
Journal of Porphyrins and Phthalocyanines
18
Journal of Porphyrins and Phthalocyanines J. Porphyrins Phthalocyanines 2009; 13: 429–445 Published at http://www.worldscinet.com/jpp/ Copyright © 2009 World Scientific Publishing Company INTRODUCTION Small stones may be the all important corner stones however, more often they are simply discreet pieces in the wall, just as useful. Our pioneering interest in the study of tetrapyrrolic macrocycle synthetic chemistry in Portugal began when we applied for a PhD under the supervision of the late Professor George W. Kenner, whom we thankfully remember here with great admi- ration. To date, our research over the past forty years have produced some interesting results in the area of synthesis of tetrapyrrolic structures, in their application for different purposes and, above all, to attract youth to be interested in chemistry and, in several cases, become competent researchers. DEVELOPMENT OF SYNTHETIC METH- ODOLOGIES OR IMPROVEMENTS IN PORPHYRIN SYNTHESIS The PhD project in Liverpool was directed at the preparation of specific deuterium labeled samples of protoporphyrin-IX designed to support NMR studies on reconstructed samples of hemoproteins [1, 2], and allowed the development of proper methodologies for early selected deuteration and significant improvements in the synthesis of dipyrrilmethanes and protoporphyrin- IX [3–6]. In Coimbra, we decided to focus on widening the use of the old Rothemund concept, attempting to make it far more useful than it had been before. After much study on the Rothemund synthesis, at the time more than forty years old [7–9], many relevant aspects were still unknown and the utility of the simple porphyrins was a matter of controversy. When we submitted our work on the syn- thesis of meso-tetraalkylporphyrins we first introduced the approach of performing the Rothemund synthesis of The small stones of Coimbra in the huge tetrapyrrolic chemistry building António M.d’A. Rocha Gonsalves* , Arménio C. Serra and Marta Pineiro Departamento de Química, Universidade de Coimbra, Rua Larga, 3004-535 Coimbra, Portugal Received 5 September 2008 Accepted 23 September 2008 ABSTRACT: Improvements over the Rothemund classical reaction methods have allowed the development of capacity to obtain a wide range of structures that were earlier unavailable. Performing the reaction in a mixed nitrobenzene/carboxylic acid medium allowed improvements of yield and purity of the porphyrin obtained, and in some cases the control of the oxidation level of the macrocycle. More recently, novel microwave synthetic methodologies were exploited to achieve important improvements in simple porphyrin chemistry. Tidy sulphonations of simple porphyrins opened the way to diverse desired, inexpensive structures in a very simple manner. The option to concentrate interests on improvements of synthetic methods to obtain simple and diversified tetrapyrrolic structures proved to be advantageous. Interesting biomimetic oxidation and photooxidation systems and PDT sensitizers were developed and significant knowledge was achieved in these specific and related areas. This review presents a view of our own results in the synthetic area, and outlines the contributions therefrom to the study of biomimetic and therapeutic processes. KEYWORDS: metalloporphyrins, synthesis, catalysis, photocatalysis, photophysical properties, Langmuir-Blodgett films, photodynamic therapy. SPP full member in good standing *Correspondence to: António M.d’A. Rocha Gonsalves, email: [email protected], tel: +351 239854479, fax: +351 2398- 26068
Transcript of Journal of Porphyrins and Phthalocyanines

Journal of Porphyrins and PhthalocyaninesJ. Porphyrins Phthalocyanines 2009; 13: 429–445
Published at http://www.worldscinet.com/jpp/
Copyright © 2009 World Scientific Publishing CompanyCopyright © 2009 World Scientific Publishing Company
INTRODUCTION
Small stones may be the all important corner stones however, more often they are simply discreet pieces in the wall, just as useful. Our pioneering interest in the study of tetrapyrrolic macrocycle synthetic chemistry in Portugal began when we applied for a PhD under the supervision of the late Professor George W. Kenner, whom we thankfully remember here with great admi-ration. To date, our research over the past forty years have produced some interesting results in the area of synthesis of tetrapyrrolic structures, in their application for different purposes and, above all, to attract youth to be interested in chemistry and, in several cases, become competent researchers.
DEVELOPMENT OF SYNTHETIC METH-ODOLOGIES OR IMPROVEMENTS IN PORPHYRIN SYNTHESIS
The PhD project in Liverpool was directed at the preparation of specifi c deuterium labeled samples of protoporphyrin-IX designed to support NMR studies on reconstructed samples of hemoproteins [1, 2], and allowed the development of proper methodologies for early selected deuteration and signifi cant improvements in the synthesis of dipyrrilmethanes and protoporphyrin-IX [3–6].
In Coimbra, we decided to focus on widening the use of the old Rothemund concept, attempting to make it far more useful than it had been before. After much study on the Rothemund synthesis, at the time more than forty years old [7–9], many relevant aspects were still unknown and the utility of the simple porphyrins was a matter of controversy. When we submitted our work on the syn-thesis of meso-tetraalkylporphyrins we fi rst introduced the approach of performing the Rothemund synthesis of
The small stones of Coimbra in the huge tetrapyrrolic chemistry building
António M.d’A. Rocha Gonsalves* , Arménio C. Serra and Marta Pineiro
Departamento de Química, Universidade de Coimbra, Rua Larga, 3004-535 Coimbra, Portugal
Received 5 September 2008Accepted 23 September 2008
ABSTRACT: Improvements over the Rothemund classical reaction methods have allowed the development of capacity to obtain a wide range of structures that were earlier unavailable. Performing the reaction in a mixed nitrobenzene/carboxylic acid medium allowed improvements of yield and purity of the porphyrin obtained, and in some cases the control of the oxidation level of the macrocycle. More recently, novel microwave synthetic methodologies were exploited to achieve important improvements in simple porphyrin chemistry. Tidy sulphonations of simple porphyrins opened the way to diverse desired, inexpensive structures in a very simple manner. The option to concentrate interests on improvements of synthetic methods to obtain simple and diversifi ed tetrapyrrolic structures proved to be advantageous. Interesting biomimetic oxidation and photooxidation systems and PDT sensitizers were developed and signifi cant knowledge was achieved in these specifi c and related areas. This review presents a view of our own results in the synthetic area, and outlines the contributions therefrom to the study of biomimetic and therapeutic processes.
KEYWORDS: metalloporphyrins, synthesis, catalysis, photocatalysis, photophysical properties, Langmuir-Blodgett fi lms, photodynamic therapy.
SPP full member in good standing
*Correspondence to: António M.d’A. Rocha Gonsalves, email: [email protected], tel: +351 239854479, fax: +351 2398-26068
00060.indd 100060.indd 1 7/2/2009 12:44:44 PM7/2/2009 12:44:44 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 430–445
430 A.M.d’A.R. GONSALVES ET AL.
meso porphyrins in two independent steps. We selected in the fi rst place the most favorable conditions to get the porphyrinogen, followed by fi nding appropriate con-ditions to oxidize this porphyrinogen to the porphyrin oxidation level. Photooxidation conditions or chemi-cal oxidation with high potential quinones were used to achieve this oxidation [10] (Table 1). Later, similar con-ditions were used by Lindsey and co-workers to extend this methodology to meso-tetraarylporphyrins [11].
The two-step approach to performing the Rothemund classical reaction expanded the utility of this procedure while opening the way for broader studies of porphyrin chemistry, demonstrating that, after all, simple meso- tetrasubstituted porphyrins can perform the role of struc-turally far more complex porphyrins and that in many cases they can even be more convenient when we intend to use them for biological and therapeutic roles.
Observations to better understand and attempt to extend and improve the usefulness of the Rothemund porphyrin synthesis led us, however, to fi nd another novel approach to this reaction. We fi rst observed that nitrated aryl aldehydes tend to give better yields of the corresponding porphyrin than other aldehydes. From the attempt to clarify this observation, we ended up with a very useful way to perform the Rothemund reaction. In many cases, the reaction can be advantageously carried out, as originally, in a single step, provided we use a mix-ture of a carboxylic acid and nitrobenzene as reaction medium [12]. We later demonstrated [13] that the best conditions require the use of acetic or propionic acid and of nitrobenzene in an approximate ratio of 70:30. Par-ticular bonuses of these “one-pot” reaction conditions are the fact that frequent direct crystallization of the porphy-rin occurs in a very pure state at the end of the reaction,
giving porphyrins uncontaminated with chlorin, contrary to those obtained using the previous classical Rothemund reaction conditions which require oxidation with a high-potential quinone to eliminate the chlorin [14]. Even in some cases where the overall porphyrin yield is lower than other synthetic methods, our one-pot specifi c condi-tions may be preferred due to the extremely easy direct isolation of very pure porphyrin (Table 2).
Our most recent developments on exploiting the nitrobenzene-assisted Rothemund porphyrin synthesis involve a modifi cation: performing the reaction under microwave irradiation (Table 3). Under such conditions, the process became even more convenient due to the sig-nifi cant reduction of the amount of solvents required and the shortening of reaction time while maintaining all the previous advantages [15]. We compared this new process with the one under classical conditions to demonstrate its high advantages from both economical and environmen-tal perspectives.
Developments made on the synthesis of meso- tetraarylporphyrins via the two-step procedure proposed by Lindsey and co-workers require the use of quinones with high oxidative potential. We studied the possibil-ity of using the same methodology but substituting the quinones for hydrogen peroxide, a much less expensive, readily available and environmental friendly oxidant [13] (Table 4).
In the area of tetrapyrrolic macrocycles synthesis, only for porphyrins were truly direct synthetic routes established in the sense that the structure is built all the way from small molecules, to end up in the required por-phyrin. From the vast fi eld of tetrapyrrolic macrocycles, chlorin and bacteriochlorin structures have intermediate oxidation states between porphyrinogen and porphyrin.
Table 1. Yields of meso-tetraalkylporphyrins obtained by photooxidation or chemical oxidation
N
NH N
HN
R
R
R
RNH
NH HN
HN
R
R
R
R
NO2C6H5/Acetic Acid (8:2)
O2, hν, ca. 3 h
CHCl3, quinone
60 °C, 30 min
CCl4/TFA(1%)
Ar, 60 °C, 16 hNH
+ RCH(OCH3)2
Yields, %
R Photooxidation Chemical oxidation
methyl 8 –
ethyl 5 11
n-propyl 10 18
iso-propyl – 5
iso-butyl 8 15.9
benzyl 6.4 17
n-undecyl 9 8.5
00060.indd 200060.indd 2 7/2/2009 12:44:45 PM7/2/2009 12:44:45 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 431–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 431
However, being structures that play important roles in nature, they have only been synthesized through reduc-tive processes applied to the corresponding porphyrin. Certainly, in all syntheses of porphyrins a diversity of traces of macrocycles in different oxidation levels are generated. Also, in all the old reaction conditions of the Rothemund synthesis of meso-tetrasubstituted porphy-rins, chlorin is formed as a contaminant of the porphyrin in clearly identifi able and often highly signifi cant quanti-ties. In a work on porphyrin synthesis by Ibers and co-workers [16] it was claimed that in the presence of some transition metal cations, the chlorin/porphyrin ratio could favor the formation of chlorin leading, in specifi c cases, to the exclusive formation of that species. However, the claimed overall yield of the cyclic product in these reac-tions was of the order of one per cent and we were unable to confi rm the results [12]. Exploiting the oxidation con-ditions created by the presence of nitrobenzene in our own specifi c conditions of Rothemund porphyrin syn-thesis, which led to uncontaminated porphyrin, we were
surprised with another peculiar capacity of such oxida-tion medium. If we select these reaction conditions to oxidize the porphyrinogen, we can in certain cases obtain substantial amounts of chlorin [13] (Table 5).
The preceding observation must fi nd some rationality if we consider the results of a study of a collaborative work with Robert A.W. Johnstone concerning the role of nitrobenzene as a cosolvent in the Rothemund reaction [17]. Such study allowed a primary understanding of the mechanism of action of nitrobenzene as a mild dehydro-genating agent and may well deserve further extension, taking into consideration these more recent results.
Having extended the capacity of the Rothemund reac-tion to produce a diversity of porphyrins in good yields and high purity under very convenient work up, and even with environmental advantage, we paid some atten-tion to the improvement of derivatizations of the basic Rothemund products which were made available. Sulpho-nation of aromatic compounds with sulfuric acid under the classic conditions had been applied to meso-tetraphe-nylporphyrin (TPP) but the isolation of a pure sample of the required tetrasulphonic acid product was diffi cult and ineffi cient [18]. We decided to exploit on TPP (P8) the adequacy of the well known chlorosulphonation reaction [19], something which strangely was never attempted before, to the best of our knowledge. Curiously the chlo-rosulphonated derivative was usually obtained from the acid derivative. Our method allows for obtaining the acid derivative from the chlorosulphonated one using a more
Table 2. Yields of porphyrins obtained by the nitrobenzene method [12, 13]
N
NH N
HN
R
R
R
RAcetic Acid/Nitrobenzene
NH
+120 °C, 1 hR
O
H
R Yield, %a
phenyl 20
2-nitrophenyl 20
3-nitrophenyl 9
4-nitrophenyl 25
4-biphenyl 40(46)
2-methoxyphenyl 15
4-methoxyphenyl 56b(78)
3,4-dimethoxyphenyl 18
3,4-dimethylenedioxyphenyl 27
2-chlorophenyl 8.5
3-chlorophenyl 22
4-chlorophenyl 49(56)
2,4-dichlorophenyl 9
2,6-dichlorophenyl 5
2,6-difluorophenyl 9
4-methylthiophenyl 37
1-naphthyl 10
n-propyl 12
n-undecyl 13
aYields using simplex optimization are shown in brackets. bUsing propionic acid instead of acetic acid.
Table 3. Yields of meso-tetraarylporphyrins obtained by micro-wave-assisted synthesis
N
NH N
HN
R
R
R
R
Propionic Acid/Nitrobenzene(70:30)5 mL
NH
+640 W, 5 minR
O
H
R Yield, %
phenyl 20
3-nitrophenyl 8
3-methoxyphenyl 15
4-methoxyphenyl 20
2-chlorophenyl 8
4-chlorophenyl 21
2,6-dichlorophenyl 4
2-bromophenyl 5.5
4-bromophenyl 12
3-hydroxyphenyl 12
4-t-butylphenyl 25
2,4,6-trimethylphenyl 1.5
3,4,5-trimethoxyphenyl 13
00060.indd 300060.indd 3 7/2/2009 12:44:45 PM7/2/2009 12:44:45 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 432–445
432 A.M.d’A.R. GONSALVES ET AL.
appropriate methodology. A signifi cant advantage of a quantitative preparation of the chlorosulphonated deriva-tive is the capacity of the chlorosulphonyl group to react effi ciently with different functionalities to generate, for example, amides, esters, thioesters, or sulfones.
STUDIES OF PHYSICAL AND PHOTO-PHYSICAL PROPERTIES OF MESO-TETRA ARYLPORPHYRINS AND THEIR APPLICATIONS
The chlorosulphonation of TPP (P8) developed by us opened the way for building a considerable diversity of structures in the periphery of the macrocycle. The screen-ing of a series of aminosulfamoylporphyrins with lipo-philic and hydrophilic substituents (Chart 1) disclosed that porphyrins with long alkyl chains, P3 and P4, were the most adequate to form Langmuir-Blodgett (LB) fi lms [20]. The phase behavior and the capability for
self-assembly were studied using analogous structures. Differential scanning calorimetry showed the presence of two polymorphic phases between crystal and isotro-pic liquid and the formation of metastable phases from the melting process. The modifi cations in their UV-vis absorption and fl uorescence spectra compared with solu-tion have small red shifts and split interpreted in terms of inter-porphyrin interactions due to stacking [21]. The environment affects the electronic structure and hence the optical properties of the porphyrins in the LB fi lm. The use of the optical absorption spectrum as a sensing param-eter is attractive, since sample preparation is extremely straightforward, does not involve the fabrication of intri-cate interdigitated electrode patterns, and demands lower electrical power than its resistive counterpart, thus offering great potential for application as chemical sensors.
Table 4. Yields of meso-tetraarylporphyrins obtained by porphyrinogen oxidation with H2O2
N
NH N
HN
R
R
R
RNH
NH HN
HN
R
R
R
RH2O2/Acetic Acid
30 °C, 1 hrt, 3−4 hNH
+R
O
H
BF3· Et2O, CHCl3
R Yield, %
phenyl 14
4-biphenyl 12
4-methoxyphenyl 21
3,4-dimethoxyphenyl 23
3,4,5-trimethoxyphenyl 30
3,4-dimethylenedioxyphenyl 20
3-chlorophenyl 19
4-chlorophenyl 27
2,6-dichlorophenyl 23
2,4,6-tribromo-3-methoxyphenyl 10
4-methylsulphoxyphenyl 40
Table 5. Isolated yields and porphyrin:chlorin ratio
Isolated yield, %Porphyrin meso-substituent (porphyrin:chlorin)
4-methoxyphenyl 36 (25:11)
4-biphenyl 35 (20:15)
3,4-methylenedioxophenyl 32 (18:14)
2,6-dichlorophenyl 18 (5:13)
2,4,6-tribromo-3-methoxyphenyl 15 (3:12)
N
NH N
HNSO
OR
S OOR
SO
OR
S OOR
P1 R = NH2
P2 R = NHC4H9
P3 R = NHC8H17
P4 R = NHC12H25
P5 R = NHC(CH2OH)3
P6 R = N(CH2COOH)2
P7 R = N(CH(CH3)2)2
Chart 1. Structures of meso-tetra(4-N-aminosulfamoylphenyl)porphyrins used to form LB fi lms
00060.indd 400060.indd 4 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 433–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 433
The effect of the environment depends on the struc-ture [22]. The application in sensing was studied with LB fi lms of meso-tetra(4-N-dodecylsulfamoylphenyl)porphyrin, P4, which exhibit intense optical absorption bands in the region of 400–700 nm. The change in the molecular environment of the porphyrin, a result of expo-sure to the sensor gas, is refl ected in substantial changes to the optical absorption. This effect is reversible in the absence of the sensor gas. In situ optical measurements were performed to quantify the response of the fi lms in a stream of chlorine gas of concentration in the ppm range and their subsequent recovery in air. Factors affecting the response are: orientation of the porphyrin within the LB fi lm (the lower the deposition surface pressure, the faster and greater the response to chlorine gas); concen-tration of the sensor gas (strong concentration-dependent optical response in the lower ppm range); temperature (a small increase in the temperature of the fi lm reduces the response and recovery time) [23]. Aging studies show that the basic response of the monolayer is not affected over a time period of at least fi ve months [24].
The interesting physical-chemical properties of the porphyrins P1 and P4 made them useful for studies con-cerning the solvent effect on absorption spectra of cen-trosymmetric compounds. These compounds are used for the development of a quadrupolar dielectric model which includes the quadrupolar interaction between the solute quadrupole moment and polarized solvent molecules [25]. Together with P2, P1 and P4 were used in the study of self-aggregation processes in reverse micelles of aero-sol OT [26]. Control of the dye association in a special organized geometry is one of the goals of the scientifi c efforts to mimetize light-harvesting units in photosyn-thesis. The type of aggregation that the dye assumes in a given supramolecular assembly is mainly determined by the type of non-covalent interactions. The difference in the number of carbons in the alkyl chains of porphy-rins P1, P2 and P4 made these porphyrins form different aggregates in reverse micelles. P1 and P2 form J-aggre-gates whereas with P4 H-aggregates are likely to be pres-ent. These results indicate that small modifi cations in the alkyl chain modify the type of aggregation (allowing for the control of the type of aggregation in the supramolecu-lar assembly).
A bichromophoric system is obtained when the mol-ecule covalently bonded to the porphyrin through chloro-sulfonation is a chromofore. The study of these systems is particularly relevant to the development of electronic and photonic devices, such as those used for tuning the color of organic light-emitting devices. The bichromophoric system formed by TPP and anthracene were studied, giving evidence of the energy transfer between the two chromophoric systems and showing that the major con-tribution to the excited state decay comes from the radia-tionless process [27].
The diverse applications developed for porphyrin compounds are based on their role in nature. The
photophysical and photochemical properties are essen-tial for the development of some of these applications. The measurement of photophysical and photochemical parameters as well as the development of new structures with improved properties is essential for the development of the applications. The capability of synthesizing a large amount of porphyrins with diverse substituents on the phenyl ring open the way to series of porphyrins with slight modifi cations and, subsequently, to the study of the relationship between the structural modifi cation and the photophysical and photochemical characteristics.
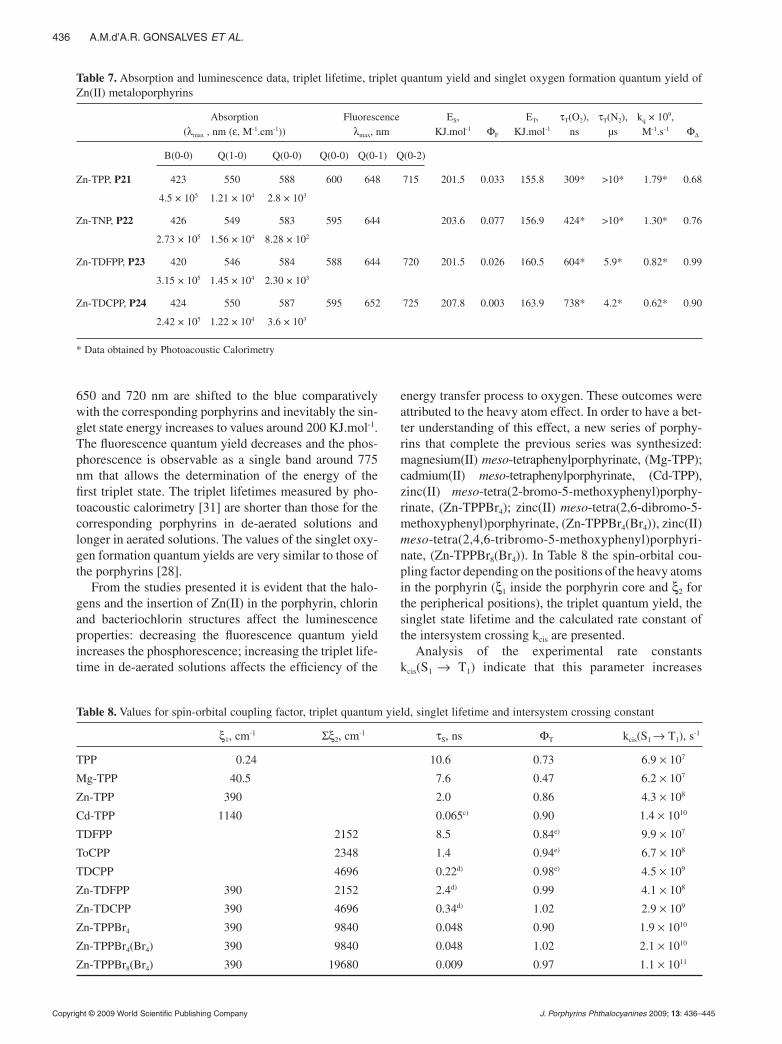
During the last ten years porphyrins and related com-pounds necessary to study the infl uence of the modifi ca-tion of the aryl group at the meso position, the insertion of a central metal, the substitution of hydrogen atoms for halogen atoms at the phenyl ring in the meso position and the reduction of the porphyrin core on the photophysi-cal and photochemical parameters of these compounds were synthesized and the relevant parameters measured [28–30]. All the synthesized compounds are presented in Chart 2. In Table 6 the absorption and luminescence data of porphyrins, chlorins and bacteriochlorins, the triplet lifetime, triplet quantum yield and singlet oxygen forma-tion quantum yield in toluene solutions are presented. The same parameters for the Zn(II) metalloporphyrins are presented in Table 7.
Porphyrins with aromatic substituents at the meso position present characteristic absorption bands between 420 and 650 nm and fl uorescence bands at 655 and 720 nm. The energy of the singlet state is around 180 KJ.mol-1 and fl uorescence quantum yields from 0.10 to 0.16. The triplet lifetime in de-aerated solutions is longer than 10 µs and in aerated solutions about 300 ns. The singlet oxygen formation quantum yields from 0.67 to 0.97 indicate a high effi ciency in the energy transfer process from the porphyrin to the oxygen molecule.
The substitution of hydrogen atoms at the ortho posi-tion of the phenyl ring for halogen brings very few modi-fi cations of the absorption and fl uorescence bands and consequently in the energy of the states. Furthermore, the fl uorescence quantum yield decreases with the number and atomic number of halogens. The triplet lifetime in aerated solutions increases and the singlet oxygen forma-tion quantum yield increases [28, 29].
The reduction to chlorin increases the absorption of the last Q band to approximately the same wavelength as the corresponding porphyrin, thus increases the fl uo-rescence quantum yield, decreasing the triplet lifetime τT(O2) and τT(N2), and decreases the singlet oxygen formation quantum yield [29].
The reduction to bacteriochlorin has a dramatic effect on the UV-vis spectrum. The Soret band of porphyrins is split into two bands around 350 and 378 nm. The Q band at higher wavelength is shifted to the red (around 745 nm) with absorption coeffi cients rising 1.4 × 105 M-1.cm-1. The shift in this band is accompanied by a decrease of the energy of the singlet state. The fl uorescence
00060.indd 500060.indd 5 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 434–445
434 A.M.d’A.R. GONSALVES ET AL.
quantum yield decreases with respect to the correspond-ing chlorin reaching values closer to the correspond-ing porphyrin. The singlet oxygen formation quantum yields are also very similar to those for the corresponding porphyrins [30].
The insertion of Zn(II) in the porphyrin core changes the symmetry of the compound. As a result, the Qx and Qy absorption bands degenerate and the absorption spectrum merely presents the B and two Q bands around 425, 550 and 585 nm, respectively. The fl uorescence bands at 600,
N
N N
N
R
R
R
R
R =P8
P9
P10
R =
R =
R =P11
P12
P13
F
F
R =
Cl
R =
Cl
Cl
N
NH N
HN
R
R
R
RM
M = 2H
M = 2H
M = 2H
M = 2H
M = 2H
M = 2H
N
NH N
HN
R
R
R
R
R =
R =
M = Zn
M = Zn
R=
F
F
M = Zn
R =
Cl
Cl
M = Zn
P21
P22
P23
P24
P14
P15
P16
P17
R =
R =
F
F
R =
Cl
R =
Cl
Cl
R =
F
F
R =
Cl
R =
Cl
Cl
P18
P19
P20
Chart 2. Porphyrins, chlorins and bacteriochlorins synthesized for the systematic study of the photophysical and photochemical properties
00060.indd 600060.indd 6 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 435–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 435
Tabl
e 6.
Abs
orpt
ion
and
lum
ines
cenc
e da
ta, t
ripl
et q
uant
um y
ield
, tri
plet
life
time
and
sing
let o
xyge
n fo
rmat
ion
quan
tum
yie
ld o
f po
rphy
rins
, chl
orin
s an
d ba
cter
ioch
lori
ns
E
S,
τ T
(O2)
, τ T
(N2)
, k q
× 1
09 ,
Abs
orpt
ion
(λm
ax, n
m (
ε, M
-1.c
m-1))
Fl
uore
scen
ce λ
max
, nm
K
J.m
ol-1
ΦF
ns
µs
M-1.s
-1
Φ∆
B
(0-0
) Q
y(1-
0)
Qy(
0-0)
Q
x(1-
0)
Qx(
0-0)
Q
(0-0
) Q
(0-1
)
TPP
, P8
418
514
548
592
650
652
719
43.6
0 0.
10
349
43
1.43
0.
67
2.72
× 1
05 1.
81 ×
104
1.24
× 1
04 1.
02 ×
104
9.62
× 1
03
TN
P, P
9 42
3 51
4 54
8 58
9 65
4 65
4 71
5 43
.66
0.16
48
0 30
1.
04
0.97
3.
78 ×
105
2.41
× 1
04 7.
70 ×
103
8.63
× 1
03 5.
17 ×
103
TfP
, P10
42
7 51
5 54
8 59
2 65
6 65
7 72
1 43
.36
0.14
56
5 54
0.
88
0.72
1.
48 ×
105
1.94
× 1
04 4.
18 ×
103
5.53
× 1
03 2.
39 ×
103
TD
FPP,
P11
41
6 50
9 53
9 58
7 65
5 65
7 71
3 43
.64
0.06
4 49
2 16
.6
1.12
0.
84
2.80
× 1
05 2.
11 ×
104
2.27
× 1
03 5.
89 ×
103
5.28
× 1
03
ToC
PP, P
12
418
512
541
588
655
657
716
43.6
6 0.
023
495
11.6
1.
12
0.94
3.
14 ×
105
2.13
× 1
04 4.
99 ×
103
6.49
× 1
03 4.
09 ×
103
TD
CPP
, P13
41
8 51
3 54
0 58
9 66
0 66
1 71
9 43
.33
0.00
5 64
1 16
.5
0.86
0.
98
1.18
× 1
05 7.
08 ×
103
3.10
× 1
03 3.
30 ×
103
2.05
× 1
03
B
(0-0
) Q
x(1-
0)
Qx(
0-0)
Q
y(1-
0)
Qy(
0-0)
TN
C, P
14
423
517
543
602
652
657
721
43.7
1 0.
36
545
1.39
0.
62
0.54
1.
57 ×
105
1.02
× 1
04 4.
48 ×
103
1.91
× 1
03 3.
73 ×
104
TD
FPC
, P15
41
0 51
0 53
6 60
1 65
5 65
8 71
9 43
.60
0.12
30
5 5.
0 1.
80
0.85
1.
34 ×
105
1.11
× 1
04 3.
92 ×
103
4.40
× 1
03 3.
99 ×
104
ToC
PC, P
16
418
515
539
602
655
657
718
43.6
6 0.
09
352
6.1
1.57
0.
92
1.20
× 1
05 1.
17 ×
104
5.46
× 1
03 4.
15 ×
103
3.89
× 1
04
TD
CPP
, P17
41
8 51
3 54
0 60
4 66
0 66
1 70
6 43
.36
0.04
39
3 8.
2 1.
41
0.93
7.
82 ×
104
7.46
× 1
03 2.
61 ×
103
2.02
× 1
03 2.
58 ×
104
B
y(0-
0)
Nx(
0-0)
Q
x(1-
0)
Qx(
0-0)
Q
y(1-
0)
Qy(
0-0)
TD
FPB
, P18
34
9 37
6 48
1 51
0 68
1 74
4 74
5 38
.4
0.06
8 21
6 33
2.
6 0.
78
1.62
× 1
05 1.
67 ×
105
5.70
× 1
03 7.
64 ×
104
6.02
× 1
03 1.
41 ×
105
ToC
PB, P
19
353
378
484
516
685
744
745
38.5
0.
048
265
44
2.0
0.95
1.
48 ×
105
1.71
× 1
05 3.
72 ×
103
8.03
× 1
04 3.
71 ×
103
1.42
× 1
05
TD
CPB
, P20
35
3 37
9 48
2 51
6 68
2 74
7 74
8 38
.3
0.01
2 25
4 32
2.
1 1.
06
1.32
× 1
05 1.
44 ×
105
2.42
× 1
03 7.
44 ×
104
2.51
× 1
03 1.
26 ×
105
00060.indd 700060.indd 7 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM
Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 436–445
436 A.M.d’A.R. GONSALVES ET AL.
650 and 720 nm are shifted to the blue comparatively with the corresponding porphyrins and inevitably the sin-glet state energy increases to values around 200 KJ.mol-1. The fl uorescence quantum yield decreases and the phos-phorescence is observable as a single band around 775 nm that allows the determination of the energy of the fi rst triplet state. The triplet lifetimes measured by pho-toacoustic calorimetry [31] are shorter than those for the corresponding porphyrins in de-aerated solutions and longer in aerated solutions. The values of the singlet oxy-gen formation quantum yields are very similar to those of the porphyrins [28].
From the studies presented it is evident that the halo-gens and the insertion of Zn(II) in the porphyrin, chlorin and bacteriochlorin structures affect the luminescence properties: decreasing the fl uorescence quantum yield increases the phosphorescence; increasing the triplet life-time in de-aerated solutions affects the effi ciency of the
energy transfer process to oxygen. These outcomes were attributed to the heavy atom effect. In order to have a bet-ter understanding of this effect, a new series of porphy-rins that complete the previous series was synthesized: magnesium(II) meso-tetraphenylporphyrinate, (Mg-TPP); cadmium(II) meso-tetraphenylporphyrinate, (Cd-TPP), zinc(II) meso-tetra(2-bromo-5-methoxyphenyl)porphy-rinate, (Zn-TPPBr4); zinc(II) meso-tetra(2,6-dibromo-5-methoxyphenyl)porphyrinate, (Zn-TPPBr4(Br4)), zinc(II) meso-tetra(2,4,6-tribromo-5-methoxyphenyl)porphyri-nate, (Zn-TPPBr8(Br4)). In Table 8 the spin-orbital cou-pling factor depending on the positions of the heavy atoms in the porphyrin (ξ1 inside the porphyrin core and ξ2 for the peripherical positions), the triplet quantum yield, the singlet state lifetime and the calculated rate constant of the intersystem crossing kcis are presented.
Analysis of the experimental rate constants kcis(S1 → T1) indicate that this parameter increases
Table 7. Absorption and luminescence data, triplet lifetime, triplet quantum yield and singlet oxygen formation quantum yield of Zn(II) metaloporphyrins
Absorption Fluorescence ES, ET, τT(O2), τT(N2), kq × 109,
(λmax , nm (ε, M-1.cm-1)) λmax, nm KJ.mol-1 ΦF KJ.mol-1 ns µs M-1.s-1 Φ∆
B(0-0) Q(1-0) Q(0-0) Q(0-0) Q(0-1) Q(0-2)
Zn-TPP, P21 423 550 588 600 648 715 201.5 0.033 155.8 309* >10* 1.79* 0.68
4.5 × 105 1.21 × 104 2.8 × 103
Zn-TNP, P22 426 549 583 595 644 203.6 0.077 156.9 424* >10* 1.30* 0.76
2.73 × 105 1.56 × 104 8.28 × 102
Zn-TDFPP, P23 420 546 584 588 644 720 201.5 0.026 160.5 604* 5.9* 0.82* 0.99
3.15 × 105 1.45 × 104 2.30 × 103
Zn-TDCPP, P24 424 550 587 595 652 725 207.8 0.003 163.9 738* 4.2* 0.62* 0.90
2.42 × 105 1.22 × 104 3.6 × 103
* Data obtained by Photoacoustic Calorimetry
Table 8. Values for spin-orbital coupling factor, triplet quantum yield, singlet lifetime and intersystem crossing constant
ξ1, cm-1 Σξ2, cm-1 τS, ns ΦT kcis(S1 → T1), s-1
TPP 0.24 10.6 0.73 6.9 × 107
Mg-TPP 40.5 7.6 0.47 6.2 × 107
Zn-TPP 390 2.0 0.86 4.3 × 108
Cd-TPP 1140 0.065c) 0.90 1.4 × 1010
TDFPP 2152 8.5 0.84e) 9.9 × 107
ToCPP 2348 1.4 0.94e) 6.7 × 108
TDCPP 4696 0.22d) 0.98e) 4.5 × 109
Zn-TDFPP 390 2152 2.4d) 0.99 4.1 × 108
Zn-TDCPP 390 4696 0.34d) 1.02 2.9 × 109
Zn-TPPBr4 390 9840 0.048 0.90 1.9 × 1010
Zn-TPPBr4(Br4) 390 9840 0.048 1.02 2.1 × 1010
Zn-TPPBr8(Br4) 390 19680 0.009 0.97 1.1 × 1011
00060.indd 800060.indd 8 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 437–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 437
gradually with the increase of the atomic number of the halogen atoms at ortho positions of the phenyl ring and with the atomic number of the internal metal, increasing the formation of the fi rst triplet state in relation to the other process for the deactivation of the singlet state. The triplet lifetime is somewhat less shortened with the peripheral substitution than with the insertion of the central metal. Therefore, the heavy atoms at the periph-ery are more favorable for the formation of the triplet state, without much decrease to the lifetime of this state. Using the Tunnel Effect Theory [32, 33] modi-fi ed to account for spin-orbit coupling [34], the relation between the intersystem crossing rates and the position of the heavy atom were rationalized for our porphyrins, free-base octaalkylporphyrins having halogen atoms at the alkyl chain, at the pyrrol and in the meso positions. The halogen atoms in the ortho position were the most appropriate to generate long-lived triplet states in high yields. Such compounds found application as singlet oxygen sensitizers, as drugs for photodynamic therapy (PDT) [35–37] and as photocatalysts [38].
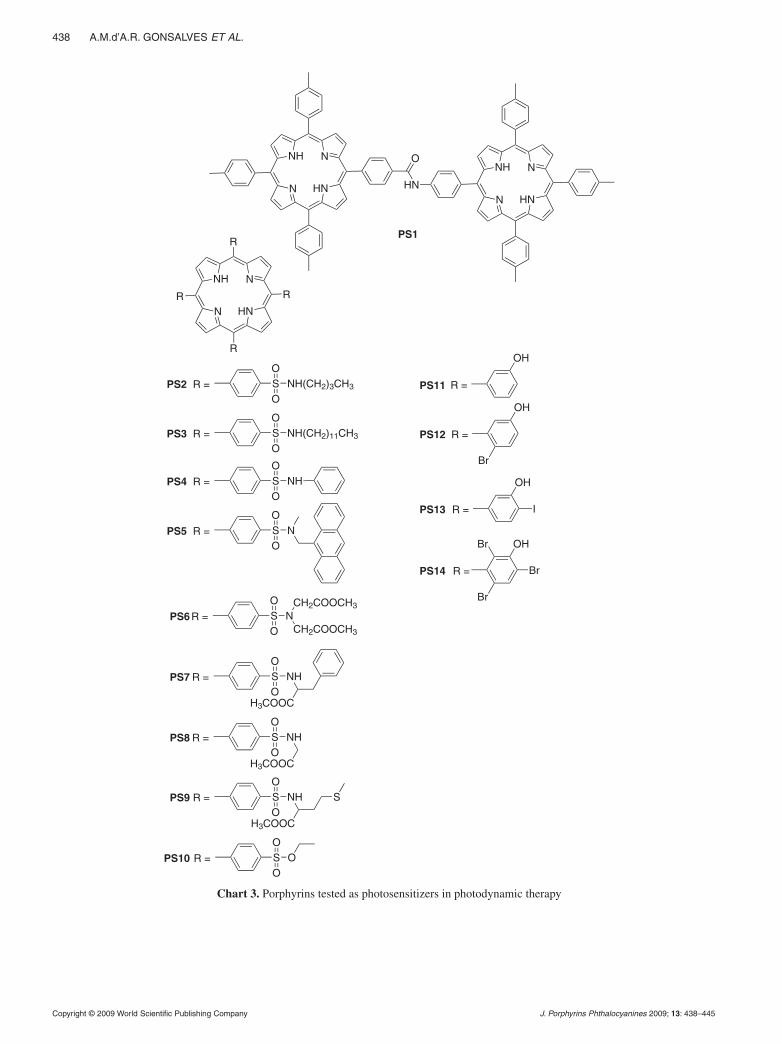
The porphyrins tested as photosensitizers for PDT are presented in Chart 3. PS1 was tested against human melanoma cells (SKMEL 188) and mouse melanoma cells (S91). Comparing with hematoporphyrin deriva-tives and Photofrin® (approved drugs for use in PDT) we obtained a comparable photokilling effect with 30 times less concentration and 2 times less irradiation dose. Among the sulphonated photosensitizers, PS2–PS10, only PS8, PS9 and PS10 have some photodynamic effect against adenocarcinoma cells HT29 (ATCC HTB 387) derived from a human colorectal adenocarcinoma. The IC50 values found for PS8, PS9 and PS10 were 2 µg/ml, 4.5 µg/ml and 3 µg/ml, respectively, values very similar to those of the approved photosensitizers Photofrin® and Foscan®. The photosensitizers PS11–PS14 protected by patent [36] have halogen atoms at the ortho position of the phenyl ring — as indicated from the previous studies to improve the photophysical and photochemical proper-ties — and a hydroxyl at the meta position to improve the amphiphilicity of the porphyrin core. The IC50 val-ues from dose/response curves for WiDr human colon adenocarcinoma cells and melanoma A375 are reported in Table 9 together with the values obtained for Photof-rin® as reference compound. The results showed that the porphyrin with four bromine atoms (PS12) presents an IC50 of 113 nM, that is about 6 times less than the IC50 of the Photofrin® and 25% less than the non-brominated analog (PS11) against WiDr cell line. In melanoma cells PS12 has an even lower IC50 (52 nM), that is, about3 times lower than the IC50 for Photofrin® and two times lower than that of PS11.
The results show that the halogen at the ortho posi-tion affects the capability of formation of singlet oxy-gen and also the cellular uptake and photostability. The best balance of these properties was obtained with two
bromine atoms at the ortho position, photosensitizer PS12.
CATALYTIC OXIDATIONS, METAL-LOPORPHYRIN-BASED OXIDATIVE SYSTEMS USING H2O2 AND NaOCL AS OXIDANTS
P450 cytochrome is a monooxygenase enzyme that contains an iron complex of a porphyrin as a prosthetic group and has the ability to catalyze epoxidation and hydroxylation reactions of many kinds of substrates using several oxygen donors instead of molecular oxy-gen, as in natural circumstances. Our studies on this topic began with the use of metalloporphyrins to mimic the action of this enzyme and have always been guided by the selection of systems that could possibly be used in large-scale applications. Environmental concerns, waste products and costs were important factors lead-ing to our choice of hydrogen peroxide and sodium hypochlorite as the source oxidants, in spite of their being very demanding of the catalyst structure. Robust-ness of the catalyst is a critical characteristic because the main problem is its easy degradation, particularly when using excess of oxidant relatively to substrate or with substrates having low reactivity. We also preferred that the porphyrin selected to prepare the metallocomplex be obtained from easily available, inexpensive starting materials via straightforward and simple synthetic pro-cesses [39–41].
The problem of the use of hydrogen peroxide as oxi-dant is related to its activation, particularly the critical step of the cleavage of the oxygen–oxygen bond. The role of manganese complexes of porphyrins is to pro-vide an effi cient heterolytic cleavage of this bond from a hydroperoxo metalloporphyrin complex (1) to generate a formally high valent manganese porphyrin complex (2) which is the functional oxidant. The process is helped by acid-base catalysis and equally important by the presence of a fi fth ligand (L) attached to the porphyrin nucleus (Scheme 1).
Imidazoles and pyridines are frequently used as axial ligands, acting also as bases, but suffer from rapid destruction and so need to be used in excess relatively to the metalloporphyrin. We demonstrated fi rstly that inor-ganic bases such as sodium carbonate and sodium acetate or tetrabutylammonium hydroxide and N-oxides as axial ligands (L) can replace imidazole although the reaction goes much slower (Table 10) [42]. Latter Mansuy et al. proved the viability of this approach developing an effi -cient catalytic system with inorganic salts as base and an axial ligand in acetonitrile [43].
Another approach which was exploited is the use of adducts of hydrogen peroxide as a way of maintaining the concentration of the peroxide low enough to avoid
00060.indd 900060.indd 9 7/2/2009 12:44:46 PM7/2/2009 12:44:46 PM
Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 438–445
438 A.M.d’A.R. GONSALVES ET AL.
N
NH N
HN
R
R
R
R
R =PS2
PS3
PS4
PS5
PS6
PS7
PS8
PS9
PS10
R =PS11
PS12
PS13
PS14
R =
Br
R =
R =
SO
ONH(CH2)3CH3
R = SO
ONH(CH2)11CH3
R = SO
ONH
R = SO
ON
R = SO
ON
CH2COOCH3
CH2COOCH3
R = SO
ONH
H3COOC
R = SO
ONH
H3COOC
R = SO
ONH
H3COOC
S
R = SO
OO
OH
OH
OH
Br
Br
I
Br
OH
N
NH N
HN HNN
NH N
HN
O
PS1
Chart 3. Porphyrins tested as photosensitizers in photodynamic therapy
00060.indd 1000060.indd 10 7/2/2009 12:44:47 PM7/2/2009 12:44:47 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 439–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 439
the destruction of the catalyst or the axial ligand [42]. In the absence of metalloporphyrins some adducts showed excellent epoxidation abilities when combined with acyl or nitrile compounds to generate peroxycarboxylic acids or peroxymidic acids respectively (Table 11) [44].
Halogenation of the β-positions of the porphyrin mac-rocycle was described by others as originating much more active and stable catalysts when iodosylbenzene is the source oxidant or when an excess of substrate in rela-tion to oxidant is present [45–47].
This kind of change in porphyrin structure was studied by us in oxidations with hydrogen peroxide but without using excess substrate. Contrary to previous observa-tions the manganese complex of porphyrin with eight chlorine atoms in the β-positions oxidizes substrates very quickly, promoting very fast oxidation reactions. However, the catalyst shows very poor stability, reveal-ing that in this case halogen substitution actually does not bring any advantage. Radical processes of destruc-tion of the macrocycle derived from interaction of this type of metalloporphyrin with hydrogen peroxide were implicated, as proved by stabilization of the catalyst in the presence of 2,6-di-tert-butylphenol [48]. The same manganese porphyrin P25 showed better activity when tested in epoxidation reactions with sodium hypochlo-rite as oxidant. Even so, activity was lower than when the porphyrin has no halogen atoms P26 (Table 12) [49].
In the case of catalyst P25 some amounts of chlorinated products were identifi ed, pointing to the occurrence of radical processes.
The relation between catalyst stability and porphyrin structure in these oxidation systems was further studied by us. Manganese complexes of meso-diarylporphyrins were prepared (P27–P30) (Chart 4) and tested in NaOCl oxidations [50]. Catalyst P27 presented much better per-formance than catalyst P28 due to its greater resistance to degradation. The presence of the two ortho chlorine atoms increases the bulkiness around the meso position of the macrocycle and diminishes the possibility of attack at this position, confi rming the suggestion made by oth-ers [51, 52]. Introducing bulky substituents around the macrocycle but in the β-positions, such as phenyl groups as in catalysts P29 and P30, do not bring any increase in the porphyrin resistance.
Several studies showed that the presence of elec-tron-withdrawing groups in the macrocycle periphery increases the activity of the catalyst. By chlorosulphona-tion of the meso phenyl groups with chlorosulphonic acid and subsequent reaction with amines we could introduce stable chlorosulphonamide groups in different positions of the macrocycle structure [19] (Chart 5).
It was reported that catalytic epoxidations could be performed in an aqueous/organic two-phase system with H2O2 (30%) in the presence of benzoic acid as
Table 9. The IC50 values from dose/response curves for WiDr human colon adenocarcinoma cells and melanoma A375
IC50
Porphyrin WiDr cells A375 cells
THPP, PS11 143 nM 115 nM
TBr4HPP, PS12 113 nM 52 nM
TI4HPP, PS13 > 10 µM 6.8 µM
TBr12HPP, PS14 3.1 µM 2.3 µM
Photofrin 666 nM 156 nM
Table 10. Effects of base and ligand combinations on the metalloporphyrin-catalyzed cis-cyclooctene epoxidation with H2O2 as oxidant
Base Ligand t, h Epoxide yield, %
Imidazole imidazole 0.20 99
Bu4N+OH- TO 5 98
Na2CO3 TO 5 74
Na(OAC)2 IO 2 96
TO; triethylamine N-oxide. IO; diisopropylethylamine N-oxide.
N
N
N
NMn Ar
Ar
Ar
Ar
OO
HHB
N
N
N
NMn Ar
Ar
Ar
Ar
OO
H
H B+
1
N
N
N
NMn Ar
Ar
Ar
Ar
O
2L L L
(V)
O+
N
N
N
NMn Ar
Ar
Ar
Ar
L
H2O2
(III)
+
+
Scheme 1. Role of acid-base catalysis in the catalytic cycle for hydrogen peroxide oxidations
00060.indd 1100060.indd 11 7/2/2009 12:44:47 PM7/2/2009 12:44:47 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 440–445
440 A.M.d’A.R. GONSALVES ET AL.
Table 11. Epoxidation of alkenes with hydrogen peroxide adducts and acyl or nitrile compounds
Acyl or nitrile compound Alkene H2O2 adduct Time, h Epoxide yield, %
p-O2NC6H4COCl Cis-cyclooctene SPC 3 92
Ac2O Cis-cyclooctene PO 3 98
Ac2O 1-methylcyclohexene U 4 85
Ac2O α-pinene U 3 78
Cl3CCN 1-methylcyclohexene U 0.3 92
Cl3CCN 1-methylcyclohexene PO 0.08 87
SPC; Na2CO3.1½H2O2. PO; Ph3PO · ½H2O2. U; H2NCONH2. H2O2.
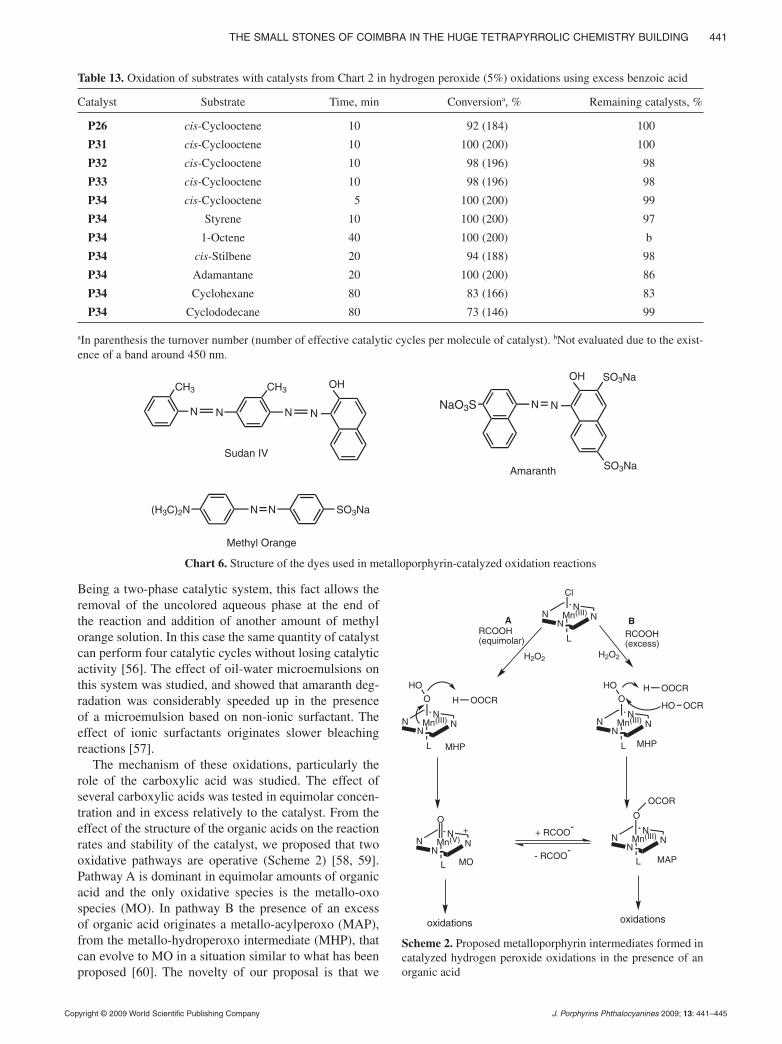
cocatalyst [53]. We further exploited this system, changing reaction conditions to more diluted hydrogen peroxide (5%) and an excess of benzoic acid. With these modifi ca-tions the system is very effi cient for alkene epoxidations and alkane hydroxylations and surprisingly the catalysts showed better stability [54, 55]. The more relevant results for epoxidation and hydroxylation reactions are presented in Table 13. In general, the presence of the sulphonamide groups originates more active catalysts than P26, which is known to be one of the more active porphyrins. Met-alloporphyrin P34 with one more diethylsulphonamido group in a β-position was found to be the best catalyst.
The same two-phase oxidative system was tried in the degradation of azo dyes such as sudan IV, amaranth and methyl orange (Chart 6).
Very fast bleaching of methyl orange solutions (140 mg.L-1) was observed using metalloporphyrin P31 as catalyst and diluted hydrogen peroxide solutions.
Table 12. Oxidation of alkenes using NaOCl as oxygen donor and metalloporphyrins P25 and P26 as catalysts
Ar=
Cl
Cl
N
N
N
NMn Ar
Ar
Ar
ArX
X XX
X
XXX (P25) X=Cl
(P26) X=H
Catalyst Alkene Time, h Epoxide yield, %
P25 styrene 2.5 70a
P26 styrene 0.3 88
P25 1-octene 4 10b
P26 1-octene 1 90
aMonochlorinated products detected by mass spectra. bThere was a 30% conversion of alkene into products of which the dominant one was 1,2-dichlorooctane.
N
N
N
NMn ArAr
P27 Ar =
Cl
Cl
NO2P28 Ar =
N
N
N
NMn ArAr
R
RR
R
NO2Ar =
P29 R =
P30 R = H2C
Chart 4. Structures of the manganese complexes of meso-diphenylporphyrins tested in NaOCl oxidations
N
N
N
NMn ArAr
Ar
Ar
P26 Ar =
Cl
Cl
P31 Ar =
Cl
Cl SO2N(C2H5)2
P32 Ar =
Cl
Cl SO2NHC4H9
P33 Ar =
Cl SO2NHC12H25
RR = H R = SO2N(C2H5)2
P34 Ar =
Cl
Cl SO2N(C2H5)2
Chart 5. Structures of manganese complexes of alkylsulphamoyl meso-arylporphyrins tested in hydrogen peroxide oxidations
00060.indd 1200060.indd 12 7/2/2009 12:44:47 PM7/2/2009 12:44:47 PM
Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 441–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 441
Being a two-phase catalytic system, this fact allows the removal of the uncolored aqueous phase at the end of the reaction and addition of another amount of methyl orange solution. In this case the same quantity of catalyst can perform four catalytic cycles without losing catalytic activity [56]. The effect of oil-water microemulsions on this system was studied, and showed that amaranth deg-radation was considerably speeded up in the presence of a microemulsion based on non-ionic surfactant. The effect of ionic surfactants originates slower bleaching reactions [57].
The mechanism of these oxidations, particularly the role of the carboxylic acid was studied. The effect of several carboxylic acids was tested in equimolar concen-tration and in excess relatively to the catalyst. From the effect of the structure of the organic acids on the reaction rates and stability of the catalyst, we proposed that two oxidative pathways are operative (Scheme 2) [58, 59]. Pathway A is dominant in equimolar amounts of organic acid and the only oxidative species is the metallo-oxo species (MO). In pathway B the presence of an excess of organic acid originates a metallo-acylperoxo (MAP), from the metallo-hydroperoxo intermediate (MHP), that can evolve to MO in a situation similar to what has been proposed [60]. The novelty of our proposal is that we
Table 13. Oxidation of substrates with catalysts from Chart 2 in hydrogen peroxide (5%) oxidations using excess benzoic acid
Catalyst Substrate Time, min Conversiona, % Remaining catalysts, %
P26 cis-Cyclooctene 10 92 (184) 100
P31 cis-Cyclooctene 10 100 (200) 100
P32 cis-Cyclooctene 10 98 (196) 98
P33 cis-Cyclooctene 10 98 (196) 98
P34 cis-Cyclooctene 5 100 (200) 99
P34 Styrene 10 100 (200) 97
P34 1-Octene 40 100 (200) b
P34 cis-Stilbene 20 94 (188) 98
P34 Adamantane 20 100 (200) 86
P34 Cyclohexane 80 83 (166) 83
P34 Cyclododecane 80 73 (146) 99
aIn parenthesis the turnover number (number of effective catalytic cycles per molecule of catalyst). bNot evaluated due to the exist-ence of a band around 450 nm.
N N SO3Na(H3C)2N
Methyl Orange
N N N N
OHCH3 CH3
Sudan IV
N N
OH
Amaranth
SO3Na
SO3Na
NaO3S
Chart 6. Structure of the dyes used in metalloporphyrin-catalyzed oxidation reactions
Scheme 2. Proposed metalloporphyrin intermediates formed in catalyzed hydrogen peroxide oxidations in the presence of an organic acid
NN
NN
NN
NN
NN
NN
+
RCOOH (equimolar)
RCOOH (excess)
OCOR
L
L L
(III)Mn
(V)Mn
O
Cl
BA
(III)Mn
O
H2O2 H2O2
NN
NN
O
L
(III)Mn
HO
H OOCR
oxidations
NN
NN
O
L
(III)Mn
HO H OOCR
HO OCR
oxidations
+ RCOO-
MHP MHP
MO MAP- RCOO-
00060.indd 1300060.indd 13 7/2/2009 12:44:47 PM7/2/2009 12:44:47 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 442–445
442 A.M.d’A.R. GONSALVES ET AL.
point out that, under conditions B, the MAP intermediate is also able to oxidize the substrate as suggested by oth-ers [61, 62] and this is the main mechanism when excess organic acid is present.
In oxidation catalysis, besides the normal catalytic cycles that originate products and regenerate the catalyst, some harmful non-productive processes occur, as illus-trated in Scheme 3. The highly active oxidation inter-mediates that are generated during catalytic cycles have the ability to destroy other molecules of porphyrin via intramolecular or intermolecular processes (A or B) or interact with oxidant, destroying its oxidative power.
Our studies conclude that catalyst deactivation pro-cesses A and B are both operative in hydrogen peroxide oxidation catalysis [63] and that these process have a rad-ical nature. Catalytic reactions in the presence of a radi-cal inhibitor such as 2,6-di-tert-butyl-4-methoxyphenol present less metalloporphyrin degradation, particularly in reaction conditions that favor the formation of metallo-oxo intermediates.
Light-promoted singlet oxygen reactions are clean oxidation processes and attractive because the oxidant is environmentally friendly. The use of porphyrins as pho-tosensitizers (PS) for this process was also studied by us. In homogeneous media porphyrin (13) seems very effi -cient in the formation of singlet oxygen reaction products
without signifi cant degradation [38], a common problem in these reactions (Scheme 4).
Limitations of these reactions using homogeneous sensitizers include the diffi culty in isolating the prod-uct from the catalyst and the poor catalyst stability in large-scale reactions. To circumvent these operational limitations we prepared heterogeneous photosensitizers by covalent immobilization of a porphyrin on Merrifi eld modifi ed-polymers through a selective chlorosulphona-tion of porphyrin (P35) [64]. From the several immobi-lized photocatalysts prepared, the most active was PSim, whose synthesis is outlined in Scheme 5.
PSim proved to be very effi cient in photooxida-tion reactions, as shown by the results with 1,5-dihy-droxynaphthalene (3), α-terpinene (4) and citronellol (5) presented in Table 14 [64, 65].
The advantage in the use of supported catalysts was tested in consecutive reactions where PSim was removed from solution by fi ltration and then used in another experiment [65] (Table 15).
The results showed that PSim was able to carry out 3 consecutive reactions with no signifi cant loss of activity
Highly active oxidation metalloporphyrin intermediates
A BC
Intramolecular macrocycle decomposition Interaction with
oxidant
Reaction with another metalloporphyrin molecule
Scheme 3. Possible pathways for the inactivation of the metal-loporphyrin intermediates in oxidations
N
HN
NH
NArAr
Ar
Ar
P13
S SOPS, O2
CH3CN
S SO (Yield)
OH
OH
OH O
O
O
OH O
94%
93%
Ar=
Cl
Cl
Scheme 4. Synthesis of quinones through photooxidations catalyzed by P13
N
NH N
HN
R1
R1
R1
R1=
Cl
Cl
HClSO3
N
NH N
HN
R1
R1
R1
ClO2S
CH2NHC12H24NH2
Merrifield-modifiedresin
N
NH N
HN
R1
R1
R1PSim
CH2NHC12H24NHSO2
Merrifield resin
P35
Scheme 5. Synthetic procedure for the immobilization of a porphyrin on Merrifi eld modifi ed-polymers
00060.indd 1400060.indd 14 7/2/2009 12:44:48 PM7/2/2009 12:44:48 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 443–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 443
and maintaining the selectivity for the addition prod-uct. Signifi cant catalyst inactivation was not observed as shown by the TOF values. With the higher substrate ratio, a total of 45,000 catalytic cycles were carried out and this corresponds to the synthesis of 4.6 g of product using only 35 mg of PSim.
Acknowledgements
The authors gratefully acknowledge Chymiotechnon, Fundação para a Ciência e Tecnologia for fi nancial sup-port and Elisa Serra for highly useful discussions on the presentation of this manuscript.
REFERENCES
1. Cavaleiro JAS, Rocha Gonsalves AMdA, Kenner GW, Smith KM, Shulman RG, Mayer A and Yamane T. J. Chem. Soc., Chem. Comm. 1974: 392–393.
2. Mayer A, Ogawa S, Shulman RG, Yamane T, Cava-leiro JAS, Rocha Gonsalves AMdA, Kenner GW and Smith KM. J. Mol. Biol. 1974; 86: 749–756.
3. Rocha Gonsalves AMdA, Kenner GW and Smith KM. Tetrahedron Lett 1972: 2203–2206.
4. Cavaleiro JAS, Rocha Gonsalves AMdA, Kenner GW and Smith KM. J. Chem. Soc., Perkin I 1973: 2471–2478.
5. Cavaleiro JAS, Rocha Gonsalves AMdA, Kenner GW and Smith KM. J. Chem. Soc., Perkin I 1974: 1771–1781.
6. Kenner GW and Smith KM. Ann. N.Y. Acad. Sci. 1973; 206: 138–150.
7. Rothemund P. J. Am. Chem. Soc. 1935; 57: 2010–2011.
8. Rothemund P. J. Am. Chem. Soc. 1939; 61: 2912–2915.
9. Rothemund P and Menotti AR. J. Am. Chem. Soc. 1941; 63: 267–270.
10. Rocha Gonsalves AMdA and Pereira MM. J. Het-erocyclic Chem. 1985; 22: 931–933.
11. Lindsey JS, Hsu HC and Schreiman IC. Tetrahe-dron Lett 1986; 27: 4969.
12. Rocha Gonsalves AMdA, Varejão JMTB and Pereira MM. J. Heterocyclic Chem. 1991; 28: 635–640.
13. Johnstone RAW, Nunes MLPG, Pereira MM, Rocha Gonsalves AMdA and Serra AC. Heterocycles 1996; 43: 1423–1437.
Table 15. Photooxidation of α-terpinene in consecutive experiments with different PSim/α-terpinene ratios
R = nph/nterpa Reaction Time, h Product, %b TOFc
1st 3.3 86 (14) 13031/5000 2nd 3.3 86 (14) 1303 3rd 2.5 87 (13) 1740 1st 4 82 (18) 30751/15,000 2nd 8 78 (22) 1463 3rd 8.5 81 (19) 1429
a PSim/substrate ratio. b In parenthesis figures the amount of p-cymene. c Moles of product/moles of photo-sensitizer x h.
Table 14. Results for photooxidations using supported catalyst PSim
Reaction R = nph/nterpa Time, h Products, %
1/100 5 75 1/600 15 58
1/5000 3.5 86 (14)b
1/15,000 4 82 (18)b
1/30,000 10 65 (35)b
1/600 6.5 98
6(47):7(53)
1/5000 10 99
6(47):7(53)
a PSim/substrate ratio. b In parenthesis figures the amount of p-cymene.
O
OH O
OH
OH3
HO
5
HO
7
HOOHO
6
HOO+
OO
4
00060.indd 1500060.indd 15 7/2/2009 12:44:48 PM7/2/2009 12:44:48 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 444–445
444 A.M.d’A.R. GONSALVES ET AL.
14. Barnett GH and Smith KM. Tetrahedron Lett 1973: 2887–2888.
15. Nascimento BFO, Pineiro M, Rocha Gonsalves AMdA, Ramos Silva M, Matos Beja A and Paixão JA. J. Porphyrins Phthalocyanines 2007; 11: 77–84.
16. Ibers JA, Ulman A, Gallucci J and Fisher D. J. Am. Chem. Soc. 1980; 102: 6852–6854.
17. Cristiano MLS, Gago DJP, Rocha Gonsalves AMdA, Johnstone RAW, McCarron M and Varejão JMTB. Org. Biomol. Chem. 2003; 1: 565–574.
18. Fleischer EB, Palmer JM, Srivastava TS and Chat-terjee A. J. Am. Chem. Soc. 1971; 93: 3162–3167.
19. Rocha Gonsalves AMdA, Johnstone RAW, Pereira MM, SantAna AMP, Serra AC, Sobral AFNL and Stocks PA. Heterocycles 1996; 43: 829–838.
20. Hudson AJ, Richardson T, Trirtle JP, Johnstone RAW, Sobral AJFN and Rocha Gonsalves AMdA. Mol. Cryst. 1993; 253: 103.
21. Burrows HD, Rocha Gonsalves AMdA, Leitão MLP, Miguel MG and Pereira MM. Supramolecu-lar Sci. 1997; 4: 241–246.
22. Grieve MB, Richardson T, Johnstone RAW, Sobral AFNL and Rocha Gonsalves AMdA. Thin Solid Films 1994; 243: 581.
23. Smith VC, Batty SV, Richardson T, Foster KA, Johnstone RAW, Sobral AFNL and Rocha Gon-salves AMdA. Thin Solid Films 1996; 284–285: 911–914.
24. Richardson T, Smith VC, Johnstone RAW, Sobral AFNL and Rocha Gonsalves AMdA. Thin Solid Films 1998; 327–329: 315–320.
25. Togashi DM, Costa SMB, Sobral AFNL and Rocha Gonsalves AMdA. Chem. Phys. 2004; 300: 267–275.
26. Togashi DM, Costa SMB, Sobral AFNL and Gon-salves AMdAR. J. Phys. Chem. B 2004; 108: 11344–11356.
27. Seixas de Melo SJ, Sobral AFNL, Rocha Gonsalves AMdA and Burrows HD. J. Photochem. Photobiol. A 2005; 172: 151–160.
28. Pineiro M, Carvalho AL, Pereira MM, Gonsalves AMdAR, Arnaut LG and Formosinho SJ. Chem. Eur. J. 1998; 4: 2299–2307.
29. Pineiro M, Pereira MM, Rocha Gonsalves AMdA, Arnaut LG and Formosinho SJ. J. Photochem. Pho-tobiol. A 2000; 138: 147–157.
30. Pineiro M, Rocha Gonsalves AMdA, Pereira MM, Formosinho SJ and Arnaut LG. J. Phys. Chem. A 2002; 106: 3787–3795.
31. Arnaut LG, Caldwell RA, Elbert JE and Melton LA. Rev. Sci. Instrum. 1992; 63: 5381–5389.
32. Formosinho SJ. J. Chem. Soc. Faraday Trans. 2 1974; 70: 605–620.
33. Formosinho SJ. Mol. Photochem. 1976; 7: 41–45. 34. Azenha EG, Serra AC, Pineiro M, Pereira MM,
Seixas de Melo SJ, Arnaut LG, Formosinho SJ and
Rocha Gonsalves AMdA. Chem. Phys. 2002; 280: 177–190.
35. Pineiro M, Arnaut LG, Formosinho SJ and Rocha Gonsalves AMdA. Pol. J. Med. Phys. Eng. 2001; 6: 177–184.
36. Rocha Gonsalves AMdA, Pineiro M and Serra AC. Tetrapyrrolic Macrocycles as Photodynamic Agents; Portugal Patent, 2003.
37. Serra AC, Pineiro M, Rocha Gonsalves AMdA, Abrantes M, Laranjo M, Santos AC and Botelho MF. J. Photochem. Photobiol. B 2008; 92: 61–67.
38. Murtinho D, Pineiro M, Pereira MM, Gonsalves AMdAR, Arnaut LG, Graça Miguel M and Bur-rows HD. J. Chem. Soc., Perkin Trans. 2 2000: 2441–2447.
39. Serra AC. In Recent Research Developments in Het-erocyclic Chemistry, Pinho e Melo TMVD, Rocha Gonsalves AMdA. (Eds.) Research Signpost: Ker-ala, India, 2007; pp 81–127.
40. Rocha Gonsalves AMdA. J. Heterocyclic Chem. 2002; 39: 499–509.
41. Rocha Gonsalves AMdA and Pereira MM. J. Mol. Catal. A: Chem. 1996; 113: 209–211.
42. Rocha Gonsalves AMdA, Johnstone RAW, Pereira MM and Shaw J. J. Chem. Soc. Perkin Trans. I 1991: 645–649.
43. Bartoli JF, Battioni P, DeFoor WR and Mansuy D. 1994: 23–25.
44. Rocha Gonsalves AMdA, Johnstone RAW, Pereira MM and Shaw J. J. Chem. Res.(M) 1991: 2101–2118.
45. Wisejekera T, Matsumoto A, Dolphin D and Lexa A. Angew. Chem. Int. Ed. 1990; 29: 1028–1030.
46. Traylor TG and Tsuchiya S. Inorg. Chem. 1987; 26: 1338–1339.
47. Hoffmann P, Robert A and Meunier B. Bull. Soc. Chim. Fr. 1992; 129: 85–97.
48. Rocha Gonsalves AMdA, Johnstone RAW, Pereira MM, Shaw J and Sobral AFNL. Tetrahedron Lett 1991; 32: 1355–1358.
49. Rocha Gonsalves AMdA, Pereira MM, Serra AC, Johnstone RAW and Nunes MLPG. J. Chem. Soc. Perkin Trans. 1 1994: 2053–2057.
50. Sobral AFNL and Rocha Gonsalves AMdA. J. Por-phyrins Phthalocyanines 2001; 5: 861–866.
51. Banfi S, Montanari F and Quici S. J. Org. Chem. 1988; 53: 2863–2866.
52. Traylor PS, Dolphin D and Traylor TG. J. Chem. Soc. Chem. Commun. 1984: 279–280.
53. Anelli L, Banfi S, Montanari F and Quici S. J. Chem. Soc. Chem. Commun. 1989: 779–780.
54. Rocha Gonsalves AMdA, Pereira MM and Serra AC. An. Quim. Int. Ed. 1996; 92: 375–380.
55. Rocha Gonsalves AMdA and Serra AC. J. Porphy-rins Phthalocyanines 2000; 4: 586–603.
56. Serra AC, Docal C and Rocha Gonsalves AMdA. J. Mol. Catal. A: Chem. 2005; 238: 192–198.
00060.indd 1600060.indd 16 7/2/2009 12:44:48 PM7/2/2009 12:44:48 PM

Copyright © 2009 World Scientific Publishing Company J. Porphyrins Phthalocyanines 2009; 13: 445–445
THE SMALL STONES OF COIMBRA IN THE HUGE TETRAPYRROLIC CHEMISTRY BUILDING 445
57. Hager M, Holmberg K, Rocha Gonsalves AMdA and Serra AC. Colloids Surf. A 2001; 183–185: 247–257.
58. Rocha Gonsalves AMdA and Serra AC. J. Mol. Catal. A: Chem. 2001; 168: 25–32.
59. Rocha Gonsalves AMdA and Serra AC. J. Chem. Soc. Perkin Trans. 2 2002: 715–719.
60. Banfi S, Maiocchi A and Quici S. Gazz. Chim. Ital. 1990; 120: 123–130.
61. Machii K, Watanabe Y and Morishima I. J. Am. Chem. Soc. 1995; 117: 6691–6697.
62. Kamaraj K and Bandyopadhyay D. J. Am. Chem. Soc. 1997; 119: 8099–8100.
63. Serra AC, Marçalo EC and Rocha Gonsalves AMdA. J. Mol. Catal. A: Chem. 2004; 215: 17–21.
64. Ribeiro SM, Serra AC and Rocha Gonsalves AMdA. Tetrahedron 2007; 63: 7885–7891.
65. Ribeiro SM, Serra AC and Rocha Gonsalves AMdA. J. Catal. 2008; 256: 331–337.
00060.indd 1700060.indd 17 7/2/2009 12:44:48 PM7/2/2009 12:44:48 PM

Copyright of the works in this Journal is vested with World Scientific Publishing. Thearticle is allowed for individual use only and may not be copied, further disseminated, orhosted on any other third party website or repository without the copyright holder’swritten permission.




















