induced thrombocytopenia (HIT) is a potentially fatal immune-mediated disorder characterized by reduced platelet counts (thrombo-cytopenia) and a seemingly paradoxical increase in thrombotic events. The underlying cause is development of antibodies against heparin–platelet protein complexes. These antibodies activate platelets and damage the vascular endothelium, thereby promoting both thrombosis and a rapid loss of circulating platelets. Thrombus formation poses a risk of DVT, PE, cerebral thrombosis, and MI. Ischemic injury secondary to thrombosis in the limbs may require amputation of an arm or leg. Coronary thrombosis can be fatal. The primary treatment for HIT is discontinuation of heparin and, if anticoagulation is still needed, substitution of a nonheparin anticoagulant (e.g., argatroban). The incidence of HIT is between 0.2% and 5% among patients who receive heparin for more than 4 days.
HIT should be suspected whenever platelet counts fall significantly or when thrombosis develops despite adequate anticoagulation. Accordingly, to reduce the risk of HIT, patients should be monitored for signs of thrombosis and for reductions in platelets. Platelet counts should be determined frequently (2 to 3 times a week) during the first 3 weeks of heparin use, and monthly thereafter. If severe thrombocytopenia develops (platelet count below 100,000/mm3), heparin should be discontinued.
Hypersensitivity Reactions. Because commercial heparin is extracted from animal tissues, these preparations may be contaminated with antigens that can promote allergy. Possible allergic responses include chills, fever, and urticaria. Anaphy-lactic reactions are rare.
Other Adverse Effects. Subcutaneous dosing may produce local irritation and hematoma. Vasospastic reactions that persist for several hours may develop after 1 or more weeks of treatment. Long-term, high-dose therapy may cause osteoporosis.
UNFRACTIONATEDHEPARIN
Pentasaccharidesequence
Thrombin
Antithrombin
Factor Xa
LMW HEPARIN
Pentasaccharidesequence
FONDAPARINUX
Pentasaccharidesequence
Fig. 52.3 ■ Mechanism of action of heparin, LMW heparins, and fondaparinux. All three drugs share a pentasaccharide sequence that allows them to bind with—and activate—antithrombin, a protein that inactivates two major clotting factors: thrombin and factor Xa. All three drugs enable antithrombin to inactivate factor Xa, but only heparin also facilitates inactivation of thrombin. Upper Panel: Unfractionated heparin binds with antithrombin, causing a conformational change in antithrombin that greatly increases its ability to interact with factor Xa and thrombin. When the heparin-antithrombin complex binds with thrombin, heparin changes its conformation so that both heparin and antithrombin come in contact with thrombin. Formation of this ternary complex is necessary for thrombin inactivation. Inactivation of factor Xa is different: It requires contact only between activated antithrombin and factor Xa; contact between heparin and factor Xa is unnecessary. Middle Panel: Low-molecular-weight (LMW) heparins have the same pentasaccharide sequence as unfraction-ated heparin and can bind with and activate antithrombin. However, in contrast to unfractionated heparin, most molecules of LMW heparin can inactivate only factor Xa. They are unable to inactivate thrombin because most molecules of LMW heparin are too small to form a ternary complex with thrombin and antithrombin. Lower Panel: Fondaparinux is a synthetic pentasac-charide identical in structure to the antithrombin binding sequence found in unfractionated heparin and LMW heparins. Being even smaller than LMW heparins, fondaparinux is too small to form a ternary complex with thrombin and can only inactivate factor Xa.
Safety Alert
ANTICOAGULANT, ANTIPLATELET, AND THROMBOLYTIC DRUGS
All the drugs discussed in this chapter increase the risk of patient bleeding. Careful assessment of mental status, blood pressure, heart rate, and mucous membranes should be com-pleted to assess for internal bleeding.

610
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
Warnings and ContraindicationsWarnings. Heparin must be used with extreme caution in
all patients who have a high likelihood of bleeding. Among these are individuals with hemophilia, increased capillary permeability, dissecting aneurysm, peptic ulcer disease, severe hypertension, or threatened abortion. Heparin must also be used cautiously in patients with severe disease of the liver or kidneys.
Contraindications. Heparin is contraindicated for patients with thrombocytopenia and uncontrollable bleeding. In addition, heparin should be avoided both during and immediately after surgery of the eye, brain, or spinal cord. Lumbar puncture and regional anesthesia are additional contraindications.
Drug Interactions. In heparin-treated patients, platelet aggregation is the major remaining defense against hemor-rhage. Aspirin and other drugs that depress platelet function or affect coagulation will weaken this defense, and hence must be employed with caution.
Protamine Sulfate for Heparin Overdose. Protamine sulfate is an antidote to severe heparin overdose. Protamine is a small protein that has multiple positively charged groups. These groups bond ionically with the negative groups on heparin, thereby forming a heparin-protamine complex that is devoid of anticoagulant activity. Neutralization of heparin occurs imme-diately and lasts for 2 hours, after which additional protamine may be needed. Protamine is administered by slow IV injection (no faster than 20 mg/min or 50 mg in 10 minutes). Dosage is based on the fact that 1 mg of protamine will inactivate 100 units of heparin. Hence, for each 100 units of heparin in the body, 1 mg of protamine should be injected.
Laboratory Monitoring. The objective of anticoagulant therapy is to reduce blood coagulability to a level that is low enough to prevent thrombosis but not so low as to promote spontaneous bleeding. Because heparin levels can be highly variable, achieving this goal is difficult and requires careful control of dosage based on frequent tests of coagulation. There are two tests used to monitor the effects of heparin.
Activated Partial Thromboplastin Time. The labora-tory test employed most commonly is the activated partial thromboplastin time (aPTT). The normal value for the aPTT is 40 seconds. At therapeutic levels, heparin increases the aPTT by a factor of 1.5 to 2, making the aPTT 60 to 80 seconds. Since heparin has a rapid onset and brief duration, if an aPTT value should fall outside the therapeutic range, coagulability can be quickly corrected through an adjustment in dosage: if the aPTT is too long (more than 80 seconds), the dosage should be lowered; conversely, if the aPTT is too short (less than 60 seconds), the dosage should be increased. Measurements of aPTTs should be made frequently (every 4 to 6 hours) during the initial phase of therapy. Once an effective dosage has been established, measuring aPTT once a day will suffice.
Anti-Factor Xa Heparin Assay. A newer way to monitor heparin levels is being used in many facilities. Unlike aPTT, the anti-Xa assay directly measures heparin and its activity. In addition, aPTT levels can be affected by other physiologic variables, such as high levels of factor VIII. Anti-Xa levels are not altered by these variables, making the test potentially more accurate. Antithrombin binding to Xa is increased in patients receiving heparin. The anti-Xa assay measures the amount of Xa activity and is, therefore, directly inversely proportional to heparin activity in the bloodstream. Much like aPTT, anti-factor Xa levels guide titration of IV heparin. Levels
of 0.3 to 0.7 IU/mL are considered within therapeutic range for anticoagulation with unfractionated heparin. One drawback of measuring anti-Xa levels is an increase in cost compared with monitoring aPTT.
Prescription and PreparationsPrescription. Heparin is prescribed in units, not in mil-
ligrams. The heparin unit is an index of anticoagulant activity. Heparin dosage is titrated on the basis of laboratory monitoring, and hence dosage can be adjusted as needed based on test results.
Preparations. Heparin sodium is supplied in single-dose vials; multidose vials; and unit-dose, preloaded syringes that have their own needles. Concentra-tions range from 1000 to 20,000 units/mL. Heparin sodium for use in heparin locks is supplied in dilute solutions (10 and 100 units/mL) that are too weak to produce systemic anticoagulant effects.
Dosage and AdministrationGeneral Considerations. Heparin is administered by
injection only. Two routes are employed: intravenous (either intermittent or continuous) and subcutaneous. Intramuscular injection causes hematoma and must not be done. Heparin is not administered orally because heparin is too large and too polar to permit intestinal absorption.
Dosage varies by indication. Postoperative prophylaxis of thrombosis, for example, requires relatively small doses. In other situations, such as open heart surgery, much larger doses are needed. The dosages given here are for “general antico-agulant therapy.” Because heparin is formulated in widely varying concentrations, you must read the label carefully to ensure that dosing is correct.
Continuous IV Infusion. Intravenous infusion provides steady levels of heparin. Dosing may consist of an initial weight-based bolus followed by a weight-based infusion titrated to laboratory results. Whether a bolus is indicated depends on the indication for treatment and the facility policy. During the initial phase of treatment, the aPTT or anti-Xa level should be measured once every 6 hours and the infusion rate adjusted accordingly. Heparin should be infused using an infusion pump, and the rate should be checked every 30 to 60 minutes.
Low-Dose Therapy. Heparin in low doses is given for prophylaxis against thromboembolism in hospitalized patients. Doses of 5000 units are given every 8 to 12 hours for the duration of hospitalization. Low-dose heparin is also employed as adjunctive therapy for patients with MI. During low-dose therapy, monitoring of the aPTT is not usually required.
Low-Molecular-Weight HeparinsGroup Properties. LMW heparins are simply heparin
preparations composed of molecules that are shorter than those found in unfractionated heparin. LMW heparins are as effective as unfractionated heparin and are easier to use because they can be given using a fixed dosage and don’t require aPTT monitoring. As a result, LMW heparins can be used at home, whereas unfractionated heparin must be given in a hospital when administering intravenously. Because of these advantages, LMW heparins are now considered first-line therapy for preven-tion and treatment of DVT. In the United States, two LMW heparins are available: enoxaparin [Lovenox] and dalteparin [Fragmin]. Differences between LMW heparins and unfraction-ated heparin are shown in Table 52.2.
Production. LMW heparins are made by depolymerizing unfractionated heparin (i.e., breaking unfractionated heparin into smaller pieces). Molecular weights in LMW preparations range between 1000 and 9000, with a mean of 4000 to 5000. In comparison, molecular weights in unfractionated heparin range between 3000 and 30,000, with a mean of 12,000 to 15,000.
Mechanism of Action. Anticoagulant activity of LMW heparin is mediated by the same active pentasaccharide sequence

611
CHAPTER 52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
available for anticoagulant effects. Half-lives are prolonged (up to 6 times longer than that of unfractionated heparin) because LMW heparins undergo less binding to macrophages, and hence undergo slower clearance by the liver. Because of increased bioavailability, plasma levels of LMW heparin are highly predictable. As a result, these drugs can be given using a fixed dosage, with no need for routine monitoring of coagulation. Because of their long half-lives, LMW heparins can be given just once or twice a day.
Administration, Dosing, and Monitoring. All LMW heparins are administered subQ. Dosage is sometimes based on body weight, depending on indication. Because plasma levels of LMW heparins are predictable for any given dose, these drugs can be employed using a fixed dosage without laboratory monitoring. This contrasts with unfractionated heparin, which requires dosage adjustments on the basis of aPTT measurements. Because LMW heparins have an extended half-life, dosing can be done once or twice daily. For prophylaxis of DVT, dosing is begun in the perioperative period and continued 5 to 10 days.
Adverse Effects and Interactions. Bleeding is the major adverse effect. However, the incidence of bleeding complications is less than with unfractionated heparin. Despite the potential for bleeding, LMW heparins are considered safe for outpatient use. Like unfractionated heparin, LMW heparins can cause immune-mediated thrombocytopenia. As with unfractionated heparin, overdose with LMW heparins can be treated with protamine sulfate.
Like unfractionated heparin, LMW heparins can cause severe neurologic injury, including permanent paralysis, when given to patients undergoing spinal puncture or spinal or epidural anesthesia. The risk of serious harm is increased by concurrent use of antiplatelet drugs (e.g., aspirin, clopidogrel) or antico-agulants (e.g., warfarin, dabigatran). Patients should be moni-tored closely for signs of neurologic impairment.
Individual Preparations. In the United States, two LMW heparins are available: enoxaparin and dalteparin. Additional LMW heparins are available in other countries. Each preparation is unique, so clinical experience with one may not apply fully to the other.
Enoxaparin. Enoxaparin [Lovenox] was the first LMW heparin available in the United States. The drug is prepared by depolymerization of unfractionated porcine heparin. Molecular weights range between 2000 and 8000.
Enoxaparin is approved for prevention of DVT following hip and knee replacement surgery or abdominal surgery in patients considered at high risk of thromboembolic complica-tions (e.g., obese patients, those over age 40, and those with malignancy or a history of DVT or pulmonary embolism [PE]). The drug is also approved for treatment of DVT or PE, and for preventing ischemic complications in patients with unstable angina, non–Q-wave MI, or STEMI.
In the event of overdose, hemorrhage can be controlled with protamine sulfate. The dosage is 1 mg of protamine sulfate for each milligram of enoxaparin administered.
ADMINISTRATION AND DOSAGE. Enoxaparin is administered by deep subQ injection. For patients with normal renal function (or moderate renal impairment), dosages are as follows:
• Prevention of DVT after hip or knee replacement surgery—30 mg every 12 hours, starting 12 to 24 hours after surgery and continuing 7 to 10 days.
• Prevention of DVT after abdominal surgery—40 mg once daily, beginning 2 hours before surgery and continuing 7 to 10 days.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Anticoagulants
Life Stage Patient Care Concerns
Infants Heparin is commonly used in infants needing anticoagulation. Argatroban has been used successfully in infants with HIT. Warfarin is also administered to infants.
Children/adolescents
Many anticoagulants can be used safely in children, just in smaller doses. Side effect profiles are similar to that of adults.
Pregnant women Warfarin is classified in FDA Pregnancy Risk Category Xa and is contraindicated in pregnancy. LMW heparins and unfractionated heparin are commonly used in pregnancy. In pregnant women with HIT, argatroban is a safe alternative.
Breast-feeding women
Data are lacking regarding safety of these medications in breast-feeding. Warfarin and heparin are both safe to use.
Older adults Atrial fibrillation becomes more common with age. In older adults, benefit must outweigh risk of bleeding secondary to falls, decreased renal function, or polypharmacy.
aAs of 2020, the FDA will no longer use Pregnancy Risk Categories. Please refer to Chapter 9 for more information.
that mediates anticoagulant action of unfractionated heparin. However, because LMW heparin molecules are short, they do not have quite the same effect as unfractionated heparin. Specifically, whereas unfractionated heparin is equally good at inactivating factor Xa and thrombin, LMW heparins preferen-tially inactivate factor Xa, being much less able to inactivate thrombin. Why the difference? To inactivate thrombin, a heparin chain must not only contain the pentasaccharide sequence that activates antithrombin, it must also be long enough to provide a binding site for thrombin. This binding site is necessary because inactivation of thrombin requires simultaneous binding of thrombin with heparin and antithrombin (see Fig. 52.3). In contrast to unfractionated heparin chains, most (but not all) LMW heparin chains are too short to allow thrombin binding, and hence LMW heparins are less able to inactivate thrombin.
Therapeutic Use. LMW heparins are approved for (1) prevention of DVT following abdominal surgery, hip replace-ment surgery, or knee replacement surgery; (2) treatment of established DVT, with or without PE; and (3) prevention of ischemic complications in patients with unstable angina, non–Q-wave MI, and ST-elevation MI (STEMI). In addition, these drugs have been used extensively off label to prevent DVT after general surgery and in patients with multiple trauma and acute spinal injury. When used for prophylaxis or treatment of DVT, LMW heparins are at least as effective as unfractionated heparin, and possibly more effective.
Pharmacokinetics. Compared with unfractionated heparin, LMW heparins have higher bioavailability and longer half-lives. Bioavailability is higher because LMW heparins do not undergo nonspecific binding to proteins and tissues, and hence are more

612
UNIT VII Drugs That Affect the Heart, Blood Vessels, and Blood
in 3% of patients. Platelet counts should be monitored, and if they fall below 100,000/mm3, fondaparinux should be discontinued.
In patients undergoing anesthesia using an epidural or spinal catheter, fondaparinux (as well as other anticoagulants) can cause spinal or epidural hematoma, which can result in permanent paralysis. However, in clinical trials, when fondaparinux was administered no sooner than 2 hours after catheter removal, no hematomas were reported.
Preparations, Dosage, and Administration. Fondaparinux [Arixtra] is available in single-dose, pre-filled syringes (2.5, 5, 7.5, and 10 mg). Dosing is done once a day by subQ injection.
For prevention of DVT, the recommended dosage is 2.5 mg once a day, starting 6 to 8 hours after surgery. The usual duration is 5 to 9 days.
For treatment of acute DVT or acute PE, dosage is based on body weight as follows: for patients under 50 kg, 5 mg once daily; for patients 50 to 100 kg, 7.5 mg once daily, and for patients over 100 kg, 10 mg once daily. The usual duration is 5 to 9 days.
Warfarin, a Vitamin K AntagonistWarfarin [Coumadin, Jantoven], a vitamin K antagonist, is our oldest oral anticoagulant. The drug is similar to heparin in some respects and quite different in others. Like heparin, warfarin is used to prevent thrombosis. In contrast to heparin, warfarin has a delayed onset, which makes it inappropriate for emergencies. However, because it doesn’t require injection, warfarin is well suited for long-term prophylaxis. Like heparin, warfarin carries a significant risk of hemorrhage, which is amplified by the many drug interactions to which warfarin is subject.
HistoryThe history of warfarin underscores its potential for harm. Warfarin was discovered after a farmer noticed that his cattle bled after eating spoiled clover silage. The causative agent was identified as bishydroxycoumarin (dicumarol). Research into derivatives of dicumarol led to the synthesis of warfarin. When warfarin was first developed, clinical use was ruled out, owing to concerns about hemorrhage. So, instead of becoming a medicine, warfarin was used to kill rats. The drug proved especially effective in this application and remains one of our most widely used rodenticides. Clinical interest in warfarin was renewed following the report of a failed suicide attempt using huge doses of a warfarin-based rat poison. The clinical trials triggered by that event soon demonstrated that warfarin could be employed safely to treat humans.
Mechanism of ActionWarfarin suppresses coagulation by decreasing production of four clotting factors, namely, factors VII, IX, X, and prothrom-bin. These factors are known as vitamin K–dependent clotting factors, because an active form of vitamin K is needed to make them. Warfarin works by inhibiting vitamin K epoxide reductase complex 1 (VKORC1), the enzyme needed to convert vitamin K to the required active form. Because of its mechanism, warfarin is referred to as a vitamin K antagonist, a term that is somewhat misleading because it implies antagonism of vitamin K actions, not antagonism of vitamin K activation. In therapeutic doses, warfarin reduces production of vitamin K–dependent clotting factors by 30% to 50%.
PharmacokineticsAbsorption, Distribution, and Elimination. Warfarin is
readily absorbed after oral dosing. Once in the blood, about 99% of warfarin binds to albumin. Warfarin molecules that remain free (unbound) can readily cross membranes, including those of the placenta and milk-producing glands. Warfarin is
• Treatment of established DVT—1 mg/kg every 12 hours for 7 days.• Patients with unstable angina or non–Q-wave MI—1 mg/kg every 12
hours (in conjunction with oral aspirin, 100 to 325 mg once daily) for 2 to 8 days.
• Patients with acute STEMI—30 mg/kg by IV bolus plus 1 mg/kg subQ, followed by 1 mg/kg subQ every 12 hours for up to 8 days.
For patients with severe renal impairment, dosage should be reduced.Dalteparin. Dalteparin [Fragmin] was the second LMW heparin approved
in the United States. The drug is prepared by depolymerization of porcine heparin. Molecular weights range between 2000 and 9000, the mean being 5000. Approved indications are prevention of DVT following hip replacement surgery or abdominal surgery in patients considered at high risk of thrombo-embolic complications, prevention of ischemic complications in patients with unstable angina or non–Q-wave MI, and management of symptomatic venous thromboembolism (VTE). Administration is by deep subQ injection. Dosages are as follows:
• Prevention of DVT after hip replacement surgery—2500 units 1 or 2 hours before surgery, 2500 units that evening (at least 6 hours after the first dose), and then 5000 units once daily for 5 to 10 days.
• Prevention of DVT after abdominal surgery—2500 units once daily for 5 to 10 days, starting 1 to 2 hours before surgery. In high-risk patients, this dose is increased to 5000 units once daily, starting the night before surgery.
• Patients with unstable angina or non–Q-wave MI—120 units/kg (but not more than 10,000 units total) every 12 hours for 5 to 8 days. Concurrent therapy with aspirin (75 to 165 mg/day) is required.
• Patients with symptomatic VTE—200 units/kg (but not more than 18,000 units total) once daily for 1 month, then 150 units/kg (but not more than 18,000 IU total) once daily for months 2 through 6.
Overdose is treated with 1 mg of protamine sulfate for every 100 units of dalteparin administered.
FondaparinuxActions. Fondaparinux [Arixtra] is a synthetic subQ anticoagulant that
enhances the activity of antithrombin to cause selective inhibition of factor Xa. The result is reduced production of thrombin, and hence reduced coagulation. Note that fondaparinux differs from the heparin preparations, which cause inactivation of thrombin as well as factor Xa.
Fondaparinux is closely related in structure and function to heparin and the LMW heparins. Structurally, fondaparinux is a pentasaccharide identical to the antithrombin-binding region of the heparins. Hence, like the heparins, fondaparinux is able to induce a conformational change in antithrombin, thereby increasing the activity of antithrombin—but only against factor Xa, not against thrombin. Why is fondaparinux selective for factor Xa? Because the drug is quite small—even smaller than the LMW heparins. As a result, it is too small to form a complex with both antithrombin and thrombin, and hence cannot reduce thrombin activity (see Fig. 52.3).
Fondaparinux has no effect on prothrombin time, aPTT, bleeding time, or platelet aggregation.
Therapeutic Use. Fondaparinux is approved for (1) preventing DVT following hip fracture surgery, hip replacement surgery, knee replacement surgery, or abdominal surgery; (2) treating acute PE (in conjunction with warfarin); and (3) treating acute DVT (in conjunction with warfarin). The drug is somewhat more effective than enoxaparin (an LMW heparin) at preventing DVT, but may cause slightly more bleeding. Anticoagulation may persist for 2 to 4 days after the last dose. Fondaparinux is administered using a fixed dosage, and does not require routine laboratory monitoring.
Pharmacokinetics. Fondaparinux is administered subQ. Bioavailability is 100%. Plasma levels peak 2 hours after dosing. The drug is eliminated by the kidneys with a half-life of 17 to 21 hours. The half-life is increased in patients with renal impairment.
Adverse Effects. As with other anticoagulants, bleeding is the biggest concern. The risk is increased by advancing age and renal impairment. Fondaparinux should be used with caution in patients with moderate renal impairment, defined as creatinine clearance (CrCl) of 30 to 50 mL/min, and avoided in patients with severe renal impairment, defined as CrCl below 30 mL/min. The drug should also be avoided for prophylactic use in patients weighing less than 50 kg because low body weight increases bleeding risk. Following surgery, at least 6 hours should elapse before starting fondaparinux. Aspirin and other drugs that interfere with hemostasis should be used with caution. In contrast to overdose with heparin or LMW heparins, overdose with fondaparinux cannot be treated with protamine sulfate.
Fondaparinux does not promote immune-mediated HIT, although it still can lower platelet counts. During clinical trials, thrombocytopenia developed
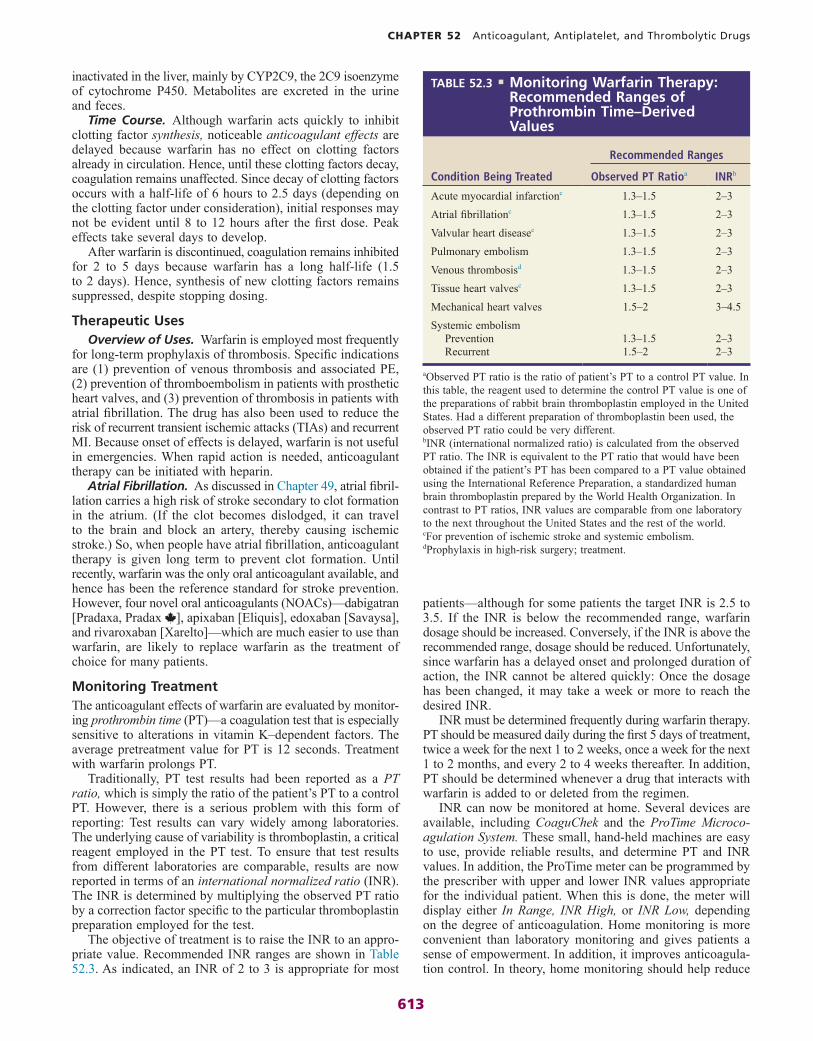
613
CHAPTER 52 Anticoagulant, Antiplatelet, and Thrombolytic Drugs
patients—although for some patients the target INR is 2.5 to 3.5. If the INR is below the recommended range, warfarin dosage should be increased. Conversely, if the INR is above the recommended range, dosage should be reduced. Unfortunately, since warfarin has a delayed onset and prolonged duration of action, the INR cannot be altered quickly: Once the dosage has been changed, it may take a week or more to reach the desired INR.
INR must be determined frequently during warfarin therapy. PT should be measured daily during the first 5 days of treatment, twice a week for the next 1 to 2 weeks, once a week for the next 1 to 2 months, and every 2 to 4 weeks thereafter. In addition, PT should be determined whenever a drug that interacts with warfarin is added to or deleted from the regimen.
INR can now be monitored at home. Several devices are available, including CoaguChek and the ProTime Microco-agulation System. These small, hand-held machines are easy to use, provide reliable results, and determine PT and INR values. In addition, the ProTime meter can be programmed by the prescriber with upper and lower INR values appropriate for the individual patient. When this is done, the meter will display either In Range, INR High, or INR Low, depending on the degree of anticoagulation. Home monitoring is more convenient than laboratory monitoring and gives patients a sense of empowerment. In addition, it improves anticoagula-tion control. In theory, home monitoring should help reduce
inactivated in the liver, mainly by CYP2C9, the 2C9 isoenzyme of cytochrome P450. Metabolites are excreted in the urine and feces.
Time Course. Although warfarin acts quickly to inhibit clotting factor synthesis, noticeable anticoagulant effects are delayed because warfarin has no effect on clotting factors already in circulation. Hence, until these clotting factors decay, coagulation remains unaffected. Since decay of clotting factors occurs with a half-life of 6 hours to 2.5 days (depending on the clotting factor under consideration), initial responses may not be evident until 8 to 12 hours after the first dose. Peak effects take several days to develop.
After warfarin is discontinued, coagulation remains inhibited for 2 to 5 days because warfarin has a long half-life (1.5 to 2 days). Hence, synthesis of new clotting factors remains suppressed, despite stopping dosing.
Therapeutic UsesOverview of Uses. Warfarin is employed most frequently
for long-term prophylaxis of thrombosis. Specific indications are (1) prevention of venous thrombosis and associated PE, (2) prevention of thromboembolism in patients with prosthetic heart valves, and (3) prevention of thrombosis in patients with atrial fibrillation. The drug has also been used to reduce the risk of recurrent transient ischemic attacks (TIAs) and recurrent MI. Because onset of effects is delayed, warfarin is not useful in emergencies. When rapid action is needed, anticoagulant therapy can be initiated with heparin.
Atrial Fibrillation. As discussed in Chapter 49, atrial fibril-lation carries a high risk of stroke secondary to clot formation in the atrium. (If the clot becomes dislodged, it can travel to the brain and block an artery, thereby causing ischemic stroke.) So, when people have atrial fibrillation, anticoagulant therapy is given long term to prevent clot formation. Until recently, warfarin was the only oral anticoagulant available, and hence has been the reference standard for stroke prevention. However, four novel oral anticoagulants (NOACs)—dabigatran [Pradaxa, Pradax ], apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]—which are much easier to use than warfarin, are likely to replace warfarin as the treatment of choice for many patients.
Monitoring TreatmentThe anticoagulant effects of warfarin are evaluated by monitor-ing prothrombin time (PT)—a coagulation test that is especially sensitive to alterations in vitamin K–dependent factors. The average pretreatment value for PT is 12 seconds. Treatment with warfarin prolongs PT.
Traditionally, PT test results had been reported as a PT ratio, which is simply the ratio of the patient’s PT to a control PT. However, there is a serious problem with this form of reporting: Test results can vary widely among laboratories. The underlying cause of variability is thromboplastin, a critical reagent employed in the PT test. To ensure that test results from different laboratories are comparable, results are now reported in terms of an international normalized ratio (INR). The INR is determined by multiplying the observed PT ratio by a correction factor specific to the particular thromboplastin preparation employed for the test.
The objective of treatment is to raise the INR to an appro-priate value. Recommended INR ranges are shown in Table 52.3. As indicated, an INR of 2 to 3 is appropriate for most
Condition Being Treated
Recommended Ranges
Observed PT Ratioa INRb
Acute myocardial infarctionc 1.3–1.5 2–3Atrial fibrillationc 1.3–1.5 2–3Valvular heart diseasec 1.3–1.5 2–3Pulmonary embolism 1.3–1.5 2–3Venous thrombosisd 1.3–1.5 2–3Tissue heart valvesc 1.3–1.5 2–3Mechanical heart valves 1.5–2 3–4.5Systemic embolism
Prevention 1.3–1.5 2–3Recurrent 1.5–2 2–3
TABLE 52.3 ■ Monitoring Warfarin Therapy: Recommended Ranges of Prothrombin Time–Derived Values
aObserved PT ratio is the ratio of patient’s PT to a control PT value. In this table, the reagent used to determine the control PT value is one of the preparations of rabbit brain thromboplastin employed in the United States. Had a different preparation of thromboplastin been used, the observed PT ratio could be very different.bINR (international normalized ratio) is calculated from the observed PT ratio. The INR is equivalent to the PT ratio that would have been obtained if the patient’s PT has been compared to a PT value obtained using the International Reference Preparation, a standardized human brain thromboplastin prepared by the World Health Organization. In contrast to PT ratios, INR values are comparable from one laboratory to the next throughout the United States and the rest of the world.cFor prevention of ischemic stroke and systemic embolism.dProphylaxis in high-risk surgery; treatment.




















