
Integrin D2 Is Dynamically Expressed by Inflamed Macrophages and Alters the Natural History of...
12
of December 2, 2013. This information is current as Natural History of Lethal Systemic Infections Inflamed Macrophages and Alters the Is Dynamically Expressed by 2 β D α Integrin Zimmerman Andrew S. Weyrich, Hugo C. Castro-Faria-Neto and Guy A. Danielle de Oliveira Nascimento, Adriana Vieira-de-Abreu, M. Prescott, Valber S. Frutuoso, Fabio C. Amendoeira, Stafforini, Estelle S. Harris, Thomas M. McIntyre, Stephen Yasunari Miyazaki, Michaeline Bunting, Diana M. http://www.jimmunol.org/content/180/1/590 2008; 180:590-600; ; J Immunol References http://www.jimmunol.org/content/180/1/590.full#ref-list-1 , 35 of which you can access for free at: cites 62 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2008 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on December 2, 2013 http://www.jimmunol.org/ Downloaded from by guest on December 2, 2013 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Integrin D2 Is Dynamically Expressed by Inflamed Macrophages and Alters the Natural History of...

of December 2, 2013.This information is current as
Natural History of Lethal Systemic InfectionsInflamed Macrophages and Alters the
Is Dynamically Expressed by2βDαIntegrin
ZimmermanAndrew S. Weyrich, Hugo C. Castro-Faria-Neto and Guy A.Danielle de Oliveira Nascimento, Adriana Vieira-de-Abreu, M. Prescott, Valber S. Frutuoso, Fabio C. Amendoeira,Stafforini, Estelle S. Harris, Thomas M. McIntyre, Stephen Yasunari Miyazaki, Michaeline Bunting, Diana M.
http://www.jimmunol.org/content/180/1/5902008; 180:590-600; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/1/590.full#ref-list-1
, 35 of which you can access for free at: cites 62 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 2, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from

Integrin �D�2 Is Dynamically Expressed by InflamedMacrophages and Alters the Natural History of LethalSystemic Infections1
Yasunari Miyazaki,2,3* Michaeline Bunting,2,4* Diana M. Stafforini,†‡ Estelle S. Harris,*‡
Thomas M. McIntyre,5‡§ Stephen M. Prescott,6†‡ Valber S. Frutuoso,¶ Fabio C. Amendoeira,¶
Danielle de Oliveira Nascimento,¶ Adriana Vieira-de-Abreu,¶ Andrew S. Weyrich,*‡
Hugo C. Castro-Faria-Neto,¶ and Guy A. Zimmerman7*‡
The leukocyte integrins have critical roles in host defense and inflammatory tissue injury. We found that integrin �D�2, a novel butlargely uncharacterized member of this family, is restricted to subsets of macrophages and a small population of circulating leukocytesin wild-type mice in the absence of inflammatory challenge and is expressed in regulated fashion during cytokine-induced macrophagedifferentiation in vitro. �D�2 is highly displayed on splenic red pulp macrophages and mediates their adhesion to local targets, iden-tifying key functional activity. In response to challenge with Plasmodium berghei, a malarial pathogen that models systemic infection andinflammatory injury, new populations of �D
� macrophages evolved in the spleen and liver. Unexpectedly, targeted deletion of �D
conferred a survival advantage in P. berghei infection over a 30-day observation period. Mechanistic studies demonstrated that theincreased survival of �D
�/� animals at these time points is not attributed to differences in magnitude of anemia or parasitemia or toalterations in splenic microanatomy, each of which is a key variable in the natural history of P. berghei infection, and indicated that analtered pattern of inflammatory cytokines may contribute to the difference in mortality. In contrast to the outcome in malarial challenge,death of �D
�/� animals was accelerated in a model of Salmonella sepsis, demonstrating differential rather than stereotyped roles for�D�2 in systemic infection. These studies identify previously unrecognized and unique activities of �D�2, and macrophages that expressit, in host defense and injury. The Journal of Immunology, 2008, 180: 590–600.
I ntegrins are plasma membrane heterodimers that are broadlydistributed on metazoan cells and mediate critical functions,including adhesion, homing, signaling, and gene expression.
The leukocyte integrin subfamily (also called �2 or CD11/CD18integrins) is expressed on myeloid and lymphoid leukocytes andtheir precursors and consists of four members (1): �L�2
(CD11aCD18; LFA-1), �M�2 (CD11b/CD18; MAC-1; CR3),�x�2 (CD11c/CD18), and �D�2 (CD11d/CD18). Leukocyte inte-grins are required for host defense against invasion by many patho-gens and for wound surveillance and repair, demonstrated byheritable deficiency syndromes that cause recurrent and often life-threatening infections in humans and domesticated animals (2).Experimental models in which leukocyte integrins are geneticallydeleted or are blocked demonstrate critical and complex activitiesrelevant to this function (1, 3–6). In contrast, unregulated accu-mulation and signaling of leukocytes mediated by �2 integrins alsocontribute to inflammatory tissue injury (1, 2, 6, 7).
Integrin �D�2 is the most recently discovered �2 integrin (1, 8).Molecular characterization of the �D subunit suggested that �D�2
has novel activation pathways and modes of regulation (8). �D�2
recognizes ICAM-3, VCAM-1, and several other ligands in assayswith primary and surrogate cells (8–11), although adhesive inter-actions in vivo have not been identified. Sequence similarity of theI domains of �D and �M (8) predicted this diversity in ligandrecognition: the I domain is critical in binding interactions of in-tegrins, and �M�2 also has multiple ligand partners in experimentsusing transfected cell lines (1, 10, 11). Limited observations sug-gest that �D�2 is preferentially expressed by macrophages in nor-mal human tissues and in clinical syndromes of dysregulated in-flammation, including atherosclerosis, rheumatoid arthritis, andacute lung inflammation (Refs. 8, 12, and 13 and our unpublished
*Program in Human Molecular Biology and Genetics, †Huntsman Cancer Institute,‡Department of Internal Medicine, and §Department of Pathology, University of Utah,Salt Lake City, UT 84112; and ¶Laboratorio de Immunofarmacologia, Departmentode Fisiologia e Farmacodinamica, Instituto Oswaldo Cruz, Fundacao Oswaldo Cruz,Rio de Janeiro, Brazil
Received for publication March 17, 2007. Accepted for publication October 26, 2007.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grant HL44525 (toG.A.Z.), a National Institutes of Health Special Center of Research in Acute Respi-ratory Distress Syndrome (P50 H150153), an Asthma Research Center Award fundedby the American Lung Association, the Margolis Foundation, and an EstablishedInvestigator Award from the American Heart Association (to A.S.W.).2 Y.M. and M.B. contributed equally to this work. M.B. accomplished cloning of themouse �D gene, generation of �D-deficient animals, initial genotype-phenotype cor-relation, and development of in vitro models of �D expression. Y.M. performed ex-tensive characterization of �D�2 in murine tissues and additional genotype-phenotypecorrelation, developed in vitro models of cellular adhesion, characterized �D expres-sion in in vivo disease models, and compiled major portions of the manuscript text.3 Current address: Department of Integrated Pulmonology Medicine, Tokyo Medicaland Dental University, 1-5-45 Yushima Bunkyo-Ku, Tokyo, Japan, 233-0015.4 Current address: Invitrogen Corporation, 1610 Faraday Avenue, Carlsbad, CA92008.5 Current address: Department of Cell Biology, NN1-28, Lerner Research Institute,Cleveland, OH 44195.6 Current address: Oklahoma Medical Research Foundation, 825 Northeast 13thStreet, Oklahoma City, OK 73104.
7 Address correspondence and reprint requests to Dr. Guy A. Zimmerman, Programin Human Molecular Biology and Genetics, University of Utah, 15 North 2030 East,Building 533, Room 4220, Salt Lake City, UT 84112. E-mail address: [email protected]
Copyright © 2007 by The American Association of Immunologists, Inc. 0022-1767/07/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

data), and that it is involved in immune regulation and inflamma-tory tissue injury in rodent models (14–16). Nevertheless, the dis-tribution, regulation, and activities of �D�2 in physiologic andpathologic inflammation are largely undefined, and phenotypes inmice made genetically deficient in �D have not been reported (1,3, 16). In this study, we examined the expression and functions of�D�2 in naive conditions and in response to inflammatory stimu-lation in models of pathologic infectious syndromes. We foundthat it is selectively expressed by subsets of splenic red pulp, thy-mic, and marrow macrophages in wild-type (WT)8 animals in thebasal state. This pattern of expression suggested roles in systemicinfectious challenge. We found that new populations of �D
� mac-rophages emerge in response to infection with Plasmodiumberghei, a blood-borne malarial protozoan recognized by splenicred pulp macrophages, demonstrating that �D�2 is dynamicallymodulated in systemic inflammation. Targeted deletion of �D didnot cause a spontaneous phenotype but, unexpectedly, conferred asurvival advantage in lethal P. berghei infection. In contrast, de-letion of �D�2 reduced survival in animals infected with Salmo-nella typhimurium, an intracellular bacterial pathogen that alsocauses a lethal systemic infection in mice and is recognized bysplenic marginal zone macrophages and other macrophage subsets.Our findings demonstrate unique functional roles for �D�2 and themacrophage populations that express it in innate responses to mi-crobial invasion and systemic infectious syndromes.
Materials and MethodsTargeting vector construction, functional disruption of �D byhomologous recombination, and generation of �D
�/� mice
Genomic clones were isolated from a murine 129Sv genomic library usingprobes specific for the �D I domain, which is critical for ligand recognition(1, 10, 11), and targeted deletion of �D was accomplished using previouslydescribed strategies (17–19) (Fig. 1).
Immunohistochemistry
The following were used as primary mAbs in immunocytochemical reac-tions: �D, 205c, and 279F (hamster IgG; ICOS) (14); F4/80, A3-1 (ratIgG2; Serotec); MARCO, ED31 (rat IgG1; Serotec); and MOMA-1 (ratIgG2; Serotec) (20). Tissues from various organs, usually from 8- to 12-wk-old animals in studies of distribution of �D�2 in WT mice, were frozenin Tissue-Tek (Sakura Finetek) and 8-�m cryostat sections were depositedon Superfrost Plus slides (Fisher Scientific). The sections were then fixedin cold acetone for 10 min. Immunoperoxidase staining was based on apreviously described method (21) (Fig. 2). For immunofluorescent staining,sections were blocked with 5% normal goat serum, 1% BSA, and avidinsolution (Vector Laboratories) for 30 min and subsequently incubated withprimary Abs overnight at 4°C. Biotin-conjugated goat anti-hamster Ab wasapplied at a 1/1000 dilution and incubated for 30 min followed by incu-bation with Alexa 488-conjugated streptavidin and Alexa 568-conjugatedanti-rat IgG (Molecular Probes) for 45 min. The sections were then layeredwith anti-fade medium (Biomeda) and sealed with a coverslip. Fluorescentstaining was analyzed using a FluoView 300 microscope (Olympus) in theUniversity of Utah Health Science Center’s Core Cell Imaging Facility.
Isolation of mouse splenocytes and F4/80-positive splenicmacrophages
Single-cell suspensions from freshly isolated spleen were obtained as de-scribed previously (22), with minor modifications. F4/80-positive splenicmacrophages were further isolated using an AutoMACS cell sorter andpositive selection using PE-conjugated anti-F4/80 Ab (Serotec) and
8 Abbreviations used in this paper: WT, wild type; PRBC, parasitized RBC; iNOS,inducible NO synthase.
FIGURE 1. Functional disruption of �D by homologous recombination. A, The murine �D genomic locus containing exons 1–10 is represented (WT).Genomic clones were isolated from a 129Sv mouse genomic � DNA library using oligonucleotide probes specific to the �D I domain. The largest (clone3.1) was 14 kb in length and contained exons 1–6 as determined by Southern blot analysis and nucleic acid sequencing. A second 10-kb genomic clone(4.1) that overlapped with the 3� end of 3.1 and contained exons 6–10 was also isolated. A 7.5-kb genomic fragment containing exons 1–5 and a 5� regionof exon 6 were cloned from 3.1 and inserted upstream of three in-frame stop codons in a polylinker modified pBlueScript vector. A 3-kb Xmn1-BglIIfragment containing the 3� end of exons 6 and 7 was cloned downstream of this fragment to generate a novel �D allele leading to termination of translationin the I domain following residue 153(Ile). The novel �D allele was cloned between two herpes simplex TK genes and a floxed neomycin selection cassettewas inserted 3� to the stop codons in exon 6 to generate the targeting vector. The targeting vector was linearized with XhoI and electroporated into TC1embryonic stem cells. Recombinant embryonic stem cells were selected in the presence of G418 and FIAU as previously described (18) and were identifiedby Southern blot analysis using BglII and a 5� flanking BamHi/BgIII probe located outside the targeted region. Positive cell lines were confirmed using XmnIdigestion and hybridization to a probe corresponding to the neomycin gene. A single recombinant cell line (1e-1) was used for blastocyst injection to createrecombinant mice. The floxed neomycin selection cassette was removed by mating to a transgenic mouse line that expresses Cre recombinase (19). Micecontaining �D
loxP and WT alleles were genotyped by PCR using isolated tail DNA. The forward (5�-GGACCCCAGGACACAGTTGAG-3�) and reverse(5�-CACAGGCCACAGTGTACAGTATT-3�) primers amplify a 268-bp band including the inserted loxP site from the recombinant allele and 215 bp fromthe WT allele. The recombinant mouse line was subsequently backcrossed and maintained on a C57BL/6 background. B, Southern blot analysis of genomicDNA isolated from WT embryonic stem cells and the �D-recombinant embryonic stem cell line (1e-1). Genomic DNA digested with BglII was probed witha 600-bp BamH1-BglII fragment from the �D genomic clone. This 5� probe hybridized to both the 11-kb fragment derived from the WT allele and the 8-kbfragment corresponding to the recombinant allele. The genomic structure of the 3� end of the �Dneo allele was confirmed in the 1e-1 clone by hybridizationof the targeting vector-specific neomycin probe to the 6.5-kb XmnI genomic fragment.
591The Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

anti-PE magnetic beads according to the manufacturer’s instructions(Miltenyi Biotec).
Flow cytometric analysis
Isolated F4/80-positive splenocytes or cultured M1 cells were incubated onice for 30 min in the presence of Alexa 488-conjugated mAb 205c or acontrol Ab. Cells were washed with FACS medium (HBSS without Ca2�
and Mg2�/0.1% NaN3/0.5% human serum albumin) and fixed in 1% para-formaldehyde. They were then analyzed by flow cytometry as previouslydescribed (23), with minor modifications.
Culture of murine M1 cells
The murine M1 myeloid leukemic cell line was obtained from AmericanType Culture Collection. Macrophage differentiation was induced as de-scribed elsewhere (24). PCR assays for �D mRNA were done essentially asdescribed previously (17). The primers used (5�-CAATCCCTGCTAC
CATGCCAGA-3� and 5�-CTCCTGTAGGGCAGTGGGTTC-3�) ampli-fied a 490-bp product. Immunocytochemical analysis was accomplished aspreviously outlined (25).
Modified Stamper-Woodruff adhesion assay
An assay for in vitro binding of splenic macrophages to freshly isolatedspleen sections was developed based on previously described methods (26,27). All steps were conducted in a cold room or on ice. Incubations weredone using freshly cut, unfixed, 10-�m sections of WT or �D knockoutspleen prepared using a cryostat (Leica CM 3050). F4/80-positive spleno-cytes were prepared and labeled with PE, suspended in RPMI 10 at a finalconcentration of 1 � 107 cells/ml, and layered over each section. The slideswere immediately placed on a rotating platform at 60 rpm. After incubationfor 30 min, the sections were washed by dipping the slides repeatedly incold PBS and fixed for 10 min in 2% paraformaldehyde at 4°C. The sec-tions were then washed again and mounted on glass slides with anti-fademedium. Images of the adherent PE-positive cells detected by fluorescentmicroscopy were captured and analyzed using a FluoView 300 microscopeand the number of PE-positive cells bound to each section was countedusing NIH ImageJ.
Characterization of �D�2-deficient and WT mice in models ofsystemic infectious challenge
The experiments in these studies were approved by the Animal WelfareCommittee of the Oswaldo Cruz Institute and by the University of UtahAnimal Care and Use Committee. C57BL/6 WT and �D
�/�mice from theOswaldo Cruz Foundation breeding unit weighing 20–25g were used formost studies. A/J, SJL/J, and Swiss mice from B&K were also used insome experiments. The animals were kept at constant temperature (25°C)with free access to chow and water in a room with a 12-h light/dark cycle.
C57BL/6 mice were infected by an i.p. injection of 200 �l of PBScontaining 105 RBC parasitized with the Pasteur strain of P. bergheiANKA (28). In parallel experiments, we established that this inoculum islethal in a high percentage of animals after 20–30 days of infection, con-sistent with studies using other P. berghei strains (29). Samples of spleenand liver were obtained at different times postinfection of WT mice aftersacrificing the animals in a CO2 chamber. The tissue was frozen and lateranalyzed by immunohistochemistry. For survival studies, groups of 10 WTand 10 �D
�/� mice were studied together. Surviving animals were sacri-ficed at 30 days postinfection. The numbers of parasitized RBC and thehematocrits were determined as previously described (28) in separategroups of animals.
For studies of Salmonella infection, Salmonella enterica serovar typhi-murium ISC 5302/2004, a generous gift from the Department of Microbi-ology of the Instituto Oswaldo Cruz, was cultured in Luria-Bertani broth(Guria Broth Miller; Sigma-Aldrich) for 16–18 h at 37°C to obtain sta-tionary growth phase cultures. The bacteria were then centrifuged (200rpm) for 10 min at 4°C and the pellets were resuspended in PBS to an ODof 0.1 at 660 nm, corresponding to 108 CFU/ml. WT and �D
�/� mice wereinfected by i.p. injection of 200 �l of bacterial suspension (105 CFU/mouse). Control WT mice were sham injected (saline alone) in parallel.Survival was monitored for 10 or 15 days, at which time animals remainingalive were sacrificed.
Log rank tests were used for comparison of Kaplan-Meier survival plotsin studies of P. berghei and Salmonella infection. A p � 0.05 was con-sidered statistically significant for differences in the mortality data.
Multiplex cytokine and IL-12 determinations
Initial screening analysis of multiple cytokines in the serum of three �D�/�
and three WT mice was done using a Luminex system (Bio-Plex; Bio-Rad).Subsequent measurements of IL-12(p40) levels in samples from largergroups of �D
�/� and WT animals were done by commercial ELISA ac-cording to the manufacturer’s instructions (R&D Systems). Differences inmean IL-12 levels were examined by one-way ANOVA.
ResultsGeneration and characterization of �D knockout mice
�D-deficient (�D�/�) mice were generated by targeted deletion of
the I domain and excision of the “floxed” selection cassette toavoid spurious in vivo consequences (Ref. 17 and Fig. 1). Progenycontaining the �D
loxP allele were backcrossed and maintained onthe C57BL/6 background. Heterozygous and homozygous animalswere born at expected Mendelian ratios and did not display grossphysical abnormalities. Homozygous animals were fertile and did
FIGURE 2. �D�2 is expressed by distinct populations of macrophagesin bone marrow, thymus, and spleen. Frozen sections of freshly isolatedtissues were stained with mAb 205c using an immunoperoxidase detectionmethod. Tissues from 8- to 12-wk-old C57BL/6 mice were frozen in Tis-sue-Tek (Sakura Finetek) and 8-�m cryostat sections were deposited onSuperfrost Plus slides (Fisher Scientific). The sections were then fixed incold acetone for 10 min. Endogenous tissue peroxidase was eliminated byincubation with 0.01% hydrogen peroxide for 15 min. The sections wereblocked by incubating with 5% normal goat serum, 1% BSA and avidinsolution (Vector Laboratories) for 30 min, and primary Abs (hamster mAbs205c or 279f raised against mouse �D) were applied to the slides andincubated overnight at 4°C. A secondary Ab (biotin-conjugated goat anti-hamster; Jackson ImmunoResearch Laboratories) was applied at a 1/1000dilution and incubated for 30 min. Negative controls were performed byomitting the primary Ab or by applying an irrelevant isotype-matched pri-mary Ab as a control. ABC reagent was prepared according to the manu-facturer’s instructions (Vector Laboratories) and applied to the sections for30 min. Staining was developed using a NovaRed kit (Vector Laborato-ries), followed by counterstaining with Gill’s hematoxylin, dehydration,and mounting using Cytoseal 60 medium (VWR). In each pair of figures,the panel on the left is magnified �40 and on the right �800. Thesedistributions of �D are representative of multiple studies. A and B, Bonemarrow; C and D, thymus; E and F, spleen; WP, white pulp; RP, red pulp.
592 �D�2, MACROPHAGES, AND SYSTEMIC INFECTIONS
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

not have an increased incidence of spontaneous infection. Leuko-cyte counts and morphology were similar between WT and ho-mozygous mutant animals. The mean weight of spleens from 10-to 11-wk-old �D
�/� mice was greater than that of WT animals(105 vs 70 mg, n � 5 of each genotype), but the spleens were ofsimilar size at 17–18 wk of age (90 vs 85 mg, n � 4).
Integrin �D�2 is constitutively expressed by limited subsets ofleukocytes in the absence of infectious challenge
A small population (�1%) of peripheral blood leukocytes fromWT mice of C57BL/6, A/J, SJL/J, and Swiss backgrounds stainedpositively for �D�2. Splenic macrophages were used as controls inthe staining reactions (see below and Fig. 2, E and F). This featurewas unexpected since other �2 integrins are highly expressed oncirculating mouse leukocytes and are involved in their emigrationfrom the blood in response to inflammatory stimuli (5, 6) andbecause �D�2 is more broadly displayed on human leukocytes(Ref. 8 and our unpublished observations). In previous studies, �D
was reported to be absent from circulating murine leukocytes (16)and to be present on only a small fraction of circulating canineleukocytes (30).
When we examined tissues, �D staining was restricted to cells ofthe splenic red pulp, thymus, and bone marrow in adult WTC57BL/6 mice (Figs. 2 and 3 and Table I). By flow cytometry,�D
� splenocytes were positive for �2 (CD18), as expected (1, 3).�D�2 is also expressed in rat splenic red pulp (data not shown) andin the red pulp of canine and human spleen (8, 30), indicatingthat its localization in this domain is conserved across species.Scattered groups of cells in the bone marrow and thymus of WTmice express �D at lower staining intensity compared with thatin spleen but with similar cellular morphology (Table I and Fig.2). These cells likely represent specific macrophage populations(see below). There was no staining of tissues from �D
�/� mice,documenting specificity of the immunohistochemical reactionsand null phenotype of the �D
�/� animals (Table I, Fig. 3F, anddata not shown).
Specific populations of splenic macrophages differentiallyexpress integrin �D�2
When splenocytes were first sorted based on their display of F4/80,a determinant that identifies murine monocytes and subpopulationsof macrophages in normal and inflamed tissues (20, 31, 32), �80%were also positive for �D (Fig. 3C). Consistent with the flowcytometric results, the majority of red pulp splenocytescostained for �D and F4/80 (Fig. 3, A, B, and K), although asmall number of cells were brightly positive for one or the othermarker. We detected F4/80 in the absence of �D�2 in WT liverKupffer cells, alveolar macrophages, and peripheral bloodmonocytes (data not shown) and in red pulp splenocytes from�D�2
�/� mice (Fig. 3F), demonstrating differential regulationof the two surface factors.
The majority of �D� red pulp splenocytes also stained for mac-
rosialin (Fig. 3D), the murine ortholog of human CD68, confirm-ing their identity as macrophages (20). CD68 and �D are alsocoexpressed on a subset of macrophages in human atherosclerotictissue (8). Costaining for MARCO, a scavenger receptor that iden-tifies the splenic marginal zone macrophage, and MOMA-1, whichidentifies the marginal metallophilic macrophage (20, 33), demon-strated distinct populations of cells and localization patterns (Fig.3, G and I). Thus, �D�2 is expressed by a specific population ofmacrophages in the red pulp that is distinct from the marginal zonemacrophage subsets. Each macrophage subtype has particular roles inblood surveillance, antimicrobial defense, and organization of splenicmicroarchitecture, and is influenced by unique differentiation and re-
tention signals (34). Analysis of expression of other � subunits(�M,�L,�X) indicated that �D�2 is the dominant �2 integrin het-erodimer on red pulp macrophages (Fig. 3E and data not shown).
FIGURE 3. �D�2 is expressed by a unique population of splenic redpulp macrophages that is altered in P. berghei infection. Frozen sections offreshly isolated spleen from WT mice were costained using anti-�D mAb(mAb 205c, green fluorescent labeling) and mAb against F4/80 (A and B),macrosialin (D), or �m (E; red labeling). F, Spleen sections from �D
�/�
mice were costained for �D and F4/80 as in A and B. The staining patternsshown are representative of tissue from multiple WT and �D
�/� mice. C,Splenocytes were selected based on binding of PE-labeled anti-F4/80 andexamined for expression of �D by flow cytometry using mAb 205c con-jugated to Alexa 488. A similar pattern was seen in two additional exper-iments. G–L, Frozen sections of spleen from control (G, I, and K) or P.berghei-infected (7 days postinfection; H, J, and L) WT mice werecostained for �D and MARCO, MOMA-1, or F4/80. In each set of panels,�D was detected by green fluorescence and the second marker was detectedby red fluorescence. The right panels (H�, J�, and L�) show higher mag-nification detail. The staining patterns were similar in tissue from threeinfected animals. RP, Red pulp; WP, white pulp.
593The Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

�D�2 mediates adhesive interactions of splenic red pulpmacrophages
The functions of �D�2 in vivo are uncharacterized, although it isassumed to mediate adhesive interactions (1, 3, 6, 8, 10, 11, 16).We established a modified Stamper-Woodruff assay system (26,27) using sections of spleen from WT and �D-deficient mice andsuspensions of isolated splenocytes. Suspended F4/80-positivemacrophages from spleens of �D
�/� animals were much less ad-herent than those from WT mice (Fig. 4). In parallel, incubation ofsuspended F4/80-positive splenocytes from WT mice with Absagainst �D inhibited adhesion to WT spleen sections by 65–90%;in addition, an Ab against VCAM-1, a ligand for human �D�2 (9,10), reduced adhesion by a mean of 22% (10–43%; n � 2, data notshown). These results demonstrate that �D�2 mediates adhesiveinteractions of red pulp macrophages and that it may influencetheir distribution. Furthermore, although VCAM-1 appears to be aligand for �D�2 in mouse tissues, these experiments also indicatethat there are additional binding partners on target spleen cellsand/or matrix. Although �D�2 mediates binding of a critical subsetof splenic macrophages (Figs. 2–4), it is not required for estab-lishment of the basal topography of the red and white pulp (34) ofthe adult murine spleen or for retention of macrophage subsets inthe splenic red pulp (Fig. 3F).
�D�2 expression is dynamically altered on inflamedmacrophages in response to P. berghei challenge and isregulated by cytokine signaling
Because red pulp macrophages have major effector activities inblood-borne infection and antigenemia (33, 34), we chose modelsof systemic infectious challenge to further characterize regulationand functions of �D�2. The spleen, an anatomic interface betweeninnate and acquired immune effector cells (34), is critical in ex-perimental malaria in all animals studied and red pulp macro-phages have unique roles in recognition and clearance of infectedRBC (35, 36). Macrophages in other organs are also activated as aconsequence of recognition of PBRC and by inflammatory signals
generated by the systemic infection (36–38). Therefore, we firstexamined �D�2 distribution and function in experimental malaria.
The distribution of splenic macrophages was dramatically al-tered by challenge with intraerythrocytic P. berghei, a lethal rodentmalarial strain that induces pathophysiologic features similar tothose in severe human malaria (28, 37–39). Spleens of the infectedanimals were also enlarged (data not shown). Serial examinationsover 5–20 days after infection demonstrated progressive reorgani-zation of the splenic architecture and extensive loss of geographiclocalization of �D
� and MARCO� macrophages (Fig. 3, G, H, H�,K, L, and L�, and data not shown). MARCO� cells were widelydistributed in the red pulp of P. berghei-infected animals, in con-trast to their strict compartmentalization in the marginal zone ofthe uninfected spleen (Fig. 3, G, H, and H�). Linear arrays ofdoubly stained �D
� and MARCO� macrophages were present inred pulp (Fig. 3, H and H�), whereas expression of �D andMARCO was restricted under basal conditions (Fig. 3G). Func-tional characteristics of the new population of �D
�MARCO�
macrophages are not yet completely defined. Nevertheless, thesecells contained heme pigment deposits, consistent with erythroph-agocytosis (data not shown). The topography of MOMA-1� mac-rophages was also altered (Fig. 3, J and J�). Redistribution ofsplenic leukocytes also occurs in infection with other malarialpathogens (40).
FIGURE 4. Integrin �D�2 mediates adhesive interactions of splenic redpulp macrophages. Suspensions of PE-labeled F4/80-positive macrophages(suspended cells) from WT or �D
�/� (knockout (KO)) mice were incu-bated with spleen sections from WT or �D
�/� animals, and the adherentsplenocytes were imaged (A) and counted (B) as described in Materials andMethods. A and B, Representative of three separate experiments.
Table I. �D�2 is restricted to macrophages in a limited number oftissues in WT C57BL/6 mice in the absence of infection or inflammationand is absent in �D
�/� animalsa
TissueSamples from
WT MiceSamples from�D
�/� Mice
Lung 0 0Thoracic lymph node 0 0Liver 0 0Kidney 0 0Peyer’s patch (gut) 0 0Alveolar macrophages 0 NDBrain 0 0Skin 0 0Spleen ���� 0Thymus �� 0Bone marrow �� 0Blood leukocytes 0–� 0Peritoneal macrophages 0–� ND
a The levels of �D�2 expression analyzed by immunocytochemistry were esti-mated based on staining intensity as strong (����), intermediate (��), weak (0–�), or absent (0). In tissues with positive staining, �D�2 was expressed by specificcells with morphologic characteristics of macrophages (see text and Figs. 2 and 3).These results summarize the findings in analysis of samples from multiple animals.Alveolar macrophages were obtained by bronchoalveolar lavage (n � 2). Residentperitoneal macrophages collected in the absence of agonist stimulation were largelynegative for �D�2, a feature that was confirmed by flow cytometry (0.23–4.7% ofcells positive for �D, n � 4). Expression of �D was not increased in peritonealmacrophages at 48 h after thioglycolate injection into the peritoneal cavity as assayedby flow cytometry (n � 3).
594 �D�2, MACROPHAGES, AND SYSTEMIC INFECTIONS
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
Parallel but distinct changes occurred in the liver, which is atarget organ in P. berghei infection (29, 41). No �D
� cells weredetected in the livers of uninfected animals (Table I, Fig. 5A, anddata not shown), indicating that murine Kupffer cells, a macro-phage subset (20), do not basally express �D. �D is also absentfrom human and canine Kupffer cells (8, 30). By day 10 ofinfection with P. berghei, a minor population of small �D
� cellswas detected, primarily in the portal vein areas (Fig. 5B). Thesecells were macrosialin� but negative for F4/80, �M�2, and�X�2. By day 20, large �D
� cells with morphology similar tothat of splenic red pulp macrophages were numerous, werefound in both the portal and central vein areas (Fig. 5C), andwere again negative for F4/80, �M�2, and �X�2 while beingpositive for macrosialin. They were associated with deposits ofheme pigment (Fig. 5, B and C, right panels, and data notshown), indicating that these cells participate in erythrophago-cytosis as do splenic macrophages. There was also progressive
hepatocyte necrosis and hepatic inflammation in the 10- to 20-day interval (data not shown).
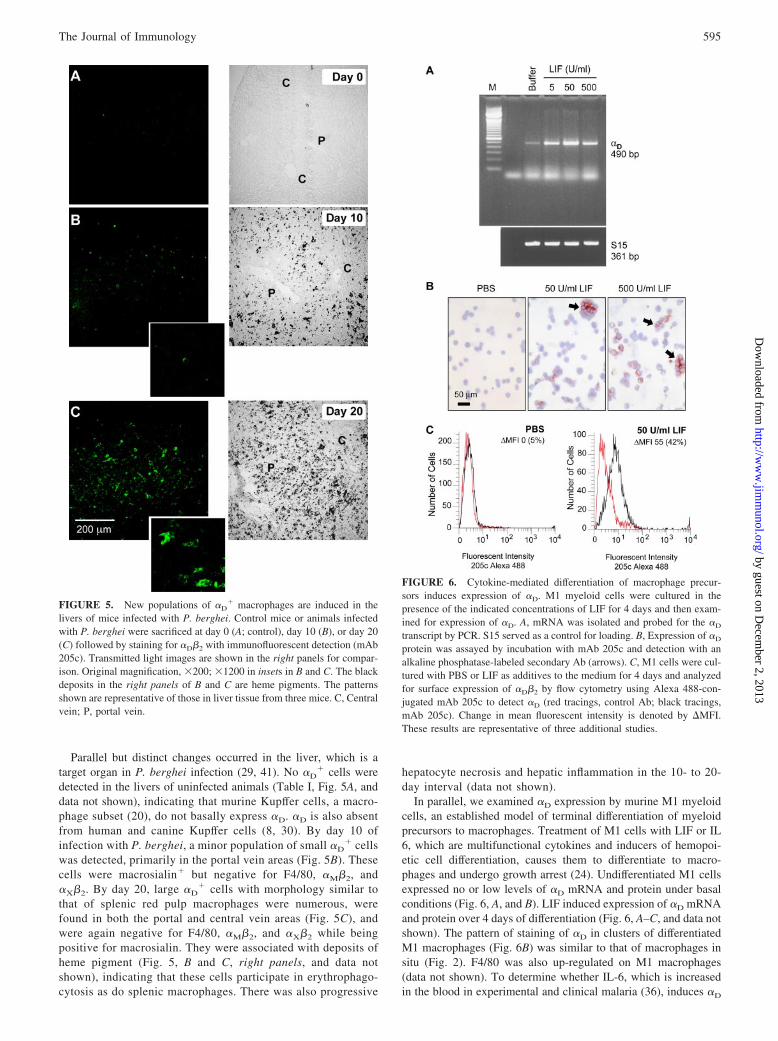
In parallel, we examined �D expression by murine M1 myeloidcells, an established model of terminal differentiation of myeloidprecursors to macrophages. Treatment of M1 cells with LIF or IL6, which are multifunctional cytokines and inducers of hemopoi-etic cell differentiation, causes them to differentiate to macro-phages and undergo growth arrest (24). Undifferentiated M1 cellsexpressed no or low levels of �D mRNA and protein under basalconditions (Fig. 6, A, and B). LIF induced expression of �D mRNAand protein over 4 days of differentiation (Fig. 6, A–C, and data notshown). The pattern of staining of �D in clusters of differentiatedM1 macrophages (Fig. 6B) was similar to that of macrophages insitu (Fig. 2). F4/80 was also up-regulated on M1 macrophages(data not shown). To determine whether IL-6, which is increasedin the blood in experimental and clinical malaria (36), induces �D
FIGURE 5. New populations of �D� macrophages are induced in the
livers of mice infected with P. berghei. Control mice or animals infectedwith P. berghei were sacrificed at day 0 (A; control), day 10 (B), or day 20(C) followed by staining for �D�2 with immunofluorescent detection (mAb205c). Transmitted light images are shown in the right panels for compar-ison. Original magnification, �200; �1200 in insets in B and C. The blackdeposits in the right panels of B and C are heme pigments. The patternsshown are representative of those in liver tissue from three mice. C, Centralvein; P, portal vein.
FIGURE 6. Cytokine-mediated differentiation of macrophage precur-sors induces expression of �D. M1 myeloid cells were cultured in thepresence of the indicated concentrations of LIF for 4 days and then exam-ined for expression of �D. A, mRNA was isolated and probed for the �D
transcript by PCR. S15 served as a control for loading. B, Expression of �D
protein was assayed by incubation with mAb 205c and detection with analkaline phosphatase-labeled secondary Ab (arrows). C, M1 cells were cul-tured with PBS or LIF as additives to the medium for 4 days and analyzedfor surface expression of �D�2 by flow cytometry using Alexa 488-con-jugated mAb 205c to detect �D (red tracings, control Ab; black tracings,mAb 205c). Change in mean fluorescent intensity is denoted by �MFI.These results are representative of three additional studies.
595The Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

expression, we incubated M1 cells with IL-6 (1–100 ng/ml) or LIF(50 U/ml) for 4 days. IL-6 induced �D mRNA and protein expres-sion in a concentration-dependent fashion with a maximal effect at10 ng/ml. By immunocytochemistry, the staining pattern and num-ber of cells positive for �D were equivalent when M1 were stim-ulated with 10 ng/ml IL-6 or 50 U/ml LIF. These experimentsdemonstrate that �D expression is regulated by cytokine-inducedmacrophage differentiation, providing a mechanism for its localinduction on macrophage subsets in inflamed tissues such as theliver (Fig. 5).
Targeted deletion of �D differentially influences survival insystemic infection with P. berghei and Salmonella
We found that targeted deletion of �D alters survival in P. bergheiinfection in an unexpected fashion. In three separate experiments,all of the WT animals infected with P. berghei were dead at 30days (Fig. 7A). In two of the experiments, each of 10 WT micesuccumbed by days 27 and 21, respectively, whereas a total of 5�D
�/� animals (3 in experiment 1 and 2 in experiment 2) survivedto day 30. A third experiment demonstrated an even more malig-nant course, with 9 of 10 WT animals dead by day 14 comparedwith 7 of 10 �D
�/� animals; 1 �D�/� mouse survived until day 30.
The difference between the cumulative survival curves for eachgenotype in these experiments (Fig. 7A) was highly significant( p � 0.0059). Thus, targeted deletion of �D alters the naturalhistory and increases survival in the first 30 days of infection inthis model, a period in which all WT animals succumbed. To con-firm that the survival advantage during this time span represents abiologic feature selectively imposed by deletion of �D�2, we per-formed two experiments in which other genetically modified micewere examined as controls. In the first, mice deficient in TLR2 (10animals) were infected with P. berghei in parallel with WT mice(10 animals). In the second, mice deficient in inducible NO syn-thase (iNOS) (15 animals) and WT mice (24 animals) were stud-ied. All iNOS and TLR2 animals were dead by days 18 and 19,respectively, whereas mortality of the WT mice was similar to thatshown in Fig. 7A, with a progressive decrease in the number ofsurviving animals to day 28. Thus, deletion of neither iNOS norTLR2, innate immune factors that influence the natural history ofmalaria (39), reproduced the mortality pattern imposed by geneticdeletion of �D in these studies (Fig. 7A).
The survival curves of WT and �D�/� mice diverged at days
8–10 after P. berghei infection and significantly more �D�/� an-
imals were alive over the succeeding 3 wk (Fig. 7A). To begin toexamine the mechanisms involved, we focused on the period ofdivergence and first examined erythrocyte parasite burden andmagnitude of anemia, which are key determinants of outcome insevere malaria (36–39). Between days 10 and 21, the mice becamehyperparasitemic and profoundly anemic but there were no con-sistent differences in samples from �D
�/� and WT mice (Fig. 7, Band C), excluding differences in these two central mechanisms (39)as causes for the difference in survival (Fig. 7A). In addition, theseresults indicate that �D�2 is not required for clearance of PRBC.The splenic microarchitecture is essential for rapid resolution ofprimary infection with Plasmodium species and influences key as-pects of the host response (35, 40). We found that the organizationof splenic red and white pulp is dramatically altered in P. bergheiinfection (Fig. 3, H, J, and L). Nevertheless, this rearrangement ofsplenic architecture also occurred in �D
�/� animals with loss ofdistinct boundaries between the red pulp and marginal zone. Thisindicated that differences in splenic macroarchitecture, whichmight occur due to alteration in adhesive properties of red pulpmacrophages (Fig. 4) as a result of deletion of �D�2, do not ac-count for the differences in survival (Fig. 7A).
Systemic cytokines are proposed to be key regulators of thepathogenesis of complicated malaria (35, 40). Therefore, we nextperformed an initial survey of multiple cytokines in the blood ofthree animals of each genotype at 10 days of infection using amultiplex screening assay. This analysis suggested diverse
FIGURE 7. Deletion of �D provides a survival advantage in P. bergheiinfection. A, Mice were infected by i.p. challenge with P. berghei PRBC.Three separate experiments, each involving 10 WT and 10 �D
�/� animals,were accomplished and the pooled results were analyzed. The differencebetween the cumulative survival curves was significant (p � 0.0059). Band C, The percentage of RBC infected by P. berghei was determined bymicroscopy and the hematocrit by RBC sedimentation. B, Results from oneof three experiments and C, results from one of two experiments. Samplesfrom variable numbers of surviving animals were studied at each timepoint. D, Serum samples were collected from WT control mice (n � 5),WT animals infected with P. berghei (n � 10), �D
�/� control mice (n �8), and P. berghei-infected �D
�/� animals (n � 7) at 10 days after i.p.inoculation with PRBC. IL-12(p 40) concentrations were measured byELISA. The differences between levels in samples from infected WT and�D
�/� mice (�) was highly significant (p � 0.0001).
596 �D�2, MACROPHAGES, AND SYSTEMIC INFECTIONS
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

alterations in systemic levels of cytokines in �D�/� and WT
mice, including variations in IL-12, IFN-�, IL-6, MCP-1, andKC. To explore this initial finding further, we examined levelsof IL-12 in serum samples from a second group of animals studiedon day 10 of infection. We chose IL-12 as an initial candidate forfurther analysis because it is implicated in the pathogenesis ofsevere human malaria and has been identified as a mediator oftissue injury in lethal P. berghei infection (29, 41). Consistent withthe preliminary analysis, we found that IL-12 concentrations werelower in the blood of infected �D
�/� mice compared with levels insamples from WT mice (Fig. 7D). Thus, alterations in the patternand levels of inflammatory cytokines may contribute to the sur-vival advantage in �D
�/� animals in this phase of the systemicinfection (Fig. 7A).
To further define the phenotype of �D-deficient mice and todetermine whether deletion of �D has a stereotyped, or conversely,differential impact on systemic infections, we examined �D
�/�
and WT mice in a model of S. typhimurium sepsis. This model waschosen for comparison because, although it is a bacterial pathogen,it also involves blood-borne phases and clearance by splenic, he-patic, and marrow macrophages (35, 44), features that are similarto P. berghei infection. A preliminary experiment (eight mice ofeach genotype) with 105 bacteria per inoculum suggested greatermortality in �D
�/� animals. A second experiment with 10 animalsof each genotype but with a 10-fold lower inoculum supported theinitial result ( p � 0.008). We then performed three additionalexperiments with 10 WT and 10 �D
�/� mice, an inoculum of 105
Salmonella, and a 10-day study period in each. (The change instudy period was required because of animal housing constraints.)These studies replicated the original finding and again documentedgreater mortality in the �D�2-deficient animals (Fig. 8; p �0.0004). Thus, survival patterns of �D
�/� mice were clearly dif-ferent in systemic infection with P. berghei (Fig. 7A) and S. ty-phimurium (Fig. 8). This difference in natural history demonstratesthat the effect of genetic deletion of �D in murine models of sys-temic infection varies with the nature of the pathogen, indicatingspecialized functions of integrin �D�2 and the immune effectorcells that express it in host defense against invading microbes.
DiscussionLeukocyte integrins have essential but incompletely defined activ-ities in host defense and conserved functions in mice and humans(1–6). Our observations provide the first detailed characterizationof �D�2 in vivo, information that is currently lacking in the field(1, 3), along with new insights into its regulation and functions.
The studies clearly demonstrate that it has unique features in in-flammation and host defense. Interestingly, although other �2 in-tegrins are highly expressed on circulating murine leukocytes andare critical in their trafficking from blood to tissues, �D�2 ispresent on only a minor and as yet uncharacterized population ofthese cells in WT animals in naive conditions (see Results; Y.Miyazaki and G. A. Zimmerman, unpublished data). In contrast,�D�2 is constitutively expressed on macrophages of splenic redpulp, bone marrow, and thymus of WT mice in the absence ofinfectious or inflammatory challenge (Table I and Figs. 2 and 3)indicating that, in this species, �D�2 has major functions apartfrom emigration of leukocytes from the blood. We found that �D�2
mediates adhesion of red pulp macrophages, identifying one of itsextravascular activities and demonstrating a functional role for the�D�2 heterodimer on macrophages for the first time. Previously,adhesive functions for �D�2 have only been observed in humaneosinophils and surrogate cell lines (8–11). We also found that inresponse to erythrocytic infection with a malarial pathogen, P.berghei, new and previously unrecognized populations of �D
�
macrophages emerge in the spleen and liver, demonstrating dy-namic regulation of �D�2 in this cell type and illustrating thedaedal nature of macrophage heterogeneity (42, 43). In contrast,bacterial and immune challenge to the murine lung, another mac-rophage-rich organ, did not induce expression of �D under severalexperimental conditions (Y. Miyazaki, H. Castro Faria Neto, andG. A. Zimmerman, unpublished data) and �D was not increased onperitoneal macrophages in response to thioglycolate instillation(Table I). This indicates that selective signals are required for itsdisplay on macrophage subpopulations, pointing to context-spe-cific functions of �D�2. Targeted deletion of �D unexpectedlyyielded a survival advantage in lethal malarial infection. Con-versely, deletion of �D had the opposite effect in a model of Sal-monella sepsis, indicating complex roles for �D�2 in defenseagainst systemic pathogens. The latter findings are consistent withboth beneficial and injurious activities of leukocyte integrins (2, 7)and macrophages (42, 44) in innate immune responses.
The roles of leukocyte integrins in responses to microbial inva-sion are incompletely characterized, although this is clearly a chiefdefensive function of these specialized effector proteins (1, 2, 6).Targeted deletion of �2, which causes deficiency of all leukocyteintegrin heterodimers, increases susceptibility to many bacterialpathogens but affords resistance to Listeria monocytogenes andimproves control of local infection by Leishmania major, a pro-tozoan parasite of macrophages, in murine models (5, 6). Thesepatterns of susceptibility and resistance demonstrate complexfunctions of leukocyte integrins in innate and acquired immuneresponses to infection (6), a feature further indicated by variablecontributions of individual heterodimers in specific experimentalmodels (45, 46). Participation of integrins in the pathobiology ofcomplicated malaria is suggested (37), but largely unexplored.mAbs against �L improved survival in models of cerebral malaria,suggesting a role for �L�2 (LFA-1), whereas Abs against �m orICAM-1, a ligand for �L�2, did not protect (47, 48). In contrast toAb administration, targeted deletion of ICAM-1 increased survivalin a cerebral malaria model (49). A �3 integrin (3) regulates thepattern of cytokines released by human macrophages and myeloiddendritic cells activated by PRBC (36); nevertheless, contributionsof �2 integrins to this process are undefined. Thus, the regulationand activities of leukocyte integrins in malarial infection are un-characterized. We found extensive display of �D on red pulp mac-rophages of the WT spleen (Figs. 2 and 3), suggesting that �D�2
mediates responses to systemic pathogens (34), potentially includ-ing malarial parasites (35). We then explored this possibility using
FIGURE 8. Mortality is accelerated in �D�2-deficient mice infectedwith S. typhimurium. WT or �D
�/� mice were infected with S. typhi-murium by the i.p. route (105 bacteria/animal). Groups of 10 �D
�/� and 10WT animals were studied in each of three separate experiments. Thepooled survival data for 30 animals of each genotype are shown. The dif-ference between the cumulative survival plots was significant (p �0.0004). Control sham-infected mice that received vehicle alone were stud-ied in parallel; no sham-infected animals died.
597The Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

a model that mimics many features of severe human malaria, P.berghei infection (28, 29, 37–39).
Challenge of WT mice with intraerythrocytic P. berghei resultedin a dramatic alteration in the microarchitecture of the splenic redpulp and marginal zone with loss of demarcation of these regionsand appearance of a new subset of �D
�MARCO� macrophages(Fig. 3 and data not shown). The distribution and activities ofsplenic macrophages and other leukocytes influence the inflamma-tory and immune responses to malarial invaders and may altersusceptibility to concomitant infection by bacterial and viral patho-gens (35, 36, 39, 40, 50, 51). In lethal P. falciparum infection, themarginal zone is dramatically altered with almost complete histo-logic “dissolution” (50), similar to the microarchitectural disrup-tion in P. berghei infection (Fig. 3). In parallel with changes insplenic microarchitecture and macrophage populations, a new sub-set of �D
� macrosialin� macrophages emerged in the livers of WTmice infected with P. berghei (Fig. 5). It is likely that these newmacrophage subpopulations in spleen and liver mediate responsesto malarial infection that are in some cases injurious or maladap-tive (36, 37, 42, 43). In addition, they clearly demonstrate dynamicexpression of �D�2 induced by pathogen challenge and inflamma-tory signals.
It is not clear whether new populations of �D�2-expressing mac-rophages in the spleen and liver in P. berghei infection are due tomigration, proliferation, and differentiation of blood leukocytesthat express �D or, conversely, to induction of �D on residentmacrophages or macrophage precursors. To begin to explore thisissue, we used the murine M1 myeloid cell model, in which ago-nist-induced macrophage differentiation can be dissociated fromcellular proliferation (24). LIF and IL-6, which are multifunctionalcytokines released into the blood in murine endotoxemia and, inthe case of IL-6, malaria (37, 38), trigger terminal differentiation ofM1 cells to macrophages (24). We found that LIF and IL-6 in-duced new expression of �D on M1 cell-derived macrophages,providing a potential mechanism for dynamic regulation of �D�2
on specific macrophage subsets in tissues such as the liver orspleen in response to inflammatory signals (Figs. 3 and 5). Asnoted previously, unique patterns of cytokines or other signalingfactors are likely required for the highly restricted expression of�D�2 on individual macrophage subsets in healthy animals (TableI and Figs. 2 and 3) and changes in expression in response toinflammatory challenge (Figs. 3 and 5). Additional characteriza-tion of the minor subpopulation of �D
� blood leukocytes will berequired to further define regulation of �D on this subset andwhether it is altered by signals for migration and/or proliferation.
Improved survival of �D�/� mice infected by P. berghei was
unexpected and indicated that pathophysiologic events mediatedby �D�2 have deleterious consequences in lethal malarial infec-tion. Although at first glance the survival advantage at 30 daysappears relatively small, significant differences over an approxi-mate 3-wk period represent a substantial benefit in a syndromewith 100% mortality (Fig. 7A). Mice infected with P. berghei diesecondary to cerebral, hepatic, pulmonary, renal, and cardiac com-plications that are superimposed on hyperparasitemia and severemalarial anemia (28, 29, 37–39, 41, 52, 53). The pathobiologicmechanisms that mediate systemic inflammation and critical organinjury in experimental and clinical malaria are complex and mul-tifactorial, and their precise identification has remained elusive(36–39). One or multiple organ-specific or multiorgan componentscould be influenced by deletion of �D�2. To begin to explore thesepossibilities, we first examined two key determinants of malarialpathogenesis, severe malarial anemia, and intraerythrocyte parasiteburden (39). Splenic red pulp macrophages, which are rich in �D�2
(Figs. 2 and 3 and Table I), are critical for removal of PRBC and
for parasite killing, and changes in splenic function and splenec-tomy alter anemia parasitemia and outcomes (35, 50, 51). Pro-found anemia in murine malaria is due in part to hemolysis andhyperphagocytosis of infected and uninfected RBC by splenic redpulp and/or hepatic macrophages and to ineffective erythropoiesis(39, 52, 53). The latter process is influenced by marrow macro-phages (54, 55) and, therefore, potentially by �D�2 (Table I andFig. 2). Nevertheless, we found similar numbers of PRBC anddegrees of anemia in �D
�/� and WT mice (Fig. 7, B and C),indicating that differences in intraerythrocytic parasite burden,clearance of PBRC by macrophage subsets, and ineffective eryth-ropoiesis are not the pivotal mechanisms that determined alteredsurvival of �D
�/� animals. In addition, we evaluated differences insplenic microarchitecture, which influences early and late host re-sponses to malarial pathogens (36, 40, 50, 51) (also see above). Wefound that dramatic alterations in red pulp and marginal zone to-pography, which were histologically similar to those reported in P.falciparum infection in humans (50), occurred in both WT and�D
�/� animals (Fig. 3 and data not shown). Therefore, differencesin this variable do not account for the survival advantage of �D
�/�
animals. In addition to hyperparasitemia, severe anemia, andsplenic reorganization, cytoadhesive vasculopathy, in whichPRBC adhere to endothelium and occlude microvessels of criticalorgans, is also a proposed mechanism of injury and death in severemalarial syndromes (37–39). We have not observed expression of�D�2 on murine erythrocytes or endothelial cells (Table I and datanot shown), consistent with the highly restricted distribution ofleukocyte integrins (1, 3), making it unlikely that altered interac-tions of PRBC with endothelium account for improved survival of�D
�/� animals. Therefore our studies, in aggregate, excluded sev-eral major determinants of mortality in P. berghei infection andpointed to other mechanisms.
Cytokine signaling is central to the systemic manifestations ofexperimental and clinical malaria (37–39, 53), and outside-in sig-nals mediated by integrins both trigger and modify synthesis ofcytokines by macrophages and monocytes (1, 3, 56–59). In pre-liminary studies, we found altered patterns of plasma cytokines in�D
�/� mice infected with P. berghei compared with those in WTanimals at 10 days after challenge. In additional assays to validatethese preliminary data, we found that IL-12 levels in the blood ofinfected �D�2-deficient mice were substantially lower than thosein samples from WT controls (Fig. 7D). IL-12 signaling induceshepatic injury in lethal erythrocytic P. berghei infection (35, 40),providing a rationale for examining this cytokine as a sentinelproinflammatory mediator under the conditions of our experiment.(Studies of additional cytokines are in progress.) Furthermore, syn-thesis of IL-12 by monocytes and macrophages is suppressed byengagement of �2 integrins (57–59), suggesting a mechanism forits regulation under these conditions, although this has not yet beenexamined for �D�2. Nevertheless, we do not propose decreasedplasma levels of IL-12 as the sole mechanism for improved sur-vival in �D
�/� animals, since excesses of individual cytokinesseem unlikely to account for all multiorgan and systemic manifes-tations of severe malarial syndromes (38, 39). Furthermore, addi-tional cellular complexity may be involved. �D�2 influences re-sponses of T cells, which are immune effectors in severeexperimental malaria (37–39, 60), and is reported to determinefunctional ontogeny of T lymphocytic subsets and their responsesto microbial Ags (16).
To further characterize the phenotype of �D�/� animals in sys-
temic infectious models and to determine whether the activities of�D�2 are stereotyped regardless of the nature of the pathogen, weexamined �D-deficient mice in S. typhimurium infection. Macro-phage activity, cytokine signaling, and immune cell interactions
598 �D�2, MACROPHAGES, AND SYSTEMIC INFECTIONS
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

are central in responses to Salmonella invasion (44), as they are inmalarial infection (36–39). Deletion of �D�2 accelerated mortalityin mice infected with S. typhimurium, in contrast to the survivalpattern in infection with P. berghei (see Results and Fig. 8). Thedifference in outcomes in the P. berghei and Salmonella modelsmay in part be due to recognition of Salmonella by splenic mar-ginal zone macrophages rather than �D�2-rich red pulp macro-phages (35). Furthermore, endogenous IL-12 augments protectiveimmune responses against Salmonella species (61), whereas it con-tributed to immune injury in P. berghei infection (29, 41). Thus,the altered levels of IL-12 in �D
�/� mice may partially account forthe differences in survival in the two infectious challenges. In ad-dition, however, different patterns of survival in the malaria andSalmonella models may also be due to different functional rolesplayed by �D�2
� macrophage subsets in systemic infection byintracellular bacteria vs malarial parasites. It is not yet clear howeffector characteristics of macrophages (42, 43) govern the balancebetween defense and injury in P. berghei and S. typhimurium in-fections (44, 62) or how �D�2 influences activation patterns ofspecific macrophage populations. Further delineation of mecha-nisms by which �D�2 and other leukocyte integrins regulate thelocalization and functional repertoires of macrophage subsets (42)will clarify this issue. This information may then be useful in un-derstanding the effects of innate immune events in humans infectedby malaria and Salmonella, which together kill �2.5 million sub-jects each year worldwide (36, 39, 44, 62). �D�2 is expressed byleukocytes in human splenic red pulp (8) and is displayed in reg-ulated fashion by primary human macrophages and dendritic cells(Y. Miyazaki, E. Harris, A. Shah, and G. A. Zimmerman, manu-script in preparation), suggesting that it has key roles in these andother human infectious syndromes.
AcknowledgmentsWe thank our laboratory staffs for technical assistance, Andressa Almeidafor preparation of infectious agents for in vivo experiments, and KateAnderson for skilled animal husbandry. Liping Chen made important con-tributions to the development of �D-deficient mice before her untimelydeath. Diana Lim prepared the figures and graphic art and Mary Madsen,Adrienne Triplett, and Linn Steele contributed invaluable effort in prepa-ration of this manuscript. We thank Leslie Ostler for performing specificexperiments, Kelley Murphy for advice regarding immunocytochemicalanalysis, and Marga Massey for samples of rat spleen. Monica Van derVieren, Patricia Hoffman, and Michael Gallatin of ICOS Corporation gen-erously provided critical reagents, including key anti-�D Abs and a murine�D probe template for genomic library screening. We also thank colleaguesin our group and Dr. Merle Sande for valuable discussions.
DisclosuresThe authors have no financial conflict of interest.
References1. Harris, E. S., T. M. McIntyre, S. M. Prescott, and G. A. Zimmerman. 2000. The
leukocyte integrins. J. Biol. Chem. 275: 23409–23412.2. Bunting, M., E. S. Harris, T. M. McIntyre, S. M. Prescott, and G. A. Zimmerman.
2002. Leukocyte adhesion deficiency syndromes: adhesion and tethering defectsinvolving �2 integrins and selectin ligands. Curr. Opin. Hematol. 9: 30–35.
3. Hynes, R. O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell110: 673–687.
4. Scharffetter-Kochanek, K., H. Lu, K. Norman, N. van Nood, F. Munoz,S. Grabbe, M. McArthur, I. Lorenzo, S. Kaplan, K. Ley, et al. 1998. Spontaneousskin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 188:119–131.
5. Etzioni, A., C. M. Doerschuk, and J. M. Harlan. 1999. Of man and mouse:leukocyte and endothelial adhesion molecule deficiencies. Blood 94: 3281–3288.
6. Lee, S. H., and D. B. Corry. 2004. Homing alone? CD18 in infectious and allergicdisease. Trends Mol. Med. 10: 258–262.
7. Yonekawa, K., and J. M. Harlan. 2005. Targeting leukocyte integrins in humandiseases. J. Leukocyte Biol. 77: 129–140.
8. Van der Vieren, M., H. Le Trong, C. L. Wood, P. F. Moore, T. St. John,D. E. Staunton, and W. M. Gallatin. 1995. A novel leukointegrin, �d�2, bindspreferentially to ICAM-3. Immunity 3: 683–690.
9. Grayson, M. H., M. Van der Vieren, S. A. Sterbinsky, W. Michael Gallatin,P. A. Hoffman, D. E. Staunton, and B. S. Bochner. 1998. �d�2 integrin is ex-pressed on human eosinophils and functions as an alternative ligand for vascularcell adhesion molecule 1 (VCAM-1). J. Exp. Med. 188: 2187–2191.
10. Van der Vieren, M., D. T. Crowe, D. Hoekstra, R. Vazeux, P. A. Hoffman,M. H. Grayson, B. S. Bochner, W. M. Gallatin, and D. E. Staunton. 1999. Theleukocyte integrin �D�2 binds VCAM-1: evidence for a binding interface be-tween I domain and VCAM-1. J. Immunol. 163: 1984–1990.
11. Yakubenko, V. P., S. P. Yadav, and T. P. Ugarova. 2006. Integrin �D�2, anadhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood 107: 1643–1650.
12. El-Gabalawy, H., J. Canvin, G. M. Ma, M. Van der Vieren, P. Hoffman,M. Gallatin, and J. Wilkins. 1996. Synovial distribution of �d/CD18, a novelleukointegrin: comparison with other integrins and their ligands. Arthritis Rheum.39: 1913–1921.
13. Noti, J. D. 2002. Expression of the myeloid-specific leukocyte integrin geneCD11d during macrophage foam cell differentiation and exposure to lipoproteins.Int. J. Mol. Med. 10: 721–727.
14. Shanley, T. P., R. L. Warner, L. D. Crouch, G. N. Dietsch, D. L. Clark, M. M.O’Brien, W. M. Gallatin, and P. A. Ward. 1998. Requirements for �d in IgGimmune complex-induced rat lung injury. J. Immunol. 160: 1014–1020.
15. Oatway, M. A., Y. Chen, J. C. Bruce, G. A. Dekaban, and L. C. Weaver. 2005.Anti-CD11d integrin antibody treatment restores normal serotonergic projectionsto the dorsal, intermediate, and ventral horns of the injured spinal cord. J. Neu-rosci. 25: 637–647.
16. Wu, H., J. R. Rodgers, X. Y. Perrard, J. L. Perrard, J. E. Prince, Y. Abe,B. K. Davis, G. Dietsch, C. W. Smith, and C. M. Ballantyne. 2004. Deficiency ofCD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cellresponse and T cell phenotypic changes. J. Immunol. 173: 297–306.
17. Bunting, M., K. E. Bernstein, J. M. Greer, M. R. Capecchi, and K. R. Thomas.1999. Targeting genes for self-excision in the germ line. Genes Dev. 13:1524–1528.
18. Mansour, S. L., K. R. Thomas, and M. R. Capecchi. 1988. Disruption of theproto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy fortargeting mutations to non-selectable genes. Nature 336: 348–352.
19. Schwenk, F., U. Baron, and K. Rajewsky. 1995. A cre-transgenic mouse strain forthe ubiquitous deletion of loxP-flanked gene segments including deletion in germcells. Nucleic Acids Res. 23: 5080–5081.
20. McKnight, A. J., and S. Gordon. 1998. Membrane molecules as differentiationantigens of murine macrophages. Adv. Immunol. 68: 271–314.
21. Cao, Y., K. J. Murphy, T. M. McIntyre, G. A. Zimmerman, and S. M. Prescott.2000. Expression of fatty acid-CoA ligase 4 during development and in brain.FEBS Lett. 467: 263–267.
22. Ohteki, T., C. Maki, and S. Koyasu. 2001. Overexpression of Bcl-2 differentiallyrestores development of thymus-derived CD4�8� T cells and intestinal intraepi-thelial T cells in IFN-regulatory factor-1-deficient mice. J. Immunol. 166:6509–6513.
23. Kessel, J. M., J. Hayflick, A. S. Weyrich, P. A. Hoffman, M. Gallatin,T. M. McIntyre, S. M. Prescott, and G. A. Zimmerman. 1998. Coengagement ofICAM-3 and Fc receptors induces chemokine secretion and spreading by myeloidleukocytes. J. Immunol. 160: 5579–5587.
24. Hoffman-Liebermann, B., and D. A. Liebermann. 1991. Interleukin-6- and leu-kemia inhibitory factor-induced terminal differentiation of myeloid leukemiacells is blocked at an intermediate stage by constitutive c-myc. Mol. Cell. Biol.11: 2375–2381.
25. Pabla, R., A. S. Weyrich, D. A. Dixon, P. F. Bray, T. M. McIntyre, S. M. Prescott,and G. A. Zimmerman. 1999. Integrin-dependent control of translation: engage-ment of integrin �IIb�3 regulates synthesis of proteins in activated human plate-lets. J. Cell Biol. 144: 175–184.
26. Stamper, H. B., Jr., and J. J. Woodruff. 1976. Lymphocyte homing into lymphnodes: in vitro demonstration of the selective affinity of recirculating lympho-cytes for high-endothelial venules. J. Exp. Med. 144: 828–833.
27. Yednock, T. A., C. Cannon, L. C. Fritz, F. Sanchez-Madrid, L. Steinman, andN. Karin. 1992. Prevention of experimental autoimmune encephalomyelitis byantibodies against �4�1 integrin. Nature 356: 63–66.
28. Cordeiro, R. S., F. Q. Cunha, J. A. Filho, C. A. Flores, H. N. Vasconcelos, andM. A. Martins. 1983. Plasmodium berghei: physiopathological changes duringinfections in mice. Ann. Trop. Med. Parasitol. 77: 455–465.
29. Adachi, K., H. Tsutsui, S. Kashiwamura, E. Seki, H. Nakano, O. Takeuchi,K. Takeda, K. Okumura, L. Van Kaer, H. Okamura, et al. 2001. Plasmodiumberghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J. Immunol. 167:5928–5934.
30. Danilenko, D. M., P. V. Rossitto, M. Van der Vieren, H. Le Trong,S. P. McDonough, V. K. Affolter, and P. F. Moore. 1995. A novel canine leu-kointegrin, �d�2, is expressed by specific macrophage subpopulations in tissueand a minor CD8� lymphocyte subpopulation in peripheral blood. J. Immunol.155: 35–44.
31. Geissmann, F., S. Jung, and D. R. Littman. 2003. Blood monocytes consist of twoprincipal subsets with distinct migratory properties. Immunity 19: 71–82.
32. Randolph, G. J., S. Beaulieu, S. Lebecque, R. M. Steinman, and W. A. Muller.1998. Differentiation of monocytes into dendritic cells in a model of transendo-thelial trafficking. Science 282: 480–483.
33. Van der Laan, L. J., E. A. Dopp, R. Haworth, T. Pikkarainen, M. Kangas,O. Elomaa, C. D. Dijkstra, S. Gordon, K. Tryggvason, and G. Kraal. 1999. Reg-ulation and functional involvement of macrophage scavenger receptor MARCOin clearance of bacteria in vivo. J. Immunol. 162: 939–947.
599The Journal of Immunology
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

34. Mebius, R. E., and G. Kraal. 2005. Structure and function of the spleen. Nat. Rev.Immunol. 5: 606–616.
35. Yadava, A., S. Kumar, J. A. Dvorak, G. Milon, and L. H. Miller. 1996. Traf-ficking of Plasmodium chabaudi adami-infected erythrocytes within the mousespleen. Proc. Natl. Acad. Sci. USA 93: 4595–4599.
36. Urban, B. C., and D. J. Roberts. 2002. Malaria, monocytes, macrophages, andmyeloid dendritic cells: sticking of infected erythrocytes switches off host cells.Curr. Opin. Immunol. 14: 458–465.
37. de Souza, J. B., and E. M. Riley. 2002. Cerebral malaria: the contribution ofstudies in animal models to our understanding of immunopathogenesis. MicrobesInfect. 4: 291–300.
38. Hunt, N. H., and G. E. Grau. 2003. Cytokines: accelerators and brakes in thepathogenesis of cerebral malaria. Trends Immunol. 24: 491–499.
39. Schofield, L. 2007. Intravascular infiltrates and organ-specific inflammation inmalaria pathogenesis. Immunol. Cell Biol. 85: 130–137.
40. Beattie, L., C. R. Engwerda, M. Wykes, and M. F. Good. 2006. CD8� T lym-phocyte-mediated loss of marginal metallophilic macrophages following infec-tion with Plasmodium chabaudi chabaudi AS. J. Immunol. 177: 2518–2526.
41. Yoshimoto, T., Y. Takahama, C. R. Wang, T. Yoneto, S. Waki, and H. Nariuchi.1998. A pathogenic role of IL-12 in blood-stage murine malaria lethal strainPlasmodium berghei NK65 infection. J. Immunol. 160: 5500–5505.
42. Gordon, S. 2002. Alternative activation of macrophages. Nat. Immunol. 3: 23–35.43. Noel, W., G. Raes, G. Hassanzadeh Ghassabeh, P. De Baetselier, and A. Beschin.
2004. Alternatively activated macrophages during parasite infections. TrendsParasitol. 20: 126–133.
44. Wijburg, O. L., C. P. Simmons, N. van Rooijen, and R. A. Strugnell. 2000. Dualrole for macrophages in vivo in pathogenesis and control of murine Salmonellaenterica var. Typhimurium infections. Eur. J. Immunol. 30: 944–953.
45. Guerau-de-Arellano, M., J. Alroy, D. Bullard, and B. T. Huber. 2005. AggravatedLyme carditis in CD11a�/� and CD11c�/� mice. Infect. Immun. 73: 7637–7643.
46. Ghosh, S., A. A. Chackerian, C. M. Parker, C. M. Ballantyne, and S. M. Behar.2006. The LFA-1 adhesion molecule is required for protective immunity duringpulmonary Mycobacterium tuberculosis infection. J. Immunol. 176: 4914–4922.
47. Grau, G. E., P. Pointaire, P. F. Piguet, C. Vesin, H. Rosen, I. Stamenkovic,F. Takei, and P. Vassalli. 1991. Late administration of monoclonal antibody toleukocyte function-antigen 1 abrogates incipient murine cerebral malaria. Eur.J. Immunol. 21: 2265–2267.
48. Falanga, P. B., and E. C. Butcher. 1991. Late treatment with anti-LFA-1 (CD11a)antibody prevents cerebral malaria in a mouse model. Eur. J. Immunol. 21:2259–2263.
49. Favre, N., C. Da Laperousaz, B. Ryffel, N. A. Weiss, B. A. Imhof, W. Rudin,R. Lucas, and P. F. Piguet. 1999. Role of ICAM-1 (CD54) in the development ofmurine cerebral malaria. Microbes Infect. 1: 961–968.
50. Urban, B. C., T. T. Hien, N. P. Day, N. H. Phu, R. Roberts, E. Pongponratn,M. Jones, N. T. Mai, D. Bethell, G. D. Turner, et al. 2005. Fatal Plasmodiumfalciparum malaria causes specific patterns of splenic architectural disorganiza-tion. Infect. Immun. 73: 1986–1994.
51. Weiss, L. 1989. Mechanisms of splenic control of murine malaria: cellular re-actions of the spleen in lethal (strain 17XL) Plasmodium yoelii malaria inBALB/c mice, and the consequences of pre-infective splenectomy. Am. J. Trop.Med. Hyg. 41: 144–160.
52. Evans, K. J., D. S. Hansen, N. van Rooijen, L. A. Buckingham, and L. Schofield.2006. Severe malarial anemia of low parasite burden in rodent models resultsfrom accelerated clearance of uninfected erythrocytes. Blood 107: 1192–1199.
53. McDevitt, M. A., J. Xie, G. Shanmugasundaram, J. Griffith, A. Liu,C. McDonald, P. Thuma, V. R. Gordeuk, C. N. Metz, R. Mitchell, et al. 2006. Acritical role for the host mediator macrophage migration inhibitory factor in thepathogenesis of malarial anemia. J. Exp. Med. 203: 1185–1196.
54. Yoshida, H., K. Kawane, M. Koike, Y. Mori, Y. Uchiyama, and S. Nagata. 2005.Phosphatidylserine-dependent engulfment by macrophages of nuclei from ery-throid precursor cells. Nature 437: 754–758.
55. Soni, S., S. Bala, B. Gwynn, K. E. Sahr, L. L. Peters, and M. Hanspal. 2006.Absence of erythroblast macrophage protein (Emp) leads to failure of erythro-blast nuclear extrusion. J. Biol. Chem. 281: 20181–20189.
56. Rosales, C., and R. L. Juliano. 1995. Signal transduction by cell adhesion recep-tors in leukocytes. J. Leukocyte Biol. 57: 189–198.
57. Marth, T., and B. L. Kelsall. 1997. Regulation of interleukin-12 by complementreceptor 3 signaling. J. Exp. Med. 185: 1987–1995.
58. Mosser, D. M., and C. L. Karp. 1999. Receptor mediated subversion of macro-phage cytokine production by intracellular pathogens. Curr. Opin. Immunol. 11:406–411.
59. Sutterwala, F. S., G. J. Noel, R. Clynes, and D. M. Mosser. 1997. Selectivesuppression of interleukin-12 induction after macrophage receptor ligation.J. Exp. Med. 185: 1977–1985.
60. Jacobs, T., T. Plate, I. Gaworski, and B. Fleischer. 2004. CTLA-4-dependentmechanisms prevent T cell induced-liver pathology during the erythrocyte stageof Plasmodium berghei malaria. Eur. J. Immunol. 34: 972–980.
61. Kincy-Cain, T., J. D. Clements, and K. L. Bost. 1996. Endogenous and exoge-nous interleukin-12 augment the protective immune response in mice orally chal-lenged with Salmonella dublin. Infect. Immun. 64: 1437–1440.
62. Stevenson, M. M., and E. M. Riley. 2004. Innate immunity to malaria. Nat. Rev.Immunol. 4: 169–180.
600 �D�2, MACROPHAGES, AND SYSTEMIC INFECTIONS
by guest on Decem
ber 2, 2013http://w
ww
.jimm
unol.org/D
ownloaded from




















