
Investigating the factors that influence the purchase behaviour of ...
Upload
bioacademyCategory
view
1download
0
proteinsSTRUCTURE O FUNCTION O BIOINFORMATICS
Investigating the structural stability of theTup1-interaction domain of Ssn6: Evidencefor a conformational change on the complexMaria Palaiomylitou,1 Athanassios Tartas,1 Dimitrios Vlachakis,1 Dimitris Tzamarias,2 and
Metaxia Vlassi1*1 Institute of Biology, National Centre for Scientific Research ‘‘Demokritos’’, 15310 Ag. Paraskevi Attikis, Greece
2 Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology, 71110 Heraklion, Greece
INTRODUCTION
Transcriptional repression of a wide variety of gene families in
Saccharomyces cerevisiae requires the function of two physically asso-
ciated proteins, Ssn6 and Tup1. The Ssn6-Tup1 complex consists of
one Ssn6 and four Tup1 molecules1 and is recruited to target pro-
moters by interacting with gene-specific DNA-binding proteins.2–5
Transcription regulators resembling Ssn6 and Tup1, both in amino
acid sequence and function, have also been found in worms, flies,
plants, and mammals, suggesting that transcriptional repression by
an Ssn6-Tup1-like system is evolutionary conserved.2,6
The N-terminal domain of the 966 amino acid Ssn6 protein contains
10 tandem tetratricopeptide repeats, known as TPRs (Fig. 1). The TPR
constitutes a degenerate recurrent sequence motif, the consensus
sequence of which is defined by a pattern of small and large hydropho-
bic amino acids.7,11 TPR mediated protein–protein interactions are
involved in a diverse spectrum of cellular functions.12–14 On the basis
of the functional diversity and importance of TPR containing proteins,
Blatch and Lassle14 proposed that the TPR motif may represent an an-
cient general protein–protein interaction module adopted by function-
ally different proteins and adapted for specific functions. The minimal
Tup1 interaction domain of Ssn6 includes its three N-terminal TPRs
(Fig. 1), whereas various combinations of TPR4 to TPR10 mediate
interactions with structurally distinct DNA-binding repressor proteins
specific for each gene family regulated by the Ssn6-Tup1 complex.15
The structure of Ssn6 is unknown. However, the crystal structures
of several natural16–26 as well as of designed TPR containing pro-
teins8 have been determined so far demonstrating that each TPR
motif is highly a-helical composed of a pair of antiparallel a-helices,termed helices A and B, as first determined by the crystal structure of
the three-TPR containing protein Pp5.16 The packing of helices
within and between adjacent TPR motifs is identical so that each
a-helix interacts with its two immediate a-helix neighbors8,16 in a
The Supplementary Material referred to in this article can be found online at http://www.
interscience.wiley.com/jpages/0887-3585/suppmat/
Grant sponsor: General Secretariat for Research and Technology of Greece, NCSR ‘‘Demoritos’’.
Maria Palaiomylitou and Athanassios Tartas contributed equally to this work.
*Correspondence to: Metaxia Vlassi, Institute of Biology, National Centre for Scientific Research
‘‘Demokritos’’, 15310 Ag. Paraskevi Attikis, Greece. E-mail: [email protected]
Received 30 December 2006; Revised 23 February 2007; Accepted 6 March 2007
Published online 16 July 2007 in Wiley InterScience (www.interscience.wiley.com).
DOI: 10.1002/prot.21489
ABSTRACT
Ssn6, a tetratricopeptide repeat (TPR) containing
protein, associates with the Tup1 repressor to
form a global transcriptional co-repressor com-
plex, which is conserved across species. The three
N-terminal TPR repeats of Ssn6, out of a total of
10, are involved in this particular interaction.
Our previously reported 3D-modeling and muta-
genesis data suggested that the structural integrity
of TPR1 and its correct positioning relatively to
TPR2 are crucial for Tup1 binding. In this study,
we first investigate the structural stability of the
Tup1 binding domain of Ssn6, in pure form,
through a combination of CD spectroscopy and
limited proteolysis mapping. The obtained data
were next combined with molecular dynamics sim-
ulations and disorder/order predictions. This com-
bined study revealed that, although competent to
fold, in the absence of Tup1, TPR1 is partially
unfolded with its helix B being highly dynamic
exposing an apolar surface to the solvent. Subse-
quent CD spectroscopy on this domain complexed
with a Tup1 fragment comprising its Ssn6 binding
region provided strong evidence for a conforma-
tional change consisting of acquisition of a-helical
structure with simultaneous stabilization of a
coiled-coil configuration upon complex formation.
We propose that this conformational change
occurs largely in the TPR1 of Ssn6 and is in
accord with the concept of folding coupled to
binding, proposed for other TPR domains. A
possible implication of the structural flexibility of
Ssn6 TPR1 in Tup1 recognition is discussed and a
novel mode of interaction is proposed for this par-
ticular TPR-mediated complex.
Proteins 2008; 70:72–82.VVC 2007 Wiley-Liss, Inc.
Key words: tetratricopeptide repeat; structural flexi-
bility; coupled folding and binding; circular dichro-
ism; limited proteolysis; molecular dynamics.
72 PROTEINS VVC 2007 WILEY-LISS, INC.

three-stranded coiled-coil manner resulting in super-helical
architectures, the size and shape of which depends on the
number of tandem TPR motifs. A significant consequence
of the TPR superhelicity is the generation of a concave sur-
face formed by helices A of tandem TPRs and a convex sur-
face formed by both A and B helices.16 The concave surface
has been proposed to be involved in ligand binding16 and
this hypothesis was indeed confirmed experimentally in the
case of several Hsp-binding TPR proteins.17,22,24,27 The
crystal structure of the p67phox/Rac complex, however,
demonstrated that binding to the TPR concave groove is
not a general structural principle of ligand binding by TPR-
containing proteins, suggesting a versatility of TPR domains
in both target recognition and their general role as scaffolds
for protein assembly.18
Using the canonical TPR structure as template we have
previously modeled the 3D-structure of the TPR1-3
domain of Ssn6 involved in Tup1 binding. The model
combined with mutagenesis data indicated that the struc-
tural integrity of TPR1 and its correct positioning relatively
to TPR2 are essential for the Ssn6-Tup1 interaction.9 Con-
sistently, an independent mutagenesis work also attributed
a major role of the structural integrity of TPR1 in this par-
ticular interaction of Ssn6.10 In our previous study we
have also suggested that the Ssn6-Tup1 interaction may
have a hydrophobic character and in this respect it is rather
unique among known TPR-mediated interactions, making
its structural analysis of major interest.
The present study is a starting point of our efforts
towards an understanding of the structural basis of this
particular interaction of Ssn6. Towards this end, we
investigate the structural stability of the Tup1 interaction
domain of Ssn6, both in isolation and in complex with a
Tup1 fragment containing the Ssn6 interaction domain.
Circular dichroism (CD) spectroscopy, limited proteolysis
mapping, and molecular dynamics (MD) simulations
combined with disorder/order predictions show that, in
isolation, TPR1 is partially unfolded with its helix B
being highly dynamic exposing an apolar surface to the
solvent. Subsequent CD spectroscopy on an Ssn6-Tup1
complex provides strong evidence for a conformational
change involving acquisition of a-helical coiled structure.
We propose that this conformational change occurs
mainly in the TPR1 of Ssn6 and is in accord with the
idea of folding coupled to binding, proposed in other
TPR domains. A possible role of the structural flexibility
of TPR1 in Tup1 recognition is discussed. In addition, a
novel mode of association involving the convex TPR sur-
face and hydrophobic rather than polar residues is pro-
posed for this particular TPR-mediated interaction of
Ssn6 in contrast to modes of interaction observed so far
in other TPR-mediated complexes.
MATERIALS AND METHODS
Construction of plasmids
The minimal Tup1 interaction domain of S. cerevisiae
Ssn6 protein (Sc3T; residues 1–175) that includes its
three and a half N-terminal TPRs was cloned using the
pGEX-2T expression vector (Amersham Biosciences) as
described previously.15
A plasmid encoding the entire TPR domain of Ssn6
(Sc10T; residues 1–415) tagged to 6xHis was constructed
by inserting a PCR generated fragment between the NdeI
and BamHI sites of pET15b (Novagen) expression vector.
The PCR product was generated using the 50-TCATCCCGGGATGAATCCGGGCGGTGAAC-30 and 50-GTAAGGATCCTTACTATGGGTTTTCTAAC-30 forward and
reverse primers, respectively and was subsequently puri-
fied and digested with XmaI and BamHI. The NdeI site
of the vector was filled in with Klenow polymerase prior
to ligation with the PCR product.
A fragment of S. cerevisiae Tup1 protein (Tn108; resi-
dues 1–108) including the Ssn6 interaction domain5 was
cloned as a XmaI-AvaII blunt fragment at the XmaI-
EcoRI blunt sites of PGEX-2T expression vector. This par-
ticular molecule was chosen based on its stability and
solubility compared to shorter Tup1 protein fragments.
The sequence of the coding region of all constructs used
in this study was confirmed by nucleotide sequencing.
Protein expression and purification
Sc3T was expressed as a GST fusion protein in Esche-
richia coli. BL21(DE3) cells were grown at 378C in LB
Figure 1A: Schematic representation of the 10-TPR containing region of Ssn6 protein
(aa: 1–415). The two fragments used in this study (Sc3T and Sc10T) are
indicated. B: Sequence alignment of TPR repeats 1 to 3 involved in Tup1
binding. Predicted TPR a-helices are depicted as cylinders above the sequence.
Residues of the TPR signature7 are grey shaded. Amino acid preferences for each
TPR position8 are indicated below the aligned sequences. Hydrophobic residues
not-matching the TPR consensus character are black shaded. A hydrophobic
patch is boxed. Point mutation sites disrupting interaction with Tup19,10 are
indicated by open circles and asterisks (see discussion). Black and open arrows
denote recognized and predicted chymotrypsin sites, respectively. Chymotrypsin
recognition sites to Ala are not included for clarity reasons. C: Model of the
three-dimensional structure of the TPR1-3 region of Ssn6.9
Structural Stability of a Domain of Ssn6
DOI 10.1002/prot PROTEINS 73

medium containing ampicillin (100 lg/mL) to an OD600
of 0.7 and induced with 0.7 mM isopropyl-b-D-thiogalac-topyranoside (IPTG) at 258C for 3 h. The bacterial cell
pellet was lysed by sonication in lysis buffer A (20 mM
Tris-HCl pH 8.0, 50 mM NaCl, 20% (v/v) glycerol, 0.1%
(v/v) Triton X-100, 5 mM EDTA, and 1 mM DTT). The
lysate was centrifuged at 12,000g for 1 h at 48C. Proteaseinhibitors were included in all purification steps. The
GST-Sc3T fusion protein was purified by affinity chro-
matography on a Glutathione Sepharose 4B (GS-H)
(Amersham Biosciences) affinity column equilibrated
with lysis buffer A. After extensive washing of the col-
umn the bound protein was digested with thrombin pro-
tease (using a molar ratio of 1:500) for 2 h at 168C to
remove the GST moiety. The reaction was stopped by the
addition of benzamidine. The resulting purified protein
was subsequently loaded on a Sephacryl S200 (Amersham
Biosciences) gel-filtration column equilibrated with
20 mM Tris-HCl (pH 8.0) and 200 mM NaCl. Purified
protein not used immediately was stored at 2208C after
addition of 20% (v/v) glycerol.
Sc10T was expressed as a 6xHis-tagged protein using
E. coli BL21(DE3) cells. Cells were grown to an OD600 of
0.5 at 308C and induced with 1 mM IPTG at 208C for 16
h. Harvested cells were lysed by sonication in lysis buffer
B (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 3 mM imidaz-
ole, 0.5 mM DTT, 20% (v/v) glycerol, and 1% (v/v) NP-
40). After clarification, the His-tagged protein was puri-
fied to near homogeneity by affinity chromatography on
a Ni-nitrilotriacetic acid column (Qiagen) equilibrated
with buffer B and eluted with an imidazole gradient.
Tn108 was expressed as a GST-fused protein as
described above for Sc3T. The protein was purified to
near homogeneity using GS-H affinity chromatography
followed by removal of the GST moiety using thrombin
cleavage.
The Sc3T-Tn108 complex was produced in vitro by
incubating purified Tn108 and GH-S beads with pre-
bound GST-Sc3T. A fourfold excess of Tn108 over Sc3T
(in molar basis) was used to ensure complete saturation
of Sc3T. A control experiment using GH-S beads with
prebound GST was carried out, in order to test out for
any nonspecific binding of Tn108 on the matrix or on
GST. After incubation with Tn108 for 3 h at room tem-
perature, the GH-S beads were extensively washed to
remove any unbound Tn108 protein. Beads carrying the
bound GST-Sc3T-Tn108 complex were subsequently
incubated with thrombin to remove the GST moiety.
The resulting Sc3T-Tn108 complex was obtained by
centrifugation.
Molecular weight and Stokes radiusdetermination of Sc3T
The molecular weight and Stokes radius of Sc3T were
determined by gel-filtration chromatography. The chro-
matographic separation on the Sephacryl S200 column
was calibrated using the protein standards: bovine serum
albumin (66 kDa), ovalbumin (45 kDa), carbonic anhy-
drase (29 kDa), cytochrome c (12.4 kDa), and potassium
dicromate (approximately 200 Da). Blue dextran (� 2
MDa) was used to determine the void volume (V0) of
the column. The relative molecular weight of the elution
peak of Sc3T was then estimated using a standard curve
obtained by plotting the logarithm of the molecular
weight of the known proteins against Kav 5(Ve 2 V0)/
(Vt 2 V0), with Vt being the total bed volume}. Estima-
tion of the Stokes radius (RST) of Sc3T was obtained by a
modified Laurent and Killander method28,29 using a lin-
ear calibration plot of RST values versus (2log Kav).
Limited proteolysis and peptide mapping
Limited proteolysis mapping was performed on the
Sc10T fragment of Ssn6. Purified Sc10T protein, as eluted
from Ni-NTA column, was digested with a-chymotrypsinII
protease (Sigma–Aldrich) at a molar ratio of 1:200 (w/w,
protease:target protein). Chymotrypsin was selected for
its property to recognize exposed hydrophobic residues.
The reaction proceeded at room temperature for 2 h and
aliquots were collected at various times and analyzed by
SDS-PAGE. The gel was subsequently transferred onto a
polyvinylidene difluoride (PVDF) membrane. Protein
bands corresponding to proteolytically resistant fragments
were excised from the PVDF membrane and subjected to
N-terminal sequencing (Biocentrum Services, Krakow,
Poland).
Circular dichroism
Far-UV (180–260 nm) CD experiments were per-
formed on a JASCO J-715 spectropolarimeter equipped
with a Peltier element for temperature control. All scans
were performed using a 0.1 mm path-length quartz cell.
The wavelength scans were made with the following
parameter settings: 0.2 nm step resolution, 20 nm scan
speed, 1 s response time, 1 s bandwidth, and 8 accumula-
tions. The CD signal from the buffer was subtracted
from the CD signals of the proteins. Conversion of the
observed ellipticities to molar ellipticities and deconvolu-
tion of the CD spectra were made using the CDNN
program.30,31 The expected CD spectrum of the
Sc3T-Tn108 complex was calculated by a summation of
the individual Sc3T and Tn108 CD spectra using the
equation: [y]theor 5 (NresSc3T 3 [y]Sc3T 1 NresTn108 3[y]Tn108)/NresSc3/Tn108, where [y]i and Nresi are the mean
residue ellipticity and residue number of protein i,
respectively. Thermal unfolding was recorded by moni-
toring changes in the CD signals at 222 nm correspond-
ing to the loss of the a-helical structure during thermal
denaturation with a scan rate of 908C/h. The melting
temperature (TM) for each protein fragment was taken as
M. Palaiomylitou/A. Tartas et al.
74 PROTEINS DOI 10.1002/prot

the temperature at which the fraction folded equals 0.5.
The protein concentrations used for the CD experiments
were 15–30 lM for Sc3T, 34 lM for Tn108, and 2.5 lMfor the Sc3T-Tn108 complex.
Sequence analysis for intrinsic disorder
Analysis of the Ssn6 amino acid sequence for intrinsic
disorder was performed using various disorder prediction
methods among which, the IUPred32 (http://iupred.
enzim.hu), RONN33 (http://www.strubi.ox.ac.ukRONN),
and PONDR1 VL-XT34,35 (http://www.pondr.com) pre-
dictors. The above predictors were chosen among avail-
able disorder predictors (reviewed in Ref. 36) because
they differ both methodologically and conceptually.
IUPRED is expected to recognize novel types of disorder
as it is not trained on disordered proteins. RONN can be
used to identify short regions of disorder with an
improved accuracy,33 whereas the VL-XT predictor of
PONDR1 may be used to predict short segments of
disorder that become ordered upon specific binding to a
biologically meaningful partner, indicating potential
binding sites.37,38
MD simulations
MD simulations were performed using MOE (The
Molecular Operating Environment) Version 2005.06 and
its built-in MD module using an NVT ensemble (N:
number of atoms, V: volume, T: temperature). A 3D-
model of the TPR1 to TPR3 region of Sc3T produced as
described previously9 was used as the initial structure.
The model was first optimized by energy minimization
using the MMFF94x molecular mechanics forcefield of
MOE. The protein was then solvated in a box of explicit
water molecules extending 8–12 A from the protein
atoms. Periodic boundaries were used in order to mini-
mize edge effects. The water structure was first equili-
brated for 100 ps keeping the whole protein fixed fol-
lowed by a 100 ps equilibration with fixed the protein
backbone. The equilibration phase was followed by a
2 ns MD simulation at 300 K with a time step for
integration of the potential function of 2 fs. A nonbonded
cutoff of at least 8 A was used.
Analysis of the MD simulation
The evaluation of the MD trajectory was done by ana-
lyzing the conformational changes over simulation time.
Coordinate files were extracted from the MD trajectory
every 0.5 ps for this purpose. The conformational analy-
sis focused on monitoring changes of the a-helical struc-ture during the simulation. Residues were assigned an a-helical conformation according to Kabsch and Sander.39
The percent simulation time during which a particular
residue followed the a-helical conformation was subse-
quently calculated. Moreover, a Ramachandran plot was
produced for all the MD structures in order to assess the
dynamics of the backbone conformational angles (u,w)during the simulation.
RESULTS
In isolation the Tup1-interaction domain ofSsn6 is partially unfolded
The minimal Tup1-interaction domain of Ssn615
(Sc3T; aa:1–175), including its three N-terminal TPR
repeats [Fig. 1(A)], was first expressed in recombinant
form and purified to homogeneity [Fig. 2(A)] in order to
study its structure in isolation. Sc3T eluted from the size
exclusion chromatography column at a volume equivalent
to a 23.2 kDa protein [Fig. 2(B)], which is close to its
deduced monomeric molecular mass (21.7 kDa). The sec-
ondary structure of this protein fragment was subse-
quently studied by CD. The far-UV CD spectrum of Sc3T
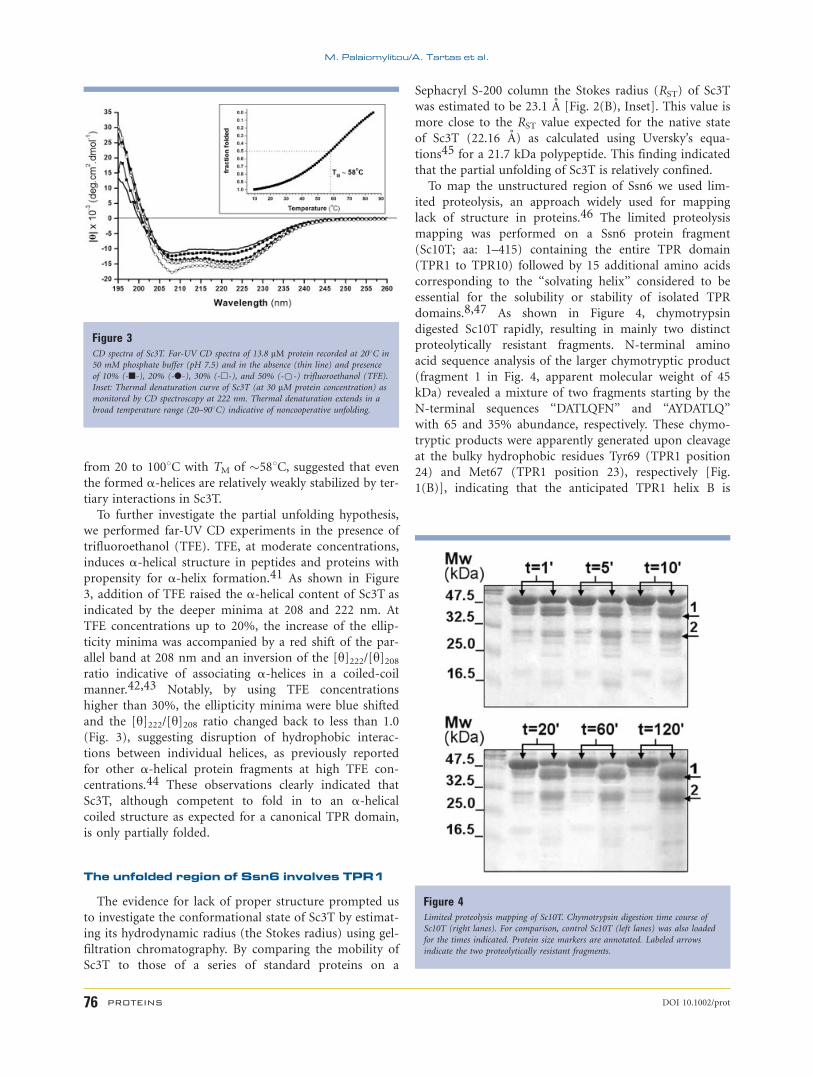
(Fig. 3, black line) revealed a predominately a-helicalstructure as indicated by the characteristic minima at 208
and 222 nm. However, deconvolution of the CD data gave
an estimate for the a-helical content of 34%, which is sig-
nificantly lower than the estimated one (�51%) based on
the expected TPR structure of this domain9 (see also
Fig. 1) possibly reflecting a partial unfolding of this Ssn6
fragment. Consistent with this possibility, the melting of
the a-helical structure of Sc3T extended from 20 to 908Cwith midpoint of the thermal denaturation (TM) of 588C(Fig. 3, Inset) as opposed to cooperative unfolding transi-
tions with high TM (838C) observed for ideal three-TPR
containing domains.8 This observation in conjunction
with the similarity of the thermal unfolding of Sc3T with
that of isolated a-helices in water,40 which also extends
Figure 2Purification and characterization of recombinant Sc3T. A: SDS-PAGE of purified
Sc3T stained with Coomassie Blue. Protein size markers are annotated. B:
Elution profile of Sc3T from the Sephacryl S200 gel-filtration column relative to
the protein standards: bovine serum albumin (BSA), ovalbumin (OVA),
carbonic anhydrase (CAH), and cytochrom c (CYT). Inset: Determination of the
Stokes radius (RST) using the same protein standards (BSA: RST 5 35.6 A,
OVA: RST 5 29.8 A, CAH: RST 5 23.5 A, CYT: RST 5 17.7 A). Sc3T is
indicated by an arrow.
Structural Stability of a Domain of Ssn6
DOI 10.1002/prot PROTEINS 75
from 20 to 1008C with TM of �588C, suggested that even
the formed a-helices are relatively weakly stabilized by ter-
tiary interactions in Sc3T.
To further investigate the partial unfolding hypothesis,
we performed far-UV CD experiments in the presence of
trifluoroethanol (TFE). TFE, at moderate concentrations,
induces a-helical structure in peptides and proteins with
propensity for a-helix formation.41 As shown in Figure
3, addition of TFE raised the a-helical content of Sc3T as
indicated by the deeper minima at 208 and 222 nm. At
TFE concentrations up to 20%, the increase of the ellip-
ticity minima was accompanied by a red shift of the par-
allel band at 208 nm and an inversion of the [y]222/[y]208ratio indicative of associating a-helices in a coiled-coil
manner.42,43 Notably, by using TFE concentrations
higher than 30%, the ellipticity minima were blue shifted
and the [y]222/[y]208 ratio changed back to less than 1.0
(Fig. 3), suggesting disruption of hydrophobic interac-
tions between individual helices, as previously reported
for other a-helical protein fragments at high TFE con-
centrations.44 These observations clearly indicated that
Sc3T, although competent to fold in to an a-helicalcoiled structure as expected for a canonical TPR domain,
is only partially folded.
The unfolded region of Ssn6 involves TPR1
The evidence for lack of proper structure prompted us
to investigate the conformational state of Sc3T by estimat-
ing its hydrodynamic radius (the Stokes radius) using gel-
filtration chromatography. By comparing the mobility of
Sc3T to those of a series of standard proteins on a
Sephacryl S-200 column the Stokes radius (RST) of Sc3T
was estimated to be 23.1 A [Fig. 2(B), Inset]. This value is
more close to the RST value expected for the native state
of Sc3T (22.16 A) as calculated using Uversky’s equa-
tions45 for a 21.7 kDa polypeptide. This finding indicated
that the partial unfolding of Sc3T is relatively confined.
To map the unstructured region of Ssn6 we used lim-
ited proteolysis, an approach widely used for mapping
lack of structure in proteins.46 The limited proteolysis
mapping was performed on a Ssn6 protein fragment
(Sc10T; aa: 1–415) containing the entire TPR domain
(TPR1 to TPR10) followed by 15 additional amino acids
corresponding to the ‘‘solvating helix’’ considered to be
essential for the solubility or stability of isolated TPR
domains.8,47 As shown in Figure 4, chymotrypsin
digested Sc10T rapidly, resulting in mainly two distinct
proteolytically resistant fragments. N-terminal amino
acid sequence analysis of the larger chymotryptic product
(fragment 1 in Fig. 4, apparent molecular weight of 45
kDa) revealed a mixture of two fragments starting by the
N-terminal sequences ‘‘DATLQFN’’ and ‘‘AYDATLQ’’
with 65 and 35% abundance, respectively. These chymo-
tryptic products were apparently generated upon cleavage
at the bulky hydrophobic residues Tyr69 (TPR1 position
24) and Met67 (TPR1 position 23), respectively [Fig.
1(B)], indicating that the anticipated TPR1 helix B is
Figure 3CD spectra of Sc3T. Far-UV CD spectra of 13.8 lM protein recorded at 208C in
50 mM phosphate buffer (pH 7.5) and in the absence (thin line) and presence
of 10% (-n-), 20% (-l-), 30% (-h-), and 50% (-*-) trifluoroethanol (TFE).
Inset: Thermal denaturation curve of Sc3T (at 30 lM protein concentration) as
monitored by CD spectroscopy at 222 nm. Thermal denaturation extends in a
broad temperature range (20–908C) indicative of noncooperative unfolding.
Figure 4Limited proteolysis mapping of Sc10T. Chymotrypsin digestion time course of
Sc10T (right lanes). For comparison, control Sc10T (left lanes) was also loaded
for the times indicated. Protein size markers are annotated. Labeled arrows
indicate the two proteolytically resistant fragments.
M. Palaiomylitou/A. Tartas et al.
76 PROTEINS DOI 10.1002/prot
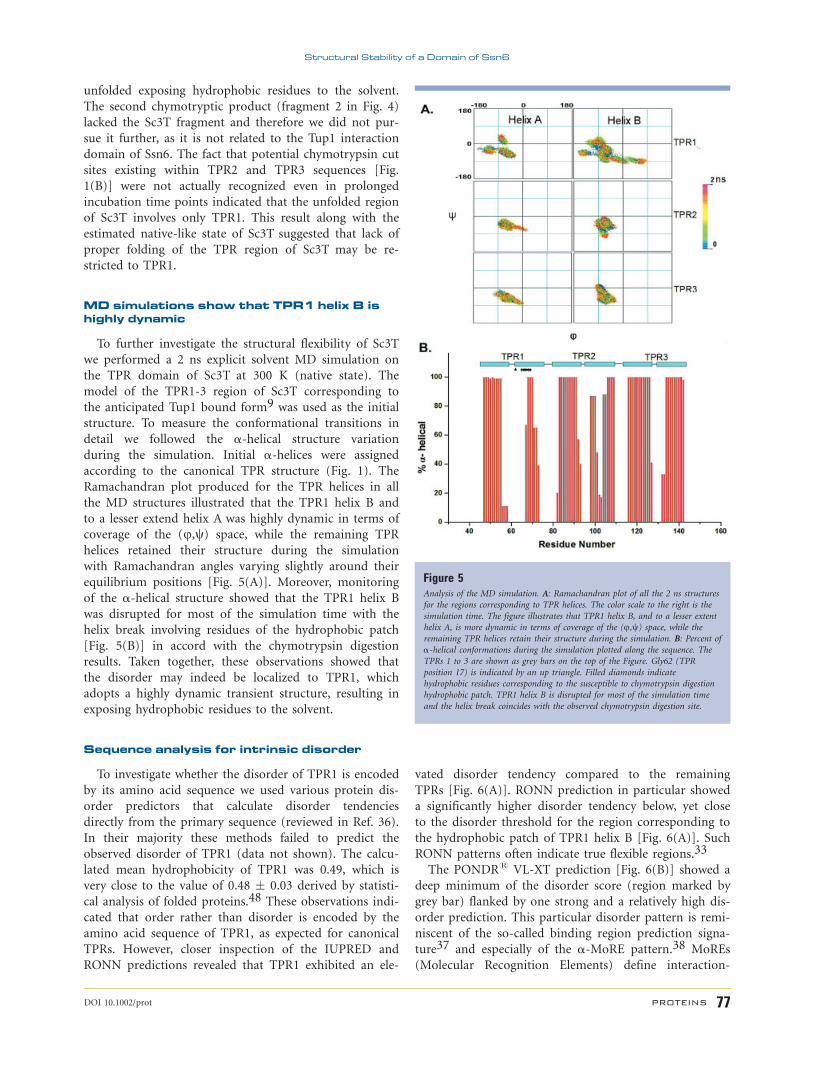
unfolded exposing hydrophobic residues to the solvent.
The second chymotryptic product (fragment 2 in Fig. 4)
lacked the Sc3T fragment and therefore we did not pur-
sue it further, as it is not related to the Tup1 interaction
domain of Ssn6. The fact that potential chymotrypsin cut
sites existing within TPR2 and TPR3 sequences [Fig.
1(B)] were not actually recognized even in prolonged
incubation time points indicated that the unfolded region
of Sc3T involves only TPR1. This result along with the
estimated native-like state of Sc3T suggested that lack of
proper folding of the TPR region of Sc3T may be re-
stricted to TPR1.
MD simulations show that TPR1 helix B ishighly dynamic
To further investigate the structural flexibility of Sc3T
we performed a 2 ns explicit solvent MD simulation on
the TPR domain of Sc3T at 300 K (native state). The
model of the TPR1-3 region of Sc3T corresponding to
the anticipated Tup1 bound form9 was used as the initial
structure. To measure the conformational transitions in
detail we followed the a-helical structure variation
during the simulation. Initial a-helices were assigned
according to the canonical TPR structure (Fig. 1). The
Ramachandran plot produced for the TPR helices in all
the MD structures illustrated that the TPR1 helix B and
to a lesser extend helix A was highly dynamic in terms of
coverage of the (u,w) space, while the remaining TPR
helices retained their structure during the simulation
with Ramachandran angles varying slightly around their
equilibrium positions [Fig. 5(A)]. Moreover, monitoring
of the a-helical structure showed that the TPR1 helix B
was disrupted for most of the simulation time with the
helix break involving residues of the hydrophobic patch
[Fig. 5(B)] in accord with the chymotrypsin digestion
results. Taken together, these observations showed that
the disorder may indeed be localized to TPR1, which
adopts a highly dynamic transient structure, resulting in
exposing hydrophobic residues to the solvent.
Sequence analysis for intrinsic disorder
To investigate whether the disorder of TPR1 is encoded
by its amino acid sequence we used various protein dis-
order predictors that calculate disorder tendencies
directly from the primary sequence (reviewed in Ref. 36).
In their majority these methods failed to predict the
observed disorder of TPR1 (data not shown). The calcu-
lated mean hydrophobicity of TPR1 was 0.49, which is
very close to the value of 0.48 � 0.03 derived by statisti-
cal analysis of folded proteins.48 These observations indi-
cated that order rather than disorder is encoded by the
amino acid sequence of TPR1, as expected for canonical
TPRs. However, closer inspection of the IUPRED and
RONN predictions revealed that TPR1 exhibited an ele-
vated disorder tendency compared to the remaining
TPRs [Fig. 6(A)]. RONN prediction in particular showed
a significantly higher disorder tendency below, yet close
to the disorder threshold for the region corresponding to
the hydrophobic patch of TPR1 helix B [Fig. 6(A)]. Such
RONN patterns often indicate true flexible regions.33
The PONDR1 VL-XT prediction [Fig. 6(B)] showed a
deep minimum of the disorder score (region marked by
grey bar) flanked by one strong and a relatively high dis-
order prediction. This particular disorder pattern is remi-
niscent of the so-called binding region prediction signa-
ture37 and especially of the a-MoRE pattern.38 MoREs
(Molecular Recognition Elements) define interaction-
Figure 5Analysis of the MD simulation. A: Ramachandran plot of all the 2 ns structures
for the regions corresponding to TPR helices. The color scale to the right is the
simulation time. The figure illustrates that TPR1 helix B, and to a lesser extent
helix A, is more dynamic in terms of coverage of the (u,w) space, while theremaining TPR helices retain their structure during the simulation. B: Percent of
a-helical conformations during the simulation plotted along the sequence. The
TPRs 1 to 3 are shown as grey bars on the top of the Figure. Gly62 (TPR
position 17) is indicated by an up triangle. Filled diamonds indicate
hydrophobic residues corresponding to the susceptible to chymotrypsin digestion
hydrophobic patch. TPR1 helix B is disrupted for most of the simulation time
and the helix break coincides with the observed chymotrypsin digestion site.
Structural Stability of a Domain of Ssn6
DOI 10.1002/prot PROTEINS 77

prone short segments of disorder that become ordered
upon specific binding to their biological partner.49 a-MoREs in particular define those MoREs that form a-helical structures upon binding. Interestingly, the chymo-
trypsin sensitive region (see above) overlaps with the pre-
dicted a-MoRE, suggesting that TPR1 may indeed be
part of a molecular recognition element that undergoes a
disorder-to-helix transition upon binding to a protein
partner. Given the requirement of the structural integrity
of TPR1.5 for the Ssn6-Tup1 interaction,9 we reasoned
that such a conformational transition might be possibly
induced by Tup1, the biological partner of Ssn6.
Tup1 interaction with Ssn6 is accompaniedby a conformational change
To test whether conformational changes occur upon
the interaction of Ssn6 with Tup1 we produced an Ssn6-
Tup1 protein complex in vitro [Fig. 7(A), see legend for
details] and studied its secondary structure by CD [Fig.
7(B)]. The complex was formed by Sc3T and a Tup1
derivative (Tn108; aa:1–108) comprising the Ssn6 inter-
action domain. The far UV CD spectrum of the Sc3T-
Tn108 complex [Fig. 7(B), filled squares] exhibited a
maximum at 195 nm and deep minima at 209 and 222
nm indicative of a highly a-helical structure.Towards the calculation of the expected CD spectrum
of the Sc3T-Tn108 complex, the individual far-UV CD
spectrum of Tn108 was also recorded [Fig. 7(B), open
circles]. Deconvolution of this spectrum gave a high a-helical content (64%) for Tn108. This observation com-
Figure 6Order/disorder predictions on various fragments of Ssn6 sequence comprising the
Tup1 interaction domain. A: Disorder tendencies predicted by the IUPred and
RONN disorder predictors. The threshold 0.5 distinguishing ordered from
disordered regions is marked by a dotted line. B: PONDR1 VL-XT predictions.
A predicted a-MoRE (see text) is indicated by a grey bar. The black bar
indicates the sensitive to chymotrypsin digestion hydrophobic patch.
Figure 7In vitro production and characterization of the Sc3T-Tn108 complex. A:
Production of the complex. Lane 1: Glutathione Sepharose (GH-S) beads with
bound GST-Sc3T. Lane 2: purified Tn108. Lanes 3 and 4: GH-S beads with
prebound GST-ScB and flow-through, respectively, obtained by centrifugation
after interaction of the beads with Tn108 followed by extensive washing. Tn108
is present on the beads indicating that formation of the GST-Sc3T-Tn108
complex has been achieved. The presence of unbound Tn108 in the flow-through
indicates that the saturation of Sc3T is complete. Lane 5: Supernatant obtained
by centrifugation of the beads with bound GST-Sc3T-Tn108 following thrombin
digestion. Only the bands corresponding to Sc3T and Tn108 are present in the
supernatant, indicating a high purity of the resulting Sc3T-Tn108 complex.
Lanes 6 and 7: Flow-through and GH-S beads following interaction of Tn108
with beads with prebound GST. Tn108 is fully recovered on the flow-through
indicating that the protein does not interact with control beads or the GST. B:
CD spectra. Far-UV CD spectrum of the Sc3T-Tn108 complex recorded at 208Cin phosphate buffer (pH 7.5). The calculated spectrum of the complex (see
Methods) as well as the individual Sc3T (as in Fig. 3 without TFE) and Tn108
(in 50 mM phosphate buffer, pH 7.5 and 150 mM NaCl) far-UV CD spectra
used for its calculation are also shown for comparison. The difference between
the observed and expected CD spectra of the complex indicates that a
conformational change accompanies the Ssn6-Tup1 interaction.
M. Palaiomylitou/A. Tartas et al.
78 PROTEINS DOI 10.1002/prot

bined with the [y]222/[y]208 > 1 ratio [Fig. 7(B), open
circles] indicated that Tn108 adopts a predominant a-helical coiled-coil structure, consistent with previous
studies on a similar Tup1 fragment.50 This observation
provided additional evidence that in contrast to Sc3T, the
Tup1 fragment is completely folded in isolation.
Comparison of the far-UV CD spectrum of the Sc3T-
Tn108 complex with the spectrum produced by a summa-
tion of the individual Sc3T and Tn108 CD spectra revealed
major changes: deepening of the ellipticity minima of the
observed spectrum relatively to the expected accompanied
by a red shift of the parallel band (at 208 nm) and inversion
of the [y]222/[y]208 ratio [Fig. 7(B)]. This change of the CD
spectrum pattern reminiscent of the effects produced by
TFE (at moderate concentrations) provided strong evidence
that a conformational change involving acquisition of a-helical structure with simultaneous stabilization of an a-helical coiled-coil configuration occurs upon the Ssn6-
Tup1 interaction. The above result supports the idea of an
induced fit mechanism in agreement with both the TFE
experiments and the PONDR VL-XT predictions.
DISCUSSION
Ssn6 TPR1 structure flexibility, in isolation
In a previous study9 we suggested that the Ssn6-Tup1
interaction may have a hydrophobic character and in this
respect it is rather unique among known TPR-mediated
interactions. In the present study we initiated a structural
analysis of the Tup1 interaction domain of Ssn6 by using CD
spectroscopy. First, we found that, in isolation this domain
of Ssn6 is only partially folded, adopting a loose flexible
structure. Subsequent limited proteolysis experiments using
the entire TPR domain of Ssn6 showed that the unstructured
region involves TPR1. This observation is consistent with
hierarchical unfolding with faster exchange in the terminal
repeats than in the central ones observed in TPR51 and
other repeat (ankyrin) containing proteins.52,53 However,
the importance of the structural integrity of TPR1 for Tup1
binding9,10 raised the possibility that proper TPR1 structure
might be induced only upon binding to Tup1. Consistent
with this, there is now increasing experimental evidence for
structural flexibility of repeat domains in the absence of their
ligand.52,54,55 Cliff et al.55 provided evidence for a confor-
mational change upon ligand binding for the TPR domain
of the Pp5 protein and proposed that folding coupled to
binding might be a common mechanism of ligand recogni-
tion by TPR domains although this idea has been debated.56
The disorder of TPR1 may be necessary forthe recognition of Ssn6 by intrinsichydrophobic coiled-coil residues of Tup1
Intrinsic disorder has been now recognized to many
protein domains and has been proposed to be essential
for biological function57–59 and especially for molecular
recognition.60 A signature of a probable intrinsically dis-
ordered region is the presence of low sequence complex-
ity coupled with an amino acid composition bias, charac-
terized by a low content of bulky hydrophobic amino
acids.34,61 However, our chymotrypsin digestion experi-
ments in conjunction with MD simulations showed that
the disordered domain of TPR1 includes a region rich in
hydrophobic residues corresponding to its anticipated he-
lix B. PONDR VL-XT disorder/order predictions identi-
fied this region as being part of a potential molecular
recognition element that may undergo disorder-to-order
transition upon binding to a specific biological partner.
The apolar character of this region supports our previous
suggestion that the Ssn6-Tup1 binding may be mediated
by hydrophobic interactions.9 In further support of
this idea, hydrophobic cluster analysis62 on the Tup1
sequence revealed that Leu62, mutation of which (L62R)
disrupts the interaction with Ssn6,63 is part of a heptad
repeat occupying intrinsic hydrophobic position d (Sup-
plementary Fig. S1). Notably, the L62R mutation does
not affect the coiled-coil structure of Tup1 or its ability
to form tetramers50,63 implying an important role of
hydrophobic heptad positions of Tup1 around Leu62 in
Ssn6 interaction rather than in self association.
The observation that Leu62 occupies an intrinsic
hydrophobic coiled-coil position combined with the non-
native structure of TPR1 raised the possibility that the
structural flexibility of this Ssn6 region may be necessary
for exposing an apolar surface to the solvent in order to
be recognized by Tup1 hydrophobic coiled-coil residues.
Such recognition mechanism has been revealed for the
first time in the case of a coiled-coil chaperone and has
been proposed to extend to other coiled-coil proteins
that interact with molecules exposing an apolar sur-
face.64 Consistent with this postulation, the aminoacid
sequence of the TPR1 helix B does not conform to the
consensus at various positions (black shaded in Fig. 1).
Namely, position 17 although occupied by bulky hydro-
phobic residues in canonical TPRs,8 is occupied by a Gly
in TPR1 [Fig. 1(B)] probably for facilitating the struc-
tural flexibility of this region and the exposure of hydro-
phobic residues to the solvent. Moreover, the enrichment
of the TPR1 helix B in nonconsensus alanine residues
[Fig. 1(B)] results in a relatively low complexity of this
region probably adding to its ability to exhibit the pre-
sumably necessary structural plasticity. Finally, the struc-
tural flexibility of TPR1 may be facilitated further by the
flexible, low complexity (poly-Gln) N-terminus (Fig. 1).
Possible involvement of the convex TPRsurface of Ssn6 in Tup1 binding
The present study suggested a direct role of TPR1 he-
lix B in Tup1 binding. In further support to this postu-
lation, two disruptive mutations of Tup1 interaction
Structural Stability of a Domain of Ssn6
DOI 10.1002/prot PROTEINS 79

concern Ala68,9 which is located at TPR1 helix B [indi-
cated with asterisks in Fig. 1(B)]. This site corresponds
to an external TPR position (position 23)8 and therefore
a mutation at this position is anticipated to affect Tup1
binding directly than the structural integrity of Ssn6.
Involvement of the TPR1 helix B in turn suggests for
the first time involvement of the convex TPR surface
(Fig. 8) in binding of this particular partner of Ssn6
consistent with the notion that interaction through the
concave surface is not a common feature of TPR-medi-
ated interactions.18 Dedication of the convex surface to
Tup1 binding may leave the concave TPR surface avail-
able for other partners of Ssn6 that interact with this
region, as is for example a2.65
Coupled folding and binding
More importantly and consistent with the idea of
coupled folding and binding, we provided evidence that
the formation of the Ssn6-Tup1 (Sc3T-Tn108) complex is
accompanied by a conformational change involving ac-
quisition of a-helical structure and stabilization of a
more compact a-helical coil structure. The increased hel-
icity detected upon complex formation combined with
the observation that similar Tup1 fragments comprising
its Ssn6 interaction domain are fully folded in isolation
(Ref. 50 and this study) suggested that the a-helix forma-
tion in the complex occurs largely within Ssn6. This ob-
servation strongly supports the idea of coupled folding
and binding in Ssn6 suggesting that such a mechanism may
indeed be more common in TPR domains as proposed by
Cliff et al.55 Moreover, coupled folding and binding is now
becoming recognized as a relatively common feature in
diverse regulatory proteins.58,60 Disorder-to-helix transi-
tions, in particular, have been predicted to be quite frequent
in various protein interaction networks.66
CONCLUSIONS
The structural flexibility of the TPR domain of Ssn6
we observed in this study may be crucial for its function.
We propose that TPR1 needs to display some flexibility
in isolation in order to be recognized by Tup1 among
the wide range of biological partners available to Ssn6.
This structural versatility may provide the means for an
easy turnover of the repression mechanism by the Ssn6-
Tup1 complex.
Moreover, the induced fit mechanism we observed
provides additional support of the idea of coupled fold-
ing and binding being a more common feature of TPR
mediated interactions.
ACKNOWLEDGMENTS
M.P and A.T acknowledge support from NCSR
‘‘Demokritos’’ Fellowship Programs. The use of the facili-
ties of the Centre for Crystallographic Studies of Macro-
molecules of NCSR ‘‘Demokritos’’ is acknowledged as
well as N. Gounalaki for plasmid construction. PONDR
was provided by Molecular Kinetics, 6201 La Pas Trail,
Suite 160, Indianapolis, IN 46268 (main@molecularkine-
tics.com). The MOE software was available from Chemi-
cal Computing Group Inc., 1010 Sherbrooke Street
West, Suite 910, Montreal, Canada H3A 2R7, http://www.
chemcomp.com.
REFERENCES
1. Varanasi US, Klis M, Mikesell PB, Trumbly RJ. The Cyc8 (Ssn6)-
Tup1 corepressor complex is composed of one Cyc8 and four Tup1
subunits. Mol Cell Biol 1996;16:6707–6714.
2. Smith RL, Redd MJ, Johnson AD. The tetratricopeptide repeats of
Ssn6 interact with the homeo domain of a 2. Genes Dev 1995;
9:2903–2910.
3. Williams FE, Varanasi U, Trumbly RJ. The CYC8 and TUP1 pro-
teins involved in glucose repression in Saccharomyces cerevisiae are
associated in a protein complex. Mol Cell Biol 1991;11:3307–3316.
4. Keleher CA, Redd MJ, Schultz J, Carlson M, Johnson AD. Ssn6-
Tup1 is a general repressor of transcription in yeast. Cell 1992;
68:709–719.
5. Tzamarias D, Struhl K. Functional dissection of the yeast Cyc8-
Tup1 transcriptional co-repressor complex. Nature 1994;369:758–
761.
6. Grbavec D, Lo R, Liu Y, Greenfield A, Stifani S. Groucho/transducin-
like enhancer of split (TLE) family members interact with the yeast
transcriptional co-repressor SSN6 and mammalian SSN6-related
proteins: implications for evolutionary conservation of transcription
repression mechanisms. Biochem J 1999;337 (Part 1):13–17.
Figure 8Molecular surface of a 3D model of the Ssn6 TPR domain. The model was
produced based on the canonical structure of tandem TPRs and is illustrated as
a ribbon model. Residues, mutations of which affect the TPR structure of Ssn6
[see also Fig. 1(B)], are depicted as ball and sticks. A disruptive mutation site
[Ala68, see also Fig. 1(B)] anticipated to affect Tup1 binding directly is colored
in red and indicated by an arrow. The susceptible to chymotrypsin digestion
hydrophobic patch is colored in green. Both the Tup1 interaction disruptive
mutation and the chymotrypsin site map on the convex surface of the TPR
structure of Ssn6 suggesting a direct role of this surface in Tup1 binding, in
contrast to TPR-mediated interaction modes reported so far.
M. Palaiomylitou/A. Tartas et al.
80 PROTEINS DOI 10.1002/prot

7. Sikorski RS, Boguski MS, Goebl M, Hieter P. A repeating amino
acid motif in CDC23 defines a family of proteins and a new rela-
tionship among genes required for mitosis and RNA synthesis. Cell
1990;60:307–317.
8. Main ER, Xiong Y, Cocco MJ, D’Andrea L, Regan L. Design of sta-
ble a-helical arrays from an idealized TPR motif. Structure 2003;
11:497–508.
9. Gounalaki N, Tzamarias D, Vlassi M. Identification of residues in
the TPR domain of Ssn6 responsible for interaction with the Tup1
protein. Febs Lett 2000;473:37–41.
10. Limbach MP, Zitomer RS. The isolation and characterization of
missense mutants in the general repressor protein Ssn6 of Saccharo-
myces cerevisiae. Mol Gen Genet 2000;263:455–462.
11. Magliery TJ, Regan L. Beyond consensus: statistical free energies
reveal hidden interactions in the design of a TPR motif. J Mol Biol
2004;343:731–745.
12. Goebl M, Yanagida M. The TPR snap helix: a novel protein repeat
motif from mitosis to transcription. Trends Biochem Sci 1991;
16:173–177.
13. Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interac-
tions: to TPR or not to TPR? Trends Biochem Sci 1995;20:257–259.
14. Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif
mediating protein-protein interactions. Bioessays 1999;21:932–939.
15. Tzamarias D, Struhl K. Distinct TPR motifs of Cyc8 are involved in
recruiting the Cyc8-Tup1 corepressor complex to differentially regu-
lated promoters. Genes Dev 1995;9:821–831.
16. Das AK, Cohen PW, Barford D. The structure of the tetratricopep-
tide repeats of protein phosphatase 5: implications for TPR-medi-
ated protein-protein interactions. EMBO J 1998;17:1192–1199.
17. Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bar-
tunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide
complexes: critical elements in the assembly of the Hsp70-Hsp90
multichaperone machine. Cell 2000;101:199–210.
18. Lapouge K, Smith SJ, Walker PA, Gamblin SJ, Smerdon SJ, Rittinger
K. Structure of the TPR domain of p67phox in complex with
Rac.GTP. Mol Cell 2000;6:899–907.
19. Gatto GJ, Jr, Geisbrecht BV, Gould SJ, Berg JM. Peroxisomal target-
ing signal-1 recognition by the TPR domains of human PEX5. Nat
Struct Biol 2000;7:1091–1095.
20. Taylor P, Dornan J, Carrello A, Minchin RF, Ratajczak T, Walkinshaw
MD. Two structures of cyclophilin 40: folding and fidelity in the TPR
domains. Structure 2001;9:431–438.
21. Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith
DF, Clardy J. Structure of the large FK506-binding protein FKBP51,
an Hsp90-binding protein and a component of steroid receptor
complexes. Proc Natl Acad Sci USA 2003;100:868–873.
22. Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B, Rao
Z. 3D structure of human FK506-binding protein 52: implications for
the assembly of the glucocorticoid receptor/Hsp90/immunophilin
heterocomplex. Proc Natl Acad Sci USA 2004;101:8348–8353.
23. Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti
E. The superhelical TPR-repeat domain of O-linked GlcNAc trans-
ferase exhibits structural similarities to importin a. Nat Struct Mol
Biol 2004;11:1001–1007.
24. Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou
C, Pearl LH. Chaperoned ubiquitylation—crystal structures of the
CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a com-
plex. Mol Cell 2005;20:525–538.
25. Wilson CG, Kajander T, Regan L. The crystal structure of NlpI. A
prokaryotic tetratricopeptide repeat protein with a globular fold.
FEBS J 2005;272:166–179.
26. Wu Y, Sha B. Crystal structure of yeast mitochondrial outer mem-
brane translocon member Tom70p. Nat Struct Mol Biol 2006;13:
589–593.
27. Cliff MJ, Harris R, Barford D, Ladbury JE, Williams MA. Confor-
mational diversity in the TPR domain-mediated interaction of pro-
tein phosphatase 5 with Hsp90. Structure 2006;14:415–426.
28. Siegel LM, Monty KJ. Determination of molecular weights and fric-
tional ratios of proteins in impure systems by use of gel filtration
and density gradient centrifugation. Application to crude prepara-
tions of sulfite and hydroxylamine reductases. Biochim Biophys
Acta 1966;112:346–362.
29. Zeev-Ben-Mordehai T, Rydberg EH, Solomon A, Toker L, Auld VJ,
Silman I, Botti S, Sussman JL. The intracellular domain of the Dro-
sophila cholinesterase-like neural adhesion protein, gliotactin, is
natively unfolded. Proteins 2003;53:758–767.
30. Bohm G, Muhr R, Jaenicke R. Quantitative analysis of protein far
UV circular dichroism spectra by neural networks. Protein Eng
1992;5:191–195.
31. Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary
structure of proteins from UV circular dichroism spectra using an
unsupervised learning neural network. Protein Eng 1993;6:383–390.
32. Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for
the prediction of intrinsically unstructured regions of proteins
based on estimated energy content. Bioinformatics 2005;21:3433–
3434.
33. Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis
function neural network technique applied to the detection of
natively disordered regions in proteins. Bioinformatics 2005;21:
3369–3376.
34. Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK.
Sequence complexity of disordered protein. Proteins 2001;42:38–48.
35. Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting pro-
tein disorder for N-, C-, and Internal Regions. Genome Inform Ser
Workshop Genome Inform 1999;10:30–40.
36. Ferron F, Longhi S, Canard B, Karlin D. A practical overview of
protein disorder prediction methods. Proteins 2006;65:1–14.
37. Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predict-
ing binding regions within disordered proteins. Genome Inform Ser
Workshop Genome Inform 1999;10:41–50.
38. Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker
AK. Coupled folding and binding with a-helix-forming molecular
recognition elements. Biochemistry 2005;44:12454–12470.
39. Kabsch W, Sander C. Dictionary of protein secondary structure:
pattern recognition of hydrogen-bonded and geometrical features.
Biopolymers 1983;22:2577–2637.
40. Privalov PL. Stability of proteins. Proteins which do not present a
single cooperative system. Adv Protein Chem 1982;35:1–104.
41. Dyson HJ, Rance M, Houghten RA, Wright PE, Lerner RA. Folding
of immunogenic peptide fragments of proteins in water solution. II.
The nascent helix. J Mol Biol 1988;201:201–217.
42. Cooper TM, Woody RW. The effect of conformation on the CD of
interacting helices: a theoretical study of tropomyosin. Biopolymers
1990;30:657–676.
43. Gibney BR, Johansson JS, Rabanal F, Skalicky JJ, Wand AJ, Dutton
PL. Global topology & stability and local structure & dynamics in a
synthetic spin-labeled four-helix bundle protein. Biochemistry
1997;36:2798–2806.
44. Zhou NE, Kay CM, Hodges RS. Synthetic model proteins. Positional
effects of interchain hydrophobic interactions on stability of two-
stranded a-helical coiled-coils. J Biol Chem 1992;267:2664–2670.
45. Uversky VN. What does it mean to be natively unfolded? Eur J Bio-
chem 2002;269:2–12.
46. Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin
M. Probing protein structure by limited proteolysis. Acta Biochim
Pol 2004;51:299–321.
47. D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends
Biochem Sci 2003;28:655–662.
48. Uversky VN, Gillespie JR, Fink AL. Why are ‘‘natively unfolded’’
proteins unstructured under physiologic conditions? Proteins 2000;
41:415–427.
49. Bourhis JM, Johansson K, Receveur-Brechot V, Oldfield CJ, Dunker
KA, Canard B, Longhi S. The C-terminal domain of measles virus
nucleoprotein belongs to the class of intrinsically disordered pro-
Structural Stability of a Domain of Ssn6
DOI 10.1002/prot PROTEINS 81

teins that fold upon binding to their physiological partner. Virus
Res 2004;99:157–167.
50. Jabet C, Sprague ER, VanDemark AP, Wolberger C. Characterization
of the N-terminal domain of the yeast transcriptional repressor
Tup1. Proposal for an association model of the repressor complex
Tup1 x Ssn6. J Biol Chem 2000;275:9011–9018.
51. Main ER, Stott K, Jackson SE, Regan L. Local and long-range sta-
bility in tandemly arrayed tetratricopeptide repeats. Proc Natl Acad
Sci USA 2005;102:5721–5726.
52. Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA. Biophysi-
cal characterization of the free IjBa ankyrin repeat domain in solu-
tion. Protein Sci 2004;13:1767–1777.
53. Bradley CM, Barrick D. Limits of cooperativity in a structurally mod-
ular protein: response of the Notch ankyrin domain to analogous
alanine substitutions in each repeat. J Mol Biol 2002;324:373–386.
54. Beddoe T, Bushell SR, Perugini MA, Lithgow T, Mulhern TD,
Bottomley SP, Rossjohn J. A biophysical analysis of the tetratricopep-
tide repeat-rich mitochondrial import receptor, Tom70, reveals an
elongated monomer that is inherently flexible, unstable, and unfolds
via a multistate pathway. J Biol Chem 2004;279:46448–46454.
55. Cliff MJ, Williams MA, Brooke-Smith J, Barford D, Ladbury JE.
Molecular recognition via coupled folding and binding in a TPR
domain. J Mol Biol 2005;346:717–732.
56. Cortajarena AL, Regan L. Ligand binding by TPR domains. Protein
Sci 2006;15:1193–1198.
57. Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assess-
ing the protein structure-function paradigm. J Mol Biol 1999;293:
321–331.
58. Dyson HJ, Wright PE. Coupling of folding and binding for unstruc-
tured proteins. Curr Opin Struct Biol 2002;12:54–60.
59. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z.
Intrinsic disorder and protein function. Biochemistry 2002;41:6573–
6582.
60. Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic
disorder as an ID for recognition, regulation and cell signaling.
J Mol Recognit 2005;18:343–384.
61. Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein
disorder. Proteins 2003;52:573–584.
62. Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier
J, Henrissat B, Mornon JP. Deciphering protein sequence informa-
tion through hydrophobic cluster analysis (HCA): current status
and perspectives. Cell Mol Life Sci 1997;53:621–645.
63. Carrico PM, Zitomer RS. Mutational analysis of the Tup1 general
repressor of yeast. Genetics 1998;148:637–644.
64. Lundin VF, Stirling PC, Gomez-Reino J, Mwenifumbo JC, Obst JM,
Valpuesta JM, Leroux MR. Molecular clamp mechanism of sub-
strate binding by hydrophobic coiled-coil residues of the archaeal
chaperone prefoldin. Proc Natl Acad Sci USA 2004;101:4367–
4372.
65. Smith RL, Johnson AD. A sequence resembling a peroxisomal tar-
geting sequence directs the interaction between the tetratricopeptide
repeats of Ssn6 and the homeodomain of a 2. Proc Natl Acad Sci
USA 2000;97:3901–3906.
66. Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN.
Flexible nets. The roles of intrinsic disorder in protein interaction
networks. FEBS J 2005;272:5129–5148.
M. Palaiomylitou/A. Tartas et al.
82 PROTEINS DOI 10.1002/prot
Copyright © 2022 FDOKUMEN




















