
Secreted β3-integrin enhances natural killer cell activity against acute myeloid leukemia cells
Upload
soauniversityCategory
view
2download
0
lable at ScienceDirect
Biochimie 101 (2014) 168e182
Contents lists avai
Biochimie
journal homepage: www.elsevier .com/locate/b iochi
Research paper
Insights into the conformational perturbations of novel agonists withb3-adrenergic receptor using molecular dynamics simulations
Parul Tewatia a, Nikhil Agrawal b, Mahendra Gaur b, Shakti Sahi b,*aAmity Institute of Biotechnology, Amity University, Sec-125, Noida 201 303, UP, Indiab School of Biotechnology, Gautam Buddha University, Greater Noida 201310, UP, India
a r t i c l e i n f o
Article history:Received 5 August 2013Accepted 13 January 2014Available online 6 February 2014
Keywords:Molecular dynamics simulationBeta 3-adrenergic receptorMolecular docking
Abbreviations: DPPC, 2-dipalmitoyl-sn-phosphocb1-AR, beta 1-adrenergic receptors; b2-AR, beta 2-beta 3-adrenergic receptors; MD, molecular dynamireceptor; QSAR, quantitative structure activity relationOPLS, optimized potentials for liquid simulations; MMwith generalized born and surface area solvation; Å,square deviations.* Corresponding author. Tel.: þ91 120 2344275.
E-mail addresses: [email protected], shaktisahi@g
http://dx.doi.org/10.1016/j.biochi.2014.01.0160300-9084/� 2014 Elsevier Masson SAS. All rights re
a b s t r a c t
Beta 3-adrenergic receptors (b3-AR), belonging to the G-protein coupled receptor family, are known to beinvolved in important physiological functions as intestinal smooth muscle relaxation, glucose homeo-stasis etc. Detailed insight into the mechanistic mode of b3-AR is not known. Molecular dynamic sim-ulations (100 ns) were performed on the 3-D molecular model of b3-AR and complexes of b3-AR withpotential agonists embedded in 2-dipalmitoyl-sn-phosphocholine (DPPC) bilayer-water system usingOPLS (Optimized Potentials for Liquid Simulations) force field to gain structural insight into b3-AR. Thedetailed structural analysis of the molecular dynamic trajectories reveal that the helical bundle con-formations remain well preserved to maintain a conformation similar to the other X-ray solved G-proteincoupled receptors, whereas significant flexibility is observed in intracellular and the extracellular loopsregion. The formation of extensive intra helical and water mediated H-bonds, and aromatic stackinginteractions play a key role in stabilizing the transmembrane helical bundles. These interactions might bespecific to the functional motifs such as D(E)RY, CWxP, S(N)LAxAD, SxxxS and NPxxY motifs whichprovide structural constraints on the b3-AR. The compound 3, 4 and 6 are proposed to act as scaffolds forpotential agonists for b3-AR based on stereochemical and energetic considerations. In lieu of the lack ofthe crystal structure available, the findings of the simulation study provides more comprehensive pictureof the functional properties of the b3-AR.
� 2014 Elsevier Masson SAS. All rights reserved.
1. Introduction
In humans and other forms of life, integral membrane proteinsplay an important role in processes such as electron transfer, iontranslocation, and transduction of extracellular signals, thus mak-ing G-protein coupled receptor (GPCR) super family significantclass of transmembrane (TM) proteins [1]. GPCRs play importantrole in signal transduction pathways across cell membranes and aremajor drug targets. Although the GPCR super family comprises of3e4% of the human genome, malfunctions in GPCRs play role indiseases such as ulcers, allergies, migraine, anxiety, psychosis,
holine; 3D, 3 dimensional;adrenergic receptors; b3-AR,cs; GPCR, G-protein coupledship; RMS, root mean square;eGBSA, molecular mechanicsangstrom; RSMD, root mean
mail.com (S. Sahi).
served.
nocturnal heartburn, hypertension, asthma, prostatic hypertrophy,congestive heart failure, Parkinson’s, schizophrenia, and glaucoma,thus making the GPCR super family one of the most importantfamilies of drug targets [1]. GPCRs consist of seven membrane-spanning a-helical segments separated by alternating intracel-lular and extracellular loop regions [1,2].
In silico models of the 3D structures of GPCRs have becomeimportant tools for studying and analyzing these receptors for drugdiscovery. In addition to the pioneering X-ray crystallographicstructures of rhodopsin, squid rhodopsin and bovine opsin, thestructures that are available of family A have recently beenexpanded to include the beta adrenergic receptors. Currently thereare about 30 crystal structures available of the inverse or partialagonist bound beta adrenergic receptors in the literature with atleast 17 crystal structures belonging to b1-adrenergic receptor and13 of b2-adrenergic receptor [2,3]. With the inactive-state struc-tures of these receptors the chemists now have experimental datafor several active therapeutic targets to guide the development ofligands. GPCRs exhibit range of conformations depending onnumber of factors such as the presence of a bound ligand, the

P. Tewatia et al. / Biochimie 101 (2014) 168e182 169
membrane environment and the presence of other interactingproteins, owing to their high flexibility [4]. GPCRs, like other inte-gral membrane proteins, require a lipid membrane environment toremain folded. Thus, it is challenging to find conditions to overexpress and purify them [5].
The Beta 3-adrenergic receptor (b3-AR) belongs to the betaadrenergic receptor subfamily Class A GPCR. The b3-AR is known tomediate a wide array of physiological and pharmacological re-sponses including lipolysis and thermogenesis in human and ro-dent adipocyte tissues, smooth muscle relaxation in intestine andincrease in the oxidation of fat. They are also known to play sig-nificant role in effective insulin mediated uptake of glucose andincrease in the energy dissipation [6e8]. These effects of b3-ARhave made it a suitable target for obesity and type II diabetes.Thus there is a dire need to design selective b3-AR agonists.
Two generations of b3-AR agonists have been introduced byvarious groups. First generation b3-AR agonists include ZD7114,BRL26830, BRL37344, CL-316243 and CGP12177A. These agonistsinclude the phenyl ethanolamines derivatives that produced verygood anti diabetic and anti obesity effects on rodent models but inhuman clinical trials they failed to have no thermogenic effect. Theb3-AR agonists second generation includes the aryl oxypropanol-amines which include Solabegron (GW-427353), Amibegron (SR58611A), L-770644, YM 178, BMS-201620, BMS-196085, L 796568and L755507 [9e12].
Pharmacophore hypothesis and 3D-QSAR model was developed[13] undertaking molecular modeling studies to explain the differ-ence in b3-AR agonistic activity in terms of physicochemical prop-erties and chemical features. Structure activity relationship studieshavebeen reported using ligand-based and receptor-basedmethodsbased on 144 b3-AR agonists to identify new lead compounds acti-vatingb3-AR [14]. Usingmolecularmodeling studieswereported3Dmodel of b3-AR obtained using b2-AR as the template. Structureactivity relationship studies were carried out with known b3-ARagonists to understand the binding mode of the receptor. Recently,energetic analysis, ligand-and structure-baseddrugdesignmethodswere used in tandem to identify novel selective compounds to act asb3-AR agonists. The integration of these methods yielded 45 com-pounds that could act as potential b3-AR agonists [15,16].
Agonist and antagonist binding is a dynamic process involvingvarious conformational intermediates [17e20]. The high conforma-tional flexibility of G protein coupled receptors poses hindrance inextracting structural information on agonist binding. To understandthe binding mode of agonists to adrenergic receptors classical mo-lecular dynamics simulations of b1- and b2-AR bound to the fullagonist isoprenaline and in their unliganded form have been per-formed [21]. These simulation studies considered the inherent re-ceptorflexibility and explicit solvation factorswhich are known to besignificant for functioning of GPCR [21]. Molecular dynamics (MD)simulations are known to be crucial in understanding the structuralbiology of membrane proteins. It allows probing the conformationaldynamic behavior within its native environment [22].
In the present study we performed seven all atom MD simula-tions of native b3-AR and b3-AR in complex with the docked po-tential agonists obtained from both ligand based and receptorbased screening [15,16]. The top six ranked compounds from thepool of 45 compounds were subjected to MD simulation studies.The native b3-AR and the complexes were embedded in a hydratedlipid bilayer environment to explore the dynamic perturbations inthe intracellular, extracellular loops and helical bundle structureand to determine the factors responsible for inter and intrahelicalstabilization. The dynamic perturbations induced in the confor-mations of ligand and receptor by the binding of agonists to b3-ARwere also analyzed. Each simulation ranged from 1 to 150 ns with atotal duration of w0.75 ms.
2. Materials and methods
The 3Dmodel of b3-AR wasmodeled using homologymodeling,fold recognition and threading techniques. The predicted structurewas validated using energetics, stereochemical considerations,tertiary structure analysis, docking and simulation studies [15].
2.1. 3D model building and validation
The b3-AR protein sequence with a length of 408 amino acidswas obtained from UNIPROT [23]. A PDBBLAST search of thissequence was performed for selection of templates based onsequence identity and similarity. The crystal structures with asequence identity greater than 45% were chain A of crystal structureof human beta 2 adrenoceptor (PDB ID: 2R4R, identity: 51%), ChainA, Crystal structure of A methylated beta 2 adrenergic receptor-fabcomplex (PDB ID: 3KJ6, identity: 50%), Chain A, turkey beta1adrenergic receptor with stabilizing mutations and bound cyano-pindolol (PDB ID: 2VT4, identity: 50%) and Chain A, high resolutioncrystal structure of human b2-Adrenergic G protein-coupled re-ceptor (PDB ID: 2RH1, identity: 46%). The multiple sequence align-ment of the three subtypes was carried out using CLUSTALW.ITASSER (Iterative implementation of the Threading ASSEmblyRefinement) which is ranked highest in CASPwas used formodelingof the b3-AR. Loop and side chain refinement was achieved byPRIME module of the Schrodinger software suite. Further refine-ment of the b-AR model was done by energy minimization withOptimized Potentials for Liquid Simulations 2005 (OPLS 2005) forcefield implemented in Schrodinger software. PROCHECK [24] andVERIFY-3D were used for stereochemical analysis of models [15].
2.2. Electrostatic charge distribution
The electrostatic charge distribution of b3-AR and crystalstructures of turkey b1-AR (PDB ID: 2VT4), and human b2-AR (PDBID: 2RH1) were generated by solving the PoissoneBoltzmannequations using Schrodinger software suite [25]. The partialcharges of the input structure were used to generate iso-surfacesand a mapping to the molecular surface. Electrostatic charge dis-tributionwas also mapped. A comparison of theoretical structure ofb3-AR was done with respect to extracellular surface charge pre-sent on the proteins.
2.3. Ligand structure preparation and docking studies
The three dimensional structure of the b3-AR potential agonists(Fig. 1) C23H23ClN2O3, compound 1 (C23H24N2O6), compound 2(C15H10FN7O2S), compound 3 (C16H19N5O3), compound 4(C22H22N5O2S1), compound 5 (C18H20N5O1S1) and compound 6(C19H17Cl2N3O4S2) which were obtained by virtual screening andstructure based pharmacophoremodeling [16] weremodeled usingbuild application of Maestro 8.0. The optimization of the geometrywas done by molecular mechanics using impact in a dynamicenvironment using standard TIP4P water model [26]. OptimizedPotentials for Liquid Simulations 2005 (OPLS 2005) force field wasused for energy minimization using Polak-Ribier conjugategradient and Truncated Newton conjugate gradient algorithms.RMS gradient of 0.01 was used as the convergence threshold. Afterthe generation of appropriate conformational models, the ligandswere then prepared by ligprep [27] with Epik [28] at 7.0 � 2.0 pHunits to expand protonation and tautomeric states.
The 3D model of b3-AR was prepared by assigning bond orders,adding hydrogens and treating metals. Exhaustive sampling optionwas used to optimize the hydrogens. A constrained refinement ofthe receptor models were performed until the average root mean

P. Tewatia et al. / Biochimie 101 (2014) 168e182170
square deviation of non-hydrogen atoms reached 0.30 �A usingImpref module of Impact with OPLS 2005. The first minimizationwas performed constraining the heavy atoms in order to allow freerotation of the hydrogens. Subsequently, minimizations were per-formed by decreasing the constraints on the heavy atoms.
The docking studies were carried out between the 3D model ofb3-AR and the six potential agonists. The proteineligand dockingwas carried out using extra precision (XP) method called Glide(Grid-based Ligand Docking with Energetics) [29]. The receptorgrid generation for docking was done using the centroid of selectedactive site residue Asp 117 as well as blind docking. The differentconformations of the compounds were docked flexibly and 1000poses per compound were generated. The analysis of the poses,complexes and the binding affinities between the receptor and li-gands were analyzed using Schrodinger’s suite.
2.4. System preparation
Preparation of the system and all MD simulations were per-formed with the GROMACS package [30,31], employing the OPLS-AA force field [32,33]. The energy of the molecular system wasderived as the sum of bond stretching, bond bending, torsional, andnonbonded terms. The bond stretching and bending parameterswere mostly taken from OPLS-AA. The torsional parameters for thesix ligands were computed from ab initio methods. The restrictedHartree-Fock (RHF) method and the 6-31G* basis set were used tooptimize molecular geometries. All the atomic charges for the sixbound ligands were derived by RESP (Restricted electrostatic po-tential) [34e36] fitting using HF/6-31G* optimized structures andelectrostatic potentials. The RESP charge model consists of a least-squares fit of the charges to the electrostatic potential but withhyperbolic restraints on heavy atom charges. This was followed by asecond fitting stage, needed to fit methyl groups which requireequivalent charges on hydrogen atoms which are not equivalent by
Fig. 1. Chemical structures of th
molecular symmetry. Lennard Jones 1e4 interactions with scalingfactor 0.5 were used for non-bonded function. Coulomb forces wereevaluated with the particleemesh Ewald method. The equations ofmotion were integrated with a 2-fs time step, using the r-RESPAalgorithm to update short- and long-range contributions atdifferent frequencies.
The b3-AR was placed in the pre-equilibrated DPPC lipid bilayer[37,38] and the overlapping lipids were removed using simple cutoff distance of 0.5�A between the protein and the lipid. The resultingsystem consisted of 408 amino acid residues, 121 DPPC mole-cules,9867 water molecules and 14 chlorine ions for a total of39,546 atoms and measured w78 � 79 � 100 �A3 and in additionagonist molecule for the bound form of b3-AR [39,40].
Prior to the MD simulations, the system was energy minimizedand equilibrated using (i) a simulated annealing protocol where thesystem was gradually heated from 0 to 323 K followed by coolingfrom 323 to 0 K and reheating to 323 K. The Berendsen weakcoupling algorithm [41] was applied with a cut off of 1.2 nm forcalculation of Coulomb and van der Waals interactions. (ii) this wasfollowed by equilibration under constant volume (NVT) at 323 Kusing V-rescale (modified Berendsen thermostat) [42] with a cut offof 0.9 and 1.4 nm (iii) Then the system was equilibrated at 323 Kunder constant pressure (NPT) conditions at 1 bar with a time stepof 2 fs using NoseeHoover thermostat [43,44] and ParrinelloeRahman barostat algorithm [45] with a cut off of 1.2 nm.
During the minimization and equilibration the heavy proteinatoms were restrained using a force constant of 1000 kJmol�1 nm�2. Finally a 150.0 ns of dynamics simulation was per-formed for unbound form and 100 ns each for the bound form ofreceptor in order to explore dynamic perturbations of the receptorproteinwith a pressure of 1 atm using the extended ensemble NoseHoover thermostat and extended ensemble ParinelloeRahmanpressure coupling. The restraints on protein heavy atoms wereremoved.
e b3-AR potential agonists.

P. Tewatia et al. / Biochimie 101 (2014) 168e182 171
All simulations were carried out at a constant temperature of323 K. The relaxation times were set to constant sT of 0.5 ps. Thesemi isotropic pressure coupling was used with a constant pressureof 1 bar and a coupling constant sp of 2.0 ps. An estimatedisothermal compressibility of 4.575 � 10�4 (kJ mol�1 nm�3)�1 wasused. A time step of 2 fs was used for the integration of the equa-tions of motions and all bond distances involving hydrogen atomswere constrained using LINCS [46]. Van der Waals and short rangeelectrostatic interactions were cut off at 12 �A and the pairlist wasupdated at every 10 fs. Particle Mesh Ewald (PME) method wasused to compute long range electrostatic interactions [47]. Afterevery 50 ps snapshots were recorded to the trajectory file forsubsequent analysis. All the molecular graphics were producedusing Visual molecular dynamics (VMD) [48].
2.5. Estimation of binding free energy
Prime MMeGBSA [35] was used to calculate the binding-freeenergy (DGbind) of each potential agonist, using the followingequation:
DGbind ¼ DEMM þ DGsolv þ DGSA
where DEMM is the difference in the minimized energies betweenthe b3-AR receptoreligand complex and the sum of the energies ofthe un-liganded b3-AR and ligands used in this study. DGsolv is thedifference in the GBSA solvation energy of the b3-AR receptoreligand complex and the sum of the solvation energies for the un-liganded b3-AR. DGSA is the difference in surface area energies forthe complex and the sum of the surface area energies for the un-liganded b3-AR and ligands [49].
For this simulations were performed using two differentprotocols:
(1) The 6 input complex structures were obtained from molec-ular docking results when compounds 1e6 were dockedwith the modeled b3-AR using GLIDE XP. Only the bestdocked pose in each case was considered.
(2) The average structures collected from the production phaseof the potential agonist in complexes with the b3-AR ob-tained after performing MD simulations were subjected toPrime MMeGBSA for free energy calculations. The ligandposes obtained were minimized using the local optimizationfeature in Prime using the OPLS-2005 force field.
2.6. Binding pockets
Site-Map ofMaestrowas used for binding site predictions in boththe bound and unbound state of b3-AR. To identify possible bindingsites, physical descriptors such as size, degree of enclosure/exposure,hydrophobic/-philic character, tightness and hydrogen-bondingpossibilities were considered [50]. The detection of channels thatconnect the binding pockets to the extracellular surface in the un-bound and the bound state of the receptor with the most active andleastputative ligands inour studywasdone. Randomsnapshotsweretaken along equilibration and production stages of simulation to findout the dynamic perturbation in the binding pockets.
3. Results and discussion
3.1. Molecular modeling of b3-AR
Homology modeling was used to predict the 3D structure of b3-AR bymaking use of the structural information from the previouslyresolved X-Ray structures with significant similarity [15]. Out of thecrystallographic structures obtained after BLAST search, 2RH1 and
2VT4 with a sequence identity of 46% and 51% respectively werechosen as templates for the homology modeling. These structureswere chosen as templates keeping in mind the high resolution ofthese two structures viz. 2.4 �A for 2RH1 and 2.7 �A for 2VT4. 2RH1was majorly used in the homology modeling owing to its higherresolution and as it is from human source. The multiple sequencealignment of the three receptor subtypes is shown in Fig. 2, wherethe motifs S(N)LAxAD D(E)RY, CWxP, two SxxxS and NPxxY aremarked in tapered rectangles and the active site residues Trp 113,Asp 117, Phe 198, Ser 208, Ser 209, Ser 212, Trp 305, Phe 308, Phe309, Asn 312 and Asn 332 of b3-AR, conserved across the threespecies aremarkedwith red stars. Amajor difference is in the activebinding site residue Arg 315 of b3-AR which corresponds to theresidue His 296 of b2-AR, Asn 313 of b1-AR. The shared motifs andfeatures owing to the relatively high identity between b3-AR andother beta adrenergic receptor subtypes are listed in Table 1. Thesequence alignment of the three subtypes of b-ARs reveals thepresence of three set of serine residues Ser 203, Ser 204 and Ser 207of b2-AR that are highly conserved and belong to the SxxxS motif inTM5 typical to all b-AR subtypes, Ser 208, Ser 209 and Ser 212 of b3-AR and Ser 211, Ser 212 and Ser 215 of b1-AR respectively. It hasbeen previously reported that these serines are conserved in theupper region of TM5 of b2-AR and are directly involved in receptoractivation and agonist binding. The native b3-AR Ser 208 and Ser209 are located at the interface of TM5 and form strong hydrogenbond with Thr 122 of TM3 which corresponds to the Thr 118 of b2-AR. This is in agreement with the observation that SxxxS motifsoften mediate helix interactions by forming interhelical hydrogenbonds. Moreover, mutational studies of Ser 207 of b2-AR havesuggested that serine is not exposed to the polar binding crevice inthe ligand free receptor. Ser 212 of b3-AR corresponds to Ser 207 inb2-AR, Ser 215 in b1-AR and His 211 in rhodopsin respectively,whereas Thr 122 of b3-AR corresponds to Thr 118 of b2AR, Thr 126of b1AR and to Glu-122 in rhodopsin respectively [51].
I-TASSER generated five models and the 3D model, which had aconfidence score (C-score: measure of the quality of the predictedmodel) of �0.56 was selected for further study. Loop refinementand side chain prediction of the 3Dmodel obtained by ITASSER wascarried out by PRIME [49] To alleviate steric clashes furtherrefinement of the predicted structure was done. The convergencecriteria of 0.3 �A RMSD for non hydrogen atoms was used duringminimization. The validation of the model was done by PROCHECKand VERIFY-3D. PDBsum [52] was used to gather the secondarystructural information for the protein which revealed the trans-membrane helices of b3-AR as TM1 (39e61), TM2 (74e96), TM3(111e133), TM4 (153e175), TM5 (211e230), TM6 (291e313) andTM7 (323e345) [15].
3.2. Electrostatic charge distribution
Electrostatic Charge distributionwas mapped for all the three b-AR subtypes to find specific surface differences. The electrostaticcharge distribution analysis showed difference in the electrostaticcharge of the binding site cavities of all the three subtypes. Strikingdifferences were observed in the extracellular surface charge of thethree subtypes. The surface charge was in order of b1-AR > b2-AR > b3-AR i.e. the negative charge was highest in case of b1-AR,and lowest in case of b3-AR (Fig. 3). The difference in the surfacecharge could be attributed to the presence of non conserved resi-dues Asn 313 in b1-AR, His 296 in b2-AR and Arg 315 in b3-AR.There is considerable difference in terms of overall size, density,charge and potential in these specific residues where Arg 315 in b3-AR is positive ionizable residue, His 296 of b2-AR is slightly positiveand Asn 313 of b1-AR is uncharged. Additionally, Ala 197 in theECL2 of b3-AR corresponds to the aspartic acid residue in both b1-

Fig. 2. Multiple sequence alignment of human b3-AR with human b2-AR (PDB ID: 2RH1) and turkey b1-AR (PDB ID: 2VT4). Dark blue color indicates the identical residues, mediumblue color indicates strong similarity and light blue color indicates the weak similarity. Conserved motifs across the three b-AR species are shown in boxes, while the residuesinvolved in ligand binding are marked with a red “*”.
Table 1Comparison of functionally important residues in b3-AR and related GPCR receptors.
b3-AR (residuenumber andstructural domain)
GPCR Shared features
C110eC196C189eC195
All GPCRs Disulphide bridge TM3-ECL3Disulphide bridge ECL3-ECL3
N55eI59TM1
Beta Adrenergicreceptors
NxLVI
D134eY136TM3
All GPCRs D/E-R-Y/W motif
S78eD83TM2
Beta Rhodopsin andAdrenergic receptors
S(N)LAxAD motif
S165eS169TM4
Beta Rhodopsin andAdrenergic receptors
SxxxS motif
S208eS212TM5
Beta Rhodopsin andAdrenergic receptors
SxxxS motif
L294eM298TM6
Beta Adrenergicreceptor
LxxIM
C304eP307TM6
All GPCRs CWxP motif
N342eY346TM7
All GPCRs N/DPxxY motif
D351eF356Helix8
Beta Adrenergicreceptor
DFRxAF
P. Tewatia et al. / Biochimie 101 (2014) 168e182172
AR (Asp 200) and b2-AR (Asp 192). The presence of these acidicresidues imparts more negative potential to b1-AR and b2-AR.These differences may be playing a significant role in the recogni-tion and initial binding of the ligands in the three subtypes of b-AR.The intracellular loops of the b3-AR have more positive potentialthan the extracellular loops.
3.3. Molecular dynamics
3.3.1. Dynamic behavior of the solvated b3-AR embedded in themembrane complexed with potential agonists
Overall, seven simulations were run, with a total duration of0.75 ms. The effects of the presence/absence of ligandwere exploredto understand the dynamic perturbations in the ligand unboundand bound complexes of b3-AR. Briefly simulations were per-formed starting with equilibrated systems:
1. One simulation (150 ns) for unbound form of human b3-AR.2. Six simulations each 100 ns of bound form of human b3-ARwith
6 different compounds identified to be potential agonists byvirtual screening and pharmacophore detection techniques.
The trajectories for each simulation were analyzed. A slow andsteady decrease in the potential energy was observed during thecourse of simulation for all of the seven runs. One of the importantcriterions to judge the conformational stability of the protein wascalculation of root mean square deviations (RSMDs) of Ca atomsfrom their initial coordinates.
Unbound form of b3-AR showed larger rearrangements (0.8 �A)as compared to the bound form. Moreover, in the different com-plexes of bound form of b3-AR the RSMD of the protein backbone aswell as ligand showed smaller re-arrangements (max. 0.5 �A) indi-cating that the equilibration procedure was sufficient to stabilizethe complexes (Fig. 4a and b). In the ligand-bound form, RSMD ofthe compound 1 showed the most variability and fluctuations inbinding pocket of b3-AR, thus resulting in more flexibility of theligand as compared to other compounds. In case of compound 2RSMD of ligand showed rearrangements for the first 20 ns ofsimulation and then remained stable upto 90 ns with minor rear-rangements until 100 ns. For compound 3 the RSMD of ligand
shows rearrangements for the first 30 ns and then was observed tobe fairly stable but shows some fluctuations in the last 10 ns ofsimulation. In compound 4 the ligand RSMD did not show majorfluctuations and remains fairly stable at the last 10 ns of simulationrun. Also the backbone RSMD of b3-AR bound to compound 4showed a smooth decline to become stable at the end of thesimulation time. In compound 5 the lowest RSMDwas observed butthere were notable fluctuations in the backbone RSMD of b3-AR.Though for compound 6 there was increase in the RSMD of theligand at start of the simulation but the RSMD remains fairly stablethereafter. The backbone RSMD of b3-AR in compound 6 also re-mains stable during the course of the simulation.
3.3.2. Extracellular loop and intracellular loopThe Intracellular loops (ICLs) and Extracellular loops (ECLs) i.e.
ECL1: 1e38, ICL1: 62e73, ECL2: 97e110, ICL2: 134e152, ECL3: 176e

Fig. 3. Electrostatic charge distribution of three subtypes of b1-AR, b2-AR and b3-AR respectively. Red color indicates negative charge, blue indicates positive charge and whiteindicates neutral charge.
P. Tewatia et al. / Biochimie 101 (2014) 168e182 173
210 ICL3: 231e289, ECL4: 314e320 and ICL4: 346e408 regionsshowed markedly different dynamics behavior.
In the simulation of unbound b3-AR, ECL2 is displaced from thebinding site and moves away from the ligand interaction region.The intracellular domains of the b-ARs are similar except for ICL2.The cytoplasmic loop (ICL 2) domain of the b1-AR has a short a-helix which is absent in b2-AR, b3-AR and rhodopsin. The ECL2domain of b-AR subtypes comprise of a short a-helix stabilized bythe disulfide bonds, whereas the ECL2 of rhodopsin has a short betasheet [53]. In ECL3 there is an intra loop disulfide bond between Cys189 and Cys 195 which may be helpful in stabilizing the ECL2. Asecond disulfide bond is formed between Cys 110 of TM3 and Cys196 which might be effective in holding the ECL2 to the trans-membrane core. The E(D)RY motif which forms a network of polarinteractions that bridges the two transmembrane domains stabi-lizing the inactive state conformations has been reported to behighly conserved amino acids amongmost of the GPCRs. It has beenreported that the salt bridge between Arg (3.50)1 of the conservedDRYmotif in TM3 and Glu (6.30)1 at the cytoplasmic side of the twohelices e TM3 and TM6 of the rhodopsin is termed as the ‘ioniclock’ which maintains the inactive state of the GPCRs mainlythrough TM6 stabilization [53e55]. The biophysical studies haverevealed that the structure around the ionic lock can be modulatedby partial and full agonists. During the course of simulation no ioniclock formation occurred in b3-AR.
During the course of the simulation dynamic perturbationsoccurred in the receptor conformation. One of the interesting per-turbations was the formation of two beta sheets in ECL2 (His 102 toPro 104) and ECL3 (Cys 195 to Ala 197) anti parallel to each otherand in close proximity to the ligand binding domain. These betasheets formed for short duration and were observed at 10.0 ns,17.0 ns, 45.0 nse46.0 ns, and 90.0 ns.
3.3.3. TM helicesAn important criterion during analysis of trajectories was to
check whether the helical structure of the seven TM domains waspreserved or not. To measure the structural stability of the b3-ARreceptor model the root mean square deviations (RMSDs) of Caatoms were calculated from the initial coordinates (Table 2).Overall, higher fluctuations were observed in the case of TM4whereas least fluctuations were observed in TM1 for both thebound and unbound states of the b3-AR.
The analysis of the binding site residues in both rhodopsin andthe beta adrenergic receptors show that the interactions with hy-drophobic and polar amino acids from transmembrane TM3, TM5,TM6 and TM7 are involved in ligand binding. The selectivity is also
1 Ballesteros Weinstein numbering.
based on the specific conformational preferences in neighboringamino acids. The corresponding conserved amino acid Trp 305 ofthe modeled b3-AR is proposed to undergo important conforma-tional transitions in the process of activation.
3.3.4. Hydrogen bond network in the unbound and bound state ofthe receptor
The role of hydrogen bond interactions in stabilizing the helicalbundles of b3-AR was examined during the course of simulation.Hydrogen bonds were calculated for a donor acceptor distance ofmaximum 3.2�A and angle cut off of 20�. The average numbers of H-bonds during the course of simulation of unbound b3-AR were 135.In the bound state the compound 4 in complex with b3-AR showedan average of 143 hydrogen bonds. Complex 1 and Complex 5 madean average of 73 hydrogen bonds whereas complex 4 made on anaverage 68 hydrogen bonds. Complex 2 and 6 on an average made66 hydrogen bonds each. The results showed that the residuesfrequently involved in these intrahelical H-bonds were Asn 55/Ala340 and Asn 55/Tyr 336 for TM1/TM7; Ser 78/Thr 127, Asp 83/Ser124 and Glu 126/Ser 165 for TM3/TM4; Thr 300/Asn 342 and Thr300/Asn 338 for TM6/TM7.
3.3.5. Salt bridgesThe trajectory analysis showed that a total of 25 salt bridges
were formed in the unbound form of b3-AR whereas in the boundforms of b3-AR the average salt bridges formed were 17. Thealignment of b3-AR with Rhodopsin, b1-AR and b2-AR revealedthat residues Arg 135 and Glu 247 are highly conserved in all thefour receptor classes. These conserved residues in Rhodopsin (Arg135 and Glu 247) form a salt bridge termed as ionic lock to keep thereceptor in the inactive state, though the corresponding conservedresidues in b1-AR (Arg 139 and Glu 285) and b2-AR (Arg 131 andGlu 268) show no evidence of the ionic lock formation [56,57].Similar trend is observed in b3-AR where the homologous residueArg 135 of the DRYmotif is not involved in any salt bridge formationwith Glu 287. However, Glu 287 forms three salt bridges in thecourse of simulation namely with Arg 239 of the ICL3, Arg 348 andArg 376 of ICL4. To monitor salt bridge flexibility, the distance be-tween atom Cg of residue Arg 135 and Nz of Glu 287 was calculatedfor both the bound and unbound states of b3-AR. In unbound statethe distance between residues Arg 135 and Glu 287 that wasplotted over the course of the simulation appears to be separatedby an average distance of 15 �A whereas the average distance wasapprox 10 �A in the bound state.
3.4. Binding of potential agonists with b3-AR
The dynamic perturbations in the receptor-compound com-plexes were obtained by MD runs of 100 ns each starting from the
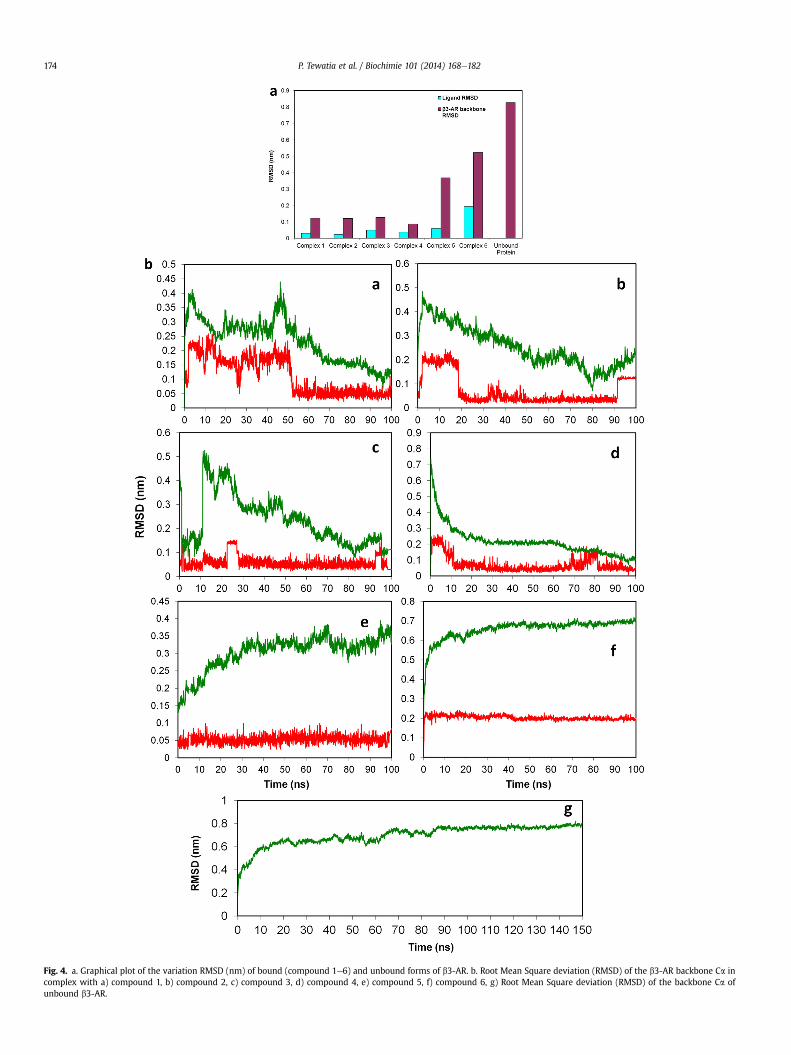
Fig. 4. a. Graphical plot of the variation RMSD (nm) of bound (compound 1e6) and unbound forms of b3-AR. b. Root Mean Square deviation (RMSD) of the b3-AR backbone Ca incomplex with a) compound 1, b) compound 2, c) compound 3, d) compound 4, e) compound 5, f) compound 6, g) Root Mean Square deviation (RMSD) of the backbone Ca ofunbound b3-AR.
P. Tewatia et al. / Biochimie 101 (2014) 168e182174
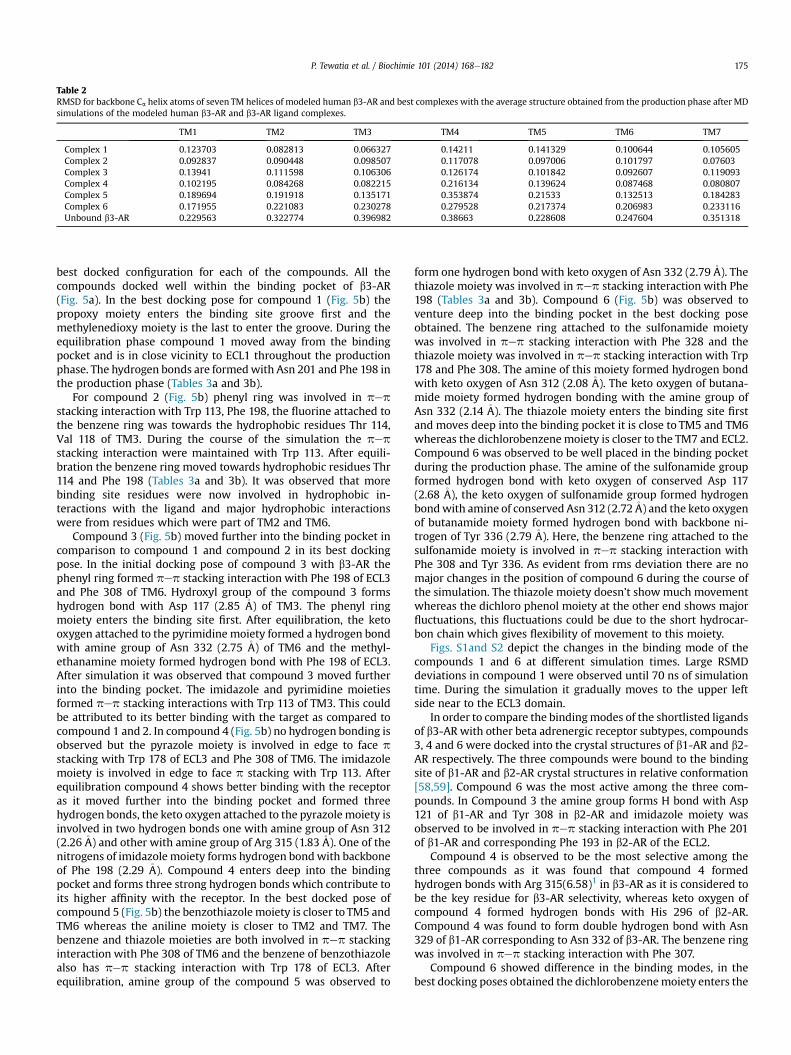
Table 2RMSD for backbone Ca helix atoms of seven TM helices of modeled human b3-AR and best complexes with the average structure obtained from the production phase after MDsimulations of the modeled human b3-AR and b3-AR ligand complexes.
TM1 TM2 TM3 TM4 TM5 TM6 TM7
Complex 1 0.123703 0.082813 0.066327 0.14211 0.141329 0.100644 0.105605Complex 2 0.092837 0.090448 0.098507 0.117078 0.097006 0.101797 0.07603Complex 3 0.13941 0.111598 0.106306 0.126174 0.101842 0.092607 0.119093Complex 4 0.102195 0.084268 0.082215 0.216134 0.139624 0.087468 0.080807Complex 5 0.189694 0.191918 0.135171 0.353874 0.21533 0.132513 0.184283Complex 6 0.171955 0.221083 0.230278 0.279528 0.217374 0.206983 0.233116Unbound b3-AR 0.229563 0.322774 0.396982 0.38663 0.228608 0.247604 0.351318
P. Tewatia et al. / Biochimie 101 (2014) 168e182 175
best docked configuration for each of the compounds. All thecompounds docked well within the binding pocket of b3-AR(Fig. 5a). In the best docking pose for compound 1 (Fig. 5b) thepropoxy moiety enters the binding site groove first and themethylenedioxy moiety is the last to enter the groove. During theequilibration phase compound 1 moved away from the bindingpocket and is in close vicinity to ECL1 throughout the productionphase. The hydrogen bonds are formedwith Asn 201 and Phe 198 inthe production phase (Tables 3a and 3b).
For compound 2 (Fig. 5b) phenyl ring was involved in pepstacking interaction with Trp 113, Phe 198, the fluorine attached tothe benzene ring was towards the hydrophobic residues Thr 114,Val 118 of TM3. During the course of the simulation the pepstacking interaction were maintained with Trp 113. After equili-bration the benzene ring moved towards hydrophobic residues Thr114 and Phe 198 (Tables 3a and 3b). It was observed that morebinding site residues were now involved in hydrophobic in-teractions with the ligand and major hydrophobic interactionswere from residues which were part of TM2 and TM6.
Compound 3 (Fig. 5b) moved further into the binding pocket incomparison to compound 1 and compound 2 in its best dockingpose. In the initial docking pose of compound 3 with b3-AR thephenyl ring formed pep stacking interaction with Phe 198 of ECL3and Phe 308 of TM6. Hydroxyl group of the compound 3 formshydrogen bond with Asp 117 (2.85 �A) of TM3. The phenyl ringmoiety enters the binding site first. After equilibration, the ketooxygen attached to the pyrimidine moiety formed a hydrogen bondwith amine group of Asn 332 (2.75 �A) of TM6 and the methyl-ethanamine moiety formed hydrogen bond with Phe 198 of ECL3.After simulation it was observed that compound 3 moved furtherinto the binding pocket. The imidazole and pyrimidine moietiesformed pep stacking interactions with Trp 113 of TM3. This couldbe attributed to its better binding with the target as compared tocompound 1 and 2. In compound 4 (Fig. 5b) no hydrogen bonding isobserved but the pyrazole moiety is involved in edge to face pstacking with Trp 178 of ECL3 and Phe 308 of TM6. The imidazolemoiety is involved in edge to face p stacking with Trp 113. Afterequilibration compound 4 shows better binding with the receptoras it moved further into the binding pocket and formed threehydrogen bonds, the keto oxygen attached to the pyrazole moiety isinvolved in two hydrogen bonds one with amine group of Asn 312(2.26�A) and other with amine group of Arg 315 (1.83�A). One of thenitrogens of imidazole moiety forms hydrogen bondwith backboneof Phe 198 (2.29 �A). Compound 4 enters deep into the bindingpocket and forms three strong hydrogen bonds which contribute toits higher affinity with the receptor. In the best docked pose ofcompound 5 (Fig. 5b) the benzothiazolemoiety is closer to TM5 andTM6 whereas the aniline moiety is closer to TM2 and TM7. Thebenzene and thiazole moieties are both involved in pep stackinginteraction with Phe 308 of TM6 and the benzene of benzothiazolealso has pep stacking interaction with Trp 178 of ECL3. Afterequilibration, amine group of the compound 5 was observed to
form one hydrogen bond with keto oxygen of Asn 332 (2.79�A). Thethiazole moiety was involved in pep stacking interaction with Phe198 (Tables 3a and 3b). Compound 6 (Fig. 5b) was observed toventure deep into the binding pocket in the best docking poseobtained. The benzene ring attached to the sulfonamide moietywas involved in pep stacking interaction with Phe 328 and thethiazole moiety was involved in pep stacking interaction with Trp178 and Phe 308. The amine of this moiety formed hydrogen bondwith keto oxygen of Asn 312 (2.08 �A). The keto oxygen of butana-mide moiety formed hydrogen bonding with the amine group ofAsn 332 (2.14 �A). The thiazole moiety enters the binding site firstand moves deep into the binding pocket it is close to TM5 and TM6whereas the dichlorobenzene moiety is closer to the TM7 and ECL2.Compound 6 was observed to be well placed in the binding pocketduring the production phase. The amine of the sulfonamide groupformed hydrogen bond with keto oxygen of conserved Asp 117(2.68 �A), the keto oxygen of sulfonamide group formed hydrogenbondwith amine of conserved Asn 312 (2.72�A) and the keto oxygenof butanamide moiety formed hydrogen bond with backbone ni-trogen of Tyr 336 (2.79 �A). Here, the benzene ring attached to thesulfonamide moiety is involved in pep stacking interaction withPhe 308 and Tyr 336. As evident from rms deviation there are nomajor changes in the position of compound 6 during the course ofthe simulation. The thiazole moiety doesn’t showmuch movementwhereas the dichloro phenol moiety at the other end shows majorfluctuations, this fluctuations could be due to the short hydrocar-bon chain which gives flexibility of movement to this moiety.
Figs. S1and S2 depict the changes in the binding mode of thecompounds 1 and 6 at different simulation times. Large RSMDdeviations in compound 1 were observed until 70 ns of simulationtime. During the simulation it gradually moves to the upper leftside near to the ECL3 domain.
In order to compare the bindingmodes of the shortlisted ligandsof b3-AR with other beta adrenergic receptor subtypes, compounds3, 4 and 6 were docked into the crystal structures of b1-AR and b2-AR respectively. The three compounds were bound to the bindingsite of b1-AR and b2-AR crystal structures in relative conformation[58,59]. Compound 6 was the most active among the three com-pounds. In Compound 3 the amine group forms H bond with Asp121 of b1-AR and Tyr 308 in b2-AR and imidazole moiety wasobserved to be involved in pep stacking interaction with Phe 201of b1-AR and corresponding Phe 193 in b2-AR of the ECL2.
Compound 4 is observed to be the most selective among thethree compounds as it was found that compound 4 formedhydrogen bonds with Arg 315(6.58)1 in b3-AR as it is considered tobe the key residue for b3-AR selectivity, whereas keto oxygen ofcompound 4 formed hydrogen bonds with His 296 of b2-AR.Compound 4 was found to form double hydrogen bond with Asn329 of b1-AR corresponding to Asn 332 of b3-AR. The benzene ringwas involved in pep stacking interaction with Phe 307.
Compound 6 showed difference in the binding modes, in thebest docking poses obtained the dichlorobenzenemoiety enters the
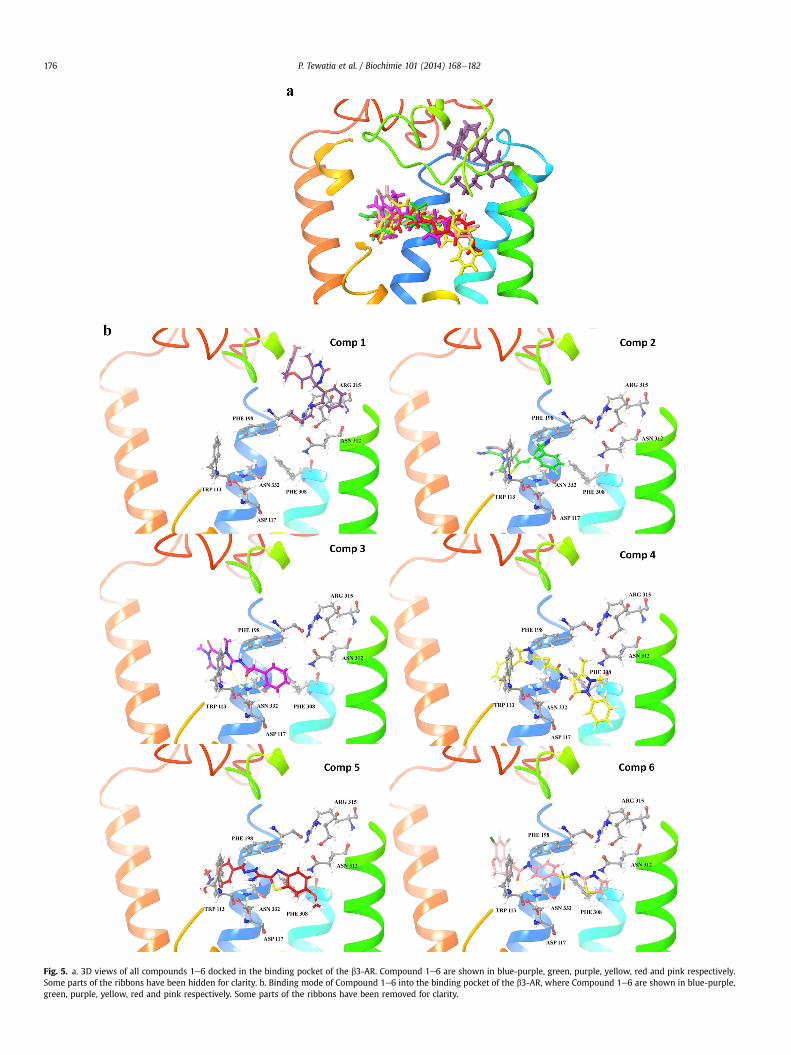
Fig. 5. a. 3D views of all compounds 1e6 docked in the binding pocket of the b3-AR. Compound 1e6 are shown in blue-purple, green, purple, yellow, red and pink respectively.Some parts of the ribbons have been hidden for clarity. b. Binding mode of Compound 1e6 into the binding pocket of the b3-AR, where Compound 1e6 are shown in blue-purple,green, purple, yellow, red and pink respectively. Some parts of the ribbons have been removed for clarity.
P. Tewatia et al. / Biochimie 101 (2014) 168e182176
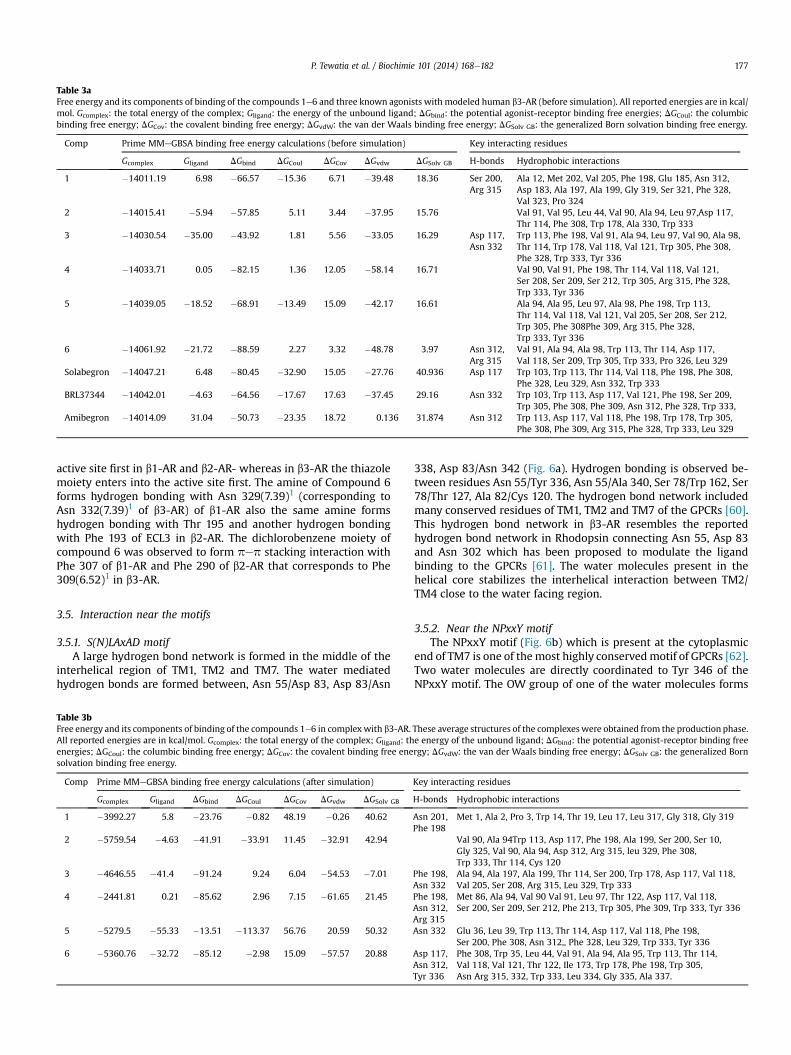
Table 3aFree energy and its components of binding of the compounds 1e6 and three known agonists with modeled human b3-AR (before simulation). All reported energies are in kcal/mol. Gcomplex: the total energy of the complex; Gligand: the energy of the unbound ligand; DGbind: the potential agonist-receptor binding free energies; DGCoul: the columbicbinding free energy; DGCov: the covalent binding free energy; DGvdW: the van der Waals binding free energy; DGSolv GB: the generalized Born solvation binding free energy.
Comp Prime MMeGBSA binding free energy calculations (before simulation) Key interacting residues
Gcomplex Gligand DGbind DGCoul DGCov DGvdw DGSolv GB H-bonds Hydrophobic interactions
1 �14011.19 6.98 �66.57 �15.36 6.71 �39.48 18.36 Ser 200,Arg 315
Ala 12, Met 202, Val 205, Phe 198, Glu 185, Asn 312,Asp 183, Ala 197, Ala 199, Gly 319, Ser 321, Phe 328,Val 323, Pro 324
2 �14015.41 �5.94 �57.85 5.11 3.44 �37.95 15.76 Val 91, Val 95, Leu 44, Val 90, Ala 94, Leu 97,Asp 117,Thr 114, Phe 308, Trp 178, Ala 330, Trp 333
3 �14030.54 �35.00 �43.92 1.81 5.56 �33.05 16.29 Asp 117,Asn 332
Trp 113, Phe 198, Val 91, Ala 94, Leu 97, Val 90, Ala 98,Thr 114, Trp 178, Val 118, Val 121, Trp 305, Phe 308,Phe 328, Trp 333, Tyr 336
4 �14033.71 0.05 �82.15 1.36 12.05 �58.14 16.71 Val 90, Val 91, Phe 198, Thr 114, Val 118, Val 121,Ser 208, Ser 209, Ser 212, Trp 305, Arg 315, Phe 328,Trp 333, Tyr 336
5 �14039.05 �18.52 �68.91 �13.49 15.09 �42.17 16.61 Ala 94, Ala 95, Leu 97, Ala 98, Phe 198, Trp 113,Thr 114, Val 118, Val 121, Val 205, Ser 208, Ser 212,Trp 305, Phe 308Phe 309, Arg 315, Phe 328,Trp 333, Tyr 336
6 �14061.92 �21.72 �88.59 2.27 3.32 �48.78 3.97 Asn 312,Arg 315
Val 91, Ala 94, Ala 98, Trp 113, Thr 114, Asp 117,Val 118, Ser 209, Trp 305, Trp 333, Pro 326, Leu 329
Solabegron �14047.21 6.48 �80.45 �32.90 15.05 �27.76 40.936 Asp 117 Trp 103, Trp 113, Thr 114, Val 118, Phe 198, Phe 308,Phe 328, Leu 329, Asn 332, Trp 333
BRL37344 �14042.01 �4.63 �64.56 �17.67 17.63 �37.45 29.16 Asn 332 Trp 103, Trp 113, Asp 117, Val 121, Phe 198, Ser 209,Trp 305, Phe 308, Phe 309, Asn 312, Phe 328, Trp 333,
Amibegron �14014.09 31.04 �50.73 �23.35 18.72 0.136 31.874 Asn 312 Trp 113, Asp 117, Val 118, Phe 198, Trp 178, Trp 305,Phe 308, Phe 309, Arg 315, Phe 328, Trp 333, Leu 329
P. Tewatia et al. / Biochimie 101 (2014) 168e182 177
active site first in b1-AR and b2-AR- whereas in b3-AR the thiazolemoiety enters into the active site first. The amine of Compound 6forms hydrogen bonding with Asn 329(7.39)1 (corresponding toAsn 332(7.39)1 of b3-AR) of b1-AR also the same amine formshydrogen bonding with Thr 195 and another hydrogen bondingwith Phe 193 of ECL3 in b2-AR. The dichlorobenzene moiety ofcompound 6 was observed to form pep stacking interaction withPhe 307 of b1-AR and Phe 290 of b2-AR that corresponds to Phe309(6.52)1 in b3-AR.
3.5. Interaction near the motifs
3.5.1. S(N)LAxAD motifA large hydrogen bond network is formed in the middle of the
interhelical region of TM1, TM2 and TM7. The water mediatedhydrogen bonds are formed between, Asn 55/Asp 83, Asp 83/Asn
Table 3bFree energy and its components of binding of the compounds 1e6 in complex with b3-AR.All reported energies are in kcal/mol. Gcomplex: the total energy of the complex; Gligand: thenergies; DGCoul: the columbic binding free energy; DGCov: the covalent binding free enesolvation binding free energy.
Comp Prime MMeGBSA binding free energy calculations (after simulation)
Gcomplex Gligand DGbind DGCoul DGCov DGvdw DGSolv GB
1 �3992.27 5.8 �23.76 �0.82 48.19 �0.26 40.62
2 �5759.54 �4.63 �41.91 �33.91 11.45 �32.91 42.94
3 �4646.55 �41.4 �91.24 9.24 6.04 �54.53 �7.01
4 �2441.81 0.21 �85.62 2.96 7.15 �61.65 21.45
5 �5279.5 �55.33 �13.51 �113.37 56.76 20.59 50.32
6 �5360.76 �32.72 �85.12 �2.98 15.09 �57.57 20.88
338, Asp 83/Asn 342 (Fig. 6a). Hydrogen bonding is observed be-tween residues Asn 55/Tyr 336, Asn 55/Ala 340, Ser 78/Trp 162, Ser78/Thr 127, Ala 82/Cys 120. The hydrogen bond network includedmany conserved residues of TM1, TM2 and TM7 of the GPCRs [60].This hydrogen bond network in b3-AR resembles the reportedhydrogen bond network in Rhodopsin connecting Asn 55, Asp 83and Asn 302 which has been proposed to modulate the ligandbinding to the GPCRs [61]. The water molecules present in thehelical core stabilizes the interhelical interaction between TM2/TM4 close to the water facing region.
3.5.2. Near the NPxxY motifThe NPxxY motif (Fig. 6b) which is present at the cytoplasmic
end of TM7 is one of the most highly conservedmotif of GPCRs [62].Two water molecules are directly coordinated to Tyr 346 of theNPxxY motif. The OW group of one of the water molecules forms
These average structures of the complexes were obtained from the production phase.e energy of the unbound ligand; DGbind: the potential agonist-receptor binding freergy; DGvdW: the van der Waals binding free energy; DGSolv GB: the generalized Born
Key interacting residues
H-bonds Hydrophobic interactions
Asn 201,Phe 198
Met 1, Ala 2, Pro 3, Trp 14, Thr 19, Leu 17, Leu 317, Gly 318, Gly 319
Val 90, Ala 94Trp 113, Asp 117, Phe 198, Ala 199, Ser 200, Ser 10,Gly 325, Val 90, Ala 94, Asp 312, Arg 315, leu 329, Phe 308,Trp 333, Thr 114, Cys 120
Phe 198,Asn 332
Ala 94, Ala 197, Ala 199, Thr 114, Ser 200, Trp 178, Asp 117, Val 118,Val 205, Ser 208, Arg 315, Leu 329, Trp 333
Phe 198,Asn 312,Arg 315
Met 86, Ala 94, Val 90 Val 91, Leu 97, Thr 122, Asp 117, Val 118,Ser 200, Ser 209, Ser 212, Phe 213, Trp 305, Phe 309, Trp 333, Tyr 336
Asn 332 Glu 36, Leu 39, Trp 113, Thr 114, Asp 117, Val 118, Phe 198,Ser 200, Phe 308, Asn 312,, Phe 328, Leu 329, Trp 333, Tyr 336
Asp 117,Asn 312,Tyr 336
Phe 308, Trp 35, Leu 44, Val 91, Ala 94, Ala 95, Trp 113, Thr 114,Val 118, Val 121, Thr 122, Ile 173, Trp 178, Phe 198, Trp 305,Asn Arg 315, 332, Trp 333, Leu 334, Gly 335, Ala 337.

P. Tewatia et al. / Biochimie 101 (2014) 168e182178
Hydrogen bond with hydroxyl group of Tyr 346. This bonding leadsto the TM1 and TM7 in this region to be closely packed. The otherwater molecule forms one hydrogen bond with Tyr 336 and onewith Asp 134.
In complex 1 hydrogen bonds are formed between Thr 300 ofTM6 with Arg 348 of ICL4 and Asn 338 of TM7 near to the NPxxYmotif. Asp 83 of SLAxAD motif forms hydrogen bond with Asn 342of NPxxY motif. In complex 2 no major interactions were observedwith the NPxxY motif. In complex 3, hydrogen bonding is observedbetween Val 48 of TM1 and Arg 348 of ICL4, Asn 338 of TM7 withThr 300 of TM6 and Asn 342 of TM7. Water mediated hydrogenbond is formed between Ser 349 of ICL4 and Val 71 of ICL1 and Ile345 of TM7. In complex 4 hydrogen bonding is observed betweenThr 300 of TM6 and Asn 342 of TM7. In complex 5 direct hydrogenbonding is observed between Thr 293 of TM6 with Ser 349 of ICL4near to NPxxY motif. In complex 6 Thr 51 forms hydrogen bondwith Ser 339.
3.5.3. Near the CWxP motifA water molecule was trapped in the space around Trp 305
and Phe 309 as shown in Fig. 6c. The keto oxygen of Thr 122forms a water mediated hydrogen bond with the amine (NA) ofTrp 305. Cys 304 of the CWxP motif forms hydrogen bond withLeu 334 of TM 7. This highly conserved CWxP motif that isconserved in all the GPCR families is proposed to undergo majorconformational transitions in the GPCR activation, referred to asthe ‘rotamer toggle switch’ [63]. In the process of activation theconserved residue Trp 305 of the CWxP in the b3-AR appears toundergo conformational transitions. The ligand binding foradrenergic receptors and rhodopsin is mediated by polar andhydrophobic residues from the transmembrane domains TM3,TM5, TM6 and TM7 [64].
For complex 1 hydrogen bonding was observed between Cys304 (TM6) and Gly 335 (TM7), keto oxygen of Asp 117 is involved inhydrogen bonding with Trp 305 and Asn 332. Water mediatedhydrogen bond was formed between Trp 305 and Asp 117. Forcomplex 2 hydrogen bonding was observed between Asp 83 (TM1)and Asn 342, Asp 83 and Ser 339, Gly 299 and Leu 303, Thr 302 andGly 299 and Thr 300 with Asn 342. In complex 3 hydrogen bondingis observed between Cys 304 and Leu 334. One water moleculedisplayed hydrogen bonding with Cys 304, Phe 308 and Leu 331. In
Fig. 6. a) H bond network viewed from the intracellular side in b3-AR after simulation. For cyellow lines. The S(N)LAxAD residues are From Ser 78 to Asp 83, all the residues are shown inred and other residues are shown in gray. The Key H-bonds are highlighted by dotted yellowKey H-bonds are highlighted by dotted yellow lines. For clarity only important residues are
complex 4, hydrogen bonding is observed between Phe 301 and Val121. In complex 5 there is no specific interhelical hydrogen bondingnear to the CWxP motif but compound 5 being close to the CWxPmotif is involved in hydrophobic interactions with the residuesnear to the motif such as Phe 308, Tyr 336. In complex 6, near toCWxPmotif water mediated hydrogen bond is formed between Thr300 and Gly 335, Gly 335 and Tyr 336. Also, hydrogen formed be-tween compound 6 and Asn 312 and hydrophobic interactions ofthe compound 6werewith Trp 305 of CWxPmotif and few residuesnear to CWxP motif.
3.6. Lipid protein interaction
The DRY motif in GPCRs has been proposed to play an importantrole inGPCR activation. In unbound state of the receptor it is revealedthat near to this motif (Fig. 7a) Thr 140 of the ICL2 is involved inhydrogen bonding interactions with lipid molecule which occupiesthis position for most of the simulation time thus providing stability.In complex 1, Tyr 136 of DRY motif is involved in direct hydrogenbonding with Arg 226 and both these residues are also involved inhydrogen bonding interactions with a lipid molecule.
The study reveals that TM5 and TM6 in the middle are looselypacked due to the kink of TM7 induced by Pro 307 of the CWxPmotif (Fig. 7b) and they form a hydrophobic pocket involving res-idues Val 205, Leu 206 and Leu 207, Val 211 of TM5 and Phe 308,Phe 309 and Leu 310 and Ala 311 of TM6. As a result, lipid can reachthe ligand binding pocket which is revealed by a lipid tail presentnear to the binding pocket. Ser 210 of TM5 (Fig. 7c) is involved inhydrogen bond interaction with another lipid head group and awater molecule. The stabilization of TM5/TM6 is likely to be ach-ieved by this lipid tail present. The two conserved serines Ser 165and Ser 169 on TM4, belonging to SxxxS motif, are involved in helixpacking and may be responsible for the stability of b3-AR.
In Rhodopsin the NPxxYxF motif has been proposed as one ofkey structural motifs to stabilize the TM7 and the cytoplasmic helix8 junction area through the aromatic stacking interaction betweenTyr 306 and Phe 316 [65]. This aromatic stacking is also observedbetween the corresponding residues Tyr 346 and Phe 352 in boththe bound and unbound states of the b3-AR. Phe 308 in rhodopsinis substituted with Arg 348 in b3-AR thus making the pocket lesshydrophobic in nature. However, in both the unbound and bound
larity only important residues are reported. The Key H-bonds are highlighted by dottedgray and water molecules as red and white. b) The NPxxY motif residues are shown in
lines. c) The CWxP motif (Cys 304 to Pro 307) and other residues are shown in gray. Thereported.

P. Tewatia et al. / Biochimie 101 (2014) 168e182 179
state Arg 348 makes hydrogen bond with a lipid head group andthis interaction is well maintained during the course of simulationthus contributes to the stability. A strong hydrogen bond network isformed by the neighboring residues with the lipids. Amino group ofArg 353 is involved in hydrogen bonding interactions with a lipidhead group in the unbound state whereas in bound state it isobserved to make hydrogen bonds with a hydroxyl group of thelipid molecule. In the unbound state Ile 345, Arg 358 and Arg 362are involved in hydrogen bonding with the lipid. Whereas in boundstate Arg 289, Arg 365 Arg 366 and Arg 357, Glu 370 are involved inhydrogen bonding with the lipid. Our analysis reveal that a hy-drophobic pocket is formed by residues Ile 345, Asn 338, Phe 341,Asn 342, Leu 344, Met 71, Val 149, Thr 150, Thr 72, Val 58 at the TM7and Helix 8 junction (Fig. 7d). This hydrophobic pocket attractshydrophobic tails of the lipids surrounding this junction whichretains strong hydrophobic interactions during the course ofsimulation. The lipid protein interactions with the hydrophobicresidues at the TM7 and Helix 8 junction play a major role in sta-bilizing Helix 8 at the membraneewater interface in both thebound and unbound state.
3.7. Estimation of binding free energy of the receptor ligandcomplexes
In order to understand the molecular recognition of proteinsurfaces the binding free energies (DGbind) of the initial complexesand the average structure of the complexes (collected from theproduction phase) were calculated using PrimeMMeGBSAmodule.
The charged centers of the ligand and the oppositely chargedresidues of the receptor contribute to the ligandereceptor binding,other contributions being the distant and localized hydrophobic pep stacking and cationep interactions.
Fig. 7. Interaction of lipid with b3-AR after simulation a) near the DRY motif. b) near CWxPintersection. The residues are shown in gray and lipid is shown in orange color.
The binding free energies were calculated along with Liaisondescriptors for the shortlisted compounds 1 to 6 forming the b3-ARcomplexes both for the initial docked poses and average structuresobtained from the production phase. The calculations are summa-rized in Tables 3a and 3b. The estimated binding affinity of com-pound 6 is the highest for both the initial compound 6-b3-ARcomplex (�85.12 kcal/mol) as well as the average compound 6-b3-AR complex (�88.59 kcal/mol) obtained from the productionphase. The second best binding affinity was of compound 4 wherethe initial compound 4-b3-AR complex had a binding energyof �82.15 kcal/mol and the average compound 4-b3-AR complexhad a binding energy of �85.62 kcal/mol. The binding affinity ofcompound 3 which was �43.92 kcal/mol for the initial compound3-b3-AR complex was �87.24 kcal/mol for the average structurecompound 3-b3-AR complex, the better binding energy of theaverage structure of the compound 3-b3-AR complex is due DGsolv
GB (�7.01 kcal/mol) and the DGvdw (�54.53 kcal/mol). Thoughcompound 2 has comparable binding energies in both the initial aswell as average structure of the compound 2-b3-AR complex, itshows no hydrogen bonding and is majorly involved in nonbondedinteractions with the receptor such as pep stacking and hydro-phobic interactions. Also the compound does not fit deep into thebinding pocket during the course of simulation. Though the com-pound 5 does not go deep into the binding pocket of b3-AR ascompound 4 and 6 but is still involved in major hydrogen bonding.
To check the validity of our calculations, a total of 11 known b3-AR agonists [66e75] were docked into the active site of the receptorand their binding free energies were calculated. The experimentalbinding affinities were correlated with the calculated binding en-ergies (Table 4). Parameters studied for correlationwere EC50 of theknown agonists with docking score, emodel and free energy ofbinding on different receptoreagonist complexes obtained using
motif. c) with Ser 210 of the SxxxS motif. d) at the Cytoplasmic end of TM7 and helix 8

P. Tewatia et al. / Biochimie 101 (2014) 168e182180
Glide and Prime MMGBSA. The study suggests that the best cor-relation was observed with Prime complex energy with a correla-tion coefficient of 0.924. The other parameters with a goodcorrelation coefficient with the EC50 of different b3-selective ago-nists are DGbind (R ¼ 0.763), docking score (R ¼ 0.773) and glideemodel (R ¼ 0.750). All these parameters calculated of all theknown agonists were in good agreement with the experimentalvalues of EC50.
Solabegron (b3-AR EC50 ¼ 3.98 nM) [66] a known agonist wasdocked into the native and the refined receptor model obtainedfrom the production phase. The comparison of glide emodel andbinding energy of the best initial docked complexes showed thatsolabegron docked with the energy of�45.23 kcal/mol and the freebinding energy of �80.45 kcal/mol. To further validate the results,simulation of one known agonist BRL37344 (EC50 ¼ 17 nM) com-plexedwith receptor model from production phasewas carried out.BRL37344 showed binding affinity of �50.03 kcal/mol (beforesimulation) and �54.13 kcal/mol (after simulation) which is inagreement with the EC50 value. The best compounds 4 and 6docked with the energy of �80.504 kcal/mol and �88.44 kcal/moland free binding energy of �82.15 kcal/mol and �88.59 kcal/molrespectively. The interaction energies of the compounds fromthe average structures obtained from the production phaserevealed that compound 3, 4 and 6 display better free energy ofbinding of �91.24 kcal/mol, �85.62 kcal/mol, �88.594 kcal/molwhich was better than the binding energy of known agonistsolabegron �80.45 kcal/mol. This shows that Compound 3, 4 and 6could be better agonists of b3-AR compared to other known ago-nists. Compound 1 was the least favorable compound as it movesout of the binding pocket during the course of the simulation.
3.8. Binding pockets
The dynamic changes in the shape, size and volume of bindingpocket were analyzed during simulation and compared with theunbound and bound state of the receptor using Sitemap (Fig. 8). Forthe starting unbound b3-AR structure the predicted binding pocketvolume was 1234.4 �A3. In the absence of any ligand the volume ofthe binding pocket was observed to increase during the course ofthe simulation, it was maximum around 50e70 ns (1693.0 �A3) anddecreases in the last 50 ns. With compound 1 and 6 the volume ofthe binding pocket decreases during the length of the simulationwhich may be attributed to the movement of transmembrane he-lices. However, in compound 1 there is a sharp decrease in volumeand the hydrophobicity of the binding pocket. The steep drop involume is observed until 55.0 ns of simulation time. The volumereduces from 808.7�A3 at the start of the simulation time to 238.0�A3
by 100 ns. The acceptor surface area remains fairly constantwhereas there is sharp decrease in the hydrophobic surface area.
Table 4Free energy and its components for the binding of the eleven known b3-AR agonists. All renergies. Correlation coefficient between individual forces and the observed EC50 for ele
Title EC50 (nM) Prime MMGBSA DGbind Dock
BRL37344 17 �64.56 �12.Solabegron 3.98 �80.45 �11.Amibegron 20 �50.73 �10.BRL35135 1.7 �75.17 �14.L-750355 10 �58.94 �11.755507 10 �52.94 �10.BRL26830 1.5 �72.37 �13.AJ9677 0.062 �93.17 �13.LY-377604 4 �77.89 �9.Kul 7211 56.3 �35.99 �6.Tetrahydroisoquinoline 6 �52.10 �10.Correlation (R) 0.763 0.
The larger size of compound 1 (425.6�A3) hinders the ligand to enterdeep into the binding pocket and with ECL2 showing larger fluc-tuations andmoving over the interface of the cavity opening duringsimulation prevents the ligand to have favorable interactions withthe receptor.
In compound 6 overall volume of the binding pocket decreasesfrom 985.0 �A3 to 558.4 �A3 but the acceptor surface area increasesduring the simulation whereas the donor surface area remainsconstant. A small decrease in the hydrophobic surface area isobserved. ECL2 is also observed to move towards the interface ofcavity opening, however the pocket size is sufficient enough forcompound 6 (325.7 �A3) to enter deep into the pocket and makefavorable interactions with the receptor. This may attribute to thebetter ligand affinity observed in case of compound 6.
4. Conclusion
In this study dynamic perturbations in unbound and boundform of b3-AR in an explicit membrane environment have beenprobed by means of MD simulations ranging between 100 ns and150 ns. The complexes for simulation comprised of receptorstructures bound to the shortlisted potential agonists derived frompharmacophore and virtual screening techniques. The simulationswere carried out to have an insight into the binding mechanism ofthe potential agonists within the active site of b3-AR and to un-derstand the conformational changes occurring in the bound andunbound receptor. The comparative analysis of dynamic trajec-tories in bound and unbound form of b3-AR revealed that therewere no major deviations in the ligand binding site. The minorconformational changes observed may be attributed to the localrearrangements within the receptor in the presence of a ligand. Thebinding affinity of the agonists is dependent on interactionwith keyresidues Asp 117 in TM3, Phe 198 in ECL3, Asn 312 and Arg 315 inTM6 and Asn 332 in TM7. Other than this strong hydrogen bondingwas also observed with Tyr 336 of TM7 in case of compound 6which imparts it higher binding affinity as compared to compound3 and 4. Although compound 1 had hydrogen bond interactionwithPhe 198 it made no interaction with other key residues such as Asp117, Arg 315 and Asn 332, this could be the reason of its very lowbinding affinity for the b3-AR receptor.
MD simulation studies were also used to study the behavior ofhighly conserved functional motifs such as SLAxAD, SxxxS, DRY,CWxP and NPxxY. They are involved in several of the discrete typesof molecular interactions involving mainly residues Ser 78-Asp 83,Ser 208-Ser 212, Asp 134-Arg 136, Cys 304-Pro 307 and Asn 342-Tyr346 which provide a stable conformation of b3-AR. No major dis-tortions were observed for the helices whereas large RMS de-viations were observed in the loop regions and N and C terminus.The helices move to some extent with respect to each other but
eported energies are in kcal/mol. DGbind: the potential agonist-receptor binding freeven compounds have been listed.
ing score Glide emodel (kcal/mol) Prime MMeGBSA Gcomplex energy
42 �50.03 �14042.0103 �45.24 �14047.2128 �31.60 �14014.0906 �67.46 �14090.9151 �43.44 �14135.8896 �49.66 �14129.7828 �82.11 �14058.9406 �85.75 �14075.6380 �44.49 �14102.2595 �18.95 �13055.9639 �58.34 �14037.86772 0.750 0.925

Fig. 8. Comparison of binding pocket in native and complex b3-AR. a) Unbound form of b3-AR, b) compound 1 in complex with b3-AR c) compound 6 in complex with b3-AR. Theligands are shown in green and binding pocket displayed in the context of the hydrophobic, ligand acceptor and ligand donor maps (yellow, red, and blue, respectively). Some partsof the ribbon have been removed for clarity.
P. Tewatia et al. / Biochimie 101 (2014) 168e182 181
does not include large instabilities. There exists strong watermediated hydrogen bond network, interhelical hydrogen bonding,van der waal and aromatic stacking interactions which stabilize thehelical bundles of the protein. There were several lipid protein in-teractions observed at the hydrophobic core of TM5/TM6 and TM7/Helix 8.
The compound 3, 4 and 6 are proposed to act as potential ago-nists for b3-AR based on their structural consistency in terms of freebinding energies, RMSD, analysis of binding pockets and molecularinteractions. Their binding free energies and van der waal inter-action energies were found to be better than the known agonists ofthe b3-AR. The interaction energies of compound 3, 4 and 6 displaybetter binding free energy of �91.24 kcal/mol, �85.62 kcal/moland �88.59 kcal/mol respectively which was better than thebinding energy of known agonist solabegron �80.45 kcal/mol. Thisshows that compound 3, 4 and 6 could be better lead agonists of b3-AR compared to other known agonists and their scaffold can beused for further developing of these compounds as potential drugmolecules.
In the lieu of the non availability of crystal structure moleculardynamics study provide a reliable model which could be used forstructure activity studies such as molecular docking and virtualscreening to advance our understanding of the function of GPCRsand thus contribute in the discovery of better therapeutic agents.
Acknowledgments
The author, Shakti Sahi, acknowledges the financial assistanceprovided by Science and Engineering Research Board, Department ofScience & Technology, Govt. of India for carrying out the researchwork. Parul Tewatia acknowledges Council of Scientific and Indus-trial Research (CSIR), New Delhi, India for providing Senior ResearchFellowship (SRF). The authors also thank Prof. B. Jayaram andresearch team at the Supercomputing Facility for Bioinformatics andComputational Biology, at Indian Institute of Technology, New Delhifor providing additional facility to complete the study.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biochi.2014.01.016.
References
[1] R.J. Trabanino, S.E. Hall, N. Vaidehi, W.B. Floriano, V.W.T. Kam, W.A. Goddard,First principles predictions of the structure and function of g-protein-coupledreceptors: validation for bovine rhodopsin, Biophys. J. 86 (2004) 1904e1921.
[2] T. Yarnitzky, A. Levit, M.Y. Niv, Homology modeling of G-protein-coupledreceptors with X-ray structures on the rise: modern homology modeling of
GPCR which structure template to choose, Curr. Opin. Drug. Discov. Dev. 13(2010) 317e325.
[3] A.L. Parrill, D.L. Bautista, Review GPCR conformations: implications for rationaldrug design, Pharmaceuticals 4 (2011) 7e43.
[4] D.M. Rosenbaum, S.G.F. Rasmussen, B.K. Kobilka, The structure and function ofG-protein-coupled receptors, Nature 459 (2009) 356e363.
[5] A. Grossfield, Review: recent progress in the study of G protein-coupled re-ceptors with molecular dynamics computer simulations, Biochim. Biophys.Acta 1808 (2011) 1868e1878.
[6] J.G. Baker, Evidence for a secondary state of the human b3-adrenoceptor, Mol.Pharmacol. 68 (2005) 1645e1655.
[7] L.J. Emorine, S. Marullo, M.M. Briend-Sutren, G. Patey, K. Tate, C. Delavier-Klutchko, A.D. Strosberg, Molecular characterization of the human b3-adrenergic receptor, Science 245 (1989) 1118e1121.
[8] M.G. Ursinoa, V. Vasinaa, E. Raschia, F. Cremab, F. De Pontia, The b3-adrenoceptor as a therapeutic target: current perspectives, Pharm. Res. 59(2009) 221e234.
[9] M.G. Perrone, A. Scilimati, b3-Adrenoceptor agonists and (antagonists as) in-verse agonists: history, perspective, constitutive activity, and stereospecificbinding, in: P.M. Conn (Ed.), Methods in Enzymology, Academic Press, Bur-lington, 2010, pp. 197e230.
[10] M. Imanishi, Y. Tomishima, S. Itou, H. Hamashima, Y. Nakajima, K. Washizuka,M. Sakurai, S. Matsui, E. Imamura, K. Ueshima, T. Yamamoto, N. Yamamoto,H. Ishikawa, K. Nakano, N. Unami, K. Hamada, Y. Matsumura, F. Takamura,K. Hattori, Discovery of a novel series of biphenyl benzoic acid derivatives aspotent and selective human b 3-Adrenergic receptor agonists with good oralbioavailability. Part I, J. Med. Chem. 51 (2008) 1925e1944.
[11] J.R.S. Arch, A.T. Ainsworth, Thermogenic and antiobesity activity of a novel b-adrenoceptor agonist (BRL 26830A) in mice and rats, Am. J. Clin. Nutr. 38(1983) 549e558.
[12] J.R. Arch, A.T. Ainsworth, M.A. Cawthorne, V. Piercy, M.V. Sennitt, V.E. Thod,C. Wilson, S. Wilson, Atypical b-adrenoceptors on brown adpocytes as targetfor anti-obesity drugs, Nature 309 (1983) 163e165.
[13] P. Prathipati, A.K. Saxena, Characterization of b3-adrenergic receptor: deter-mination of pharmacophore and 3D QSAR model for b3 adrenergic receptoragonism, J. Comput. Aided Mol. Des. 19 (2005) 93e110.
[14] F. Jin, C. Lu, X. Sun, W. Li, G. Liu, Y. Tang, Insights into the binding modes ofhuman b3-adrenergic receptor agonists with ligand-based and receptor-basedmethods, Mol. Diversity 15 (4) (2011) 817e831.
[15] S. Sahi, P. Tewatia, B.K. Malik, Modeling and simulation of human b3 adren-ergic receptor and its interactions with agonists, Curr. Comput. Aided DrugDes. 8 (2012) 283e295.
[16] Tewatia, B.K. Malik, S. Sahi, Identification of novel b3-adrenoceptor agonistsusing energetic analysis, structure based pharmacophores and virtualscreening, Comb. Chem. High Throughput Screen. 15 (2012) 623e640.
[17] G. Liapakis, J.A. Ballesteros, S. Papachristou, W.C. Chan, X. Chen, J.A. Javitch, Theforgotten serine e a critical role for Ser-203(5.42) in ligand binding to and acti-vationof thebeta (2)-adrenergic receptor, J. Biol. Chem. 275 (2000) 37779e37788.
[18] G. Swaminath, X. Deupi, T.W. Lee, W. Zhu, F.S. Thian, T.S. Kobilka, B. Kobilka,Probing the beta (2) adrenoreceptor binding site with catechol reveals dif-ferences in binding and activation by agonists and partial agonists, J. Biol.Chem. 280 (2005) 22165e22171.
[19] X.J. Yao, C. Parnot, X. Deupi, V.R.P. Ratnala, G. Swaminath, D.L. Farrens,B.K. Kobilka, Coupling ligand structure to specific conformational switches inthe beta(2)-adrenoreceptor, Nat. Chem. Biol. 2 (2006) 417e422.
[20] P. Ghanouni, Z. Gryczynski, J.J. Steenhuis, T.W. Lee, D.L. Farrens, B. Kobilka,Functionally different agonists induce distinct conformations in the G proteincoupling domain of the beta (2) adrenergic receptor, J. Biol. Chem. 276 (2001)24433e24436.
[21] S. Vanni, M. Neri, U. Rothlisberger, Predicting novel binding modes of agoniststo b adrenergic receptors using all-atom molecular dynamics simulations,PLoS Comput. Biol. 7 (2011) e1001053.

P. Tewatia et al. / Biochimie 101 (2014) 168e182182
[22] P.J. Stansfeld, M.S. Sansom, Molecular simulation approaches to membraneproteins, Structure 19 (2011) 1562e1572.
[23] E. Jain, A. Bairoch, S. Duvaud, I. Phan, N. Redaschi, B.E. Suzek, M.J. Martin,P. McGarvey, E. Gasteiger, Infrastructure for the life sciences: design andimplementation of the UniProt website, BMC Bioinform. 10 (2009) 136.
[24] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, PROCHECK: aprogram to check the stereochemical quality of protein structures, J. Appl.Crystallogr. 26 (1993) 283e291.
[25] Schrödinger, Version 9.3, Schrödinger, Inc, New York, NY, 2012.[26] J.L. Banks, H.S. Beard, Y.X. Cao, A.E. Cho, W. Damm, R. Farid, A.K. Felts,
T.A. Halgren, D.T. Mainz, J.R. Maple, R. Murphy, D.M. Philipp, M.P. Repasky,L.Y. Zhang, B.J. Berne, R.A. Friesner, E. Gallicchio, R.M. Levy, Integratedmodeling program, applied chemical theory (IMPACT), J. Comput. Chem. 26(2005) 1752e1780.
[27] LigPrep, Version 2.5, Schrödinger, LLC, New York, NY, 2012.[28] Epik, Version 2.3, Schrödinger, LLC, New York, NY, 2012.[29] R.A. Friesner, R.B. Murphy, M.P. Repasky, L.L. Frye, J.R. Greenwood,
T.A. Halgren, P.C. Sanschagrin, D.T. Mainz, Extra precision glide: docking andscoring incorporating a model of hydrophobic enclosure for protein-ligandcomplexes, J. Med. Chem. 49 (2006) 6177e6196.
[30] H.J.C. Berendsen, D. van der Spoel, R. van Drunen, GROMACS: a message-passing parallel molecular dynamics implementation, Comput. Phys. Com-mun. 91 (1995) 43e56.
[31] D. van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark, H.J. Berendsen,GROMACS: fast, flexible, and free, J. Comput. Chem. 26 (2005) 1701e1718.
[32] W.L. Jorgensen, D.S. Maxwell, J. Tirado-Rives, Development and testing of theOPLS all-atom force field on conformational energetics and properties oforganic liquids, J. Am. Chem. Soc. 118 (45) (1996) 11225e11236.
[33] G.A. Kaminski, R.A. Friesner, J. Tirado-Rives, W.L. Jorgensen, Evaluation andreparametrization of the OPLS-AA force field for proteins via comparison withaccurate quantum chemical calculations on peptides, J. Phys. Chem. B 105 (28)(2001) 6474e6487.
[34] W.D. Cornell, P. Cieplak, C.I. Bayly, I.R. Gould, K.M. Merz, et al., A 2nd gener-ation force-field for the simulation of proteins, nucleic-acids, and organicmolecules, J. Am. Chem. Soc. 117 (1995) 5179e5197.
[35] C.I. Bayly, P. Cieplak, W.D. Cornell, P.A. Kollman, A well-behaved electrostaticpotential based method using charge restraints for deriving atomic charges ethe RESP model, J. Phys. Chem. 97 (1993) 10269e10280.
[36] J.M. Wang, P. Cieplak, P.A. Kollman, How well does a restrained electrostaticpotential (RESP) model perform in calculating conformational energies oforganic and biological molecules? J. Comput. Chem. 21 (2000) 1049e1074.
[37] D.P. Tieleman, S.J. Marrink, H.J.C. Berendsen, A computer perspective ofmembranes: molecular dynamics studies of lipid bilayer systems, Biochem.Biophys. Acta 1331 (1997) 235e270.
[38] D.P. Tieleman, H.J.C. Berendsen, Molecular dynamics simulations of fully hy-drated DPPC with different macroscopic boundary conditions and parameters,J. Chem. Phys. 105 (1996) 4871e4880.
[39] Biocomputing at the University of Calgary, http://moose.bio.ucalgary.ca(accessed 20.07.11).
[40] H.J.C. Berendsen, J.P.M. Postma, W.F. Van Gunsteren, J. Hermans, Interactionmodels for water in relation to protein hydration, in: B.R. Pullman (Ed.),Intermolecular Forces, 1981, pp. 331e342. Dordrecht.
[41] H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. Dinola, J.R. Haak,Molecular dynamics with coupling to an external bath, J. Chem. Phys. 81(1984) 3684e3690.
[42] G. Bussi, D. Donadio, M. Parrinello, Canonical sampling through velocityrescaling, J. Chem. Phys. 126 (2007) 014101.
[43] S. Nose, M.L. Klein, Constant pressure molecular dynamics for molecularsystems, Mol. Phys. 50 (1983) 1055e1076.
[44] W.G. Hoover, Canonical dynamics: equilibrium phase-space distributions,Phys. Rev. A 3 (1985) 1695e1697.
[45] M. Parrinello, A. Rahman, Polymorphic transitions in single crystals: a newmolecular dynamics method, J. Appl. Phys. 52 (1981) 7182e7190.
[46] B. Hess, H. Bekker, H.J.C. Berendsen, J.G.E.M. Fraaije, LINCS: a linear constraintsolver for molecular simulations, J. Comput. Chem. 18 (1997) 1463e1472.
[47] T. Darden, D. York, L. Pedersen, Particle mesh Ewald: an N-log (N) method forEwald sums in large systems, J. Chem. Phys. 98 (1993) 10089e10092.
[48] W. Humphrey, A. Dalke, K. Schulten, VMD e visual molecular dynamics, J. Mol.Graphics 14 (1996) 33e38.
[49] Prime, Version 3.1, Schrödinger, LLC, New York, NY, 2012.[50] T. Halgren, New method for fast and accurate binding-site identification and
analysis, Chem. Biol. Drug. Des. 69 (2007) 146e148.[51] P. Chelikani, V. Hornak, M. Eilers, P.J. Reeves, S.O. Smith, U.L. RajBhandary,
H.G. Khorana, Role of group-conserved residues in the helical core of b2-adrenergic receptor, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 7027e7032.
[52] R.A. Laskowski, PDBsum new things, Nucleic Acids Res. 37 (2009) 355e359.[53] T. Okada, M. Sugihara, A.N. Bondar, M. Elstner, P. Entel, V. Buss, The retinal
conformation and its environment in the light of new 2.2 Å crystal structure,J. Mol. Biol. 342 (2004) 571e581.
[54] K. Palczewski, T. Kumasaka, T. Hori, C.A. Behnke, H. Motoshima, B.A. Fox, I. LeTrong, D.C. Teller, T. Okada, R.E. Stenkamp, M. Yamamoto, M. Miyano, Crystal
structure of rhodopsin: a G-protein coupled receptor, Science 289 (2000)739e745.
[55] J.A. Ballesteros, A.D. Jensen, G. Liapakis, S.G. Rasmussen, L. Shi, U. Gether,J.A. Javitch, Activation of the b2-adrenergic receptor involves disruption of anionic lock between the cytoplasmic ends of transmembrane segments 3 and 6,J. Biol. Chem. 276 (2001) 29171e29177.
[56] T. Warne, M.J. Serrano-Vega, J.G. Baker, R. Moukhametzianov, P.C. Edwards,R. Henderson, A.G. Leslie, C.G. Tate, G.F. Schertler, Structure of a beta1-adrenergic G-protein-coupled receptor, Nature 454 (2008) 486e491.
[57] D.M. Rosenbaum, V. Cherezov, M.A. Hanson, S.G. Rasmussen, F.S. Thian,T.S. Kobilka, H.J. Choi, X.J. Yao, W.I. Weis, C. Stevens, B.K. Kobilka, GPCR en-gineering yields high-resolution structural insights into beta2-adrenergic re-ceptor function, Science 318 (2007) 1266e1273.
[58] A. Warne, R. Moukhametzianov, J.G. Baker, R. Nehme, P.C. Edwards,A.G.W. Leslie, G.F.X. Schertler, C.G. Tate, The structural basis for agonistand partial agonist action on a b(1)-adrenergicreceptor, Nature 469 (2011)241e244.
[59] V. Cherezov, D.M. Rosenbaum, M.A. Hanson, S.G. Rasmussen, F.S. Thian,T.S. Kobilka, H.J. Choi, P. Kuhn, W.I. Weis, B.K. Kobilka, R.C. Stevens, Highresolution crystal structure of an engineered human beta2-adrenergic Gprotein-coupled receptor, Science 318 (2007) 1258e1265.
[60] P.L. Yeagle, G. Choi, A.D. Albert, Studies on the structure of the G-protein-coupled receptor rhodopsin including the putative G-protein binding site inunactivated and activated forms, Biochemistry 40 (2001) 11932e11937.
[61] D. Schneider, Rendezvous in a membrane: close packing, hydrogen bonding,and the formation of transmembrane helix oligomers, FEBS Lett. 577 (2004)5e8.
[62] J.F. Hunt, T.N. Earnest, O. Bousché, K. Kalghatgi, K. Reilly, C. Horváth,K.J. Rothschild, D.M. Engelman, A biophysical study of integral membraneprotein folding, Biochemistry 36 (1997) 15156e15176.
[63] L. Shi, G. Liapakis, R. Xu, F. Guarnieri, J.A. Ballesteros, J.A. Javitch, Beta 2adrenergic receptor activation. Modulation of the proline kink in trans-membrane 6 by a rotamer toggle switch, J. Biol. Chem. 277 (2002) 40989e40996.
[64] D.M. Rosenbaum, S.G.F. Rasmussen, B.K. Kobilka, The structure and function ofG-protein coupled receptors, Nature 459 (2009) 356e363.
[65] O. Fritze, S. Filipek, V. Kuksa, K. Palczewski, K.P. Hofmann, O.P. Ernst, Role ofthe conserved NPxxY(x)5,6F motif in the rhodopsin ground state and duringactivation, Proc. Natl. Acad. Sci. U.S.A. 100 (2003) 2290e2295.
[66] A. Hicks, G.P. McCafferty, E. Riedel, N. Aiyar, M. Pullen, C. Evans, T.D. Luce,R.W. Coatney, G.C. Rivera, T.D. Westfall, J.P. Hieble, GW427353 (Solabegron), anovel, selective b3-adrenergic receptor agonist, evokes bladder relaxation andincreases micturition reflex threshold in the dog, J. Pharmacol. Exp. Ther. 323(2007) 202e209.
[67] H. Kato, M. Ohue, K. Kato, A. Nomura, K. Toyosawa, Y. Furutani, S. Kimura,T. Kadowaki, Mechanism of amelioration of insulin resistance by b 3-Adrenoceptor agonist AJ-9677 in the KK-Ay/Ta diabetic obese mouse model,Diabetes 50 (2001) 113e122.
[68] T. Yoshida, The antidiabetic b3-adrenoceptor agonist BRL 26830A works byrelease of endogenous insulin, Am. J. Clin. Nutr. 55 (1992) 237e241.
[69] F.D. King, A.W. Oxford, Progress in Medicinal Chemistry, vol. 41, ElsevierScience B.V., Amsterdam, 2003.
[70] D.H. Overstreet, J. Stemmelin, G. Griebel, Confirmation of antidepressant po-tential of the selective b3 adrenoceptor agonist amibegron in an animal modelof depression, Pharmacol. Biochem. Behav. 89 (2008) 623e626.
[71] M. Sato, D.S. Hutchinson, B.A. Evans, R.J. Summers, The b3-adrenoceptoragonist 4-[[(hexylamino)carbonyl]amino]-N-[4-[2-[[(2S)-2-hydroxy-3-(4-hydroxyphenoxy)propyl]amino]ethyl]-phenyl]-benzenesulfonamide(L755507) and antagonist (S)-N-[4-[2-[[3-[3-(acetamidomethyl)phenoxy]-2-hydroxypropyl]amino]-ethyl]phenyl]benzenesulfonamide (L748337)activate different signaling pathways in Chinese hamster ovary-K1 cellsstably expressing the human b 3-adrenoceptor, Mol. Pharmacol. 74 (2008)1417e1428.
[72] M.A. Cawthorne, M.V. Sennitt, J.R.S. Arch, S.A. Smith, BRL 35135, a potent andselective atypical b3-adrenoceptor, Am. J. Clin. Nutr. 55 (1992) 252Se257S.
[73] D.E. Uehling, K.H. Donaldson, D.N. Deaton, C.E. Hyman, E.E. Sugg, D.G. Barrett,R.G. Hughes, B. Reitter, K.K. Adkison, M.E. . Lancaster, F. Lee, R. Hart, M.A. .Paulik, B.W. . Sherman, T. True, C. Cowan, Synthesis and evaluation of potentand selective b3 adrenergic receptor agonists containing acylsulfonamide,sulfonylsulfonamide, and sulfonylurea carboxylic acid isosteres, J. Med. Chem.45 (2002) 567e583.
[74] Y. Tomiyama, M. Murakami, K. Hayakawa, K. Akiyama, Y. Yamazaki,M. Kojima, N. Shibata, M. Akahane, Pharmacological profile of KUL-7211, aselective e adrenoceptor agonist, in isolated ureteral smooth muscle,J. Pharm. Sci. 9 (2003) 411e419.
[75] M.J. Forrest, G. Hom, T. Bach, M.R. Candelore, M.A. Cascieri, C. Strader, L. Tota,M.H. Fisher, J. Szumiloski, H.O. Ok, A.E. Weber, M. Wyvratt, P. Vicario,O. Marko, L. Deng, C. Cioffe, B. Hegarty-Friscino, E. MacIntyre, L-750355, ahuman b3-adrenoceptor agonist; in vitro pharmacology and profile of activityin vivo in the rhesus monkey, Eur. J. Pharmacol. 407 (2000) 175e181.
Copyright © 2022 FDOKUMEN



















