
Investigating SPTLC1 mutations on protein and lipid profiles in ...
179
Investigating mitochondrial and ER protein profiles of cells expressing SPTLC1 mutations Scott Stimpson Thesis submitted for the award of Doctor of Philosophy Supervisor: Dr. Simon Myers Associate Supervisor: Prof. Jens Coorssen Associate Supervisor: Assoc. Prof. Paul Witting Neuro-Cell Biology Laboratory Molecular Medicine Research Group School of Science and Health Western Sydney University Australia
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Investigating SPTLC1 mutations on protein and lipid profiles in ...

Investigating mitochondrial and ER protein profiles of cells expressing
SPTLC1 mutations
Scott Stimpson
Thesis submitted for the award of
Doctor of Philosophy
Supervisor: Dr. Simon Myers
Associate Supervisor: Prof. Jens Coorssen
Associate Supervisor: Assoc. Prof. Paul Witting
Neuro-Cell Biology Laboratory
Molecular Medicine Research Group
School of Science and Health
Western Sydney University
Australia

ii | P a g e
STATEMENT OF AUTHENTICATION
I Scott Stimpson declare that this thesis contains no material that has been accepted
for the award of any other degree or diploma and that, to the best of my knowledge
and belief, this thesis contains no material previously published or written by another
person, except where due reference has been made in the text of this thesis.
August 2015
S.E. Stimpson BMedSci (Hons)

iii | P a g e
ABSTRACT Axonal degeneration is the final common path in many neurological disorders. It is
seen in its pure form in hereditary axonal neuropathies. The hereditary neuropathies
are the most common group of diseases. Subsets of neuropathies involving the
sensory neuron are known as hereditary sensory neuropathies (HSNs). Hereditary
sensory neuropathy type I (HSN-I) (the most common subtype of HSNs) is an
autosomal dominant inherited disorder, characterised by the progressive degeneration
of the dorsal root ganglion and with onset of clinical symptoms occurring between the
second or third decade of life. Heterozygous mutations in the serine
palmitoyltransferase (SPT) long chain subunit 1 (SPTLC1) have been identified as the
cause of HSN-I.
In Paper I, we optimised an isolation method of mitochondria to allow the production
of a full and in-depth proteomic profile to elucidate the molecular mechanisms
underlying mitochondrial (dys) function in HSN-I. Paper II, detailed examinations of a
small sub-set of proteins that were found to be altered in abundance within harvested
mitochondria from HSN-I mutant SPTLC1 cells. Comparison of mitochondrial protein
isolates from control and patient lymphoblasts, showed an increased abundance of
Ubiquinol Cytochrome C Reductase Core Protein 1, an electron-transport chain
protein, as well as the immunoglobulin, Ig Kappa Chain C. In, Paper III, endoplasmic
reticulum (ER) protein lysates from HSN-I patient and control lymphoblasts, were
examined leading to identification of changes in expression of five proteins; Hypoxia
Up regulated Protein 1, Chloride Intracellular Channel Protein 1, Ubiqutin-40s
Ribosomal Protein S27a, Coactosin and Ig Kappa chain C.

iv | P a g e
Further investigations into mitochondrial and ER protein profiles were carried out in
Paper IV, which showed a number of proteins that were altered in their relative
abundance using membrane and soluble isolation techniques. Further analyses of
these identified changes were carried out and replicated in Paper V, which revealed
and confirmed the changes in protein expression and abundance of proteins earlier
identified in Papers I and II. Changes were identified in V144D mutations, as well as
C133W and C133Y mutations. All of which are implicated to be casual of HSN-I.
Lipid droplets and alterations of lipid metabolism are hallmarks of a variety autosomal
dominant neurodegenerative diseases, including Alzheimer’s and Parkinson’s
disease. Paper VI, revealed significant increases in the presence of lipid droplets in
HSN-I patient-derived lymphoblasts, indicating a potential connection between lipid
droplet formation and the molecular mechanisms of HSN-I.
In conclusion, this study has shown alteration in mitochondrial and ER protein profiles
in patient-derived lymphoblasts and in transfected neuronal cells expressing the
mutations V144D, C133W and C133Y. This investigation has contributed to the field
by identifying protein alterations which has yielded a more detailed and in-depth
analysis of the cellular and molecular mechanisms involved in HSN-I.

v | P a g e
THESIS STRUCTURE The work presented in this thesis provides an investigation into mitochondrial and
endoplasmic reticulum proteome changes caused by mutations in the SPTLC1 gene.
These investigations are provided as a series of six papers (listed below). These
papers are either published (Paper I, II, III, IV & VI), or currently submitted to journals
for peer-review (Paper V), and adverted to in the thesis text by their Roman numerals.
I. Stimpson, SE. Coorssen, JR. Myers, SJ. Optimal isolation of mitochondria for
proteomic analyses. Analytical Biochemistry: Methods in Biological Sciences,
2015. doi:10.1016/j.ab.2015.01.005
II. Stimpson, SE. Coorssen, JR. Myers, SJ. Mitochondrial protein alterations in a
familial peripheral neuropathy caused by mutations in the sphingolipid protein,
SPTLC1. J Chem Biol, 2014. 8 (1):25-35. Doi: 10.1007/s12154-014-0125-x.
III. Stimpson, SE. Lauto, A. Coorssen, JR. Myers, SJ. Isolation and identification of
ER associated proteins with unique expression changes specific to the V144D
SPTLC1 mutations in HSN-I. Biochem and Anal Biochem. 2016; 5 (1)
IV. Stimpson, SE. Coorssen, JR. Myers, SJ. Proteome alterations associated with the
V144D SPTLC1 mutation that causes Hereditary Sensory Neuropathy-I. Electronic
J Biol, 2015; 11 (4): 176-186
V. Stimpson, SE. Coorssen, JR. Myers, SJ. Identifying unique protein alterations
caused by SPTLC1 mutations in a transfected neuronal cell model. Proteomes.
2015 (Manuscript under Review).
VI. Marshall, LL*. Stimpson, SE*, Coorssen, JR and Myers, SJ. Increased lipid droplet
accumulation associated with a peripheral sensory neuropathy. J Chem Biol. 2014;
7(2):67-76. Doi: 10.1007 (* Co-first authors).

vi | P a g e
TABLE OF CONTENTS Abbreviated list of contents………………………………………..……………………… vi
Comprehensive list of contents………………….……………………..………………... vii
List of Figures………….………………………………………..……………..…….….. viii
List of Tables ...…………………..…………………………..………………….............viii
Acknowledgements..................................................................................................ix
List of Abbreviations………………..……..................................................................x

vii | P a g e
TABLE OF CONTENTS 1.1 Hereditary Sensory Neuropathies ............................................................................................... 1
1.1.1 What are hereditary sensory neuropathies? ...................................................................... 1
1.1.2 Clinical features of HSN-I ...................................................................................................... 5
1.1.3 Pathological features of HSN-I ............................................................................................. 5
1.1.4 Genetics of HSN-I .................................................................................................................. 6
1.2 Intracellular Lipid Functions and Interactions ............................................................................ 7
1.2.1 The role of lipids in biological membranes ......................................................................... 7
1.2.2 Interactions of lipids with intracellular proteins and organelles ....................................... 9
1.2.3 Sphingolipids, their role in normal cellular homeostasis and in the neuronal cell ...... 11
1.3 Serine Palmitoyltransferase ....................................................................................................... 13
1.3.1 The structure and regulation of SPT and its subunits ..................................................... 13
1.3.2 The role of the SPTLC1 protein in neurodegenerative disease .................................... 14
1.4 Protein Functions and Interactions ........................................................................................... 17
1.4.1 Altered protein expression and the disease state ........................................................... 17
1.5 Mitochondria in neurodegenerative diseases .......................................................................... 19
1.5.1 Mitochondrial Dynamics ...................................................................................................... 19
1.5.2 Mitochondrial fusion and fission ......................................................................................... 20
1.5.3 Mitochondrial changes in inherited peripheral neuropathies ......................................... 27
1.5.4 Examples of Mitochondrial changes in inherited peripheral .......................................... 29
Neuropathies ................................................................................................................................... 29
1.5.5 Novel link to Mitochondria in HSN-I ................................................................................... 32
1.6 The Role of ER in neurodegenerative diseases ..................................................................... 33
1.6.1 Protein processing in the ER .............................................................................................. 33
1.6.2 UPR: Unfolded protein response pathway ....................................................................... 37
1.7 Mitochondria-Associated Membranes ...................................................................................... 41
1.7.1 MAM Calcium Regulation .................................................................................................... 41
1.7.2 Autophagy formation at MAMs ........................................................................................... 42
1.7.3 Lipid transportation via MAMs ............................................................................................ 44
1.7.4 MAMs and Neurodegeneration .......................................................................................... 46
1.8 Aims of this Thesis ...................................................................................................................... 49
Paper I .................................................................................................................................................. 51
Paper II ................................................................................................................................................ 52
Paper III ............................................................................................................................................... 67

viii | P a g e
Paper IV ............................................................................................................................................... 76
Paper V ................................................................................................................................................ 88
Paper VI ............................................................................................................................................. 121
General Discussion .......................................................................................................................... 132
Future Directions .............................................................................................................................. 149
References ........................................................................................................................................ 156
LIST OF FIGURES
Figure 1.1: Molecular structure of the plasma membrane.......................................... 8
Figure 1.2: The pathway of sphingolipid metabolism............................................... 12
Figure 1.3: Theoretical model of SPT complex structure..........................................14
Figure 1.4: Schematic model of mitochondrial fusion................................................22
Figure 1.5: Schematic diagram for the proposed new model of fission.................... 24
Figure 1.6: Schematic diagram of mitosis-specific mitochondrial fission ................ 25
Figure 1.7: Overview of mitochondrial dynamics and homeostasis......................... 26
Figure 1.8: Cotranslational targeting of secretory proteins to the ER....................... 34
Figure 1.9: Posttranslational translocation of proteins into the ER........................... 35
Figure 1.10: ER stress and the unfolded protein response....................................... 39
LIST OF TABLES Table 1.1: Hereditary sensory neuropathies (HSNs) ................................................. 4
Table 1.2: The three subunits of SPT; SPTLC1, SPTLC2 and SPTLC3 .................. 14
Table 1.3: Neurodegenerative diseases that interfere with mitochondrial function ..31

ix | P a g e
ACKNOWLEDGEMENTS This thesis is the cumulative work of not only myself, but also of the numerous people
who have supported me and were supportive of me, during my degree. It has been
both whirlwind and arduous journey, of which I could not have travelled, if not for the
presence of my mentors and friends.
Firstly, I would like to thank my supervisors, Dr Simon Myers and Prof. Jens Coorssen.
Thank you both for all your time and effort throughout this entire project. Thanks also
go to Dr Chandra Malladi and Dr Elise Wright for their technical assistance on two-
dimensional gel electrophoresis and to Dr Cathy Luxford for your enduring guidance
and support.
Secondly, I owe large “thank yous” to the following people for their moral support,
intellectual discussions, technical assistance, motivation and friendship, Noor Jwad
and Melissa Partridge. Thirdly, to Chris, thank you for always believing in me and
supporting me through the good and bad times, for being my rock and guiding light.
To others whom I may have forgotten, I am sorry, but thank you for your contributions.
Finally, to my family, especially Mum and Dad, my gratitude extends beyond words.
Scott Stimpson August 2015

x | P a g e
ABBREVIATIONS 2DGE Two Dimensional Gel Electrophoresis
AD Alzheimer’s disease
ALS Amyotrophic Lateral Sclerosis
AMPIB Amresco Mitochondrial Protein Isolation Buffer
APP ß-Amyloid Precursor Protein
ATF4 Activating Transcription Factor 4
ATF6 Activating Transcription Factor 6
ATL3 Atlastin GTPase 3
ATG Autophagy-Related Proteins
ATP Adenosine Triphosphate
BiP Immunoglobulin Heavy Chain Binding Protein
C133W Cytosine to Tryptophan at Position 133
Ca2+ Calcium ion
CAG Cytosine-Adenine-Guanine
CDases Ceramidases
CH2 Collagen Homology Domain
CHO Chinese Hamster Ovary
CoA Coenzyme A
DCFP1 Double FYVE-Containing Protein 1
DNA Deoxyribonucleic Acid
DNMT1 DNA Methyl Transferase 1
DRG Dorsal Root Ganglion
DRP Dynamin-related Protein
DSBs Deoxy-Sphingoid Bases
eIF2a Eukaryotic Translation Initiation Factor 2 Subunit a
eIF5A Eukaryotic Translation Initiation Factor 5A-1
ER Endoplasmic Reticulum
ERAD Endoplasmic Reticulum Associated Degradation
FCCP Carbonyl Cyanide p-trifluoromethoxyphenylhydrazone

xi | P a g e
Fis1 Mitochondrial Fission Protein 1
GED GTPase Effector Domain
GPI Glycosylphosphatidylinositol
GRB2 Growth Factor Receptor-Binding Protein 2
GSL Glycosphingolipid
GTPase Guanine Triphosphatase
hFis1 Mitochondrial Fission Protein 1
HIV-1 Human Immunodeficiency Virus
HSANs Hereditary Sensory and Autonomic Neuropathies
HSNs Hereditary Sensory Neuropathies
HSN-I Hereditary Sensory Neuropathy Type 1
HSN-II Hereditary Sensory Neuropathy Type 2
HSN-III Hereditary Sensory Neuropathy Type 3
HSN-IV Hereditary Sensory Neuropathy Type 4
HSN-V Hereditary Sensory Neuropathy Type 5
Hsp70 Heat Shock Protein 70
IKBKAP Inhibitor of Kappa Light Polypeptide Enhancer in B-cells, Kinase Complex Associated Protein
IL-1b Interleukin 1b
IMM Inner Mitochondrial Membrane
IMS Inner Membrane Space
IP3 1, 4, 5- Trisphosphate
iPS Induced Pluripotent Stem Cells
IRE1 Inositol-Requiring Enzyme 1
LCB1 Long-Chain Base 1
MAM Mitochondrial Associated Membrane
Mff Mitochondrial Fusion Factor
Mfn2 Mitofusin 2
MIB Mitofusin Binding Protein
MiD49 Mitochondrial Dynamics Protein 49
MiD51 Mitochondrial Dynamics Protein 51

xii | P a g e
MiEF1 Mitochondrial Elongation Factor 1
mTOR Mechanistic Target of Rapamycin
NGF Nerve Growth Factor
NGFB Nerve Growth Factor Beta
NLRs NOD-like Receptors
OCR Oxygen Consumption Rate
OMM Outer Mitochondrial Membrane
PDI Protein Disulphide Isomerase
PE Phosphatidylethanolamine
PERK Protein Kinase-Like ER Kinase
PINK1 PTEN-induced Putative Kinase 1
PKA Cyclic-AMP-Dependent Protein Kinase
PS Phosphatidylserine
PSD PS Decarboxylase
RALA Ras-Related Protein Ral-A
ROS Reactive Oxygen Species
RER Rough Endoplasmic Reticulum
RNase Endoribonuclease
ScaMc-1 Calcium Binding Mitochondrial Carrier Protein
SL Sphingolipids
SKs Sphingosine Kinases
SOD1 Superoxide Dismutase-1
S1P Sphingosine-1-Phosphate
SPT Serine Palmitoyltransferase
SPTLC1 Serine Palmitoyltransferase Long Chain Subunit 1
SPTLC2 Serine Palmitoyltransferase Long Chain Subunit 2
SPTLC3 Serine Palmitoyltransferase Long Chain Subunit 3
SR Sarcoplasmic Reticulum
SREBP Sterol Response Element Binding Protein
SRP Signal Recognition Particle
TCA Tricarboxylic Acid Cycle

xiii | P a g e
TEM Transmission Electron Micrograph
TRKA Tyrosine Kinase A Receptor
TT Transiently Transfect
UCH-L1 Ubiquitin C-Terminal Esterase L1
UPR Unfolded Protein Response Pathway
UVB Ultraviolet Light B
VDAC Voltage Dependent Anion Selective Channel Protein 1
VAPB Vesicle-Associated Membrane Protein B
V144D Valine to Aspartate at position 144
WNK With No Lysine Kinase
WNK1 Lysine Deficient Protein Kinase 1
XBP1 X-Box-Binding Protein

1 | P a g e
1.1 Hereditary Sensory Neuropathies 1.1.1 What are hereditary sensory neuropathies?
Subsets of neuropathies involving the sensory neuron are categorised as hereditary
sensory neuropathies (HSNs) (Dyck et. al., 2005). These inherited nerve disorders in
which the sensory dysfunction of the neuron is most prevalent and involvement of the
autonomic system are referred to as hereditary sensory and autonomic neuropathies
(HSANs) (Dyck et. al., 2005). HSNs are associated with a range of clinical
presentations, pathologic alterations, electrophysiological abnormalities and
increasingly specific biochemical, molecular and/or genetic abnormalities,
summarised in Table 1.1 (Dyck et. al., 2005). HSNs have also been described with
sensorineural deafness, keratitis (inflamed corneas), ataxia (dysfunction of motor
coordination), spasticity (altered skeletal muscle performance), dementia and mental
retardation (Cavanagh, 1979; Janzer, 1986; Donaghy, 1978; Kherbaoui-Redouani,
2004). HSNs are a clinically and genetically heterogeneous group of disorders which
are classified into five different HSN subtypes labelled HSN I-V. With the exception of
HSN type I, which has an autosomal dominant trait, HSN II-V are autosomal recessive
traits (Verhoeven et. al., 2006). Molecular genetics research has shown that at least
eight loci and six genes are associated with HSNs (Verhoeven et. al., 2006).
Hereditary sensory neuropathy type I (HSN-I) is the most common subtype of HSNs
(Dyck et. al., 2005). As stated HSN-I is autosomal dominant inheritance and is
characterised by progressive degeneration of the dorsal root ganglion (DRG) and an
onset of clinical symptoms between the second or third decade of life (Verhoeven et.
al., 2004). HSN-I is rarely fatal but imposes lifelong disability with the disease initially
manifesting with sensory loss in the feet, followed by distal muscle wasting and

2 | P a g e
weakness and subsequent positive sensory phenomena (such as lancinating or
'shooting' pains). Heterozygous mutations in the serine palmitoyltransferase (SPT)
long chain subunit 1 (SPTLC1) were identified as the pathogenic cause of HSN-I
(Bejaoui et. al., 2001; Dawkins et. al., 2001).
HSN-II, also known as Morvan disease, is an autosomal recessive, early onset and
very severe disease with clinical symptoms appearing in early infancy (Verpoorten et.
al., 2006). It is manifested with sensory loss affecting all modalities but with touch
being most severely affected (Verpoorten et. al., 2006). A well-conserved 434- amino-
acid open reading frame located within intron 8 of the WNK (With no-lysine-kinase)
lysine deficient protein kinase 1 gene (WNK1) gene for HSN-II has been identified
(Rivie`re et. al., 2004; Roddier et. al., 2005).The mutations identified to date predict
truncation of the protein, suggesting a complete loss of function (Verpoorten et. al.,
2006).
HSN-III, also known as Familial Dysautonomia or Riley–Day syndrome, is an
autosomal recessive disorder that affects the development and survival of sensory,
sympathetic and some parasympathetic neurons (Houlden et. al., 2004). HSN-III
manifests a variety of symptoms, including decreased sensitivity to pain, vibration and
temperature, cardiovascular instability, recurrent pneumonias, vomiting crises and
gastrointestinal dysfunction (Houlden et. al., 2004). The HSN-III mutations were
located to chromosome 9q31, with sequencing identifying two mutations causing HSN-
III with the major haplotype mutation located on intron 20 involving the inhibitor of
kappa light polypeptide enhancer in B-lymphocytes, kinase complex associated
protein (IKBKAP) (Houlden et. al., 2004).

3 | P a g e
HSN-IV, Congenital Insensitivity to Pain with Anhidrosis, is an autosomal recessive
disorder manifesting with insensitivity to pain, recurrent febrile episodes, anhidrosis,
self-mutilating behaviour and mental retardation. The tyrosine kinase A receptor
(TRKA) gene was identified as causal for HSN-IV. Additional mutations were identified
and found to be caused by a 1926- ins-T mutation in the TRKA gene (Houlden et. al.,
2004). The mechanism underlying the development of HSN-IV in families with TRKA
mutations is currently unknown, but there are some in-vitro data implicating deficient
TRKA phosphorylation in neuronal and non-neuronal cells caused by mutations
(Houlden et. al., 2004).
HSN-V is a rare disorder, with very few reported cases. The disease is characterised
by loss of deep pain perception and impaired temperature sensitivity, ulcers, and in
some cases self-mutilation, with most other neurological functions including sweating
intact (Einarsdottir et. al., 2004). The genetic background of HSN-V is still unclear;
however Einarsdottir et. al., (2004) located a potential causative mutation on a
conserved region of the nerve growth factor beta gene (NGF) that produces the HSN-
V phenotype (Einarsdottir et. al., 2004).

4 | P a g e
HSN-I SPTLC1 9q22.2 Autosomal
Dominant Adulthood Sensory loss in the feet, followed by distal
muscle wasting and weakness and subsequent positive sensory phenomena, such as lancinating or 'shooting' pains.
(Bejaoui et. al., 2001; Dawkins et. al., 2001)
HSN-II HSN2 12p13.3 Autosomal Recessive
Childhood Distal sensory loss affecting all modalities, with touch most severely affected leading to amputations.
(Verpoorten et. al., 2006)
HSN-III IKBKAP 9q31 Autosomal Recessive
Congenital Impairment of development and survival of sensory, sympathetic and some parasympathetic neurons manifests a variety of symptoms, including decreased sensitivity to pain, vibration and temperature, cardiovascular instability, recurrent pneumonias, vomiting crises and gastrointestinal dysfunction
(Houlden et. al., 2004)
HSN-IV TRKA 1q21-22 Autosomal Recessive
Congenital Insensitivity to pain and temperature sensation with frequent bone and joint fractures.
(Houlden et. al., 2004)
HSN-V NGF 1p13.1 Autosomal Recessive
Congenital Distal loss of pain and temperature sensation leading to ulcers and self-mutilation.
(Einarsdottir, et. al., 2004)
Table 1.1 Hereditary sensory neuropathies (HSNs). HNSs are inherited nerve disorders in which the sensory dysfunction of the neuron is most prevalent with occasional involvement of the autonomic system. HSN-I is the most prevalent of these diseases. Adapted from (Verpoorten et. al., 2006)

5 | P a g e
1.1.2 Clinical features of HSN-I
HSN-I is the most common and best characterised of the degenerative sensory
disorders. Degeneration of the DRG and ventral horn neurons is typical with one report
of amyloid-like material accumulation within the DRG (Denny-Brown, 1951). Loss of
sensation results in painless injuries that when left untreated display slow wound
healing and can develop into osteomyelitis, often requiring amputation (Dyck et. al.,
2005; Auer-Grumbach et. al., 2003).
As the disease progresses into advanced stages, motor involvement with distal muscle
weakness and wasting becomes apparent and symptoms spread to the proximal limbs
(Auer-Grumbach et. al., 2008). These complications cause long-term disablement with
economic and social repercussions. Currently there is no cure for HSN-I and treatment
is entirely symptom-focused (Auger-Grumbach et. al., 2003).
1.1.3 Pathological features of HSN-I
The pathological features of HSN-I are associated with axonal degeneration ('dying
back') of the peripheral sensory fibres (Dyck et. al., 2005). Significant reductions in all
of the peripheral sensory fibres (small/large, myelinated/non-myelinated) in the distal
extremities have been observed through post mortem and electrophysiological
studies. Individual neurons undergo axonal atrophy sufficiently slowly that myelin
wrinkling and remodelling occur and eventually these axons further degenerate into
linear rows of myelin ovoids and balls (Dyck et. al., 2005). A decrease in corresponding
cell bodies in the DRG and atrophy of the dorsal spinal tract together with lumbosacral
spinal ganglion neurons, accompanies fibre loss (Denny-Brown, 1951). As the
peripheral sensory axons progressively retract from their peripheral targets over time,

6 | P a g e
it reaches their cell body in the DRG resulting in death of the neuronal cells (Dyck et.
al., 2005).
1.1.4 Genetics of HSN-I
As stated earlier, HSN-I is an autosomal dominant inherited disease with the genetic
locus being mapped to chromosome 9q22.1-q22.3 in 1996 using 4 large Australian
kindreds (Verhoeven et. al., 2006; Nicholson et. al., 1996). Further studies confirmed
the region that was narrowed and located to a missense mutation in the open reading
frame of SPTLC1 as the pathogenic cause of HSN-I. This mutation causes a change
in a single base resulting in an aberrant amino acid being incorporated, ultimately
changing the final protein structure. The most common mutation in HSN-I as seen in
eight Australian/English families was a single DNA base mutation 399T→ G in exon 5
of the SPTLC1 coding region, resulting in an amino acid substitution of cysteine to
tryptophan at position 133 (C133W) (Dawkins et. al., 2001). A second mutation in 431T
→ A was identified in two families and a third mutation in 398G → A observed in
another family which resulted in a substitution of valine to aspartate at position 144
(V144D) and cysteine to tyrosine at position 133, respectively (Dawkins et. al., 2001).
A fourth mutation was identified in twin sisters by Verhoeven et. al., (2004) at 387G →
A, changing glycine for alanine. Recently, three more mutations in, SPTLC2, DNA
methyl transferase 1 (DNMT1) and atlastin GTPase 3 (ATL3) have been observed to
cause HSN-I (Rotthier et al. 2010; Klein et al. 2011 and Kornak et al. 2014)

7 | P a g e
1.2 Intracellular Lipid Functions and Interactions 1.2.1 The role of lipids in biological membranes
Biological membranes are lipid structures that define cells and cellular organelle
structure. They divide the interior of eukaryotic cells into distinct compartments and
provide surfaces for the localisation of metabolic enzymes, transport proteins,
receptors and various substrates (Fenske et. al., 1995). Membranes are
semipermeable barriers which regulate the transport of water, ions and other
metabolites, thereby providing a means of controlling the internal cellular environment.
In 1972, Singer and Nicholson first proposed the fluid-mosaic model of biological
membranes with the basic understanding of membrane structure changing little since
(Fenske et. al., 1995).
On average 98% of the molecules in membranes are lipids. The main classes of lipids
found in eukaryotic biological membranes include the glycerophospholipids, the
sphingolipids and cholesterol (Cullis and Hope, 1991; Saladin, 2007). The molecules
are amphiphilic and arrange themselves into a liquid-crystalline bilayer with their
hydrophilic phosphate-containing heads facing the water on each side of the
membrane with their hydrophobic tails directed towards the centre of the membrane.
The phospholipids can drift laterally, able to spin on their axes and move their tails,
keeping the membrane fluid (Saladin, 2007). The membrane also consists of
cholesterol molecules, membrane-bound and transmembrane proteins as shown in
Figure 1.1. Historically, the lipid portion of the membrane was viewed as a convenient
barrier and environment for enzymes (Fenske et. al., 1995). However, many studies
have been undertaken to show that biological membranes contain a wide diversity of
lipids, far more than are needed to perform structural functions, with these lipids
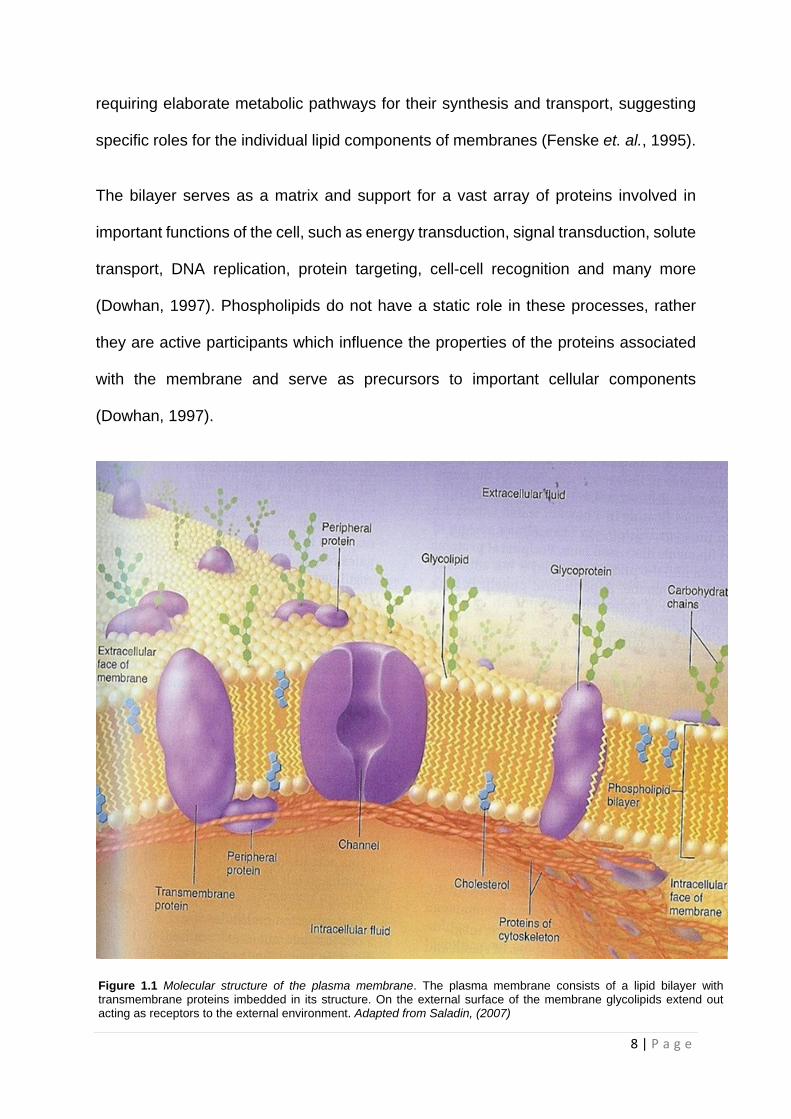
8 | P a g e
requiring elaborate metabolic pathways for their synthesis and transport, suggesting
specific roles for the individual lipid components of membranes (Fenske et. al., 1995).
The bilayer serves as a matrix and support for a vast array of proteins involved in
important functions of the cell, such as energy transduction, signal transduction, solute
transport, DNA replication, protein targeting, cell-cell recognition and many more
(Dowhan, 1997). Phospholipids do not have a static role in these processes, rather
they are active participants which influence the properties of the proteins associated
with the membrane and serve as precursors to important cellular components
(Dowhan, 1997).
Figure 1.1 Molecular structure of the plasma membrane. The plasma membrane consists of a lipid bilayer with transmembrane proteins imbedded in its structure. On the external surface of the membrane glycolipids extend out acting as receptors to the external environment. Adapted from Saladin, (2007)

9 | P a g e
1.2.2 Interactions of lipids with intracellular proteins and organelles
Lipids in eukaryotic membranes are fluid (Van Meer and Sprong, 2004). Lipids in the
ER are generally unsaturated, whereas the saturated lipids in the other membranes
are “fluidised” by the presence of cholesterol (Van Meer and Sprong, 2004). Most
membranes are composed mainly of phospholipids, glycosphingolipids and
cholesterol. Phospholipids are loosely packed in bilayers, forming liquid disordered
membranes. In contrast, sphingolipids have longer and more saturated acyl chains
than phospholipids and exhibit stronger lateral cohesion, generating tightly packed
regions (Van Meer and Sprong, 2004). Cholesterol preferentially interacts with
sphingolipids and occupies the space between the acyl chains. The combination of
sphingolipids and cholesterol in small domains is responsible for the formation of lipid
rafts, important in transportation of proteins, influencing membrane fluidity and
regulating neurotransmission and receptor trafficking (Van Meer and Sprong, 2004).
Several membrane proteins have been shown to reside in lipid rafts, either
permanently or temporarily, and this association can be facilitated by covalent lipid
modification of the protein molecule (Van Meer and Sprong, 2004). In fact, addition of
a lipid chain (N-myristoylation, palmitoylation) to proteins increases their
hydrophobicity and therefore their propensity to associate with cellular membranes
(Van Meer and Sprong, 2004). Lipid rafts on the plasma membrane can serve as
scaffolding platforms where different receptors with similar affinity for the lipid domains
meet and orchestrate various signalling events (Van Meer and Sprong, 2004). It has
also been shown that lipids can exchange in the cell membrane to alter kinase
signalling events (Myers and Stanley, 1999). Moreover, the existence of lipid domains
also on the membrane of cell organelles seems to be responsible for appropriate

10 | P a g e
protein-lipid sorting to specific cell compartments, thus enabling vesicles to arrive at
the correct destination and facilitating secretion of cellular products (Van Meer and
Sprong, 2004).
Eukaryotic cells use the physical properties of their individual membrane lipid classes
at specific steps in vesicle traffic. The local concentration of these lipids is regulated
by a multitude of enzymes and translocators and appears to be one parameter in
regulating the membrane flux through the various pathways (Van Meer and Sprong,
2004).
The occurrences of two glycosylphosphatidylinositol proteins (GPI) located in separate
plasma membrane domains of different lipid composition, and with the finding of lipid-
anchored proteins in cholesterol independent microdomains on the cytosolic surface,
illustrate the dynamic organisation of biomembranes and how they cannot be
explained by the mere notion of sphingolipid/cholesterol rafts (Van Meer and Sprong,
2004). A specific function of sphingolipids on cytosolic surfaces is suggested by the
cytosolic protein FAPP2, which contains a glycolipid-binding domain that plays a role
in regulating membrane flow between the Golgi and the plasma membrane (Van Meer
and Sprong, 2004).
Cholesterol transport out of endosomes and lysosomes requires both the soluble
cholesterol-binding protein NPC2 in the lumen and the putative cholesterol transporter
NPC1 in the membrane, which work together in concert (Van Meer and Sprong, 2004).
Lipids and proteins have been well-studied but a lot remains to be learnt about how
they ‘act in concert’ in cell membranes (Van Meer and Sprong, 2004).

11 | P a g e
1.2.3 Sphingolipids, their role in normal cellular homeostasis and in the neuronal cell
Sphingolipids (SLs) are a ubiquitous class of lipids present in all higher order
organisms. A large number of individual sphingolipid species exist, resulting from
differences in both the hydrophobic ceramide moiety and in the polar head group
(Buccoliero et. al., 2003). SLs form a complex family of molecules which all contain a
long-chain amino alcohol, known as the sphingoid base to which a variety of fatty acids
are attached via N-acylation (Buccoliero et. al., 2003). The polar head group consists
of phosphorylcholine, sugar or sulfatide residues. Many different combinations of
sphingoid long chain bases, fatty acids and head group moieties lead to a vast array
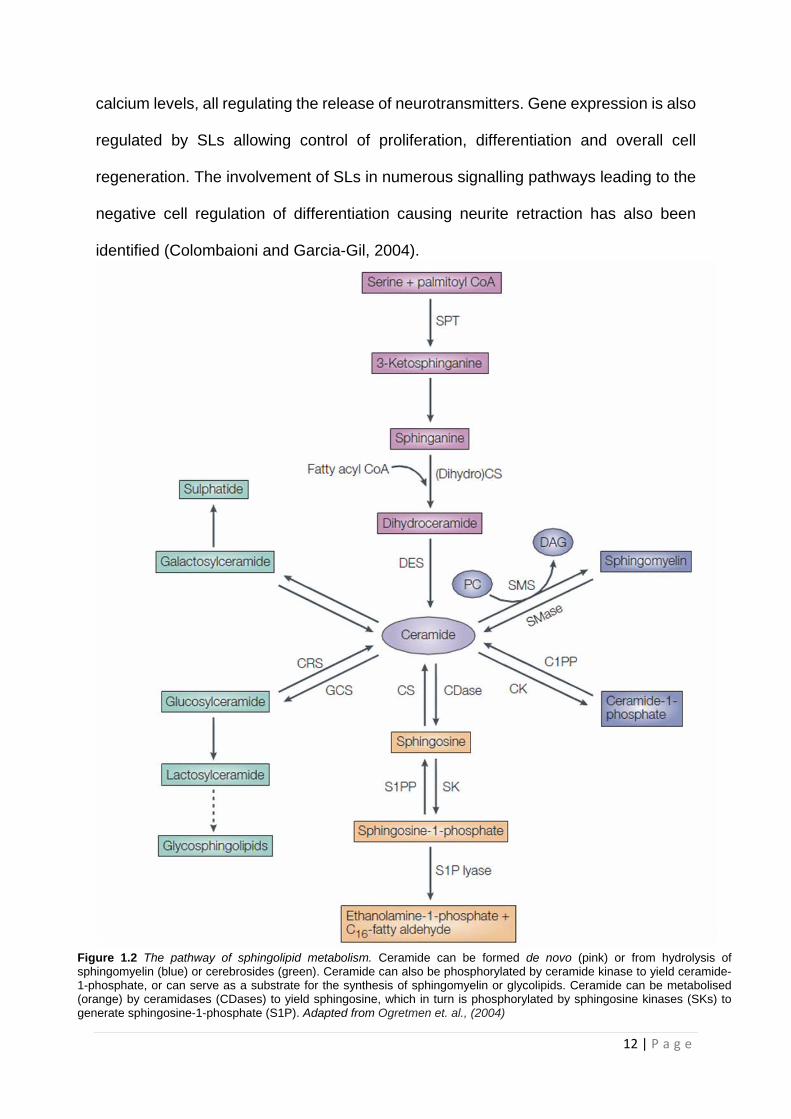
of possible SL and glycosphingolipid (GSL) structures, which is seen in Figure 1.2
(Buccoliero et. al., 2003; Sabourdy et.al., 2008).
Complex sphingolipids such as ceramide, sphingomyelin, glucosylceramide,
galactosylceramide, sphingosine, sphingosylphospho-choline, psychosine and
sphingosine-1-phosphate (S1P), glycosphingolipids and phosphosphingolipids have
essential roles in many aspects of cell biology. Some of these role include,
inflammatory responses, cell proliferation and apoptosis to cell migration,
differentiation and senescence. Many sphingolipids carry out structural roles in cell
membranes, particularly the plasma membrane (Ogretmen et. al., 2004).
In the neuronal cell, SLs have been shown to participate in neuronal development,
proliferation, survival, migration, differentiation and plasticity. SLs play an essential
role in cell regeneration after cell damage has occurred (Colombaioni and Garcia-Gil,
2004). Neurotransmitter release has also been shown to be regulated by SLs, with
SLs binding to certain receptors that control ion channels, enzymes and intracellular
12 | P a g e
calcium levels, all regulating the release of neurotransmitters. Gene expression is also
regulated by SLs allowing control of proliferation, differentiation and overall cell
regeneration. The involvement of SLs in numerous signalling pathways leading to the
negative cell regulation of differentiation causing neurite retraction has also been
identified (Colombaioni and Garcia-Gil, 2004).
Figure 1.2 The pathway of sphingolipid metabolism. Ceramide can be formed de novo (pink) or from hydrolysis of sphingomyelin (blue) or cerebrosides (green). Ceramide can also be phosphorylated by ceramide kinase to yield ceramide-1-phosphate, or can serve as a substrate for the synthesis of sphingomyelin or glycolipids. Ceramide can be metabolised (orange) by ceramidases (CDases) to yield sphingosine, which in turn is phosphorylated by sphingosine kinases (SKs) to generate sphingosine-1-phosphate (S1P). Adapted from Ogretmen et. al., (2004)

13 | P a g e
1.3 Serine Palmitoyltransferase
1.3.1 The structure and regulation of SPT and its subunits
SPT is a pyridoxal 5'-phosphate-dependent multimeric enzyme (Figure 1.3) that
catalyses the first step of the biosynthesis of sphingolipids (SL), ceramide and
sphingomyelin (Hornemann et. al., 2006; Breslow. D, 2013). SPT is a rate determining
enzyme in the de novo sphingolipid synthesis pathway. It is a key enzyme in regulation
of cellular sphingolipid content by the condensation of palmitoyl coenzyme A (CoA)
with L-serine to form 3-ketodihydrosphingosine. SPT is comprised of a combination of
two known subunits; SPTLC1, SPTLC2 (also known as LCB1 and LCB2 respectively)
and SPTLC3 which are summarised in Table 1.2 (Breslow. D, 2013).
SPT is a type 1 integral membrane protein with a single highly hydrophobic domain in
the amino-terminal region of the SPTLC1 subunit, which represents a transmembrane
domain that anchors the enzyme to the ER membrane (Wei et. al., 2007; Lowther et.
al., 2012). SPTLC2 contains an active lysine residue required for PLP-binding while
SPTLC1 lacks this lysine and other key catalytic residues which has led to speculation
as to whether this lysine residue plays a regulatory role in the SPT dimer (Yard et. al.,
2007; Lowther et. al., 2012). SPTLC1 and SPTLC2 subunits associate in a 1:1 ratio
with the active site residing between the interfaces of these two subunits (Hanada,
2003). Both of these subunits are essential to produce the functionally active,
heterodimeric SPT; however, both subunits are membrane-associated and this has
encumbered their isolation and characterisation (Yard et. al., 2007; Lowther et. al.,
2012). Previously the third subunit, SPTLC3, was thought to be a functionally
redundant isoform of SPTLC2. However, SPTLC3 has been shown to generate two

14 | P a g e
Table 1.2 The three subunits of SPT; SPTLC1, SPTLC2 and SPTLC3 (Dawkins et. al., 2002; Verhoeven et. al., 2006; Hornemann et. al., 2006 and Hanada, 2003)
new sphingoid base metabolites, C (16)-sphinganine and C (16)-sphingosine
(Hornemann et. al., 2009).
SPT activity is regulated at both a transcriptional and post-translational level. SPT is
a ubiquitously expressed protein with its subunits expressed at varying levels (Wei et.
al., 2007; Breslow. D, 2013). SPT has been shown to be upregulated in response to a
variety of extracellular stimuli such as inflammatory cytokines, skin barrier
requirements, ultraviolet light B (UVB), excess fatty acids and other factors that induce
apoptosis (Hanada, 2003).
1.3.2 The role of the SPTLC1 protein in neurodegenerative disease
SPT is the crucial enzyme in the complex metabolic pathway of sphingolipid
metabolism, with mutations in the SPT subunits resulting in potential dysfunction and
perturbations in sphingolipid synthesis and metabolism causing a variety of diseases,
in particular HSN-I (Wei et. al., 2007).
Subunit Chromosome Gene size (kb) Protein size (kDa)
SPTLC1 9q21.1-q22.3 85 55
SPTLC2 14q24.3-q31 110 65
SPTLC3 20p12.1-12.3 1.7 63
Figure 1.3 Theoretical model of SPT complex structure. A) SPTLC2 or SPTLC3 join SPTLC1 to form a dimer resulting in formation of the basic functional unit of SPT which then forms a final octameric state. B) Model of the octameric SPT complex with two SPTLC1–SPTLC2 and two SPTLC1–SPTLC3 dimers assembled to form an octameric circular structure. C) Cytosolic view of the SPT enzyme. Adapted from (Hornemann et. al., 2007).

15 | P a g e
SPTLC1 encodes the long-chain base one (LCB1) subunit of SPT. Mutations in this
gene identified in HSN-I occur at single amino acids that are highly conserved
throughout different species and are likely to interfere with the functionality and
structure of SPT (Verhoeven et. al., 2006). SPT mutations causing HSN-I are
autosomal dominant, and have a late age of onset with only missense mutations
occurring. This is most consistent with two current hypotheses for the mechanism of
HSN-I: either a gain of function or a dominant negative effect (Verhoeven et. al., 2006).
The 'gain of function' hypothesis implies that mutations confer one or more toxic
properties on SPTLC1. A 'gain of toxic function' is the accumulation of mutant protein,
that then forms an insoluble aggregate that eventually leads to cell death (Verhoeven
et. al., 2006). Alternatively, peripheral neurons may be sensitive to a perturbation of
the sphingolipid metabolism (decrease in functional levels) caused by a mutation-
induced reduction in SPT enzyme activity (Verhoeven et. al., 2006). This hypothesis
has been shown to be consistent with studies on C133W and V144D, demonstrating
that both mutations reduce normal SPT activity in various cell types, including cultured
patient lymphoblasts (Verhoeven et. al., 2006). A concomitant change in the
membrane lipid composition would be expected to be seen but data has been
contradictory. Initially, an increase in glucosylceramide synthesis was reported,
however a decrease in ceramide levels and sphingomyelin synthesis yielded no
change in the lipid composition (Verhoeven et. al., 2006).
Bejaoui et. al., (2002) and Dedov et. al., (2004) studied SPT activity using patient
lymphoblasts that endogenously expressed the SPTLC1 mutation and reported
greater than 50% reduction of SPT activity. While the mechanism by which SPT
activity is reduced is yet to be confirmed, Bejaoui et. al., (2002) observed that the

16 | P a g e
mutation did not directly affect the stability of the protein translated and postulated that
it may directly interfere with enzyme function. As SPTLC1 mutations have a direct
effect on the activity of SPT this supports the dominant negative effect theory. Thus,
competition possibly arising between mutated and wild type SPTLC1 for interaction
with SPTLC2 may be a potential molecular mechanism, with the mutated SPTLC1
possessing a higher affinity than wild type (Bejaoui et. al., 2002). As seen by
Verhoeven et. al., (2006), the SPTLC1 mutation does not reduce SL levels despite
SPT activity being reduced by more than half. Therefore, it suggests that the remaining
50% of SPT activity is sufficient to maintain the normal sphingolipid homeostasis in
these cells, presumably because the cell may have more total SPT activity than is
required as seen by in-vivo SPT down-regulation (Dedov et. al., 2004).
In contrast, higher overexpression of SPTLC1-like subunit mutations in yeast and
Chinese hamster ovary (CHO) cells can result in cell death (Dedov et. al., 2004). As a
50% reduction in SPT activity appears to have little impact on sphingolipid metabolism,
resulting normal cell proliferation and viability may explain why HSN-I is a late on-set
disorder (Dedov et. al., 2004). It is possible that the expression of SPTLC1 and
SPTLC2 are not as balanced in ageing neurons with a dominant negative effect
manifesting in these cells more profoundly than in lymphoblasts (Dedov et. al., 2004).
It is also plausible that subtle changes in sphingolipid metabolism are involved, such
as the accumulation of abnormal metabolites from dysregulation of these pathways
(Dedov et. al., 2004). There may also be accumulation of mutant SPT leading to the
formations of insoluble aggregates, as demonstrated to be the common pathogenic
mechanism of other common neurodegenerative disorders such as Alzheimer’s or
Huntington's diseases (Dedov et.al., 2004). Although no SPTLC1 aggregates have yet

17 | P a g e
been discovered in HSN-I, elevated levels of deoxysphingoid bases (Deoxy-LCBs)
arising from condensation of alanine and glycine with palmitoyl-CoA has been
observed in HSN-I transgenic mice (Penno et. al., 2010). Such evidence would support
the 'toxic gain of function' theory and may also explain the late-onset of the disease.
1.4 Protein Functions and Interactions 1.4.1 Altered protein expression and the disease state
In biological structures such as proteins, 'form follows function' and 'form is function'
(Lodish et. al., 2008). This highlights the importance of correctly synthesised protein
primary structure and folding into the proper three-dimensional conformation, essential
for correct protein functioning. Recent evidence has shown that a protein may fold into
an alternative three-dimensional structure or have a resulting post-translational
modification that arises from mutations or inappropriate covalent modifications (Lodish
et. al., 2008). This misfolding or modification leads to a loss of normal function and
often marks the protein for proteolytic degradation (Lodish et. al., 2008). When
degradation is incomplete or not sufficient to keep up with the amount of misfolding
occurring, then subsequent accumulation of the proteolytic fragments or misfolded
proteins occurs. This has been shown to contribute to certain degenerative diseases
that are characterised by the presence of insoluble protein plaques in various organs
including the liver and brain (Lodish et. al., 2008). Diseases like Alzheimer's,
Parkinson’s and Huntington's disease are all neurodegenerative diseases that have
manifested from the resulting increase in proteolytic fragments and misfolded proteins
(Lodish et. al., 2008).

18 | P a g e
Between 60 and 80% of all dementia-related illness is due to Alzheimer's disease
(Goldman et. al., 2008). Alzheimer's disease is marked by the presence of increasing
numbers of β-amyloid peptide-containing deposits in neuritic plaques within the
neocortex, and is associated with increasing severity of dementia (Goldman et. al.,
2008). It has been assumed for many years that the plaques were themselves toxic;
instead the soluble aggregates of β-amyloid in oligomeric forms (i.e. consisting of a
small number of monomers) are the key pathogenic molecules (Goldman et. al., 2008).
Neurofibrillary tangles are intracellular aggregates of the microtubule-associated
protein tau. The neurofibrillary tangles represent a nonspecific response to β-amyloid
(Goldman et. al., 2008). Tamboli et. al., (2005) have shown that glycosphingolipid
inhibition reduces transport of β-amyloid precursor protein (APP) and β-amyloid, thus
highlighting the importance of sphingolipids and potential treatment in the future.
Parkinson's disease is the second most common neurodegenerative disorder
occurring in approximately 1 in 1000 in the general population and in 1% of persons
older than 65 years (Goldman et. al., 2008). Both autosomal dominant and recessive
genes have been implicated in classic Parkinson's disease (Goldman et. al., 2008).
The protein α-synuclein, which is the major constituent of the cytoplasmic inclusion
known as the Lewy body, is critical in the disease pathogenesis (Goldman et. al.,
2008). Abnormal aggregation of the protein, either from mutations in the α-synuclein
gene or excessive production of the normal protein due to gene duplications or
triplications, is associated with the varying disease phenotypes observed in
Parkinson's (Goldman et.al., 2008).
Huntington's disease is an autosomal dominant disorder caused by unstable cytosine-
adenine-guanine (CAG) repeat expansions on chromosome 4 (Schapira et. al., 2007).

19 | P a g e
Afflicted individuals have 37 to 86 repeats, compared to normal individuals that have
11 to 34 repeats. The normal protein, huntingtin, serves a role in intracellular trafficking
and membrane recycling (Schapira et. al., 2007). The trinucleotide CAG codes for
glutamine, with an increase in the polyglutamine tract preventing normal protein
turnover, thereby resulting in protein aggregation (Schapira et. al., 2007). The aberrant
protein product is ubiquitinated (marked for degradation) but fails to be efficiently
degraded, leading to the formation of intranuclear inclusions that may disrupt
mitochondrial processes and other functions. The mutant huntingtin is cleaved into
fragments, with these fragments playing a primary role in huntingtin toxicity, including
transcriptional dysregulation (Schapira et. al., 2007). Mutant huntingtin associates with
an array of proteins. These “huntingtin-interacting proteins” are involved with
numerous important cellular functions, including transcription, trafficking, signalling
and metabolism (Schapira et. al., 2007). It is therefore essential that proteins are
folded correctly and that any misfolded proteins are successfully degraded and
removed to maintain normal cellular function.
1.5 Mitochondria in neurodegenerative diseases 1.5.1 Mitochondrial Dynamics
Mitochondria are the most recognisable membrane-bound organelle, responsible for
several biological processes, such as oxidative phosphorylation, lipid metabolism,
tricarboxylic acid (TCA) cycle, iron-sulphur cluster formation and apoptosis. They are
interconnected and form a highly dynamic network shaped by constant fusion and
fission events. They are usually scattered throughout the cytoplasm of most cells, but
they often concentrate in specific areas of high energy utilisation. Their number and

20 | P a g e
size vary with metabolic activity and cell type: mature erythrocytes have none; a
hepatocyte has up to 2500. They are 1-10 μm in size and may be elongated, spherical,
or pleomorphic. These very dynamic organelles show constant motion, fusion, and
division in cells (Ovalle and Nahireny, 2008).
1.5.2 Mitochondrial fusion and fission
Mitochondrial fission contributes not only to the proper distribution of mitochondria in
response to the local demand for ATP, but also to the elimination of damaged
mitochondrial fragments through mitophagy (autophagy for mitochondria).
Mitochondrial fusion facilitates the exchange of mtDNA and other vital components
between mitochondria for the maintenance of normal function (van de Bliek et. al.,
2013). Mitochondrial fusion and fission are controlled by four high molecular weight
GTPases conserved from yeast to mammals: mitofusins, Mfn1 and Mfn2, in
mitochondrial outer membrane (OMM) fusion; Opa1 (a gene product of optic atrophy
type I) in mitochondrial inner membrane (IMM) fusion and cristae organisation; and
Drp1 (Dynamin related protein 1) in mitochondrial fission, indicating that the
fundamental mechanisms controlling mitochondrial dynamics have been maintained
throughout evolution (Figure 1.4). The morphology of the mitochondria results from a
balance between these opposing processes (van de Bliek et. al., 2013).
1.5.2.1 Mitochondrial Fusion
Mitochondrial fusion between closely apposed mitochondria is a complex regulatory
process involving multiple proteins which fuse both the OMM and IMM of each
mitochondrion (van de Bliek et. al., 2013). Although fusion reactions between OMMs

21 | P a g e
of apposed mitochondria and the subsequent fusion between IMMs are normally
highly synchronised, the two processes can be functionally uncoupled (Otera and
Mihara, 2011). Mfn1 and Mfn2 are anchored to the OMM with a large N-terminal
GTPase domain and a C-terminal coiled-coil domain that is exposed to the cytosol
and they mediate OMM fusion in a GTPase-dependent manner (Otera and Mihara,
2011).
Mfn1 and Mfn2 form homo- or hetero-protein complexes. These interactions between
the mitofusins on opposing mitochondria serve to tether and fuse the OMMs (van de
Bliek et. al., 2013). Mitofusin 1 and 2 appear to have distinct roles within mitochondrial
OMM fusion; Mfn1 is thought to be responsible for the initial GTP-dependent OMM
tethering (Figure 1.4) (Otera and Mihara, 2011). Mfn2 is enriched in the mitochondria-
associated membranes (MAM) of the ER, where it interacts with Mfn1 and Mfn2 on
the mitochondria to form interorganellar bridges (Otera and Mihara, 2011).
Opa1 is another key molecule essential for mitochondrial IMM fusion and cristae
remodelling. There are eight Opa1 splice variants, which are all synthesised as
precursor proteins with the mitochondrial localisation sequence in the N-termini and
the following hydrophobic stretches that are responsible for sorting the protein into the
IMM (Otera and Mihara, 2011). During mitochondrial import, the mitochondrial
localisation sequence of Opa1 precursors is removed by mitochondrial processing
peptidase to form L-forms (Ishihara et. al., 2006). These Opa1 L-forms are anchored
to the IMM with the GTPase domain exposed to the mitochondrial intermembrane
space (IMS), and are subsequently processed either in the IMS to produce S-forms by
the intermembrane space AAA protease (i-AAA protease) YME1L or in the matrix by
Afg3L2 and Paraplegin, depending on where the process site localises in the IMS.
22 | P a g e
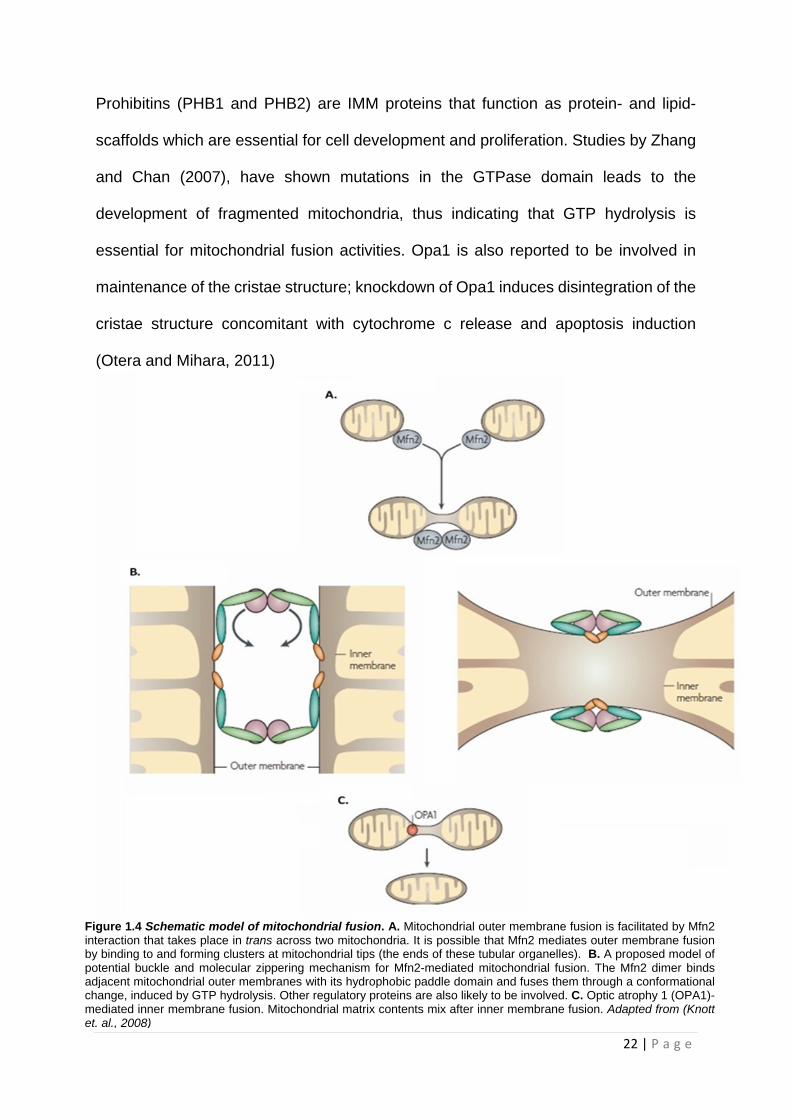
Prohibitins (PHB1 and PHB2) are IMM proteins that function as protein- and lipid-
scaffolds which are essential for cell development and proliferation. Studies by Zhang
and Chan (2007), have shown mutations in the GTPase domain leads to the
development of fragmented mitochondria, thus indicating that GTP hydrolysis is
essential for mitochondrial fusion activities. Opa1 is also reported to be involved in
maintenance of the cristae structure; knockdown of Opa1 induces disintegration of the
cristae structure concomitant with cytochrome c release and apoptosis induction
(Otera and Mihara, 2011)
Figure 1.4 Schematic model of mitochondrial fusion. A. Mitochondrial outer membrane fusion is facilitated by Mfn2 interaction that takes place in trans across two mitochondria. It is possible that Mfn2 mediates outer membrane fusion by binding to and forming clusters at mitochondrial tips (the ends of these tubular organelles). B. A proposed model of potential buckle and molecular zippering mechanism for Mfn2-mediated mitochondrial fusion. The Mfn2 dimer binds adjacent mitochondrial outer membranes with its hydrophobic paddle domain and fuses them through a conformational change, induced by GTP hydrolysis. Other regulatory proteins are also likely to be involved. C. Optic atrophy 1 (OPA1)-mediated inner membrane fusion. Mitochondrial matrix contents mix after inner membrane fusion. Adapted from (Knott et. al., 2008)

23 | P a g e
1.5.2.2 Mitochondrial Fission
The mitochondrial division process requires at least two conserved proteins, hFis1
(human mitochondrial fission protein 1) and Drp1, and several other proteins, including
MTP18 in mammals. Drp1 and hFis1 localise to discrete points at future sites of
scission on the mitochondrial outer membrane (Marchi et. al., 2014). Drp1 is a
cytosolic protein with an N-terminal GTPase domain thought to provide mechanical
force with a dynamin-like middle domain and a GTPase effector domain (GED) located
in the C-terminal region. Drp1 mainly localises in the cytosol, and during mitochondrial
fission, translocates from the cytosol to prospective fission sites on the mitochondria
(Marchi et. al., 2014).
These spiral higher-order structures are thought to constrict and eventually sever the
mitochondrial membrane by a GTP hydrolysis dependent mechanism. Intermolecular
interactions between the N-terminal GTPase domain and C-terminal GED are also
important for Drp1 self-assembly and functional regulation (Marchi et. al., 2014). In
mammals, Fis1 has also been identified in mitochondria and is thought to be involved
in recruiting Drp1 to mitochondria (Marchi et. al., 2014). Mitochondrial fission factor
(Mff) is able to recruit Drp1 to mitochondria and form a complex with Drp1 and promote
mitochondrial fission (Figure 1.5). Mff linked to the plasma membrane results in Drp1
recruitment to exactly that membrane. Mitochondrial dynamics proteins 49 and 51
(MiD49 and MiD51 respectively) and MIEF1 (mitochondrial elongation factor 1) are
also capable of recruiting Drp1 to mitochondria (Figure 1.5). Importantly, mitochondrial
fission is blocked rather than promoted by sequestering Drp1. MIEF1/MiD51 are able
to form two different protein complexes which, in an apparent mutually exclusive
manner, bind to either Drp1 or Fis1 (Palmer et. al., 2011;Zhao et. al., 2011)

24 | P a g e
Figure 1.5 Schematic diagram for the proposed new model of fission. Classical model: hFis1 acts as the mitochondrial receptor for Drp1 promoting fission. Extended model: hFis1, Mff and/or MIEF1/MiD51/Mid49 recruit Drp1 to the mitochondria. The Mff–Drp1 complex promotes mitochondrial fission. In contrast, the MIEF1/Mid49/51–Drp1 sequesters Drp1, inhibits Drp1 function and promotes fusion in an Mfn2-independent manner. In an apparently mutually exclusive manner, MIEF1/MiD51 can also form a complex with hFis1. The inhibitory effect of MIEF1/MiD51 on Drp1 function is reduced and hence mitochondrial fission is indirectly promoted by Fis1. Active Drp1: green. Inhibited Drp1: red. Adapted from (Dikov and Reichert, 2011).
Mitotic kinase Aurora A and the small Ras-like GTPase RALA also controls the
recruitment of Drp1 to mitochondria Kashatus et. al., (2011). Aurora A is known as a
serine/threonine kinase and has a pivotal role in many aspects of cell division, such
as mitotic entry, chromosome segregation and spindle assembly. On the other hand,
RALA, a small G protein belonging to the Ras superfamily, controls and participates
in cellular processes such as vesicle sorting, cell morphology and gene expression by
cycling between active GTP-bound and inactive GDP-bound conformations (Kashatus
et. al., 2011). Kashatus et. al., (2011) found that phosphorylated RALA accumulates
preferentially on mitochondria, they also demonstrated that Drp1 protein levels in the

25 | P a g e
mitochondria are reduced both in cells with diminished levels of RALA thus inhibiting
mitochondrial fission and induces a more highly interconnected mitochondrial network
(Kashatus et. al., 2011). RALBP1, a multifunctional effector of RALA, has also been
shown to affect the recruitment of Drp1. RALBP1 and cyclinB–Cdk1 interact with each
other and phosphorylate Drp1 (Dikov and Reichert, 2011; Kashatus et. al., 2011). This
suggests that RALBP1, following recruitment to mitochondria by RALA, may form a
mitosis-specific complex with cyclin B–CDK1, and that this interaction potentiates
cyclin B–Cdk1 kinase activity towards Drp1 and promotes Drp1 oligomerisation and
subsequent mitochondrial fission (Figure 1.6) (Dikov and Reichert, 2011; Kashatus et.
al., 2011).
Despite the growing insight into mitochondrial fission, it remains unclear whether the
phosphorylation of Drp1 is always essential for membrane scission. If so, cyclin B–
CDK1 can phosphorylate Drp1 during mitosis, but other kinases would be required for
the fission activity of DRP1. Furthermore, cyclic-AMP-dependent protein kinase (PKA)
has been identified to phosphorylate different, but nearby, serine residues during
Figure 1.6 Schematic diagram of mitosis-specific mitochondrial fission. Mitosis-specific mitochondrial fission involves the phosphorylation of DRP1. Mitochondrial morphology is coordinated with the cell cycle. During metaphase, in which condensed chromosomes align in the middle of the cell, mitotic kinase Aurora A phosphorylates RALA, which relocates to mitochondria. This encourages the formation of a complex consisting of phosphorylated RALA, RALBP1and cyclin B–CDK1 on mitochondria, and mediates the phosphorylation of DRP1. Finally, oligomeric Drp1 wraps around the mitochondrial surface and mitochondrial division ensues. Adapted from (Yamano and Youle, 2011).

26 | P a g e
Figure 1.7 Overview of mitochondrial dynamics and homeostasis. Mitochondrial morphology is maintained by fusion and fission. Excessive mitochondrial fission often causes the generation of depolarised mitochondria. Dysfunctional mitochondria return to the cell soma and are eliminated by the autophagy system, named mitophagy. Disruption of the mitochondrial dynamics or mitochondrial quality control system leads to the accumulation of dysfunctional mitochondria and causes a collapse of the cellular environment followed by cell death. Adapted from (Otera and Mihara, 2011).
starvation (Dikov and Reichert, 2011; Kashatus et. al., 2011). A surmounting body of
research indicates that mitochondrial division responds to cellular stimuli in highly
regulated ways where a central role is the modulation of Drp1, not only by
phosphorylation, but potentially by other post-translational modifications including
ubiquitylation, Sumoylation and S-nitrosylation (Figure 1.7) (Dikov and Reichert, 2011;
Kashatus et. al., 2011). Thus, post-translational modifications could play a major role
in the regulation of Drp1 activity and thereby controlling mitochondrial morphology
(Otera and Mihara, 2011).

27 | P a g e
1.5.3 Mitochondrial changes in inherited peripheral neuropathies
Neurodegenerative diseases are a clinically heterogeneous group of chronic
progressive illnesses with varying, but distinct, clinical manifestations. The genetic
cause of disorders such as Huntington’s disease, amyotrophic lateral sclerosis (ALS),
Charcot-Marie-Tooth syndrome Type II (CMT II), Parkinson’s disease, Friedreich’s
ataxia and Alzheimer’s disease have been well established. Despite their obvious
differences in primary aetiologies, the role for mitochondrial dysfunction is evident in
the pathogenesis of these diseases (Manfredi and Flint Beal, 2000; Kwong et. al., 2006
and Duffy et. al., 2011).
The nervous system is especially susceptible to mitochondrial dysfunction due to the
high metabolic activity of neurons, in particular bioenergetic failure through the loss of
ATP production. Mitochondrial dysfunction in neurons can lead to a myriad of different
effects such as apoptosis, oxidative stress, excitotoxicity and destructive rises in
intracellular calcium levels that contribute to several pathologies of the nervous system
(Hollenbeck and Saxton, 2005). Mitochondrial transport is also intimately dependent
upon the functional state of the cell and of mitochondria themselves, and is essential
for neurons due to their long axonal processes and high demand for energy (Kwong
et. al., 2006).
Mitochondrial membrane depolarisation and inhibition of ATP synthesis, has been
shown to inhibit movement of organelles, with 80% of slightly depolarised mitochondria
in DRG neurons move retrogradely, implying unhealthy mitochondria are returned to
the cell body for repair or removal, reducing the number of mitochondria that are
transported in the anterograde direction (Miller and Sheetz, 2004).

28 | P a g e
Mitochondrial dysfunction is likely to play an important role in several major
neurodegenerative diseases. However, despite an ever-expanding body of literature
on the mitochondrial involvement in neurodegeneration, many crucial questions
remain to be answered. How do mutations in neurodegenerative genes result in
mitochondrial dysfunction? In some cases mutant proteins localised in multiple cell
compartments (often including mitochondria) may cause dysfunction, such as aberrant
aggregates in cellular compartments, as observed in Huntington’s disease and
Alzheimer’s disease (Duffy et. al., 2011). In addition to how mutations cause
mitochondrial dysfunction, what are the consequences of mitochondrial dysfunction?
With the multiplicity of mitochondrial functions (including intermediate metabolism,
energy production and apoptosis) there are many, nonmutually exclusive, potential
avenues whereby damaged mitochondria can sabotage the survival of a neuronal cell
(Kwong et. al., 2006, Duffy et. al., 2011).
While extensive research efforts are currently underway to answer these questions,
the consequence of mitochondrial dysfunction in very large and metabolically active
cells such as peripheral sensory neurons are especially adverse (Duffy et. al., 2011).
Differential sensitivity of distal neuronal regions to dysfunctions in mitochondrial
energy productions, axonal transport of mitochondria, or a combination of both are
now thought to be a major cause for axonal degeneration in peripheral neuropathies
(Duffy et. al., 2011). Until further research is thoroughly conducted the precise
involvement of mitochondria in inherited peripheral neuropathies remains to be
elucidated.

29 | P a g e
1.5.4 Examples of Mitochondrial changes in inherited peripheral
Neuropathies
Charcot-Marie-Tooth type IIA is an inherited peripheral neuropathy with both motor
and sensory involvement that exhibits dying back of the axon, with similar clinical
presentation to HSN-I. This disease has been identified as a gene mutation involved
in mitochondrial function, known as mitofusin 2 (Zuchner et. al., 2004). While the exact
cause of axonal degeneration of CMT type IIA is undefined, recent studies have
postulated that impairment in anterograde axonal transportation of mitochondria and
consequent bioenergetic failure at the distal ends of neurons may be responsible
(Zuchner et. al., 2004; Baloh et. al, 2007 and Warner and Hammans, 2009).
Mitochondrial membrane potential, oxidative respiration and ATP production have
been found to be normal in neurones from patients with CMT type IIA. However
observed mitochondrial aggregates were postulated to ineffectively attach to the motor
proteins involved in mitochondrial transport (Baloh et. al., 2007).
While axonal degeneration may be caused by impairment of mitochondrial
transportation, neuronal die back could also be caused by proteins not normally
associated with mitochondria. Amyotrophic lateral sclerosis (ALS) is a fatal
neurodegenerative disorder, which is pathologically defined by the progressive loss of
motor neuron groups in the spinal cord, brain stem and motor cortex. ALS clinically
presents within lower motor neuron with signs of progressive weakening and wasting
of voluntary muscles and upper motor neuron spasticity and hyper-reflexia (Duffy et.
al., 2011).

30 | P a g e
ALS results from mutations in the gene encoding superoxide dismutase 1 (SOD1), the
ubiquitous enzyme that mediates conversion of a superoxide anion, derived from
oxidative phosphorylation, into hydrogen peroxide, which is an imperative role in
antioxidant defence. SOD1 was previously thought to be solely a cytoplasmic enzyme,
but studies have shown that it partially localises to mitochondria, with mutant SOD1
observed to be more abundant in nervous tissue of transgenic mice expressing the
ALS-associated mutants G93A-SOD1, when compared to tissue derived from the liver
or heart (Duffy et. al., 2011).
Proportions of mutant SOD1 have been shown to localise to the mitochondrial IMS,
the site of reactive oxygen species (ROS) generation and with mutant SOD1 identified
in proteinaceous aggregates. This evidence suggests that mutant SOD1 is
preferentially recruited to the IMS, where it acts to increase production of toxic ROS
(Duffy et. al., 2011).
Mutant SOD1 associated with mitochondria has an increased propensity to form cross-
linked oligomers, similar to those formed by ß-amyloid protein in Alzheimer’s disease
(Duffy et. al., 2011). The cross-linking enables mutant SOD1 to bind to the IMM,
shifting the redox state of the mitochondria. The shift in the redox state causes a
change to an oxidising environment, impairing the activity of the respiratory
complexes, with the presence of oxidative stress; SOD1 becomes insoluble,
increasing the potential to form aggregation upon oxidation (Duffy et. al., 2011).
Studies have also predicted that the formation of mutant SOD1 aggregates in both the
mitochondrial matrix, and associating with the cytosolic-facing outer mitochondrial
membrane, may induce stress in mitochondria, potentially damaging the mitochondrial
import machinery, dramatically impairing protein import as well as disturbing ionic

31 | P a g e
homeostasis and dynamic regulation of mitochondria (Liu et. al., 2004; Vijayvergiya
et. al., 2005). Other neurodegenerative diseases involving inference with
mitochondrial function are summarised in Table 1.3.
Functional changes in mitochondria are often accompanied by disturbance in
mitochondrial morphology with changes to membrane integrity, disintegration of
cristae and massive swelling of the mitochondria (Kwong et. al., 2006). Mitochondrial
morphological changes have also been recently associated with HSN-I (Myers. et. al.,
2014).
Disease Mutated Gene Product
Mitochondrial Functions
Charcot-Marie-Tooth type IIA Mitofusin 2
Outer membrane GTPase involved in mitochondrial fusion,
regulation of apoptosis
Amyotrophic Lateral Sclerosis SOD1
Cytosolic ROS scavenging enzyme found to localise to IMS
and matrix when mutated
Alzheimer’s Disease
Amyloid ß Precursor Protein
Mutated protein and its product ß-amyloid localises to mitochondria
where they increase ROS production
Parkinson’s Disease α-synuclein
Cytosolic protein that causes mitochondrial dysfunction leading
to oxidative stress
Huntington’s Disease Huntingtin
Mutant protein binds the outer mitochondrial membrane and
reduces mitochondrial uptake of calcium
Friedreich Ataxia Frataxin
Mitochondrial protein involved in heme biosynthesis, formation of iron sulphur clusters, and iron
detoxification
Table 1.3 Neurodegenerative diseases that interfere with mitochondrial function. Mitochondrial dysfunction in neurodegenerative diseases are not all caused by mutations in mitochondrial-related proteins, such as ALS, AD and HD. Adapted from (Kwong et. al., 2006).

32 | P a g e
1.5.5 Novel link to Mitochondria in HSN-I
Despite the numerous studies of SPTLC1 mutations and the effect upon cellular
functions by sphingolipid metabolism, other mechanisms have not been studied in
detail. Investigations may provide the missing link in our understanding of the
pathogenesis of HSN-I. Mitochondrial morphology and localisation in HSN-I patient-
derived lymphoblasts were examined by transmission electron micrographs (TEMs)
and changes have been reported (Myers et. al., 2014). Mitochondria within the patient-
derived lymphoblasts carrying the C133W and V144D mutations cluster to the
perinuclear area of the cell with the ER seen to be flanking mitochondrial aggregations.
Mitochondrial morphology was also altered with the mitochondrial membrane
displaying discontinuous breakages. The inner matrix exhibited a swollen appearance
with a significant loss of cristae. Perturbation to the mitochondria appear to be mutant-
specific especially within the C133W mutant lymphoblasts.
These striking morphological changes occurring in the mitochondria have not been
previously explored. The discontinuous and abnormal appearance of the
mitochondrial membranes is suggestive of membrane depolarisation, potentially
leading to deficiencies in ATP production and transport. As a result, these changes
may in turn have consequences on axonal transport of mitochondria in neurons, with
a potential for increased apoptosis resulting from the mitochondrial dysfunction, as
demonstrated in ALS. As mitochondria are extremely important in all cells, and with
known adverse effects of mitochondrial dysfunction in peripheral sensory
neuropathies, these changes in mitochondrial function may provide an exciting new
prospect in the understanding of the HSN-I molecular mechanisms.

33 | P a g e
1.6 The Role of ER in neurodegenerative diseases
1.6.1 Protein processing in the ER
The mechanism by which secretory proteins are targeted to the ER during their
translation (the Cotranslational pathway) is well understood (Figure 1.8). The signal
sequences span approximately 20 amino acids, including a stretch of hydrophobic
residues usually at the amino terminus of the polypeptide chain (Cooper and
Hausman, 2007). As the polypeptide chains emerge from the ribosome, signal
sequences are recognised and bound by a signal recognition particle (SRP) consisting
of six polypeptides and a small cytoplasmic RNA (7SL RNA). SRP binds the ribosome
as well as the signal sequence inhibiting further translation and targeting the entire
complex (the SRP, ribosome, and growing polypeptide chain) to the rough ER by
binding to the SRP receptor on the ER membrane (Cooper and Hausman, 2007).
Binding to the receptor releases the SRP from both the ribosome and the signal
sequence of the growing polypeptide chain. The ribosome then binds to a protein
translocation complex in the ER membrane and the signal sequence is inserted into a
membrane channel (Cooper and Hausman, 2007).
The translocation channels through the ER membrane are complexes of three
transmembrane proteins, called the Sec61 proteins. Transfer of the ribosome from the
SRP to the Sec61 complex allows translation to resume and the growing polypeptide
chain is transferred directly into the Sec61 channel and across the ER membrane
(Cooper and Hausman, 2007). As translocation proceeds, the signal sequence is
cleaved by signal peptidase and the polypeptide is released into the lumen of the ER
(Cooper and Hausman, 2007).

34 | P a g e
Figure 1.8 Cotranslational targeting of secretory proteins to the ER. As the signal sequence emerges from the ribosome, it is recognised and bound by the SRP. The SRP escorts the complex to the ER membrane, where it binds to the SRP receptor. The SRP is released, the ribosome binds to a membrane translocation complex of Sec61 proteins and the signal sequence is inserted into a membrane channel. Translation resumes, and the growing polypeptide chain is translocated across the membrane. Cleavage of the signal sequence by signal peptidase releases the polypeptide into the lumen of the ER. Adapted from (Cooper and Hausman, 2007).
Many proteins are targeted to the ER after their translation is complete
(posttranslational translocation) rather than being transferred into the ER during
synthesis on membrane-bound ribosomes (Cooper and Hausman, 2007). These
proteins are synthesised on free cytosolic ribosomes and their posttranslational
incorporation into the ER does not require SRP (Figure 1.9). Cytosolic chaperones are
required to maintain the polypeptide chains in an unfolded conformation so they can
enter the Sec61 channel. BiP (immunoglobulin heavy chain-binding protein), is
required to “pull” the polypeptide chain through the channel and into the ER (Cooper
and Hausman, 2007).

35 | P a g e
Figure 1.9 Posttranslational translocation of proteins into the ER. Proteins destined for posttranslational import to the ER are synthesised on free ribosomes and maintained in an unfolded conformation by cytosolic chaperones. Their signal sequences are recognised by the Sec62/63 complex, which is associated with the Sec61 translocation channel in the ER membrane. The Sec63 protein is also associated with a chaperone protein (BiP), which acts as a molecular ratchet to drive protein translocation into the ER. Adapted from (Cooper and Hausman, 2007).
Protein chaperones facilitate protein folding in the ER, but amino acid posttranslational
modifications such as asparagine (N)-linked glycosylation and disulphide bond
formation are also involved (Cooper and Hausman, 2007). In contrast to the highly
reducing environment of the cytosol, where disulphide bonds do not typically form, the
lumen of the ER is very oxidising, resulting in the rapid formation of disulphide bonds
(Cooper and Hausman, 2007).

36 | P a g e
Proteins destined for secretion or residence within the lumen of the ER, Golgi
apparatus or lysosomes are translocated across the ER membrane and released into
the lumen of the ER. However, proteins destined for incorporation into membranes are
initially inserted into the ER membrane instead of being released into the lumen. From
the ER membrane, they proceed to their final destination through budding and
intracellular vesicular transport. (Cooper and Hausman, 2007).
The ER is also the site of protein folding, assembly of multisubunit proteins, disulphide
bond formation, the initial stages of glycosylation and the addition of glycolipid anchors
to some plasma membrane proteins. Indeed, the primary role of luminal ER proteins
is to catalyse the folding and assembly of newly translocated polypeptides (Cooper
and Hausman, 2007).
As previously mentioned, proteins are translocated across the ER membrane as
unfolded polypeptide chains while their translation is still in progress. These
polypeptides, therefore, fold into their three-dimensional conformations within the ER,
assisted by the molecular chaperones that facilitate the folding of polypeptide chains
(Cooper and Hausman, 2007). Immunoglobulin Binding protein (BiP) binds to the
unfolded polypeptide chain as it crosses the membrane and then mediates protein
folding and the assembly of multisubunit proteins within the ER. Correctly assembled
proteins are released from BiP and are available for transport to the Golgi apparatus.
Abnormally folded or improperly assembled proteins, however, remain bound to BiP
and are consequently retained within the ER or degraded rather than being
transported further along the secretory pathway (Cooper and Hausman, 2007).

37 | P a g e
1.6.2 UPR: Unfolded protein response pathway
As a membranous compartment associated with the critical cellular functions
mentioned, the ER is extremely sensitive to changes that affect its structure, integrity
and function. As such any changes in calcium homeostasis leading to calcium
depletion from the ER lumen, inhibitors of protein glycosylation, inhibitors of
disulphide-bond formation, virus infection, hypoxia, ischemia and growth factor
depletion can all disrupt protein synthesis, translation and folding, resulting in unfolded
or misfolded proteins. The accumulation of unfolded and/or misfolded proteins causes
an imbalance between the synthesis of new proteins and the ER’s ability to process
newly synthesised proteins, resulting in the failure of the ER to cope with the excess
protein load, which is termed ER stress. Cells in turn activate an integrated intracellular
signalling cascade, known as the unfolded protein response (UPR) to avert ER stress
(Rao and Bredesen, 2004).
UPR activation results initially with a transient attenuation in the rate of protein
synthesis. The next event is an upregulation of genes encoding chaperones and other
proteins that prevent polypeptide aggregation and participate in polypeptide folding,
followed by retro translocation and degradation of ER-localised proteins. These
cellular responses minimise the accumulation and aggregation of misfolded proteins
by increasing the capacity of the ER machinery for folding and degradation (Malhotra
and Kaufman, 2007).
During this phase, nascent-folding-competent polypeptides are maintained in a
soluble form by interaction with ER luminal chaperones. BiP/GRP78 (glucose-
regulated protein) are one of the most highly expressed ER resident chaperones. BiP
is a member of the heat-shock protein (Hsp70) family (Malhotra and Kaufman, 2007).

38 | P a g e
Currently there are three identified components of the UPR; the double stranded RNA-
activated protein kinase-like ER kinase (PERK), the eukaryotic translation initiation
factor 2 (eIF2a); the activating transcription factor 6 (ATF6) and the inositol-requiring
enzyme 1 (IRE1). All of these proteins associate with BiP in their inactive states
(Malhotra and Kaufman, 2007). It has been postulated that when ER homeostasis is
perturbed, BiP preferentially binds to and is sequestered by unfolded/misfolded
proteins that accumulate within the ER lumen. As a consequence, BiP dissociates
from the UPR transducers to permit their signalling. Although the UPR pathway is
simultaneously activated upon severe ER stress, the immediate response occurs
through the PERK/eIF2a pathway (Malhotra and Kaufman, 2007).
PERK is a type I ER transmembrane protein kinase. Upon release from BiP, PERK
dimerises to promote self autophosphorylation and activation. Activated PERK
phosphorylates eIF2a, thus attenuating the rate of general translation initiation (Shen
et. al., 2004). In conjunction with PERK activation upon BiP release, the release of BiP
from IRE1 allows its dimerisation and autophosphorylation to activate its site-specific
endoribonuclease (RNase) activity. The RNase activity of IRE1 initiates splicing of a
26-base intron from the X-box-binding protein 1 (XBP1) mRNA, altering the C-terminus
of the protein to create a potent transcription factor (Malhotra and Kaufman, 2007).
ATF6 is a type II transmembrane protein of the ER. BiP release allows ATF6 to transit
to the cis-Golgi compartment, where it is cleaved by site-1 protease and site-2
protease (the same enzymes that process the sterol response element binding protein
(SREBP) upon cholesterol deprivation). The cleaved cytosolic N-terminal fragment of
ATF6 migrates to the nucleus and acts as an active transcription factor (together with
ATF4 and spliced XBP1) to increase the expression of the genes encoding proteins

39 | P a g e
Figure 1.10 ER stress and the unfolded protein response. Stress in the ER stimulates the activation of the three stress receptors; PERK, ATF6 and Ire1, which are involved in the unfolded protein response (UPR). PERK phosphorylates eIF2α which inhibits general protein translation, allowing eIF2α-independent translation of ATF4, which activates transcription of chaperones such as GRP78. ATF6 undergoes specific proteolysis in the Golgi apparatus which leads to activation. IRE1 catalyses the alternative splicing of XBP1 mRNA leading to expression of the active XBP1 transcription factor. Together the three arms of the UPR block protein translation, increase chaperone expression and enhance ER-associated protein degradative pathways. Adapted from (Fulda et. al., 2010).
that function to augment the ER protein folding capacity (Malhotra and Kaufman,
2007). In addition, UPR activated genes stimulate ER biogenesis to compensate for
the increased demand for the protein-folding machinery and accelerate endoplasmic
reticulum associated degradation (ERAD) to remove terminally misfolded proteins
(Malhotra and Kaufman, 2007).
If protein repair by the ER chaperones is unsuccessful, aberrant proteins are cleared
from the ER by ERAD. During ERAD, aberrant proteins are translocated back to the
cytosol and degraded by the ubiquitin-proteasome system (Li et. al., 2011). Disposal
by ERAD involves the retro translocation of aberrant proteins across the ER
membrane to the cytosol, where they are ubiquitinated prior to degradation by the
proteasome (Figure 1.10) (Li et. al., 2011). Proteins that are ubiquitinated are
degraded by cytosolic proteasomes, with the free amino acids reused in protein
synthesis (Li et. al., 2011).

40 | P a g e
Misfolded proteins, and associated ER stress, appears to be an implicated feature of
neurodegenerative diseases. Misfolded proteins, UPR and ER stress induced cell
death could thus be involved in the pathogenesis of several neurodegenerative
disorders. Since these neurodegenerative disorders may be caused by specific mutant
proteins that accumulate as misfolded proteins and escape degradation, it is likely that
ER stress plays an important pathogenetic role.
Studies have suggested the following: firstly, a role for presenilin-1 in the activation of
IRE1 and induction of the UPR; secondly, a role for BiP in binding and limiting the
production of beta-amyloid peptide; and thirdly, reduced cytotoxicity of the ß-amyloid
peptide in caspase-12 deficient mice, suggesting a link between the role of the UPR
and ER stress in Alzheimer’s disease (Rao and Bredesen, 2004). Oxidative stress and
protein misfolding play critical roles in the pathogenesis of these neurodegenerative
diseases that are characterised by fibrillar aggregates composed of misfolded proteins
(Malhotra and Kaufman, 2007). At the cellular level, neuronal death or apoptosis may
be mediated by oxidative stress and/or ER stress. Upregulation of ER stress markers
has been demonstrated in post-mortem brain tissues and cell culture models of many
neurodegenerative disorders, including Parkinson’s disease, Alzheimer’s disease,
ALS and expanded polyglutamine diseases such as Huntington’s disease and
spinocerebellar ataxias (Malhotra and Kaufman, 2007).
It seems likely that the protein chaperone BiP uses ATP/ADP exchange that is
essential to preserve ER function and prevent activation of the UPR. A reduced
efficiency of ATP-dependent BiP-mediated chaperone function may predispose it to
unfolded protein accumulation in the ER, activate the UPR and contribute to cellular
damage and degeneration (Malhotra and Kaufman, 2007).

41 | P a g e
1.7 Mitochondria-Associated Membranes
Cellular processes require the proper communication between mitochondria and the
ER, this communication is facilitated by the mitochondria-associated endoplasmic
reticulum membrane (MAM) (Vance, J 2014). The MAM is characterised by direct
apposition to a mitochondrion, a unique lipid profile, and the expression of a unique
set of proteins involved in Ca2+ signaling, phospholipid biosynthesis, protein folding,
and membrane tethering (Vance, J 2014). The association of the MAM with a
mitochondrion is partially cytoskeletal independent and dynamically changes when
cytosolic Ca2+ level become elevated. The MAM is the centre for intermembrane
transportation of phospholipids and Ca2+ transmission to mitochondria that activates
the tricarboxylic acid cycle (TCA) (Vance, J 2014). However, MAM is also involved in
the interorganellar transport of cholesterol, ceramides, ATP, as well as in proteasomal
protein degradation and lipid droplet formation. Recently the importance of
interorganellar communication in the pathophysiology of neurodegenerative disorders
has come more to light (Paillusson, et. al., 2016).
1.7.1 MAM Calcium Regulation
Calcium exchange via ER–mitochondrial contacts is facilitated following its release
from ER stores via inositol 1, 4, 5- trisphosphate (IP3) receptors (Paillusson, et. al.,
2016). Ca2+ is required by mitochondria to generate ATP via the TCA. Some of the
mitochondrial enzymes involved in ATP synthesis, such as some dehydrogenases,
are regulated by Ca2+ (Vance, J 2014). The concentrations of Ca2+ that is required to
elicit a response are high, however close contacts with the ER allows Ca2+ to be
released from the ER and can be achieved with high local concentrations capable of

42 | P a g e
driving an effect (Paillusson, et. al., 2016). Excessive uptake of Ca2+ by mitochondria
can lead to opening of the mitochondrial permeability transition pore and apoptotic
signalling occurs. Phospholipid exchange also occurs within the MAMs, this is
important because the enzymes involved in some lipid biosynthesis are present in both
organelles and so exchange is required for their production (Vance, J 2014).
MAMs are also known to be a specialised type of lipid raft linking intracellular trafficking
between the mitochondria and ER. Both ER and mitochondria are transported by
kinesin 1 and cytoplasmic dynein molecular motors (Paillusson, et. al., 2016). Kinesin
1 drives anterograde transport within neurons through the axons. Attachment of
mitochondria to kinesin 1 involves the outer mitochondrial membrane (OMM) protein
Miro, which acts as a Ca2+ sensor; elevated Ca2+ levels alters their transport. Miro has
been shown to localise to MAM contact sites and areas of ER have been shown to be
co-transported with mitochondria (Paillusson, et. al., 2016). Potentially some ER may
be transported with mitochondria through axons and Miro may sense Ca2+ exchange
between the two organelles to regulate this transport in response to physiological
stimuli (Paillusson, et. al., 2016).
1.7.2 Autophagy formation at MAMs
Autophagy is the process whereby damaged proteins and organelles are cleared from
the cell by sequestering them in a double membrane-bound vesicle termed the
autophagosome (Krols et.al. 2016). Subsequent delivery to the lysosome allows
proteasomal breakdown and recycling of the substrates. Mitochondria contain many
autophagy-related (ATG) proteins and regulators and have been proposed as an origin
for phagophore formation, Many ATG proteins have been found to accumulate

43 | P a g e
specifically at ER-mitochondria contact sites in conditions of starvation, and there is
mounting evidence that MAMs might be the site of autophagosome formation (Krols
et.al. 2016). Lipid transfer from the ER to mitochondria during autophagosome
biogenesis is crucial. In mouse cells lacking MFN2, autophagy induction was disturbed
due to a decrease in lipid transfer towards mitochondria as a consequence of ER-
mitochondrial uncoupling and lipids transferred from the ER accumulate in the OMM,
from where they are trafficked to the expanding phagophore (Krols et.al. 2016).
In addition, during the selective autophagy of mitochondria, known as mitophagy, ER-
mitochondrial contact sites appear to constitute a platform promoting mitophagosome.
Mitochondria destined for degradation recruit the E3 ubiquitin ligase, Parkin, to the
OMM through PTEN-induced putative kinase 1 (PINK1) kinase activity (Krols et.al.
2016). Ubiquitination of Parkin substrates subsequently recruits autophagy proteins.
Accumulation of Double FYVE-containing protein 1 (DFCP1) along mitochondria that
are labelled for degradation with Parkin suggests a role for the ER/MAM as a
membrane source for the mitophagosome (Krols et.al. 2016). Drp1-mediated fission,
is in part mediated by contacts with the ER suggesting that it promotes mitophagy by
breaking off pieces of mitochondria thus enhancing engulfment by the
mitophagosome. Depolarisation of the IMM via Ca2+ over-loading could be involved in
PINK1 and Parkin translocation to the OMM, activating mitophagy (Krols et.al. 2016).
The presence at the MAMs of several important players in metabolism, such as
mechanistic target of rapamycin (mTOR), suggest that ER-mitochondrial contact could
represent a site of crosstalk between sensors of the cells energetic needs and the
early players in autophagy (Vance, J 2014). Additionally, through interorganellar
communication, dysfunctional mitochondria may be detected at MAMs, followed by

44 | P a g e
mitophagy induction and targeted removal of these unhealthy mitochondria (Vance, J
2014).
1.7.3 Lipid transportation via MAMs
Significant alterations in membrane lipid content is usually not tolerated by cells or
organelles, thus lipid composition of organelle membranes are highly regulated. The
synthesis of the majority of membrane phospholipids in eukaryotic cells occurs on ER
membranes and these lipids are subsequently distributed (Krols et.al. 2016). However
not much is understood about the molecular mechanisms that are involved in
interorganellar protein transport including lipid trafficking (Vance, J 2014).
Spontaneous diffusion of phospholipids between organelles through the cytosol is an
energetically unfavourable process and therefore occurs at very slow rates (Krols et.al.
2016). Lipids are transported between organelles by well characterised vesicle
mediated processes that are used for protein trafficking, but mitochondria are not
known to be connected to the ER by vesicular trafficking pathways (Krols et.al. 2016).
Non-vesicular mechanisms are important for the trafficking of lipids between
organelles. Although the cytosol contains several lipid transfer proteins that can
mediate phospholipid transfer between membranes, the net interorganellar transfer of
lipids mediated by these soluble transport proteins is yet to be demonstrated (Krols
et.al. 2016).
Compared to other organelles, mitochondria, particularly their inner membranes, are
enriched in phosphatidylethanolamine (PE) (Vance, J 2014). PE is synthesised in
mammalian cells by two major pathways that operate in spatially distinct organelles;
in the ER via the CDP–ethanolamine pathway, and in mitochondria by the

45 | P a g e
decarboxylation of phosphatidylserine (PS) (Vance, J 2014). The PS decarboxylation
pathway provides the majority of PE in at least some cell types. For PE production by
this pathway, PS must be imported from its site of synthesis in the ER/MAM to IMM
where PS decarboxylase (PSD) is located (Vance, J 2014). Thus, two distinct pools of
PE can potentially be generated: one in the ER, the other in mitochondria. In
mammalian cells, the PE that is synthesised by PSD in mitochondria is essential for
the normal functioning of mitochondria and cell survival (Krols et.al. 2016). A reduction
in the PE content of mitochondria can profoundly alter their morphology and reduce
cell growth, oxygen consumption and ATP production via the mitochondrial electron
transport chain (Vance, J 2014). The mechanism by which PS is imported into
mitochondria has been extensively studied in mammalian cells. The two mammalian
base-exchange enzymes that synthesise PS (PS synthase-1 and -2) are highly
enriched in MAM compared to the ER. The rate-limiting step in PE production from PS
is the transfer of PS from the ER/MAM to the OMM (Krols et.al. 2016).
Sterols and sphingolipids are minor constituents of mitochondrial membranes. The
processes by which these lipids are transported to, and imported into, mitochondria
have yet to be fully elucidated. MAMs have been reported to be enriched in cholesterol
relative to that of the ER (Krols et.al. 2016). Some studies with MAMs and
mitochondria have suggested that the depletion of cholesterol from MAMs promotes
the association between them and mitochondria, thereby increasing the conversion of
PS to PE (Krols et.al. 2016). Another functionally significant, sphingolipid constituent
of mitochondria is ceramide which can induce mitochondria-mediated apoptosis by
permeabilising the OMM (Vance, J 2014). However, the origin of the mitochondrial
pool of ceramide is not entirely clear. Although a large amount of cellular ceramide is

46 | P a g e
probably synthesised in the ER, some ceramide appears to be made in the MAM, and
some ceramide is also synthesised in mitochondria (Krols et.al. 2016). However,
despite evidence that PS is imported into mitochondria via the MAM, more
investigation is required to elucidate the exact role attributed to MAMs in the import of
other lipids into mitochondria from the ER (Vance, J 2014).
1.7.4 MAMs and Neurodegeneration
A major function of mitochondria is the generation of energy as ATP through oxidative
phosphorylation via the electron transport chain. Several disorders exhibit alterations
in mitochondrial morphology and/or calcium homeostasis, both of which can be altered
by MAMs (Vance, J 2014). Neurons are highly polarised cells that rely heavily upon
energy generated by mitochondria, particularly mitochondria that are recruited to
dendrites and axons at sites far removed from the cell bodies (Krols et.al. 2016). Thus,
the distribution of mitochondria within axons and dendrites is crucial for determining
neuronal survival since mitochondria and elements of the ER are present in these
neuronal processes, it is likely that axons and dendrites contain zones of contact
between MAM and mitochondria (Krols et.al. 2016). Impaired regulation of
mitochondrial calcium homeostasis provides one potential link between neuronal
dysfunction and disruption of MAM–mitochondria contact sites since mitochondrial
calcium modulates neurotransmitter release at the synapse (Vance, J 2014).
Alterations in mitochondrial dynamics and morphology are common features of
neurodegenerative diseases such as Alzheimer's, Parkinson's and Huntington's
disease (Krols et.al. 2016). Whether alterations in mitochondrial fragmentation

47 | P a g e
highlight a response to the disease pathogenesis or directly contribute to the disease
has not yet been established. Nevertheless, loss of either fusion or fission of
mitochondria can severely impair mitochondrial function and neuronal survival (Vance,
J 2014). Several neurodegenerative diseases are characterised by either an
abnormally high or low level of the mitochondrial fission factor, DRP1. DRP1 activity
and regulation of mitochondrial fragmentation have been linked to the stability of
mitochondrial–MAM contact sites (Krols et.al. 2016).
Excessive mitochondrial fragmentation and slower axonal transport of mitochondria
are prominent features of Charcot–Marie–Tooth disease type 2A, a peripheral
neuropathy that is caused by mutations in the mitochondrial fusion protein MFN2
(Vance, J 2014). Similarly, the neuropathy, dominant optic atrophy, is caused by
mutations in another mitochondrial fusion factor, OPA1, resulting in diminished
mitochondrial fusion. Consequently, since MAMs have been implicated in regulating
mitochondrial fusion and fission, reduced formation of contacts between MAMs and
mitochondria might contribute to the pathogenesis of these disorders (Vance, J 2014).
ER–mitochondrial contact have been linked to ER stress and increased UPR. The ER
UPR is an intracellular signalling pathway that is activated by the accumulation of
unfolded proteins in the ER, which then stimulates transcriptional responses to
modulate the protein-folding capacity of the ER (Krols et.al. 2016). Two of the known
tethering proteins involved in connecting ER with mitochondria, MFN2 and vesicle-
associated membrane protein-associated protein B (VAPB), have roles in the UPR. A
variety of ER chaperones involved in protein folding, such as BiP, calnexin, calreticulin,
ERp44, ERp57, and the Sigma 1 receptor, are also present in MAMs, and structural

48 | P a g e
uncoupling of ER from mitochondria induces ER stress (Krols et.al. 2016). Thus,
crosstalk between ER and mitochondria at MAM contact sites may have a role in
facilitating stress responses and the UPR (Krols et.al. 2016).
MAMs have also been linked to formation of the inflammasome. Tissue damage and
cell stresses, such as those that occur in neurodegenerative diseases, are sensed by
the innate immune system through recognition receptors (Krols et.al. 2016). One class
of these is the NOD-like receptors (NLRs), which sense abnormal cytosolic changes.
Upon activation, some NLRs, including NLRP3, form multiprotein complexes function
to initiate proteolytic maturation of the proinflammatory cytokine interleukin 1b (IL-1b)
(Krols et.al. 2016). Reactive oxygen species from mitochondria are one signal for
activation of the NLRP3 inflammasome. Recently, ROS was shown to cause the
relocation of NLRP to MAMs, and this may provide a mechanism whereby NLRP
senses damage to mitochondria to activate the inflammasome (Krols et.al. 2016).

49 | P a g e
1.8 Aims of this Thesis
SPTLC1 mutations reduce the activity of SPT in HSN-I patient-derived lymphoblasts.
However, the mechanism by which this occurs is yet to be identified. Notably,
conflicting evidence has arisen in the literature. While SPT is inhibited in the disease
state and thus should reduce or stop sphingolipid production, some studies have
reported no detectable changes in sphingolipid metabolism (Dedov et. al., 2004;
Verhoeven et. al., 2006). Why this occurs is still unknown but some studies suggest
that the overall reduction in SPT is not enough to globally reduce sphingolipid
metabolism, yet axonal degeneration is still observed in HSN-I (Penno et. al., 2010).
Recent studies suggest that these mutations cause a deleterious gain of function with
the production of toxic sphingolipids (Hornemann et. al., 2009).
Myers et. al., 2014, have observed by transmission electron microscopy (TEM) in
HSN-I patient-derived lymphoblasts that the mitochondrial morphology is completely
altered and that the ER, wraps these morphologically challenged mitochondria,
showing physical contact between the two organelle membranes.

50 | P a g e
We hypothesise that SPTLC1 mutations in HSN-I cause alterations in mitochondrial
and ER proteins, and potentially cause alterations to lipids within the cells.
To investigate this hypothesis, this study aims to:
• Isolate clean mitochondrial protein fractions and to separate them into soluble
and membrane fractions.
• Determine by 2-dimensional gel electrophoresis (2DGE) alterations in the
protein profiles as a result of the SPTLC1 mutations.
• Identify the altered proteins observed.
• Isolate and determine the exact protein-protein interactions occurring due to
the altered proteins.
• Transiently transfect neuronal cells to produce the SPTLC1 mutations and
then confirm changes observed in patient-derived lymphoblast cell model in a
neuronal cell
• Determine whether other cellular functions are altered due to the disease state
and protein alterations.
• Identify microsomal protein alterations and interactions caused by the disease
state.
A patient-derived lymphoblast cell model was chosen as they endogenously express
the SPTLC1 mutation; furthermore lymphoblasts are easily isolated and cultured
lending themselves as a useful cell culture system (Verhoeven et. al., 2006).

51 | P a g e
Paper I
Published in Analytical Biochemistry
Contributions
SES carried out all experimentation, data analysis and formatted data.

Analytical Biochemistry 475 (2015) 1–3
Contents lists available at ScienceDirect
Analytical Biochemistry
journal homepage: www.elsevier .com/locate /yabio
Notes & Tips
Optimal isolation of mitochondria for proteomic analyses
http://dx.doi.org/10.1016/j.ab.2015.01.0050003-2697/� 2015 Elsevier Inc. All rights reserved.
⇑ Corresponding authors at: Molecular Medicine Research Group, School ofMedicine, University of Western Sydney, Campbelltown, NSW 2560, Australia. Fax:+61 4620 3890 (J.R. Coorssen). Neuro-Cell Biology Laboratory, School of Science andHealth, University of Western Sydney, Campbelltown, NSW 2560, Australia. Fax:+61 4620 3025 (S.J. Myers).
E-mail addresses: [email protected] (J.R. Coorssen), [email protected](S.J. Myers).
1 Abbreviations used: 2DE, two-dimensional gel electrophoresis; AMPIB, AmrescoMitochondrial Protein Isolation Buffer; ER, endoplasmic reticulum; PBS, phosphate-buffered saline; EDTA, ethylenediaminetetraacetic acid; 2D, two-dimensional.
Scott E. Stimpson a,b, Jens R. Coorssen b,c,⇑, Simon J. Myers a,b,⇑a Neuro-Cell Biology Laboratory, School of Science and Health, University of Western Sydney, Campbelltown, NSW 2560, Australiab Molecular Medicine Research Group, School of Medicine, University of Western Sydney, Campbelltown, NSW 2560, Australiac Molecular Physiology Unit, School of Medicine, University of Western Sydney, Campbelltown, NSW 2560, Australia
a r t i c l e i n f o a b s t r a c t
Article history:Received 31 October 2014Received in revised form 23 December 2014Accepted 8 January 2015Available online 14 January 2015
Keywords:MitochondriaProteomicsTwo-dimensional gel electrophoresis
Considering the key role of mitochondria in cellular (dys)functions, we compared a standard isolationprotocol, followed by lysis in urea/detergent buffer, with a commercially available isolation buffer thatrapidly yields a mitochondrial protein fraction. The standard protocol yielded significantly better overallresolution and coverage of both the soluble and membrane mitochondrial proteomes; although the kitwas faster, it resulted in recovery of only approximately 56% of the detectable proteome. The qualityof ‘‘omic’’ analysis depends on sample handling; for large-scale protein studies, well-resolved proteomesare highly dependent on the purity of starting material and the rigor of the extraction protocol.
� 2015 Elsevier Inc. All rights reserved.
Considering the key role of mitochondria in cellular (dys)functions, we compared a standard isolation protocol, followed bylysis in urea/detergent buffer, with a commercially available isola-tion buffer that rapidly yields a mitochondrial protein fraction. Thestandard protocol yielded significantly better overall resolution andcoverage of both the soluble and membrane mitochondrial proteo-mes; although the kit was faster, it resulted in recovery of onlyapproximately 56% of the detectable proteome. The quality of any‘‘omic’’ analysis depends on sample handling; for large-scaleprotein studies, well-resolved proteomes are highly dependent onthe purity of the starting material and the rigor of the extractionprotocol. In many neurodegenerative disorders, mitochondria areknown to play key roles in disease etiology [1]; because theseintracellular powerhouses have critical roles in the maintenanceand stability of cellular homeostasis, there are catastrophic conse-quences with mitochondrial dysfunction. To better understandthe molecular mechanisms underlying mitochondrial function, weroutinely analyzed the proteome of this organelle [2] using high-resolution two-dimensional gel electrophoresis (2DE)1 [3] stainedusing a modified colloidal Coomassie blue protocol [4].
In efforts to optimize ongoing proteomic analyses, we thoughtto switch from well-established protocols for intact mitochondrialisolation and protein extraction to the promise of more rapid pro-tein isolation using a commercially available kit. Thus, we carriedout a detailed assessment of mitochondrial proteins harvestedusing the Amresco Mitochondrial Protein Isolation Buffer (AMPIB)relative to those extracted by 2DE sample buffer following stan-dard isolation of mitochondria by sucrose gradient fractionation.A comparison of resulting total mitochondrial proteomes indicated510 ± 3 resolved protein species in the AMPIB extracts relative tothe extracts of isolated mitochondria, 583 ± 7 (Fig. 1A and D). Nota-bly, however, there was 87% overlap between the datasets. Toassess this further, we subjected isolated mitochondria to a well-established protocol to recover separate total membrane and solu-ble protein fractions prior to protein extraction and analysis. Theresulting 2DE analyses were consistently of markedly lower qualityfor the AMPIB extracts (Fig. 1B and C) relative to those using stan-dard 2DE sample buffer (Fig. 1E and F); there was a markedincrease in the resolution of proteins in the low- to mid-molecu-lar-weight region (i.e., < �75 kDa) across the full range of 3 to10 pI using the standard method as opposed to AMPIB. Essentially,no membrane proteins of less than approximately 75 kDa and nosoluble proteins of less than approximately 25 kDa were resolvedfrom the AMPIB isolates. To determine what low-molecular-weightproteins were absent from the AMPIB isolation methods, a fewprotein species were identified from membrane and solublefractions—from the membrane annexin V (20 kDa, 5 pI) andvoltage-dependent anion channel 1 (30 kDa, 8.7 pI) and from thesoluble 60-kDa heat shock protein (60 kDa, 6 pI) and inorganic

Total Membrane SolubleA
3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
510 ± 3
B3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
346 ±6
C3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
280 ± 4
D 3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
583 ± 7
E3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
550 ± 9
F3 pI 10
MW
(kD
a)
250150
100
7537
25
20
1510
576 ± 6
Fig.1. Average union two-dimensional gel images of mitochondrial protein isolates from human lymphoblasts. Protein fractions isolated using AMPIB: (A) totalmitochondrial proteins; (B, C) membrane and soluble mitochondrial proteins. Protein fractions isolated using sucrose density gradients: (D) total mitochondrial proteins; (E,F) membrane and soluble mitochondrial proteins. Here, 100 lg of protein was used for each 2DE analysis (n = 3) [3]. 2DE was carried out, and resulting gels were imaged andanalyzed using Delta2D software as described previously [4,7,9]. MW, molecular weight.
MTCO260 kDa
TOMM 2215 kDa
1 2 3 4
Fig.2. Representative immunoblot images of MTCO2 and TOMM 22 from AMPIBand sucrose gradient isolated mitochondrial proteins (n = 3). Lanes 1 and 2represent AMPIB isolated mitochondrial proteins, and lanes 3 and 4 representsucrose gradient isolated mitochondrial proteins. MTCO2 was present in bothAMPIB and sucrose gradient mitochondrial fractions; however, the lower molecularweight protein TOMM 22 was present only in the sucrose gradient mitochondrialfraction.
2 Notes & Tips / Anal. Biochem. 475 (2015) 1–3
pyrophosphatase 2 (39 kDa, 7 pI). This is in contrast to the manu-facturer’s information indicating that, following cell lysis, the cyto-solic fraction yielded by buffer extraction is separated bycentrifugation from an enriched mitochondrial fraction to increasethe likelihood of detecting low-abundance proteins. Immunoblotanalysis (Fig. 2) revealed the high-molecular-weight proteinMTCO2 present in both AMPIB and sucrose gradient isolated mito-chondrial proteins; however, the low-molecular-weight proteinTOMM 22 was present only in the sucrose gradient isolated pro-teins correlating with Fig. 1. This apparent loss of low-molecular-weight mitochondrial proteins from the AMPIB kit may result fromthe kit not isolating intact mitochondria; such disruption of mem-branes and loss of soluble proteins in the multiple centrifugationsteps could be the cause. Endoplasmic reticulum (ER) is a sourceof contamination during subcellular fractionations; to ensure thatboth isolation methods were free from contamination, we carriedout immunoblot analyses with specific ER markers, such as calnex-in and BiP, with immunoblot analysis revealing that there was nocontamination in either isolation method. It would appear thatAMPIB yields quantitatively different extraction of mitochondrial(and perhaps some cytosolic) proteins from cell lysates; in contrastto what might be expected, this selective extraction is even morepronounced when starting with isolated mitochondria. However,if total mitochondria isolated are to be quantitatively analyzed,then the AMPIB method would seem to have quite limited capacityin terms of genuinely reflecting the mitochondrial proteome. Ourdata unequivocally show that to obtain a full protein profile formitochondria (i.e., either the total protein pool, the mitochondrialmembrane pool, or the soluble mitochondrial protein pool), a tra-ditional subcellular fractionation method using sucrose densitygradient centrifugation is most optimal.
In the Amresco mitochondrial protein isolation protocol(according to the manufacturer’s instructions), briefly, cells
(1 � 106) were centrifuged at 1500g for 5 min at 4 �C, and theresulting pellet was washed in 10 ml of cold 1� phosphate-bufferedsaline (PBS). The supernatant was removed, and the cell pellet wassuspended in 1 ml of ice-cold 1� PBS, transferred to a 1.5-ml micro-centrifuge tube, and centrifuged at 1500g for 5 min at 4 �C. Theresulting pellet was resuspended in Mitochondrial Protein IsolationBuffer (400 ll for 1 � 106 cells). The cells were homogenized on iceby passaging 20 times through a 1cc syringe with a 26.5-gaugeneedle, followed by centrifugation at 1000g for 10 min at 4 �C.The supernatant was collected and transferred to a fresh 1.5-mltube, and the pellet (e.g., whole cells and nuclei) was discarded.The collected supernatant was centrifuged at 14,000g for 15 minat 4 �C, and the resulting supernatant was collected and transferredinto a new tube labeled ‘‘cytosolic proteins.’’ The residual pellet(containing mitochondrial proteins) was resuspended in Mitochon-drial Protein Isolation Buffer (1 ml for 1 � 106 cells), which wasthen centrifuged at 14,000g for 1 min at 4 �C. The supernatantwas discarded, and the pellet was resuspended in 40 ll of

Notes & Tips / Anal. Biochem. 475 (2015) 1–3 3
Mitochondrial Protein Isolation Buffer (i.e., as per 1 � 106 cells ofstarting material).
In mitochondrial isolation using sucrose density gradientcentrifugation [5,6], briefly, cells (1 � 106) were pelleted at 1500gfor 5 min at 4 �C and then washed in 10 ml of ice-cold 1� PBS priorto suspension in 10 ml of ice-cold CaSRB buffer (10 mM NaCl,1.5 mM CaCl2, and 10 mM Tris–HCl, pH 7.5) and incubation onice for 10 min. Cells were homogenized using a Douncehomogenizer (Kimble–Chase, USA), and 7 ml of 2.5� MS buffer(210 mM mannitol, 70 mM sucrose, 5 mM ethylenediaminetetra-acetic acid [EDTA], and 5 mM Tris–HCl, pH 7.6) was added torestore isotonicity. Homogenate was centrifuged at 700g for5 min to remove nuclei and unbroken cells. The resulting superna-tant was centrifuged at 15,000g for 10 min to pellet the crudemitochondria. Sucrose gradients were made in 4-ml high-speedcentrifuge tubes (Beckman Coulter, USA) by overlaying 1 ml of1.7 M sucrose buffer (1.7 M sucrose, 10 mM Tris base, and0.1 mM EDTA, pH 7.6) with 1.6 ml of 1.0 M sucrose buffer (1.0 Msucrose, 10 mM Tris base, and 0.1 mM EDTA, pH 7.6). The mito-chondrial pellet was resuspended in 1.6 ml of 1� MS buffer, lay-ered on top of the sucrose gradient, and centrifuged at 40,000gfor 30 min at 4 �C. The mitochondrial band in the middle of the gra-dient was gently removed using a 20-gauge needle, transferred to a1.5-ml tube, and centrifuged at 16,000g for 15 min. The resultingpellet was resuspended in two-dimensional (2D) solubilizationbuffer containing 8 M urea, 2 M thiourea, 4% (w/v) CHAPS, and acocktail of protease inhibitors [7–9] for total proteome analysisor resuspended in PBS for further separation into membrane andsoluble fractionations.
In membrane and soluble protein fractionation, harvested mito-chondrial proteins from both the AMPIB and sucrose gradient wereseparated into membrane and soluble protein fractions asdescribed previously [10]. Briefly, isolated mitochondria wereplaced in 20 mM Hepes for 3 min on ice with an equal volume of
2� PBS subsequently added. Membranes were collected at125,000g for 3 h. The supernatant was collected, and the mem-brane pellet was resuspended in 1� PBS and spun at 125,000gfor a further 3 h. Washed membranes were solubilized in 2D solu-bilization buffer containing 8 M urea, 2 M thiourea, and 4% (w/v)CHAPS. Soluble protein fractions for both isolation methods wereconcentrated using 3-kDa cutoff Millipore Amicon Ultra Centrifu-gal filters and resuspended in 4 M urea.
References
[1] P.J. Hollenbeck, W.M. Saxton, The axonal transport of mitochondria, J. Cell Sci.118 (2005) 5411–5419.
[2] S. Myers, C. Malladi, R. Hyland, T. Bautista, R. Boadle, P. Robinson, G. Nicholson,Mutations in the SPTLC1 protein cause mitochondrial structural abnormalitiesand endoplasmic reticulum stress in lymphoblasts, DNA Cell Biol. 33 (2014)399–407.
[3] M. Churchward, R.H. Butt, J. Lang, K. Hsu, J. Coorssen, Enhanced detergentextraction for analysis of membrane proteomes by two-dimensional gelelectrophoresis, Proteome Sci. 3 (2005) 5.
[4] V.J. Gauci, M.P. Padula, J.R. Coorssen, Coomassie blue staining for highsensitivity gel-based proteomics, J. Proteomics 90 (2013) 96–106.
[5] P. Bozidis, C.D. Williamson, A.M. Colberg-Poley, Isolation of endoplasmicreticulum, mitochondria, and mitochondria-associated membrane fractionsfrom transfected cells and from human cytomegalovirus-infected primaryfibroblasts, Curr. Protoc. Cell Biol. chap. 3 (2007). unit 3.27.
[6] A.V. Vaseva, U.M. Moll, Identification of p53 in mitochondria, Methods Mol.Biol. 962 (2013) 75–84.
[7] R.H. Butt, M. Lee, S.A. Pershahid, P. Backlund, S. Wood, J.R. Coorssen, An initialproteomic analysis of human preterm labour: placental membranes, J.Proteome Res. 5 (2006) 3161–3172.
[8] R.H. Butt, T.A. Pfeifer, A. Delaney, T.A. Grigliatti, W.G. Tetzlaff, J.R. Coorssen,Enabling coupled quantitative genomic and proteomic analyses from rat spinalcord samples, Mol. Cell. Proteomics 6 (2007) 1574–1588.
[9] E.P. Wright, M.A. Partridge, M.P. Padula, V.J. Gauci, C.S. Malladi, J.R. Coorssen,Top-down proteomics: enhancing 2D gel electrophoresis from tissueprocessing to high-sensitivity protein detection, Proteomics 14 (2014) 872–889.
[10] R.H. Butt, J.R. Coorssen, Postfractionation for enhanced proteomic analyses:routine electrophoretic methods increase the resolution of standard 2D-PAGE,J. Proteome Res. 4 (2005) 982–991.

55 | P a g e
Paper II
Published in Journal of Chemical Biology
Contributions
SES carried out all experimentation, data analysis and wrote first draft manuscript.

ORIGINAL ARTICLE
Mitochondrial protein alterations in a familial peripheralneuropathy caused by the V144D amino acid mutationin the sphingolipid protein, SPTLC1
Scott E. Stimpson & Jens R. Coorssen & Simon J. Myers
Received: 17 July 2014 /Accepted: 29 October 2014 /Published online: 14 November 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract Axonal degeneration is the final common path inmany neurological disorders. Subsets of neuropathies involv-ing the sensory neuron are known as hereditary sensory neu-ropathies (HSNs). Hereditary sensory neuropathy type I(HSN-I) is the most common subtype of HSN with autosomaldominant inheritance. It is characterized by the progressivedegeneration of the dorsal root ganglion (DRG) with clinicalsymptom onset between the second or third decade of life.Heterozygous mutations in the serine palmitoyltransferase(SPT) long chain subunit 1 (SPTLC1) gene were identified
as the pathogenic cause of HSN-I. Ultrastructural analysis ofmitochondria from HSN-I patient cells has displayed uniquemorphological abnormalities that are clustered to theperinucleus where they are wrapped by the endoplasmic re-ticulum (ER). This investigation defines a small subset ofproteins with major alterations in abundance in mitochondriaharvested from HSN-I mutant SPTLC1 cells. Using mito-chondrial protein isolates from control and patient lympho-blasts, and a combination of 2D gel electrophoresis, immuno-blotting and mass spectrometry, we have shown the increasedabundance of ubiquinol-cytochrome c reductase core protein1, an electron transport chain protein, as well as the immuno-globulin, Ig kappa chain C. The regulation of these proteinsmay provide a new route to understanding the cellular andmolecular mechanisms underlying HSN-I.
Keywords Hereditary sensory neuropathy type 1 . Serinepalmitoyltransferase long chain subunit 1 . Mitochondria .
Ubiquinol-cytochrome c reductase core protein 1
Introduction
Subsets of neuropathies involving the sensory neuron areknown as hereditary sensory neuropathies (HSNs). HSNsare associated with a range of clinical presentations, path-ologic alterations, electrophysiological abnormalities andincreasingly specific biochemical or molecular geneticabnormalities [10].
Hereditary sensory neuropathy type I (HSN-I) is the mostcommon subtype of the HSN [10]. With autosomal dominantinheritance, it is characterised by the progressive degenerationof the dorsal root ganglion (DRG) and an onset of clinicalsymptoms between the second or third decade of life [25].HSN-I is rarely fatal but imposes lifelong disability with thedisease initially manifesting with sensory loss in the feet,
S. E. Stimpson : S. J. MyersNeuro-Cell Biology Laboratory, University of Western Sydney,Penrith, Australia
J. R. CoorssenMolecular Physiology, University of Western Sydney, Penrith,Australia
S. E. Stimpson : J. R. Coorssen : S. J. MyersMolecular Medicine Research Group, University ofWestern Sydney,Penrith, Australia
S. E. Stimpson : J. R. Coorssen : S. J. MyersSchool of Science and Health, University of Western Sydney,Penrith, Australia
J. R. Coorssen : S. J. MyersSchool of Medicine, University of Western Sydney, Locked Bag1797, Penrith, NSW 2751, Australia
S. J. Myers (*)University of Western Sydney, Office 21.1.05, Campbelltowncampus, Locked Bag 1797, Penrith, NSW 2751, Australiae-mail: [email protected]
J. R. Coorssen (*)School of Medicine, University of Western Sydney, Office 30.2.15,Campbelltown campus, Locked Bag 1797, Penrith, NSW 2751,Australiae-mail: [email protected]
J Chem Biol (2015) 8:25–35DOI 10.1007/s12154-014-0125-x

followed by distal muscle wasting and weakness, and subse-quent positive sensory phenomena such as lancinating or ‘shoot-ing’ pains. Heterozygous mutations in the serinepalmitoyltransferase (SPT) long chain subunit 1 (SPTLC1) wereidentified as the pathogenic cause of HSN-I [1, 6]. The associ-atedmutations in this gene occur at single amino acids which arehighly conserved throughout different species and are thereforelikely to interfere with SPT functionality and structure [25].
SPT is a pyridoxal 5′-phosphate-dependent multimeric en-zyme that catalyses the first step in the biosynthesis ofsphingolipids, ceramide and sphingomyelin [14]; mutationsin the SPT subunits thus result in potential dysfunction andperturbations in sphingolipid synthesis and metabolism linkedto a variety of diseases, in particular HSN-I [26]. As the rate-determining enzyme in the de novo sphingolipid synthesispathway, SPT is therefore a key enzyme in the regulation ofcellular sphingolipid content by condensation of palmitoylcoenzyme A (CoA) wi th L - s e r i ne to fo rm 3-ketodihydrosphingosine. SPT is composed of three knownsubunits: SPTLC1, SPTLC2 and SPTLC3 [12]. SPT is a type1 integral membrane protein with a single highly hydrophobicdomain in the amino-terminal region that anchors the enzymeto the endoplasmic reticulum (ER) membrane [18, 26, 29, 30].
Neurodegenerative diseases are a clinically heterogeneousgroup of chronic progressive illnesses with varying, but dis-tinct, clinical manifestations. Many of the genetic causes ofsuch disorders, including Huntington’s disease, some forms offamilial amyotrophic lateral sclerosis (ALS), Charcot-Marie-Tooth syndrome type II (CMT II), Parkinson’s disease,Friedreich’s ataxia and Alzheimer’s disease, are wellestablished; notably, despite obvious differences in underlyingaetiologies, a role for mitochondrial dysfunction is evident inthe pathogenesis of all these diseases [9, 17, 19].
Mitochondrial dysfunction in neurons can lead to a myriadof different effects such as apoptosis, oxidative stress,excitotoxicity and destructive rises in intracellular calciumlevels that contribute to several pathologies of the nervoussystem [13]. Mitochondrial transport is also intimately depen-dent upon the functional state of the cell and of mitochondriathemselves. Functioning mitochondria are essential for neuro-nal survival due to their long axonal processes and high de-mand for energy [17]. Mitochondrial membrane depolarisationand inhibition of ATP synthesis have been shown to altermovement of organelles; 80 % of slightly depolarised mito-chondria in DRG neurons undergo retrograde movement, im-plying that unhealthymitochondria are returned to the cell bodyfor repair or removal, reducing the number of mitochondria thatare transported in the anterograde direction [22].
Recently, it has been shown that mitochondria from HSN-Ipatient cells, expressing the V144D SPTLC1 mutant, haveexceptionally electron-dense cristae [23]. Considering thisfinding, in this study, we have investigated altered proteinexpression changes from the mitochondria of HSN-I patient
cells using an integrated cell biology and proteomic method-ology. This combined approach yields a detailed profile ofmitochondrial proteins using coupled 2DE and mass spectro-metric technologies. The resulting proteomic analyses identi-fied, for the first time, a statistically significant increase in theabundance of ubiquinol-cytochrome c reductase core protein Iand the immunoglobulin protein, Ig kappa chain C protein.
Materials
All cell culture stock solutions, including RPMI-1640, foetalbovine serum (FBS), penicillin (100 U/mL), streptomycin(100 μg/mL), L-glutamine (2 mM), 4(2-hydroxyethyl)-1-piperazineethane sulfonic acid (HEPES) (1 M) andphosphate-buffered saline (PBS), were purchased from GibcoInvitrogen (Australia). Cell culture consumables were pur-chased from BD Falcon (Greiner, USA). MTCO2, Tomm22, ubiquinol-cytochrome c reductase core protein 1, Ig kappachain C and GAPDH primary antibodies were purchased fromAbcam (USA). Secondary HRP mouse antibodies and 4', 6-diamidino-2-phenylindole (DAPI) stains were purchased fromSigma-Aldrich (Australia).
Methods
EBV-transformed lymphoblasts
Epstein-Barr virus (EBV)-transformed control and V144DHSN-I patient lymphoblasts were kindly provided by Prof.Garth Nicholson (Molecular Medicine Laboratory, Anzac Re-search Institute, Sydney) [7].
Lymphoblast cultures
Lymphoblasts were cultured in RPMI-1640 media (Gibco),supplemented with FBS (10 % v/v), penicillin (1 U/mL),streptomycin (1 μg/mL), L-glutamine (2 mM) and HEPES(1 M) at 37 °C in a humidified atmosphere of 5 % CO2, usingT75 cm2 culture flasks (Greiner, Interpath). Prior to use inbiochemical assays, lymphoblasts were collected by centrifu-gation at 1500×g (5 min at RT) and washed in PBS. Cellcounts were obtained using the Countess Automated CellCounter (Invitrogen, Australia).
Isolation of mitochondrial proteins
Briefly, mitochondria were isolated using a sucrose den-sity gradient [2, 24]. Lymphoblasts were first centrifugedat 1500×g for 5 min, and the cells were then washed in10 mL of ice-cold 1× PBS prior to suspension in 10 mLice-cold CaSRB buffer (10 mM NaCl, 1.5 mM CaCl,
26 J Chem Biol (2015) 8:25–35

10 mM Tris–HCl, pH 7.5) and left on ice for 10 min.Cells were homogenised using a Dounce homogenizer(Kimble-Chase, USA), and 7 mL of 2.5× MS buffer(210 mM mannitol, 70 mM sucrose, 5 mM ethylenedi-aminetetraacetic acid (EDTA), 5 mM Tris–HCl, pH 7.6)was added to restore isotonicity. Homogenate was centri-fuged at 700×g for 5 min to remove nuclei and unbrokencells. The resulting supernatant was centrifuged at15,000×g for 10 min to pellet the crude mitochondria.Sucrose gradients were made in 4-mL high-speed centri-fuge tubes (Beckman Coulter, USA) by adding 1 mL of1.7 M sucrose buffer (1.7 M sucrose, 10 mM Tris-base,0.1 mM EDTA, pH 7.6) overlayed with 1.6 mL of 1.0 Msucrose buffer (1.0 M sucrose, 10 mM Tris-base, 0.1 mMEDTA, pH 7.6). The mitochondrial pellet was resuspend-ed in 1.6 mL of 1× MS buffer and overlayed on top of thesucrose gradient and centrifuged at 40,000×g for 30 min.The mitochondrial band, in the middle of the gradient,was gently removed using a 20-G needle, transferred to a1.5-mL tube and centrifuged at 16,000×g for 15 min. Theresulting pellet was resuspended in 2D solubilisation buff-er containing 8 M urea, 2 M thiourea, 4 % (w/v) CHAPSand a cocktail of protease inhibitors.
Protein concentration
Determination of total cellular protein was performed usingthe EZQ Protein Estimation Assay (Invitrogen, Australia) aspreviously described by Churchward et al. [4].
2D gel electrophoresis
Protein concentration estimations (EZQ assay) were per-formed on patient and control mitochondrial protein fractions;a total of 100 μg protein was used for each 2DE analysis. 2DEwas carried out as previously described [3, 11, 27]; briefly,mitochondrial proteins were reduced and alkylated in solu-tions containing total protein extraction buffer (containing8M urea, 2M thiourea and 4%CHAPSwithout ampholytes),total extraction buffer with 2 % ampholytes, TBP/dithiothreitol (DTT) disulphide reduction buffer (2.3 mMtributyl phosphine and 45 mM DTT) and alkylation buffer(230 mM acrylamide monomer).
After incubation, the treated samples were added to 7-cmnon-linear pH 3–10 immobilised pH gradient (IPG) strips(Bio-Rad ReadyStrip) and left to rehydrate for 16 h at RT.Isoelectric focusing was then carried out at 20 °C using theProtean IEF Cell (Bio-Rad, USA); the initial 15 min at 250 Vfollowed by linear ramping to 4,000 V at 50 μA/gel for afurther 2 h. After 2 h, isoelectric focusing was continued at4000 V (constant) for a total of 37,500 Vh.
After IEF, the IPG strips were incubated in IEF equilibra-tion buffer with 2 % DTT and 350 mM acrylamide monomer
to ensure complete reduction and alkylation. The equilibratedIPG strips were then resolved in the second dimension usingthe MiniProtean II (Bio-Rad).
A 12.5 % T, 2.6 % C polyacrylamide gel was buff-ered with 375 mM Tris buffer (pH 8.8), 0.1 % (w/v)sodium dodecyl sulphate and polymerised with 0.05 %(w /v) ammonium persulphate and 0.05 % (v /v)tetramethylethylenediamine (TEMED). A stacking gelcontaining a 5 % T, 2.6 % C polyacrylamide bufferedwith 375 mM Tris buffer (pH 8.8), 0.1 % (w/v) sodiumdodecyl su lpha te (SDS) and inc luded 0 .1 %bromophenol blue was added to the resolving gel. TheIPG strips were placed onto the stacking gel and over-laid with 0.5 % (w/v) low melting agarose dissolved in375 mM Tris (pH 8.8), with 0.1 % (w/v) SDS. Electro-phoresis was carried out at 4 °C using pre-chilled Tris-glycine-SDS electrode buffer; 150 V was initially usedto rapidly drive proteins out of the IPG strips and intothe stacking gel for 5–10 min, and the voltage reducedto 90 V for 2–3 h. The gels were fixed with 10 %methanol and 7 % acetic acid for 1 h. The gels werewashed three times with distilled water for 20 min.
The gels were stained with colloidal Coomassie Blue(0.1 % (w/v) CCB G-250, 2 % (v/v) phosphoric acid, 10 %(w/v) ammonium sulphate, 20% (v/v) methanol) for 20 h, withconstant shaking at RT [11] and subsequently de-stained fivetimes with 0.5 MNaCl, 15 min each. Imaging of CBB-stainedgels on the FLA-9000 imager (FUJIFILM, Tokyo, Japan) wascarried out at 685/750 excitation/emission with aphotomultiplier tube (PMT) setting of 600 V and pixel reso-lution set to 100 μm [11]. Analysis of 2D gel images wasperformed using Delta 2D software with automated spot de-tection (local background region, 96; average spot size, 32;and sensitivity in percentage, 20.0) (version 4.0.8;DECODON GmbH, Gerifswald, Germany).
SDS-PAGE and immunoblotting
Control and patient mitochondrial protein fractions (25 μgtotal protein) were subjected to SDS-PAGE on 12.5 % resolv-ing gels and transferred to PVDF membrane. The membraneswere blocked with 5 % skim milk in TBS buffer containing0.1 % Tween-20. Whole membranes were blocked and incu-bated with anti-Tomm22, anti-ubiquinol-cytochrome c reduc-tase core protein 1, anti-Ig kappa chain C, anti-GAPDH, andanti-MTCO2 at 1:1,000, for 16 h at 4 °C. The membrane wasthen incubated with secondary horse radish peroxidase anti-body (1:2,000 dilution) for 1 h at RT. Blots were developedusing an enhanced chemiluminescence (ECL) detection kit(Pierce Thermo Scientific, USA). The membrane was devel-oped on CL-Xposure Film (Thermo Fisher Scientific, USA)using an AGFA X-ray developer.
J Chem Biol (2015) 8:25–35 27

Mass spectrometry
2D gels were analysed for uniquely present or absent proteinspots in control versus V144D mutant (i.e. all-or-none chang-es). The protein spots of interest were excised from gels andde-stained overnight with 1:1 absolute acetonitrile and 25mMammonium bicarbonate. The gel pieces were then reduced andalkylated in 10 mM dithiothreitol (DTT) and 15mM idoaceticacid (IAA) and subsequently incubated with trypsin solution(10 ng/μL, pH 7.4) for 16 h at 37 °C. The digested peptideswere concentrated in a speedy vac and resuspended in 0.1 %formic acid for subsequent analysis. LC-MS/MS analysis wascarried out on a nanoAquity UPLC (Waters Corp., Milford,MA, USA) linked to a Xevo QToF mass spectrometer fromWaters (Micromass, UK). Three microlitres of digested pep-tides was loaded onto a nanoAquity C18 BEH130 column(1.7 μm, 75 μm×150 mm) and then resolved and eluted fromthe column using a binary gradient program at a flow rate of0.4 μL/min: mobile phase Awas 0.1 % formic acid in water,and mobile phase B was 0.1 % formic acid in acetonitrile. Thenano-UPLC gradient was as follows: 0 min, 99:1 A/B; 1 min,99:1 A/B; 31 min, 50:50 A/B; 33 min, 15:85 A/B; 36 min,15:85 A/B; and 37min, 99:1 A/B. The mass spectrometer wasoperated in positive ESI mode with capillary voltage of2.3 kV, cone voltage of 25 V and source temperature of80 °C. Targeted MS/MS data or data-dependent acquisition(DDA) was acquired with collision energy ramping from 30 to40 eVon the eight most intense peaks of MS mode with massranges of 350–1,500 Da. The data were acquired usingMasslynx software (version 4.1, Micromass, UK).
The acquired DDA data from Masslynx were converted toPKL files by Protein Lynx Global Server (Waters, UK). TheMS/MS data files were searched against SwissProt databasewith semi-trypsin as the enzyme. The following parameterswere used in Mascot for identification of the peptides: maxi-mummissed cleavage of 2; positive peptide charge of 2, 3 and4; peptide mass tolerant of 0.5 Da in MS and MS/MS database; fixed modification,carbamidomethyl (C); and variablemodifications,oxidation (M).
Immunofluorescence
Lymphoblasts (1×106 cells) were suspended in 1 mL of warmPBS. After centrifugation at 1,000×g for 5 min, at RT, the cellpellet was resuspended in 4 % paraformaldehyde for 15 min.Cells were then placed in 0.5 % TritonX-100 and incubated at37 °C for 30 min. The cells were then centrifuged and blockedin 5 % BSA solution at 37 °C for 30 min. After washing inPBS, the cells were resuspended in primary antibody, MTCO2(Abcam, 1:50), ubiquinol-cytochrome c reductase core pro-tein 1 and Ig kappa chain C (Abcam, 1:100) and incubated for1 h at RT. The cells were subsequently washed and resuspend-ed in secondary antibody, anti-mouse rhodamine (Millipore,
1:200), and incubated for 1 h at RT. DAPI (1 μg/μL) wasadded to the cell suspension, and after 2 min, the cells werecentrifuged and washed two times with PBS. Aliquots(300 μL) were added to six-well culture plates containingcoverslips coated in Histogrip (Invitrogen, USA) and centri-fuged at 500×g for 10 min. The coverslips were washed inwarm PBS, left overnight to dry and mounted onto glass slidesprior to confocal imaging. The LSM 5 confocal microscopecomprising the LSM 5 exciter laser scanning microscope withAxiovert 200M inverted optical microscope (Carl Zeiss, Jena,Germany) was used for the acquisition of immunofluores-cence images. For all acquisitions, a plan-Apochromat 63×/1.40 oil DIC objective was usedwith an excitationwavelengthof 405 and 543 nm. Fluorescence was detected in a bandwidthof 460–560 nm. Data was analysed using the Carl Zeiss Zen2009 Software.
Flow cytometry
For flow cytometry analyses, lymphoblasts were isolated asabove; the cells were then suspended in 1 mL of 4 % parafor-maldehyde in PBS and incubated for 15 min at RT. Thereafter,the cell suspension was centrifuged at 1,000×g for 5 min at RTand then resuspended in 0.3 % Triton X-100 for 15 min at37 °C. Cells were incubated in primary antibody for 1 h at RT.After incubation, the cell suspension was centrifuged at1,000×g for 5 min and the pellet resuspended in secondaryantibody, anti-mouse FITC (Millipore, 1:200), for 1 h at RT.The cell suspension was washed two times in PBS andanalysed using the MACSQuant flow cytometer (MiltenyiBiotech). Live cells were gated, and the mean of fluorescencewas obtained per 10,000 cellular events with an excitationwavelength of 488 nm and emission filter of 525/50 nm. Datawas analysed with MACSQuant software.
Results
Expression of GAPDH and mitochondrial markers in HSN-Ipatient-derived lymphoblasts
Quantitative immunoblotting was used to determine whetherboth Tomm 22 (translocase of the outer mitochondrial mem-brane) and MTCO2 (cytochrome c oxidase subunit II) wereexpressed in protein lysates from isolated mitochondrial frac-tions and in the total cell extracts from control and V144Dmutant HSN-I patient-derived lymphoblasts (V144D cells)(Fig. 1a, b). Analyses indicated that there was no statisticallysignificant change in expression of these proteins in eitherprotein sample (Fig. 1d), with values of 1.74 and 4.24 forcontrol and V144D Tomm 22mitochondrial fractions, and 5.6and 11.30, and 1.46 and 2.06 from control and V144D
28 J Chem Biol (2015) 8:25–35

mitochondrial and total fractions respectively. As a compara-tive housekeeping protein for quantitation, as well as a proteinloading control, analysis of GAPDH was carried out in orderto establish relative protein expression levels (Fig. 1c). De-spite some variability, 3,167.06 and 1,973.15 for control andV144Dmitochondrial fractions and 7,766.75 and 7,091.37 forcontrol and V144D total fractions, there were no statisticallysignificant changes in the expression of GAPDH in either thecontrols or V144D cells (Fig. 1e).
2D gel images of mitochondrial proteins from controland patient-derived lymphoblasts
Total isolated mitochondrial proteins were resolved and quan-titatively assessed using a refined 2DE protocol (Fig. 2) [3, 11,27]. Mitochondrial proteins from control and V144D cellswere resolved using mini gel format; image analysis indicated
583±7 and 571±6 detectable proteins in the control andV144D cell mitochondria, respectively. Further analysis ofthe total mitochondrial protein profiles from control andV144D cells revealed three consistent ‘all-or-none’ proteinchanges in the V144D cells relative to control lymphoblasts.These protein species were located at pI/MW (kDa) coordi-nates of 5.7/55, 6.6/24 and 8.3/24 (Table 1). Subsequent LC/MS analysis identified these proteins to be ubiquinol-cytochrome c reductase core protein 1 and Ig kappa chain C.
Expression of ubiquinol-cytochrome c and Ig kappafrom HSN-I patient-derived lymphoblasts
Immunoblot analysis was performed on isolated mitochondriaand total cell lysates from control and V144D cells in order tofurther quantitatively assess changes in protein abundance.Blots were normalised to GAPDH as shown in Fig. 1c. These
Fig. 1 Immunoblot analysis ofTomm 22, MTCO2 and GAPDH.Expression of GAPDH andmitochondrial markers in HSN-Ipatient-derived lymphoblasts. aImmunoblot of Tomm 22; bimmunoblot of MTCO2; cimmunoblot of GAPDH. Lanes 1and 2 represent controlmitochondrial proteins; lanes 3and 4 represent control totalproteins; lanes 5 and 6 representV144D mitochondrial proteins;and lanes 7 and 8 representV144D total proteins.Representative graphs showingno statistically significant(p>0.05) difference betweenmitochondrial control and patientlymphoblast lysates (n=3) ofTomm 22 (blue) and MTCO2(red) (d) and GAPDH (e) areshown. All blots were normalisedto GAPDH. Error bars depict SEof means
J Chem Biol (2015) 8:25–35 29

30 J Chem Biol (2015) 8:25–35

data showed a concomitant increase in the amount ofubiquinol-cytochrome c (5.01 compared to 2.59) in the proteinsamples from V144D cells compared to the control samples(p<0.05; Fig. 3a, c). In contrast, immunoblotting confirmed asignificant decrease in the amount of Ig kappa protein in thetotal lysates of V144D cells compared to controls (1.47 com-pared to 2.71) (p<0.05; Fig. 3b, d).
SPTLC1 mutations cause no change to intracellularlocalisation
In order to establish the intracellular localisation and abun-dance of ubiquinol-cytochrome c, immunofluorescence stud-ies were performed on control and V144D cells. There wereno detectable changes in intracellular localisation of MTCO2or ubiquinol-cytochrome c protein in control and V144Dcells, whereby both proteins were peripherally localised.When the intracellular localisation of Ig kappa protein wasassessed, there was also no detectable change between thecontrol or V144D cells and the localisation of the protein wasalso unchanged in the periphery (Fig. 4).
Relative quantification of MTCO2, ubiquinol-cytochrome cand Ig kappa in V144D cells
ImmunostainedMTCO2, ubiquinol-cytochrome c and Ig kap-pa control and V144D cells were analysed using fluorescence-assisted cell sorting (FACS) to determine the total fluores-cence per cell (Fig. 5a, b). There was no overall shift orincrease in the fluorescence histograms from control andV144D mutant cell populations with respect to the MTCO2protein. However, there was a marked increase in the relativefluorescence intensity of ubiquinol-cytochrome c in theV144D cells compared to that of control lymphoblasts withan increase in relative fluorescence of 80±1.5 OD (a 2.2-fold
increase) respectively, relative to the stained controls (Fig. 5c).There was an increase in Ig kappa peak intensity in the V144Dcells (an ∼1.5-fold increase) with an observed peak widthincrease in the control cells.
Discussion
HSN-I is an autosomal dominant sensory neuropathy resultingin the dying back of the peripheral sensory neurons and aprogressive degeneration of the dorsal root ganglia [21]. HSN-I is caused bymissense mutations in the SPTLC1 gene, but theactual cellular and molecular mechanisms underlying the dis-ease remain poorly understood. A recent study has shownmitochondrial ultrastructural changes to be linked with ERstress in HSN-I cells [23]. Using an integrated proteomic andcell biology approach, we have identified a significant in-crease in the amount of ubiquinol-cytochrome c reductasecore protein 1 in the mitochondria from HSN-I (V144D)patient-derived lymphoblasts relative to control lymphoblasts.Of further interest, there is a decreased amount of the immu-noglobulin protein, Ig kappa chain C, in the V144D cells aswell as a change in the pI of this protein.
Mitochondria are the intracellular energy producing organ-elles where substrates are metabolised to fuel oxidative phos-phorylation through the electron transport chain within theirinner membrane [31]. The electron transport chain consists offour multimeric enzyme complexes. These complexes facili-tate the flow of electrons from the reducing substrates tooxygen to build a proton gradient required for ATP generation[5]. Ubiquinol-cytochrome c reductase core protein 1 (alsoknown as cytochrome b-c1 complex subunit I) is a centralcomponent of the electron transport chain, catalysing theoxidization of ubiquinol (ubihydroquinone) and reduction ofcytochrome c [5].
In order to test whether there were protein changes dueto the HSN-I SPTLC1 mutation, we assessed theproteomes of mitochondria isolated from control andV144D cells using high-resolution 2DE, to enable quan-titative assessments (Fig. 2a). Tomm 22 and MTCO2,
�Fig. 2 Representative images of 2D gels and regions of mitochondrialproteins from control and patient-derived lymphoblasts. a Control andV144Dmitochondrial proteins; b resolved protein species having a vastlyaltered abundance as indicated (red arrow). The molecular weights are inkilodaltons (kDa), and the IEF dimension is in pH units
Table 1 Mass spectrometry
Spotnumber
Protein identified Accessionnumber
Number of uniquepeptides matched
Sequencecoverage
Mascotproteinscore
PredictedpI
PredictedMW (kDa)
MascotpI
MascotMW (kDa)
I Ubiquinol-cytochrome creductase core protein 1
P31930 4 18 % 208 5.7 55.00 5.9 53.3
II Ig kappa chain C P01834 14 88 % 908 6.6 22.00 5.5 11.7
III Ig kappa chain C P01834 12 86 % 1280 8.3 22.00 5.5 11.7
Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified ubiquinol-cytochrome c reductase core protein 1and Ig kappa chain C from V144D patient-derived lymphoblast and Ig kappa chain C from control lymphoblasts isolated mitochondria
J Chem Biol (2015) 8:25–35 31

Fig. 3 Immunoblot blot analysisof ubiquinol-cytochrome c and Igkappa chain C. Expression ofubiquinol-cytochrome c and Igkappa chain C from HSN-Ipatient-derived lymphoblasts. aImmunoblot detection ofubiquinol-cytochrome c. bImmunoblot detection of Ig kappachain C. Lanes 1 and 2 representcontrol mitochondrial proteins;lanes 3 and 4 represent controltotal proteins; lanes 5 and 6represent V144D mitochondrialproteins; and lanes 7 and 8represent V144D total proteins. c,d Representative graphs showingstatistically significant (*p<0.05)difference between control patientlymphoblasts and the mutantV144D lymphoblasts ofubiquinol-cytochrome c and Igkappa chain C respectively (n=3).Blots were normalised toGAPDH (Fig. 1c). Error barsdepict SE of means
Fig. 4 Immunofluorescence ofubiquinol-cytochrome c, MTCO2and Ig kappa chain C. SPTLC1mutations cause no change to theintracellular localisation ofubiquinol-cytochrome c, MTCO2and Ig kappa chain C.Representative confocalmicrographs showingubiquinol-cytochrome c, MTCO2and Ig kappa chain C stainedlymphoblasts (red) and DAPInuclear stain (blue). Scale bar=5 μm
32 J Chem Biol (2015) 8:25–35

mitochondrial marker proteins, confirmed the quality ofthe isolated mitochondrial fraction used for analysis.There were no statistically significant changes in expres-sion of these mitochondrial markers, suggesting that theseproteins turn over at a constant rate in both the controland V144D cells. Analysis of GAPDH from control andV144D cells also indicated no significant changes in thisprotein (Fig. 1e). There was a protein selectively detectedin the V144D cells at pI 5.7 and molecular weight55 kDa; this protein was undetectable in the controlprotein profile (Fig. 2b). This protein proved to beubiquinol-cytochrome c reductase core protein 1. Quanti-tative immune-blotting confirmed a significant (i.e. 2-fold) increase in the amount of ubiquinol-cytochrome c
reductase core protein 1 in V144D cells relative to controlcells.
Ubiquinol-cytochrome c reductase core protein 1 functionsto ensure that the electron transfer rate is optimal and that fastdissociation of electrons occurs following transfer [8]. Theseprocesses are essential to maintain the electron flow and toprevent any potential electron leaks or break down of therespiratory chain [8]. Ubiquinol-cytochrome c reductase coreprotein 1 is also involved in free radical generation, producingreactive oxygen species (ROS) within mitochondria. ROSproduction can disrupt the homeostasis and interactions withinthe mitochondrial matrix resulting in the loss of the oxidativephosphorylation, along with the disruption of mitochondrialfunctions and physiology leading to cell death [5].
Fig. 5 Flow cytometry analysis of ubiquinol-cytochrome c,MTCO2 andIg kappa chain C. Relative quantification of ubiquinol-cytochrome c,MTCO2 and Ig kappa chain C in HSN-I patient-derived lymphoblastsexpressing the V144D mutant SPTLC1 genes. Flow cytometry analysis
of the relative fluorescence intensity of ubiquinol-cytochrome c (a),MTCO2 (b) and Ig kappa chain C (c) in control and V144D patient-derived lymphoblasts. Red histogram represents the V144D patient lym-phoblasts, and blue histogram represents control lymphoblasts (n=3)
J Chem Biol (2015) 8:25–35 33

Further analysis was performed using immunostaining(Fig. 4) and FACS (Fig. 5) to determine cellular localisationand expression of ubiquinol-cytochrome c reductase coreprotein 1 and MTCO2. Immunostaining revealed no distinctdifference between the localisation of ubiquinol-cytochrome creductase core protein 1 in the control versus V144D cells.FACS analysis correlated with the previous expression data,exhibiting a concomitant 2.2-fold increase in fluorescenceintensity of ubiquinol-cytochrome c reductase core protein 1in the V144D cells relative to controls. In contrast, theMTCO2 protein displayed no increase in fluorescence inten-sity in control versus patient (V144D) lymphoblasts.
Furthermore, in the comparison of the control and V144Dcell proteomes, we identified two other marked protein chang-es. Both of these proteins were located in the 24-kDa molec-ular weight region but were located in different pI (6.6 and 8.3respectively) regions. Both proteins were identified as Igkappa chain C. The cause of the apparent shift in pI is yet tobe determined; however, it seems likely due to an as yetunidentified posttranslational modification (potentially a gly-cosylation, phosphorylation or methylation, or any combina-tion of the three). This is the first study to identify a change inan immunoglobulin due to the SPTLC1 mutation causingHSN-I. The Ig kappa light-chain constant region undergoeslittle to no variation in human immunoglobulins and formspart of the five immunoglobulin classes produced in mature Bcells [16]. While B cells inherently produce Ig kappa, we havedetermined a statistically significant decrease in the amount ofIg kappa chain C in the total cell lysate of V144D cells relativeto control lymphoblasts. Immunoglobulin light chains havebeen implicated in and are biomarkers of diseases such asmultiple myeloma and primary systemic or amyloid light-chain (AL) amyloidosis [28]. In these diseases, it has beenshown that the free Ig kappa chain associates withsphingomyelin on the plasma membrane of the myeloma cellsforming aggregates that are required for intercalation withmembranes [15]. This further suggests the important rolesphingolipids play in these disease processes.
The novel findings in this study thus suggest a link tooxidative phosphorylation, via ubiquinol-cytochrome c reduc-tase core protein 1 (perhaps through regulation of ROS pro-duction), and that ensuing interference with energy productionultimately leads to axonal retraction that is the hallmark char-acteristic of hereditary sensory neuropathy. Clearly, if ROSproduction increases with the increased levels of ubiquinol-cytochrome c reductase core protein 1, the ensuing potentialcellular damagewould only further contribute to a progressingavalanche of damage that could well characterize axonalretraction at both the molecular and cellular levels. In thatregard, it is notable that our proteomic analyses did not iden-tify marked alterations in the levels of known antioxidant,chaperone or other repair proteins; do the mutations stymiesuch responses? The data also indicate for the first time that
there is also a potential immunological component to thisneurodegenerative disorder, characterised by significantly de-creased amounts of the immunoglobulin protein, Ig kappachain C. The exact role of this protein and its relationship toHSN-I will be the aim of further investigations. Are reducedlevels of this protein responsible for apparent reductions incellular repair responses? Clearly, far more work is required tofully elucidate the mechanisms underlying peripheral neurop-athies, but the novel findings arising from this first coupled,quantitative proteomic cell biological analysis provide criticalnew directions not even previously hypothesised. The find-ings in this study, coupled with the findings of Marshall et al.[20] and Myers et al. [23], suggest that there may well beunderlying molecular alterations broadly common toneurodegenerations as a whole, linked to both mitochondriaand lipids.
Acknowledgments We are grateful to Prof. Garth Nicholson (Molecu-lar Medicine Laboratory and Northcott Neuroscience Laboratory AnzacResearch Institute, Sydney) for providing all EBV-transformed lympho-blast lines used in this study. SES was supported by APA ResearchScholarship and the UWS School of Science and Health Postgraduateresearch fund. SJM notes the continuing support of an anonymous privatefoundation. JRC acknowledges the support of the UWS School ofMedicine.
References
1. Bejaoui K,Wu C, SchefflerMD, Haan G, Ashby P,Wu L, De Jong P,Brown RH Jr (2001) SPTLC1 is mutated in hereditary sensoryneuropathy, type 1. Nat Genet 27:261–262
2. Bozidis P, Williamson CD, Colberg-Poley AM (2007) Isolation ofendoplasmic reticulum, mitochondria, and mitochondria-associatedmembrane fractions from transfected cells and from humancytomegalovirus-infected primary fibroblasts. Curr Protoc CellBiol. doi:10.1002/0471143030.cb0327s37
3. Butt RH, Coorssen JR (2005) Postfractionation for enhanced prote-omic analyses: routine electrophoretic methods increase the resolu-tion of standard 2D-PAGE. J Proteome Res 4:982–991
4. Churchward M, Butt RH, Lang J, Hsu K, Coorssen J (2005)Enhanced detergent extraction for analysis of membrane proteomesby two-dimensional gel electrophoresis. Proteome Sci 3:5
5. Crofts AR (2004) The cytochrome bc1 complex: function in thecontext of structure. Annu Rev Physiol 66:689–733
6. Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M,Nicholson GA (2001) Mutations in SPTLC1, encoding serinepalmitoyltransferase, long chain base subunit-1, cause hereditarysensory neuropathy type I. Nat Genet 27:309–312
7. Dedov V, Dedova I, Merrill A, Nicholson G (2004) Activity ofpartially inhibited serine palmitoyltransferase is sufficient for normalsphingolipid metabolism and viability of HSN1 patient cells.Biochim Biophys Acta 1688(2):168–175
8. Drose S, Brandt U, WittigI (2014) Mitochondrial respiratory chaincomplexes as sources and targets of thiol-based redox-regulation.Biochim Biophys Acta 1844(8):1344–1354. doi:10.1016/j.bbapap.2014.02.006
9. Duffy LM, Chapman AL, Shaw PJ, Grierson AJ (2011) Review: therole of mitochondria in the pathogenesis of amyotrophic lateralsclerosis. Neuropathol Appl Neurobiol 37:336–352
34 J Chem Biol (2015) 8:25–35

10. Dyck PJ, Thomas PK (2005) Dyck: peripheral neuropathy, 4th edn.Mosby Elsevier, Philadelphia
11. Gauci VJ, Padula MP, Coorssen JR (2013) Coomassie blue stainingfor high sensitivity gel-based proteomics. J Proteomics 90:96–106
12. Hanada K (2003) Serine palmitoyltransferase, a key enzyme ofsphingolipid metabolism. Biochim Biophys Acta 1632:16–30
13. Hollenbeck PJ, Saxton WM (2005) The axonal transport of mito-chondria. J Cell Sci 118:5411–5419
14. Hornemann T, Richard S, Rutti M, Wei Y, Von-Eckardstein A (2006)Cloning and initial characterization of a new subunit for mammalianserine-palmitoyltransferase. J Biol Chem 281(49):37275–37281
15. Hutchinson AT, Ramsland PA, Jones DR, Agostino M, Lund ME,Jennings CV, Bockhorni V, Yuriev E, Edmundson AB, Raison RL(2010) Free Ig light chains interact with sphingomyelin and are foundon the surface of myeloma plasma cells in an aggregated form. JImmunol 185:4179–4188
16. Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007) Kuby immu-nology. W.H. Freeman, New York
17. Kwong JQ, Beal MF, Manfredi G (2006) The role of mitochondria ininherited neurodegenerative diseases. J Neurochem 97:1659–1675
18. Mandon EC, Ehses I, Rother J, Van Echten G, Sandhoff K (1992)Subcellular localization and membrane topology of serinepalmitoyltransferase, 3-dehydrosphinganine reductase, andsphinganine N-acyltransferase in mouse liver. J Biol Chem 267:11144–11148
19. Manfredi G, Beal MF (2000) The role of mitochondria in thepathogenesis of neurodegenerative diseases. Brain Pathol 10:462–472
20. Marshall LL, Stimpson SE, Hyland RA, Coorssen JR, Myers SJ(2014) Increased lipid droplet accumulation associated with a periph-eral sensory neuropathy. J Chem Biol 7:67–76
21. McCampbell A, Truong D, Broom D, Allchorne A, Gable K, CutlerRG, Mattson M, Woolf C, Frosch M, Harmon J, Dunn T, Brown R(2005) Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase
activity in vivo and confers an age-dependent neuropathy. Hum MolGenet 14(22):3507–3521
22. Miller KE, Sheets MP (2004) Axonal mitochondrial transport andpotential are correlated. J Cell Sci 117:2791–2804
23. Myers S, Malladi C, Hyland R, Bautista T, Boadle R, Robinson P,Nicholson G (2014) Mutantions in the SPTLC1 protein cause mito-chondrial structual abnormalisites and endoplasmic reticulum stressin lymphoblasts. DNA Cell Biol 33(7):399–407
24. Vaseva AV, Moll UM (2013) Identification of p53 in mitochondria.Methods Mol Biol 962:75–84
25. Verhoeven K, Timmerman V, Mauko B, Pieber TR, DeJonghe P, Auer-Grumbach M (2006) Recent advances in he-reditary sensory and autonomic neuropathies. Curr OpinNeurol 19:474–480
26. Wei J, Yerokun Y, Liepelt M, Momin A, Wang E, Hanada K, MerrilAH Jr. (2007) 2–1 Serine palmitoyltransferase. Sphingolipid Biology.Springer, Japan, p 25–27
27. Wright EP, Partridge MA, Padula MP, Gauci VJ, Malladi CS,Coorssen JR (2014) Top-down proteomics: enhancing 2D gel elec-trophoresis from tissue processing to high-sensitivity protein detec-tion. Proteomics 14:872–889
28. Yamamoto K, Yagi H, Lee YH, Kardos J, Hagihara Y, Naiki H, GotoY (2010) The amyloid fibrils of the constant domain of immuno-globulin light chain. FEBS Lett 584:3348–3353
29. Yard B, Carter L, Johnson K, Overton I, Dorward M, Liu H,McMahon S, Oke M, Puech D, Barton G, Naismith J, CampopianoD (2007) The structure of serine palmitoyltransferase; gateway tosphingolipid biosynthesis. J Mol Biol 370(5):870–886
30. Yasuda S, Nishijima M, Hanada K (2003) Localization, topology,and function of the LCB1 subunit of serine palmitoyltransferase inmammalian cells. J Biol Chem 278:4176–4183
31. Zhu Y, Li M, Wang X, Jin H, Liu S, Xu J, Chen Q (2012) Caspasecleavage of cytochrome c1 disrupts mitochondrial function and en-hances cytochrome c release. Cell Res 22:127–141
J Chem Biol (2015) 8:25–35 35

67 | P a g e
Paper III
Published in Biochemistry and Analytical Biochemistry
Contributions
SES carried out all experimentation, data analysis and wrote first draft manuscript

Research Article Open Access
Biochemistry & Analytical BiochemistryBi
oche
mist
ry & Analytical Biochem
istry
ISSN: 2161-1009
Stimpson et al., Biochem Anal Biochem 2016, 5:1http://dx.doi.org/10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
*Corresponding author: Simon J. Myers, Western Sydney University, Campbelltown campus, Locked Bag 1797, Penrith, Australia, Tel: +61 02 4620 3383; E-mail: [email protected]
Jens Coorssen, Professor, Western Sydney University, Campbelltown campus, Locked Bag 1797, Penrith, Australia, Tel: +61 4620 3802; E-mail: [email protected]
Received: February 06, 2016; Accepted: February 15, 2016; Published February 18, 2016
Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Copyright: © 2016 Stimpson SE, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
AbstractAxonal degeneration is the final common path in many neurological disorders. Hereditary sensory neuropathies
(HSN) are a group of neuropathies involving the sensory neurons. The most common subtype is autosomal dominant hereditary sensory neuropathy type I (HSN-I). Progressive degeneration of the dorsal root ganglion (DRG) neuron with an onset of clinical symptoms between the second or third decade of life characterises HSN-I. Mutations in the serine palmitoyltransferase (SPT) long chain subunit 1 (SPTLC1) gene cause HSN-I. The endoplasmic reticulum (ER) is a dynamic organelle that houses the SPTLC1 protein. Ultra structural analysis has shown the ER in the HSN-I mutant cells to wrap around dysfunctional mitochondria and tethers them to the perinucleus.
This investigation establishes that the V144D mutant of SPTLC1 alters the expression of and potentially interacts with a set of proteins within the ER. Using ER protein lysates from HSN-I patient and control lymphoblasts: we have identified a change in regulation of five proteins; Hypoxia Up regulated Protein 1: Chloride intracellular channel protein 1: Ubiqutin-40s Ribosomal protein S27a: Coactosin and Ig Kappa chain C. The expression and regulation of these proteins may help to establish a link between the ER and the ‘dying back’ process of the DRG neuron.
Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-IScott E Stimpson1,3,4, Antonio Lauto4,6, Jens R Coorssen2,3,4,5* and Simon J Myers1,3,4,5*1Neuro-Cell Biology Laboratory, Western Sydney University, Australia2Molecular Physiology, Western Sydney University, Australia3Molecular Medicine Research Group, Western Sydney University, Australia4School of Science and Health, Western Sydney University, Australia5School of Medicine, Western Sydney University, Australia6Biomedical Engineering and Neuroscience (BENS), Western Sydney University, Australia
Keywords: Hereditary sensory neuropathy type 1; Serine palmitoyltransferase long chain sub-unit 1; Endoplasmic reticulum; Oxidative stress
Abbreviations: HSN: Hereditary Sensory Neuropathies; HSN-I: Hereditary Sensory Neuropathy Type I; DRG: Dorsal Root ganglion; SPT: Serine palmitoyl transferase; SPTLC1: Serine palmitoyl transferase long chain subunit 1; ER: Endoplasmic Reticulum; LCB1: Long-Chain Base One; UPR: Unfolded Protein Response; UPS: Ubiquitin Proteasome System; ROS: Reactive Oxygen Species; ORP-150: Hypoxia up Regulated Protein 1; CLIC1: Chloride Intracellular Channel Protein 1; RPS27a: Ubiqutin-40s Ribosomal Protein s27a; COTL1: Coactosin; HRP: Horse Radish Peroxidase; IEF: Isoelectric Focusing; TEMED: Tetramethylethylenediamine; PMT: Photomultiplier Tube; 2DE: Two Dimensional Gel Electrophoresis; IAA: Idoacetic acid; LC/MS: Liquid Chromatography/Mass Spectrometry; ECL: Enhanced Chemiluminescence; FACS: Fluorescence’s Assisted Cell Sorting; Grp170: Glucose related protein 170; G-actin: Globular actin; F-actin: Filamentous actin
IntroductionThe ER is an intracellular organelle which supports and maintains a
plethora of functions critical for cellular survival. The ER plays a crucial role in many aspects of protein compartmentalisation which include membrane translocation, folding, post-translational modification, and transport of both membrane and soluble proteins [1]. In addition, the ER is involved in the synthesis of phospholipids and steroids and also in the regulation of Ca2+ homeostasis. The ER is organised in a complex: continuous network of tubules and sheets that includes the nuclear envelope and extends throughout the cytosol into the cell periphery [1]. Nascent polypeptide chains fold, acquire further modifications like glycosylation and disulphide bonds and often assemble with other subunits before traversing further along the secretory pathway with in the ER. These processes are both assisted and monitored by molecular chaperones [1].
SPT is an ER bound and key rate determining enzyme in the
complex sphingolipid metabolic pathway consisting of 3 subunits; SPTLC1: SPTLC2 and SPTLC3 [2]. Mutations within SPTLC1 result in potential dysfunction and perturbations in sphingolipid synthesis and metabolism causing HSN-I [2]. These mutations are single amino acid changes in the SPTLC1 gene that encodes the long-chain base one (LCB1) subunit [3]. HSN-I is the most common subtype of HSN [4]. HSN-I is an autosomal dominant disorder characterised by degeneration of the DRG neuron and with a clinical onset between the second or third decades of life.
The ER carries out extensive quality control of proteins to enable normal cellular function. Disruption to the function of the ER or loss of its integrity leads to ER stress [5]. ER stress can be characterised by the accumulation of unfolded proteins and changes in calcium homeostasis within the ER with stress activating the unfolded protein response (UPR) [6]. The UPR and its signalling components can change the expression of specific proteins such as, those designated for the ER chaperones; the enhancement of degradation of misfolded (mutant or unfolded) proteins; and the inhibition of protein synthesis to decrease the load within the ER [6].

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 2 of 8
Disturbed functions of the ubiquitin proteasome system (UPS): responsible for the degradation of cytosolic: ER and synaptic proteins: can contribute to ER stress [5]. A common occurrence in many neurodegenerative disorders is the accumulation and deposits of misfolded proteins that affects various cell signalling systems: as well as neuronal connectivity and cell death such as in Alzheimer’s: Parkinson’s and Huntington’s disorders.
The activity of the UPS is mitigated in degenerative disorders by the protein aggregation or by enhanced oxidative stress with other toxic products [5]. Dysfunctional UPS in turn causes increased accumulation of proteins in the cell leading to ER stress and the aggravation of the disorder [5]. Additionally the effect of environmental toxins, reactive oxygen species (ROS) and other signals that influence mitochondria lead to the activation of the caspase family of cysteine proteases causing cell death [7]. This negative cycle of increasing ‘stressors’ within the cell often leads to the cells inability to function properly and eventually leading to cell death [7].
It has recently been shown that HSN-I patient cells, expressing the V144D SPTLC1 mutant have a marked increase in ER stress in comparison to healthy control cells [8]. Considering this finding: in this study we have investigated altered protein changes in the ER membrane of these HSN-I patient cells. The resulting proteomic and cell biology analyses have identified for the first time, increases in the Hypoxia up regulated Protein 1 (ORP-150), Chloride intracellular channel protein 1 (CLIC1), Ubiqutin-40s ribosomal protein S27a (RPS27a), Coactosin (COTL1) and Ig Kappa chain C protein expression in HSN-I in this membrane.
Materials and Methods All cell culture stock solutions, including RPMI-1640, Foetal
bovine serum (FBS), Penicillin (100 U/mL), Streptomycin (100 µg/mL), L-glutamine (2 mM), HEPES (1 M): and phosphate buffered saline (PBS) were purchased from GIBCO Invitrogen (Australia). Cell culture consumables were purchased from BD Falcon (Greiner, USA). DAPI stains were purchased from Sigma-Aldrich (Australia).
EBV Transformed lymphoblasts
EBV transformed control and V144D HSN-I patient lymphoblasts were kindly provided by Prof. Garth Nicholson (Molecular Medicine Laboratory: Anzac Research Institute: Sydney) [9].
Lymphoblast culturesLymphoblasts were cultured in RPMI-1640 media (GIBCO),
supplemented with FBS (10% v/v), Penicillin (1 U/mL), Streptomycin (1 µg/mL), L-glutamine (2 mM), and HEPES (1 M) at 37°C in a humidified atmosphere of 5% CO2, using T75 cm2 culture flasks (Greiner: Interpath). Prior to use in biochemical assays, lymphoblasts were collected by centrifugation at 1,500 × g (5 min at RT) and washed in PBS. Cell counts were obtained using the Countess Automated Cell Counter (Invitrogen, Australia).
Isolation of ER proteins
Briefly, ER proteins were isolated using a sucrose density gradient [10,11]. Lymphoblasts were first centrifuged at 1,500 × g for 5 min, and the cells were then washed in 10 mL of ice cold 1 × PBS prior to suspension in 10 mL ice cold CaSRB Buffer (10 mM NaCl, 1.5 mM CaCl, 10 mM Tris-HCL, pH 7.5) and left on ice for 10 min. Cells were homogenised using a Dounce homogenizer (Kimble-Chase, USA) and 7 mL of 2.5 × MS buffer (210 mM Mannitol, 70 mM sucrose, 5
mM EDTA, 5 mM Tris-HCl, pH 7.6) was added to restore isotonicity. Homogenate was centrifuged at 700 x g for 5 min to remove nuclei and unbroken cells. The resulting supernatant was centrifuged at 15,000 x g for 10 mins to remove mitochondria. A sucrose gradient was made in 15 mL high speed centrifuge tubes (Beckman Coulter, USA) by adding 2 mL of 2.0 M sucrose buffer (2.0 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) overlayed with 3.0 mL of 1.5 M sucrose buffer (1.5 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) and 3.0 mL of 1.3 M sucrose (1.3 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) ER containing supernatant was loaded on top of the sucrose gradient and spun at 152,000 x g for 70 min. The ER band, the interface of the 1.5 M and 1.3 M sucrose, was gently removed using a 20 G needle, transferred to a 4 mL high speed centrifuge tubes (Beckman Coulter, USA) and centrifuged at 100,000 x g for 35 min. The resulting pellet was resuspended in 2D solubilisation buffer containing 8 M urea, 2 M thiourea, 4% (w/v) CHAPS and a cocktail of protease inhibitors.
Protein concentration
Determination of total cellular protein was performed using the EZQ protein estimation assay (Invitrogen, Australia) as previously described by Churchward [12].
Two dimensional gel electrophoresis
Protein concentration estimations (EZQ assay) were performed on patient and control ER protein fractions; a total of 100 µg protein was used for each 2DE analysis. 2DE was carried out as previously described [13-16]. Briefly, ER proteins were reduced and alkylated in solutions containing total protein extraction buffer (containing 8 M urea, 2 M thiourea and 4% CHAPS without ampholytes), total extraction buffer with 2% ampholytes, TBP/DTT disulphide reduction buffer (2.3 mM Tributyl phosphine and 45 mM DTT) and alkylation buffer (230 mM acrylamide monomer).
The treated samples were added to 7 cm Non-Linear pH 3-10 IPG strips (Bio-Rad ReadyStrip), and rehydrated for 16 hrs at RT. Isoelectric focusing (IEF) was then carried out at 20°C using the Protean IEF Cell (Bio-Rad, USA). After IEF, IPG strips were then resolved in the second dimension using a 12.5% T, 2.6% C polyacrylamide gel buffered with 375 mM Tris buffer (pH 8.8), 0.1% (w/v) sodium dodecyl sulphate and polymerised with 0.05% (w/v) ammonium persulphate and 0.05% (v/v) tetramethylethylenediamine (TEMED). A stacking gel containing a 5% T, 2.6% C polyacrylamide buffered with 375 mM Tris buffer (pH 8.8), 0.1% (w/v) SDS and included 0.1% bromophenol blue was added to the resolving gel. The IPG strips were placed onto the stacking gel and overlaid with 0.5% (w/v) low melting agarose dissolved in 375 mM Tris (pH 8.8): with 0.1% (w/v) SDS. Electrophoresis was carried out at 4°C; 150 V initially for 10 min then reduced to 90 V for 2.5 h. The gels were placed in fixative containing 10% methanol and 7% acetic acid for 1 hr. The gels were washed with distilled water for 20 min, 3 times and subsequently stained with colloidal coomassie blue (0.1% (w/v) CCB G-250, 2% (v/v) phosphoric acid, 10% (w/v) ammonium sulphate, 20% (v/v) methanol) for 20 hr, with constant shaking at RT [14], the gels were then de-stained 5 times with 0.5 M NaCl, 15 min each. Imaging of CBB-stained gels on the FLA-9000 imager (FUJIFILM, Tokyo, Japan) was carried out at 685/750 excitation/emission with a photomultiplier tube (PMT) setting of 600 V and pixel resolution set to 100 µm [14]. Analysis of 2D gel images was performed using Delta 2D software with automated spot detection (Local background region, 96; Average spot size, 32 and sensitivity in percentage, 20.0) (version 4.0.8; DECODON GmbH, Greifswald, Germany).

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 3 of 8
Mass spectrometry
2D gels were analysed for uniquely present or absent protein spots in control versus V144D mutant, (i.e. all or none changes) as previously described 25 and 18. Briefly, the protein species of interest were excised from gels and de-stained overnight. The gel pieces were then reduced and alkylated in 10 mM Dithiothreitol (DTT) and 15 mM Idoacetic acid (IAA), and subsequently incubated with trypsin solution (10 ng/µL, pH 7.4) for 16 hours at 37°C. LC-MS/MS analysis was carried out on a nanoaquity UPLC (Waters Corp, Milford, MA, USA) linked to a Xevo QToF mass spectrometer from Waters (Micromass: UK). The data were acquired using Masslynx software (Version 4.1, Micromass UK). The MS/MS data files were searched against SwissProt databases with semi-trypsin as the enzyme.
SDS-PAGE and immunoblottingControl and patient ER protein fractions (25 µg total protein)
were subjected to SDS-PAGE on 12.5% resolving gels and transferred to PVDF membrane. The membranes were blocked with 5% skim milk in TBS buffer containing 0.1% Tween-20. Whole membranes were blocked and incubated with anti-Calnexin (Cell Signalling Cat# 2679S, RRID:AB_2228381), anti-SPTLC1 (Santa Cruz Biotechnology Cat# sc-32916, RRID,AB_2195864), anti-Ig kappa chain C (Abcam Cat# ab1050, RRID:AB_297240), anti-GAPDH (Abcam Cat# ab9485, RRID:AB_307275), anti-ORP-150 (Abcam Cat# ab124884, RRID:AB_10973544), anti-CLIC1 (Abcam Cat# ab77214, RRID:AB_1566060), anti-RPS27a (Abcam Cat# ab57646, RRID:AB_2180587) and anti-COTL1 (Proteintech Group Cat# 10781-1-AP, RRID:AB_2084785) at 1:1000, for 16 h. The membrane was then incubated with secondary HRP antibodies (Sigma-Aldrich Cat# A9044, RRID:AB_258431 and Cat# A0545, RRID:AB_257896) (1:2000 dilution) for 1 hr at RT. Blots were developed using an enhanced chemiluminescence (ECL) detection kit (Pierce Thermo Scientific, USA). The membrane was developed on CL-Xposure Film (Thermo Fisher Scientific, U.S.A) using an AGFA X-ray developer.
Immunofluorescence
Immunofluorescence was carried out as previously described by Stimpson [16]. Briefly: Lymphoblasts (1 × 106 cells) were suspended in 4% paraformaldehyde for 15 min. Cells were then placed in 0.5% TritonX-100 and incubated at 37°C for 30 min. The cells were then blocked in 5% BSA solution at 37°C for 30 min: then resuspended in primary antibody, SPTLC1, ORP-150, CLIC1, RPS27a, COTL1, and stained for 1 hr at RT. The cells were subsequently washed
and resuspended in secondary antibody: anti-mouse Rhodamine (Millipore, 1:200), and incubated for 1 hour at RT. DAPI (1 µg/µL) was added to the cell suspension for 2 min, the cells were centrifuged and washed twice with PBS. Aliquots (300 µL) were added to 6 well culture plates containing coverslips coated in Histogrip (Invitrogen, USA) and centrifuged at 500 x g for 10 min. The coverslips were washed in warm PBS, left overnight to dry and mounted onto glass slides prior to confocal imaging using the LSM 5 confocal microscope comprising the LSM 5 exciter laser scanning microscope with Axiovert 200 M inverted optical microscope (Carl Zeiss: Jena: Germany).
Flow cytometry
FACS analysis was carried out as previously described by 18. Lymphoblasts were isolated as above; the cells were then suspended in 4% paraformaldehyde and incubated for 15 min at RT and then resuspended in 0.3% Triton X-100 for 15 min at 37°C. After incubation the cell suspension was centrifuged at 1,000 x g for 5 min and the pellet resuspended in primary antibody for 1 hr at RT. Cell suspension was centrifuged, washed in PBS and resuspended in secondary antibody, anti-mouse FITC (Millipore, 1:200) for 1 hr at RT. The cell suspension was then analysed using the MACSQuant flow cytometer (Miltenyi Biotech).
2D gel images of ER proteins from control and patient de-rived lymphoblasts
Total isolated ER proteins were resolved using a refined 2DE protocol [13,14] (Figure 1). Control and V144D total ER proteins were resolved using mini gel format. Standard spot counts indicated 656 ± 5 and 675 ± 3 protein species were resolved in control and V144D mutant ER fractions respectively. Further analysis of the total ER protein profiles from control and HSN-I patient derived lymphoblasts revealed five ‘all or none’ protein changes in the V144D mutant and control lymphoblasts (Figure 2). These proteins were located in the pI of 5.5, 5.6, 8.0, 5.8, 6.6 and 8.3 and molecular weight of 120, 30, 15, 17, 22 and 22 respectively (kDa) (Table 1). Subsequent LC/MS analysis identified the protein to be ORP-150, CLIC1, RPS27a, Coactosin (COTL1) and Ig Kappa Chain C.
Expression of identified ER proteins from HSN-I patient-de-rived lymphoblasts
In order to determine protein expression changes, immunoblot analysis was carried out on isolated ER and total cell lysates from control and HSN-I patient derived lymphoblasts. These data showed a
Figure 1: Representative images of 2D gels following resolution of ER proteins from control and patient derived lymphoblasts. (A) Control ER proteins; (B) V144D ER proteins. The molecular weights are in kilodaltons (kDa) and the IEF dimension is in pH units.

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 4 of 8
concomitant increase in the amount of ORP-150, CLIC1, and COTL1 in the V144D mutant protein samples compared to the control samples with a p value of < 0.05 (Figures 3A-3J). RPS27a was slightly increased in the V144D mutant; however this change was not statistically significant. Whereas quantitative analysis of Ig kappa protein displayed significant increase in the amount of Ig Kappa protein being expressed in the control total protein lysates compared to that of the protein isolated from the V144D mutant samples (p < 0.05) (Figure 3J).
The intracellular localisation and abundance of SPTLC1, ORP-150, CLIC1, RPS27a and COTL1 was established using immunofluorescence studies on control and patient-derived lymphoblasts. It was observed that there was no apparent change in intracellular localisation of the SPTLC1
protein in control and V144D mutant HSN-I patient lymphoblasts. However, whilst there appeared to be an increase in the abundance of ORP-150, CLIC1, RPS27a and COTL1 in the V144D (patient) lymphoblasts, there was no change in intracellular localisation (Figure 4).
Control and patient-derived lymphoblasts were immuno stained for ORP-150, CLICL1, RPS27a and COTL1 were analysed by fluorescence assisted cell sorting (FACS) to determine the total fluorescence per cell (Figure 5). There was a marked increase in the relative fluorescence intensity of all the proteins in the V144D cells compared to that of control lymphoblasts with an increase in relative fluorescence of 1.4, 1.25, 2.4, 1.5 OD (fold increase) respectively, relative to the stained controls.
Spot Number Protein Identified Accession
Number
Number of Unique peptides
matched
Sequence Coverage
Mascot Protein Score Predicted pI
Predicted Mw (kDa) Mascot pI
Mascot Mw (kDa)
I. Hypoxia Up Regulated Protein 1 QY94L1 42 49% 1741 5.5 120 5.2 112
II. Chloride Intracellular Channel Protein 1 O00299 15 50% 448 5.6 30 5.1 27.3
III.Ubiquitin-40s
Ribosomal protein S27a
P62979 10 33% 182 8.0 15 9.7 18.3
IV. Coactosin Q14019 19 73% 190 5.8 17 5.6 16V. Ig Kappa Chain C P01834 9 71% 587 6.6 22 5.5 12VI. Ig Kappa Chain C P01834 10 80% 985 8.3 22 5.5 12
Table 1: Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified Hypoxia up Regulated Protein 1, Chloride Intracellular Channel Protein 1, Ubiquitin 40s ribosomal protein s27a, Coactosin and Ig Kappa Chain C from V144D patient derived lymphoblast control lymphoblasts isolated ER.
Figure 2: Representative images of 2D gel regions of ER proteins from control and patient derived lymphoblasts. Resolved protein species having a vastly altered abundance is indicated (Red Arrow). The molecular weights are in kilodaltons (kDa) and the IEF dimension is in pH units.

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 5 of 8
ORP-150 150 kDa
A. 1 2 3 4 5 6 7 8
CLIC1 30 kDa
B. 1 2 3 4 5 6 7 8
RPS27a 16 kDa
C. 1 2 3 4 5 6 7 8
COTL1 16 kDa
D. 1 2 3 4 5 6 7 8
Figure 3: Expression of five ER proteins from HSN-I patient-derived lymphoblasts. (A) Immunoblot detection of ORP-150. (B) Immunoblot detection of CLIC1. (C) Immunoblot detection of RPS27a. (D) Immunoblot detection of COTL1. (E) Immunoblot detection of Ig Kappa Chain C. Lanes 1 and 2 represent control ER proteins, 3 and 4 represent control total proteins, 5 and 6 represent V144D ER proteins, 7 and 8 represent V144D total proteins. Figures (F-J) are representative graph showing statistical significant (*) (p < 0.05) difference between control patient lymphoblasts compared to the mutant V144D lymphoblasts of ORP-150, CLIC1, RPS27a, COTL1 and Ig Kappa Chain C respectively (n=3). Blots normalised to GAPDH. Errors bar depict SE of means.
Figure 4: Representative immunofluorescence images of the intracellular localisation of five proteins. Representative confocal micrographs showing SPTLC1, CLIC1, ORP-150, RPS27a and COTL1 stained lymphoblasts (red) and DAPI nuclear stain (blue). Scale bar = 5 µm.

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 6 of 8
DiscussionSPT is an ER bound and key rate determining enzyme in sphingolipid
metabolism. Mutations within the SPT subunits result in potential dysfunction with possible perturbations in sphingolipid synthesis and metabolism causing HSN-I [2]. The ER plays a crucial role in many aspects of protein compartmentalisation which include membrane translocation, protein folding, post-translational modifications of proteins, transport of both membrane and soluble proteins, as well as monitoring protein synthesis and degradation [1]. These processes are both assisted and monitored by molecular chaperones. This investigation has identified several proteins that change in expression in the V144D SPTLC1 mutant lymphoblasts. Whilst this investigation only analysed one patient derived lymphoblast sample: we have conducted further investigation using a tranfected neuronal model, data in follow-up manuscript, with the data here correlating with the transfect neuronal system, thus indicating the changes observed here are not due to patient-patient variations. The proteins that have major increases in expression are ORP-150, CLIC1, COTL1, Ig Kappa Chain C, and with an increase in RPS27a in the ER.
To elucidate if protein changes in the proteomes of the ER fractions occur due to mutations in SPTLC1 causing HSN-I ER membranes were isolated from control and V144D patient lymphoblasts and lysed proteins were subjected to high resolution 2DE (Figure 1). Calnexin, a marker for the ER: confirmed the quality of the isolated fraction used for analysis. In a study by 14, ER stress markers increased in V144D lymphoblasts: here we detected no statistically significant changes in expression of calnexin, suggesting that this protein is constant in both the control and V144D cells (Figures 6A-6F) Expression of the SPTLC1 protein in the V144D fractions analysed, in comparison to the control and revealed no significant increase in expression (Figure 6E) indicating a constant expression state of SPTLC1 in the diseased state. GAPDH (a
house keeping protein) analysis showed no significant changes in in the V144D compared to that of the control lymphoblasts (Figure 6F).
2DE analysis revealed 5 proteins species only detected in the V144D ER fractions in the pI of 5.5, 5.6, 8.0, 5.8, 6.6 and molecular weight of 120, 30, 15, 17, 24 respectively (kDa) (Figure 2). Subsequent LC/MS analysis identified these protein species to be ORP-150, CLIC1, RPS27a, COTL1, and Ig Kappa Chain C (Table 1). A protein species was detected in the control ER fractions in the pI region of 8.3 and molecular weight range of 24 kDa. This protein species was identified as Ig Kappa Chain C. Quantitative immunoblot analysis was carried out to determine the expression of these 5 proteins: it was shown that ORP-150, CLIC1 and COTL1 had statistically significant increases in the V144D mutant (Figures 3A-3J), however RPS27a showed slight increase in the V144D mutant ER fraction: but was not statistically significant (Figure 3I).
Oxygen regulated proteins are overexpressed under conditions of hypoxia. The heat shock protein, oxygen-regulated protein of 150 kDa (ORP-150) also known as Glucose related protein 170 (Grp170), serves as an important molecular chaperone of the endoplasmic reticulum during stress [17]. Notably, hypoxia mediated up-regulation of ORP-150 suppresses programmed cell death driven by oxygen deprivation [18]. Neurons with increased ORP-150 expression demonstrated suppressed caspase-3-like activity [19].
Chloride intracellular channel protein 1 (CLIC1), is small in size and exists in both soluble cytoplasmic and integral membrane forms [20]. CLIC1 exists usually in a soluble form in the cytoplasm and nucleoplasm, but following stimuli undergoes major structural changes and inserts in lipid membranes, where it acts as a chloride-selective ion channel. Cell oxidation seems to be the most important stimulus controlling the transition of CLIC1 between these two forms [21].
A.
Relativ
e Fl
uore
scen
ce In
tens
ity
(Arb
itrar
y U
nits
)
ORP-150
Relativ
e Fl
uore
scen
ce In
tens
ity
(Arb
itrar
y U
nits
)
B. CLIC1
C.
Relativ
e Fl
uore
scen
ce In
tens
ity
(Arb
itrar
y U
nits
)
RPS27a D.
Relativ
e Flu
ores
cenc
e In
tens
ity
(Arb
itrar
y Uni
ts)
COTL1
Figure 5: Relative quantification of ORP-150, CLICL1, RPS27a and COTL1 HSN-I patient-derived lymphoblasts expressing the V144D mutant SPTLC1 genes. Flow cytometry analysis of the relative fluorescence intensity of (A) ORP-150, (B) CLICL1, (C) RPS27a and (D) COTL1 in control and V144D patient-derived lymphoblasts. (A) Blue histogram represents the V144D patient lymphoblasts and Red histogram represents control lymphoblasts. (n=3).

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 7 of 8
While many of the processes that require ubiquitin: are common to all cell types, ubiquitin also has distinct roles in protein degradation. For example; the ubiquitin proteasome system: protein ubiquitylation is also responsible for regulating cell signalling by controlling the endocytosis of plasma membrane receptors. Ubiquitin: is highly conserved and is involved in processes of signal transduction, endocytosis, and DNA repair [22].
Cells are able to move and extend dynamically which is facilitated by actin dynamics. Coactosin (COTL1) is an actin binding protein, and has been shown to associate with F-actin [23]. Under normal cellular conditions, monomeric globular actin (G-actin) is in a state of equilibrium with filamentous actin (F-actin), forming the actin cytoskeleton and is responsible for maintaining and modifying cell shape in motility, phagocytosis, and cytokinesis [24]. The actin cytoskeleton is regulated by numerous actin-binding proteins that interact with actin and regulate the cytoskeleton in cells [23]. COTL1 was also found to directly interact with the filamentous, F-actin but does not form a stable complex with globular, G-actin [23].
Immunostaining (Figure 3) and FACS (Figure 4) analyses yielded the cellular localisation and expression of the following 5 proteins SPTLC1, ORP-150, CLIC1, RPS27a and COTL1 was established. It was observed that there was no apparent change in intracellular localisation of the SPTLC1 protein in control and V144D mutant HSN-I patient lymphoblasts. ORP-150, CLIC1, RPS27a and COTL1 displayed an intracellular localisation change to the cellular periphery and increased abundance. FACS analysis of ORP-150, CLIC1, RPS27a and COTL1 was used to determine the total fluorescence per cell, revealing a marked increase in the relative fluorescence intensity of all the proteins in the V144D cells compared to that of control lymphoblasts with an increase in relative fluorescence of 1.4, 1.25, 2.4, 1.5 OD (fold increase) respectively: relative to the stained controls.
It was identified that there were two other proteins with a marked change in protein expression. Both proteins were located at 24 kDa, but each had a different pI (6.6 and 8.3 respectively). Following mass
spectral analysis: both proteins were identified as Ig Kappa Chain C. Ig kappa Chain C was found to significantly increase in the control lymphoblasts compared to that of the V144D mutant lymphoblasts (Figure 3J). This finding correlates with our previous studies in the V144D patient lymphoblasts [15].
ConclusionThe novel findings in this study thus suggest a link to increased
oxidative stress within the V144D lymphoblasts. Previous studies [8,15] have shown there is an increase in both ER stress and potential oxidative phosphorylation (via a potential change to ROS) changes in V144D. It is evident that there is an increase in oxidative stress within the V144D patient lymphoblasts demonstrated by the increased expression of ORP-150, CLIC1, COTL1 and RPS27a. While these proteins are functionally independent from one and another: together they help establish a strong connection that mutations in SPTLC1 cause oxidative stress within the cell. This increase in oxidative stress could be linked to the increase in Ubiquinol Cytochrome C expression from the mitochondria of V144D mutant cells previously observed: thus an increase in ORP-150 is observed to compensate and protect the cell from an increase in ROS production. Actin function is highly regulated by the association of actin binding proteins. Studies have shown that actin oxidation generally inhibits the association of actin binding proteins with actin [25]. As COTL1 is an actin binding partner its upregulation could be due to the increased oxidative stress upon the cellular cytoskeletal system. Oxidative stress can cause actin remodelling and potential axonal retraction in the neuron [22]. Under the conditions of stress the UPR is activated to ensure misfolded proteins are targets for destruction [22]. RPS27a has a major role in targeting cellular proteins for destruction as such its apparent increase in the V144D mutant demonstrates that there is a possible increase in misfolded protein either directly due to ER stress, oxidative stress or by another mechanism that affects protein conformation. The findings in this study, coupled with others [8,15] suggest that there is a probable underlying mechanism that is common to sensory neurodegenerations.
Figure 6: Expression of ER resident proteins and GAPDH in HSN-I patient-derived lymphoblasts. (A) Immunoblot of Calnexin, (B) Immunoblot of SPTLC1, (C) Immunoblot of GAPDH. Lanes 1 and 2 represent control ER proteins, 3 and 4 represent control total proteins, 5 and 6 represent V144D ER proteins, 7 and 8 represent V144D total proteins. A representative graph showing no statistical significant (p > 0.05) difference between ER control and patient lymphoblasts lysates (n=3) of Calnexin (D), SPTLC1 (E) and GAPDH (F). Blots were normalised to GAPDH. Errors bar depict SE of means.

Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248
Volume 5 • Issue 1 • 1000248Biochem Anal BiochemISSN: 2161-1009 Biochem, an open access journal
Page 8 of 8
ResultsExpression of GAPDH and ER markers in HSN-I patient-de-rived lymphoblasts
In order to assess the levels of calnexin and SPTLC1 expression in isolated ER fractions and total cell lysates from control and V144D mutant HSN-I patient-derived lymphoblasts: immunoblotting was carried out (Figures 6A-6F). Purity of the ER fraction was determined by immunoblotting for membrane, mitochondrial and Golgi complex markers: all of which were absent from the isolated ER fractions, indicating the isolated ER fraction was devoid of other organelles. There were no expression differences observed between the control and V144D samples in either isolated ER isolations or total cell lysates. Quantitation of the immunoblots of the isolated ER and total cell lysate fractions from control and HSN-I patient-derived lymphoblasts (Figures 6D and 6E): confirmed that there was no statistically significant change in expression of these proteins. Expression analysis of GAPDH (Figure 6C) established there was no statistically significant change in the expression of GAPDH (Figure 6F).
Acknowledgements
We are grateful to Prof Garth Nicholson (Molecular Medicine Laboratory and Northcott Neuroscience Laboratory Anzac Research Institute: Sydney) for providing all EBV transformed lymphoblast lines used in this study. SS was supported by APA Research Scholarship: and the UWS School of Science and Health Postgraduate research fund. SM notes the continuing support of an anonymous Private Foundation. JC acknowledges the support of the UWS School of Medicine.
References
1. Pendin D, Mcnew JA, Daga A (2011) Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr Opin Cell Biol 2: 435-442.
2. Wei J, Yerokun Y, Liepelt M, Momin A, Wang E, et al. (2007) 2-1 Serine Palmitoyltransferase. Sphingolipid Biology 25-27.
3. Verhoeven K, Timmerman V, Mauko B, Pieber TR, De Jonghe P, et al. (2006) Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol 19: 474-480.
4. Dyck PJ, Thomas PK (2005) Dyck: Peripheral Neuropathy, 4th Edition, Mosby Elsevier, Philadelphia.
5. Lindholm D, Wootz H, Korhonen L (2006) ER stress and neurodegenerative diseases. Cell Death Differ 1: 385-392.
6. Rao RV, Bredesen DE (2004) Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 16: 653-662.
7. Fulda S, Gorman AM, Hori O, Samali A (2010) Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010: 1-23.
8. Myers S, Malladi C, Hyland R, Bautista T, Boadle R, et al. (2014) Mutantions in the SPTLC1 protein cause mitochondrial structual abnormalisites and endoplasmic reticulum stress in lymphoblasts. DNA and Cell Biology 3: 7.
9. Dedov V, Dedova I, Merrill A, Nicholson G (2004) Activity of partially inhibited
serine palmitoyltransferase is sufficient for normal sphingolipid metabolism and viability of HSN1 patient cells. Biochimica et biophysica acta 1688: 168-175.
10. Bozidis P, Williamson CD, Colberg-Poley AM (2007) Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr Protoc Cell Biol Chapter 3: Unit 3: 27.
11. Vaseva AV, Moll UM (2013) Identification of p53 in mitochondria. Methods Mol Biol 962: 75-84.
12. Churchward MA, Butt RH, Lang JC, Hsu KK, Coorssen JR (2005) Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci 3: 5.
13. Butt RH, Coorssen JR (2005) Postfractionation for enhanced proteomic analyses: routine electrophoretic methods increase the resolution of standard 2D-PAGE. J Proteome Res 4: 982-991.
14. Gauci VJ, Padula MP, Coorssen JR (2013) Coomassie blue staining for high sensitivity gel-based proteomics. J Proteomics 90: 96-106.
15. Stimpson S, Coorssen J, Myers S, (2014) Mitochondrial protein alterations in a familial peripheral neuropathy caused by the V144D amino acid mutation in the sphingolipid protein, SPTLC1. Journal of Chemical Biology 8: 25-35.
16. Wright EP, Partridge MA, Padula MP, Gauci VJ, Malladi CS, et al. (2014) Top-down proteomics: enhancing 2D gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics 14: 872-889.
17. Behnke J, Hendershot LM (2014) The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem 289: 2899-2907.
18. Stojadinovic A, Hooke JA, Shriver CD, Nissan A, Kovatich AJ, et al. (2007) HYOU1/Orp150 expression in breast cancer. Med Sci Monit 13: BR231-239.
19. Wu YB, Li HQ, Ren MS, Li WT, Lv XY, et al. (2013) CHOP/ORP150 ratio in endoplasmic reticulum stress: a new mechanism for diabetic peripheral neuropathy. Cell Physiol Biochem 32: 367-379.
20. Warton K, Tonini R, Fairlie WD, Matthews JM, Valenzuela SM, et al. (2002) Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J Biol Chem 277: 26003-26011.
21. Averaimo S, Milton RH, Duchen MR, Mazzanti M (2010) Chloride intracellular channel 1 (CLIC1): Sensor and effector during oxidative stress. FEBS Lett 584: 2076-2084.
22. Hallengren J, Chen PC, Wilson SM (2013) Neuronal ubiquitin homeostasis. Cell Biochem Biophys 67: 67-73.
23. Provost P, Doucet J, Stock A, Gerisch G, Samuelsson B, et al. (2001) Coactosin-like protein, a human F-actin-binding protein: critical role of lysine-75. Biochem J 359: 255-263.
24. Carlier MF, Laurent V, Santolini J, Melki R, Didry D, et al. (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol 136: 1307-1322.
25. Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC (2011) Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken) 68: 340-354.
OMICS International: Publication Benefits & Features Unique features:
• Increased global visibility of articles through worldwide distribution and indexing• Showcasing recent research output in a timely and updated manner• Special issues on the current trends of scientific research
Special features:
• 700 Open Access Journals• 50,000 editorial team• Rapid review process• Quality and quick editorial, review and publication processing• Indexing at PubMed (partial), Scopus, EBSCO, Index Copernicus and Google Scholar etc• Sharing Option: Social Networking Enabled• Authors, Reviewers and Editors rewarded with online Scientific Credits• Better discount for your subsequent articles
Submit your manuscript at: http://www.omicsonline.org/submission
Citation: Stimpson SE, Lauto A, Coorssen JR, Myers SJ (2016) Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem Anal Biochem 5: 248. doi:10.4172/2161-1009.1000248

76 | P a g e
Paper IV
Published in the Electronic Journal of Biology
Contributions
SES carried out all experimentation, data analysis and wrote first draft manuscript.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 176 -
Proteome Alterations Associated With the V144D SPTLC1 Mutation That Causes Hereditary Sensory Neuropathy-IScott E. Stimpson1, 3, 4, Jens R. Coorssen2,3,4,5,*, Simon J. Myers1,3,4,5,*
1 Neuro-Cell Biology Laboratory, Western Sydney University, Australia;2 Molecular Physiology, Western Sydney University, Australia;
3 Molecular Medicine Research Group, Western Sydney University, Australia;4 School of Science and Health, Western Sydney University, Australia;
5 School of Medicine, Western Sydney University, Australia.*Corresponding author. Tel: 61 02 4620 3383, 61 4620 3802; E-mail: [email protected]; [email protected]
Citation: Stimpson SE, Coorssen JR, Myers SJ, Proteome Alterations Associated With the V144D SPTLC1 Mutation That Causes Hereditary Sensory Neuropathy-I. Electronic J Biol, 11:4
Received: November 16, 2015; Accepted: December 11, 2015; Published: December 17, 2015
Abstract
Background: Hereditary sensory neuropathy type I is the most common subtype and presents with clinical onset in the second to third decade of life with progressive degeneration of the dorsal root ganglion neurons. Three different missense mutations in the gene encoding for serine palmitoyltransferase long chain subunit 1 have been linked to HSN-I. Here we quantitatively assess the proteomes and identify marked protein alterations in both mitochondria and endoplasmic reticulum from HSN-I patient lymphoblasts which harbour the V144D mutation.
Methods: Mitochondria and endoplasmic reticulum were fractionated and lysed from control and patient-derived lymphoblasts. Protein samples were separated into total soluble and total membrane fractions and analysed using a well-established top-down proteomic protocol. Altered protein species were identified by LC MS/MS.
Results: Using a detailed proteomic approach, we identified 36 proteins that were completely altered in abundance in cells harbouring the V144D SPTLC1 mutation relative to normal controls.
Conclusion: The data establish that major protein alterations occur in both the endoplasmic reticulum, where the SPTLC1 protein resides, and in the mitochondria from V144D patient lymphoblasts. These proteins potentially play a major role in disease pathogenesis and may thus help to further elucidate the molecular mechanism(s) underlying hereditary sensory neuropathy type I and might also prove to be potential therapeutic targets.
Keywords: Mitochondria; Endoplasmic reticulum; SPTLC1; HSN-I; Proteomics.
1. Introduction
Hereditary sensory neuropathy type I (HSN-I) is the most common subtype of the HSNs [1], characterised by the progressive degeneration of the dorsal root ganglion (DRG). Onset of clinical symptoms is between the second and third decade of life [2]. Heterozygous mutations in the serine palmitoyltransferase (SPT) long chain subunit 1 (SPTLC1) have been identified as the cause of HSN-I [3,4]. The associated mutations in this gene occur at single amino acids which are highly conserved throughout different species and are therefore likely to interfere with SPT functionality and structure [5].
SPT is a pyridoxal 5'- phosphate dependent multimeric enzyme that catalyses the first step in the biosynthesis of sphingolipids, ceramide and sphingomyelin [6]. Mutations in the SPT subunits thus result in potential dysfunction and perturbations in sphingolipid synthesis and metabolism linked to a variety of diseases, in particular HSN-I [7]. As the rate determining enzyme in the de novo sphingolipid synthesis pathway, SPT is therefore a key enzyme in the regulation of cellular sphingolipid content by condensation of palmitoyl coenzyme A (CoA) with L-serine to form 3-ketodihydrosphingosine [8-10].
We recently noted altered protein expression in the mitochondria and ER from HSN-I (SPTLC1 V144D) mutant lymphoblasts [11,12]. In order to improve characterisation, a more detailed (i.e. ‘deeper’) top down analysis of the total membrane and total soluble proteomes from the mitochondria and ER of control and HSN-I (SPTLC1 V144D) patient lymphoblasts was carried out.
Numerous protein species were found to change markedly in abundance in the mitochondria and ER from the HSN-I (SPTLC1 V144D) patient
Research Article

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 177 -
lymphoblasts; these proteins are involved in energy metabolism, catalytic activity, protein transport, oxidative stress and the cytoskeleton. These protein alterations reflect the changing cellular events that underlie HSN-I.
2. Results
2.1 Gel images of mitochondrial and ER membrane and soluble proteins from control and patient derived lymphoblasts
All membrane and soluble protein samples were well-resolved proteomes covering the entire MW and pI range of the gels. The total numbers of resolved protein species for mitochondrial and ER membrane soluble samples are summarised in Table 1.
Analysis of the membrane and soluble mitochondrial and ER protein profiles from control and HSN-I (SPTLC1 V144D) patient lymphoblasts revealed protein species alterations, change in abundance greater than or equal to 2.0 fold, in the V144D cells relative to control lymphoblasts. The analysis revealed 36 protein species that were located at varying pI / MW (kDa) coordinates. These proteins were excised; digested and LC/MS/MS analysis was carried out to identify these proteins (protein identifications are summarized in Tables 2-5).
2.2 Functions of the identified proteins in mitochondrial and ER fractions
Proteins identified from mitochondrial and ER
Membrane SolubleOrganelle Control V144D Control V144DMitochondrial 550 ± 9 561 ± 9 576 ± 6 562 ± 7Endoplasmic Reticulum 558 ± 6 577 ± 5 623 ± 4 627 ± 4
Table 1. Total protein species resolved by 2D from mitochondrial and ER membrane and soluble fractions obtained from controls and HSN-I (SPTLC1 V144D) patient lymphoblast.
Spot Number
Protein Identified
Accession Number
Unique peptides matched
Sequence Coverage
Mascot Protein Score
Predicted pI
Predicted Mw (kDa)
Mascot pI
Mascot Mw (kDa)
Fold Change in
V144D
1
Succinate Dehydrogenase
Flavoprotein Subunit,
Mitochondrial
P31040 23 24% 617 6.5 70 7.06 73.7 6.1 Fold ↑
2Aldehyde
Dehydrogenase X, Mitochondrial
P30837 20 26% 340 6.1 50 6.36 57.6 2.2 Fold ↑
3
Calcium Binding Mitochondrial Carrier Protein
SCaMc-1
Q6NUK1 19 22% 98 5.7 45 6.22 53.5 2.6 Fold ↓
4
Cytochrome B-C1 Complex
Subunit 1, Mitochondrial
P31930 28 38% 971 5.5 50 5.94 53.3 4.0 Fold ↑
5
Pyruvate Dehydrogenase E1 Component Subunit Alpha, Somatic Form, Mitochondrial
P08559 7 13% 23 7.9 38 8.17 44 3.2 Fold ↑
6
Voltage-Dependent
Anion-Selective Channel Protein
1
P45880 10 26% 265 8.2 30 8.62 31 2.1 Fold ↓
7 Ig Kappa Chain C P01834 18 70% 802 8.3 22 5.58 12 Absent in
V144D
8 Ig Kappa Chain C P01834 15 63% 704 6.6 22 5.58 12
Only present in
V144D
Table 2. Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified a number of proteins from control and V144D lymphoblasts isolated mitochondrial membrane proteins.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 178 -
has been used to provide a visual analysis of protein changes in the HSN-I (SPTLC1 V144D) disease state
membrane and soluble fractions were grouped based upon their biological functionality; a pie graph
Spot Number
Protein Identified
Accession Number
Unique peptides matched
Sequence Coverage
Mascot Protein Score
Predicted pI
Predicted Mw (kDa)
Mascot pI
Mascot Mw
(kDa)
Fold Change in
V144D1 Ezrin P15311 55 55% 1021 5.5 67 5.94 69.5 3.9 Fold ↓
2Prolyl 4-hydroxylase subunit Alpha 1
P13674 40 58% 1188 5.4 60 5.70 61.3 3.0 Fold ↑
360 kDa heat shock protein, Mitochondrial
P10809 26 31% 694 5.2 58 5.70 61.2 2.7 Fold ↑
4 Dipeptidyl Peptidase 1 Q9Y2B0 14 16% 534 7.9 43 8.35 44 2.6 Fold ↑
5
Pyruvate dehydrogenase E1 component subunit alpha, somatic form, Mitochondrial
P08559 26 38% 634 6.4 49 6.54 52.6 2.5 Fold ↑
6Inorganic Pyrophosphatase 2, Mitochondrial
Q9H2U2 36 59% 1265 7.0 32 7.07 38.4 2.2 Fold ↑
7 Pro-Cathepsin H P09668 21 48% 581 6.1 30 8.35 38 2.2 Fold ↑8 Peroxiredoxin-4 Q13162 36 69% 1350 5.6 25 5.86 30.7 2.1 Fold ↑
9 Ig Kappa Chain C P01834 31 88% 1354 8.3 22 5.58 12 Absent V144D
10 Ig Kappa Chain C P01834 27 76% 998 6.6 22 5.58 12 Only present in V144D
Table 3. Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified a number of proteins from control and V144D lymphoblasts isolated mitochondrial soluble proteins.
Spot Number Protein Identified Accession
Number
Unique peptides matched
Sequence Coverage
Mascot Protein Score
Predicted pI
Predicted Mw (kDa)
Mascot pI
Mascot Mw
(kDa)
Fold Change
in V144D
1
Heterogeneous nuclear ribonucleoprotein D-like
O14979 8 14% 230 8.1 40 9.59 46.6 3.5 Fold ↓
2
Serine/Threonine-protein Phosphatase PP1-Beta Catalytic Subunit
P62140 11 40% 418 5.2 35 5.84 38 2.0 Fold ↑
3 Apolipoprotein L2 Q9BQE5 11 42% 210 5.7 36 6.28 37.1 2.8 Fold ↓
4 UPF0568 Protein Cl4orf166 Q9Y224 18 56% 315 6.1 25 6.19 28.2 3.3 Fold ↓
5 Elongation Factor 1-Beta P24534 9 46% 243 4.1 24 4.50 25 4 Fold ↓
6 Serine/Arginine-rich Splicing Factor 3 P84103 6 36% 152 5.8 17 9.64 19.5 2.9 Fold ↓
7 Ig Kappa Chain C P01834 26 80% 1303 6.6 22 5.58 12Only
present in V144D
8 Ig Kappa Chain C P01834 22 80% 1065 8.8 22 5.58 12Absent
from V144D
Table 4. Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified a number of proteins from control and V144D lymphoblasts isolated ER membrane proteins.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 179 -
(Figure 1). This analysis indicates that the majority of the protein alterations in mitochondria are involved in catalytic activity, cytoskeleton, transport, oxidative stress, calcium binding and energy metabolism. However, whist there is some overlap in terms of alterations to mitochondrial and ER proteins (Figure 2), many of those identified from the ER function in the areas of protein biosynthesis, apoptosis, cell proliferation, protein binding and lipid binding (Figure 1).
3. Discussion
SPT is the key rate determining enzyme in sphingolipid metabolism. Mutations within the SPTLC1 subunit thus result in potential perturbations in sphingolipid synthesis and metabolism that may be the underlying causative effects of HSN-I [7]. In initial studies, we showed that a number of mitochondrial and ER proteins are altered in abundance, correlating with the SPT mutations in patient derived cells [11-13]. Here we have carried out a more detailed top-down proteomic analysis and identified 36 protein species that change in abundance in the mitochondria and ER of HSN-I patient cells.
To identify potentially critical protein changes, mitochondria and ER were first isolated from control and HSN-I (SPTLC1 V144D) patient lymphoblasts
and further separated into total membrane and total soluble protein fractions prior to high resolution top-down proteomic analyses using 2DE [14-16]. These analyses revealed numerous protein changes in both the membrane and soluble protein fractions from the control and HSN-I (SPTLC1 V144D) patient lymphoblasts. Mitochondrial protein species that changed in abundance were involved in catalytic activity, cytoskeleton, protein transport, oxidative stress, calcium binding and energy metabolism (Figure 1). The proteins identified in the ER fractions were involved in catalytic activity, cytoskeleton, and lipid binding (Figure 1). While there were a number of non-related protein species that changed in the mitochondria compared to the ER, there were a number of similarities in biological processes, most notably catalytic activity, cytoskeleton, protein transport and oxidative stress [17-19] (Figure 2).
Mitochondria are known to play a role in neurodegeneration, and structural alterations have been characterised in V144D patient lymphoblasts [20], with further studies identifying changes at the protein level within isolated mitochondria [11]. The higher resolution analyses here provide more detailed information still. Oxidative stress can have an impact upon the cell, causing severe and
Spot Number Protein Identified Accession
Number
Unique peptides matched
Sequence Coverage
Mascot Protein Score
Predicted pI
Predicted Mw (kDa)
Mascot pI
Mascot Mw (kDa)
Fold Change in
V144D1 Peroxiredoxin-2 P32119 19 65% 453 5.0 20 5.66 45.3 2.4 Fold ↑
2 Lymphocyte-Specific Protein 1 P33241 24 49% 901 3.5 50 4.69 37.4 3.2 Fold ↑
3Proteasome Activator Complex Subunit 1
Q06323 10 37% 357 6.0 35 5.78 28.9 4.2 Fold ↑
4Proteasome Subunit Alpha Type-3
P25788 18 41% 372 5.2 30 5.19 28.6 2.6 Fold ↑
5 Protein CDV3 Homolog Q9UKY7 14 64% 578 6.2 60 6.06 27.3 2.3 Fold ↓
6Chloride intracellular channel protein 1
O00299 38 68% 1174 5.2 30 5.09 27.2 2.7 Fold ↑
7Adenine Phosphoribos yltransferase
P07741 24 72% 876 5.1 17 5.78 19.8 3.0 Fold ↓
8
Eukaryotic Translation Initiation Factor 5A-1
P63241 26 70% 898 4.3 15 5.08 17 2.1 Fold ↑
9 Ig Kappa Chain C P01834 21 80% 1278 6.6 22 5.58 12Only
present in V144D
10 Ig Kappa Chain C P01834 29 86% 1609 8.3 22 5.58 12 Absent in V144D
Table 5. Summary table of mascot protein identification. LC-MS/MS and Mascot Database searching identified a number of proteins from control and V144D lymphoblasts isolated ER soluble proteins.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 180 -
Figure 1. Pie graph of the identified proteins function in mitochondrial and ER fractions. Representative pie graph of proteins identified in both membrane and soluble mitochondrial and ER fractions grouped into their biological functions.
Figure 2. Similarities in protein changes identified in Mitochondria and ER. Representative graph revealing four common protein changes occurring in the mitochondria and the ER.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 181 -
3 pI 10
MW
(kD
a)
250 150 100 75 37 25 20 15 10
C. 1
2
3
5 4 6
7 8 9 10
3 pI 10
MW
(kD
a)
250 150 100 75 37 25 20 15 10
D. 1
2
3
5 4
6
7 8 9 10
Figure 3. Representative 2D gels images of mitochondrial membrane and soluble proteomes from control and patient derived lymphoblasts. (A) Gel of control mitochondrial membrane proteins. (B) Gel of V144D mitochondrial membrane proteins. (C) Gel of control mitochondrial soluble proteins. (D) Gel of V144D mitochondrial soluble proteins. Red circles represent identified protein species. The molecular weights are in kilodaltons (kDa) and the IEF dimension is in pH units.
Figure 4. Representative 2D gels images of ER membrane and soluble proteomes from control and patient derived lymphoblasts. (A) Gel of control ER membrane proteins (B) Gel of V144D ER membrane proteins. (C) Gel of control ER soluble proteins. (D) Gel of V144D ER soluble proteins. Red circles represent identified protein species. The molecular weights are in kilodaltons (kDa) and the IEF dimension is in pH units.

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 182 -
extensive damage including protein aggregation and impaired ion transport [21]. Previously, ubiquinol cytochrome C subunit 1 was found to increase in abundance in V144D patient lymphoblasts [11]; here this protein was found to increase in abundance (i.e. 4-fold), and this was accompanied by a 2.1-fold increase in the abundance of Peroxiredoxin-4, a protein with antioxidant functions, that reduces the build-up of hydrogen peroxide via a thiol-dependent cycle [21]. These findings correlate with a potential increase in reactive oxygen species (ROS) within the disease cells, which could lead to further disruption to mitochondria.
Perturbations to energy production within neurons, having high metabolic demands, can have catastrophic consequences. Succinate dehydrogenase flavoprotein and pyruvate dehydrogenase E1 subunit are both part of the electron transport chain, and both are increased in abundance (i.e. 6.1 and 3.2 fold, respectively), likely highlighting an energy metabolism issue within the mitochondria [22]. In addition to a potential need to increase energy output, increased levels of succinate dehydrogenase could also potentially increase superoxide formation [22]. Whether these proteins apparently increased abundance is due to a direct need for increased energy or a compensatory effect due to an increase in ROS production and oxidative stress disrupting the electron transport chains ability to produce energy remains unclear, but a destructive spiral would seem a distinct possibility.
Ca2+ is also required for energy production within mitochondria, but increased Ca2+ levels can lead to free radical generation [23]. The data identifies a 2.6-fold decrease in the Ca2+ binding mitochondrial carrier protein (ScaMc-1). This decrease might be a protective mechanism due to the already high levels of ROS but will also cause a decrease in ATP production within the mitochondria. Voltage dependent anion selective channel protein 1 (VDAC) allows mitochondrial influx/efflux of metabolites such as ATP, and may also have a role in regulating Ca2+ in mitochondria [24]. A decrease in VDAC in the V144D mutant, in conjunction with the reduction of SCaMc-1 could result in an overall decrease in intracellular Ca2+ levels in mitochondria and thus decreased ATP production, again strengthening the possibility for a destructive circle of cellular events.
Dipeptidyl peptidase 1, also known as Cathepsin C and Pro-cathepsin H has been shown to be pro-apoptotic by cleaving Bid and Blc-2 family proteins released by mitochondria; greatly increasing the cascade of caspase apoptotic factors to be released [25,26]. The abundance of these proteins are increased in the mutant cells indicating a link to
increased mitochondrial apoptotic process occurring (2.6 and 2.2 fold increase respectively).
Eukaryotic translation initiation factor 5A-1 (eIF5A) has been shown to regulate the Bcl-2 binding protein P53 and the P53 apoptosis pathway. In addition, eIF5A has a regulatory function in protein synthesis. Its increase in abundance of 2.1 fold in the V144D mutant may possibly be the cells response to stabilise uncontrolled protein misfolding due to ER stress [27]. Peroxiredoxin-2 was detected in the ER with an increase in abundance of 2.4 fold, peroxiredoxin-2 just like peroxiredoxin-4 found in the mitochondrial fraction is an antioxidant [28], potentially increased in the mutant state in response to increased ROS production occurring within the ER and throughout the cell.
ER stress may decrease mRNA to reduce the protein load upon the ER to help reduce the amount of misfolded proteins being produced [29], thus we see reduced levels of the serine/threonine phosphatase PP-1, serine/arginine-rich splicing factor and Elongation factor 1-beta within HSN-I (SPTLC1 V144D) patient lymphoblasts. With known ER stress occurring [20] the reduction in abundance of these proteins could be as a result of a compensatory effect reducing the load of protein synthesis occurring in the stressed ER.
Lymphocyte specific protein 1 is an F-actin binding protein [30], it’s 3.2 fold increase in abundance, correlates with other cytoskeletal changes observed in the HSN-I (SPTLC1 V144D) patient lymphoblasts suggesting that maintenance of the cytoskeleton is being increased potentially due to increasing amount of ROS, known to cause actin remodelling and potential axonal retraction in the neuron [31].
Proteasome activator complex 1 (PSME1) and proteasome subunit alpha type 3 (PSMA3), degrade misfolded proteins, in an ubiquitin dependent process [32,33]. Both these proteins are increased in abundance, 4.2 and 2.6 fold respectively in the V144D patient derived lymphoblasts. Previous studies have identified Ubiqutin-40s Ribosomal Protein S27a [12], as such we see here the increase in proteasomes correlating a potential increase in the number misfolded proteins directly due to ER stress, oxidative stress or by another mechanism that affects protein conformation.
Bcl-2 family proteins are regulators of mitochondrial derived apoptosis. Bcl-2 proteins can illicit or inhibit cell death. Apolipoprotein L2 (ApoL2) has a potential apoptotic role being a BH3- protein, localising to mitochondria. This region, known as the ‘BH3-domain’, is essential for the apoptotic function of Bcl-2 autophagy, while the exact role of ApoL2 remains

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 183 -
to be determined, a reduction in the V144D diseased state may cause dysregulation of authopghy [34].
Interestingly, the Chloride intracellular channel protein 1 was identified with a 2.7 fold increased abundance in the HSN-I (SPTLC1 V144D) patient lymphoblasts. We have previously reported this proteins increased expression within the HSN-I (SPTLC1 V144D) patient lymphoblasts [12]. It acts as a chloride-selective ion channel and usually exists in a soluble form in the cytoplasm and nucleoplasm [35], but following stimuli undergoes major structural changes and inserts in lipid membranes, where cell oxidation appears to be an important stimuli determining the transition of Chloride intracellular channel protein 1 between these two forms [36].
It was identified that there were four other proteins with a marked absence or presence in all mitochondrial and ER fractions. These protein species were located at 24 kDa, but each had a different pI (6.6 and 8.3 respectively). Following mass spectral analysis, these proteins identified as Ig Kappa Chain C. This finding correlates with our previous studies in the HSN-I (SPTLC1 V144D) patient lymphoblasts [11,12].
4. Conclusion
This investigation has shown a correlation between previous studies revealing an increase in proteins induced by oxidative stress and mitochondrial electron transport chain proteins. This study also identified changes in calcium channel proteins, cytoskeletal proteins, energy transport proteins. Some of these findings reflect previous studies carried out, providing more evidence for a link of increased misfolded proteins, oxidative stress, and cytoskeleton remodelling and potential changes in Ca2+ signalling within the mitochondria. With mounting discoveries into protein alterations in the V144D mutation it may provide a greater in-sight into the molecular mechanisms that are occurring in HSN-I.
5. Materials
All cell culture stock solutions, including RPMI-1640, Foetal Bovine Serum (FBS), Penicillin (100 U/mL), Streptomycin (100 µg/mL), L-glutamine (2 M), HEPES (1 M), and phosphate buffered saline (PBS) were purchased from GIBCO Invitrogen (Australia). Cell culture consumables were purchased from BD Falcon (Greiner, USA).
6. Methods
6.1 EBV transformed lymphoblasts
EBV transformed control and V144D HSN-I patient
lymphoblasts were kindly provided by Prof. Garth Nicholson (Molecular Medicine Laboratory, Anzac Research Institute, Sydney) [13].
6.2 Lymphoblast cultures
Lymphoblasts were cultured in RPMI-1640 media (GIBCO), supplemented with FBS (10% v/v), Penicillin (1 U/mL), Streptomycin (1 µg/mL), L-glutamine (2 mM), and HEPES (1 mM) at 37oC in a humidified atmosphere of 5% CO2, using T75 cm2 culture flasks (Greiner, Interpath). Prior to use in biochemical assays, lymphoblasts were collected by centrifugation at 1,500 x g (5 min at RT) and washed in PBS. Cell counts were obtained using the Countess Automated Cell Counter (Invitrogen, Australia).
6.3 Isolation of mitochondrial proteins
Briefly, mitochondria were isolated using a sucrose density gradient [14,15]. Lymphoblasts were first centrifuged at 1,500 x g for 5 min, and the cells were then washed in 10 ml of ice cold 1X PBS prior to suspension in 10 ml ice cold CaSRB Buffer (10 mM NaCl, 1.5 mM CaCl, 10 mM Tris-HCL, pH 7.5) and left on ice for 10 min. Cells were homogenised using a Dounce homogenizer (Kimble-Chase, USA) and 7 mL of 2.5X MS buffer (210 mM Mannitol, 70 mM sucrose, 5 mM EDTA, 5 mM Tris-HCl, pH 7.6) was added to restore isotonicity. Homogenates were centrifuged at 700 x g for 5 min to remove nuclei and unbroken cells. The resulting supernatant was centrifuged at 15,000 x g for 10 min to pellet the crude mitochondria. Sucrose gradients were made in 4 mL high speed centrifuge tubes (Beckman Coulter, USA) by adding 1 mL of 1.7 M sucrose buffer (1.7 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) overlayed with 1.6 mL of 1.0 M sucrose buffer (1.0 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6). The mitochondrial pellet was resuspended in 1.6 mL of 1x MS buffer and overlayed on top of the sucrose gradient and centrifuged at 40,000 x g for 30 min. The mitochondrial band, in the middle of the gradient, was gently removed using a 20 G needle, transferred to a 1.5 mL tube, and centrifuged at 16,000 x g for 15 min. The resulting pellet was resuspended in 2D solubilisation buffer containing 8 M urea, 2 M thiourea, 4% (w/v) CHAPS and a cocktail of protease inhibitors.
6.4 Isolation of ER proteins
Briefly, [14,15] Lymphoblasts were first centrifuged at 1,500 x g for 5 min, and the cells were then washed in 10 ml of ice cold 1X PBS prior to suspension in 10 mL ice cold CaSRB Buffer (10 mM NaCl, 1.5 mM CaCl, 10 mM Tris-HCL, pH 7.5) and left on ice for 10 min. Cells were homogenised using a Dounce homogenizer (Kimble-Chase, USA) and 7 mL of 2.5 X

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 184 -
MS buffer (210 mM Mannitol, 70 mM sucrose, 5 mM EDTA, 5 mM Tris-HCl, pH 7.6) was added to restore isotonicity. Homogenates were centrifuged at 700 x g for 5 min to remove nuclei and unbroken cells. The resulting supernatant was centrifuged at 15,000 x g for 10 mins to remove mitochondria. A sucrose gradient was made in 15 mL high speed centrifuge tubes (Beckman Coulter, USA) by adding 2 mL of 2.0 M sucrose buffer (2.0 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) overlayed with 3.0 mL of 1.5 M sucrose buffer (1.5 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) and 3.0 mL of 1.3 M sucrose (1.3 M sucrose, 10 mM Tris-base, 0.1 mM EDTA, pH 7.6) ER containing supernatant was loaded on top of the sucrose gradient and spun at 152,000 x g for 70 min. The ER band, the interface of the 1.5 M and 1.3 M sucrose, was gently removed using a 20 G needle, transferred to a 4mL high speed centrifuge tubes (Beckman Coulter, USA) and centrifuged at 100,000 x g for 35 min. The resulting pellet was resuspended in 2D solubilisation buffer containing 8 M urea, 2 M thiourea, 4% (w/v) CHAPS and a cocktail of protease inhibitors.
6.5 Membrane and soluble protein fractionation
Harvested mitochondrial and ER proteins were separated into membrane and soluble protein fractions as previously described [16]. Briefly; isolated proteins were placed in 20mM HEPES for 3 min on ice with an equal volume 2X PBS subsequently added. Membranes were collected at 125 000 x g for 3 h. The supernatant was collected and membrane pellet resuspended in 1X PBS and spun at 125 000 x g for a further 3 h. Washed membranes were solubilised in 2D solubilisation buffer containing 8 M urea, 2 M thiourea, 4% (w/v) CHAPS. Soluble protein fractions were concentrated using a 3 kDa cut-off Millipore Amicon Ultra Centrifugal filters and resuspended in 4M Urea.
6.6 Protein concentration
Determination of total cellular protein was performed using the EZQ Protein Estimation Assay (Invitrogen, Australia) as previously described [17].
6.6.1 Two dimensional gel electrophoresis
Protein concentration estimations (EZQ assay) were performed on patient and control mitochondrial and ER protein fractions; a total of 100 µg protein was used for each 2DE analysis. 2DE was carried out as previously described [11,16,18,19]; briefly, proteins were reduced and alkylated in solutions containing total protein extraction buffer (containing 8M urea, 2M thiourea and 4% CHAPS without ampholytes), total extraction buffer with 2% ampholytes, TBP/DTT disulphide reduction buffer (2.3 mM Tributyl
phosphine and 45 mM DTT) and alkylation buffer (230 mM acrylamide monomer).
The treated samples were added to 7 cm Non-Linear pH 3-10 IPG strips (Bio-Rad ReadyStrip), and rehydrated for 16 h at RT. Isoelectric focusing (IEF) was then carried out at 20° C using the Protean IEF Cell (Bio-Rad, USA). After IEF, IPG strips were then resolved in the second dimension using a 12.5% T, 2.6% C polyacrylamide gel buffered with 375 mM Tris buffer (pH 8.8), 0.1% (w/v) sodium dodecyl sulphate and polymerised with 0.05% (w/v) ammonium persulphate and 0.05% (v/v) tetramethylethylenediamine (TEMED). A stacking gel containing a 5% T, 2.6% C polyacrylamide buffered with 375 mM Tris buffer (pH 8.8), 0.1% (w/v) SDS and included 0.1% bromophenol blue was added to the resolving gel. The IPG strips were placed onto the stacking gel and overlaid with 0.5% (w/v) low melting agarose dissolved in 375 mM Tris (pH 8.8), with 0.1% (w/v) SDS. Electrophoresis was carried out at 4° C; 150V initially for 10 min then reduced to 90V for 2.5 h. The gels were placed in fixative containing 10% methanol and 7% acetic acid for 1 h. The gels were washed with distilled water for 20 min, 3 times and subsequently stained with colloidal coomassie blue (0.1% (w/v) CCB G-250, 2% (v/v) phosphoric acid, 10% (w/v) ammonium sulphate, 20% (v/v) methanol) for 20 h, with constant shaking at RT [18]; the gels were the de-stained 5 times with 0.5 M NaCl, 15 min each. Imaging of CBB-stained gels on the FLA-9000 imager (FUJIFILM, Tokyo, Japan) was carried out at 685/750 excitation/emission with a photomultiplier tube (PMT) setting of 600 V and pixel resolution set to 100 µm [18]. Analysis of 2D gel images was performed using Delta 2D software (version 4.0.8; DECODON GmbH, Gerifswald, Germany) with automated spot detection (Local Background Region: 96; Average Spot Size: 32 and sensitivity in percentage: 20.0) (Figure 3 and 4).
6.6.2 Mass spectrometry
For analysis a selection criteria was applied. For inclusion, changes in mean normalised spot volume (the abundance of resolved protein species) had to be greater than or equal to a 2.0 fold increase or decrease between control and HSN-I (SPTLC1 V144D) patient lymphoblast, have a p-value <0.05 and be present in all replicate gels [18,19]. The protein species of interest were excised from gels and de-stained overnight. The gel pieces were then reduced and alkylated in 10 mM Dithiothreitol (DTT) and 15 mM Idoacetic acid (IAA), and subsequently incubated with trypsin solution (10 ng/µL, pH 7.4) for 16 h at 37ºC. LC-MS/MS analysis was carried out on a nanoAquity UPLC (Waters Corp., Milford, MA, USA) linked to a Xevo QToF mass spectrometer from

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 185 -
Waters (Micromass, UK). The data were acquired using Masslynx software (Version 4.1, Micromass UK). The MS/MS data files were searched against SwissProt databases with semi-trypsin as the enzyme. The following parameters were used in Mascot for identification of the peptides: maximum missed cleavage of 2, positive peptide charge of 2, 3 and 4, peptide mass tolerant of 0.5 Da in MS and MS/MS data base, fixed modification: carbamidomethyl (C) and variable modifications: oxidation (M).
7. Competing InterestsThe authors have no competing interests.
8. Author Contributions
SES carried out all experimentation, data analysis and wrote an initial draft of the manuscript; JRC participated in design of study, provided access to the proteomics facility in which the bulk of the work was carried out, and re-drafted substantial portions of the draft; SJM conceived the study, participated in design of study, re –drafted substantial sections of the drafts. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Prof Garth Nicholson (Molecular Medicine Laboratory and Northcott Neuroscience Laboratory Anzac Research Institute, Sydney) for providing all EBV transformed lymphoblast lines used in this study. SES was supported by an APA Research Scholarship, and the UWS School of Science and Health Postgraduate research fund. SJM notes the continuing support of an anonymous Private Foundation. JRC acknowledges the support of the UWS School of Medicine.
List of Abbreviations
2DE, two dimensional gel electrophoresis; Apol2, Apolipoprotein L2.; DRG, dorsal root ganglion; DTT, Dithiothreitol; eIF2A, Eukaryotic translation factor 5A-1; ER, endoplasmic reticulum; FBS, Foetal Bovine Serum; HSN, Hereditary sensory neuropathies; HSN-I, Hereditary sensory neuropathy type I; IAA, Idoacetic acid; IEF, Isoelectric focusing; kDa, Kilodaltons; LCB1, long-chain base one; LC/MS, liquid chromatography/ mass spectrometry; PBS, phosphate buffered saline; PMT, photomultiplier tube; PSME1, Proteasome activator complex subunit 1; PSMA3, Proteasome subunit alpha type-3; ROS, reactive oxygen species; SCaMc-1, Calcium binding mitochondrial carrier protein 1; SPT, serine palmitoyltransferase; SPTLC1, serine palmitoyltransferase long chain subunit 1; TEMED, tetramethylethylenediamine; VDAC, Voltage dependent anion-selective channel protein 1.
References[1] Dyck PJ, Thomas PK.( 2005). Dyck: Peripheral
Neuropathy, 4th Edition. Philadelphia, Mosby Elsevier.
[2] Verhoeven K, Coen K, De Vriendt E, et al. (2004). SPTLC1 mutation in twin sisters with hereditary sensory neuropathy type I. Neurology. 62: 1001-2.
[3] Bejaoui K, Wu C, Scheffler MD, et al. (2001). SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. 27: 261-2
[4] Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson G A. (2001). Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Gene. 27: 309-12.
[5] Verhoeven K, Timmerman V, Mauko B, et al. (2006). Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol. 19: 474-80.
[6] Hornemann T, Richard S, Rutti M, Wei Y, Von-Eckardstein A. (2006) ‘Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase’. The Journal of biological chemistry. 49: 37275-37281.
[7] Wei J, Yerokun Y, Liepelt M, et al. (2007). 2-1 Serine Palmitoyltransferase. Sphingolipid Biology. Springerlink. 25-27.
[8] Mandon EC, Ehses I, Rother J, Van Echten G, Sandhoff K. (1992). Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem. 267: 111-448.
[9] Yard B, Carter L, Johnson K, et al. (2007). The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. Journal of molecular biology. 370: 870-886.
[10] Yasuda S, Nishijima M, Hanada K. (2003). Localization, topology, and function of the LCB1 subunit of serine palmitoyltransferase in mammalian cells. J Biol Chem. 278: 4176-83.
[11] Stimpson SE, Coorssen JR, Myers SJ. (2014). Mitochondrial protein alterations in a familial peripheral neuropathy caused by mutations in the sphingolipid protein, SPTLC1. J Chem Biol.
[12] Stimpson, SE, Coorssen JR, Myers SJ. (2015). Isolation and identification of ER associated proteins with unique expression changes specific to the V144D SPTLC1 mutations in HSN-I. BMC Neuroscience.
[13] Dedov V, Dedova I, Merrill A, Nicholson G. (2004). Activity of partially inhibited serine palmitoyltransferase is sufficient for normal sphingolipid metabolism and viability of HSN1 patient cells. Biochimica et biophysica acta.1688: 168-175.
[14] Bozidis P, Williamson CD, Colberg-Poley AM. (2007). Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from

Electronic Journal of Biology, 2015, Vol.11(4): 176-186
ISSN 1860-3122 - 186 -
transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr Protoc Cell Biol. 27.
[15] Vaseva AV, Moll UM. (2013). Identification of p53 in mitochondria. Methods Mol Biol. 962: 75-84.
[16] Butt RH, Coorssen JR. (2005). Postfractionation for enhanced proteomic analyses: routine electrophoretic methods increase the resolution of standard 2D-PAGE. J Proteome Res. 4: 982-91.
[17] Churchward M, Butt RH, Lang J, Hsu K, Coorssen J. (2005). Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 3: 5.
[18] Gauci VJ, Padula MP, Coorssen JR. (2013). Coomassie blue staining for high sensitivity gel-based proteomics. J Proteomics. 90: 96-106.
[19] Wright EP, Partridge MA, Padula MP, et al. (2014). Top-down proteomics: Enhancing 2D gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics. 14: 872-89.
[20] Myers S, Malladi C, Hyland R, et al. (2014). Mutantions in the SPTLC1 protein cause mitochondrial structual abnormalisites and endoplasmic reticulum stress in lymphoblasts. DNA and Cell Biology. 33: 7.
[21] Tavender TJ, Bulleid NJ. (2010). Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J Cell Sci. 123: 2672-9.
[22] Guzzo G, Sciacovelli M, Bernardi P, Rasola A. (2014). Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget. 5: 11897-908.
[23] Feissner RF, Skalska J, Gaum WE, Sheu SS. (2009). Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci (Landmark Ed). 14: 1197-218.
[24] Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 287: C817-33.
[25] Droga-Mazovec G, Bojic L, Petelin A, et al. (2008). Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2.
[26] Turk V, Stoka V, Vasiljeva O, et al. (2012). Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 1824: 68-88.
[27] Huang Y, Higginson DS, Hester L, Park MH, Snyder SH. (2007). Neuronal growth and survival mediated by eIF5A, a polyamine-modified translation initiation factor. Proc Natl Acad Sci U S A. 104: 4194-9.
[28] Ogasawara Y, Ohminato T, Nakamura Y, Ishii K. (2012). Structural and functional analysis of native peroxiredoxin 2 in human red blood cells. Int J Biochem Cell Biol. 44: 1072-7.
[29] Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M. (2004). Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol Cell Biol. 24: 6773-87.
[30] Liu L, Cara DC, Kaur J, et al. (2005). LSP1 is an endothelial gatekeeper of leukocyte transendothelial migration. J Exp Med. 201: 409-18.
[31] Hallengren J, Chen PC, Wilson SM. (2013). Neuronal ubiquitin homeostasis. Cell Biochem Biophys. 67: 67-73.
[32] Johnston SC, Whitby FG, Realini C, Rechsteiner M, Hill CP. (1997). The proteasome 11S regulator subunit REG alpha (PA28 alpha) is a heptamer. Protein Sci. 6: 2469-73.
[33] Shi Z, Li Z, Li ZJ, et al. (2014). Cables1 controls p21/Cip1 protein stability by antagonizing proteasome subunit alpha type 3. Oncogene.
[34] Galindo-Moreno J, Iurlaro R, El Mjiyad N, et al. (2014). Apolipoprotein L2 contains a BH3-like domain but it does not behave as a BH3-only protein. Cell Death Dis. 5: e1275.
[35] Warton K, Tonini R, Fairlie WD, et al. (2002). Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J Biol Chem. 277: 26003-11.
[36] Averaimo S, Milton RH, Duchen MR, Mazzanti M. (2010). Chloride intracellular channel 1 (CLIC1): Sensor and effector during oxidative stress. FEBS Lett. 584: 2076-84.

88 | P a g e
Paper V
Presented in Proteomes format. Submitted to Proteomes
Contributions
SES carried out all experimentation, data analysis and wrote first draft
manuscript.

89 | P a g e
Title: Identifying unique protein alterations caused by SPTLC1 mutations in a transfected neuronal cell model.
Running title:
Scott E. Stimpson, 1, 3, 4 Jens R. Coorssen†2,3,4,5 and Simon J. Myers†1,3,4,5
University of Western Sydney 1Neuro-Cell Biology Laboratory 2Molecular Physiology 3Molecular Medicine Research Group 4School of Science and Health 5School of Medicine
Locked Bag 1797, NSW 2751, Australia
† Co-corresponding authors: Dr. Simon Myers [To communicate with Editorial and Production Offices]
Address: University of Western Sydney, Office 21.1.05, Campbelltown campus, Locked Bag 1797, Penrith, NSW 2751, Australia
Phone: +61 02 4620 3383 Email: [email protected] Facsimile: +61 4620 3025
Professor Jens Coorssen
Address: University of Western Sydney, Office 30.2.15, Campbelltown campus, Locked Bag 1797, Penrith, NSW 2751, Australia
Phone: +61 4620 3802 Email: [email protected] Facsimile: +61 4620 3890

90| P a g e
ABSTRACT
Hereditary sensory neuropathy type I is an autosomal dominant disorder that affects the sensory neurons.
Three missense mutations in serine palmitoyltransferase long chain subunit 1 cause hereditary sensory
neuropathy type I. The endoplasmic reticulum, where the serine palmitoyltransferase long chain subunit
1 protein resides, and mitochondria are both altered in hereditary sensory neuropathy type I mutant cells.
Utilising a transfected neuronal cell line (ND15) we have identified and confirmed, as previously described
in a lymphoblast model [6-7], altered protein expression levels of Ubiquinol Cytochrome C, Hypoxia Up
regulated Protein 1, Chloride Intracellular Channel Protein 1, Ubiqutin-40s Ribosomal Protein S27a, and
Coactosin. Additionally, a further 14 new proteins that exhibiting altered expression within V144D, C133W
and C133Y mutants were identified. These data have shown that mutations in SPTLC1 alters the
expression of a set of proteins that may help to establish a causal link between the mitochondria and ER
and the ‘dying back’ process of dorsal root ganglion neurons observed in HSN-I.
Keywords: Hereditary sensory neuropathy type 1, ND15, Transient Transfection, V144D, C133W, C133Y.

91| P a g e
Background
Hereditary sensory neuropathy type I (HSN-I) is an autosomal dominant inherited neurodegenerative
disorder. It is caused by the missense mutations in the open reading frame of serine palmitoyltransferase
(SPT) long chain subunit 1 (SPTLC1) [1]. SPTLC1 mutations are amino acid substitutions of cysteine to
tryptophan at position 133 (C133W), valine to aspartate at position 144 (V144D) and cysteine to tyrosine
at position 133 (C133Y) [1]. SPT is an endoplasmic reticulum (ER)-bound, key rate-determining enzyme in
the complex sphingolipid metabolic pathway [3]. HSN-I is characterised by a degeneration in the dorsal
root ganglion (DRG) neuron and presents with a clinical onset between the second or third decades of life
[2-5].
In previous studies we investigated altered protein profile changes in the mitochondria and ER of HSN-I
patient cells (SPTLC1 V144D mutation) [6-8]. Changes in protein expression were identified in both the
isolated mitochondria and ER subcellular fractions. Ubiquinol cytochrome C was most notably altered in
expression within the mitochondria of the HSN-I patient cells. Within the ER fractions; Hypoxia up
regulated Protein 1 (ORP-150), Chloride Intracellular Channel Protein 1 (CLIC1), Ubiqutin-40s Ribosomal
Protein S27a (RPS27a), and Coactosin (COTL1) protein expression was increased within the HSN-I patient
cells (SPTLC1 V144D mutation). In addition to these findings, a further 36 proteins were identified from
both the mitochondria and ER of HSN-I patients cells using top-down proteomic analyses [6-8]. Of these
36 proteins, the observed alterations were in proteins related to oxidative stress and cytoskeleton.
Based on these earlier findings, this investigation utilised a ND15 cell line (hybrid of rat dorsal root
ganglion neurone and a mouse neuroblastoma) which had been transiently transfected (TT) to
overexpress the three SPTLC1 missense mutations; V144D, C133W and C133Y. The data obtained from
this neuronal cell model confirmed previous results identified from the HSN-I patient lymphoblast, while
notably identifying changes exhibited in the C133W and C133Y mutations. We have also identified an
additional 14 proteins that are altered in abundance within the transfected ND15 cells. Together these
findings offer a greater insight into the molecular mechanisms occurring in the three known mutations
causing HSN-I.

92| P a g e
Materials
All cell culture stock solutions, including DMEM, Foetal Bovine Serum (FBS), Penicillin (100 U/mL),
Streptomycin (100 µg/mL), L-glutamine (2 M), NEAA (1 M) and phosphate buffered saline (PBS) were
purchased from GIBCO Invitrogen (Australia). Cell culture consumables were purchased from BD Falcon
(Greiner, USA). ORP-150, CLIC1, RPS27a, Calnexin, Ubiquinol Cytochrome C, MTCO2, and GAPDH primary
antibodies were purchased from Abcam (USA). SPTLC1 primary antibody was purchased from Santa Cruz
Biotechnology (USA). COTL1 primary antibody was purchased from Protein SciTech (USA). Kif2A and GFP
primary antibodies were purchased from Merck Millipore (USA). Secondary horse radish peroxidase (HRP)
labelled anti-mouse antibodies and DAPI stains were purchased from Sigma-Aldrich (Australia).
Methods
ND15 Cultures
ND15 cell lines were cultured in DMEM media (GIBCO), supplemented with FBS (10 % v/v), Penicillin (1
U/mL), Streptomycin (1 µg/mL), L-glutamine (2 mM), and NEAA (1 mM) at 37 oC in a humidified
atmosphere of 5 % CO2, using T75 cm2 culture flasks (Greiner, Interpath). Prior to use in biochemical
assays, ND15 cells were collected by centrifugation at 1,500 x g (5 min at RT) and washed in PBS. Cell
counts were obtained using the Countess Automated Cell Counter (Invitrogen, Australia).
Transient Transfection
ND15 cells were transiently transfected (TT) with plasmid constructs (wild type, V144D, C133W and C133Y
(GFP) using Lipofectamine 2000 (L2K; Invitrogen, USA). Cells were plated at a density of 2x 105 per well in
6 well plates. Transfections were carried out when cells were 90-95 % confluent (approximately 24 hours
after plating, at 37 oC and 5 % CO2). Per well, both DNA plasmid constructs and L2K reagent were diluted
in 250 µL of Opti-MEM I Reduced Serum Media (Invitrogen, USA). DNA constructs were diluted to 16 μg/ml
and L2K to 40 μL/ml. Within 5 min of each dilution, the DNA construct diluents and L2K diluents were
combined and incubated for 30 min at 25 oC. After incubation, DNA-L2K complexes (500 μL) were then
added into each well, as required. The cells were then incubated at 37 oC in a humidified atmosphere of
5 % CO2 for 6 h. Cells then had media replaced with fresh media and were cultured for a further 48 h
before being assessed using the LSM 5 confocal microscope (comprising the LSM 5 exciter laser scanning
microscope with Axiovert 200M inverted optical microscope [Carl Zeiss, Jena, Germany]).

93| P a g e
Protein Concentration
Determination of total cellular protein was carried out using the EZQ Protein Estimation Assay (Invitrogen,
Australia) as previously described [9].
SDS-PAGE and Immunoblotting
Wild type and mutant protein fractions (25 µg total protein) were subjected to SDS-PAGE on 12.5%
resolving gels and transferred to PVDF membrane. The membranes were blocked with 5% skim milk in
TBS buffer containing 0.1% Tween-20 for 1 h and then incubated with anti-SPTLC1, anti-GAPDH, anti-ORP-
150, anti-CLIC1, anti-RPS27a, anti-COTL1, anti-MTCO2, anti-GFP and anti-Kif2A at 1:1000, for 16 h.
Membrane were then incubated with secondary HRP antibody (1:2000 dilution) for 1 h at RT. Blots were
developed using an enhanced chemiluminescence (ECL) detection kit (Pierce Thermo Scientific, USA). All
membranes were developed on CL-Xposure Film (Thermo Fisher Scientific, U.S.A) using an AGFA X-ray
developer.
Immunofluorescence
Immunofluorescence was carried out as previously described by [6]. Briefly, ND15 cells (1 x106 cells) were
grown on sterile glass coverslips in 6-well plates 24 h prior to transfection. Forty-eight hours post
transfection, 4% paraformaldehyde was added to the cells for 15 min. Cells were then placed in 0.5%
Triton X-100 and incubated at 37 oC for 20 min. The cells were then blocked in 5% BSA solution at 37 oC
for 30 min, then resuspended in primary antibody, SPTLC1, Kif2A, Cytochrome C, RPS27a, CLIC1, ORP-150,
COTL1, Calnexin and MTCO2, and stained for 1 h at RT. The cells were subsequently washed and
resuspended in secondary antibody, anti-mouse Rhodamine (Millipore, 1:200), and incubated for 1 h at
RT. DAPI (1µg/µL) was added for 2 min and then the cells were washed twice with PBS. The coverslips
were left overnight to dry and mounted onto glass slides prior to confocal imaging using the LSM 5
confocal microscope comprising the LSM 5 exciter laser scanning microscope with Axiovert 200M inverted
optical microscope (Carl Zeiss, Jena, Germany).
Flow Cytometry
FACS analyses were carried out as previously described by [6]. ND15 cells were transfected as above and
then cells were then suspended in 4% paraformaldehyde and incubated for 15 min at RT and then
resuspended in 0.3% Triton X-100 for 15 min at 37 oC. After incubation the cell suspension was centrifuged
at 1,000 x g for 5 min and the pellet resuspended in primary antibody for 1 h at RT. Cell suspension was

94| P a g e
centrifuged, washed in PBS and resuspended in secondary antibody, anti-mouse or anti-rabbit Rhodamine
(Millipore, 1:200) for 1 h at RT. The cell suspension was then analysed using the MACSQuant flow
cytometer (Miltenyi Biotech, Germany).
Two Dimensional Gel Electrophoresis
Protein concentration estimations (EZQ assay) were carried out on wild type and mutant protein fractions;
a total of 100 µg protein was used for each 2DE analysis. 2DE was carried out as previously described [10-
12] and [6]; briefly, whole ND15 proteins were reduced and alkylated in solutions containing total protein
extraction buffer (containing 8M urea, 2M thiourea and 4% CHAPS without ampholytes), total extraction
buffer with 2% ampholytes, TBP/DTT disulphide reduction buffer (2.3 mM Tributyl phosphine and 45 mM
DTT) and alkylation buffer (230 mM acrylamide monomer).
The treated samples were added to 7 cm Non-Linear pH 3-10 IPG strips (Bio-Rad ReadyStrip) and
rehydrated for 16 h at RT. Isoelectric focusing (IEF) was then carried out at 20 °C using the Protean IEF Cell
(Bio-Rad, USA). After IEF, IPG strips were then resolved in the second dimension using a 12.5% T, 2.6%, C
polyacrylamide gel buffered with 375 mM Tris buffer (pH 8.8), 0.1% (w/v) sodium dodecyl sulphate and
polymerised with 0.05% (w/v) ammonium persulphate and 0.05% (v/v) tetramethylethylenediamine
(TEMED). A stacking gel containing a 5% T, 2.6% C polyacrylamide buffered with 375 mM Tris buffer (pH
8.8), 0.1% (w/v) SDS and included 0.1% bromophenol blue was added to the resolving gel. The IPG strips
were placed onto the stacking gel and overlaid with 0.5% (w/v) low melting agarose dissolved in 375 mM
Tris (pH 8.8), with 0.1% (w/v) SDS. Electrophoresis was carried out at 4 °C; 150V initially for 10 min, then
reduced to 90 V for 2.5 h. The gels were placed in fixative containing 10% methanol and 7% acetic acid for
1 h. The gels were washed with distilled water for 20 min, 3 times and subsequently stained with colloidal
coomassie blue (0.1% (w/v) CCB G-250, 2% (v/v) phosphoric acid, 10% (w/v) ammonium sulphate, 20%
(v/v) methanol) for 20 h, with constant shaking at RT (Gauci et. al., 2013). Gels were the de-stained by
washing 5 times with 0.5 M NaCl for 15 min each wash. Imaging of CBB-stained gels on the FLA-9000
imager (FUJIFILM, Tokyo, Japan) was carried out at 685/750 excitation/emission with a photomultiplier
tube (PMT) setting of 600 V and pixel resolution set to 100 µm (Gauci et. al., 2013). Analysis of 2D gel
images was performed using Delta 2D software with automated spot detection (Local Background Region:
96; Average Spot Size: 32 and sensitivity in percentage: 20.0) (version 4.0.8; DECODON GmbH, Gerifswald,
Germany).

95| P a g e
Mass Spectrometry
For analyses a selection criteria was applied. For inclusion, changes in mean normalised spot volume (the
abundance of resolved protein species) had to be greater than a 1.0 fold difference between samples from
wild type versus V144D, C133W and C133Y mutants and be present in all replicate gels [11-12]. Briefly,
the protein species of interest were excised from gels and de-stained overnight. The gel pieces were then
reduced and alkylated in 10 mM dithiothreitol (DTT) and 15 mM Iodoacetic acid (IAA), and subsequently
incubated in trypsin solution (10 ng/µL, pH 7.4) for 16 hours at 37 oC. LC-MS/MS analysis was carried out
on a nanoAquity UPLC (Waters Corp., Milford, MA, USA) linked to a Xevo QToF mass spectrometer from
Waters (Micromass, UK). The data were acquired using Masslynx software (Version 4.1, Micromass UK).
The MS/MS data files were searched against SwissProt databases with semi-trypsin as the enzyme.
Calcium imaging
ND15 cells were grown for 24 h in 35 mm glass bottom size 0 dishes (MatTek, USA) and transfected as
previously described. Molecular Probes Rhod-3 calcium imaging kit (Molecular Probes, USA) was used to
stain the cells for imaging. Briefly, cells were incubated at RT in the dark for 1 h in 10 µM Rohd-3 AM, 2.5
mM probenecid and 1x Powerload. Cells were briefly washed in calcium-free PBS and incubated for a
further 1 h at RT with 2.5 mM probenecid. To obtain low and high intracellular calcium images, non-
transfected (NT) ND15 cells were infused with PBS without calcium, containing 5 mM EGTA and 2 µM
ionomycin to facilitate intracellular calcium to efflux from the cell. High intracellular calcium images were
obtained by infusing the cells in PBS containing calcium and 2 µM ionomycin. Cells were ready for imaging
after a further two washes in calcium-free PBS and imaged on the LSM 5 confocal microscope comprising
the LSM 5 exciter laser scanning microscope with Axiovert 200M inverted optical microscope (Carl Zeiss,
Jena, Germany).

96| P a g e
Results
Expression of proteins identified in HSN-I transfected ND15 cells
In order to assess the level of expression of proteins previously reported as altered, total cellular protein
fractions from wild type and mutant HSN-I TT ND15 cells were isolated and quantitative immunoblot
analyses were carried out (Figure 1). Quantitation of isolated total cell lysate immunoblots of wild type
and mutant ND15 cells (Figures 2 A-J) confirmed that there were statistically significant (p < 0.05) changes
in expression of COTL1 (Figure 2G), Cytochrome C (Figure 2H) and ORP-150 (Figure 2I) in all mutants
compared to the wild type. RPS27a (Figure 2F) was significantly increased in V144D and C133Y.
Quantitation analysis showed there was no statistically significant increase in expression of CLIC1 (Figure
2E), however there was an increase in expression in all mutants compared to the wild type. Expression
analysis determined that there were no statistically significant changes in the expressions of MTCO2
(Figure 2A), GFP (Figure 2D), SPTLC1 (Figure 2C), GAPDH (Figure 2J) and Kif2A (Figure 2B).
Intracellular localisation analyses of SPTLC1 and proteins within transiently transfected ND15 cells.
The intracellular localisation and abundance of the proteins SPTLC1, Kif2A, Cytochrome C, RPS27a, CLIC1,
ORP-150, COTL1 and MTCO2 were established using immunostained wild type and mutant TT ND15 cells.
There were no apparent changes in intracellular localisation of the SPTLC1 when transfected, with GFP-
labelled SPTLC1 localising to the perinuclear region where the ER resides (Figure 3A). Kif2A is a
microtubule associated protein distributed evenly across the cytoskeleton [14]. Kif2A displayed consistent
cytoskeletal patterns throughout the cell in both wild type and mutants (Figure 3B). MTCO2 is classically
found to be distributed across the mitochondrial inner membrane. MTCO2 was distributed evenly
throughout the mitochondria of the cells, indicating no change in the localisation within the mitochondria
of mutant cells compared to the wild type cells (Figure 3H).
Cytochrome C is typically located within the mitochondrial inner membrane [15]. Interestingly,
Cytochrome C showed a more perinuclear clustering in the mutant cells compared to that of the wild type
cells, where the proteins were found to be more evenly distributed throughout the periphery of the cells
(Figure 3C). RPS27a is located within the cytoplasm and nucleoplasm of cells [16]. With these proteins
being evenly distributed throughout the cells in the wild type and mutants indicating no clustering of
ubiquinated proteins occurring (Figure 3D). CLIC1 exists in a soluble and membrane bound form, typically
distributed evenly within the cells [17]. CLIC1 was localised in the cell periphery in the wild type and
mutant cells. However the mutants displayed a larger localisation towards the perinuclear region,

97| P a g e
indicating that CLIC1 may be present more in the membrane form in the mutants. ORP-150 is a chaperon
protein localised throughout the cell [18] and was found to be distributed throughout the wild type and
mutant cells. ORP-150 was also found to be more abundant within the mutants compared to the wild type
cells (Figure 3F). COTL1 is a cytoskeletal associated protein interacting with the cytoskeleton [19]. COTL1
exhibited an even cytoskeletal pattern through all the cells. Interestingly, there appeared to be a more
perinuclear clustering and potential co-localisation occurring in the C133Y mutant in comparison to the
wild type cells (Figure 3G).
FACS analyses of Wild Type, V144D-, C133W- and C133Y-transfected ND15 cells reveal changes in fluorescence intensity of CLIC1, Cytochrome C, ORP-150 and RPS27a.
Fluorescence assisted cell sorting (FACS) was used to determine the total fluorescence per cell of TT ND15
immunostained cells for the proteins SPTLC1, Kif2A, Cytochrome C, RPS27a, CLIC1, ORP-150, COTL1,
MTCO2 and GFP (Figure 4). There was a marked increase in the relative fluorescence intensity of CLIC1
(Figure 4E), Cytochrome C (Figure 4C), ORP-150 (Figure 4F) and RPS27a (Figure 4D) in the mutant cells
compared to that of wild type. There were no changes to SPTLC1 (Figure 4A), Kif2A (Figure 4B) and GFP
(Figure 4I) between the wild type and mutants. These results correlate with the quantitative immunoblot
data presented in Figure 2.
Resolution of total cellular proteins using 2D gels from SPTLC1-transfected ND15 Cells
Total isolated wild type and mutant ND15 proteins were resolved and quantitatively assessed using a
refined two dimensional gel electrophoresis (2DE) protocol [10-11]. The samples resolved covering the
entire MW and pI range in all triplicate gels (Figure 5). Standard spot counts indicated 674 ± 7, 669 ± 4,
663 ± 9 and 655 ± 5 protein species were resolved in wild type, V144D, C133W and C133Y mutant fractions
respectively. LC/MS data coupled with Mascot Daemon searches of SwissProt database resulted in
identification of a further 14 protein alterations in the three mutant samples relative to the wild type, as
summarised in Table 1.
Alterations within the intracellular calcium levels of V144D-, C133W-, C133Y-transfected ND15 cells
compared to that of wild type and non-transfected controls.
Wild type and mutant ND15 cells were analysed for total intracellular calcium using the Rhod-3 Am
calcium stain (Figure 6). Cell images were analysed using ImageJ (NIH, USA) and corrected total cellular
fluorescence obtained. Analyses revealed a marked decrease in intracellular calcium in C133W and C133Y

98| P a g e
mutant cells. V144D mutant cells however, showed an increase in intracellular calcium when compared
against basal levels determined in wild type and non-transfected (NT) cells (Figure 7).

99| P a g e
Discussion:
Mutations in the SPTLC1 subunit are known to be causal in HSN-I. Molecular and cellular studies of cells
over-expressing the SPTLC1 mutations have identified potential dysfunction in sphingolipid biosynthesis
and metabolic activity [4]. This investigation has correlated the previous findings from the lymphoblast
cell model [6-8, 13] with a neuronal model. In addition to the V144D mutation, we have also investigated
changes within the C133W and C133Y mutations causing HSN-I.
Ubiquinol cytochrome C Reductase Core Protein 1 is a central component of the electron transport chain,
catalysing the oxidisation of ubiquinol and reduction of cytochrome C [15]. Here, we have shown in both
quantitative protein expression (Figure 2G) and FACS analyses (Figure 4C) that cytochrome C abundance
was increased significantly in the TT ND15 cells containing the individual mutations. These finding thus
strengthen the potential link to oxidative phosphorylation, via ubiquinol cytochrome C, and altered energy
production ultimately leading to axonal degeneration.
Further quantitative analyses were carried out which confirmed that the protein expression of RPS27a,
COTL1, and ORP-150 (Figure 2F-2I) were significantly increased in the V144D TT ND15 cells. These finding
correlated with results previously identified in the lymphoblast model [6-7]. In addition, COTL1 was
significantly increased in the C133W and C133Y mutation. ORP-150 and RPS27a were found to be
increased significantly in the C133Y mutation, however these proteins were increased in comparison to
the wild type in C133W. FACS (Figure 4) analyses confirmed the altered expression of RPS27a (Figure 4D),
CLIC1 (Figure 4E), ORP-150 (Figure 4F), and COTL1 (Figure 4G) in the TT mutant cells compared to that of
the wild type.
The novel findings in this study indicate links to dysfunction in oxidative phosphorylation, via Ubiquinol
Cytochrome C Reductase Core Protein 1 in all three mutations causing HSN-I. The increased expression of
Cytochrome C results in the interference of energy production and oxidative stress upon the ER,
eventually causing axonal retraction, a characteristic hallmark of HSN-I. Additionally, Stress-70
mitochondrial protein levels were identified in the C133Y mutant as being increased 2.3 fold relative to
the wild type (Figure 5). When mitochondria are under stress, Stress-70 protein levels increase
compensating for increased oxidative damage and maintain normal protein import and synthesis [20].
Thus, if mitochondrial oxidative stress is increased (potentially via ROS production), further cellular
damage would occur, ultimately leading to a demise in ER efficiency eventually resulting in ER stress.

100| P a g e
It is evident that there is an increase in oxidative and ER stress within the cells containing HSN-I mutations.
This is demonstrated by the increased expression of ORP-150, CLIC1, COTL1 and RPS27a. ORP-150 is an
important molecular chaperone of the ER during stress [18]. CLIC1 usually exists in a soluble form in the
cytoplasm but during times of stress undergoes structural changes and inserts into lipid membranes [17].
COTL1 is an actin binding partner with upregulation in response to stress upon the cytoskeletal system
[19]. Alterations in expression of these proteins strongly indicate that oxidative stress could be linked to
increases ubiquinol cytochrome C. Thus, an increase in ORP-150 is potentially observed to compensate
and protect the cell from an increase in ROS production, causing a shift in CLIC1 expression and
stabilisation of the cytoskeleton via COTL1. RPS27a is responsible for targeting misfolded proteins for
destruction, with an apparent increase highlighting potential increases in misfolded proteins and protein
aggregation due to oxidative stress [16].
Further strengthening the connection between ER stress and HSN-I; peptidyl-prolyl cis-trans isomerase
was found to be increased in abundance by 1.7-fold in the C133W mutant. This protein ensures newly
synthesised proteins are folded into their correct conformation [21]. The 26s proteasome is responsible
for regulating the proteome through degradation of ubiquitin-tagged substrates [22]. 26s proteasome
regulatory subunit 8 was found to be increased in abundance by 1.7-fold in the V144D mutation [22]. The
increase in abundance of these two proteins coupled with the increased expression of RPS27a and ORP-
150 highlights the possible increased oxidative stress affecting protein folding conformation.
Calcium is an important signalling molecule involved in the regulation of many cellular functions.
Mitochondrial calcium uptake has been shown to lead to free radical production, with a delicate balance
existing between moderate ROS production to modulate physiological signalling. Overproduction of ROS
can ultimately lead to oxidative and ER stress [23-24]. Decreases in calcium are believed to be a cellular
response to increased stress, serving as a mechanism to limit further damage and increase cell survival
[24]. As part of this study we examined the intracellular levels of calcium in wild type, V144D, C133W, and
C133Y. Whilst intracellular calcium is decreased within C133W and C133Y, calcium within the V144D
mutation is increased. Hence, is the increased level of calcium a correlation of ER stress and mitochondrial
dysfunction occurring, and the result of the V144D-mutation being unable to reduce intracellular calcium
to compensate and protect the cell? Could this difference give insight into how the three mutations differ,
ultimately causing HSN-I? Further investigation into intracellular and mitochondrial calcium levels is
required to delineate the differences within the three mutations.

101| P a g e
Conclusion
This investigation has shown a correlation between previous studies revealing an increase in a
mitochondrial electron transport chain protein, increases in proteins induced by oxidative stress and
changes in the intracellular calcium levels in all three SPTLC1 mutations causing HSN-I. These findings
provide further evidence for mitochondrial and ER dysfunction occurring as a result of mutations in
SPTLC1. Further analysis is required, however, the novel findings provide critical new directions in
understanding the underlying molecular and cellular alterations broadly common (and specific) to all
mutations causing HSN-I and neurodegenerations as a whole.

102| P a g e
References
[1] Dawkins, J. L., Hulme, D. J., Brahmbhatt, S. B., Auer-Grumbach, M. & Nicholson, G. A. 2001. Mutations
in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory
neuropathy type I. Nat Genet, 27, 309-12.
[2] Verhoeven, K., Coen, K., De Vriendt, E., Jacobs, A., Van Gerwen, V., Smouts, I., Pou-Serradell, A., Martin,
J. J., Timmerman, V. & De Jonghe, P. 2004. SPTLC1 mutation in twin sisters with hereditary sensory
neuropathy type I. Neurology, 62, 1001-2.
[3] Hanada, K. 2003. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim
Biophys Acta, 1632, 16-30.
[4] Wei,J., Yerokun, Y., Liepelt, M., Momin, A., Wang, E., Hanada, & K. Merril, Jr A. H., 2007. 2-1 Serine
Palmitoyltransferase. Sphingolipid Biology. Springerlink. 25-27.
[5] Dyck, P .J. & Thomas, P .K. 2005. Dyck: Peripheral Neuropathy, 4th Edition. Philadelphia, Mosby
Elsevier.
[6] Stimpson, SE. Coorssen, JR. & Myers, SJ. 2014. Mitochondrial protein alterations in a familial peripheral
neuropathy caused by mutations in the sphingolipid protein, SPTLC1. J Chem Biol. Doi: 10.1007/s12154-
014-0125-x
[7] Stimpson, SE. Coorssen, JR. & Myers, SJ. 2015. Isolation and identification of ER associated proteins
with unique expression changes specific to the V144D SPTLC1 mutations in HSN-I. BB Reports.
(Manuscript Submitted).
[8] Stimpson, SE. Coorssen, JR. & Myers, SJ. 2015. Proteome alterations associated with the V144D SPTLC1
mutation that causes Hereditary Sensory Neuropathy-I. BMC Clinical Proteomics (Manuscript Submitted).
[9] Churchward, M., Butt, R. H., Lang, J., Hsu, K., & Coorssen, J., 2005. Enhanced detergent extraction for
analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 3, 5.
[10] Butt, R. H. & Coorssen, J. R. 2005. Postfractionation for enhanced proteomic analyses: routine
electrophoretic methods increase the resolution of standard 2D-PAGE. J Proteome Res, 4, 982-91.
[11] Gauci, V. J., Padula, M. P. & Coorssen, J. R. 2013. Coomassie blue staining for high sensitivity gel-based
proteomics. J Proteomics, 90, 96-106.

103| P a g e
[12] Wright, E. P., Partridge, M. A., Padula, M. P., Gauci, V. J., Malladi, C. S. & Coorssen, J. R. 2014. Top-
down proteomics: Enhancing 2D gel electrophoresis from tissue processing to high-sensitivity protein
detection. Proteomics, 14, 872-89.
[13] Myers, S., Malladi, C., Hyland, R., Bautista, T., Boadle, R., Robinson, P., & Nicholson, G. 2014.
Mutantions in the SPTLC1 protein cause mitochondrial structual abnormalisites and endoplasmic
reticulum stress in lymphoblasts. DNA and Cell Biology, vol.33, no. 7.
[14] Jang, C. Y., Wong, J., Coppinger, J. A., Seki, A., Yates, J. R., 3rd & Fang, G. 2008. DDA3 recruits
microtubule depolymerase Kif2a to spindle poles and controls spindle dynamics and mitotic chromosome
movement. J Cell Biol, 181, 255-67.
[15] Crofts, A. R. 2004. The cytochrome bc1 complex: function in the context of structure. Annu Rev
Physiol, 66, 689-733.
[16] Hallengren, J., Chen, P. C. & Wilson, S. M. 2013. Neuronal Ubiquitin Homeostasis. Cell Biochem
Biophys, 67, 67-73.
[17] Averaimo, S., Milton, R. H., Duchen, M. R. & Mazzanti, M. 2010. Chloride intracellular channel 1
(CLIC1): Sensor and effector during oxidative stress. FEBS Lett, 584, 2076-84.
[18] Behnke, J. & Hendershot, L. M. 2014. The large Hsp70 Grp170 binds to unfolded protein substrates
in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem, 289, 2899-907.
[19] Provost, P., Doucet, J., Stock, A., Gerisch, G., Samuelsson, B. & Radmark, O. 2001. Coactosin-like
protein, a human F-actin-binding protein: critical role of lysine-75. Biochem J, 359, 255-63.
[20] Herrmann, J. M., Stuart, R. A., Craig, E. A. & Neupert, W. 1994. Mitochondrial Heat Shock Protein 70,
A Molecular Chaperone For Proteins Encoded By Mitochondrial Dna. J Cell Biol, 127, 893-902.
[21] Shaw, P. E. 2002. Peptidyl-Prolyl Isomerases: A New Twist To Transcription. Embo Rep, 3, 521-6.
[22] Matyskiela, M. E., Lander, G. C. & Martin, A. 2013. Conformational Switching Of The 26s Proteasome
Enables Substrate Degradation. Nat Struct Mol Biol, 20, 781-8
[23] Glancy, B. & Balaban, R. S. 2012. Role Of Mitochondrial Ca2+ In The Regulation Of Cellular Energetics.
Biochemistry, 51, 2959-73.

104| P a g e
[24] Feissner, R. F., Skalska, J., Gaum, W. E. & Sheu, S. S. 2009. Crosstalk Signaling Between Mitochondrial
Ca2+ And Ros. Front Biosci (Landmark Ed), 14, 1197-218.

105| P a g e
Figure 1
Figure 1. Immunoblots of proteins identified in HSN-I-transfected ND15 cells. Representative
Immunoblots of MTCO2, Kif2A, SPTLC1, GFP, CLIC1, RPS27a, COTL1, Cytochrome C, ORP-150, and GAPDH
from wild type, V144D, C133W and C133Y transfected ND15 cells. 25 µg of protein loaded per lane (n=3).

106| P a g e
Figure 2

107| P a g e
F.

108| P a g e

109| P a g e
Figure 2. Expression of proteins and GAPDH in HSN-I-transfected ND15 cells. Representative graphs
showing the difference between wild type and mutant ND15 proteins. (A) MTCO2, (B) Kif2A, (C) SPTLC1,
(D) GFP, (E) CLIC1, (F) RPS27a, (G) COTL1, (H) Cytochrome C, (I) ORP-150, and (J) GAPDH (*) p <0.05
statistically significant increase (n=3). Errors bar depict SE of means. Blots were normalised to GAPDH.
J.

110| P a g e
Figure 3

111| P a g e

112| P a g e

113| P a g e
Figure 3. Representative immunofluorescence images of the intracellular localisation of proteins in transfected ND15 cells. Representative confocal micrographs showing (A) SPTLC1, (B) Kif2A, (C) Cytochrome C, (D) RPS27a, (E) CLIC1, (F) ORP-150, (G) COTL1 and (H) MTCO2 stained transfected (green) ND15 cells (red) DAPI nuclear stain (blue). Scale bar = 5 µm

114| P a g e
Figure 4

115| P a g e
Figure 4. Relative quantification of wild type, V144D-, C133W- and C133Y-transfected ND15 cells. Flow
cytometric analysis of the relative fluorescence intensity of (A) SPTLC1, (B) Kif2A (C) Cytochrome C (D)
RPS27a (E) CLIC1 (F) ORP-150 (G) COTL1 (H) MTCO2 and (I) GFP. Gold histogram represents Wild Type,
Blue histogram represents V144D, Green histogram represents C133W and Red histogram represents
C133Y (n=3).

116| P a g e
Figure 5

117| P a g e
Figure 5. Representative images of 2D gels following resolution of total cellular proteins from HSN-I-
transfected ND15 Cells. (A) wild Type proteins; (B) V144D proteins (C) C133W proteins and (D) C133Y
proteins. The molecular weights are in kilodaltons (kDa) and the IEF dimension is in pH units.

118| P a g e
Table 1. Summary table of mascot protein identifications. LC-MS/MS and Mascot Database searching identified 14 altered proteins from V144D, C133W
and C133Y transiently transfected ND15 cells.
Spot Number Protein Identified Accession
Number
Unique peptides matched
Sequence Coverage
Mascot Protein Score
Predicted
pI
Predicted Mw (kDa)
Mascot pI
Mascot Mw (kDa)
Fold increase or decrease within the mutant
1. Protein Disulphide Isomerase P09103 35 42% 1468 4.2 90 4.77 57.01 V144D- 1.3 Fold
2. Alpha-Enolase P17182 52 67% 1901 6.5 65 6.37 47.11 V144D- 1.6 Fold
3. Long-chain specific acyl-CoA dehydrogenase
P51174 20 50% 1188 8.3 69 8.53 48.2 V144D- 1.7 Fold
4. 26s protease regulatory subunit 8
P62196 44 63% 1287 7.0 60 7.11 45.2 V144D- 1.7 Fold
5. RPS27a P62983 18 42% 193 9.2 16 8.00 9.68 C133W- 2.6 Fold
6. Peptidyl-prolyl cis-trans isomerase
P30416 39 41% 576 5.4 90 5.54 57.6 C133W- 1.7 Fold
7. Stress-70 protein, Mitochondrial
P38647 33 38% 884 4.3 75 5.81 73.70 C133Y- 2.3 Fold
8. 10 kDa heat shock protein, Mitochondrial
Q64433 3 37% 90 7.6 15 7.93 10.96 C133Y- 3.5 Fold
9. Voltage-dependent anion-selective channel protein 2
Q60930 7 29% 229 7.9 30 7.44 32.34 C133Y- 1.6 Fold
10. Long-chain specific acyl-CoA dehydrogenase, Mitochondrial
P51174 45 43% 1117 8.2 50 8.53 48.37 C133Y- 2.1 Fold
11. 26s Proteasome non-ATPase regulatory subunit 14
O35593 25 39% 580 5.8 30 6.06 34.77 C133Y- 1.5 Fold
12. STIP1 homology and U box-containing protein 1
Q9WUD1 27 58% 366 5.9 35 5.71 35.34 C133Y- 2.1 Fold
13. Eukaryotic Translation Initiator Factor 2 Subunit 1
Q6ZWX6 47 64% 944 4.5 37 5.02 36.37 C133Y- 2.4 Fold
14. Triosephosphate Isomerase P48500 34 79% 1162 8.5 26 6.89 27.4 C133Y- 2.7 Fold

119| P a g e
Figure 6
Figure 6. Representative calcium images of wild Type, V144D-, C133W-, C133Y-, Non-Transfected,
Low and High Calcium in ND15 cells. Representative confocal micrographs showing (A) Wild Type, (B)
V144D, (C) C133W (D) C133Y (E) NT (F) Low Calcium (G) High Calcium intracellular calcium stained.
GFP (Green) ND15 cells (red) N=25.
E.
F.
G.
A.
B.
C.
D.

120| P a g e
* *
Figure 7
Figure 7. Relative intensity of intracellular calcium of wild Type, V144D-, C133W-, C133Y-, Non-
Transfected, Low and High Calcium in ND15 cells. A graph showing the difference between NT, wild
type and mutant ND15 proteins (n=25). Table shows p-values of mutants compared to WT and to NT
stained ND15 cells. (*) p <0.05 statistically significant decrease. (**) p <0.05 statistically significant
increase. Errors bar depict SE of means.
p-values Wild-Type Non-Transfected
V144D 0.27139 0.01551**
C133W 0.00001* 0.00023*
C133Y 0.00002* 0.00148*

121| P a g e
Paper VI
Published in Journal of Chemical Biology
Author Contributions
SES assisted in experimentation, data analysis and finalised preliminary draft manuscript.

ORIGINAL ARTICLE
Increased lipid droplet accumulation associated with a peripheralsensory neuropathy
Lee L. Marshall & Scott E. Stimpson & Ryan Hyland &
Jens R. Coorssen & Simon J. Myers
Received: 3 December 2013 /Accepted: 3 March 2014 /Published online: 23 March 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract Hereditary sensory neuropathy type 1 (HSN-1) isan autosomal dominant neurodegenerative disease caused bymissensemutations in the SPTLC1 gene. The SPTLC1 proteinis part of the SPT enzyme which is a ubiquitously expressed,critical and thus highly regulated endoplasmic reticulumboundmembrane enzyme that maintains sphingolipid concen-trations and thus contributes to lipid metabolism, signalling,and membrane structural functions. Lipid droplets are
dynamic organelles containing sphingolipids and membranebound proteins surrounding a core of neutral lipids, and thusmediate the intracellular transport of these specific molecules.Current literature suggests that there are increased numbers oflipid droplets and alterations of lipid metabolism in a varietyof other autosomal dominant neurodegenerative diseases, in-cluding Alzheimer’s and Parkinson’s disease. This study es-tablishes for the first time, a significant increase in the pres-ence of lipid droplets in HSN-1 patient-derived lymphoblasts,indicating a potential connection between lipid droplets andthe pathomechanism of HSN-1. However, the expression ofadipophilin (ADFP), which has been implicated in the regu-lation of lipid metabolism, was not altered in lipid dropletsfrom the HSN-1 patient-derived lymphoblasts. This appears tobe the first report of increased lipid body accumulation in aperipheral neuropathy, suggesting a fundamental molecularlinkage between a number of neurodegenerative diseases.
Keywords Hereditary sensory neuropathy type 1 . Serinepalmitoyltransferase . Serine palmitoyltransferase long chainsubunit 1 . Lipid droplets . Nile red . ADFP
Introduction
Serine palmitoyltransferase (SPT) is a critical, ubiquitouslyexpressed, and highly regulated endoplasmic reticulum boundmembrane enzyme that maintains cellular sphingolipid con-centrations [1–3]. SPT is a pyridoxal-5′-sphosphate (PLP)-dependent multimeric enzyme that catalyses the first and ratelimiting step in the de novo synthesis of sphingolipids [1, 2].
SPT is a multimeric enzyme composed of three similarsubunits: serine palmitoyltransferase long chain subunit 1(SPTLC1), SPTLC2, and SPTLC3. SPTLC2 and SPTLC3both contain a lysine residue at the active site required forPLP binding; therefore, these two subunits are essential for
Lee L. Marshall and Scott E. Stimpson contributed equally to this work.
L. L. Marshall : S. E. Stimpson : R. HylandNeuro-Cell Biology Laboratory, University of Western Sydney,Locked Bag 1797, Penrith South DC, NSW 1797, Australia
J. R. CoorssenMolecular Physiology, University of Western Sydney, Locked Bag1797, Penrith South DC, NSW 1797, Australia
L. L. Marshall : S. E. Stimpson : R. Hyland : J. R. Coorssen :S. J. MyersMolecular Medicine Research Group, University ofWestern Sydney,Locked Bag 1797, Penrith South DC, NSW 1797, Australia
L. L. Marshall : S. E. Stimpson : R. Hyland : J. R. Coorssen :S. J. MyersSchool of Science and Health, University ofWestern Sydney, LockedBag 1797, Penrith South DC, NSW 1797, Australia
J. R. Coorssen : S. J. MyersSchool of Medicine, University of Western Sydney, Locked Bag1797, Penrith South DC, NSW 1797, Australia
S. J. Myers (*)University of Western Sydney, Office 21.1.05, Campbelltowncampus, Locked Bag 1797, Penrith South DC, NSW 1797, Australiae-mail: [email protected]
J. R. Coorssen (*)University of Western Sydney, Office 30.2.15, Campbelltowncampus, Locked Bag 1797, Penrith South DC, NSW 1797, Australiae-mail: [email protected]
J Chem Biol (2014) 7:67–76DOI 10.1007/s12154-014-0108-y

activating the SPT enzyme [2, 4]. SPTLC1, however, lacksthis PLP binding site and other key catalytic residues, sug-gesting that SPTLC1 plays a more regulatory role in the SPTcomplex [2, 3].
The SPTLC1 gene is located on chromosome 9p22, andpositional cloning has identified three missense mutationsassociated with Hereditary sensory neuropathy type 1(HSN-1), an autosomal dominant sensory neuropathy affect-ing peripheral sensory neurons [1, 5]. These mutations resultin a single amino acid substitution of cysteine to tryptophan atposition 133 (C133W), cysteine to tyrosine at position 133(C133Y), and valine to aspartic acid at position 144 (V144D)[6]. HSN-1 is the most common HSN subtype resulting in theprogressive degeneration and dying back of neurons in thedorsal root ganglia. Despite its initial characterisation over50 years ago [7] and the identification of critical mutationsin the SPTLC1 gene, the molecular mechanisms underlyingdisease development and progression still remain poorly un-derstood [8, 9].
There are currently two main hypotheses as to thepathomechanism(s) in HSN-1, suggesting either a ‘gain oftoxic function’ of the SPT enzyme or a dominant negativeeffect [1, 10, 11]. Peripheral neurons may be sensitive to theperturbation of sphingolipid metabolism (i.e. decrease in func-tional levels) caused by a mutation-induced reduction in SPTenzyme activity [11]. This hypothesis has been shown to beconsistent with recent studies on C133W and V144D, dem-onstrating that both mutations reduce normal SPT activity invarious cell types, including cultured patient lymphoblasts[11]. A concomitant change in the membrane lipid composi-tion would be expected to be seen but recent data has beencontradictory. Initially, an increase in glucosylceramide syn-thesis was reported; however, a decrease in ceramide levelsand sphingomyelin synthesis yielded no change in the overallsphingolipid composition [11].
Studies of SPT activity using patient lymphoblaststhat endogenously expressed the SPTLC1 mutation re-ported greater than 50 % reduction of SPT activity [8,10]. While the mechanism by which SPTactivity is reduced isyet to be confirmed, Bejaoui et al. [8] observed that themutation did not directly affect the stability of the protein astranslated but may interfere with the function of the enzyme.As SPTLC1 mutations have a direct effect on the activity ofSPT, this supports the dominant negative effect theory. Thus,competition possibly arising between mutated and wild typeSPTLC1 for interaction with SPTLC2 may represent an un-derlying disease mechanism, with the mutated SPTLC1possessing a higher affinity then wild type [8]. Nonetheless,the SPTLC1 mutation does not reduce SL levels despiteSPT activity being reduced by more than half [11].Therefore, the remaining 50 % of SPT activity may besufficient to maintain normal sphingolipid homeostasisin these cells, presumably because the total SPT activity
is normally than sufficient; indeed, this is suggested byin vivo SPT downregulation [10].
The alternative theory is that mutations in the SPTLC1gene cause a gain of toxic function. Mutations in theSPTLC1 gene are thought to induce a shift in the substratespecificity of the SPT enzyme resulting in the production ofone or more toxic lipid species [1]. The production of twoatypical deoxysphingoid bases (DSB) has been linked to themutant SPT enzyme [1]. These DSB metabolites can neitherbe converted to sphingolipids nor degraded and thereforeaccumulate in the cell, producing a neurotoxic affect. Thisgain of toxic function occurs in many other autosomal dom-inant, inherited neurodegenerative disorders, includingAlzheimer’s disease and Parkinson’s disease [12, 13].
Increases in the number of lipid droplets and changes inlipid metabolism have also been identified in a variety ofautosomal dominant neurodegenerative diseases, includingAlzheimer’s and Parkinson’s [12–14]. Lipid droplets are or-ganelles which contribute to cellular homeostasis by regulat-ing lipid metabolism and the transport of proteins and lipids(including sphingolipids) throughout the cell [15–19]. Currentresearch into Alzheimer’s disease emphasises that accumula-tion of lipid droplets and abnormalities in lipid metabolismmay cause or exacerbate the disease phenotype [13].Similarly, in Parkinson’s disease, the dysregulation of intra-cellular lipid droplet interactions and expressions, as well aschanges in lipid metabolism, may also contribute to the dis-ease phenotype [12, 14].
Considering the potential linkages between central andperipheral neurodegenerative conditions [13], including theaccumulation of lipid droplets and alterations of lipid metab-olism in other autosomal dominant neurodegenerative dis-eases [12], we have tested the hypothesis that an accumulationof lipid droplets would also be associated with HSN-1 disease.The data confirm a significantly increased number of lipiddroplets in lymphoblasts from HSN-1 patients expressing theC133Wand V144Dmutant SPTLC1 proteins. This appears tobe the first report of increased lipid body accumulation in anautosomal dominant sensory neuropathy. We discuss thesefindings in terms of probable molecular mechanisms underly-ing the development and progression of HSN-1.
Results
SPTLC1 mutations do not alter cellular morphologydespite increases in lipid droplet accumulation
EBV transformed, patient-derived lymphoblasts endogenous-ly express the mutant SPTLC1 enzymes associated with HSN-1 [10]. Detailed analysis indicated no gross morphologicalchanges in lymphoblasts derived from healthy controls andHSN-1 patients expressing the C133W and V144D mutant
68 J Chem Biol (2014) 7:67–76
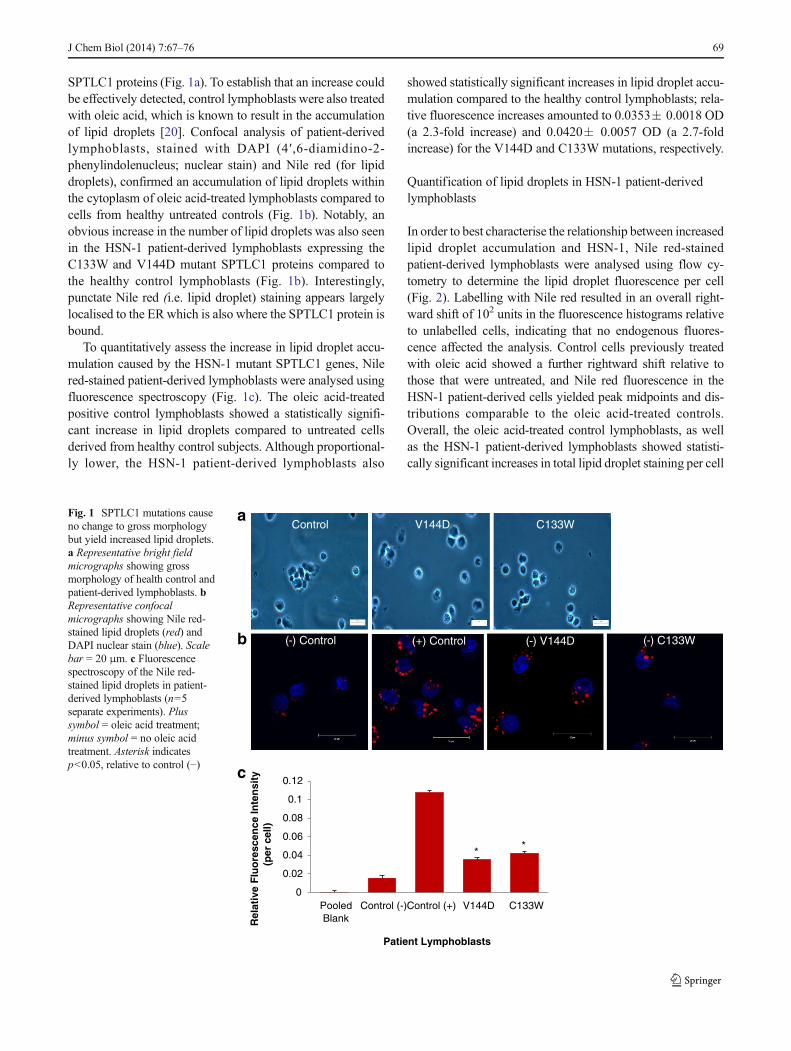
SPTLC1 proteins (Fig. 1a). To establish that an increase couldbe effectively detected, control lymphoblasts were also treatedwith oleic acid, which is known to result in the accumulationof lipid droplets [20]. Confocal analysis of patient-derivedlymphoblasts, stained with DAPI (4′,6-diamidino-2-phenylindolenucleus; nuclear stain) and Nile red (for lipiddroplets), confirmed an accumulation of lipid droplets withinthe cytoplasm of oleic acid-treated lymphoblasts compared tocells from healthy untreated controls (Fig. 1b). Notably, anobvious increase in the number of lipid droplets was also seenin the HSN-1 patient-derived lymphoblasts expressing theC133W and V144D mutant SPTLC1 proteins compared tothe healthy control lymphoblasts (Fig. 1b). Interestingly,punctate Nile red (i.e. lipid droplet) staining appears largelylocalised to the ER which is also where the SPTLC1 protein isbound.
To quantitatively assess the increase in lipid droplet accu-mulation caused by the HSN-1 mutant SPTLC1 genes, Nilered-stained patient-derived lymphoblasts were analysed usingfluorescence spectroscopy (Fig. 1c). The oleic acid-treatedpositive control lymphoblasts showed a statistically signifi-cant increase in lipid droplets compared to untreated cellsderived from healthy control subjects. Although proportional-ly lower, the HSN-1 patient-derived lymphoblasts also
showed statistically significant increases in lipid droplet accu-mulation compared to the healthy control lymphoblasts; rela-tive fluorescence increases amounted to 0.0353� 0.0018 OD(a 2.3-fold increase) and 0.0420� 0.0057 OD (a 2.7-foldincrease) for the V144D and C133W mutations, respectively.
Quantification of lipid droplets in HSN-1 patient-derivedlymphoblasts
In order to best characterise the relationship between increasedlipid droplet accumulation and HSN-1, Nile red-stainedpatient-derived lymphoblasts were analysed using flow cy-tometry to determine the lipid droplet fluorescence per cell(Fig. 2). Labelling with Nile red resulted in an overall right-ward shift of 102 units in the fluorescence histograms relativeto unlabelled cells, indicating that no endogenous fluores-cence affected the analysis. Control cells previously treatedwith oleic acid showed a further rightward shift relative tothose that were untreated, and Nile red fluorescence in theHSN-1 patient-derived cells yielded peak midpoints and dis-tributions comparable to the oleic acid-treated controls.Overall, the oleic acid-treated control lymphoblasts, as wellas the HSN-1 patient-derived lymphoblasts showed statisti-cally significant increases in total lipid droplet staining per cell
0
0.02
0.04
0.06
0.08
0.1
0.12
PooledBlank
Control (-)Control (+) V144D C133W
Rel
ativ
e F
luo
resc
ence
Inte
nsi
ty
(per
cel
l)
Patient Lymphoblasts
Control
(-) Control
V144D
(-) V144D
C133W
(-) C133W(+) Control
* *
a
b
c
Fig. 1 SPTLC1 mutations causeno change to gross morphologybut yield increased lipid droplets.a Representative bright fieldmicrographs showing grossmorphology of health control andpatient-derived lymphoblasts. bRepresentative confocalmicrographs showing Nile red-stained lipid droplets (red) andDAPI nuclear stain (blue). Scalebar = 20 μm. c Fluorescencespectroscopy of the Nile red-stained lipid droplets in patient-derived lymphoblasts (n=5separate experiments). Plussymbol = oleic acid treatment;minus symbol = no oleic acidtreatment. Asterisk indicatesp<0.05, relative to control (−)
J Chem Biol (2014) 7:67–76 69

aControl (+)
C133W (-)
Control (#)
PI/PE-Cy5-A
Cel
lula
r E
ven
ts
PI/PE-Cy5-A
PI/PE-Cy5-A PI/PE-Cy5-A
PI/PE-Cy5-A
Cel
lula
r E
ven
ts
Cel
lula
r Eve
nts
Cel
lula
r E
ven
ts
Cel
lula
r E
ven
ts
Control (-) V144D (-)
b
0
20
40
60
80
100
120
Pooled Blank Control (-) Control (+) V144D C133W
Rel
ativ
e F
luo
resc
ence
Inte
nsi
ty
(per
cel
l)
Patient Lymphoblasts
**
70 J Chem Biol (2014) 7:67–76

compared to the untreated healthy controls; for the C133Wand V144D, mutants this amounted to increases in relativefluorescence of 46.004� 1.563 OD (a 1.6-fold increase) and40.89� 1.099 OD (a 1.8-fold increase), respectively, relativeto the stained controls.
Expression of lipid droplet marker protein ADFP in HSN-1patient-derived lymphoblasts
Adipophilin (ADFP) is a membrane-associated protein pres-ent in mature lipid droplets, and is thus widely used as amarker for lipid droplets. Western blot analysis was performedfor ADFP on total cell lysates from oleic acid-treated controland on untreated control and HSN-1 patient-derived lympho-blasts (Fig. 3a), revealing ADFP expression is below the levelof detection in the untreated samples. To confirm this, westernblot analysis was performed on total cell lysates of oleic acid-treated control and HSN-1 patient-derived lymphoblasts(Fig. 3c). This western blot analyses show that ADFP proteinexpression is relatively abundant in the oleic acid-treatedhealthy control and patient lymphoblasts, thus highlightingthe need for oleic acid treatment for total cell lysate analysis.
Further western blot analyses of SPTLC1 (Fig. 3b) andADFP (Fig. 3c) were carried out, normalising to the house-keeping protein glyceraldehyde-3-phosphate dehydrogenase(GAPDH) to establish relative protein expression levels ofSPTLC1 and ADFP from cells treated with oleic acid.Despite some variability in expression levels, there were nostatistically significant changes in the expression of SPTLC1(Fig. 3e) or ADFP (Fig. 3f) in the HSN-1 patient-derivedlymphoblasts compared to those derived from healthy controlsubjects.
Lipid droplets show no co-localisation with mitochondria
Previous ultrastructural analysis has shown lipid droplet pres-ence in close proximity to the ER and mitochondria mem-branes in the patient lymphoblasts (Myers, S.J, unpublishedobservations). In order to determine if the lipid droplets co-localise with mitochondria, oleic acid treated-healthy controland HSN-1 patient-derived lymphoblasts were fractionatedinto mitochondrial and cytosolic fractions and immunoblottedfor ADFP and MTCO2; the latter is part of Cytochrome c
oxidase subunit 2 of complex IV, located in the inner mem-brane of mitochondria. Fluorescence micrographs of patient-derived lymphoblasts, stained with DAPI (nucleus) and anti-MTCO2 antibody, confirmed normal healthy mitochondrialstructures within the cytoplasm (Fig. 4a). The mitochondrialmarker MTCO2 was detected solely in the mitochondrialfractions, and the lipid droplet marker, ADFP, only in thecytosolic fractions, suggesting that the mitochondria and lipiddroplets do not co-localise and are thus less likely to interact.
Protein expression of ADFP, enriched from a lipid dropletfractionation
In order to identify whether an isolated untreated lipid dropletfraction expressed SPTLC1 and ADFP, lipid droplets wereisolated using a well-established protocol [20] whereby, froman equivalent number of lymphoblasts lipid droplets wereisolated and an equal concentration of protein from each lipiddroplet fraction (7 μg) was loaded onto an SDS-PAGE andassessed by western blot analysis. SPTLC1 was never detect-ed in the isolated lipid droplet fraction; however, ADFP wasconsistently detected in the lipid droplet fractions from boththe healthy control and the patient lymphoblasts. Thishighlighted the successful isolation of an enriched lipid drop-let fraction from untreated lymphoblasts, indicating that oleicacid treatment was not required for this level of analysis(Fig. 5). Most importantly, there was no change in the amountof ADFP in control vs. patient lipid droplets.
Discussion
HSN-1 is an autosomal dominant neuropathy resulting in theprogressive degeneration and dying back of the peripheralsensory neurons in the dorsal root ganglia [5]. HSN-1 is causedby missense mutations in the SPTLC1 gene; however, theactual cellular and molecular mechanisms underlying the dis-ease still remain poorly understood [10]. This study quantifiedincreases in lipid droplets in HSN-1 patient-derived lympho-blasts, and also determined that there was no association be-tween these inclusions and mitochondria, the latter being oftenalso effected in neurodegenerative diseases [12, 13].
Increased numbers of lipid droplets and changes inlipid metabolism have been seen in a variety of autoso-mal dominant neurodegenerative disorders such asAlzheimer’s disease and Parkinson’s disease [12, 13].Current research into Alzheimer’s disease emphasisesthat accumulation of lipid droplets and abnormalities inlipid metabolism may cause or exacerbate the diseasephenotype [13]. Parkinson’s disease research has identi-fied a deregulation of intracellular lipid droplet interac-tions and expressions, and also changes in lipid metab-olism, which may contribute to the disease phenotype.
�Fig. 2 Relative quantification of lipid droplets in HSN-1 patient-derivedlymphoblasts expressing the C133Wand V144D mutant SPTLC1 genes.a Representative flow cytometry scatter plots of Nile red-stained lipiddroplets in control and patient-derived lymphoblasts. Number symbol =unstained, non-treated; plus symbol = oleic acid treated, stained; andminus symbol = non-treated, stained lymphoblasts. b Flow cytometryanalysis of the relative fluoresence intensity of Nile red-stained lipiddroplets in patient-derived lymphoblasts. Plus symbol = oleic acid treat-ment. Minus symbol = no oleic acid treatment (n=5 separate experi-ments). Asterisk indicates p<0.05, relative to control (−)
J Chem Biol (2014) 7:67–76 71

The data presented here establish a significant increasein lipid droplets in lymphoblasts from patients withHSN-1, a peripheral neurodegenerative disorder. With abreadth of research identifying accumulation of lipiddroplets and changes in lipid metabolism in other auto-somal dominant neurodegenerative diseases, it is nowreasonable to suggest that the increased presence oflipid droplets may cause or exacerbate the HSN-1 dis-ease phenotype. Indeed, such alterations suggest a morecentral connection with the pathomechanism(s) underly-ing a host of neurodegenerative disease, both centraland peripheral.
Bright field micrographs indicated no gross morphologicalchanges between healthy control and HSN-1 patient-derivedlymphoblasts; however, confocal micrographs of Nile redstained lipid droplets showed an increase of these inclusionswithin the oleic acid-treated positive controls compared to thehealthy controls and patient-derived lymphoblasts (Fig. 1).Therefore, oleic acid-induced lipid droplet formation [21]was used as a positive control. More importantly the resultsindicated a significant quantitative increase of lipid dropletswithin the patient-derived lymphoblasts compared to thehealthy controls. This increase in lipid droplet abundance inHSN-1 patient-derived lymphoblasts was confirmed usingfluorescence spectroscopy. The confocal and fluorescencespectroscopy analyses thus indicated a possible connectionbetween increased lipid droplet numbers and the cellularmechanism of HSN-1. The data confirmed using Nile redstaining and flow cytometric analysis indicating a significant
increase in lipid droplet numbers within individual patient-derived lymphoblasts (Fig. 2).
ADFP is a lipid droplet-associated membrane protein, andhas thus been used as a marker for these inclusions [19].Immunoblotting revealed that only the oleic acid-treated sam-ples showed detectable ADFP protein from total cell lysates(Fig. 3a). Analysis of SPTLC1 from oleic acid-treated healthycontrol and patient-derived lymphoblasts revealed no signifi-cant difference in expression (Fig. 3f), comparable to theresults of another study that found no change in the levels ofSPTLC1 in HSN-1 patient lymphoblasts [10]. By both con-focal and immunoblotting analyses, the mitochondrial mem-brane marker MTCO2 was detected only in the mitochondrialfraction, and not in the cytoplasmic fraction isolated fromoleic acid-treated healthy control patient-derived lympho-blasts (Fig. 4b). In contrast, immunoblotting of the lipiddroplet marker ADFP identified it only in the cytosolic frac-tion, and not in the mitochondrial fraction, suggesting no co-localisation between the mitochondrial and lipid dropletmembranes.
Immunoblotting for ADFP in lipid droplet fractionsisolated from healthy control and HSN-1 patient-derivedlymphoblasts expressing the C133W and V144D mutantSPTLC1 proteins indicated no detectable changes in pro-tein expression. ADFP is currently thought to be involvedin lipid homeostasis and lipolysis by protecting triacylglyc-erol, within the lipid droplets, from cytosolic lipases [17,19]. These results suggest that the increase in lipid drop-lets within the HSN-1 patient lymphoblasts, without a
a
0
0.5
1
1.5
2
2.5
3
3.5
Control V144D C133W
Rel
ativ
e O
pti
cal D
ensi
ty
(Arb
itar
y U
nit
s)
SPTLC1
0123456789
Control V144D C133W
Rel
ativ
e O
pti
cal D
ensi
ty
(Arb
itar
y U
nit
s)
ADFP
b
c
d
1 2 3 4 5 6 7 8 9
ADFP
SPTLC1
GAPDH
55 kDa
48 kDa
40 kDa
e f
1 2 3 4ADFP48 kDa
Fig. 3 Expression of lipid dropletmarker protein ADFP in HSN-1patient-derived lymphoblasts. aImmunoblot detection of ADFPin oleic acid-treated and oleicacid-untreated total cell lysates; 1represents treated controls; 2,untreated controls; 3, untreatedV144D mutant; and 4, untreatedC133W mutant. Immunoblots oftotal protein lysates from oleicacid-treated cells probed forSPTLC1 (b), ADFP (c), andGAPDH (d); 1–3 representstreated controls; 4–6, treatedV144D mutants; and 7–9, treatedC133W mutants. Histograms ofthe immunoblotting results ofoleic acid-treated controls and ofSPTLC1 (e) and ADFP (f),normalised to GAPDH (n=3)
72 J Chem Biol (2014) 7:67–76

change in ADFP, may cause abnormalities in lipid metab-olism or be indicative of a protein trafficking defect thatinvolve elements of the cytoskeleton [17]. Previously,abnormalities in lipid metabolism have been linked to avariety of other autosomal dominant neurodegenerativediseases, including Alzheimer’s disease and Parkinson’sdisease, suggesting that changes in lipid metabolism mayeither cause or exacerbated the disease phenotype.Therefore, comparable changes in lipid metabolism, alongwith a trafficking defect, may also cause or exacerbate theHSN-1 disease phenotype, a peripheral neurodegenerativedisorder. Further analyses are warranted to determine ifthere is a deregulation of lipid metabolism within theHSN-1 patient lymphoblasts and to identify its link withHSN-1 disease and other autosomal dominant neurode-generative diseases.
This is the first study to investigate lipid droplet formationin an autosomal dominant sensory neuropathy. These dataindicate a possible connection between increased lipid dropletabundance and the cellular mechanism underlying HSN-1.The increase in lipid droplets without a parallel increase inthe expression of ADFP in the HSN-1 patient-derived
lymphoblasts may well indicate a deregulation of lipid metab-olism which may exacerbate the HSN-1 phenotype. The datathus also suggest a more common and/or central role for thesemolecular alterations in specific types of central and periph-eral neurodegeneration.
Materials
All cell culture stock solutions, including RPMI-1640, fetalbovine serum (FBS), penicillin (100 U/mL), streptomycin(100 μg/mL), L-glutamine (2 mM), HEPES (1 M), and phos-phate buffered saline (PBS) were purchased from GibcoInvitrogen (Australia). Cell culture consumables were pur-chased from BD Falcon (Greiner, USA). MTCO2 andGAPDH primary antibodies were purchased from Abcam(USA); SPTLC1 primary antibody was purchased fromSanta Cruz Biotechnology (USA). ADFP primary antibody,secondary HRPMouse and Rabbit antibodies, oleic acid, Nilered, and DAPI stains were purchased from Sigma-Aldrich(Australia).
aControl V144D C133W
b 1 2 3 4 5 6 7 8 9
1 2 3 4 5 6 7 8 9
MTCO260 kDa
c
MTCO260 kDa
ADFP48 kDa
ADFP48 kDa
Mitochondria
Cytosolic
Mitochondria
Cytosolic
Fig. 4 Mitochondria show no co-localisation with lipid droplets. aRepresentative confocal micrographs showing MTCO2 stained mito-chondria (red) and DAPI nuclear stain (blue). Scale bar = 20 μm. bImmunoblot detection of MTCO2 at 60 kDa from mitochondrial and
cytosolic fractions, 1–3 represents oleic acid-treated controls; 4–6, treatedV144D mutants; and 7–i, treated C133W mutants. c Immunoblot detec-tion of ADFP at 48 kDa from oleic acid-treated mitochondrial andcytosolic fractions
J Chem Biol (2014) 7:67–76 73

Methods
EBV-transformed lymphoblasts
Epstein-Barr Virus (EBV)-transformed control and HSN-1patient lymphoblasts were graciously provided by Prof.Garth Nicholson (Molecular Medicine Laboratory, AnzacInstitute, Sydney) [10].
Lymphoblast cultures
Lymphoblasts were cultured in RPMI-1640 media (Gibco),supplemented with FBS (10 % v/v), penicillin (1 U/mL),streptomycin (1 μg/mL), L-glutamine (2 mM), and HEPES(1 M) at 37 °C in a humidified atmosphere of 5 % CO2,using T75 cm2 culture flasks (Greiner, Interpath). Prior touse in biochemical assays, lymphoblasts were collected bycentrifugation at 1,000×g (3 min at RT) and washed in PBS.Normalisation of cell count and viability was obtainedusing the Countess Automated Cell Counter (Invitrogen,Australia).
Oleic acid treatment
For positive controls, lymphoblasts from normal healthy do-nors were treated (in culture) with a 400 μM solution of oleicacid (Sigma, Australia) for 24 h prior to isolation. This is astandardised and routine protocol to induce lipid droplet for-mation [21].
Fluorescence microscopy
For fluorescence microscopy analyses, glass cover slips wereprepared in advance (i.e. overnight). The cover slips weredipped 20 times into a 1:50 dilution of HistoGrip(Invitrogen, Australia) in pure acetone. The cover slips werethen dipped 10 times in dH2O to wash off excess HistoGripand dried overnight. Lymphoblasts were collected by centri-fugation at 1,000×g for 5 min at RT, and resuspended in PBS.Cell suspensions (containing 1×106 to 2×106 cells) werecentrifuged at 1,000×g for 5 min at RT, resuspended in1 mL of 4 % paraformaldehyde in PBS and incubated for15 min at RT.
For lipid droplet analysis, the cell suspension was cen-trifuged at 1,000×g for 5 min at RT, resuspended in 1 mL
of 320 nM Nile red (Sigma, Australia) in Hank’s BufferedSalt Solution (HBSS; prewarmed to 37 °C) and incubatedin the dark for 15 min at RT. For mitochondrial analysisthe cell suspension was permiabilised with a 0.3 % TritonX-100 for 10 min at 37 °C. The cells were centrifuged at1,000×g for 5 min, resuspended in 1 mL of 1 % BSA inPBS, and incubated for 30 min at 37 °C. The cells weresubsequently centrifuged at 1,000×g for 5 min, resuspend-ed in 200 μL of a solution of 1 % BSA in PBS that alsocontained primary antibody (MTCO2 at 1:50 dilution), andincubated for 1 h at RT. The cells were then centrifugedat 1,000×g for 5 min, washed in 1 mL of PBS, centri-fuged again, and resuspended in 200 uL of a solution of1 % BSA in PBS that also contained secondary antibody(1:200 dilution, anti-mouse, rhodamine conjugated) for 1 hat RT. Thereafter, the cell suspension was centrifuged at1,000×g for 5 min at RT, resuspended in 1 mL of DAPI(10 mg/mL stock) in PBS, and incubated for 2 min at RT.The cell suspension was then washed twice in 1 mL ofPBS then suspended in 2 mL of PBS, aliquoted intoindividual wells on 6-well plates, and centrifuged at500×g (10 min, at RT) to pellet the cells onto the coverslips at the bottom of each well. The PBS and non-adherent cells were then aspirated from the wells and thecover slips allowed to dry for 20 min at RT. Thereafter,the cover slips were mounted using 20 μL of DAKOsolution (Dako, Australia) and sealed with nail polish priorto assessment using a LSM-5 Exciter ConfocalMicroscope (Carl Zeiss, Australia).
Fluorescence detection
For fluorescence detection, lymphoblasts were isolated asabove; the cells were then suspended in 1 mL of 4 % parafor-maldehyde in PBS, and incubated for 15 min at RT.Thereafter, the cell suspension was centrifuged at 1,000×gfor 5 min at RT, and the resulting pellet suspended in 1 mLof 320 nM Nile red (Sigma, Australia), (10 mM stock) inHank’s Buffered Salt Solution (HBSS), and incubated in thedark for 15 min at RT. After incubation, the cell suspensionwas centrifuged at 1,000×g for 5 min and resuspended in 1mLof PBS at RT. The cell suspension was then analysed using aBMG Polar Star Omega Fluorescence Plate Reader (BMGLabtech, Germany) and a MACS Quant Flow Cytometry(Miltenyi Biotech, Australia).
ADFP
SPTLC1
48 kDa
1 2 3 4 5 6 7 8 9
55 kDa
Fig. 5 Expression of ADFP in an enriched lipid droplet fraction. Immunoblot detection of ADFP and SPTLC1 proteins from untreated lipid dropletenriched fractions, 1–3, untreated controls; 4–6, untreated V144D mutants; and 7–9, untreated C133W mutants (n=3)
74 J Chem Biol (2014) 7:67–76

Isolation of mitochondrial and cytosolic proteins
Briefly, mitochondria were isolated using a MitochondrialProtein Isolation kit (Amresco Scientific, USA).Lymphoblasts were first centrifuged at 1,000×g for 5 min,and the cells were then washed in 10 ml of ice cold 1X PBSprior to suspension in 1 ml ice cold 1X PBS. Cells weretransferred to a 1.5 ml microcentrifuge tube, centrifuged at1,000×g (4 °C for 5 min) and then suspended in theMitochondrial Protein Isolation Buffer (including 1XProtease Inhibitor cocktail). Cells were then homogenizedon ice by 20 passages through a 26 ½G needle attached to a1 cm3 syringe prior to centrifugation at 1,000×g (4 °C for10 min). The supernatant was collected, transferred to a fresh1.5-ml tube, and centrifuged at 14,000×g (15 min at 4 °C).The supernatant was collected and transferred into a new tube;this fraction contained the cytosolic proteins. The pellet, con-taining mitochondrial proteins, was suspended in 1 mlMitochondrial Protein Isolation Buffer and centrifuged at14,000×g (1 min at 4 °C). The supernatant was discardedand the pellet suspended in 100 μl of Mitochondrial ProteinIsolation Buffer.
Lipid droplet isolation
Lipid droplets were isolated essentially as previouslydescribed [20]. Briefly, lymphoblasts were collected bycentrifugation at 1,000×g (5 min at 4 °C) to yield anequivalent number of lymphoblasts, suspended in 30 mLof dissociation buffer (25 mM Tris-HCl pH 7.4, 1 mMEGTA, 1 mM EDTA, and 100 mM KCl). This cellsuspension was then centrifuged at 1,000×g (5 min at4 °C); the pellet was then suspended in 10 mL ofdissociation buffer and transferred to 15-mL tubes. Thecell suspension was then centrifuged at 1,000×g (5 minat 4 °C), the cells suspended in 750 μL of dissociationbuffer containing 1× protease inhibitors, and transferredto 1.5 mL microcentrifuge tubes. The cell suspensionwas then placed on ice and homogenised using a 26 ½G needle, as described above. The resulting cell lysatewas then centrifuged at 15,000×g (10 min at 4 °C) toremove whole cells, nuclei, and large organelles. Thesupernatant was collected and transferred into new1.5 mL microcentrifuge tubes. An equal volume of1.08 M sucrose solution (approximately 300 μL) wasadded to the supernatant and the total volume then trans-ferred to an ultracentrifuge tube. The supernatant/sucrosemixture was overlayed sequentially with 700 μL of0.27 M sucrose, 500 μL 0.135 M sucrose, and 500 μLof top solution (25 mM Tris-HCl pH 7.4, 1 mM EGTA,and 1 mM EDTA). The gradient was then centrifuged at149,711×g (60 min at 4 °C). The middle and bottomlayers were subsequently removed using a fine glass
pipette, leaving only the white top layer containing lipiddroplets.
Total protein preparation
For a positive control, normal healthy donor lymphoblastswere treated with a 400 μM solution of oleic acid (Sigma-Aldrich, Australia) in standard media, 24 h prior to isolation.Lymphoblasts were collected by centrifugation at 1,000×g for3 min at RT and resuspended in 30 mL of PBS. The cellsuspension was then centrifuged at 500×g for 3 min at RT,resuspended in 1 mL of PBS, and 10 μL was aliquoted for cellcounts. The cell suspension was then centrifuged at 500×g for3 min at RT, resuspended in 300 μL of NDRM (non-detergentresistant membrane lysis buffer; 10 mM Tris-HCl pH 8.0,150 mM NaCl, 1 % Triton X-100, and 1× protease inhibitorsin PBS), and lysed for 20 min on ice. Following lysis, the celllysate was centrifuged at 18,000×g for 15 min at 4 °C. Thesupernatant containing total cell proteins were collected andtransferred to fresh 1.5 mL microcentrifuge tubes ready forprotein concentration and analysis. The pellet containingwhole cells and detergent resistant membranes were resus-pended in 200 μL PBS containing 1× protease inhibitors.
Protein concentration
Determination of total cellular protein was performed usingthe bicinchoninic acid (BCA) protein assay (Sigma-Aldrich,Australia).
SDS-PAGE and immunoblotting
Cell lysates and lipid droplet fractions (50 μg total protein)were subjected to SDS-PAGE on 15 % resolving gels andtransferred to PVDF membrane. The membranes wereblocked with 5 % skim milk in TBS buffer containing 0.1 %Tween-20. The blocked membranes were incubated with anti-ADFP, anti-SPTLC1, and anti-MTCO2 at 1:1,000 and anti-GAPDH at 1:5,000, for 16 h. The membrane was then incu-bated with secondary horse radish peroxidase antibody(1:2,000 dilution) for 1 h at RT. Blots were developed usingan enhanced chemiluminescence (ECL) detection kit (WEST-ZOL, Biotech, Korea).
Acknowledgments We are grateful to Prof Garth Nicholson (Molecu-lar Medicine Laboratory and Northcott Neuroscience Laboratory AnzacResearch Institute, Sydney) for providing all EBV-transformed lympho-blast lines [10] used in this study. LLM was supported by a UWSHonours Scholarships, the School of Science andHealth Honours supportand an anonymous private foundation; SES was supported by APAResearch Scholarship, the UWS School of Science and Health Postgrad-uate research fund; RH was supported by a UWS Postgraduate ResearchAward and anonymous private foundation. SJM notes the support of ananonymous Private Foundation. JRC acknowledges the support of theUWS School of Medicine.
J Chem Biol (2014) 7:67–76 75

References
1. Penno A, ReillyM, Houlden H, LauraM, Rentsch K, Niederkofler V,Stoeckli E, Nicholson G, Eichler F, Brown R, Von-Eckardstein A,Hornemann T (2010) Hereditary sensory neuropathy type 1 is causedby the accumulation of two neurotoxic sphingolipids. J Biol Chem285(15):11178–11187
2. Hornemann T, Richard S, Rutti M, Wei Y, Von-Eckardstein A (2006)Cloning and initial characterization of a new subunit for mammalianserine-palmitoyltransferase. J Biol Chem 281(49):37275–37281
3. Yard B, Carter L, Johnson K, Overton I, Dorward M, Liu H,McMahon S, Oke M, Puech D, Barton G, Naismith J, CampopianoD (2007) The structure of serine palmitoyltransferase; gateway tosphingolipid biosynthesis. J Mol Biol 370(5):870–886
4. Han G, Gupta S, Gable K, Niranjanakumari S, Moitra P, Eichler F,Brown R, Harmon J, Dunn T (2009) Identification of small subunitsof mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc Natl Acad Sci U S A 106(20):8186–8191
5. McCampbell A, Broom D, Truong D, Allchorne A, Gable K, CutlerRG, Mattson M, Woolf C, Frosch M, Harmon J, Dunn T, Brown R(2005) Mutan t SPTLC1 dominan t ly inh ib i t s se r inepalmitoyltransferase activity in vivo and confers an age-dependentneuropathy. Hum Mol Genet 14(22):3507–3521
6. Hornemann T, Penno A, Richard S, Nicholson G, Van-Dijk F,Rotthier A, Timmerman V, Von-Eckardstein A (2009) A systematiccomparison of all mutations in hereditary sensory neuropathy type I(HSAN I) reveals that the G387A mutation is not disease associated.Neurogenetics 10(2):135–143
7. Houlden H, King R, Blake J, Groves M, Love S, Woodward C,Hammans S, Nicoll J, Lennox G, O'Donovan DG, Gabriel C,Thomas PK, Reilly MM (2006) Clinical, pathological and geneticcharacterization of hereditary sensory and autonomic neuropathytype 1 (HSAN I). Brain 129:411–425
8. Bejaoui Y, Uchida Y, Yasuda S, Ho M, Nishijima M, Brown RH Jr,HolleranWM, Hanada K (2002) Hereditary sensory neuropathy type1 mutations confer dominant negative effects on serinepalmitoyltransferase, critical for sphingolipid synthesis. J ClinInvest 110:1301–1308
9. Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M,Nicholson GA (2001) Mutations in SPTLC1, encoding serine
palmitoyltransferase, long chain base subunit-1, cause hereditarysensory neuropathy type I. Nat Genet 27:309–312
10. Dedov V, Dedova I, Merrill A, Nicholson G (2004) Activityof partially inhibited serine palmitoyltransferase is sufficientfor normal sphingolipid metabolism and viability of HSN1patient cells. Biochim Biophys Acta 1688(2):168–175
11. Verhoeven K, Timmerman V, Mauko B, Pieber TR, De Jonghe P,Auer-GrumbachM (2006) Recent advances in hereditary sensory andautonomic neuropathies. Curr Opin Neurol 19:474–480
12. Cole N, Murphy D, Grider T, Rueter S, Brasaemle D, Nussbaum R(2002) Lipid droplet binding and oligomerization properties of theParkinson’s disease protein alpha-synuclein. J Biol Chem 277(8):6344–6352
13. Lane R, FarlowM (2005) Lipid homeostasis and apolipoprotein E inthe development and progression of Alzheimer’s disease. J Lipid Res46(5):949–968
14. Gitler A, Chesi A, Geddie M, Strathearn K, Hamamichi S, Hill K,Caldwell K, Caldwell G, Cooper A, Rochet J, Lindquist S (2008) α-Synuclein is part of a diverse and highly conserved interactionnetwork that includes PARK9 and manganese toxicity. Nat Genet41(3):308–315
15. Beller M, Thiel K, Thul P, Jackle H (2010) Lipid droplets: a dynamicorganelle moves into focus. FEBS Lett 584(11):2176–2182
16. Farese R,Walther T (2009) Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139(5):855–860
17. Ducharme N, Bickel P (2008) Minireview: lipid droplets in lipogen-esis and lipolysis. Endocrinology 149(3):942–949
18. Zehmer J, Huang Y, Peng G, Pu J, Anderson R, Liu P (2009) A rolefor lipid droplets in inter-membrane lipid traffic. Proteomics 9(4):914–921
19. Hodges B, Wu C (2010) Proteomic insights into an expandedcellular role for cytoplasmic lipid droplets. J Lipid Res 51(2):262–273
20. Lay, S.L, Hajduch, E, Linday, M.R, Liepvre, X.L, Thiele, C, Ferre, P,Parton, R.G, Kurzchalia, T, Simons, K & Dugail, I. 2006.Cholesterol-Induced Caveolin Targeting to Lipid Droplets inAdipocytes: A Role for Caveolar Endocytosis. Traffic, vol. 7, pp.549–561. Journal of Biological Chemistry. Vol. 280, No. 52, pp.42841–42847
21. Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR,Londos C (2005) Post-translational regulation of adipose differenti-ation related protein by the ubiquitin/proteasome pathway
76 J Chem Biol (2014) 7:67–76

132 | P a g e
General Discussion The research presented in this thesis has been undertaken to provide greater insight
into the molecular and cellular mechanisms of the neurodegenerative disorder, HSN-
I, with the identification of significantly altered protein profiles within isolated
mitochondria and ER fractions from patient derived lymphoblasts. There is an
increasing number of studies highlighting the essential and critical role mitochondria
play in many disease processes and in particular neurodegenerative diseases. As
stated earlier, HSN-I is one such disease that exhibits altered mitochondrial
morphology in patient derived lymphoblasts carrying mutations in SPTLC1, although
the mechanism by which this occurs is yet to be fully elucidated. Previous studies have
suggested that a reduction in SPT could not account for the global reduction of
sphingolipid metabolism, yet axonal degeneration is still observed in HSN-I (Penno et.
al., 2010). Other studies have proposed that these mutations cause a deleterious gain
of function with the production of toxic sphingolipids (Hornemann et. al., 2009).
Recently, investigations utilising transmission electron microscopy (TEM) have shown
that the ER wraps around morphologically-challenged mitochondria in a perinuclear
fashion (Myers et. al., 2014). From these studies, we hypothesised that SPTLC1
mutations in HSN-I cause alterations to both mitochondrial and ER proteins, and alters
cellular lipids. These alterations can cause perturbations in neuronal homeostasis as
neurons are sensitive to changes in homeostasis (particularly energy production).
It is known that mitochondria are membrane-bound organelles responsible for
numerous essential biological processes (including, but not limited to, oxidative
phosphorylation, lipid metabolism, and apoptosis). They are inter-connected, forming
highly dynamic networks throughout the cytoplasm of most cells, but are often

133 | P a g e
concentrated in specific areas of high energy utilisation. In order to investigate the
functionality of mitochondria and their critical role within the disease HSN-I, it was
imperative to isolate these organelles at the highest quality and purity. Thus,
comparative analyses were undertaken to determine the most efficient and optimal
isolation method of mitochondria, specifically comparing a commercial kit against
traditional sucrose methods. We carried out detailed assessments of mitochondrial
proteins harvested using the Amresco Mitochondrial Protein Isolation Buffer (AMPIB)
relative to those extracted by 2DE sample buffer following isolation of mitochondria by
sucrose gradient fractionation.
Analyses of the profiles obtained from total mitochondrial proteomes indicated there
was greater than 87% overlap between the two isolation methods. To further assess
these methods, isolated mitochondria were subjected to a well-established protocol to
recover separate total membrane and soluble protein fractions. 2DE analyses showed
consistently lower quality proteome isolates from the AMPIB extracts relative to those
extracted using standard sucrose fractionation in 2DE sample buffer. A marked
increase in protein resolution within the low to mid molecular weight region was
observed using the standard isolation method as opposed to AMPIB. This data
unequivocally showed that to obtain a full protein profile for mitochondria the traditional
subcellular fractionation method using sucrose density gradient centrifugation yielded
higher resolution of protein profiles, allowing isolated mitochondria to be quantitatively
analysed and thus more accurately reflect the mitochondrial proteome.
Successful optimisation of mitochondrial isolation then enabled the investigation and
comparison of total protein profiles of mitochondria isolated from both control and
V144D patient lymphoblasts using 2DE. We observed one protein that was selectively

134 | P a g e
detected in the V144D cells at pI 5.7 and molecular weight 55 kDa which was
undetectable in the control protein profiles. This protein was subsequently determined
to be ubiquinol-cytochrome c reductase core protein 1. We were able to confirm this
significant increase in the amount of ubiquinol cytochrome c reductase core protein 1
in V144D cells relative to control cells using quantitative immune-blotting. Additionally,
comparison of control and V144D cell proteomes revealed two additional marked
protein changes. Both of these proteins were located in the 24-kDa molecular weight
region although were isolated in different pI regions (6.6 and 8.3 respectively). Both
proteins were subsequently identified as Ig kappa chain C.
As described earlier, mitochondria function as intracellular energy producing
organelles, where substrates are metabolised to fuel oxidative phosphorylation
through the electron transport chain within their inner membranes. Specific protein
complexes within the mitochondria facilitate the flow of electrons from the reducing
substrates to oxygen and concomitantly build a proton gradient required for ATP
generation (Crofts, AR 2004). Ubiquinol-cytochrome c reductase core protein 1 is a
central component of this electron transport chain, catalysing the oxidisation of
ubiquinol (ubihydroquinone) and reduction of cytochrome c. These processes are
essential in maintaining the electron flow and preventing potential electron leaks
and/or break down of the respiratory chain. (Crofts, AR 2004). Ubiquinol cytochrome
c reductase core protein 1 is known to be involved in free radical generation, primarily
by the production of reactive oxygen species (ROS), within mitochondria. ROS
production has been shown to disrupt the homeostasis of and interactions within the
mitochondrial matrix resulting in the perturbation of oxidative phosphorylation, leading
to the disruption of mitochondrial function and physiology and resulting in cell death
(Drose et. al., 2014).

135 | P a g e
The novel findings in this study provide a strong link between alterations in oxidative
phosphorylation, via ubiquinol cytochrome c reductase core protein 1 (possibly
through regulation of ROS production), and the ensuing interference of energy
production. These alterations are hypothesised to ultimately lead to axonal retraction
that is the characteristic hallmark of HSN-I. If ROS production increased as a result of
increased levels of ubiquinol cytochrome c reductase core protein 1, such as the levels
observed in mitochondria derived from HSN-I patient cells, this could contribute to the
progression of damage attributed to the observed axonal retraction.
These initial analyses did not detect alterations in known antioxidant, chaperone or
other repair proteins that would typically be expected to be modified or up-regulated
in response to such perturbations in ROS production. The possibility that mutations
within the SPTLC1 protein hindered such a response needs to be investigated. It will
be important to further investigate whether this potential decrease or lack of regulation
of ROS production may have been a cause for the observed morphological alterations
within the mitochondria from the HSN-I affected cells.
This study identified a change in an immunoglobulin potentially due to the SPTLC1
mutation causing HSN-I. The Ig kappa light-chain constant region undergoes little to
no variation in human immunoglobulins and forms part of the five immunoglobulin
classes produced in mature B cells. We have determined a statistically significant
decrease in the amount of Ig kappa chain C in the total cell lysates isolated from
V144D cells relative to control lymphoblast cell lysates. While the mechanism leading
to the apparent shift in pI is yet to be fully elucidated, it seems likely to be due to an
as yet unidentified post-translational modification (potentially glycosylation,
phosphorylation or methylation, or possibly some combination of the three).

136 | P a g e
Immunoglobulin light chains have been implicated in, and are biomarkers of, diseases
such as multiple myeloma and primary systemic or amyloid light chain (AL)
amyloidosis (Yamamoto et. al., 2010). In these diseases, it has been shown that the
free Ig kappa chain associates with sphingomyelin on the plasma membrane of the
myeloma cells forming aggregates that are required for intercalation with membranes
(Hutchinson et. al., 2010). This suggests an important role for sphingolipids in these
disease processes. The exact role of Ig kappa chain C and its relationship to HSN-I
could form the basis of a future investigation, as reduced levels of this protein may be
responsible for apparent reductions in cellular repair responses observed within cells
from HSN-I patients.
The ER is an intracellular organelle and is critical for cellular survival. The ER plays a
crucial role in many aspects of protein maturation that includes membrane
translocation, folding, post-translational modification and transport of both membrane
and soluble proteins (Pendin et. al., 2011). In addition, the ER is involved in the
synthesis of phospholipids and steroids and also in the regulation of Ca2+
homeostasis. The results presented earlier in this study indicated that if altered protein
profiles were observable within mitochondria isolated from HSN-I patient
lymphoblasts, then there could be subsequent detectable protein alterations within the
ER isolated from the same cells. As stated previously, the ER has been shown to be
wrapped around the damaged mitochondria in HSN-I lymphoblasts and thus we
hypothesised that there would also be significantly altered expression of proteins
caused by the SPTLC1 mutations.
Mutations within SPTLC1 result in potential dysfunction and perturbations in
sphingolipid synthesis and metabolism causing HSN-I (Wei et. al., 2007). Disruption

137 | P a g e
to the function of the ER or loss of its integrity leads to ER stress (Lindholm et. al.,
2006), and ER stress is characterised by the accumulation of unfolded proteins and
changes in calcium homeostasis within the ER termed the unfolded protein response
(UPR) (Rao et. al., 2004).
To elucidate if protein changes in the proteomes of the ER fractions occur due to
mutations in SPTLC1 causing HSN-I, ER membranes were isolated from control and
V144D patient lymphoblasts and proteins from the lysed cells were subjected to 2DE.
Analysis of the amount of the SPTLC1 protein fractions from cells carrying the
V144D mutation compared with the control cell fractions showed no significant
difference in expression, indicating a constant expression of SPTLC1 in the diseased
state.
2DE analyses revealed five protein species present only in the ER fractions of cells
carrying the V144D mutation, with subsequent LC/MS analyses identifying these
protein species to be Hypoxia Up Regulated Protein 1 (ORP-150), Chloride
Intracellular Channel Protein 1 (CLIC1), Ubiqutin-40s Ribosomal Protein S27a
(RPS27a), Coactosin (COTL1) and Ig Kappa Chain C. Quantitative immunoblot
analyses of these isolated proteins were then carried out to determine their expression
levels and we were able to determine that ORP-150, CLIC1 and COTL1 were
increased significantly in the V144D mutant cells when compared to the control.
Oxygen-regulated proteins are known to be overexpressed under conditions of
hypoxia. The heat shock protein, ORP-150 serves as an important molecular
chaperone of the ER during stress (Behnke and Hendershot, 2014). Notably, hypoxia-
mediated up-regulation of ORP-150 suppresses programmed cell death driven by
oxygen deprivation (Stojadinovic et. al., 2007). CLIC1 is a small protein and exists in

138 | P a g e
both soluble cytoplasmic and integral membrane forms (Warton et. al., 2002). CLIC1
exists primarily in a soluble form within the cytoplasm and nucleoplasm, but following
cell oxidation undergoes major structural changes and becomes inserted in lipid
membranes, where it acts as a chloride-selective ion channel (Averaimo et. al., 2010).
The ability of cells to move and extend dynamically is facilitated by actin dynamics.
Coactosin (COTL1) is an actin binding protein and has been shown to associate with
F-actin (Provost et. al., 2001). These findings, coupled with the alterations observed
in the isolated mitochondrial proteomes, provide a strong evidence for increased
oxidative stress within the V144D lymphoblasts. It is evident that there is an increase
in oxidative stress within the V144D patient lymphoblasts demonstrated by the
increased expression of ORP-150, CLIC1, COTL1 and RPS27a. While these proteins
are functionally independent from each other, together they help establish a strong
connection to mutations in SPTLC1 causing oxidative stress within the cell. This
increase in oxidative stress could be linked to the increase in ubiquinol cytochrome c
expression from the mitochondria, thus an increase in ORP-150 is observed to
compensate and protect the cell from an increase in ROS production.
Actin function is highly regulated by the association of actin binding proteins. Studies
have shown that actin oxidation generally inhibits the association of actin binding
proteins with actin (Farah et. al., 2011). As COTL1 is an actin binding partner its
upregulation could be due to the increased oxidative stress upon the cellular
cytoskeletal system. Oxidative stress can cause actin remodelling and potential axonal
retraction in the neuron (Hallengren et. al., 2013). Under the conditions of stress the
UPR is activated to ensure misfolded proteins are targets for destruction (Hallengren
et. al., 2013). RPS27a has a major role in targeting cellular proteins for destruction as

139 | P a g e
such its apparent increase in the V144D mutant demonstrates that there is a possible
increase in misfolded proteins either directly due to ER stress, oxidative stress or by
another mechanism that affects protein conformation. The findings presented in this
study suggest that there is a strong underlying mechanism of oxidative damage
occurring in the mitochondria of the V144D lymphoblasts.
Given the evidence that altered protein species were identified within the total
mitochondrial and ER proteomes it was clear that greater, more in-depth analyses of
these two organelles were required to examine any further alterations. We then carried
out detailed, high resolution top-down proteomic analyses, with mitochondria and ER
initially isolated from control and V144D lymphoblasts that were subjected to further
separations into total membrane and total soluble protein fractions prior to analyses
by 2DE.
These analyses revealed a further 36 protein changes in both the membrane and
soluble protein fractions from the control and V144D lymphoblasts. Mitochondrial
protein species that changed in abundance were identified as being involved in
catalytic activity, cytoskeleton, protein transport, oxidative stress, calcium binding and
energy metabolism. The changed expression of the proteins identified within the ER
fractions were involved in catalytic activity, cytoskeleton and lipid binding.
While there were a number of nonrelated protein species that were found to be altered
in the mitochondria compared to the ER, there were a number of similarities in
biological processes, most notably catalytic activity, cytoskeleton, protein transport
and oxidative stress.

140 | P a g e
In the previous analyses on total mitochondrial proteomes we showed that ubiquinol
cytochrome c subunit 1 increased in abundance in V144D lymphoblasts. We again
determined it to be increased in abundance in the ER fraction and this was
accompanied by an increase in the abundance of Peroxiredoxin-4, a protein with
antioxidant functions that reduces the build-up of hydrogen peroxide via a thiol-
dependent cycle (Travender and Bullied, 2010). These findings again strengthen the
correlation of potential increase in ROS within the V144D lymphoblasts that could lead
to further disruption of mitochondrial homeostasis.
Ca2+ is also required for energy production within mitochondria, but increased Ca2+
levels can lead to free radical generation (Feissner et. al., 2009). The in-depth analysis
identified a decrease in the Ca2+ binding mitochondrial carrier protein (ScaMc-1). This
decrease might be a protective mechanism due to the already (potentially) high levels
of ROS but will also cause a decrease in ATP production within the mitochondria.
Voltage dependent anion selective channel protein 1 (VDAC) allows mitochondrial
influx/efflux of metabolites such as ATP, and may also have a role in regulating Ca2+
in mitochondria (Brooks et. al., 2004). A decrease in VDAC in the V144D mutant, in
conjunction with the reduction of SCaMc-1 could result in an overall decrease in
intracellular Ca2+ levels in mitochondria and thus decreased ATP production, again
strengthening the possibility for perturbation of their mitochondrial homeostasis.
Dipeptidyl peptidase 1, also known as Cathepsin C and Pro-cathepsin H has been
shown to be proapoptotic by cleaving Bid and Blc-2 family proteins released by
mitochondria; greatly increasing the cascade of caspase apoptotic factors released
(Droga-Mazovec et. al., 2008). The abundance of these proteins is increased in the
mutant cells indicating a link to increased occurrence of mitochondrial apoptotic

141 | P a g e
processes (Turk et. al., 2012). Eukaryotic translation initiation factor 5A-1 (eIF5A) has
been shown to regulate the Bcl-2 binding protein P53 and the P53 apoptosis pathway
(Huang et. al., 2007). In addition, eIF5A has a regulatory function in protein synthesis.
The increase in abundance of elF5A in the V144D mutant may possibly be the result
of the cell’s response to stabilise uncontrolled protein misfolding due to ER stress
(Ogasawara et. al., 2012). Interestingly, CLIC1 was also identified with an increased
abundance in the V144D lymphoblasts. We previously identified this change in the
isolated total ER proteome. Cell oxidation appears to be the important stimuli
determining the transition of CLIC1 between soluble and membrane bound forms
(Averaimo et. al., 2010). Four other proteins were identified with a marked absence or
presence in all mitochondrial and ER fractions.
These analyses show a correlation between previous investigations on the total
proteome revealing an increase in proteins induced by oxidative stress and
mitochondrial electron transport chain proteins. We also identified further changes in
calcium channel proteins, cytoskeletal proteins, and energy transport proteins. This
provided more evidence for a link of increased misfolded proteins, oxidative stress,
and cytoskeleton remodelling and potential changes in Ca2+ signalling within the
mitochondria.
The investigations so far have revealed many important changes that are occurring in
the V144D mutation in the patient derived lymphoblasts. While these alterations are
significant this model has its limitations. As HSN-I is a neurodegenerative disease, we
wanted to extend these investigations and examine a neuronal cell model. To carry
out these analyses we utilised a ND15 cell line (hybrid of rat dorsal root ganglion
neurone and a mouse neuroblastoma) which was transiently transfected (TT) to

142 | P a g e
overexpress not only the V144D mutation but two other known missense mutations
that cause HSN-I; C133W and C133Y. The data obtained from this neuronal cell model
confirmed our previous results identified from the lymphoblast model, while notably
identifying changes exhibited in the C133W and C133Y mutations. We also identified
an additional 14 proteins that were altered in abundance within the transfected ND15
cells
Quantitative protein expression and FACS analyses enabled identification of
alterations in ubiquinol cytochrome c abundance. The ubiquinol cytochrome c levels
were increased significantly in the TT ND15 cells containing the individual mutations.
These findings further strengthened the potential link between oxidative
phosphorylation, via ubiquinol cytochrome c, and altered energy production ultimately
leading to axonal degeneration. Further to this finding, quantitative analyses confirmed
that the protein expressions of RPS27a, COTL1, and ORP-150 were significantly
increased in the V144D TT ND15 cells compared to the levels measured from non-
transfected ND15 cell controls.
Interestingly, Stress-70 mitochondrial protein levels were identified in the C133Y
mutant as being increased relative to the wild type. When mitochondria are under
stress, Stress-70 protein levels increase compensating for increased oxidative
damage and maintain normal protein import and synthesis. Thus, if mitochondrial
oxidative stress is increased (potentially via ROS production), further cellular damage
would occur, ultimately leading to a demise in ER efficiency eventually resulting in ER
stress.
Alterations in expression of these proteins strongly indicated that oxidative stress
could be linked to increases in ubiquinol cytochrome c. Thus, an increase in ORP-150

143 | P a g e
could be potentially explained to compensate and protect the cell from an increase in
ROS production, causing a shift in CLIC1 expression and stabilisation of the
cytoskeleton via COTL1. RPS27a is responsible for targeting misfolded proteins for
destruction, with an apparent increase highlighting potential increases in misfolded
proteins and protein aggregation due to oxidative stress (Hallengren et. al., 2013).
Further strengthening the connection between ER stress and HSN-I; peptidyl-prolyl
cis-trans isomerase was found to be increased in abundance in the C133W mutant.
This protein ensures newly synthesised proteins are folded into their correct
conformation. The 26s proteasome is responsible for regulating the proteome through
degradation of ubiquitin-tagged substrates (Shaw, PE 2002). 26s proteasome
regulatory subunit 8 was found to be increased in abundance in the cells containing
the V144D mutation. The increase in abundance of these two proteins coupled with
the increased expression of RPS27a and ORP-150 highlights the possible increased
oxidative stress affecting protein folding conformation.
As described previously, mitochondrial calcium uptake has been shown to lead to free
radical production, with a delicate balance existing between moderate ROS production
to modulate physiological signalling (Glancy and Balaban, 2012). Overproduction of
ROS can ultimately lead to oxidative and ER stress. A decrease in intracellular calcium
is believed to be a cellular response to increased stress, serving as a mechanism to
limit further damage and increase cell survival (Feissner et. al., 2009). As part of this
study we examined the intracellular levels of calcium in wild type, V144D, C133W, and
C133Y ND15 cells. Intracellular calcium was found to be decreased within cells
containing C133W and C133Y mutations, whilst intracellular calcium within the cells
with the V144D mutation was increased.

144 | P a g e
Although this investigation has revealed substantial amounts of information not
previously known about intracellular calcium levels in SPTLC1 mutant cells, the
following questions remain unanswered; is the increased level of calcium due to a
correlation of ER stress and mitochondrial dysfunction occurring, and the result of the
V144D mutation being unable to reduce intracellular calcium to compensate and
protect the cell? Could this difference give insight into how the three mutations differ,
ultimately causing HSN-I? While much remains to be elucidated, the findings
described within this thesis broadens our knowledge of what is occurring as a
consequence of the individual known mutations that cause HSN-I.
Increases in the number of lipid droplets and changes in lipid metabolism have been
identified in a variety of autosomal dominant neurodegenerative diseases, including
Alzheimer’s and Parkinson’s (Cole et. al., 2002; Lane and Farkow, 2005). Considering
the potential accumulation of lipid droplets and alterations of lipid metabolism in HSN-
I, we set out to determine if an accumulation of lipid droplets was associated with HSN-
I.
Confocal micrographs of Nile red-stained lipid droplets identified an increase of these
inclusions within the oleic acid-treated positive controls compared to the healthy
control and patient-derived lymphoblast cells. A significant increase of lipid droplets
was observed within the patient-derived lymphoblasts compared to the healthy control
cells. This increase in lipid droplet abundance in HSN-I patient-derived lymphoblasts
provided a possible connection between increased lipid droplet numbers and the
cellular mechanism of HSN-I.
Immunoblotting revealed that only the oleic acid treated samples showed detectable
ADFP protein from total cell lysates. Analysis of SPTLC1 from oleic acid treated

145 | P a g e
healthy control and patient-derived lymphoblasts revealed no significant difference in
expression. ADFP is thought to be involved in lipid homeostasis and lipolysis by
protecting triacylglycerol, within the lipid droplets, from cytosolic lipases (Ducharme
and Bickel, 2008; Hodges and Wu, 2010). These results suggest that the increase in
lipid droplets within the HSN-I patient lymphoblasts, without a change in ADFP, may
cause abnormalities in lipid metabolism or be indicative of a protein-trafficking defect
that involved elements of the cytoskeleton. The increase in lipid droplets without a
parallel increase in the expression of ADFP in the HSN-I patient-derived lymphoblasts
may well indicate a deregulation of lipid metabolism which may exacerbate this
phenotype.
Normal cellular processes require the proper communication between mitochondria
and the ER, which is facilitated by the mitochondria-associated endoplasmic reticulum
membrane (MAM) (Vance, J 2014). MAM is involved in the inter-organellar transport
of cholesterol, ceramides, ATP, as well as in proteasomal protein degradation and lipid
droplet formation. Recently the importance of inter-organellar communication in the
pathophysiology of neurodegenerative disorders has been identified (Paillusson, et.
al., 2016).
An interesting example of a MAMs resident reactive oxygen species (ROS) generating
protein is the p66Shc protein. Under physiological conditions, this growth factor
adaptor protein is involved in signal transduction via the RAS protein (Wieckowski et.
al., 2009). However, under oxidative stress, p66Shc can participate in the signalling
pathway leading to apoptosis. The p66Shc protein contains an N-terminal proline-rich
collagen homology domain (CH2) containing a serine phosphorylation site (Ser36) that
is important for its ‘proapoptotic’ properties as well as a functional region that is
responsible for its interaction with cytochrome c (Wieckowski et. al., 2009). Studies

146 | P a g e
have revealed that mitochondrial p66Shc is present in the IMS where it interacts with
cytochrome c and, as a redox enzyme producing hydrogen peroxide (Wieckowski et.
al., 2009).
As we have identified numerous proteins that are upregulated during times of oxidative
stress it would be interesting to know if the p66Shc protein was also altered in HSN-I.
If this protein is indeed altered it would further strengthen the correlation between ROS
production occurring from the cytochrome c subunit of the mitochondria and
subsequent apoptotic events.
Calcium exchange via ER–mitochondrial contacts is facilitated following its release
from ER stores via inositol 1, 4, 5- trisphosphate (IP3) receptors (Paillusson, et. al.,
2016). Ca2+ is required by mitochondria to generate ATP via the TCA. Some of the
mitochondrial enzymes involved in ATP synthesis, such as some dehydrogenases,
are regulated by Ca2+ (Vance, J 2014). Excessive uptake of Ca2+ by mitochondria can
lead to opening of the mitochondrial permeability transition pore and apoptotic
signalling occurs. During hypoxic conditions, the ER and MAM resident proteins Ero1-
La and ERp44 interact and modulate ER-mitochondria Ca2+ levels. In addition,
elevated levels of Ero1-La may promote activation of the UPR system with a
consequent increase in ROS production (Gilady et. al., 2010).
Disorganisation of the ER mitochondrial interface is relevant to the progression of
Alzheimer's Disease (AD) (Marchi et. al., 2014). Up-regulation of MAM associated
proteins were observed in sections of brains from human AD patients, and both
amyloid β-peptide and the over-expression of presenilin 2, which is mutated in some
cases of familial AD, elevates the number of ER mitochondria contact sites, favouring

147 | P a g e
Ca2+ transfer between the two organelles (Marchi et. al., 2014). A link between Ca2+
signalling and ER mitochondria connections also appears in the context of ER stress-
mediated apoptosis. The RNA dependent protein kinase (PKR)-like ER kinase
(PERK), a key ER stress sensor in the unfolded protein response, is enriched at MAMs
and is important for maintaining ER mitochondrial contact sites (Marchi et. al., 2014).
Our in-depth analyses identified decreases in ScaMc-1 and VDAC, both of which are
involved in regulating Ca2+. These decreases might be a protective mechanism due to
the already high levels of ROS but will also cause a decrease in ATP production within
the mitochondria. However this decrease may also be part of ER-stress-mediated
processes that could be occurring during the cross talk between the ER and
mitochondria. This is strengthened by the identification of UPR proteins that are
altered in the disease state.
A clear picture can be seen of direct communication via MAM with ER and
mitochondria altering Ca2+ regulation, generating ROS and upregulating the UPR
pathway. It is also clear that both of these organelles are altered in their morphology
and function in all mutant forms of HSN-I. However while we know that mutant SPTLC1
protein is causing this change the exact mechanism of action is still unclear. It may be
that the ER-bound SPT complex is directly connecting the two organelles via MAMs.
Sterols and sphingolipids are minor constituents of mitochondrial membranes. The
processes by which these lipids are transported to, and imported into, mitochondria
may occur via MAMs (Krols et.al. 2016). Ceramide can induce mitochondria mediated
apoptosis by permeabilising the OMM, with a large amount of cellular ceramide being
synthesised in the ER, MAM and mitochondria (Krols et.al. 2016, Vance, J 2014).
Given this knowledge it is possible that the mutant SPTLC1, being involved in the

148 | P a g e
sphingolipids synthesis pathway, is altering the homeostasis within the MAM that is
then causing a chain of events that results in changes in Ca2+ signalling and generation
of ROS. Another possibility is the toxic deoxy-sphingolipids that are known to be
produced as a result of the mutation may also be impairing the communication
between the ER and mitochondria as seen in other neurodegenerations where protein
aggregations cause a shift in the cellular homeostasis.
Overall, a detailed picture is emerging that MAMs have a large impact on how
organelles communicate and maintain their functions and may help to bridge the
connection of how SPTLC1 mutation cause axonal degeneration. Alteration in one or
more of the organelles causes downstream perturbations that ultimately lead to
impaired signalling, toxic product formation, unregulated protein synthesis and
disruptions to energy production that terminate in the loss of neuronal function
eventually causing axonal degeneration and cell death.
In conclusion, the results presented within this study provide novel and important
information, insights and directionality to ultimately uncover the mechanism(s) within
peripheral neuropathies. These findings suggest that there may be underlying
molecular alterations broadly common to neurodegenerative diseases as a whole,
linked to mitochondria, ER and lipids, that maybe facilitated by MAMs.
With mounting evidence into the nature of the protein alterations in HSN-I we have
provided new and novel in-sights into the molecular and cellular mechanisms that are
occurring within these diseased cells which could ultimately aid in identification of
disease-specific bio-markers and facilitate future drug development to assist patient
treatment, care and a cure.

149 | P a g e
Future Directions
The research presented in this thesis has identified significant alterations within the
protein profiles of purified mitochondria and ER fractions isolated from HSN-I cells.
Uncovering the essential and critical roles of both mitochondria and the ER in
neurodegenerative disease processes is undoubtedly of the utmost importance. We
have also highlighted the potential communication network via MAMs between
mitochondria and ER and how mutant SPTLC1 may cause mitochondrial disturbance.
Many cellular events such as intracellular signalling of important pathways, including
the synthesis of cholesterol and phospholipids, calcium homeostasis, ROS generation
and activity occur within MAMs (Giorgi et. al., 2009). There are techniques available
to isolate mitochondria, as we have determined in this investigation, however, only few
specifically isolate MAMs. The isolation of MAMs containing the unique regions of ER
membranes attached to the outer mitochondrial membrane requires it to be without
contamination from other organelles such as pure mitochondria.
For Example, Wieckowski et. al., 2009 used a procedure to study the proposed role of
MAMs in cellular responses to oxidative stress connected with the phosphorylation of
p66Shc protein. They indicated that a significant portion of p66Shc is present in the
MAM fraction. Briefly, Wieckowski et. al., 2009 designed the procedure for MAM
isolation by first separating the cellular contaminants from a crude mitochondrial
fraction and then further purifying this via differential centrifugation resulting in isolation
of a highly purified MAM fraction.
Once the purified MAM fraction has been obtained a detailed proteomic analysis can
then be carried out. The methods employed in this thesis have yielded high quality, in

150 | P a g e
depth analyses and these protocols should be utilised to assess total proteomes to
determine the spectrum of proteins altered with the MAM. It would be of great interest
to further examine other proteins upregulated during times of stress, in particular
proteins affected by ROS activation. In addition to proteins that might be upregulated,
the possibility exists that SPT may be present. Considering SPT is housed within the
ER and that the ER and mitochondria communicate via the MAM this would be a highly
unique finding and would give much greater insight into how an ER-bound protein
facilitates such alterations.
Calcium homeostasis occurs through ‘cross-talk’ between the ER and mitochondria at
MAMs (Giorgi et al., 2015; Marchi et. al., 2014). The Ca2+ signalling apparatus consists
of the Sarcoplasmic Reticulum (SR) Ca2+ release channel, the Troponin protein
complex that mediates the Ca2+ effect to the myofibrillar structures leading to
contraction, the Ca2+ pump responsible for its reuptake into the SR, and Calsequestrin,
the Ca2+ storage protein in the SR (Hofer, AM 2005). When calcium signalling is
stimulated in a cell, Ca2+ enters the cytoplasm from one of two general sources: it is
released from intracellular stores, or it enters the cell across the plasma membrane
(Putney et. al., 2001). We have shown that intracellular calcium levels are increased
in cells carrying the V144D mutation and decreased in cells carrying the C133W and
C133Y mutations. Thus isolating MAMs from cells carrying the different mutations and
investigating the calcium “cross-talking” between the ER and mitochondria due to
SPTLC1 mutations should be explored.
We have tested the intracellular calcium levels of the TT ND15 neuronal cell models
using the technique of fluorescent-based imaging. However future investigations could
incorporate other sensitive assays, such as ratiometric analysis of intracellular calcium

151 | P a g e
levels using Fura-2 AM (Thermofisher). Ratiometric methods are based on the use of
a ratio between two fluorescence intensities allowing for correction of artefacts due to
bleaching, changes in focus, and variations in laser intensity. This would ensure a
more accurate assessment of calcium levels within TT ND15 cells as well as
measuring the levels within the patient-derived lymphoblasts.
In addition to intracellular calcium levels, the expression of SR protein should be
assessed to see if there are any changes within the calcium storage and channel
proteins that are potentially causing mitochondrial alterations in the SPTLC1 mutant
cells.
While an assessment of calcium signalling is a good assessment of mitochondrial
function, it can also be measured in four additional ways: basal respiration rate, ATP-
linked respiration, proton leak, and reserve capacity (Choi et. al., 2009). Bioenergetic
capacity can be used to determine whether a response to stress is dysregulated,
therefore testing the capacity of mitochondria in cells carrying the SPTLC1 mutation
to respond to increasing energy demand under basal conditions as measured by
oxygen consumption rate (OCR). If the energetic reserves are depleted under normal
conditions due to the SPTLC1 mutation(s), respiration will fail and cell death occurs
(Choi et. al., 2009). Thus, measuring the bioenergetic reserve capacity may be an
effective way to assess or predict the ability of cells to manage and overcome stress,
such as that encountered during acute oxidative insults. This can be done by exposing
the cells to oligomycin (complex V inhibitor), FCCP (carbonyl cyanide p-
trifluoromethoxyphenylhydrazone) and antimycin A sequentially. The OCR will
increase in a concentration-dependent manner in an effort to counteract the increased
energy demand caused by the oxidative damage (Choi et. al., 2009). The OCR would

152 | P a g e
reveal if there is increase in the leak of protons (thereby decreasing mitochondrial
efficiency) and therefore increasing energy demand (Choi et. al., 2009).
Also of interest would be to assess the expression of the individual mitochondrial
complexes (I-V). While we have determined that a subunit of the complex III is altered,
determining if the expression of other complexes are altered would be the obvious
next stage for investigation. Using the antibody preparation called OXPHOS (a cocktail
containing 5 antibodies (Abcam, USA) would allow a comparison of the level of
expression of the mitochondrial complexes (I-V) in normal cells and in cells carrying
SPTLC1 mutation.
We have also shown that there is increased oxidative stress occurring within cells
expressing the SPTLC1 mutations, possibly via ROS production. An assay that can
be carried out to assess if/what species are being produced is called MitoSox Red
(Thermofisher Scientific). MitoSox Red is a fluorescent dye that becomes highly
oxidised when it comes into contact with superoxide, resulting in an increase in
fluorescence. Additionally, Amplex Red (Thermofisher Scientific) is another dye that
when reduced by hydrogen peroxide molecules produce a red fluorescent product. If
these assays reveal ROS is produced, then further assessment would need to be
undertaken to determine the source of the ROS production and to ensure that any
ROS generated is not part of normal cellular functions. While the determination of a
ROS species being present is informative, this would need to be further investigated
as to how this may alter proteins within the mitochondria. Additional assessment could
use a ROS-scavenging molecule to see if the reduction in ROS changes protein
expression, especially with regard to the proteins identified in this investigation.

153 | P a g e
Although the methodology for quantifying protein abundance has been in use for many
years, methods for assessing and analysing mRNA expression are becoming
essential (Greenbaum et. al., 2003). The quantification of both mRNA and protein is
not an exercise in redundancy; measurements taken from mRNA and protein levels
are complementary and both provide a more complete understanding of how the cell
works. Also, as mRNA is translated into protein, it can be assumed that there should
be a correlation between the level of mRNA and that of protein (Greenbaum et. al.,
2003). mRNA profiles should be analysed from patient-derived lymphoblasts cells and
would add to the assessment of the expression of the major proteins described in this
study. Of particular interest would be the analysis of the mRNA expression for the
proteins ubiquinol cytochrome c, ORP-150, RPS27a, Coactosin and CLIC1, to assess
whether there is evidence for any changes in mRNA expression due to the mutant
SPTLC1 protein. A technique that could be used to assess mRNA levels and profiles
with high throughput is RT-PCR. The information obtained from such future
investigations would strengthen and build upon the numerous protein expression
profiles occurring in HSN-I detailed within this thesis.
The proper functioning of quality control systems like autophagy is essential to
maintain cellular homeostasis. Autophagy dysfunction has been implicated in many
neurodegenerative disorders (Martinez-Vincente, M. 2015; Menzies et. al., 2015).
Dysfunction can occur at several steps of the autophagy machinery and can contribute
to the formation of intracellular aggregates and ultimately to neuronal death (Menzies
et. al., 2015). While studies have shown that protein aggregation does not occur in
HSN-I, two atypical neurotoxic deoxy-sphingoid bases (DSBs) are produced in HSN-I
(Penno et. al., 2010; Ernst et. al., 2015). Dysfunctional mitochondria release pro-death
proteins resulting in activation of cell death pathways. Aberrant mitochondria must be

154 | P a g e
removed to limit damage to the cell. It would be of interest to determine if normal
autophagy processes are occurring in SPTLC1 mutant cells and whether the deoxy-
sphingoid bases potentially interact or impair the autophagosome/mitophagy
formation and thus removal of dysfunctional mitochondria.
In addition to potentially altering autophagy processes, the neurotoxic DSBs have
been shown to interact and affect the cytoskeleton of neurites (Penno et. al., 2010).
We have shown that Coactosin is upregulated within the SPTLC1 mutant cells. As
such, examination of whether this change is caused by the increased formation of
DSBs or a combination of increased oxidative stress, neurotoxic DSBs accumulation
and ER stress should be explored.
Production of induced pluripotent stem (iPS) cells by cellular reprogramming somatic
cells can be achieved by ectopic expression of specific transcription factors (Singh et.
al., 2009). The reprogramming factors are introduced into cultured somatic cells and
the cells are then grown under embryonic stem cell conditions. After approximately 3
weeks, iPS cells emerge. These induced pluripotent stem cells may be differentiated
into various cell types (Singh et. al., 2009).
In attempting to answer the question of ‘why mutation of the SPTLC1 protein subunit
causes HSN-I’, the next step could be to generate human DRGs. Somatic cells can
be harvested from HSN-I patients harbouring SPTLC1 mutations and be differentiated
into DRGs. Such patient-derived iPS cells would provide greater insight into the
molecular and cellular mechanisms of the neurodegenerative disease process,
enabling the study of patient-specific SPTLC1 mutations and testing of candidate
drugs. This would be essential in understanding the cellular mechanisms of how HSN-
I affects human DRGs (Singh et. al., 2009).

155 | P a g e
Ultimately, any future investigations into the molecular and cellular mechanisms of the
neurodegenerative disease processes of HSN-I must expand on the findings
presented within this thesis and include investigations into the pathogenic
mechanisms of SPTLC1 mutations and the links to mitochondrial and ER localisation,
MAMs and the subsequent disturbances and axonal degeneration that is a defining
characteristic of HSN-I.

156 | P a g e
REFERENCES
AUER-GRUMBACH, M. 2008. Hereditary sensory neuropathy type I. Orphanet J Rare Dis, 3, 7.
AUER-GRUMBACH, M., DE JONGHE, P., VERHOEVEN, K., TIMMERMAN, V., WAGNER, K., HARTUNG, H. P. & NICHOLSON, G. A. 2003. Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: a review. Arch Neurol, 60, 329-34.
AVERAIMO, S., MILTON, R. H., DUCHEN, M. R. & MAZZANTI, M. 2010. Chloride intracellular channel 1 (CLIC1): Sensor and effector during oxidative stress. FEBS Lett, 584, 2076-84
BALOH, R. H., SCHMIDT, R. E., PESTRONK, A. & MILBRANDT, J. 2007. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci, 27, 422-30.
BEHNKE, J. & HENDERSHOT, L. M. 2014. The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem, 289, 2899-907.
BEJAOUI, K., UCHIDA, Y., YASUDA, S., HO, M., NISHIJIMA, M., BROWN, R. H., JR., HOLLERAN, W. M. & HANADA, K. 2002. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest, 110, 1301-8.
BEJAOUI, K., WU, C., SCHEFFLER, M. D., HAAN, G., ASHBY, P., WU, L., DE JONG, P. & BROWN, R. H., JR. 2001. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet, 27, 261-2.
BRESLOW, D. K. 2013. Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb Perspect Biol, 5, a013326.
BUCCOLIERO, R. & FUTERMAN, A. H. 2003. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol Res, 47, 409-19.
CAVANAGH, N. P., EAMES, R. A., GALVIN, R. J., BRETT, E. M. & KELLY, R. E. 1979. Hereditary sensory neuropathy with spastic paraplegia. Brain, 102, 79-94.
CHOI, SW, GERENCSER, AA & NICHOLLS, DG 2009, 'Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure', J Neurochem, vol. 109, no. 4, pp. 1179-91.
COLE N, MURPHY D, GRIDER T, RUETER S, BRASAEMLE D, NUSSBAUM R (2002) Lipid droplet binding and oligomerization properties of the Parkinsons disease protein alpha-synuclein. J Biol Chem 277(8): 6344–6352.

157 | P a g e
COLOMBAIONI, L. & GARCIA-GIL, M. 2004. Sphingolipid metabolites in neural signalling and function. Brain Res Rev, 46, 328-55.
COOPER, G. M. & HAUSMAN, R. E. 2007. The cell : a molecular approach, Washington, D.C.Sunderland, Mass., ASM Press ; Sinauer Associates.
CROFTS AR (2004) The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol 66:689–733
CULLIS, P .R., HOPE, M .J.,1991. Physical properties and functional roles of lipids in membranes. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier Science Publishing Co. Inc
DAWKINS, J. L., BRAHMBHATT, S., AUER-GRUMBACH, M., WAGNER, K., HARTUNG, H. P., VERHOEVEN, K., TIMMERMAN, V., DE JONGHE, P., KENNERSON, M., LEGUERN, E. & NICHOLSON, G. A. 2002. Exclusion of serine palmitoyltransferase long chain base subunit 2 (SPTLC2) as a common cause for hereditary sensory neuropathy. Neuromuscul Disord, 12, 656-8.
DAWKINS, J. L., HULME, D. J., BRAHMBHATT, S. B., AUER-GRUMBACH, M. & NICHOLSON, G. A. 2001. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet, 27, 309-12.
DEDOV, V. N., DEDOVA, I. V., MERRILL, A. H., JR. & NICHOLSON, G. A. 2004. Activity of partially inhibited serine palmitoyltransferase is sufficient for normal sphingolipid metabolism and viability of HSN1 patient cells. Biochim Biophys Acta, 1688, 168-75.
DENNY-BROWN, D. 1951. Hereditary sensory radicular neuropathy. J Neurol Neurosurg Psychiatry, 14, 237-52.
DIKOV, D. & REICHERT, A. S. 2011. How to split up: lessons from mitochondria. EMBO J, 30, 2751-3.
DONAGHY, M., HAKIN, R. N., BAMFORD, J. M., GARNER, A., KIRKBY, G. R., NOBLE, B. A., TAZIR-MELBOUCY, M., KING, R. H. & THOMAS, P. K. 1987. Hereditary sensory neuropathy with neurotrophic keratitis. Description of an autosomal recessive disorder with a selective reduction of small myelinated nerve fibres and a discussion of the classification of the hereditary sensory neuropathies. Brain, 110 ( Pt 3), 563-83.
DOWHAN, W. 1997. The role of phospholipids in cell function. Advances in Lipobiology, 2, 79-107.
DROGA-MAZOVEC, G., BOJIC, L., PETELIN, A., IVANOVA, S., ROMIH, R., REPNIK, U., SALVESEN, G. S., STOKA, V., TURK, V. & TURK, B. 2008. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 283

158 | P a g e
DROSE S, BRANDT U, WITTIGI .2014. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim Biophys Acta 1844(8):1344–1354. doi:10.1016/j.bbapap. 2014.02.006
DUCHARME N, BICKEL P (2008) Minireview: lipid droplets in lipogenesis and lipolysis. Endocrinology 149(3):942–949.
DUFFY, L. M., CHAPMAN, A. L., SHAW, P. J. & GRIERSON, A. J. 2011. Review: The role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol, 37, 336-52.
DYCK, P .J. & THOMAS, P .K. 2005. Dyck: Peripheral Neuropathy, 4th Edition. Philadelphia, Mosby Elsevier.
EINARSDOTTIR, E., CARLSSON, A., MINDE, J., TOOLANEN, G., SVENSSON, O., SOLDERS, G., HOLMGREN, G., HOLMBERG, D. & HOLMBERG, M. 2004. A mutation in the nerve growth factor beta gene (NGFß) causes loss of pain perception. Hum Mol Genet, 13, 799-805.
ERNST, D., MURPHY, S. M., SATHIYANADAN, K., WEI, Y., OTHMAN, A., LAURA, M., LIU, Y. T., PENNO, A., BLAKE, J., DONAGHY, M., HOULDEN, H., REILLY, M. M. & HORNEMANN, T. 2015. Novel HSAN1 mutation in serine palmitoyltransferase resides at a putative phosphorylation site that is involved in regulating substrate specificity. Neuromolecular Med, 17, 47-57.
FARAH, M. E., SIROTKIN, V., HAARER, B., KAKHNIASHVILI, D. & AMBERG, D. C. 2011. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken), 68, 340-54
FENSKE, D. B., MYRNA A. M., HOPE, M. J., & CULLIS, P. R. 1995. The functional roles of lipids in biological membranes. Biomembranes, 1, 1-28.
FEISSNER, R. F., SKALSKA, J., GAUM, W. E. & SHEU, S. S. 2009. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci (Landmark Ed), 14, 1197-218
FULDA, S., GORMAN, A. M., HORI, O. & SAMALI, A. 2010. Cellular stress responses: cell survival and cell death. Int J Cell Biol, 2010, 214074.
GLANCY, B. & BALABAN, R. S. 2012. Role of Mitochondrial Ca2+ In the Regulation of Cellular Energetics. Biochemistry, 51, 2959-73.
GILADY, SY, BUI, M, LYNES, EM, BENSON, MD, WATTS, R, VANCE, JE & SIMMEN, T 2010, 'Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM)', Cell Stress Chaperones, vol. 15, no. 5, pp. 619-29.
GIORGI, C., DE STEFANI, D., BONONI, A., RIZZUTO, R. & PINTON, P. 2009. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol, 41, 1817-27.

159 | P a g e
GIORGI, C., MISSIROLI, S., PATERGNANI, S., DUSZYNSKI, J., WIECKOWSKI, M. R. & PINTON, P. 2015. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal, 22, 995-1019.
GOLDMAN, L., CECIL, R. L. & AUSIELLO, D. A. 2008. Cecil medicine, Philadelphia, Saunders Elsevier.
GREENBAUM, D, COLANGELO, C, WILLIAMS, K & GERSTEIN, M 2003, 'Comparing protein abundance and mRNA expression levels on a genomic scale', Genome Biol, vol. 4, no. 9, p. 117.
HALLENGREN, J., CHEN, P. C. & WILSON, S. M. 2013. Neuronal ubiquitin homeostasis. Cell Biochem Biophys, 67, 67-73.
HANADA, K. 2003. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta, 1632, 16-30.
HERRMANN, J. M., STUART, R. A., CRAIG, E. A. & NEUPERT, W. 1994. Mitochondrial Heat Shock Protein 70, a Molecular Chaperone for Proteins Encoded By Mitochondrial DNA. J Cell Biol, 127, 893-902.
HISAHARA, S. & SHIMOHAMA, S. 2010. Toxin-induced and genetic animal models of Parkinson's disease. Parkinsons Dis, 2011, 95, 1709.
HODGES B, WU C (2010) Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. J Lipid Res 51(2): 262–273.
HOFER, AM 2005, 'another dimension to calcium signaling: a look at extracellular calcium', J Cell Sci, vol. 118, no. 5, pp. 855-62
HOLLENBECK, P. J. & SAXTON, W. M. 2005. The axonal transport of mitochondria. J Cell Sci, 118, 5411-9.
HOLLENBECK, P. J. 1996. The pattern and mechanism of mitochondrial transport in axons. Front Biosci, 1, d91-102.
HORNEMANN, T., PENNO, A., RUTTI, M. F., ERNST, D., KIVRAK-PFIFFNER, F., ROHRER, L. & VON ECKARDSTEIN, A. 2009. The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. J Biol Chem, 284, 26322-30.
HORNEMANN, T., WEI, Y. & VON ECKARDSTEIN, A. 2007. Is the mammalian serine palmitoyltransferase a high-molecular-mass complex? Biochem J, 405, 157-64.
HORNEMANN, T., RICHARD, S., RUTTI, M. F., WEI, Y. & VON ECKARDSTEIN, A. 2006. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem, 281, 37275-81.

160 | P a g e
HOULDEN, H., KING, R., BLAKE, J., GROVES, M., LOVE, S., WOODWARD, C., HAMMANS, S., NICOLL, J., LENNOX, G., O'DONOVAN, D. G., GABRIEL, C., THOMAS, P. K. & REILLY, M. M. 2006. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain, 129, 411-25.
HOULDEN, H., BLAKE, J. & REILLY, M. M. 2004. Hereditary sensory neuropathies. Curr Opin Neurol, 17, 569-77.
HUANG, Y., HIGGINSON, D. S., HESTER, L., PARK, M. H. & SNYDER, S. H. 2007. Neuronal growth and survival mediated by eIF5A, a polyamine-modified translation initiation factor. Proc Natl Acad Sci U S A, 104, 4194-9.
HUTCHINSON AT, RAMSLAND PA, JONES DR, AGOSTINO M, LUND ME, JENNINGS CV, BOCKHORNI V, YURIEV E, EDMUNDSON AB, RAISON RL (2010) Free Ig light chains interact with sphingomyelin and are found on the surface of myeloma plasma cells in an aggregated form. J Immunol 185:4179–4188
ISHIHARA, N., FUJITA, Y., OKA, T. & MIHARA, K. 2006. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J, 25, 2966-77.
JANZER, R. C., SPYCHER, M. A., BOLTSHAUSER, E. & CSERHATI, M. 1986. A case of congenital non-progressive sensory neuropathy with tonic pupils. Clin Neuropathol, 5, 209-16.
KASHATUS, D. F., LIM, K. H., BRADY, D. C., PERSHING, N. L., COX, A. D. & COUNTER, C. M. 2011. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol, 13, 1108-15.
KHERBAOUI-REDOUANI, L., PLOTON, D., ABELY, M., BEDNAREK, N., STOURBE, A., SABOURAUD, P. & MOTTE, J. 2004. Hereditary sensory neuropathy with spastic paraplegia. Eur J Paediatr Neurol, 8, 95-9.
KLEIN, C. J., BOTUYAN, M. V., WU, Y., WARD, C. J., NICHOLSON, G. A., HAMMANS, S., HOJO, K., YAMANISHI, H., KARPF, A. R., WALLACE, D. C., SIMON, M., LANDER, C., BOARDMAN, L. A., CUNNINGHAM, J. M., SMITH, G. E., LITCHY, W. J., BOES, B., ATKINSON, E. J., MIDDHA, S., PJ, B. D., PARISI, J. E., MER, G., SMITH, D. I. & DYCK, P. J. 2011. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet, 43, 595-600.
KORNAK, U., MADEMAN, I., SCHINKE, M., VOIGT, M., KRAWITZ, P., HECHT, J., BARVENCIK, F., SCHINKE, T., GIESSELMANN, S., BEIL, F. T., POU-SERRADELL, A., VILCHEZ, J. J., BEETZ, C., DECONINCK, T., TIMMERMAN, V., KAETHER, C., DE JONGHE, P., HUBNER, C. A., GAL, A., AMLING, M., MUNDLOS, S., BAETS, J. & KURTH, I. 2014. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain, 137, 683-92.

161 | P a g e
KNOTT, A. B., PERKINS, G., SCHWARZENBACHER, R. & BOSSY-WETZEL, E. 2008. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci, 9, 505-18.
KROLS, M, VAN ISTERDAEL, G, ASSELBERGH, B, KREMER, A, LIPPENS, S, TIMMERMAN, V & JANSSENS, S 2016, 'Mitochondria-associated membranes as hubs for neurodegeneration', Acta Neuropathol, vol. 131, no. 4, pp. 505-23.
KWONG, J. Q., BEAL, M. F. & MANFREDI, G. 2006. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem, 97, 1659-75.
LANE R, FARLOW M (2005) Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimers disease. J Lipid Res 46(5):949–968.
LEYNE, M., MULL, J., GILL, S. P., CUAJUNGCO, M. P., ODDOUX, C., BLUMENFELD, A., MAAYAN, C., GUSELLA, J. F., AXELROD, F. B. & SLAUGENHAUPT, S. A. 2003. Identification of the first non-Jewish mutation in familial Dysautonomia. Am J Med Genet A, 118A, 305-8?
LI, X., ZHANG, K. & LI, Z. 2011. Unfolded protein response in cancer: the physician's perspective. J Hematol Oncol, 4, 8.
LINDHOLM, D., WOOTZ, H. & KORHONEN, L. 2006. ER stress and neurodegenerative diseases. Cell Death Differ, 13, 385-92.
LIU, Y. C., ZHAI, X. Y., OHSATO, K., FUTAMATA, H., SHIMADA, O. & ATSUMI, S. 2004. Mitochondrial accumulation in the distal part of the initial segment of chicken spinal motoneurons. Brain Res, 1026, 235-43.
LODISH, H. F. 2008. Molecular cell biology, New York, W.H. Freeman.
LOWTHER, J., NAISMITH, J. H., DUNN, T. M. & CAMPOPIANO, D. J. 2012. Structural, mechanistic and regulatory studies of serine palmitoyltransferase. Biochem Soc Trans, 40, 547-54.
MALHOTRA, J. D. & KAUFMAN, R. J. 2007. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol, 18, 716-31.
MANFREDI, G. & BEAL, M. F. 2000. The role of mitochondria in the pathogenesis of neurodegenerative diseases. Brain Pathol, 10, 462-72.
MARCHI, S., PATERGNANI, S. & PINTON, P. 2014. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta, 1837, 461-9.
MARTINEZ-VICENTE, M. 2015. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin Cell Dev Biol, 40, 115-26.

162 | P a g e
MATSUOKA, K., ORCI, L., AMHERDT, M., BEDNAREK, S. Y., HAMAMOTO, S., SCHEKMAN, R. & YEUNG, T. 1998. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell, 93, 263-75.
MENZIES, F. M., FLEMING, A. & RUBINSZTEIN, D. C. 2015. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci, 16, 345-57.
MIHARA, K. 2000. Targeting and insertion of nuclear-encoded preproteins into the mitochondrial outer membrane. Bioessays, 22, 364-71.
MILLER, K. E. & SHEETZ, M. P. 2004. Axonal mitochondrial transport and potential are correlated. J Cell Sci, 117, 2791-804.
MYERS S, MALLADI C, HYLAND R, BAUTISTA T, BOADLE R, ROBINSON P, & NICHOLSON G (2014) Mutations in the SPTLC1 protein cause mitochondrial structural abnormalities and endoplasmic reticulum stress in lymphoblasts. DNA Cell Biol 33(7):399–407
MYERS, S. J. & STANLEY, K. K. 1999. Src family kinase activation in glycosphingolipid-rich membrane domains of endothelial cells treated with oxidised low density lipoprotein. Atherosclerosis, 143, 389-97.
NICHOLSON, G. A., DAWKINS, J. L., BLAIR, I. P., KENNERSON, M. L., GORDON, M. J., CHERRYSON, A. K., NASH, J. & BANANIS, T. 1996. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet, 13, 101-4.
OGASAWARA, Y., OHMINATO, T., NAKAMURA, Y. & ISHII, K. 2012. Structural and functional analysis of native peroxiredoxin 2 in human red blood cells. Int J Biochem Cell Biol, 44, 1072-7
OGRETMEN, B. & HANNUN, Y. A. 2004. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer, 4, 604-16.
OTERA, H. & MIHARA, K. 2011. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem, 149, 241-51.
OVALLE, W. K., NAHIRNEY, P. C. & NETTER, F. H. 2008. Netter's essential histology, Philadelphia, Saunders/Elsevier.
PALMER, C. S., OSELLAME, L. D., LAINE, D., KOUTSOPOULOS, O. S., FRAZIER, A. E. & RYAN, M. T. 2011. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep, 12, 565-73.
PAILLUSSON, S, STOICA, R, GOMEZ-SUAGA, P, LAU, DH, MUELLER, S, MILLER, T & MILLER, CC 2016, 'There's Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases', Trends Neurosci, vol. 39, no. 3, pp. 146-57.

163 | P a g e
PENDIN, D., MCNEW, J. A. & DAGA, A. 2011. Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr Opin Cell Biol, 23, 435-42.
PENNO, A., REILLY, M. M., HOULDEN, H., LAURA, M., RENTSCH, K., NIEDERKOFLER, V., STOECKLI, E. T., NICHOLSON, G., EICHLER, F., BROWN, R. H., JR., VON ECKARDSTEIN, A. & HORNEMANN, T. 2010. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem, 285, 11178-87.
PUTNEY, JW, JR., BROAD, LM, BRAUN, FJ, LIEVREMONT, JP & BIRD, GS 2001, 'Mechanisms of capacitative calcium entry', J Cell Sci, vol. 114, no.12, pp. 2223-9.
PROVOST, P., DOUCET, J., STOCK, A., GERISCH, G., SAMUELSSON, B. & RADMARK, O. 2001. Coactosin-like protein, a human F-actin-binding protein: critical role of lysine-75. Biochem J, 359, 255-63.
RAO, R. V. & BREDESEN, D. E. 2004. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol, 16, 653-62.
RIVIERE, J. B., VERLAAN, D. J., SHEKARABI, M., LAFRENIERE, R. G., BENARD, M., DER KALOUSTIAN, V. M., SHBAKLO, Z. & ROULEAU, G. A. 2004. A mutation in the HSN2 gene causes sensory neuropathy type II in a Lebanese family. Ann Neurol, 56, 572-5.
RODDIER, K., THOMAS, T., MARLEAU, G., GAGNON, A. M., DICAIRE, M. J., ST-DENIS, A., GOSSELIN, I., SARRAZIN, A. M., LARBRISSEAU, A., LAMBERT, M., VANASSE, M., GAUDET, D., ROULEAU, G. A. & BRAIS, B. 2005. Two mutations in the HSN2 gene explain the high prevalence of HSAN2 in French Canadians. Neurology, 64, 1762-7.
ROTTHIER, A., AUER-GRUMBACH, M., JANSSENS, K., BAETS, J., PENNO, A., ALMEIDA-SOUZA, L., VAN HOOF, K., JACOBS, A., DE VRIENDT, E., SCHLOTTER-WEIGEL, B., LOSCHER, W., VONDRACEK, P., SEEMAN, P., DE JONGHE, P., VAN DIJCK, P., JORDANOVA, A., HORNEMANN, T. & TIMMERMAN, V. 2010. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet, 87, 513-22.frontiers. Biochim Biophys Acta, 1824, 68-88.
SABOURDY, F., KEDJOUAR, B., SORLI, S. C., COLIE, S., MILHAS, D., SALMA, Y. & LEVADE, T. 2008. Functions of sphingolipid metabolism in mammals--lessons from genetic defects. Biochim Biophys Acta, 1781, 145-83.
SALADIN, K. S. 2007. Anatomy & physiology: the unity of form and function, New York, N.Y., McGraw-Hill.
SCHAPIRA, A. H. V. & BYRNE, E. 2007. Neurology and clinical neuroscience, Philadelphia, Mosby Elsevier.

164 | P a g e
SHAW, P. E. 2002. Peptidyl-Prolyl Isomerases: A New Twist to Transcription. Embo Rep, 3, 521-6.
SHEN, X., ZHANG, K. & KAUFMAN, R. J. 2004. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat, 28, 79-92.
SINGH, RP, CHENG, YH, NELSON, P & ZHOU, FC 2009, 'Retentive multipotency of adult dorsal root ganglia stem cells', Cell Transplant, vol. 18, no. 1, pp. 55-68
STOJADINOVIC, A., HOOKE, J. A., SHRIVER, C. D., NISSAN, A., KOVATICH, A. J., KAO, T. C., PONNIAH, S., PEOPLES, G. E. & MORONI, M. 2007. HYOU1/Orp150 expression in breast cancer. Med Sci Monit, 13, BR231-239.
SWANSON, A. G. 1963. Congenital insensitivity to pain with anhydrosis. A unique syndrome in two male siblings. Arch Neurol, 8, 299-306.
TAMBOLI, I. Y., PRAGER, K., BARTH, E., HENEKA, M., SANDHOFF, K. & WALTER, J. 2005. Inhibition of glycosphingolipid biosynthesis reduces secretion of the beta-amyloid precursor protein and amyloid beta-peptide. J Biol Chem, 280, 28110-7.
TAVENDER, T. J. & BULLEID, N. J. 2010. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J Cell Sci, 123, 2672-9.
TURK, V., STOKA, V., VASILJEVA, O., RENKO, M., SUN, T., TURK, B. & TURK, D. 2012. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta, 1824, 68-88.
VAN DER BLIEK, A. M., SHEN, Q. & KAWAJIRI, S. 2013. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol, 5.
VANCE, JE 2014, 'MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond', Biochim Biophys Acta, vol. 1841, no. 4, pp. 595-609
VAN MEER, G. & SPRONG, H. 2004. Membrane lipids and vesicular traffic. Curr Opin Cell Biol, 16, 373-8.
VERHOEVEN, K., TIMMERMAN, V., MAUKO, B., PIEBER, T. R., DE JONGHE, P. & AUER-GRUMBACH, M. 2006. Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol, 19, 474-80.
VERHOEVEN, K., COEN, K., DE VRIENDT, E., JACOBS, A., VAN GERWEN, V., SMOUTS, I., POU-SERRADELL, A., MARTIN, J. J., TIMMERMAN, V. & DE JONGHE, P. 2004. SPTLC1 mutation in twin sisters with hereditary sensory neuropathy type I. Neurology, 62, 1001-2.
VERPOORTEN, N., DE JONGHE, P. & TIMMERMAN, V. 2006. Disease mechanisms in hereditary sensory and autonomic neuropathies. Neurobiol Dis, 21, 247-55.

165 | P a g e
VIJAYVERGIYA, C., BEAL, M. F., BUCK, J. & MANFREDI, G. 2005. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci, 25, 2463-70.
WARNER, T. T., HAMMANS, S. R. & ECCLES, D. M. 2009. Practical guide to neurogenetics, Philadelphia, PA, Saunders/Elsevier.
WARTON, K., TONINI, R., FAIRLIE, W. D., MATTHEWS, J. M., VALENZUELA, S. M., QIU, M. R., WU, W. M., PANKHURST, S., BAUSKIN, A. R., HARROP, S. J., CAMPBELL, T. J., CURMI, P. M., BREIT, S. N. & MAZZANTI, M. 2002. Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J Biol Chem, 277, 26003-11.
WEI, J., YEROKUN, Y., LIEPELT, M., MOMIN, A., WANG, E., HANADA, K. & MERRIL, Jr A. H., 2007. 2-1 Serine Palmitoyltransferase. Sphingolipid Biology. Springerlink. 25-27.
WIECKOWSKI, MR, GIORGI, C, LEBIEDZINSKA, M, DUSZYNSKI, J & PINTON, P 2009, 'Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells', Nat Protoc, vol. 4, no. 11, pp. 1582-90.
YAMANO, K. & YOULE, R. J. 2011. Coupling mitochondrial and cell division. Nat Cell Biol, 13, 1026-7.
YAMAMOTO K, YAGI H, LEE YH, KARDOS J, HAGIHARA Y, NAIKI H, GOTOY (2010) The amyloid fibrils of the constant domain of immunoglobulin light chain. FEBS Lett 584:3348–3353
YARD, B. A., CARTER, L. G., JOHNSON, K. A., OVERTON, I. M., DORWARD, M., LIU, H., MCMAHON, S. A., OKE, M., PUECH, D., BARTON, G. J., NAISMITH, J. H. & CAMPOPIANO, D. J. 2007. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol, 370, 870-86.
YOULE, R. J. & STRASSER, A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol, 9, 47-59.
YOUNG, A. R., CHAN, E. Y., HU, X. W., KOCHL, R., CRAWSHAW, S. G., HIGH, S., HAILEY, D. W., LIPPINCOTT-SCHWARTZ, J. & TOOZE, S. A. 2006. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci, 119, 3888-900.
YOUNG, P. 2005. Architecture of the peripheral nerve. Hereditary Peripheral Neuropathies. 3-12. Steinkopff.
YU, L, DING, Y, SPENCER, A, MA, J, LU, R, RUDKIN, BB & YUAN, C 2012, 'Dorsal root ganglion progenitors differentiate to gamma-aminobutyric acid- and choline acetyltransferase-positive neurons', Neural Regen Res, vol. 7, no. 7, pp. 485-91.

166 | P a g e
ZHANG, Y & CHAN, D, .2007. New Insight to Mitochondrial Fusion. FEBS Lett... 581(11). 2168-2173.
ZHAO, J., LIU, T., JIN, S., WANG, X., QU, M., UHLEN, P., TOMILIN, N., SHUPLIAKOV, O., LENDAHL, U. & NISTER, M. 2011. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J, 30, 2762-78.
ZUCHNER, S., MERSIYANOVA, I. V., MUGLIA, M., BISSAR-TADMOURI, N., ROCHELLE, J., DADALI, E. L., ZAPPIA, M., NELIS, E., PATITUCCI, A., SENDEREK, J., PARMAN, Y., EVGRAFOV, O., JONGHE, P. D., TAKAHASHI, Y., TSUJI, S., PERICAK-VANCE, M. A., QUATTRONE, A., BATTALOGLU, E., POLYAKOV, A. V., TIMMERMAN, V., SCHRODER, J. M. & VANCE, J. M. 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet, 36, 449-51




















