
Intradermal NKT cell activation during DNA priming in heterologous prime-boost vaccination enhances...
14
Intradermal NKT cell activation during DNA priming in heterologous prime-boost vaccination enhances T cell responses and protection against Leishmania Blaise Dondji 1 , Eszter Deak 1 , Karen Goldsmith-Pestana 1 , Eva Perez-Jimenez 2 , Mariano Esteban 2 , Sachiko Miyake 3 , Takashi Yamamura 3 and Diane McMahon-Pratt 1 1 Department of Epidemiology & Public Health, Yale University School of Medicine, New Haven, USA 2 Centro Nacional de Biotecnologȷa, CSIC, Madrid, Spain 3 National Center of Neurology and Psychiatry, Tokyo, Japan Heterologous prime-boost vaccination employing DNA-vaccinia virus (VACV) modality using the Leishmania homologue of receptors for activated C kinase (LACK) (p36) antigen has been shown to elicit protective immunity against both murine cutaneous and visceral leishmaniasis. However, DNA priming is known to have limited efficacy; therefore in the current study the effect of NKT cell activation using a-galactosyl- ceramide (aGalCer) during intradermal DNAp36 priming was examined. Vaccinated mice receiving aGalCer + DNAp36 followed by a boost with VVp36 appeared to be resolving their lesions and had at ten- to 20-fold higher reductions in parasite burdens. NKT cell activation during aGalCer + DNAp36 priming resulted in higher numbers of antigen-reactive effector CD4 + and CD8 + T cells producing granzyme and IFN-c, with lower levels of IL-10. Although immunodepletion studies indicate that both CD4 and CD8 T cells provide protection in the vaccinated mice, the contribution of CD4 + T cells was significantly increased in mice primed with DNAp36 together with aGalCer. Notably 5 months after boosting, mice vaccinated with DNAp36 + aGalCer continued to show sustained and heightened T cell immune responses. Thus, heterologous prime- boost vaccination using aGalCer during priming is highly protective against murine cutaneous leishmaniasis, resulting in the heightened activation and development of CD4 and CD8 T cells (effector and memory T cells). Introduction The leishmaniases are a group of vector-borne parasitic diseases caused by protozoa of the genus Leishmania. The disease is endemic in the tropics, subtropics and southern Europe [1]. The clinical forms range from the self-healing cutaneous leishmaniasis to visceral leish- maniasis in which the parasite spreads into the reticuloendothelial system with fatal outcome. Although drug treatment exists for leishmaniasis [2, 3], alter- native approaches for the control of this disease (vector control, immunotherapeutic, chemotherapeutic and vaccine) are still needed [4–6]. Over the past decade, DNA vaccination has been shown to be a promising tool to combat intracellular infectious agents since it induces cell-mediated immu- nity [7] as well as humoral responses. LACK, a 36-kDa (p36) antigen from Leishmania, is a homologue of the receptors for activated C kinase. Immunofluorescence Correspondence: Diane McMahon-Pratt, Ph.D., Yale University School of Medicine, Department of Epidemiology & Public Health, 60 College Street, LEPH 711, P.O. Box 208034, New Haven, CT 06520-8034, USA Fax: +1-203-737-2921 e-mail: [email protected] Received 11/7/07 Revised 28/10/07 Accepted 10/12/07 [DOI 10.1002/eji.200737660] Key words: Adjuvant Leishmania Memory NKT cells Vaccine Abbreviations: aGalCer: a-galactosyl-ceramide LACK: Leish- mania homologue of receptors for activated C kinase SLA: soluble leishmanial antigen VACV: vaccinia virus Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719 706 f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Intradermal NKT cell activation during DNA priming in heterologous prime-boost vaccination enhances...

Intradermal NKT cell activation during DNA primingin heterologous prime-boost vaccination enhancesT cell responses and protection against Leishmania
Blaise Dondji1, Eszter Deak1, Karen Goldsmith-Pestana1, Eva Perez-Jimenez2,Mariano Esteban2, Sachiko Miyake3, Takashi Yamamura3 andDiane McMahon-Pratt1
1 Department of Epidemiology & Public Health, Yale University School of Medicine,New Haven, USA
2 Centro Nacional de Biotecnolog�a, CSIC, Madrid, Spain3 National Center of Neurology and Psychiatry, Tokyo, Japan
Heterologous prime-boost vaccination employing DNA-vaccinia virus (VACV) modalityusing the Leishmania homologue of receptors for activated C kinase (LACK) (p36)antigen has been shown to elicit protective immunity against both murine cutaneousand visceral leishmaniasis. However, DNA priming is known to have limited efficacy;therefore in the current study the effect of NKT cell activation using a-galactosyl-ceramide (aGalCer) during intradermal DNAp36 priming was examined. Vaccinatedmice receiving aGalCer + DNAp36 followed by a boost with VVp36 appeared to beresolving their lesions and had at ten- to 20-fold higher reductions in parasite burdens.NKT cell activation during aGalCer + DNAp36 priming resulted in higher numbers ofantigen-reactive effector CD4+ and CD8+ T cells producing granzyme and IFN-c, withlower levels of IL-10. Although immunodepletion studies indicate that both CD4 andCD8 T cells provide protection in the vaccinated mice, the contribution of CD4+ T cellswas significantly increased in mice primed with DNAp36 together with aGalCer.Notably 5 months after boosting, mice vaccinated with DNAp36 + aGalCer continuedto show sustained and heightened T cell immune responses. Thus, heterologous prime-boost vaccination using aGalCer during priming is highly protective against murinecutaneous leishmaniasis, resulting in the heightened activation and development ofCD4 and CD8 T cells (effector and memory T cells).
Introduction
The leishmaniases are a group of vector-borne parasiticdiseases caused by protozoa of the genus Leishmania.The disease is endemic in the tropics, subtropics and
southern Europe [1]. The clinical forms range from theself-healing cutaneous leishmaniasis to visceral leish-maniasis in which the parasite spreads into thereticuloendothelial systemwith fatal outcome. Althoughdrug treatment exists for leishmaniasis [2, 3], alter-native approaches for the control of this disease (vectorcontrol, immunotherapeutic, chemotherapeutic andvaccine) are still needed [4–6].
Over the past decade, DNA vaccination has beenshown to be a promising tool to combat intracellularinfectious agents since it induces cell-mediated immu-nity [7] as well as humoral responses. LACK, a 36-kDa(p36) antigen from Leishmania, is a homologue of thereceptors for activated C kinase. Immunofluorescence
Correspondence: DianeMcMahon-Pratt, Ph.D., Yale UniversitySchool of Medicine, Department of Epidemiology & PublicHealth, 60 College Street, LEPH 711, P.O. Box 208034, NewHaven, CT 06520-8034, USAFax: +1-203-737-2921e-mail: [email protected]
Received 11/7/07Revised 28/10/07
Accepted 10/12/07
[DOI 10.1002/eji.200737660]
Key words:Adjuvant � Leishmania� Memory � NKT cells
� Vaccine
Abbreviations: aGalCer: a-galactosyl-ceramide � LACK: Leish-mania homologue of receptors for activated C kinase � SLA:soluble leishmanial antigen � VACV: vaccinia virus
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719706
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

experiments indicate that LACK localizes to the cellcytoplasm (near the kinetoplast disc) [8], and is highlyconserved among Leishmania species; LACK is expressedby both the promastigote and amastigote forms of theparasite [9]. Studies indicate that DNA encoding for theLACK antigen can provide protection against L. majorand that this is due to the induction of T cell responsesand IFN-c production [10–14]. However, DNA vaccina-tion with LACK has not proven to provide protectionacross Leishmania species causing both visceral andcutaneous forms of the disease [15–17]; consequently,new modalities have been sought.
The antigen delivery system is a critical component indetermining antigenic efficacy. Vaccinia virus (VACV)and fowlpox vectors have been shown to be a goodantigen delivery system for the control of infectiousdiseases in animal model studies [18] and, importantly,are known to provide long-term memory [19]. A DNAprime-poxvirus boost regime has been shown to induceprotection against Plasmodium bergheimalaria [18] andto induce immunological control of an immunodef-iciency virus infection in monkeys [20]. A prime-boostregime using DNA and VACV expressing the leishmanialLACK antigen has been shown to be immunogenic andprotective against murine L. major infection [21–23].Further, the prime-boost modality is effective ininducing protection against visceral leishmaniasiscaused by L. infantum, while DNA vaccination alonefails to protect [16, 24]. However, although significantreductions in parasite burdens were observed, protec-tion was partial. Consequently, methods for enhancingthe protection provided by the DNA-VACV prime-boostmodality have been sought.
It is known that the potency of DNA priming is limitedand this could potentially restrict the efficacy of boostingwith recombinant VACVor other attenuated viral vectors.Consequently, adjuvants (cytokines, costimulatory mol-ecules, delivery vehicles) with potential to enhance DNAvaccinationhavebeenevaluated[21,25].BothCD4(Th1)and CD8 Tcells have been demonstrated to be importantfor the control of L. major infection [12,13, 26–28]. NKTcell activationhas been reported to lead to enhancedCD4and CD8 T cell responses [29, 30] as well as inducedendritic cell maturation [31]; further, NKT cells havebeen shown to contribute to the control of L. majorinfection [32, 33]. Interestingly, TNF-a (induced byvarious pathogen-associated molecular patterns/TLR)has been shown to be responsible for the recruitment ofCD1d/NKT cells [34]. Further, CpG has been shown toenhance NKT cell activation through interplay betweenplasmacytoid and myeloid dendritic cells [35, 36].Consequently, it might be anticipated that a vaccineemploying DNA priming (TLR9) together with NKT cellactivation (a-galactosyl-ceramide; aGalCer) wouldfurther amplify a developing Th1/Tc1 immune response.
However, aGalCer can drive both Th1 and Th2responses [37, 38] and can induce the proliferation ofTreg populations [39, 40]; further, the use of aGalCer asan adjuvant in vaccine studies has met with mixedresults. Disease exacerbation and impairment ofCD8 T cell function have been observed for systemicaGalCer-mediated NKT cell activation during DNAvaccination against Trypanosoma cruzi [41] infection.In contrast, studies have shown that the systemicactivation of NKT cells (using the ligand aGalCer)during vaccination using viral vectors or irradiatedsporozoites can lead to enhanced CD8 T cell responsesand protection against murine malaria [42–44].
In this study, we have examined the effect ofintradermal NKT cell activation during DNA primingimmunization on vaccine efficacy in controlling murineleishmaniasis due to L. major. Intradermal priming withDNA-LACK together with aGalCer significantly en-hances protection induced by a heterologous DNA-VACVprime-boost vaccination. This is due to increased levelsof antigen-specific T cells (CD4 and CD8); in particularCD4+ T cells responses are increased. Further, theenhancement of T cell responses is sustained and isevident 5 months post-vaccination, indicating that NKTcell activation during DNA vaccination can contribute tothe appropriate development of both T cell effector andmemory populations.
Results
aGalCer enhances the protection inducedby heterologous prime-boost (DNA-VACV)vaccination
We have previously demonstrated in amouse model [22,24, 45] that heterologous prime-boost DNA-VACVvaccination with each vector expressing the leishmanialLACK antigen can provide protection against bothcutaneous and visceral leishmaniasis. Although signifi-cant reductions in parasite burdens are achieved usingthis modality, protection observed was partial, overallresulting in a delay in onset of infection and less severedisease. Consequently, approaches were pursued forenhancing the protection provided by the DNA-VACVprime-boost modality.
NKT cell activation has been reported to lead toenhanced CD4 and CD8 Tcell effector responses [29, 30,46] as well as induce dendritic cell maturation [31] andhence might be expected to act to increase T cellresponses during vaccination. As both CD4 (Th1) andCD8 T cell responses have been demonstrated to beimportant for the control of L. major infection [12, 13,26–28], we evaluated the efficacy of aGalCer as anadjuvant in DNA priming.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 707
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Experimentally, mice were primed intradermallywith p36 (LACK) plasmid DNA (DNAp36) togetherwith or without a-GalCer. Mice were then boosted 2 wklater with VVp36 (1�107 PFU). Control groups of micewere inoculated with either DNA empty plasmid vectorfollowed by 1�107 PFU VV-Luc (virus control) orreceived PBS alone. Three weeks after boosting, micewere infected in the right rear hind foot with 5�104 late-stationary-phase L. major promastigotes. As shown inFig. 1A, mice that received aGalCer during DNA primingdeveloped smaller lesions over time than mice receivingDNAp36 alone. In fact, the lesions of the mice in theaGalCer-DNAp36 group appeared to be resolving at
13 wk post-infection. Evaluation of parasite burdens at13 wk post-infection (Fig. 1B) confirmed lesion devel-opment results and revealed that mice primed withaGalCer together with DNAp36 had tenfold furtherreduction in their parasite burdens than those receivingDNAp36 alone.
Examinations of the cellular immune responses ofthe mice either before infection (3 wk after the finalVVp36 boost) (Fig. 2) or at 13 wk post-infection (datanot shown) were comparable and indicated thatvaccinated mice receiving aGalCer together withDNAp36 produced higher levels of IFN-c and nitricoxide and reduced levels of IL-10 in response toleishmanial antigen [soluble leishmanial antigen(SLA) or recombinant LACK]. In addition, the antibodyresponses of the vaccinated mice were evaluated at13 wk post-infection against both SLA (whole leishma-nial lysate) and LACK antigen. As shown in Fig. 3, miceprimed with aGalCer together with DNAp36 in compar-ison to mice receiving DNAp36 alone, had elevatedlevels of IgG2a against LACK antigen, but comparablelevels of IgG1 antibody. However, it should be noted thatthe overall antibody responses against SLA (whichwould be representative of the ongoing response to theparasite) were comparable for both groups of vaccinatedmice as well as vaccine control mice (receiving pCINeoand VVLuc). Hence, overall the adjuvant aGalCerappeared to enhance a cellular Th1-like response inthe vaccinated mice, which correlated with increasedprotection against L. major infection.
a-GalCer-enhanced responses duringheterologous prime-boost (DNA-VACV)vaccination are long-lived
In order to evaluate the long-term effect of aGalCer onthe overall immune response, several vaccinated mice(three per group) were left unchallenged. Five monthsafter the final VVp36 boost, the immune response toleishmanial antigens and concanavalin A were exam-ined in these mice. As seen in Table 1, the IFN-cresponses of the mice receiving DNAp36 together withaGalCer continued to be elevated in comparison to thoseof mice receiving DNAp36 alone. Further, the ratio ofIFN-c/IL-10 remained higher (twofold) in the micereceiving DNAp36 together with aGalCer. Interestinglyand consistent with this observation, the mice receivingDNAp36 together with aGalCer also maintained higherlevels of IgG2a LACK antibody (and higher IgG2a/IgG1)at 5 months post-boost than mice not receiving aGalCer(data not shown). The enhanced immunological resp-onses of mice vaccinated with DNAp36 together withaGalCer were sustained. Therefore, NKT cell activationduring DNA vaccination led not only to the development
Figure 1. The aGalCer adjuvant during DNA priming enhancesprotection against L. major infection. Mice were primedintradermally with p36 (LACK) plasmid DNA (DNAp36,100 lg) with or without 2 lg aGalCer. Mice were boosted2 wk later with VVp36 (1�107 PFU). Control groups of micewere vaccinated with either control DNA followed by1�107 PFU VV-Luc (vaccine controls) or received PBS alone(infection controls). Three weeks after boosting, mice wereinfected in the right rear hind foot with 5�104 late-stationary-phase L. major promastigotes. (A) Lesion development withtime post-infection in mice receiving PBS (control, diamonds),pCINeoDNA + aGalCer + VVluc (vector control, circles),DNAp36 + VVp36 (triangles) or p36DNA + aGalCer + VVp36(asterisks); p=0.03 for p36DNA versus p36DNA + aGalCer. (B)Evaluation of parasite burdens at 13 wk post-infection in micereceiving PBS (control, black), pCINeoDNA + VV-Luc (vectorcontrol, grey), DNAp36 + VVp36 (horizontally hatched) orp36DNA + aGalCer + VVp36 (vertically hatched); *p=0.02 forp36DNA versus p36DNA + aGalCer. The results presented arerepresentative of three independent experiments.
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719708
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

of higher numbers of T cell effectors and protection, butalso to long-lived T cell immunological memory.
NKT cells contribute to protection in micevaccinated using aGalCer during DNA priming
To verify the role of NKT cells in the aGalCerenhancement of protection induced during DNAp36priming, groups of NKT-deficient BALB/c mice werevaccinated and challenged with L. major (Fig. 4). NKT-deficient mice were protectively vaccinated using theheterologous prime-boost modality. However, the lesiondevelopment over the 13-wk period of observation didnot show a reduction in lesion size for the groupreceiving aGalCer + DNAp36 in comparison to thegroup receiving DNAp36 alone; further, the parasiteburdens were identical for these two groups ofvaccinated mice. These results paralleled the immuneresponses (Table 2) found in these two groups ofvaccinated mice. NKT-deficient mice receiving
DNAp36 + aGalCer or DNAp36 alone displayed com-parable IFN-c, granzyme, nitric oxide and IL-10responses. It should be noted that this experimentwas performed together with the experiment shown inFig. 5 (below). Hence an enhanced protection wasobserved for BALB/c mice vaccinated with p36DNA +aGalCer. Consequently, the effect of aGalCer on DNApriming was mediated through the activation of NKTcells.
aGalCer during DNA priming leads to enhancedCD4 and CD8 T cell IFN-c and granzymeresponses
Both CD4 and CD8 T cells are known to contribute toprotection against murine leishmaniasis in vaccinatedmice (reviewed in [47]). It is well established that VACVcan induce both CD8 and CD4 Tcell responses; however,a generally dominant CD8 T cell response is observed[19]. Further, vaccination with DNAp36 alone or using a
Figure 2. Immune responses of vaccinatedmice. Micewere primed intradermally with p36 (LACK) plasmid DNA (DNAp36, 100 lg)with or without 2 lg aGalCer (GalCer) and were boosted 2 wk later with VVp36 (1�107 PFU), as indicated in Fig. 1. Shown are theresults from ELISA analyses of splenic cells stimulated with either recombinant LACK antigen (p36) or SLA, 3 wk after the finalVVp36 boost: p36 antigen (hatched), SLA (black). PBSwas used as the control; PCINeowas used as the vector control. These resultsare representative of three independent experiments.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 709
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

DNA-VACV prime-boost modality has been shownprimarily to lead to the development of a dominantCD8 T cell response [12, 13, 48]. However, activation ofNKT cells has been shown to lead to dendritic cellmaturation and CD4 T cell responses [49–52]. Conse-quently, given the enhanced protection observed, the
impact of aGalCer on the development of CD4 andCD8 T cell responses was of interest.
To quantitatively evaluate the response of CD8 andCD4 T effector cell populations to the ongoing infection,the numbers of splenic CD4 and CD8 T cells respondingin WT vaccinated and control mice after 13 wk ofinfection were compared using FACS (Table 3). Pro-liferative responses (blast cells) together with theproduction of IFN-c or granzyme were employed asmarkers of activated T cells [53, 54]. Further, the effectof in vitro blocking of either the CD4 or CD8 T cellresponse on the overall ongoing response to SLA wasdetermined for the vaccinated mice at 13 wk post-infection (Table 4).
FACS analyses indicate that NKT cell activationduring DNA priming leads to higher numbers of antigen-reactive CD4 and CD8 Tcells, as the numbers of antigen-activated T cells (as determined by the production ofeither IFN-c or granzyme) increased (1.5- to 5.8-fold) inthe mice receiving DNAp36 + aGalCer in comparison tothose receiving DNAp36 alone. The overall increases inDNAp36 + aGalCer group in the responding CD4 orCD8 T cells producing IFN-c were comparable (1.5- and2.0-fold, respectively), as were the corresponding meanintensities of fluorescence (data not shown). However,consistently (in both LACK-vaccinated groups) signifi-cantly (twofold) higher levels of CD4 than CD8 T cellswere found to produce IFN-c, indicating that CD4 Tcellsare the predominant source of IFN-c. Overall thecontribution of CD4 T cell IFN-c was higher in the micereceiving DNAp36 + aGalCer. Further, the levels ofcytolytic (granzyme-producing) CD4 and CD8 T cellswere also increased in the mice receiving DNAp36 +aGalCer. Although the level of cytolytic CD4+ gran-zyme+ T cells increased 5.8-fold after vaccination withDNAp36 + aGalCer (compared to DNAp36 alone) incomparison to the increase in CD8+ granzyme+ T cells(twofold), CD8+ T cells (2.6%) were the predominantgranzyme-producing cells (Table 3).
Table 1. T cell responses to LACK antigen in vaccinated mice at 5 months after the VVp36 boosta)
Vaccine group IFN-c (ng/mL) IL-10 (pg/mL)
Concanavalin A SLA LACK Concanavalin A SLA LACK
PBS 12 � 4.1 15 � 0.5 15 � 0.1 152 � 2.8 ND 6.8 � 2.9
Control DNA + VV-Luc 14 � 6.2 27 � 0.4 52 � 0.6 176.6 � 4.3 ND 127.2 � 4.6
DNAp36 + VVp36 160 � 2.6 82 � 1.2 199 � 2.8 420 � 1.7 ND 634 � 15.0
DNAp36 + aGalCer + VVp36 190 � 4.1* 111 � 4.0* 272 � 6.0** 46.8 � 4.9**** ND 459 � 15.6***
a) Additionalmice (three per group), vaccinated as part of the experiment shown in Fig. 1, were allowed to rest for 5.5 months afterthe final VVp36 boost. Subsequently, splenic cellswere isolated and stimulatedwith concanavalin A, rLACK antigen or SLA, andthe levels of IFN-c and IL-10 determined. In addition, antibody responses were evaluated. These results indicate a sustainedenhanced immunologic response in themice receivingDNAp36 togetherwith aGalCer; *p<0.05, **p<0.01, ***p<0.001, ***p<0.0001for DNAp36 + VVp36 versus DNAp36 + aGalCer + VVp36; ND = not determined.
Figure 3. Antibody responses of vaccinatedmice. The antibodyresponses of thevaccinated and controlmicewere evaluated at13 wk post-infection against (A) LACK antigen (1:500) and (B)SLA (whole leishmanial lysate, 1:1000). (See the legends to Fig. 1and Fig. 2 for more information.) The levels of total IgG as wellas specific IgG1 and IgG2 were determined. The antibodyresponses of mice receiving aGalCer + DNAp36 versus DNAp36were significantly different for LACK: total IgG (p=0.0001), IgG1(p=0.04), IgG2a (p=0.00001); SLA: total IgG (p=0.03), IgG2a(p=0.01), IgG1 (not significant). These results are representativeof three independent experiments.
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719710
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

FACS results were consistent with antibody blockingexperiments (Table 4) of vaccinated mice that indicateda predominant contribution of CD4 T cells to theproduction of IFN-c. The level of inhibition of CD8 alonewas partial, while blocking the response of CD4 T cellsresulted in almost complete ablation of the IFN-cresponse. These results suggest that co-operationbetween CD4 and CD8 T cells is likely involved in anoptimal secondary response, as has been observed inother systems [55–57].
Interestingly, blocking experiments indicated thatCD4 Tcells were also the predominant source of IL-10, asinhibition of CD4 cells extensively reduced the devel-opment of IL-10 responses. IL-10 CD4 Treg as well asCD4+ CD25– Foxp3– Th1 populations have beenreported to modulate leishmaniasis pathogenesis innon-vaccinated (naive) mice [58–60] as well as tomodulate protection elicited in response to prime-boostvaccination [61]. Although the precise CD4 T cellsubpopulation(s) responsible for the IL-10 productionremain to be determined, consistently in all groups ofmice (control and vaccine), CD4 blockage led to lowerlevels of IL-10 produced, suggesting that CD4 T cells aregenerally the primary source of IL-10 during infection.As mice depleted of either CD4 or CD8 T cells (seebelow) before infection have levels of IL-10 comparableto those of non-vaccinated controls, vaccination im-portantly leads to overall reductions in IL-10 levels inresponse to infection; notably both T cell populationsappear critical for this effect.
CD8 and CD4 T cells contribute to protectionin mice vaccinated using aGalCer during DNApriming
To further evaluate the immunological consequences ofNKT cell activation and evaluate the contributions ofCD4 and CD8 T cells, we asked which the actualpopulations of effector cells induced by vaccinationwere
Figure 4. NKT cells are required for increased protectioninduced by aGalCer. To verify the role of NKT cells in theaGalCer enhancement of protection induced during DNAp36priming, groups of NKT-deficient BALB/c micewere vaccinatedand challenged with L. major: PBS (control, squares),pCINeoDNA + VV-Luc (vector control, circles), DNAp36 +VVp36 (triangles), p36DNA + aGalCer + VVp36 (asterisks); alsoshown is the course of infection for control WT BALB/c mice(PBS, diamonds); p=0.45 for DNAp36 versus DNAp36 + aGalCer.(B) Evaluation of parasite burdens at 13 wk post-infection; WTBALB/c PBS (control, black), NKT-deficient PBS (control, white),NKT-deficient pCINeoDNA + VV-Luc (vector control, diagonallyhatched), NKT-deficient DNAp36 + VVp36 (vertically hatched),NKT-deficient DNAp36 + aGalCer + VVp36 (grey); p=0.65 forDNAp36 versus DNAp36 + aGalCer. These results are represen-tative of two independent experiments. It should be noted thatthis experiment was performed together with the experimentshown inFig. 5.Henceanenhancedprotectionwasobserved forBALB/c mice vaccinated with p36DNA + aGalCer.
Table 2. Immune responses at 13 wk post-infection in NKT-deficient micea)
Vaccination group
Control (PBS) pCINeo + aGalCer + VV-Luc DNAp36 + VVp36 DNAp36 + aGalCer + VVp36
IFN-c (pg/mL) 19.1 � 6.4 56.6 � 5.1 5153 � 107b) 5958 � 88b)
IL-10 (pg/mL) 835 � 81 735 � 70 202 � 40b) 165 � 50b)
Granzyme (ng/mL) 0.00 � 0.00 0.03 � 0.0 1.4 � 0.7b) 1.26 � 0.3b)
Nitric oxide (lM) 0.06 � 0.01 0.12 � 0.03 9.12 � 0.83b) 8.02 � 1.11b)
a) Shown are the responses of splenic lymphocytes fromNKT-deficient mice vaccinated and then infectedwith L. major. At 13 wkpost-infection, splenic lymphocytes were stimulated with SLA and the levels of IFN-c, IL-10, granzyme and nitric oxidemeasured. NKT-deficientmice receiving DNAp36 + aGalCer, or DNAp36 alone displayed comparable IFN-c, IL-10, granzyme andnitric oxide responses. Consequently, the effect of aGalCer on DNA priming was mediated through the activation of NKT cells.
b) None of the responses found were significantly different between the mice receiving DNAp36 + VVp36 and those vaccinatedwith DNAp36 + aGalCer + VVp36; p>0.05. These results are representative of two independent experiments.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 711
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

and whether this did significantly change in thepresence of aGalCer during DNA priming. Cellulardepletion (CD4 or CD8) was employed. Briefly, micewere vaccinated as indicated above and CD4 orCD8 T cells were depleted as previously described[62, 63] immediately prior to infection with L. major.Depletion at this time in vaccinated mice wouldprimarily target antigen-specific memory cells. Lesiondevelopment was monitored and immune responses toinfection determined. At 13 wk post-infection, theparasite levels at the cutaneous site of infection wereenumerated.
As shown in Fig. 5, lesion size development andparasite burdens were significantly increased in LACK-vaccinated mice depleted of either CD4 or CD8 Tcells. InFig. 5A, although there was no significant differencebetween control mice (pCINeoDNA + VV-Luc) andthose vaccinated with DNAp36 + VVp36 and depletedof CD8 T cells (p>0.05), vaccinated mice depleted ofCD4 T cells were still significantly protected (p=0.01).Vaccinated mice depleted of either CD4 or CD8 T cellswere significantly less protected than their non-depletedcounterparts (p=0.001 and 0.0001, respectively). Invaccinated mice receiving aGalCer together withDNAp36 (Fig. 5B), mice depleted of either CD4 orCD8 T cells were significantly less protected than theirnon-depleted counterparts (p=0.01 and 0.001, respec-tively). Overall parasite burden analyses (Fig. 5C)confirmed these results. Protection in mice receivingaGalCer together with DNAp36 was better than in micevaccinated with DNAp36 alone (p=0.0001, ANOVA).Further, the parasite burdens in mice depleted ofCD8 T cells in either group (aGalCer together withDNAp36, or DNAp36 alone) were significantly higherthan in mice depleted of CD4 Tcells (p=0.01 and 0.001,respectively).
Overall these results reveal a larger contribution/roleof CD8 T cells in protection induced by DNA-VACVprime-boost vaccination. In particular in the DNAp36 +VVp36-vaccinated group, the reversal of protection wasmore pronounced for mice depleted of CD8 T cells.Parasite numbers at the site of infection in CD8-depletedmice were increased 65-fold, while in CD4-depletedmice parasite levels were increased only 2.4-fold.However, for the mice receiving aGalCer during DNApriming, the effect of CD4 cell depletion was moresubstantial and approached that found after depletion ofCD8 cells, with 156- (CD4) and 875-fold (CD8) increasesin parasite levels at the site of infection. These datasuggest that NKT cell activation increased the antigen-specific T cell responses (CD4 and CD8) during DNApriming. However, it appeared that NKT cell activationpreferentially facilitated the development of antigen-specific CD4 Tcells, leading to an enhanced contributionof CD4 T cells to protection.
Figure 5. Immunodepletion of CD4 or CD8 T cells reversesprotection in vaccinated mice. Mice were vaccinated asindicated in the Figure and CD4 or CD8 T cells were depletedas previously described [62, 63] immediately prior to infectionwith L. major. (A, B) Lesion development was monitored asdescribed in the Materials and methods. (C) At 13 wk post-infection, the parasite levels in the cutaneous site of infectionwere enumerated; PBS (control, black), pCINeoDNA + VV-Luc(vector control, white), DNAp36 + VVp36 (horizontallyhatched), DNAp36 + aGalCer + VVp36 (vertically hatched),DNAp36 + VVp36, CD4-depleted (grey), DNAp36 + VVp36, CD8depleted (rhomboid-patterned), DNAp36 + aGalCer + VVp36,CD4-depleted (diagonally hatched), DNAp36 + aGalCer +VVp36, CD8-depleted (checked). These results represent theaveraged values of five mice per group.
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719712
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
To further examine the effects of cellular depletionon the ongoing immune response and effect of the cellscontributing to protection, cytokine responses wereevaluated in the T cell-depleted mice at 14 wk post-infection; splenic lymphocytes were stimulated usingSLA and IFN-c, and granzyme, nitric oxide and IL-10levels were determined. Overall the effect of CD4 orCD8 T cell depletion resulted in the ablation of IFN-c,nitric oxide and granzyme production in response toinfection. In contrast, either CD4 or CD8 depletion justprior to infection led to increased IL-10 production,which approached that of control mice (Table 5).
IL-10+ CD4+ Tcell populations have been reported tobe critical for parasite persistence in murine leishma-niasis caused by L. major [58–61]. As mice depleted ofeither CD4 or CD8 T cells before infection have levels ofIL-10 comparable to those of non-vaccinated controls, itcan be concluded that vaccination importantly leads tooverall reductions in IL-10 levels induced duringinfection and that interactions between CD4 andCD8 T cells may be critical to the ablation of the IL-10
response. Given the decrease in IL-10 levels observed inthe vaccinated mice receiving aGalCer during DNApriming, these results suggest that NKTcell activation inthe context of TLR9 activation by CpG may prevent theknown NKT enhancement of Treg populations [39, 40].
Overall, experimental evidence indicates that NKTcell activation by aGalCer during intradermal DNAvaccination leads to enhanced curative Th1-like/Tc1CD4 and CD8 Tcell responses which result in significantincreases in protection against infection. Further, thesestudies appear to reveal that CD4-CD8 interactions arecritical/key to this protection during the memory/recallresponse, as the absence of either Tcell subpopulation atthe time of infection severely impacts on the develop-ment of the host immune response. Hence, CD4 andCD8 Tcells are important as memory populations as wellas their direct effector functions to the development of aprotective response to infection.
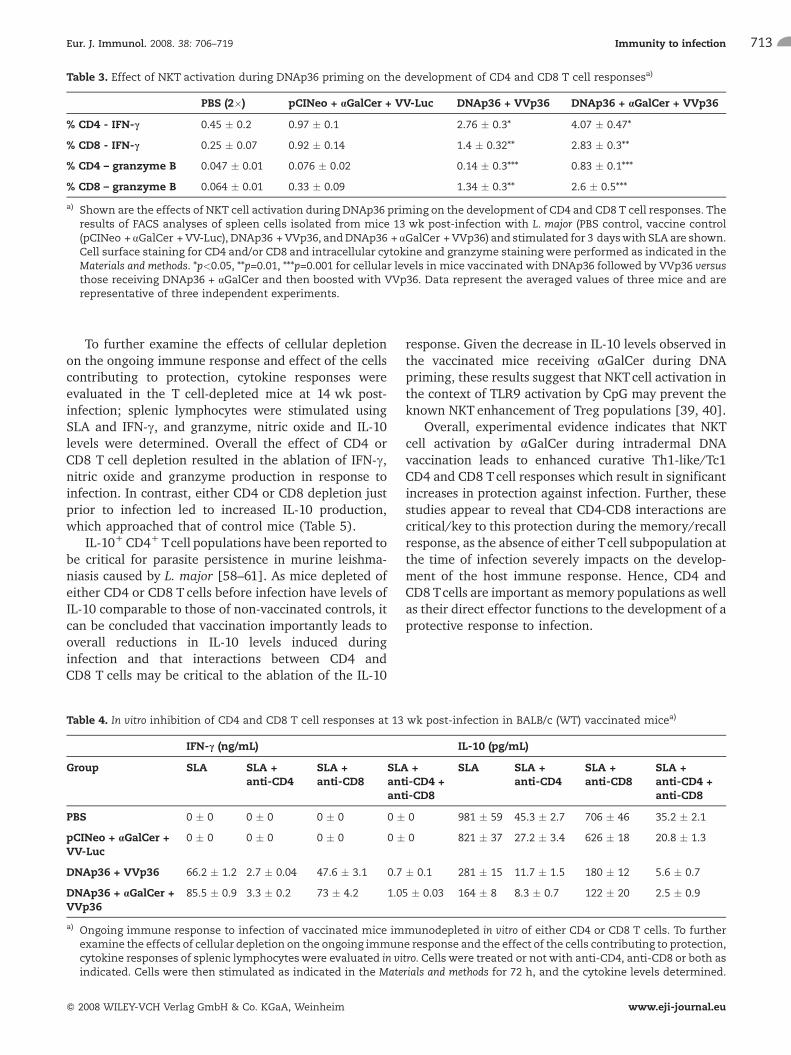
Table 3. Effect of NKT activation during DNAp36 priming on the development of CD4 and CD8 T cell responsesa)
PBS (2�) pCINeo + aGalCer + VV-Luc DNAp36 + VVp36 DNAp36 + aGalCer + VVp36
% CD4 - IFN-c 0.45 � 0.2 0.97 � 0.1 2.76 � 0.3* 4.07 � 0.47*
% CD8 - IFN-c 0.25 � 0.07 0.92 � 0.14 1.4 � 0.32** 2.83 � 0.3**
% CD4 – granzyme B 0.047 � 0.01 0.076 � 0.02 0.14 � 0.3*** 0.83 � 0.1***
% CD8 – granzyme B 0.064 � 0.01 0.33 � 0.09 1.34 � 0.3** 2.6 � 0.5***
a) Shown are the effects of NKT cell activation during DNAp36 priming on the development of CD4 and CD8 T cell responses. Theresults of FACS analyses of spleen cells isolated from mice 13 wk post-infection with L. major (PBS control, vaccine control(pCINeo + aGalCer + VV-Luc), DNAp36 + VVp36, andDNAp36 + aGalCer + VVp36) and stimulated for 3 dayswith SLA are shown.Cell surface staining for CD4 and/or CD8 and intracellular cytokine and granzyme staining were performed as indicated in theMaterials and methods. *p<0.05, **p=0.01, ***p=0.001 for cellular levels in mice vaccinated with DNAp36 followed by VVp36 versusthose receiving DNAp36 + aGalCer and then boosted with VVp36. Data represent the averaged values of three mice and arerepresentative of three independent experiments.
Table 4. In vitro inhibition of CD4 and CD8 T cell responses at 13 wk post-infection in BALB/c (WT) vaccinated micea)
IFN-c (ng/mL) IL-10 (pg/mL)
Group SLA SLA +anti-CD4
SLA +anti-CD8
SLA +anti-CD4 +anti-CD8
SLA SLA +anti-CD4
SLA +anti-CD8
SLA +anti-CD4 +anti-CD8
PBS 0 � 0 0 � 0 0 � 0 0 � 0 981 � 59 45.3 � 2.7 706 � 46 35.2 � 2.1
pCINeo + aGalCer +VV-Luc
0 � 0 0 � 0 0 � 0 0 � 0 821 � 37 27.2 � 3.4 626 � 18 20.8 � 1.3
DNAp36 + VVp36 66.2 � 1.2 2.7 � 0.04 47.6 � 3.1 0.7 � 0.1 281 � 15 11.7 � 1.5 180 � 12 5.6 � 0.7
DNAp36 + aGalCer +VVp36
85.5 � 0.9 3.3 � 0.2 73 � 4.2 1.05 � 0.03 164 � 8 8.3 � 0.7 122 � 20 2.5 � 0.9
a) Ongoing immune response to infection of vaccinated mice immunodepleted in vitro of either CD4 or CD8 T cells. To furtherexamine the effects of cellular depletion on the ongoing immune response and the effect of the cells contributing to protection,cytokine responses of splenic lymphocytes were evaluated in vitro. Cells were treated or not with anti-CD4, anti-CD8 or both asindicated. Cells were then stimulated as indicated in the Materials and methods for 72 h, and the cytokine levels determined.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 713
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Discussion
A heterologous DNA-VACV prime-boost antigen deliverysystem has been shown to be efficacious for the controlof infectious diseases in animal model studies and inhuman trials [18, 20, 64–73]. A prime-boost regimeusing DNA and VACV expressing the leishmanial LACKantigen has been shown to be protective against murineL. major infection [21–23] and is effective in inducingprotection against visceral leishmaniasis caused byL. infantum, while DNAvaccination alone fails to protect[16, 24]. However, the efficacy of DNA priming is knownto be limited; consequently, adjuvants (cytokines,costimulatory molecules, delivery vehicles) with poten-tial to enhance DNA priming have been sought [21, 25].
The use of recombinant protein cytokines or DNAencoding cytokines (IL-12, IL-18) has been demon-strated to enhance DNA priming, leading to increasedprotection against leishmanial infection [21]. However,in terms of vaccine development, the use of syntheticimmunomodulatory compounds, such as aGalCer, offerssimilar advantages to DNA in terms of stability andproduction, while having direct immediate effects on thetarget cell population. Additionally, structural modifica-tion of the parent molecule has been shown to producelonger-lived NKT cell activation or the induction ofspecific cytokine profiles [51, 74]. Further, aGalCer hasalready been utilized in the treatment of human tumors[75, 76]. In terms of potential vaccine use, NKT cellactivation has been reported to lead to enhanced CD4and CD8 T cell effector responses [29, 30, 46] as well asinduce dendritic cell maturation [31].
However, the precise response (Th1 versus Th2) thatdevelops as a consequence to aGalCer activation of NKT
cells is not always predictable [38]. For example,aGalCer provides protection in a murine model ofmalaria [42, 44], but disease exacerbation was found inT. cruzi vaccine studies in mice [41]. However, CpG hasbeen shown to enhance NKT cell activation throughinterplay between plasmacytoid and myeloid dendriticcells [35, 36], which may be in part dependent uponTNF-a [35]. Recent studies indicate that activated NKTcells increase dendritic cell migration and enhanceCD8 T cell responses in the skin [77].
Consequently, the interaction of adjuvants (anddeveloping inflammatory milieu) in a vaccine employ-ing intradermal DNA priming (TLR9) together with NKTcell (aGalCer) activation could selectively amplify thedeveloping Th1/Tc1 immune response. Results fromanalyses of vaccination of mice indicate that this hasoccurred; mice vaccinated with aGalCer showed higherlevels of both CD4 and CD8 Tcells upon infection. Thesecells produced higher levels of IFN-c/granzyme andlower levels of IL-10 in response to infection. In addition,depletion experiments revealed that both populations(CD4 and CD8) were important for protection, while inmice not receiving aGalCer, a predominant role forCD8 T cells was apparent. Notably, these effects(elevated IFN-c/granzyme, lower IL-10 and increasedprotection) were not observed in vaccinated NKT-deficient mice.
CD4 and CD8 T cells producing IFN-c have beendemonstratedtobeimportantfor thecontrolofcutaneousleishmaniasiscausedbyL. major infection[12,13,26–28]as well as visceral disease [78–80]. In the current study,depletion experiments indicate that neither population issufficient for protection, in that immunodepletion ofeither CD4 or CD8 T cells prior to infection for mice
Table 5. Immune responses of vaccinated and CD4- and CD8-deficient/depleted BALB/c mice in response to L. major infectiona)
Group IFN-c, ng/mL � SD Granzyme,ng/mL � SD
IL-10, pg/mL � SD Nitric oxide,lM � SD
PBS (2�) 0 � 0 0 � 0 981 � 60 0.7 � 0.2
pCINeo + aGalCer + VV-Luc 0 � 0 0 � 0 822 � 37 1.5 � 0.5
DNAp36 + VVp36 66.2 � 1.2 1.5 � 0.3 282 � 15 15.6 � 0.3
DNAp36 + aGalCer + VVp36 85.5 � 1 3.5 � 0.6 164 � 8 24 � 0.5
DNAp36 + VVp36 CD4-depleted 5.7 � 0.7 0.8 � 0.2 470 � 21 6.3 � 0.1
DNAp36 + VVp36 CD8-depleted 2.1 � 0.3 0.4 � 0.1 650 � 25 2.6 � 0.1
DNAp36 + aGalCer + VVp36CD4-depleted
7 � 0.8 1 � 0.1 343 � 14 8.6 � 0.2
DNAp36 + aGalCer + VVp36CD8-depleted
1.3 � 0.7 0.5 � 0.2 640 � 60 4 � 0.6
a) To examine the effects of in vivoCD4 and CD8T cell depletion on the ongoing immune response to infection, cytokine responseswere evaluated in vitro of splenic lymphocytes. Cells were treated or not with anti-CD4 or anti-CD8 as indicated in theMaterialsand methods. Cells were then stimulated for 72 h and the cytokine levels determined.
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719714
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

receiving aGalCer results in reversal of protection.Interestingly, aGalCer/NKT cell activation duringDNAp36 priming leads to enhanced CD4 T cell con-tribution to protection in comparison to DNAp36vaccination alone. These results suggest that theinteractions between CD4 and CD8 T cells are requiredfor the activation and development of effector popula-tions.
Previous studies have demonstrated that the con-tribution of CD8 IFN-c was critical to vaccinationinduced by DNAp36 alone [12–14] and that this ledto heightened CD4 Tcell activation. Although somewhatcontroversial [81], the interaction between T cellsubpopulations (and a requirement of both populationsduring chronic infection or secondary responses) hasbeen documented in other systems and recently forL. major infection [55–57, 82]. It is unclear in thecurrent study what the role of CD8 T cells might be inprotection; however, it is clear that overall these cells arenot the dominant source of IFN-c. Previous leishma-niasis vaccine studies have shown that CD8 IFN-c [13,64] can be critical in the development of CD4 T cellresponses and indeed, such mechanisms may also beinvolved in the prime-boost-vaccinated mice.
However, VACV vaccination is known to lead to thedevelopment of cytolytic CD8 T cells. The role of CD8CTL in vaccine-mediated protection has not beenextensively studied. A role for perforin andCD8 T cells has been documented only in the case ofvaccine studies against L. amazonensis using a murinemodel and the P-8 proteoglycolipid complex antigen[63]. However, in mice primed with DNAp36 alone,little if any consequence of CD4 T cell depletion wasfound, suggesting that CD8 T cells alone can providesignificant protection and the question arises as to theCD8 mechanisms pertinent to this protection. We foundincreased levels of granzyme B as a consequence ofvaccination in both CD4 and CD8 populations, suggest-ing that these CTL populations could contribute todisease control. Further studies are required to deter-mine this point. However, these could point toadditional effector targets for vaccine development.
The ideal vaccine against leishmaniasis would be onethat protected against the various species and forms ofdisease (visceral, cutaneous and mucocutaneous).However, the literature suggests that the ability toprovide cross-protection is elusive [16, 78, 83]. We havepreviously demonstrated that heterologous prime-boostvaccination (DNA-VACV) can provide protection againstboth cutaneous and visceral leishmaniasis. We havedemonstrated herein that the development of protectivememory and effector populations are enhanced throughthe activation of NKT populations during DNA priming.Consequently, amultiple-subunit vaccine employing thismodality could provide for an effective protection across
Leishmania species, and the development of a pan-leishmaniasis vaccine. However, work is required todetermine whether this modality represents a vaccinemodality for humans. Given the known effects ofaGalCer in humans [29–31, 46, 75, 76], these resultsare encouraging.
Materials and methods
Parasite strains and animals
L. major strainWR309 (MHOM/IL/79/LRC-L251) were grownas promastigotes at 24�C in Schneider's Drosophila medium(Sigma, Saint Louis, MO) supplemented with 15% heat-inactivated fetal bovine serum and antibiotics, as previouslydescribed [84]. The virulence of the strain was preserved byperiodic passage through BALB/c mice. All mice used in thisstudy were of BALB/c background (WT or NKT-deficient).Breeding pairs of BALB/c NKT-deficient mice were kindlyprovided by Dr. Malcolm Duthie (Infectious Disease ResearchInstitute, Seattle, WA). BALB/c mice (4–6 wk of age) werepurchased from the NIH National Cancer Institute (Frederick,MD). All mice were housed in the Yale University School ofMedicine, an American Association for Accreditation ofLaboratory Animal Care-approved animal facility (FederalAnimal Welfare Assurance number A3230-01). Sentinel micewere periodically checked for the presence of viruses in thecolony.
Plasmids, recombinant p36 protein and recombinantvaccinia viruses
The gene encoding for L. infantum p36 proteinwas obtained asdescribed [22] and inserted downstream of the cytomegalo-virus promoter into the SmaI site of the pCINeo vector(Promega, Madison, WI). The empty plasmid pCINeo (Pro-mega) was used as control (DNA control). Plasmid DNA waspurified using the Qiagen Endofree Plasmid Maxi Kit (QiagenInc., Valencia, CA) and eluted in pyrogen-free deionized water.The endotoxin level of the purified plasmid was tested usingQCL-1000 chromogenic Limulus amebocyte lysate test (LonzaWalkersville Inc., Walkersville, MD) before immunization.Preparations had a maximum of 0.1 ng LPS/100 lg DNA. VV-LACK (VVp36) was derived from the WR strain and wasprepared using standard methods previously described [22,45, 85, 86].
Prime-boost vaccination and infection of mice withL. major
Mice (seven to ten per group) were primed intradermally withp36 (LACK) plasmid DNA or control vector DNA (100 lg/mouse in 100 lL of PBS) together with or without a-GalCer(2 lg/mouse). Mice were boosted intraperitoneally 2 wk later(14 days post-immunization) with VVp36 (1�107 PFU/mouse). Mice immunized earlier with the control DNA wereboosted with 1�107 PFU of the control virus encoding for theluciferase gene (VV-Luc). Infection control mice received PBS
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 715
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

alone during the two immunizations. Three weeks afterboosting, three of the mice from each group were sacrificedand spleen and draining lymph node were collected forimmunological analyses. At the same time, other mice(minimally four per group) were infected in the right rearhind foot with 5�104 late-stationary-phase L. major promas-tigotes. The course of infection was monitored by measuringthe lesion development; using a dial gauge caliper (StarrettThickness Gauge), the ratio of the infected compared with the(left) uninfected foot was determined. At the end ofmonitoring lesion development, the mice were sacrificedand the parasites were enumerated in the cutaneous tissue(site of infection) and draining lymph node by a limitingdilution assay, as reported previously [87].
Immunodepletion of CD4+ or CD8+ T cells
Anti-CD4 (L3T4) or anti-CD8 (Ly-2) monoclonal antibodyobtained from eBioscience (San Diego, CA) was employed forcellular immunodepletion of vaccinated mice. For T celldepletion, mice were injected intraperitoneally with 100 lg ofanti-CD4 (clone GK1.5) or anti-CD8 (clone 53-6.7) antibodyon days –3 and –1 of infection. Efficacy of depletion wasmonitored by flow cytometry and showed that more than 95%of the target cell population was depleted.
Cytokine assays
Splenic lymphocytes and lymph node cells from control andimmunizedmice were harvested. Single-cell preparations weremade in RPMI medium supplemented with 10% fetal bovineserum, 2-mercaptoethanol, antibiotics and L-glutamine. Cells(5�106 per well) were plated in 24-well plates (Costar,Cambridge, MA) and stimulated at 37�C with purifiedrecombinant LACK antigen (5 lg/mL), SLA (25 lg/mL),concanavalin A (1 lg/mL) or left unstimulated. For antibodyblocking experiments, cells were cultured in the presence of20 lg/mL of inhibitory anti-CD4 and/or anti-CD8 antibody (asindicated above for depletion experiments) or isotype control(eBiosciences, San Diego, CA). Preliminary experimentsindicated that maximal blocking was achieved under theseconditions. Supernatants were collected after 72 h of incuba-tion and stored at –20�C until used. Cytokine levels weremeasured from culture supernatants by sandwich ELISA,according to the manufacturer's specifications, as previouslydescribed [84, 87]. Background cytokine levels were deter-mined using supernatant from unstimulated cell populations.
FACS analyses were performed as previously described[63]. Cell surface staining for CD4 and/or CD8 was performedusing specific FITC or CyChrome antibody conjugates (BDPharmingen, San Jose, CA). For intracellular cytokine staining,cells were treated with Golgi Plug (BD Pharmingen) for 4 hbefore harvesting, stained for surface markers CD4 (L3T4 ofH129.19 clone) and CD8 (Ly-2 of 53-6.7 clone), and then fixedand permeabilized [63]. Cells were subsequently stained foreither IFN-c (BD Pharmingen) or granzyme B (eBioSciences)using a PE-conjugated specific antibody. Cell fluorescence wasmeasured using a FACScan (Becton Dickinson); data wereanalyzed using FlowJo software (Tree Star, Inc.).
Measurement of nitrite/nitric oxide
Nitric oxide production was measured as nitrite using theGreiss reaction as previously described [88]. Briefly, to eachwell 96-well flat-bottom microtiter plate (Nunc) containing50 lL of culture supernatant equal volumes of the Greissreagents (1% sulfanilamide and 0.1% naphthylethylenedi-amine dichloride in 2.5% phosphoric acid) were added andincubated at room temperature for 10 min. Standards ofNaNO2 using concentrations from 1.56 to 100 lMNaNO2 wereemployed with each assay. Absorbance was measured at540 nm with the Bio-tek plate reader (Bio-tek Instruments,Winooski, VT). Background concentrations were assessedusing supernatant from unstimulated splenocytes.
Antibody response
Antibody levels to LACK (p36) antigen and SLA were assessedby ELISA. Total IgG as well as IgG1 and IgG2a isotype responseswere evaluated. This was conducted on sera collected fromeach group of animals before challenge and at 1 month post-challenge. The ELISA was performed as previously described[21] with some modifications. Briefly, 96-well Maxisorp plates(Nunc) were coated overnight at 4�C with 100 lL of purifiedrecombinant p36 protein (5 lg/mL). The following day, plateswere washed four times with PBS/0.05% Tween-20 andblocked with 3%BSA in PBS/0.05% Tween-20 (blockingbuffer) for 2 h at 4�C. Sera were diluted in blocking bufferat 1:500 and 1:1000 for pre- and post-challenge sera respec-tively, and incubated overnight at 4�C. Then, plates werewashed and peroxidase-conjugated goat anti-mouse total IgG(H+L) (Pierce, Rockford, IL), IgG1 or IgG2a (SouthernBiotechnology, Birmingham, AL) was added. Reactions weredeveloped using ATBS (Kirkegaard Perry Laboratories,Gaithersburg, MD). Absorbance was read at 405 nm withthe Bio-tek plate reader (Bio-tek Instruments).
Evaluation of the parasite burden
The total number of parasites in the various infected tissues(cutaneous site, draining lymph node) was evaluated bylimiting dilution analysis using Schneider's Drosophila me-dium, as previously described [87]. The individual parasiteburdens from four mice per groupwere determined; results areexpressed as the averaged value � standard errors of theparasite burdens obtained from each group.
Statistical analysis of data
Data are presented as mean � standard error. The Mann–Whitney test was performed to assess the statistical signifi-cance between data acquired from mice primed with p36(LACK) with and without aGalCer. For multiple groupcomparisons, Kruskal–Wallis one-way analysis of variance(ANOVA) was performed followed by a Dunn's post-test. For allthese statistical analyses, p values less than or equal to 0.05were considered significant.
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719716
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Acknowledgements: This work was supported bygrants from the NIH (AI 27811 and AI 06787 toD.Mc.-P.). Eva Perez-Jimenez is a recipient of predoctoralfellowship from the Ministerio de Educacion y Ciencia,Spain (BIO2004-03954 to M.E).
Conflict of interest: The authors declare no financial orcommercial conflict of interest.
References
1 Herwaldt, B. L., Leishmaniasis. Lancet 1999. 354: 1191–1199.
2 Guerin, P. J., Olliaro, P., Sundar, S., Boelaert, M., Croft, S. L., Desjeux, P.,Wasunna, M. K. and Bryceson, A. D., Visceral leishmaniasis: Current statusof control, diagnosis, and treatment, and a proposed research anddevelopment agenda. Lancet Infect. Dis. 2002. 2: 494–501.
3 Sundar, S., Jha, T. K., Thakur, C. P., Engel, J., Sindermann, H., Fischer,C., Junge, K. et al., Oral miltefosine for Indian visceral leishmaniasis.N. Engl. J. Med. 2002. 347: 1739–1746.
4 Tesh, R. B., Control of zoonotic visceral leishmaniasis: Is it time to changestrategies? Am. J. Trop. Med. Hyg. 1995. 52: 287–292.
5 Dye, C., The logic of visceral leishmaniasis control. Am. J. Trop. Med. Hyg.1996. 55: 125–130.
6 Gramiccia, M. and Gradoni, L., The current status of zoonoticleishmaniases and approaches to disease control. Int. J. Parasitol. 2005.35: 1169–1180.
7 Gurunathan, S., Klinman, D. M. and Seder, R. A., DNA vaccines:Immunology, application, and optimization. Annu. Rev. Immunol. 2000. 18:927–974.
8 Gonzalez-Aseguinolaza, G., Taladriz, S., Marquet, A. and Larraga, V.,Molecular cloning, cell localization and binding affinity to DNA replicationproteins of the p36/LACK protective antigen from Leishmania infantum. Eur.J. Biochem. 1999. 259: 909–916.
9 Mougneau, E., Altare, F., Wakil, A. E., Zheng, S., Coppola, T., Wang, Z. E.,Waldmann, R. et al., Expression cloning of a protective Leishmania antigen.Science 1995. 268: 563–566.
10 Ahmed, S. B., Bahloul, C., Robbana, C., Askri, S. and Dellagi, K., Acomparative evaluation of different DNA vaccine candidates againstexperimental murine leishmaniasis due to L. major. Vaccine 2004. 22:1631–1639.
11 Bourreau, E., Prevot, G., Gardon, J., Pradinaud, R., Hasagewa, H.,Milon,G. and Launois, P., LACK-specific CD4(+) T cells that induce gammainterferon production in patients with localized cutaneous leishmaniasisduring an early stage of infection. Infect. Immun. 2002. 70: 3122–3129.
12 Gurunathan, S., Sacks, D. L., Brown, D. R., Reiner, S. L., Charest, H.,Glaichenhaus, N. and Seder, R. A., Vaccination with DNA encoding theimmunodominant LACK parasite antigen confers protective immunity tomice infected with Leishmania major. J. Exp. Med. 1997. 186: 1137–1147.
13 Gurunathan, S., Stobie, L., Prussin, C., Sacks, D. L., Glaichenhaus, N.,Iwasaki, A., Fowell, D. J. et al., Requirements for the maintenance of Th1immunity in vivo following DNA vaccination: A potential immunoregulatoryrole for CD8+ T cells. J. Immunol. 2000. 165: 915–924.
14 Gurunathan, S., Prussin, C., Sacks, D. L. and Seder, R. A., Vaccinerequirements for sustained cellular immunity to an intracellular parasiticinfection. Nat. Med. 1998. 4: 1409–1415.
15 Dumonteil, E., Maria Jesus, R. S., Javier, E. O. and Maria del Rosario, G.M., DNA vaccines induce partial protection against Leishmania mexicana.Vaccine 2003. 21: 2161–2168.
16 Melby, P. C., Yang, J., Zhao, W., Perez, L. E. and Cheng, J., Leishmaniadonovani p36(LACK) DNA vaccine is highly immunogenic but not protectiveagainst experimental visceral leishmaniasis. Infect. Immun. 2001. 69:4719–4725.
17 Coelho, E. A., Tavares, C. A., Carvalho, F. A., Chaves, K. F., Teixeira, K. N.,Rodrigues, R. C., Charest, H. et al., Immune responses induced by theLeishmania (Leishmania) donovani A2 antigen, but not by the LACK antigen,
are protective against experimental Leishmania (Leishmania) amazonensisinfection. Infect. Immun. 2003. 71: 3988–3994.
18 Anderson, R. J., Hannan, C. M., Gilbert, S. C., Laidlaw, S. M., Sheu, E. G.,Korten, S., Sinden, R. et al., Enhanced CD8+ T cell immune responses andprotection elicited against Plasmodium berghei malaria by prime boostimmunization regimens using a novel attenuated fowlpox virus. J. Immunol.2004. 172: 3094–3100.
19 Amanna, I. J., Slifka, M. K. and Crotty, S., Immunity and immunologicalmemory following smallpox vaccination. Immunol. Rev. 2006. 211: 320–337.
20 Ramsburg, E., Rose, N. F., Marx, P. A., Mefford, M., Nixon, D. F., Moretto,W. J., Montefiori, D. et al., Highly effective control of an AIDS viruschallenge in macaques by using vesicular stomatitis virus and modifiedvaccinia virus Ankara vaccine vectors in a single-boost protocol. J. Virol.2004. 78: 3930–3940.
21 Tapia, E., Perez-Jimenez, E., Lopez-Fuertes, L., Gonzalo, R., Gherardi, M.M. and Esteban, M., The combination of DNA vectors expressing IL-12 +IL-18 elicits high protective immune response against cutaneous leishma-niasis after priming with DNA-p36/LACK and the cytokines, followed by abooster with a vaccinia virus recombinant expressing p36/LACK. MicrobesInfect. 2003. 5: 73–84.
22 Gonzalo, R. M., del Real, G., Rodriguez, J. R., Rodriguez, D.,Heljasvaara, R., Lucas, P., Larraga, V. and Esteban, M., A heterologousprime-boost regime using DNA and recombinant vaccinia virus expressingthe Leishmania infantum P36/LACK antigen protects BALB/c mice fromcutaneous leishmaniasis. Vaccine 2002. 20: 1226–1231.
23 Lopez-Fuertes, L., Perez-Jimenez, E., Vila-Coro, A. J., Sack, F., Moreno,S., Konig, S. A., Junghans, C. et al., DNA vaccination with linearminimalistic (MIDGE) vectors confers protection against Leishmania majorinfection in mice. Vaccine 2002. 21: 247–257.
24 Dondji, B., Perez-Jimenez, E., Goldsmith-Pestana, K., Esteban, M. andMcMahon-Pratt, D., Heterologous prime-boost vaccination with the LACKantigen protects against murine visceral leishmaniasis. Infect. Immun. 2005.73: 5286–5289.
25 Babiuk, S., Babiuk, L. A. and van Drunen Littel-van den Hurk, S., DNAvaccination: A simple concept with challenges regarding implementation.Int. Rev. Immunol. 2006. 25: 51–81.
26 Stager, S., Alexander, J., Kirby, A. C., Botto, M., Rooijen, N. V., Smith, D.F., Brombacher, F. and Kaye, P. M.,Natural antibodies and complement areendogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat. Med.2003. 9: 1287–1292.
27 Mendez, S., Gurunathan, S., Kamhawi, S., Belkaid, Y., Moga, M. A.,Skeiky, Y. A., Campos-Neto, A. et al., The potency and durability of DNA-and protein-based vaccines against Leishmania major evaluated using low-dose, intradermal challenge. J. Immunol. 2001. 166: 5122–5128.
28 Scott, P., Immunologic memory in cutaneous leishmaniasis. Cell. Microbiol.2005. 7: 1707–1713.
29 Nishimura, T., Kitamura, H., Iwakabe, K., Yahata, T., Ohta, A., Sato, M.,Takeda, K. et al., The interface between innate and acquired immunity:Glycolipid antigen presentation by CD1d-expressing dendritic cells to NKTcells induces the differentiation of antigen-specific cytotoxic T lymphocytes.Int. Immunol. 2000. 12: 987–994.
30 Eberl, G. andMacDonald, H. R., Selective induction of NK cell proliferationand cytotoxicity by activated NKTcells. Eur. J. Immunol. 2000. 30: 985–992.
31 Silk, J. D., Hermans, I. F., Gileadi, U., Chong, T. W., Shepherd, D., Salio,M., Mathew, B. et al., Utilizing the adjuvant properties of CD1d-dependentNK T cells in T cell-mediated immunotherapy. J. Clin. Invest. 2004. 114:1800–1811.
32 Ishikawa, H., Hisaeda, H., Taniguchi, M., Nakayama, T., Sakai, T.,Maekawa, Y., Nakano, Y. et al., CD4(+) v(alpha)14 NKTcells play a crucialrole in an early stage of protective immunity against infection withLeishmania major. Int. Immunol. 2000. 12: 1267–1274.
33 Mattner, J., Donhauser, N., Werner-Felmayer, G. and Bogdan, C., NKTcells mediate organ-specific resistance against Leishmania major infection.Microbes Infect. 2006. 8: 354–362.
34 Mempel, M., Ronet, C., Suarez, F., Gilleron, M., Puzo, G., Van Kaer, L.,Lehuen, A. et al., Natural killer T cells restricted by the monomorphic MHCclass 1b CD1d1molecules behave like inflammatory cells. J. Immunol. 2002.168: 365–371.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 717
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

35 Montoya, C. J., Jie, H. B., Al-Harthi, L., Mulder, C., Patino, P. J., Rugeles,M. T., Krieg, A. M. et al., Activation of plasmacytoid dendritic cells withTLR9 agonists initiates invariant NKT cell-mediated cross-talk with myeloiddendritic cells. J. Immunol. 2006. 177: 1028–1039.
36 Marschner, A., Rothenfusser, S., Hornung, V., Prell, D., Krug, A.,Kerkmann, M., Wellisch, D. et al., CpG ODN enhance antigen-specific NKTcell activation via plasmacytoid dendritic cells. Eur. J. Immunol. 2005. 35:2347–2357.
37 Brutkiewicz, R. R., CD1d ligands: The good, the bad, and the ugly.J. Immunol. 2006. 177: 769–775.
38 Kronenberg, M., Toward an understanding of NKT cell biology: Progressand paradoxes. Annu. Rev. Immunol. 2005. 23: 877–900.
39 La Cava, A., Van Kaer, L. and Fu Dong, S., CD4+CD25+ Tregs and NKTcells: Regulators regulating regulators. Trends Immunol. 2006. 27: 322–327.
40 Jiang, S., Game, D. S., Davies, D., Lombardi, G. and Lechler, R. I.,Activated CD1d-restricted natural killer T cells secrete IL-2: Innate help forCD4+CD25+ regulatory T cells? Eur. J. Immunol. 2005. 35: 1193–1200.
41 Miyahira, Y., Katae, M., Takeda, K., Yagita, H., Okumura, K., Kobayashi,S., Takeuchi, T. et al., Activation of natural killer T cells by alpha-galactosylceramide impairs DNA vaccine-induced protective immunityagainst Trypanosoma cruzi. Infect. Immun. 2003. 71: 1234–1241.
42 Gonzalez-Aseguinolaza, G., de Oliveira, C., Tomaska, M., Hong, S.,Bruna-Romero, O., Nakayama, T., Taniguchi, M. et al., a-Galactosylcer-e-activated Va14 natural killer T cells mediate protection against murinemalaria. Proc. Natl. Acad. Sci. USA 2000. 97: 8461–8466.
43 Schmieg, J., Yang, G., Franck, R. W. and Tsuji, M., Superior protectionagainst malaria and melanoma metastases by a C-glycoside analogue of thenatural killer Tcell ligand alpha-galactosylceramide. J. Exp. Med. 2003. 198:1631–1641.
44 Gonzalez-Aseguinolaza, G., Van Kaer, L., Bergmann, C. C., Wilson, J. M.,Schmieg, J., Kronenberg, M., Nakayama, T. et al., Natural killer T cellligand alpha-galactosylceramide enhances protective immunity induced bymalaria vaccines. J. Exp. Med. 2002. 195: 617–624.
45 Gonzalo, R. M., Rodriguez, J. R., Rodriguez, D., Gonzalez-Aseguinolaza,G., Larraga, V. and Esteban, M., Protective immune response againstcutaneous leishmaniasis by prime/booster immunization regimens withvaccinia virus recombinants expressing Leishmania infantum p36/LACK andIL-12 in combination with purified p36. Microbes Infect. 2001. 3: 701–711.
46 Eberl, G., Brawand, P. and MacDonald, H. R., Selective bystanderproliferation of memory CD4+ and CD8+ T cells upon NK T or T cellactivation. J. Immunol. 2000. 165: 4305–4311.
47 Kedzierski, L., Zhu, Y. and Handman, E., Leishmania vaccines: Progressand problems. Parasitology 2006. 133 Suppl: S87–S112.
48 Perez-Jimenez, E., Kochan, G., Gherardi, M. M. and Esteban, M., MVA-LACK as a safe and efficient vector for vaccination against leishmaniasis.Microbes Infect. 2006. 8: 810–822.
49 Munz, C., Steinman, R.M. and Fujii, S.,Dendritic cell maturation by innatelymphocytes: Coordinated stimulation of innate and adaptive immunity.J. Exp. Med. 2005. 202: 203–207.
50 Hermans, I. F., Silk, J. D., Gileadi, U., Salio, M., Mathew, B., Ritter, G.,Schmidt, R. et al., NKT cells enhance CD4+ and CD8+ T cell responses tosoluble antigen in vivo through direct interaction with dendritic cells.J. Immunol. 2003. 171: 5140–5147.
51 Fujii, S., Shimizu, K., Hemmi, H., Fukui, M., Bonito, A. J., Chen, G.,Franck, R. W. et al., Glycolipid alpha-C-galactosylceramide is a distinctinducer of dendritic cell function during innate and adaptive immuneresponses of mice. Proc. Natl. Acad. Sci. USA 2006. 103: 11252–11257.
52 Chen, Y. G., Choisy-Rossi, C. M., Holl, T. M., Chapman, H. D., Besra, G. S.,Porcelli, S. A., Shaffer, D. J. et al., Activated NKT cells inhibit autoimmunediabetes through tolerogenic recruitment of dendritic cells to pancreaticlymph nodes. J. Immunol. 2005. 174: 1196–1204.
53 Jabbari, A. andHarty, J. T., Simultaneous assessment of antigen-stimulatedcytokine production and memory subset composition of memoryCD8 T cells. J. Immunol. Methods 2006. 313: 161–168.
54 Masopust, D., Murali-Krishna, K. and Ahmed, R., Quantitating themagnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cellresponse: It is even bigger than we thought. J. Virol. 2007. 81: 2002–2011.
55 Lutjen, S., Soltek, S., Virna, S., Deckert, M. and Schluter, D., Organ- anddisease-stage-specific regulation of Toxoplasma gondii-specific CD8-T-cellresponses by CD4 T cells. Infect. Immun. 2006. 74: 5790–5801.
56 Hu, H. M., Winter, H., Urba, W. J. and Fox, B. A., Divergent roles for CD4+
T cells in the priming and effector/memory phases of adoptive immu-notherapy. J. Immunol. 2000. 165: 4246–4253.
57 Janssen, E. M., Lemmens, E. E., Wolfe, T., Christen, U., von Herrath, M.G. and Schoenberger, S. P., CD4+ T cells are required for secondaryexpansion and memory in CD8+ T lymphocytes. Nature 2003. 421:852–856.
58 Anderson, C. F., Oukka, M., Kuchroo, V. J. and Sacks, D.,CD4(+)CD25(–)Foxp3(–) Th1 cells are the source of IL-10-mediatedimmune suppression in chronic cutaneous leishmaniasis. J. Exp. Med.2007. 204: 285–297.
59 Belkaid, Y., Hoffmann, K. F., Mendez, S., Kamhawi, S., Udey, M. C.,Wynn, T. A. and Sacks, D. L., The role of interleukin (IL)-10 in thepersistence of Leishmania major in the skin after healing and the therapeuticpotential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 2001.194: 1497–1506.
60 Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M. and Sacks, D. L.,CD4+CD25+ regulatory T cells control Leishmania major persistence andimmunity. Nature 2002. 420: 502–507.
61 Stober, C. B., Lange, U. G., Roberts,M. T., Alcami, A. and Blackwell, J. M.,IL-10 from regulatory T cells determines vaccine efficacy in murineLeishmania major infection. J. Immunol. 2005. 175: 2517–2524.
62 Kar, S., Metz, C. andMcMahon-Pratt, D., CD4+ Tcells play a dominant rolein protection against New World leishmaniasis induced by vaccination withthe P-4 amastigote antigen. Infect. Immun. 2005. 73: 3823–3827.
63 Colmenares, M., Kima, P. E., Samoff, E., Soong, L. and McMahon-Pratt,D., Perforin and gamma interferon are critical CD8+ T-cell-mediatedresponses in vaccine-induced immunity against Leishmania amazonensisinfection. Infect. Immun. 2003. 71: 3172–3182.
64 Gurunathan, S., Wu, C. Y., Freidag, B. L. and Seder, R. A., DNA vaccines:A key for inducing long-term cellular immunity. Curr. Opin. Immunol. 2000.12: 442–447.
65 Kirman, J. R. and Seder, R. A., DNA vaccination: The answer to stable,protective T-cell memory? Curr. Opin. Immunol. 2003. 15: 471–476.
66 Melby, P. C., Recent developments in leishmaniasis. Curr. Opin. Infect. Dis.2002. 15: 485–490.
67 Melby, P. C., Vaccination against cutaneous leishmaniasis: Current status.Am. J. Clin. Dermatol. 2002. 3: 557–570.
68 van Drunen Littel-van den Hurk, S., Babiuk, S. L. and Babiuk, L. A.,Strategies for improved formulation and delivery of DNA vaccines toveterinary target species. Immunol. Rev. 2004. 199: 113–125.
69 Hill, A. V., Reece, W., Gothard, P., Moorthy, V., Roberts, M., Flanagan, K.,Plebanski, M. et al.,DNA-based vaccines for malaria: A heterologous prime-boost immunisation strategy. Dev. Biol. (Basel) 2000. 104: 171–179.
70 Huygen, K., Plasmid DNA vaccination. Microbes Infect. 2005. 7: 932–938.
71 Gherardi, M. M., Ramirez, J. C. and Esteban, M., Towards a newgeneration of vaccines: The cytokine IL-12 as an adjuvant to enhance cellularimmune responses to pathogens during prime-booster vaccination regi-mens. Histol. Histopathol. 2001. 16: 655–667.
72 Hanke, T., McMichael, A. J. and Dorrell, L., Clinical experience withplasmid DNA- and modified vaccinia virus Ankara-vectored humanimmunodeficiency virus type 1 clade A vaccine focusing on T-cell induction.J. Gen. Virol. 2007. 88: 1–12.
73 Gilbert, S. C., Moorthy, V. S., Andrews, L., Pathan, A. A., McConkey, S. J.,Vuola, J. M., Keating, S. M. et al., Synergistic DNA-MVA prime-boostvaccination regimes for malaria and tuberculosis. Vaccine 2006. 24:4554–4561.
74 Parekh, V. V., Singh, A. K., Wilson, M. T., Olivares-Villagomez, D.,Bezbradica, J. S., Inazawa, H., Ehara, H. et al., Quantitative andqualitative differences in the in vivo response of NKT cells to distinct alpha-and beta-anomeric glycolipids. J. Immunol. 2004. 173: 3693–3706.
75 Okai, M., Nieda, M., Tazbirkova, A., Horley, D., Kikuchi, A., Durrant, S.,Takahashi, T. et al., Human peripheral blood Valpha24+ Vbeta11+ NKT
Blaise Dondji et al. Eur. J. Immunol. 2008. 38: 706–719718
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

cells expand following administration of alpha-galactosylceramide-pulseddendritic cells. Vox Sang. 2002. 83: 250–253.
76 Giaccone, G., Punt, C. J., Ando, Y., Ruijter, R., Nishi, N., Peters, M., vonBlomberg, B. M. et al., A phase I study of the natural killer T-cell ligandalpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin.Cancer Res. 2002. 8: 3702–3709.
77 Gorbachev, A. V. and Fairchild, R. L., Activated NKTcells increase dendriticcell migration and enhance CD8+ T cell responses in the skin. Eur.J. Immunol. 2006. 36: 2494–2503.
78 Rachamim, N. and Jaffe, C. L., Pure protein from Leishmania donovaniprotects mice against both cutaneous and visceral leishmaniasis. J. Immunol.1993. 150: 2322–2331.
79 Stager, S., Smith, D. F. and Kaye, P. M., Immunization with a recombinantstage-regulated surface protein from Leishmania donovani induces protec-tion against visceral leishmaniasis. J. Immunol. 2000. 165: 7064–7071.
80 Squires, K. E., Schreiber, R. D., McElrath, M. J., Rubin, B. Y., Anderson, S.L. and Murray, H. W., Experimental visceral leishmaniasis: Role ofendogenous IFN-gamma in host defense and tissue granulomatous response.J. Immunol. 1989. 143: 4244–4249.
81 Castellino, F. and Germain, R. N., Cooperation between CD4+ and CD8+
T cells: When, where, and how. Annu. Rev. Immunol. 2006. 24: 519–540.
82 Herath, S., Kropf, P. and Muller, I., Cross-talk between CD8(+) andCD4(+) Tcells in experimental cutaneous leishmaniasis: CD8(+) Tcells arerequired for optimal IFN-gamma production by CD4(+) T cells. ParasiteImmunol. 2003. 25: 559–567.
83 Dumonteil, E., Andrade-Narvarez, F., Escobedo-Ortegon, J., Ramirez-Sierra, M. J., Valencia-Pacheco, G., Flores-Serrano, A., Canto-Lara, S.and Arjona-Torres, A., Comparative study of DNA vaccines encodingvarious antigens against Leishmania mexicana. Dev. Biol. (Basel) 2000. 104:135–141.
84 Ahmed, S., Colmenares, M., Soong, L., Goldsmith-Pestana, K., Muns-termann, L., Molina, R. and McMahon-Pratt, D., Intradermal infectionmodel for pathogenesis and vaccine studies of murine visceral leishmaniasis.Infect. Immun. 2003. 71: 401–410.
85 Chakrabarti, S., Brechling, K. and Moss, B., Vaccinia virus expressionvector: Coexpression of beta-galactosidase provides visual screening ofrecombinant virus plaques. Mol. Cell. Biol. 1985. 5: 3403–3409.
86 Gomez, C. E., Rodriguez, D., Rodriguez, J. R., Abaitua, F., Duarte, C. andEsteban, M., Enhanced CD8+ T cell immune response against a V3 loopmulti-epitope polypeptide (TAB13) of HIV-1 Env after priming with purifiedfusion protein and booster with modified vaccinia virus Ankara (MVA-TAB)recombinant: A comparison of humoral and cellular immune responses withthe vaccinia virus Western Reserve (WR) vector. Vaccine 2001. 20: 961–971.
87 Soong, L., Duboise, S. M., Kima, P. and McMahon-Pratt, D., Leishmaniapifanoi amastigote antigens protect mice against cutaneous leishmaniasis.Infect. Immun. 1995. 63: 3559–3566.
88 Green, S. J., Aniagolu, J., and Raney, J. J.,Oxidativemetabolism of murinemacrophages. In Coligan J. E. (Ed.) Current protocols in immunology. JohnWiley & Sons, New York 1994, pp 14.5.1–14.5.11.
Eur. J. Immunol. 2008. 38: 706–719 Immunity to infection 719
f 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu












![Poly[di(carboxylatophenoxy)phosphazene] is a potent adjuvant for intradermal immunization](https://static.fdokumen.com/doc/165x107/6335c6c4a1ced1126c0af097/polydicarboxylatophenoxyphosphazene-is-a-potent-adjuvant-for-intradermal-immunization.jpg)







