
Global Reaction Kinetics for Oxidation and Storage in Diesel ...
Upload
khangminh22Category
view
0download
0
KINETICS AND MECHANISM OF OXIDATION OF
4-OXOACIDS BY N-BROMO COMPOUNDS
A thesis submitted to the
Bharathidasan University, Tiruchirappalli
for the award of the degree of
DOCTOR OF PHILOSOPHY IN CHEMISTRY
By
A. AFROOS BANU, M.Sc.,M.Phil.,B.Ed., (Ref No.42426/Ph.D.1/Chemistry/Full-Time/January-2012)
Under the guidance of Dr. N.A.Mohamed Farook, M.Sc., M.Phil., P.G.D.E-Com., Ph.D.,
Research Advisor & Associate Professor P.G & Research Department of Chemistry
Khadir Mohideen College Adirampattinam-614 701
P.G. and Research Department of Chemistry
KHADIR MOHIDEEN COLLEGE
(Nationally Reaccredited with B by NAAC)
ADIRAMPATTINAM-614 701
INDIA
September - 2015

Declaration
This is to declare that the thesis entitled “Kinetics and Mechanism of
Oxidation of 4-Oxoacids by N-Bromo Compounds” submitted by
me to the Bharathidasan University, Tiruchirappalli for the award of
the degree of Doctor of Philosophy in Chemistry is a bonafide record
of the research work carried out by me under the guidance of
Dr. N.A.Mohamed Farook, Associate Professor, Khadir Mohideen
College, Adirampattinam. The contents of the thesis have not been
submitted and will not be submitted to any other University or
Institute for the award of any degree or diploma.
Signature of the Research Scholar
Adirampattinam
Date:
Forwarded
Research Advisor
(Dr. N.A.Mohamed Farook)

Phone No. 242236
KHADIR MOHIDEEN COLLEGE ADIRAMPATTINAM Thanjavur- District
Date___________
Dr. N. A. Mohamed Farook, Ph.D., Research Supervisor
CERTIFICATE This is to certify that the thesis entitled “KINETICS AND
MECHANISM OF OXIDATION OF 4–OXOACIDS BY N–
BROMO COMPOUNDS” submitted to the Bharathidasan University
for the award of the degree of DOCTOR OF PHILOSOPHY, is a
bonafide record of the research work carried out by A. AFROOS
BANU, M.Sc., M. Phil., B.Ed., (Ref No.42426/Ph.D.1/Chemistry/Full-
Time/January-2012) in the Department of Chemistry, Khadir
Mohideen College, Adirampattinam, during the period 2012– 2015
under my guidance and supervision. The work is original and has not
previously formed the basis for the award of any Diploma, Degree,
Associateship or Fellowship in this or any other university.
N.A.MOHAMED FAROOK

ACKNOWLEDGEMENT
I express my deep sense of gratitude and heartfelt thanks to my
research supervisor Dr. N. A. Mohamed Farook, Associate Professor
of Chemistry, Khadir Mohideen College, Adirampattinam for his
inspiring guidance, ingenious suggestions and valuable discussions. I
am very indebted to him for his constant encouragement.
I wish to record my respectful thanks to Dr. A. Jalal, Principal,
Khadir Mohideen College, Adirampattinam for his encouragement to
complete this work.
I express my heartfelt thanks to Dr. A. M. Uduman Mohideen,
Head of the Department of Chemistry, Khadir Mohideen College,
Adirampattinam for his kind gesture.
My sincere thanks are due to my learned colleagues and the non-
teaching staff of the department for their co-operation and support.
Finally, I want to place on record the sustained encouragement
offered by my husband Mr. G. Basul Khan, family members, friends
and students.
Above all, my thanks are due to the Almighty for the successful
completion of this work.
A. Afroos Banu

List of Abbreviations
NBA - N-Bromoacetamide
NBB - N-Bromobenzamide
NBBS - N-Bromobenzenesulphonamide
NBP -N-Bromophthalimide
NBS -N-Bromosuccinimide
NBSac -N-Bromosaccharin
NCSA -N-Chlorosaccharin
NCS -N-Chlorosuccinimide
NCA -N-Chloroacetamide
PTA -Phosphotungstic acid
KA -Keto acid
TA - Tartaric acid
MA - Malic acid
CAB -Chloramine - B
CAT -Chloramine - T
TMG - Trimethylene glycol
AcOH -Acetic Acid
CTAB - Cetyltrimethylammonium bromide
LFER - Linear Free Energy Relationship
r - Regression Coefficient

CONTENTS
Chapter 1 Page No.
Introduction 1.1 Chemical Kinetics 1
1.2 Significance of N-Halo compounds 2
1.3 Review of Literature 4
1.3.1 N-Halo compounds as oxidizing agents 4
1.3.2 N- Halo compounds in acid medium 5
1.3.3 Studies with N-Bromo Compounds
1.3.3.1 N-Bromophthalimide 6
1.3.3.2 N-Bromoacetamide 10
1.3.3.3 N-Bromobenzamide 13
1.3.3.4 N-Bromosaccharin 15
1.3.3.5 N-Bromosuccinimide 21
1.3.3.6 N-Bromobenzene-sulphonamide 25
1.3.3.7 N-Bromoanisamide 26
1.4 Glimpses of Oxoacids
1.4.1 General features 28
1.4.2 Reported methods of preparation of 4-oxoacids 29
1.4.3 Biological importance of oxoacids 31
1.5 Oxidation Studies with Oxoacids 32
1.6 Structure-Reactivity Relationships 36
1.7 Scope of the Present Investigation 39

Chapter 2
Experimental Methods
2.1 Materials 42
2.1.1 Preparation of N-Bromobenzamide 42
2.1.2 Chemicals 42
2.1.3 Preparation of 4-Oxoacids 42
2.1.3.1 4-Oxo-4-phenylbutanoic acid 43
2.1.3.2 4- Oxo-4-biphenylbutanoic acid 44
2.1.3.3 4- Oxo-4- (4’-bromophenyl)butanoic acid 44
2.1.3.4 4- Oxo-4-(3’-nitrophenyl)butanoic acid 44
2.1.4 Melting points of 4-oxoacids 44
2.1.5 Purification of solvents 45
2.2 Instrumentation 46
2.3 Methods 46
2.3.1 Oxidation of 4-oxoacids by NBB 46
2.3.1.1 Rate measurements 46
2.3.1.2 Product analysis 47
2.3.1.3 Stoichiometry 48
2.3.2 Oxidation of 4-oxoacids by NBSac 49
2.3.2.1 Rate measurements 49
2.3.2.2 Product analysis 49
2.3.2.3 Stoichiometry of oxidation 50

Chapter 3
Results and Discussion
4-Oxoacids and N-Bromobenzamide System
3.1 Structure of 4-oxoacid and N-Bromobenzamide 51
3.2 List of Substituted 4-oxoacids 51
3.3 Kinetics of oxidation of 4-oxo-4-phenylbutanoic acid
by N-bromobenzamide 52
3.3.1 Effect of varying [S1]0 53
3.3.2 Effect of varying [NBB]0 57
3.3.3 Effect of varying [H+]0 58
3.3.4 Comparison of rates in presence and
absence of H+ ion 61
3.3.5 Effect of varying ionic strength on reaction rate 62
3.3.6 Effect of added benzamide 64
3.3.7 Effect of added acrylonitrile 66
3.3.8 Effect of solvent polarity on reaction rate 67
3.3.9 Rate of enolization by bromination method 70
3.4 Studies with Substituted Oxoacids 71
3.5 Effect of Substituents and Applicability of LFER 83
3.6 Mechanism of Oxidation 88
3.7 Derivation of Rate Law 91
3.8 Structure-Reactivity Correlations 94
3.9 Activation Parameters 95
3.10 Isokinetic Relationship 99

Chapter 4
4-Oxoacids and N-Bromosaccharin System
4.1 Kinetics of Oxidation of 4-Oxo-4-Phenylbutanoic acid
by N-Bromosaccharin 104
4.1.1 Effect of varying [reactants]0 105
4.1.2 Effect of varying [H+] 109
4.1.3 Effect of varying ionic strength 112
4.1.4 Effect of added Saccharin 112
4.1.5 Effect of free-radical inhibitor 115
4.1.6 Effect of solvent composition 117
4.1.7 Rate of enolization of substrate 119
4.2 Studies with Substituted Oxoacids 119
4.3 Effect of Substituents and Applicability of LFER 129
4.4 Mechanism of Oxidation 135
4.5 Derivation of Rate Law 137
4.6 Structure-Reactivity Correlations 140
4.7 Activation Parameters 142
4.8 Isokinetic Relationship 145
4.9 Comparative Study of NBB / 4-Oxoacids and
NBSac / 4-Oxoacids Systems 148
SUMMARY 151
REFERENCES 155

CHAPTER 1
INTRODUCTION

1
CHAPTER 1
INTRODUCTION
1.1 Chemical Kinetics
Chemical kinetics, also known as reaction kinetics, is the study
of rates of chemical processes. Chemical kinetics includes
investigations of how different experimental conditions can influence
the speed of a chemical reaction and yield information about
the reaction's mechanism and transition states, as well as the
construction of mathematical models that can describe the
characteristics of a chemical reaction.
The processes of oxidation and reduction are common in
chemistry. The knowledge of oxidative path way may be very useful in
understanding the phenomena in nature and synthetic situations. The
oxidation processes are many, varied and are manifested in a variety of
net effects. The rate of chemical processes and their dependence on
different experimental parameters have been studied for many years.
These studies are useful for understanding the behavior of different
reaction mechanisms.
There is no limit to the number of possible organic reactions and
mechanisms1,2. However, certain general patterns are observed that
can be used to describe many common or useful reactions. Each
reaction has a stepwise reaction mechanism that explains how it

2
happens, although this detailed description of steps is not always clear
from a list of reactants alone. Organic reactions can be organized into
several basic types3-9. Some reactions fit into more than one category.
For example, some substitution reactions follow an addition-elimination
pathway. This overview isn't intended to include every single organic
reaction. Rather, it is intended to cover the basic reactions.
1.2 Significance of N-Halo Compounds
The N-haloamides or imides are generally named by putting the prefix,
e.g., N-bromo, before the name of the parent amide or imide. The
halogen when linked to oxygen or nitrogen acquires a positive oxidation
state. The electro-negativity of nitrogen is further enhanced by linking it to
a certain electron-withdrawing groups, e.g., acyl groups. Thus N-
Substituted haloimides are referred to as “positive halgen compounds”. A
large number of N-halo compounds have been prepared and tested as
reagents for allylic halogeneation and oxidation of organic compounds.
Some of the commonly used ones are listed it the table below10.
Some N-Halo imides and amides
Name and formula Active halogen %
N-Bromoacetamide (NBA)
CH3CONHBr 58
N-Bromobenzamide (NBB)
C6H5CONHBr 40
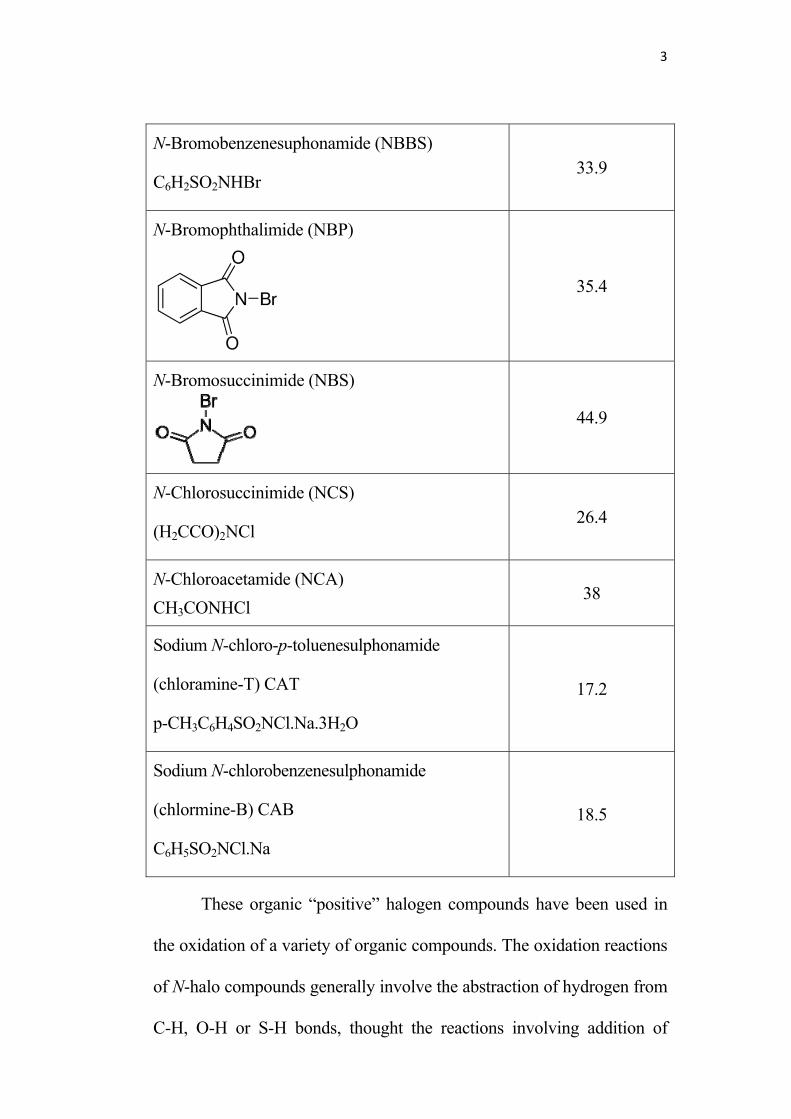
3
These organic “positive” halogen compounds have been used in
the oxidation of a variety of organic compounds. The oxidation reactions
of N-halo compounds generally involve the abstraction of hydrogen from
C-H, O-H or S-H bonds, thought the reactions involving addition of
N-Bromobenzenesuphonamide (NBBS)
C6H2SO2NHBr 33.9
N-Bromophthalimide (NBP)
35.4
N-Bromosuccinimide (NBS)
44.9
N-Chlorosuccinimide (NCS)
(H2CCO)2NCl 26.4
N-Chloroacetamide (NCA)
CH3CONHCl 38
Sodium N-chloro-p-toluenesulphonamide
(chloramine-T) CAT
p-CH3C6H4SO2NCl.Na.3H2O
17.2
Sodium N-chlorobenzenesulphonamide
(chlormine-B) CAB
C6H5SO2NCl.Na
18.5

4
oxygen have also been observed. These reactions have found extensive
applications in the estimation of a variety of organic compounds.
Though hypohalite solutions have frequently been used to
bring about oxidations, N-halo compounds have been found to
possess certain specific advantages as they are available in a high
state of purity, they can, therefore, be used as primary standards and
the solid reagents are fairly stable.
A variety of reactions conditions have been employed to affect
such oxidations and the ease of reactions and the selectivity is often
dependent on the solvent and pH of the medium. Under suitable
conditions, these N-halo compounds also react with defines to add
halogen to the double bond or act as a source of hypohalous acid in
aqueous solution. These compounds have been used successfully not
only as halogenating agents for oxidation and dehydrogenation.
1.3 Review of Literature
1.3.1 N-Halo Compounds as Oxidizing Agents
N-Halo compound forms a separate branch in chemistry, which is of
great synthetic importance11-48. N-Halo amides have been extensively
employed as oxidizing agents for organic substrates49-91. In the
recent development, N-halo amides are the sources of positive halogen
and have been exploited as oxidant for a variety of substrates in both
the acidic and alkaline media. The nature of active oxidizing species

5
and mechanism depends on the nature of the halogen atom, the groups
attached to the nitrogen and the reaction conditions.
The various N-halo compounds extensively used as reagents in
organic chemistry are N-bromophthalimide92,93, N-bromoacetamide94-113,
N-chloroacetamide114, N-chlorobenzenesulphonamide115, N-bromo-
benzenesulphonamide116-117, N-chlorobenzamide118-121, N-bromobenz-
amide122-125, N-chloro-p-toluensulphonamide126, N-chloronicotin-
amide127,128, N-chlorosuccinimide129-149, N-bromosaccharin150-158,
N-bromo-3,5-dinitrobenzamide159, N-chlorosaccharin160-175, N-bromo-
succinimide176-211 and N-bromoanisamide212,213.
1.3.2 N-Halo compounds in acid medium
It has been reported earlier in the case of N-halo compounds that in the
absence of mineral acids, HOX is the reactive oxidant species87,102. In
the oxidation with N-bromo compounds such as N-bromoacetamide,
“positive” bromine is the effective oxidant90,201. Further Mukerji and
Banerji96 have proposed HOBr as oxidizing species in the study of
oxidation of primary alcohol by N-bromoacetamide in the absence of
mineral acid.
The probable reactive species74 of N-halo amides in acid solution
are >NX, HOX, >N+HX or H2O
+X and the reactive species in alkaline
solution are >NX, HOX and OX- For example, in the case of N-bromo-
benzamide the actual reacting species in acid medium are as follows.

6
NBB + H2O HOBr + Benzamide (1)
HOBr + H+ H2O
+Br (2)
NBB + H+ NBBH
+ (3)
NBBH+ + H2O H2O
+Br + Benzamide (4)
The protonation of HOBr results in a hypobromous acidium ion
H2O+Br, a prime cationic and remote profile of the choice.
Some of the mechanisms that have been reported for the
oxidation of a number of organic substrates by N-bromo compounds
are outlined in the following pages to get a broad view on the subject.
1.3.3 Studies with N-Bromo Compounds
1.3.3.1 N-Bromophthalimide
The kinetics of oxidation of glycine by N-bromophthalimide (NBP)
were studied92a in the presence of an anionic surfactant, sodium
dodecyl sulfate, in acidic medium at 308 K. The rate of reaction was
found to have first-order dependence on [NBP] and fractional-order
dependence on [glycine] and [H+]. The addition of reduced product of
the oxidant had no significant effect on the rate of reaction. Increasing
[Hg(OAc)2] and [Br−] increased the rate of reaction, whereas a change
in ionic strength (μ) of the medium had no effect on oxidation velocity.
The rate of reaction decreased with a decrease in dielectric constant of
the medium. HCN was identified as the main oxidation product of the
reactions. The various activation parameters have been computed. A

7
suitable mechanism consistent with the experimental findings has been
proposed. The index of cooperativity and the micelle binding constant
have been calculated.
The kinetics and mechanism of the oxidation of lactose by N-
bromophthalimide in the absence and presence of cetyltrimethyl-
ammonium bromide and sodium dodecyl sulfate micelles was
investigated by Katre et al.92b in the presence of sulfuric acid medium.
Under pseudo-first-order conditions reaction rate agreed with a first-,
fractional- and negative fractional-order kinetics in N-
bromophthalimide, lactose and sulfuric acid, respectively. In the
presence of additives, the critical micellar concentration values were
lower than those given in the literature. The catalytic role of cationic
micelles was explained by the Berezin model. The anionic micelles
showed slightly inhibitory effect. The influence of salts, phthalimide and
mercuric acetate on the reaction rate was also studied. Using the kinetic
data, the rate constant, binding constants, and corresponding activation
parameters were evaluated. A possible reaction mechanism, which is
based on the kinetic results and the product analysis, is proposed.
Kinetics of oxidation of aniline by N-bromophthalimide (NBP) in
acetonitrile-water solvent mixture at 303 K in the presence of perchloric
acid has been studied92c. The reaction is first order with respect to both
aniline and NBP and is catalyzed by H+ ion and the order of the reaction

8
with respect to [H+] is also one. It has been found that the reaction rate is
not affected by changes in ionic strength of the reaction medium or by
the addition of acrylonitrile and potassium bromide. However, addition
of phthalimide causes a decrease in the rate of reaction. An increase in
the water content of the solvent mixture decreases the rate of reaction.
Thermodynamic and activation parameters have also been evaluated.
NBr
O
O
H+ H2OK1
H2O+BrNH
O
O
H2O+Br
NH2 NH+
HBr H2O
NH2NH+
NH HN
+
slow
k
+ +
+ + +
+fast
H++
N N 2H++NH HNoxidation
The rate law was given as
Rate = [NHP]
]ine][H[NBP][Anil1kK
This rate law explains the first-order dependence of the reaction on
[aniline], [NBP] and [H+] and the retarding effect of phthalimide.
kobs = [NHP]
]H[Aniline][1kK
k2 = [NHP]
][H1kK
(5)
(6)
(7)
(8)
(9)
(10)
(11)

9
Kinetic investigations in Keggin-type phosphotungstic acid
catalyzed oxidation of benzhydrol and p-substituted benzhydrols by N-
bromophthalimide (NBP) in aqueous acetic acid medium in presence of
mercuric(II) acetate as a scavenger have been studied by Jagdish et
al.,92d. In absence of mineral acids, the oxidation kinetics of benzhydrols
by NBP in presence of PTA (Phosphotungstic acid) shows a first order
dependence on NBP and fractional order on benzhydrols and PTA. The
variation of ionic strength, Hg(OAC)2, H+ and phthalimide (reaction
product) have insignificant effect on reaction rate. Activation parameters
for the reaction have been evaluated from Arrhenius plot by studying the
reaction at different temperature. A mechanism involving transfer of
hydride ion in rate determining step is suggested.
The kinetics of oxidation of some α-hydroxy acids viz. Tartaric acid
(TA) and Malic acid (MA) by N-bromophthalimide (NBP) were studied
by Sangeeta et al.92f in the presence of a cationic surfactant,
cetyltrimethylammonium bromide (CTAB), in perchloric acid medium at
313 K. The oxidation of TA and MA by N-bromophthalimide in the
presence of CTAB is faster than in the absence of surfactant. The rate of
oxidation of hydroxy acids was found to be in the order: TA > MA. First
order kinetics with respect to NBP was observed in the oxidation of both
hydroxy acids. The kinetics results indicate that the first order kinetics in
hydroxy acids at lower concentrations tends towards a zero order at its

10
higher concentrations. Inverse fractional order in [H+] and [phthalimide]
were noted throughout its tenfold variation. With a progressive increase
in [CTAB], the rate of reaction increased, reaches a maximum value and
then constancy in k Ψ was observed. Variation of [Hg(OAc)2] and ionic
strength (μ) of the medium did not bring about any significant change in
the rate of reaction. The applicability of different kinetic models viz. the
Piszkiewicz cooperative model, the Raghvan and Srinivasan model, and
the Menger–Portnoy model were tested to explain the observed micellar
effects. The effect of [CTAB] on the activation parameters was explored
to rationalize the micellar effect. The values of rate constants observed at
four different temperatures were utilized to calculate the activation
parameters. A suitable mechanism consistent with the experimental
findings has been proposed. The index of cooperativity and the micelle
binding constant have been calculated.
1.3.3.2 N-Bromoacetamide
The kinetics of oxidation of 3-benzoylpropionic acid (KA) with N-
bromoacetamide (NBA) have been studied by Farook et al.94a
potentiometrically in 50:50 (v/v) aqueous acetic acid medium at 298 K
The reaction was first order each with respect to [KA], [NBA] and
[H+]. The main product of the oxidation is the corresponding
carboxylic acid. The rate decreases with the addition of acetamide, one
of the products of the reaction. Variation in ionic strength of the

11
reaction medium has no significant effect on the rate of oxidation. But
the rate of the reaction is enhanced by lowering the dielectric constant
of the reaction medium. A mechanism consistent with observed results
have been proposed and the related rate law was deduced.
The oxidation of 2-ketoglutaric acid in the presence of N-
bromoacetamide have been studied by singh et al.94b in alkaline
medium in temperature range 30-40 oC shows first order kinetics with
respect to N-bromoacetamide (NBA) and zero order kinetics with
respect to 2-ketoglutaric acid. Hydroxide ions variations show negative
effect while acetamide and sodium perchlorate additions show
insignificant effect on oxidation rate. Addition of mercuric acetate
(used as Br- scavenger) increases the rate which shows that probably
Hg(II) acts as catalyst. NBA as such is the reactive species. Products
identified are oxalic and malonic acids. Various activation parameters
have been calculated and recorded on the basis of the experimental
findings, and a suitable mechanism has been proposed.
The kinetics of oxidation of the sugars d(+)Melibiose (mel) and
Cellobiose (cel) by N-bromoacetamide (NBA) in the presence of
Rh(III) chloride as homogeneous catalyst in acidic medium at 45o C
have been investigated by Srivastava et al.95a. The reactions are first-
order with respect to [NBA], [Rh(III)] and [substrate]. The rate is
proportional to [H+]. No effects of [Hg(II)], [NHA] or [Cl-] on the rates

12
were observed. Ionic strength and dielectric constant also have little
effect. The observed kinetic data, available literature and spectroscopic
evidence lead us to conclude that NBAH+ and [RhCl5(H2O)]2- are the
reactive species of NBA and Rh(III) chloride, respectively. The rate-
determining step of the proposed reaction path common for both sugars
gives an activated complex by the interaction of a charged complex
species and neutral sugar molecule, which in the subsequent steps
disproportionates into the reaction products with the regeneration of
catalyst. The reactions have been studied at four different temperatures
and with the help of first-order rate constant values, various activation
parameters have been calculated. The main oxidation products of the
reactions were identified as arabinonic acid, formic acid and lyxonic
acid in the case of mel and arabinonic acid and formic acid in the
case of cel.
The kinetic oxidation of trimethylene glycol (TMG) with Os(VIII)
in alkaline N-bromoacetamide (NBA) in the presence of mercuric
acetate as Br- ions scavenger has been studied by Singh et al.99a. The
reaction is first order in NBA, Os(VIII) and OH- while zero order
dependence of the reaction on trimethylene glycol was observed. The
rate of reaction was independent on addition of acetamide and sodium
perchlorate. A solvent isotope effect (K-0(D2O)/K-0(H2O)=2.3-2.7 and
2.4-2.8 for trimethylene glycol) has been observed at 35 oC. Various

13
thermodynamic parameters have been computed and the corresponding
trimethylene glycol was found to be product. A mechanism consistent
with the kinetic data has been proposed.
1.3.3.3 N-Bromobenzamide
Kinetics Studies of the oxidation of ethanol by p-methoxy-N-
bromobenzamide in aqueous acetic acid medium in the presence of
mercuric acetate have been investigated by Badole et al.122c. The reaction
was first order with respect to both, the oxidant and near about are with
respect to the substrates. The order with respect to perchloric acid was
fractional or first order depending upon the substrate concentration. The
reaction was retarded by the initial addition of benzamide and was
enhanced by the added potassium bromide. The activation parameters
have been calculated and a suitable mechanism has been proposed.
Kinetics of oxidation of ethylene glycol (EG) by NBB (N-
bromobenzamide) has been studied122d in aq. HClO4 with PdCl2 as
catalyst and in the presence of Hg (OAc)2 to ensure oxidation by pure
NBB. The order of reaction with respect to NBB was unity. However
the rate decreased with the increasing concentration of [NBB]0. The
rate was directly proportional to Pd(II) for EG. The retarding effects of
HClO4 benzamide, Cl- and AcOH on the rate of oxidation were
observed. A mechanism consistent with the observed kinetic data in
proposed.

14
The kinetics of the oxidation of twelve ortho-substituted
benzaldehydes by N-bromobenzamide (NBB) to the corresponding
benzoic acids have been studied123c. The reaction was first order with
respect to NBB, the aldehyde and hydrogen ions. The addition of
benzamide has no effect on the reaction rate. (PhCONH2Br)+ has been
postulated as the reactive oxidising species. The correlation of rates with
the single substituent-parameter equations is poor. The correlation with
Charton’s equation of inductive, resonance and steric parameters was
satisfactory. However, excellent correlations were obtained, when
Charton’s steric parameter was used along with Taft’s σ1; and σR+
substituent constants. The polar reaction constants have negative values.
The reaction is subject to steric hindrance by the ortho-substituents.
The oxidation of six aliphatic aldehydes by N-bromobenzamide
(NBB) in 1:1 (v/v) acetic acid-water leads to the formation of the
corresponding carboxylic acids have been investigated by Banerji
et al.,123d. The reaction was first order with respect to both NBB and
aldehyde and was catalysed by hydrogen ions. The observed hydrogen
ion dependence indicates that both NBB and its protonated form are
reactive oxidizing species. The oxidation of MeCDO exhibits a
substantial kinetic isotope effect. With an increase in the proportion of
acetic acid in the solvent mixture of acetic acid and water, the rate
decreases. Addition of benzamide has no effect on the rate. The

15
reaction fails to induce the polymerization of acrylonitrile. The role of
aldehyde hydrate in the oxidation process is discussed. The rates
correlate well with Taft's sigma substituent constants, with negative
reaction constant. A mechanism involving transfer of a hydride ion to
the oxidant in the rate determining step has been proposed.
1.3.3.4 N-Bromosaccharin
N-Bromosaccharin (NBSac) is a strong oxidizing and chlorinating
agent. It is a white powder, easy to handle, with its melting point at 160-
170 °C. It is soluble in organic solvents, for example in acetic acid,
alcohols, acetonitrile, tetrachloromethane, ethyl acetate, trichloro-
methane, acetone, and 1,4-dioxane. N-Bromosaccharin has been
proven to be a useful and alternative reagent for diverse organic
transformations, such as halogenation of aromatic compounds, co-
halogenation of alkenes, oxidation of alcohols, halogenation of
benzylic and carbonylic positions, etc. N-Bromosaccharin150a can
easily be prepared by bromination of the sodium salt of saccharin
which is commonly available, non-corrosive, and non-toxic.
The kinetics of bromination of some substituted 4-piperidones
and 4-selenanones by N-bromosaccharin in the presence of perchloric
acid in aqueous acetic acid have been investigated150b. The bromination
is first order in both substrate and H3O+ and zero order in NBSA.
A plausible mechanism based on these observations is proposed.

16
The effects of the various substituents on the rates of bromination have
been rationalized on the basis of their inductive and steric effects. The
effect of solvent polarity on the rate has also been studied.
N-Bromo and N-iodosaccharins in the presence of triphenyl-
phosphine convert alcohols into the corresponding bromides and
iodides in good to excellent yields at room temperature under neutral
conditions151a.
ROH RX (12)
X = Br.I
Chemoselective oxidation of thiols to their corresponding
disulfides with N-bromosaccharin in a very short time and with high
yields in dichloromathane under microwave irradiation conditions are
described by Khazaei et al.151c.
Mechanism 1
NXSac/PPh3
CH2Cl2 / r.t.
(13)
(14)
(15)
(16)

17
Mechanism 2
N-Chloro- and N-bromosaccharins react with electron rich
aromatic compounds (anisole, acetanilide, N,N-dimethylaniline)
producing halogenated compounds152a. The reaction with N-bromo-
saccharin gives para- substituted compounds only, whereas N-chloro-
saccharin produces ortho and para mixtures (para isomer
predominantly, ca. 4-5 : 1). The reactions of the N-halosaccharins with
alkenes (cyclohexene, styrene, α-methylstyrene, and 1-hexene) give the
corresponding halohydrins.
A new method for the direct conversion of various oximes into
aldehydes and ketones by treatment with N-bromosaccharin is
described153a. N-Bromosaccharin was used for an effective, selective
and mild oxidizing agent for the regeneration of carbonyl compounds
from oximes in good yield.
The kinetics of oxidation of acetophenone and substituted
acetophenones by N-bromosaccharin has been investigated158 in
aqueous acetic acid medium in the temperature range 308–323 K. The
reaction is found to be first order with respect to acetophenone and
(17)
(18)

18
zero order with respect to the reaction does not induce polymerisation
of added acrylonitrile. A positive catalytic effect has been noticed on
the addition of A-cyclodextrin. Thermodynamic parameters such as
ΔS#, ΔH# and ΔG# have been evaluated and presented.
The oxidation of -hydroxy acids by N-bromosaccharin has been
studied by Vijaya Mohan et al.158b. On the basis of observed first order
with respect to [substrate], [oxidant] and [H+] and the inverse effect of
[saccharin] on the reaction rate, they proposed a mechanism involving
protonated hydroxyl acid and HOBr in the rate-determining step.
The kinetics of oxidation of benzaldehyde by N-bromosaccharin
and N-bromophthalimide has been investigated158c in aqueous acetic acid
medium in the presence of mercuric acetate over a temperature range of
308-328 K. The rate was first order with respect to both the substrate and
oxidants. The stoichiometry of the process is 1 : 1, substrate : oxidant.
(19)
(20)
(21)
(22)

19
The effect of ionic strength on the rate was negligible, but the dielectric
constant of the medium has a positive influence. The thermodynamic
parameters were calculated. Saccharin and H+ were found to inhibit the
rate, whereas, phthalimide did not inhibit the rate.
The kinetics of oxidation of acetophenone and substituted
acetophenones by N-bromosaccharin has been investigated158d in
aqueous acetic acid medium in the temperature range 308–323 K. The
reaction is found to be first order with respect to acetophenone and
zero order with respect to the reaction does not induce polymerisation
of added acrylonitrile. A positive catalytic effect has been noticed on
the addition of -cyclodextrin. Thermodynamic parameters such as
ΔS#, ΔH#, and ΔG# have been evaluated and presented.
Kinetics of oxidation of some aldoses viz. D- ribose, D-xylose,
L– arabinose and D- glucose by N-bromosaccharin in aqueous acetic
medium have been studied158e in the presence of mercuric acetate as a
scavenger for Br-, exhibits first order dependence in [NBSA] and
[HC1O4]. The order with respect to aldose varies from 1 to 0. The
reaction rate is retarded by the addition of saccharin. Effect of variation
of composition of acetic acid-water binary mixture was also studied.
Various activation parameters have been computed. These results
points to a polar mechanism involving the formation of hypobromite

20
ester in pre- equilibrium step which disproportionates into products via
rate limiting attack of water molecule.
The effect of sodium lauryl sulphate on the oxidation of glycolic
and tactic acid by N-bromosaccharin in aqueous-acetic acid in the
presence of Hg(II) acetate has been investigated158f. The reactions are
first order with respect to oxidant in the presence as well as in absence
of sodium lauryl sulphate (NaLS). The Michaelis-Menten kinetics is
observed in the substrate. The reactions exhibit complex kinetics in H+.
Change in polarity of the medium, effect of addition of saccharin and
Hg(II) acetate have also been investigated, Sodium lauryl sulphate
exhibits an inhibition effect. These effects are discussed on the basis of
interactions of hydroxy acids with the micelle. Binding parameters
have been calculated by analyzing the data using the model suggested
by Piszkiewicz. Influence of surfactant on the activation parameters of
the reaction has also been discussed.
The rates of oxidation of benzilic acid by N-bromosaccharin were
measured158g in aqueous acetic acid medium in the absence and in
presence of cationic surfactant, cetyl trimethylammonium bromide
(CTAB) and anionic surfactant, sodium lauryl sulphate (NaLS). Kinetic
observations indicate first order in [NBSA] and fractional to [BA]. The
reaction rate is retarded in the presence of perchloric acid and by addition
of the reaction product, saccharin. Addition of NaClO4 and Hg(II) acetate

21
has no effect. The rates were also found to be sensitive to solvent
polarity. Formation of an intermediate complex between NBSA and BA
in pre-equilibrium step and subsequent decomposition in a slow step has
been proposed as probable mechanism. The inhibitory effect of CTAB
and NaLS is analyzed on the basis of Piszkiewicz model.
1.3.3.5 N-Bromosuccinimide
Dipeptides (DP), namely valyl–glycine (Val–Gly), alanyl–proline (Ala–
Pro), and valyl–proline (Val–Pro) were synthesized by classical solution
phase methods and characterized. The kinetics of oxidation of amino acids
(AA) and DP by N-bromosuccinimide (NBS) was studied by Gowda et
al.188a in the presence of perchlorate ions in acidic medium at 28°C. The
reaction was followed spectrophoto-metrically at λmax = 240 nm. The
reactions follow identical kinetics, being first order each in [NBS], [AA],
and [DP]. No effect on [H+], reduction product [succinimide], and ionic
strength was observed. Effects of varying dielectric constant of the
medium and addition of anions such as chloride and perchlorate were
studied. Activation parameters have been computed. The oxidation
products of the reaction were isolated and characterized. The proposed
mechanism is consistent with the experimental results. An apparent
correlation was noted between the rate of oxidation of AA and DP.
The kinetics of oxidation of gabapentin (GBP) by N-
bromosuccinimide (NBS) in an alkaline medium has been investigated

22
by Alaa Eldin et al.,189a. The oxidation reaction showed unique kinetics
that greatly differed on going from acid to base medium. In an acid
medium (pH=2.52), the reaction rate showed first order dependence on
[NBS], fractional order dependence on both [GBP] and [H+] and
increased with temperature over (303–321oK) range. In an alkaline
medium, the rate showed first order dependence on [GBP], fractional
order on [H+] over (1.99-39.80) x10-9 range and zero order dependence
on [NBS]. It is noteworthy that the reaction rate decreased with
temperature over the range studied. An inner- sphere mechanism for the
oxidation pathway supported by free radicals intervention was proposed.
Kinetics and mechanism of micellar catalyzed N-
bromosuccinimide oxidation of dextrose in H2SO4 medium was
investigated by Minu Singh195a under pseudo-first-order condition
temperature of 40 °C. The results of the reactions studied over a wide
range of experimental conditions show that NBS shows a first order
dependence, fractional order, on dextrose and negative fractional order
dependence on sulfuric acid. The determined stoichiometric ratio was
1 : 1 (dextrose : N-bromosuccinimide). The variation of Hg(OAC)2 and
succinimide (reaction product) has insignificant effect on reaction rate.
Effects of surfactants, added acrylonitrile, added salts, and solvent
composition variation have been studied. The Arrhenius activation
energy and other thermodynamic activation parameters are evaluated.

23
The rate law has been derived on the basis of obtained data. A plausible
mechanism has been proposed from the results of kinetic studies,
reaction stoichiometry, and product analysis. The role of anionic and
nonionic micelle was best explained by the Berezin’s model.
The oxidation of diazepam (DZ) by N-Bromosuccinimide (NBS)
have been studied by Nanda et al.,196a in aqueous acid medium follows a
first-order kinetics in [NBS] and a fractional-order each on [HCl] and
[DZ]. The reaction stoichiometry involves one mol NBS consumed by
one mol DZ. The rate of the reaction increases with the decrease in
dielectric constant of the medium. Added products and the variation of
ionic strength have no significant effect on the rate of the reaction. The
oxidation products were identified by spectral analysis. A mechanism
involving the formation of an intermediate NBS-DZ complex has been
proposed. The solvent effect is consistent with the charge dispersion
going into the transition state. The activation parameters for the
reaction have been determined. The negative entropy of activation
suggests the formation of a rigid, associative transition state involving
loss of degrees of freedom.
Kinetics of oxidation of glycine (gly) and valine (val) by N-
bromosuccinimide (NBS) using chloro complex of Rh(III) in its nano-
concentration range as homogeneous catalyst have been investigated by
Singh et al.,197a at 35 oC . The reaction shows first order kinetics with

24
respect to NBS and Rh(III) in the oxidation of both the amino acids. The
first order kinetics with respect to amino acid obtained at its lower
concentration changes to zero order at its higher concentration. Inverse
fractional order with respect to [H+] was obtained in Rh(III)-catalysed
oxidation of gly and val. Variation in [Hg(II)], [NHS], [Cl-], ionic
strength and dielectric constant of the medium has no effect on the rate of
oxidation of both the amino acids. NBS itself and [RhCl5(H2O)]2- have
been postulated as the reactive species of NBS and Rh(III) chloride in
acidic medium, respectively. Various activation parameters have been
calculated with the pseudo-first-order rate constant values observed at
four different temperatures. The main oxidation products of the reactions
have been identified as formaldehyde and ammonia in the case of gly
and isobutaldehyde and ammonia in the case of val. The proposed
reaction mechanism is well supported by kinetic data, spectrophoto-
metric evidence and positive entropy of activation.
A kinetic study of oxidation of 2-phenylethylamine (PEA), a
bioactive compound, with potent oxidant, N-bromosuccinimide (NBS)
has been investigated by Mohana et al.198a in HCl and NaOH media at
313 K. The experimental rate laws obtained are:
-d[NBS] = k[NBS][PEA][H+] dt
in hydrochloric acid medium and
-d[NBS] = k[NBS][PEA]x [OH-]y dt
(23)
(24)

25
in alkaline medium where x and y are less than unity. Accelerating effect
of [Cl-], and retardation of the added succinimide on the reaction rate
have been observed in acid medium. Variation of ionic strength of the
medium shows negligible effect on rate of reaction in both media.
Decrease in dielectric permittivity of the medium decreased the rate in
both media. The stoichiometry of the reaction was found to be 1:1 in acid
medium and 1:2 in the case of alkaline medium. The oxidation products
of PEA were identified as the corresponding aldehyde and nitrile in acid
and alkaline medium, respectively. The reactions were studied at different
temperatures and the activation parameters have been evaluated. The
reaction constants involved in the proposed mechanisms were computed.
The reaction was found to be faster in alkaline medium in comparison
with the acid medium, which is attributed to the involvement of different
oxidizing species. The proposed mechanisms and the derived rate laws
are consistent with the observed experimental results.
1.3.3.6 N-Bromobenzene-sulphonamide
The kinetics of oxidation of 2-propanol, 2-butanol, 2-pentanol, 2-hexanol
and 2-heptanol to the respective ketones by sodium N-bromobenzene-
sulphonamide (bromamine-B) in presence of HCl was studied by Mohan
et al.116a at 40°C. The rate shows a first-order dependence on both
[oxidant]0 and [alcohol]0 and is fractional in [H+] and [Cl−]. The proposed
mechanism assumes the formation of a hypobromite in the rate-limiting

26
step followed by a fast reaction to form products. The magnitude of the
solvent isotope effect, k′H2O/k′D2O is 0–90. The rates do not correlate
satisfactorily with Taft’s substituent constants. An isokinetic relation is
observed with β=331 K indicating enthalpy as a controlling factor.
Kinetics of oxidation of cysteine in the presence of H2SO4 and
HClO4 by sodium N-bromobenzene sulfonamide (Bromamine-B or BAB)
has been investigated by Rangaswamy et al.117a at 30 °C. The reactions
follow identical kinetics, and obeys the rate law, rate = k [BAB] [S]
[H+]x- where x is less than unity. Addtion of [SO42-] and [ClO4-] in the
form of Na2SO4 and NaClO4 had no effect on the reaction rate. The
reaction product, benzene sulfonamide had no effect on the reaction rate.
Variation of ionic strength and dielectric constant of the medium on the
rate of reaction has been studied. Thermodynamic parameters have been
evaluated by studying the kinetics at various temperatures. The
protonation constant of monobromamine-B is found to be 36.50 in H2SO4
medium and 65.06 in HClO4 medium. Suitable mechanism has been
proposed in consistency with the kinetic results.
1.3.3.7 N-Bromoanisamide
The kinetics of the oxidation of the mandelic by N-bromoanisamide has
been studied by Siriah et al211. in 40% acetic acid medium in the
presence HC1O4 and of [Hg(OAc)2]. The reactions exhibit a first order
rate dependence with respect to oxidant and fractional order with respect

27
to substrate. The reaction rate decreases slightly with increasing the
concentration of [H+] and retarded by the addition of anisamide, (as one
of the oxidation product of oxidant). The decrease in dielectric constant
of the medium decreases rate of the reaction. Increase in ionic strength,
by the addition of sodium perchlorate has no effect on the rate constant.
The effect of temperature on the reaction has been investigated in the
temperature range 308-323 K. The activation parameters were
calculated and a possible operative mechanism was proposed.
From the mechanisms, the following rate equation was derived.
The kinetics of the oxidation of the malic and by N-bromo-
anisamide in HC1O4 and in the presence of Hg(OAc)2 have been studied
by Malviya et al.212. The reactions exhibit a first order rate dependence
with respect to the oxidant and substrate. The reactions are acid catalyzed
and retarded by the addition of anisamide, a byproduct of reaction. The
rate of oxidation decreases with decrease in dielectric constant of the
(25)
(26)
(27)
(29)
(28)

28
medium. The effect of temperature on the reaction has been investigated
in the temperature range 313-328 K. The stoichiometric studies revealed
1:1 mole ratio. Various thermodynamic parameters have been computed
and a possible operative mechanism is proposed.
1.4 Glimpses of Oxoacids
1.4.1 General features
In 2-oxoacids (R-CO-COOH), due to the interaction of the pi electron
clouds of the carbonyl and carboxyl groups in the 2 and 1 position
respectively, each group influences the characteristics of the other
group. The studies of the 2-oxoacids are found widely in the
literature214-217. For instance, 2-oxopropionic acid commonly called
pyruvic acid (CH3-CO-COOH) is involved in the biochemical
processes like respiration. Among 3-oxoacids, the well known is the
ester of 3-oxobutanoic acid, namely acetoacetic ester which is most
useful in the synthesis of various organic compounds218.
In 4-oxoacids, the carbonyl and the carboxyl groups are
separated by two carbon atoms and so they possess the characteristics
of both compounds without the direct influence of the other group.
However, intramolecular catalysis (carboxylic acid group can catalyze
the reactions of oxo group) has been reported in the halogenation of
4-oxoacids219. Among the 4-oxoacids, the reaction of levulinic acid
(CH3-CO-CH2-CH2-COOH) has been studied extensively220,221.

29
1.4.2 Reported methods of preparation of 4-oxoacids
The preparation of 4-oxo-4-phenylbutanoic acid, commonly known as β-
benzoylpropionic acid, by the Friedel-Craft’s reaction between benzene
and succinic anhydride in the presence of anhydrous aluminium chloride
was reported222 as early as 1882. Haworth synthesized naphthalene from
4-oxo-4-phenylbutanoic acid.223 The Friedel-Craft’s reaction of succinic
anhydride with toluene, xylenes, mesitylene and a number of
alkylated benzenes were reported to give the corresponding phenyl
substituted 4-oxo-4-phenylbutanoic acids224,225. Phenanthrene
derivatives226,227 were obtained from 4-oxo-4-phenylbutanoic acids
which were synthesized by the condensation between succinic
anhydride and naphthalene derivatives. Tetralin, p-cymene,
phenanthrene and anthracene were reacted with succinic anhydride
to get the corresponding 4-oxoacids228-230. 4-Oxo-4-(4’-bromo-
phenyl)butanoic acid was prepared from bromobenzene and succinic
anhydride under drastic conditions231. The structure of the oxoacid
was established by heating it with alkaline potassium permanganate
and identifying the resulting 4-bromobenzoic acid. 4-Oxo-4-(3’-nitro-
phenyl)butanoic acid was prepared232 by the nitration of 4-oxo-4-
phenylbutanoic acid. Aromatic ethers like anisole, phenetole, p-
methylanisole, o-methylanisole, p-chloroanisole and veratrole were
condensed with succinic anhydride to get the corresponding 4-oxoacids233.

30
Methyl ethers of dihydric phenols were also condensed with succinic
anhydride234. The Friedel-Craft’s succinolylation of 1,2-dichlorobenzene
yielded 4-oxo-4-(3’,4’-dichlorophenyl)butanoic acid235.
The following mechanism has been proposed for the
condensation between succinic anhydride and aromatic compounds in
the presence of anhydrous aluminium chloride236. For the complete
reaction, one molecule of succinic anhydride (I) requires two
molecules of anhydrous aluminium chloride. It is suggested that one
molecule of aluminium chloride is used to open the anhydride ring
with the formation of the carboxylic acid salt from one half of the
anhydride group and the other half is converted into a carbonyl
chloride group (II). This reacts in the usual manner with the second
molecule of aluminium chloride to give the complex (III) which then
reacts with the nucleophilic component to form the 4-oxoacid (VI) via
the intermediates IV and V.
I II III
IV V
(30)
(31)

31
VI
4-Oxo-4-phenylbutanoic acid (4-oxoacid)
1.4.3 Biological importance of oxoacids
Many of the 4-oxoacids and their esters possess fungicidal,
antibacterial, microbial237 and anti-inflammatory activities238.
For example, 4-oxo-4-phenylbutanoic acid is involved in human
metabolism239. The 4-oxoacids are also used for protecting the
hydroxyl functions in nucleosides. The esters and salts of the 4-
oxoacids are also utilized widely in the industrial preparation of insect
repellents and plastics240.
The 4-oxoacids are very useful in the synthesis of several
carboxylic acid and heterocyclic compounds. For instance241, the
bromo oxoacids are used for preparing imidazothiazoles and
pyridazinones. They are the starting compounds in the preparation of
β-benzoylacrylic esters.
Structure-activity studies242 of 3-benzoylpropionic acid
derivatives establish the fact that these acids possess immunodulative
activity and suppress adjuvant arthritis.
3-Benzoylpropionic acid (4-oxoacid) and its derivatives play an
important role in the pharmaceutical chemistry243-251. 3-Benzoyl-
(32)

32
propionic acid derivatives are used as antirheumatic agents243. A study
with three types of 3-benzoylpropionic acid derivatives having a
mercapto moiety in their structures shows that substitution on the
phenyl ring contributes to the antirheumatic activity.
1.5 Oxidation Studies with Oxoacids
Although a lot of work has been reported on the -ketoesters, hydroxyl
acids, aldehyde and aromatic ketones, a very little work has been
reported so far on the oxidation of oxoacids252-260.
Kinetics of oxidation of 4-oxoacids by permanganate in buffer
media have been reported253. Oxidation of 4-oxo-4-phenylbutanoic
acid and its phenyl substituted compounds by permanganate in
different buffer media is first order each in [oxoacid] and [MnO4-].
The reactions undergo general acid catalysis. Addition of
electrolytes has no significant effect on the reaction rate. Electron
releasing substituents in aromatic ring enhance the reaction rates,
while electron withdrawing substituents retard the rate. The
reaction constant is – 1.08 at 303 KThe oxidation products have
been identified and activation parameters are computed. A mechanism
consistent with the kinetic results has been proposed.
The oxidation of substituted and unsubstituted 4-oxoacids by
alkaline hexacyanoferrate(III) in sodium carbonate-bicarbonate buffer

33
has been studied254. The reaction is zero order in oxidant, first order
with respect to Os(VIII), first order at higher concentrations with
respect to both substrate and alkali. The reaction products are identified
as benzoic acid and malonic acid by comparing the Rf values with that
of the authentic samples. The presence of electron releasing groups in
the benzene ring decreases the rate of oxidation and the presence of
electron withdrawing groups enhances the rate. The observed rate
constants increase with temperature for all the compounds. The
reaction constants are positive and decrease with increasing
temperature. Based on the kinetic results and observations, an
oxidation mechanism is formulated as given in eqs. (33) – (39).
(33)
(34)
(35)
I

34
+ H2O (36)
C1 C2
(37) C3
(38)
(39)
Kinetics of oxidation of unsubstituted and substituted
oxoacids by acid permanganate in aqueous acetic acid medium have
been studied255 at high and low [H3O+]. At high [H3O
+], the reaction is
first order each in [H3O+] and the [oxoacid]. Variation in ionic strength
of the reaction medium has no significant effect on the rate of the
reaction. But the rate of the reaction is enhanced by lowering the
dielectric constant of the reaction medium. Electron releasing
substituents in the aromatic ring accelerate the reaction rates and
electron withdrawing substituents retard them. The value of the at
303 K (at [H3O+] = 1 M) obtained from the Hammett’s plot is -1.49. A
mechanism involving the attack of permanganic acid on the enol form
of the substrate in the rate determining step has been proposed. The
protonation of permanganate ion leads to the formation of permanganic
acid. The enolization is proposed to be the necessary step prior to the
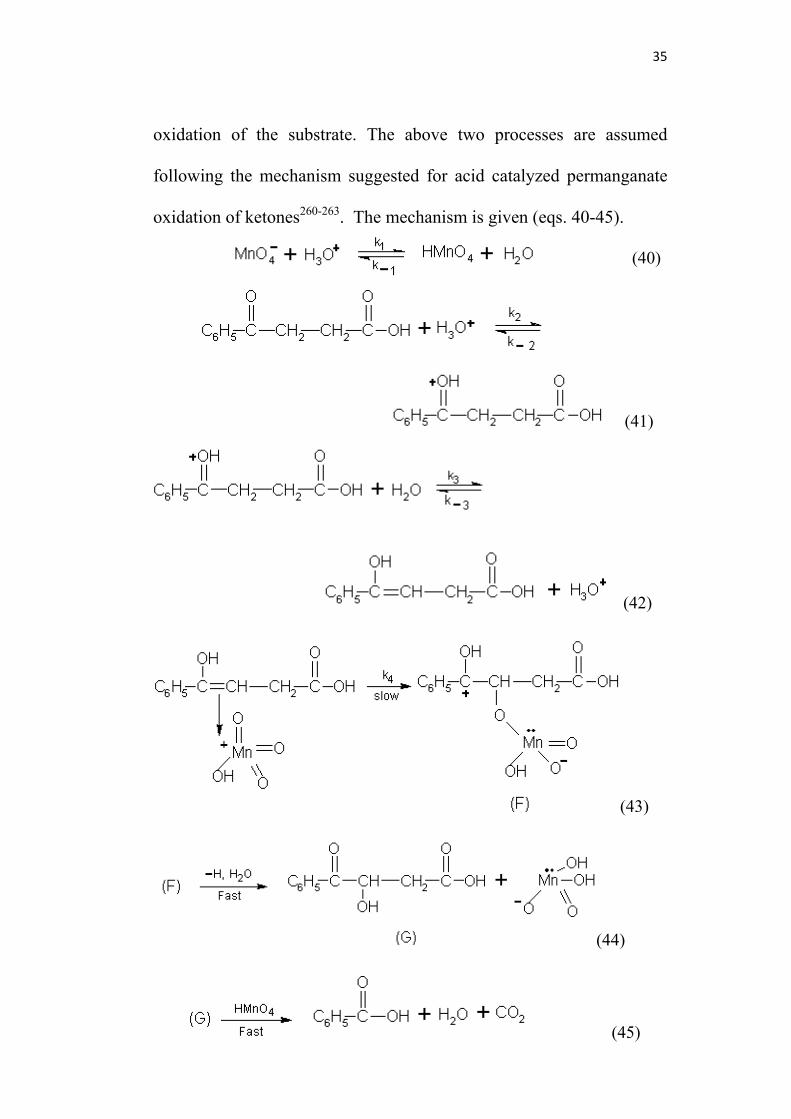
35
oxidation of the substrate. The above two processes are assumed
following the mechanism suggested for acid catalyzed permanganate
oxidation of ketones260-263. The mechanism is given (eqs. 40-45).
(40)
(41)
(42)
(43)
(44)
(45)

36
The kinetics of oxidative decarboxylation of benzoylpropionic
acid by manganese(III) acetate in aqueous acetic acid medium have been
studied259. The reaction is first order each in [substrate], [oxidant] and
[H+] ion. The effects of solvent polarity and temperature on the rate of
oxidation have been studied. A suitable mechanism consistent with the
experimental results has been proposed.
1.6 Structure-Reactivity Relationships
For justifying and generalizing a particular reaction mechanism for
similar reactions, almost all the kinetic studies invoke structure-
reactivity relationships which depend on the empirical and qualitative
rule that like substances react similarly and that similar change in
structure produce similar changes in reactivity264. The most successful
quantitative correlation between structure and reactivity is given by the
Hammett equation265, a linear free- energy relationship.
log k = log k0 +
loglog K0
where k or K is the rate or equilibrium constant respectively, for a side-
chain reaction of meta- or para- substituted benzene derivatives. The k0
or K0 denotes the corresponding quantity for the parent compound. The
substituents constant is independent of the nature of the reaction and
gives a measure of the polar effect of replacing H by a given substituents

37
(in the m- or p- position). The reaction constant depends on the nature
of the reaction and its conditions (reagent, catalyst or temperature) and
is independent of substituents. For evaluating for a given substituents
the ionization of benzoic acid in water at 25o C is chosen as the standard
process for which was arbitrarily defined as 1.00.
Hammett equation is applied to a given reaction by getting a best
straight line, by the method of least squares, between log k or log K and
and the slope of that line gives the value of the reaction constant .
The success of the Hammett equation is commonly assessed266-270 in
terms of correlation coefficient (r) and the standard deviation (s).
To account for the failure of Hammett equation in reactions
where the conjugation involving substituents and reaction center is
substantially more marked than in the ionization of benzoic acids
(cross-conjugation)271,272 ‘exalted’ constants (and) are
introduced. The values are used for +R substituents (e.g. NO2, CN,
COOH, COOMe, and SO2Me) and are based on the ionization of
anilinium ions or of phenols in water. The values, introduced by
Brown and Okamato273 based on the solvolysis of t-cumyl chloride in
90% acetone-water at 250C, are used for –R substituents (e.g. OMe,
Me, OH, NH2, SMe, and Hal ). The differences (and
(give a measure of conjugative ability of a given acceptor and

38
donor respectively. Both and
values are sometimes used in the
same correlation for a reaction in which an electron-deficient /
electron-rich reaction centre can directly conjugate with electron-
donating / electron-withdrawing substituents.
On the view that the contribution of the resonance effect of a
substituents must vary continuously as the electron-demanding quality
of the reaction centre is varied, Wepster274, 275 introduced a ‘sliding
scale’ of (unexalted) values, called n. Taft276 also evaluated similar
set of unexalted constants, called , on the basis of ionization of
phenylacetic and phenylpropionic acids.
To deal with the influence of –R and +R substituents respectively
on reactions that are more or less electron-demanding than the ionization
of benzoic acid, Yukawa and Tsuno277 and Yasioka278 formulated the
following eqs. (48) and (49) known as Yukawa-Tsuno equations.
log k = log k0 + r
R
log K = log K0 + r
R
where
R = and
R =
r ±
gives a measure of the
extent to which cross-conjugation of substituents with reaction centers
stabilizes the transition state or product relative to the initial state. The
r+ in eq. (48) can have values varying from 0 to unity and values
greater than one is also possible for r- in eq. (49).

39
A quantitative separation of substituents effect into inductive
and resonance contributions by Taft279, 280 led to the possibility
of a ‘dual substituents parameters’ (DSP) treatment of reaction
series, where simple correlations based on Hammett equation fail,
in the form of eq. (50)
log ( k / k0 ) = R (50)
where and R are the inductive and resonance substituent constants,
and I and R are the corresponding reaction constants.
1.7 Scope of the Present Investigation
A thorough literature survey reveals that only few works on the
oxidation of 4-oxoacids have been reported so far252-259. Although the
N-bromo compounds oxidation of a large variety of organic
compounds have been studied, there seems to be no report on a
systematic kinetic study of the oxidation of 4-oxoacids by N-
bromobenzamide and N-bromosaccharin.
The present investigation employs N-bromo compounds such as
N-bromobenzamide and N-bromosaccharin as oxidants, perchloric acid
as catalyst and 4-oxoacids as substrates. The choice of this system may
be rationalized on the following aspects.

40
Of the many efficient oxidation systems known, those involving
positive halogen species as an oxidant are among the most numerous
and well studied. The 4-oxoacid has much biological significance
associated with it and plays an essential role in the pharmaceutical
chemistry. Few examples are; the 4-oxoacids and its derivatives act as
antirheumatic agents for human being242. It plays an important role in
suppressing adjuvant arthritis243.
Mutations in components of the extraordinarily large -ketoacid
dehydrogenase multienzyme complexes can lead to serious and often
fatal disorders in humans, including maple syrup urine disease246.
In view of these facts, 4-oxoacid is a suitable substrate to be
employed in the oxidation studies. This study makes interesting and
useful findings in elucidating the mechanism of the 4-oxoacid
oxidation process.
A detailed study of the oxidation of 4-oxoacids by N-bromo
compounds is undertaken with a view to propose the mechanism for
the oxidation. The experiments have been focused to explore the
following aspects:
i) To determine the order of the reaction with respect to the
reactants namely, the oxoacids and N-bromo compounds.

41
ii) To determine the catalytic activity of hydrogen ions in the
oxidation.
iii) To determine whether the reaction is general acid
catalyzed.
iv) To determine the rate of enolization by bromination
method.
v) To determine the effect of dielectric constant of the
reaction medium on the rate of reaction.
vi) To determine the effect of ionic strength on the reaction
rate.
vii) To determine whether the reaction involves the formation
of polar (or) free radical intermediates.
viii) To determine the stoichiometry of the reaction and to
study the product analysis.
ix) To determine the effect of substituents on the reaction
rate and to apply the linear free energy relationship.
x) To determine the reaction constant and isokinetic
temperature.
xi) To determine the activation parameters for the oxidation
and finally and
xii) To propose a suitable mechanism.

CHAPTER 2
EXPERIMENTAL

42
CHAPTER 2
Experimental Methods 2.1 Materials
2.1.1 Preparation of N-Bromobenzamide281
690 mg of solid sodium bromide (6.7mmol) was slowly added to a stirred
solution of benzamide (1.21g, 10 mmol), sodium bromate (760 mg, 5
mmol), and conc. H2SO4 (740 mg, 7.5 mmol) in 70% aqueous acetic acid
(7 ml), and the mixture was stirred for 20 min at room temperature. The
resulting precipitate was collected by filtration, washed with cold H2O
and dried to give colorless solid, yield 1.55 g (mp. 124-126 oC).
2.1.2 Chemicals
All the chemicals used were of AR grade. Acetic acid (BDH) was
first refluxed over chromic acid for 6 h and then distilled. Solutions
of sodium perchlorate, perchloric acid were prepared in double
distilled water. Double distilled water (conductivity <10 S.cm-1) was
employed in all kinetic runs.
2.1.3 Preparation of 4-Oxoacids
The 4-oxo-4-(4'-methoxyphenyl)butanoic acid (S2), 4-oxo-4-(4'-
methyl-phenyl)-butanoic acid (S3) and 4-oxo-4-(4'-chlorophenyl)-
butanoic acid (S5) were obtained from Sigma-Aldrich Chemical
Co. The remaining 4-oxoacids (S1, S4, S6 and S7) were prepared by

43
Friedel-Crafts acylation of the substituted benzene with succinic
anhydride282.
All the 4-oxo acids used in this study were crystallized twice
from water and their purity was checked by their melting points.
2.1.3.1 4-Oxo-4-phenylbuatnoic acid (S1)
Succinic anhydride, 9 g was mixed with sodium dried benzene, 45 ml in
a round bottomed flask fitted with a reflux condenser protected by a
drying tube containing calcium chloride. The mixture was added with
anhydrous aluminium chloride, 25 g all at once. The mixture was
refluxed on a water bath with continued stirring for an hour. After
cooling, it was added with 50 ml of water and 20 ml of ice cold con.
HCl. The contents were then steam distilled to separate the
unreacted benzene and cooled in freezing mixture. The colorless
crystals of 4-oxo-4-phenylbutanoic acid separated from the solution
were filtered and washed with a cold dilute solution of HCl and
then with cold water. The crude product was dissolved in sodium
carbonate solution, boiled for 10 minutes and filtered. The hot filtrate
was treated with decolorizing carbon, 1 g, stirred and filtered. The
filtrate was cooled, acidified with con. HCl, 30 ml and further cooled in
a freezing mixture. The oxoacid obtained was filtered, washed
thoroughly with cold water and dried. Finally, the oxoacid was
recrystallised in boiling water and dried for 12 h at 45-500 C.

44
2.1.3.2 4-Oxo-4-biphenylbutanoic acid (S4)
Biphenyl, 40 g was mixed with succinic anhydride, 18 g and nitro
benzene, 50 ml in a round bottomed flask. Anhydrous aluminium
chloride, 50 g, was added in several portions and the mixture was
refluxed in a water bath for two hours. The contents after cooling
were added with ice cold HCl, 50 ml. then the reaction mixture was
steam distilled and cooled. The crude product obtained was purified
and recrystallised from alcohol.
2.1.3.3 4-Oxo-4-(4’-bromophenyl)butanoic acid (S6)
Bromobenzene was condensed with succinic anhydride. About 10 ml
of nitrobenzene was also added to the reaction mixture and refluxed
for a long time. The oxoacid was recrystallised from hot water.
2.1.3.4 4-Oxo-4-(3’-nitrophenyl)butanoic acid (S7)
4-Oxo-4-phenylbutanoic acid, 5 g was added in small quantities to a
ice cooled (< 50 C) mixture of fuming nitric acid, 10 ml and Con.
H2SO4, 2 ml with constant stirring for about 30 minutes. The
temperature of the reaction mixture was allowed to rise to 15 0C,
during the addition of the oxoacid. A clear solution was obtained and it
was added slowly to crushed ice, with constant stirring. The product
was formed as a precipitate. It was filtered and washed thoroughly with
cold water. The oxoacid was purified and recrystallised in water.

45
2.1.4 Melting points of 4-Oxoacids The melting points of all the 4-oxoacids synthesized correspond to the
values reported in the literature. These values along with their
percentage yield are presented in following Table
S.No 4-oxoacids Lit. [Ref]
Melting Point 0C
Observed Reported
S1 4-Oxo-4-phenylbutanoic acid 282 116 116
S2 4-Oxo-4-(4’-methoxyphenyl)butanoic acid
283 146 147
S3 4-Oxo-4-(4’-methylphenyl)butanoic acid
224 128 129
S4 4-Oxo-4-biphenylbutanoic acid 284 184 185
S5 4-Oxo-4-(4’-chlorophenyl)butanoic acid
231 132 133
S6 4-Oxo-4-(4’-bromophenyl)butanoic acid
231 148 149
S7 4-Oxo-4-(3’-nitrophenyl)butanoic acid
232 163 164
2.1.5 Purification of solvents Analar acetic acid (BDH) was refluxed for three hours with chromic
acid. The acid was then distilled in a glass distillation apparatus and the
fraction distilling at 117-118 0C was collected. The fraction collected
has a boiling point of 118 0C.
Water purified, by a permutit ion exchanger, was distilled with a
few crystals of potassium permanganate in a glass distillation apparatus.
The distillate collected was used for preparing all the reagents and
solutions. Fresh solutions were used for each kinetic runs

46
2.2 Instrumentation The reaction was followed potentiometrically by setting up a cell made
up of the reaction mixture into which a platinum electrode and a
standard calomel electrode were dipped. The emf of the cell was
measured periodically using an Equip-Tronics potentiometer, while the
reaction mixture was continuously stirred using a magnetic stirrer. An
electrically operated thermostat was used to maintain the desired
temperature with an accuracy of ±0.10 C. A double walled 100 ml
beaker with inlet-outlet water circulation facility, specially designed
for this experiment, was used as reaction vessel.
2.3 Methods
2.3.1 Oxidation of 4-oxoacids by N-Bromobenzamide
N-Bromobenzamide (NBB) was employed as oxidant for the oxidation
of unsubstituted and substituted 4-oxoacids. The kinetics of oxidation
of 4-oxoacids by NBB was studied by potentiometric method.
2.3.1.1 Rate measurements
The reactions were carried out in binary mixtures of acetic acid and
water. Requisite amounts (appropriate volume of known concentration)
of separately thermostated oxoacid, NBB, acetic acid, sodium
perchlorate, perchloric acid solutions and water were pipetted out into
the reaction vessel. The total volume of the reaction mixture being 50 ml,

47
after the addition of all the solutions. The vessel was kept at the desired
temperature (30 0C). The emf values of the reaction mixture were
determined at definite intervals of time. The pseudo-first order rate
constants were computed from the plots of ln (Et - E∞) versus time.
The following precautions were taken:
i) When the reaction was followed potentiometrically and
iodometrically both the methods gave the same values.
ii) Duplicate experiments were conducted in an atmosphere of
nitrogen and without nitrogen. But there was no difference in the
results obtained. All experiments reported in this thesis were
done in air.
iii) All reactions were carried out under pseudo-first order
conditions with substrate concentration in large excess. The rate
constants were computed from linear plots of log(Et-E) versus
time by least square method using linear regression
method(r>0.98).
All experiment was carried out in duplicate and the velocity constants
were reproducible within ±2% error. All pseudo first order rate
constant (kobs) are expressed in s-1.
2.3.1.2 Product analysis A typical product analysis was carried out as follows: 4-Oxo-4-
phenylbutanoic acid (0.1 M), perchloric acid (1.0 M) and N-

48
bromobenzamide (0.5 M) were mixed together in 50 percent aqueous
acetic acid so that the total volume of the mixture was 100 ml. The
reaction mixture was allowed to stand for about 24 h to ensure the
completion of the reaction. The gas evolved during the reaction was
found out to be carbon dioxide. The solution was then shaken well with
ether and aqueous layer was discarded. The ether layer was washed with
distilled water several times, dried over anhydrous sodium sulphate and
evaporated. The products were extracted with ether, dried and analyzed.
Benzoic acid was identified by its m.pt. (121 oC) and estimated
quantitatively with a standard curve at λmax = 235 nm and also tested
with its characteristic spot test. Identification of the products,
namely, benzoic acid, were also made by comparing the Rf values of the
authentic samples.
2.3.1.3 Stoichiometry
Different sets of reaction mixtures containing different quantities of
NBB and 4-oxoacids at constant concentration of perchloric acid and
sodium perchlorate were allowed to react for 24 h at 30o C and then
analyzed. The remaining NBB was estimated. The results show that
one mole of oxoacid consumes one mole of NBB.
C6H5COCH2CH2COOH + C6H5CONHBr + 5H2OH
+
C6H5COOH C6H5CONH2 3CO2 6H2 HBr+ + + +

49
2.3.2 Oxidation of 4-Oxoacids by N-Bromosaccharin
N-Bromosaccharin (NBSac) was purchased from Sigma Aldrich
Chemical Co. and employed as oxidant for the oxidation of
unsubstituted and substituted 4-oxoacids. The kinetics of oxidation of
4-oxoacids by NBSac was studied by potentiometric method.
2.3.2.1 Rate measurements
The reactions were carried out in binary mixtures of acetic acid and water.
Requisite amounts (appropriate volume of known concentration) of
separately thermostated oxoacid, NBSac, acetic acid, sodium perchlorate,
perchloric acid solutions and water were pipette out into the reaction
vessel. The total volume of the reaction mixture being 50 ml, after the
addition of all the solutions. The vessel was kept at the desired
temperature (300 C). The emf values of the reaction mixture were
determined at definite intervals of time. The pseudo-first order rate
constants were computed from the plots of ln (Et - E∞) versus time.
2.3.2.2 Product analysis
A typical product analysis was carried out as follows: 4-Oxo-4-
phenylbutanoic acid (0.1 M), perchloric acid (1.0 M) and NBSac (0.5 M)
were mixed together in 50 percent aqueous acetic acid so that the total
volume of the mixture was 100 ml. The reaction mixture was allowed
to stand for about 24 h to ensure the completion of the reaction.

50
The gas evolved during the reaction was found out to be carbon
dioxide. The solution was then shaken well with ether and aqueous
layer was discarded. The ether layer was washed with distilled water
several times, dried over anhydrous sodium sulphate and evaporated.
The products were extracted with ether, dried and analyzed. Benzoic
acid was identified by its m.pt. (121 oC) and estimated quantitatively
with a standard curve at λmax = 235 nm and also tested with its
characteristic spot test. Identification of the products, namely,
benzoic acid, was also made by comparing the Rf values of the
authentic samples285.
2.3.2.3 Stoichiometry of oxidation
The stoichiometry of the reaction was determined by equilibrating
reaction mixture of various [NBSac]/[4-oxoacid] ratios at 30 oC for 12 h,
keeping all other reagent concentrations constant. Estimation of
unconsumed NBSac revealed that one mole of 4-oxoacid consumed
one mole of NBSac.
C6H5COCH2CH2COOH C6H4SO2CONBr 5H2O
C6H5COOH C6H4SO2CONH 3CO2 6H2 HBr
+ +
+ ++
H+
+

CHAPTER 3
RESULTS AND DISCUSSION
4-Oxoacids and N-Bromobenzamide system

51
CHAPTER 3
Results and Discussion
4-Oxoacids and N-Bromobenzamide System
Unsubstituted and substituted 4-oxoacids were taken as substrates and N-
bromobenzamide (NBB) as oxidant. The oxidation was carried out in the
presence of perchloric acid. The results obtained on the oxidation of 4-
oxoacids with N-bromobenzamide in aqueous acetic acid medium are
presented and discussed here.
3.1 Structure of 4-Oxoacid and N-Bromobenzamide
4-Oxo-4-phenylbutanoic acid (S1)
Br
O NH
N-Bromobenzamide (NBB)
3.2 List of Substituted 4-Oxoacids The various substituted 4-oxoacids employed in the present study are
listed below (Table 1).

52
Table 1. Name of the unsubstituted and substituted 4-oxoacids used in
this study
Compound Code Name
S1 4-Oxo-4-phenylbutanoic acid
S2 4-Oxo-4-(4’-methoxyphenyl)butanoic acid
S3 4-Oxo-4-(4’-methylphenyl)butanoic acid
S4 4-Oxo-4-biphenylbutanoic acid
S5 4-Oxo-4-(4’-chlorophenyl)butanoic acid
S6 4-Oxo-4-(4’-bromophenyl)butanoic acid
S7 4-Oxo-4-(3’-nitrophenyl)butanoic acid
3.3 Kinetics of Oxidation of 4-Oxo-4-phenylbutanoic Acid
(S1) by N-Bromobenzamide
All the experiments were carried out in aqueous acetic acid medium. The
kinetic runs were conducted under pseudo-first order conditions keeping
the concentration of oxoacid at least ten times greater than that of NBB.
The reaction was followed potentiometrically by setting up a cell made up
of the reaction mixture into which a platinum electrode and a standard
calomel electrode were dipped. The emf of the cell was measured
periodically using an Equip-Tronics potentiometer, while the reaction
mixture was continuously stirred.
The kinetics of oxidation of 4-oxoacids by N-bromobenzamide
has been studied potentiometrically in aqueous acetic acid medium

53
in the presence of perchloric acid at constant ionic strength. The ionic
strength of the medium was maintained by the addition of NaClO4. The
reaction was carried out in the absence of perchloric acid or other
mineral acids, it was found that the reaction proceeded extremely slow
(Table 3). The same trend was observed in the earlier reports285-288.
Hence the reaction was carried out in the presence of perchloric acid.
The pseudo-first order rate constants k1 computed from the plots of
ln(Et-E) against time were reproducible within 3%. Experiments
were carried out at 30o C
3.3.1 Effect of varying [S1]0
The order of the reaction with respect to the concentration of 4-
oxoacid was determined by studying the rate of the reaction at different
initial concentrations of the oxoacid. The rate constants were
determined at various initial concentrations of 4-oxo-4-phenylbutanoic
acid (0.02-0.08 M) at constant concentration of N-bromobenzamide
(0.002 M), perchloric acid (0.5 M) and sodium perchlorate (0.5 M) in
50% (v/v) aqueous acetic acid medium. The values are given in
Table 2. The plots of ln(Et - E∞) versus time (sec) for the various
concentrations of the oxoacid are linear (Figure 1). The second order
rate constants k2, were calculated by dividing the observed pseudo first
order rate constants with the initial concentrations of the oxoacids.

54
The values are found to be constant. These results indicate the first
order dependence of the oxoacid on the rate of the reaction. This is
further supported by the fact that the plot log k1 against log [S1] is
linear (r2 = 0.989) with a slope value of 0.996 (Figure 2) and the plot
of k1 versus [S1] gives a straight line passing through the origin
(Figure 3).
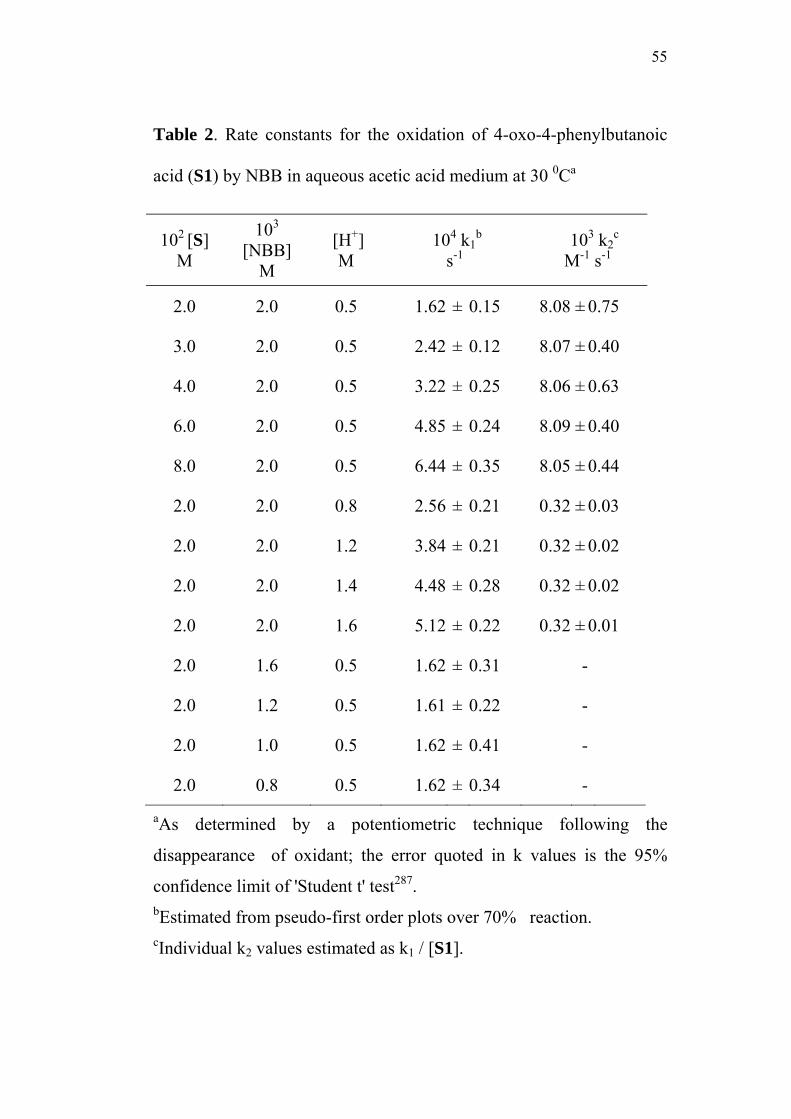
55
Table 2. Rate constants for the oxidation of 4-oxo-4-phenylbutanoic
acid (S1) by NBB in aqueous acetic acid medium at 30 0Ca
aAs determined by a potentiometric technique following the
disappearance of oxidant; the error quoted in k values is the 95%
confidence limit of 'Student t' test287. bEstimated from pseudo-first order plots over 70% reaction. cIndividual k2 values estimated as k1 / [S1].
102 [S] M
103 [NBB]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 1.62 ± 0.15 8.08 ± 0.75
3.0 2.0 0.5 2.42 ± 0.12 8.07 ± 0.40
4.0 2.0 0.5 3.22 ± 0.25 8.06 ± 0.63
6.0 2.0 0.5 4.85 ± 0.24 8.09 ± 0.40
8.0 2.0 0.5 6.44 ± 0.35 8.05 ± 0.44
2.0 2.0 0.8 2.56 ± 0.21 0.32 ± 0.03
2.0 2.0 1.2 3.84 ± 0.21 0.32 ± 0.02
2.0 2.0 1.4 4.48 ± 0.28 0.32 ± 0.02
2.0 2.0 1.6 5.12 ± 0.22 0.32 ± 0.01
2.0 1.6 0.5 1.62 ± 0.31 -
2.0 1.2 0.5 1.61 ± 0.22 -
2.0 1.0 0.5 1.62 ± 0.41 -
2.0 0.8 0.5 1.62 ± 0.34 -
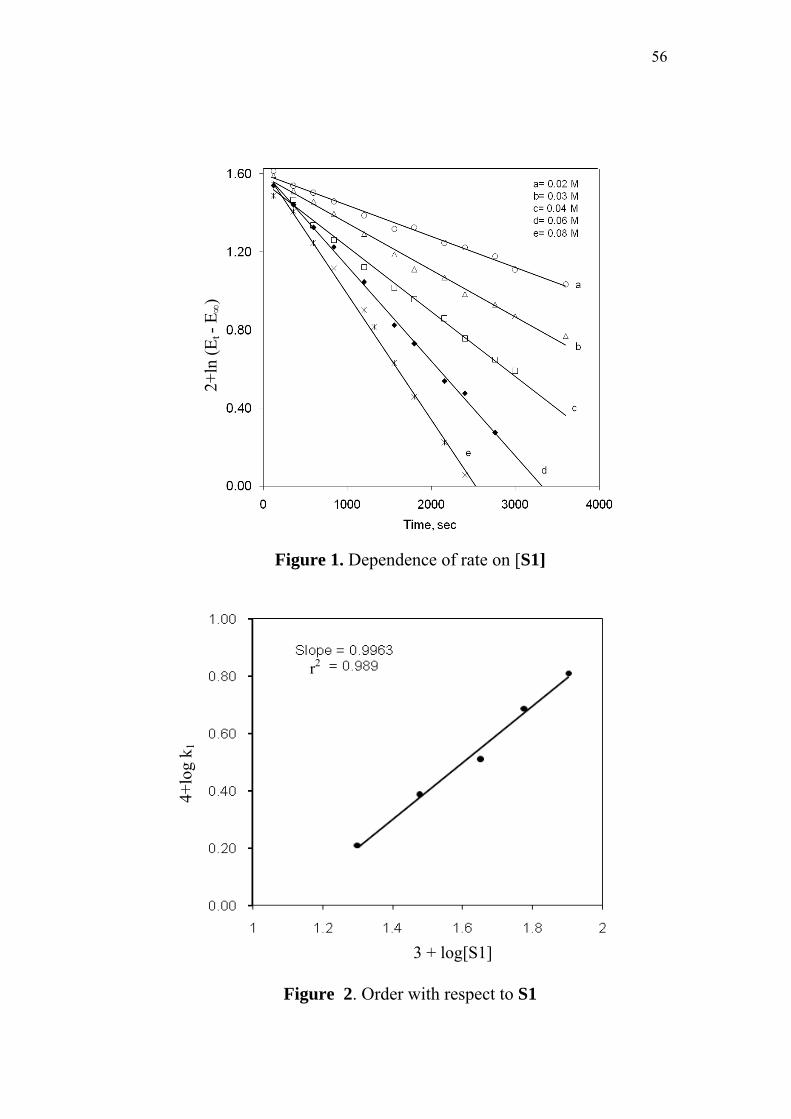
56
Figure 1. Dependence of rate on [S1]
Figure 2. Order with respect to S1
4+lo
g k 1
3 + log[S1]
2+ln
(E
t - E
)
r2
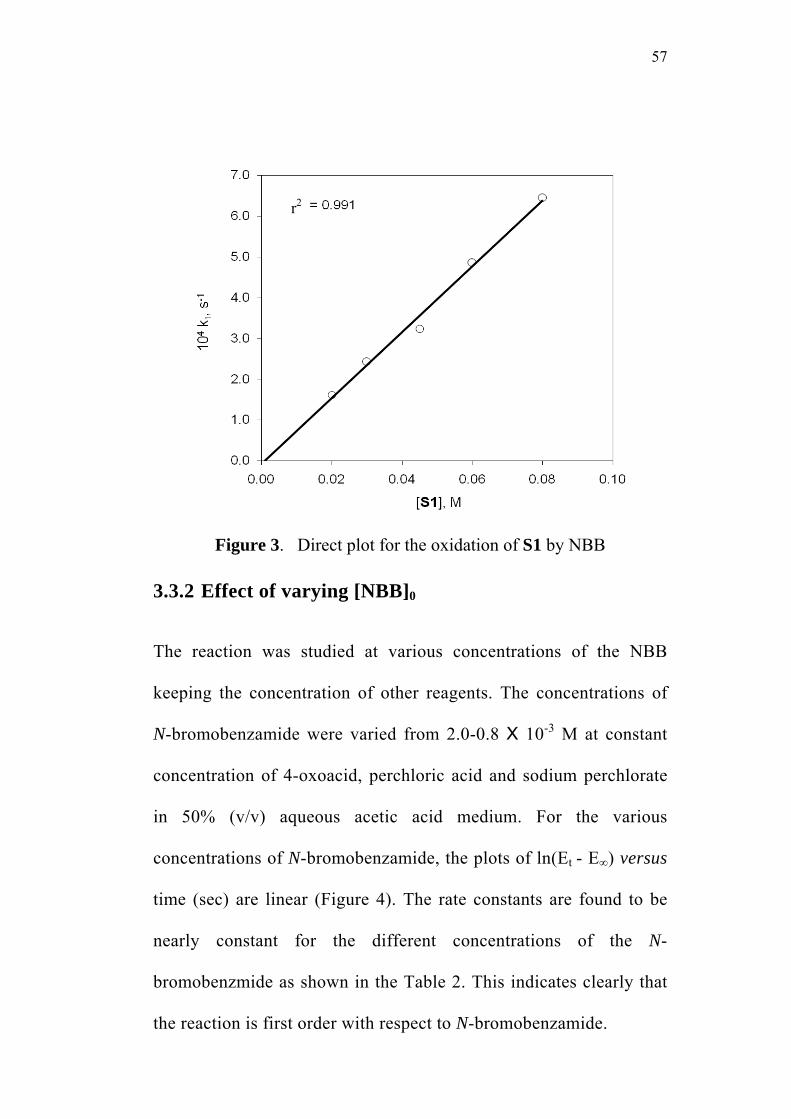
57
Figure 3. Direct plot for the oxidation of S1 by NBB
3.3.2 Effect of varying [NBB]0
The reaction was studied at various concentrations of the NBB
keeping the concentration of other reagents. The concentrations of
N-bromobenzamide were varied from 2.0-0.8 X 10-3 M at constant
concentration of 4-oxoacid, perchloric acid and sodium perchlorate
in 50% (v/v) aqueous acetic acid medium. For the various
concentrations of N-bromobenzamide, the plots of ln(Et - E∞) versus
time (sec) are linear (Figure 4). The rate constants are found to be
nearly constant for the different concentrations of the N-
bromobenzmide as shown in the Table 2. This indicates clearly that
the reaction is first order with respect to N-bromobenzamide.
r2
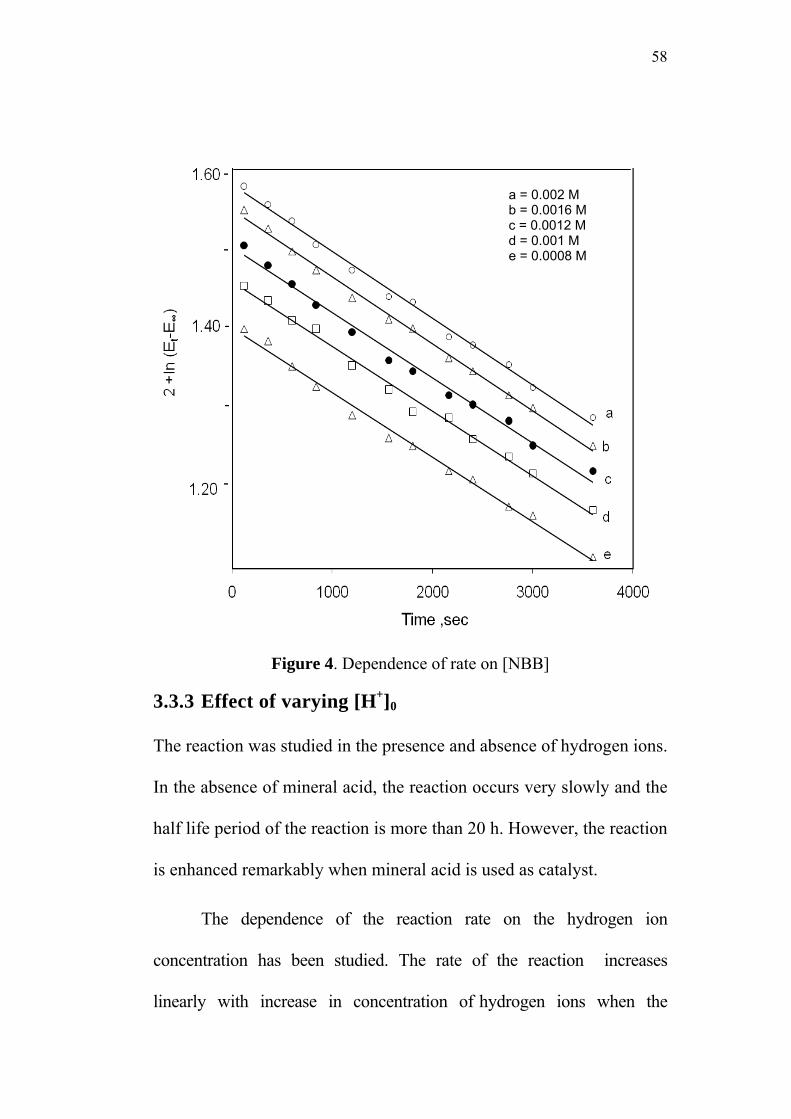
58
Figure 4. Dependence of rate on [NBB]
3.3.3 Effect of varying [H+]0
The reaction was studied in the presence and absence of hydrogen ions.
In the absence of mineral acid, the reaction occurs very slowly and the
half life period of the reaction is more than 20 h. However, the reaction
is enhanced remarkably when mineral acid is used as catalyst.
The dependence of the reaction rate on the hydrogen ion
concentration has been studied. The rate of the reaction increases
linearly with increase in concentration of hydrogen ions when the
a = 0.002 M b = 0.0016 M c = 0.0012 M d = 0.001 M e = 0.0008 M
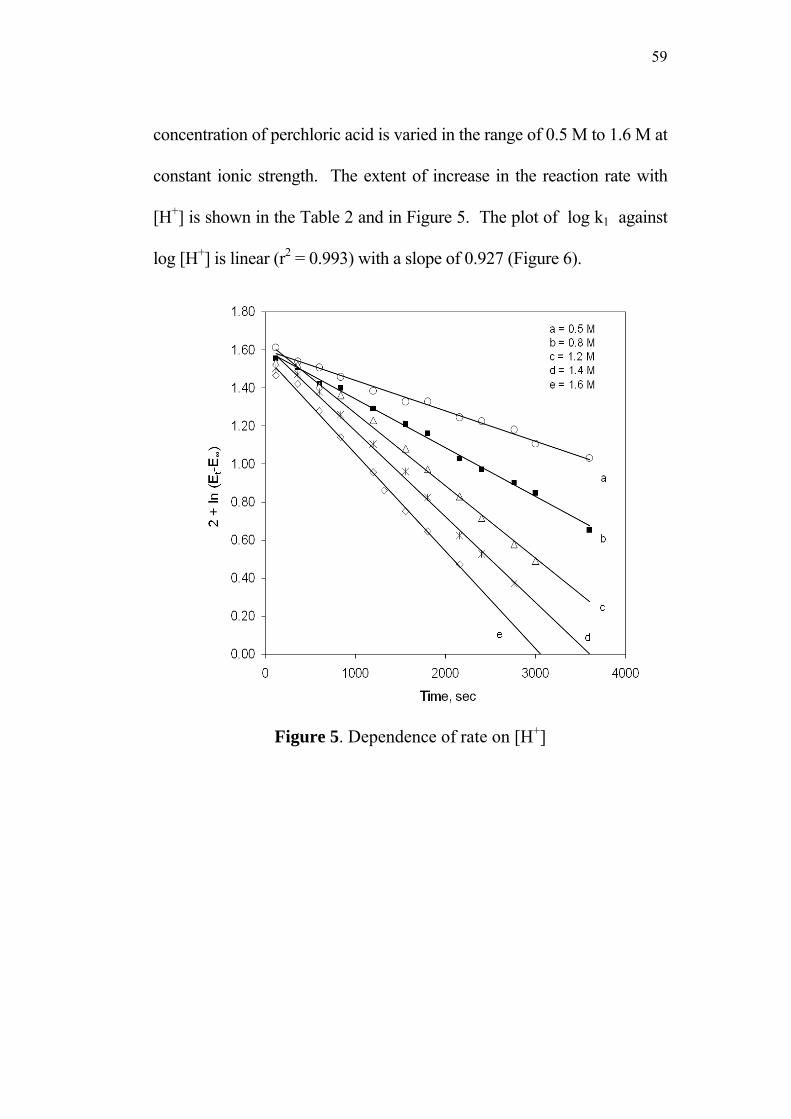
59
concentration of perchloric acid is varied in the range of 0.5 M to 1.6 M at
constant ionic strength. The extent of increase in the reaction rate with
[H+] is shown in the Table 2 and in Figure 5. The plot of log k1 against
log [H+] is linear (r2 = 0.993) with a slope of 0.927 (Figure 6).
Figure 5. Dependence of rate on [H+]
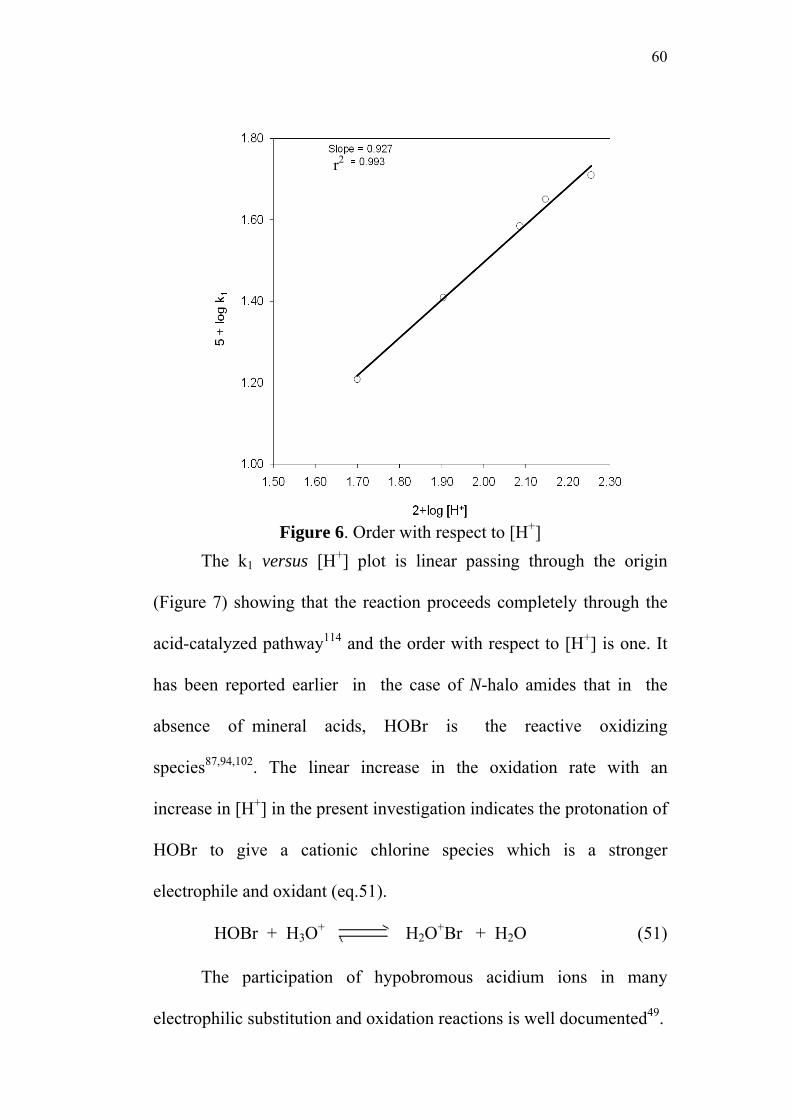
60
Figure 6. Order with respect to [H+]
The k1 versus [H+] plot is linear passing through the origin
(Figure 7) showing that the reaction proceeds completely through the
acid-catalyzed pathway114 and the order with respect to [H+] is one. It
has been reported earlier in the case of N-halo amides that in the
absence of mineral acids, HOBr is the reactive oxidizing
species87,94,102. The linear increase in the oxidation rate with an
increase in [H+] in the present investigation indicates the protonation of
HOBr to give a cationic chlorine species which is a stronger
electrophile and oxidant (eq.51).
HOBr + H3O+ H2O
+Br + H2O (51)
The participation of hypobromous acidium ions in many
electrophilic substitution and oxidation reactions is well documented49.
r2
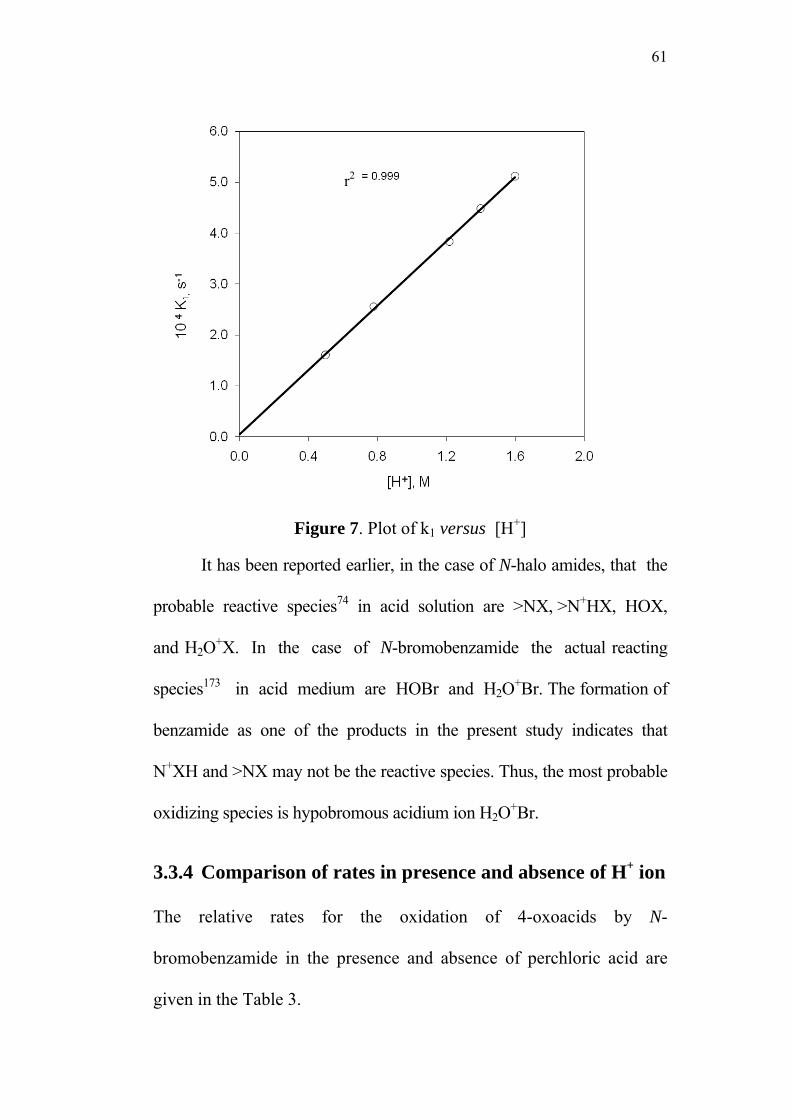
61
Figure 7. Plot of k1 versus [H+]
It has been reported earlier, in the case of N-halo amides, that the
probable reactive species74 in acid solution are >NX, >N+HX, HOX,
and H2O+X. In the case of N-bromobenzamide the actual reacting
species173 in acid medium are HOBr and H2O+Br. The formation of
benzamide as one of the products in the present study indicates that
N+XH and >NX may not be the reactive species. Thus, the most probable
oxidizing species is hypobromous acidium ion H2O+Br.
3.3.4 Comparison of rates in presence and absence of H+ ion
The relative rates for the oxidation of 4-oxoacids by N-
bromobenzamide in the presence and absence of perchloric acid are
given in the Table 3.
r2

62
Table 3. Comparative rates in the presence and absence of H+ iona.
S 104 k1b, s-1
In the absence of HClO4 In the presence of 0.5 M HClO4
S1 0.14 ± 0.03 1.62 ± 0.15
S2 0.28 ± 0.06 4.55 ± 0.32
S3 0.24 ± 0.08 3.22 ± 0.26
S4 0.23 ± 0.04 1.88 ± 0.17
S5 0.18 ± 0.06 1.24 ± 0.23
S6 0.15 ± 0.04 0.89 ± 0.12
S7 0.11 ± 0.03 0.23 ± 0.09
a General conditions: [S] = 0.02 M, [NBB] = 0.002 M, Temp: 30 0C,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots.
3.3.5 Effect of varying ionic strength on reaction rate
The ionic strength of the reaction medium was changed by the addition
of anhydrous sodium perchlorate and the influence of ionic strength on
the reaction rate has been studied. The values of the rate constants at
different ionic strengths are given for several 4-oxoacids in Table 4. It
is found that the ionic strength of the reaction medium has no
significant effect on the reaction rate. Similar result was obtained by
Harihar et al.,176 in the oxidation of L-arginine by N-bromosuccinimide
in aqueous acetic acid medium.

63
Table 4. Effect of ionic strength for the oxidation of 4-oxoacids by
NBB in aqueous acetic acid medium at 30 0Ca
S [NaClO4], M 104 k1b, s-1
S1 0.5 1.62 ± 0.15
1.0 1.60 ± 0.13
1.5 1.61 ± 0.22
2.0 1.61 ± 0.12
S2 0.5 4.55 ± 0.32
1.0 4.65 ± 0.34
1.5 4.45 ± 0.31
2.0 4.54 ± 0.44
S7 0.5 0.23 ± 0.09
1.0 0.23 ± 0.14
1.5 0.23 ± 0.17
2.0 0.23 ± 0.13
Thimme Gowda and Ishwara Bhat178 have reported that the
increase in ionic strength of the medium slightly decreased the rates for
both N-chlorosuccinimide and N-bromosuccinimide oxidation of
thiosemicarbazide (TSC) in the absence of mercuric acetate in aqueous
medium. The effect of ionic strength was considerable in NCS
oxidation in 1:1 (v/v) aqueous acetic acid medium. In the case of NBS
oxidation the rate increased with increase in ionic strength of the
medium. In the present study, it is observed that the rate of the reaction
is independent of ionic strength of the medium.

64
3.3.6 Effect of added benzamide
The reversibility or otherwise of the electron transfer step is usually
tested by the effect of adding one of the products. As benzamide is
one of the products of oxidation in the present study, the oxidation
reaction is carried out in the presence of benzamide. The addition of
benzamide reduced the rate of oxidation of oxoacids (Table 5).
Thus the retardation of reaction rate on the addition of
benzamide suggests128 a pre-equilibrium step involving a process in
which benzamide is one of the products.
NBB + H3O+ H2O
+Br + Benzamide (52)
If this equilibrium is involved in the oxidation process, the
rate should be an inverse function of benzamide concentration,
which is borne out by the observation that the inverse of the rate
constant gives a linear (r2 = 0.984) plot against [benzamide] (Figure 8).
Similar conclusions have been arrived at in the N-chloronicotinamide
oxidation of amino acids128, and N-bromoacetamide oxidation of some
hydroxy acids93.
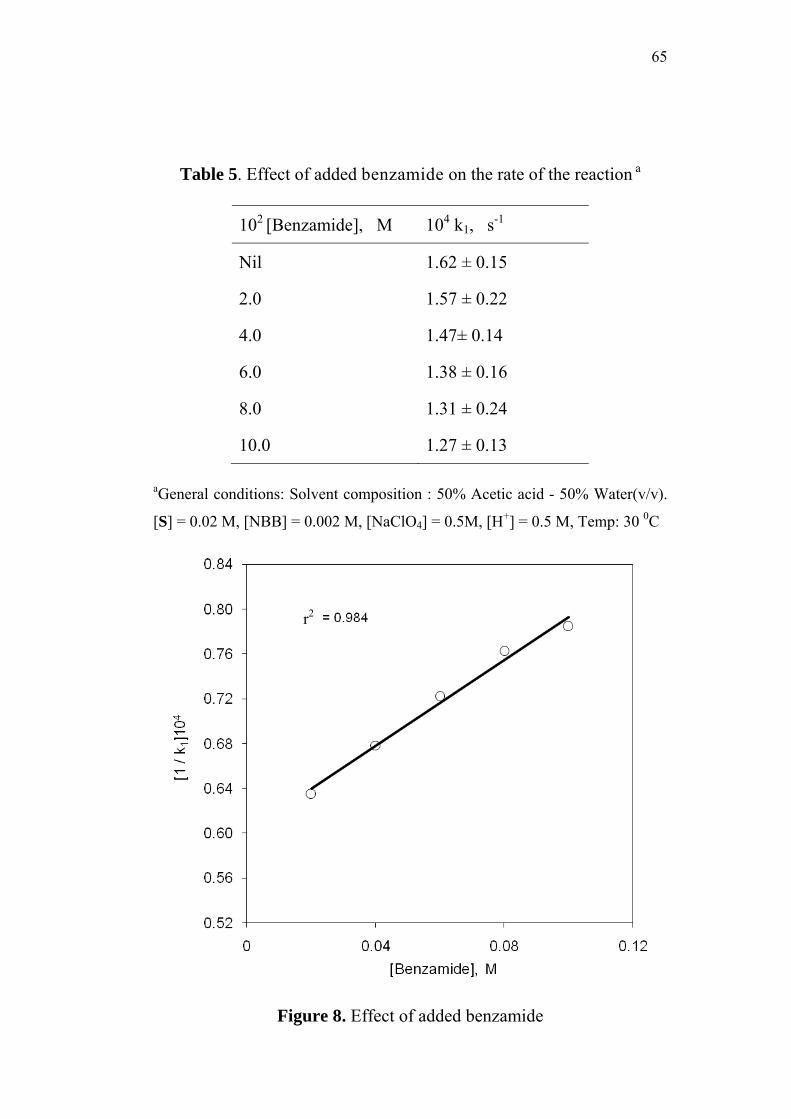
65
Table 5. Effect of added benzamide on the rate of the reaction a
102 [Benzamide], M 104 k1, s-1
Nil 1.62 ± 0.15
2.0 1.57 ± 0.22
4.0 1.47± 0.14
6.0 1.38 ± 0.16
8.0 1.31 ± 0.24
10.0 1.27 ± 0.13
aGeneral conditions: Solvent composition : 50% Acetic acid - 50% Water(v/v).
[S] = 0.02 M, [NBB] = 0.002 M, [NaClO4] = 0.5M, [H+] = 0.5 M, Temp: 30 0C
Figure 8. Effect of added benzamide
r2

66
3.3.7 Effect of adding acrylonitrile
Vinyl monomers like acrylonitrile are added to the reaction mixture
under nitrogen atmosphere to find out whether the reaction under
investigation involves the formation of free radicals as the
reaction intermediates.92
In the present study, freshly distilled acrylonitrile freed from
inhibitor is added to the deaerated reaction mixture containing 0.2 M
perchloric acid, since the presence of high concentration of
perchloric acid may prevent precipitation of the polymer. After the
completion of the reaction, the reaction mixture is diluted with
methanol to observe the formation of polymer. It is observed that the
oxidation reaction does not induce the polymerization. Similarly, the
polymer formation is not obtained when ethyl acrylate is added to the
reaction mixture. It reveals the absence of any free radical intermediate
during the oxidation of 4-oxoacid by N-bromobenzamide. Further it is
confirmed by carrying out the oxidation of S1 at different initial
concentrations of acrylonitrile. The results are tabulated (Table 6). It
shows that the rate of the reaction has not been affected by the addition
of acrylonitrile.

67
Table 6. Effect of added acrylonitrile on the rate of the reactiona
103 [Acrylonitrile], M 104 k1, s-1
Nil 1.62 ± 0.15
2.0 1.61 ± 0.28
4.0 1.61 ± 0.23
6.0 1.61 ± 0.11
8.0 1.61 ± 0.25
a General conditions: Solvent composition: 50% Acetic acid - 50% Water (v/v).
[S] = 0.02 M, [NBB] = 0.002 M, [NaClO4] = 0.5M, [H+] = 0.5 M, Temp: 300 C.
3.3.8 Effect of solvent polarity on reaction rate
The oxidation of 4-oxoacids by N-bromobenzamide has been studied in
the binary mixture of acetic acid and water as the solvent medium. For
the oxidation of all the 4-oxoacids, the reaction rate increased
remarkably with the increase in the proportion of acetic acid in the
solvent medium. These results are presented in Table 7.
The enhancement of the reaction rate with an increase in the
amount of acetic acid may generally be attributed to two factors, viz, (i)
increase in acidity at constant [perchloric acid] and (ii) decrease in
dielectric constant with increase in acetic acid content. The plots of log
k1 against the inverse of the dielectric constant are linear with positive
slopes (Figure 9), indicating an interaction between a positive ion and
dipolar molecule287-290 .This supports the postulation of H2O+Br as the
reactive species.

68
Similar results and conclusions are arrived at in the oxidation
of primary alcohols by N-bromo-3,5-dinitrobenz-amide159. Sikkandar
and Basheer Ahamed have observed255 that as acetic acid
percentage is increased three times, the reaction rate also
increases three times and have attributed this trend i) to the
lowering of dielectric constant of the medium which favors reactions
involving protonation and ii) to the enolization of the oxoacid which
may be catalyzed by acetic acid.
In the present study, the significant rate enhancement with
the lowering of dielectric constant of the solvent medium may
be attributed to the enolization of the ketoacid group in the 4-
oxoacid. This trend is observed for all the substituted 4-oxoacids taken
for investigation. The enolization of the keto group of the oxoacid
is facilitated by the increase in percentage of acetic acid in the
solvent medium and this may also favors the rate enhancement. This
is in agreement with the observation made by Banerji et al. during the
oxidation of cyclohexanone and several methyl ketones291.
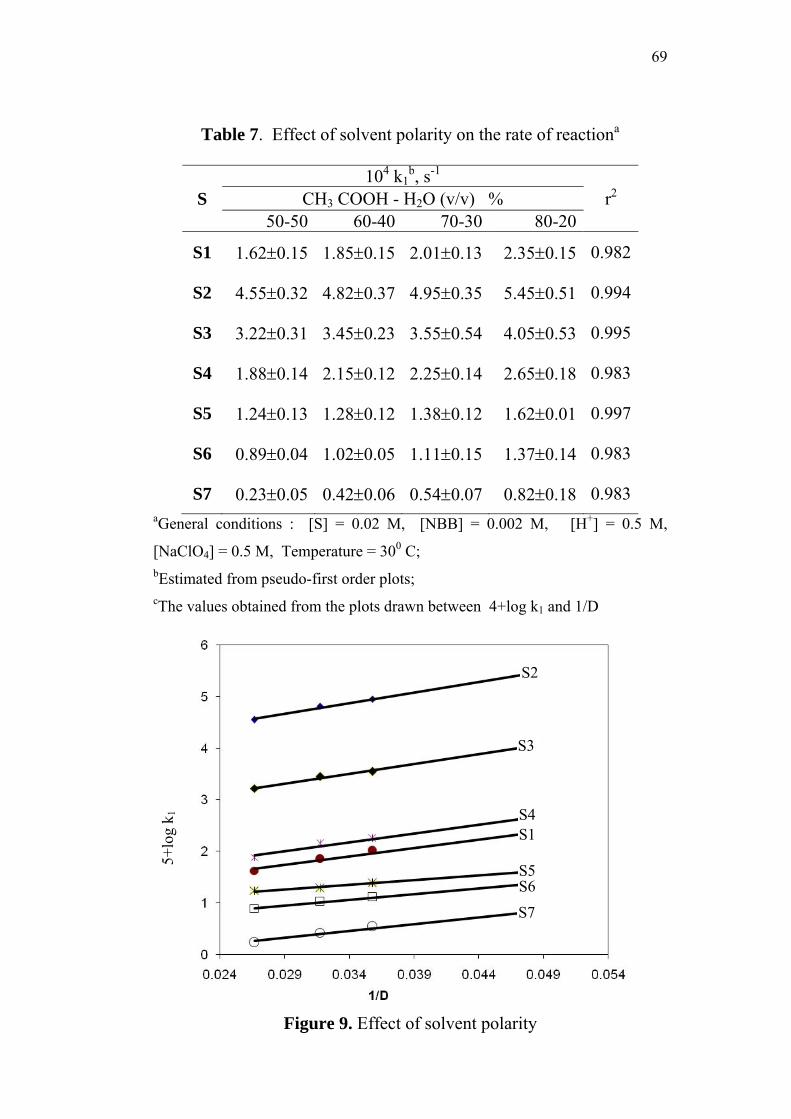
69
Table 7. Effect of solvent polarity on the rate of reactiona
S 104 k1
b, s-1 r2 CH3 COOH - H2O (v/v) %
50-50 60-40 70-30 80-20
S1 1.620.15 1.850.15 2.010.13 2.350.15 0.982
S2 4.550.32 4.820.37 4.950.35 5.450.51 0.994
S3 3.220.31 3.450.23 3.550.54 4.050.53 0.995
S4 1.880.14 2.150.12 2.250.14 2.650.18 0.983
S5 1.240.13 1.280.12 1.380.12 1.620.01 0.997
S6 0.890.04 1.020.05 1.110.15 1.370.14 0.983
S7 0.230.05 0.420.06 0.540.07 0.820.18 0.983 aGeneral conditions : [S] = 0.02 M, [NBB] = 0.002 M, [H+] = 0.5 M,
[NaClO4] = 0.5 M, Temperature = 300 C; bEstimated from pseudo-first order plots; cThe values obtained from the plots drawn between 4+log k1 and 1/D
Figure 9. Effect of solvent polarity
5+lo
g k 1
S2
S3
S4S1
S5S6
S7

70
3.3.9 Rate of enolization by bromination method
It has been reported earlier in the case of oxidation of keto compounds
that the oxidation proceeds via enolization of the keto compounds291-295,298.
The rate of enolization of keto compound is found to be faster than
the rate of oxidation. The reactive species of the substrate may be
determined by enolization, which is an acid as well as base
catalyzed reaction and proceeds by a concerted or push-pull
mechanism. The rate of enolization was determined by bromination
method299 for the system under investigation.
The order for the bromination reaction with respect to the 4-
oxoacid, bromine and H+ has been determined. The experimental
results are presented in Table 8. These data indicate that the
bromination of the oxoacid is first order each with respect to the
oxoacid and H+ ion but zero order with respect to bromine.
From the values, it is obvious that the rate of enolization
is faster than the rate of oxidation of the oxoacid. It is interesting
to note that similar trend has been reported by Marigangaiah and
Banerji in the oxidation of methyl aryl ketones288. Hence enol form
of the substrate is probably participating in the rate determining step.
Enol as a reactive species of the substrate has also been reported
in oxidation by Tl(III)300,301.
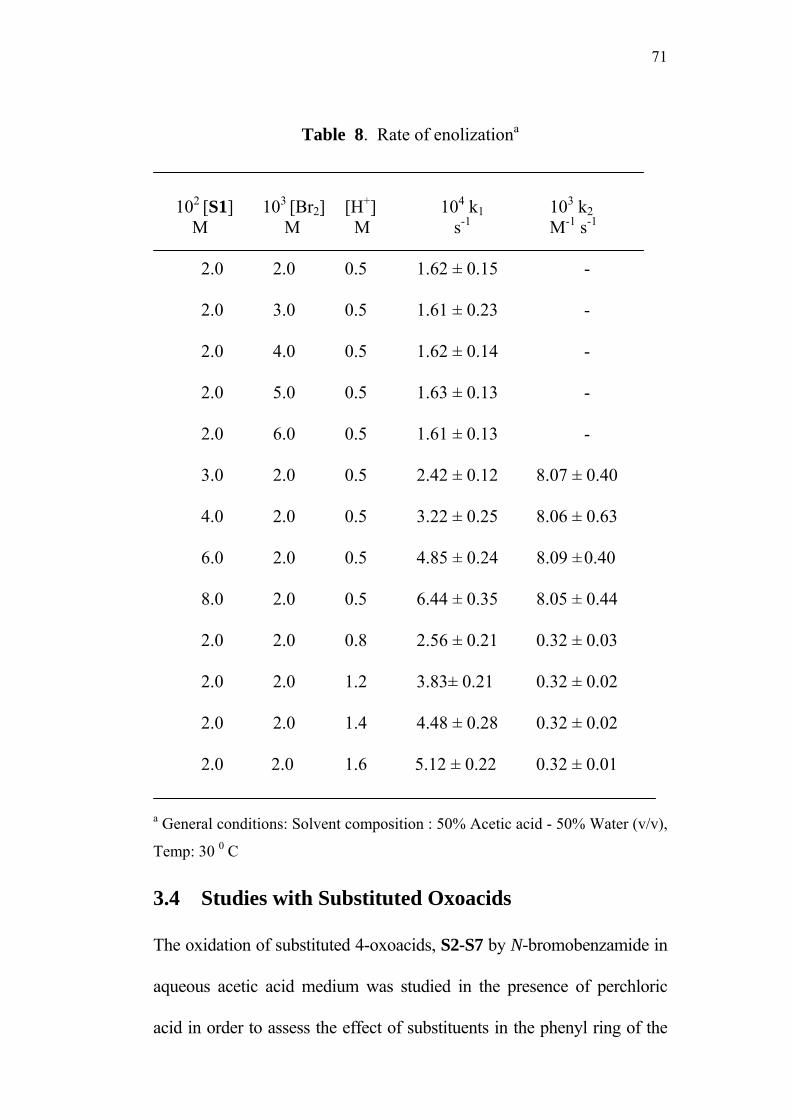
71
Table 8. Rate of enolizationa
102 [S1] 103 [Br2] [H+] 104 k1 103 k2
M M M s-1 M-1 s-1
2.0 2.0 0.5 1.62 ± 0.15 -
2.0 3.0 0.5 1.61 ± 0.23 -
2.0 4.0 0.5 1.62 ± 0.14 -
2.0 5.0 0.5 1.63 ± 0.13 -
2.0 6.0 0.5 1.61 ± 0.13 -
3.0 2.0 0.5 2.42 ± 0.12 8.07 ± 0.40
4.0 2.0 0.5 3.22 ± 0.25 8.06 ± 0.63
6.0 2.0 0.5 4.85 ± 0.24 8.09 ± 0.40
8.0 2.0 0.5 6.44 ± 0.35 8.05 ± 0.44
2.0 2.0 0.8 2.56 ± 0.21 0.32 ± 0.03
2.0 2.0 1.2 3.83± 0.21 0.32 ± 0.02
2.0 2.0 1.4 4.48 ± 0.28 0.32 ± 0.02
2.0 2.0 1.6 5.12 ± 0.22 0.32 ± 0.01
a General conditions: Solvent composition : 50% Acetic acid - 50% Water (v/v),
Temp: 30 0 C
3.4 Studies with Substituted Oxoacids
The oxidation of substituted 4-oxoacids, S2-S7 by N-bromobenzamide in
aqueous acetic acid medium was studied in the presence of perchloric
acid in order to assess the effect of substituents in the phenyl ring of the

72
oxoacid on reaction rate. The pseudo first order rate constants were
measured at different initial concentrations of oxoacids. The second
order rate constants k2 were found to be constant. The values are given in
the Tables 9-14. The plots of log k1 against log [S] are linear (Figure 10).
These results indicate the first order dependence of the oxoacids
on the rate of the reaction. This is further supported by the fact that
the plot of k1 versus [S] gives a straight line passing through the origin
(Figure 11).
The oxidation was carried out at different initial concentrations of
N-bromobenzamide. The rate constants are found to be nearly constant
(Tables 9-14). This shows that the reaction is first order with respect to N-
bromobenzamide.
The dependence of the reaction rate on the hydrogen ion
concentration has been studied for the various oxoacids (S2-
S7). The rate of the reaction increases linearly with increase in the
concentration of hydrogen ion. The plots of log k1 against log [H+] is
linear with slope values equal to one (Figure 12). This establishes that
the reaction is first order with respect to hydrogen ion concentration.
Also the plots of k1 versus [H+] for the oxidation of various oxoacids
are linear passing through the origin (Figure 13) showing that the
reaction proceeds completely through the acid catalyzed pathway and
the order is one.
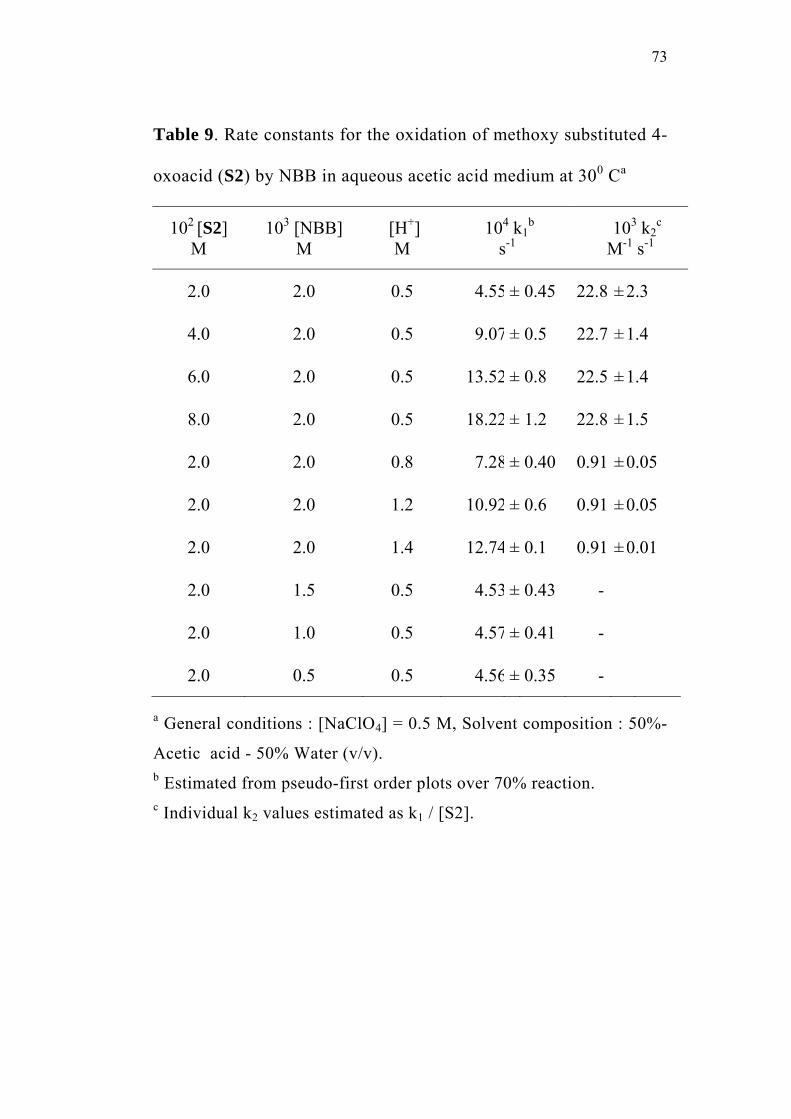
73
Table 9. Rate constants for the oxidation of methoxy substituted 4-
oxoacid (S2) by NBB in aqueous acetic acid medium at 300 Ca
102 [S2] M
103 [NBB] M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 4.55 ± 0.45 22.8 ± 2.3
4.0 2.0 0.5 9.07 ± 0.5 22.7 ± 1.4
6.0 2.0 0.5 13.52 ± 0.8 22.5 ± 1.4
8.0 2.0 0.5 18.22 ± 1.2 22.8 ± 1.5
2.0 2.0 0.8 7.28 ± 0.40 0.91 ± 0.05
2.0 2.0 1.2 10.92 ± 0.6 0.91 ± 0.05
2.0 2.0 1.4 12.74 ± 0.1 0.91 ± 0.01
2.0 1.5 0.5 4.53 ± 0.43 -
2.0 1.0 0.5 4.57 ± 0.41 -
2.0 0.5 0.5 4.56 ± 0.35 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].
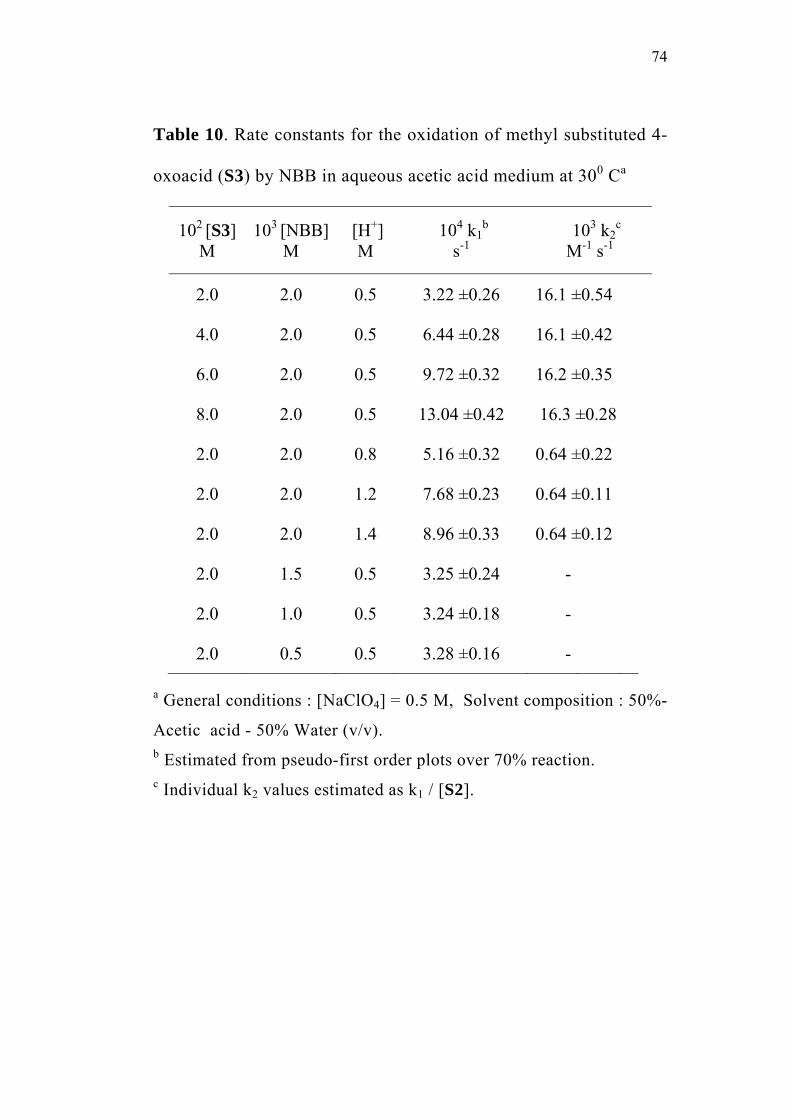
74
Table 10. Rate constants for the oxidation of methyl substituted 4-
oxoacid (S3) by NBB in aqueous acetic acid medium at 300 Ca
102 [S3] M
103 [NBB] M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 3.22 ±0.26 16.1 ±0.54
4.0 2.0 0.5 6.44 ±0.28 16.1 ±0.42
6.0 2.0 0.5 9.72 ±0.32 16.2 ±0.35
8.0 2.0 0.5 13.04 ±0.42 16.3 ±0.28
2.0 2.0 0.8 5.16 ±0.32 0.64 ±0.22
2.0 2.0 1.2 7.68 ±0.23 0.64 ±0.11
2.0 2.0 1.4 8.96 ±0.33 0.64 ±0.12
2.0 1.5 0.5 3.25 ±0.24 -
2.0 1.0 0.5 3.24 ±0.18 -
2.0 0.5 0.5 3.28 ±0.16 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].
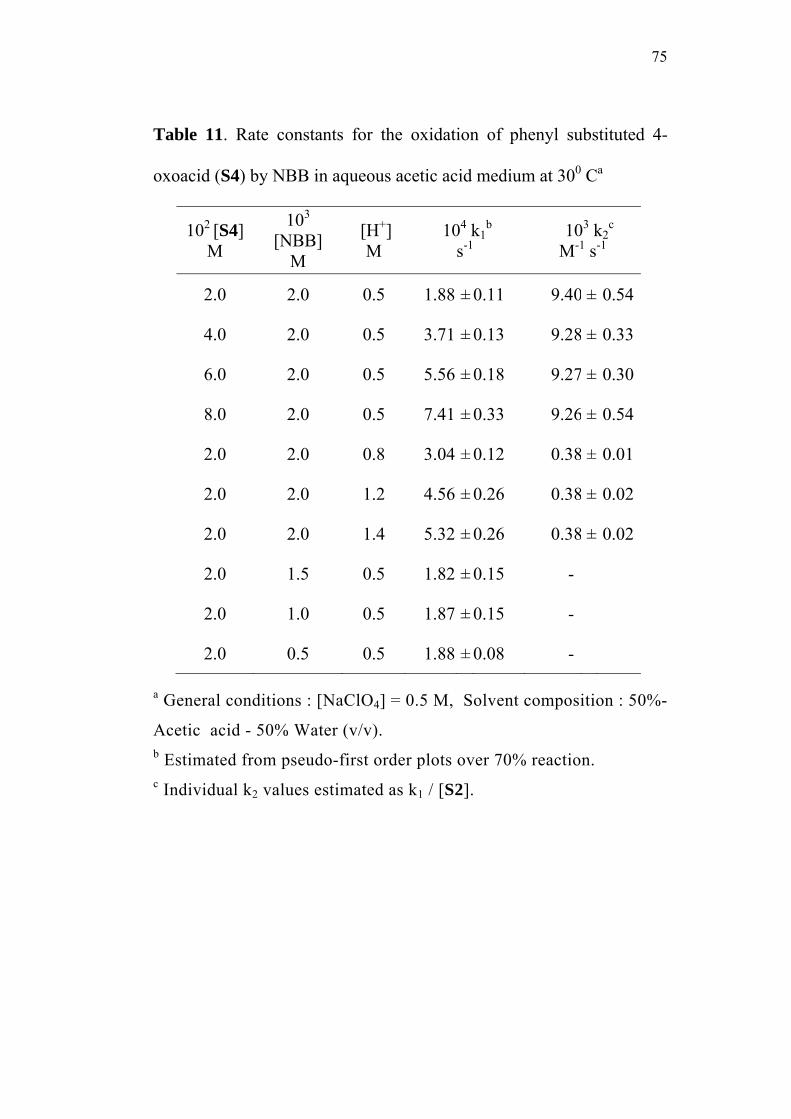
75
Table 11. Rate constants for the oxidation of phenyl substituted 4-
oxoacid (S4) by NBB in aqueous acetic acid medium at 300 Ca
102 [S4] M
103 [NBB]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 1.88 ± 0.11 9.40 ± 0.54
4.0 2.0 0.5 3.71 ± 0.13 9.28 ± 0.33
6.0 2.0 0.5 5.56 ± 0.18 9.27 ± 0.30
8.0 2.0 0.5 7.41 ± 0.33 9.26 ± 0.54
2.0 2.0 0.8 3.04 ± 0.12 0.38 ± 0.01
2.0 2.0 1.2 4.56 ± 0.26 0.38 ± 0.02
2.0 2.0 1.4 5.32 ± 0.26 0.38 ± 0.02
2.0 1.5 0.5 1.82 ± 0.15 -
2.0 1.0 0.5 1.87 ± 0.15 -
2.0 0.5 0.5 1.88 ± 0.08 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].

76
Table 12. Rate constants for the oxidation of chloro substituted 4-
oxoacid (S5) by NBB in aqueous acetic acid medium at 300 Ca
102
[S5] M 103 [NBB]
M [H+]
M 104 k1
b s-1
103 k2c
M-1 s-1
2.0 2.0 0.5 1.24 ±0.17 6.20 ± 0.85
4.0 2.0 0.5 2.10 ±0.20 5.25 ± 0.50
6.0 2.0 0.5 3.13 ±0.17 5.21 ± 0.28
8.0 2.0 0.5 4.19 ±0.22 5.24 ± 0.28
2.0 2.0 0.8 2.04 ±0.09 0.25 ± 0.01
2.0 2.0 1.2 2.83 ±0.07 0.24 ± 0.02
2.0 2.0 1.4 3.35 ±0.12 0.24 ± 0.01
2.0 1.5 0.5 1.05 ±0.07 -
2.0 1.0 0.5 1.03 ±0.13 -
2.0 0.5 0.5 1.06 ±0.14 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].

77
Table 13. Rate constants for the oxidation of bromo substituted 4-
oxoacid (S6) by NBB in aqueous acetic acid medium at 30 0Ca
102 [S6] M
103
[NBB] M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 0.89 ± 0.04 4.45 ± 0.20
4.0 2.0 0.5 1.77 ± 0.10 4.44 ± 0.25
6.0 2.0 0.5 2.67 ± 0.12 4.45 ± 0.20
8.0 2.0 0.5 3.57 ± 0.23 4.46 ± 0.29
2.0 2.0 0.8 1.43 ± 0.10 0.18 ± 0.01
2.0 2.0 1.2 2.14 ± 0.13 0.18 ± 0.01
2.0 2.0 1.4 2.49 ± 0.12 0.18 ± 0.01
2.0 1.5 0.5 0.88 ± 0.11 -
2.0 1.0 0.5 0.87 ± 0.11 -
2.0 0.5 0.5 0.89 ± 0.10 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].

78
Table 14. Rate constants for the oxidation of nitro substituted 4-
oxoacid (S7) by NBB in aqueous acetic acid medium at 30 0Ca
102 [S7] M
103 [NBB] M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 0.23 ± 0.05 1.15 ± 0.15
4.0 2.0 0.5 0.45 ± 0.06 1.13 ± 0.15
6.0 2.0 0.5 0.68 ± 0.06 1.13 ± 0.10
8.0 2.0 0.5 0.91 ± 0.09 1.14 ± 0.11
2.0 2.0 0.8 0.36 ± 0.05 0.04 ± 0.01
2.0 2.0 1.2 0.54 ± 0.06 0.04 ± 0.01
2.0 2.0 1.4 0.63 ± 0.11 0.05 ± 0.01
2.0 3.0 0.5 0.24 ± 0.02 -
2.0 5.0 0.5 0.23 ± 0.01 -
2.0 7.0 0.5 0.25 ± 0.02 -
a General conditions : [NaClO4] = 0.5 M, Solvent composition : 50%-
Acetic acid - 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].
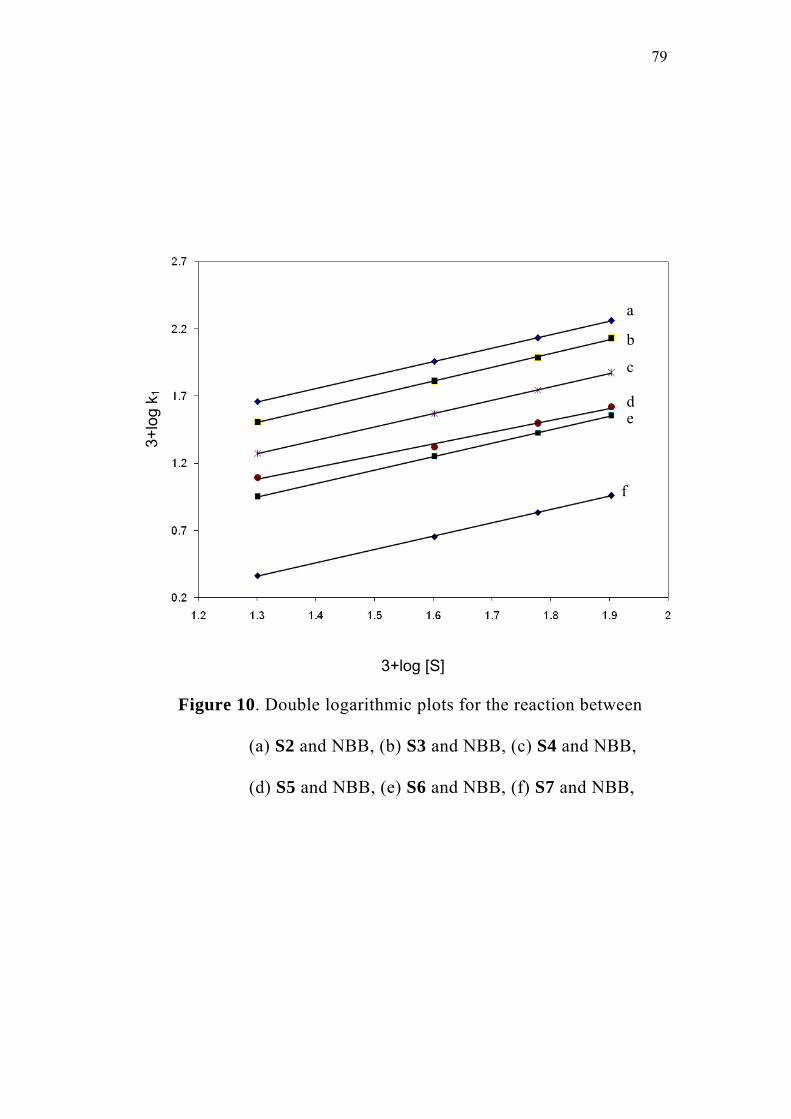
79
Figure 10. Double logarithmic plots for the reaction between
(a) S2 and NBB, (b) S3 and NBB, (c) S4 and NBB,
(d) S5 and NBB, (e) S6 and NBB, (f) S7 and NBB,
3+log [S]
3+lo
g k 1
a
b
c
d e
f
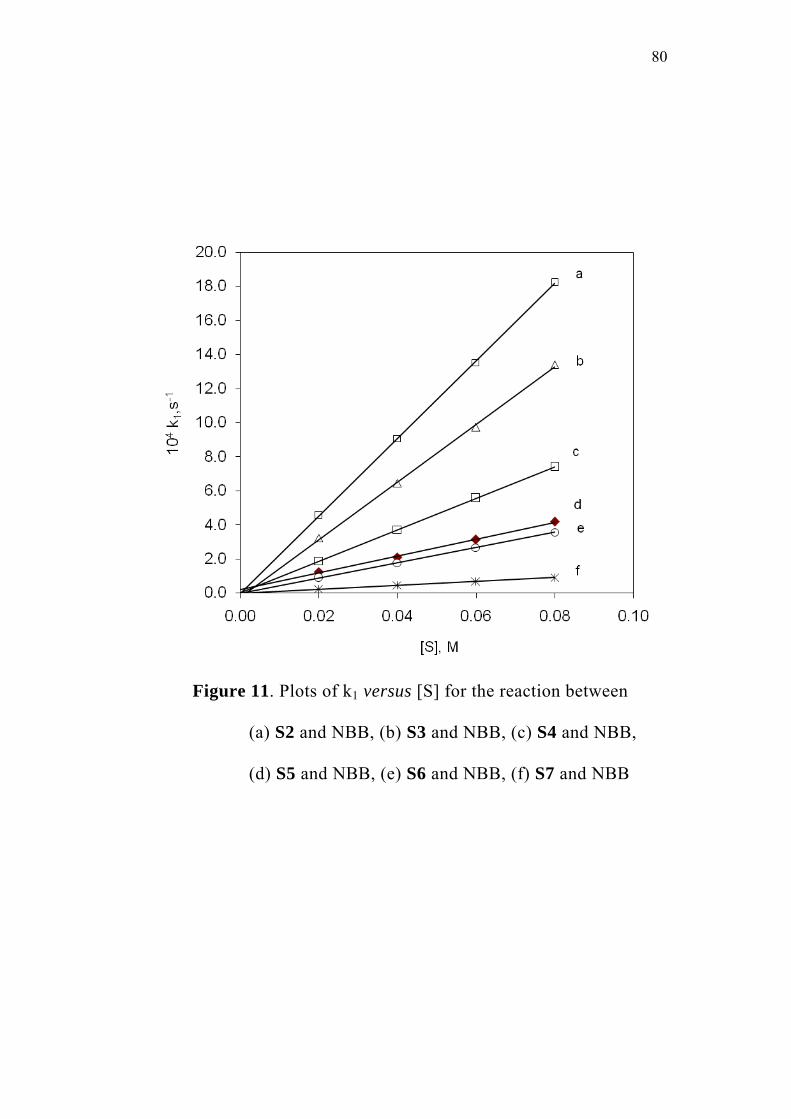
80
Figure 11. Plots of k1 versus [S] for the reaction between
(a) S2 and NBB, (b) S3 and NBB, (c) S4 and NBB,
(d) S5 and NBB, (e) S6 and NBB, (f) S7 and NBB
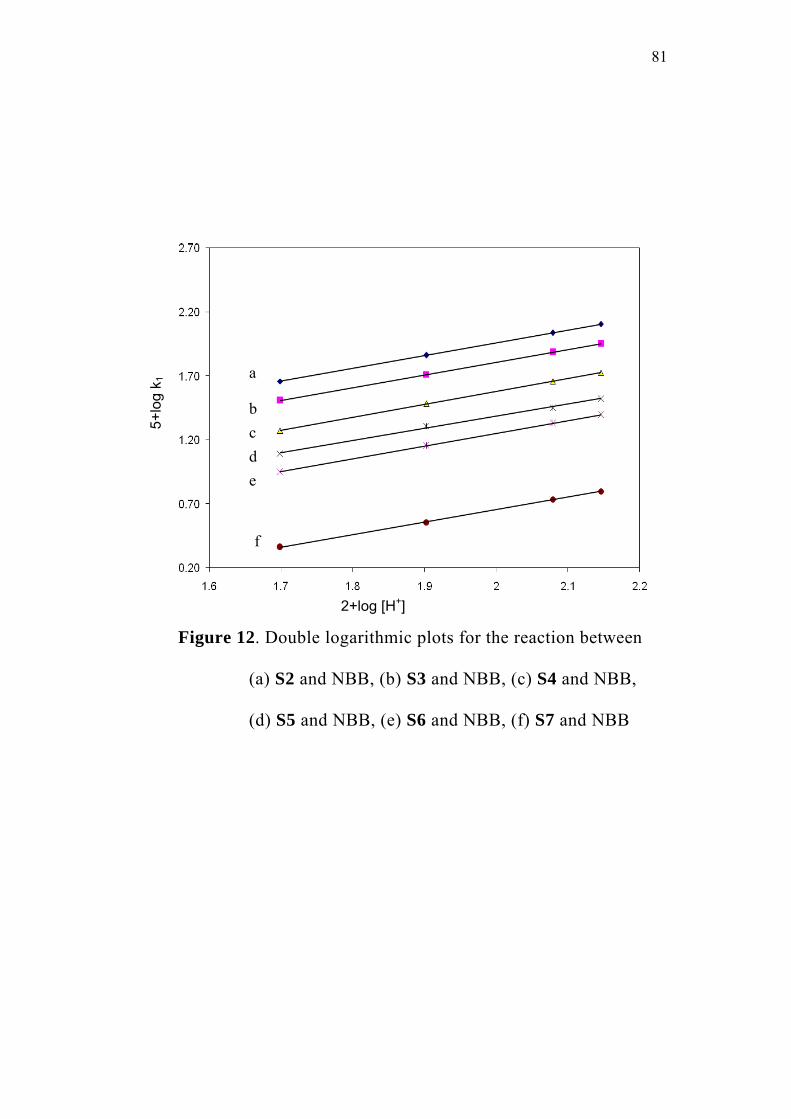
81
Figure 12. Double logarithmic plots for the reaction between
(a) S2 and NBB, (b) S3 and NBB, (c) S4 and NBB,
(d) S5 and NBB, (e) S6 and NBB, (f) S7 and NBB
2+log [H+]
5+lo
g k 1
a
b
c
d
e
f

82
Figure 13. Plots of k1 versus [H+] for the reaction between
(a) S2 and NBB, (b) S3 and NBB, (c) S4 and NBB,
(d) S5 and NBB, (e) S4 and NBB, (f) S7 and NBB
[H+], M
104 k
1, s
-1

83
3.5 Effect of Substituents and Applicability of LFER
All the seven 4-oxoacids with mono substituents in the phenyl ring
are subjected to oxidation by NBB in 50% aqueous acetic acid
medium in the presence of 0.5 M perchloric acid at four different
temperatures from 303 K to 323 K.
It has been observed that the presence of electron-releasing
groups in the phenyl ring enhances the reaction rate while the electron
withdrawing groups retard the rate. The second order rate constants
for the oxidation of all of the substituted 4-oxoacids are presented
in Table 15.
The reactivity decreases for substituents in the order 4-Methoxy >
4-Methyl > 4-Phenyl > 4- H > 4-Cl > 4-Br > 3-NO2
To correlate the effect of the substituents on the reaction rate
and to find out the validity of linear free energy relationship in the
present series, Hammett’s equation
log k = log k0 +
is applied. The Hammett’s plot for the oxidation of 4-oxoacids by NBB
at various temperatures is found to be linear. The Hammett’s plots are
shown in Figures 14-17. The reaction constant values obtained
from the plots are negative and are presented in Table 16.

84
Table 15. Second order rate constants for the oxidation of 4-
oxoacidsa
S 10 2 k2
b, M-1 s-1
303 K 308 K 313 K 323 K
S1 0.81 ± 0.04 1.13 ± 0.85 1.26 ± 0.11 1.71 ± 0.32
S2 2.27 ± 0.30 2.60 ± 0.30 2.94 ± 0.41 3.45 ± 0.29
S3 1.61 ± 0.29 1.78 ± 0.03 1.92 ± 0.20 2.17 ± 0.25
S4 0.94 ± 0.06 1.14 ± 0.05 1.32 ± 0.01 1.71 ± 0.08
S5 0.62 ± 0.28 0.78 ± 0.54 0.93 ± 0.08 1.22 ± 0.11
S6 0.44 ± 0.02 0.57 ± 0.01 0.74 ± 0.01 0.99 ± 0.01
S7 0.12 ± 0.02 0.23 ± 0.01 0.31 ± 0.08 0.51 ± 0.04
a General conditions : [S] = 0.02 M, [NBB] = 0.002 M, [H+] = 0.5 M,
[NaClO4] = 0.5 M, Solvent composition is 50%-Acetic acid - 50%
Water (v/v); b Individual k2 values estimated as k1 / [S].
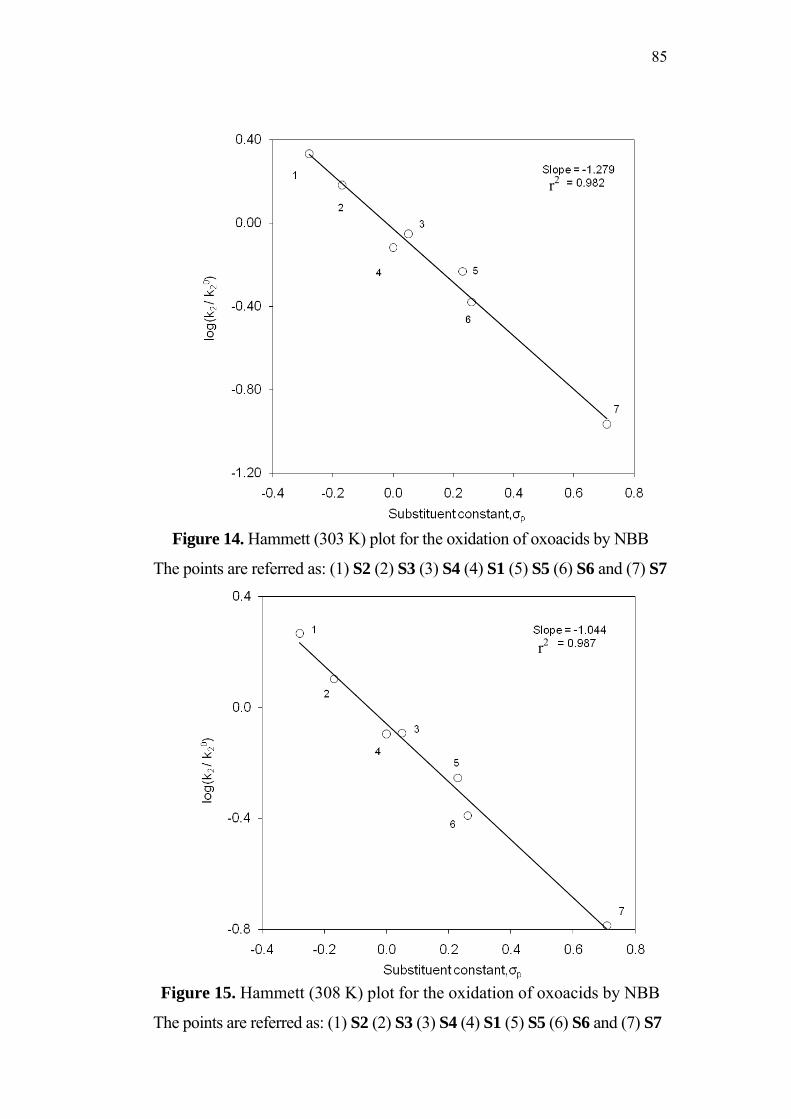
85
Figure 14. Hammett (303 K) plot for the oxidation of oxoacids by NBB
The points are referred as: (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Figure 15. Hammett (308 K) plot for the oxidation of oxoacids by NBB
The points are referred as: (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
r2
r2
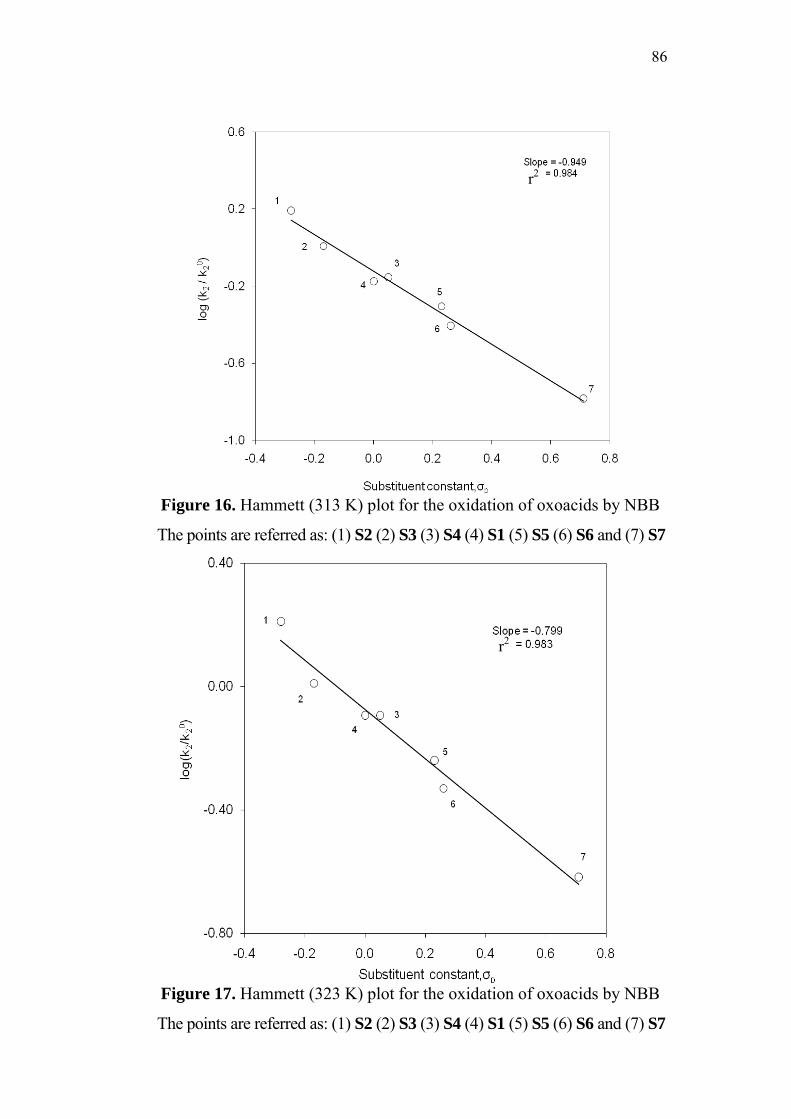
86
Figure 16. Hammett (313 K) plot for the oxidation of oxoacids by NBB
The points are referred as: (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Figure 17. Hammett (323 K) plot for the oxidation of oxoacids by NBB
The points are referred as: (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
r2
r2

87
Table 16. Reaction constant values at different temperatures a
Temperature
K
Reaction constant b
Correlation
coefficient
303 -1.28 0.982
308 -1.04 0.987
313 -0.95 0.984
323 -0.80 0.983 a values were taken from reported works302-304.
b The values were obtained by correlating log (k2 / k20) with p for the reactions
of S1-S7 with NBB.
The values indicate the sensitivity of a reaction to the effects
of electronic perturbation. It also provides information about the nature
of the transition state involved during the reaction. A reaction
involving a development of positive charge in the transition state is
aided by electron-releasing substituents and the valueis negative305-307.
In the present investigation, the acceleration of reaction rate
with the electron-releasing substituents and the negative value of the
reaction constant, indicate explicitly that the mechanism of oxidation
involves the development of positive charge in the transition state.
The Hammett correlations were also made for the rate constants
obtained at different solvent compositions (c.f. Table 7) and the
values are reported in the following Table 17 .

88
Table 17. Reaction constant values at different solvent compositionsa
Solvent
CH3COOH-H2O (v/v) %
Reaction constant b
Correlation
coefficient
50-50 -1.28 0.982
60-40 -1.06 0.986
70-30 -0.97 0.988
80-20 -0.84 0.987 a values were taken from reported works302-304.
b The values were obtained by correlating log (k2 / k20) with p for the reactions
of S1-S7 with NBB.
The data reveal that the selectivity of the oxidant towards
oxoacids decreases with increase in the acetic acid content in the
solvent mixture.
3.6 Mechanism of Oxidation
A probable mechanism for the oxidation of 4-oxoacids by N-
bromobenzamide has been proposed based on the experimental results
and in analogy with the oxidation of oxo compounds with other
oxidants. The results obtained in the kinetic study are briefly
summarized below.
i) The reaction is first order each with respect to the 4-
oxoacid, NBB, and H+ ion.
ii) The linear increase in the reaction rate with the increase
in [H+] ion is attributed to the formation of hypobromous

89
acidium ion (H2O+Br) and to the enolization of the 4-
oxoacid.
iii) The formation of hypobromous acidium ion and the
enolization of the 4-oxoacid is facilitated at lower
dielectric constant of the medium.
iv) The rate of enolization is found to be greater than the rate
of oxidation.
v) The course of the oxidation does not involve any free
radical intermediate.
vi) The retarding effect of the addition of benzamide
suggests that the pre-equilibrium step involves a process
in which benzamide is one of the products.
vii) The reaction rate is enhanced by electron-releasing
substituents in the phenyl ring while the rate is retarded
by electron-withdrawing substituents.
viii) The reaction constant is negative which indicates the
electron deficient transition state.
Considering these facts and findings a suitable mechanism
has been proposed for the oxidation (scheme 1).
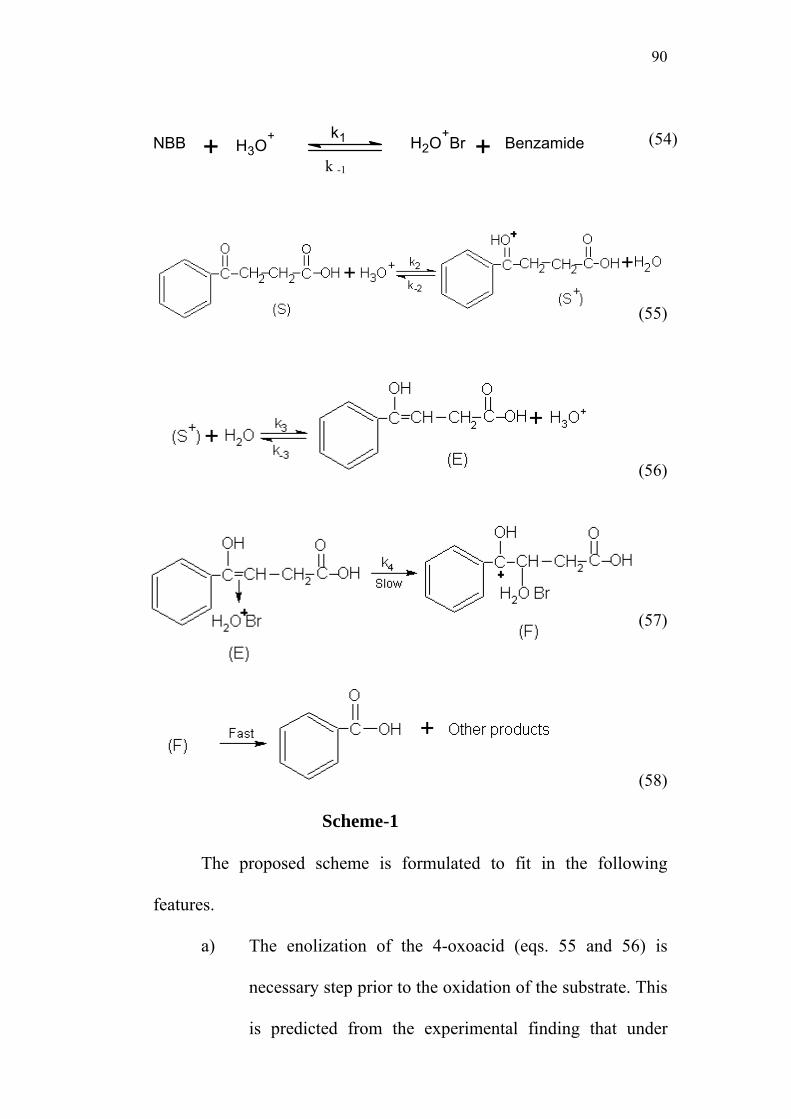
90
NBB + H3O+ H2O
+Br Benzamide+
k1
k-1
(55)
(56)
(57)
(58)
Scheme-1
The proposed scheme is formulated to fit in the following
features.
a) The enolization of the 4-oxoacid (eqs. 55 and 56) is
necessary step prior to the oxidation of the substrate. This
is predicted from the experimental finding that under
k -1
(54)

91
identical conditions, the rate of enolization is greater than
the rate of oxidation. Similar conclusions have been
arrived at in the oxidation of several ketones291,292,294 by
acid permanganate.
b) The interaction of oxidizing species H2O+Br with the
enol form of the 4-oxoacid in the rate determining step
(eq.57) leads to the formation of intermediate (F). A
similar type of intermediate has been proposed in the
oxidation of many organic and inorganic compounds308-310.
The intermediate then undergoes further oxidative
cleavage to give the products.
3.7 Derivation of Rate Law
Based on kinetic observations and the mechanism proposed, the
following rate expression can be derived applying steady-state
approximation,
The rate of the reaction is given by
-d [NBB] = k4 [E] [H2O+Br] (59)
dt Applying steady state approximation for [E] k -3 [H3O
+] [E] + k4 [H2O+Br] [E] = k3 [S
+] [H2O] k3 [S
+] [H2O] Therefore, [E] = (60) k-3 [H3O
+] + k4 [H2O+Br]

92
Applying steady state approximation for [S+] k -2 [H2O] [S+] + k 3 [H2O] [S+] = k 2 [H3O
+] [S] + k -3 [H3O+] [E]
k 2 [H3O
+] [S] + k -3 [H3O+] [E]
Therefore, [S+] = k -2 [H2O] + k 3 [H2O]
[H3O
+] { k 2 [S] + k -3 [E]} i.e. [S+] = (61) [H2O] (k -2 + k 3 ) Using the value of [S+] in eq. (60). k 3 [H3O
+] { k 2 [S] + k -3 [E] } [E] = ( k -2 + k 3 ) { k -3 [H3O
+] + k 4 [H2O+Br] }
[E] (k -2 + k 3 ) {k -3 [H3O
+] + k 4 [H2O+Br] } = k 3 [H3O
+] { k 2 [S] + k -3 [E] }
[E](k -2 + k 3 ){ k -3 [H3O+]+k 4[H2O
+Br]}= k 3 k 2 [H3O
+][S]+k3 k -3 [H3O+][E]
[E]{(k -2 +k 3 ) k -3 [H3O
+] + k 4 [H2O+Br] – k 3 k -3 [H3O
+]}= k 3 k2 [H3O+] [S]
k 3 k 2 [H3O
+] [S] [E] = { (k -2 +k 3 ) k -3 [H3O
+] + k 4 [H2O+Br]} – k 3 k -3 [H3O
+] k 3 k 2 [H3O
+] [S] [E] =
k -2 k -3 [H3O+] + k -2 k 4 [H2O
+Br] + k 3 k 4 [H2O
+Br]
k 3 k 2 [H3O
+] [S] [E] = (62)
k -2 k -3 [H3O+] + k 4 (k -2 + k 3) [H2O
+Br]

93
Substituting the value of [E] in eq. (59)
-d [NBB] k 2 k 3 k 4 [H3O+] [S] [H2O
+Br]
= (63) dt k -2 k -3 [H3O
+] + k 4 (k -2 + k 3) [H2O+Br]
At high concentration of [H3O+] = 0.5 M
k -2 k -3 [H3O
+] > > k 4 (k -2 + k 3) [H2O+Br]
So the eq. (63) simplifies to the form
-d [NBB] k 2 k 3 k 4 [H3O+] [S] [H2O
+Br]
= dt k -2 k -3 [H3O
+] k 2 k 3 k 4 [S] [H2O
+Br]
= (64)
k -2 k -3
The value of [H2O+Br] can be obtained from eq. (54) in
the scheme of mechanism
k -1 [NBB] [H3O+]
K a = = k 1 [H2O
+Br] [Benz]
[NBB] [H3O+]
Therefore, [H2O+Br] =
Ka [Benz] Using the value of [H2O
+Br] in eq. (64) -d [NBB] k 2 k 3 k 4 [S] [H3O
+] [NBB] = (65) dt k -2 k -3 Ka [Benz]

94
Hence, at higher concentration of mineral acid, the
reaction is first order each with respect to the oxoacid (S), [NBB]
and [H3O+].
The observed rate constant at high [H3O+] is
k 2 k 3 k 4 kobs = (66)
k -2 k -3 K a
3.8 Structure –Reactivity Correlations
The effect of the substituents present in the phenyl ring of the 4-
oxoacids (S1-S7), on the rates of oxidation has been correlated
quantitatively by the application of Hammett equation. Plots of
log (k2 / k20) versus p are excellently linear with negative slopes
(Figures 14-17). The ρ values for the oxidation range between -1.28 and
-0.80 at different temperatures.
It is generally recognized that oxidations lead to electron
deficient species which are either radical cations, radicals or
carbocations. These reactions normally have a negative ρ value and the
magnitude of values depends on the extent of electron deficiency.
Oxidation reactions involving free radical formation in the rate
controlling step usually have a small negative ρ value and the
oxidations involving the formation of carbocation have a large negative
ρ values. Based on these arguments we expect a large value but the

95
measured ρ value is in the range -1.28 to -0.84. The low value may
be attributed to the nature of observed rate constant. The observed rate
constant is composite of several terms and is shown in eq.(66).
The terms shown in eq.(66) deserve comment. The rate constant
k3 depends on the concentration of the protonated substrate (S+) and
the electron donating substituents tend to delocalize the positive charge
on S+ and hence favors the formation of this positive species. The
value for the enolization of the 4-oxoacids, determined by bromination
method has been reported to be -0.75311. In the rate limiting step
(eq.57) of the scheme, the formation of the carbocation is facilitated by
electron releasing substituents.
Thus we get a slightly low negative value of -1.28 (at 303 K)
because kobs is composite of the enolization as well as the oxidation of the
4-oxoacids. Hence in the present investigation the measured value and
other findings fit in with the formulation of mechanism outlined in the
scheme 1.
3.9 Activation Parameters
The dependence of reaction rate on the structure of the reacting
molecule is related to activation parameters. The decisive term
concerning the dependence on the structure is neither free energy nor
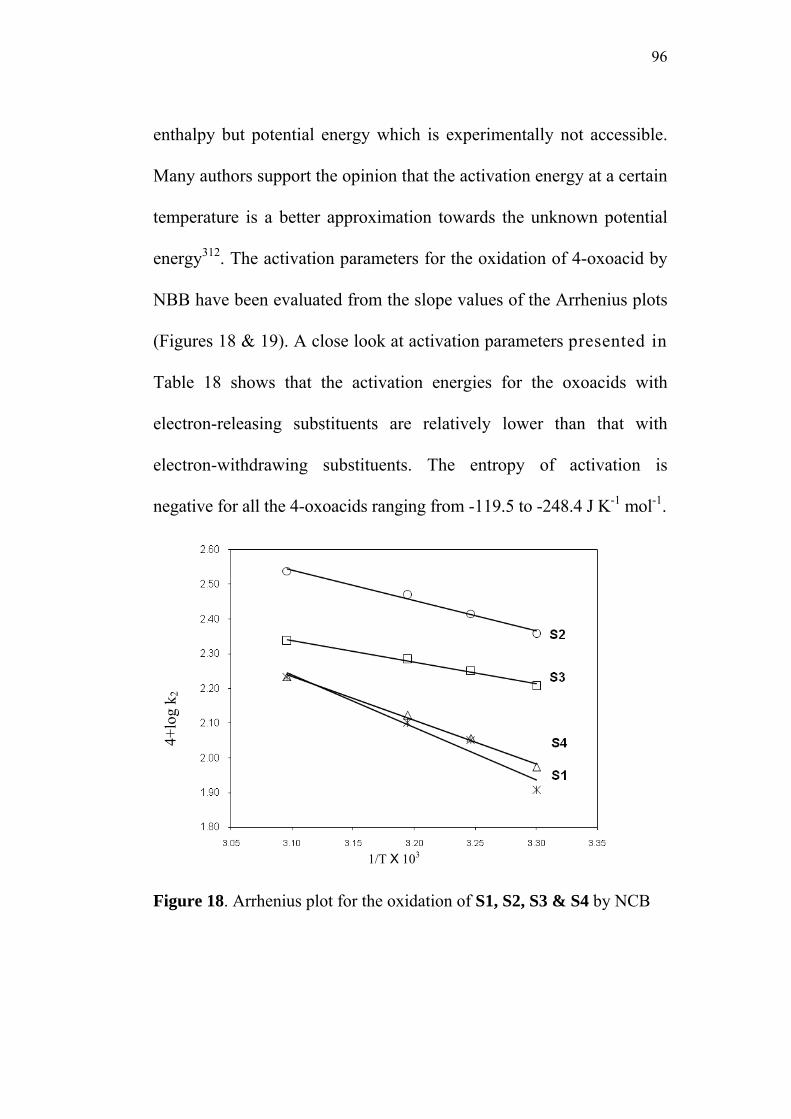
96
enthalpy but potential energy which is experimentally not accessible.
Many authors support the opinion that the activation energy at a certain
temperature is a better approximation towards the unknown potential
energy312. The activation parameters for the oxidation of 4-oxoacid by
NBB have been evaluated from the slope values of the Arrhenius plots
(Figures 18 & 19). A close look at activation parameters presented in
Table 18 shows that the activation energies for the oxoacids with
electron-releasing substituents are relatively lower than that with
electron-withdrawing substituents. The entropy of activation is
negative for all the 4-oxoacids ranging from -119.5 to -248.4 J K-1 mol-1.
Figure 18. Arrhenius plot for the oxidation of S1, S2, S3 & S4 by NCB
1/T X 103
4+lo
g k 2
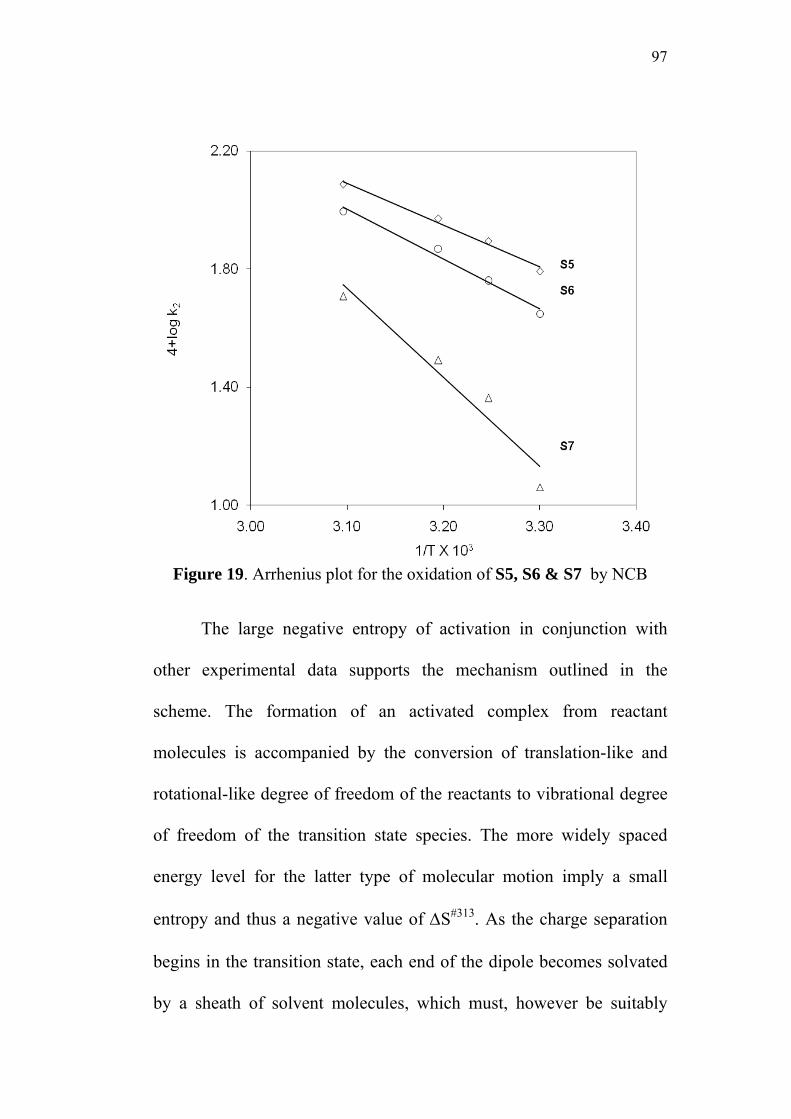
97
Figure 19. Arrhenius plot for the oxidation of S5, S6 & S7 by NCB
The large negative entropy of activation in conjunction with
other experimental data supports the mechanism outlined in the
scheme. The formation of an activated complex from reactant
molecules is accompanied by the conversion of translation-like and
rotational-like degree of freedom of the reactants to vibrational degree
of freedom of the transition state species. The more widely spaced
energy level for the latter type of molecular motion imply a small
entropy and thus a negative value of S#313. As the charge separation
begins in the transition state, each end of the dipole becomes solvated
by a sheath of solvent molecules, which must, however be suitably
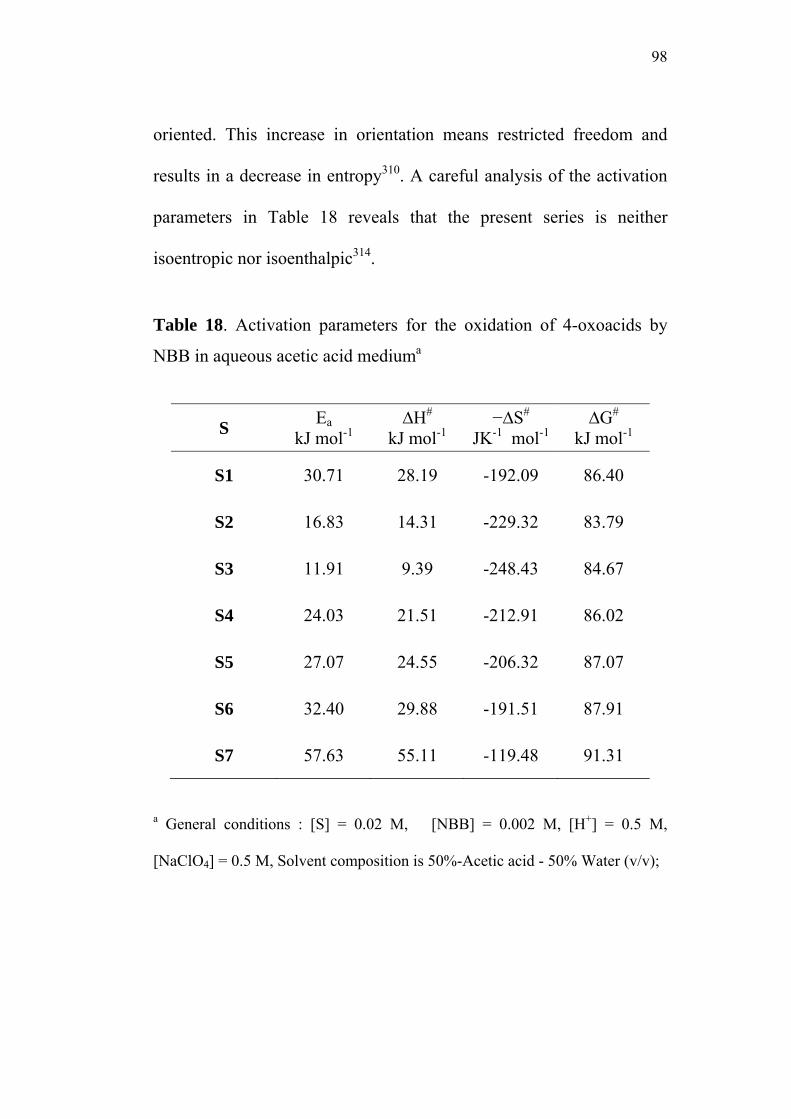
98
oriented. This increase in orientation means restricted freedom and
results in a decrease in entropy310. A careful analysis of the activation
parameters in Table 18 reveals that the present series is neither
isoentropic nor isoenthalpic314.
Table 18. Activation parameters for the oxidation of 4-oxoacids by
NBB in aqueous acetic acid mediuma
S Ea
kJ mol-1 ∆H#
kJ mol-1 −∆S#
JK-1 mol-1∆G#
kJ mol-1
S1 30.71 28.19 -192.09 86.40
S2 16.83 14.31 -229.32 83.79
S3 11.91 9.39 -248.43 84.67
S4 24.03 21.51 -212.91 86.02
S5 27.07 24.55 -206.32 87.07
S6 32.40 29.88 -191.51 87.91
S7 57.63 55.11 -119.48 91.31
a General conditions : [S] = 0.02 M, [NBB] = 0.002 M, [H+] = 0.5 M,
[NaClO4] = 0.5 M, Solvent composition is 50%-Acetic acid - 50% Water (v/v);

99
3.10 Isokinetic Relationships
Leffler’s isokinetic equations
Ea = E0 + 2.303 R log A (67)
H# = H0 + S# (68)
hold good in a series of related reactions. The validity of the isokinetic
relation can be tested graphically by plotting either H# versus S# or
Ea versus log A. This linear relationship between activation enthalpies
and activation entropies or activation energies and frequency factors in
a series of related reactions is known as isokinetic relationship315,316.
The validity of the isokinetic plot is questionable317,318 because
the quantities H# and S# are mutually dependent, both being derived
from the same experimental rate constants. An alternative graphical
method for finding out the isokinetic temperature is suggested by
Exner. A plot of the rate constants measured at two different
temperatures [log k2 (T2) versus log k2 (T1)] is known as Exner plot.
From the slope b, of the Exner plot, the isokinetic temperature , can
be calculated using the equation.
= T1 T2 (1- b) (69)
T1 – b T2
Based on the values of b, the reaction series (Table 19) have been
characterized as follows317.

100
Table 19. b values of the reaction series
Series b
Isoentropic ∞ T2 / T1
Isoenthalpic 0 1
Compensation > Texp <1
The Exner plots drawn for the reaction by choosing k2 values at
any two different temperatures are shown in Figures 20-22 . The values
of b are less than unity in all the plots. This indicates that the present
reaction series is neither isoentropic nor isoenthalpic but involves
compensation. The lsokinetic temperature evaluated from the Exner
plots is found to be 347 K. As the experiment has been carried at a
temperature far away from the isokinetic temperature the application of
Hammett equation to the observed kinetic data is valid. The validity
of isokinetic relationship in the present study implies that all the 4-
oxoacids undergo oxidation by the same mechanism310, 319, 320.
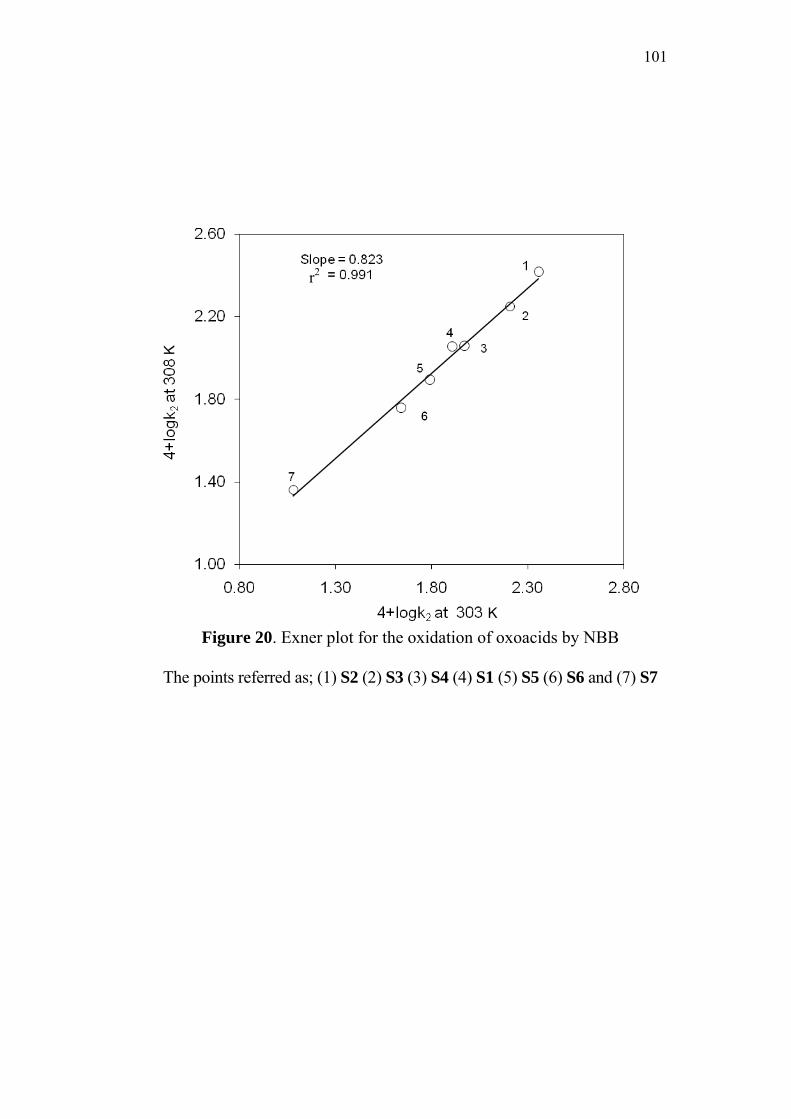
101
Figure 20. Exner plot for the oxidation of oxoacids by NBB
The points referred as; (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
K
K
r2

102
Figure 21. Exner plot for the oxidation of oxoacids by NBB
The points referred as; (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
K
K
r2
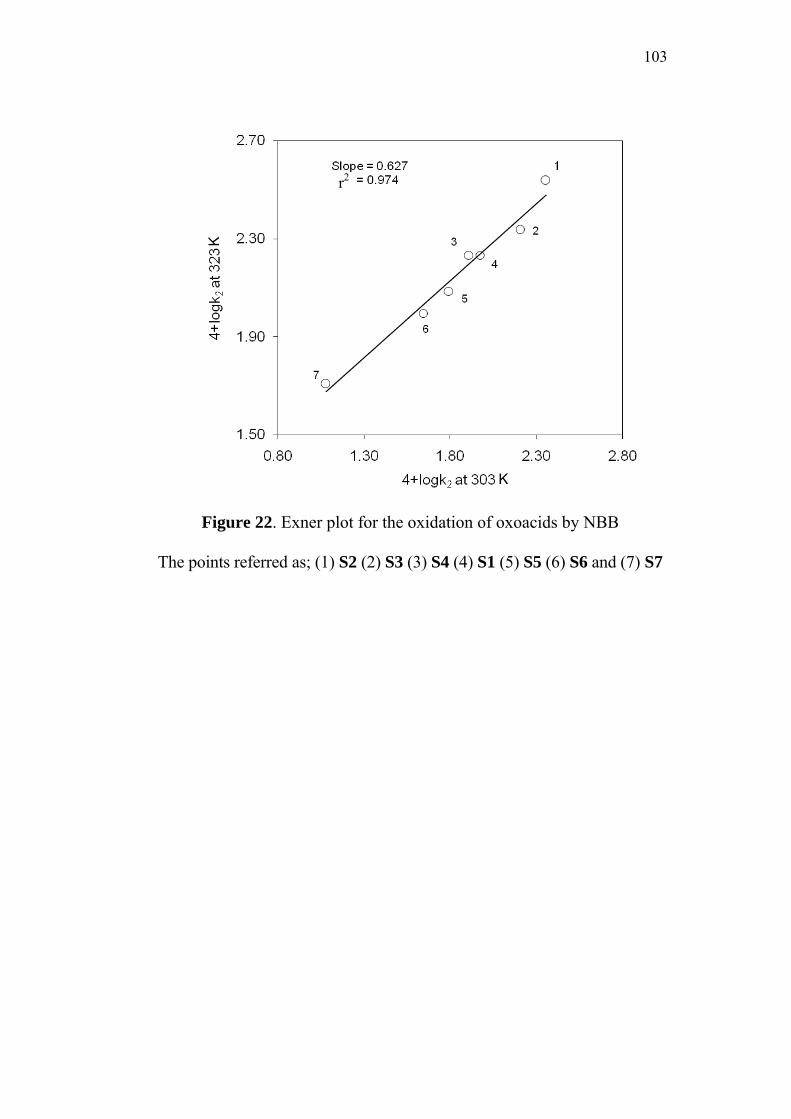
103
Figure 22. Exner plot for the oxidation of oxoacids by NBB
The points referred as; (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
K
K
r2

CHAPTER 4
4-Oxoacids and N-Bromosaccharin system

104
CHAPTER 4
4-Oxoacids and N-Bromosaccharin System
Studies of oxidation of various organic compounds by N-halo amides
in the presence of perchloric acid have attracted considerable
attention176-213. N-Bromosaccharin (NBSac) is a source of positive
halogen and have been exploited as oxidant for a variety of substrates
in both the acidic and alkaline media.
Moreover with the same 4-oxoacids no detailed kinetic study on
oxidation with N-bromosaccharin (NBSac) has been attempted so far.
It is also interesting to know whether the oxidation of unsubstituted and
substituted 4-oxoacids by NBSac follow similar type of mechanism or
not. In this chapter the results obtained from a detailed kinetic study on
the oxidation of 4-oxoacids with NBSac in the presence of perchloric
acid in aqueous acetic acid medium are analyzed.
4.1 Kinetics of Oxidation of 4-Oxo-4-phenylbutanoic
Acid (S1) by N-Bromosaccharin
The kinetics of oxidation of 4-oxoacids by N- bromosaccharin (NBSac)
has been studied potentiometrically in aqueous acetic acid medium in the
presence of perchloric acid at constant ionic strength. The ionic strength
of the medium was maintained by the addition of NaClO4. Using 10 – 50
fold excess of oxoacid over oxidant, excellent ln (Et - E∞) versus time

105
plots were obtained; from these, the pseudo first order rate constants k1,
and hence k2 were determined. (cf. chapter 3).
4.1.1 Effect of varying [reactants]0
The pseudo first order rate constants (k1) obtained at different initial
concentrations of one reactant, keeping the concentration of other
reactant constant for the oxidation of 4-oxo-4-phenylbutanoic acid
with NBSac in the presence of perchloric acid are listed in Table 20.
The plots of ln(Et - E∞) versus time (sec) for the various concentrations
of the oxoacids are linear (Figure 23).
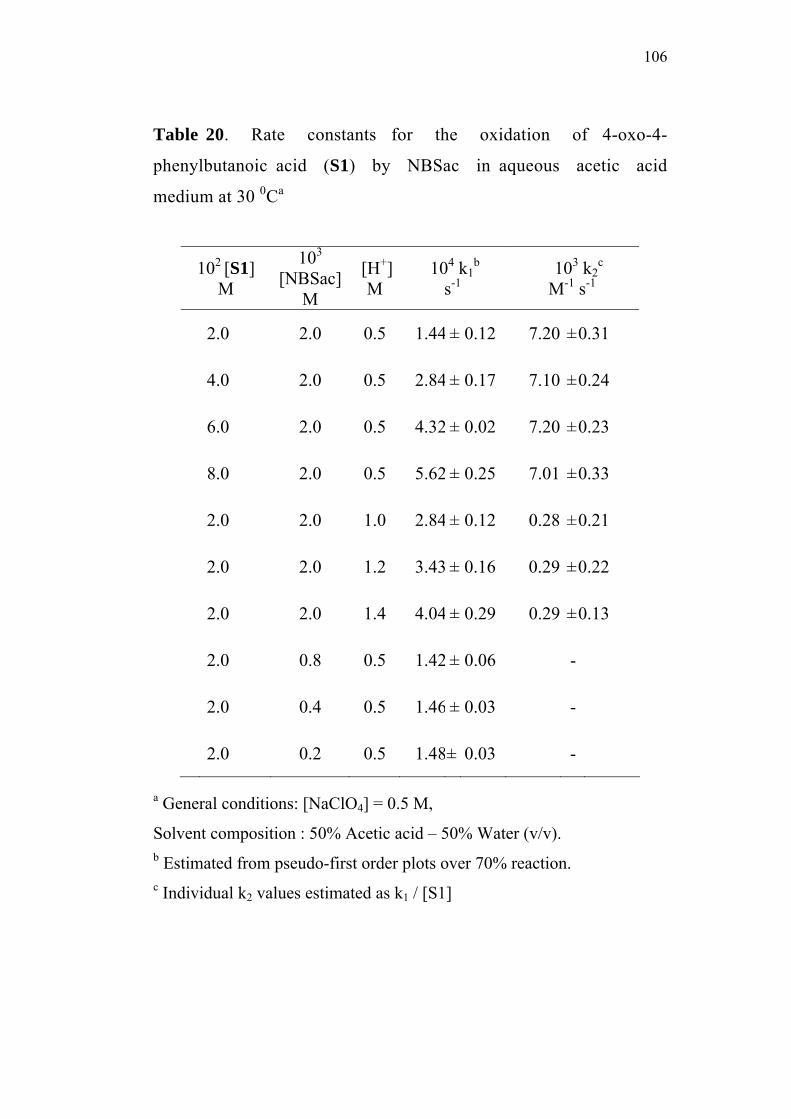
106
Table 20. Rate constants for the oxidation of 4-oxo-4-
phenylbutanoic acid (S1) by NBSac in aqueous acetic acid
medium at 30 0Ca
102 [S1] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 1.44 ± 0.12 7.20 ± 0.31
4.0 2.0 0.5 2.84 ± 0.17 7.10 ± 0.24
6.0 2.0 0.5 4.32 ± 0.02 7.20 ± 0.23
8.0 2.0 0.5 5.62 ± 0.25 7.01 ± 0.33
2.0 2.0 1.0 2.84 ± 0.12 0.28 ± 0.21
2.0 2.0 1.2 3.43 ± 0.16 0.29 ± 0.22
2.0 2.0 1.4 4.04 ± 0.29 0.29 ± 0.13
2.0 0.8 0.5 1.42 ± 0.06 -
2.0 0.4 0.5 1.46 ± 0.03 -
2.0 0.2 0.5 1.48± 0.03 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S1]
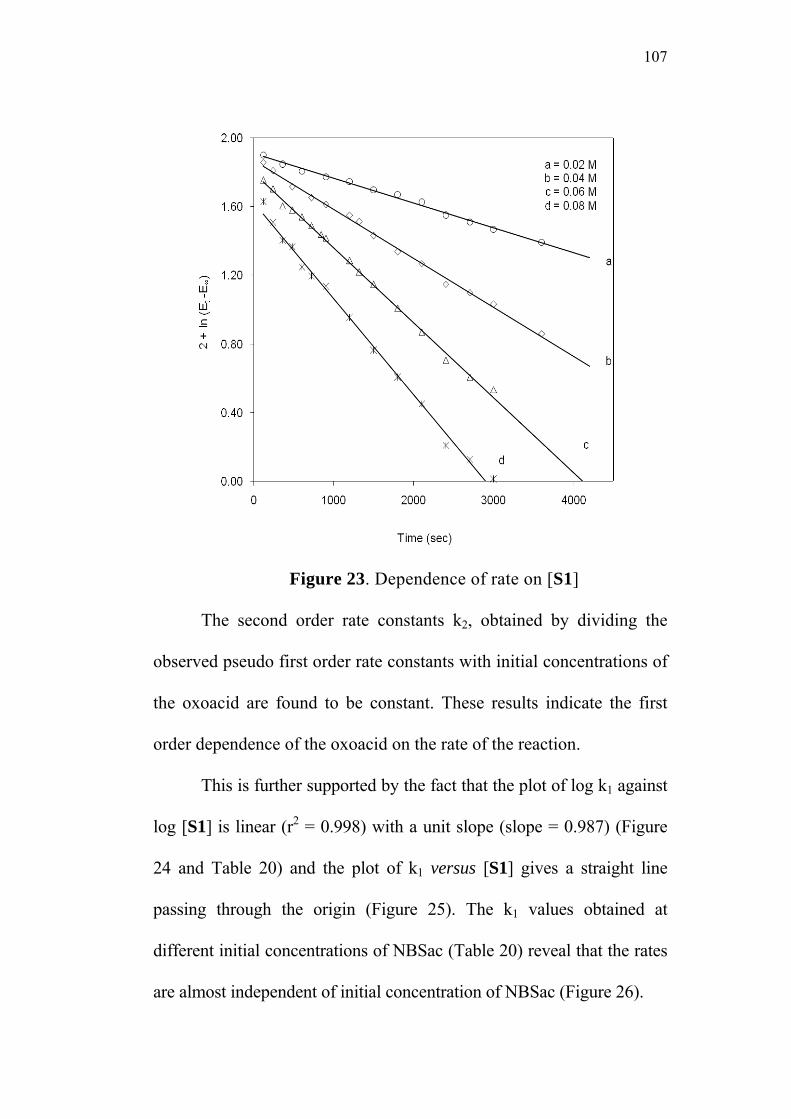
107
Figure 23. Dependence of rate on [S1]
The second order rate constants k2, obtained by dividing the
observed pseudo first order rate constants with initial concentrations of
the oxoacid are found to be constant. These results indicate the first
order dependence of the oxoacid on the rate of the reaction.
This is further supported by the fact that the plot of log k1 against
log [S1] is linear (r2 = 0.998) with a unit slope (slope = 0.987) (Figure
24 and Table 20) and the plot of k1 versus [S1] gives a straight line
passing through the origin (Figure 25). The k1 values obtained at
different initial concentrations of NBSac (Table 20) reveal that the rates
are almost independent of initial concentration of NBSac (Figure 26).

108
Figure 24. Order with respect to [S1]
Figure 25. Plot of k1 versus [S1]
r2
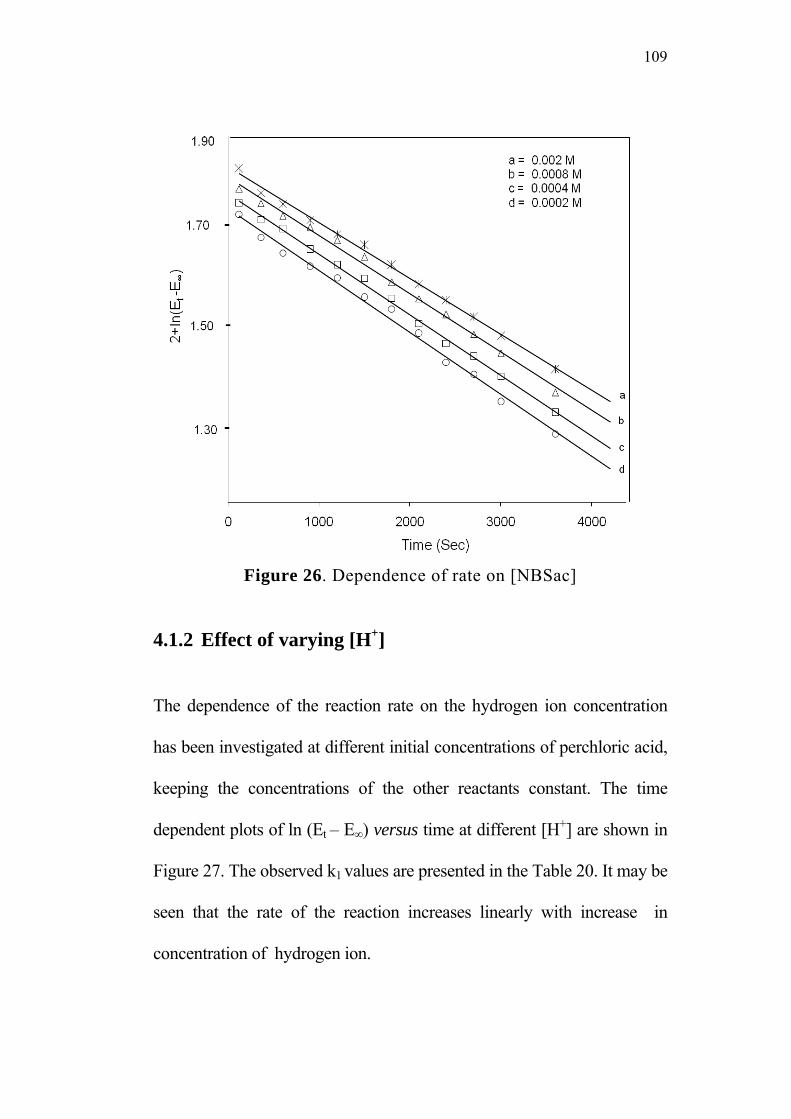
109
Figure 26. Dependence of rate on [NBSac]
4.1.2 Effect of varying [H+]
The dependence of the reaction rate on the hydrogen ion concentration
has been investigated at different initial concentrations of perchloric acid,
keeping the concentrations of the other reactants constant. The time
dependent plots of ln (Et – E∞) versus time at different [H+] are shown in
Figure 27. The observed k1 values are presented in the Table 20. It may be
seen that the rate of the reaction increases linearly with increase in
concentration of hydrogen ion.
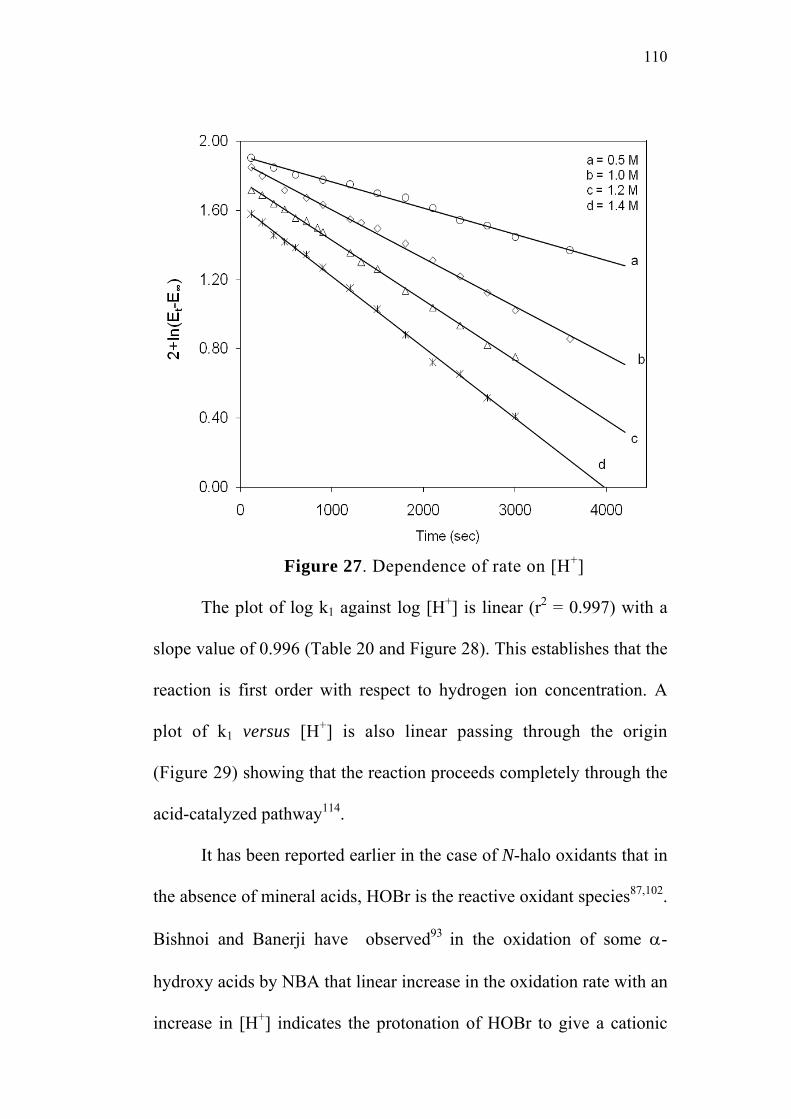
110
Figure 27. Dependence of rate on [H+]
The plot of log k1 against log [H+] is linear (r2 = 0.997) with a
slope value of 0.996 (Table 20 and Figure 28). This establishes that the
reaction is first order with respect to hydrogen ion concentration. A
plot of k1 versus [H+] is also linear passing through the origin
(Figure 29) showing that the reaction proceeds completely through the
acid-catalyzed pathway114.
It has been reported earlier in the case of N-halo oxidants that in
the absence of mineral acids, HOBr is the reactive oxidant species87,102.
Bishnoi and Banerji have observed93 in the oxidation of some -
hydroxy acids by NBA that linear increase in the oxidation rate with an
increase in [H+] indicates the protonation of HOBr to give a cationic
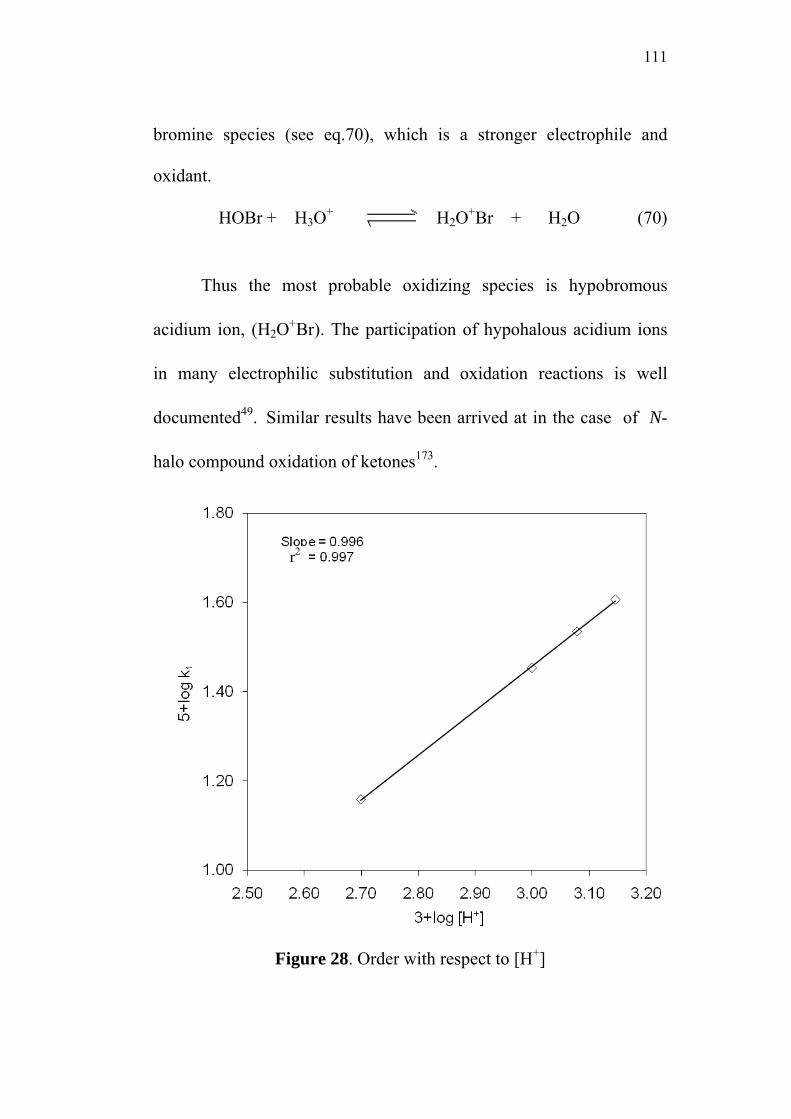
111
bromine species (see eq.70), which is a stronger electrophile and
oxidant.
HOBr + H3O+ H2O
+Br + H2O (70)
Thus the most probable oxidizing species is hypobromous
acidium ion, (H2O+Br). The participation of hypohalous acidium ions
in many electrophilic substitution and oxidation reactions is well
documented49. Similar results have been arrived at in the case of N-
halo compound oxidation of ketones173.
Figure 28. Order with respect to [H+]
r2
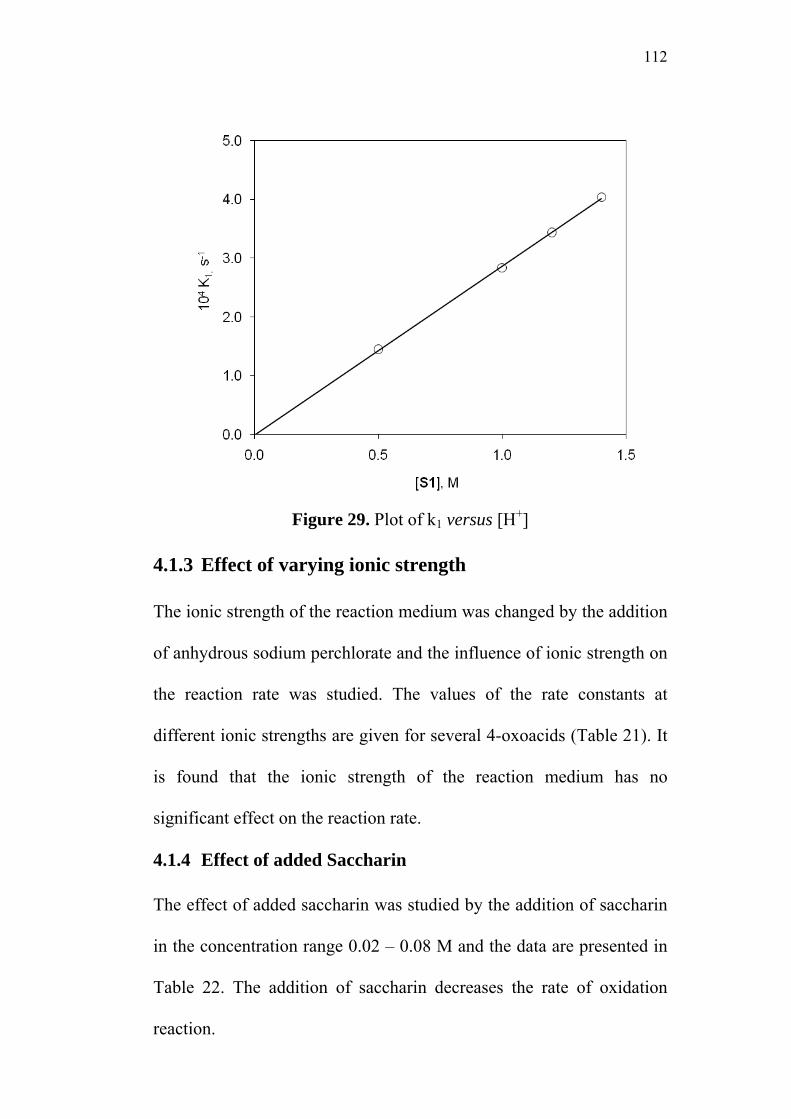
112
Figure 29. Plot of k1 versus [H+]
4.1.3 Effect of varying ionic strength
The ionic strength of the reaction medium was changed by the addition
of anhydrous sodium perchlorate and the influence of ionic strength on
the reaction rate was studied. The values of the rate constants at
different ionic strengths are given for several 4-oxoacids (Table 21). It
is found that the ionic strength of the reaction medium has no
significant effect on the reaction rate.
4.1.4 Effect of added Saccharin
The effect of added saccharin was studied by the addition of saccharin
in the concentration range 0.02 – 0.08 M and the data are presented in
Table 22. The addition of saccharin decreases the rate of oxidation
reaction.

113
Thus the retardation of reaction rate on the addition of saccharin
suggests128 a pre-equilibrium step involving a process in which
saccharin is one of the products.
NBSac + H3O+ H2O
+Br + Saccharin (71)
Table 21. Effect of ionic strength for the oxidation of 4-oxoacids by
NBSac in aqueous acetic acid medium at 30 0C a
S [NaClO4]
M
104 k1b
s-1
S1 0.5 1.44 ± 0.12
1.0 1.41 ± 0.16
1.5 1.43 ± 0.17
2.0 1.42 ± 0.21
S2 0.5 5.25 ± 0.42
1.0 5.22 ± 0.33
1.5 5.21 ± 0.24
2.0 5.20 ± 0.21
S7 0.5 0.18 ± 0.03
1.0 0.19 ± 0.13
1.5 0.17 ± 0.12
2.0 0.20 ± 0.12
a General conditions: [S1] = 0.02 M, [NBSac] = 0.002M, [H+] = 0.5 M,
Solvent composition: 50% Acetic acid-50% Water (v/v). Temp:30 0C. b Estimated from pseudo-first order plots.

114
Table 22. Effect of added saccharin on the rate of the reactiona
S 102 [Saccharin] 104 k1b
M s-1
S1 Nil 1.44 ± 0.12
2.0 1.28 ± 0.11
4.0 1.07 ± 0.12
6.0 1.01 ± 0.13
8.0 0.87 ± 0.12
a General conditions: [S1] = 0.02 M, [NBSac] = 0.002M, [H+] = 0.5 M,
Solvent composition: 50% Acetic acid-50% Water (v/v). Temp:30 0C. b Estimated from pseudo-first order plots.
If this equilibrium is involved in the oxidation process, the
rate should be an inverse function of saccharin concentration, which
is borne out by the observation that the inverse of the rate constant
(Figure 30) gives a linear (r2 = 0.982) plot against [Saccharin].
Similar conclusions have been arrived at in the N-
chloronicotinamide oxidation of amino acids128, and N-
bromoacetamide oxidation of some - hydroxy acids93.
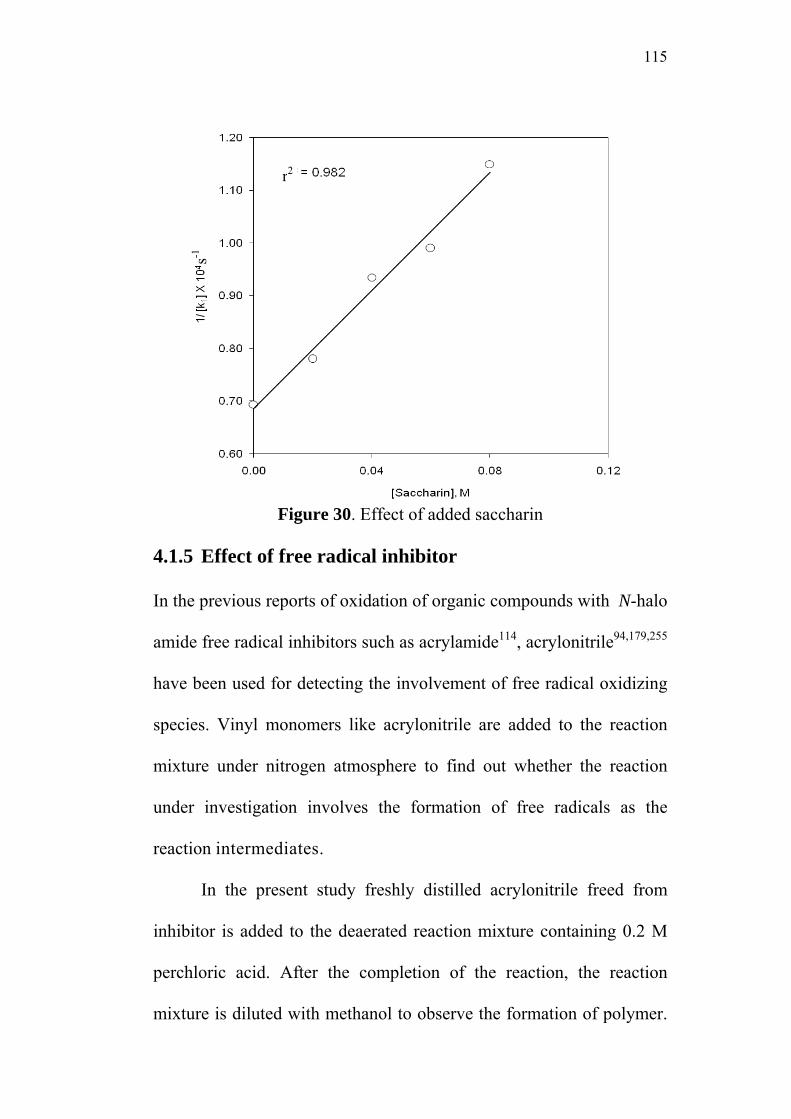
115
Figure 30. Effect of added saccharin
4.1.5 Effect of free radical inhibitor
In the previous reports of oxidation of organic compounds with N-halo
amide free radical inhibitors such as acrylamide114, acrylonitrile94,179,255
have been used for detecting the involvement of free radical oxidizing
species. Vinyl monomers like acrylonitrile are added to the reaction
mixture under nitrogen atmosphere to find out whether the reaction
under investigation involves the formation of free radicals as the
reaction intermediates.
In the present study freshly distilled acrylonitrile freed from
inhibitor is added to the deaerated reaction mixture containing 0.2 M
perchloric acid. After the completion of the reaction, the reaction
mixture is diluted with methanol to observe the formation of polymer.
s-1
r2

116
It is observed that the oxidation reaction does not induce the
polymerization.
Further it is confirmed by carrying out the oxidation reaction at
different initial concentrations of the inhibitor for the reaction of S1
with NBSac catalyzed by perchloric acid and the data are collected in
the Table 23. These data illustrate that the rate of the reaction is almost
unaffected in the presence of inhibitor. Consequently it may be inferred
that free radicals are not involved in the rate controlling step of the
present reaction.
Table 23. Effect of added acrylonitrile on the rate of the reaction a
S 103 [Acrylonitrile] 104 k1b
M s-1
S1 Nil 1.44 ± 0.12
3.0 1.45 ± 0.13
4.0 1.44 ± 0.11
6.0 1.43 ± 0.13
8.0 1.45 ± 0.12
a General conditions: [S1] = 0.02 M, [NBSac] = 0.002M, [H+] = 0.5 M,
Solvent composition: 50% Acetic acid-50% Water (v/v). Temp:300 C. b Estimated from pseudo-first order plots.

117
4.1.6 Effect of solvent composition
The effect of changing solvent composition on the reaction rate was
studied by varying the concentration of acetic acid from 50% - 80%.
The pseudo first-order rate constants for the oxidation reactions of all
oxoacids, S1-S7 with NBSac were estimated in the presence of
perchloric acid at constant ionic strength. The rate constants listed in
Table 24 suggest that the rate of the reaction increases with increase in
acetic acid content of the solvent mixture. A plot of log k1 versus 1/D
is linear with positive slope.
Table 24. Effect of solvent polarity on the rate of reactiona
S 104 k1b, s-1
rc CH3 COOH - H2O (v/v) % 50-50 60-40 70-30 80-20
S1 1.440.12 2.240.24 3.120.14 5.280.29 0.998
S2 5.250.42 6.830.34 7.850.48 9.350.60 0.983
S3 3.820.41 4.910.22 6.310.24 7.520.34 0.985
S4 1.560.11 2.110.07 2.920.14 4.230.18 0.998
S5 0.980.14 1.320.06 2.320.11 3.010.01 0.998
S6 0.910.02 1.070.05 1.680.12 2.360.15 0.994
S7 0.180.03 0.310.03 0.610.07 0.920.08 0.998
a General conditions: [S1] = 0.02 M, [NBSac] = 0.002M, [H+] = 0.5 M, b Estimated from pseudo-first order plots. c The values obtained from the plots drawn between 4+log k1 and 1/D

118
The observed effect is similar to those reported in the oxidation
of other organic compounds by NBA110. In the solution containing
acetic acid, one can not exclude the possibility of (AcO+HBr) acting as
a reactive oxidizing species.
The enhancement of the reaction rate with an increase in the
amount of acetic acid may generally be attributed to two factors, viz
(i) increase in acidity at constant [perchloric acid] and (ii) decrease in
dielectric constant with increase in HOAc content. The plots of log k1
against the inverse of dielectric constant are linear with positive slopes,
indicating an interaction between a positive ion and a dipole
molecule287-290. This supports the postulation of H2O+Br as the reactive
species.
But, in the oxidation of some amino acids by NBSac in aqueous
acetic acid medium, variation of solvent composition shows negligible
effect on the rate of the reaction110. The negligible effect of changing
the dielectric constant of the medium might be due to the involvement
of neutral species in the reaction.
In the present system, the significant rate enhancement, with the
lowering of dielectric constant of the solvent medium may be
attributed to the enolization of the ketoacid group in the 4-oxoacid.
This trend is observed for all the substituted 4-oxoacids taken for
investigation. The enolization of the keto group of the oxoacid is

119
facilitated by the increase in percentage of acetic acid in the solvent
medium and this may also favor the rate enhancement. This is in
agreement with the observations made by Basheer Ahamed and
Sikkandar255 during the oxidation of 4-oxoacids by acid permanganate.
4.1.7 Rate of enolization of substrate
The rate of enolization was studied by bromination method and it has
been discussed in the chapter 3.3.9. It is known that the rate of
enolization was much faster than the rate of oxidation (cf. Table 8).
Hence in the present system also we can infer that the enol forms of the
substrate probably participate in the rate determining step.
4.2 Studies with Substituted Oxoacids
The oxidation of substituted oxoacids, S2-S7 by NBSac in aqueous acetic
acid medium was studied in the presence of perchloric acid in order to
assess the effect of substituents in the phenyl ring of the oxoacid on
reaction rate. The pseudo first order rate constants were measured at
different initial concentrations of oxoacids. The second order rate
constants k2 were found to be constant. The values are given in the Tables
25-30. The plots of log k1 against log [S] is linear and the slope is close to
unity. These results indicate the first order dependence of the
oxoacids on the rate of the reaction (Figure 31). This is further
supported by the fact that the plot of k1 versus [S] gives a straight line
passing through the origin (Figure 32).
120
The oxidation was carried out at different initial
concentrations of N-bromosaccharin. The rate constants are found to
be nearly constant (Table 25-30). This shows that the reaction is first
order with respect to N-bromosaccharin.
Table 25. Rate constants for the oxidation of methoxy substituted
4–oxoacid (S2) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S2] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 5.25± 0.42 26.3 ± 1.5
4.0 2.0 0.5 10.41± 0.60 26.1 ± 1.7
6.0 2.0 0.5 15.7± 1.2 26.2 ± 1.8
8.0 2.0 0.5 20.70± 2.1 25.8 ± 1.6
2.0 2.0 1.0 10.5± 0.50 1.05 ± 0.02
2.0 2.0 1.2 12.4± 0.60 1.03 ± 0.04
2.0 2.0 1.4 14.2± 0.1 1.10 ± 0.06
2.0 0.8 0.5 5.21± 0.06 -
2.0 0.4 0.5 5.25± 0.07 -
2.0 0.2 0.5 5.23± 0.16 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2]
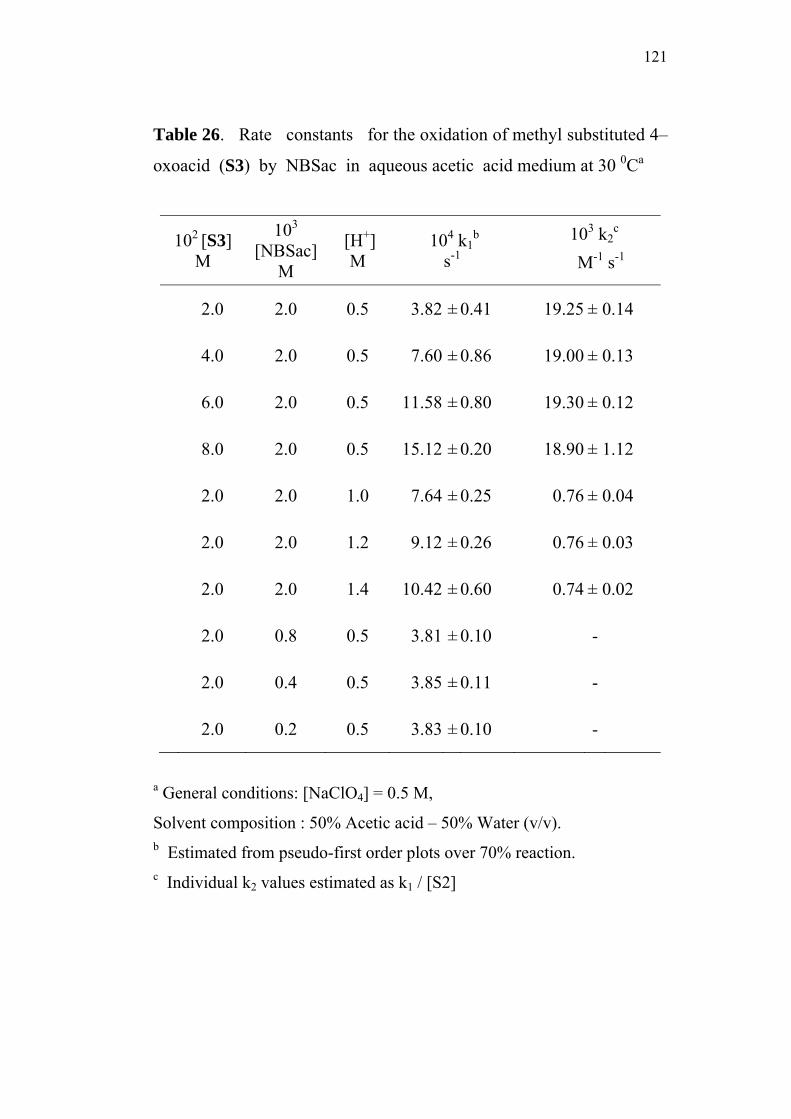
121
Table 26. Rate constants for the oxidation of methyl substituted 4–
oxoacid (S3) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S3] M
103 [NBSac]
M
[H+] M
104 k1b
s-1
103 k2c
M-1 s-1
2.0 2.0 0.5 3.82 ± 0.41 19.25 ± 0.14
4.0 2.0 0.5 7.60 ± 0.86 19.00 ± 0.13
6.0 2.0 0.5 11.58 ± 0.80 19.30 ± 0.12
8.0 2.0 0.5 15.12 ± 0.20 18.90 ± 1.12
2.0 2.0 1.0 7.64 ± 0.25 0.76 ± 0.04
2.0 2.0 1.2 9.12 ± 0.26 0.76 ± 0.03
2.0 2.0 1.4 10.42 ± 0.60 0.74 ± 0.02
2.0 0.8 0.5 3.81 ± 0.10 -
2.0 0.4 0.5 3.85 ± 0.11 -
2.0 0.2 0.5 3.83 ± 0.10 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2]
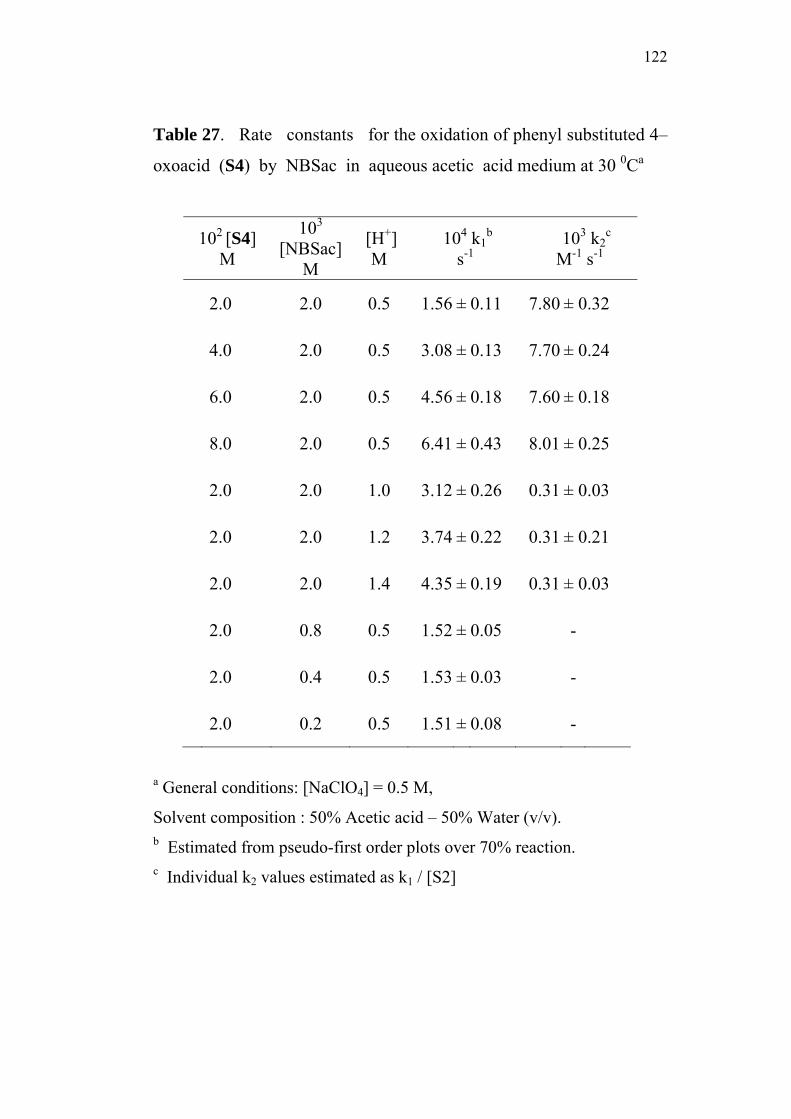
122
Table 27. Rate constants for the oxidation of phenyl substituted 4–
oxoacid (S4) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S4] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 1.56 ± 0.11 7.80 ± 0.32
4.0 2.0 0.5 3.08 ± 0.13 7.70 ± 0.24
6.0 2.0 0.5 4.56 ± 0.18 7.60 ± 0.18
8.0 2.0 0.5 6.41 ± 0.43 8.01 ± 0.25
2.0 2.0 1.0 3.12 ± 0.26 0.31 ± 0.03
2.0 2.0 1.2 3.74 ± 0.22 0.31 ± 0.21
2.0 2.0 1.4 4.35 ± 0.19 0.31 ± 0.03
2.0 0.8 0.5 1.52 ± 0.05 -
2.0 0.4 0.5 1.53 ± 0.03 -
2.0 0.2 0.5 1.51 ± 0.08 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2]
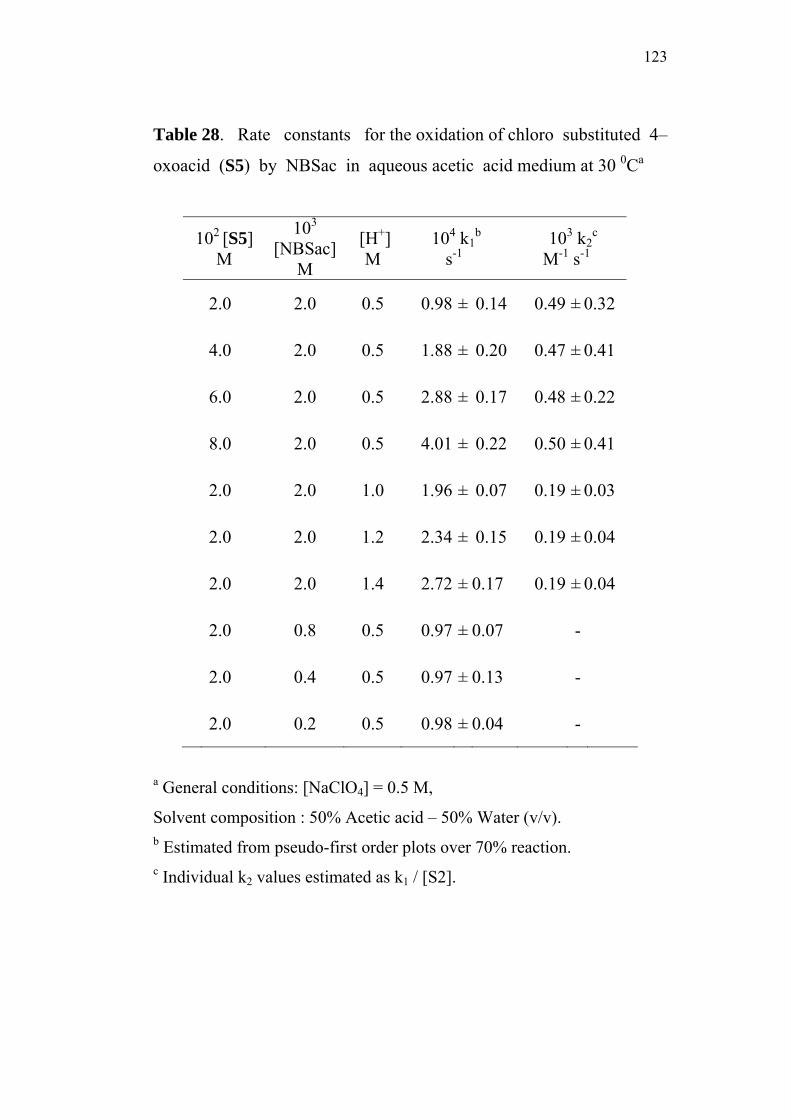
123
Table 28. Rate constants for the oxidation of chloro substituted 4–
oxoacid (S5) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S5] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c M-1 s-1
2.0 2.0 0.5 0.98 ± 0.14 0.49 ± 0.32
4.0 2.0 0.5 1.88 ± 0.20 0.47 ± 0.41
6.0 2.0 0.5 2.88 ± 0.17 0.48 ± 0.22
8.0 2.0 0.5 4.01 ± 0.22 0.50 ± 0.41
2.0 2.0 1.0 1.96 ± 0.07 0.19 ± 0.03
2.0 2.0 1.2 2.34 ± 0.15 0.19 ± 0.04
2.0 2.0 1.4 2.72 ± 0.17 0.19 ± 0.04
2.0 0.8 0.5 0.97 ± 0.07 -
2.0 0.4 0.5 0.97 ± 0.13 -
2.0 0.2 0.5 0.98 ± 0.04 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2].
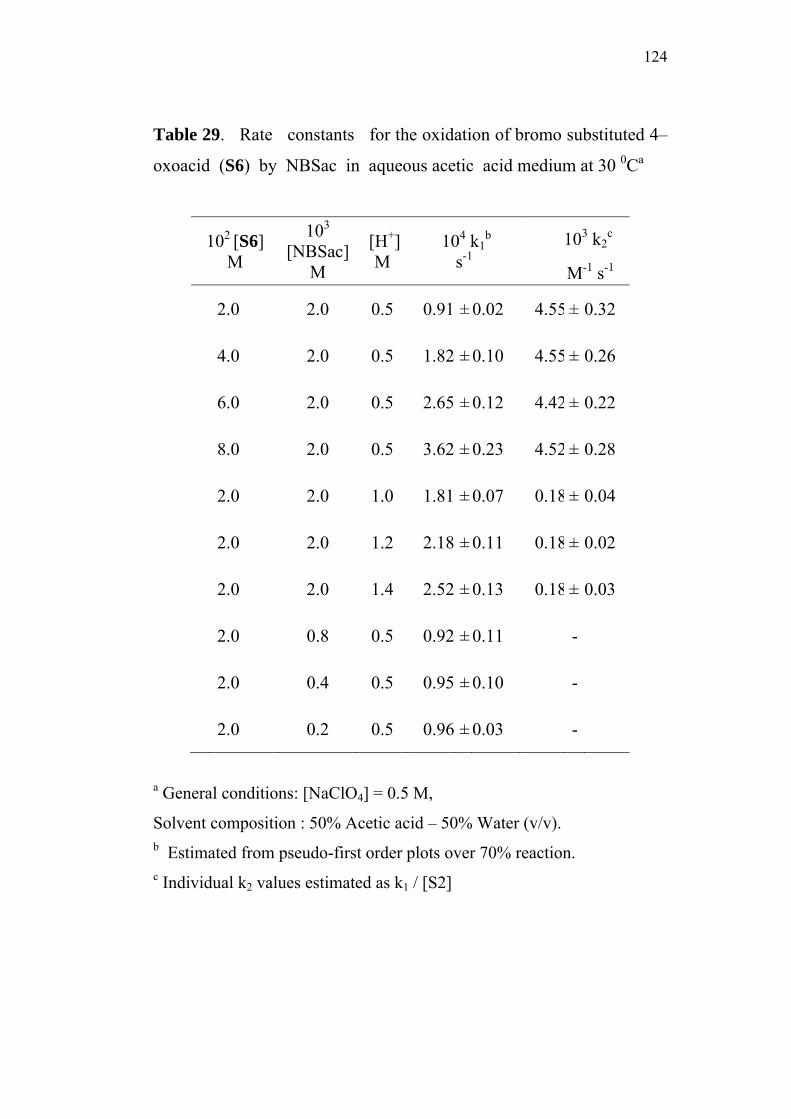
124
Table 29. Rate constants for the oxidation of bromo substituted 4–
oxoacid (S6) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S6] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c
M-1 s-1
2.0 2.0 0.5 0.91 ± 0.02 4.55 ± 0.32
4.0 2.0 0.5 1.82 ± 0.10 4.55 ± 0.26
6.0 2.0 0.5 2.65 ± 0.12 4.42 ± 0.22
8.0 2.0 0.5 3.62 ± 0.23 4.52 ± 0.28
2.0 2.0 1.0 1.81 ± 0.07 0.18 ± 0.04
2.0 2.0 1.2 2.18 ± 0.11 0.18 ± 0.02
2.0 2.0 1.4 2.52 ± 0.13 0.18 ± 0.03
2.0 0.8 0.5 0.92 ± 0.11 -
2.0 0.4 0.5 0.95 ± 0.10 -
2.0 0.2 0.5 0.96 ± 0.03 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2]
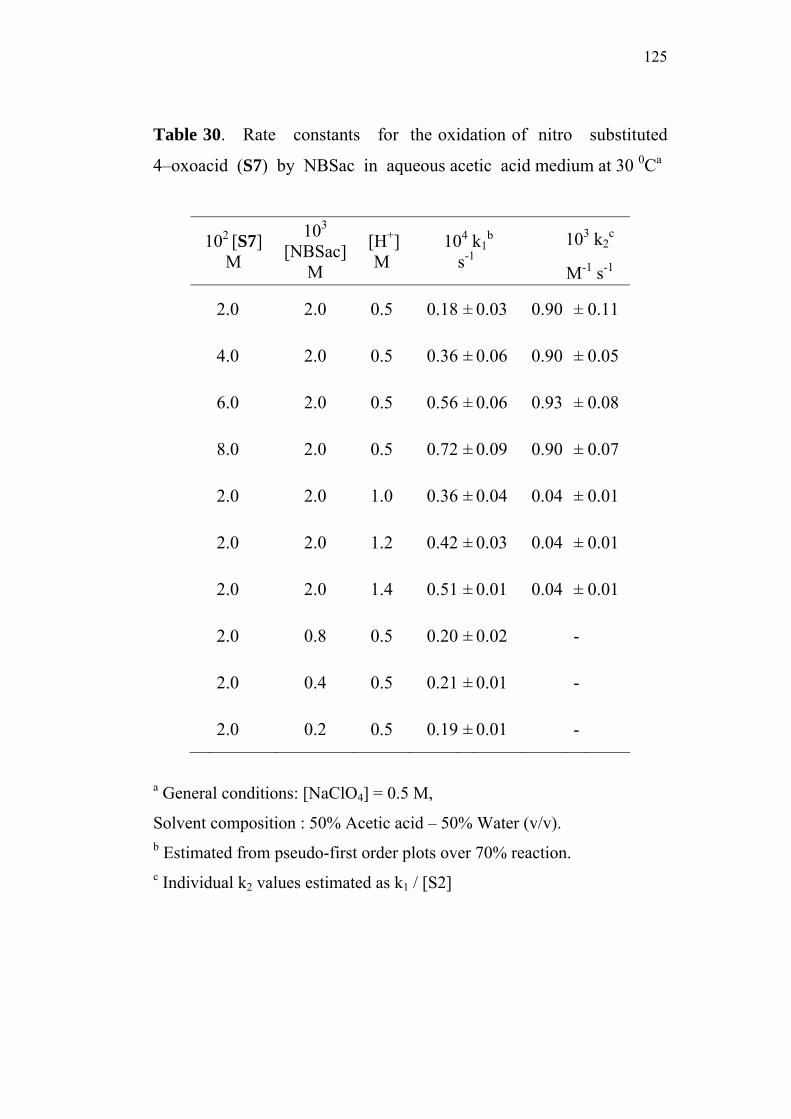
125
Table 30. Rate constants for the oxidation of nitro substituted
4–oxoacid (S7) by NBSac in aqueous acetic acid medium at 30 0Ca
102 [S7] M
103 [NBSac]
M
[H+] M
104 k1b
s-1 103 k2
c
M-1 s-1
2.0 2.0 0.5 0.18 ± 0.03 0.90 ± 0.11
4.0 2.0 0.5 0.36 ± 0.06 0.90 ± 0.05
6.0 2.0 0.5 0.56 ± 0.06 0.93 ± 0.08
8.0 2.0 0.5 0.72 ± 0.09 0.90 ± 0.07
2.0 2.0 1.0 0.36 ± 0.04 0.04 ± 0.01
2.0 2.0 1.2 0.42 ± 0.03 0.04 ± 0.01
2.0 2.0 1.4 0.51 ± 0.01 0.04 ± 0.01
2.0 0.8 0.5 0.20 ± 0.02 -
2.0 0.4 0.5 0.21 ± 0.01 -
2.0 0.2 0.5 0.19 ± 0.01 -
a General conditions: [NaClO4] = 0.5 M,
Solvent composition : 50% Acetic acid – 50% Water (v/v). b Estimated from pseudo-first order plots over 70% reaction. c Individual k2 values estimated as k1 / [S2]
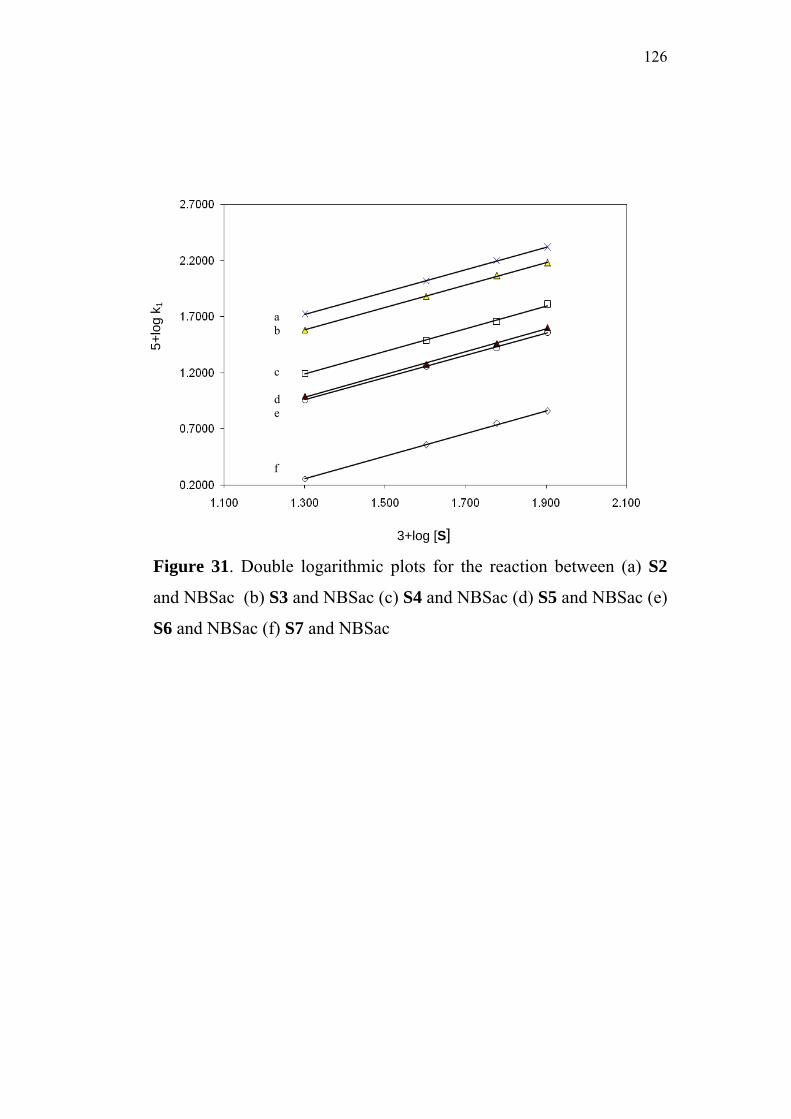
126
Figure 31. Double logarithmic plots for the reaction between (a) S2
and NBSac (b) S3 and NBSac (c) S4 and NBSac (d) S5 and NBSac (e)
S6 and NBSac (f) S7 and NBSac
3+log [S]
5+lo
g k 1
a b c d e f

127
Figure 32. Plots of k1 versus [S] for the reaction between (a) S2 and
NBSac (b) S3 and NBSac (c) S4 and NBSac (d) S5 and NBSac (e) S6
and NBSac (f) S7 and NBSac
The dependence of the reaction rate on the hydrogen ion
concentration has been studied for the various oxoacids (S2-S7).
The rate of the reaction increases linearly with increase in the
concentration of hydrogen ion. The plot of log k1 against log [H+]
is linear (shown in Figure 33) and the slope is unity. This establishes
that the reaction is first order with respect to hydrogen ion
concentration. Also the plots of k1 versus [H+] for the oxidation of
various oxoacids are linear passing through the origin (Figure 34)
showing that the reaction proceeds completely through the acid
catalyzed pathway and the order is one.
[S], M
104 , k
1, s
-1
a
b
c
d e
f
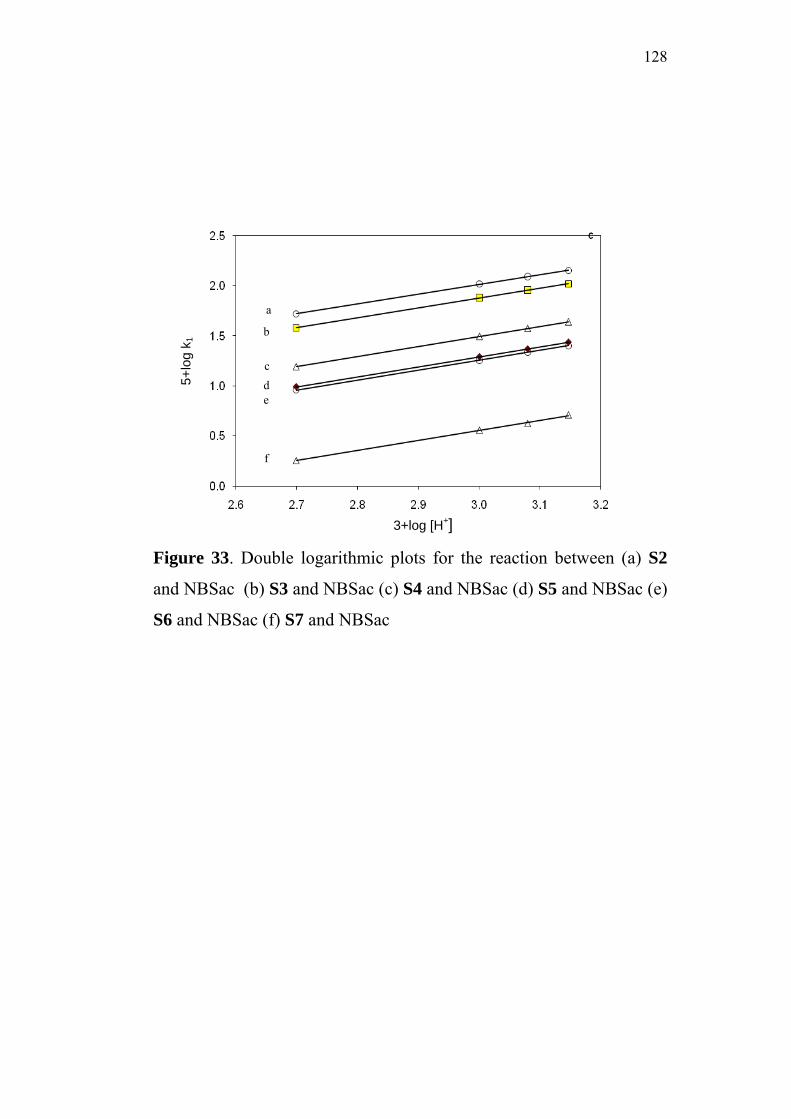
128
Figure 33. Double logarithmic plots for the reaction between (a) S2
and NBSac (b) S3 and NBSac (c) S4 and NBSac (d) S5 and NBSac (e)
S6 and NBSac (f) S7 and NBSac
3+log [H+]
5+lo
g k 1
a
b
c
d e
f
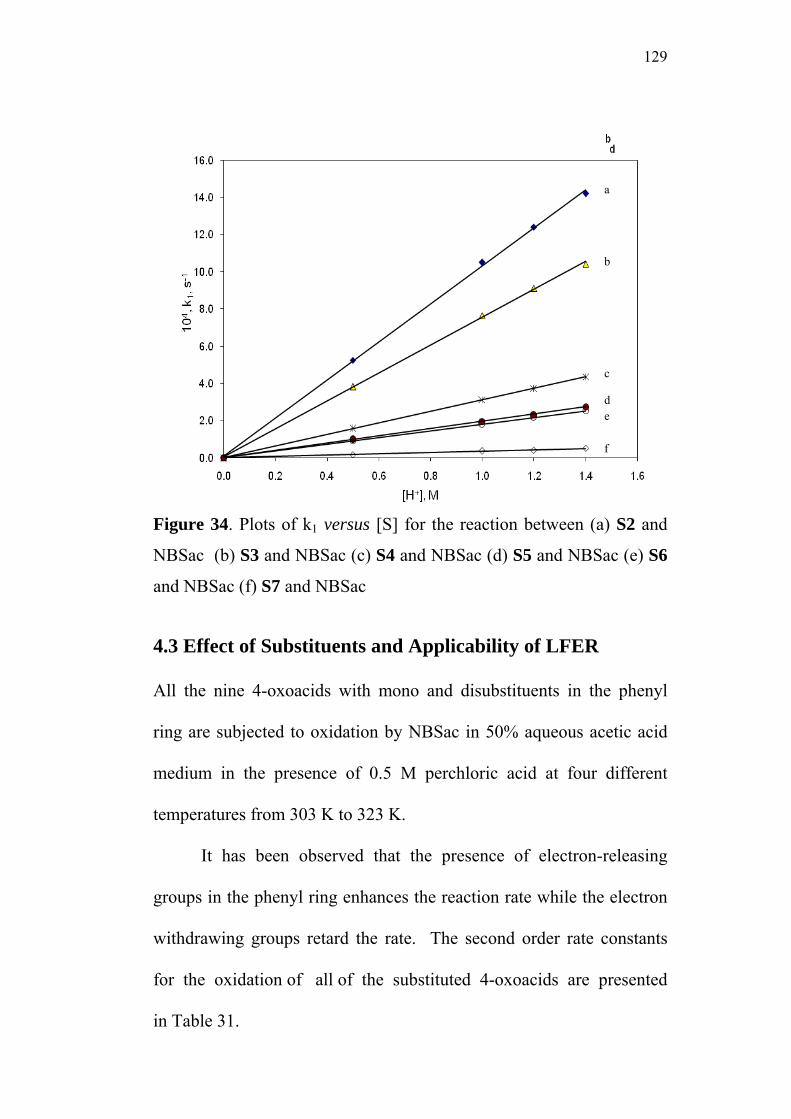
129
Figure 34. Plots of k1 versus [S] for the reaction between (a) S2 and
NBSac (b) S3 and NBSac (c) S4 and NBSac (d) S5 and NBSac (e) S6
and NBSac (f) S7 and NBSac
4.3 Effect of Substituents and Applicability of LFER
All the nine 4-oxoacids with mono and disubstituents in the phenyl
ring are subjected to oxidation by NBSac in 50% aqueous acetic acid
medium in the presence of 0.5 M perchloric acid at four different
temperatures from 303 K to 323 K.
It has been observed that the presence of electron-releasing
groups in the phenyl ring enhances the reaction rate while the electron
withdrawing groups retard the rate. The second order rate constants
for the oxidation of all of the substituted 4-oxoacids are presented
in Table 31.
a
b
c
d e
f
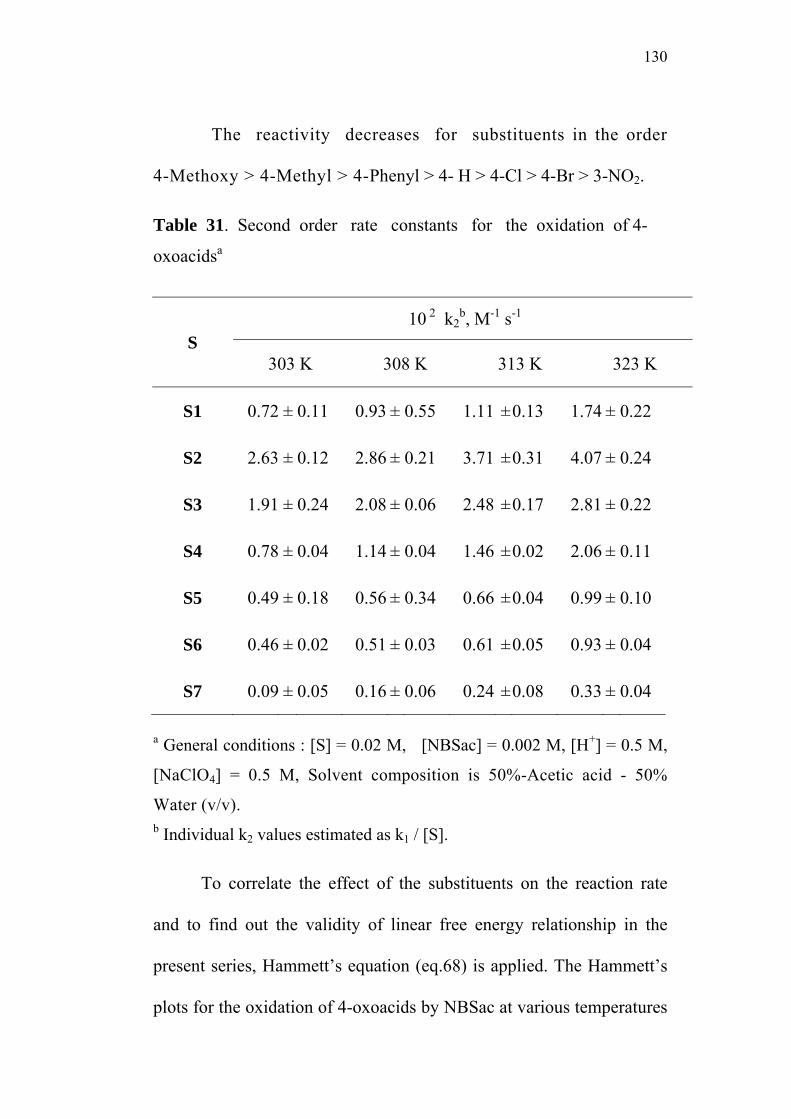
130
The reactivity decreases for substituents in the order
4-Methoxy > 4-Methyl > 4-Phenyl > 4- H > 4-Cl > 4-Br > 3-NO2.
Table 31. Second order rate constants for the oxidation of 4-
oxoacidsa
S 10 2 k2
b, M-1 s-1
303 K 308 K 313 K 323 K
S1 0.72 ± 0.11 0.93 ± 0.55 1.11 ±0.13 1.74 ± 0.22
S2 2.63 ± 0.12 2.86 ± 0.21 3.71 ±0.31 4.07 ± 0.24
S3 1.91 ± 0.24 2.08 ± 0.06 2.48 ±0.17 2.81 ± 0.22
S4 0.78 ± 0.04 1.14 ± 0.04 1.46 ±0.02 2.06 ± 0.11
S5 0.49 ± 0.18 0.56 ± 0.34 0.66 ±0.04 0.99 ± 0.10
S6 0.46 ± 0.02 0.51 ± 0.03 0.61 ±0.05 0.93 ± 0.04
S7 0.09 ± 0.05 0.16 ± 0.06 0.24 ±0.08 0.33 ± 0.04
a General conditions : [S] = 0.02 M, [NBSac] = 0.002 M, [H+] = 0.5 M,
[NaClO4] = 0.5 M, Solvent composition is 50%-Acetic acid - 50%
Water (v/v). b Individual k2 values estimated as k1 / [S].
To correlate the effect of the substituents on the reaction rate
and to find out the validity of linear free energy relationship in the
present series, Hammett’s equation (eq.68) is applied. The Hammett’s
plots for the oxidation of 4-oxoacids by NBSac at various temperatures

131
are found to be linear. The Hammett’s plots are shown in Figures 35- 38.
The reaction constant ( values obtained from the Hammett’s plots
are negative and are presented in Table 32.
Table 32. Reaction constant values at different temperatures a
Temperature
K
Reaction constant b
Correlation
coefficient
303 -1.41 0.983
308 -1.32 0.992
313 -1.20 0.988
323 -0.98 0.996 a values were taken from reported works302-304.
b The values were obtained by correlating log (k2 / k20) with p for the reactions
of oxidations S1-S7 with NBSac.
The value indicates the sensitivity of a reaction to the
effects of electronic perturbation. It also provides information about
the nature of the transition state involved during the reaction. A
reaction involving a development of positive charge in the transition
state is aided by electron-releasing substituents and the value is
negative305-307.
In the present investigation, the acceleration of reaction rate
with the electron-releasing substituents and the negative value of the
reaction constant, indicate explicitly that the mechanism of oxidation
involves the development of positive charge in the transition state.

132
The Hammett correlations were also made for the rate constants
obtained at different solvent compositions (c.f. Table 24) and the
values are reported in the following Table 33.
Table 33. Reaction constant values at different solvent compositions a
Solvent
CH3COOH-H2O (v/v) %
Reaction constant b
Correlation
coefficient
50-50 -1.41 0.982
60-40 -1.35 0.983
70-30 -1.12 0.978
80-20 -1.03 0.993 a values were taken from reported works302-304.
b The values were obtained by correlating log (k2 / k20) with p for the reactions
of S1-S7 with NBSac.
The data reveal that the selectivity of the oxidant towards oxoacids
decreases with increase in the acetic acid content in the solvent mixture.
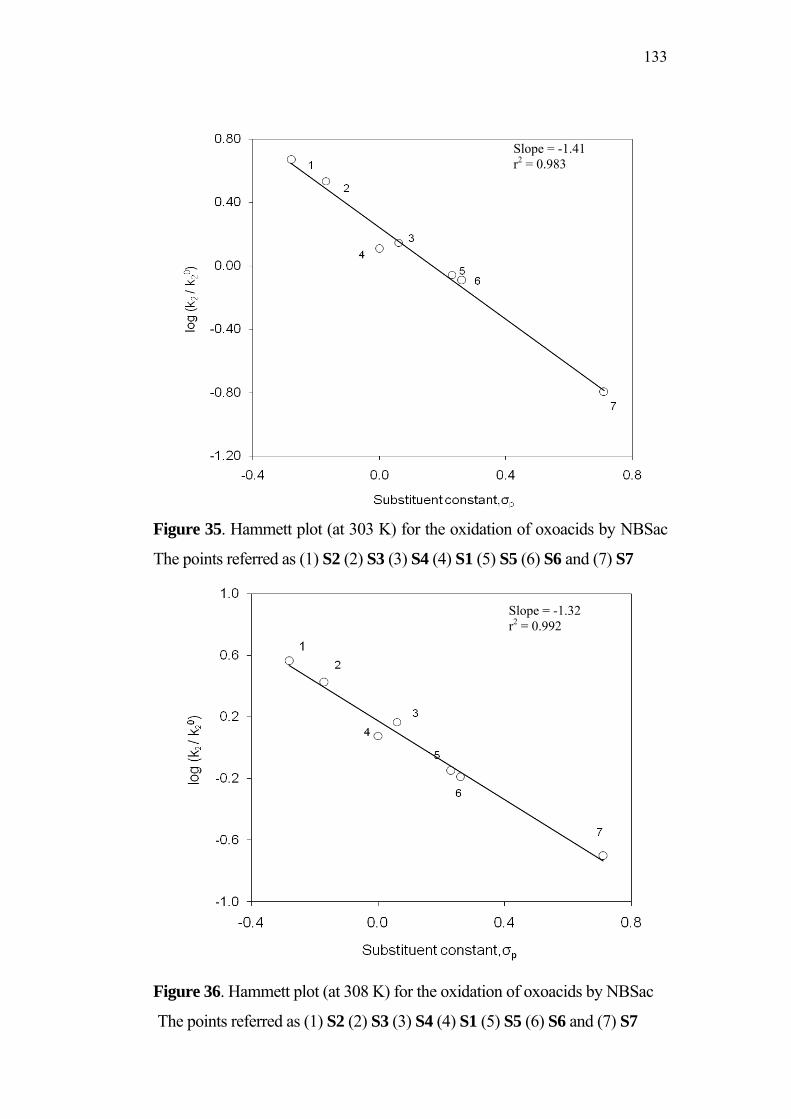
133
Figure 35. Hammett plot (at 303 K) for the oxidation of oxoacids by NBSac
The points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Figure 36. Hammett plot (at 308 K) for the oxidation of oxoacids by NBSac
The points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Slope = -1.41 r2 = 0.983
Slope = -1.32 r2 = 0.992
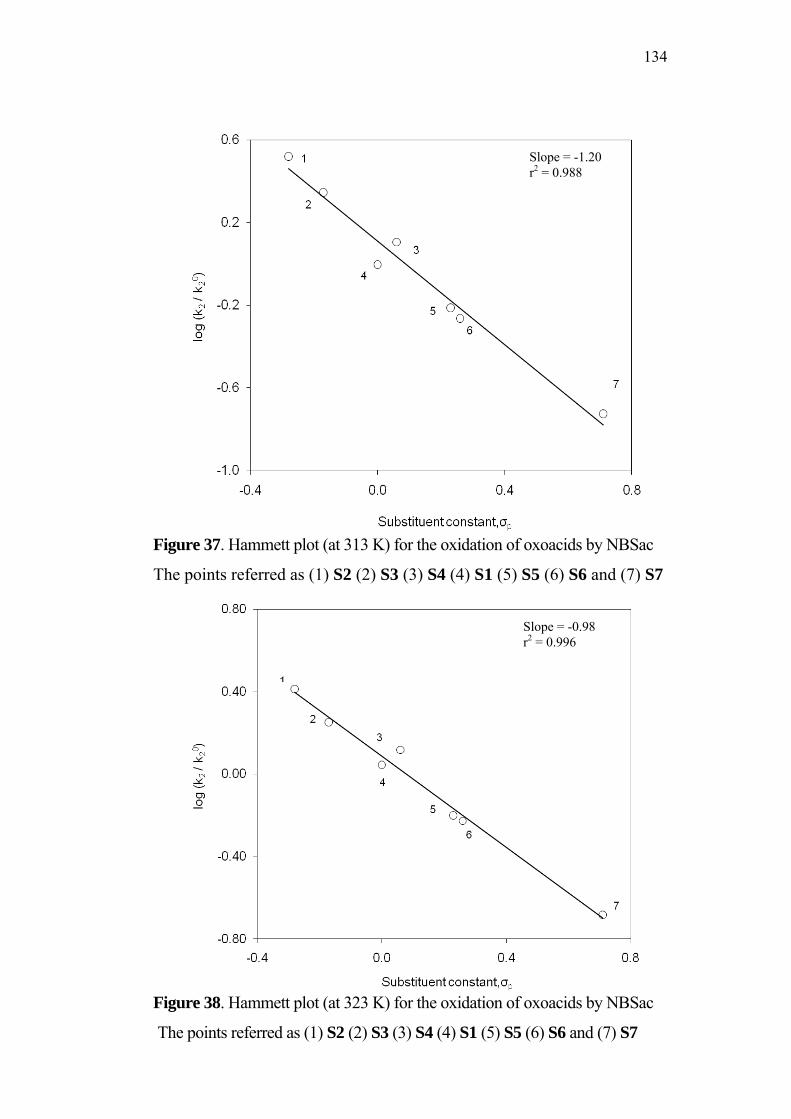
134
Figure 37. Hammett plot (at 313 K) for the oxidation of oxoacids by NBSac
The points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Figure 38. Hammett plot (at 323 K) for the oxidation of oxoacids by NBSac
The points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Slope = -1.20 r2 = 0.988
Slope = -0.98 r2 = 0.996

135
4.4 Mechanism of Oxidation
A probable mechanism for the oxidation of 4-oxoacids by NBSac has
been proposed based on the experimental results and in analogy with
the oxidation of oxo compounds with other oxidants. The results
obtained in the kinetic study are briefly summarized below.
i) The reaction is first order each with respect to the 4-
oxoacid, NBSac, and H+ ion.
ii) The linear increase in the reaction rate with the increase
in [H+] ion is attributed to the formation of hypobromous
acidium ion (H2O+Br) and to the enolization of the 4-
oxoacid.
iii) The formation of hypobromous acidium ion and the
enolization of the 4-oxoacid is facilitated at lower
dielectric constant of the medium.
iv) The rate of enolization is found to be greater than the rate
of oxidation.
v) The course of the oxidation does not involve any free
radical intermediate.
vi) The retarding effect of the addition of saccharin suggests
that the pre-equilibrium step involves a process in which
saccharin is one of the products.
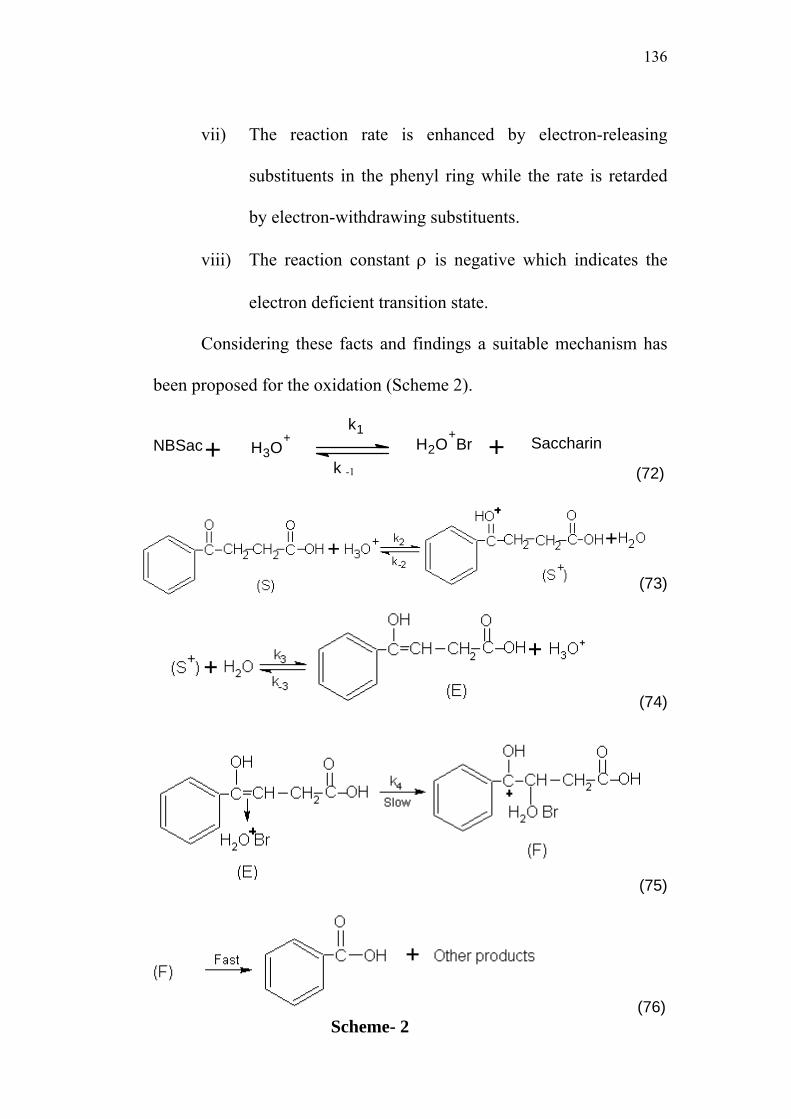
136
vii) The reaction rate is enhanced by electron-releasing
substituents in the phenyl ring while the rate is retarded
by electron-withdrawing substituents.
viii) The reaction constant is negative which indicates the
electron deficient transition state.
Considering these facts and findings a suitable mechanism has
been proposed for the oxidation (Scheme 2).
NBSac+ H3O+ H2O
+Br Saccharin+
k1
k-1 (72)
(73)
(74)
(75)
(76) Scheme- 2
k -1

137
The proposed scheme is formulated to fit in the following
features.
a) The enolization of the 4-oxoacid (eqs.73 and 74) is a
necessary step prior to the oxidation of the substrate. This
is predicted from the experimental finding that under
identical conditions, the rate of enolization is greater than
the rate of oxidation. Similar conclusions have been
arrived at in the oxidation of several ketones by acid
permanganate291, 292, 294.
b) The interaction of oxidizing species (H2O+Br) with the
enol form of the 4-oxoacid in the rate determining step
(eq.75) leads to the formation of intermediate (F). Similar
type of intermediate has been proposed in the oxidation
of many organic and inorganic compounds308-310. The
intermediate then undergoes further oxidative cleavage to
give the products.
4.5 Derivation of Rate Law
Based on kinetic observations and the mechanism proposed, the
following rate expression can be derived applying steady-state
approximation,
The rate of the reaction is given by
-d [NBSac] = k4 [E] [H2O+Br] (77)
dt

138
Applying steady state approximation for [E]
k -3 [H3O+] [E] + k4 [H2O
+Br] [E] = k3 [S
+] [H2O] k3 [S
+] [H2O] Therefore, [E] = (78)
k -3 [H3O+] + k4 [H2O
+Br]
Applying steady state approximation for [S+] k -2 [H2O] [S+] + k 3 [H2O] [S+] = k 2 [H3O
+] [S] + k -3 [H3O+] [E]
k 2 [H3O
+] [S] + k -3 [H3O+] [E]
Therefore, [S+] = k -2 [H2O] + k 3 [H2O]
[H3O
+] { k 2 [S] + k -3 [E]} i.e. [S+] = (79)
[H2O] (k -2 + k 3 ) Using the value of [S+] in eq. (78) k 3 [H3O
+] { k 2 [S] + k -3 [E] } [E] =
( k -2 + k 3 ) { k -3 [H3O+] + k 4 [H2O
+Br] }
[E] (k -2 + k 3 ) {k -3 [H3O+] + k 4 [H2O
+Br] } = k 3 [H3O
+] { k 2 [S] + k -3 [E] }
[E](k -2 + k 3 ){ k -3 [H3O+]+k 4[H2O
+Br]}= k 3 k 2 [H3O
+][S]+k3 k -3 [H3O+][E]
[E]{(k -2 +k 3 ) k -3 [H3O+] + k 4 [H2O
+Br] – k 3 k -3 [H3O
+]}= k 3 k2 [H3O+] [S]
k 3 k 2 [H3O
+] [S] [E] = { (k -2 +k 3 ) k -3 [H3O
+] + k 4 [H2O+Br]} – k 3 k -3 [H3O
+] k 3 k 2 [H3O
+] [S] [E] =
k -2 k -3 [H3O+] + k -2 k 4 [H2O
+Br] + k 3 k 4 [H2O
+Br]

139
k 3 k 2 [H3O+] [S]
[E] = (80)
k -2 k -3 [H3O+] + k 4 (k -2 + k 3) [H2O
+Br]
Substituting the value of [E] in eq. (77)
-d [NBSac] k 2 k 3 k 4 [H3O+] [S] [H2O
+Br]
= (81)
dt k -2 k -3 [H3O+] + k 4 (k -2 + k 3) [H2O
+Br]
At high concentration of [H3O+] = 0.5 M
k -2 k -3 [H3O+] > > k 4 (k -2 + k 3) [H2O
+Br]
So the eq. (81) simplifies to the form
-d [NBSac] k 2 k 3 k 4 [H3O+] [S] [H2O
+Br]
= dt k -2 k -3 [H3O
+]
k 2 k 3 k 4 [S] [H2O+Br]
= (82)
k -2 k -3
The value of [H2O+Br] can be obtained from eq. (72) given in
the Scheme 2 of mechanism
k -1 [NBSac] [H3O+]
K a = =
k 1 [H2O+Cl] [Saccharin]
[NBSac] [H3O+]
Therefore, [H2O+Br] =
Ka [Saccharin] Using the value of [H2O
+Br] in eq. (82)
-d[NBSac] k 2 k 3 k 4 [S] [H3O+] [NBSA]
= (83) dt k -2 k -3 K a [Saccharin]

140
Hence, at higher concentration of mineral acid, the reaction is
first order each with respect to the oxoacid (S), [NBSac] and [H3O+].
The observed rate constant at high [H3O+] is
k 2 k 3 k 4 kobs = (84)
k -2 k -3 K a
4.6 Structure –Reactivity Correlations
The effect of the substituents present in the phenyl ring of the 4-
oxoacids (S1-S7) on the rates of oxidation has been correlated
quantitatively by the application of Hammett equation. Plots of log
(k2/k20) versus P are excellently linear with negative slopes
(Figures 34-37). The reaction constant (ρ) for the oxidation ranges
between -1.41 and -0.98 at different temperatures.
It is generally recognized that oxidations lead to electron
deficient species which are either radical cations, radicals or
carbocations. These reactions normally have a negative ρ value and the
magnitude of value depends on the extent of electron deficiency.
Oxidation reactions involving free radical formation in the rate
controlling step usually have a small negative ρ value and the
oxidations involving the formation of carbocation have a large negative
ρ values. Based on the these arguments we expect a large ρ value but
the measured value is in the range -1.41 to -0.98. The low value

141
may be attributed to the nature of observed rate constant. The observed
rate constant is composite of several terms and is shown in eq.(85).
(85)
The terms shown in eq.(85) deserve comment. The rate constant
k3 depends on the concentration of the protonated substrate (S+) and
the electron donating substituents tend to delocalize the positive charge
on S+ and hence favors the formation of this positive species. The
value for the enolization of the title compounds, determined by
bromination method has been reported311 to be -0.75. In the rate
limiting step (eq.77) of the scheme, the formation of the carbocation is
facilitated by electron releasing substituents.
Thus we get a slightly low negative value of -1.41 (at 303 K)
because kobs is composite of the enolization as well as the oxidation of
the 4-oxoacids. Hence in the present investigation the measured
value and other findings fit in with the formulation of mechanism
outlined in the scheme 2.
4.7 Activation Parameters
The dependence of reaction rate on the structure of the reacting
molecule is related to activation parameters. The decisive term
concerning the dependence on the structure is neither free energy

142
nor enthalpy but potential energy which is experimentally not
accessible. Many authors support the opinion that the activation
energy at a certain temperature is a better approximation towards
the unknown potential energy312. The activation parameters for the
oxidation of 4-oxoacid by NBSac have been evaluated from the
slope values of the Arrhenius plots (Figures 39 & 40). A close look
at activation parameters presented in Table 34 shows that the
activation energies for the oxoacids with electron-releasing
substituents are relatively lower than that with electron-
withdrawing substituents. The entropy of activation is negative for
all the 4-oxoacids ranging from -126.2 to -232.6 J K-1 mol-1.
The large negative entropy of activation in conjunction with other
experimental data supports the mechanism outlined in the scheme 2. The
formation of an activated complex from reactant molecules is
accompanied by the conversion of translation-like and rotational-like
degree of freedom of the reactants to vibrational degree of freedom of the
transition state species. The more widely spaced energy level for the latter
type of molecular motion imply a small entropy and thus a negative
value313 of S#. As the charge separation begins in the transition state,
each end of the dipole becomes solvated by a sheath of solvent molecules,
which must, however be suitably oriented. This increase in orientation
means restricted freedom and results in a decrease in entropy310. A careful
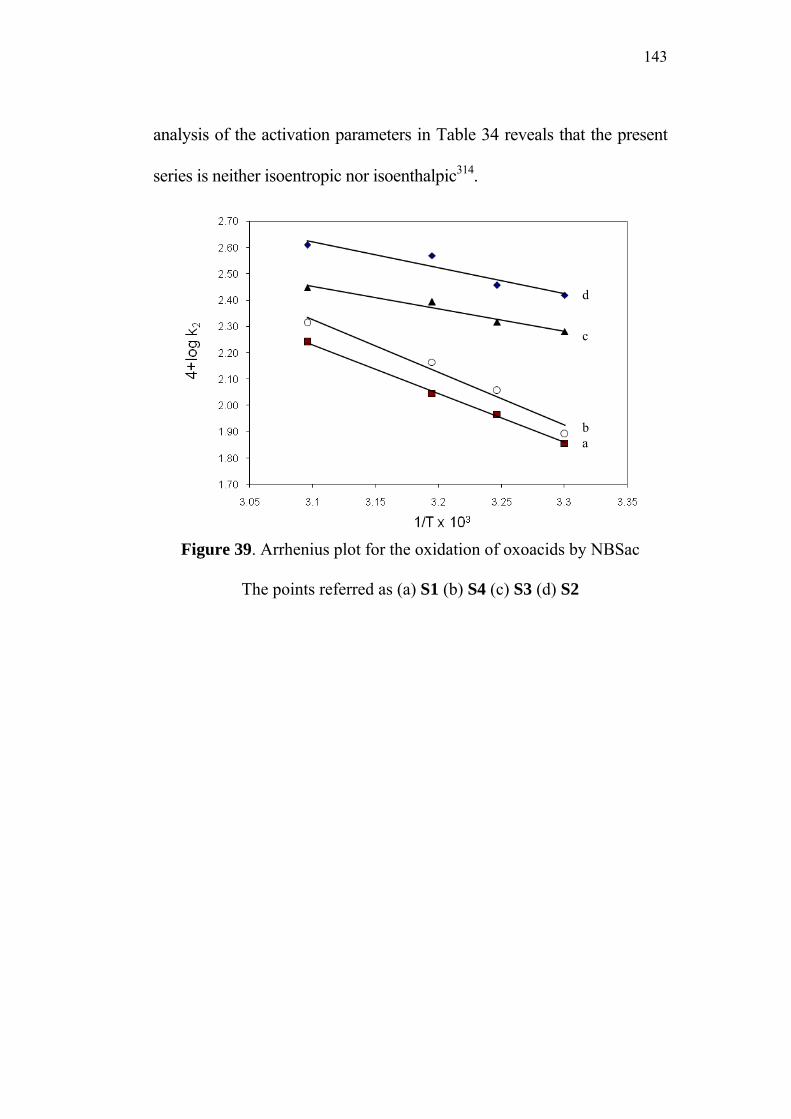
143
analysis of the activation parameters in Table 34 reveals that the present
series is neither isoentropic nor isoenthalpic314.
Figure 39. Arrhenius plot for the oxidation of oxoacids by NBSac
The points referred as (a) S1 (b) S4 (c) S3 (d) S2
a b
c
d
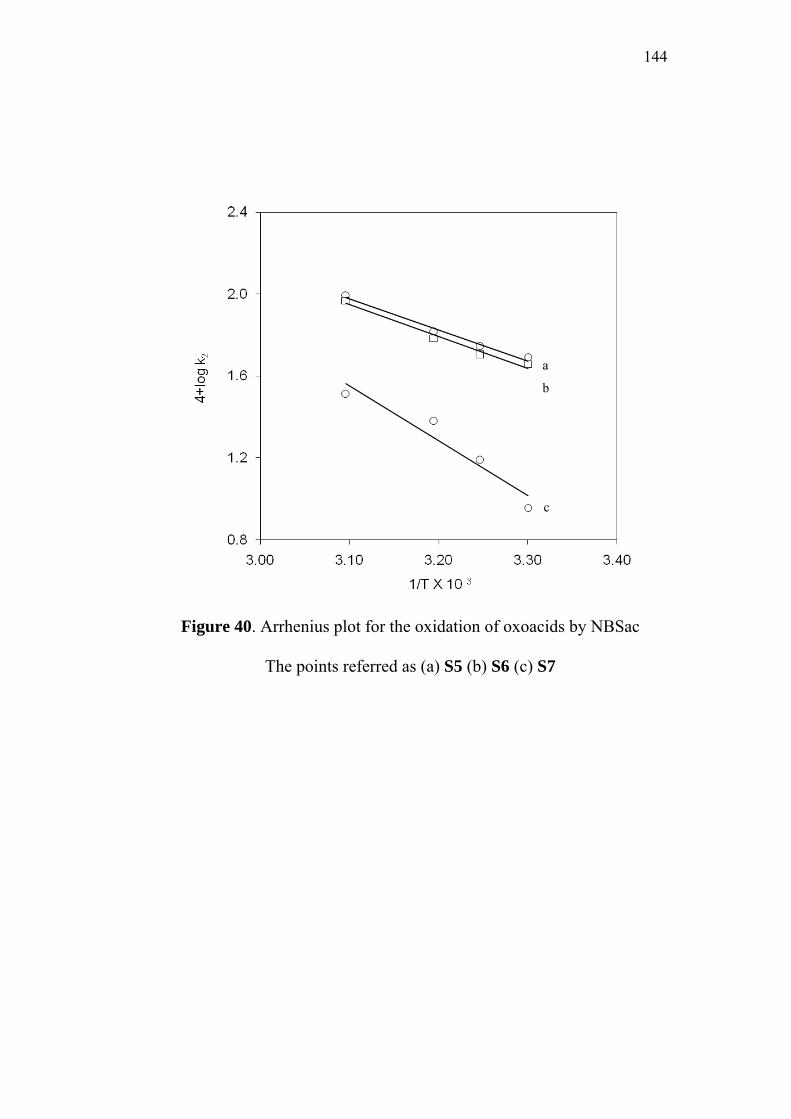
144
Figure 40. Arrhenius plot for the oxidation of oxoacids by NBSac
The points referred as (a) S5 (b) S6 (c) S7
a
b
c
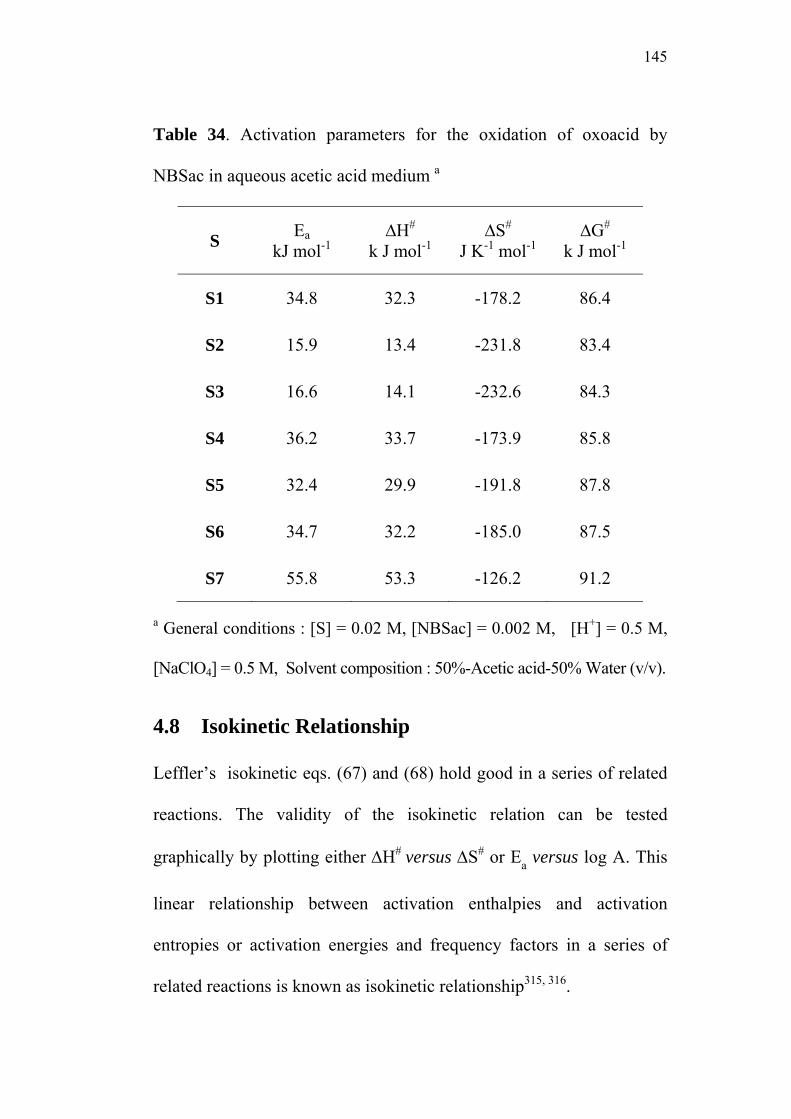
145
Table 34. Activation parameters for the oxidation of oxoacid by
NBSac in aqueous acetic acid medium a
S Ea
kJ mol-1 ∆H#
k J mol-1 ∆S#
J K-1 mol-1 ∆G#
k J mol-1
S1 34.8 32.3 -178.2 86.4
S2 15.9 13.4 -231.8 83.4
S3 16.6 14.1 -232.6 84.3
S4 36.2 33.7 -173.9 85.8
S5 32.4 29.9 -191.8 87.8
S6 34.7 32.2 -185.0 87.5
S7 55.8 53.3 -126.2 91.2
a General conditions : [S] = 0.02 M, [NBSac] = 0.002 M, [H+] = 0.5 M,
[NaClO4] = 0.5 M, Solvent composition : 50%-Acetic acid-50% Water (v/v).
4.8 Isokinetic Relationship
Leffler’s isokinetic eqs. (67) and (68) hold good in a series of related
reactions. The validity of the isokinetic relation can be tested
graphically by plotting either H# versus S# or Ea versus log A. This
linear relationship between activation enthalpies and activation
entropies or activation energies and frequency factors in a series of
related reactions is known as isokinetic relationship315, 316.

146
The validity of the isokinetic plot is questionable317, 318 because
the quantities H# and S# are mutually dependent, both being derived
from the same experimental rate constants. An alternative graphical
method for finding out the isokinetic temperature is suggested by
Exner. A plot of the rate constants measured at two different
temperatures [log k2 (T2) versus log k2 (T1)] is known as Exner plot.
From the slope b, of the Exner plot, the isokinetic temperature , can
be calculated using the eq.(84).
Based on the values of b, the reaction series have been
characterized as mentioned in the Table 19.
The Exner plots drawn for the reaction by choosing k2 values at
any two different temperatures are shown in Figures 41 - 43. The
value of b is less than unity for all the plots. This indicates that the
present reaction series is neither isoentropic nor isoenthalpic but
involves compensation. The lsokinetic temperature evaluated from the
Exner plots is found to be 333.3 K (from Figure 43). As the experiment
has been carried out at a temperature far away from the isokinetic
temperature the application of Hammett equation to the observed
kinetic data is valid. The validity of isokinetic relationship in the
present study implies that all the 4-oxoacids undergo oxidation by the
same mechanism310, 319, 320.
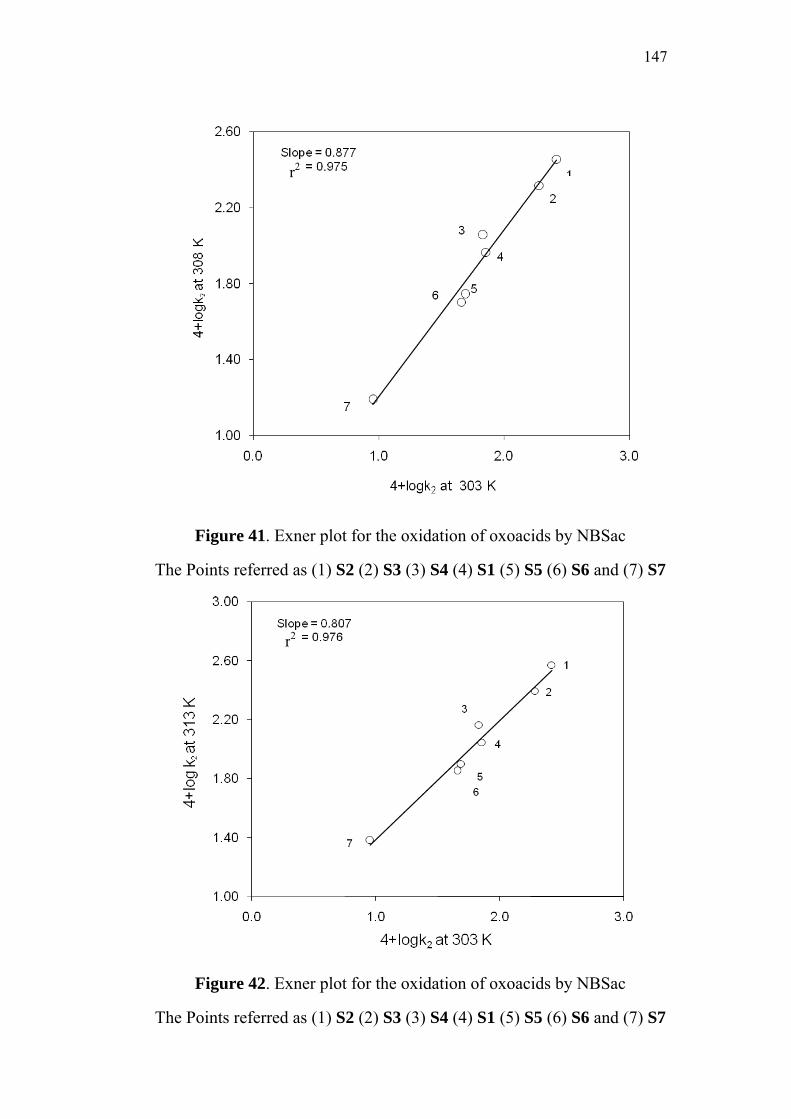
147
Figure 41. Exner plot for the oxidation of oxoacids by NBSac
The Points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
Figure 42. Exner plot for the oxidation of oxoacids by NBSac
The Points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
r2
r2
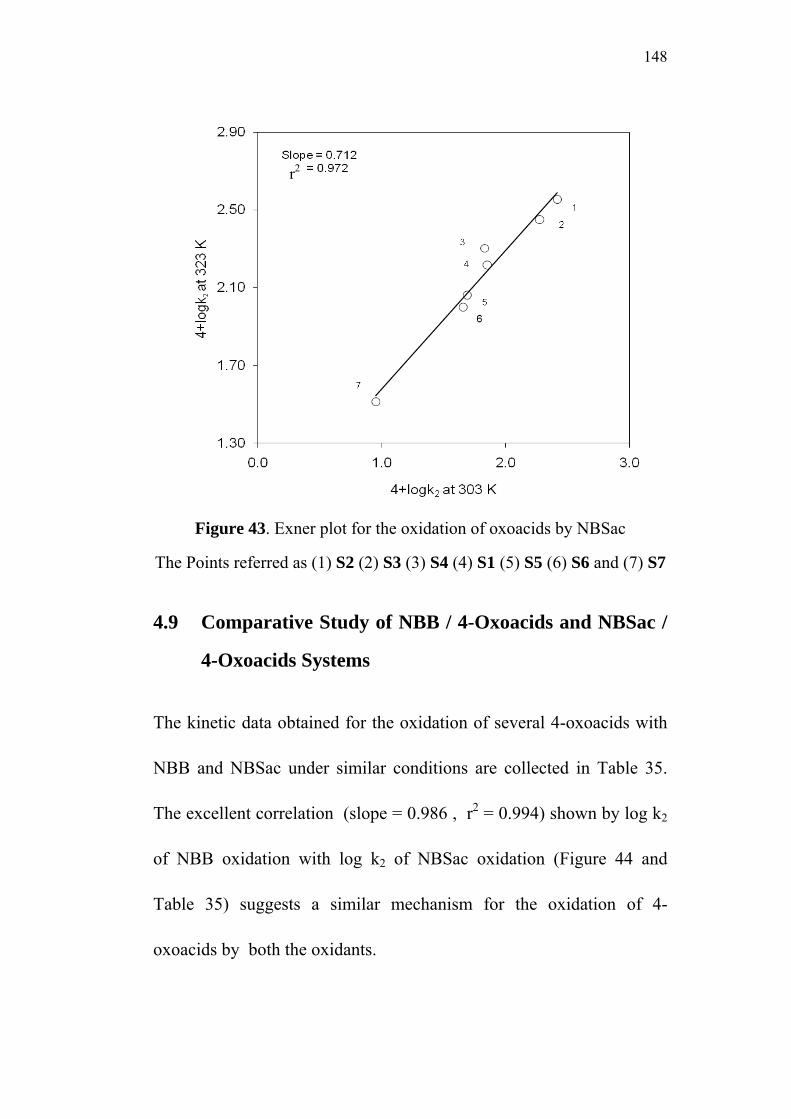
148
Figure 43. Exner plot for the oxidation of oxoacids by NBSac
The Points referred as (1) S2 (2) S3 (3) S4 (4) S1 (5) S5 (6) S6 and (7) S7
4.9 Comparative Study of NBB / 4-Oxoacids and NBSac /
4-Oxoacids Systems
The kinetic data obtained for the oxidation of several 4-oxoacids with
NBB and NBSac under similar conditions are collected in Table 35.
The excellent correlation (slope = 0.986 , r2 = 0.994) shown by log k2
of NBB oxidation with log k2 of NBSac oxidation (Figure 44 and
Table 35) suggests a similar mechanism for the oxidation of 4-
oxoacids by both the oxidants.
r2
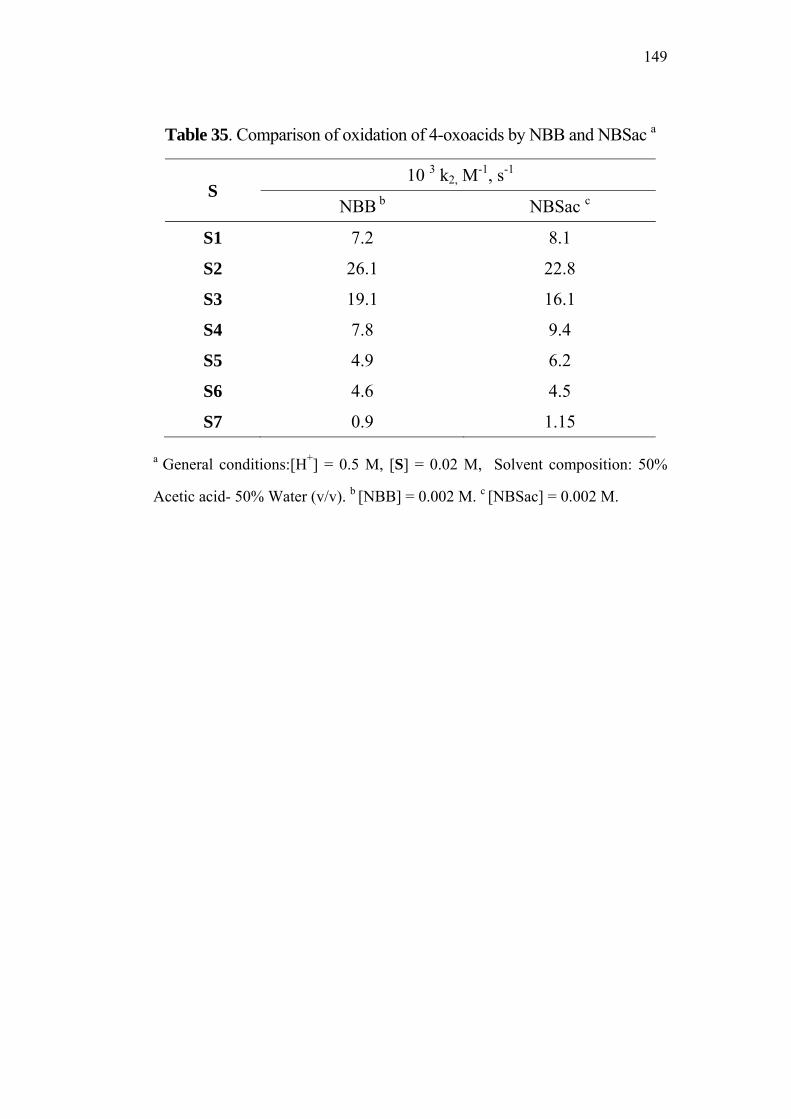
149
Table 35. Comparison of oxidation of 4-oxoacids by NBB and NBSac a
S 10 3 k2, M
-1, s-1
NBB b NBSac c
S1 7.2 8.1
S2 26.1 22.8
S3 19.1 16.1
S4 7.8 9.4
S5 4.9 6.2
S6 4.6 4.5
S7 0.9 1.15
a General conditions:[H+] = 0.5 M, [S] = 0.02 M, Solvent composition: 50%
Acetic acid- 50% Water (v/v). b [NBB] = 0.002 M. c [NBSac] = 0.002 M.
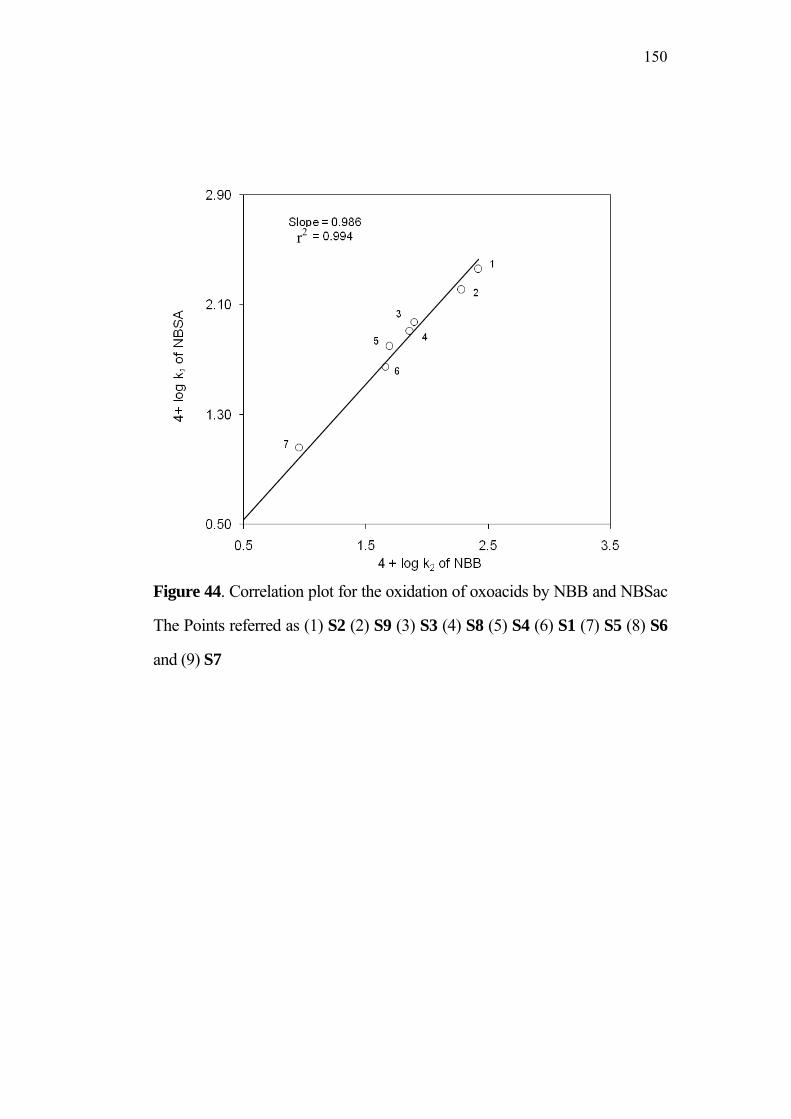
150
Figure 44. Correlation plot for the oxidation of oxoacids by NBB and NBSac
The Points referred as (1) S2 (2) S9 (3) S3 (4) S8 (5) S4 (6) S1 (7) S5 (8) S6
and (9) S7
r2

SUMMARY

151
SUMMARY
4-Oxoacids / NBB – System
The hypobromous acidium ion (H2O+Br), a prime cationic bromine
species is responsible for the oxidation of 4-oxoacids in the present
system. The kinetics of oxidation of 4-oxoacids have been investigated
in aqueous acetic acid medium in the presence of perchloric acid
potentiometrically at 30 0C.
The oxidation of 4-oxoacids by N-bromobenzamide is first
order each with respect to the 4-oxoacid, NBB and hydrogen ion.
The oxidation is observed to be a general acid catalyzed reaction.
The linear increase in the reaction rate with the increase in [H+] ion
is attributed to the formation of hypobromous acidium ion and the
formation of enol form of the substrate.
The lowering of dielectric constant of reaction medium
enhances the reaction rate significantly. This favors the formation of
H2O+Br and the enolization of the 4-oxoacid has been studied by
bromination method. The enolization of the oxoacid is found to be first
order each with respect to the 4-oxoacid and hydrogen ion but zero
order with respect to bromine. The rate of enolization is greater than
the rate of oxidation of the 4-oxoacid. Hence the enolization of the
oxoacid is proposed to occur prior to the oxidation. The ionic strength

152
of the reaction medium does not affect the rate of the reaction. The
reaction does not induce the polymerization, which indicates the
absence of free radical intermediate in the oxidation.
The effect of substituents on the reaction rate has been studied
with all the 4-oxoacids. The electron-releasing group in the phenyl ring
accelerates the reaction rate while the electron-withdrawing group
retards the rate. The Hammett plot is linear (r = 0.987) and the value of
the reaction constant () is -1.279. The negative value indicates the
development of positive charge in the transition state.
The activation parameters for the oxidation reaction are
evaluated from the Arrhenius plot. The Exner plot is linear and the
isokinetic temperature ( = 347 K) is evaluated from the plot. The
validity of Exner plot indicates that the oxidation of all the 4-oxoacids
by NBB in aqueous acetic acid medium proceeds by the same
mechanism. A rate law consistent with the experimental results has
been derived. Based on the kinetic results, a suitable mechanism has
been proposed for the oxidative cleavage.
4-Oxoacids / NBSac – System
The kinetics of oxidation of 4-oxoacids (S1- S7) in aqueous acetic acid
medium have been followed potentiometrically at 30 0C. The reaction

153
is first order each in NBSac, 4-oxoacid and perchloric acid. The
hypobromous acidium ion, H2O+Br is the reactive oxidizing species,
which interacts with the enolic form of substrate.
The present reaction is observed to be a general acid catalyzed
reaction. The oxidation reaction does not induce the polymerization,
which indicates the absence of free radical intermediates in the
oxidation.
The effect of changes in the electronic nature of the substrate
has been studied with substituted oxoacids (S1-S7). The electron-
releasing group in the phenyl ring accelerates the reaction rate while
the electron-withdrawing group retards the rate. An excellent
correlation exists between log k2 and values ( = -1.41, r2 = 0.983).
The negative value of the reaction constant indicates the development
of positive charge in the transition state. The activation parameters for
the oxidation are evaluated from the Arrhenius plot. The Exner plot is
linear and the isokinetic temperature ( = 333 K) is evaluated from the
plot. The validity of Exner plot points out that all oxoacids follows
similar mechanism towards NBSac. A rate law consistent with the
experimental results has been derived. Based on the kinetic results, a
suitable mechanism has been proposed for the oxidation of 4-oxoacids
by NBSac.

154
A comparative study of NBB / 4-oxoacids and NBSac / 4-
oxoacids system have been made. In both the systems the
hypobromous acidium ion (H2O+Br) is the reactive oxidizing species,
which interacts with the enolic form of substrate. The excellent
correlation (slope = 0.986, r2 = 0.994) shown by log k2 of NBB
oxidation with log k2 of NBSac oxidation suggests a similar
mechanism for both the oxidants and for all the 4-oxoacids. The rate of
the oxidation reaction of oxoacids with NBS is slower compared to
NBB. The lower value measured in NBB oxidation is also in
consistent with the rate data for the two system, i.e., NBB has less
selectivity towards oxoacids than NBSac.

REFERENCES

155
References
1. Robert B. Grossman, The Art of Writing Reasonable Organic
Reaction Mechanisms, 2nd Edn., 2003, Springer, ISBN:
0-387-95468-6.
2. P.S. Kalsi, Organic Reactions and Their Mechanisms, New
Age International, 2009, ISBN: 8122425968,
9788122425963.
3. Micheal B. Smith and Jerry March, March’s Advanced
Organic Chemistry, Sixth Edn., Willey Interscience, 2007,
ISBN: 978-0-471-72091-7.
4. Adam Jacobs, Understanding Organic Reaction Mechanisms,
Cambridge University Press, 1997, ISBN: 0521467764,
9780521467766.
5. M. Gomez Gallego, M. A. Sierra, Organic Reaction
Mechanism, Springer, Berlin, 2004, ISBN: 3-540-00352-5.
6. Michael Edenborough, Organic reaction mechanisms: a step
by step approach, Taylor & Francis, 1999; ISBN:
0748406417, 9780748406418.
7. A. C. Knipe, Organic Reaction Mechanisms, 2009, Volume
141, John Wiley and Sons, 2011; ISBN: 1119972485,
9781119972488.

156
8. P. N. Mukherjee, Organic Reaction Mechanism, Dominant
Publishers & Distribuors, 2003; ISBN: 8178881101,
9788178881102.
9. B. Capon and C. W. Rees, Organic Reaction Mechanisms,
1967, Volume 105, John Wiley & Sons, 2008; ISBN:
0470067012, 9780470067017.
10. Kuldeep singh and A.K.Agrawal, kinetics and mechanism of
the oxidation of organic compounds, by N-
bromophthalimide, Ph.D Thesis, Ch. Charan Singh
University, Meerut, India
11. F. A. Patrocinio and J. S. Paulo Moran, J. Organomet.
Chem., 603, 220, 2000.
12. A. Graven, K. A. Joergensen, S. Dahl and A. Stanczak,
J. Org. Chem., 59, 3543, 1994.
13. S. Duan, J. Turk, J. Speigle, J. Corbin, J. Masnovi and
R.J. Baker, J. Org. Chem., 65, 3005, 2000.
14. P. S. Dhurn, N. U. Mohe and M. M. Salunkhe, Synth.
Commun., 31, 3653, 2001.
15. V. Canibano, J. F. Rodriguez, M. Santos, M. A. Sanz-
Tejedor, M. C. Carreno, G. Gonzalez and J. L. Garcia-
Ruano, Synthesis, 14, 2175, 2001.

157
16. B. P. Bandgar, L.S. Uppalla and V.S. Sadavarte, Syn. Lett.,
11, 1715, 2001.
17. M. K. Hossain, T. Kameyama, T. Shibata and K. Takagi,
Bull. Chem. Soc. Jpn., 74, 2415, 2001.
18. A. K. Tehrani, D. Borremans and N. De Kimpe,
Tetrahedron, 55, 4133, 1999.
19. C. Brindaban Ranu and U. Jana, J. Org. Chem., 64,
6380, 1999.
20. J. He and Jr. U. Charles Pittman, Synth. Commun., 29,
855, 1999.
21. R. Bartnik, D. Cal, P. Alan Marchand, S. Alihodzic and
A. Devasagayaraj, Synth. Commun., 28, 3949, 1998.
22. P. Maganus and D. Jennifer Kreisberg, Tetrahedron Lett.,
40, 451, 1999.
23. Pinho e Melo , M. V. D. Teresa, M. A. R Gonsloves, M. A.
Antonio, M. S. Sara Carrapato and M. O. M. P. Tabordo,
Molecules, 40, 217, 1999.
24. A. Olofson, K. Yakushijin and A. David Horne, J. Org.
Chem., 63, 1248, 1998.
25. A. Panayiotis Koutentis and W. Charles Rees, J. Chem. Soc.,
Perkin Trans. 1 , 2505, 1998.

158
26. R. P. Litrinovskaya, S.V. Drach and V. A. Kripach, Russ. J.
Org. Chem., 33, 172, 1997.
27. R. Adam Renslo and R. L. Danheiser, J. Org. Chem., 63,
7840, 1998.
28. B. Touaux, F. Texier-Boullet and J. Hamelin, Heteroat.
Chem., 9, 351, 1998.
29. M. Sergio Bones and R. Erra-Balsells, J. Heterocycl. Chem.,
34, 877, 1997.
30. M. Serge Ivono, J. Juston Rockwell, M. Susie Miller,
P. Oren Anderson, A. Konstantin Solntsev and H. Steven
Strauss, Inorg. Chem., 35, 7882, 1996.
31. K. John Gallos, V. Theoharis Koftis, I. Vassiliki Karamitrou
and C. Alexandros Koumbis, J. Chem. Soc., Perkin Trans. 1,
17, 2461, 1997.
32. M. Werner, S. David Stephenson and G. Szeimies, Liebigs
Ann., 11, 1705, 1996.
33. I. Janigova and J. Rychly, Chem. Pap., 45, 845, 1991.
34. (a) V. Ozola, N. Ramzaeva, Y. Maurinsh and M. Lidaks,
Nucleosides Nucleotides, 12, 479, 1993; (b) K. Ardeshir,
M. Tajbakhsh and S. Habibzadeh, Asian J. Chem.,12, 291,
2000.

159
35. Y. Msuyama, Y. Kobayashi and Y. Kurusu, J. Chem. Soc.
Chem. Commun., 9, 1123, 1994.
36. K. Takagi, H. Tkachi and N. Hayama, Chem. Lett., 3,
509, 1992.
37. J. M. Antelo, F. Arce, J. Florencio, L. Garcia, C. Maria,
M. Sanchez and A. Varela, Int. J. Chem. Kinet., 20, 397,
1988.
38. (a) A. Madjdabadi, R. Beugelmans and A. Lechvallier,
Synthesis, 826, 1986; (b) S. D. Bhatta Mishra and S. K.
Mishra, Oxid. Commun., 10, 235, 1987.
39. (a) B. M. Sasmal, D. P. Patnaik and P. S. N. Subudhi,
Acta. Cienc. Indica. Chem., 12, 55, 1986; (b) A. Khazaei,
E. Mehdipour and A. Roodpeima, Iran J. Chem., and Chem.
Eng., 2, 14, 1995.
40. (a) C. Tetzlaff, E. Vilsmaier and R. Wolf Schlag,
Tetrahedron, 6, 8117,1990. (b) A. Khazaei, E. Mehdipour
and M. Sadri, Iran Poly. J., 2, 5, 1996.
41. J. Garin, C. Guillen, E. Melendez, F. L. Merchand J. Orduna
and T. Tejero, Heterocycles, 26, 2371, 1987.
42. M. V. D. Terasa, M. A. Rocha Antonio, S. J. Glaudia Lopes
and L. Thomas Gilchrist, Tetrahedron Lett., 40, 789, 1999.

160
43. L. Eberson, P. Michael Hartshorn, F. Radner and O. Persson,
Perkin Trans. 2, 59, 1998.
44. R. Hyoung Kim, I. Seung Shin, Ju. H. Park, Ju. Dong Jeon
and K. Eung Ryn, Syn. lett., 7, 789, 1998.
45. M. Uma, R. Gurumurthy and C. S. Kalavathi, Acta. Cienc.
Inidca. Chem., 12, 88, 1989.
46. A. Deagostino, B. Paolo Tivola, C. Prandi and F. Venturello,
Syn.lett., 11, 1841, 1999.
47. M. M. Cambell and G. Jhonson, Chem. Rev., 78, 65,1978.
48. L. Horner and E. H. Winkelmann in Newer Methods of
Preparation of Organic Chemistry, edn. W. FOERSI
Academic , New York, 1964.
49. E. Berliner, J. Chem. Educ., 43, 124, 1966.
50. V. A. Morab and S. T. Nandibewoor, React. Kinet. Catal.
Lett., 53, 25, 1994.
51. S. V. Rao and V. Jagannadham, React. Kinet. Catal. Lett.,
27, 239, 1985.
52. S. C. Negi, I. Bhatia and K. K. Banerji, J. Chem. Res.,
360, 1981.
53. R. C. Gupta and S. P. Srivastava, Indian J. Chem., 10,
706, 1972.

161
54. S. P. Srivastava, R. C. Gupta and A. K. Shukla, Indian J.
Chem., 15A, 605, 1977.
55. E. Boyland and P. Sims, J. Chem. Soc., Part I, 980, 1954.
56. A. Lal and M. C. Agarwal, Indian J. Chem., 26A, 696,
1987.
57. G. P. Panigrahi and D. D. Mahapatro, Int. J. Chem. Kinet.,
14, 977, 1982.
58. P. S. Radhakrishnamoorthy and R. Panda, Indian J. Chem.,
9, 1247, 1971.
59. R. Vedavrath, B. Sethuram and T. Navaneeth Rao, Proc.
Natl. Acad. Sci. Inida, 51A, 443, 1981.
60. M. D. Prasada Rao and J. Padmanabha, Indian J. Chem.,
19A, 984, 1980.
61. Vijayalakshmi and E. V. Sundaram, Indian J. Chem., 17A,
495, 1979.
62. A. Lal and M. C. Agarwal, Indian J. Chem., 23A, 411,
1984.
63. C. Subas Pati, B. R. Dev, B. Narasingh and M. Minakshi,
Proc. Ind. Natl. Sci. Acad., 49A, 538, 1983.
64. M. Prasada Rao, B. Sethuram and T. Navaneeth Rao,
Indian J. Chem., 17A, 52, 1979.

162
65. P. V. Sreenivasulu, M. Adinarayana, B. Sethuram and
T. Navaneeth Rao, Oxid. Commun., 8, 199, 1985.
66. A. Lal and M. C. Agarwal, J. Indian Chem. Soc., 67, 164,
1990.
67. A. Agarwal, S. Mittal and K. K. Banerji, Indian J. Chem.,
26A, 339, 1987.
68. M. K. Reddy, C. H. S. Reddy and E. V. Sundaram,
Tetrahedron, 41, 307, 1985.
69. M. L. Bishnoi and K. K . Banerji, Tetrahedron., 41, 6047,
1985.
70. P. Manikyamba and E. V. Sundaram, Indian J. Chem., 19A,
1122, 1980.
71. P. S. Radhakrishnamoorthy and J. C. Panigrahi, Oxid.
Commun., 12, 25, 1989.
72. S. C. Hiremath, S. Mayanna and N. Venkatasubramanian,
J. Chem. Soc., Perkin Trans. 2, 1569, 1987.
73. S. Kandlikar, B. Sethuram and T. Navaneeth Rao, Indian J.
Chem., 17A, 264, 1979.
74. S. C. Negi and K. K. Banerji, Indian J. Chem., 21B, 946,
1982.
75. K. Vijayamohan, K. Rao and E.V. Sundaram, J. Indian
Chem. Soc., 61, 225, 1984.

163
76. B. Thimme Gowda and J. Iswara Bhat, Tetrahedron, 43,
2119, 1987.
77. (a) V. Thiagarajan and N. Venkatasubramanian, Indian J.
Chem., 6, 809, 1967; (b) B. Thimme Gowda and R. V.
Rao, J. Chem Soc., Perkin Trans. 2, 355, 1988.
78. (a) S. C. Negi and K. K. Banerji, Indian J. Chem., 21B, 846,
1982; (b) B. Thimme Gowda and B. S. Sherigara, Int. J.
Chem. Kinet., 21, 31, 1989.
79. B. Thimme Gowda and P. Ramachandra, J. Chem Soc.,
Perkin Trans. 2, 1067,1989.
80. F. Ruff and A. Kucsman, J. Chem. Soc., Perkin Trans. 2,
1075,1982.
81. D. M. Rosa, C. G. Nieto and F. F. Gago, J. Org. Chem., 54,
5437, 1989.
82. X . L. Armesto, M. Canle , M. V. Carcia and J. A.
Santaballa, Chem. Soc. Rev., 27, 453, 1998.
83. W. Stummn and J. J. Morgan, Aquatic Chemistry, Chemical
Equilibria and Rates in Natural Waters, John Wiley, New
York, 3rd edn., 1996.
84. R. L. Jolley, L. W. Condie, J. D. Johnson, S. Katz, R. A.
Minear, J. S. Matice and V. A. Jacobs, eds., Water

164
Chlorination: Chemistry, Environmental and Health Effects,
Lewis Publishers, Michigam, 6, 1990.
85. P. Kovacic, M. K. Lowery and K. W. Field, Chem. Rev., 70,
639, 1970.
86. J. Arotsky and M. C. R. Symons, Quart. Rev. Chem. Soc.,
16, 282, 1962.
87. P. S. Radhakrishnamurthi and M. D. Prasad Rao, Indian J.
Chem., 14B, 790, 1976.
88. N. S. Shrinivasan and N. Venkatasubramanian, Tetrahedron,
30, 419, 1974.
89. P. Saroja, K. B. Kishore and S. Kandlikar, Indian J. Chem.,
28A, 501, 1989.
90. M. Puttaswamy, T. M. Anuradha, R. Ramachandrappa, and
N. M. Made Gowda, Int. J. Chem. Kinet., 32, 221, 2000.
91. P. S. Radhakrishnamurti and L. D. Sarangi, Indian J. Chem.,
20A, 30, 1981.
92. (a) Ghanat K. Joshi, Y. R. Katre, and Ajaya K. Singh,
Journal of Surfactants and Detergents, 9(3), 231-235, 2006,
DOI:10.1007/s11743-006-5002-3; (b) Yokraj Katre ,
Minu Singh and Ajaya K. Singh, Colloid Journal, 74(3),
391-400, 2012, DOI:10.1134/S1061933X12030167; (c) N.
M. I. Alhaji, G. K. Ayyadurai and A. Shajahan, Chemical

165
Science Transactions , 2(2), 467-472, 2013,
DOI:10.7598/cst2013.409; (d) Jagdish V. Bharad, Balaji R.
Madje and Milind B. Ubale, International Journal of
ChemTech Research, 2(1), 346-353, 2010; (e) N.M.I.Alhaji
and Sowfiya Lawrence Mary, E-J Chem., 8(4),1472-1477,
2011.; (f) P. Sangeeta, K. Y Katre and Singh Ajaya,
Journal Surfactants and Detergents, 10(3), 175-184, 2007.
93. N.M.I.Alhaji and Sowfiya Lawrence Mary, E-J Chem.,
8(4),1728-1733, 2011.
94. (a) N. A. Mohamed Farook and G. A. Seyed Dameem, E-
Journal of Chemistry, 8(2), 479-482, 2011, DOI:
10.1155/2011/173749; (b) B. Singh, S. Sahai and D. Gupta,
Oxidation Communications, 34(4), 741-745, 2011; (c) P. R.
Sharadamani and V. Jagannadham, Indian J. Chem., 29A,
700, 1990.
95. (a) R. K. Singh, A.K. Singh, R. Srivastava, S. Srivastava, J.
Srivastava and Rahmani, Transition Metal Chemistry, 35(3),
349-355, 2010, DOI: 10.1007/s11243-010-9334-5; (b) M.
Komal Reddy, Ch. Sanjeeva Reddy and E.V. Sundaram,
Indian J. Chem., 23A, 197,1984.
96. S. Bajpai, M. Saxena, B. Singh and A. K. Singh, J Indian
Chem Soc., 68(9), 497-499, 1991.

166
97. (a) R. K. Singh, A.K. Singh, R. Srivastava, S. Srivastava, J.
Srivastava and Rahmani, Abstracts of Papers Of The
American Chemical Society, 238, Meeting Abstract: 608-
ORGN, Published: AUG 16 2009; (b) B. T. Gowda and P. J.
Rao, Proceedings of the Indian Academy of Sciences-
Chemical Sciences, 103(1), 55-68, 1991.
98. (a) A. K. Singh, S. Srivastava, J. Srivastava and Rahmani,
Chinese Journal of Chemistry, 26(6), 1057-1067, 2008,
DOI: 10.1002/cjoc.200890188; (b) N. Gupta, N. Chaurasia,
V. Singh and A. K. Singh, Oxid. Commun., 23(1), 42-49,
2000.
99. (a) R. A. Singh, V. Yadav, R. A. Singh, and K. Singh,
Oxidation Communications, 31(1), 160-166, 2008;
(b) S. Kabilan, S. Kirubasankar, and K. Krishnasamy,
Oxid. Commun., 29(2), 335-342, 2006.
100. A. Moondra, A. Mathur and K.K. Banerji, Int J Chem Kinet.,
23(8), 669-681, 1991.
101. M. S. Ramachandran, D. Easwaramoorthy and R. P. M.
Maniraj, Int J Chem Kinet., 28(7), 545-551, 1996.
102. B.B.L. Saxena, S. Trivedi, C. Waqar, A. Kumar and
B. Singh, J Indian Chem Soc., 67(3), 202-203, 1990.

167
103. M. Saxena, R. Gupta, A. Singh, B. Singh and A. K. Singh, J.
Mol. Catal., 65(3), 317-327. 1991.
104. P. R. Sharadamani and V. Jagannadham, Indian J Chem., A,
30(6), 518-521, 1991.
105. P. Shardamani and V. Jagannadham, National Academy
Science Letters-India, 14(8), 331-335, 1991.
106. A. K. Singh, A. Srivastava, A. Kumar and B. Singh,
Transition Metal Chemistry, 18(4), 427-430, 1993.
107. A. K. Singh, V. Singh, N. Gupta and B. Singh, Carbohydrate
Research, 334(4), 345-351, 2002.
108. A. K. Singh, V. Singh, S. Rahmani and Singh, J Mol Catal.,
A-Chemical, 197(1-2), 91-100, 2003.
109. A. K. Singh, S. Rahmani, B. Singh, R. K. Singh, and M.
Singh, J Phys Org Chem., 17(3), 249-256, 2004.
110. a) A. K. Singh, V. Singh and Ashish Srivastava, J Indian J
Chem., A, 45(3), 599-608, 2006; b) R. A. Singh, V. Yadav, A.
K. Singh, K. Singh, Oxid. Commun., 38(1), 160-166, 2008; c)
R.A.Singh, V. K. Srivastava, A. Srivastava and V. Yadav,
Oxid Commun., 27(3), 643-649, 2004; d) B. Singh, S. Sahai
and D. Gupta, Oxid. Commun., 33(2), 295-299, 2010;
e) B. Singh and S. Sahai, J Indian Chem Soc., 68(4), 208-209,

168
1991; f) B. Singh, D. Singh, S. Bajpai and A Kumar,
Transition Metal Chem., 16(6), 610-613, 1991.
111. P. K. Tandon, J. P. Singh and M.P. Singh, Oxid. Commun.,
22(3), 424-431, 1999.
112. S. Srivastava, V. Srivastava, V. Gupta and L. Chaudhary,
Oxid. Commun., 30, 553-560, 2007.
113. a) S. Srivastava and S. Singh, Oxid. Commun., 29(3), 708-
714, 2006; b) S. Srivastava, A. Awasthi and K. Singh, Int J
Chem Kinet., 37(5), 275-281, 2005: c) S. Srivastava,
A. Awasthi and V. Srivastava, Oxid. Commun., 26(3), 426-
431, 2003.; d) S. Srivastava, V. Srivastava, and A. Awasthi,
Oxid Commun., 26(3), 378-384, 2003.
114. S. Perumal and M. Ganesan, Indian J. Chem., 28A, 961,
1989.
115. (a) D. S. Mahadevappa, M. Sayeed Ahmed, N. M. Made
Gowda and B. Thimme Gowda, Int J of Chem Kinet.,
15(8), 775 – 793, 2004; (b) S. Meenakshisundaram and M.
Selvaraju, Int. J. Chem. Kinet., 34, 27, 2002; (c) H.
Ramachandra, K. S. Rangappa and D. S. Mahadevappa,
Chinese Journal of Chemistry, 26(3), 536 – 542, 2008; (d)
K. S. Rangappa, H. Ramachandra and D. S. Mahadevappa,
Int J Chem Kinet., 37 (9), 572 – 582, 2005; (e) Puttaswamy

169
and R. V. Jagadeesh, Central European Journal of
Chemistry, 3(3), 482-501, 2005.
116. (a) K. Mohan, Mahadevappa and Puttaswamy, Chemical
Science, 102(1), 65-72, 1990, DOI:10.1007/BF02861572; (b)
Puttaswamy, Nirmala Vaz and N. M. Made Gowda, Int J
Chem Kinet., 37(11), 700-709, 2005; (c) B. Thimme Gowda,
V. Pardhasarathi and P. Ramachandra, Indian J. Chem., 29A,
1192, 1990.
117. (a) M. Rangaswamy and Ananda, Journal of Indian Council
of Chemists, 18(2), 12-19, 2001; (b) Mallamma, S. Ananda,
Rangaswamy and N. M. Made Gowda, Synthesis and
Reactivity in Inorganic, Metal-Organic, and Nano-Metal
Chemistry, 33(9), 1555 – 1568, 2003; (b) M. Puttaswamy,
V. Nirmala and N. M. Made Gowda, Int. J. Chem. Kinet., 33
480, 2001.
118. (a) M.C.Agarwal and A. Lal, Journal of the Indian
Chemical Society, 62(2), 164-166, 1990; (b) A.N. Mohamed
Kassim and K.A. Basheer Ahamed, Indian J Chem., A 38(6),
533-540, 1999; (c) N.A. Mohamed Farook and G.A. Seyed
Dameem, Journal of Chemistry, 8(2), 561-564, 2011.
119. (a) Suresh P. Nayak and Thimme Gowda, Journal of
Physical Organic Chemistry, 5(11), 755-763, 2004,

170
DOI: 10.1002/poc.610051108; (b) S. P. Nayak and B.T.
Gowda, J Indian Chem Soc., 69(10), 637-641, 1992.
120. (a) D. Anjali, Indian Journal of Chemical Society, 67, 164-
166, 1990; (b) S. P. Nayak and B. T. Gowda, J Phys Org
Chem., 92, 755-763, 1992, DOI: 10.1002/poc.610051108.
121. (a) B. T. Gowda and P. J. M. Rao, Indian J Chem., A, 30(2),
148-153, 1991; (b) B. T. Gowda and P. J. M. Rao,
Zeitschrift Fur Physikalische Chemie Neue Folge, 68, 183-
192, 1990; c) K. A. B. Ahamed, Indian J Chem., A, 36(3),
222-224, 1997.
122. (a) K. Chowdhury and K. K. Banerji, J. Org. Chem., 55,
5391, 1990; (b) V. K. Siriah and N. Mohan Gupta, Asian J.
Chem., 12, 1071, 2000; c) M.K. Badole, L.N. Malviya, K.S.
Sariya and V.K. Siriah, Oriental Journal of Chemistry, 28,
1433-1436, 2012; d) Rachna Prakash and Shashi Agarwal,
Journal of Engineering, Science and Management
Education, 6(1), 2013;
123. (a) K. K. Banerji, J. Org. Chem., 51, 4764, 1986;
(b) P. Archana and V. K. Seeriya, Asian J. Chem., 4, 744,
1992; (c) Kalyan K Banerji, Indian Academy of Science
(Chem. Sci), 100(5), 397-403, 1988; (d) Vyas V.K, Kothari S

171
and Banerji K.K, Indian Journal of Chemistry, Section A,
35(2), 112-115, 1996.
124. V. K. Seeriya, V. R. Chourey, R. S Varmaa, S. K. Solankee
and Vi. Raa. Shaastry, J. Indian Chem. Soc., 63, 847,1986.
125. (a) V. K. Viyas, S. Kothari and K. K. Banerji, Indian J.
Chem., 35A, 112, 1996; (b) O. N. Choubey , V. K.
Seeriya and S. K. Aswathi, Asian J. Chem., 12, 302, 2000.
126. K. S. Rangappa, K. Manjunathaswamy, M. P. Ragahavendra
and N. M. Made Gowde, Int. J. Chem. Kinet., 34, 49, 2002.
127. K. Vivekanandan and K. Nambi, Indian J. Chem., 35B,
1117, 1996.
128. K. Vivekanandan and K. Nambi, J. Indian Chem. Soc., 76,
198, 1999.
129. G. F. Hambly and T. H. Chan, Tetrahedron Lett., 27, 2563,
1986.
130. K. K. Banerji, J. Chem. Soc., Perkin Trans. 2, 1015, 1988.
131. D. Thenraja, P. Subramaniam and C. Srinivasan, J. Chem.
Soc., Perkin Trans. 2, 2125, 2002.
132. T. Mukaiyama, J. Matsuo, D. Iida and H. Kitagawa, Chem.
Lett., 8, 846, 2001.
133. J. Einborn, C. Einborn, F. Ratajczak and J. Pierre, J. Org.
Chem., 61, 7452, 1996.

172
134. J. M. Antelo, F. Arce, J. O. Crugeiras and M. Parajo, J. Phys.
Org. Chem., 10, 631, 1997.
135. L. Fieser and S. Rajagopalan, J. Am. Chem. Soc., 71, 3935,
1979.
136. N. N. Hallingudi, S. M. Desai and S. T. Nandibewoor, Oxid.
Commun., 23, 117, 2000.
137. S. T. Nandibewoor, S. M .Desai and N. N. Hallingudi, Indian
J. Chem., 38A, 943, 1999.
138. M. S. Ramachandran, R. Santhanalakshmi and D. Easwara-
moorthy, Oxid. Commun., 21, 230, 1998.
139. K. Takaki, M. Ysumura and K. Negoro, J.Org. Chem., 48,
54, 1983.
140. M. S. Ramachandran, D. Easwaramoorthy, V. Rajasingh and
T. S. Venkatasundaram, Bull. Chem. Soc. Jpn., 63, 2397,
1990.
141. E. J. Barry, M. Finkelstein, W. M. Moore and D. S.
Ross, J. Org. Chem., 50, 528, 1985.
142. S. M. Farook, S. Sivakamasundari and S. Viswanathan,
Indian J. Chem., 23A, 239, 1984.
143. Y. Nagao and S. Katagiri , Bull. Chem. Soc., Jpn, 61, 1393,
1998.

173
144. K. S. Kim, I. H. Cho, K. B. Yoo, Y. H. Song and C. S.
Hahn, J. Chem. Soc., Chem. Commun., 12, 762, 1984.
145. (a) N. Mathiyalagan and R. Sridharan, Indian J Chem., A,
44(10), 2044-2047, 2005; (b) P. S. Radhakrishnamurthy and
B .M. Sasmal, Indian J. Chem., 19A, 880, 1980.
146. J. M. Antelo, F. Arce, J. Crugeiras, C. Pastoriza and R. A.
Cristina, J. Chem. Res. Synop., 1, 20, 2001.
147. T. Ikemoto and M. Wakimasu, Heterocycles, 55, 99, 2001.
148. B. Thimme Gowda and P. S. Krishna Kumar, Z. Phys. Chem.
(Munich) , 167, 151, 1990.
149. N. Venkatasubramanian and V. Thiagarajan, Can. J. Chem.,
47, 694, 1969.
150. (a) Pandey, K. L., Synlett., 947, 2008; (b) M. Jambulingam,
P. Nagarajan, P. Arulvani and K. Ramarajan, J. Chem. Soc.,
Perkin Trans., 2, 957-959, 1986,
DOI: 10.1039/P29860000957; (c) V. K. Sharma, K. Sharma,
H.D. Gupta and O. P. Gupta, O. P. Oxidation
Communications, 18(4), 395-406, 1995.
151. (a) H. Firouzabadi, N. Iranpoor and F. Ebrahimzadeh,
Tetrahedron Lett., 47, 1771, 2006, DOI:10.1016/
j.tetlet.2006.01.033; (b) U. Mishra, K. Sharma and V.K.
Sharma, Carbohydr. Res., 147, 155-159, 1986; (c) Khazaei

174
A, Rostami A and Aminimanesh A, J. Chin. Chem. Soc., 53,
437-441, 2006.
152. (a) S. P. L. De Souza, J. F.Da Silva and M.C.S De Mattos,
J. Braz., Chem. Soc., 14, 832, 2003, DOI:10.1590/S0103-
50532003000500021; (b) K. Mohan, R. Vijaya, P.
Rahgunath and E. V. Sundaram, J. Indian. Chem. Soc., 65,
91, 1988.
153. (a) A.Khazaei, A.Aminimanesh and A.Rostami, Phosphorus
Sulfur Relat. Elem., 179, 2483, 2004, DOI:10.1080/
10426500490485471; (c) C. M. Das and P. Indrasenan,
Indian J. Chem., 26A, 55, 1987, ibid., 26A, 717, 1987.
154. (a) E.I.Shchez and M.J.Fumarola, J. Org. Chem., 47, 1588,
1982; (b) S. Meenakshisundaram and M. Selvaraju, Int. J.
Chem. Kinet., 34, 1, 2002.
155. (a) D.Yang, Ye X Y, M. Xu, K.W.Pang and K.K.Cheung, J.
Am. Chem. Soc., 122, 1658, 2000; (b) U. Mishra, K. Sharma
and V. K. Sharma, J. Indian Chem. Soc., 63, 586, 1986.
156. (a) J.T.Tateiwa and A.Hosomi, Eur. J. Org. Chem., 1445,
2001; (b) D.Yang, Y.Yan and B.Lui, J. Org. Chem., 67,
7429, 2002; (c) V. Manoharan and N. Venkatasubramanian,
Indian J. Chem., 23A, 389, 1984.

175
157. (a) A. K. Tiwari, O.P. Gupta, K. Sharma and V.K.Sharma,
Oxidation Communications, 22, 591-598, 1999; (b)
K. Vijayamohan, R. Rao and E. V. Sundaram, J. Indian
Chem. Soc., 61, 225, 1984.
158. (a) B. Ramkumar, Asian J. Chem., 14, 463, 2002; (b)
K. Vijaya Mohan, P. Raghunatha Rao and E.V. Sundaram,
Proc. Nat. Acad. Sci. India, 58(A), I, 49, 1988; (c) Zachariah
A, Asian Journal of Chemistry, 15, 1567-1571, 2003; (d) T.
D. R. Nair, and A. Zachariah, Asian Journal of Chemistry,
14, 117-120, 2002; (e) P. C. Shukla, P. S. Tiwari and Deepa
Khare, International Journal of Pharmacy & Life Sciences,
3(1), 1380-1384, 2012; (f) V.K. Sharma, K. Sharma, A.
Pandey and P.F.Hashmi, Oxidation Communications, 23(1),
62-70, 2000; (g) V.K. Sharma, K. Sharma and P.F.Hashmi,
Oxidation Communications, 23(1), 71-77, 2000.
159. S. Kothari, A. Agrawal and K. K. Banerji, Indian J. Chem.,
25A, 722, 1986.
160. K. Nagarajan, V. Balasubramanian and N. Venkatasubra-
manian, Indian J. Chem., 19A, 576, 1980.
161. K. A. Basheer Ahamed, S. Sheik Dawood, B. Baskaran and
K. Nambi, J. Indian Chem. Soc., 73, 687, 1996.

176
162. (a) N. A. Mohamed Farook, G. A. Seyed Dameem,
A. Murugesan and M. Kanagaraj, E-J Chem., 1(2), 132-
136, 2004; (b) V. K. Mohan Rao, P. Ragunatha and E.
V. Sundaram, J. Indian Chem. Soc., 63, 698,1986.
163. J. M. Bacchawat and N. C. Mathur, Indian J. Chem., 9, 1335,
1993.
164. M. U. Khan, J. K. Verma, V. R. Singh and H. P. Dwivedi,
Oxid. Commun., 16, 235, 1993.
165. K. Vijayamohan, P. Raghunath Rao and E. V. Sundaram,
Proc. Indian Acad. Sci., 58A, 49, 1988.
166. (a) N. A. Mohamed Farook and N. M. I. Alhaji, Int. J. Chem.
Sci, 2(1), 76-80, 2004; (b) P. K. Tiwari, J. K. Verma and H.
D. Gupta, Oxid. Commun., 20, 117, 1997.
167. M. U. Khan, J. K. Verma, S. K. Nigam and S. S. Parihar,
Oxid. Commun., 21, 362, 1998.
168. M. U. Khan, S. K. Nigam, J. K. Verma and A. Nigam, Oxid.
Commun., 18, 304, 1995.
169. N. Jayasree and P. Indrasenen, Indian J. Chem., 26, 714,
1987.
170. V. P. Singh, M. U. Khan, D. B. S. Chauhen and J. K. Verma,
Oxid. Commun., 20 , 124, 1997.

177
171. (a) N. A. Mohamed Farook, G. A. Seyed Dameem, A.
Murugesan and M. Kanagaraj, E-J Chem., 2(1), 132-136,
2004; (b) M. U. Khan, S. K. Nigam, and O. P Gupta Oxid.
Commun., 22, 259, 1999.
172. (a) N. A. Mohamed Farook, R. Prabaharan, S. Rahini,
R. Senthil Kumar, G. Rajamahendran, and B. Gopala
krishnan , E-J Chem., 1(2), 127- 131, 2004; (b) M. U. Khan,
J. K. Verma, S. K. Nigam, S. S. Pariher and H. P. Dwivedi,
Oxid. Commun., 21, 362, 1998.
173. S. Khan, M. U. Khan, S. K. Singh, H. D. Gupta and P. K.
Singh, Asian J. Chem., 15, 595, 2003.
174. (a) N. A. Mohamed Farook, J Solution Chem., 36(3), 345-
356, 2007; (b) H. L. Riley, J. F. Moreley and N. A. Friend, J.
Chem. Soc., 1875, 1932.
175. S. Tiwari, M. U. Khan, B. M. L. Tiwari, K. S. Tiwari and
N. D. Valechha, Oxid. Commun., 22, 259-267, 1999.
176. (a) B. L. Hiran1, R. K. Malkani1 and N. Rathore, Kinetics
and Catalysis, 46(3), 334-339, 2005; (b) A. L. Harihar, M.
R. Kembhavi and S. T. Nandibewoor, J. Indian Chem.
Soc., 76, 128, 1999.
177. (a) Abdel-Hady, Alaa Eldin Mokhtar, Farag, Rabie S, Abu-
Khadra and Ahmad S. Transition Metal Chemistry,35(5),

178
571-576, 2010; (b) A. K. Singh, S. Rahmani, K. V. Singh, V
.Gupta, D. Kesarwani and B. Singh, Indian J. Chem., 40,
519, 2001.
178. (a) N. A. Mohamed Farook, J Iran Chem Soc., 3(4),378-386,
2006; (b) B. Thimme Gowda and J. Ishwara Bhat, Indian J.
Chem. 28A, 211, 1989.
179. (a) Katre Y, Singh M and A.K. Singh, Tenside Surfactants
Detergents, 47(2), 98-105, 2010; (b) C. Karunakaran and
K. Ganapathy, Indian J. Chem., 29A, 133, 1990.
180. (a) B. Thimme Gowda and J. Ishwara Bhat, Indian J.
Chem., 28A, 43, 1989; (b) R. Saxena, S. Gupta and K. S.
Upadhyay, Indian J. Chem., 29, 847,1990.
181. (a) M. N. Kumara, N.S. Gowda, Linge and Mantelingu K et
al., Journal of Molecular Catalysis A-Chemical, 309(1-
2), 172-177, 2009; (b) P. G. Reddy, T. Kistayya, J. A. Khan
and S. Kandlikar, Z. Phys. Chem. (Leipzig), 269, 1253,
1988.
182. (a) Abdel-Hady, Alaa Eldin M, Transition Metal Chemistry,
33(7), 887-892, 2008; (b) P. S. Radhakrishnamurti, B. M.
Sasmal and D. P. Patnaik, Indian J. Chem., 25A, 69, 1986.

179
183. (a) M. Alaa Eldin, Abdel-Hady, Transition Metal Chemistry,
33(7), 887-892, 2008; (b) P. Saroja and S. Kandlikar,
Indian J. Chem., 27A, 632, 1988.
184. (a) P. Shalini and U. Santosh, J Colloid and Interface
Science 285(2), 789-794, 2005; (b) A. Sukla and S. K.
Upadhyay, J. Indian Chem. Soc., 11, 745, 1992.
185. (a) Patil, B.Dilip, Chachere and D.Sunil, Asian Journal of
Chemistry, 20(2), 1539-1543, 2008; (b) B. Singh, V. K.
Singh, A. K. Singh and M. B. Singh, J. Indian Chem. Soc.,
63, 1049, 1986.
186. (a) A. Ewais Hassan, A. Nagdy Mohamed and A. Abdel-
Khalek Ahmed, Journal of Coordination Chemistry, 60(22-
24), 2471-2483, 2007; (b) R. Saxena and S. K. Upadhyay,
Indian J. Chem., 32A, 1060, 1993; (c) Ewais Hassan A,
Nagdy Mohamed A and Abdel-Khalek Ahmed A, Journal of
Coordination Chemistry, 60(22-24), 2471-2483, 2007.
187. (a) A. Ewais Hassan, E. Ahmed Ahmed and A. Abdel-
Khalek Ahmed, Int J Chem Kinet., 36(7), 394-400, 2004; (b)
B. Bhargava, B. Sethuram and T. N. Rao, Indian J. Chem.,
16A, 651, 1978.
188. (a) N.S. Linge Gowda, M.N.Kumara, D.C.Gowda and
Rangappa, International Journal of Chemical Kinetics,

180
38(6), 376-385, 2006; (b) G. S. Sundaram and N.
Venkatasubramanian, J. Chem. Soc., Perkin Trans. 2, 949,
1983.
189. (a) M.A.Alaa Eldin, International Research Journal of Pure
and Applied Chemistry, 4(1), 76-87, 2014; (b) R. T. De
Mattos, E. R. Lachter and A. C. Neto, Quim. Nova, 1, 17,
1985 (Chem. Abstr., 107, 1987).
190. (a) K. N. Mohana and K. R. Ramya, J Molr Catal A,
Chemical, 302(1/2), 80-85, 2009; (b) J. P. Sharma, R. N. P.
Singh, A. K. Singh and B. Singh, Tetrahedron, 42, 2739,
1986.
191. N. K. Mathu and C. K. Narang, The Determination of
Organic Compounds with N-Bromosuccinimide and Allied
Reagents. Academic Press, New York, 15, 1975.
192. (a) R. Ramachandrappa, Puttaswamy, S. M. Mayanna and N.
M. Made Gowda, Int J Chem Kinet., 36(7), 394-400, 2004;
(b) S. Perumal, K. Syed Abbas and R. Chandrasekaran,
Indian J. Chem., 28A, 592, 1989.
193. (a) S. Pandey and S. K. Upadhyay, Langmuir., 21(24),
10983-10991, 2005; (b) D. L. Kamble and S. T.
Nandibewoor, Pol. J. Chem., 71, 91, 1997.

181
194. R. B. Chougale, D. L . Kamble and S. T. Nandibewoor, Pol.
J. Chem., 71, 986, 1997.
195. (a) Minu Singh, International Journal of Carbohydrate
Chemistry, 2014, Article ID 783521, 9 pages,
DOI:10.1155/2014/783521; (b) D. L. Kamble, G.H.
Hugar and S. T. Nandibewoor, Indian J. Chem., 35, 144,
1996.
196. (a) N. Nanda, S. Malini, Puneeth Kumar and Netkal M.
Gowda, International Research Journal Pure and Applied
Chemistry, 4(6), 834-845, 2014; (b) K. Meenal and
D. Vimala Devi, J. Indian Chem. Soc., 73, 391, 1996.
197. (a) A. K. Singh, R. K. Singh, Srivastava, J. Rahmani and Y.
Sarita, Indian Journal of Chemistry, 51A, 681-689, 2012; (b)
D. L. Kamble, R. B. Chougale and S. T. Nandibewoor,
Indian J. Chem., 35, 865, 1996.
198. (a) K. N. Mohana and P. M. Ramdass, Journal of the
Chinese Chemical Society, 54, 1223-1232, 2007; (b)
R. Ramachandrappa, M. Puttaswamy, S. M. Mayanna and
N. M. Made Gowda, Int. J. Chem. Kinet., 30, 407, 1998.
199. A. A. Abdel-Khalek, M. El -Said Sayyah and S. H. E.
Khaled, Indian J. Chem., 37, 489, 1998.

182
200. A. K Singh, D. Chopra, S. Rahmani and B. Singh,
Carbohyd. Res., 314, 157, 1998.
201. (a) A. A. Abdel-Khalek, H. A. Ewais, E. S. H. Khaled and
A. Abdel-Hamied, Transition Metal Chemistry, 28(6), 635-
639, 2003; (b) V. Thiagarajan and N. Venkatasubramanian,
Tetrahedron Lett., 3349, 1967.
202. T. Kistayya, M. S. Reddy and S. Kandlikar, Indian J. Chem.,
25A, 905, 1986.
203. (a) B. Hiran, R. Malkani, and N. Rathore, Kinetics and
Catalysis, 46(3), 334-339, 2005; (b) G. Gopalakrishnan and
L. H. John, J. Org. Chem., 50, 1206, 1985.
204. S. Bharat, P. Lalji, J. Sharma and S. M. Pandey,
Tetrahedron, 38, 169, 1982.
205. K. Ganapathy and C. Karunakaran, Monatsh. Chem., 113,
1239, 1982.
206. S. P. Srinivasan and N. S. Gnanapragasam, J. Indian Chem.
Soc., 60, 953, 1983.
207. L. S. A. Dikshitulu, P. Vani and B. Vasantha Kumar, J.
Indian Chem. Soc., 61, 385, 1984.
208. M. Vijayasree, K. Srinivas and P.V. Subba Roa, Indian J.
Chem., 22A, 806, 1983.

183
209. S. M. Pandey, J. Sharma, O. Prakash and S. P. Mushran,
Indian J. Chem., 20A, 1021, 1981.
210. (a) S. P. Mushran, L. Pandey and S. Kameshwar, Monatsh.
Chem., 111, 1135, 1980; (b) G. Gopalakrishnan, B. R. Pai
and N. Venkatasubramanian, Indian J. Chem., 19B, 293,
1980; (c) P. S. Radhakrishnamurti and S. N. Sahu, Indian J.
Chem., 15A, 785, 1977.
211. (a) V. Siriah and M. K. Badole, International Journal of
Scientific Research Publications, 3(9), 1-5, 2013.
212. L. N. Malviah, V. K. Siriah and M.K.Badole, Oriental
Journal of Chemistry, 29(2), 767-770, 2013.
213. R. Natarajan and N. Venkatasubramanian, Indian J. Chem.,
13A, 261, 1975.
214. (a) A. Mamoru and K. Ohdan, Chem. Lett., 5, 405, 1995;
(b) F. V. Bagrov, G. P. Parlov and D. F. Bagrov, Russ. J.
Org. Chem., 36, 117, 2000.
215. (a) P. Ruoff and G. Nevda, J. Phys. Chem., 93, 7802, 1989;
(b) M. A. El-Maghraby and A. I. M. Koraiem, Indian J.
Chem., 20B, 73, 1981.
216. I. Tapia, V. Alcazar, J. R. Moran and M. Grande, Bull.
Chem. Soc. Jpn., 63, 2408, 1990.

184
217. P. C. Samal, B. B. Pattnaik, S. C. Dharma Rao and S. N.
Mahapatro, Tetrahedron, 39, 143, 1983.
218. (a) I. D. Komaritsa, Chem. Heterocycl. Compd., 25, 1297,
1988; (b) B. Zaleska and S. Lis, Synth. Commun., 31,
189,2001.
219. R. P. Bell and D. Covington, J. Chem. Soc., Perkin Trans. 2,
1343, 1975.
220. (a) S. Prasad and R. K. Prasad, Indian J. Chem., 17A, 491,
1979; (b) D. Vlaovic, G. Cetkovic, I. Juranic, J. Balaz,
S. Lajsic and D. Djokovic, Monatsh. Chem., 121, 931,
1990; (c) J. S. F. Pade and W. A. Waters, J. Chem. Soc.,
712, 1956; (d) E. B. Hughes, H. B. Watson and E. D. Yates.
J. Chem. Soc., 3318, 1931.
221. (a) B. Singh, B. B. Singh and S. Singh, J. Indian Chem. Soc.,
57, 662, 1980; (b) U. R. Joshi and P. A. Limaye, Indian J.
Chem., 21B, 1122, 1982; (c) P. L. Huang and P. Williams, J.
Chem. Soc., 2637, 1958.
222. E. Burcher, Ann. Chim., 26, 435, 1882.
223. R. D. Haworth, J. Chem. Soc., 1125, 1932.
224. E. Barnet, De Barry and F. G. Sanders, J. Chem. Soc., 434.
1933.
225. G. Levy, Ann. Chim., 9, 5, 1938.

185
226. L. F. Fieser and M.A. Peters, J. Am. Chem Soc., 54, 4347,
1937.
227. W. E. Bachman and W. S. Struve, J. Org. Chem., 4, 472,
1939.
228. J. D. Reinheimer and S. Taylor, J. Org. Chem., 19, 802,
1954.
229. F. Muhr, Ber., 28, 3215, 1895.
230. N. W. Fadnavis and G. Bagavant, Indian J. Chem., 17B, 518,
1979.
231. L. F. Fieser and A. M. Seligman, J. Am. Chem. Soc., 60, 170,
1938.
232. E. L. Martin, J. Am. Chem. Soc., 58, 1439, 1936.
233. J. D. Reinheimer and S. Taylor, J. Org. Chem., 17, 1505,
1952.
234. G. A. Dalal and K. S. Nargund, J. Indian Chem. Soc., 14,
406, 1937.
235. Syed Sheriff, Can. J. Chem., 39, 2563, 1961.
236. M. A. Saboor, J. Chem. Soc., 922, 1945.
237. C. Forzato, R. Gandolfi, F. Molinari, P. Nitti, G.Pittiaco and
E. Valentin, Tetrahedron, Asymmetry., 12, 1039, 2001.
238. F. Reisch, W. Spitzner and K. E. S. Arzneim, Forsch., 17,
816, 1967.

186
239. O. Helia, T. Autonin and M. Senionsky, Prange (Zech),
Neoplauma, 16, 597,1969.
240. H. Adolphi and G. Gerhard (BASF. A-G), Ger. Offen. 070,
2530, 1975.
241. G. Sikkandar, Ph. D. Thesis, Bharathidasan University,
Tiruchy, 58, 1992.
242. K. Kameo, K. Orgawa, K. Takeshita, S. Nakaike, K. Tomi-
sawa and K. Sota, Chem. Pharm. Bull., 36, 2050, 1988
243. Y. Kawashima, K. Kameo, M. Kato, M. Hasegawa, and
K. Tomisawa, Chem. Pharm. Bull., 40, 774, 1992.
244. F. V. Bagrov, Russ. J. Org. Chem., 37, 15, 2001.
245. T. N. Mandar, Yu-Chu Chang and Tai-huang Huang, FEBS
Lett., 530, 133, 2002.
246. A. Avarsson, J. L Chaung, R. Max Wynn, S. Turley, D. T
Chauang and W. G. J. Hol, FEBS Lett., 8, 277, 2000.
247. C. A. Aspiotis Renee, B. Cameron, G. Andre, L. Grimm
Erich and H. Yongxin, Merck Forsst Canada Inc. C.A.,
(Patent No. US 6525025), 2003.
248. D. Denoya Claudio and J. Stutzman-Engwall kim, Pfizer
Inc., (Patent No. US 6399324), 2002.
249. A. Renee, B. Christopher, B. Cameron, F. Sabastien, G.
Andre, G. Erich, H. Yongxin, M. Dan, P. Petpiboon and Z.

187
Robert, Merch Frosst-Canada Inc., C. A, ( Patent No. US
6225288), 2001.
250. J. Van Scott Eugene and J. Yu. Ruey, FEBS Lett., (Patent
No. US 6225288), 1996.
251. B. Zaleska and S. Lis, Synth. Commun., 31, 189, 2001.
252. G. Sikkandar, Asian J. Chem., 12, 1037, 2000; ibid., 12,
1337, 2000.
253. K. A. Basheer Ahamed, G. Sikkandar and S. Kannan, Indian
J. Chem., 38A, 183,1999.
254. D. Freeda Gnana Rani, F. J. Maria Pushparaj, I. Alphones
and K. S. Rangappa, Indian J. Chem., 41B, 2153, 2002.
255. G. Sikkandar and K. A. Basheer Ahamed, Indian J. Chem.,
31A, 845, 1992.
256. D. V. Hertzler, J. M. Berdahl and E. J. Eisenbraun, J. Org.
Chem., 33, 2008, 1968.
257. P. P. Sane, K. J. Divakar and A. S. Rao, Synthesis, 541,
1973.
258. M. Krishna Pillai and K. Banumathi, J. Chem. Research,
225, 1997.
259. M. V. Bhat, M. S. Ravindranathan and G. V. Rao, J. Org.
Chem., 49, 3170, 1984.

188
260. P. Nath and K. K. Banerji, Bull. Chem. Soc., Jpn, 42, 2038,
1969.
261. J. S. Littler, G. R. Quick and D. Woznik, J. Chem. Soc.,
Perkin Trans. 2, 657, 1980.
262. J. Shorter and C. N. Hinshelwood, J. Chem. Soc., 3425,
1962.
263. N. P. Marigangaiyah and K. K. Banerji, Indian J. Chem.,
14A, 85, 1976.
264. L. P. Hammett, Physical Organic Chemistry, II edn.
McGraw- Hill, Tokyo, chapter 11, 348, 1970.
265. L. P. Hammett, J. Am. Chem. Soc., 59, 96, 1937.
266. R. Dubois, M. Ruasse and A. Argile, J. Am. Chem. Soc., 106,
4840, 1984.
267. J. Shorter, Correlation Analysis of Organic Reactivity,
Research Studies Press, England, chapter 3 & 7, 1982.
268. R. P. Wells, Chem. Rev., 63, 171, 1963.
269. M. Charton, Progr. Phys. Org. Chem., 8, 235, 1971.
270. R. W. Taft, in Steric Effects in Organic Chemistry, edn.
M. S. Newman, John-Wiley and Sons, Inc., New York,
Chapter 13, 1956.
271. J. Shorter, Correlation Analysis in Organic Chemistry,
Clarendon Press, Oxford, Chapter 2, 1973.

189
272. C. D. Johnson, The Hammett Equation, Cambridge
University Press, Chapter 1 & 2, 1973.
273. H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 80, 4979,
1958.
274. H. Van Bekkum, P. E. Verkade and B. M. Wepster, Rect.
Trav. Chim., 78, 815, 1959.
275. B. M. Webster, J. Am. Chem. Soc., 95, 102, 1973.
276. R. W. Taft Jr, S. Ehrenson, I. C. Lewis and R. E. Glick, ibid.,
81, 5352, 1959.
277. Y. Yukawa and T. Tsuno, Bull. Chem. Soc., Jpn, 32, 971,
1959.
278. M. Yoshioka, K. Hamamoto and T. Kubota, ibid., 35, 1723,
1961.
279. R. W. Taft, J. Phys. Chem., 64, 1805, 1960.
280. S. Ehrenson, R.T.C. Brownlee and R. W. Taft, Progr. Phys.
Org. Chem., 10, 1, 1973.
281. (a) Weinreb S. M. J and Antoline, Science of Synthesis,
George Theime Verlag KG, Theime Newyork, p. 854, 2005,
ISBN: 3-13-118721-2; (b) Shizuo Fujisaki, Satoshi Hamura,
Hisao Eguchi and Akiko Nishida, Bulletin of Chemical
Society of Japan, 66(8), 2426-2428, 1993; DOI:
10.1246/bcsj.66.2426.

190
282. A. I. Vogel, A Text book of Practical Organic Chemistry,
Long mans Green, London, 737, 1962.
283. W. G. Douben, and R. E. Adams, J. Am. Chem. Soc., 70,
1559, 1948.
284. D. H. Hey and R. Wilkinson, J. Chem. Soc., 1030, 1940.
285. F. Feigl, Spot Tests in organic Analysis, Elsevier, New York,
1975.
286. G. P. Panigrahi and P. K Misro, Indian J. Chem., 15A,
1066, 1977.
287. S. B. Chapman, Advances in Linear Free Energy
Relationships, Shorter, J., Ed.; Plenum Press: New York,
1972.
288. K. B. Wiberg and R. Evans, J. Am. Chem. Soc., 80, 3019,
1958.
289. K. K. Banerji, Indian J. Chem., 16A, 595, 1978.
290. E. S. Amis, Solvent effects on reaction rates and
mechanisms, Academic Press, New York, 1967.
291. N. P. Marigangaiah and K. K. Banerji, Indian J. Chem., 14A,
660, 1976.
292. S. Rao and V. Pulugurtha, Z. Phys. Chem (Leipzig), 255,
382, 1974.
293. J. S. Littler, J. Chem. Soc., 827, 1962.

191
294. N. P. Marigangaiah, and K K. Banerji, Aust. J. Chem., 29,
1939, 1976.
295. J. Shorter and C. N. Hinshelwood, J. Chem. Soc., 3276,
1950.
296. P. S. Radhakrishnamurti and M. D. Prasad Rao, Indian J.
Chem., 15A, 524, 1977.
297. M. Krishnapillay and A. Thirunavukkarasu, Indian J. Chem.,
20B, 583, 1981.
298. J. Shorter, J. Chem. Soc., 1868, 1962.
299. W. S. Nathan and H. B. Waston, J. Chem. Soc., 217, 1933.
300. A. Meenakshi and M. Santhappa, Indian J. Chem., 11, 393,
1973.
301. K. B. Wiberg and H. Schaefer, J. Am. Chem. Soc., 91, 933,
1969.
302. H. H. Jaffe, Chem. Rev., 53, 191, 1953.
303. K. B. Wiberg, Physical Organic Chemistry, John Wiley,
New York, 416, 1964.
304. E. S. Gould, Mechanism and Structure in Organic
Chemistry, Holt Riehart and Winston, New York, 181, 1964.
305. A. Y. Richard Johnes, Physical and Mechanistic Organic
Chemistry, Cambridge Univ. Press, New York, II edn. 42,
1984.

192
306. F. Ruff and A. Kucsman, J. Chem. Soc., Perkin Trans. 2,
683, 1985.
307. F. Ruff, A. Komoto, N. Furukawa and S. Oae, Tetrahedron,
32, 2763, 1976.
308. F. A. Cotton and G. Wilkinson, Advanced Inorganic
Chemistry, John Wiley, 5th edn. 707, 1988.
309. N. N. Greenwood and A. Earnshaw, Chemistry of the
Elements, Pergamon Press, Oxford, 1222, 1984.
310. Freeman. Fillmore, Rev. React. Species. Chem. React., 1,
179, 1976.
311. K. Banumathi, M.Phil. Dissertation, Bharathidasan
University, 1987.
312. S Neil Issacs, Physical Organic Chemistry, Longman
Scientific & Technical, New York, 1987.
313. P. S. Radhakrishnamurti and B. V. Damoder Rao,, Indian J.
Chem., 20A, 473, 1981.
314. B. A. Blackaddar and C. Hinshelwood, J. Chem. Soc., 2720,
1978.
315. W. Good, D. B. Ingham and J. Stone, Tetrahedron, 31, 257,
1975.
316. J. E. Leffler, J. Chem. Phys., 27, 981, 1957.

193
317. O. Exner, Colln. Czech. Commun., 29, 1094, 1964 ; ibid., 37,
1425, 1972.
318. O. Exner, J. Chem. Soc., Perkin Trans. 2, 973, 1993.
319. J. E. Leffler and E. Grunwald, Rates and Equilibrium of
Organic Reactions, Wiley, New York, 1963.
320. J. E. Leffler, J. Org. Chem., 20, 1202, 1955.
321. D. G. Peters, J. M. Hayes and G. M. Hiefitje, Chemical
Separations and Measurements-Theory and Practice of
Analytical Chemistry, Saunders Golden Series, London,
32, 1974.

PUBLISHED PAPERS

http://www.e-journals.in Chemical Science Transactions DOI:10.7598/cst2015.1054 2015, 4(2), 638-641
Kinetics of Oxidation of 3-Benzoylpropionic Acid by N-Bromobenzamide in Aqueous Acetic Acid Medium
N. A. MOHAMED FAROOK* and A. AFROOS BANU
P. G & Research Department of Chemistry Khadir Mohideen College, Adirampattinam - 614 701, India [email protected]
Received 10 January 2015/ Accepted 15 February 2015
Abstract: The kinetics of the oxidation of 3-benzoylpropionic acid (KA) by N-bromobenzamide (NBB)to yield the corresponding carboxylic acid were studied potentiometrically in 50:50 (v/v) aqueous acetic acid medium at 298 K. The effects of temperature, composition of solvent medium and concentration of added mineral acid on the rate of reaction were also studied. The reaction was first order with respect to the [KA], [NBB] and hydrogen ions. There is no effect of added benzamide. Protonated NBB has been postulated as the reactive oxidizing species.
Keywords: Kinetics, Oxidation, N-Bromobenzamide, 4-Oxoacids, 3-Benzoylpropionic acid.
Introduction
The chemistry of reactions of N-halo compounds forms a separate branch, which is of great synthetic importance1-4. N-halo compounds have been extensively employed as oxidizing agents for organic substrates5,6. N-Halo compounds are the source of positive halogen and have been exploited as oxidant for a variety of substrates in both acidic and alkaline media. The species responsible for such oxidizing character may be different depending on the pH of the medium7,8. Although a lot of works have been reported on the oxidation of organic compounds by N-halo compounds9-12 it is to be noted that no systematic kinetic investigation on the oxidation of 3-benzoylpropionic acid by N-bromobenzamide has yet been reported in the literature. Here we report the results of the kinetics of the oxidation of 3-benzoylpropionic acid (KA) with N-bromobenzamide (NBB) in aqueous acetic acid medium in the presence of perchloric acid.
Experimental All the chemicals used were of p.a.grade. Their purity was checked by comparing their boiling or melting points with the literature values. Acetic acid was refluxed over chromic oxide for 6 h and then fractionated. Solutions of sodium perchlorate and perchloric acid were prepared in double-distilled water. Double-distilled water was employed in all kinetic runs.
RESEARCH ARTICLE

NBB H 3O H 2O Brk1
-1+
k Benzamide+
Chem Sci Trans., 2015, 4(2), 638-641 639
The reaction was followed potentiometrically by setting up a cell containing the reaction mixture, into which a platinum electrode and a standard calomel electrode were dipped. The emf of the cell was measured periodically using a Equip-Tronic potentiometer, while the reaction mixture was continuously stirred. The pseudo-first order rate constants computed from the plots of log (Et-E∞) against time were reproducible within ± 3%.
Results and Discussion Reaction order The reaction orders were determined from the slopes of log k1 versus log (concentration) plots by varying the concentration of substrate (KA) and perchloric acid in turn while keeping others constant. The plot log k1 against log [KA] is linear (r = 0.989) with a slope value of 0.988 and the plot log k1 against log [H+] is also linear (r = 0.996) with a unit slope. This is further supported by the fact that the plots of k1 versus [KA] and k1 versus [H+] gives a straight line passing through the origin, the linearity of the plots of log [NBB] versus time indicates the order in [NBB] as unity, this is also confirmed by constant values of k1 at varying [NBB] (Table 1). This indicates clearly that the reaction is first order with respect to [KA], [NBB] and [H+].
Table 1. Rate constant for the oxidation of 3-benzoylpropionic acid by NBB in aqueous acetic acid medium at 30 0Ca
102 [KA] mol dim-3 103 [NBB] mol dm-3 [H+]mol dm-3 104 k1b,s-1 103 k2
c dm3 mol-1 s-1
2.0 2.0 0.5 1.615 8.08 3.0 2.0 0.5 2.422 8.07
4.0 2.0 0.5 3.224 8.06
6.0 2.0 0.5 4.854 8.09
8.0 2.0 0.5 6.441 8.05 2.0 2.0 0.8 2.562 0.32
2.0 2.0 1.2 3.839 0.32
2.0 2.0 1.4 4.482 0.32
2.0 2.0 1.6 5.121 0.32
2.0 1.6 0.5 1.621 -
2.0 1.2 0.5 1.624 -
2.0 1.0 0.5 1.618 -
2.0 0.8 0.5 1.621 -
aGeneral conditions: [NaClO4] = 0.5 mol dm-3,Solvent composition: 50% Acetic acid - 50% Water (v/v). bEstimated from pseudo-first order plots, the error quoted in k1 values is the 95% confidential limit of ‘Student t’ test.13 cIndividual k2 values estimated as k1 / [KA] or k1 / [H+]
Effect of products The effect from adding benzamide was studied, which caused a decrease in the oxidation rate. Thus, retardation of the reaction rate upon addition of benzamide suggests that there is a pre-equilibrium step involving a process in which benzamide is a product. (1)
The effect of dielectric constant in the reaction medium was studied by adding acetic acid (40%-80%) in the reaction medium at constant concentrations of other reactants. The reaction rate increased remarkably with the increase in the proportion of acetic acid in the solvent medium. The effect of ionic strength was studied by varying the concentration of NaClO4 in the reaction medium. It was found that the rate of reaction is independent of ionic strength of the medium. The reaction mixture was kept for 24 h with acrylonitrile in an inert atmosphere. Test for free radical was negative.

NBB H3O H2O Brk1
-1+
k Benzamide+
C CH 2 CH 2 C OH
O O
+ H3O
C CH 2 CH 2 C OH
OH O
H2O+
k2
k -2
(S)
(S )
C CH CH2 C OH
OOH
H3O+H2O+(S )k3
k -3
(E)
C CH CH 2 C OH
OOHk4
slow
H2O Br
C CH CH 2 C OH
OOH
H2O Br
(F)
640 Chem Sci Trans., 2015, 4(2), 638-641
Effect of temperature The rate of reaction was measured at different temperatures. The activation parameters for the oxidation of keto acid by NBB have been evaluated from the slope of the Arrhenius plots.
Mechanism It is known that14 the probable reactive species of NCA in acid solution. H2O
+Br. The reaction is first order in [NBB], [KA] and [H+]. The reaction rate increases with increase in [H+] at constant ionic strength, showing that the reaction proceeds completely through the acid-catalyzed pathway. The change in the polarity of the medium has a marked effect on the reaction rate. The trend in the rate observed may be due to more than one factor. It may be attributed to the lowering of dielectric constant of the medium which favors reaction involving protonation. Further, the enolization of the keto acid may be catalyzed by acetic acid and this may also contribute to rate enhancement. The plot of log k1 versus 1/D is linear (r = 0.988) with positive slope, indicating an interaction between a positive ion and a dipole molecule. This supports the postulation of (H2O
+Br) as the reactive species. The retardation of reaction rate on the addition of saccharin suggests15 a pre-equilibrium step involves a process in which benzamide is one of the products.
If this equilibrium is involved in the oxidation process, the retardation should be an inverse function of benzamide concentration, which is borne out by observation that the inverse of the rate constant gives a linear (r = 0.987) plot against [benzamide].
A mechanism has been proposed involving the attack of H2O+Br on the enol form of the
substrate (E) in the rate determining step. It is known14 that the enolization is proposed to be the necessary step prior to the oxidation of the substrate
(2)
(3) .
(4)
(5)

(F)C
O
OHOther products+
fast
+ C6H5COCH2CH2COOH C6H5CONHBr 5 H2O
C6H5CONH2C6H5 COOH 6H23CO2 HBr
H+
+
+ + + +
Chem Sci Trans., 2015, 4(2), 638-641 641
(6)
Scheme 1
Scheme 1 leads to rate law (7)
][
]][][[][
32
23432
Benzamidekkk
BrOHOHSkkk
dt
NBBd
a
(7)
Equation (7) clearly points out the observed results i.e. first order in [KA], [NBB], [H+] and inverse order in [Benzamide] on the rate of the oxidation.
Stoichiometry and reaction products Different sets of reaction mixtures containing different quantities of NBB and KA at constant [H+] and ionic strength were reacted for 24 h at 30 0C and then analyzed. The remaining NBB was estimated. The oxidation products were identified as benzoic acid and benzamide. It was confirmed by noting the mixed melting point, chemical methods and TLC techniques. The results are in good agreement with 1:1 stoichiometry.
(8)
Reference 1. Amauri F Patrocino and Paulo J S Moran., Organomet Chem., 2000, 603, 220-224. 2. Sameer P Dhuru, Nikhil U Mohe and Manikrao M Salunkhe, Synth Commun., 2001,
31(23), 3653-3657. 3. Canibano V, Rodriguez J F, Santose M, Sanz-Tejedor M A, Carreno M C, Gonzalez
G and Garcia-Ruano J L, Synthesis, 2001, 14, 2175-2179. 4. Bandgar B P, Uppalla L S and Sadavarte V S, Syn Lett., 2001, 11, 1715-1718. 5. Duraisamy Thenraja, Perumal Subramaniam and Chockalingam Srinivasan, J Chem
Soc Perkin Trans 2, 2002, 2125-2129. 6. Teruaki Mukaiyama, Jun-ichi Matsuo, Daisuke Lida and Hideo Kitagawa, Chem
Lett., 2001, 8 846-847. 7. Thenraja D, Subramaniam P and Srinivasan C, J Chem Soc Perkin Trans 2, 2002, 2125. 8. Mukaiyama T, Mastsuo J I, Lida D and Kitagawa H, Chem Lett., 2001, 8, 846. 9. Hambly G F and Chan T H, Tetrahedron Lett., 1986, 27, 2563. 10. Antelo J M, Arce F, Crugeiras J O and Parajo M, J Phys Org Chem., 1997, 10, 631-636. 11. Karunakaran C and K Ganapathy K, Indian J Chem., 1990, 29A, 133. 12. Harihar A L, Kembhavi M R and Nandibewoor S T, J Indian Chem Soc., 1999, 76,
128-130. 13. Shorter J, Correlation Analysis in Organic Chemistry, Clarendon Press, Oxford,
Chapter 2, 1973. 14. Shahnaz Khan, Khan M U, Singh S K, Gupta H D and Singh P K, Asian J Chem.,
2003, 15, 595. 15. Vivekanandan K and Nambi K, J Indian Chem Soc., 1999, 76, 198-201.

1 23
Journal of Solution Chemistry ISSN 0095-9782Volume 42Number 1 J Solution Chem (2013) 42:239-250DOI 10.1007/s10953-012-9942-0
Kinetics and Mechanism of Oxidationof Substituted 4-Oxoacids by N-Bromosaccharin
N. A. Mohamed Farook, S. Manochitra &A. Afroos Banu

1 23
Your article is protected by copyright and all
rights are held exclusively by Springer Science
+Business Media New York. This e-offprint is
for personal use only and shall not be self-
archived in electronic repositories. If you
wish to self-archive your work, please use the
accepted author’s version for posting to your
own website or your institution’s repository.
You may further deposit the accepted author’s
version on a funder’s repository at a funder’s
request, provided it is not made publicly
available until 12 months after publication.

Kinetics and Mechanism of Oxidation of Substituted4-Oxoacids by N-Bromosaccharin
N. A. Mohamed Farook • S. Manochitra • A. Afroos Banu
Received: 29 November 2011 / Accepted: 11 March 2012 / Published online: 3 January 2013� Springer Science+Business Media New York 2012
Abstract The kinetics of the oxidation of substituted 4-oxoacids by N-bromosaccharin
(NBSac) has been studied in aqueous acetic acid medium at 30 �C. The reactions follow
first-order kinetics in the 4-oxoacids, NBSA and H?. Variation in the ionic strength has no
effect on the reaction rate. The order of reactivity among the studied 4-oxoacids is:
4-methoxy [ 4-methyl [ 4-phenyl [ 4-H [ 4-Cl [ 4-Br [ 3-NO2. The effect of changes
in the electronic nature of the substrate revealed that there is a development of positive
charge in the transition state. The activation parameters were computed from an Arrhenius
plot. Based on the kinetic results, a suitable mechanism has been proposed. The mechanism
involves the attack of the oxidizing species hypobromous acidium ion, (H2O?Br).
Keywords Kinetics � Oxidation � 4-Oxoacids � NBSac
1 Introduction
4-Oxoacid is an attractive substrate in terms of its enolization. In strong acid medium the
substrate undergoes enolization. The reactive species of the substrate has been reported in
the literature to be the enol form [1].
N-Bromosaccharin is a source of positive halogen and this reagent has been exploited as
oxidant for a variety of substrates in both acidic and alkaline medium [2]. The nature of
active oxidizing species and mechanism depends on the nature of halogen atom, the groups
attached to the nitrogen and the reaction condition. The species responsible for such
oxidizing character may be different depending on the pH of the medium. The probable
reactive species [3] of NBSac in acid solution are[ NX, HOX, [ N?HX, or H2OX? and
the reaction species in alkaline solutions are [ NH, HOX, and OX-.
In recent years, studies of the oxidation of various organic compounds by N-halo
compounds, in the presence of perchloric acid, have attracted considerable attention. A
through literature survey reveals that relatively little work on the oxidation of 4-oxoacid
N. A. Mohamed Farook (&) � S. Manochitra � A. Afroos BanuDepartment of Chemistry, Khadir Mohideen College, Adirampattinam 614 701, Indiae-mail: [email protected]
123
J Solution Chem (2013) 42:239–250DOI 10.1007/s10953-012-9942-0
Author's personal copy
have been reported so far [4–7]. Although the N-bromosaccharin oxidations of organic
compounds have been studied, there seems to be no report on a systematic kinetic study of
the oxidation of 4-oxoacids by N-bromosaccharin.
The main objectives of the present study are to ascertain the reactive species of the
substrate and oxidant, elucidate a plausible mechanism, deduce an appropriate rate law,
identify the oxidation products and evaluate the kinetic parameters.
2 Experimental
2.1 Materials
All the chemicals used were of AR grade. Acetic acid (BDH) was first refluxed over
chromic acid for 6 h and then distilled. Solutions of sodium perchlorate and perchloric acid
and were prepared in double distilled water. Double distilled water (conductivity \10 lS�cm-1) was employed in all kinetic runs. All the chemicals used were 99.8 % pure.
The 4-oxo-4-(40-methoxyphenyl)butanoic acid (S2), 4-oxo-4-(40-methylphenyl)-buta-
noic acid (S3), 4-oxo-4-(40-chlorophenyl)butanoic acid (S5) and N-bromosaccharin
(NBSac) were obtained from Sigma-Aldrich Chemical Co. The remaining 4-oxoacids (S1,S4, S6, S7) were prepared by Friedel–Crafts acylation of the substituted benzene with
succinic anhydride [5–12].
All the 4-oxo acids used in this study (Table 1) were crystallized twice from water and
their purity was checked by their melting points and UV, IR and NMR spectra. Solutions of
the reagents were prepared either in doubly distilled water or in purified acetic acid and
were standardized by iodometric methods [10] using standard potassium dichromate and
sodium thiosulfate solutions. Fresh solutions were used for each kinetic run.
2.2 Kinetic Studies
The reaction mixtures, containing 4-oxo acid, acetic acid and mercuric acetate solutions
were thermally equilibrated for an hour at the desired temperature. The reaction was
initiated by the addition of temperature-equilibrated NBSac solution of the requisite
concentration. The rate of the reaction was followed by estimating the amount of unreacted
NBSac iodometrically. All the reactions were carried out under pseudo-first order
Table 1 List of substituted 4-oxoacids
S.no. 4-oxoacids Code Melting point �C
Observed Reported5
1 4-Oxo-4-phenylbutanoic acid S1 116 116
2 4-Oxo-4-(40-methoxyphenyl)butanoic acid S2 146 147
3 4-Oxo-4-(40-methylphenyl)butanoic acid S3 128 129
4 4-Oxo-4-biphenylbutanoic acid S4 184 185
5 4-Oxo-4-(40-chlorophenyl)butanoic acid S5 132 133
6 4-Oxo-4-(40-bromophenyl)butanoic acid S6 148 149
7 4-Oxo-4-(30-nitrophenyl)butanoic acid S7 163 164
240 J Solution Chem (2013) 42:239–250
123
Author's personal copy

conditions by keeping an excess (910 or greater) of [4-oxo acid] over [NBSac]. The
pseudo-first order rate constants were computed from linear plots of log10 [NBSac]t against
time up to 90 % completion of the reaction, the rate constants (k) were reproducible within
5 %.
The precision of the rate constant values is given in terms of 95 % confidence limit of
the student’s t test [13]. Freshly prepared solutions of 4-oxo acids in purified acetic acid
were used. The stoichiometry of the reaction was determined by equilibrating reaction
mixture of various [NBSac]/[4-oxo acid] ratios at 30 �C for 12 h, keeping all other reagent
concentrations constant. Estimation of unconsumed NBSac revealed that one mole of
4-oxo acid consumed one mole of NBSac.
C6H5COCH2CH2COOHþ C6H4SO2CONBrþ 5H2O
! C6H5COOHþ C6H4SO2CONHþ 3CO2 þ 6H2 þ HBr ð1Þ
The products were extracted with ether, dried and analyzed. Benzoic acid was identified by
its melting point (121 �C). Then it was estimated quantitatively using UV–Vis spectro-
photometry with a standard curve at kmax = 235 nm and also tested with its characteristic
spot test. Identification of benzoic acid was also made by comparing the Rf values of
authentic samples.
3 Results and Discussion
3.1 Order of Reaction
The rate of oxidation was found to be first order each in [NBSac] and [S]. Linear plots of log10k1
versus log10 [S] with unit slope (S1: slope = 1.03 ± 0.02, r = 0.994; S2: slope = 1.01 ±
0.03, r = 0.996; S3: slope = 1.04 ± 0.01, r = 0.989; S4: slope = 1.04 ± 0.03, r = 0.999;
S5: slope = 0.995 ± 0.02, r = 0.991; S6: slope = 0.993 ± 0.07, r = 0.994; S7: slope =
1.10 ± 0.08, r = 0.992) show first order dependences of the rate on [S]. The k1 values at
different [S] are given in Table 2. The k1 values obtained at different initial concentrations of
NBSac reveal that the rates are almost independent of the initial concentration of NBSac (Table 2).
The dependence of the reaction rate on the hydrogen ion concentration has been
investigated at different initial concentrations of perchloric acid, keeping the concentra-
tions of the other reactants constant. The observed k1 values are presented in the Table 2. It
may be seen that the rate of the reaction increases linearly with increase in concentration of
hydrogen ion. This establishes that the reaction is first order with respect to hydrogen ion
concentration. A plot of k1 versus [H?] is linear, passing through the origin (Fig. 1),
showing that the reaction proceeds completely through the acid-catalyzed pathway [14].
It was reported earlier in the case of N-halo oxidants that in the absence of mineral
acids, HOBr is the reactive oxidant species. Farook et al. [14] have observed for the
oxidation of some a-hydroxy acids by NBSac that the linear increase in the oxidation rate
with an increase in [H?] indicates the protonation of HOBr to give a cationic bromine
species (Eq.2), which is a stronger electrophile and oxidant:
HOBrþ H3Oþ � H2OþBrþ H2O ð2Þ
Thus the most probable oxidizing species is the hypobromous acidium ion, (H2O?Br). The
participation of hypohalous acidium ions in many electrophilic substitution and oxidation
reactions is well documented [15].
J Solution Chem (2013) 42:239–250 241
123
Author's personal copy
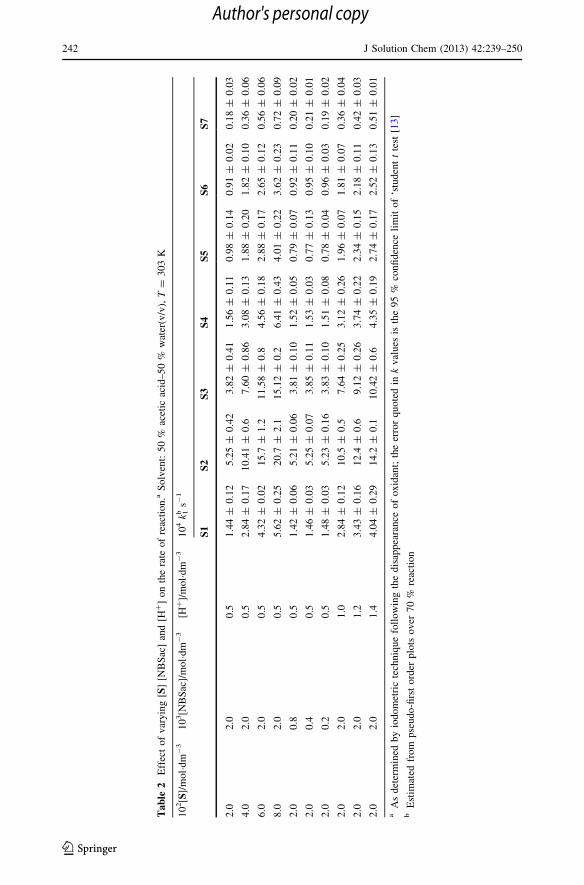
Ta
ble
2E
ffec
to
fvar
yin
g[S
][N
BS
ac]
and
[H?
]o
nth
era
teof
reac
tion.a
Solv
ent:
50
%ac
etic
acid
–50
%w
ater
(v/v
),T
=3
03
K
10
2[S
]/m
ol�d
m-
31
03[N
BS
ac]/
mol�d
m-
3[H
?]/
mo
l�dm
-3
10
4k 1b
s-1
S1
S2
S3
S4
S5
S6
S7
2.0
2.0
0.5
1.4
4±
0.1
25
.25
±0
.42
3.8
2±
0.4
11
.56
±0
.11
0.9
8±
0.1
40
.91
±0
.02
0.1
8±
0.0
3
4.0
2.0
0.5
2.8
4±
0.1
71
0.4
1±
0.6
7.6
0±
0.8
63
.08
±0
.13
1.8
8±
0.2
01
.82
±0
.10
0.3
6±
0.0
6
6.0
2.0
0.5
4.3
2±
0.0
21
5.7
±1
.21
1.5
8±
0.8
4.5
6±
0.1
82
.88
±0
.17
2.6
5±
0.1
20
.56
±0
.06
8.0
2.0
0.5
5.6
2±
0.2
52
0.7
±2
.11
5.1
2±
0.2
6.4
1±
0.4
34
.01
±0
.22
3.6
2±
0.2
30
.72
±0
.09
2.0
0.8
0.5
1.4
2±
0.0
65
.21
±0
.06
3.8
1±
0.1
01
.52
±0
.05
0.7
9±
0.0
70
.92
±0
.11
0.2
0±
0.0
2
2.0
0.4
0.5
1.4
6±
0.0
35
.25
±0
.07
3.8
5±
0.1
11
.53
±0
.03
0.7
7±
0.1
30
.95
±0
.10
0.2
1±
0.0
1
2.0
0.2
0.5
1.4
8±
0.0
35
.23
±0
.16
3.8
3±
0.1
01
.51
±0
.08
0.7
8±
0.0
40
.96
±0
.03
0.1
9±
0.0
2
2.0
2.0
1.0
2.8
4±
0.1
21
0.5
±0
.57
.64
±0
.25
3.1
2±
0.2
61
.96
±0
.07
1.8
1±
0.0
70
.36
±0
.04
2.0
2.0
1.2
3.4
3±
0.1
61
2.4
±0
.69
.12
±0
.26
3.7
4±
0.2
22
.34
±0
.15
2.1
8±
0.1
10
.42
±0
.03
2.0
2.0
1.4
4.0
4±
0.2
91
4.2
±0
.11
0.4
2±
0.6
4.3
5±
0.1
92
.74
±0
.17
2.5
2±
0.1
30
.51
±0
.01
aA
sdet
erm
ined
by
iodom
etri
cte
chniq
ue
foll
ow
ing
the
dis
appea
rance
of
oxid
ant;
the
erro
rquote
din
kv
alu
esis
the
95
%co
nfi
den
celi
mit
of
‘stu
den
tt
test
[13]
bE
stim
ated
from
pse
udo-fi
rst
ord
erplo
tsover
70
%re
acti
on
242 J Solution Chem (2013) 42:239–250
123
Author's personal copy
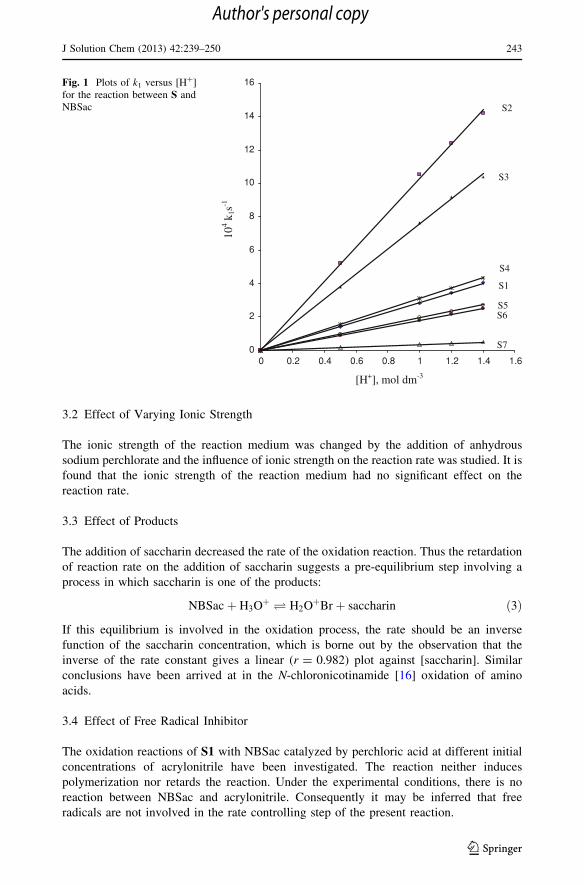
3.2 Effect of Varying Ionic Strength
The ionic strength of the reaction medium was changed by the addition of anhydrous
sodium perchlorate and the influence of ionic strength on the reaction rate was studied. It is
found that the ionic strength of the reaction medium had no significant effect on the
reaction rate.
3.3 Effect of Products
The addition of saccharin decreased the rate of the oxidation reaction. Thus the retardation
of reaction rate on the addition of saccharin suggests a pre-equilibrium step involving a
process in which saccharin is one of the products:
NBSacþ H3Oþ � H2OþBrþ saccharin ð3Þ
If this equilibrium is involved in the oxidation process, the rate should be an inverse
function of the saccharin concentration, which is borne out by the observation that the
inverse of the rate constant gives a linear (r = 0.982) plot against [saccharin]. Similar
conclusions have been arrived at in the N-chloronicotinamide [16] oxidation of amino
acids.
3.4 Effect of Free Radical Inhibitor
The oxidation reactions of S1 with NBSac catalyzed by perchloric acid at different initial
concentrations of acrylonitrile have been investigated. The reaction neither induces
polymerization nor retards the reaction. Under the experimental conditions, there is no
reaction between NBSac and acrylonitrile. Consequently it may be inferred that free
radicals are not involved in the rate controlling step of the present reaction.
0
2
4
6
8
10
12
14
16
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
[H+], mol dm-3
104 k
1s-1
S1
S2
S3
S4
S5S6
S7
Fig. 1 Plots of k1 versus [H?]for the reaction between S andNBSac
J Solution Chem (2013) 42:239–250 243
123
Author's personal copy

3.5 Effect of Solvent Composition
The effect of changing solvent composition on the reaction rate was studied by varying
the concentration of acetic acid from 50 to 80 %. The pseudo first-order rate constants
for the oxidation reactions of all oxoacids, S1–S7 with NBSac, were estimated in the
presence of perchloric acid at constant ionic strength. The rate of the reaction increases
remarkably with increase in the proportion of acetic acid in the medium (Table 3).
When the acetic acid content increases in the medium, the acidity of the medium is
increased while the dielectric constant of the medium is decreased. These two effects
cause the rate of oxidation to increase remarkably. The observed effect is similar to
those reported in the oxidation of other organic compounds by N-chlorosaccharin [17].
When the acetic acid content of the medium is increased from 50 to 80 %, the pH of the
medium changes, leading to the increase in [H?] and hence catalysis by acetate ion is
untenable.
The enhancement of the reaction rate with an increase in the amount of acetic acid may
generally be attributed to two factors: (i) increase in acidity at constant [perchloric acid]
and (ii) decrease in dielectric constant with increase in HOAc content. The plots of log10 k1
against the inverse of dielectric constant are linear with positive slopes, indicating an
interaction between a positive ion and a dipole molecule [18, 19]. This supports the
postulation of H2O?Br as the reactive species.
3.6 Rate of Enolization by the Bromination Method
It has been reported, in the case of oxidation of keto compounds, that the oxidation
proceeds via enolization of the keto compounds [20]. The rate of enolization of keto
compound was found to be faster than the rate of oxidation. The reactive species of the
substrate may be determined by enolization, which is an acid as well as base catalyzed
reaction and proceeds by a concerted or push–pull mechanism. The rate of enolization was
determined by the bromination method for the system under investigation.
Table 3 Effect of solvent polarity on the rate of reaction, where [S] = 2.0 9 10-2 mol�dm-3,[NBSac] = 2.0 9 10-3 mol�dm-3, [H?] = 0.5 mol�dm-3, and T = 303 K
S 104 k1a/ s-1 ra
CH3COOH–H2O (v/v) %
50–50 60–40 70–30 80–20
S1 1.44 ± 0.12 2.24 ± 0.24 3.12 ± 0.14 5.28 ± 0.29 0.998
S2 5.25 ± 0.42 6.83 ± 0.34 7.85 ± 0.48 9.35 ± 0.60 0.983
S3 3.82 ± 0.41 4.91 ± 0.22 6.31 ± 0.24 7.52 ± 0.34 0.985
S4 1.56 ± 0.11 2.11 ± 0.07 2.92 ± 0.14 4.23 ± 0.18 0.998
S5 0.98 ± 0.14 1.32 ± 0.06 2.92 ± 0.11 3.01 ± 0.01 0.998
S6 0.91 ± 0.02 1.07 ± 0.05 1.68 ± 0.12 2.36 ± 0.15 0.994
S7 0.18 ± 0.03 0.31 ± 0.03 0.61 ± 0.07 0.92 ± 0.08 0.998
a Estimated from pseudo first-order plots
244 J Solution Chem (2013) 42:239–250
123
Author's personal copy

3.7 Effect of Substituents
The oxidation of 4-oxo-4-phenylbutanoic acid (S1) and substituted 4-oxoacids (S2–S7)
were carried out in the temperature range of 303–323 K. The observed rate constants
increase with temperature for all of the compounds. The activation parameters for the
oxidation of 4-oxoacid by NBSac have been evaluated from the slope values of the
Arrhenius plots.
A close look at activation parameters presented in Table 4 shows that the activation
energies for the oxoacids with electron-releasing substituents are relatively lower than
those with electron-withdrawing substituents. The entropy of activation is negative for all
the 4-oxoacids ranging from -126 to -232 J � K-1 � mol-1. The large negative entropy of
activation in conjunction with other experimental data supports the mechanism outlined in
Scheme-1. The formation of an activated complex from reactant molecules is accompanied
by the conversion of translation-like and rotational-like degrees of freedom of the reactants
to vibrational degrees of freedom of the transition state species. The more widely spaced
energy level for the latter type of molecular motion implies a small entropy and thus a
negative value of DS# [21].
It is interesting to note that the reactivity decreases for substituents in the order:
4-methoxy [ 4-methyl [ 4-phenyl [ 4-Cl [ 4-Br [ 3-NO2 ð4ÞThe Hammett’s plot for the oxidation of S by NBSac at various temperatures was found
to be linear. The Hammett’s plot is shown in Fig. 2. The values of reaction constants (q)
are shown in Table 5.
The points are referenced as (1) S2, (2) S3, (3) S4, (4) S1, (5) S5, (6) S6, (7) S7.
The q value indicates the sensitivity of a reaction to the effects of electronic pertur-
bation. It also provides information about the nature of the transition state involved in the
reaction. A reaction involving a development of positive charge in the transition state is
aided by electron-releasing substituents and the q coefficient is negative [21–26].
In the present investigation, the acceleration of reaction rate with the electron-releasing
substituents and the negative value of the reaction constant, q, indicates explicitly that the
mechanism of oxidation involves the development of positive charge in the transition state.
It is generally recognized that oxidations lead to electron deficient species which are
radical cations, radicals or carbocations. These reactions normally have a negative q value
and the magnitude of q depends on the extent of electron deficiency. Oxidation reactions
involving free radical formation in the rate controlling step usually have a small negative qvalue and the oxidations involving the formation of a carbocation have large negative qvalues. Based on these arguments we expect a large q value but the measured q value is in
the range -1. 41 to -0.98. The low q values may be attributed to the nature of observed
rate constant. The observed rate constant is a composite of several terms and is shown in
Eq. 12.
The terms shown in Eq. 12 deserve comment. The rate constant k3 depends on the
concentration of the protonated substrate (S?) and the electron donating substituents tend
to delocalize the positive charge on S? and hence favor the formation of this positive
species. In the rate limiting step (Eq. 6) of the Scheme-1, the formation of the carbocation
is facilitated by electron releasing substituents.
Thus we get a slightly low negative q value of -1.41 (at 303 K) because kobs is
composite of the enolization as well as the oxidation of the 4-oxoacids. Hence in the
present investigation the measured q value and other findings fit in with the formulation of
mechanism outlined in the Scheme-1.
J Solution Chem (2013) 42:239–250 245
123
Author's personal copy
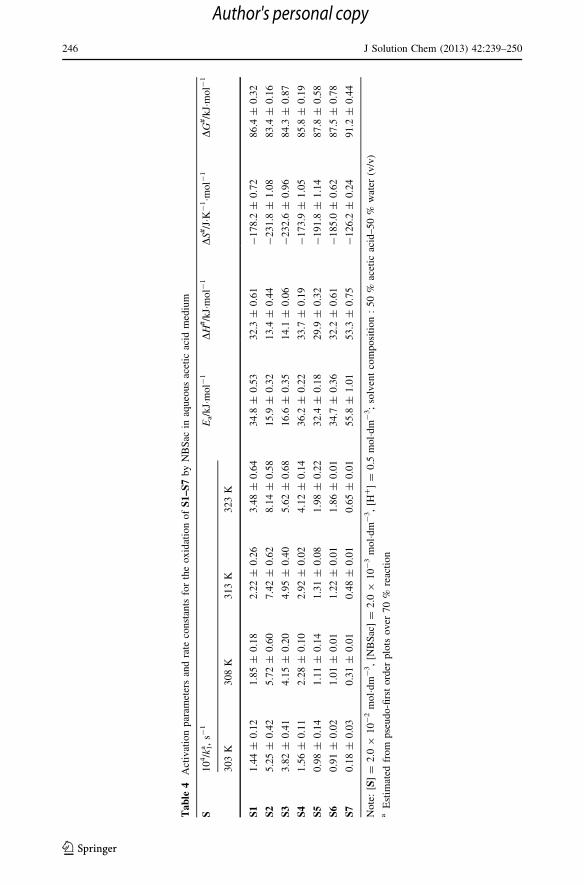
Ta
ble
4A
ctiv
atio
np
aram
eter
san
dra
teco
nst
ants
for
the
ox
idat
ion
of
S1
–S
7b
yN
BS
acin
aqu
eous
acet
icac
idm
ediu
m
S1
04/k
1a,
s-1
Ea/k
J�mol-
1D
H#/k
J�mo
l-1
DS
#/J�K
-1�m
ol-
1D
G#/k
J�mo
l-1
30
3K
30
8K
31
3K
32
3K
S1
1.4
4±
0.1
21
.85
±0
.18
2.2
2±
0.2
63
.48
±0
.64
34
.8±
0.5
33
2.3
±0
.61
-1
78
.2±
0.7
28
6.4
±0
.32
S2
5.2
5±
0.4
25
.72
±0
.60
7.4
2±
0.6
28
.14
±0
.58
15
.9±
0.3
21
3.4
±0
.44
-2
31
.8±
1.0
88
3.4
±0
.16
S3
3.8
2±
0.4
14
.15
±0
.20
4.9
5±
0.4
05
.62
±0
.68
16
.6±
0.3
51
4.1
±0
.06
-2
32
.6±
0.9
68
4.3
±0
.87
S4
1.5
6±
0.1
12
.28
±0
.10
2.9
2±
0.0
24
.12
±0
.14
36
.2±
0.2
23
3.7
±0
.19
-1
73
.9±
1.0
58
5.8
±0
.19
S5
0.9
8±
0.1
41
.11
±0
.14
1.3
1±
0.0
81
.98
±0
.22
32
.4±
0.1
82
9.9
±0
.32
-1
91
.8±
1.1
48
7.8
±0
.58
S6
0.9
1±
0.0
21
.01
±0
.01
1.2
2±
0.0
11
.86
±0
.01
34
.7±
0.3
63
2.2
±0
.61
-1
85
.0±
0.6
28
7.5
±0
.78
S7
0.1
8±
0.0
30
.31
±0
.01
0.4
8±
0.0
10
.65
±0
.01
55
.8±
1.0
15
3.3
±0
.75
-1
26
.2±
0.2
49
1.2
±0
.44
No
te:
[S]
=2
.09
10
-2
mo
l�dm
-3,
[NB
Sac
]=
2.0
91
0-
3m
ol�d
m-
3,
[H?
]=
0.5
mol�d
m-
3;
solv
ent
com
po
siti
on
:5
0%
acet
icac
id–5
0%
wat
er(v
/v)
aE
stim
ated
from
pse
udo-fi
rst
ord
erplo
tsover
70
%re
acti
on
246 J Solution Chem (2013) 42:239–250
123
Author's personal copy

The dependence of the reaction rate on the structure of the reacting molecule is related
to activation parameters. The decisive term concerning the dependence on the structure is
neither Gibbs energy nor enthalpy but potential energy, which is experimentally not
accessible. Many authors support the opinion that the activation energy at a certain tem-
perature is a better approximation towards the unknown potential energy [27].
The validity of the isokinetic relation can be tested graphically by plotting with Exner
plots. The isokinetic temperature evaluated from the Exner plots is found to be 378 K. As
the experiment has been carried out at a temperature far away from the isokinetic tem-
perature, application of the Hammett equation to the observed kinetic data is valid. The
validity of isokinetic relationship in the present study implies that all the 4-oxoacids
undergo oxidation by the same mechanism [28].
NBSac H3O H2O Brk1
-1+
k Saccharin+
C CH2 CH2 C OH
O O
+ H3O
C CH2 CH2 C OH
OH O
H2O+
k2
k -2
(S)
(S )
C CH CH2 C OH
OOH
H3O+H2O+(S )k3
k -3
(E)
C CH CH2 C OH
OOHk4
slow
H2O Br
C CH CH2 C OH
OOH
H2O Br
(F)
(F)C
O
OHOther products+
fast
(5)
(3)
(4)
(7)
(6)
Scheme 1 Reaction mechanism
J Solution Chem (2013) 42:239–250 247
123
Author's personal copy
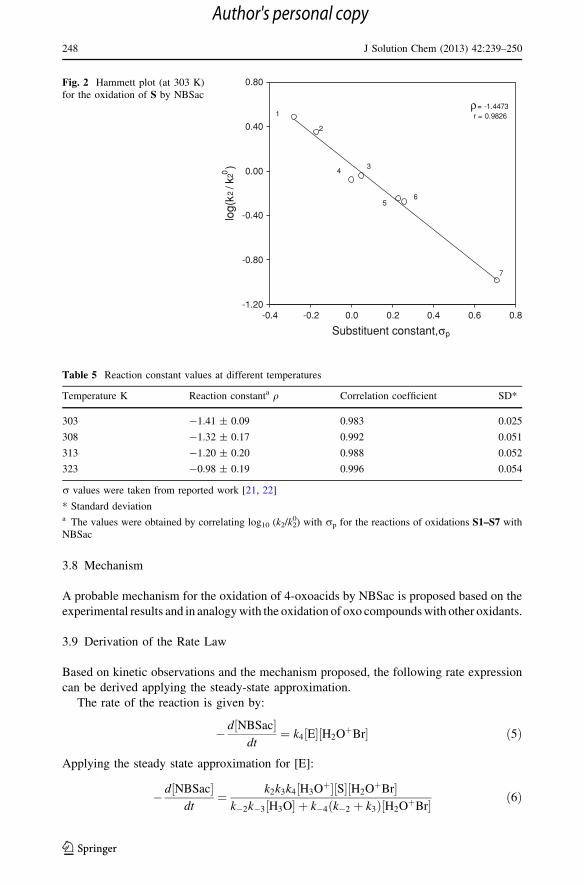
3.8 Mechanism
A probable mechanism for the oxidation of 4-oxoacids by NBSac is proposed based on the
experimental results and in analogy with the oxidation of oxo compounds with other oxidants.
3.9 Derivation of the Rate Law
Based on kinetic observations and the mechanism proposed, the following rate expression
can be derived applying the steady-state approximation.
The rate of the reaction is given by:
� d NBSac½ �dt
¼ k4 E½ � H2OþBr½ � ð5Þ
Applying the steady state approximation for [E]:
� d NBSac½ �dt
¼ k2k3k4 H3Oþ½ � S½ � H2OþBr½ �k�2k�3 H3O½ � þ k�4 k�2 þ k3ð Þ H2OþBr½ � ð6Þ
Table 5 Reaction constant values at different temperatures
Temperature K Reaction constanta q Correlation coefficient SD*
303 -1.41 ± 0.09 0.983 0.025
308 -1.32 ± 0.17 0.992 0.051
313 -1.20 ± 0.20 0.988 0.052
323 -0.98 ± 0.19 0.996 0.054
r values were taken from reported work [21, 22]
* Standard deviationa The values were obtained by correlating log10 (k2/k2
0) with rp for the reactions of oxidations S1–S7 withNBSac
ρ = -1.4473r = 0.9826
-1.20
-0.80
-0.40
0.00
0.40
0.80
-0.4 -0.2 0.0 0.2 0.4 0.6 0.8
Substituent constant,σp
log(
k2 /
k20)
1
2
3 4
5 6
7
Fig. 2 Hammett plot (at 303 K)for the oxidation of S by NBSac
248 J Solution Chem (2013) 42:239–250
123
Author's personal copy

At high concentration of [H3O?] = 0.5 mol�dm-3,
k�2k�3 H3Oþ½ �[ [ k4 k�2 þ k3ð Þ H2OþBr½ � ð7Þ
So Eq. 9 simplifies to the form:
� d NBSac½ �dt
¼ k2k3k4 H3Oþ½ � S½ � H2OþBr½ �k�2k�3 H3O½ �
¼ k2k3k4 S½ � H2OþBr½ �k�2k�3
ð8Þ
The value of [H2O?Br] can be obtained from Eq. 3 given in Scheme-1:
Ka ¼k�1
k1
¼ NBSac½ � H3Oþ½ �H2OþBr½ � saccharin½ � ð9Þ
Therefore,
H2OþBr½ � ¼ NBSac½ � H3Oþ½ �Ka saccharine½ � ð10Þ
Using the value of [H2O?Br] in Eq. 10:
� d NBSac½ �dt
¼ k2k3k4 S½ � NBSac½ �k�2k�3Ka saccharine½ � ð11Þ
Hence, at higher concentrations of mineral acid, the reaction is first order each with respect
to the oxoacid (S), [NBSac] and [H3O?].
The observed rate constant at high [H3O?] is:
kobs ¼k2k3k4
k�2k�3Ka
ð12Þ
4 Conclusion
The above study shows that oxidizing species is the hypobromous acidium ion, (H2O?Br),
which reacts with the enol form of 4-oxo acid in the rate determining step, giving the
product. This experimental protocol suggests that this reaction could find utility as a
regioselective route for the synthesis of benzoic acids. The order of reactivity among the
studied 4-oxoacids is: 4-methoxy [ 4-methyl [ 4-phenyl [ 4-H [ 4-Cl [ 4-Br [ 3-NO2.
References
1. Reddy, C.S., Manjari, P.S.: Kinetics and mechanism of acid bromate oxidation of substituted 4-oxo-acids. Indian J. Chem. 49A, 418–424 (2010)
2. Gupta, M.-K., Rekha, L., Sharma, P.-K., Sharma, K., Sharma, V.-K.: Oxidation of glycolic acid byN-bromosaccharin–A kinetic and mechanistic study in presence and absence of cationic surfactant.Asian J. Chem. 18, 1340–1346 (2006)
3. Mohamed Farook, N.A.: Kinetics of oxidation of 4-oxo acids by N-bromosuccinimide in aqueous aceticacid medium. J. Iranian Chem. Soc. 3, 378–386 (2006)
4. Mohamed Farook, N.-A.: Kinetics of oxidation of 4-oxo acids by N-chlorosaccharin in aqueous aceticacid. J. Solution Chem. 36, 345–356 (2007)
J Solution Chem (2013) 42:239–250 249
123
Author's personal copy

5. Mohamed Farook, N.-A., Seyed Dameem, G.-A.: Kinetics of oxidation of 3-benzoylpropionic acid byN-chlorobenzamide in aqueous acetic acid medium. J. Chem. 8, 561–564 (2011)
6. Freeda Gnana Rani, D., Maria Pushparaj, F.-J., Alphones, I., Rangappa, K.-S.: Kinetics and mechanismof oxidation of 4-oxoacids by hexacyanoferrate(III) catalysed by Os(VIII). Indian J. Chem. 41B,2153–2159 (2002)
7. Sikkandar, G.: Kinetics of oxidatative decarboxylation of b-benzoylpropionic acid by manganese(III)acetate. Asian J. Chem. 12, 1037–1040 (2000)
8. De Barry, E.B., Sanders, F.G.: The synthesis of some homologous naphthalenes. J. Chem. Soc. 434–437(1933)
9. Fieser, L.F., Seligman, A.M.: 10-Methyl- and 10,10-dimethyl-1,2-benzanthracene. J. Am. Chem. Soc. 60,170–176 (1938)
10. Vogel, A.I.: A Text Book of Practical Organic Chemistry. Longmans Green, London (1962)11. Russell, R.R., Van der Werf, C.A.: The malonic ester synthesis with styrene oxide and with butadiene
oxide. J. Am. Chem. Soc. 69, 11–13 (1947). doi:10.1021/ja01193a00312. Hey, D.H., Wilkinson, R.: The preparation of 2-phenylnapthalene from diphenyl. J. Chem. Soc. 1030
(1940)13. Chapman, N.B., Shorter, J.: Advances in linear free energy relationships. Plenum Press (1972)14. Mohamed Farook, N.-A., Dameem, S.: Kinetics of oxidation of 3-benzoylpropionic acid by N-bro-
moacetamide in aqueous acetic acid medium. J. Chem. 8, 479–482 (2011)15. Mohamed Farook, N.-A., Prabhkaran, R., Rahini, S., Senthil Kumar, R., Raja Mahendran, G., Gopala
Krishnan, G.: Kinetics of oxidation of some amino acids by N-chlorosaccharin in aqueous acetic acidmedium. J. Chem. 2, 127–131 (2004)
16. Vivekanandan, K., Nambi, K.: Kinetics and mechanistic study of amino acids by N-chloronicotinimidein acid medium. J. Indian Chem. Soc. 76, 198–202 (1999)
17. Mohamed Farook, N.-A., Alhaji, N.-M.-I.: Kinetics of oxidation of substituted c-ketoacid by N-chlo-rosaccharin in aqueous acetic acid medium. Int. J. Chem. Sci. 2, 76–80 (2004)
18. Mohamed Farook, N.-A., Seyed Dameem, G.-A., Murugesan, A., Kanagaraj, M.: Kinetics of oxidationof some essential amino acids by N-chlorosaccharin in aqueous acetic acid medium. J. Chem. 2,132–136 (2004)
19. Kothari, S., Agarwal, A., Banerji, K–.K.: Kinetics and mechanism of oxidation of primary alcohols byN-bromo-3-5-dinitrobenzamide. Indian J. Chem. 25A, 722–724 (1986)
20. Nath, M.-P., Banerji, K.-K.: Kinetics and mechanisms of the oxidation of methyl aryl ketones by acidpermanganate. Aust. J. Chem. 29, 1939–1945 (1976)
21. Vanangamudi, G., Srinivasan, S.: Kinetic studies on the oxidation of some para and meta-substitutedcinnamic acids by pyridinium bromochromate in the presence of oxalic acid (a co-oxidation study).J. Chem. 6, 920–927 (2009)
22. Gould, E.-S.: Mechanism and Structure in Organic Chemistry, pp. 181–185. Holt Riehart and Winston,New York (1964)
23. Richard Johnes, A.-Y.: Physical and Mechanistic Organic Chemistry. Cambridge University Press, NewYork (1984)
24. Ruff, F., Kucsman, A.: Mechanism of the oxidation of sulphides with sodium periodate. J. Chem. Soc.Perkin Trans. II, 683–688 (1985)
25. Cotton, F.-A., Wilkinson, G.: Advanced Inorganic Chemistry. John Wiley, 5th edn. (1988)26. Greenwood, N-.N., Earnshaw, A.: Chemistry of the Elements, p. 1222. Pergamon Press, Oxford (1984)27. Issacs, S.N.: Physical Organic Chemistry. Longman Scientific & Technical, New York (1987)28. Leffler, J.-F., Grunwald, E.: Rates and Equilibrium of Organic Reactions. Wiley, New York (1963)
250 J Solution Chem (2013) 42:239–250
123
Author's personal copy
Copyright © 2022 FDOKUMEN




















