
Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a...
13
The Journal of Immunology Induction of Immunogenic Apoptosis by Blockade of Epidermal Growth Factor Receptor Activation with a Specific Antibody Greta Garrido,* ,1 Ailem Rabasa,* ,1 Belinda Sa ´nchez,* Marı ´a Victoria Lo ´pez,* Rances Blanco, † Armando Lo ´pez, ‡ Diana Rosa Herna ´ndez,* Rolando Pe ´rez,* ,‡,x and Luis Enrique Ferna ´ndez x Despite promising results in the use of anti-epidermal growth factor receptor (EGFR) Abs for cancer therapy, several issues remain to be addressed. An increasing emphasis is being placed on immune effector mechanisms. It has become clear for other Abs directed to tumor targets that their effects involve the adaptive immunity, mainly by the contribution of Fc region-mediated mechanisms. Given the relevance of EGFR signaling for tumor biology, we wonder whether the oncogene inhibition could contribute to Ab- induced vaccine effect. In a mouse model in which 7A7 (an anti-murine EGFR Ab) and AG1478 (an EGFR-tyrosine kinase inhibitor) displayed potent antimetastatic activities, depletion experiments revealed that only in the case of the Ab, the effect was dependent on CD4 + and CD8 + T cells. Correspondingly, 7A7 administration elicited a remarkable tumor-specific CTL response in hosts. Importantly, experiments using 7A7 F(ab9) 2 suggested that in vivo Ab-mediated EGFR blockade may play an important role in the linkage with adaptive immunity. Addressing the possible mechanism involved in this effect, we found quantitative and qualitative differences between 7A7 and AG1478-induced apoptosis. EGFR blocking by 7A7 not only prompted a higher pro- apoptotic effect on tumor metastases compared with AG1478, but also was able to induce apoptosis with immunogenic potential in an Fc-independent manner. As expected, 7A7 but not AG1478 stimulated exposure of danger signals on tumor cells. Subcutaneous injection of 7A7-treated tumor cells induced an antitumor immune response. This is the first report, to our knowledge, of a tumor- specific CTL response generated by Ab-mediated EGFR inhibition, suggesting an important contribution of immunogenic apo- ptosis to this effect. The Journal of Immunology, 2011, 187: 4954–4966. A mong the molecular targets that are currently in clinical evaluation for cancer treatment, the epidermal growth factor receptor (EGFR) has been widely validated. EGFR is a cell membrane growth factor receptor characterized by tyrosine kinase activity that plays a crucial role in the control of key cellular transduction pathways in both normal and tumor cells. EGFR is overexpressed in a variety of human epithelial tumors (1). Ligand binding to EGFR results in stabilization of the receptor active conformation that allows its dimerization and autophosphorylation (2). EGFR tyrosine kinase activation ultimately leads to stimula- tion of cell proliferation, invasion, angiogenesis, and the blocking of apoptosis (3). In addition, recent studies have demonstrated that EGFR signaling is involved not just in the malignant behavior of tumor cells, but also in the modification of the tumor microenvi- ronment to favor cancer progression (4). Two pharmacological approaches have been successfully used in cancer treatment to inhibit EGFR functions: neutralizing mAbs and small-molecule tyrosine kinase inhibitors (TKI) (5). Anti-EGFR Ab therapy has been reported to mediate tumor regression by interrupting oncogenic signals and inducing Fc- mediated innate mechanisms (6). However, the capacity of anti- EGFR mAbs to activate cell populations of adaptive immunity has not yet been explored. Recent evidence has shown the possibility that a vaccine effect could be generated in animals and patients with tumors after Ab-based immunotherapy. DC101, an Ab spe- cific for vascular endothelial growth factor receptor-2, induced T cell-dependent tumor regression in mice in addition to mediat- ing an antiangiogenic effect (7). Immunotherapy using anti-CD20 Abs provoked the generation of tumor-specific T cell response in mice (8) and patients (9) with hematological malignances. Also, dependence on adaptive immunity for the therapeutic effect of an anti-HER2 Ab has been recently documented (10, 11). The ca- pacity of some Abs to induce a specific antitumor immunity, not restricted to the target Ag but against several unknown tumor- derived Ags, reinforces the relevance of exploring the connec- tion between EGFR targeting and adaptive immunity activation. Considering that preclinical reports of the mechanisms involved in an Ab-induced vaccine effect have demonstrated an Fc-de- pendence (8, 10), the most relevant question would be whether EGFR blockade contribute to this effect. *Tumor Immunology Direction, Molecular Immunology Institute, Center of Molec- ular Immunology, Havana 11600, Cuba; † Quality Control Department, Quality Di- rection, Center of Molecular Immunology, Havana 11600, Cuba; ‡ System Biology Direction, Molecular Immunology Institute, Center of Molecular Immunology, Ha- vana 11600, Cuba; and x Innovative Direction, Molecular Immunology Institute, Cen- ter of Molecular Immunology, Havana 11600, Cuba 1 G.G. and A.R. contributed equally to this work. Received for publication October 22, 2010. Accepted for publication September 1, 2011. This work was supported by the Cuban Government. Address correspondence and reprint requests to Dr. Greta Garrido, Tumor Immunol- ogy Direction, Molecular Immunology Institute, Center of Molecular Immunology, 216 Street and 15th Avenue, Atabey, Siboney, Playa, P.O. Box 16040, Havana 11600, Cuba. E-mail address: [email protected] The online version of this article contains supplemental material. Abbreviations used in this article: CRT, calreticulin; DC, dendritic cells; DX, doxo- rubicin; EGFR, epidermal growth factor receptor; eIF2a, eukaryotic initiation factor 2a; FSC, forward-scattered; HSP, heat shock protein; MC, mitomycin C; MFI, mean fluorescence intensity; MHC II, MHC class II; PI, propidium iodide; TKI, tyrosine kinase inhibitor. Copyright Ó 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00 www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003477
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a...

The Journal of Immunology
Induction of Immunogenic Apoptosis by Blockade ofEpidermal Growth Factor Receptor Activation with a SpecificAntibody
Greta Garrido,*,1 Ailem Rabasa,*,1 Belinda Sanchez,* Marıa Victoria Lopez,*
Rances Blanco,† Armando Lopez,‡ Diana Rosa Hernandez,* Rolando Perez,*,‡,x and
Luis Enrique Fernandezx
Despite promising results in the use of anti-epidermal growth factor receptor (EGFR) Abs for cancer therapy, several issues remain
to be addressed. An increasing emphasis is being placed on immune effector mechanisms. It has become clear for other Abs directed
to tumor targets that their effects involve the adaptive immunity, mainly by the contribution of Fc region-mediated mechanisms.
Given the relevance of EGFR signaling for tumor biology, we wonder whether the oncogene inhibition could contribute to Ab-
induced vaccine effect. In amousemodel in which 7A7 (an anti-murine EGFRAb) and AG1478 (an EGFR-tyrosine kinase inhibitor)
displayed potent antimetastatic activities, depletion experiments revealed that only in the case of the Ab, the effect was dependent on
CD4+ and CD8+ T cells. Correspondingly, 7A7 administration elicited a remarkable tumor-specific CTL response in hosts.
Importantly, experiments using 7A7 F(ab9)2 suggested that in vivo Ab-mediated EGFR blockade may play an important role
in the linkage with adaptive immunity. Addressing the possible mechanism involved in this effect, we found quantitative and
qualitative differences between 7A7 and AG1478-induced apoptosis. EGFR blocking by 7A7 not only prompted a higher pro-
apoptotic effect on tumor metastases compared with AG1478, but also was able to induce apoptosis with immunogenic potential in
an Fc-independent manner. As expected, 7A7 but not AG1478 stimulated exposure of danger signals on tumor cells. Subcutaneous
injection of 7A7-treated tumor cells induced an antitumor immune response. This is the first report, to our knowledge, of a tumor-
specific CTL response generated by Ab-mediated EGFR inhibition, suggesting an important contribution of immunogenic apo-
ptosis to this effect. The Journal of Immunology, 2011, 187: 4954–4966.
Among the molecular targets that are currently in clinicalevaluation for cancer treatment, the epidermal growthfactor receptor (EGFR) has been widely validated. EGFR
is a cell membrane growth factor receptor characterized by tyrosinekinase activity that plays a crucial role in the control of key cellulartransduction pathways in both normal and tumor cells. EGFR isoverexpressed in a variety of human epithelial tumors (1). Ligandbinding to EGFR results in stabilization of the receptor activeconformation that allows its dimerization and autophosphorylation(2). EGFR tyrosine kinase activation ultimately leads to stimula-
tion of cell proliferation, invasion, angiogenesis, and the blockingof apoptosis (3). In addition, recent studies have demonstrated thatEGFR signaling is involved not just in the malignant behavior oftumor cells, but also in the modification of the tumor microenvi-ronment to favor cancer progression (4). Two pharmacologicalapproaches have been successfully used in cancer treatment toinhibit EGFR functions: neutralizing mAbs and small-moleculetyrosine kinase inhibitors (TKI) (5).Anti-EGFR Ab therapy has been reported to mediate tumor
regression by interrupting oncogenic signals and inducing Fc-mediated innate mechanisms (6). However, the capacity of anti-EGFR mAbs to activate cell populations of adaptive immunity hasnot yet been explored. Recent evidence has shown the possibilitythat a vaccine effect could be generated in animals and patientswith tumors after Ab-based immunotherapy. DC101, an Ab spe-cific for vascular endothelial growth factor receptor-2, inducedT cell-dependent tumor regression in mice in addition to mediat-ing an antiangiogenic effect (7). Immunotherapy using anti-CD20Abs provoked the generation of tumor-specific T cell response inmice (8) and patients (9) with hematological malignances. Also,dependence on adaptive immunity for the therapeutic effect of ananti-HER2 Ab has been recently documented (10, 11). The ca-pacity of some Abs to induce a specific antitumor immunity, notrestricted to the target Ag but against several unknown tumor-derived Ags, reinforces the relevance of exploring the connec-tion between EGFR targeting and adaptive immunity activation.Considering that preclinical reports of the mechanisms involvedin an Ab-induced vaccine effect have demonstrated an Fc-de-pendence (8, 10), the most relevant question would be whetherEGFR blockade contribute to this effect.
*Tumor Immunology Direction, Molecular Immunology Institute, Center of Molec-ular Immunology, Havana 11600, Cuba; †Quality Control Department, Quality Di-rection, Center of Molecular Immunology, Havana 11600, Cuba; ‡System BiologyDirection, Molecular Immunology Institute, Center of Molecular Immunology, Ha-vana 11600, Cuba; and xInnovative Direction, Molecular Immunology Institute, Cen-ter of Molecular Immunology, Havana 11600, Cuba
1G.G. and A.R. contributed equally to this work.
Received for publication October 22, 2010. Accepted for publication September 1,2011.
This work was supported by the Cuban Government.
Address correspondence and reprint requests to Dr. Greta Garrido, Tumor Immunol-ogy Direction, Molecular Immunology Institute, Center of Molecular Immunology,216 Street and 15th Avenue, Atabey, Siboney, Playa, P.O. Box 16040, Havana 11600,Cuba. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: CRT, calreticulin; DC, dendritic cells; DX, doxo-rubicin; EGFR, epidermal growth factor receptor; eIF2a, eukaryotic initiation factor2a; FSC, forward-scattered; HSP, heat shock protein; MC, mitomycin C; MFI, meanfluorescence intensity; MHC II, MHC class II; PI, propidium iodide; TKI, tyrosinekinase inhibitor.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003477

Absence of appropriate preclinical models has constituted animportant limitation to determine the involvement of adaptive im-mune response in the antitumor activity of anti-EGFR Ab-basedtherapies. Because anti-human EGFR Abs do not cross-react withthe murine molecule, in vivo experiments studying their antitumoreffect have been conducted with xenograft tumors in nude mice.Use of immunocompetent mice in a complete autologous scenariois required to evaluate this phenomenon. Description of murinetumor models with EGFR expression has been quite limited. In thatsense, we have generated an Ab specific for the extracellular do-main of murine EGFR named 7A7 (12). Preliminary experimentsusing a syngeneic metastasis model have suggested that 7A7 mAb,in addition to conventional mechanisms associated with the anti-EGFR Ab effects, is able to induce a T cell response (CD4+ andCD8+) that is indispensable to its antimetastatic effect (13).The goal of the current study was to determine the role of EGFR
signal interference in the generation of an antitumor adaptiveimmunity. We characterized tumor-specific T cell response after7A7 treatment and explored whether this ability was exclusive foranti-EGFR Abs or a TKI specific for this receptor was also capableof inducing a T cell response. For this purpose, we used AG1478 asan inhibitor of EGFR tyrosine kinase activity, which has demon-strated its therapeutic potential in preclinical studies (14). Weconducted experiments to evaluate whether the apoptosis inducedby EGFR inhibition contributes to CTL response generation.
Materials and MethodsCell line and reagents
C57BL/6-derived D122 metastatic clone of the Lewis lung carcinoma (15)and B16F10 metastatic clone of the B16 melanoma (16) were cultured inRPMI 1640 medium (Life Technologies, Gaithersburg, MD) supplementedwith 10% FCS and penicillin-streptomycin (Life Technologies). 7A7, an Abspecific for the extracellular domain of murine EGFR (12), was obtained atthe Center of Molecular Immunology (Havana, Cuba). 7A7 F(ab9)2 wasobtained by pepsin digestion (17). AG1478, a small-molecule EGFR TKI,was purchased from LC Laboratories (Woburn, MA). AG1478 was dis-solved in DMSO for in vitro studies and in 0.05% (v/v) Tween 80 solutionfor in vivo use. Abs to phospho-eukaryotic initiation factor 2a (eIF2a) andtotal eIF2a were used for immunoblot experiments and obtained from CellSignaling Technology (Beverly, MA). Abs specific for mouse moleculesCD11c, I-A/I-E, CD40, CD80, CD86, CD3, CD8, CD4, CD69, CD44,granzyme B, and IFN-g were used for flow cytometry analyses and pur-chased from eBioscience (San Diego, CA) or BD Biosciences (San Fran-cisco, CA). Abs specific for calreticulin (CRT), ERp57 (Abcam, Paris,France), cleaved caspase-3, heat shock protein (HSP) 70, and HSP 90 (CellSignaling Technology) were also used for flow cytometry studies.
Experimental and spontaneous metastasis models
All animal studies were done according to protocols approved by the In-stitutional Animal Care and Use Committee of the Center of MolecularImmunology. D122 cells were grown to 70–85% confluence before beingharvested for cell counting. For experimental metastasis assays, 2.5 3 105
cells were injected into tail lateral veins of 6–8-wk-old female C57BL/6mice (Center for Laboratory Animal Production, Havana, Cuba). 7A7 mAb(56 mg i.v.), 7A7 F(ab9)2 (373.3 mg i.v.), and AG1478 TKI (1 mg orally)administration began on day 6 after tumor challenge and continued atthree doses per week. On day 21, mice were euthanized, and lungs wereremoved. The number of D122 lung metastases was counted or lungswere subjected to histological examination as described below. The anti-metastatic effect of one dose (day 6) or two doses (days 6 and 13) of 7A7mAb or AG1478 TKI was measured. For spontaneous metastasis assays,C57BL/6 mice were challenged with D122 cells (5 3 104) s.c. into dor-solumbar regions. The evolution of primary tumors was monitored. Thelargest perpendicular diameters of the resulting tumors were measuredwith caliper, and tumor volume was calculating using the formula: p/6 3length 3 width2. Two weeks later when tumors reached ∼0.3 cm3, theadministration of 7A7 mAb (56 mg i.v.) or AG1478 TKI (1 mg orally)began and continued every 3 d. On day 35, mice were euthanized, andlungs were removed. The number of D122 lung metastases was counted.Cell population depletions were achieved by i.p. injections of 0.1 mg anti-CD8+ or anti-CD4+ Abs [obtained from rat anti-mouse hybridomas
YTS169 or YTS191 (American Type Culture Collection, Manassas, VA)and purified as previously described (18)]. In both scenarios, depletionschedule began at the same time as anti-EGFR agents and continued every4 d. Mice injected with PBS or 0.05% (v/v) Tween 80 in water were usedas controls.
Assessment of specific T cell response
Mice receiving six doses of 7A7 mAb, 7A7 F(ab9)2, or AG1748 TKI asdescribed above were euthanized 16 d after tumor inoculation by i.v. in-jection. Cells from axillary lymph nodes were isolated and restimulatedin vitro during 7 d with IL-2 (200 U/ml; R&D Systems, Minneapolis, MN)in the presence of irradiated D122 cells (previously treated with 120 U/mlIFN-g to increase H-2Kb expression). Cytotoxic activity was determinedin 4 h in vitro lactate dehydrogenase assay using IFN-g–treated D122 orIFN-g–treated B16F10 as target cells. CTL response against D122tumor-associated Ag H-2Kb–restricted peptide [Mut 1 (FEQNTAQP) (19)]and EGFR H-2Kb–restricted peptide [DLHAFENL; SYFPEITHI score: 22(20)] was determined. H-2Kb–binding OVA257–264 peptide (SIINFEKL)was used as irrelevant. Percentage of specific lysis was calculated as:([experimental release 2 effector cell release 2 spontaneous release]/[maximum release 2 spontaneous release]) 3 100. Maximum releasewas obtained by adding 1% Triton X-100 to target cells, and spontaneousrelease was determined by incubating target cells with medium alone.
Analyses of lung mononuclear cells
To evaluate lung-infiltrating CD8+ T cells, D122 experimental metastasis-bearing mice treated with 7A7 mAb or AG1478 TKI were euthanized16 d after tumor injection. Lung mononuclear cells were isolated as de-scribed previously using a collagenase/DNAse enzymatic digestion (21).Mononuclear cells were collected, and surface expression of CD3, CD8,CD69, and CD44 molecules was determined by flow cytometry using thespecific Abs described above. Granzyme B and IFN-g intracellular de-tection was performed using BD Cytofix/Cytoperm Plus kit with BDGolgiPlug kit following the protocol recommended by the manufacturer(BD Biosciences).
Apoptosis measurements
D122 cells were plated (5 3 105) in 10% FCS RPMI 1640 in six-wellplates (Costar, Cambridge, MA). Twenty-four hours later, 7A7 mAb (10mg/ml), 7A7 F(ab9)2 (6.66 mg/ml), or AG1478 TKI (10 mM) were added in1% FCS RPMI 1640 and cells were incubated for an additional 24, 48, or72 h. Control cells were grown without treatment or cultured in mediumwith the appropriate concentration of DMSO. To measure phosphatidyl-serine exposure (24 h), an Annexin V-propidium iodide (PI) doublestaining was carried out according to manufacturer’s protocol (BenderMedSystems, Vienna, Austria). To detect caspase activation (48 h), per-meabilized cells were stained with an Ab specific to cleaved form ofcaspase 3. To analyze DNA fragmentation (72 h), cells were fixed with ice-cold methanol/acetone (4:1) and stained by incubation with a solutioncontaining 100 mg/ml PI (Sigma-Aldrich, St. Louis, MO) and 50 mg/mlRNase (Sigma-Aldrich). All analyses were performed on an FACScan flowcytometer (BD Biosciences) by collecting a minimum of 20,000 events andanalyzed using WinMDI 2.8 and ModFit 3.0 software packages. D122cells treated with mitomycin C (MC; 50 mg/ml) or doxorubicin (DX; 1mM) were used as negative and positive controls of immunogenic apo-ptosis, respectively. To determine in vivo apoptosis induction, lung spec-imens were shock frozen and stored in liquid nitrogen until been ana-lyzed. Immunostaining was performed using 5-mm tissue sections placedon glass slides. TUNEL was done following manufacturer instructions(Roche Diagnostic, Mannheim, Germany). The number of apoptotic cellsand total cell number per high-power microscopic field (original magni-fication 310) in metastatic foci were counted, and eight high-poweredfields were analyzed. Metastasis areas were defined by H&E routinestaining. Results are expressed as percentages of TdT-positive cells of thetotal tumor cells counted.
Immunogenic apoptosis assays
A total of 1.5 3 106 D122 cells treated with 7A7 mAb, 7A7 F(ab9)2,AG1478 TKI, MC, or DX during 24 h as described above were inoculateds.c. into one flank of mice. Despite the fact that ∼75% of the 7A7, 7A7F(ab9)2, and AG1478-treated cells and ∼30% of the MC, DX-treated cellswere still nonapoptotic (PI2An2), treated cells injected rarely inducedtumors, presumably because these nonapoptotic cells were already pro-grammed to die later. Seven days later, mice received a challenge withD122 or B16F10 live cells (5 3 104) onto the contralateral flank. Theevolution of these tumors was monitored. In mice that did not receive the
The Journal of Immunology 4955

second challenge, cells from afferent lymph nodes were isolated 10 d afterinjection of dying cells and restimulated in vitro for 7 d as described above.Cytotoxic activity was determined in 4 h in vitro lactate dehydrogenaseassay using IFN-g–treated D122 as target cells. Depletion of CD8+ orCD4+ cells was achieved by i.p. injection of specific Abs 3 d beforechallenge with dying [7A7, 7A7 F(ab9)2, DX-treated] tumor cells and3 d before injection with live tumor cells. Animals that bore tumors inexcess of 20–25% of the body mass or that were necrotic were killed.
Flow cytometric analysis of chaperones on the cell surface
Flow cytometry was used to detect CRT, ERp57, HSP 70, and HSP 90exposure in cells treated (preincubated or not with 10 mM brefeldin A) with7A7 mAb, 7A7 F(ab9)2, AG1478 TKI, MC, or DX. D122 cells were col-lected after treatment at several times (4, 6, 12, and 48 h), washed twicewith PBS, and fixed with 0.25% paraformaldehyde in PBS for 5 min. Afterwashing twice in cold PBS, cells were incubated with primary Abs fol-lowed by washing and incubation with the FITC-conjugated secondary Ab.Isotype-matched IgG Abs or second Ab alone were used as controls. ForCRT and ERp57 exposure, cells analyzed were gated from PI-negativecells.
Immunoblot analyses
Serum-starved D122 cells were treated with 7A7 mAb or DX for 6 h. Celllysates were prepared in RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodiumdeoxycholate, and 0.1% SDS) with 50 mM NaF, 1 mM Na3VO4, 5 mMEDTA, and 1 mM PMSF that were freshly added to the lysis solutionbefore each experiment. Cell extracts were applied to SDS-PAGE gels andtransferred to polyvinylidine difluoride membranes (Gelman, Ann Arbor,MI). Membranes were blocked with NEGT buffer (0.15 M NaCl, 5 mMEDTA, 500 mM Tris-HCl [pH 7.5], 0.02% Tween 20, and 0.04% gelatin)and incubated with the primary Abs described above. Protein content wasvisualized using HRP-conjugated secondary Abs (Cell Signaling Tech-nology) followed by Chemiluminescent Substrate (Pierce, Rockford, IL).
Generation of bone marrow-derived dendritic cells and culturewith apoptotic cells
As previously described (22), bone marrow cells were harvested fromfemurs and tibias of normal C57BL/6 mice and filtered through a Falcon40 mM cell strainer (Thomas Scientific, Swedesboro, NJ). Whole bonemarrow cells were seeded (0.6 3 106) in six-well plates in RPMI 1640supplemented with 10% FCS and 20 ng/ml recombinant murine GM-CSF(R&D Systems). Cultures were fed at day 3, adding an equal amount offresh, growth factor-supplemented medium. On day 6 of culture, immaturedendritic cells (DC) were incubated with apoptotic cells for 16 h at a 1:1ratio. DC surface phenotype was analyzed by flow cytometry using thespecific Abs described above. Changes in cell size were also studied. LPSstimuli were used to monitor the maturation potential of DC generated inour experimental conditions. DX- and MC-treated D122 cells were used aspositive and negative controls of DC maturation, respectively.
Statistical analyses
All statistical analyses were performed using SPSS software (SPSS).To analyze data from DC maturation experiments, parametric statisticalmethods (unpaired t test and one-way ANOVAwith Dunnet posttest) wereused. For comparisons from CTL experiments, a two-way ANOVA wasperformed using treatment group and target as factors, followed by meanmultiple comparisons by Bonferroni test. These statistical methods werealso used for tumor volume comparisons (factors: treatment group andday). Log-rank tests were used for Kaplan–Meier curves of tumor-freesurvival. When variables studied were not normally distributed (numberof lung metastases and percentage of TdT+, cleaved caspase-3+, CRT+, andERp57+ cells), nonparametric statistical methods were applied. Mann–Whitney U test was used to compare variables between two groups. Whenthree or more groups were compared, the Kruskal–Wallis test with Dunnposttest was used. Data were considered significant when p , 0.05.
ResultsT cells are essentials for 7A7 mAb but not for AG1478 TKItherapeutic effect
Previous experiments had shown that treatment with 7A7 mAbinhibited cellular functions associated with EGFR signaling onD122 cells and impaired the metastatic spread of this tumor in anexperimental metastasis model (13). Therefore, we examined theability of EGFR-specific TKI AG1478 to induce an antimetasta-
tic effect on D122 cells. First, we analyzed the capacity of thisdrug to inhibit EGFR activation. AG1478 inhibited EGF-inducedEGFR tyrosine phosphorylation (Supplemental Fig. 1A). We alsodetermined the effect of AG1478 on D122 cell cycle progres-sion. An increase in the proportion of cells in G0-G1 phase withthe corresponding decrease of cells in S and G2-M phases wasfound after AG1478 treatment (Supplemental Fig. 1B). When weconducted cell viability experiments, AG1478 was able to reducethe percentages of viable D122 cells in a dose-dependent man-ner (Supplemental Fig. 1C). To determine the in vivo effect ofAG1478 on D122 tumor, we first carried out an experimentalmetastasis assay in which 7A7 used in therapeutic setting dem-onstrated a potent antimetastatic effect (13). Administration ofAG1478 provoked a significant reduction in D122 lung metastasesnumber similar to that induced by the treatment with the Ab (Fig.1A).Our previous results had also suggested a dependence on CD8+
and CD4+ T cells for the antimetastatic effect of 7A7 on D122experimental metastasis assays (13). Thus, the next step was todefine the contribution of T cell response for the AG1478 effect onthis model. With this aim, anti-EGFR agent-treated mice wereinoculated with CD8- or CD4-depleting Abs. Neither CD4 norCD8 depletion affected the potent antimetastatic effect of AG1478(Fig. 1B, upper panel). However, in accordance with our previousreport, in vivo injection of CD4/CD8-specific Abs completelyabolished 7A7 antimetastatic effect (Fig. 1B, lower panel). Ad-ditional data suggesting the differential contribution of T cells for7A7 and AG1478 antimetastatic activity were obtained when theadministration frequency of anti-EGFR agents was decreased inthe experimental metastasis assay. A single 7A7 dose given at day6 after tumor cell inoculation or two doses at days 6 and 13retained the same effect of six doses of the Ab (Fig. 1C, leftpanel). In contrast, the reduction of AG1478 administration fre-quency provoked a significant decrease in its antimetastatic effect(Fig. 1C, right panel).Relevance of CD8+ T cells for the antimetastatic activity of 7A7
was also verified using a D122 spontaneous metastasis model asdescribed in Fig. 2A. In this scenario, 7A7 and AG1478 decreasedboth primary tumor growth as well as the appearance of metas-tases (Fig. 2B, 2C). Depletion of CD8+ cells had no detectableimpact on AG1478 action. In contrast, this treatment provoked theabrogation of 7A7 antimetastatic activity (Fig. 2C). In primarytumors, a complete dependence on CD8+ cells for 7A7 effect wasnot found. In mice treated with anti-CD8 depleting Ab togetherwith 7A7, an effect on tumor growth was achieved when com-pared with control group. This effect was similar to that inducedby AG1478 groups (Fig. 2B).
7A7 mAb treatment induces a potent CD8+ T response on D122metastasis-bearing mice
We explored the capacity of 7A7 versus AG1478 to generate atumor-specific CTL activity on D122 experimental metastasis-bearing mice. First, we examined whether D122 cells express asufficient amount of H-2Kb molecules that could stimulate CD8+
T cells from treated mice. In agreement with a previous report(15), D122 cells expressed very low levels of H-2Kb molecules,which could be increased by IFN-g treatment (data not shown).Thus, IFN-g–treated D122 cells were used for in vitro restim-ulation of lymphocytes from axillary lymph nodes and as targetcells in cytotoxic experiments. C57BL/6 mice were i.v. inoculatedwith D122 cells and treated with anti-EGFR passive agents. Six-teen days after tumor inoculation, in vitro CTL assays were per-formed (Fig. 3A). Previously, we had detected that 7A7 treatmentpromotes a significant mobilization of T cells into metastatic sites
4956 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS
between days 12 and 17 after tumor inoculation (13). As expected,administration of AG1478 did not stimulate measurable lytic ac-tivity against IFN-g–treated D122 cells. In contrast, 7A7 treatmentstimulated a potent CTL activity that eliminated a high percentageof IFN-g–treated D122 target cells (∼75% using an E:T ratio of100:1). This response was not present when the IFN-g–treatedmurine melanoma cell line B16F10, which is negative for EGFRexpression (23), was used as target cell.To determine the presence of EGFR-specific clones in the 7A7-
induced CTL repertoire, in vitro CTL assays were carried out usingas a target D122 cells pulsed with an EGFR-derived peptide (Fig.3B). Mut 1, a peptide described as immunodominant for MHCclass I in Lewis lung carcinoma (19), was used as a positivecontrol. We found that 7A7-stimulated CD8+ T cells recognizedand killed not only mut 1-pulsed D122 cells, but also EGFR-derived peptide-pulsed D122 cells. In contrast, no CTL activitywas detected in 7A7-treated mice against D122 cells pulsed withan irrelevant OVA peptide.
To exert a therapeutic antimetastatic effect, effector T cells mustinfiltrate lungs to destroy implanted metastases. At day 16 aftertumor inoculation, we found a 5-fold increase in CD8+ T cellinfiltration into lungs following 7A7 administration when com-pared with AG1478-treated mice (Fig. 3C, left panel). The phe-notype of these CD8+ T cells corresponded with mature and Ag-experienced lymphocytes, measured by CD44 and CD69 cellmarkers, which were accordingly augmented in lung-infiltratingCD8+ T cells from 7A7-treated mice when compared with micethat received AG1478 (Fig. 3C, right panel). Given the low per-centage of CD8+CD69+ T cells in lungs from AG1478-treatedmice, we did not analyze the effector phenotype of these cells.Instead, CD8+CD69+ T cells from 7A7-treated mice were positivefor granzyme B and IFN-g staining, suggesting the functionalactivity of these cells in the metastatic site (Fig. 3D).We also evaluated the induction of an antitumor humoral re-
sponse in 7A7-treated mice comparing to control mice. Sera of7A7-treated mice did not show IgG binding to D122 cells (Sup-
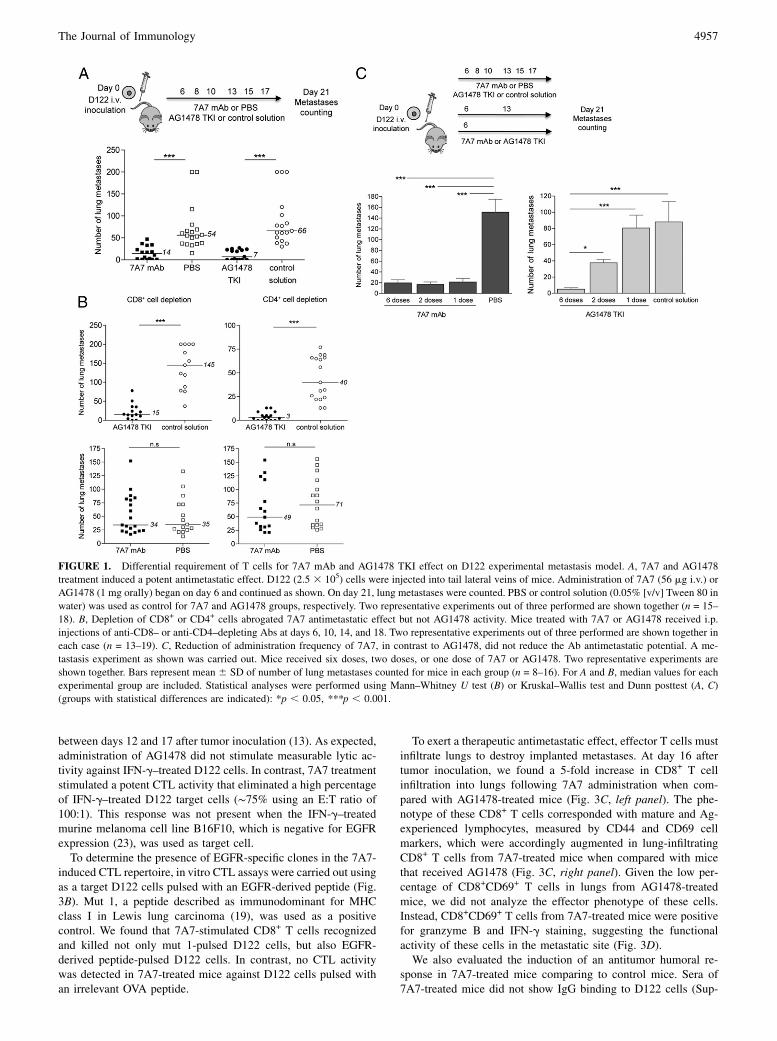
FIGURE 1. Differential requirement of T cells for 7A7 mAb and AG1478 TKI effect on D122 experimental metastasis model. A, 7A7 and AG1478
treatment induced a potent antimetastatic effect. D122 (2.5 3 105) cells were injected into tail lateral veins of mice. Administration of 7A7 (56 mg i.v.) or
AG1478 (1 mg orally) began on day 6 and continued as shown. On day 21, lung metastases were counted. PBS or control solution (0.05% [v/v] Tween 80 in
water) was used as control for 7A7 and AG1478 groups, respectively. Two representative experiments out of three performed are shown together (n = 15–
18). B, Depletion of CD8+ or CD4+ cells abrogated 7A7 antimetastatic effect but not AG1478 activity. Mice treated with 7A7 or AG1478 received i.p.
injections of anti-CD8– or anti-CD4–depleting Abs at days 6, 10, 14, and 18. Two representative experiments out of three performed are shown together in
each case (n = 13–19). C, Reduction of administration frequency of 7A7, in contrast to AG1478, did not reduce the Ab antimetastatic potential. A me-
tastasis experiment as shown was carried out. Mice received six doses, two doses, or one dose of 7A7 or AG1478. Two representative experiments are
shown together. Bars represent mean 6 SD of number of lung metastases counted for mice in each group (n = 8–16). For A and B, median values for each
experimental group are included. Statistical analyses were performed using Mann–Whitney U test (B) or Kruskal–Wallis test and Dunn posttest (A, C)
(groups with statistical differences are indicated): *p , 0.05, ***p , 0.001.
The Journal of Immunology 4957

plemental Fig. 2A). IgM binding to tumor cells was detected in theAb-treated group at lower sera dilution (1:100), but a similar re-cognition capacity was achieved in control and naive mice (Sup-plemental Fig. 2B), suggesting that 7A7 administration was notable to induce a tumor-specific humoral response.
7A7 F(ab9)2 elicits a tumor-specific cytotoxic T cell response
To evaluate whether the EGFR blockade by 7A7mAbwas involvedin its capacity to stimulate antitumor CD8+ T cells, we conductedin vivo experiments as described in Fig. 1A using 7A7 F(ab9)2.First, we demonstrated in vitro biological activity of this mole-cule. We tested the capacity of 7A7 F(ab9)2 to recognize the mu-rine EGFR extracellular domain recombinant protein by ELISA.A similar recognition pattern was detected for 7A7 F(ab9)2 and7A7 at equimolar concentrations (Supplemental Fig. 3A). Similarin vitro activity was also achieved in Western blot analyses ofEGFR phosphorylation status and in cell viability experimentsusing 7A7 F(ab9)2 and 7A7-treated D122 cells (Supplemental Fig.3B, 3C).Due to differences in the m.w. of both molecules and the de-
scribed differences in clearance by the reticuloendothelial systemand excretion by the kidneys (24), we previously had conducted anstudy of pharmacokinetic and biodistribution for 7A7 and 7A7F(ab9)2 (our unpublished data). When equimolar concentrations ofboth agents were injected i.v. into C57BL/6 mice, we found thatF(ab9)2 were cleared 10-fold more rapidly than the whole mole-cule. The dose used for in vivo treatment was then adjusted basedon the t1/2 achieved for 7A7 mAb and 7A7 F(ab9)2, similar tostrategies followed by several groups (17, 25). Ten-fold largeramounts of the F(ab9)2 were administered [56 mg of 7A7 and37.33 mg 3 10 of 7A7 F(ab9)2].Results from this experimental setup demonstrated the capacity
of 7A7 F(ab9)2 to reduce metastases number when compared with
PBS group, suggesting that EGFR inhibition is sufficient to me-diate an in vivo effect (Fig. 4A). This response pattern was ob-served in three independent experiments. With this schedule of Aband bivalent fragment administration, 7A7 F(ab9)2 appeared toproduce less complete metastasis inhibition than 7A7 mAb, sug-gesting the involvement of Fc-mediated mechanisms in the anti-metastatic activity of the complete Ab. As multiple factors otherthan t1/2 could impact on the biological efficacy of F(ab9)2 frag-ment, further experiments testing different doses should be con-ducted to define the relative contribution of the antitumormechanisms induced by F(ab9)2 versus Fc region to 7A7-mediatedantimetastatic effect.Next, we determined whether CD8+ cells were involved in the
in vivo effect of 7A7 F(ab9)2 (Fig. 4B). Mice treated with thebivalent fragment were inoculated with a CD8-specific depletingAb. CD8+ cell depletion completely abrogated the antimetastaticeffect of 7A7 F(ab9)2. Finally, the induction of CD8+ T cells after7A7 F(ab9)2 treatment was evaluated (Fig. 4C). These experimentsrevealed that 7A7 F(ab9)2 was able to stimulate a CTL activity.However, the percentages of specific lysis after 7A7 F(ab9)2 ad-ministration were slightly lower than in 7A7-treated mice (29.6 68.0% versus 38.6 6 4.4%), suggesting a possible contribution of7A7 Fc region to CTL response generation.
EGFR inhibition by 7A7 mAb induces a potent proapoptoticeffect on D122 lung metastases
In an attempt to understand the mechanisms involved in thedifferential activation of T cells for 7A7 versus AG1478, weexamined the contribution of apoptosis induction. The capacityof cells dying by apoptosis to elicit an effective antitumor im-mune response has been recently demonstrated for anthracy-clines and UVC irradiation (26). In response to 7A7 or AG1478,D122 cells underwent apoptosis, as indicated by staining with
FIGURE 2. Dependence on CD8+ cells to 7A7 mAb effect on D122 primary tumor and spontaneous metastasis. A, Schematic representation of D122
spontaneous metastasis model. Mice were challenged s.c. with D122 (5 3 104) cells. Administration of 7A7 (56 mg i.v.), AG1478 (1 mg orally), or PBS
(i.v.) began on day 14 when tumors reached ∼0.3 cm3 and continued as shown. Each experimental group was separated into two subgroups that received
i.p. injections of anti-CD8–depleting Abs or PBS at days 14, 18, 22, 26, and 30. Primary tumor growth was monitored, and lung metastases were counted on
day 35. One representative experiment out of two performed is shown (n = 7). B, 7A7 effect on primary tumor, in contrast to AG1478, was partially affected
by CD8+ cell depletion. Each point represents mean 6 SD of tumor volume for mice in each group. Analyses were performed according to two-way
ANOVA test and Bonferroni posttest. Statistical differences between groups are indicated in the graph. C, Depletion of CD8+ cells completely abrogated
7A7 antimetastatic effect but not AG1478 activity. Statistical analyses were performed using Kruskal–Wallis test and Dunn posttest (different letters in-
dicate statistical differences): a versus b, p , 0.01. Median values for each experimental group are included.
4958 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS
Annexin V-PI. A 4-fold increase of phosphatidyl serine-positiveD122 cells was detected for both agents (Fig. 5A). In 7A7- andAG1478-treated D122 cells, we also found similar levels ofcaspase-3 activation (Fig. 5B) and DNA fragmentation (Fig.5C).However, differences in the capacity of both therapeutic agents
to induce apoptosis in vivo were detected. Histological analyses oflung samples revealed that 7A7 treatment, in contrast to AG1478administration, induced a potent proapoptotic effect on metasta-tic areas (Fig. 5D). Percentages of apoptotic (TdT-positive) cellsin 7A7-treated mice were significantly higher with respect tothe control group (12.5 6 2.7% versus 1.0 6 0.7%), whereasAG1478 treatment induced only a moderate increase in apoptoticcells (4.0 6 1.2%) (Fig. 5D). It is noteworthy that differences inapoptotic index were detected using 7A7 and AG1478 regimesthat induced similar antimetastatic activity, suggesting that apo-ptosis could be a mechanism particularly relevant for 7A7 in vivoeffect.
To evaluate the contribution of EGFR inhibition to the generationof 7A7-induced apoptosis, the experiments were conducted usingthe bivalent fragment of the Ab. In vitro 7A7 F(ab9)2 treatment ofcells revealed an identical capacity of the fragment compared withthe whole Ab to induce apoptosis (Fig. 5A–C). TUNEL studies inlung sections showed the capacity of 7A7 F(ab9)2 to provokea significant higher proapoptotic effect on remaining D122 me-tastases (9.8 6 2.3%) when compared with AG1478-treated andcontrol groups. However, the percentages of apoptotic cells in 7A7F(ab9)2-treated mice were slightly lower than in 7A7-treated mice(Fig. 5D). These results confirmed that EGFR blockade is relevantfor the in vivo apoptosis induction by 7A7, but, in addition, sug-gested that Ab Fc region could also be involved. As previouslysuggested by Kono and Rock (27), quantitative differences inapoptosis induction by 7A7 and AG1478 could contribute to theexclusive capacity of the Ab to generate an effective antitumorCTL response. However, differential release of endogenous dan-ger signals could also be involved.
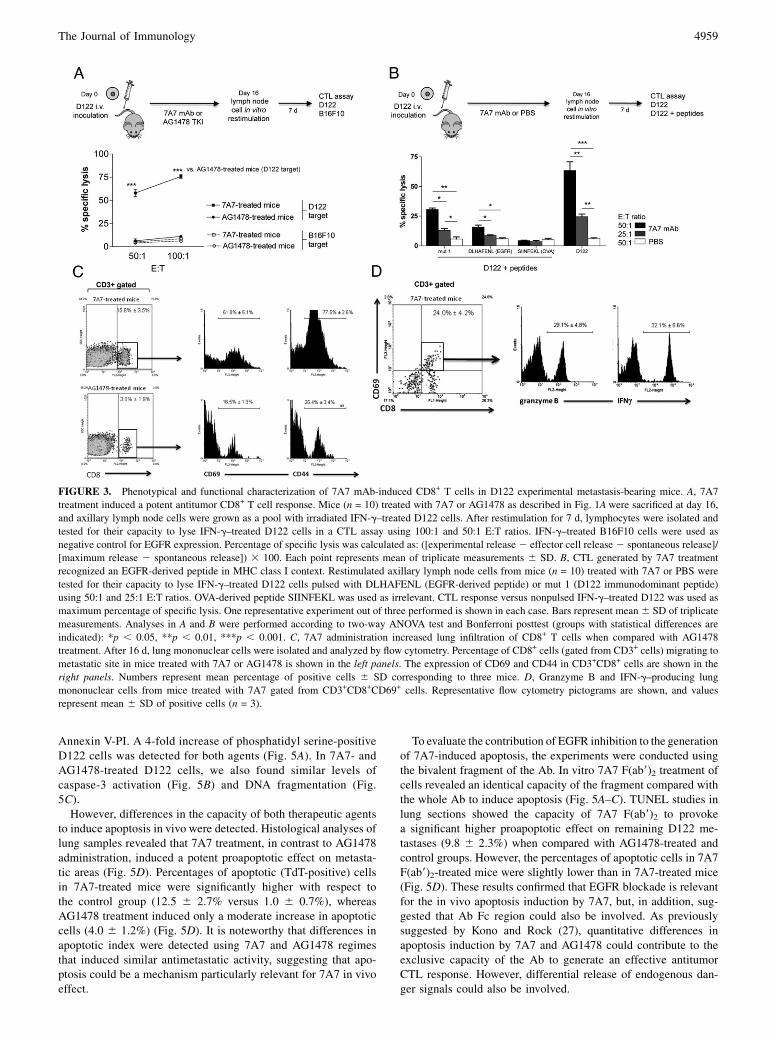
FIGURE 3. Phenotypical and functional characterization of 7A7 mAb-induced CD8+ T cells in D122 experimental metastasis-bearing mice. A, 7A7
treatment induced a potent antitumor CD8+ T cell response. Mice (n = 10) treated with 7A7 or AG1478 as described in Fig. 1Awere sacrificed at day 16,
and axillary lymph node cells were grown as a pool with irradiated IFN-g–treated D122 cells. After restimulation for 7 d, lymphocytes were isolated and
tested for their capacity to lyse IFN-g–treated D122 cells in a CTL assay using 100:1 and 50:1 E:T ratios. IFN-g–treated B16F10 cells were used as
negative control for EGFR expression. Percentage of specific lysis was calculated as: ([experimental release2 effector cell release 2 spontaneous release]/
[maximum release 2 spontaneous release]) 3 100. Each point represents mean of triplicate measurements 6 SD. B, CTL generated by 7A7 treatment
recognized an EGFR-derived peptide in MHC class I context. Restimulated axillary lymph node cells from mice (n = 10) treated with 7A7 or PBS were
tested for their capacity to lyse IFN-g–treated D122 cells pulsed with DLHAFENL (EGFR-derived peptide) or mut 1 (D122 immunodominant peptide)
using 50:1 and 25:1 E:T ratios. OVA-derived peptide SIINFEKL was used as irrelevant. CTL response versus nonpulsed IFN-g–treated D122 was used as
maximum percentage of specific lysis. One representative experiment out of three performed is shown in each case. Bars represent mean 6 SD of triplicate
measurements. Analyses in A and B were performed according to two-way ANOVA test and Bonferroni posttest (groups with statistical differences are
indicated): *p , 0.05, **p , 0.01, ***p , 0.001. C, 7A7 administration increased lung infiltration of CD8+ T cells when compared with AG1478
treatment. After 16 d, lung mononuclear cells were isolated and analyzed by flow cytometry. Percentage of CD8+ cells (gated from CD3+ cells) migrating to
metastatic site in mice treated with 7A7 or AG1478 is shown in the left panels. The expression of CD69 and CD44 in CD3+CD8+ cells are shown in the
right panels. Numbers represent mean percentage of positive cells 6 SD corresponding to three mice. D, Granzyme B and IFN-g–producing lung
mononuclear cells from mice treated with 7A7 gated from CD3+CD8+CD69+ cells. Representative flow cytometry pictograms are shown, and values
represent mean 6 SD of positive cells (n = 3).
The Journal of Immunology 4959
7A7 mAb induces immunogenic apoptosis on D122 tumormodel with independence on its Fc region
To explore the putative intrinsic immunogenicity of cells dying bytreatment with anti-EGFR agents, D122 cells were treated in vitroto program them for cell death and s.c. injected into the right flankof mice. Inoculation of equivalent percentages of apoptotic cells forthe groups of anti-EGFR agents was verified by Annexin V/PIstaining (Fig. 6A). D122 cells treated with DX or MC were usedas positive and negative controls of immunogenic apoptosis in-duction, respectively (28). Seven days later, these animals werechallenged with live D122 cells into the opposite flank, and tumorgrowth was monitored (Fig. 6B). The absence or delay of tumorgrowth was then scored as an indication of antitumor immuneresponse elicited by dying cells (29).Similar to animals that received DX-treated D122, inoculation of
7A7-treated D122 cells reduced the frequency of tumor developingfrom live cells (∼60% of tumor-free mice for DX group and ∼50%for 7A7 group on day 39). In contrast, all mice injected withAG1478-treated D122 developed tumors on day 15. An identicaltumor incidence was found in mice injected with MC-treated anduntreated cells (Fig. 6B, left panel). Furthermore, in mice injectedwith 7A7-treated D122 cells that developed tumors, a significantdelay in tumor growth was achieved. This effect was not detectedin tumor-bearing mice from the AG1478 group (Fig. 6B, leftpanel). Antitumor immune response induced by injection with7A7-treated D122 was specific because mice were not protectedagainst a challenge with an unrelated tumor (Fig. 6B, right panel).Taken together, these results suggested that 7A7, in contrast toAG1478, was able to induce apoptosis with molecular determi-nants of immunogenicity.Next, we wondered whether 7A7 F(ab9)2 induced apoptosis with
immunogenic features. As shown in Fig. 6B, an identical degreeof protection was obtained in mice inoculated with 7A7 F(ab9)2or 7A7 mAb-treated D122 cells, suggesting that Fc-independentmechanisms are associated to the immunogenic apoptosis inducedby 7A7. In accordance with these experimental results, we alsofound that 7A7 or 7A7 F(ab9)2-treated D122 cells elicited a similarcytotoxic T cell response after in vivo inoculation (Fig. 6C). Thisresponse was not detected in mice treated with cells dying byAG1478 treatment. In addition, depletion experiments revealed
that CD4+ and CD8+ cells were required for the 7A7 F(ab9)2 and7A7 mAb-elicited immunogenic effect (Fig. 6D).We therefore studied the expression of molecular determinants
for immunogenic apoptosis in D122 cells treated with anti-EGFRagents (30). Similar to DX, we found that 7A7 and 7A7 F(ab9)2induced an early (12 h) preapoptotic exposure of CRT and ERp57on the plasma membrane (Fig. 7A). Using identical conditions,AG1478 failed to elicit early CRT and ERp57 exposure. A widerange of AG1478 concentrations was proved with similar results(data not shown). Our experimental data suggested that mecha-nisms behind CRT/ERp57 translocation induced by 7A7 mAb aresimilar to those reported by Kroemer’s group for anthracyclinesand UVC irradiation (29, 31). In 7A7-treated D122 cells, the CRT/ERp57 translocation to the cell surface is a rapid process thatoccurs within hours, well before phosphatidyl-serine exposure,which manifests in ∼24 h (Fig. 7B). Similar to DX, the treatmentof D122 cells with 7A7 in presence of brefeldin A, an inhibitor ofanterograde endoplasmic reticulum–Golgi traffic, did not induceCRT/ERp57 exposition (Fig. 7C). We also found that 7A7 pro-voked an early (6 h) phosphorylation of eIF2a (Fig. 7D), a keystep for anthracycline-induced immunogenic apoptosis (31).In addition, D122 exposed to 7A7 or 7A7 F(ab9)2 exhibited an
increase in the expression of HSP 70 and HSP 90 in plasmamembrane, whereas AG1478 failed to provoke this effect, asmeasured 48 h after stimulation (Fig. 7E). In accordance within vivo immunogenic apoptosis experiments, these results sug-gested that EGFR inhibition by 7A7 is sufficient to induce thetranslocation of molecules with immunogenic properties to thecell surface. Differential capacity of 7A7 versus AG1478 to elicitthese danger signals could contribute to the divergence in thein vivo induction of immunogenic apoptosis by these anti-EGFRpassive agents.
D122 cells dying by 7A7 mAb treatment posses the capacity ofinducing DC maturation
To explore the capacity of 7A7-treated dying tumor cells to stim-ulate DC maturation, we challenged DC with dying D122 cells andmeasured their maturation. DC maturation has been commonlyassociated with an increase in cell size; to detect this morphologicalchange, we measured in CD11c+ cells the forward-scattered (FSC)
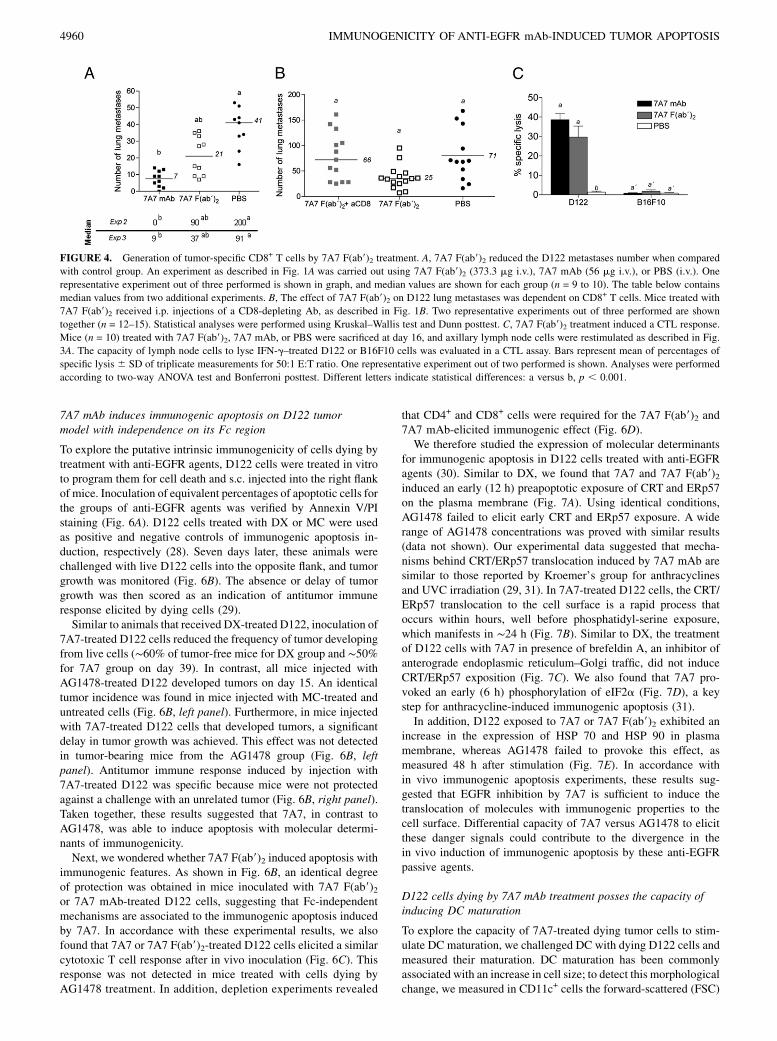
FIGURE 4. Generation of tumor-specific CD8+ T cells by 7A7 F(ab9)2 treatment. A, 7A7 F(ab9)2 reduced the D122 metastases number when compared
with control group. An experiment as described in Fig. 1A was carried out using 7A7 F(ab9)2 (373.3 mg i.v.), 7A7 mAb (56 mg i.v.), or PBS (i.v.). One
representative experiment out of three performed is shown in graph, and median values are shown for each group (n = 9 to 10). The table below contains
median values from two additional experiments. B, The effect of 7A7 F(ab9)2 on D122 lung metastases was dependent on CD8+ T cells. Mice treated with
7A7 F(ab9)2 received i.p. injections of a CD8-depleting Ab, as described in Fig. 1B. Two representative experiments out of three performed are shown
together (n = 12–15). Statistical analyses were performed using Kruskal–Wallis test and Dunn posttest. C, 7A7 F(ab9)2 treatment induced a CTL response.
Mice (n = 10) treated with 7A7 F(ab9)2, 7A7 mAb, or PBS were sacrificed at day 16, and axillary lymph node cells were restimulated as described in Fig.
3A. The capacity of lymph node cells to lyse IFN-g–treated D122 or B16F10 cells was evaluated in a CTL assay. Bars represent mean of percentages of
specific lysis 6 SD of triplicate measurements for 50:1 E:T ratio. One representative experiment out of two performed is shown. Analyses were performed
according to two-way ANOVA test and Bonferroni posttest. Different letters indicate statistical differences: a versus b, p , 0.001.
4960 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS

light intensity (Fig. 8A). DC cocultured with 7A7 mAb or DX-treated D122 cells showed an increased FSC signal when com-pared with DC cocultured with untreated D122. In contrast, nei-ther in DC cocultured with AG1478-treated D122 nor in DCcocultured with MC-treated D122 was the intensity of this pa-rameter increased. The exclusive capacity of 7A7 to induce DCmaturation was confirmed when the expression of costimulatorymolecules CD80, CD86, and CD40, as well as MHC class II(MHC II), was studied in CD11c+ cells cocultured with 7A7-treated or AG1478-treated D122 cells (Fig. 8B, 8C). These resultssuggested that 7A7-treated D122 cells are particularly effectivein stimulating DC maturation.As expected, dying D122 cells by treatment with 7A7 F(ab9)2
also induced DC maturation (Fig. 8). However, in DC coculturedwith 7A7 F(ab9)2-treated cells, we detected a slight decrease inMHC II mean fluorescence intensity (MFI) values when comparedwith 7A7-treated D122 cells. We did not find these differencesbetween 7A7 and 7A7 F(ab9)2 in in vitro and in vivo immunogenic
apoptosis assays. Further experiments must be conducted to clar-ify the relative contribution of cell death induced by EGFR in-hibition to DC maturation.
DiscussionDevelopment of anti-EGFR agents might represent a major break-through in cancer therapy, but several issues still need to be clarifiedto achieve better clinical results. These include understandingof the mechanisms involved in the antitumor effect of anti-EGFR passive therapeutic agents. In this regard, main inves-tigations have been focused in the capacity of Abs and TKI tointerfere with EGFR signaling. Additionally, recent preclinical andclinical data have suggested the relevance of Ab-dependent cellularcytotoxicity as an immunological mechanism of anti-EGFR Abs(32–34). However, there are clinical observations suggesting thatshort-term mechanisms such as EGFR signaling inhibition andAb-dependent cellular cytotoxicity are not the only ones involved.Some studies have indicated that maximal clinical and molecular
FIGURE 5. Significant proapoptotic effect in D122 metastatic niches by 7A7 mAb and 7A7 F(ab9)2 treatment. A–C, 7A7, 7A7 F(ab9)2, and AG1478
induced similar levels of apoptosis on D122 cells. Tumor cells were treated as indicated. A, Twenty-four hours later, D122 were double-stained with
Annexin V-FITC and PI and analyzed by flow cytometry. Percentages of Annexin V-positive cells from three independent experiments are shown as mean 6SD. B, Forty-eight hours later, the expression of cleaved caspase-3 was detected by flow cytometry. Dot plot graphs show the results of one representative
experiment indicating the percentages of cleaved caspase-3–positive cells for each group. A quantitative analysis of four independent experiments is
included. C, Seventy-two hours later, cells were fixed with ice-cold methanol/acetone and stained with PI. Percentages of cells with DNA fragmentation
(M1: sub G0-G1 phase) from three independent experiments are shown as mean 6 SD. D, 7A7 or 7A7 F(ab9)2 treatment induced a higher apoptotic index
than AG1478 administration in D122 lung metastases. An experiment as described in Fig. 1A was carried out (n = 10). After 21 d, lung sections were
immunostained with TUNEL system. Bars represent mean 6 SD of deoxinucleotidyl transferase enzyme (TdT)-positive cells from eight fields randomly
counted. Microscope images show the results of one representative field (original magnification 310) for each experimental group. Black arrows show
representative apoptotic cells. Statistical analyses were performed using Kruskal–Wallis test and Dunn posttest (different letters indicate statistical dif-
ferences). For B: a versus b, p , 0.001. For D: a versus b, p , 0.01; a versus c, p , 0.001; c versus b, p , 0.05.
The Journal of Immunology 4961
responses to anti-EGFR Abs may take several months (35–37).Mechanisms underlying the induction of long-time disease stabi-lization by EGFR Ab treatment have not been formally inves-tigated.To address this idea, using an animal model, we previously
generated anAb that recognizes murine EGFR named 7A7 (12). Ourprevious results indicated the involvement of T cells in the 7A7effect on D122 experimental metastasis (13). In the present report,we confirmed that 7A7 antimetastatic activity was dependent onT cells in D122 experimental and spontaneous metastasis assays,whereas the activity of AG1478 was not affected in CD4+ andCD8+ cell-depleted mice. Consistent with these findings, one ortwo doses of 7A7 Ab were able to induce an effective eliminationof D122 experimental metastasis. On the contrary, the reduction ofAG1478 inoculation frequency provoked a significant decrease onits antimetastatic potential, as expected for passive drugs. Takinginto consideration t1/2 values for both molecules, 23.94 h for 7A7(our unpublished data) and 30 min for AG1478 (14), these datasuggest that, in contrast to AG1478, the antimetastatic effect of7A7 mAb occurs through target-indirect mechanisms.
Taking into consideration the reliance on CD8+ and CD4+ cellsto the antimetastatic activity of 7A7, we evaluated whether mAbtherapy could lead to tumor-specific CTL and humoral responses.We found that 7A7 treatment of D122 experimental metastasis-bearing mice, in contrast to AG1478 administration, was an ef-fective strategy to stimulate a cytotoxic CD8+ T cell responseagainst D122 tumor Ags, including EGFR. Moreover, 7A7 ad-ministration markedly increased CD8+ T cell infiltration into lungmetastases. When we examined the phenotype of lung-infiltratingCD8+ T cells, an upregulation of CD69 molecule, an activationmarker, was verified in 7A7-treated mice when compared with thegroup that received AG1478. Importantly, 7A7-stimulated CD8+
T cells were able to produce IFN-g and granzyme B in the absenceof ex vivo stimulation, suggesting that lung-infiltrating T cells arefunctional in the metastasis microenvironment. Given that T celldysfunction is an event frequently induced by tumors (38), furtherexperiments must be conducted to confirm the lytic function oflung-infiltrating CD8+ T cells. In contrast, we did not detect atumor-specific humoral response in 7A7-treated mice. A relevantissue that needs to be explored to complete the characterization of
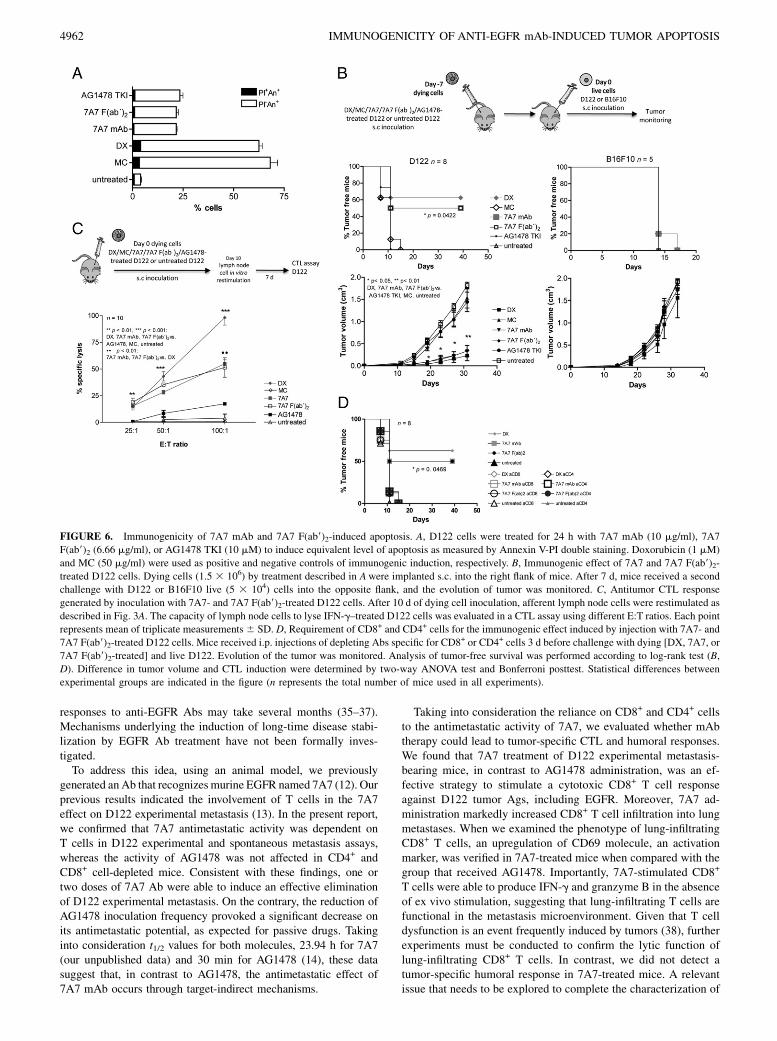
FIGURE 6. Immunogenicity of 7A7 mAb and 7A7 F(ab9)2-induced apoptosis. A, D122 cells were treated for 24 h with 7A7 mAb (10 mg/ml), 7A7
F(ab9)2 (6.66 mg/ml), or AG1478 TKI (10 mM) to induce equivalent level of apoptosis as measured by Annexin V-PI double staining. Doxorubicin (1 mM)
and MC (50 mg/ml) were used as positive and negative controls of immunogenic induction, respectively. B, Immunogenic effect of 7A7 and 7A7 F(ab9)2-
treated D122 cells. Dying cells (1.5 3 106) by treatment described in Awere implanted s.c. into the right flank of mice. After 7 d, mice received a second
challenge with D122 or B16F10 live (5 3 104) cells into the opposite flank, and the evolution of tumor was monitored. C, Antitumor CTL response
generated by inoculation with 7A7- and 7A7 F(ab9)2-treated D122 cells. After 10 d of dying cell inoculation, afferent lymph node cells were restimulated as
described in Fig. 3A. The capacity of lymph node cells to lyse IFN-g–treated D122 cells was evaluated in a CTL assay using different E:T ratios. Each point
represents mean of triplicate measurements6 SD. D, Requirement of CD8+ and CD4+ cells for the immunogenic effect induced by injection with 7A7- and
7A7 F(ab9)2-treated D122 cells. Mice received i.p. injections of depleting Abs specific for CD8+ or CD4+ cells 3 d before challenge with dying [DX, 7A7, or
7A7 F(ab9)2-treated] and live D122. Evolution of the tumor was monitored. Analysis of tumor-free survival was performed according to log-rank test (B,
D). Difference in tumor volume and CTL induction were determined by two-way ANOVA test and Bonferroni posttest. Statistical differences between
experimental groups are indicated in the figure (n represents the total number of mice used in all experiments).
4962 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS
the adaptive immune response induced by this Ab is the capacityof 7A7 to induce a long-lasting antitumor response. In this regard,the involvement of CD4+ T cells in the antimetastatic effect of7A7 could suggest the generation of a memory CD8+ T cell re-sponse in this model (39). In line with this idea, we found in Ab-treated mice a high percentage of lung-infiltrating CD8+ T cellsthat expressed the CD44 molecules (∼75%), which is a markerassociated to memory phenotype. However, in-depth experimentsshould be performed to formally demonstrate the induction ofa memory T cell response by 7A7 treatment.An obvious scenario could be envisaged in which the differential
capacity to stimulate an effective CTL response for both anti-EGFRagents is due to the effector functions mediated by Ab Fc region.Several studies have demonstrated the relevance of the Fc-FcgRsystem in adaptive immunity. Ag uptake in form of opsonized tu-mor cells has been associated with enhanced Ag presentation by DCand the generation of Ag-specific CD4+ and CD8+ T cell responses(40). It has been described that some Abs approved for cancertreatment, including those specific to EGFR, promote trogocytosisin vitro upon binding to their target cells (41). In this process, FcgRon acceptor cells (e.g., DC) remove and internalize cognate ligandsand cell membrane fragments from tumor cells (42). In addition, ithas been proposed recently that the Fc region of cetuximab, anEGFR-directed Ab used in the clinic, could contribute to adaptiveimmune activation by the induction of an NK/DC cross talk (43).Therefore, we decided to test whether not only Fc region but also
the EGFR signaling inhibition by Abs could contribute to the
generation of a tumor-specific T cell immunity. To address thisissue, we used the bivalent fragment of 7A7. Experiments toevaluate the effect of 7A7 F(ab9)2 on D122 experimental metas-tases demonstrated its capacity to reduce the metastases number,although the fragment activity was less complete (∼50%) than thatof the native Ab. This result suggested the relevance of the Fcregion for the in vivo effect of 7A7 mAb. The possibility that dualactivities involving both pharmacological receptor blockade andFc-mediating mechanisms may contribute to antitumor activity ofanti-EGFR Abs has been suggested previously by Fan and col-leagues (17). We used a similar schedule for bivalent fragmentadministration to that reported by these authors. Our next step wasto determine the involvement of CD8+ T cells in the 7A7 F(ab9)2in vivo activity. The antimetastatic potential of 7A7 F(ab9)2 wascompletely reliant on the presence of this cell population. In vitroCTL experiments showed that 7A7 F(ab9)2 was able to inducea tumor-specific CTL response. However, a slight decrease in thecytotoxic activity was achieved in 7A7 F(ab9)2-treated mice whencompared with mice that received the whole molecule, suggestingthat 7A7 Fc region contribute to CTL generation. These datademonstrated that EGFR-mediated signaling inhibition by 7A7,and not by AG1478, plays an important role in establishing a linkwith adaptive immunity.To understand why EGFR signaling inhibition by 7A7 but not by
AG1478 induced a CTL response, we focused on the apoptosisinduction. It has been shown in recent reports that therapy with Absprovoking apoptosis either by direct damage to tumor cells (44) or
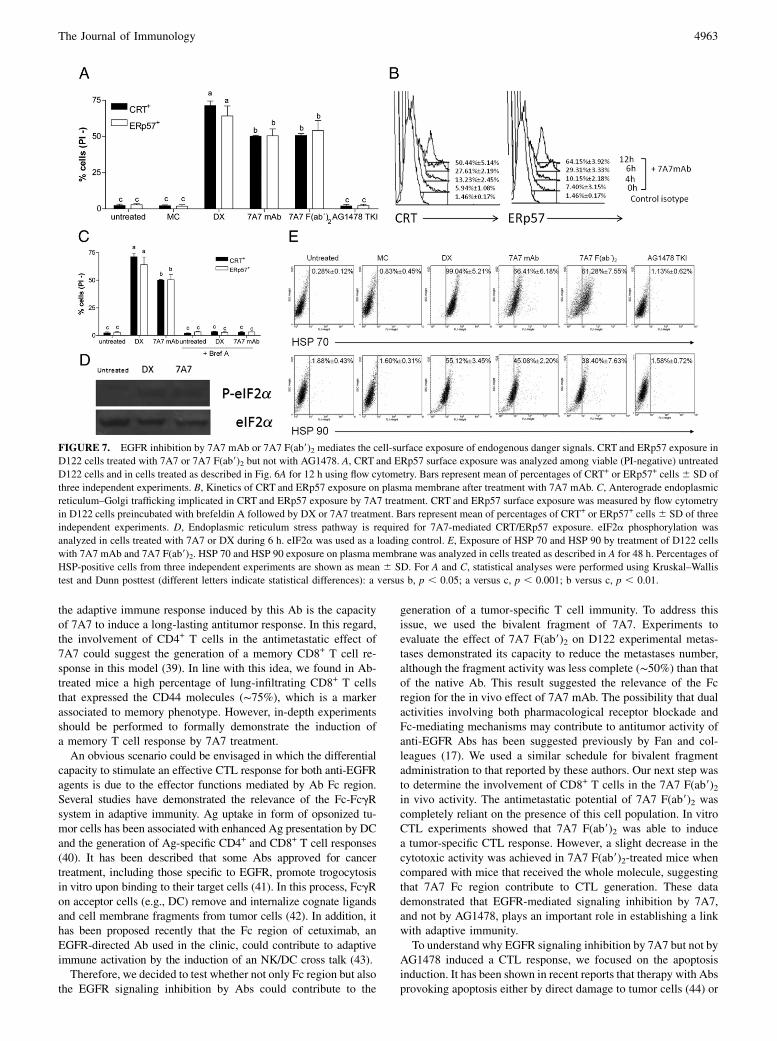
FIGURE 7. EGFR inhibition by 7A7 mAb or 7A7 F(ab9)2 mediates the cell-surface exposure of endogenous danger signals. CRT and ERp57 exposure in
D122 cells treated with 7A7 or 7A7 F(ab9)2 but not with AG1478. A, CRT and ERp57 surface exposure was analyzed among viable (PI-negative) untreated
D122 cells and in cells treated as described in Fig. 6A for 12 h using flow cytometry. Bars represent mean of percentages of CRT+ or ERp57+ cells 6 SD of
three independent experiments. B, Kinetics of CRT and ERp57 exposure on plasma membrane after treatment with 7A7 mAb. C, Anterograde endoplasmic
reticulum–Golgi trafficking implicated in CRT and ERp57 exposure by 7A7 treatment. CRT and ERp57 surface exposure was measured by flow cytometry
in D122 cells preincubated with brefeldin A followed by DX or 7A7 treatment. Bars represent mean of percentages of CRT+ or ERp57+ cells6 SD of three
independent experiments. D, Endoplasmic reticulum stress pathway is required for 7A7-mediated CRT/ERp57 exposure. eIF2a phosphorylation was
analyzed in cells treated with 7A7 or DX during 6 h. eIF2a was used as a loading control. E, Exposure of HSP 70 and HSP 90 by treatment of D122 cells
with 7A7 mAb and 7A7 F(ab9)2. HSP 70 and HSP 90 exposure on plasma membrane was analyzed in cells treated as described in A for 48 h. Percentages of
HSP-positive cells from three independent experiments are shown as mean 6 SD. For A and C, statistical analyses were performed using Kruskal–Wallis
test and Dunn posttest (different letters indicate statistical differences): a versus b, p , 0.05; a versus c, p , 0.001; b versus c, p , 0.01.
The Journal of Immunology 4963
through antiangiogenic effects (7) can activate CTL responses.Apoptotic cells are an especially attractive source of Ag for cross-presentation (45–47). The immunological consequence of the in-gestion of apoptotic cellular material by DC has been controver-sial. In general, apoptosis is thought to be intrinsically tolerogenic,whereas necrosis is inherently immunogenic and elicits inflam-matory reactions (48, 49). However, in-depth investigations haveshown that cells dying by apoptosis can be highly immunogenic(50). For example, the apoptosis of tumor cells induced by somechemotherapeutic agents (28, 51) or radiotherapy (52) has beenshown to prime a tumor-specific immune response.Why, then, is apoptosis sometimes silent and other times
proinflammatory? A likely key factor is how rapidly the apoptoticcells are cleared by phagocytes. This is because, over time, apo-ptotic cells undergo a process known as secondary necrosis inwhich their membranes become permeable to proinflammatorycomponents (27). Another important element is the nature of themolecules exposed and/or released by dying cells even at earlystages. It is now assumed that apoptosis can elicit an immuneresponse only if dying cells emit specific danger signals thatmediate their efficient phagocytosis by DC and the consequent DCmaturation (26).Previous reports have described apoptosis induction on tumor
cells using anti-EGFR passive agents. Apoptosis associated with
EGFR blockade has been characterized by an increase of moleculeswith proapoptotic functions (53, 54) and the activation of thecaspase cascade (55, 56). Importantly, the capacity of anti-EGFRtherapies to promote immunogenic apoptosis has not yet beenexplored. In the current study, we found that both agents, 7A7 andAG1478, were able to induce apoptosis in lung metastatic lesions,but quantitative differences were detected. The percentage of apo-ptotic cells in remaining D122 metastases was markedly increasedin 7A7-treated mice when compared with the AG1478-treatedgroup. In addition, our experimental data showed an importantcontribution of EGFR interference to the apoptosis induced by7A7 in metastatic niches. Interestingly, apoptosis levels detectedwith 7A7 and 7A7 F(ab9)2 treatment were similar to those reportedfor murine carcinomas treated in vivo with chemotherapeuticagents (57). Based on the considerations about factors contribut-ing to immunogenic apoptosis, 7A7’s capacity to generate higherapoptosis rates than AG1478 could contribute to the exclusiveability of the Ab to induce a vaccine effect.However, results from our experiments demonstrated that not
only quantitative differences can be found between 7A7 andAG1478-induced apoptosis. Despite their similar capacity to in-duce the apoptosis routine (phosphatidyl-serine exposure, caspase-3 activation, and detectable DNA fragmentation), 7A7 and AG1478differed in their ability to elicit apoptosis with immunogenic
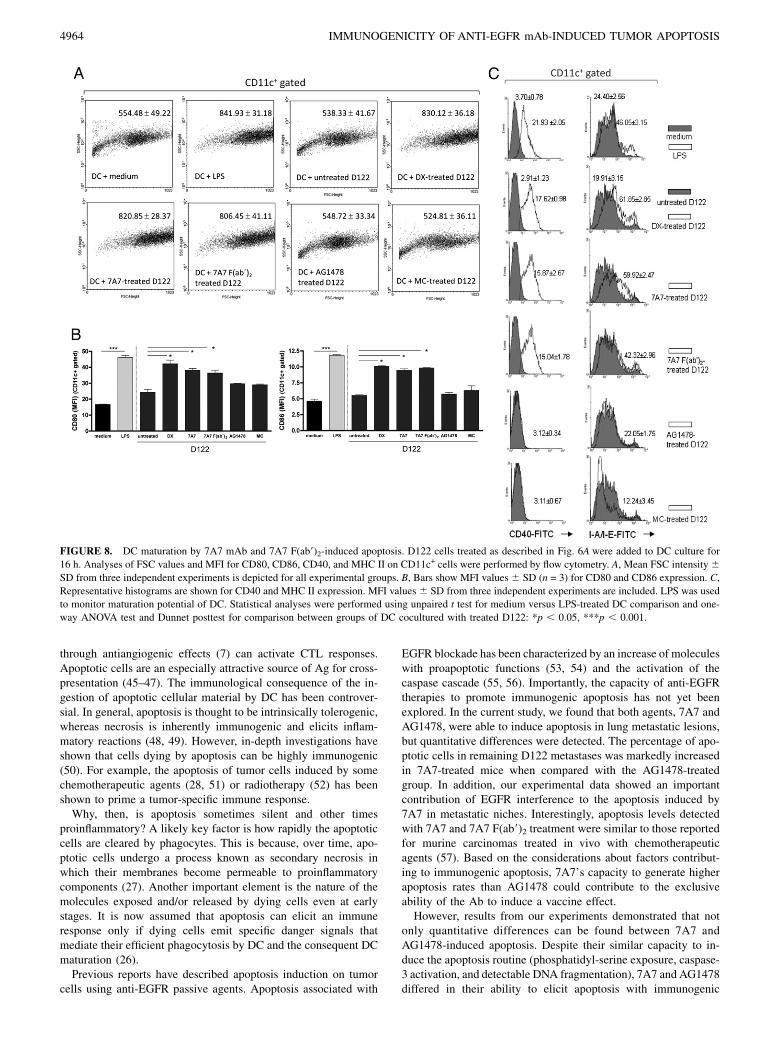
FIGURE 8. DC maturation by 7A7 mAb and 7A7 F(ab9)2-induced apoptosis. D122 cells treated as described in Fig. 6A were added to DC culture for
16 h. Analyses of FSC values and MFI for CD80, CD86, CD40, and MHC II on CD11c+ cells were performed by flow cytometry. A, Mean FSC intensity6SD from three independent experiments is depicted for all experimental groups. B, Bars show MFI values 6 SD (n = 3) for CD80 and CD86 expression. C,
Representative histograms are shown for CD40 and MHC II expression. MFI values 6 SD from three independent experiments are included. LPS was used
to monitor maturation potential of DC. Statistical analyses were performed using unpaired t test for medium versus LPS-treated DC comparison and one-
way ANOVA test and Dunnet posttest for comparison between groups of DC cocultured with treated D122: *p , 0.05, ***p , 0.001.
4964 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS

features. When mice were challenged s.c. with 7A7- or AG1478-treated D122 cells having similar percentages of apoptosis, in theabsence of an adjuvant, only 7A7 was successful to mediatea specific protection against a second challenge with live tumorcells. This effect was dependent on CD8+ and CD4+ cells. Also, weconfirmed in this scenario the exclusive capacity of 7A7-treatedcells to prime a tumor-specific CTL response. At the biochemicallevel, we found differences between 7A7- and AG1478-triggeredapoptosis that could explain their differential capacity to induceimmunogenic apoptosis in vivo. 7A7 stimulated the translocationof CRT and ERp57 from the endoplasmic reticulum to the cellsurface, whereas AG1478 failed to do so. These data underscore thenotion from immunogenic chemotherapy that immunogenic apo-ptosis correlates with the early CRT/ERp57 exposure. In fact, theonly biochemical difference between classical nonimmunogenicand immunogenic apoptosis inducers is the early wave of CRTexposure (29). We also identified for 7A7- and AG1478-inducedapoptosis differences in HSP exposure. This event has also beenassociated with immunogenic chemotherapy (58). Importantly,our experiments demonstrated that the Fc region of 7A7 was notinvolved in the immunogenic apoptosis induction measuredby in vivo injection of 7A7 F(ab9)2-treated cells and in vitrocharacterization of endogenous danger signals in F(ab9)2-treatedcells.Induction of chaperone exposure by 7A7 could suggest an in-
volvement of DC in the Ab capacity to induce an effective T cellresponse. In vitro and in vivo studies have demonstrated that CRT isan eat-me signal allowing engulfment of tumor cells by DC (29). Ithas been suggested that HSP 70 and HSP 90 can capture tumorAgs and facilitate their presentation following uptake by DC (59).They have also been associated with DC maturation (30). We needto study the impact of DC on the immune response induced in vivoby injection with 7A7-treated cells. However, we found evidencesuggesting that apoptosis induced by 7A7, but not AG1478, waseffective to induce DC maturation.Recent data reported by Weihua et al. (60) could contribute to
understanding the mechanisms involved in the differential capa-city of Abs and TKIs specific for EGFR to induce immunogeniccell death. This group demonstrated that EGFR, independentlyof its kinase activity, maintains basal intracellular glucose levelsthrough interaction and stabilization of the sodium/glucosecotransporter 1, thereby preventing cells from autophagy (60).Interestingly, it has been demonstrated that autophagy might alsobe involved in Ag cross-presentation within the Ag donor cells(61). Moreover, several reports have revealed that autophagicfeatures could in fact be essential for exposing the eat-me signalson apoptotic cells (62, 63). Thus, it is possible that the inductionof cell death by anti-EGFR Abs, and not by TKIs, follows a bio-chemically distinct subroutine characterized by apoptosis accom-panied by autophagy.In summary, using 7A7 as a model treatment of EGFR-positive
tumor cells, we described a novel antitumor mechanism for anti-EGFR Abs based on the induction of immunogenic apoptosis.We provide evidence regarding how EGFR inhibition by 7A7, inan Fc-independent manner, leads to molecular events defined ashallmarks of immunogenic apoptosis. Our data suggested that theinduction of apoptosis with immunogenic features by 7A7 couldhave an important contribution to its capacity to generate a tumor-specific response. Thus, the next step will be to define the capacity ofanti-EGFR Abs approved for cancer treatment to trigger an im-munogenic apoptosis, and to evaluate if CTL responses are criticallyinvolved in the clinical response after anti-EGFRAb administration.These data could be relevant in the design of new combination-based approaches for patients with EGFR-positive tumors.
DisclosuresThe authors have no financial conflicts of interest.
References1. Yarden, Y., and M. X. Sliwkowski. 2001. Untangling the ErbB signalling net-
work. Nat. Rev. Mol. Cell Biol. 2: 127–137.2. Ferguson, K. M. 2008. Structure-based view of epidermal growth factor receptor
regulation. Annu. Rev. Biophys. 37: 353–373.3. Pal, S. K., and M. Pegram. 2005. Epidermal growth factor receptor and signal
transduction: potential targets for anti-cancer therapy. Anticancer Drugs 16:483–494.
4. Shepard, H. M., C. M. Brdlik, and H. Schreiber. 2008. Signal integration:a framework for understanding the efficacy of therapeutics targeting the humanEGFR family. J. Clin. Invest. 118: 3574–3581.
5. Ciardiello, F., and G. Tortora. 2008. EGFR antagonists in cancer treatment. N.Engl. J. Med. 358: 1160–1174.
6. Martinelli, E., R. De Palma, M. Orditura, F. De Vita, and F. Ciardiello. 2009.Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy.Clin. Exp. Immunol. 158: 1–9.
7. Manning, E. A., J. G. Ullman, J. M. Leatherman, J. M. Asquith, T. R. Hansen,T. D. Armstrong, D. J. Hicklin, E. M. Jaffee, and L. A. Emens. 2007. A vascularendothelial growth factor receptor-2 inhibitor enhances antitumor immunitythrough an immune-based mechanism. Clin. Cancer Res. 13: 3951–3959.
8. Abes, R., E. Gelize, W. H. Fridman, and J. L. Teillaud. 2010. Long-lasting an-titumor protection by anti-CD20 antibody through cellular immune response.Blood 116: 926–934.
9. Hilchey, S. P., O. Hyrien, T. R. Mosmann, A. M. Livingstone, J. W. Friedberg,F. Young, R. I. Fisher, R. J. Kelleher, Jr., R. B. Bankert, and S. H. Bernstein.2009. Rituximab immunotherapy results in the induction of a lymphomaidiotype-specific T-cell response in patients with follicular lymphoma: supportfor a “vaccinal effect” of rituximab. Blood 113: 3809–3812.
10. Park, S., Z. Jiang, E. D. Mortenson, L. Deng, O. Radkevich-Brown, X. Yang,H. Sattar, Y. Wang, N. K. Brown, M. Greene, et al. 2010. The therapeutic effectof anti-HER2/neu antibody depends on both innate and adaptive immunity.Cancer Cell 18: 160–170.
11. Stagg, J., S. Loi, U. Divisekera, S. F. Ngiow, H. Duret, H. Yagita, M. W. Teng,and M. J. Smyth. 2011. Anti-ErbB-2 mAb therapy requires type I and II inter-ferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc. Natl.Acad. Sci. USA 108: 7142–7147.
12. Garrido, G., B. Sanchez, H. M. Rodriguez, P. Lorenzano, D. Alonso, andL. E. Fernandez. 2004. 7A7 MAb: a new tool for the pre-clinical evaluation ofEGFR-based therapies. Hybrid. Hybridomics 23: 168–175.
13. Garrido, G., P. Lorenzano, B. Sanchez, I. Beausoleil, D. F. Alonso, R. Perez, andL. E. Fernandez. 2007. T cells are crucial for the anti-metastatic effect of anti-epidermal growth factor receptor antibodies. Cancer Immunol. Immunother. 56:1701–1710.
14. Ellis, A. G., M. M. Doherty, F. Walker, J. Weinstock, M. Nerrie, A. Vitali,R. Murphy, T. G. Johns, A. M. Scott, A. Levitzki, et al. 2006. Preclinical analysisof the analinoquinazoline AG1478, a specific small molecule inhibitor of EGFreceptor tyrosine kinase. Biochem. Pharmacol. 71: 1422–1434.
15. Eisenbach, L., N. Hollander, L. Greenfeld, H. Yakor, S. Segal, and M. Feldman.1984. The differential expression of H-2K versus H-2D antigens, distinguishinghigh-metastatic from low-metastatic clones, is correlated with the immunogenicproperties of the tumor cells. Int. J. Cancer 34: 567–573.
16. Porgador, A., B. Brenner, E. Vadai, M. Feldman, and L. Eisenbach. 1991. Im-munization by gamma-IFN-treated B16-F10.9 melanoma cells protects againstmetastatic spread of the parental tumor. Int. J. Cancer Suppl. 6: 54–60.
17. Fan, Z., H. Masui, I. Altas, and J. Mendelsohn. 1993. Blockade of epidermalgrowth factor receptor function by bivalent and monovalent fragments of 225anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 53:4322–4328.
18. Lasarte, J. J., P. Sarobe, J. Prieto, and F. Borras-Cuesta. 1995. In vivo cytotoxicT-lymphocyte induction may take place via CD8 T helper lymphocytes. Res.Immunol. 146: 35–44.
19. Mandelboim, O., G. Berke, M. Fridkin, M. Feldman, M. Eisenstein, andL. Eisenbach. 1994. CTL induction by a tumour-associated antigen octapeptidederived from a murine lung carcinoma. Nature 369: 67–71.
20. Rammensee, H., J. Bachmann, N. P. Emmerich, O. A. Bachor, and S. Stevanovic.1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunoge-netics 50: 213–219.
21. Watanabe, T., T. Kawamura, H. Kawamura, M. Haga, K. Shirai, H. Watanabe,S. Eguchi, and T. Abo. 1997. Intermediate TCR cells in mouse lung: their ef-fector function to induce pneumonitis in mice with autoimmune-like graft-versus-host disease. J. Immunol. 158: 5805–5814.
22. Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu,and R. M. Steinman. 1992. Generation of large numbers of dendritic cells frommouse bone marrow cultures supplemented with granulocyte/macrophagecolony-stimulating factor. J. Exp. Med. 176: 1693–1702.
23. Iivanainen, E., S. Lauttia, N. Zhang, D. Tvorogov, J. Kulmala, R. Grenman,P. Salven, and K. Elenius. 2009. The EGFR inhibitor gefitinib suppresses re-cruitment of pericytes and bone marrow-derived perivascular cells into tumorvessels. Microvasc. Res. 78: 278–285.
24. Covell, D. G., J. Barbet, O. D. Holton, C. D. Black, R. J. Parker, andJ. N. Weinstein. 1986. Pharmacokinetics of monoclonal immunoglobulin G1,F(ab9)2, and Fab9 in mice. Cancer Res. 46: 3969–3978.
The Journal of Immunology 4965

25. Spiridon, C. I., S. Guinn, and E. S. Vitetta. 2004. A comparison of the in vitroand in vivo activities of IgG and F(ab9)2 fragments of a mixture of threemonoclonal anti-Her-2 antibodies. Clin. Cancer Res. 10: 3542–3551.
26. Green, D. R., T. Ferguson, L. Zitvogel, and G. Kroemer. 2009. Immunogenic andtolerogenic cell death. Nat. Rev. Immunol. 9: 353–363.
27. Kono, H., and K. L. Rock. 2008. How dying cells alert the immune system todanger. Nat. Rev. Immunol. 8: 279–289.
28. Casares, N., M. O. Pequignot, A. Tesniere, F. Ghiringhelli, S. Roux, N. Chaput,E. Schmitt, A. Hamai, S. Hervas-Stubbs, M. Obeid, et al. 2005. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp.Med. 202: 1691–1701.
29. Obeid, M., A. Tesniere, T. Panaretakis, R. Tufi, N. Joza, P. van Endert,F. Ghiringhelli, L. Apetoh, N. Chaput, C. Flament, et al. 2007. Ecto-calreticulinin immunogenic chemotherapy. Immunol. Rev. 220: 22–34.
30. Tesniere, A., T. Panaretakis, O. Kepp, L. Apetoh, F. Ghiringhelli, L. Zitvogel,and G. Kroemer. 2008. Molecular characteristics of immunogenic cancer celldeath. Cell Death Differ. 15: 3–12.
31. Panaretakis, T., O. Kepp, U. Brockmeier, A. Tesniere, A. C. Bjorklund,D. C. Chapman, M. Durchschlag, N. Joza, G. Pierron, P. van Endert, et al. 2009.Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death.EMBO J. 28: 578–590.
32. Schneider-Merck, T., J. J. Lammerts van Bueren, S. Berger, K. Rossen, P. H. vanBerkel, S. Derer, T. Beyer, S. Lohse, W. K. Bleeker, M. Peipp, et al. 2010.Human IgG2 antibodies against epidermal growth factor receptor effectivelytrigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only bycells of myeloid lineage. J. Immunol. 184: 512–520.
33. Lopez-Albaitero, A., S. C. Lee, S. Morgan, J. R. Grandis, W. E. Gooding,S. Ferrone, and R. L. Ferris. 2009. Role of polymorphic Fc gamma receptor IIIaand EGFR expression level in cetuximab mediated, NK cell dependent in vitrocytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol.Immunother. 58: 1853–1864.
34. Zhang, W., M. Gordon, A. M. Schultheis, D. Y. Yang, F. Nagashima, M. Azuma,H. M. Chang, E. Borucka, G. Lurje, A. E. Sherrod, et al. 2007. FCGR2A andFCGR3A polymorphisms associated with clinical outcome of epidermal growthfactor receptor expressing metastatic colorectal cancer patients treated withsingle-agent cetuximab. J. Clin. Oncol. 25: 3712–3718.
35. Crombet, T., O. Torres, V. Rodrıguez, A. Menendez, A. Stevenson, M. Ramos,F. Torres, R. Figueredo, I. Veitıa, N. Iznaga, et al. 2001. Phase I clinical eval-uation of a neutralizing monoclonal antibody against epidermal growth factorreceptor in advanced brain tumor patients: preliminary study. Hybridoma 20:131–136.
36. Ramos, T. C., J. Figueredo, M. Catala, S. Gonzalez, J. C. Selva, T. M. Cruz,C. Toledo, S. Silva, Y. Pestano, M. Ramos, et al. 2006. Treatment of high-gradeglioma patients with the humanized anti-epidermal growth factor receptor (EGFR)antibody h-R3: report from a phase I/II trial. Cancer Biol. Ther. 5: 375–379.
37. Bonner, J. A., P. M. Harari, J. Giralt, N. Azarnia, D. M. Shin, R. B. Cohen,C. U. Jones, R. Sur, D. Raben, J. Jassem, et al. 2006. Radiotherapy pluscetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med.354: 567–578.
38. Gabrilovich, D., and V. Pisarev. 2003. Tumor escape from immune response:mechanisms and targets of activity. Curr. Drug Targets 4: 525–536.
39. Harty, J. T., and V. P. Badovinac. 2008. Shaping and reshaping CD8+ T-cellmemory. Nat. Rev. Immunol. 8: 107–119.
40. Dhodapkar, K. M., J. Krasovsky, B. Williamson, and M. V. Dhodapkar. 2002.Antitumor monoclonal antibodies enhance cross-presentation ofcCellular anti-gens and the generation of myeloma-specific killer T cells by dendritic cells. J.Exp. Med. 195: 125–133.
41. Beum, P. V., D. A. Mack, A. W. Pawluczkowycz, M. A. Lindorfer, andR. P. Taylor. 2008. Binding of rituximab, trastuzumab, cetuximab, or mAb T101to cancer cells promotes trogocytosis mediated by THP-1 cells and monocytes. J.Immunol. 181: 8120–8132.
42. Joly, E., and D. Hudrisier. 2003. What is trogocytosis and what is its purpose?Nat. Immunol. 4: 815.
43. Lee, S. C., R. M. Srivastava, A. Lopez-Albaitero, S. Ferrone, and R. L. Ferris.2011. Natural killer (NK):dendritic cell (DC) cross talk induced by therapeuticmonoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol.Res. 50: 248–254.
44. Selenko, N., O. Maidic, S. Draxier, A. Berer, U. Jager, W. Knapp, and J. Stockl.2001. CD20 antibody (C2B8)-induced apoptosis of lymphoma cells promotesphagocytosis by dendritic cells and cross-priming of CD8+ cytotoxic T cells.Leukemia 15: 1619–1626.
45. Albert, M. L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigenfrom apoptotic cells and induce class I-restricted CTLs. Nature 392: 86–89.
46. Baselga, J., R. Herbst, P. LoRusso, D. Rischin, M. Ranson, R. Plummer,E. Raymond, A.-M. Maddox, S. Kaye, D. Kieback, et al. 2000. Continuousadministration of ZD1839 (Iressa), a novel oral epidermal growth factor re-ceptor tyrosine kinase inhibitor (EGFR-TKI), in patients with five selected tu-mor types: evidence of activity and good tolerability. Proc. Am. Soc. Clin.Oncol. 19: 2000.
47. Yrlid, U., and M. J. Wick. 2000. Salmonella-induced apoptosis of infectedmacrophages results in presentation of a bacteria-encoded antigen after uptakeby bystander dendritic cells. J. Exp. Med. 191: 613–624.
48. Savill, J., and V. Fadok. 2000. Corpse clearance defines the meaning of celldeath. Nature 407: 784–788.
49. Matzinger, P. 2002. An innate sense of danger. Ann. N. Y. Acad. Sci. 961: 341–342.
50. Zitvogel, L., N. Casares, M. O. Pequignot, N. Chaput, M. L. Albert, andG. Kroemer. 2004. Immune response against dying tumor cells. Adv. Immunol.84: 131–179.
51. Tesniere, A., F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli,L. Aymeric, M. Michaud, L. Apetoh, L. Barault, et al. 2010. Immunogenic deathof colon cancer cells treated with oxaliplatin. Oncogene 29: 482–491.
52. Obeid, M., T. Panaretakis, N. Joza, R. Tufi, A. Tesniere, P. van Endert,L. Zitvogel, and G. Kroemer. 2007. Calreticulin exposure is required for theimmunogenicity of gamma-irradiation and UVC light-induced apoptosis. CellDeath Differ. 14: 1848–1850.
53. Gilmore, A. P., A. J. Valentijn, P. Wang, A. M. Ranger, N. Bundred,M. J. O’Hare, A. Wakeling, S. J. Korsmeyer, and C. H. Streuli. 2002. Activationof BAD by therapeutic inhibition of epidermal growth factor receptor andtransactivation by insulin-like growth factor receptor. J. Biol. Chem. 277: 27643–27650.
54. Real, P. J., A. Benito, J. Cuevas, M. T. Berciano, A. de Juan, P. Coffer, J. Gomez-Roman, M. Lafarga, J. M. Lopez-Vega, and J. L. Fernandez-Luna. 2005.Blockade of epidermal growth factor receptors chemosensitizes breast cancercells through up-regulation of Bnip3L. Cancer Res. 65: 8151–8157.
55. Liu, B., M. Fang, M. Schmidt, Y. Lu, J. Mendelsohn, and Z. Fan. 2000. Inductionof apoptosis and activation of the caspase cascade by anti-EGF receptormonoclonal antibodies in DiFi human colon cancer cells do not involve the c-junN-terminal kinase activity. Br. J. Cancer 82: 1991–1999.
56. Janmaat, M. L., F. A. Kruyt, J. A. Rodriguez, and G. Giaccone. 2003. Responseto epidermal growth factor receptor inhibitors in non-small cell lung cancer cells:limited antiproliferative effects and absence of apoptosis associated with per-sistent activity of extracellular signal-regulated kinase or Akt kinase pathways.Clin. Cancer Res. 9: 2316–2326.
57. Macluskey, M., R. Baillie, L. M. Chandrachud, N. Pendleton, and A. M. Schor.2000. High levels of apoptosis are associated with improved survival in non-small cell lung cancer. Anticancer Res. 20(3B): 2123–2128.
58. Garg, A. D., D. Nowis, J. Golab, P. Vandenabeele, D. V. Krysko, andP. Agostinis. 2010. Immunogenic cell death, DAMPs and anticancer therapeu-tics: an emerging amalgamation. Biochim. Biophys. Acta 1805: 53–71.
59. Srivastava, P. K. 2006. Therapeutic cancer vaccines. Curr. Opin. Immunol. 18:201–205.
60. Weihua, Z., R. Tsan, W. C. Huang, Q. Wu, C. H. Chiu, I. J. Fidler, andM. C. Hung. 2008. Survival of cancer cells is maintained by EGFR independentof its kinase activity. Cancer Cell 13: 385–393.
61. Li, Y., L. X. Wang, G. Yang, F. Hao, W. J. Urba, and H. M. Hu. 2008. Efficientcross-presentation depends on autophagy in tumor cells. Cancer Res. 68: 6889–6895.
62. Mellen, M. A., E. J. de la Rosa, and P. Boya. 2008. The autophagic machinery isnecessary for removal of cell corpses from the developing retinal neuro-epithelium. Cell Death Differ. 15: 1279–1290.
63. Qu, X., Z. Zou, Q. Sun, K. Luby-Phelps, P. Cheng, R. N. Hogan, C. Gilpin, andB. Levine. 2007. Autophagy gene-dependent clearance of apoptotic cells duringembryonic development. Cell 128: 931–946.
4966 IMMUNOGENICITY OF ANTI-EGFR mAb-INDUCED TUMOR APOPTOSIS




















