
Relationship between Dietary Macronutrients Intake ... - MDPI

IN VITRO METHODOLOGY FOR MEASURING BIOACCESSIBLE CESIUM-137,
STRONTIUM-90, LEAD, AND MERCURY ASSOCIATED WITH DIETARY OR
NON-DIETARY INGESTION
by
CHANG HO YU
A Dissertation submitted to the
Graduate School-New Brunswick
Rutgers, The State University of New Jersey
And
University of Medicine and Dentistry of New Jersey
in partial fulfillment of the requirements
for the degree of
Doctor of Philosophy
Graduate Program in Environmental Sciences and
Graduate School of Biomedical Sciences
written under the direction of Dr. Paul J. Lioy
and approved by
________________________
________________________
________________________
________________________
New Brunswick, New Jersey
October, 2005

2005
Chang Ho Yu
ALL RIGHTS RESERVED

ii
ABSTRACT OF THE DISSERTATION
In Vitro Methodology for Measuring Bioaccessible Cesium-137, Strontium-90, Lead, and
Mercury Associated with Dietary or Non-dietary Ingestion
By CHANG HO YU
Dissertation Director:
Paul J. Lioy, Ph. D.
A methodology to determine the bioaccessibility of heavy metals and low-level
radionuclides in soil has been developed to estimate the bioavailability of each toxicant in
the digestion system of mammalians. In this thesis, it is applied as a method for different
toxicants within matrices associated with dietary or non-dietary ingestion. Three specific
cases were tested to expand the application of the bioaccessibility method.
The bioaccessible radionuclides in radioactive-contaminated soils were measured
and compared with the soil hazard class. The bioaccessibility of cesium-137 ranged from
8.4 % to 31.2 % and 8.3 % to 38.8 % in gastric and intestinal fluid, respectively. The
level of strontium-90 ranged from 29.7 to 97.1 % and 17.8 to 71.0 % for gastric and
intestinal bioaccessibility, respectively. The comparison of bioaccessibility with levels of
soil radioactivity was proportionate between the bioaccessibility of strontium-90 in soil
with the level of strontium-90 in the soil; however, the relationship of the bioaccessibility
of cesium-137 in soil did not correspond to the level of cesium-137 in that soil.
Lead present in house dust was measured in gastric and intestinal fluid, and the
association of lead bioaccessibility with three size fractions of house dust (below 75 µm,
75-150 µm, and 150-250 µm) was explored. The bioaccessible lead in the house dust

iii
ranged from 52.4 % to 77.2 % and 4.9 % to 32.1 % for gastric and intestinal
bioaccessibility, respectively. The bioaccessibility of lead within the three size fractions
was not significantly different for the gastric bioaccessibility (p = 0.7019); however, the
intestinal bioaccessibility was significantly different among three particle size fractions (p
= 0.0067).
The bioaccessibility method was applied to simulate dietary ingestion of a
contaminant using realistic food matrices. Two approaches (direct measurement and mass
balance estimation) were adopted to report the bioaccessible mercury in the gastric and
intestinal phase. Mercury bioaccessibility for tuna steak ranged between 67.0 ~ 88.8 %
and 24.4 ~ 65.4 % in gastric and intestinal phase, respectively. The gastric
bioaccessibility of mercury in canned tuna ranged from 55.0 % to 74.0 %. The intestinal
bioaccessibility for canned tuna was estimated ranging from 12.7 % to 36.6 % by the
approach of mass balance estimation. This was the first known use of the bioaccessibility
method to examine cooked food bioaccessibility.

iv
Acknowledgement
There are many people to whom I owe a deep debt of gratitude. Even though this
work is credited to me, I have received a tremendous amount of support and help during
its’ progression. To those people I will always be grateful.
First, I have to thank my father and mother. Without their love and support I
couldn’t finish this work. I also must thank my wife, Enu-Hyeong Yi, a greater supporter
and sometimes a keen-edged outside observer for my research. Because she is also a Ph.
D. student in Electrical and Computer Engineering, her understanding for graduate
student life and difficulties allowed me make excuses for fast 5 years of studying.
I am always indebted to my advisor, Dr. Paul J. Lioy. He introduced me to this
dissertation topic, bioaccessibility of inorganics, and enabled me to finish the whole work
greatly. His continuing enthusiasm for the truth and confidence in me was a driving force
to keep this long-time project rolling forward. I always thank his support mentally and
financially. I also am grateful for the help and guidance of Dr. Clifford P. Weisel, Dr.
Natalie C. G. Freeman, and Dr. Junfeng (Jim) Zhang. They always encouraged me to
finish the dissertation writing nicely, and guided me to get back on the right track when I
headed into a wrong direction. Their dedication and efforts to my dissertation were
deserved. Special thanks to Dr. Joanna Burger for her help to use a nice mercury analyzer,
Dr. Lih-Ming Yiin for his support for lead analysis and mentoring as a predecessor of Dr.
Lioy’s student, and Dr. Alan H. Stern for his valuable comments about mercury.
Several individuals must be acknowledged for all of their technical expertise,
without which none of this research could ever be possible. I am indebted to Carl

v
Schopfer for his help of radionuclides sample analyses in soil matrix, Kristie Ellickson
for her help for the lead sample analyses in house dust matrix, and to Tara Shukla with
the mercury sample analyses in fish matrix. Each of you spent countless hours unselfishly
sharing your expertise with me.
The administrative staff at EOHSI has also been invaluable in helping me to
survive in the tons of paperwork. The financial and budget-related help was from Susan
Wund and Mary Doran. Special thanks to the following secretarial staff for their help
with every task: Ann Marie McCann-Roe, Teresa Boutillette, and Martha Rajaei.
Additionally I must thank the following dear friends and laboratory colleagues at
EOHSI for helping me keep my research through this process and for cheering me
whenever I encountered with an emergency: Kyung Hwa Jung, Jaymin Kwon, In Kyu
Han, Yuri Mun, Il Yang, Kunning Zhu, Jason Herrington, Lin Zhang, Chen Zhang, Maria
Perez, Xianlei Zhu, and Xiangmei Wu. And my former laboratory colleagues and
successful students of Dr. Lioy: Dr. Kristie Ellickson, Dr. Vito Ilacqua, and Dr. Paromita
Hore.
Finally, I have to acknowledge the funding agency for this dissertation work
possible. This work was funded in part by U.S. Department of Energy through CRESP
(Consortium for Risk Evaluation with Stakeholder Participation, grant # DE-FC01-
95EW55084) project, and U.S. Housing and Urban Development through ECSC
(Evaluating Dry Steam Cleaning in Reducing Contaminants in Carpets, grant #
NJLHH0111-02).

vi
Table of Contents
Abstract………………………………………………………………………….…. ii
Acknowledgement………………………………………………………………….. iv
Table of Contents…………………………………………………………………... vi
List of Tables………………………………………………………………………. xii
List of Figures and Illustrations……………………………………………………. xv
Chapter 1. Introduction…………………………………………………………….. 1
1.1. Human Exposure via Ingestion……………………………………………. 1
1.2. Susceptible Populations……………………………….…………………... 4
1.3. Bioavailability and Bioaccessibility……………………………………….. 7
1.4. Purpose of the Study………………………………………………………. 12
1.5. Hypotheses & Specific Aims……………………………….……………... 13
1.5.1. Hypotheses………………………………………………………….. 13
1.5.2. Specific Aims………………………………………….……………. 13
1.6. Objectives of Current Work……………………………………………….. 14
1.6.1. Bioaccessible Radionuclides in Soil…………..…………………...... 14
1.6.2. Bioaccessible Lead in House Dust……………..……….…………… 15
1.6.3. Bioaccessible Mercury in Fish………………………………………. 16
Chapter 2. Bioaccessible Radionuclides in SRS Soils……………………….…….. 17
2.1. Introduction…………………………………………………………..……. 17
2.2. Background………………………………………………..………............. 18

vii
2.2.1. Radionuclides (137Cs & 90Sr)………………………………………… 18
2.2.1.1. Radiation Units………………………………………………... 19
2.2.1.2. Ionizing Radiation……………………..……………………… 21
2.2.1.2.1. Alpha Particles………………………………...………… 21
2.2.1.2.2. Beta Particles……………………………………………. 21
2.2.1.2.3. Gamma Rays………………….......................................... 22
2.2.1.3. Cesium-137……………………….…………………………… 24
2.2.1.4. Strontium-90…………………………………………………... 25
2.2.2. Savannah River Site (SRS) and CRESP Involvement………………. 26
2.2.3. Crystal Ball………………………………………………………….. 29
2.3. Methods…………..…………………………………………...................... 30
2.3.1. Test Soils ………………………………………………..................... 30
2.3.2. Soil Preparation……………………………………………………… 33
2.3.2.1. Soil Drying…………………………………………………….. 34
2.3.2.2. Soil Sieving……………………………………………………. 34
2.3.3. Total Soil Dissolution for Radionuclide Analysis…………………... 35
2.3.4. Bioaccessibility Extraction………………………………………….. 36
2.3.4.1. Artificial Fluids Preparation…………………………………… 37
2.3.4.2. In Vitro Extraction…………………………………………….. 38
2.3.5. Cesium-137 Samples: Preparation & Measurement………………… 39
2.3.6. Strontium-90 Samples: Preparation & Measurement……………….. 40
2.3.7. Statistical Analyses………………………………………………….. 42
2.3.8. SRS vs ChNPP Area….……………………………………………... 43

viii
2.3.9. Exposure / Dose Calculations……………………………………….. 45
2.4. Results and Discussion…..……………………………………………........ 53
2.4.1. Bioaccessible Cesium-137 and Strontium-90 in Two Biofluids…….. 53
2.4.2. Bioaccessibility with Soil Hazard Class…………………………….. 54
2.4.3. Bioaccessibility with Soil Physico-chemical Characteristics……….. 57
2.4.4. Exposure / Dose Estimation…………………………………………. 64
Chapter 3. Bioaccessible Lead in Carpet House Dust……………………………… 75
3.1. Introduction………………………………………………………………... 75
3.2. Background………………………………………………………………... 78
3.2.1. Lead………………………………………………………………….. 78
3.2.2. IEUBK Model……………………………………………………….. 80
3.3. Methods……………………………………………………………………. 83
3.3.1. Test House Dusts……………………………………………............. 83
3.3.2. House Dust Preparation………………………................................... 83
3.3.3. Total House Dust Dissolution for Lead Analysis…………………… 84
3.3.4. Bioaccessibility Extraction………………………………………….. 85
3.3.4.1. Artificial Fluids Preparation…………………………………… 85
3.3.4.2. In Vitro Extraction…………………………………………….. 85
3.3.5. Analysis for Total and Bioaccessible Lead…………………………. 87
3.3.6. Statistical Analyses………………………………………………….. 87
3.3.7. IEUBK Model Predictions…………………………………………... 88
3.4. Results and Discussion…..……………………………………………........ 90

ix
3.4.1. Bioaccessibility Test for Vacuumed House Dusts…………………... 90
3.4.2. Bioaccessibility / Recovery Test for Different Size Fractions………. 93
3.4.3. IEUBK Model Estimates of Dose Using Bioaccessible Lead………. 96
Chapter 4. Bioaccessible Mercury from Fish Consumption……………………….. 101
4.1. Introduction………………………………………………………………... 101
4.2. Background………………..………………………………………………. 103
4.2.1. Mercury……………………………………………………………… 103
4.2.2. Mercury Absorption…………………………………………………. 105
4.2.2.1. Elemental Mercury…………………………………………….. 105
4.2.2.2. Inorganic Mercury……………………………………………... 105
4.2.2.3. Organic Mercury………………………………………………. 106
4.2.2.4. Methylmercury in Tuna……………………………………….. 106
4.2.2.5. Digestion and Absorption of Food…………………………….. 107
4.3. Methods……………………………………………………………………. 109
4.3.1. Approach…………………………………………………………….. 109
4.3.2. Study Materials (Tuna Steak & Canned Tuna)……………………… 110
4.3.3. Synthetic Fluid Preparation………………………………………….. 110
4.3.4. Bioaccessibility Modifications………………………………………. 111
4.3.4.1. Pilot Study…………………………………………………….. 111
4.3.4.2. Changing Experimental Conditions…………………………… 112
4.3.4.3. Final Experimental Conditions…………………………..……. 113
4.3.5. Bioaccessibility Extraction………………………………………….. 114

x
4.3.6. Digestion for Total and Bioaccessible Mercury…………………….. 116
4.3.7. Mercury Analysis……………………………………………………. 116
4.3.8. Bioaccessibility Calculations………………………………………... 117
4.3.9. Statistical Analyses………………………………………………….. 119
4.4. Results……………………………………………………………………... 120
4.4.1. Bioaccessibility / Recovery for Tuna Steak (Raw & Cooked)……… 120
4.4.2. Bioaccessibility / Recovery for Canned Tuna (Light & White)…….. 122
4.5. Discussion…………………………………………………………………. 124
4.5.1. Direct vs Directadj vs Mass Balance Bioaccessibility…………….…. 124
4.5.2. Bioaccessibility Differences by Biofluids and Preparation Methods.. 126
4.5.3. Bioaccessibility Mercury for Fish Matrix…………………………… 132
Chapter 5. Summary and Conclusions……………………………………………... 135
5.1. Bioaccessible Radionuclides in SRS Soils…………………….…………... 135
5.2. Bioaccessible Lead in Carpet House Dust.…………………….………….. 136
5.3. Bioaccessible Mercury from Fish Consumption…...………….…………... 137
5.4. Conclusions………………………………………………………………... 138
Chapter 6. Recommendations for Future Research………………………………… 140
Appendices 1. CRESP SOP-001 “Soil Drying”…….……………………………… 141
Appendices 2. CRESP SOP-002 “Preparation of Soil Sub-Fractions”……….……. 153
Appendices 3. CRESP SOP-003 “Bioaccessibility Assay”………………………... 167

xi
Appendices 4. CRESP SOP-004 “Acid Digestion of Soil or House Dust for Liquid
Scintillation Counting”……………………………...……………………………... 182
Appendices 5. Data for Bioaccessible Radionuclides (137Cs & 90Sr)……………… 193
5.1. Cesium-137…………………………...…………………………………… 193
5.1. Strontium-90………………...…………………………………………….. 196
Appendices 6. Data for Bioaccessible Lead…………….…………………………. 198
6.1. Three Particle Size Fractions……………………………………………… 198
6.2. Particle Size < 75 µm Fractions…………………………………………… 201
Appendices 7. Data for Bioaccessible Mercury…………….……………………… 203
7.1. Tuna Steak…………………………………………………………………. 203
7.2. Canned Tuna………………………………………………………………. 207
References………………………………………………………………………….. 211
Curriculum Vitae…………………………………………………..……………….. 223

xii
Lists of Tables
Table 1. The Bioaccessibility of Heavy Metals and Low-level Radionuclides from
Previous Studies……………………………………………………………………. 11
Table 2. Unit for Radiation and Radiation Dose in Radiological Science…………. 20
Table 3. SRS Soil Hazard Classification, Radionuclides Activity, and Sampled
Conditions………………………………………………………………………….. 32
Table 4. Standard Efficiencies of Sample Geometry for Cesium-137 Gamma
Spectroscopy.………………………………………………………………………. 40
Table 5. Shapiro-Wilk Normality Test for the Bioaccessibility of 137Cs and 90Sr
from Soil Samples……………………………………………….…………………. 43
Table 6. Comparison of Total Radioactivity between SRS Soils and Chernobyl
Areas…………………………………………………...…………………………... 45
Table 7. Two Radionuclides Ingestion / Inhalation Absorption Parameters for
Different Aged Groups……………………………………………………………... 49
Table 8. Age-specific Input Values for Exposure / Dose Estimations………..……. 51
Table 9. Age-specific Effective Dose Coefficients for Each Exposure Pathway….. 52
Table 10. Total Cesium-137 and Strontium-90 Concentration and Gastric /
Intestinal Bioaccessibility for Analyzed SRS Berm Soil Samples………………… 53
Table 11. Non-parametric Kruskal-Wallis ANOVA Test for Each Bioaccessibility
by Soil Class and Non-parametric Post Hoc Multiple Comparison Test…………... 57
Table 12. Soil Physico-chemical Characterization Data for SRS Seepage Basin /
Berm Soils……………………………………………………..…………………… 58
Table 13. The Spearman Rank-order Correlation Tests among Six Variables…….. 59

xiii
Table 14. Non-parametric Kruskal-Wallis ANOVA Test for Each Bioaccessibility
by Sampled Depth and Non-parametric Post Hoc Multiple Comparison Test…….. 60
Table 15. Non-parametric Kruskal-Wallis ANOVA Test for Each Bioaccessibility
by Sampled Location and Non-parametric Post Hoc Multiple Comparison Test….. 61
Table 16. The Spearman Rank-order Correlation Tests between Gastric / Intestinal
Bioaccessibility and Seven Variables…………………….…………….………….. 62
Table 17. The Age-specific Variables and Converted MCL for 137Cs and 90Sr
Respect to Each Age Group………………….…………………………………….. 65
Table 18. Potential Dose Estimates from Ingestion and Inhalation Pathways for
137Cs and 90Sr……………………………………………………….……………… 70
Table 19. Effective Dose Estimates for 137Cs from Short-term Exposure…………. 71
Table 20. Effective Dose Estimates for 90Sr from Short-term Exposure.………….. 72
Table 21. Effective Dose Estimates for 137Cs from Long-term Exposure…….…… 73
Table 22. Effective Dose Estimates for 90Sr from Long-term Exposure…………... 74
Table 23. The Mass Distribution by Particle Size Fraction for 5 House Dust
Samples…………………………………………………………………………….. 84
Table 24. Shapiro-Wilk Normality Test for the Bioaccessibility of Lead from
House Dust Samples……………………………………………………………….. 88
Table 25. The Bioaccessibility Test Used for 15 Vacuumed House Dust Samples.. 93
Table 26. The Bioaccessibility / Recovery of Vacuumed House Dust Samples for
Three Sub-fractions………………………………………………………………… 95
Table 27. Non-parametric Tests for Lead Bioaccessibility by Particle Size
Fractions (Kruskal-Wallis ANOVA Test & Post Hoc Multiple Comparison Test).. 96

xiv
Table 28. The Comparison of Actual Blood Lead Levels with Predicted Blood
Lead Levels for Each Home Environment…………………………………………. 98
Table 29. The Percentile and Confidence Intervals (95 %) Corresponding to 30 %
Intestinal Bioaccessibility………………………………………………………….. 100
Table 30. Serving Size of Fish and Volumes of Biofluids Used in Mercury
Bioaccessibility Experiments………………………………………………………. 114
Table 31. Shapiro-Wilk Normality Test for the Bioaccessibility of Mercury from
Tuna Samples………………………………………………………………………. 120
Table 32. Mercury Bioaccessibility / Recovery Data for Tuna Steak……………… 121
Table 33. Mercury Bioaccessibility / Recovery Data for Canned Tuna………...…. 123
Table 34. Non-parametric Tests for Gastric Bioaccessibility by Preparation Ways
(Kruskal-Wallis ANOVA Test & Post Hoc Multiple Comparison Test).…………. 130
Table 35. Non-parametric Tests for Intestinal Bioaccessibility by Preparation
Ways (Kruskal-Wallis ANOVA Test & Post Hoc Multiple Comparison Test).…... 131

xv
List of Figures and Illustrations
Figure 1. Schematic Diagram of Dose and Exposure by Oral Route………………. 4
Figure 2. Areas of the Savannah River Site………………………...……………… 27
Figure 3. Savannah River Site Soil Sample Locations…………………………...... 33
Figure 4. Soil Sieve Unit…………………………………………………………… 35
Figure 5. Sample Processing Unit for Hydrofluoric Acid Digestion………………. 36
Figure 6. Schematic Representation of the Bioaccessibility Procedure……………. 39
Figure 7. The Example of Crystal Ball™ Cesium-137 Potential Dose Estimates for
Pica Children on Class “A” Soil via Ingestion……………...……………………... 52
Figure 8. The Example of Crystal Ball™ Cesium-137 Total Dose Predictions for
Pica Children on Class “A” Soil during One-year Residence…………………..…. 52
Figure 9. The Comparison of Cesium-137 Bioaccessibility by Soil Class………… 56
Figure 10. The Comparison of Strontium-90 Bioaccessibility by Soil Class……… 56
Figure 11. The Change of Total Bioaccessible Percentage in Dust Matrix from
Default to Gastric Bioaccessibility and Intestinal Bioaccessibility………………... 89
Figure 12. The Set-up of IEUBK Model for Predictions of Blood Lead vs Media
Concentration………………………………………………………………………. 90
Figure 13. The Comparison of Bioaccessible Lead among Three Different Test
Matrices…………………………………………………………………...………... 92
Figure 14. The Comparison of IEUBK Model Predictions Using Gastric
Bioaccessibility, Intestinal Bioaccessibility, and Model Default Value.…………... 97
Figure 15. The Comparison of Mercury Bioaccessibility Methods (Direct vs
Directadj vs Mass Balance)…………………………………………………………. 126

xvi
Figure 16. The Comparison of Mercury Bioaccessibility(MB) by Different Fish
Preparation Methods……………………………………………………………….. 131

1
I. Introduction
1.1. Human Exposure via Ingestion
Human risk is generally defined as the probability (or likelihood) of a harmful
consequence as a function of a hazard and the actual or potential contact (exposure) with
the hazard. To evaluate the risk accurately, risk assessment is evolved with a four-step of
process: hazard identification, dose-response assessment, exposure assessment, and risk
characterization, which is followed by risk management (NAS, 1983). Of the four
processes associated with risk assessment, “exposure assessment provides both
qualitative and quantitative evaluation of contact with a hazardous material including
descriptions of the intensity, frequency, and duration of exposure. Often one evaluates the
rates at which a hazardous material crosses the boundary (chemical intake and uptake
rate), the route by which a hazardous material crosses the boundary (exposure route:
dermal, oral, or respiratory), and the resulting amount of the hazardous material that
actually crosses the boundary (a potential dose) and the amount absorbed (internal dose)”
(EPA, 1992, p. 5).
A hazardous material contacting the outer boundary of a human can yield an
exposure based on the concentration at the point of contact, which is called exposure
concentration at the contact of time (EPA, 1992). “Most of the time, a toxicant is present
in air, water, soil, or a product (e.g., food). It is then transported or carried to the point of
contact where exposure can be measured. Exposure over a period of time can be
represented by a time-dependent profile of the exposure concentration” (EPA, 1992, p. 6).
“The area under the curve of this profile is the magnitude of the exposure, in
concentration-time units (Lioy, 1990, p. 939):

2
2
1
( )t
t
E C t d t= ∫
Where E is the magnitude of exposure (e.g., µg/kg-day or mg/kg-day), C(t) is the
exposure concentration as a function of time, and dt is an increment of time from t1 to t2”.
“The process by which a chemical enters the body can be described in two steps:
contact to achieve exposure, which is followed by crossing a boundary to achieve a dose”
(EPA, 1992, p5). “There are two major processes by which a chemical can cross the
boundary from outside the body to inside the body. Intake (entry) involves physically
moving the chemical through an opening in the outer boundary (usually mouth or nose),
typically via breathing, eating, or drinking. Uptake (absorption) involves absorption of
the chemical through the skin, lung, or gastrointestinal (GI) tract” (EPA, 1992, p. 5-6).
“Dose, which can result from exposure, can be represented by the integration of
exposure time contact rate and modifiers that ultimately may lead to a biologically
effective dose (Lioy, 1990, p. 940):
0 0
( ) ( ) ( ) ( ) ( )t t
D D t dt f x g y h z C t dt = = ∫ ∫
For the above, D is an integrated dose at a target tissue or cell (mass or mass/body
weight); D(t) is a time-varying function for dose; f(x) is the contact rate (e.g., m3/time for
inhalation; mass/time for ingestion; area/time for dermal exposure); g(y) is a variable
dependent on the target organ or system, and the bioavailability that affects the extent of
absorption; and h(z) is a variable dependent on the nature of a contaminant’s assimilation,
the cell repair or damage, elimination, and metabolism”. Dose can be expressed in several
different ways, and will be dependent upon the location of the agent in the body and level

3
of interaction with system. “Potential (administered) dose is simply the amount of
chemical ingested, inhaled, or material applied to the skin. Applied (external) dose is the
amount of a chemical at the absorption barrier (skin, lung, and gastrointestinal tract). The
amount of chemical that has been absorbed and is available for interaction with
biologically significant receptors is called internal (absorbed or bioavailable) dose. Once
absorbed, the chemical can undergo metabolism, storage, excretion, or transport within
the body. The amount transported to an individual organ, tissue, or fluid of interest is
termed the delivered dose. Biologically effective dose, or the amount that actually
reaches cells, sites, or membranes where adverse health effects occur, may only be a part
of the delivered dose” (EPA, 1992, p. 7-9).
Based upon the preceding, knowledge of human exposure is necessary for
understanding the continuum of human contacts with environmental toxicants and other
hazardous materials that can result in a dose and potentially lead to a human health effect.
For the ingestion route of exposure, it is important to accurately estimate bioavailable
dose for use in risk characterizations. Since childhood non-dietary ingestion of soil or
house dust can be a critical exposure pathway for radionuclides (Simon, 1998) and heavy
metals (Clayton et al., 1999), the analysis of the bioavailability for the toxicants in soil
and dust should be done prior to assessing human risk. For dietary or non-dietary
ingestion, environmental toxics present in various media will pass to the body through the
oral route (intake process via the mouth), and will be processed within the digestive
system. Subsequently, dissolvable toxics are released in the stomach and can be absorbed
through the small intestine (uptake process via gastrointestinal tract). The toxicant

4
absorbed via the small intestine can be transported to target organs (e.g., liver, kidney,
brain…etc), and can lead to health effects.
The schematic diagram for ingestion related exposure is presented in Figure 1.
Oral bioaccessibility defined as the soluble fraction in the gastrointestinal tract part of the
digestive system. Oral bioavailability defined as the fractional potential dose that reaches
the bloodstream. Both oral bioaccessibility and bioavailability can affect the internal dose.
Figure 1. Schematic diagram of dose and exposure by oral route (Modified from EPA, 1992)
1.2. Susceptible Populations
In exposure assessment, attention is currently focused on susceptible populations.
The purpose is to better protect a vulnerable group of the general population. Lead
poisoning of young children is a classical example that can assist in understanding the
issues surrounding of susceptible populations (ATSDR, 1999). After the United States
phased out lead as an additive to gasoline, the dominant exposure pathway to lead has
been the ingestion of lead-contaminated soil and house dust contaminated with lead paint
or soil, or inhalation of smaller lead-contaminated particles (ATSDR, 1999). Young
children (aged from 2 to 6 years old) are more likely to be exposed by these routes

5
because of frequent hand-to-mouth activities (HUD, 1995). Therefore, focusing on young
children has been considered an effective way to minimize the public health impact of
lead.
Children are more vulnerable to exposure to environmental toxicants than adults.
This is due to the differences in child and adult behavior patterns, physiological
characteristics, and metabolism (EPA, 2003). “For example, children and adults divide
their time differently across locations (the amount of time spent indoors versus outdoors,
or the amount of time spent on the floor versus sitting in the chair), and across activities
(e.g., playing, crawling, and mouthing objects). In many instances, children’s behavior
patterns result in exposures that are greater than those of adults over time, or result in
exposures that adults do not experience at all” (EPA, 2003, p. 7).
“Children are physiologically different from adults, and have higher basal
(resting) metabolic rates. Combined with the fact that children grow rapidly and generally
are more active than adults, their higher metabolic rate means that they eat, drink, and
breathe more than adults in proportion to their body size. As a result, children may
receive a higher dose of an environmental contaminant than the adults living in the same
house. Since the nature and severity of adverse effects of environmental exposures are
closely related to the dose, higher doses translate into increased concern for adverse
effects” (EPA, 2003, p. 7-8).
“Finally, children and adults may also metabolize environmental agents
differently. In addition, children’s metabolic capacity (i.e., their ability to chemically
process environmental agents) can be less well-developed than that of adults. These
factors contribute to differences in the persistence of environmental agents in children

6
and adult bodies and the concentration of the agents in specific organs. Concern about
children’s environmental exposures arises when agents persist in the body for longer
periods of time or are associated with higher concentrations” (EPA, 2003, p. 8).
Women are exposed to environmental toxicants, and the differences derived by
activity patterns (spend more time in indoors than men), physiological characteristics
(more body fat than men), and specific events (pregnancy and nursing a baby) may lead
to more susceptibility than men (Hatch, 2000). For women of childbearing age, the risks
from exposure to environmental toxicants are greater when they are pregnant or breast-
feed children. During such times they can become a source of a toxicant, since the unborn
child or developing infants are exposed to environmental toxicants in the womb (in utero)
during pregnancy or by consuming milk while nursing (Jacobson et al., 1990, 1996).
For example, lead from the maternal skeleton is transferred across the placenta to
the fetus during the gestation. Additional lead exposure may occur during breast feeding.
This means that lead stored in the mother’s body from prior to conception can result in
exposure to fetus or nursing neonate (ATSDR, 1999). Fetus might be exposed to
radiation from the decay of radioactive strontium: from transfer of strontium across the
placenta or from proximity to radiation emitted from the maternal body (ATSDR, 2004).
Methylmercury exposure to childbearing women is another example. After a pregnant
mother is exposed to mercury, the child developing in utero can be exposed because of
mercury’s ability to cross the placenta and reach the fetal brain (EPA, 1997). Mercury in
the mother’s body will also enter the milk, and infants who are breast-fed can be exposed
to the toxicants from breast milk (EPA, 1997). The predominant source of mercury to
women is fish (e.g., swordfish and tuna) (EPA, 1997).

7
1.3. Bioavailability and Bioaccessibility
The study of the bioavailability of chemicals in various media began around 1980
and continues to be an important topic (Paustenbach, 2000). Ruby et al. (1999) defined
the appropriate terminology for the study of bioavailability / bioaccessibility:
Oral Bioavailability: The fraction of ingested dose that reaches the central (blood)
compartment from the gastrointestinal tract.
Oral Bioaccessibility: The fraction that is soluble in the gastrointestinal environment and
is available for absorption.
For several years, simple extraction methods were used to assess the degree of
metals dissolution in a simulated gastrointestinal tract environment (Davis et al., 1993;
Ruby et al., 1993, 1996). The predecessor of these systems was developed originally to
assess the bioavailability of iron from food, for studies of nutrition (Miller et al., 1981).
In these systems, various metal salts or soils containing metals were incubated in a low
pH solution for a period of time that was intended to mimic the residence time in the
stomach. The pH was then increased to near neutral, and incubation continued for a
period of time intended to mimic the residence time in the small intestine. Enzymes and
organic acids were added to simulate gastric and small intestinal fluid.
“Bioavailability studies are used to characterize the dose of a drug or toxicants
delivered to an animal or plant receptor. Such studies are also used in human exposure
assessment to prioritize remediation by identifying areas on a contaminated site where
toxicants, such as heavy metals, are highly available to humans upon ingestion”
(Ellickson et al., 2001, p. 128). “The two principal factors limiting quantification of the

8
oral bioavailability of a heavy metal by a mammalian system are 1) dissolution in the
gastrointestinal tract and 2) absorption through the intestine” (Ellickson et al., 2001, p.
128). “The dissolution of a metal depends on the characteristics of the contaminant itself,
the environmental matrix in which it is incorporated, and the composition of the GI
(gastrointestinal) fluid. Once dissolved in the GI fluid, intestinal absorption is limited by
the speciation of the metal (e.g., particle bound, free ion, complexation interactions with
food, exogenous chemicals, or GI components) and GI motility. Inherent in the definition
of oral bioavailability is the fact that a biological membrane (intestinal mucosa) has been
crossed. Bioaccessibility will be greater than bioavailability because the latter includes
membrane transport while the former does not” (Ellickson et al., 2001, p. 128).
“Several studies have measured the oral bioavailability of metals. These studies
incorporated various leaching and dissolution techniques (Ruby et al., 1996; Davis et al.,
1993), studies of soil type and structure (Davis et al., 1997), human feeding studies
(Gargas et al., 1994; Maddaloni et al., 1998) and animal experiments (Freeman et al.,
1992, 1996; Groen et al., 1994; Clapp et al., 1991). Studies have also been performed to
compare GI dissolution techniques with in vivo animal models (swine model - Rodriquez
et al., 1999; mouse model - Sheppard et al., 1995; rat, rabbit, and monkey models - Ruby
et al., 1996). However, no clear pharmacokinetic relationship has been established
between oral bioavailability and the dissolution of contaminants within the GI system
(bioaccessibility)” (Ellickson et al., 2001, p. 128-129).
The laboratories of EMAD-EOHSI (Division of Exposure Measurement and
Assessment in the Environmental and Occupational Health Sciences Institute) have
successfully developed a method for determining bioaccessibility of heavy metals (lead,

9
arsenic, chromium, and cadmium), and low-level radionuclides (cesium-137 and
strontium-90) in soil samples (Hamel et al., 1999; Ellickson et al., 2001). Bioaccessibility
and recovery data were obtained through the mass balance calculations for heavy metals
in each set of the sequential extraction procedures that represented the entire digestive
process. The basic mass balance and bioaccessibility / recovery equations for the
analyzed toxicant in system design are:
T T E E R RC W C V C W ε× = × + × +
where, CT = total contaminant concentration of experimental sample (ng/g)
WT = total weight of each sample administered in the experiment (g)
CE = contaminant concentration in extracted fluid (ng/mL)
VE = volume of extracted fluid (mL)
CR = contaminant concentration in re-captured filter (ng/g)
WR = net weight of re-captured filter (g)
ε = error term
(%) 100
( )(%) 100
E E
T T
E E R R
T T
C VBioaccessibilityC W
C V C WRecoveryC W
× = ×
×× + ×
= ××
Mass balance of each metal is established by the summation of maximal
extractable mass in the biofluids, re-captured mass on the filter (i.e., non-extractable
portion of the metal and any residual mass precipitated during the sequential extraction),
and an error term (e.g., metal loss during processing). If one assumes the error is
negligible, one can set up the bioaccessibility and recovery calculation formulae using the
above approach to obtain a mass balance. Bioaccessibility (%) is calculated from the
extracted mass divided by total mass and multiplying by 100. Recovery (%) is also

10
obtained from the summation of extractable mass and re-captured mass divided by total
mass and multiplying by 100.
“Results of oral bioavailability and bioaccessibility measurements vary according
to the method, the soil, and contaminant characteristics. Consequently, the importance of
validation process reviewed by Ruby et al. (1999) becomes self-evident” (Ellickson et al.,
2001 p. 129). Validation of the EMAD-EOHSI bioaccessibility method for arsenic,
chromium, nickel, cadmium, and lead in soil, as a function of the liquid-to-solid ratio,
was completed by Hamel et al. (1998). The authors determined that bioaccessibility of
metals in synthetic gastric juice was affected only slightly by changes in the liquid-to-
solid ratios in the range of 100:1 to 5,000:1 (mL/g). Two methods called mass-balance
and soil recapture were used to estimate the bioaccessibility of heavy metals (Pb, As, and
Cr) in soils (Hamel et al., 1999). The study concluded that the mass-balance technique
could be employed routinely to determine bioaccessibility of heavy metals in
contaminated soils and the result could be used as an estimate of human bioavailability.
The soil recapture technique provided a reasonable estimate of bioaccessibility in soils,
allowing for rapid prioritization of remediation sites or soils within a site. The
comparison of in vitro dissolution and in vivo rat feeding techniques for oral
bioavailability of soil contaminants (Pb and As) was conducted by Ellickson et al. (2001).
Their results showed that bioaccessibility was greater than bioavailability for both metals
in both gastrointestinal compartments. The published soil bioaccessibility method was
modified to measure the bioaccessible radionuclides (cesium-137 and strontium-90) in
contaminated soils (Ellickson et al., 2002). The physico-chemical characteristics were
investigated to explore the association with the bioaccessibility. For cesium-137, the

11
bioaccessibility was negatively correlated with clay content, while strontium-90 was
significantly correlated to calcium bioaccessibility (Ellickson et al., 2002).
The previous studies modified the bioaccessibility protocols and artificial fluids
composition to each study design. More detail information about the bioaccessibility
protocols and compositions for biofluids is provided in the appendices (CRESP SOP-003,
“Bioaccessibility Assay”). The results obtained from the previous EMAD-EOHSI
bioaccessibility experiments are summarized in Table 1. They showed the differences in
bioaccessibility among target environmental toxicants and soil samples. Significant
differences in bioaccessibility were found between the gastric and intestinal fluid
extractions for cesium-137 (Mann-Whitney U test; p = 0.031) and strontium-90 (Mann-
Whitney U test; p < 0.001) (Ellickson et al., 2002).
Table 1. The bioaccessibility of heavy metals and low-level radionuclides from previous EMAD-EOHSI studies
Bioaccessibility Sample Matrix
Gastric Fluid Intestinal Fluid Lead NIST 2710 Soil (<74µm) Residential Soil, NJ (<125µm) Bunker Hill Soil, ID (<125µm) Jersey City Slag, NJ (<125µm)
62 ± 1 %
69 % 70 ± 11 % 39 ± 14 %
ND
Arsenic NIST 2710 Soil (<74µm) Residential Soil, NJ (<125µm)
66 ± 8 % 41 ± 2 %
ND
Chromium Jersey City Slag, NJ (<125µm)
34 ± 14 %
ND
Cesium-137 (Low Radioactivity Soil) SRS* Soil (<250µm) LTR** Soil (<250µm)
10.6 ~ 22.0 % 6.3 ± 0.6 %
12.0 ~ 28.7 % 8.7 ± 1.9 %
Strontium-90 (Low Radioactivity Soil) SRS* Soil (<250µm)
60.1 ~ 95.2 %
40.6 ~ 59.3 %
*SRS: Savannah River Site (see the section of 2.2.2. and 2.3.1. for SRS) **LTR: Lower Three Runs samples in SRS (see the section of 2.3.1. for LTR) ND: not determined (Taken from Hamel et al., 1999 for heavy metals and Ellickson et al., 2002 for radionuclides)

12
1.4. Purpose of the Study
Evaluation of exposure and dose received from the non-dietary ingestion of
environmentally contaminated media (e.g., soil, house dust…etc) is an important
component of the characterization of exposure for risk assessments. The bioavailability of
specific contaminants has a key role in linking the exposure to the dose absorbed by the
human digestive system. Methods that evaluate bioavailability rather than methods that
use strong acid leaching procedures for total environmental toxicants levels, can achieve
a more accurate estimation of human risks associated with dietary or non-dietary
ingestion exposure. The number of human bioavailability studies is limited, and rely
primarily on the results from animal studies. Since animals are not necessarily equivalent,
physiologically or quantitatively to humans, the results from animal studies may not
appropriate for human risk characterizations. The soluble amount of contaminants
released in artificial human gastrointestinal fluids has been measured as an alternative to
bioavailability data to define the bioaccessible fraction of contaminants present in
environmental matrices (Ruby et al., 1996; Davis et al., 1997; Oliver et al., 1999).
Bioaccessibility can be employed as a predictor of bioavailability, and can be
incorporated within calculations of the exposure / dose for risk characterization.
Previously, the methodologies measuring bioaccessible heavy metals (Pb, As, Cr,
and Cd) and low-level radionuclides (137Cs and 90Sr) in various soil samples were
successfully developed in the EMAD-EOHSI laboratory (Hamel, 1998; Ellickson, 2001).
In the present thesis, the established bioaccessibility method is expanded to important,
but different types of pollutants and matrices to which human can be exposed: high-level
radionuclides from contaminated soils, lead from ingested house dust, and mercury from

13
consumed fish. In addition, the obtained bioaccessibility data were used to calculate
exposure / dose estimations.
1.5. Hypotheses & Specific Aims
1.5.1. Hypotheses
A. The bioaccessibility of cesium-137 and strontium-90 in radioactive SRS soils is
not equivalent among radioactive soil hazard classes.
B. The lead bioaccessibility for the size fraction of below 75 µm diameter in house
dust is less than 100 %, and lead bioaccessibility from smaller particles is greater
than from the total particles present in vacuumed house dust.
C. The mercury bioaccessibility is different among raw tuna steak, cooked tuna steak,
light canned tuna, and white canned tuna.
1.5.2. Specific aims
A. Evaluate the bioaccessibility of cesium-137 and strontium-90 present in high
radioactive-contaminated soils.
B. Quantify the bioaccessibility of lead in vacuumed house dust in residences.
C. Investigate the difference in bioaccessibility among three particle size fractions
(below 75 µm, 75-150 µm, and 150-250 µm).
D. Develop a bioaccessibility method for mercury found in fish used for human
consumption.
E. Investigate the bioaccessibility of mercury from tuna steak filet (cooked and
uncooked) and canned tuna (light and white).

14
1.6. Objectives of Current Work
1.6.1. Bioaccessible radionuclides in soil
Previous experiments were successfully conducted to quantify the bioaccessible
fraction of radionuclides for low-level radioactive contaminated soil collected at the DOE
(Department of Energy) Savannah River Site (SRS; see the section of 2.2.2.). In these
experiments (Ellickson et al., 2002), the method used to determine the bioaccessible
heavy metals (Hamel, 1998) was expanded to detect the concentration of radioactivity in
contaminated soil (e.g., SRS soil samples). For safety reasons, the method to measure
bioaccessible radionuclides developed by Ellickson et al. (2002) for radionuclides in low-
level radioactive contaminated soil, required additional protocols to further minimize
potential contact with radioactive soil and radiation hazard. All procedures and analyses
performed in the EMAD-EOHSI laboratories were approved by both the University-wide
(RWJMS/UMDNJ) and internal safety committee (CRESP SOP-001 “Soil Drying”,
CRESP SOP-002 “Preparation of Soil Sub-Fractions”, CRESP SOP-003
“Bioaccessibility Assay”, and CRESP SOP-004 “Acid Digestion of Soil or House Dust
for Liquid Scintillation Counting” in the appendices). Bioaccessible radionuclides found
in low-level and high-level radioactive contaminated soils were compared with the
radioactive soil hazard class (e.g., “A”, “B”, and “C”), and each was ranked as cesium-
137 and strontium-90 radiation hazard from high to low, respectively (see the section of
2.3.1. and Table 3). The levels of bioaccessible radionuclides present in different samples
were examined to identify changes in the bioaccessibility with soil sampled depth,

15
location, as well as soil physico-chemical characteristics (pH, SOM, CEC, and clay / sand
content).
1.6.2. Bioaccessible lead in house dust
As part of this thesis, a new method for quantifying bioaccessible lead levels was
developed specifically for vacuumed house dust. Since the ingestion of house dust is a
major route of children’s non-dietary lead exposure (Lanphear et al., 1996), data on the
bioaccessible fraction of lead present in dust matrices provide valuable information that
can improve residential exposure and dose estimations. The lead bioaccessibility method
for house dust was derived from the bioaccessibility method used for heavy metals in
contaminated soils (Ellickson et al., 2001). The target of the current study is the
bioaccessibility of lead in house dust. The justification is the presence of high-levels of
lead in the dust of homes that can yield high exposure, and the parallel concern about
exposure to lead after ingestion of residential dust by young children (EPA, 1995). The
study primarily tested house dust sieved below 75 µm diameter, but included sieving the
dust samples into three particle size fractions: below 75 µm, 75-150 µm, and 150-250 µm.
The goal was to identify the relationship between the sieved particle fractions (< 75 µm,
75-150 µm, and 150-250 µm) and the bioaccessibility of lead in both biofluids. The
acquired bioaccessibility data were applied into one of the current risk assessment models
(i.e., IEUBK model) to show the feasibility of future use of lead bioaccessibility in house
dust / soil as an alternative to default model parameters. In the IEUBK model, 30% of
lead in house dust / soil matrix is assumed to be absorbed through children’s
gastrointestinal tract.

16
1.6.3. Bioaccessible mercury in fish
Tuna steak and canned tuna (light and white) purchased from the supermarket
were used to determine the bioaccessibility of mercury associated with the dietary food
consumption. The tuna sample was selected for the study because tuna is often
environmentally contaminated with mercury, and tuna (including canned tuna product) is
one of the most frequently consumed fish species by much of the U.S. population (EPA,
1997). To determine the bioaccessibility of mercury in food, the techniques associated
with the soil-based method (Ellickson, 2001) were modified. The development of a
method for bioavailability of mercury in a food matrix was not simple. Food has different
components and variables to consider than soil or house dust, and mercury can be lost via
volatilization and coagulation to organic materials in the sample or condensation on the
walls during an experiment. Thus, two approaches (Hamel et al., 1999) were proposed to
estimate the bioaccessibility: 1) direct calculation of a mercury bioaccessibility by
measuring the mercury mass released in both biofluids; 2) indirectly estimate
bioaccessible mercury by calculating a mass balance for mercury in a simulated human
digestive system. Different food matrices (raw tuna filet, cooked tuna filet, light canned
tuna, and white canned tuna) were tested to evaluate the effect of food type on
bioaccessibility.

17
II. Bioaccessible Radionuclides in SRS Soils
2.1. Introduction
Ingestion of radioactively contaminated soil is a critical pathway for human
exposure to radionuclides that are relatively immobile in surface soils and the
environments (Simon, 1998). Childhood soil ingestion is an important route of exposure
to environmentally contaminated soils for young children whether directly, as inhaled
dust, or indirectly as a constituent of contaminated house dust (Thornton et al., 1990;
Clayton et al., 1999). Young children are more susceptible to high incidence of soil
ingestion than adults because of their greater hand-to-mouth activity (Hubal et al., 2001),
and are more likely susceptible to high exposures than infants since they are more active
(Hubal et al., 2001).
Both cesium-137 and strontium-90 are radionuclides of concern for exposure and
dose assessment, because of their long half-life (almost 30 years each) and biological
activity. Cesium-137 tends to behave in a similar manner to potassium (K), causing it to
distribute pervasively throughout body. Strontium-90 is chemically similar to calcium
(Ca), and is a bone-seeking element, which causes it to deposit in bones.
Risk assessments for inorganic contaminated soils have been primarily based on
total inorganic contaminant levels, however, the strong binding properties of inorganic
compounds in soil can reduce the oral bioavailability, or the fraction of inorganics to be
able to reach systemic circulation (Ruby et al., 1999). Since oral bioavailability is
dissolution-limited, we can estimate bioavailability using human gastrointestinal
dissolution model that measures bioaccessibility (Hamel et al., 1999). Current
bioaccessibility work has focused on measurements of stable metals, but some

18
radionuclide studies were completed by Ellickson et al. (2002). Bioaccessibility of 137Cs
and 90Sr was measured for low levels of radionuclides (42.2 ~ 85.4 pCi/g of 243/244Cm) in
contaminated SRS soils (Ellickson et al., 2002). This work presents the application of
bioaccessibility method for both measurements of low and high levels of radionuclides
(776 pCi/g of 243/244Cm) in radioactive-contaminated soils.
The bioaccessibility of cesium-137 and strontium-90 was evaluated using SRS
seepage / basin soils. SRS seepage / basin was a disposal area for mixed radionuclides
wastes (tritium, strontium-89/90, cesium-137…etc) throughout the operation of the
nuclear facilities (see the section of 2.2.2.). Three soils: high-level radioactive soils (class
“A”) and low-level radioactive soils (class “B” and “C”) were compared with
bioaccessibility determined for each radionuclide level in soil. The factors that control
impact on gastric and intestinal bioaccessibility were analyzed in SRS seepage / basin
experimented soil sets. Exposure and dose levels were estimated on three soil classes
with two exposure durations (short-term and long-term for two weeks and one year,
respectively) and four sub-groups (pica young children, young children, children, adults).
2.2. Background
2.2.1. Radionuclides (137Cs & 90Sr)
At the Savannah River Site, radioactive materials were released to the
environment during the processing of radiological waste created by the previous
operation of the nuclear reactors and support facilities at SRS. Airborne emissions and
liquid discharges were the main pathways to the environment (SRS, 2000). Through the
release of more than 100 different radioisotopes to SRS, radionuclides (unstable nuclide

19
capable of spontaneous transformation into other nuclides by changing its nuclear
configuration or energy level) have accumulated in SRS soils from various sources such
as the cooling water from reactors and atmospheric deposition from airborne effluents
(SRS, 2000). The magnitude of harmful properties of radionuclides is weighed in several
ways: its persistence, measured by half-life; its physicochemical properties (biological
affinity); and the type and energy of its emissions (ATSDR, 1999).
2.2.1.1. Radiation units
One curie (Ci = 37×109 Bq) of radioactive material will have 37 billion atomic
transformations (disintegrations) in one second. One becquerel (Bq) is the radiation
caused by one disintegration per second; this is equivalent to 27.0270 picocuries (pCi).
Picocuries are 1 million millionth of a curie (1×10-12 Ci) used in measuring the typically
small amount of radioactivity in air and water. EPA has established a Maximum
Contaminant Level (MCL) of 4 millirem per year for beta particle and photon
radioactivity from man-made radionuclides in drinking water. The average concentration
of cesium-137, which is assumed to yield 4 millirem per year, is 200 picocuries per liter
(pCi/L). Also the average concentration of strontium-90, equivalent to MCL level (4
mrem/year), is 8 pCi/L (EPA, 1993).
Gray (Gy) is the SI unit of measurement for absorbed dose. It relates to the
amount of energy actually absorbed in a material, and is used for any type of radiation
and material. The dose is the amount of energy deposited per unit of mass. One gray is
defined to be the dose of one joule (J) of energy absorbed per kilogram (kg) of matter, or
100 rad. Rad (radiation absorbed dose) is the metric unit of measuring radiation dose.
One rad is equal to a dose of 0.01 J/kg.

20
Sievert (Sv) is an SI unit for measuring the effective (equivalent) dose of radiation
received by a human or some other living organism. Various kinds of radiation have
different effects on living tissue, so a simple measurement of dose as energy received,
stated as grays or rads, does not give a clear indication of the probable biological effects
of the radiation. The equivalent dose, in sieverts, is equal to the actual dose, in grays,
multiplied by a quality factor that is larger for more dangerous forms of radiation. An
effective dose of one sievert requires one gray of beta or gamma radiation but only 0.05
gray of alpha radiation or 0.1 gray of neutron radiation. The sievert is a large unit, so
radiation doses are often measured in millisieverts (mSv). One sievert equals 100 rem.
Rem (radiation equivalent in man) is a unit used for measuring effective (equivalent)
dose of radiation received by a human.
Table 2. Unit for radiation and radiation dose in radiological science
Specification Unit Explanation
Radiation Ci Bq
Curie: 37 billion atomic disintegration per second Becquerel: one disintegration per second pCi = 10-12 Ci
Radiation Absorbed Dose
Gy Rad
Gray: dose of one joule of energy absorbed per kilogram (= 1 J/kg) Radiation absorbed dose: metric unit of measuring dose (= 0.01 J/kg)
Gy = 100 rad
Radiation Equivalent Dose
Sv Rem
Sievert: equivalent dose of radiation received by human or living organism (= 1 Gy for β/γ; 0.05 Gy for α; 0.01 Gy for neutron) Radiation equivalent in men: equivalent dose of radiation received by human
Sv = 100 rem mSv = 10-3 Sv

21
2.2.1.2. Ionizing radiation
2.2.1.2.1. Alpha particles1
Alpha particles are a type of ionizing radiation ejected by the nuclei of some
unstable atoms. They are large subatomic fragments consisting of 2 protons and 2
neutrons. Each is a relatively heavy, high-energy particle, with a positive charge of +2
from its two protons. Alpha particles have a velocity in air of approximately one-
twentieth the speed of light, depending upon the individual particle’s energy.
The health effects of alpha particles depend heavily upon how exposure takes
place. External exposure is of far less concern than internal exposure, because alpha
particles lack the energy to penetrate the outer dead layer of skin. However, if alpha
emitters have been inhaled, ingested, or absorbed into the blood stream, sensitive living
tissue can be exposed to alpha radiation. The resulting biological damage increases the
risk of cancer; in particular, alpha radiation is known to cause lung cancer in humans
when alpha emitters are inhaled.
2.2.1.2.2. Beta particles2
Beta particles are subatomic particles ejected from the nucleus of some
radioactive atoms. They are equivalent to electrons. The difference is that beta particles
originate in the nucleus and electrons originate outside the nucleus. Beta particles have an
electrical charge of –1, and a mass of 549 millionths of one atomic mass unit. While beta
particles are emitted by atoms that are radioactive, beta particles themselves are not
radioactive. It is their energy, in the form of speed that causes harm to living cells. When
transferred, this energy can break chemical bonds and form ions.
1 The text was excerpted from EPA website (http://www.epa.gov/radiation/understand/alpha.htm) 2 The text was excerpted from EPA website (http://www.epa.gov/radiation/understand/beta.htm)

22
Beta particle emission occurs when the ratio of neutrons to protons in the nucleus
is too high. There is an excess neutron transforms into a proton and an electron. The
proton stays in the nucleus and the electron is ejected energetically. Strontium-90 was the
major man-made beta emitter released in the environment. Fallout from atmospheric
nuclear testing from the 1950’s to the early 1970’s has spread strontium-90 worldwide.
Beta radiation can cause both acute and chronic health effects. Acute exposures
are uncommon. Chronic effects are much more concern. Chronic effects result from fairly
low-level exposures over a long period of time. They develop relatively slowly (5 to 30
years for example). The main chronic health effect from radiation is cancer. When taken
internally, beta emitters can cause tissue damage and increase the risk of cancer. The risk
of cancer increases with increasing dose.
A number of radionuclides, including 3H, 14C, 32P, 35S, 45Ca, 89Sr, 90Sr, and 90Y,
emit only beta radiation. Liquid scintillation counting systems are widely used for the
assay of low levels of beta-emitting radionuclides and can be used to quantify the above
radionuclides (ATSDR, 1999).
2.2.1.2.3. Gamma rays3
A gamma ray is a packet of electromagnetic energy, i.e., photon. Gamma photons
are the most energetic photons in the electromagnetic spectrum. Gamma rays (gamma
photons) are emitted from the nucleus of some unstable (radioactive) atoms. Gamma
radiation is very high-energy ionizing radiation. Gamma photons have about 10,000
times as much energy as the photons in the visible range of the electromagnetic spectrum.
Gamma photons have no mass and no electrical charge. They are pure electromagnetic
3 The text was excerpted from EPA website (http://www.epa.gov/radiation/understand/gamma.htm)

23
energy. Because of their high energy, gamma photons travel at the speed of light and can
cover hundreds to thousands of meters in air before spending their energy.
Gamma rays can pass through many kinds of materials, including human tissue.
Very dense materials, such as lead, are commonly used as shielding to slow or stop
gamma photons. Cesium-137 provides an example of radioactive decay by gamma
radiation. A neutron transforms to a photon and a beta particle. The additional photon
changes the atom to barium-137. The nucleus ejects the beta particle. However, the
nucleus still has too much energy and ejects a gamma photon (gamma radiation) to
become more stable.
Both direct (external) and internal exposures to gamma rays are of concern.
Gamma rays can travel much farther than alpha or beta particles and have enough energy
to pass entirely through the body, potentially exposing all organs. A large portion of
gamma radiation largely passes through the body without interacting with tissue –the
body is mostly empty space at the atomic level and gamma rays are vanishingly small in
size. By contrast, alpha and beta particles inside the body lose all their energy by
colliding with tissue and causing damage. Gamma rays do not directly ionize atoms in
tissue. Instead, they transfer energy to atomic particles such as electrons (which are
essentially the same size as beta particles). These energized particles then interact with
tissue to form ions, the indirect ionizations they cause generally occur farther into tissue.
For environmental samples containing radionuclides that emit gamma rays,
scintillation detectors (sodium iodide) and semiconductor detectors (germanium) are
commonly used. These detectors, along with the appropriate electronics, computers and
software, can be used to simultaneously identify and quantify a number of gamma-

24
emitting radionuclides. Germanium detectors have superior resolution and are more
suitable if more than a few radionuclides are present in the sample (ATSDR, 1999).
2.2.1.3. Cesium-137
The source of cesium-137 found in the SRS region comes from two major routes:
the atmospheric and liquid release of cesium-137 to the environment. The amount of each
released was 8.15×10-3 Ci/year and 8.81×10-2 Ci/year, respectively (SRS, 2000). The
management of radiological nuclear waste from nuclear reactors and support facilities
(separation facilities, tritium facilities, Savannah River Technology Center…etc)
contributed most of cesium-137 released to the SRS environments (SRS, 2000).
Cesium-137 undergoes radioactive decay with the emission of beta particles and
relatively strong gamma radiation (EPA, 2002). Cesium-137 decays to barium-137 (a
short-lived decay product, half-life of 2.6 minutes), that in turn decays to a non-
radioactive form of barium. The half-life of cesium-137 is 30.17 years. Because of the
chemical nature of cesium, it moves easily through the environment. This makes the
cleanup of cesium-137 difficult to achieve.
Everyone is exposed to very small amounts of cesium-137 present in soil and
water as a result of atmospheric fallout (EPA, 2002). People may also be externally
exposed to gamma radiation emitted by cesium-137 by walking on contaminated sites
and coming in contact with waste materials at contaminated sites. Also, people may
ingest cesium-137 that is present in food and water, or they may inhale it as dust. If
cesium-137 enters the body, it is distributed fairly uniformly throughout the body's soft
tissues, resulting in exposure of those tissues. Compared to some other radionuclides,

25
cesium-137 remains in the body for a relatively short time and is eliminated through the
urine.
Like all radionuclides, exposure to radiation from cesium-137 results in increased
risk of cancer (EPA, 2002). If exposures are very high, serious burns and even death can
result. The magnitude of the health risk depends on exposure conditions. These include
the strength of the source, length of exposure, distance from the source, and whether
there was shielding between you and the source (such as metal plating).
2.2.1.4. Strontium-90
The presence of strontium-90 at the SRS area resulted from atmospheric and
liquid release of radioactive materials to the environments. The amount of strontium-90
release was 3.89×10-3 Ci/year and 5.44×10-2 Ci/year for atmospheric and liquid release,
respectively (SRS, 2000). The management of radiological waste from reactors and
release of radioisotopes from separation processes contributed most of strontium-90
accumulated in SRS environments (SRS, 2000). Strontium-90 is considered a hazardous
constituent of nuclear wastes.
As strontium-90 decays, it releases beta radiation and forms yttrium-90 (90Y),
which in turn decays to stable zirconium (EPA, 2002). The half-life of strontium-90 is
29.1 years, and that of yttrium-90 is 64 hours. Strontium-90 emits moderate energy of
beta particles, and yttrium-90 emits very strong (energetic) beta particles. Strontium-90
can form many chemical compounds, including halides, oxides, and sulfides, and moves
easily through the environment.
Everyone is exposed to small amounts of strontium-90, since it is also widely
dispersed in the environment and the food chain (EPA, 2002). People may inhale trace

26
amounts of strontium-90 as a contaminant in dust. But, swallowing strontium-90 with
food or water is the primary pathway of intake. When people ingest strontium-90, about
70 ~ 80 % of it passes through the body. Virtually all of the remaining 20 to 30 % is
absorbed and deposited in the bone. About 1 % is distributed among the blood volume,
extracellular fluid, soft tissue, and surface of the bone, where it may stay and decay or be
excreted from the body.
Strontium-90 is chemically similar to calcium, and tends to deposit in bone and
blood-forming tissue (bone marrow). Thus, strontium-90 is referred to as a "bone seeker".
Strontium-90 deposited onto bone is linked to bone cancer, cancer of the soft tissue near
the bone, and leukemia (EPA, 2002). The risk of cancer increases with increased
exposure to strontium-90.
2.2.2. Savannah River Site (SRS) and CRESP Involvement
The Savannah River Site (SRS), a facility in the U.S. Department of Energy
(DOE) complex, encompasses approximately 310 square miles in South Carolina and is
adjacent to the Savannah River. The site was established by an agreement between the
U.S. Atomic Energy Commission (AEC) and Du Pont in 1950 to produce plutonium and
tritium for national defense and additional special nuclear materials for other government
uses and for civilian purposes. Production of these materials continued for about 40 years.
Du Pont operated the site until March 31, 1989 at which point Westinghouse Savannah
River Company (WSRC) became the prime operator. The WSRC is still the operator of
the currently named SRS. When the Cold War ended in 1991, DOE responded to
changing world conditions and national policies by refocusing its mission. The site’s

27
priorities shifted toward waste management, environmental restoration, technology
development and transfer, and economic development (SRS, 2000).
Figure 2. Areas of the Savannah River Site (Taken from SRS Environmental Report for 2000)

28
SRS was divided into several areas (Figure 2) to achieve the site mission 4 :
Reactor Materials Area, Reactor Areas, Heavy Water Reprocessing Area, Separation
Areas, Waste Management Areas, Administration Area, and Other Areas (SRS, 2000).
The reactor materials area (M-Area) is home to three analytical laboratories, various
offices, and the vendor treatment facility. This facility, which completed its operations in
1999, processed 670,000 gallons of mixed-waste (both radioactive and hazardous) sludge
into glass beads. The Reactor Areas (C, K, L, P, and R-Area) house the site’s five heavy
water reactors. All five reactors (C, K, L, P, and R-Reactor) are permanently shut down.
Facilities in C-Area, K-Area, and L-Area are being used to store heavy water. Heavy
water was used as a coolant and moderator in the SRS reactors. P-Area and R-Area are
shut down completely. The Heavy Water Reprocessing Area (D-Area) is no longer
conducting any nuclear-related operations. The Separation Areas (F and H-Area) include
the facilities for separating and purifying the waste products, storage of spent fuel for
processing, recycling tritium remaining after the decay from nuclear weapons reservoirs,
and waste treatment. The Waste Management Areas (E, F, H, S, and Z-Area) contain the
engineered concrete facilities for disposal of low-level radioactive solid waste (E-Area);
waste tanks farms consisting of large underground storage tanks that hold high-level
liquid radioactive waste resulting primarily from the reprocessing of spent nuclear fuel (F
and H-Area); and the defense waste processing facilities to immobilize the high-level
waste sludge and the precipitate by changing it into a solid glass waste form (S-Area).
The Administration Area (A-Area) contains DOE’s Savannah River Operation Office,
Westinghouse Savannah River Company’s (WSRC’s) administrative offices, Savannah
River Technology Center (SRTC), and the Savannah River Ecology Laboratory (SREL). 4 The text for areas of Savannah River Site was excerpted from SRS Report for 2000

29
Other Areas (B, N, TNX, and G-Area) include an engineering complex and some
administrative offices (B-Area); central shops (N-Area); multipurpose pilot plant campus
and research / development area (TNX-Area); and general area, not designated for
specific purposes (G-Area).
“The DOE, South Carolina Department of Health and Environmental
Conservation (SCDHEC) and the EPA have worked with local citizens to form a
Citizen’s Advisory Board to facilitate public participation in the SRS cleanup decisions.
In consideration of the involvement of the general public with risk assessment, the
Consortium for Risk Evaluation with Stake Holder Participation (CRESP) was
established. The CRESP is a national organization and was created to provide
information necessary for risk-based cleanup of contaminated sites. CRESP has worked
to fulfill its mission by improving the scientific and technical basis of environmental
management decisions leading to advance protective and cost-effective cleanup of the
nation’s nuclear weapons and to enhance stakeholder understanding of the national
nuclear production facility waste sites (http://www.cresp.org). In the work with SRS
seepage / basin soils, our association with CRESP allowed scientific work to be
conducted on soils with high relevancy to both current public health issues and general
soil-related cleanup goals” (Ellickson, 2001, p. 5-6).
2.2.3. Crystal Ball
Crystal Ball® v7.1 (Decisioneering Inc, Denver, CO) is a forecasting and risk
analysis program that works with a spreadsheet program, specifically Microsoft Excel®.
It runs with a built model that processes combinations of data, variables, formulas, and
functions in a spreadsheet. The forecasting procedure is performed with defining each

30
input value of a model by specific probability distribution and simulating the model with
generated numbers from assumed probability distribution. The Crystal Ball® program
includes 17 built-in pre-specified probability distributions (e.g., normal, lognormal,
uniform, binomial, exponential distribution…etc) and two sampling methods. The two
sampling methods are: Monte Carlo, randomly selects any valid value that are totally
independent from each assumption’s defined distribution, and Latin Hypercube,
randomly selects values, but spreads the random values evenly over each assumption’s
defined distribution.
The Crystal Ball® program repeats the calculations up to the desired iteration
numbers with randomly chosen values from the pre-specified distribution of variables.
Then it produces the results with distribution chart and percentiles. The simulation allows
us to predict the possible exposure / dose numbers for general population using given
scenarios with site-specific variables. Detailed model adaptation and calculation is
provided in the section of 2.3.9.
2.3. Methods
2.3.1. Test soils
“Several soils known to contain elevated levels of cesium-137 were collected
along the Lower Three Runs (LTR) in the SRS. The exact sampling sites were chosen as
a result of preliminary investigations on the sampling locations using a NOMAD
(Neutrino Oscillation Magnetic Detector) portable gamma counter (EG&G Corp,
Wellesley, MA). LTR samples were collected and indicated on Figure 3 as identifiers of
SB3 (Stinson Bridge), TC3 (Tabernacle Church), and DS1 (Donora Station). The LTR is

31
a stream discharging Par pond, which was a man-made cooling water pond that had a
history of contamination” (Ellickson, 2001, p. 38). The soils were of interest in the
previous bioaccessibility study on low-level radionuclides conducted by Ellickson (2001).
“Another soil was collected from the Savannah River Laboratory (SRL) seepage /
basin sites. The samples have been used to manage low-level radioactive waste disposal
by the SREL building from 1954 to 1982 (Figure 3). The laboratory’s waste was stored in
waste tanks until levels reached lower than 100 disintegrations per minute per milliliter
(dpm/mL) alpha or 50 dpm/mL for beta. Once waste met these criteria, the liquid was
sent to basin 1 via sewer line. There are no records of overflow out of the basins or
ground surface seepage. The wastewater was allowed to evaporate leaving behind
contaminated soils and sediments; however, SRS has found that the contamination did
not travel down vertically further than 2 feet. If radionuclide levels in the soils or
sediments ever exceeded federal regulatory standards, the waste was sent by tank truck to
the 200-F Area separations facility for disposal. Final remediation of the SRL seepage /
basin site started in February 2000. The contaminated soil and vegetation was sent to
Environcare of Utah, Inc., which is a low-level radioactive waste storage facility”
(Ellickson, 2001, p. 41).
The soil samples (Table 3) were acquired from seepage / basin soils at berm
locations-1/2/3/4, and depth profiles-1/2/3/4. “This was done at the time of the final
facility remediation investigation, which also included the work on the baseline risk
assessment, corrective measurement study, and feasibility study. CRESP investigators
were able to take the samples discussed in this thesis in parallel with samples collected
for afore mentioned SRS studies. All samples contained elevated levels of many

32
radionuclides including cesium-137, strontium-90, and curium-243/244; however, the
initial risk assessment for allowable levels of intake for CRESP researchers was driven
by the curium-243/244 content soils” (Ellickson, 2001, p. 41). SRS soil samples were
distinguished from the highest to lowest soil hazard class: class “A” (1berm1 and 1berm2
with 776 pCi/kg of 243/244Cm), class “B” (2berm1, 2berm2, 2berm3, 2berm4, 4berm1, and
4berm2 with 76.5 ~ 85.4 pCi/kg of 243/244Cm), and class “C” (1berm3, 3berm1, and
3berm2 with 42.2 pCi/kg of 243/244Cm). The soil class (“A”, “B”, and “C”) determination
relied upon the levels of radionuclides found in each soil for classifying the hazard class
of SRS seepage / basin samples. In addition, the accessibility of each soil set to people
was considered for the classification of soil hazard class. The berm soil samples were
characterized as sands, and had acidic pH, very low SOM contents and CEC.
Table 3. SRS soil hazard classification, radionuclides activity, and sampled conditions
Soil ID Class Mass (kg)
Cm-243/244 (pCi/g)
Cs-137 (pCi/g)
Sr-90 (pCi/g)
Depth (ft) Location
1Berm1 0.767 776 4660 265 0.0 – 0.5 1Berm2
A 0.758 0.5 – 1.0
1Berm3 C 1.095 44.3 1.0 – 2.0
Basin 1/2
2Berm1 0.989 76.5 144 119 0.0 – 0.5 2Berm2 1.097 0.5 – 1.0 2Berm3 1.028 4.76 1.0 – 2.0 2Berm4
B
1.172 2.0 – 3.0
Basin 2/3
3Berm1 1.180 42.2 81.7 14.6 0.0 – 0.5 3Berm2
C 1.214 0.5 – 1.0
Basin 3/4
4Berm1 1.328 85.4 36.2 2.71 0.0 – 0.5 4Berm2
B 1.374 0.5 – 1.0
Basin 4

33
Figure 3. Savannah River Site soil sample locations (Taken from Ellickson et al., Health Physics, 83(4):476-484, 2002)
2.3.2. Soil preparation
Each SRS soil set was transferred from the REHS (Rutgers Environmental Health
and Safety) to the EOHSI laboratory. From one set of soil samples, a small amount of soil
(approximately 50 g) was transferred to the soil jar; the remaining soil sample was
immediately returned to REHS. All procedures and analyses performed at the EMAD-
EOHSI laboratories were approved by both the University-wide (RWJMS/UMDNJ) and
internal safety committee (CRESP SOP-001 “Soil Drying”, CRESP SOP-002
“Preparation of Soil Sub-Fractions”, CRESP SOP-003 “Bioaccessibility Assay”, CRESP
SOP-004 “Total Soil Dissolution Procedure” in the appendices).

34
2.3.2.1. Soil drying
The SRS soil samples were oven dried using a Fisher Isotemp oven at the
temperature of 70 °C to obtain a constant weight. Then the samples were labeled and
stored in a desiccator. All soil drying experiments followed CRESP SOP-001 “Soil
Drying” (appendices 1).
2.3.2.2. Soil sieving
“An important factor contributing to the estimation of exposure from hand to
mouth activity is the quantity of soil that adheres to a child’s hand. Particle size has been
shown to be a very important factor affecting soil adherence variability on a child’s hands,
with soil adherence increasing as particle size decreased (Driver et al., 1989). Although
soils of particle size < 150 µm (1.4 mg/cm2) tend to adhere to the hand at the highest
percentage, the < 250 µm size incorporates particle size range capable of adhering to a
child’s hand. Driver et al. (1989) found that a significant drop off in soil adhesion for the
particle size region > 250 µm (0.95 mg/cm2). Based upon the above, the soil used in these
bioaccessibility studies were sieved to < 250 µm in diameter” (Ellickson, 2001, p. 46-47).
Radionuclide-contaminated soils were sieved in enclosed and disposable
containers to minimize radiation contamination. The sieve unit was fabricated with high-
density polyethylene sample jars and nylon sieve sheets of 250 µm cut-off size (Ellickson
et al., 2002 and Figure 4). The entire sieve unit was fixed with laboratory tape and placed
in a plastic bag for double containment and again sealed with tape. The whole unit was
inverted and placed on the sieve shaker and performed for 20 minutes within a glove box.
Dust was settled for one hour before removing the soil collection jar for storage. All soil

35
sieving experiments followed the CRESP SOP-002 “Preparation of Soil Sub-Fractions”
(appendices 2).
SoilCollection
Jar
Soil
Soil DryingJar
Sieve
SieveShaker
SieveAnchor
ModifiedJar
ModifiedLid
CentralUnit
Heat-Sealable
Bag
Static FreePlastic
Figure 4. Soil sieve unit (Taken from Ellickson et al., Health Physics, 83(4):476-484, 2002)
2.3.3. Total soil dissolution for radionuclide analysis
The total concentration of each radionuclide in a sample was obtained by
dissolution of the soil. Total radioactivity was used as the denominator to calculate the
bioaccessibility of the radionuclides in soil. The sample refluxed on a hot plate, and the
acid was prevented from volatilization (Ellickson et al., 2002 and Figure 5).
Approximately 1 g of soil was weighed into a Teflon™ sample container and 9 mL of 1:1

36
nitric acid (HNO3) was added to the solution. The acid-soil mixture was refluxed at 95 ±
5 °C for two hours. At the end of the reflux period, 10 mL of hydrogen peroxide (H2O2)
was added, and then 1.5 mL of hydrofluoric acid (HF) was also added to the container.
After the addition of hydrofluoric acid, the mixture was heated for another one hour at 95
± 5 °C. The samples were then decanted into polyethylene bottles containing 90 mL of
4 % boric acid (H3BO3) solution at the room temperature. It was allowed to stabilize in a
refrigerator for 3 days. The total soil dissolution experiments followed the CRESP SOP-
004 “Total Soil Dissolution Procedures” (appendices 4).
Teflon
Tube
Screw Top Teflon Container
Diagram of Sample Processing
Figure 5. Sample processing unit for hydrofluoric acid digestion (Taken from Ellickson et al., Health Physics, 83(4):476-484, 2002)
2.3.4. Bioaccessibility extraction
The in vitro bioaccessibility procedure was completed using the Ellickson et al.
(2002) modified version of the method developed for elemental inorganic bioaccessibility
(Ruby et al., 1992, 1993, and 1996). The assay consisted of a sequential extraction using

37
synthetic human saliva, gastric and intestinal fluids. Each step was completed at
physiological temperature (around 37 °C) and a mixing speed of 90 rpm. All
bioaccessibility extraction experiments followed the CRESP SOP-003 “Bioaccessibility
Assay” (appendices 3).
2.3.4.1. Artificial fluids preparation
The formula for preparation of artificial saliva was a modified version of the
Fusayama’s protocol (Fusayama et al., 1963). It included 0.004 M of calcium chloride
dihydrate (CaCl2⋅2H2O), 0.4 % (w/v) of mucin, 0.005 M of potassium chloride (KCl),
0.007 M of sodium chloride (NaCl), 0.004 M of sodium phosphate dibasic (Na2HPO4),
and 0.007 M of urea (H2NCONH2). Hypothiocyanite, used as an antimicrobial
component, and sodium benzonate (C7H5O2Na), a preservative, were excluded in the
artificial saliva because neither microbial activity nor long-term storage of the artificial
saliva was of concern in this experiment. The artificial saliva was stored at 4 °C.
The synthetic gastric fluid (Hamel, 1998) was prepared following the
recommendations of the U.S. Pharmacopoeia, which is utilized in drug dissolution studies
(U.S. Pharmacopoeia, 1990). The gastric fluid was prepared on the day of use as follows:
0.03 M of sodium chloride, 0.084 M of hydrochloric acid (HCl), and 0.32 % (w/v) of
pepsin.
The synthetic intestinal fluid analogue (Hamel, 1998) was modified from the U.S.
Pharmacopoeia formulation to yield a fluid with stronger solvent properties. The artificial
intestinal fluid was a 0.2 M sodium bicarbonate (NaHCO3) solution.

38
2.3.4.2. In vitro extraction
Triplicate soil samples were prepared for gastric and intestinal bioaccessibility
test. 0.5 g for class “B” & “C” soils and 0.05 g for class “A” soil were mixed with 8 mL
of the artificial saliva. This was followed by the addition of 100ml of gastric fluid. A pH
= 1.4 ± 0.2 was maintained throughout the extraction. Subsequently, the samples were
placed into a constant temperature water bath at 37 °C and allowed to shake for two hours
at 90 rpm. After the saliva-gastric incubation, the gastric samples were filtered using a
0.45 µm of pore sized cellulose nitrate filter in a polycarbonate 250 mL filter funnel
apparatus. The intestinal samples were adjusted to a pH of 6.5 by adding 100 mL of
intestinal juice, and then the intestinal samples were digested in the incubation process at
the same conditions. After the saliva-gastric+intestinal incubation, the intestinal samples
were also filtered using the gastric sample procedures. Following the sequential
gastrointestinal extraction, the residual soil mass was re-captured on the filter to
determine the mass of soil and radionuclides not released as bioaccessible radionuclides
from the soil. Bioaccessibility was calculated by dividing the concentrations in filtered
fluids by the total radionuclides concentrations found in the soil. The schematic diagram
(Ellickson et al., 2002 and Figure 6) shows the processes of in vitro extraction step by
step.

39
Figure 6. Schematic representation of the bioaccessibility procedure (Modified from Ellickson et al., Health Physics, 83(4):476-484, 2002)
2.3.5. Cesium-137 samples: preparation & measurement
After filtration, the gastric / intestinal extracts were stored in polyethylene bottles
and sealed with parafilm™. Total soil samples and re-captured soil samples were placed
into 2.5 mL of polyethylene liquid scintillation vials. No further preparation was
necessary prior to measurement of cesium-137 concentration. Cesium-137 was measured
directly by gamma counting using a 50 % relative efficiency High-Purity Germanium
Detector (GEM, EG&G/Ortec Inc., Gaithersburg, MD) with a Digital Gamma Ray
Spectrometer (DSPEC, EG&G/Ortec Inc., Gaithersburg, MD) at 661.52 keV. The activity
of a sample was determined using efficiency values obtained from spiked solutions for
each geometry of sample. The efficiency values (cps/dps) varied and were dependent
upon the bottle size and number of samples. Standard efficiency for each geometry of

40
sample was counted at least ten times, and reported in Table 4. The over-all efficiency
calculation was based on the average of these values. Samples were blank subtracted
when the method blank was greater than the MDA (Minimum Detection Activity; the
smallest amount of activity that can be quantified for comparison). The sample counts
were divided by the efficiency values for each sample type and the mass of soil using the
following:
Sample Bq/g = {(net countssample / tseconds) – (net countsmethod blank / tseconds)} / {εcesium-137 *
soil mass (g)}
Table 4. Standard efficiencies of sample geometry for cesium-137 gamma spectroscopy
Geometry Efficiency (cps/dps)
125 mL Nalgene Bottle – single sample 0.0150
250 mL Nalgene Bottle – single sample 0.0102
2.5 mL Liquid Scintillation Vial 0.0376
125 mL Nalgene Bottle – triple sample 0.0087
250 mL Nalgene Bottle – triple sample 0.0061
2.3.6. Strontium-90 samples: preparation & measurement
The 3M™ Empore™ Sr Rad Disk (3M Bioanalytical, St. Paul, MN) is a thin
membrane consisting of strontium selective particles held in a stable inert matrix of
polytetrafluoroethylene (PTFE) fibrils. The 3M™ Sr disk is a cost-effective alternative to
conventional radiochemical sample preparation methods that use wet chemistry or packed
columns. It has advantages of a quick separation time (within 20 minutes) in aqueous
samples and the elimination of time-consuming evaporation steps (at least 2 hours for
each batch sample). Thus, the 3M™ Sr disk was used for Sr separation by modifying the

41
method suggested by the supplier (Goheen, 1997). It was fabricated from a strontium
selective chelating ligand embedded in a Teflon™ matrix.
To prepare samples for analysis, 2 M nitric acid was added to 50 mL
gastrointestinal extract or HF acid digested solution. All gastrointestinal samples were
allowed to digest in nitric acid for about 48 hours. After the initial digestion, 5 mL of
hydrogen peroxide was added to the fluids to complete the digestion. The strontium-90
separation was conducted using a filtering manifold with 3 parallel 50 mL filter funnels.
The filter funnel was equipped with a 3M™ Empore™ Rad disk and a 45 µm Nylon filter
as a pre-filter. These filters were washed with 2 mL methanol (CH3OH) and 20 mL of 2
M HNO3, respectively. After 24 hours of digestion by hydrogen peroxide, the samples
were filtered through the disks using a mild vacuum (flow rate was less than 14 mL/min).
At the end of filtering, 20 mL of nitric acid (2 M) were applied to the filter funnel to
complete filtering and cleaning. The 3M™ Empore™ Rad disks were placed in low
potassium glass liquid scintillation vials containing 20 mL of Packard Ultra LLT liquid
scintillation cocktail. The Sr disks were then allowed to equilibrate for 30 days to ensure
equilibrium of yttrium-90 with strontium-90. This growth period (30 days) was
determined by the fact that Cherenkov counting, in conjunction with liquid scintillation,
has been used to detect strontium-90 by measuring the concentration of its progeny,
yttrium-90 in solution (ATSDR, 2004). After 30 days, all Sr disks were counted in a
Liquid Scintillation Analyzer (Tricarb 2770 TR/SL, Packard Bioscience at PerkinElmer
Life Sciences, Boston, MA) for 60 ~ 240 minutes each. The alpha / beta discriminator
value was 128 and the chosen channel for β was 15.0 ~ 2,000 keV. Minimum Detection
Activity (MDA) and Lower Limit of Detection (LLD; the smallest amount of activity that

42
can be detected for comparison) were calculated for each sample. The MDA calculation
was dependent on the relative counting time of blanks and samples. The samples were
calculated using efficiency values (εstrontium-90) resulting from the counts of spiked disks of
equal geometry to the sample containing 3M™ Empore™ Rad disks. All samples were
blank subtracted using the method blank, adjusted by the dilution factor using the original
sample weight divided by the strontium sample aliquot weight, and divided by the mass
of soil used in the procedure:
εstrontium-90 = {(CPMAlaboratory spike + CPMBlaboratory spike) – (CPMAlaboratory blank +
CPMBlaboratory blank)} / spike activityCPM
Sample Bq/g = [{(CPMAsample + CPMBsample) – (CPMAmethod blank + CPMBmethod blank)} /
{εstrontium-90 * 60seconds/minute} * dilution factor] / soil mass (g)
2.3.7. Statistical analyses
A normality test is a statistical process used to determine if a sample or any group
of data fits a standard normal distribution. Because many data analysis methods (e.g., t-
test, ANOVA, regression) depend on the assumption that data were sampled from a
Gaussian distribution. Non-parametric tests are often used in place of their parametric
counterparts when certain assumptions about the underlying population are questionable.
Normality tests were completed, and the results found that the cesium-137 data
sets for gastric bioaccessibility (Shapiro-Wilk test; p = 0.0022; N = 20) and intestinal
bioaccessibility (Shapiro-Wilk test; p < 0.0001; N = 19) were neither normally nor log-
normally distributed. Similarly strontium-90 for gastric bioaccessibility data was not
normally or log-normally distributed (Shapiro-Wilk test; p < 0.0001; N = 57). Although
intestinal bioaccessibility for strontium-90 data (Shapiro-Wilk test; p = 0.1541; N = 41)

43
were normally distributed, non-parametric statistics were also performed for these data.
All results were analyzed using the Wilcoxon-Mann-Whitney test and Spearman rank
correlations. All statistical analyses were conducted by using SAS® Program v9.1 (SAS
Institute Inc., Cary, NC) and SigmaStat® Program v3.11 (Systat Software Inc., Richmond,
CA).
Table 5. Shapiro-Wilk normality test for the bioaccessibility of cesium-137 and strontium-90 from soil samples
Radionuclides Tested Group Data No. Test Statistic p-value
Gastric Bioaccessibility 20 0.82682 0.0022 137Cs Intestinal Bioaccessibility 19 0.715262 <0.0001 Gastric Bioaccessibility 57 0.870437 <0.0001 90Sr
Intestinal Bioaccessibility 41 0.959748 0.1541*
A 6 0.903342 0.3941* B 8 0.977409 0.9490* 137Cs Intestinal Bioaccessibility
C 5 0.822224 0.1215*
A 9 0.890918 0.2039*
B 20 0.964359 0.6341* 90Sr Intestinal Bioaccessibility C 12 0.854429 0.0417
*Insufficient evidence to reject the assumption of normality
2.3.8. SRS vs ChNPP area
Exposure and dose were calculated for cesium-137 and strontium-90 based on
radionuclides concentrations on SRS soils to estimate the magnitude of impact of people
living around or visiting SRS. This estimation would not be realistic, considering the very
high level of radioactivity on tested soil (331 ~ 2.49 Bq/g for cesium-137 and 24.97 ~
1.48 Bq/g for strontium-90). However, if one assumes a worst possible scenario for the
general population, one can provide probable estimates of exposure / dose from contact
with the radiation. The levels of total radioactivity on SRS soils used in the following
exposure / dose models are similar to radioactivity present in the Chernobyl surface soil

44
located within Exclusion Zone, 30 km around the Chernobyl Nuclear Power Plant
(ChNPP). The radioactivity levels, after 10 years of accident, ranged from 0.07 to 400
Bq/g and from 0.09 to 100 Bq/g for cesium-137 and strontium-90, respectively, on
undisturbed sandy / peaty soils sampled 6 km distant from Chernobyl Nuclear Power
Plant at depth profile of 0 ~ 0.5 ft (Amano et al., 2003).
After the Chernobyl catastrophe on April 26, 1986, approximately 161,000
residents were evacuated from within a 30 km radius of ChNPP (2,800 km2) and later
relocated. Since then, the site has been declared an Exclusion Zone and strictly prohibited
public access up to date (http://www.chernobyl.info). However, at least 800, mostly older,
people have returned to their former villages in the Exclusion Zone, and the governments
(Ukraine, Belarus, Russian Federation) anticipates to resettle the evacuated families in
the Resettlement Zone (15 ~ 40 Ci/km2 of cesium-137) in 20 to 30 years later. From the
view of facts, the estimated exposure / dose levels on SRS soils can be successfully
translated into Chernobyl situation. Because people are not going to live in SRS area;
however, some people are living in the Chernobyl Exclusion Zone and more may move
back to the vicinity of Chernobyl regions in the near future. Thus, this exposure / dose
estimations can provide a worst possible scenario for people contacting with very high
level of radioactivity or current residents inside the Chernobyl Exclusion Zone facing
similar total radioactivity levels on surface soils.
SRS soils were classified as high-level (“A” soils) and low-level (“B” and “C”
soils). Similarly, mean (± S.D.) cesium-137 and strontium-90 activities at two Chernobyl-
contaminated field sites (Chistogalovka; 3.5 km from the Chernobyl reactor and
Polesskoye; 60 km SW of the Chernobyl reactor) in Ukraine (Malek et al., 2002) were

45
close to the high-level of radioactive SRS soil (class “A”) and low-level of radioactive
SRS soils (class “B” and “C”), respectively (Table 6). Thus, one can assume the exposure
/ dose estimates based on SRS soils can be smoothly applied for Chernobyl radioactive-
contaminated soils (e.g., Exclusion Zone for high level of class “”A and Resettlement
Zone for low level of class “B” and “C”). Following exposure / dose estimations can
allow us to evaluate the radiological health effects from long-lived radionuclides (about
30 years of half-life for 137Cs and 90Sr) deposited on the surface soil in or around the
ChNPP Exclusion Zone.
Table 6. Comparison of total radioactivity (mean ± S.D.) between SRS soils and Chernobyl areas
SRS Chernobyl Soil Class 137Cs (Bq/g) 90Sr (Bq/g) 137Cs (Bq/g) 90Sr (Bq/g) Radius
A 331 ± 363 24.97 ± 12.27 74 ± 17 36 ± 19 < 3.5 km B 2.49 ± 2.85 1.78 ± 1.72
C 6.36 ± 1.73 1.48 ± 1.40 15 ± 3 0.13 ± 0.03 < 60 km
2.3.9. Exposure / dose calculations
The exposure / dose estimates were calculated based on SRS soil sample results,
and the case for Chernobyl soils were also conducted using the results of SRS
experiments. Two radionuclides exposure scenarios were defined: one is visiting SRS or
ChNPP area for two weeks and the other is residing there for one year. The hypothetical
populations were placed into four specific categories: 2.5-year-old pica children, 2.5-
year-old children, 6.0-year-old children and adults. Because the risk of radioactivity in
soil via non-dietary soil ingestion was expected to increase based upon the magnitude of
age-specific soil ingestion rate. The soil ingestion rate would be highest for 2.5-year-old
children and the rate for 6-year-old was reduced by one fifth. Following age groups were

46
selected for the exposure / dose calculations: pica / young children (aged from 2 to 6),
older children (aged from 6 to 11), and adults.
To estimate the exposure and dose levels for SRS areas or a similar area around
the Chernobyl Exclusion Zone, we assumed the direct contact of radionuclides in soil by
the inhalation of re-suspended soils, ingestion of non-dietary soils, and external exposure
to radionuclides in soil through the penetration of radiation within the body. The dermal
route of exposure to radionuclides is not included in the exposure / dose estimations. The
radiation exposure through skin is considered as the external exposure, since the radiation
arises “external” the body. This situation is in contrast to the intake of radionuclides by
inhalation or ingestion, where the radiation is emitted inside the body (EPA, 1993). The
modes for external exposures are: 1) submersion in a contaminated atmospheric cloud,
i.e., air submersion, 2) immersion in contaminated water, i.e., water immersion, and 3)
exposure to contamination or in the ground, i.e., ground exposure (EPA, 1993). Thus, the
dermal route was replaced with the external exposure to radionuclides on or in the ground.
The other two external exposure modes, air submersion and water immersion, were not
considered in the estimations due to the lack of reasonable data for making such
estimations. However, at a minimum, they could increase the total exposure to
radionuclides.
In this model, an exposure was displayed as the ingested or inhaled radiation
(pCi) normalized with body weight (kg) over time (day) to represent the radiation energy
potentially affecting the organs or tissues within the body. Otherwise, a dose was
displayed as pathway specific-effective dose (mSv) over lifetime to provide an effective
dose that could lead to health effects within organs or tissues. Thus, considering the

47
concept of oral route related bioaccessibility / bioavailability (provided in Figure 1), the
exposure and resulting dose represent the potential dose and (biologically) effective dose,
respectively, associated with the schematic diagram of oral contact. This work relies on
predicted effective doses to compare the calculated doses with recommendations of
NCRP (National Commission of Radiological Protection and Measurements) for public
health.
Exposure, as a potential dose from ingestion and inhalation pathways, was
calculated using:
1,
1, ( )
p g s g
p h s l h
D C R W
D C f R W
−
−
= × ×
= ⋅ × ×
where, Dp,g is the potential dose per body weight over time via ingestion (pCi/kg-day)
Dp,h is the potential dose per body weight over time via inhalation (pCi/kg-day)
Cs is the radioactivity of each soil class (Bq/g)
Rg is the soil ingestion rate for age-specific group (mg/day)
Rh is the resuspended-soil breathing rate for age-specific group (m3/day)
W is the body weight for each exposure group (kg)
fl is the mass loading factor for radioactivity in air by soil resuspension (10-5 g/m3)
Mass loading factor (fl) is obtained from Anspaugh, et al. (1975). The authors
have suggested the use of 100 µg /m3 for predictive purposes. The choice of this value is
partly based upon measurements of particulate concentration in 1966 reported by
NAPCA (National Air Pollution Control Administration) for 30 non-urban locations.
Annual arithmetic averages of particulate matter varied from 9 to 79 µg/m3 with a mean
for all 30 locations of 38 µg/m3. In this work, a value of 10 µg /m3 (= 10-5 g/m3) was
chosen for mass loading factor (fl), which is the most typical of current levels in the air.
Total soil concentration, ingestion rate, and inhalation rate were used with an assumption

48
of a lognormal distribution; while body weight was assumed to have a normal
distribution in the exposure / dose estimations.
Dose, as a biologically effective dose from ingestion, inhalation, and external
exposure to SRS soils or ChNPP surface soils, was calculated using:
, ,
, ,
,
, , ,
{ ( )} ( )
e g p g g
e h p h h
e x s i s o x a s
t e g e h e x
D D B W d
D D W d
D C d t f t fD D D D
ε
ε
ε ρ
= × × × ×
= × × ×
= × × ⋅ + × ⋅ ×
= + +
where, De,g is the biologically effective dose via ingestion (mSv)
De,h is the biologically effective dose via inhalation (mSv)
De,x is the biologically effective dose via external exposure (mSv)
Dt is the total dose from radioactive-contaminated soil (mSv)
B is the intestinal bioaccessibility for each soil class (unitless)
d is the duration of exposure (14 or 365 days)
εg is the age-dependent effective dose coefficients from ingestion (Sv/Bq)
εh is the age-dependent effective dose coefficients from inhalation (Sv/Bq)
ti is the indoor time for age-specific group (hrs/day)
fs is the shielding effect of structure (unitless)
to is the outdoor time for age-specific group (hrs/day = 24 hrs/day – ti)
εx is the effective dose coefficients for external exposure to soil contaminated to
an infinite depth (Sv⋅m3/Bq-sec)
fa is the age-specific factor for εx (unitless)
ρs is the density of soil (kg/m3)
The sum of the effective doses obtained from ingestion, inhalation, and external
exposure pathways yielded a total dose, Dt (mSv). The ingestion and inhalation dose
coefficients for each exposure pathway (εg and εh; Sv/Bq) were obtained from ICRP

49
report (No. 67 and 71). These effective dose coefficients were derived from the age-
variable GI absorption fraction and the lung model (Fast, Moderate, and Slow) that was
related to the inhaled particle distribution and absorption in lungs. The dose coefficients
are calculated in the following process: 1) Exposure → Absorbed (Internal) Dose, 2)
Absorbed Dose → Committed Equivalent Dose (the time integral of the equivalent dose
rate in a particular tissue or organ that will be received by an individual following intake
of radioactive material into the body), 3) Committed Equivalent Dose → Committed
Effective Dose (the sum of the products of the committed organ or tissue equivalent
doses and the appropriate organ or tissue weighting factors). However, these parameters
were based on human data or results of animal experiments as soluble form of cesium
(e.g., 137CsCl) and strontium (e.g., SrCl2) or dietary strontium (cow milk). The
bioaccessibility obtained in the experiments conducted on each soil class was introduced
into effective dose calculations of ingestion. For inhalation dose estimates, there was no
information for solubility of re-suspended soil in human lung; therefore the ICRP default
values were used. Here are the parameter values for each radionuclide:
Table 7. Two radionuclides ingestion / inhalation absorption parameters for different aged groups
Age Absorption Type
3 months 1 year 5 years 10 years 15 years adults
137Cs GI absorption 1.00 1.00 1.00 1.00 1.00 1.00 90Sr GI absorption 0.60 0.45 0.30 0.35 0.40 0.30
137Cs F type absorption 1.00 1.00 1.00 1.00 1.00 1.00 90Sr M type absorption 0.20 0.10 0.10 0.10 0.10 0.10
(Taken from Annals of ICRP publication No. 67 and 71)

50
The effective dose coefficients for external exposure to soil contaminated to an
infinite depth (εx; Sv⋅m3/Bq-sec) were obtained from EPA Federal Guidance Report (No.
12). The tested SRS soils have four depth profiles (profile “1”: 0-0.5 ft, “2”: 0.5-1.0 ft,
“3”: 1.0-2.0 ft, and “4”: 2.0-3.0 ft); even the same hazard class has different depth
profiles. The guidance report provides several effective dose coefficients for the radiation
source from the surface, depth of 1 cm, depth of 5 cm, depth of 15 cm (≈ 0.5 ft), and
infinite depth. Therefore, to cover all depth-profiled SRS soils (0 ~ 3 ft), we selected
effective dose coefficients for exposure to soil contaminated to an infinite depth.
The coefficient, εx, was developed for the reference adult males (height: 179 cm
and weight 73 kg). However, these coefficients were not relevant for our hypothetical
young / older children groups. Thus, age adjustment factor (fa) was obtained from the
work of Petoussi et al. (1991). The author found the discrepancy was 40 % between the
organ dose for 8 weeks old baby and man, and recommends multiplying the fa for adults
by 1.5 for young children (aged < 2 year) to modify the coefficients for young children.
Young children are more vulnerable to the effect of external radiation due to the lower
height and closer to the radiation source for developing organs than modeled man.
Another adjustment of external exposure obtained from the contaminated soil was
the shielding effects (fs) provided by the structures, buildings, or vehicles in the radiation
field. Without the shielding effect, duration of dose will be 24 hours per day. This will
cause the external dose to be overestimated. Therefore, the shielding effect was adapted
to the calculation of duration, d, using the indoor time (hours/day) for each exposed
group obtained from EPA Exposure Handbook (1999). The outdoor time (hours/day) is
calculated by subtracting an indoor time from a total of 24 hours per day. The structure

51
shielding effect is assumed to reduce the fraction of 0.1 for external radiation by residing
in single house with basement of 1 or 2 walls fully exposed case. The reduction factor of
0.1 was obtained from Burson, et al. (1977).
Effective dose forecasts using the Crystal Ball® program required an assumed
distribution for each input value. We found that the intestinal bioaccessibility for most
radionuclides was normally distributed (Table 5), and assumed normal distributions for
the effective dose coefficients from ingestion (εg), inhalation (εh), and external exposure
to contaminated soil (εx). Body weight values, used in the exposure / dose estimation,
were averaged by age and race, and were obtained from the National Center for Health
Statistics Studies (EPA, 1999). Soil ingestion and inhalation rate, and indoor time for
each age-specific group were referred from the Exposure Factor Handbook (EPA, 1999).
The model input values and effective dose coefficients for each age-specific group are
provided in the Table 8 and 9, respectively. The calculations were repeated 10,000 times,
and reported the exposure / dose estimations at distribution percentiles of 50 % and 95 %
showing the central and maximum value for each estimation. Examples of Crystal Ball®
model runs are provided in Figure 7 and 8 for pica children on class “A” soil for the
ingestion exposure and total dose of one-year residence, respectively.
Table 8. Age-specific input values for exposure / dose estimations
Subgroups Ingestion Rate (mg/day)
Inhalation Rate (m3/day)
Body Weight (kg)
Indoor Time (hours/day)
Pica Children 500 6.8 14.3 20
2.5-Year Children 110 6.8 14.3 20
6.0-Year Children 22 10 22.6 19.6
Adults 20.4 15.2 72.8 21

52
Table 9. Age-dependent effective dose coefficients for each exposure pathway Ingestion Inhalation External Exposure
Subgroups 137Cs (Sv/Bq)
90Sr (Sv/Bq)
137Cs (Sv/Bq)
90Sr (Sv/Bq)
137Cs (Sv*m3/Bq-s)
90Sr (Sv*m3/Bq-s)
Pica Children 1.2×10-8 7.2×10-8 5.4×10-9 1.1×10-7 6.0×10-21 5.7×10-21
2.5-Year Children 1.2×10-8 7.2×10-8 5.4×10-9 1.1×10-7 6.0×10-21 5.7×10-21
6.0-Year Children 9.7×10-9 4.7×10-8 3.6×10-9 6.5×10-8 4.0×10-21 3.8×10-21
Adults 1.4×10-8 2.8×10-8 4.6×10-9 3.6×10-8 4.0×10-21 3.8×10-21
Figure 7. The example of Crystal Ball™ cesium-137 potential dose estimates for pica children on class “A” soil via ingestion
Figure 8. The example of Crystal Ball™ cesium-137 total dose predictions for pica children on class “A” soil during one-year residence
Statistics Value Trials 10000Mean 1.17E-1Median 1.15E-1S.D. 1.99E-2Skewness 0.52Kurtosis 3.54C.V. 0.17Minimum 6.09E-2Maximum 2.33E-1S.E. 1.99E-490 % 1.44E-195 % 1.52E-1
Statistics Value Trials 10000Mean 316.785Median 310.703S.D. 56.420Skewness 0.64Kurtosis 3.66C.V. 0.18Minimum 165.387Maximum 666.927S.E. 0.56490 % 390.30095 % 418.487

53
2.4. Results & Discussion 2.4.1. Bioaccessible cesium-137 and strontium-90 in two biofluids
Total radioactivity and gastric / intestinal bioaccessibility for radionuclides found
within each soil sample are reported in Table 10. Bioaccessibility and total radioactivity
levels for all 2Berm, 3Berm, and 4Berm soils were acquired from Ellickson (2001). Class
“A” soil (1berm1 and 1berm2) had much higher total radioactivity for both cesium-137
and strontium-90 than class “B” or “C” soil. The total radioactivity of each radionuclide
was utilized to calculate the bioaccessibility of each soil set and to complete the exposure
/ dose calculations. The percentage of bioaccessible cesium-137 and strontium-90 is
calculated from the concentration of soluble portion (Bq/g of soil added) dividing by the
total concentration of contaminant in that soil.
Table 10. Total cesium-137 and strontium-90 concentration (mean ± S.E.) and gastric / intestinal bioaccessibility (mean ± S.D.) for analyzed SRS berm soil samples
Cesium-137 Strontium-90 Bioaccessibility (%) Bioaccessibility (%) Soil ID Total Conc.
(Bq/g) Gastric Intestinal Total Conc.
(Bq/g) Gastric Intestinal
1Berm1 588.4±4.79 9.98±0.2 13.38±0.2 33.64±1.10 93.36±6.4 58.13±4.3
1Berm2 75.38±2.12 14.63±0.8 14.15±0.7 16.29±0.30 97.05±6.7 60.52±2.4
1Berm3 8.35±0.15 12.30±0.4 16.28±0.5 3.10±0.02 78.13±5.1 46.98±7.8
2Berm1 6.71±0.28 8.36±0.6 11.82±0.8 1.76±0.13 82.22±2.5 38.26±8.5
2Berm2 0.76±0.05 11.17±2.7 8.25±1.0 4.45±1.31 81.74±13.9 30.17±6.5
2Berm3 0.43±0.05 ND ND⋅ 3.12* 90.81±3.3 53.39±3.9
2Berm4 0.09±0.01 ND ND⋅ 1.07* 92.65±3.4 71.01±4.0
3Berm1 5.52±1.12 22.42±3.4 31.18±6.8 0.71±0.04 29.69±4.7 17.83±3.1
3Berm2 5.20±0.21 31.15±1.6 38.76±2.0 0.64±0.15 72.41±3.6 56.91±3.3
4Berm1 0.72±0.05 13.86±2.4 12.69±3.2 0.14±0.01 49.63±4.6 48.00±7.2
4Berm2 1.75±0.23 8.86±1.1 15.04±1.9 0.13±0.08 74.30±13.7 46.17±1.6 *Only one value was available for the analysis; bioaccessible cesium-137 for 2berm3 / 2berm4 soils was not included in the table due to the results of lower method detection limit

54
For cesium-137, most soil samples had higher intestinal bioaccessibility than
gastric bioaccessibility; however, a few soil samples (2Berm2 and 4Berm1) gave
different results. Most samples had gastric / intestinal bioaccessibility between 8.3 % and
16.3 %; although, 3Berm1 and 3Berm2 were relatively higher values and ranged between
22.4 % and 38.8 %. The difference between gastric and intestinal bioaccessibility was not
statistically significant (Wilcoxon two-sample test; two-sided; p = 0.1002).
The gastric / intestinal bioaccessibility of strontium-90 (Table 10) was quite
different than the cesium-137 bioaccessibility: gastric samples had higher bioaccessibility
than intestinal samples. Some samples (2Berm1 and 2Berm2) had gastric bioaccessibility
at least two times higher than intestinal bioaccessibility. The difference between gastric
and intestinal bioaccessibility for strontium-90 was statistically significant (Wilcoxon
two-sample test; two-sided; p = 0.0001).
2.4.2. Bioaccessibility with soil hazard class
Comparison of bioaccessible cesium-137 by soil hazard class (Figure 9) showed
the bioaccessibility in intestinal phase was slightly higher than in gastric phase for three
soil classes (difference of gastric and intestinal bioaccessibility ranging from 1.3 % to
6.7 %), and the gastric / intestinal bioaccessibility for class “C” soil (22.0 % ~ 28.7 %)
was higher than other soils (10.6 % ~ 13.8 %). The elevated level of bioaccessibility in
class “C” soil resulted from higher bioaccessibility of 3Berm1 and 3Berm2 (22.4 % ~
38.8 %). The comparison of bioaccessibility by soil hazard class (Figure 10) also yielded
distinctive differences in strontium-90 bioaccessibility. The magnitude of bioaccessible
strontium-90 for two biofluids was ordered as the soil hazard class “A”, “B”, and “C”.
The soil hazard classification was summarized in Table 3 for each soil sample set.

55
A non-parametric one-way analysis of variance between gastric and intestinal
bioaccessibility for cesium-137 and strontium-90 was tested with respect to the variable
of soil hazard class. The Kruskal-Wallis tests were conducted to compare each
bioaccessibility group by soil class, and the results are presented in Table 11. There were
statistically significant differences among three soil classes for all categories. Further,
non-parametric post hoc multiple comparison tests (Dunn’s method for unbalanced
sample subgroups; significance level of 0.05) were conducted to identify within group
differences. The bioaccessible cesium-137 was different (p < 0.05) between soil class “C”
and soil class “B”; however, the difference was not significant between soil class “C” and
soil class “A”. The levels of bioaccessible strontium-90 in the gastric samples were
different for each soil hazard class (p < 0.05); however, the difference between soil class
“B” and soil class “C” for bioaccessible strontium-90 in intestinal fluids was not
significant.
Soil hazard class with different levels of radioactivity affected the bioaccessibility
in both biofluids for cesium-137 and strontium-90. Thus, one can expect proportionally
higher risk from strontium-90 exposure for soil that has higher level of strontium-90
since the higher radioactivity soil was also more bioaccessible. In contrast,
bioaccessibility of cesium-137 for class “C” soil was more soluble than cesium-137
bioaccessibility for class “B” soil.

56
0.0
0.1
0.2
0.3
0.4
0.5
Class "A" Class "B" Class "C"
Bio
acce
ssib
le C
esiu
m-1
37
Gastric Solubility
Intestinal Solubility
Figure 9. The comparison of cesium-137 bioaccessibility by soil class
0.0
0.2
0.4
0.6
0.8
1.0
Class "A" Class "B" Class "C"
Bio
acce
ssib
le S
tron
tium
-90
Gastric Solubility
Intestinal Solubility
Figure 10. The comparison of strontium-90 bioaccessibility by soil class

57
Table 11. Non-parametric Kruskal-Wallis ANOVA test for each bioaccessibility by soil class and non-parametric post hoc multiple comparison test
Class No. Mean Score Significance Min. Med. Max. Grouping
C 6 15.33 0.101 0.176 0.312
A 6 11.33 0.088 0.123 0.157 Cesium-137 Gastric
B 8 6.25
Pr > χ2 (<0.0001)
0.063 0.091 0.139
A A A B B B
C 5 16.40 0.152 0.173 0.388
A 6 9.67 0.131 0.139 0.143 Cesium-137 Intestinal
B 8 6.25
Pr > χ2 (<0.0001)
0.066 0.124 0.179
A A A B B B
A 12 47.50 0.858 0.955 1.060 B 30 29.07 0.461 0.829 0.958 Strontium-90
Gastric C 15 14.07
Pr > χ2 (<0.0001)
0.246 0.715 0.831
A B C
A 9 32.78 0.532 0.608 0.632 B 20 18.30 0.233 0.453 0.755 Strontium-90
Intestinal C 12 16.67
Pr > χ2 (0.0035)
0.154 0.491 0.607
A B B B
*Different letters indicate significant differences (p < 0.05) by unbalanced Dunn’s multiple comparison test; the same letter indicates no difference between median ranks
2.4.3. Bioaccessibility with soil physico-chemical characteristics
The soil characteristic data for SRS seepage basin / berm soils are provided in
Table 12. The soil physico-chemical characteristic experiments were based on only 2/3/4-
berm soils, excluding 1berm soils (1berm-1/2/3) for those experiments, due to the safety
issue of very high radioactive 1berm soils (Table 3). The detailed analytical procedures
for measuring soil physico-chemical characteristics can be obtained from Ellickson
(2001). “The cation exchange capacity (CEC) of soil was measured by Southwest
Research Institute (SwRI, San Antonio, TX). The measurement method for CEC involves
the equilibration of an ammonium solution with the soil, the subsequent measurement of
absorbed ammonium ions, and the report of CEC as centimoles of ammonium per 100
grams of soil. The CEC data were not highly variable between these soils; however, it

58
decreased from the first berm soil (2berm1) to the final berm soil (4berm2). The CEC
values ranged from 5.06 meq/100g in 2berm1 to 0.64 meq/100g in 4berm2. The soil pH
was measured using the Corning pH meter with electrode (Model 430, Cole-Parmer,
Vernon Hills, IL) at EMAD-EOHSI laboratory. The soil pH values ranged from 3.86 in
3berm1 to 5.06 in 3berm2, and did not increase or decrease dependent on their sampling
location or depth. Soil organic matter was measured using the Walkley-Black method at
EMAD-EOHSI laboratory. The method uses chronic acid to measure the oxidizable
organic carbon in a soil. The SOM percentage was low overall, with values ranging from
4.1 % in 2berm2 to 0.3 % in 3berm2. The soil particle size determination was analyzed
using a sedigraph, which measured sedimentation of particles with an X-ray beam by
Southwest Research Institute. The silt+clay percentage was 0.96 % in 2berm1 and
decreased to 0.13 % in 4berm2. These values are very low, which places their textural
classification well in the sand region” (Ellickson, 2001, p. 43-46).
Table 12. Soil physico-chemical characterization data for SRS seepage basin / berm soils
Soil Identity 2berm1 2berm2 2berm3 2berm4 3berm1 3berm2 4berm1 4berm2
Cation Exchange Capacity
(meq/100g)
5.06 4.93 2.61 3.61 1.38 1.34 1.46 0.64
Soil pH 4.59 5.12 4.69 4.78 3.86 5.06 4.21 3.94
Soil Organic Matter (%) 3.9 4.1 3.0 0.6 0.8 0.3 0.8 0.8
Sand (%) 99.04 99.49 99.41 99.62 99.64 99.54 99.72 99.87
Silt+Clay (%) 0.96 0.51 0.58 0.38 0.36 0.46 0.27 0.13
Textural Class Sand Sand Sand Sand Sand Sand Sand Sand
* Taken from Ellickson, K. M. Ph. D. Dissertation, 2001.

59
A correlation analysis examines the relationship among two or more variables,
reported usually as a correlation coefficient. A correlation coefficient is a measure of the
linear association between two variables, and the significance is a measure of error
probability in the intensity of the association. A correlation coefficient is not a measure of
quantitative change of one variable with respect to another, while it can be used to
account for variability in one factor when accounting for the variability in another. A
Spearman rank test was completed to explore the relationships among several soil
physico-chemical characteristics: SOM (Soil Organic Matter content; %), CEC
(meq/100g), clay content (%), pH, and radioactivity of each soil set (Bq/g for cesium-137
and strontium-90). The resulting correlation coefficients and statistical significance
reported in Table 13 indicated that strontium-90 radioactivity, clay content, and CEC
were strongly correlated (positively) each other in that soil set.
Table 13. The Spearman rank-order correlation tests among six variables (total radioactivity of 137Cs / 90Sr, SOM, CEC, pH, and Clay) (N = 8)
137Cs 90Sr SOM CEC pH Clay
137Cs 1.00000 -0.14286 (0.7358)
0.14639 (0.7294)
-0.09524 (0.8225)
-0.33333 (0.4198)
0.14286 (0.7358)
90Sr 1.00000 0.65873 (0.0757)
0.80952 (0.0149)
0.57143 (0.1390)
0.83333 (0.0102)
SOM 1.00000 0.63434 (0.0912)
0.07319 (0.8633)
0.51235 (0.1942)
CEC 1.00000 0.42857 (0.2894)
0.73810 (0.0366)
pH 1.00000 0.54762 (0.1600)
Clay 1.00000
The results in Table 14 show a non-parametric one-way analysis of variance for
gastric and intestinal bioaccessibility for two target radionuclides by sampling depth. The
depth index (No. “1” ~ “4”) is proportional to the depth of sampled soil (see Table 3). A

60
statistical difference was found only for the category of intestinal bioaccessibility of
strontium-90. Further, a non-parametric multiple comparison test (Dunn’s method for
unbalanced sample subgroups, α = 0.05) was conducted for intestinal bioaccessibility of
strontium-90. Soil sampled depth “1” was different (p < 0.05) with other soil depths
(“2”,”3“, and “4”) for intestinal strontium-90 bioaccessibility. The gastric and intestinal
bioaccessibility for two radionuclides generally increased with the depth index of
sampled soil. This can be described by the effect of clay in the soils that can bind the
radionuclides and make it hard to dissolve in the human gastrointestinal system. Clay
contents in tested soil samples were usually decreasing with the depth index of soil
sampling.
Table 14. Non-parametric Kruskal-Wallis ANOVA test for each bioaccessibility by soil sampled depth and non-parametric post hoc multiple comparison test
Depth No. Mean Score Significance Min. Med. Max. Grouping
2 8 11.50 0.101 0.176 0.312 1 8 10.25 0.088 0.123 0.157 Cesium-137
Gastric 3 4 9.00
Pr > χ2 (0.7788)
0.063 0.091 0.139
3 4 13.25 0.152 0.173 0.388 2 8 10.00 0.131 0.139 0.143 Cesium-137
Intestinal 1 7 8.14
Pr > χ2 (0.3505)
0.066 0.124 0.179
4 4 44.50 0.876 0.941 0.947 2 21 31.81 0.604 0.853 1.060 3 9 29.56 0.715 0.831 0.947
Strontium-90 Gastric
1 23 23.52
Pr > χ2 (0.0856)
0.246 0.821 1.015
4 3 40.00 0.679 0.696 0.755 2 14 24.57 0.233 0.565 0.632 3 9 20.11 0.377 0.503 0.571
Strontium-90 Intestinal
1 15 14.40
Pr > χ2 (0.0039)
0.154 0.440 0.609
A A A A A B
*Different letters indicate significant differences (p < 0.05) by unbalanced Dunn’s multiple comparison test; the same letter indicates no difference between median ranks
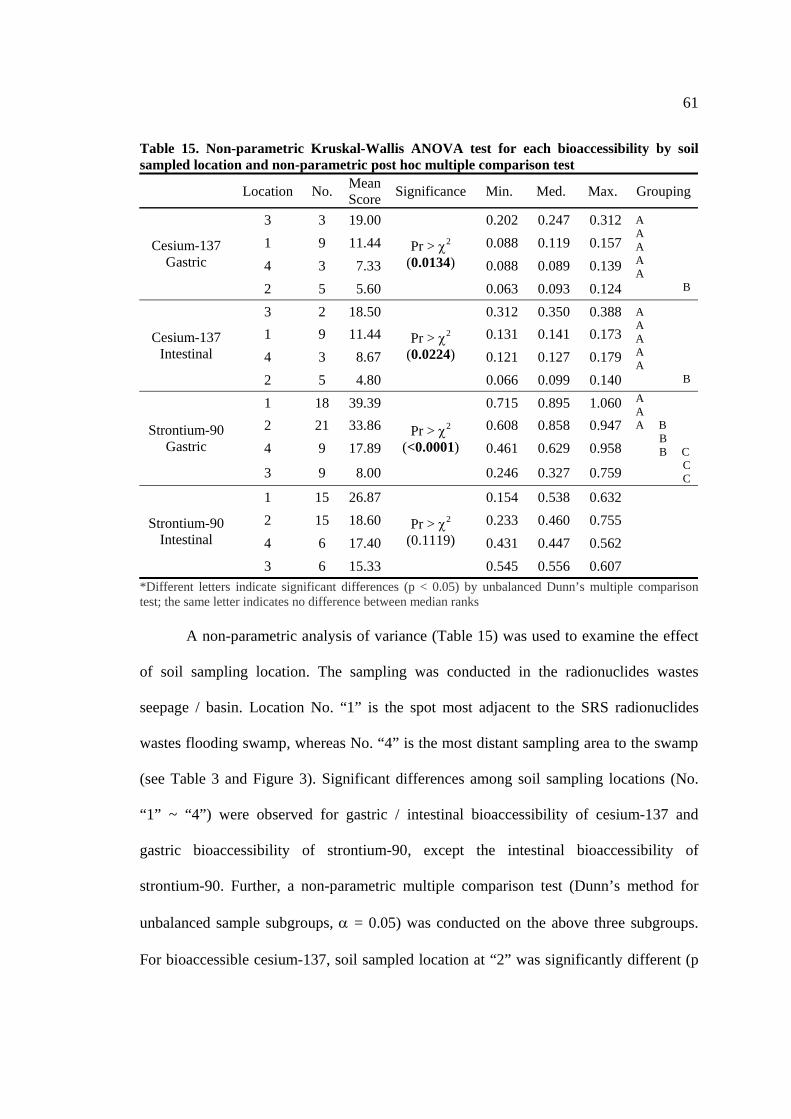
61
Table 15. Non-parametric Kruskal-Wallis ANOVA test for each bioaccessibility by soil sampled location and non-parametric post hoc multiple comparison test
Location No. Mean Score Significance Min. Med. Max. Grouping
3 3 19.00 0.202 0.247 0.312 1 9 11.44 0.088 0.119 0.157 4 3 7.33 0.088 0.089 0.139
Cesium-137 Gastric
2 5 5.60
Pr > χ2 (0.0134)
0.063 0.093 0.124
A A A A A B
3 2 18.50 0.312 0.350 0.388 1 9 11.44 0.131 0.141 0.173 4 3 8.67 0.121 0.127 0.179
Cesium-137 Intestinal
2 5 4.80
Pr > χ2 (0.0224)
0.066 0.099 0.140
A A A A A B
1 18 39.39 0.715 0.895 1.060 2 21 33.86 0.608 0.858 0.947 4 9 17.89 0.461 0.629 0.958
Strontium-90 Gastric
3 9 8.00
Pr > χ2 (<0.0001)
0.246 0.327 0.759
A A A B B B C C C
1 15 26.87 0.154 0.538 0.632 2 15 18.60 0.233 0.460 0.755 4 6 17.40 0.431 0.447 0.562
Strontium-90 Intestinal
3 6 15.33
Pr > χ2 (0.1119)
0.545 0.556 0.607
*Different letters indicate significant differences (p < 0.05) by unbalanced Dunn’s multiple comparison test; the same letter indicates no difference between median ranks
A non-parametric analysis of variance (Table 15) was used to examine the effect
of soil sampling location. The sampling was conducted in the radionuclides wastes
seepage / basin. Location No. “1” is the spot most adjacent to the SRS radionuclides
wastes flooding swamp, whereas No. “4” is the most distant sampling area to the swamp
(see Table 3 and Figure 3). Significant differences among soil sampling locations (No.
“1” ~ “4”) were observed for gastric / intestinal bioaccessibility of cesium-137 and
gastric bioaccessibility of strontium-90, except the intestinal bioaccessibility of
strontium-90. Further, a non-parametric multiple comparison test (Dunn’s method for
unbalanced sample subgroups, α = 0.05) was conducted on the above three subgroups.
For bioaccessible cesium-137, soil sampled location at “2” was significantly different (p

62
< 0.05) than the values obtained for the other sampling locations (“3”, “1”, and “4”).
However, the difference (p < 0.05) was found between sampled location “1-4”, “1-3”,
and “2-3” for gastric bioaccessibility of strontium-90. This analysis showed that a
variable such as soil sampling location could affect the bioaccessibility of two
radionuclides, and emphasized the importance of collecting site-specific data for risk
assessments.
Table 16. The Spearman rank-order correlation tests between gastric / intestinal bioaccessibility and seven variables (radioactivity, sampling depth, sampling location, SOM, CEC, pH, and Clay)
Radio-activity Depth Location SOM CEC pH Clay
137Cs Gastric
0.1286 (0.5890)
N=20
-0.0323 (0.8924)
N=20
-0.0383 (0.8727)
N=20
-0.4408 (0.1748)
N=11
-0.2569 (0.4457)
N=11
-0.1285 (0.7066)
N=11
-0.2752 (0.4127)
N=11
137Cs Intestinal
0.2022 (0.4064)
N=19
0.33040 (0.1671)
N=19
-0.1042 (0.6711)
N=19
-0.7553 (0.0115)
N=10
-0.5138 (0.1287)
N=10
-0.4159 (0.2319)
N=10
-0.2936 (0.4103)
N=10
90Sr Gastric
0.6320 (0.0001)
N=57
0.2888 (0.0294)
N=57
-0.6032 (0.0001)
N=57
0.2000 (0.2222)
N=39
0.3862 (0.0152)
N=39
0.5779 (0.0001)
N=39
0.4130 (0.0090)
N=39
90Sr Intestinal
0.2872 (0.0686)
N=41
0.4297 (0.0050)
N=41
-0.3613 (0.0203)
N=41
-0.5524 (0.0034)
N=26
-0.1637 (0.4241)
N=26
0.4104 (0.0373)
N=26
-0.0767 (0.7096)
N=26
Associations between bioaccessibility and other characteristic variables in the
tested soils were examined using a non-parametric Spearman statistical test (Table 16).
For gastric bioaccessible cesium-137, there were no significant associations with any of
the seven soil characteristic variables. For intestinal bioaccessible cesium-137, SOM
percentage in soil was the only factor correlated with the bioaccessibility (p = 0.0115),
and that was a negative correlation. Clay is a known absorbent for cesium-137 and causes
limited dissolution of cesium-137 in the extraction system (Ellickson et al., 2002). It was
expected that the clay factor would have the negative relationship between cesium-137

63
bioaccessibility and clay contents; but there was no significant correlation. The clay
content of each soil sample was minimal (below 1 %), therefore, the association between
clay content and bioaccessible cesium-137 was not found in these samples.
Conversely, both gastric and intestinal bioaccessible strontium-90 had statistically
significant associations with the following variables: sampling depth (p = 0.0294, 0.0050),
sampling location (p = 0.0001, 0.0203), and soil pH (p = 0.0001, 0.0373), respectively.
Since the soil distribution coefficient (Kd) increased with soil pH, it could be expected
that a negative association between strontium-90 bioaccessibility and soil pH would exist.
Increasing soil pH would cause cation extraction decrease, due to more binding of
strontium-90 with soil (Brady et al., 1995). However, correlation coefficient for the
bioaccessibility and soil pH for strontium-90 in seepage / basin soils was positive. A key
observation here is the positive correlations between total strontium-90 and bioaccessible
strontium-90 in gastric phase (coefficient of 0.6320; p = 0.0001) as well as soil pH
(coefficient of 0.57143; p = 0.1390). This result was associated with the relatively high
solubility of strontium-90 in gastric fluid (95.2 ~ 60.1 %) and therefore, shows little
variation with soil pH value. The index number (No. “1” ~ “4”) for soil sampling depth
and location was inversely correlated with the radioactivity of strontium-90 in soil. Total
radioactivity of strontium-90 was positively correlated with the bioaccessible strontium-
90 (coefficient of 0.36189; p = 0.0003). Therefore, the negative association of sampling
location with bioaccessible strontium-90 was associated with the higher radioactive soil.
However, a positive association between sampling depth and bioaccessible strontium-90
might be explained by the soil characteristics, especially the clay content in each soil. The

64
correlation between intestinal bioaccessibility of strontium-90 and clay contents was
negative (-0.0767), however, not significant (p = 0.7096).
2.4.4. Exposure / dose estimation
Two radionuclides exposure and dose estimations were completed on three soil
hazard classes for four hypothetical population groups by the duration of short-term and
long-term exposure. Three exposure pathways (ingestion of soil, inhalation of re-
suspended soil, and external radiation exposure from soil) were considered to estimate
exposure / dose predictions for each population group. Two exposures (ingestion and
inhalation) and three dose levels were projected along with each hypothetical group’s
physiological condition and scenario using the Monte Carlo simulation of Crystal Ball®
program.
The estimated exposure (potential dose) values for cesium-137 and strontium-90
from ingestion / inhalation pathways are presented in Table 18 for three soil classes and
four sub-groups. For each route of exposure, the median and 95 percentile for estimates
of potential dose to cesium-137 and strontium-90 are provided in the table. The potential
dose estimations are given as intake radiation (pCi) per body weight (kg) over time (day).
These estimated potential dose values for two radionuclides could be compared with the
allowable regulatory values. For example, EPA has established a Maximum Contaminant
Level (MCL) of 4 millirem per year from man-made radionuclides in drinking water. The
average concentration for equivalent cesium-137 is 200 picocuries per liter (pCi/L), and
the average concentration for equivalent strontium-90 is 8 pCi/L (EPA, 1993). Thus, the
MCL was converted to the same unit of calculated potential dose of ingestion (pCi/kg-

65
day) for two radionuclides, and provided in Table 17. For cesium-137, the potential dose
estimates surpassed the converted MCL for pica, 2.5, and 6 years old children on class
“A” soil; however, the other estimates were below the MCL. In case of strontium-90, the
potential dose estimates were above the converted MCL for pica and 2.5-year old
children on three soil classes as well as 6-year old children and adults on class “A” soil.
Table 17. The age-specific variables (dinking rate and body weight) and converted MCL for cesium-137 and strontium-90 respect to each age group
2.5-year-old Children
6.0-year-old Children Adults
Drinking* (L/day) 0.61 0.74 1.4
Weight* (kg) 14.3 22.6 72.8
Cesium-137 (pCi/kg-day) 8.53 6.55 3.85
Strontium-90 (pCi/kg-day) 0.34 0.26 0.15
*: Taken from U.S. EPA Exposure Factor Handbook, 1999
The result for estimated potential dose levels showed large differences among
class “A” soil and other soil classes (“B” and “C”). The ratio of potential dose for
cesium-137 ranged from 131 to 134 for soil class “A” / “B” and 51 to 52 for soil class
“A” / “C”, respectively. The ratio of soil class (“A” / “B” and “A” / “C”) for strontium-90
dropped to 13 and 17, respectively. The higher proportion of radionuclide potential dose
for pica and young children resulted from non-dietary ingestion rather than inhalation.
For pica children, especially the estimated potential doses were approximately five times
higher than non-pica children in the same age group (2.5 years old). The higher potential
dose values would be a result of children’s pica activity, yielding the differences in the
amount of soil ingested for pica children, 500 mg/day versus 2.5 years old non-pica
children, 110 mg/day. The elevated potential dose levels resulted from high radioactivity

66
of soil (soil hazard class of “A”), children’s specific activity (pica or hand-to-mouth), and
exposure pathway (non-dietary ingestion).
The biologically effective dose values were estimated based on the radioactivity
of each soil class, age-specific exposure variables, and durations. The predicted short-
term (two weeks duration) effective doses for cesium-137 and strontium-90 are presented
in Table 19 and 20, respectively. For each target radionuclide, the median and 95
percentile were provided for three major exposure pathways (ingestion, inhalation, and
external exposure) to the contaminated soil and subsequent total dose. The median and 95
percentile values were descriptive statistics and approached the maximum probable
values for radionuclides effective dose for age-specific groups, respectively. The
exposure duration was extended to one-year in residence (as a long-term exposure) for
Chernobyl residents, and the calculated effective dose estimates for cesium-137 and
strontium-90 are found in Table 21 and Table 22, respectively.
The NCRP, in considering the risks associated with low-level radiation exposures,
has recommended that there be some level of exposure below which no individual need
be concerned. This is called “Negligible Individual Dose” (NID). A value of 1.0 millirem
(0.01 mSv) in a year has been recommended as constituting a “boundary below which the
dose can be dismissed from consideration” (NCRP Report No. 116, 1993). Currently
NCRP recommends that “when the annual effective dose limit as a result of irradiation
attributable to a single site, the site operator should ensure that the annual exposure of the
maximally exposed individual, from all man-made exposure (excepting that individual’s
medical exposure), the dose should not exceed 1 mSv on a continuous basis” (NCRP
Report No. 116, 1993).

67
For short-term exposure to cesium-137 and strontium-90, the effective dose
estimates were below 0.01 mSv level - a negligible individual dose by the NCRP- with
the exception of pica children on class “A” soil for strontium-90. Due to the short
duration of two weeks, the cumulative amount of cesium-137 and strontium-90 was not
high enough to impact on the health of visitors.
The results for total dose calculated changed greatly for the example of long-term
exposure to these radionuclides. Total dose of cesium-137 and strontium-90 was nearly
26X higher than that of short-term exposure. For cesium-137, total doses of all sub-
groups on class “A” soil were over the 0.01 mSv level. Effective doses resulted from both
ingestion and external exposure contributed to exceed the level for pica / young children;
however, for older children and adults, effective dose from external exposure was enough
to surpass the level of 0.01 mSv. In the case of strontium-90, effective dose estimates for
pica children on three soil classes and young children on class “A” soil exceeded the
NCRP NID (0.01 mSv). It can be concluded that the effective dose for strontium-90 via
ingestion was a dominant exposure pathway, and yielded the total dose easily surpassed
the level of NCRP negligible individual dose.
Effective dose predictions were based on three hazard classes for radioactive-
contaminated soils with three exposure routes (ingestion, inhalation, and external
exposure) and two exposure scenarios (two week vacation and one year residence). The
effective dose contributed by each exposure pathway to total dose was different for each
exposed group and target radionuclide. For cesium-137, contribution of external radiation
to total dose was smaller than that of non-dietary soil ingestion for pica children (below
20 %) due to their very high amount of soil ingestion (500 mg per day). However,

68
contribution of external exposure to total dose was equal to or slightly larger than that of
ingestion to total dose for young children. The contribution of external exposure
outweighed the portion of non-dietary soil ingestion for older children and adults (over
70 %). For strontium-90, the contribution of ingestion to total dose was dominant for pica
/ young children (over 95 %), and reduced greatly for older children and adults (below
25 %). Due to high ingestion rate for pica and young children, non-dietary ingestion was
a primary exposure route for both radioactive contaminants. However, for older children
and adults, external radiation was a major exposure pathway from radioactive-
contaminated soil. The contribution of inhalation to total dose was very low (below 4 %
of contribution to total dose) for all exposure scenarios and soil classes.
Among four hypothetical population groups, pica children on class “A” soil were
a worst case for effective dose predictions. Their effective doses were close to the NCRP
acceptable dose limit (1 mSv) for public health. The NCRP considers 1 mSv dose as an
acceptable dose limit for continuous exposure to the public (NCRP, 1987, 1993). For
long-term exposure, there was a distinct gap between total doses associated with high-
level soil (class “A”) versus low-level soil (class “B” and “C”). The high-level
radionuclide effective doses were larger than the low-level effective doses by the
magnitude of 15 to 150X reflecting the high radiation emitted from class “A” soils. The
greater intestinal bioaccessibility contributed to the increased radiation dose for
strontium-90 as compared to cesium-137. For example, the total cesium-137 radiation in
class “A” soil was much higher, approximately 13 times, than the values for strontium-90.
However, the effective dose estimations for young children based upon class “A” soils
showed an opposite pattern: namely, the estimated effective doses calculated for

69
strontium-90 were two times higher than those of cesium-137. This was caused by the
much higher bioaccessibility of strontium-90 (59.3 % bioaccessible in intestinal phase)
versus cesium-137 (13.8 % of intestinal bioaccessibility) and the difference in effective
dose coefficient of 9.1*10-8 Sv/Bq for strontium-90 versus 1.1*10-8 Sv/Bq for cesium-
137 for young children.
Pica children and 2.5-year-old children, residing on highly radioactive-
contaminated sites such as Chernobyl Nuclear Power Plant Exclusion Zone and
Resettlement Zone at least one year, would be at higher risk of exposure to cesium-137
and strontium-90 than adults or older children. This would be their activities (i.e., hand-
to-mouth activity, more mobility than younger children) and potential higher possibilities
of soil ingestion. Further, the calculated risk to the general population (median value)
would be above the negligible dose (0.01 mSv) for cesium-137 as a result of exposure to
very high radioactive-contaminated soils (e.g., the surface soil of ChNPP Exclusion Zone)
for one year. Similarly pica children would be above the negligible dose established by
NCRP (0.01 mSv) for strontium-90, if they lived on the highly radioactive-contaminated
soils in the ChNPP Exclusion Zone or Resettlement Zone for one year.

70
Table 18. Potential dose estimates (50 % & 95 %) from ingestion and inhalation pathways for Cesium-137 and Strontium-90
Cesium-137 (pCi/kg-day) Strontium-90 (pCi/kg-day) Subgroup Soil
Class Ingestion Inhalation Ingestion Inhalation
A 310.703 418.487
4.24E-02 5.67E-02
23.508 31.415
3.19E-03 4.25E-03
B 2.337 3.113
3.18E-04 4.26E-04
1.668 2.227
2.27E-04 3.01E-04
Pica Children
C 5.953 7.992
8.12E-04 1.08E-03
1.390 1.860
1.89E-04 2.52E-04
A 68.361 92.297
4.22E-02 5.66E-02
5.147 6.943
3.19E-03 4.23E-03
B 0.515 0.687
3.17E-04 4.25E-04
0.365 0.491
2.26E-04 3.04E-04
2.5-Year Children
C 1.312 1.760
8.12E-04 1.09E-03
0.305 0.407
1.89E-04 2.53E-04
A 8.699 11.606
3.95E-02 5.28E-02
0.650 0.871
2.96E-03 3.94E-03
B 0.065 0.087
2.96E-04 3.93E-04
0.046 0.062
2.12E-04 2.82E-04
6.0-Year Children
C 0.166 0.221
7.57E-04 1.01E-03
0.039 0.052
1.76E-04 2.35E-04
A 2.495 3.310
1.86E-02 2.49E-02
0.188 0.250
1.39E-03 1.86E-03
B 0.019 0.025
1.40E-04 1.86E-04
0.013 0.018
9.95E-05 1.33E-04 Adults
C 0.048 0.064
3.57E-04 4.76E-04
0.011 0.015
8.31E-05 1.11E-04
*Bold means the ingestion estimate is above the EPA’s MCL (maximum contamination level) from man-made radionuclides in drinking water

71
Table 19. Effective dose estimates (50 % & 95 %) for cesium-137 from short-term exposure
Subgroup Soil Class
Ingestion (mSv)
Inhalation (mSv)
External (mSv)
Total (mSv)
A 3.47*10-3 4.79*10-3
1.69*10-6 2.24*10-6
9.64*10-4 1.21*10-3
4.44*10-3 5.85*10-3
B 2.25*10-5 3.09*10-5
1.26*10-8 1.67*10-8
7.23*10-6 9.11*10-6
2.98*10-5 3.90*10-5
Pica Children
C 1.39*10-4 1.92*10-4
3.23*10-8 4.31*10-8
1.84*10-5 2.32*10-5
1.57*10-4 2.13*10-4
A 7.62*10-4 1.06*10-3
1.69*10-6 2.24*10-6
9.62*10-4 1.20*10-3
1.73*10-3 2.17*10-3
B 4.96*10-6 6.83*10-6
1.27*10-8 1.69*10-8
7.23*10-6 9.05*10-6
1.22*10-5 1.53*10-5
2.5-Year Children
C 3.04*10-5 4.19*10-5
3.22*10-8 4.27*10-8
1.85*10-5 2.31*10-5
4.90*10-5 6.28*10-5
A 1.25*10-4 1.73*10-4
1.65*10-6 2.20*10-6
6.77*10-4 8.51*10-4
8.06*10-4 9.98*10-4
B 8.12*10-7 1.12*10-6
1.24*10-8 1.62*10-8
5.08*10-6 6.39*10-6
5.91*10-6 7.35*10-6
6.0-Year Children
C 4.98*10-6 6.91*10-6
3.17*10-8 4.18*10-8
1.30*10-5 1.64*10-5
1.81*10-5 2.25*10-5
A 1.67*10-4 2.30*10-4
3.21*10-6 4.25*10-6
5.44*10-4 6.88*10-4
7.16*10-4 8.89*10-4
B 1.09*10-6 1.50*10-6
2.40*10-8 3.18*10-8
4.09*10-6 5.11*10-6
5.21*10-6 6.43*10-6 Adults
C 6.66*10-6 9.18*10-6
6.15*10-8 8.15*10-8
1.04*10-5 1.31*10-5
1.72*10-5 2.14*10-5

72
Table 20. Effective dose estimates (50 % & 95 %) for strontium-90 from short-term exposure
Subgroup Soil Class
Ingestion (mSv)
Inhalation (mSv)
External (mSv)
Total (mSv)
A 9.31*10-3 1.24*10-2
2.59*10-6 3.41*10-6
6.81*10-5 8.55*10-5
9.38*10-3 1.25*10-2
B 5.34*10-4 7.14*10-4
1.84*10-7 2.42*10-7
4.86*10-6 6.05*10-6
5.39*10-4 7.19*10-4
Pica Children
C 3.77*10-4 5.03*10-4
1.53*10-7 2.02*10-7
4.03*10-6 5.04*10-6
3.81*10-4 5.08*10-4
A 2.04*10-3 2.73*10-3
2.58*10-6 3.42*10-6
6.78*10-5 8.53*10-5
2.11*10-3 2.81*10-3
B 1.18*10-4 1.56*10-4
1.84*10-7 2.45*10-7
4.84*10-6 6.08*10-6
1.23*10-4 1.62*10-4
2.5-Year Children
C 8.31*10-5 1.10*10-4
1.53*10-7 2.03*10-7
4.03*10-6 5.07*10-6
8.74*10-5 1.15*10-4
A 1.85*10-5 2.46*10-5
2.25*10-6 2.97*10-6
4.79*10-5 6.01*10-5
6.86*10-5 8.48*10-5
B 1.06*10-6 1.41*10-6
1.60*10-7 2.11*10-7
3.41*10-6 4.28*10-6
4.64*10-6 5.72*10-6
6.0-Year Children
C 7.49*10-7 9.94*10-7
1.33*10-7 1.76*10-7
2.85*10-6 3.56*10-6
3.74*10-6 4.60*10-6
A 1.46*10-5 1.95*10-5
1.89*10-6 2.50*10-6
3.83*10-5 4.80*10-5
5.50*10-5 6.76*10-5
B 8.40*10-7 1.12*10-6
1.35*10-7 1.79*10-7
2.74*10-6 3.44*10-6
3.72*10-6 4.59*10-6 Adults
C 5.93*10-7 7.91*10-7
1.12*10-7 1.48*10-7
2.28*10-6 2.87*10-6
3.00*10-6 3.70*10-6
*Bold number means the estimate is above the NCRP negligible annual dose level (0.01 mSv)

73
Table 21. Effective dose estimates (50 % & 95 %) for cesium-137 from long-term exposure
Subgroup Soil Class
Ingestion (mSv)
Inhalation (mSv)
External (mSv)
Total (mSv)
A 9.02*10-2 1.24*10-1
4.38*10-5 5.80*10-5
2.51*10-2 3.15*10-2
1.15*10-1 1.52*10-1
B 5.86*10-4 8.14*10-4
3.30*10-7 4.39*10-7
1.88*10-4 2.36*10-4
7.76*10-4 1.02*10-3
Pica Children
C 3.59*10-3 5.00*10-3
8.40*10-7 1.11*10-6
4.81*10-4 6.03*10-4
4.08*10-3 5.52*10-3
A 1.98*10-2 2.74*10-2
4.39*10-5 5.81*10-5
2.51*10-2 3.15*10-2
4.50*10-2 5.64*10-2
B 1.29*10-4 1.79*10-4
3.29*10-7 4.35*10-7
1.89*10-4 2.36*10-4
3.19*10-4 3.98*10-4
2.5-Year Children
C 7.90*10-4 1.10*10-3
8.43*10-7 1.12*10-6
4.82*10-4 6.01*10-4
1.28*10-3 1.65*10-3
A 3.24*10-3 4.48*10-3
4.30*10-5 5.73*10-5
1.77*10-2 2.22*10-2
2.10*10-2 2.61*10-2
B 2.11*10-5 2.90*10-5
3.22*10-7 4.26*10-7
1.32*10-4 1.66*10-4
1.54*10-4 1.92*10-4
6.0-Year Children
C 1.30*10-4 1.80*10-4
8.24*10-7 1.09*10-6
3.39*10-4 4.24*10-4
4.71*10-4 5.84*10-4
A 4.34*10-3 5.98*10-3
8.39*10-5 1.11*10-4
1.42*10-2 1.78*10-2
1.87*10-2 2.31*10-2
B 2.83*10-5 3.93*10-5
6.24*10-7 8.27*10-7
1.06*10-4 1.33*10-4
1.36*10-4 1.68*10-4 Adults
C 1.74*10-4 2.40*10-4
1.60*10-6 2.11*10-6
2.72*10-4 3.41*10-4
4.49*10-4 5.58*10-4
*Bold number means the estimate is above the NCRP negligible annual dose level (0.01 mSv)

74
Table 22. Effective dose estimates (50 % & 95 %) for strontium-90 from long-term exposure
Subgroup Soil Class
Ingestion (mSv)
Inhalation (mSv)
External (mSv)
Total (mSv)
A 2.43*10-1 3.24*10-1
6.73*10-5 8.86*10-5
1.77*10-3 2.22*10-3
2.45*10-1 3.26*10-1
B 1.39*10-2 1.85*10-2
4.81*10-6 6.36*10-6
1.26*10-4 1.59*10-4
1.40*10-2 1.87*10-2
Pica Children
C 9.84*10-3 1.31*10-2
4.00*10-6 5.30*10-6
1.05*10-4 1.32*10-4
9.95*10-3 1.32*10-2
A 5.35*10-2 7.17*10-2
6.72*10-5 8.90*10-5
1.77*10-3 2.23*10-3
5.53*10-2 7.37*10-2
B 3.06*10-3 4.08*10-3
4.79*10-6 6.33*10-6
1.26*10-4 1.58*10-4
3.19*10-3 4.23*10-3
2.5-Year Children
C 2.16*10-3 2.86*10-3
3.99*10-6 5.29*10-6
1.05*10-4 1.32*10-4
2.27*10-3 2.99*10-3
A 4.82*10-4 6.41*10-4
5.85*10-5 7.72*10-5
1.25*10-3 1.57*10-3
1.80*10-3 2.20*10-3
B 2.77*10-5 3.69*10-5
4.17*10-6 5.55*10-6
8.93*10-5 1.12*10-4
1.21*10-4 1.49*10-4
6.0-Year Children
C 1.96*10-5 2.59*10-5
3.46*10-6 4.59*10-6
7.39*10-5 9.30*10-5
9.71*10-5 1.20*10-4
A 3.79*10-4 5.07*10-4
4.93*10-5 6.52*10-5
1.00*10-3 1.25*10-3
1.43*10-3 1.76*10-3
B 2.19*10-5 2.91*10-5
3.52*10-6 4.65*10-6
7.12*10-5 8.94*10-5
9.68*10-5 1.19*10-4 Adults
C 1.54*10-5 2.05*10-5
2.93*10-6 3.89*10-6
5.93*10-5 7.46*10-5
7.78*10-5 9.61*10-5
*Bold number means the estimate is above the NCRP negligible annual dose level (0.01 mSv)

75
III. Bioaccessible Lead in Carpet House Dust
3.1. Introduction
House dust has been investigated to identify a proximate source of toxicants and
quantify levels of toxicants in an exposure pathway that can be used for the estimation of
human exposure in indoor residential settings (Lioy et al., 2002). Toxicants present in
house dust include semi-volatile and non-volatile pesticides, PAHs (polyaromatic
hydrocarbons), heavy metals, asbestos, and persistent organic compounds (Lioy et al.,
2002). The specific information has been obtained from measurements of total level of
each toxicant in house dust. The values were represented as dust loading (µg/ft2, µg/cm2)
or dust concentration (µg/g) collected by wipe and vacuum sampling (Lioy et al., 2002).
Since the ingestion of house dust can be a major route of children’s non-dietary exposure
to lead (Thornton et al., 1990), data on the bioaccessible fraction of toxicants, like lead,
present in house dust matrices can provide valuable new information on the exposure. It
should also be a better indicator of the dose available to an individual or population (e.g.,
young children) from house dust.
House dust contaminated with lead is currently a primary source of childhood
non-dietary exposure to lead in the United States (Lanphear ea al., 1996; Rhoads et al.,
1999). Hand-to-mouth activity is generally considered as the most important lead
exposure pathway for young children (EPA, 1995). It is well known that children are
especially vulnerable to the effects caused by lead exposure because their rapidly
developing nervous systems are particularly sensitive to lead (ATSDR, 1999). Moreover,
lead exposure to a fetus has been correlated with slow mental development and lower
intelligence later in life (ATSDR, 1999). Thus, exposure to lead can have a wide range of

76
effects on children’s development and behavior (EPA, 2001). Considering that after
exposure to small amounts of lead levels, children may appear inattentive, hyperactive,
and irritable. Children with high lead levels can have problems with learning and reading,
and hearing loss. At high levels, lead exposures can cause permanent brain damage and
even death.
A method for measuring bioaccessible lead in house dust used in this study was
derived from a bioaccessibility method developed for heavy metals present in
contaminated soils (Hamel et al., 1999; Ellickson et al., 2001). The laboratory of EMAD-
EOHSI has successfully developed the bioaccessibility method for heavy metals (Pb, As,
Cr, and Cd) and low-level radionuclides (137Cs and 90Sr) in various soil samples. The
method was modified for use with a different sample matrix (vacuumed house dust for in
vitro extraction). The target for the current study was bioaccessible lead in house dust,
and the levels were measured in both simulated gastric and intestinal fluids.
Based upon previous research, the levels of bioaccessible lead in vacuumed house
dust (sieved to < 75 µm) were expected to be different from lead levels obtained after
total extraction of lead from each sample (Ruby et al., 1992, 1993, 1996). Previous lead
dissolution studies showed a significant difference in bioaccessible lead levels at different
pH conditions for household dust (Oliver et al., 1999), soil (Ellickson et al., 2001; Ruby
et al., 1992, 1996; Oomen et al., 2002), and slagged aggregate (Davis et al., 1997). To
quantify the bioaccessible lead in artificially developed human fluids in the stomach and
small intestine, the bioaccessibility of lead was measured in simulated gastric and
intestinal fluids.

77
It was also expected that lead present in smaller particles had higher levels of
bioaccessibility than lead associated with larger dust particles. Barltrop and Meek (1979)
examined the relationship between lead particle size and absorption from the
gastrointestinal tract of rats, and found an inverse relationship between particle size and
lead absorption. Studies have shown that lead is generally more concentrated in the fine
fraction of dust (Duggan et al., 1985), and lead absorption into the body is inversely
related to particle size (Barltrop et al., 1979). Empirical evidence, however, has not
necessarily shown that collection and analysis of the lead content of a fine fraction of
dust (diameter sized below 250 µm) is a better predictor of children’s blood lead level
than total dust (EPA, 1995).
The size fraction of house dust that most readily adheres to a hand is less than 200
µm (Duggan et al., 1985) or 246 µm (Que Hee et al., 1985). Driver et al. (1989) dry-
sieved five soil types to obtain two size fractions of particles, one less than 250 µm and
the other less than 150 µm. The results showed that the finer soil particles adhere more
readily to hands than do coarse particles. Edwards et al. (1999) also demonstrated that
particles less than 100 µm adhere to hands longer than particles greater than 100 µm, and
Rodes et al. (2001) determined that particle size of dust that adheres best to the hand is
typically less than 40 µm. To link the bioaccessibility of lead with particle size fractions,
the bioaccessible lead in two biofluids and total lead in house dust was measured in three
particle size ranges: < 75 µm, 75-150 µm, and 150-250 µm.

78
3.2. Background
3.2.1. Lead
Lead is a naturally occurring bluish-gray metal found in small amounts in the
earth’s crust. Lead occurs naturally in the environment. However, most of the high levels
found throughout the environment come from human activities. Before leaded gasoline
was banned in the United States, most of the lead released into the environment came
from car exhaust. Other sources of lead released to the air include burning fuel, such as
coal or oil, industrial processes, and burning solid waste.
Once lead is released into the atmosphere, it may travel thousands of miles if the
lead is on small particles or if the lead compounds (e.g., organo lead vapors such as lead
alkyls) easily evaporate (ATSDR, 1999). Lead is removed from the air by rain and by
particles depositing to the ground or onto surface water. The release of lead to the air is
now lower than the ground of lead that has been deposited on / in the soil or dust
(ATSDR, 1999). Most of the lead found in inner city soils comes from both houses with
deteriorated paint containing lead and lead deposited in soil from automotive exhaust
emitted when gasoline contained lead (ATSDR, 1999). Higher levels of lead in soil can
be measured near roadways. This accumulation came from previous car exhaust
emissions. Once lead settles onto soil, it sticks to soil particles; however, it can be
redistributed slowly due to resuspension.
Lead can affect almost every organ and system in the human body (ATSDR,
1999). The most sensitive is the central nervous system, particularly in children. Lead
also damages kidneys and the reproductive system. The effects are the same whether it is
breathed or swallowed. At high levels, lead may decrease reaction time, cause weakness

79
in fingers, wrists, or ankles, and possibly affect the memory. Lead may also cause
anemia, a disorder of the blood, as well as damage the male reproductive system. The
connection between these effects and exposure to low levels of lead is uncertain (ATSDR,
1999).
Young children (< 6 years old) can be exposed to lead by eating lead-based paint
chips, chewing on objects painted with lead-based paint, or swallowing house dust or soil
that contains lead (ATSDR, 1999). A child who swallows large amounts of lead-
contaminated soil or dust may develop blood anemia, severe stomachache, muscle
weakness, and brain damage. Even at much lower levels of exposure, lead can affect a
child’s mental and physical growth. Unborn children can be exposed to lead through their
mothers (ATSDR, 1999). Lead can easily cross the placenta; therefore, exposure of
women to lead during pregnancy results in uptake by fetus. Prenatal exposure may be
related to postnatal mental retardation, impaired postnatal neurobehavioral development,
and reduced birth weight and gestational age (ATSDR, 1986).
Lead is ubiquitous in the urban environment, and has detrimental impacts on
human health. Elevated levels of lead are found in house dust; the source is mostly from
the tacked-in soil that is contaminated with lead (EPA, 1995). The ingestion of lead-
contaminated house dust by young children as a result of hand-to-mouth and object-to-
mouth behavior is recognized as an important pathway of lead exposure to children
(Lanphear et al., 1996; Rhoads et al, 1999). For these reasons, lead was chosen as the
target contaminant of concern to evaluate the bioaccessibility of a heavy metal in house
dust obtained from urban residences.

80
3.2.2. IEUBK model5
The Integrated Exposure Uptake Biokinetic Model for Lead in Children (IEUBK;
EPA, 2002) is used to predict blood lead concentrations (PbBs) for children exposed to
lead in their environment. The IEUBK model allows the user to input relevant absorption
parameters (e.g., the fraction of lead absorbed from soil / house dust) as well as intake
and exposure rates. Using these input values, the model estimates the potential
concentration of lead in the blood for a hypothetical child or population of children (6
months to 7 years of age). Measured lead concentration is not only an indication of
exposure, but also a widely-used index for discerning future health problems. The U.S.
EPA and U.S. Center for Disease Control (CDC) have determined that childhood PbB
concentrations at or above the 10 µg/dL are present risks to children’s health. The
IEUBK model calculates the probability that children’s PbB concentrations will exceed
10 µg/dL (or other user entered value). By varying the data entered into the model, the
user can evaluate how changes in environmental conditions may affect PbB levels in
exposed children.
The IEUBK model is designed to predict the probable PbB concentrations for
children between 6 months and 7 years of age who have been exposed to lead through
environmental media (air, water, soil, dust, and diet). The model has the following 4
functional components:
Exposure Component compares lead concentrations in environmental media with the
amount of lead entering a child’s body. The exposure component uses environmental
5 The text was excerpted from EPA website (http://www.epa.gov/superfund/programs/lead/ieubk.htm)

81
media-specific consumption rates and lead concentrations to estimate media-specific lead
intake rates.
Uptake Component compares lead intakes into the lungs or digestive tract with the
amount of lead absorbed into child’s blood.
Biokinetic Component shows the transfer of lead between blood and other body tissues,
or the elimination of the lead from the body altogether.
Probability Distribution Component shows the probability of certain outcome (e.g., a
PbB concentration greater than 10 µg/dL in an exposed child based on the parameters
used in the model).
The IEUBK model attempts to standardize exposure by assuming age-weighted
parameters for intake of food, water, soil, and dust. The model simulates continual
growth under constant exposure levels (on a year-to-year basis). The model also
simulates lead uptake, distribution within the body, and elimination of the body. It is
commonly thought that the IEUBK model can predict the average PbB level for an entire
community. This is misleading since many factors can vary between homes within a
single community. Instead of using community means for the environmental lead inputs,
it would be more accurate to apply the IEUBK model to individual homes or
homogeneous areas, then combine the results in order to find the mean for a
neighborhood or community. However, for individuals at risk, an individual estimate of
exposure and risk would be the appropriate approach.
The IEUBK model is intended to estimate a typical child’s long-term exposure to
lead in or out his / her residence;

82
• to provide an accurate estimate of the geometric average PbB concentrations for a
typical child aged from 6 months to 7 years
• to provide a basis for estimating the risk of elevated PbB concentrations for a
hypothetical child
• to predict likely changes in the risk of elevated PbB concentrations from exposure
to soil, dust, water, or air following concerted action to reduce such exposure
• to provide assistance in determining target cleanup levels at specific residential
sites for soil or dust containing high amounts of lead
• to provide assistance in estimating PbB levels associated with the lead
concentrations of soil or dust at undeveloped sites which may be developed at a
later date.
The site-specific risk assessments require information on soil and dust lead
concentrations for the particular location in question. Variables affecting any
consideration of lead exposure from soil and dust include: ingestion parameters for soil
and dust (e.g., how much soil or dust a typical child may ingest or inhale over a set of
period time); and the amount of lead that can be absorbed from the soil. These parameters
are quite sensitive, that is, changing one variable can significantly affect the results. The
IEUBK model is designed to facilitate calculating the elevated risk of PbB levels, and is
helpful in demonstrating how results may change when the user enters different
parameters. Overall, the IEUBK model is a tool for assessing PbB concentration in
children exposed to lead. Its advantage is that it considers the several different media
which children can come in contact with doing their days.

83
3.3. Methods
3.3.1. Test house dusts
All house dust samples were obtained from 50 homes participated in the ECSC
(Evaluation of Carpet Dry Steam Cleaning) project which was sponsored by U.S. HUD,
Department of Housing and Urban Development. The ECSC study was designed to
evaluate two different cleaning methods, HEPA vacuum only vs. HEPA vacuum + dry
steaming, for their effectiveness in removing lead and other toxicants from participant’s
carpets. Samples were taken, before and after each home was cleaned, and then the
samples were analyzed for lead and other materials in the house dust. Carpets were
located in typical northern New Jersey residences on the period of July 2003 to June 2004.
A participant had to have at least one child diagnosed with a blood lead level higher than
15 µg/dL, which had been reported to State Health Department from 2000 to 2002. Dust
samples were collected in dust mite allergen bag of HEPA vacuum cleaner (HEPA
Vacuum Cleaner Upright, Hoover, Newton, IA) from the carpets. All sampled vacuum
bags were shipped directly to EOHSI and stored in a cold room (constant temperature at
4°C and relative humidity at 35%) before the analysis.
3.3.2. House dust preparation
House dust vacuum samples collected from fifteen residences were sieved to
below the 75 µm-diameter fraction using the unit shown in Figure 4. This mass fraction
was used as the primary sample for conducting the lead bioaccessibility experiments. The
total lead and the lead that was bioaccessible in the gastric and intestinal fluids were
measured in all samples. Five of these house dust samples were also sieved into three

84
separate fractions (< 75 µm, < 150 µm, and < 250 µm). Further sieving tests for the < 150
µm and < 250 µm samples were conducted to determine the lead mass distribution by
particle size (Table 23) and to examine the lead bioaccessibility as a function of the
following particle size fractions: < 75 µm, 75-150 µm, and 150-250 µm. Analyses were
completed on each size fraction to determine which particle size fraction had the highest
amount of bioavailable lead. The dust sieving was conducted using the standard operation
procedures (CRESP SOP-002 “Preparation of Soil Sub-Fractions” in the appendices) that
currently employed by EMAD-EOHSI laboratory (Ellickson et al., 2002).
Table 23. The mass distribution by particle size fraction for 5 house dust samples Dust Size Fractions Home 001 Home 002 Home 004 Home 005 Home 006
< 75 µm 36.50 % 16.73 % 15.99 % 30.94 % 46.38 % 75-150 µm 31.87 % 27.71 % 16.27 % 34.72 % 23.37 %
150-250 µm 31.63 % 55.56 % 67.74 % 24.34 % 30.25 %
3.3.3. Total house dust dissolution for lead analysis
The total lead content in house dust was determined using microwave-assisted
digestion with atomic absorption analysis, and these data were used as the reference total
lead concentration in each dust sample. Subsequently, the total lead value was used as the
denominator in all calculations of bioaccessible fraction of lead in house dust. Two sub-
samples of house dust (0.2 g from each prepared dust sample) were transferred into 50
mL centrifuge tubes. Twenty millimeters of nitric acid (TraceMetal grade, lead < 0.1 ppb,
Fisher Scientific, Edison, NJ) and 10 mL of de-ionized water were added to each tube.
Samples were digested in microwave oven (CEM Corporation, Matthews, NC, USA)
with the three-step program (1st stage of 90 % power for 20 minutes, 2nd stage of 0 %
power for 20 minutes, and 3rd stage of 90 % power for 20 minutes). After processing in

85
the microwave oven, the samples were allowed to cool down in a hood, and then diluted
up to 45 mL.
3.3.4. Bioaccessibility extraction
The in vitro determinative of bioaccessible lead was conducted using the modified
version of the method (Hamel et al., 1999; Ellickson et al., 2001) developed for
measuring elemental bioaccessibility in soil (Ruby et al., 1992, 1993, 1996). The assay
consisted of sequential extractions using synthetic human saliva, gastric and intestinal
fluids. Each step was conducted at physiological temperature and mixing conditions. All
bioaccessibility extraction experiments followed the University-wide (RWJMS/UMDNJ)
approved standard operation procedures (CRESP SOP-003 “Bioaccessibility Assay” in
the appendices).
3.3.4.1. Artificial fluids preparation
The artificial saliva / gastric / intestinal fluids were prepared using the same
procedures as discussed previously for the soil radionuclides bioaccessibility study (see
the section of 2.3.4.1. for detailed information).
3.3.4.2. In vitro extraction
The amount of each house dust sample used in the lead bioaccessibility
experiment was based on soil / dust intake rate (0.135 g/day) for young children between
one and four years old. Lead dissolution in synthetic gastric juice was studied previously
by Hamel et al. (1998) who showed that the lead solubility was relatively consistent
within liquid-to-solid ratios of 100:1 to 5000:1 mL/g for simulated soil ingestion of 0.05
to 0.5 g. A 0.135 g of house dust sample as the amount of house dust ingested and 400:1

86
mL/g (50+4 mL / 0.135 g) liquid-to-solid ratio were used throughout the extraction
procedure.
Approximately 0.135 g sample of sieved house dust (triplicate samples for gastric
and intestinal, respectively) was mixed with 4 mL of the artificial saliva. This was
followed by the addition of 50 mL of gastric juice (pH = 1.4 ± 0.2 throughout the
extraction). Then, the samples were placed into a constant temperature water bath (MSB-
1122A-1 General Signal, Blue Island, IL) at 37 °C and shaken for two hours at 90 rpm.
After the saliva-gastric incubation, all samples were taken from the water bath.
Subsequently, the gastric samples were filtered using a 0.45 µm pore sized cellulose
nitrate filter in a polycarbonate 250 mL filter funnel apparatus. With the addition of 1 mL
of nitric acid the filtered aqueous gastric samples were preserved in polyethylene bottles.
Meanwhile the intestinal samples were adjusted to a pH 6.5 by adding 50 mL of intestinal
juice. The samples were incubated again in the water bath at the same conditions. After
the saliva-gastric+intestinal incubation, the intestinal samples were also filtered, and
preserved in polyethylene bottles with 2 mL of nitric acid. Following the sequential
gastrointestinal extraction, the house dust residue was captured on a filter. The residual
was used to determine the total recovery and obtain a mass balance of lead for gastric and
intestinal samples. The captured house dust residue samples followed the same
procedures of total lead analysis, i.e., 10 mL of nitric acid and 5 mL of de-ionized water,
before the digestion of microwave oven. The bioaccessible lead was obtained by dividing
the lead concentration of each filtered fluid with total lead concentration found in each
house dust sample. The schematic diagram (Figure 6) showed the processes of in vitro
extraction step by step (Ellickson et al., 2002).

87
3.3.5. Analysis for total and bioaccessible lead
The gastric and intestinal extracts were directly transferred into centrifuge tubes
from the polyethylene bottles, and diluted from 10 up to 100 times prior to lead analysis.
A 10 mL of microwave-digested total lead and house dust residue sample was decanted,
spun in Metpath tabletop centrifuge (906×g) for 10 min, and then diluted to properly be
within the range of calibration curve from 2.5 ppb to 25 ppb. A GFAA (Graphite Furnace
Atomic Absorption Spectrometer, Perkin Elmer Zeeman 4100ZL, Norwalk, CT) with a
detection limit of 2.5 ppb in solution was used to quantify the lead levels. The GFAA was
calibrated for each run with standards prepared in Optima grade acid (Fisher Scientific,
Edison, NJ). The National Institute of Standardized Testing (NIST, Gaithersburg, MD)
reference material 1643D (lead dissolved in water with a concentration of 18.15 ± 0.64
µg/L, density of the solution = 1.016 g/mL at 22 °C) was used for quality control at each
run of the GFAA analysis. Sample digestion blanks and NIST 2710 soil spikes were
included in all analytical runs. The allowable instrument error was ± 20 %, although most
of the quality control results were within ± 10 %.
3.3.6. Statistical analyses
The data for the bioaccessibility experiments were tested for normality using a
Shapiro-Wilk test for both the gastric and intestinal data (N = 45). The test for normality
was conducted on fifteen house dust samples sieved to < 75 µm, and five house dust
samples that were separated into particle size fractions of < 75 µm, 75-150 µm, and 150-
250 µm. The results in Table 24 showed that three out of four sets of experimented data
were not normally distributed. Therefore, further statistical analyses were completed

88
using non-parametric methods. All statistical analyses were conducted using SAS®
Program v9.1 (SAS Institute Inc., Cary, NC) and SigmaStat® Program v3.11 (Systat
Software Inc., Richmond, CA).
Table 24. Shapiro-Wilk normality test for the bioaccessibility of lead from house dust samples (N = 45)
Experiment Tested Group Test Statistic p-value
Gastric Bioaccessibility 0.900696 0.0010 15 Home Samples
Intestinal Bioaccessibility 0.767012 <0.0001 Gastric Bioaccessibility 0.965795 0.2025*
5 Home Samples Intestinal Bioaccessibility 0.910170 0.0020
*Insufficient evidence to reject the assumption of normality
3.4.7. IEUBK model predictions
The IEUBK model estimates the potential concentration of lead in the blood for a
hypothetical child or population of children (6 months to 7 years old) based upon typical
reference variables, including absorption parameters, intake and exposure rates (Lewis et
al., 1999). However, errors can occur due to assumptions made about lead bioavailability
as well as other variables. In this study, the bioaccessible fractions of lead in gastric and
intestinal fluids determined for house dust may be used to augment the default input
values used as determinants of GI/Bioavailability in the model.
Two parts of the IEUBK were modified to use the house-specific bioaccessibility
data as input data: The GI Values/Bioavailability Information option on the Parameter
Input menu allowed the user to make adjustments to the gastrointestinal absorption
coefficient to account for site-specific information on bioavailability. The Blood Pb vs
Media Concentration option on the Computation menu allowed the user to generate a
plot of the relationship between blood lead concentration and the exposure to lead in a

89
specific, user-selected, environmental medium (e.g., soil, dust, air, drinking water, or
diet).
Using the results from the lead bioaccessibility analyses, blood lead levels were
predicted by substituting the default absorption value with the values of bioaccessible
lead obtained from the house dust study. These values were 65 % for gastric
bioaccessible lead and 12 % for intestinal bioaccessible lead. The input parameters for
total lead levels in house dust were changed ranging from 209 mg/kg as the lowest lead
concentration of tested house dust to 1770 mg/kg as the highest concentration. Age of the
child was selected to be from 0 to 84 months. The model simulation was run 15 times
(maximum allowed by the program) to estimate the blood lead concentrations within a
population of children exposed to bioaccessible lead levels found in house dust (Figure
11 and Figure 12).
Figure 11. The change of total bioaccessible percentage in dust matrix from default (30 %) to gastric bioaccessibility (65 %) and intestinal bioaccessibility (12 %)

90
Figure 12. The set-up of IEUBK model for predictions of blood lead vs media concentration based on studied dust concentration from 209 mg/kg to 1770 mg/kg with 15 times of running for 0 to 7 years old children
3.4. Results & Discussion
3.4.1. Bioaccessibility test for vacuumed house dusts
Fifteen house dust samples (the sample size used for the analysis was satisfied
with the significance level, α=0.05 and power, β=0.8) were chosen from the ECSC study,
and analyzed for bioaccessible lead (Table 25). The lead that was soluble in gastric fluid
yielded bioaccessibility values that ranged from 52.4 % (Home 027) to 77.2 % (Home
009). Simulated gastric juice resulted in the highest level of bioaccessible lead because of
the strong acidity (pH = 1.4 ± 0.2 throughout the extraction) associated with the artificial

91
gastric juice. This strong acid solution is representative of a fasting child and maximum
lead that would be resulted from the house dust. This would provide an upper-bound
estimate of available lead and would apply to ingestion of small particles by children
several hours after food ingestion or under fasting conditions (Oomen et al., 2003). After
the gastric solution passed into the intestinal phase, the bioaccessibility was reduced, and
ranged from 4.9 % (Home 13) to 32.1 % (Home 10). The levels dropped abruptly below
gastric bioaccessibility values since the pH increased to 6.5 with the addition of NaHCO3
solution. Thus, part of the mobilized lead was removed from intestinal solution. The
difference between gastric bioaccessibility and intestinal bioaccessibility was statistically
significant (Wilcoxon two-sample test; two-sided; p < 0.0001). Such a pH-dependent
decrease in lead solubility (Figure 13) has been observed by previous lead
bioaccessibility tests on soil sets (Ellickson et al., 2001; Ruby et al., 1992, 1993, 1996;
Oomen et al., 2002), household dust samples (Oliver et al., 1999), and slagged aggregates
(Davis et al., 1997).
Based on studies that determined the lead solubility in soils, the lead
bioaccessibility appears to depend on chemistry (e.g., pH in the solution), particle size
distribution (Ruby et al., 1992), the mechanism of dissolution (Ruby et al., 1992), and the
geochemistry of the soils (Davis et al., 1997; Ruby et al., 1999). The sharp decrease in
lead bioaccessibility after passing the intestine may be due to one of three possibilities: 1)
readsorption of lead onto the dust particles (Ellickson et al., 2001; Ruby et al., 1992,
1996), 2) complexation by pepsin (Ellickson et al., 2001), or 3) chemical precipitation of
the lead caused by phosphate within the higher pH environment found in the intestinal
compartment (Ellickson et al., 2001; Ruby et al., 1992, 1996).

92
0%
20%
40%
60%
80%
100%
House Dust, Newark NIST 2710 Soil House Dust, PortPirie
Test Matrices
Bio
acce
ssib
ility
GastricIntestinal
Figure 13. The comparison of bioaccessible lead among three different test matrices *House dust, Newark (< 75µm; N=15), the current study results NIST 2710 Soil (< 74µm; N=3), certified soil matrix from Ellickson et al., 2001 House dust, Port Pirie (< 53µm; N=7), collected household dust in Australia study from Oliver et al., 1999
and conducted the in vitro extraction test for gastric / intestinal phase at pH 1.3 / 7.0, respectively
The lead bioaccessibility ranged from 52.4 % to 77.2 % for gastric extracts and
4.9 % to 32.1 % for intestinal extracts. This finding shows that bioaccessibility of
individual house dusts can be different. Current risk assessment models (e.g., the IEUBK
model adopted 30 % fraction of lead in house dust will be absorbed in the children’s
body) do not consider the variation of house dust bioaccessibility. The range of house-
specific bioaccessibility data found on these experiments can reduce the uncertainty in
dose estimations for children, but support the general use of 30 % lead absorption value
as a default in the IEUBK model.

93
Table 25. The bioaccessibility test used for 15 vacuumed house dust samples (mean±S.D.)
Home ID Total Pb Concentration (µg/g)
Gastric Bioaccessibility (%)
Intestinal Bioaccessibility (%)
001 1097 76.6±3.1 6.7±0.3
002 596 74.3±14.5 6.7±0.2
004 216 70.1±4.5 8.5±0.7
005 1125 68.9±0.5 17.8±0.7
006 358 71.3±3.5 7.3±1.1
007 1312 74.7±2.2 6.0±0.2
009 709 77.2±0.9 11.1±0.2
010 487 52.7±1.4 32.1±0.7
013 732 55.8±1.4 4.9±0.2
014 209 57.0±1.9 9.3±0.7
016 820 72.6±2.7 5.8±0.2
021 676 59.9±2.0 12.3±0.7
023 1770 55.1±2.1 6.4±0.2
027 813 52.4±1.1 28.8±2.1
029 541 53.7±0.3 17.7±0.9
Mean±S.D. 764.1±422.5 64.8±9.4 12.1±8.2
3.4.2. Bioaccessibility / recovery test for different size fractions Lead in house dust is generally considered to be more concentrated in the fine
fraction of dust and lead absorption into the body is inversely related to particle size
(EPA, 1995). However, data showing a relationship between lead absorption and particle
size is not strong. The in vitro lead bioaccessibility was determined for three particle size

94
fractions in five house dust samples. The analyses were designed to examine changes in
bioaccessible lead that would be associated with different particle size fractions.
Recovery tests were included as a part of the bioaccessibility procedures for artificial
fluids and three particle size fractions. The recovery tests were designed to establish that
the proposed extraction system does not lose much lead through the in vitro extraction
and to examine the behavior of dissolved lead in the extraction system. The
bioaccessibility / recovery results are provided in Table 26. The results for the recovery
tests conducted for each fluid showed that the in vitro extraction procedures did not lose
much lead during the sequential extractions.
The values obtained for gastric bioaccessible lead with each of the three particle
size fractions were not significantly different (Kruskal-Wallis test; p = 0.7019). However,
intestinal bioaccessibility obtained for each of three particle size fractions was
significantly different (Kruskal-Wallis test; p = 0.0067). Further, a non-parametric post
hoc multiple comparison test (Tukey’s method for balanced subgroups; α = 0.05) was
conducted to compare the intestinal bioaccessibility obtained for each fraction, and there
were statistical differences between the size fractions (Table 27).

95
Table 26. The bioaccessibility / recovery (mean±S.D.) of vacuumed house dust samples for three sub-fractions (< 75 µm, 75-150 µm, and 150-250 µm)
Home ID Gastric Bioaccessibility Recovery Intestinal
Bioaccessibility Recovery
<75µm 76.6±3.1 % 85.1±2.4 % 6.7±0.3 % 71.2±4.2 %
75-150µm 71.0±9.9 % 87.4±4.0 % 15.6±1.0 % 96.4±13.2 % 001
150-250µm 63.8±7.1 % 91.4±2.8 % 15.2±3.7 % 94.6±1.4 %
<75µm 74.3±14.5 % 84.6±13.3 % 6.7±0.2 % 117.3±12.6 %
75-150µm 68.5±4.2 % 83.3±4.1 % 8.8±0.6 % 115.9±7.9 % 002
150-250µm 71.2±11.0 % 82.4±8.6 % 4.7±1.0 % 96.8±10.6 %
<75µm 70.1±4.5 % 76.5±2.4 % 8.5±0.7 % 76.6±8.1 %
75-150µm 69.3±3.1 % 77.0±1.4 % 9.9±1.1 % 80.9±3.4 % 004
150-250µm 79.2±21.0 % 86.1±16.0 % 15.4±5.8 % 76.7±26.8 %
<75µm 68.9±0.5 % 75.4±4.3 % 17.8±0.7 % 98.7±3.7 %
75-150µm 85.9±7.2 % 87.7±5.5 % 27.2±4.9 % 121.8±11.1 % 005
150-250µm 90.9±7.2 % 84.4±4.6 % 26.8±4.6 % 118.9±1.8 %
<75µm 71.3±3.5 % 78.2±3.5 % 7.3±1.1 % 96.8±2.9 %
75-150µm 56.3±7.5 % 76.0±5.7 % 23.9±4.7 % 84.0±5.1 % 006
150-250µm 65.0±6.0 % 79.9±1.7 % 19.1±6.9 % 93.5±8.1 %
The bioaccessibility of particle size fraction sieved to < 75 µm was significantly
different (p < 0.05) from the values obtained for particle size fractions both the 75-150
µm and 150-250 µm. Larger particle size fractions, 75-150 µm and 150-250 µm, had
higher intestinal bioaccessibility than smaller particle size fraction (< 75 µm). The
difference in bioaccessibility obtained for each size fraction suggested that lead
dissolution in gastric fluid was not affected by the dust particle size, however lead

96
dissolution for each size fraction was influenced by contacting with the intestinal fluid.
This behavior seems to indicate that lead is dissolved uniformly in gastric fluid but can be
quickly re-adsorbed onto the surface of smaller dust particles after the addition of
artificial intestinal juice. The magnitude of adsorption of dissolved lead onto the dust
particles is inversely related to particle size, because smaller particles have larger surface
area at unit mass (EPA, 1995; Yiin et al., 2002).
Table 27. Non-parametric tests for lead bioaccessibility by particle size fractions (Kruskal-Wallis ANOVA test & post hoc multiple comparison test)
Matrix No. Mean Score p-value Min. Med. Max. Grouping
< 75 µm 15 24.600 0.577 0.719 0.848
75-150 µm 15 20.733 0.490 0.687 0.941 Gastric
150-250 µm 15 23.667
Pr < χ2 (0.7019)
0.561 0.689 1.001
< 75 µm 15 14.333 0.062 0.072 0.183
75-150 µm 15 28.467 0.081 0.157 0.319 Intestinal
150-250 µm 15 26.200
Pr < χ2 (0.0067)
0.038 0.141 0.320
A B B B
*Different letters indicate significant differences (p < 0.05) by balanced Tukey’s multiple comparison test; the same letter indicates no difference between median ranks.
Intestinal recovery data also supported the explanation that the larger difference
between gastric and intestinal bioaccessibility resulted from the absorption of dissolved
lead onto dust particles or chemical precipitation in the mixture caused by phosphates in a
high pH environment.
3.4.3. IEUBK model estimates of dose using bioaccessible lead
The IEUBK model predicted two levels of blood lead concentrations: one that
was based on gastric bioaccessible lead and the other was based upon intestinal
bioaccessibility of lead (Figure 14). It is believed that most lead is absorbed into the
upper part of the intestine (ATSDR, 1999); consequently the values obtained for

97
intestinal bioaccessible lead were more representative of the absorption fraction.
However, gastric bioaccessibility provided a maximum fraction of lead available within
the human gastrointestinal system. The IEUBK model predictions showed that blood lead
levels generally would not exceed the lower blood lead level of 11 µg/dL using intestinal
bioaccessibility (12 %) or default value (30 %) in the range of tested house dust
concentrations. However, the calculated values based on gastric bioaccessibility (65 %)
easily surpassed the level of 11 µg/dL.
House Dust Concentration (µg/g)
200 400 600 800 1000 1200 1400 1600 1800
Blo
od L
ead
Leve
l (µg
/dL)
0
5
10
15
20
25
30
35
Upper PbB
Median PbB
Lower PbB
GastricIntestinalDefault ValueUpper PbBMedian PbBLower PbB
Fig. 14. The comparison of IEUBK model predictions using gastric bioaccessibility (65 %), intestinal bioaccessibility (12 %), and model default value (30 %)
The IEUBK predictions, based on model default parameter of 30 % GI absorption,
generally did not yield blood lead levels above 11 µg/dL. This was inconsistency with
reported blood lead levels. The model predictions using the 30 % absorption were
expected to approach the reported blood lead levels ranged from 11 to 32 µg/dL; however,

98
the predictions could not reach the levels. Instead, using the gastric bioaccessibility value,
65% absorption, the predictions did yield values close to the median blood lead level of
19 µg/dL. The IEUBK model predictions were obtained using the arithmetic mean of
gastric and intestinal bioaccessibility of the house dust. However, the gastric / intestinal
bioaccessibility is site-specific (dust from each home), and each home and child has
different levels of total lead and blood lead, respectively. A comparison of actual blood
lead levels was made with predicted blood lead level obtained using the intestinal, gastric,
and default value of bioaccessibility (see Table 28). The predicted blood lead levels were
found to be below the actual blood lead levels; generally blood lead levels predicted by
gastric bioaccessibility had values that were more closely associated with children’s
blood lead levels.
Table 28. The comparison of actual blood lead levels with predicted blood lead levels for each home environment.
Bioaccessibility (%) Predicted PbB (µg/dL) Home ID Dust Conc.
(mg/kg) Intestinal Gastric
Actual PbB
(µg/dL) Intestinal Default Gastric
002 596 6.66 74.28 26 3.3 5.8 9.9 004 216 8.51 70.05 17 2.9 3.8 5.4 006 358 7.29 71.28 26 3.0 4.6 7.1 007 1312 6.00 74.66 11 4.0 9.2 16.4 009 709 11.11 77.16 21 4.1 6.4 11.3 013 732 4.87 55.82 14 3.2 6.5 9.4 014 209 9.29 57.03 32 2.9 3.7 4.8 016 820 5.75 72.63 14 3.5 7.0 12.0 027 813 28.75 52.36 21 6.8 6.9 9.7 029 541 17.68 53.74 16 4.4 5.6 7.7
This inconsistency may be derived from the difference between the time for each
child’s blood lead level measurements (at least 6 months or above) and the time at which

99
lead concentration was determined in the house dust from the carpets. A child might be
exposed to lead present in previous residences, different locations (e.g., windowsills or
window walls where house dust was contaminated with lead), different matrices (paint
chips, lead-painted toys), or doing some types of activities (e.g., ingesting lead-
contaminated paint chips). In addition, the home environment might be improved with
the vacuum cleaning of carpets or abatement of leaded paint inside the house after the
high blood lead levels was reported to State Health Department.
The default value for GI/Bioavailability (30 %) in the IEUBK model is considered
reasonable estimator for predicting the blood lead levels in a target population. It has
been supported somewhat by the results of the current bioaccessibility experiments for
house dust, but the range in values obtained from the bioaccessibility analyses should be
considered for future use in sensitivity analyses. The highest value of intestinal
bioaccessibility (32 %) is essentially equivalent to the default absorption value used by
the IEUBK model. A one sample median test was conducted to determine whether or not
a sample median differs significantly from a hypothesized value. The IEUBK model
default parameter, 30 % lead absorption from soil / dust, was assumed as a hypothesized
value. Then it was significantly different with the obtained intestinal bioaccessibility data
(Wilcoxon singed rank test; H0 = 0.3; p < 0.0001). The percentiles and confidence
intervals (95 %) corresponding to 30 % intestinal bioaccessibility were obtained (Table
29). The IEUBK model default value covers over 90 % of all intestinal bioaccessibility
values determined in our experiments, however, it still could not predict blood lead
values above 11 µg/dL which was seen in each child. Thus, based upon our work, the
default value used for IEUBK risk estimates is a reasonable estimator of absorption for

100
determining the blood lead levels in target populations, however the range of values in
sensitivity analyses should be expanded to including values up to 65 % in an effort to
account for underestimate due to the lack of site-specific in the future.
Table 29. The percentile and confidence intervals (95 %) corresponding to 30 % intestinal bioaccessibility
95% Confidence Limits Distribution Free Order Statistics
Quantile Estimate
Lower Upper LCL Rank UCL Rank Coverage
100% 0.3270 99% 0.3270 ⋅ ⋅ ⋅ ⋅ ⋅ 95% 0.3128 0.2939 0.3270 41 45 82.76 90% 0.2939 0.1801 0.3270 37 45 95.93 75% 0.1663 0.1103 0.2939 29 41 95.46 50% 0.0841 0.0660 0.1129 16 30 96.43 25% 0.0631 0.0568 0.0668 5 17 95.46 10% 0.0568 0.0463 0.0616 1 9 95.93 5% 0.0504 0.0463 0.0568 1 5 82.76 1% 0.0463 ⋅ ⋅ ⋅ ⋅ ⋅ 0% 0.0463

101
IV. Bioaccessible Mercury from Fish Consumption
4.1. Introduction
Mercury cycles in the environment as a result of natural and human
(anthropogenic) activities. The most common mercury compound present in biota is
methylmercury. Methylmercury is of particular concern because it accumulates in edible
fish tissue and can reach levels millions of times greater than surrounding water
concentrations (Watras et al., 1992). The health effects of low-level human exposure to
methylmercury include cardiovascular and immunological effects, and developmental
neurotoxicity (Jacobson et al., 1989, 1990, 1996; Sparks et al., 1994; ATSDR, 1999).
Populations that are sensitive to mercury are pregnant women, infants, and younger
children (EPA, 1997; FDA, 2001). Recently, U.S. Food and Drug Administration issued a
consumption advisory based on mercury which suggested that pregnant women and
women of childbearing age who may become pregnant should avoid eating four species
or species groups of saltwater fish: shark, swordfish, king mackerel, and tilefish (FDA,
2004).
Inorganic and metallic mercury have shown limited absorption in humans (15 %;
Rahola et al., 1973) and in rodents (20 ~ 25 %; Piotrowski et al., 1992). Organic mercury
is more easily absorbed with 95 % absorption measured in humans (Aberg et al., 1969).
The protective effect of selenium on mercury toxicity has been known for about a quarter
of a century ago (Satoh et al., 1985; Ganther et al., 1972; Berlin et al., 1978). Overall, the
past findings indicate that methylmercury bound to fish tissue is almost completely
absorbed after consumption (95 %; Miettinen et al., 1973). Animal data support this
finding (Berglund et al., 1971).

102
Understanding the driving factor of mercury bioavailability in fish is a key to
predicting the level of risk. It is also important to study environmentally contaminated
fish, the portion of the fish that is used for consumption, and the manner in which the fish
is prepared for consumption by humans (i.e., cooked). Considering that the bioavailable
portion of many metals is less than 100 % in the human gastrointestinal system, new data
of mercury bioaccessibility is valuable to the estimation of methylmercury risk in the
general population and important in establishing what type of the fish matrices (e.g., raw
tuna steak vs. cooked tuna steak) are more readily bioavailable. Towards this end, the
results from the development of a method are presented for estimating mercury
bioaccessibility in fish. The result was obtained specifically for mercury in a fish;
however, the method may be generalized for application to other food products or other
metals.
Considering that fish and seafood are important dietary sources of protein and can
contain relatively high level of mercury, bioaccessibility data on mercury for fish
products will give a better understanding of the mercury dose received by the sub-groups
of the population that consume fish. Similarly, bioaccessibility data on cooked filet and
canned tuna will be more relevant than levels in raw fish to assess the risk of exposure to
mercury when dealing with the consumption of the processed product. The current data
of mercury content in fish are based on the uncooked flesh of fish (FDA, 2001), but most
people eat fish after it is cooked in a variety of ways. Thus, a method that includes
cooked fish products is necessary to construct meaningful estimates of bioaccessibility.
Also this study is a first attempt to quantify the bioaccessible mercury from food matrices
to estimate the exposure / dose levels used for risk assessments.

103
4.2. Background
4.2.1. Mercury
Mercury is a naturally occurring element; however, human activities such as
burning coal and using mercury in manufacture have increased the amount of mercury
that is currently cycling in the atmosphere, soil, lakes, streams, and oceans (EPA, 1997).
According to the EPA National Emissions Inventory (1999), coal-fired electric power
plants are the largest source of anthropogenic mercury atmospheric emissions in the U.S.
Power plants account for about 40 % of the total U.S. anthropogenic mercury emissions.
Other large sources are industrial boilers (about 10 % of U.S. mercury emissions),
burning hazardous waste (about 5 %), and chlorine production (also about 5 %).
When mercury is deposited into the water, or runs off the ground into the water,
microorganisms convert it to methylmercury, a highly toxic form of mercury (EPA,
1997). Small organisms accumulate the mercury as they feed. The accumulation of
methylmercury is magnified as animals higher up the food chain eat those organisms. The
process known as bioaccumulation results in higher levels of mercury in the tissue of
organisms as predators move up the food chain. Fish that are higher in the food chain
(e.g., sharks and swordfish) have much higher mercury concentration than fish that are
lower on the food chain (EPA, 1997). Humans become exposed when they eat mercury-
contaminated fish.
The nervous system is affected by all forms of mercury (EPA, 1999). However,
methylmercury and metallic mercury vapors are more harmful than other forms, because
mercury in these forms can cross the blood-brain barrier. Exposure to high levels of

104
metallic, inorganic, or organic mercury can permanently damage the brain, kidneys, and a
developing fetus. Effects on brain functioning may result in irritability, shyness, tremors,
changes in vision or hearing, and memory problems (ATSDR, 1999). Short-term
exposure to high levels of metallic mercury vapors may cause effects including lung
damage, nausea, vomiting, diarrhea, increases in blood pressure or heart rate, skin rashes,
and eye irritation (ATSDR, 1999).
Very young children (< 6 months old) are more sensitive to effects caused by
mercury exposure than adults (EPA, 1997). Mercury in the mother's body can pass to a
fetus and may accumulate there. It can also pass to a nursing infant through breast milk.
Mercury's harmful effects to the fetus include brain damage, mental retardation,
incoordination, blindness, seizures, and inability to speak. Children poisoned by mercury
may develop problems in their nervous and digestive systems, and can also have kidney
damage. Since the developing fetus is highly sensitive to the effects of mercury, women
of childbearing age are the population of greatest concern (EPA, 1997). Children born to
women exposed to relatively high levels of mercury during pregnancy have exhibited a
variety of abnormalities, including delayed onset of walking and talking, reduced
neurological test scores, and delays and deficits in learning ability. In addition, there is
growing evidence that methylmercury exposure can have adverse cardiovascular effects
for adults, resulting in elevated blood pressure and incidence of heart attack (ATSDR,
1999).
Mercury is accumulated by the edible fish through biomagnification in the food
chain (EPA, 1997). The most important exposure route for mercury in the general
population is through dietary ingestion of fish (FDA, 2001). Mercury exposure to

105
pregnant women is at greater concern than exposure to others, due to mercury having
detrimental impacts on the health of developing fetus (FDA, 2001). Mercury was,
therefore chosen as a toxin of concern for this study.
4.2.2. Mercury absorption
4.2.2.1. Elemental mercury
Liquid metallic mercury is very poorly (less than 0.01 %) absorbed from the
gastrointestinal tract (Bornmann et al., 1970). The release of mercury vapor from liquid
elemental mercury in the gastrointestinal tract and the subsequent absorption of the
released vapor are limited by reaction of the mercury with sulfur to form mercuric sulfide
(Berlin, 1986).
4.2.2.2. Inorganic mercury
The absorption of mercuric mercury from the gastrointestinal tract has been
estimated at approximately 7 ~ 15 % in human volunteers following oral administration
of radiolabeled inorganic mercury. Rahola et al. (1973) examined mercury absorption and
elimination after oral administration of mercuric nitrate to five male and female
volunteers, and reported very low and variable rate of gastrointestinal absorption (8 to
25 %). Investigations (Miettinen, 1973) were carried out to determine the percentage of
absorption for protein-bound mercury using 203Hg-labeled compounds and the whole-
body counting technique. Eight subjects were fed 4 µCi to 8 µCi 203Hg in calf liver
protein and two subjects 4 µCi and 14 µCi 203Hg(NO3)2 in water solution. Within the first
4 to 5 days, 85 percent of the 203Hg was excreted, mostly in the feces (absorption of
15 %).

106
4.2.2.3. Organic mercury
Methylmercury is efficiently absorbed from the gastrointestinal tract.
Approximately 95 % of methylmercury in fish ingested by volunteers was absorbed from
the gastrointestinal tract. The oral intake of 2.6 µCi of methyl-mercuric nitrate 203Hg
(CH3203HgNO3) in aqueous solution by three volunteers and total body counting were
conducted (Aberg et al., 1969). The absorption from the gastrointestinal tract was almost
complete (over 95 %) and the dominant excretion route was via the feces. An organic
mercury absorption study (Miettinen, 1973) was conducted to estimate the absorption
percentage for protein-bound methylmercury. Fifteen subjects were fed 2 µCi Me203Hg in
fish muscle protein, then about six percent of the administered Me203Hg was excreted
mostly in feces (absorption of 94 %).
The mercury in experimental fish-meal (fish caught in polluted areas of Halifa
Bay, Israel; mercury concentration of 60.2 ppb) and commercial fish-meal (Milubar,
Mobile Post Oshrat, Israel; mercury concentration of 280.8 ppb) was approximately 93 ±
5 % and 75 ± 3 % bioavailable in rats, respectively (Yannai et al., 1993). Plant based
mercury (corn infused with 203Hg during the growth; mercury concentration of 14 ppb)
tends to be less bioavailable (51 ± 11 %) than meat based (fish) mercury in rats; and this
is explained by fiber in the plants (Yannai et al., 1993).
4.2.2.4. Methylmercury in tuna
Many studies have shown that almost all of the mercury in fish tissue is
methylmercury, and 90 % is a reasonable approximation of this proportion, which varies
somewhat among fish types and laboratories (Burger et al., 2005). Storelli et al. (2002)
reported a range of 77 ~ 100 % of the total mercury is methylmercury (average of

107
91.3 %) for albacore tuna (N = 127) and 75 ~ 100 % is methylmercury (average of 91 %)
for bluefin tuna (N = 161) from the Mediterranean Sea. Burger et al. (2004) measured the
methylmercury in light and white canned tuna sampled in New Jersey supermarket (N =
40), and reported that methylmercury was 83 ~ 89 % of the total mercury. Considering
only cans with total mercury > 0.1 ppm, the value was about 89 %.
4.2.2.5. Digestion and absorption of food6
Most food is initially consumed as large particles containing many
macromolecules, such as proteins and polysaccharides that are unable to cross the wall of
the gastrointestinal tract. Before ingested food can be absorbed, it must be dissolved and
broken down into much smaller molecules. This dissolution and breaking-down process
(digestion) is accomplished by the action of hydrochloric acid secreted by the stomach,
bile secreted by liver, and a variety of digestive enzymes that are released by the system’s
exocrine glands, a process termed secretion. The molecules produced by digestion then
moved from the lumen of the gastrointestinal tract across a layer of epithelial cells and
enter the blood or the lymph- absorption. While digestion, secretion and absorption are
taking place, contractions of smooth muscles in the gastrointestinal tract wall mix the
luminal contents with the various secretions and move them through the tract from mouth
to anus- motility.
Proteins are broken down to peptide fragments in the stomach by pepsin and in
the small intestine by trypsin and chymotrypsin, which are secreted by the pancreas
(Marieb, 1997). These fragments are further digested to free amino acids by
carboxypeptidase secreted by the pancreas and by aminopeptidase located in the luminal
epithelial membranes of the small intestine. These last two enzymes split off amino acids 6 The text was excerpted from “Essentials of Human Anatomy and Physiology, 5th Ed.” Marieb, 1997

108
from the carboxyl and amino ends of the peptide chains, respectively. The free amino
acids then undergo secondary active transport, coupled with sodium, across the intestinal
wall. Short chains of two or three amino acids are also actively absorbed. Protein
digestion and absorption are largely completed in the early portion of the small intestine.
Fat (most is in the form of triacylglycerols) digestion occurs almost entirely in the
small intestine (Marieb, 1997). The major digestive enzyme in this process is pancreatic
lipase, which catalyzes the splitting of bonds linking fatty acids to the first and third
carbon atoms of glycerol. This produces two free fatty acids and a monoglyceride as
products. The triacylglycerols entering the small intestine from the stomach are insoluble
in water and are aggregated into large lipid droplets. Since only the lipids at the surface
are accessible to the water-soluble lipase. At this point, digestion would proceed very
slowly without the solubilizing action of bile. Bile salts increase the rate of fat digestion
and absorption in two ways by 1) emulsification with preventing large lipid droplets from
aggregating into still larger ones and 2) combining with the fatty acids and
monoglycerides produced by lipase at the droplet surface to form very small, water-
soluble aggregates (micelles). In the absence of bile salts, the resulting lipid
emulsification and micelle formation, fat digestion and absorption would occur so slowly
that much ingested fat would pass to the large intestine and would be excreted in the
feces.
Carbohydrate composed of polysaccharides, disaccharides, and monosaccharides
(Marieb, 1997). About two thirds of this carbohydrate is the plant polysaccharide starch
and the remainder is mostly the disaccharides sucrose and lactose. Cellulose and certain
other complex polysaccharides found in vegetable matter cannot be broken down by the

109
enzymes in the small intestine. They are passed on to the large intestine, where they are
metabolized in bacteria. Starch is partially digested by salivary amylase in the upper part
of the stomach before the enzyme is destroyed by gastric acid, and digestion is continued
in the small intestine by pancreatic amylase. The products formed by the amylases, along
with ingested sucrose and lactose, are broken down into monosaccharides (glucose,
galactose, and lactose) by enzymes located in the plasma membranes of the small
intestinal epithelial cells. These monosaccharides are then transported across the
epithelium by facilitated diffusion, while glucose and galactose undergo secondary active
transport coupled to sodium.
The tested matrices (tuna steak and canned tuna) consist mostly of proteins, fats,
and water. For example, canned white tuna (drained 56 g) is composed of protein (15 g of
26.8 %), total fat (1.0 g of 1.8 %), total carbohydrate (0 g of 0.0 %), with the remainder
of the mass being water and minerals. The mechanism of food digestion facilitated with
enzymes and secretion fluids in human gastrointestinal tract is important in understanding
the behavior of bioavailable mercury in vitro extraction system.
4.3. Methods
4.3.1. Approach
The in vitro protocol for estimating human exposure to mercury in tuna products
developed for this work was adapted to our previous heavy metal bioaccessibility studies
in soils (Hamel et al., 1998, 1999; Ellickson et al., 2001, 2002), but with a different
procedure for recapturing the residual tuna tissues at the end of each sequential extraction.
The approach taken to recapturing tuna tissues included combining the non-extracted

110
portion of the mercury and any residual matter precipitated during the extraction. An
acid-digestion of the material in the recaptured tuna tissues was considered to be the
amount of mercury retained in the recaptured tuna tissues after sequential extraction by
the biofluids. The amount of mercury in the recaptured tuna was used for bioaccessibility
/ recovery calculation for the overall tuna extraction procedure.
4.3.2. Study materials (tuna steak & canned tuna)
Yellow-fin tuna (Thunnus Albacares) steak and commercial canned tuna (light
chunk tuna in water and white solid tuna in water) were purchased from a local grocery
store in New Brunswick, NJ. Analysis of tuna steak and canned tuna was conducted to
achieve two goals: environmentally contaminated fish and one of the most widely
consumed seafood (fresh tuna - 1.79 %, white canned tuna - 5.29 %, and light canned
tuna - 13.35 % of market share; FDA, 2001) in the United States. These particular species
were chosen because it has some of the highest mercury concentrations due to its
biomagnification and bioaccumulation. One pound of previously frozen tuna steak and
commercial tuna cans (light and white) were purchased on July 7, 2004 and returned to
the EOHSI laboratory where they were stored in a laboratory freezer at -10.7 °C.
4.3.3. Synthetic fluid preparation
The artificial saliva / gastric / intestinal fluids were prepared using the same
procedures as discussed previously for the soil radionuclides bioaccessibility study (see
the section of 2.3.4.1. for detailed information).

111
4.3.4. Method modifications
4.3.4.1. Pilot study
The measurement of bioaccessible mercury from human fish consumption was
tested at the laboratory of EMAD-EOHSI with the adaptation of bioaccessibility method
for heavy metals in soils. The first experiment to measure bioaccessible mercury from
raw tuna steak and cooked tuna steak was conducted to evaluate exposures to
childbearing-aged women who frequently consumed fish. The amount of tuna (232
g/meal) and artificial saliva / gastric / intestinal volumes used (0.6 L/day for saliva and 2
L/day for gastric and intestinal fluids) were obtained from Burger et al. (2000) and
Marieb (1997). Based on each of the above daily excretion of human fluids, artificial
saliva / gastric / intestinal fluids were determined to 10 mL, 50 mL, and 50 mL,
respectively. The amount of tuna (17.4 g) was calculated from the fish consumption (232
g/meal), multiplied by gastric fluid volume (50 mL), and divided by the gastric fluid
volume per one-meal (667 mL/meal).
The sequential extraction experiments were conducted to determine the
bioaccessibile mercury following the same experimental conditions previously conducted
to determine the bioaccessible radionuclides from a soil matrix. The bioaccessibility of
triple replicates for raw tuna steak were 8.47 ± 2.54 % and 8.35 ± 1.05 % for gastric and
intestinal samples, respectively. The ranges for cooked tuna steak were lower, between
1.34 ± 0.76 % and 0.92 ± 0.02 % in gastric and intestinal phase, respectively. The
averaged mass recovery for mercury was 60.1 % and 85 % for gastric and intestinal
sample, respectively. These bioaccessibility results are considerably lower than the
reported mercury bioavailability (i.e., 95 % in vivo; ATSDR, 1999).

112
One possible reason for the lower bioaccessibility values might be that the in vitro
extraction protocols were not sufficient to release bioaccessible mercury into simulated
gastrointestinal fluids (see the section of 4.2.2.5. for the digestion of food). Considering
much of mercury mass was still remained in the solid phase (recaptured tuna flesh on the
filter), more in vitro extraction procedures may be required.
4.3.4.2. Changing experimental conditions
In order to digest tuna samples more completely, a smaller mass of tuna was used,
and changes were made in volume of artificial fluids. The mass of tuna used for each
sequential extraction test was reduced to 4.46 gram of tuna steak, while the new volume
of artificial saliva and gastric fluid were 8 mL and 42 mL, respectively for the sequential
extraction test. The new amount of tuna used (4.46 g) was calculated from the fish
consumption (232 g/day- assuming one meal per day), multiplied by saliva+gastric fluid
volume (50 mL), and divided by the saliva+gastric fluid excretion volume per day (2,600
mL/day). The intestinal volume (50 mL) was not changed from previous values. To make
the tuna samples homogenous and reduce the size of the digestate, a laboratory blender
(7009G, Waring® Laboratory & Science, Torrington, CT) method was adopted. The food
particle size (< 2 mm) obtained by the blender represents the food mixtures right after
passing the human throat. The previously used polyethylene bottles were replaced with
Teflon® bottles to extract and store samples. The use of Teflon® bottle (FEP 500 mL
wide mouth, Nalgene Labware, Rochester, NY) was introduced into the sequential
extraction method to minimize the mercury loss via volatilization or sticking to the wall
of polyethylene bottles.

113
Using the revised method, the mercury bioaccessibility determined for tuna steak
was 88.5 ± 3.44 % and 31.0 ± 18.27 % for gastric and intestinal samples (N = 3),
respectively. Total recovery of mercury was 96.9 ± 5.69 % for gastric samples, and 48.2
± 18.38 % for intestinal samples. The bioaccessibility and recovery for intestinal samples
were not only lower, but also had much higher variability. The analysis for bioaccessible
mercury was conducted again to check the reliability of the method. The bioaccessible
mercury was 88.1 ± 9.36 % and 21.5 ± 13.87 % for gastric and intestinal samples (N= 3),
respectively. Total mercury recoveries for gastric and intestinal fluids were 96.3 ± 5.27 %
and 54.4 ± 23.83 %, respectively.
The bioaccessibility and recovery in intestinal phase were much lower than the
levels in gastric fluid. It was possible that mercury was lost during the sample storage or
mercury analysis. For example, EPA recommends that the analysis for low-levels
mercury should be conducted within one or two days to avoid mercury loss from
prepared samples (EPA, 2002). Otherwise the samples were acidified to pH 1.0 with
nitric acid and stored in the EOHSI cold room for 3 ~ 4 days prior to mercury analysis.
4.3.4.3. Final experimental conditions
Based upon the test runs for the tuna samples, the method measuring
bioaccessible mercury in a food matrix has been modified for the artificial human
biofluids. The final experimental condition is determined by changes in the amount of
tuna provided in each extraction test set and the volume of fluids added to each extraction
bottle. The reference and experimental values for serving size and volumes of biofluids
are reported in Table 30. These values were determined on the assumption that the
childbearing aged women eating tuna steaks at local restaurants. The amount of tuna

114
(2.92 g) was further reduced from previous tuna weight (4.46 g) due to the change of
serving size in reference value. This was expected to improve the digestion process by
increasing solid-to-liquid ratio. The experimental tuna weight was obtained from the
amount of fish (111 g/meal) consumed in a restaurant, multiplied by total volume of
artificial fluids (100 mL), and divided by maximal excretion of human biofluids per meal
(3,800 mL/meal = 7,600 mL/day ÷ 2). The sample preservation method was changed
from nitric acid to hydrochloric acid, and it was added in aqueous solution after filtering.
EPA Method 1631 Revision E (Mercury in Water by Oxidation, Purge, and Trap, and
Cold Vapor Atomic Fluorescence Spectrometry) recommends the use of either 5 mL/L of
pre-tested 12 N HCl or 5 mL/L of BrCl solution within 48 hours of sample collection to
preserve the sample. Acid-preserved or BrCl-preserved samples are stable for a period of
90 days. Therefore, 1 mL and 2 mL of HCl (Optima grade, Fisher Chemical, Fair Lawn,
NJ) were added to gastric and intestinal filtrate samples, respectively.
Table 30. Serving size of fish and volumes of biofluids used in mercury bioaccessibility experiments
Description Reference Value Used in Experiments Source
Serving Size 111 g/meal 2.92 g Burger et al., 2000
Saliva Fluid 0.6 ~ 1.5 L/day 8 mL Davenport, 1982
Gastric Fluid 3 ~ 3.5 L/day 46 mL Davenport, 1982
Intestinal Fluid 3.5 L/day 46 mL Vander, 1990
4.3.5. Bioaccessibility extraction
Tuna samples were analyzed for four experimental matrices (cooked tuna filet,
uncooked tuna filet, light canned tuna, and white canned tuna). Samples of cooked filet
were prepared by the following procedures: a portion of tuna was thawed, weighed, and
pan-fried in a frying pan with vegetable oils. After fully cooking the filet, it was cooled

115
and weighed again to record changes in total weight. No preliminary preparation steps
were taken for the uncooked tuna samples (i.e., raw tuna steak). Canned tuna samples
were opened and placed on a funnel with paper filter for at least 30 minutes to drain as
much water as possible.
The four different tuna samples were divided into pieces weighing 2.92 grams
each, and individually placed into a laboratory blender for processing with 8 mL of
artificial saliva. Each mixture (tuna sample + artificial saliva) was processed in the
blender for 2 ~ 3 minutes. After saliva+fish samples were completely mixed, 46 mL of
synthetic gastric juice was mixed into the Teflon® sample bottles. These samples were
shaken (125 cycles per minute) at 37 °C for two hours in a Constant General Bath,
(MSB-1122A-1 Water Bath, Blue M Electronic Co., Blue Island, IL). After the saliva-
gastric incubation, all samples were removed from the water bath.
Subsequently, 46 mL of intestinal juice was added to the intestinal samples, and
they were allowed to incubate again for another 2 hours at 37 °C with moderate shaking
(125 cycles/min). During the incubation of intestinal samples, the gastric samples were
filtered with a 0.45 µm diameter membrane filter. They were acidified by adding 1.0 mL
of Optima grade hydrochloric acid to preserve the samples and each was labeled as
“gastric liquid 1, 2 or 3”. The undigested semi-solid tuna tissue that remained on the
membrane filter was collected, weighed and labeled as “gastric solid 1, 2 or 3”.
After the saliva-gastric+intestinal incubation was completed, the intestinal
samples were removed from the water bath and filtered using the gastric sample filtering
procedure. Similarly, the intestinal samples were acidified by adding 2.0 mL of
hydrochloric acid to each fluid sample and labeled as “intestinal liquid 1, 2 or 3”. The

116
insoluble portion of intestinal bioaccessible samples were obtained and labeled as
“intestinal solid 1, 2 or 3”. All gastric and intestinal bioaccessibility samples were tightly
sealed and kept in the cold room at 4 °C for later mercury analysis.
4.3.6. Digestion for total and bioaccessible mercury
To determine the mercury levels in the experimented sample matrices (tuna steak,
white canned tuna, and light canned tuna), total mercury digestion was conducted prior to
each experiment. Duplicate tuna tissue samples (approximately 1.5 g each) were selected
from four experimental tuna samples, and these were digested using a microwave-
assisted digestion method (see the section of 4.3.7.) for total mercury. To quantify the
mercury levels in recaptured tuna tissue, it was digested with the same procedures used
for total mercury analysis. Aqueous extracted samples (5 mL) were concurrently digested
with the solid-phase tuna samples (tuna tissue and recaptured tuna tissue sample). The
acid-digestion of aqueous extracted samples was conducted to reduce the mercury in the
samples to an elemental form and prevent the generation of foams during the analytical
analysis by totally dissociating mercury from mercury-bound organics in the samples.
Four milliliter of Ultrex® II Ultrapure nitric acid (Reagent grade, J. T. Baker, Phillipsburg,
NJ) and 2 mL of de-ionized water were added into the mixture of each solid digestion
vessel. Two milliliter of Ultrex® II Ultrapure nitric acid was used for each liquid
digestion vessel.
4.3.7. Mercury analysis
Mercury was analyzed by EPA Method 7471A Cold Vapor Atomic Absorption
Spectrometry (Mercury in Solid or Semisolid Waste; EPA, 1994). This method is based

117
on the absorption of radiation at the 253.7 nm wavelength by mercury vapor. The
mercury is reduced to the elemental state and aerated from solution in a closed system.
The mercury vapor passes through a cell positioned in the light path of an atomic
absorption spectrophotometer. Absorbance is measured as a function of mercury
concentration. The method detection limit (MDL) is 0.2 ppb. The fish digestion
procedure was modified from EPA Method 3051: Microwave Assisted Acid Digestion of
Sediments, Sludges, Soils, and Oils (EPA, 1994).
Samples were digested in Ultrex® II Ultrapure nitric acid in a microwave oven
(MD 2000 CEM, Matthews, NY), using three-stage digestion protocol of ten minutes
each under 50, 100 and 150 (3.5, 7, and 10.6 kg/cm2) pounds per square inch at 70X
power. Digested samples were subsequently diluted in 25 mL of de-ionized water. All
laboratory equipment and containers were washed in 10 % HNO3 prior to each use. All
concentrations are expressed in parts per billion (ng/g) on a wet weight basis. Detection
limit was 0.2 ppb. All specimens were run in batches that included blanks, a standard
calibration curve, and spiked specimens. The accepted recoveries for spikes ranged from
85 % to 115 %; no batches were outside of these limits. Mercury was measured using a
cold vapor atomic absorption spectrometer (Lumex Mercury Analyzer RA-915+,
OhioLumex Co., Twinsburg, OH).
4.3.8. Bioaccessibility calculations
Two estimations of bioaccessible mercury were made in this study. The first was
the direct measurement of extracted mercury in two biofluids, and the second was the
estimation of bioaccessibility indirectly by obtaining the mass balance of mercury in the
system. Mercury concentrations obtained in the intestinal fluid for canned tuna were too

118
low (< 0.2 ppb) to reliably calculate bioaccessibility by the direct method for the
intestinal fluid.
The indirect estimation of bioaccessibility was previously used by Hamel et al.
(1999) as one of two novel methods, mass-balance and soil recapture, for estimating the
bioaccessibility of heavy metals in soils. The Hamel et al. (1999) study concluded that the
recaptured soil metal mass, obtained by the same way of indirect estimation for
bioaccessible mercury, was a valuable measurement since it greatly reduced analysis for
efficient processing of many soil samples and analysis of metals in each fluid; yet also
provided a reasonable estimate of bioaccessibility. It also allowed for calculating
bioaccessibility in a soil containing very low metal mass. Otherwise the analysis would
have resulted in non-detectable concentrations at the dilutions required in the synthetic
human fluid system. Therefore, the approach for estimating bioaccessibility indirectly by
mass balance was added to the conventional direct measurement of bioaccessibility for
canned tuna.
The direct measurement of bioaccessibility (BioaccessibilityD) was calculated by
multiplying the mercury concentration in the soluble portion of mercury (L-mercury) in
gastric or intestinal fluid by the volume of each fluid (around 54 and 100 ml for gastric
and intestinal samples, respectively), divided by the averaged total mercury concentration
(T-mercury) in tuna samples and the weight of tuna used in each experiment:
( ) ( )(% ) 100( ) ( )D
L m ercury ppb vo lum e m lB ioaccessib ilityT m ercury ppb w eigh t g
− ×= ×
− ×
The mass balance estimate of bioaccessibility (BioaccessibilityMB) was estimated
through the following approach. A bioaccessible percentage was estimated by subtracting
the ratio of recaptured mercury mass (S-mercury times approximately 3 g of weight for

119
recaptured filter) to total mercury mass (T-mercury times 2.92 g of weight for tuna) from
100 percent:
( ) ( )(% ) 100 100( ) ( )M B
S m ercury ppb w eight gBioaccessibilityT m ercury ppb w eight g
⎡ ⎤− ×= − ×⎢ ⎥− ×⎣ ⎦
The recovery of each set of gastric and intestinal sample was obtained from the
measured total mass, divided by total mercury in each set of samples:
[ ( ) ( ) ( ) ( )](%) 100( ) ( )
L mercury ppb volume ml S mercury ppb weight gRecoveryT mercury ppb weight g
− × + − ×= ×
− ×
4.3.9. Statistical analyses
To test for normality in the data, a Shapiro-Wilk test was completed on both the
gastric and intestinal data (N = 60). The direct measurements for intestinal
bioaccessibility were below the detection limit of 0.2 ppb, and they were not included in
the statistical analyses (N = 30). The result (Table 31) showed that direct measurements
of bioaccessibility (BioaccessibilityD) in two biofluids were normally distributed, while
mass balance estimates of bioaccessibility (BioaccessibilityMB) were not normally
distributed. Further statistical analyses (α = 0.05) were done with the parametric
approach for direct bioaccessibility estimates and non-parametric approach for mass
balance bioaccessibility estimates. All statistical analyses were provided by SAS®
Program v9.1 (SAS Institute Inc., Cary, NC) and SigmaStat® Program v3.11 (Systat
Software Inc., Richmond, CA).

120
Table 31. Shapiro-Wilk normality test for the bioaccessibility of mercury from tuna samples
Experiment Data No. Tested Group Test Statistic p-value
60 Gastric Bioaccessibility 0.984694 0.6541* Direct Measurement 30 Intestinal Bioaccessibility 0.767012 0.9129*
60 Gastric Bioaccessibility 0.944817 0.0089 Mass Balance Estimation 60 Intestinal Bioaccessibility 0.875299 <0.0001
60 Gastric Bioaccessibility 0.985059 0.6731* Recovery
60 Intestinal Bioaccessibility 0.965221 0.0849* *Insufficient evidence to reject the assumption of normality
4.4. Results
4.4.1. Bioaccessibility / recovery for tuna steak (raw & cooked)
The results obtained for bioaccessible mercury in tuna steak are reported in Table
32. The averaged total mercury levels (mean ± S.D.) found in raw and cooked tuna steak
were 688.4 ± 23.2 ppb and 1075 ± 90.9 ppb, respectively. Estimates of bioavailability
obtained from direct method ranged from 48.7 % to 76.4 % for the gastric
bioaccessibility of raw tuna steak. The gastric bioaccessibility for cooked tuna steak
ranged from 49.1 % to 70.9 %. These gastric bioaccessibility data represent the maximum
levels of mercury expected to be released in the human digestive system. After the
addition of artificial intestinal juice into the samples, the intestinal bioaccessibility of
mercury was measured in each sample. The intestinal bioaccessibility of mercury for raw
tuna steak ranged from 33.8 % to 51.9 %, while the values ranged from 22.1 % to 57.8 %
for cooked tuna steak. The difference between the gastric bioaccessibility and intestinal
bioaccessibility was statistically significant (N = 30, pooled t-test, p < 0.0001).

121
Table 32. Mercury bioaccessibility / recovery data for tuna steak (wet weight)
Test Matrix Fluid BioaccessibilityD (mean±S.D.)
BioaccessibilityMB (mean±S.D.)
Recovery (mean±S.D.)
Gastric 49.3±6.2 % 80.6±4.2 % 68.7±2.2 % Raw
Intestinal 39.2±3.7 % 41.7±14.8 % 97.6±18.1 % Gastric 59.9±3.0 % 82.6±4.4 % 77.3±4.1 %
1
Cook Intestinal 22.1±8.0 % 32.3±4.6 % 89.8±4.9 %
Gastric 68.3±7.1 % 91.4±1.5 % 77.0±8.5 % Raw
Intestinal 33.8±2.8 % 60.3±2.8 % 73.5±5.0 % Gastric 65.2±4.3 % 85.1±0.5 % 80.0±4.8 %
2
Cook Intestinal 49.0±1.9 % 66.7±2.5 % 82.3±3.2 %
Gastric 48.7±1.7 % 82.7±3.0 % 66.0±4.2 % Raw
Intestinal 33.8±12.1 % 62.3±1.5 % 71.5±12.3 % Gastric 49.1±12.3 % 74.1±19.9 % 75.0±8.8 %
3
Cook Intestinal 45.3±4.9 % 64.3±1.0 % 81.0±5.8 %
Gastric 76.4±7.9 % 86.1±2.5 % 90.2±6.7 % Raw
Intestinal 44.2±12.9 % 71.8±2.0 % 72.4±13.6 % Gastric 70.9±1.7 % 85.6±0.6 % 85.3±2.0 %
4
Cook Intestinal 57.8±7.7 % 68.1±2.5 % 89.6±8.2 %
Gastric 69.6±8.1 % 86.7±1.8 % 82.8±7.1 % Raw
Intestinal 51.9±9.2 % 73.0±3.1 % 78.9±6.3 % Gastric 61.9±8.7 % 73.2±5.3 % 88.8±3.6 %
5
Cook Intestinal 22.6±6.7 % 35.3±1.5 % 87.3±7.0 %
The mass balance method for calculating bioaccessibility was also used for the
tuna steak samples. The gastric bioaccessibility of mercury for raw tuna steak and cooked
tuna steak, using the mass balance method, ranged from 80.6 % to 91.4 % and 73.2 % to
85.6 %, respectively. The intestinal bioaccessibility of mercury for raw tuna steak ranged
from 41.7 % to 73.0 %, and the bioaccessibility of mercury for cooked tuna steak ranged
from 32.3 % to 68.1 %. Thus, bioaccessibility determined by the mass balance approach
was generally higher than the values obtained using the direct bioaccessibility approach.
The reason may be resulted from the fact that the mass balance estimation represents the

122
portion of total mercury theoretically achievable in the extraction system, while the direct
measurement only shows the portion of mercury remaining in the sample after the loss of
mercury during in vitro sequential extraction. The difference in bioaccessibility obtained
for the gastric and intestinal fluids by the mass balance method was also significant (N =
30, Wilcoxon two-sample test, p < 0.0001). The recovery test was completed to
determine if there were mercury losses during each experiment. Most recovery results
were above 70 %, ranging from 66.0 % to 97.6 % for tuna steak study.
4.4.2. Bioaccessibility / recovery for canned tuna (light & white)
The results of the mercury bioaccessibility experiment for canned tuna (light and
white) are reported in Table 33. The averaged total mercury levels (mean±S.D.) found in
light and white canned tuna were 52 ± 3.5 ppb and 231 ± 5.7 ppb, respectively. For the
direct method of calculating bioaccessibility, the gastric bioaccessibility for light canned
tuna ranged from 36.6 % to 66.7 %, while the gastric bioaccessibility for white canned
tuna ranged from 47.3 % to 65.7 %. The intestinal bioaccessibility, however, dropped to
9.4 % and 2.1 % for light and white tuna, respectively. The detection limit (0.2 ppb) is
equivalent to 40.7 % and 18.2 % of total extracted mercury in intestinal fluid for light and
white canned tuna, respectively. All intestinal bioaccessibility data were below the
method detection limit (< 0.2 ppb), thus these data are not included in further analyses.
The mass balance method for calculating bioaccessibility was also applied for
canned tuna matrices (light and white). The gastric bioaccessibility of mercury for light /
white canned tuna ranged from 70.5 % to 77.9 % and 72.1 % to 76.8 %, respectively. The
intestinal bioaccessibility of mercury ranged for 27.9 ~ 36.8 % and 12.7 ~ 21.0 % for

123
light / whit canned tuna, respectively. The recovery results were mostly above 70 %,
ranging from 61.8 to 93.8 %, and close to the tuna steak values. The difference in
mercury bioaccessibility obtained between gastric fluid and intestinal fluid was
significant (N = 30, Wilcoxon two-sample test, p < 0.0001).
Table 33. Mercury bioaccessibility / recovery data for canned tuna (wet weight)
Test Matrix Fluid BioaccessibilityD (mean±S.D.)
BioaccessibilityMB (mean±S.D.)
Recovery (mean±S.D.)
Gastric 52.5±5.6 % 77.9±3.7 % 74.6±9.1 % Light
Intestinal BDL 35.3±2.9 % 74.0±2.9 % Gastric 65.7±7.2 % 76.8±4.2 % 88.9±11.0 %
1
White Intestinal BDL 17.3±5.5 % 84.8±5.5 %
Gastric 50.3±8.1 % 71.1±2.5 % 79.2±9.2 % Light
Intestinal BDL 27.9±4.3 % 81.6±4.3 % Gastric 54.2±1.8 % 72.1±1.4 % 82.1±1.2 %
2
White Intestinal BDL 13.9±1.6 % 88.2±1.6 %
Gastric 66.7±4.5 % 72.9±0.7 % 93.8±4.4 % Light
Intestinal BDL 32.4±5.3 % 77.1±5.4 % Gastric 63.8±5.5 % 75.2±2.6 % 88.6±7.4 %
3
White Intestinal BDL 12.7±1.7 % 89.4±1.7 %
Gastric 36.6±7.4 % 70.5±2.7 % 66.1±7.9 % Light
Intestinal BDL 29.7±6.3 % 79.5±6.4 % Gastric 47.3±2.6 % 76.8±1.0 % 70.6±2.6 %
4
White Intestinal BDL 21.0±2.5 % 81.1±2.5 %
Gastric 38.4±6.3 % 76.6±2.1 % 61.8±5.0 % Light
Intestinal BDL 36.8±1.7 % 72.9±1.7 % Gastric 53.5±3.0 % 74.9±0.7 % 78.6±2.4 %
5
White Intestinal BDL 16.4±2.0 % 85.7±2.0 %
*BDL: below the detection limit (< 0.2 ppb) In an attempt to account for mercury losses prior to in vitro sequential extraction,
the level of mercury volatilized from canned tuna was measured using a simple mercury
emission test. Each of three canned solid white and chunk light tuna (6 oz.) were opened

124
and placed inside a chamber with a volume of 0.15 m3 at room temperature (18 ~ 22 °C).
It took 4 hours, after opening the lid of a can, to achieve an equilibrium status inside the
chamber. Background ambient air mercury level was subtracted from measured mercury
concentration (ng/m3). The average mercury concentrations found inside the chamber for
white and light canned tuna were 107 ng/m3 and 34 ng/m3, respectively. Thus, only a
small portion of the mercury (approximately 0.12 % and 0.18 % of total mercury for light
and white canned tuna, respectively, in 4 hours) was volatilized at room temperature. The
mercury volatilization occurred through the diffusion of mercury concentration gradient
inside of chamber without the movement of air current. The previous total mercury
analysis showed that the white canned tuna has higher mercury concentration (231 ng/g)
than light canned tuna (52 ng/g).
4.5. Discussion
4.5.1. Direct vs Directadj vs Mass Balance bioaccessibility
Two different approaches were proposed to estimate bioaccessible mercury levels
in artificial biofluids used to attempt to simulate fish consumption. One was derived from
the direct measurements of mercury in extracted biofluids. The other was indirectly
estimated by the subtraction of recaptured mercury from total mercury based on a mass
balance for mercury, assuming all extracted mercury remained stable and bioaccessible in
the bottle except the non-extracted mercury retained on the filter. This mass balance
estimation was additionally proposed because the direct measurement of bioaccessible
mercury provided very low levels of mercury especially for all canned tuna samples (0.1
~ 0.2 ppb) after intestinal extraction. The lower concentrations of mercury extracted in

125
intestinal fluids may be resulted from the lower concentrations of total mercury found in
canned tuna (52 ppb for light and 231 ppb for white tuna) versus tuna steak (688 ppb),
and the possible mercury loss in the intestinal phase via volatilization, or sticking to the
surface of experimental apparatus (filtering system, digestion bottle…etc). The mass
balance estimation provided relatively higher mercury concentrations for both tuna steak
and canned tuna samples (all above 2.0 ppb), and lower variations in triplicate samples
(mostly below 5 %).
All direct mercury bioaccessibility data determined by this research were lower
than those of mass balance bioaccessibility estimates for the two biofluids and all tested
matrices. That difference was supported by the recovery obtained for entire extraction
procedure (N = 60). The mercury recovery was 80.7 ± 8.3 % and 79.9 ± 8.3 % for tuna
steak and canned tuna, respectively. Thus, we adjusted the direct bioaccessibility (N =
60) for each sample by dividing the corresponding recovery (N = 60). For example, the
direct bioaccessibility for one sample of raw tuna steak (54.3 %) was divided by a
corresponding recovery (70.9 %) for the same sample. A new bioaccessibility (76.6% =
54.3 / 70.9) was generated, and the value was close to a mass balance estimate for the
same sample (83.4 %). This corrected bioaccessibility value (called directadj) should
compensate for the effects of mercury loss during the sample handling. The values of
adjusted direct bioaccessibility were much closer to the levels of mass balance estimated
bioaccessibility. However, directadj bioaccessibility values for canned tuna in the
intestinal phase were still lower than the values determined by the mass balance
estimation, due to very low concentrations of mercury in the intestinal fluid for canned

126
tuna. The graphical comparison among direct, directadj, and mass balance bioaccessibility
is illustrated in Figure 15.
The data obtained by the mass balance approach are appropriate for analyzing the
differences by fish preparation methods and gastrointestinal fluids; because they indicate
the maximum achievable levels of mercury in extracted biofluids and are much consistent
within the same test matrix. Thus, the discussion in following section is based only on the
mass balance estimation method.
Direct vs Directadj vs Mass Balance Bioaccessibility
0%
20%
40%
60%
80%
100%
Gastric Intestinal Gastric Intestinal Gastric Intestinal Gastric Intestinal
Raw Cooked Light White
Bio
acce
ssib
ility
(%)
DirectDirectadjMass Balance
Figure 15. The comparison of mercury bioaccessibility methods (Direct vs Directadj vs Mass Balance)
4.5.2. Bioaccessibility differences by biofluids and preparation methods
A higher bioaccessibility from gastric fluid was determined compared to intestinal
fluid in all tested sample sets. The difference was statistically significant (N = 30;
Wilcoxon two-sample test; p < 0.0001), and recovery for all experimental sample sets

127
showed the method recovered above 70 % of total mercury during the whole
experimental procedures.
In general, 95 % of methylmercury is absorbed in human gastrointestinal tracts
(Aberg et al., 1969; Miettinen, 1973). This value was also supported by several animal
tests (Yannai et al., 1993; Berglund et al., 1971; Charbonneau et al., 1976). However, our
finding was somewhat lower than the human / animal tests, especially due to the lower
bioaccessibility in the intestinal phase. Organic mercury comprises about 90 % of the
mercury in tuna tissue (Burger et al., 2005). The fraction of organic mercury and
inorganic mercury absorbed is 95 % and 7 %, respectively (Burger et al., 2005). Thus, the
total amount of mercury absorption is expected to be around 86 % from the fish. The
gastric bioaccessibility determined by mass balance approach was close to the previously
reported 86 percent. Therefore, the gastric bioaccessibility determined by mass balance
approach provided reliable levels to estimate in vitro bioaccessible mercury from two
tested tuna matrices (tuna steak and canned tuna). This suggests that the mercury in
gastric phase is dissociated from tuna muscles at the expected levels (86.2 of total
mercury as an absorption fraction in human gastrointestinal tract) due to gastric fluid’s
very strong acidity (pH = 1.4 ± 0.2) and physiological conditions (temp = 37 °C and
mixing at 90 rpm).
After the addition of intestinal juice, the acidity of intestinal sample was increased
to pH = 6.5. The bioaccessibility dropped significantly (p < 0.0001), to a range of 16.3 %
and 62.8 %. The decrease of bioaccessible mercury in intestinal phase was not expected
in the study, because more tissue-bound mercury would be dissolved in the intestinal
fluid with the completion of digestion process as the undigested tuna tissue in gastric

128
phase was transferred to the intestinal fluid. On explanation for the decline in
bioaccessible mercury in the intestinal phase, it may be 1) the transport mechanisms of
mercury in digestive fluid, and 2) biochemical interactions with proteins rich in the
experimental assay from tuna flesh (personal communication with Dr. Alan H. Stern).
The transport mechanism for mercury in biofluids is not dependent on solubility
only, especially for methylmercury. Due to organic mercury’s low water solubility (1.0
g/L at 21 °C as dimethylmercury), methylmercury is expected to be relatively lipid
soluble (NAS, 2000). The organic mercury absorbed across the small intestine can be
transported actively by the complexes of Hg-SH (monovalent or divalent mercury cations
+ sulfhydrl groups such as cysteine or glutathione due to its high affinity) through the
lumen of intestinal mucosa (NAS, 2000). The methylmercury cation can bind to the thiol
group of the amino acid cysteine, creating a chemical structure similar to that of the
essential amino acid methionine (Clarkson, 1995). In such a manner, methylmercury can
cross the blood-brain barrier "disguised" as an amino acid that is the basic form of
breakdown of proteins in biochemistry via a carrier-mediated system (i.e., transport is not
solely the result of methylmercury’s lipid solubility). Thus, some dissolved mercury
(ionized monovalent or divalent mercury in gastric phase) can bind very tightly with free
cysteine (most active one from sulfhydryl or thiol groups and abundant in the
gastrointestinal fluid with the result of tuna tissue digestion), and may re-adsorb into the
undigested tuna tissues with the condition of neutral pH.
Lawrence et al. (1999) showed that the dissolved monomethylmercury (filtration
passing) from environmentally contaminated sediments in digestive juice of deposit
feeding lugworm was decreased over time ranging from 13 % at 0.5 hour of residence

129
time to 6 % at 4 hours of residence time. The reduction of dissolved methylmercury over
time was 53.8 % for the lugworm study. To estimate how much of the dissolved mercury
would be adsorbed onto the tuna tissues from gastric fluid to intestinal fluid for this study,
the mercury reduction was calculated. The result (mean±S.D.) was 27.9 ± 13.6 % and
33.6 ± 21.3 % for raw and cooked tuna steak, respectively, and it was greater for light
and white canned tuna, 56.2 ± 3.2 % and 78.4 ± 3.9 %, respectively. The authors
concluded that the decrease in the amount of soluble methylmercury over time may be
due to partitioning and reabsorption of methylmercury to particles, coagulation of
colloidal material, or adsorption / precipitation of protein onto particulate matter rather
than decreased solubilizing power of the digestive fluid (Lawrence et al., 1999). Animal
studies also showed that the in vivo absorption of mercury in intestine was reduced by the
addition of dietary fibers (wheat bran, corn silage) with administered mercuric chloride
diets (Rowland et al., 1986; Yannai et al., 1993; Kimura et al., 2001). They explained the
effect of dietary fiber in lowering absorption of mercury by reducing gastrointestinal tract
residence time and by adsorption processes.
For this study, the undigested tuna tissues provided binding sites for mercury-
cysteine complexes similar to the role of fibers or sediments that adsorbed some
dissolved mercury in digestive fluids and consequently lowered the fraction of mercury
absorption in the experiment of in vitro and in vivo. Therefore, some portion of mercury
complexes, ranging from 23.7 % to 58.9 % of dissolved mercury in gastric fluid, could
have been adsorbed onto the surface of undigested tuna flesh, and couldn’t penetrate the
filter (pore sized of 0.45 µm) as a dissolved form; instead they came out as a part of
mercury mass precipitated on the recaptured filter. This explanation is supported by an

130
equivalent recovery percentage from gastric and intestinal samples for all four kinds of
tested tuna matrices.
The difference of bioaccessibility was also explored in four different tuna
matrices: raw tuna steak, cooked tuna steak, light canned tuna, and white canned tuna.
For gastric mass balance bioaccessibility (Table 34), there was a difference among four
test matrices (Kruskal-Wallis test; N = 15; p < 0.0001). A non-parametric post hoc
multiple comparison test (Tukey’s method for balanced subgroups, α = 0.05) was
conducted for gastric bioaccessibility by serving method. The test revealed a difference
(p < 0.05) between the tuna steak and canned tuna; however, the difference was not
significant either between raw and cooked tuna steak or between light and white canned
tuna. Therefore, the mercury bioaccessibility in the gastric phase of the human digestive
system is higher for tuna steak compared to canned tuna.
Table 34. Non-parametric tests for gastric bioaccessibility by preparation ways (Kruskal-Wallis ANOVA test and post hoc multiple comparison test)
Matrix No. Mean Score p-value Min. 25 % Med. 75 % Max. Grouping
Raw Steak 15 47.267 0.758 0.834 0.852 0.883 0.926
Cooked Steak 15 37.600 0.511 0.779 0.846 0.858 0.876
White Can 15 20.933 0.705 0.728 0.750 0.767 0.810
Light Can 15 16.200
Pr < χ2 (<0.0001)
0.675 0.716 0.736 0.758 0.819
A A A B B B
*Different letters indicate significant differences by balanced Tukey’s multiple comparison test; the same letter indicates no difference between median ranks
For intestinal mass balance mercury bioaccessibility (Table 35), there was a
difference among four tested matrices (Kruskal-Wallis test; N = 15; p < 0.0001). A non-
parametric post hoc multiple comparison test (Tukey’s method, α = 0.05) was conducted,
and it was found that white canned tuna was different (p < 0.05) from the other three

131
samples, and light canned tuna was significantly different (p < 0.05) with raw tuna steak;
however, the difference between tuna steaks (raw and cooked) was not significant.
Table 35. Non-parametric tests for intestinal bioaccessibility by preparation ways (Kruskal-Wallis ANOVA test and post hoc multiple comparison test)
Matrix No. Mean Score p-value Min. 25 % Med. 75 % Max. Grouping
Raw Steak 15 46.467 0.278 0.586 0.635 0.712 0.755 Cooked Steak 15 41.200 0.270 0.349 0.640 0.654 0.702
Light Can 15 26.267 0.229 0.290 0.332 0.366 0.386
White Can 15 8.067
Pr < χ2 (<0.0001)
0.115 0.129 0.157 0.183 0.235
A A A B B B C
*Different letters indicate significant differences by balanced Tukey’s multiple comparison test; the same letter indicates no difference between median ranks
The differences in bioaccessible fraction found for gastric or intestinal extraction
may have been affected by way the tuna was prepared (Figure 16). Greater decrease from
gastric to intestinal bioaccessibility was observed for canned tuna versus tuna steak.
Canned tuna sample had lower gastric bioaccessibility, which could provide more
adsorption capacity (i.e., undigested tuna tissue) for mercury dissolved in digestive fluids
than tuna steak.
Hg Bioaccessibility in Tuna
0%
20%
40%
60%
80%
100%
Raw Tuna Steak Cooked Tuna Steak Light Canned Tuna White Canned Tuna
Tuna Matrices
Bio
acce
sssi
bilit
y (M
B)
GastricIntestinal
Figure 16. The comparison of mercury bioaccessibilityMB by different fish preparation methods

132
4.5.3. Bioaccessible mercury for fish matrix
The soil-based bioaccessibility method was adapted for a fish matrix (tuna steak
and canned tuna) to estimate the exposure to mercury by human consumption, especially
for sub-population group such as childbearing aged women. Through the bioaccessibility
experiments for two types of tuna, we have achieved progress on the adaptation of our
bioaccessibility system for use in a different exposure pathway. However, the
bioaccessible mercury results were lower than general value of 95 % reported for
methylmercury absorption in human gastrointestinal system.
In the gastric phase, in vitro sequential extraction procedures provided relatively
comparable levels for bioaccessible mercury, using either an adjusted direct
bioaccessibility or a mass balance estimate of bioaccessibility. A bioaccessibility, at most,
of 85.5 % for mercury tightly bound to tuna muscle was determined for the sequential
extraction in gastric phase. The solubilizing power of gastric fluid (hydrochloric acid +
pepsin) dissociated the tissue-bound mercury from the tuna muscles and ionized it
(usually monovalent or divalent status) into gastric fluid. Mass balance estimation in
gastric phase was close to the expected levels of mercury bioavailability (86.2 %) from
usual tuna consumption.
Mercury bound to tuna tissue is dissolved almost completely in human
gastrointestinal tract, and then the dissolved mercury is absorbed through the lumen of
intestinal walls. For example, methylmercury is approximately 90 % of the total mercury
in tuna and is actively transported through wall of intestine as methylmercury-cysteine.
Inorganic mercury, which is approximately 10 % of the total mercury in tuna, is not

133
absorbed actively like methylmercury. Generally 95 % and 7 % of the total mercury is
absorbed through the human gastrointestinal tract as methylmercury and inorganic
mercury, respectively. Thus, the absorption mechanism of mercury in tuna is not only
dependent on solubility, but also active transport and biochemical complexes. The
proposed approach for bioaccessible mercury may underestimate the bioavailability of
mercury. Since the sequential extraction system is only based on solubility of mercury
that is bound to tuna tissue, and the artificial digestive fluid does not digest the tested
tuna matrix completely. The direct measurement data were lower than the mass balance
estimation data throughout all tested sample matrices, ranging from 50.0 % to 91.6 % of
the estimated bioaccessibility by mass balance. The proposed direct method for
calculating bioaccessibility was not appropriate, especially for low-level mercury samples
(e.g., canned tuna matrices).
In the intestinal phase, however, the methods for estimating bioaccessible
mercury either by direct measurement or mass balance estimation were not relevant for
determining mercury bioaccessibility in the extracts. All direct intestinal bioaccessibility
for canned tuna were below the method detection limit (0.2 ppb). There may be two
reasons for the lower bioaccessibility in intestinal fluid compared to gastric fluid. One is
the possible readsorption of mercury onto the undigested tuna tissues that remained in our
in vitro extraction system. The other is mercury loss during the sample preparation or
lower concentration of mercury in the sample tested.
The artificial intestinal juice used did not contain some enzymes that are
important components for breaking down of proteins and fats in human guts. Bile
secretion enzyme (bile salts) by liver can start to dissolve water-insoluble fats in food

134
matrix; pancreatic secretion enzyme (trypsin, chymotrypsin, carboxypeptidase) by
pancreas can finish digesting proteins to amino acids that are object to intestinal
absorption. The tested intestinal fluid was composed of only 0.2 M of sodium bicarbonate
to return the pH to neutral when mixed with saliva+gastric solution in test bottles. The
lack of enzymes in the prepared gastrointestinal fluids might cause the incomplete
digestion of tuna tissues in gastrointestinal system, not releasing all tissue-bound mercury
in artificial digestive fluids, and subsequently allow the readsorption of dissolved
mercury onto the undigested tuna tissues during the in vitro extraction system. Because
proteins are not completely digested in the human stomach, but rather the digestion
process requires the presence of pancreatic secretions in small intestines to be completed.
The in vitro method for measuring bioaccessible mercury from food matrix was
developed to estimate the mercury levels in simulated human gastrointestinal system.
Based upon the experiments, the gastric bioaccessibility obtained by mass balance
approach came close to the level of mercury absorption reported from in vivo
experiments (86.2 % by general knowledge); however, intestinal bioaccessibility,
physiologically more relevant for human absorption than gastric bioaccessibility, did not
reach the reported mercury absorption value. Analytical difficulties have been observed
when intestinal fluid is prepared for in vitro sequential extraction (Hamel, 1998). Thus, a
method, that only uses the levels of mercury in gastric fluid and the mass balance
approach for calculating bioaccessibility, is appropriate for estimating bioaccessible
mercury from food matrix at this time. This is actually a strong conclusion since the
method is simple, and can be used as a conservative screening tool for mercury and other
metals found in food matrices.

135
V. Summary and Conclusions
5.1. Bioaccessible Radionuclides in SRS Soils
A method to measure bioaccessibility has been successfully adapted for two
radionuclides (cesium-137 and strontium-90) for both low-level radioactive-contaminated
soils (class “B” and “C”) and high-level radioactive-contaminated soils (class “A”) from
the Savannah River Sites. The method development adopted several procedures (e.g.,
sample processing unit for hydrofluoric acid digestion, soil sieve unit, glove box, swipe
test) to minimize the radiation contamination for the radioactive soil experiments at the
EMAD-EOHSI laboratory.
For cesium-137 bioaccessibility estimates, the gastric bioaccessibility (8.9 ~
22.4 %) was lower than or equal to the levels of intestinal bioaccessibility (8.3 ~ 38.8 %).
However, the gastric bioaccessibility (29.7 ~ 97.1 %) was higher than intestinal
bioaccessibility for strontium-90 (17.8 ~ 60.5 %). The comparison of gastric and
intestinal bioaccessibility by soil hazard class showed that strontium-90 of class “A” soil
was more soluble than that of class “B” or “C” soil in simulated gastrointestinal tract (P <
0.05). The difference of bioaccessibility within soil class was also observed for cesium-
137, and the bioaccessibility of class “C” soil was higher than that of class “B” soil (p <
0.05). The sub-population group, children with pica activities, was predicted to be
potentially at the elevated risk of radiation exposure to cesium-137 and strontium-90 via
the non-dietary ingestion or external exposure to radioactive-contaminated soil such as
the surface soil of Chernobyl Nuclear Power Plant Exclusion Zone (< 30 km) or
Resettlement Zone (15 ~ 40 Ci/km2 of 137Cs). The values predicted to exceed the NCRP

136
negligible dose limit (0.01 mSv) for cesium-137 and strontium-90 when residing in the
ChNPP Exclusion Zone at least one year.
The in vitro bioaccessibility method for radionuclides in radioactive-contaminated
soils allows the estimation of radionuclides bioavailability is lower in cost and quicker to
perform than in vivo test. Further, it can be used safely to model human exposure / dose
from radionuclides-contaminated soils. The bioaccessibility measurements for
radionuclides contaminated soils can be used to improve risk prioritization of
contaminated sites as well as enhance risk assessments by increasing their accuracies.
5.2. Bioaccessible Lead in Carpet House Dust
The bioaccessibility method for lead in vacuumed house dust derived from the
method used for soils has been successfully streamlined to quantify the bioaccessible
fraction of lead in environmentally contaminated house dust. The method was optimized
for lead in house dust by a slight change in parameters (e.g., volume of artificial fluids
and amount of house dust used). Reliable values of bioaccessibility were obtained in both
gastric and intestinal fluids with overall good recoveries (above 70 %).
The gastric bioaccessibility for lead in house dust (sieved to below 75 µm-
diameter size) from an urban environment was significantly higher than the intestinal lead
bioaccessibility (p < 0.0001). This was due to the readsorption of dissolved lead onto the
surface of dust particles at a higher pH in the intestinal fluid. The level of intestinal
bioaccessibility was significantly different (p = 0.0067) among three dust particle size
fractions (< 75 µm, 75-150 µm, and 150-250 µm), but the gastric bioaccessibility was
equal for each size fraction (p = 0.7019).

137
The bioaccessibility data obtained for two biofluids (gastric and intestinal juice)
was applied to the IEUBK model, and provided a support for the 30 % absorption default
value used for dust bioavailability. However, since the model did not predict actual high
blood lead values in the children, sensitivity analyses should be expanded to include
absorption values up to 65 % to reduce the underestimation for site-specific lead on
individuals. The variability in bioaccessibility observed in these experiments could
contribute towards the uncertainty in estimations of blood lead concentrations for young
children from non-dietary ingestion of dust. In addition, the bioaccessibility method for
lead in house dust can be expanded to other toxicants present in house dust (e.g.,
cadmium, arsenic, copper…etc).
5.3. Bioaccessible Mercury from Fish Consumption
A method to measure bioaccessible mercury for fish matrix was developed based
on the current bioaccessibility method for soil. The mercury bioaccessibility study was
conducted for commercial tuna products (supermarket-purchased tuna steaks and canned
tunas). Two approaches, direct measurement and mass balance estimation, were used to
report the bioaccessible mercury from human fish consumption. Mass balance estimation
was relevant than direct measurement to calculate the bioaccessible mercury in sequential
extraction system, especially for low-level of mercury in canned tuna.
The mass balance bioaccessibility for gastric fluid ranged from 73.8 % to 85.5 %
for tuna. While for intestinal fluid, the bioaccessibility by mass balance estimation was
lower and ranged from 16.3 % to 61.8 %. Thus, the experiments concluded that the level
of mercury extracted by gastric fluid is more appropriate than intestinal fluid as an

138
indicator of bioaccessible mercury extracted from food matrix in simulated human
gastrointestinal system.
The study estimated the bioaccessible mercury obtained from various prepared
methods for tuna. The results showed that tuna steak (either cooked or uncooked) had
statistically higher bioaccessibility than canned tuna (light and white tuna) in both the
gastric and intestinal phase (p < 0.05). Additionally white canned tuna had lower
bioaccessibility than light canned tuna in the intestinal phase (p < 0.05).
The current method for measuring bioaccessible mercury from tuna consumption
has been modified, and a combination of using only the gastric fluid digestion results
with a mass balance approach for calculating bioaccessible contaminants provides a
reasonable screen tool for other elements found in food.
5.4. Conclusions
• The magnitude of strontium-90 bioaccessibility was proportional to the soil
hazard class (“A” > “B” ≥ “C”); however, the relation of cesium-137
bioaccessibility was not proportional to soil hazard class (“A” = “B” ≤ “C”).
• The dose predictions for cesium-137 and strontium-90 from the exposure to
radioactive-contaminated soil such as the surface soil of ChNPP Exclusion Zone
showed that the non-dietary ingestion was critical for pica / young children due to
their high soil ingestion amount; however, the external exposure by the
penetration of radiation was significant for older children / adults.
• The difference between gastric and intestinal bioaccessibility for lead in house
dust was significant (p < 0.0001), and explained by the readsorption onto the

139
residual house dust and / or precipitation of dissolved lead due to the increase of
pH from 1.4 to 6.5 in the intestinal phase.
• The levels of lead bioaccessibility for three particle size fractions in house dust
were not statistically different in the gastric phase (p = 0.7019), however they
were significantly different in the intestinal phase (p = 0.067).
• The intestinal bioaccessibility data supported the IEUBK default parameter
(30 %) as a reasonable estimator for children’s blood lead level predictions;
however, the gastric bioaccessibility could yield the model predictions close to the
reported blood lead levels (> 11 µg/dL), and may be useful for future use in
sensitivity analyses to reduce the underestimation of a child’s blood lead value.
• Direct measurements and mass balance estimations were tested to report the
mercury bioaccessibility from human fish consumption; the mass balance
approach was more relevant than direct measurement approach for representing
mercury levels in gastrointestinal tract, especially for low-level mercury samples
(i.e., canned tuna). However, the method is not used for field application.

140
VI. Recommendations for Future Research
Through the experiments for proposed bioaccessibility studies, we have modified
the current bioaccessibility method into various environmental media and toxicants that
are important to understand the estimation of exposure / dose by the pathway of dietary
or non-dietary ingestion in human gastrointestinal tracts. However, some research is still
required in the future. Here are some recommendations for future work:
First, the relationships between soil physico-chemical characteristics and
bioaccessibility of environmental radionuclides are not clear. Future studies for
investigating the soil physico-chemical factor(s) to determine the level of bioaccessibility
for radionuclides are necessary for risk assessments based on site-specific soil as well as
future soil remediation / prioritization processes.
Second, three possible mechanisms were mentioned in lead study to explain the
decreased lead levels in intestinal phase: readsorption of lead onto house dust particles,
complexation by pepsin, and chemical precipitation of the lead caused by phosphates in
higher pH environment. Future work is needed to explore the feasibility of each
mechanism in simulated gastrointestinal system to understand the behavior of dissolved
lead in the sequential extraction.
Third, the method for measuring bioaccessible mercury from food matrix was not
developed to relevantly report the mercury levels in tested fish samples. The method
should be reliable for various foods and comparable to in vivo study results. Future
studies are needed to develop the bioaccessibility method working for food matrices in
the simulated human digestive system for environmental toxicants.

141
Appendices 1. CRESP SOP-001 “Soil Drying” 1.0 Cover Page Author(s): Kristie Ellickson Date Robert Meeker Date Scott Petlick Date Mike Pollock Date
Carl Schopfer Principal Investigator(s): David Kosson, Ph.D. Date Paul Lioy, Ph.D. Date Robert Tata, Ph.D. Date Authoree: David Kosson, Ph.D. Date EOHSI Radiation Safety Committee Chair: Arthur Upton, M.D. Date

142
2.0 Experimental The traditional definition for a “dry soil” is one that has come to constant weight after being in an oven at a temperature between 100 and 110ºC. Soil samples dried to constant weight may provide more precise results, as some physical and physico-chemical properties of soil depend on water content. For example, pore size may vary with water content and may also affect metal availability in soil. If soils are dried to approximately the same water content (by means of constant weight analysis) this variable can be controlled. 2.1 Hypothesis A soil sample dried to constant weight will be apportioned more easily to smaller particle sizes. 2.2 Objectives Approximately 200 soil samples will be dried in preparation for sieving. Drying will be conducted in a vacuum dessicator or drying oven to a constant weight. Resultant samples will be stored in a vacuum dessicator until they can be processed further. 2.3 Experimental Design Samples will dried for a specified amount of time, weighed, dried for that same time, and then weighed again until scale measurement is constant. 2.4 Method Summary Soil samples are dried in either an oven or vacuum dessicator. The oven is used for gross drying, whereas the vacuum dessicator will be used for the final drying step. 2.5 Procedure Quality Objectives Soils will be dried to constant weight. This weight must be constant to the first decimal point. For example 100.1, 100.2, and 100.3 would be considered constant drying weights. 3.0 Radionuclides 3.1 Radionuclides present in the soils to be processed are from core samples of seepage
basins used to dispose of radioactive wastes over several decades. Other samples to be processed may include surface soil samples from areas thought to be radioactively contaminated. Also, background samples will be processed. (Background soils contain no known radionuclide contaminants other than that which may be naturally present.)

143
3.2 The EOHSI Radiation Safety committee will review radionuclide characterization data prior to releasing each sample. All samples, including background samples, are characterized for radionuclides prior to use.
3.3 The principal investigator prior to authorizing work with any sample shall consider
the possibility of the presence of other toxic contaminants, such as metals or organics. 3.4 Each principal investigator supervising persons using this procedure shall review the
known or suspected contents of each sample, the sample quantity and its physical form and may consult with members of the EOHSI Radiation Safety committee if desired.
3.5 The principal investigator shall discuss with the technician any hazards in the sample. 3.6 Technicians processing soils shall observe any quantity limits imposed by the EOHSI
Radiation Safety Committee and shall follow instructions regarding custodianship of samples and contaminant inventory.
4.0 List of Procedures This protocol includes one standard operating procedure, SOP-001, ”Soil Drying.” 4.1. Purpose Soil is dried in order to obtain an additional degree of control over an experiment. Drying allows soil to be processed with greater ease in its entirety. Drying soil is applicable to most applications, but will affect some biological, physical, and chemical characteristics. 4.2 Scope and Application 4.2.1 This procedure applies to experiments requiring constant weight dryness from soils
and sediments that are declared to be or which may be radioactive, or have been determined to require handling appropriate for radioactive hazards.
4.2.2 Approximately 200 samples will be dried using this method. Approximately 40 of
these will be samples containing substantial amounts of radioactive isotopes. One hundred fifty four of these samples will contain background or low-levels of radioactive contaminants.
4.2.3 This procedure is an approved “plug-in” procedure for CRESP personnel. 4.3 Summary Field-moist samples dried to constant weight. Constant weight will be obtained by the use of one or more of the following pieces of equipment: drying oven, vacuum oven, or vacuum desiccators.

144
4.4 Definitions 4.4.1 Glove Bag - a completely sealed, disposable, plastic containment apparatus with an
internal frame used to avoid release of dust, aerosols, or other contaminants to the laboratory.
4.4.2 Soil Drying Jar - A Wide Mouth Nalgenetm Jar used to dry the soil, and then
inverted directly into the sieving apparatus.
4.4.3 HEPA filter box - an apparatus containing two suction hoses which will be fitted into the wall of the glove box and uses a blower or fan to suction away any airborne contaminants from the glove box. The box contains a HEPA filter that traps soil particles.
4.5 Responsibilities 4.5.1 All technicians performing work under this protocol are responsible for following
all instructions as written. 4.5.2 The principal investigator supervises persons implementing this protocol. 4.5.3 The authoree, when not the principal investigator, approves the written protocol and
may provide instructions to the technician as appropriate. 4.5.4 EOHSI Radiation Safety Committee members may provide advice on protocols and
materials safety as requested. 4.5.5 Scott Petlick acts as Sample Manager. 4.5.6 The following people can provide on-the-job training (OJT) on this protocol. Kristie Ellickson – soil sieving (module OJT-002) Robert Meeker – soil sieving (module OJT-002) Michael Pollock– soil sieving (module OJT-002) Carl Schopfer - health physics surveys and instrumentation (module OJT-001) 4.6 Prerequisites All soil samples must be in 250ml Wide Mouth Nalgenetm Polypropylene Sample Jars before transfer. 4.7 Interferences

145
Interferences of concern are the cross contamination of soil samples. In order to eliminate this possibility, polypropylene bottles will not be reused and adhesives with low leaching characteristics will be used. 4.8 Materials 4.8.1 Parafilm M 4.8.2 1000ml Wide Mouth Nalgenetm Sample Jars, with lids 4.8.3 250ml Wide Mouth Nalgenetm Sample Bottles, with lids 4.8.4 Paper Towels or Kimwipes 4.8.5 laboratory tape 4.8.6 Sharpi magic marker 4.8.7 decontaminating detergent (Picoclean) 4.8.8 TORBAL* Flexible Glove Cabinet with Frame (Vertex# GC-2 VWR# 32980-
000) 4.8.9 Grade #1 Filter Paper 3.2cm diameter (Whatman # 1001-032, VWR# 28450-021) 4.8.10 Respirator with HEPA filter cartridge 4.8.11 Timer 4.8.12 personal safety equipment: elbow-length gloves, Tyvek laboratory coat, eye
protection Plastic Adhesive (3M Scotchtm Super Glue Gel)
4.9 Equipment 4.9.1 Drying Oven 4.9.2 Vacuum Oven 4.9.3 Vacuum Desiccator 4.9.4 Appropriate radiation survey instruments 4.9.5 PACE multi arm evac II fume extraction system with HEPA/Gas filtration
cartridge 4.10 Safety 4.10.1 All experiments will be carried out in compliance with safety recommendations
provided by the Radiation Safety Committee at Rutgers University, which is authorized by the Nuclear Regulatory Commission and the State of New Jersey (see reference 1.20.2.)
4.10.2 Samples will be stored at room temperature. Constant weight dried samples will
be stored in a vacuum desiccator. 4.10.3 Soil, which can be released as a dispersible dust or aerosol, represents a potential
inhalation hazard. The work being performed creates fine particles, which must be contained to avoid exposure to the worker and others in the vicinity.

146
4.10.4 Soil drying is accomplished with minimum disturbance to the media and any outflow is filtered where possible.
4.10.5 To contain the samples during sieving and sample transfers requires working in a
closed, HEPA-filtered chamber or glovebox. The entire assembly is placed in a fume hood.
4.10.6 Soil materials should be contained within and transferred between sealed vessels.
The drying takes place while shaking within a sealed assembly. Opening the assembly is done in a filtered environment, after settling has take place. Dust creation is minimized by not allowing the soil to dry completely. Static electricity is avoided by not using plastic bags as the primary containment. Double containment is used, such as a bottle within a bag, or sealing an assembly with plastic wrap within a filtered chamber.
4.10.7 Samples must be in closed containers when removed from the filtered enclosure or
fume hood. 4.10.8 The potential for radiation exposure from soils will be determined prior to starting
work. The quantities approved for use will generally be limited by the exposure potential such that exceeding regulatory limits will not be possible, or determined to be extremely unlikely.
4.10.9 Radiation dosimetry badges will be worn if necessary. 4.10.10 All samples are labeled with the sample identifier code and radiation hazard
class. A chain of custody form is kept with each container. (See reference 1.) 4.10.11 Protective eyeglasses, dust mask, long-sleeved laboratory coat, long pants, and
two pairs of disposable vinyl gloves (elbow length) will be worn when working with radioactive samples. Gloves are to be surveyed periodically during work, and when removed. Hands must be washed after gloves are removed.
4.10.12 Laboratory coats, safety glasses and other safety wear must remain in room 149.
Do not bring the items into the counting room (room 145.) 4.10.13 All items shall be surveyed for contamination before exiting the laboratory. 4.10.14 Bench surfaces shall be covered with disposable bench paper. 4.10.15 Survey the work area and the laboratory for radioactive contamination at the end
of each day. 4.10.16 Wastes are contained in closed containers for pick-up by REHS and disposed per
agreement with the Savannah River Site. (See references 2 and 3.)

147
The Drying Oven, Vacuum Oven, and Vacuum Desiccator will be equipped with traps to ensure that the sample will not be released to the laboratory environment as well as protect the vacuum pump. 4.11 Sample Collection, Handling, and Preservation Soil samples will be collected by the Department of Energy and will be stored at Rutgers Environmental Health and Safety Department’s Environmental Services Building. Samples are stored in 1-liter polyethylene bottles, wrapped in a polyethylene bag, and placed in Styrofoam coolers/shipping boxes. Samples will be requested by the researcher by completing a sample request form and samples will only be transported by REHS. A record of sample flow will be recorded with a chain of custody form that will be assigned to each soil sample (See reference 1.) 4.11.1 Quality Control Constant weight drying will be tested according to the procedures below. To eliminate cross contamination, the sample bottles and jars will not be reused rather disposed after a soil sample has been dried. Each sample will be dried to the same extent. Samples will be labeled with a batch number, the date of sieving, and the sample identification number in order to eliminate possible identification confusion. Finally, any balances used will be checked with a test weight prior to use. 4.11.2 Calibration Balances will be calibrated on an annual basis. This calibration will be checked by a test weight before use. 4.11.3 Calculations None 4.11.4 Procedure Read this entire document before beginning any work in this section. Note: When working with radioactive soils, keep materials and equipment away from the edge of the laboratory bench at all times. 4.11.4.1 Weigh a 1000ml Wide Mouth Nalgenetm sample jar and lid. 4.11.4.2 In order to transfer the soil in a closed environment, seal the lid of a 250ml wide
mouth Nalgenetm Sample bottle to the lid of a 1000ml wide mouth Nalgenetm sample jar with an epoxy adhesive. The top of the two lids should be bonded

148
facing away from one another and so the smaller (250ml) lid is in the center of the larger(1000ml) lid.
4.11.4.3 Once the epoxy adhesive has bonded, drill a 1/2” circle into the center of the two
lids. 4.11.4.4 Secure the larger lid onto a 1000ml Nalgenetm sample jar. 4.11.4.5 Invert the 1000ml jar, and screw the lid onto the 250ml wide mouth bottle. 4.11.4.6 Transfer approximately 100 grams or less of soil into the 1000ml Wide Mouth
Nalgenetm sample jar within a glove bag. This glove box must have a HEPA filter attached to capture dust.
Note: It may help to mark an approximate 100 gram point on the 1000ml sample jar. 4.11.4.7 After the transfer, turn on the HEPA vacuum for 1 minute. Note: While the HEPA vacuum is on, make sure the hood sash is down to operating
level, or lower. 4.11.4.8 Wipe the sample jar with a moistened paper towel. 4.11.4.9 Take a wipe of the sample jar before removing it from the glove box. Include the
inner accessible part of the lid, and measure the wipe with the appropriate survey equipment.
4.11.4.10 Weigh the 1000ml Wide Mouth Nalgenetm sample jar and soil, and calculate
the approximate soil weight by subtracting the soil and jar from the initial jar weight.
4.11.4.11 Once the soil has been weighed, remove the tops from the jars and place them
in the oven at 100°C -110°C to dry. 4.11.4.12 Shake the soil occasionally back and forth in order to allow the soil on the
bottom of the sample jar to dry as well. 4.11.4.13 When the soil appears dry (light in color, and a loosening of the texture), place
the soil on a balance and monitor stability of the weight. 4.11.4.14 If the weight decreases over time, the soil will need to be dried further. If the
soil is still quite humid, place the soil jar in the drying oven again at 100°C - 110°C.
4.11.4.15 If the weight of the soil remains unstable, a vacuum oven may be used to take
off the last portion of water from the soil. This final drying step can be

149
conducted using a vacuum oven with the following settings: temperature = 105°C and pressure = 15”Hg.
Note: Make sure temperature within the vacuum oven is maintained at or below 110°F. 4.11.4.16 Once the soil has dried by visual observation, test for constant weight. 4.11.4.17 To test for constant weight, place the soil sample(with lid secured) into the
vacuum desiccator for one day. 4.11.4.18 After one day weigh the soil. 4.11.4.19 Place the soil in the desiccator for another day (times may be changed, but must
remain consistent through constant weight testing) .
4.11.4.20 After the second time in the vacuum desiccator, weight the soil sample again. If it is the same as the previous weight, this sample is ready for use.
4.11.4.21 Store all dried samples in a vacuum desiccator. 4.11.5 Storage and containment 4.11.5.1 All samples will be stored in sealed polypropylene or polyethylene containers. 4.11.5.2 Polypropylene jars will then be placed inside Ziploc storage bags as a secondary
containment measure. 4.11.5.3 Samples will be stored in the dark, under conditions of reasonable humidity and
room temperature. 4.11.5.4 Dried samples will be stored within a vacuum desiccator. 4.11.6 Cleaning or disposal of glassware and equipment 4.11.6.1 All weighing papers, spatulas, soil sieve and catch basin that exhibit
radioactivity when measured with a radiation meter will be disposed of in a container marked “radioactive solid waste”.
4.11.6.2 Dispose of radioactive soils, which do not pass through the sieve in the container
for radioactive solid wastes. 4.11.6.3 Remove all other solid radioactive wastes to a container for radioactive wastes. 4.11.6.4 Rinse all non-contaminated equipment with 1% reagent grade nitric acid. Rinse
acid washed equipment into a Buchner funnel and Erlenmeyer flask (separate from the funnel and flask used for acid washed materials.

150
4.11.6.5 Follow the 1% acid rinse procedure with a deionized water rinse following step. 4.11.6.6 In the case of mixed waste (liquid with solid), a vacuum filtration apparatus will
be used to separate solid from liquid. The solids, contained on the filter will be disposed of as solid waste. The liquids, contained in the Erlenmeyer flask, will be disposed of in liquid waste (see 1.15.5).
4.11.6.7 Deionized rinse water should be stored and disposed of separately from the 1%
nitric acid rinse, or neutralized. 4.11.6.8 Remove the liquid radioactive wastes to a container for radioactive wastes (see
1.16.5) 4.11.6.9 After operations are completed, check all work surfaces, non-waste materials,
and the regular trash, with radiation probes to assure that no radioactivity is present outside of permitted storage and waste containers.
4.11.6.10 Check for contamination on all equipment and containers and record the survey
results before removal from the radioactive facility. Check the activity of the normal trash as well, to ensure that no radioactive waste has been placed there accidentally.
4.11.6.11 After all operations are completed, perform swipes on all work areas and count
in a scintillation counter. 4.11.7 Waste Minimization By enclosing the drying and transferring apparatus in a small, filtered environment, we will minimize the spread of contamination and, therefore, the amount of cleanup. In addition, it may be possible to use the waste soil for other experiments that do not need smaller, sieved fractions. 4.11.8 Waste Disposal Wastes generated by these procedures will include dry solid waste such as gloves, paper towels, dust-containment apparatus and unused soil. Liquid waste will include any aqueous waste generated for the purpose of dust minimization/collection and decontamination procedures. Acidic Liquid waste will include any acid rinse waste collected in an Erlenmeyer flask. All waste from these procedures will be collected in appropriate containers for transport to the Savannah River Site for disposal. 4.11.9 Accident Instructions 4.11.9.1 Prior to beginning work, discuss with the OJT trainers how to respond to
accidents. Accidents that might result in an unplanned external radiation

151
exposure or in any internal exposure (inhalation or ingestion) require immediate REHS notification. Review the instructions on pages 24, 25, 29 and 30 of reference 1.20.2.
4.11.9.2 Small, localized spills (with no personal contamination) will be handled by the
technician. Use a spray bottle with water to prevent the spilled material from drying out and becoming airborne. Use moist paper towels to cleanse the spill area with a blotting action to prevent the spread of radioactive material. Use decontaminating detergent to clean surfaces. Check for persistent contamination following cleaning. Contact REHS immediately (445-2550, or 5-2550 from laboratory phones) if the area cannot be decontaminated. Dispose of the contaminated cleaning materials in the radioactive waste. Report the lost material to the Sample Manager.
4.11.9.3 If there is personal contamination, remove contaminated clothing. Use a spray
bottle with water to prevent the spilled material from drying out and becoming airborne, and cover with damp paper towels. Wash contaminated skin with mild soap and water. Do not use abrasives. Limit contact with others to avoid spreading contamination into clean areas. Exit the immediate area of the spill, but do not leave the laboratory area until advised by REHS or Rutgers Police.
4.11.9.4 Contact REHS for assistance in cleaning large spills. 4.11.9.5 In the event of dispersal of radioactive material outside of the ventilated
enclosure, exit the laboratory immediately and contact REHS. Restrict access to area.
4.11.10 References 4.11.10.1 EOHSI/CRESP Radiation Safety Global Procedures Manual. 4.11.10.2 Rutgers University Radiation Safety Guide, 7th edition, 1989. 4.11.11 Attachments/Enclosures 4.11.11.1 REHS Radioisotope Disposal Form, DY 6/93 AF/disp. 4.11.11.2 Chain-of-Custody form 4.11.11.3 Waste Log 5.0 Training Task-specific training, laboratory and radiation safety training are required before this procedure is used to process soil samples. All training modules must be completed successfully before work may begin.
5.1 On-the-Job Training (SOP-001, Soil Sieving) is required.

152
5.1.1 The technician shall demonstrate to the OJT trainer familiarity and understanding of all written procedures and manuals.
5.1.2 The technician shall obtain adequate training on the use of all equipment and an
understanding of expected outcomes. 5.1.3 The technician shall demonstrate the ability to successfully operate the equipment
under normal conditions with typical samples, and to recognize abnormal situations. 5.1.4 The technician shall demonstrate proficiency by obtaining the desired product or
results while performing the procedure under the direction of the OJT trainer. 5.2 Safety training includes at a minimum, the following components: Rutgers or UMDNJ Laboratory Safety Training Right-to-Know Training Radiation Safety Training On-the-Job Training - Health Physics Surveys and Instrumentation 6.0 Facility 6.1 Location Work conducted under this procedure will be performed in room 149, EOHSI. 6.2 Major Equipment Fume hood. 6.3 Time Factors, Staffing, and Resources No critical issues. 7.0 Other Reviews A copy of this procedure has been provided to REHS. 8.0 References SOP-002, Soil Sieving, November 13, 1998. Editor - Klute, A Method of Soil Analysis Part I – Physical and Minerological Methods. Agronomy. No. 9. Part 1. 2nd Edition. Madison, Wisconsin. 1986.

153
2. CRESP SOP-002 “Preparation of Soil Sub-Fractions” 1.0 Cover Page Author(s): Kristie Ellickson Date Robert Meeker Date Scott Petlick Date Mike Pollock Date
Carl Schopfer Principal Investigator(s): David Kosson, Ph.D. Date Paul Lioy, Ph.D. Date Robert Tata, Ph.D. Date Authoree: David Kosson, Ph.D. Date EOHSI Radiation Safety Committee Chair: Arthur Upton, M.D. Date

154
2.0 Experimental Soil is sieved in order to obtain an additional degree of control over an experiment. Different particle sizes vary in biological activity, and in chemical and physical properties. For example, smaller particle sized fractions of a soil sample have higher surface area to volume ratio. Therefore, these fractions may have greater amounts of available or surface-bound contamination per unit mass. Sieving separates soil particles into different size ranges without mechanical grinding. 2.1 Hypothesis It is important to prepare soil and sediments to achieve a more uniform particle size distribution prior to experimentation. 2.2 Objectives Soils are separated to sizes appropriate for testing in a manner that will not alter bulk composition or structure of the component material. The methods described provide an efficient means to fractionate soil so as not to waste the limited amount of parent material. Fractionation and sample transfer is accomplished in such a way as to minimize the risk of exposing research personnel to airborne particles. In sieving, soil fractionation is accomplished without mechanical grinding. 2.3 Experimental Design The sieving process utilizes a stack of calibrated sieves that decrease in mesh size in a descending manner. By adding air-dried soil to the top of the stack and shaking for an appropriate amount of time, particles of various sizes are separated and collected. In addition, each sieve may be sprayed with water to minimize the creation of airborne particles, thereby minimizing the chance of exposure to workers. 2.4 Method Summary Soils are air-dried prior to sieving. Sieves of appropriate sizes are assembled in order of descending pore size. A sample of soil is added to the top of the sieve, the entire stack assembly sealed and then shaken for an appropriate amount of time. Moisture is added if necessary. The soil is allowed to settle. Soil fractions are collected individually from the bottom sieve, proceeding upward. Soils are stored in labeled polypropylene bottles. Disposal parts of the sieving apparatus are discarded as radioactive waste. Other parts will be decontaminated and surveyed prior to storage. 2.5 Procedure Quality Objectives Mechanical grinding of the soil is minimized when sieving to ensure only intact particles are used. Particle sizes may be verified by microscopic or light-scattering techniques if

155
necessary. Parts of the sieving apparatus are disposable; other parts will be decontaminated to minimize cross-contamination. 3.0 Radionuclides 3.1 Radionuclides present in the soils to be processed are from core samples of seepage
basins used to dispose of radioactive wastes over several decades. Other samples to be processed may include surface soil samples from areas thought to be radioactively contaminated. Also, background samples will be processed. (Background soils contain no known radionuclide contaminants other than that which may be naturally present.)
3.2 The Environmental and Occupational Health Sciences Institute Radiation safety
committee will review radionuclide characterization prior to releasing each sample. All samples, including background samples, are characterized for radionuclides prior to use.
3.3 The principle investigator prior to authorization work with any sample shall consider
the possibility of the presence of other toxic contaminants, such as metals or organics. 3.4 Each principle investigator supervising persons using the procedure shall review the
known or suspected contents of each sample, the sample quantity and its physical form and may consult with members of the Environmental and Occupational Health Sciences Institute Radiation Safety committee if desired.
3.5 The principle investigator shall discuss with the technician any hazards in the smaple. 3.6 Technicians processing the soils shall observe any quantity limits imposed by the
Environmental and Occupational Health Sciences Institute Radiation Safety committee and shall follow instructions regarding custodianship of samples and contaminant inventory.
4.0 List of Procedures 4.0.1 Before starting any work described in this procedure, the laboratory worker is
required to read all of this plug-in-procedure (PIP-002). 4.0.2 A prerequisite to this procedure is PIP-001, “Soil Drying”, which involves the
drying of the soil. 4.0.3 Included with this procedure is a procedure for sieving soils entitled “Preparation of
Soil Sub-fractions”. 4.1.1 Purpose Soil is sieved in order to obtain an additional degree of control over an experiment. Different particle sizes vary in biological activity, and in chemical and physical properties. For example, smaller particle sized fractions of a soil sample have higher surface area to volume ratio. These fractions may have greater amounts of available or

156
surface-bound contamination per unit mass. Sieving separates soil particles into different size ranges without aggressive mechanical grinding. 4.1.2 Scope and Application
4.1.2.1 This procedure applies to experiments requiring uniform particle sizes from soils and sediments that are declared to be or which may be radioactive, or have been determined to require handling appropriate for radioactive hazards.
4.1.2.2 Approximately 200 samples will be sieved using this method. Approximately 40
of these will be samples containing substantial amounts of radioactive isotopes. One hundred fifty four of these samples will contain background or low-levels of radioactive contaminants.
4.1.3 Summary Field-moist samples will be sieved to 2mm. Due to the lack of a viable procedure to clean sieves of radioactive material, all sieving will be performed on the same day and the sieve discarded after use. 4.1.4 Definitions 1.4.1 Sieve Mesh - a nylon mesh at the bottom containing holes of a singular and specific
size. The soil is agitated within it to allow the specific particle sizes to pass through the mesh openings and be collected in the collection bin.
1.4.2 Soil Collection Jar - A 500ml Wide Mouth Nalgene Sample Jar with modified lid to
be used to collect soil during sieving as well as store it for later use. 1.4.3 Modified Lid - A Wide Mouth Nalgene Sample Jar Lid where the inner diameter
inside the white indentation has been cut. 1.4.4 Modified Jar - A Wide Mouth Nalgene Jar with the bottom cut out to form a
cylinder. 1.4.5 Sieve Shaker - A motor, with a circular base into which fits the soil collection jar. A
small hole is located on either side of the circular base, where the fasteners from the sieve anchor fit.
1.4.6 Sieve Anchor - An “H-shaped” securing clamp with two large screws on either side
joined by a horizontal (with respect to the sieve lid) bar. Two slits are cut into the central horizontal bar, and are used to stabilize the sieve apparatus. Also, on either side of the horizontal bar are screws for tightening the circular shaped mount onto the top of the sieve lid.

157
1.4.7 Glove Bag - a completely sealed, disposable, plastic containment apparatus with an internal frame used to avoid release of dust, aerosols, or other contaminants to the laboratory.
1.4.8 Central Unit - The central part of the sieving apparatus containing: Nylon mesh
secured to a modified jar with a modified lid. 1.4.9 Soil Drying Jar - A Wide Mouth Nalgene Jar used to dry the soil, and then inverted
directly into the sieving apparatus.
1.4.10 HEPA filter box - an apparatus containing two suction hoses which will be fitted into the wall of the glove box and uses a blower or fan to suction away any airborne contaminants from the glove box. The box contains a HEPA filter that traps soil particles.
1.5 Responsibilities 1.5.1 All technicians performing work under this protocol are responsible for following
all instructions as written. 1.5.2 The principal investigator supervises persons implementing this protocol. 1.5.3 The authoree, when not the principal investigator, approves the written protocol and
may provide instructions to the technician as appropriate. 1.5.4 EOHSI Radiation Safety Committee members may provide advice on protocols and
materials safety as requested. 1.5.5 Scott Petlick acts as Sample Manager. 1.5.6 The following people can provide on-the-job training (OJT) on this protocol. Kristie Ellickson – soil sieving (module OJT-002) Robert Meeker – soil sieving (module OJT-002) Michael Pollock– soil sieving (module OJT-002) Carl Schopfer - health physics surveys and instrumentation (module OJT-001) 1.6 Prerequisites All soil samples must be dried prior to the start of any sieving operations according to PIP-001, “soil drying”. 1.7 Interferences As small particle dispersion is a concern with the sieving, water will be introduced to the sieving apparatus in order to eliminate dust. Other interferences of concern are the cross

158
contamination of soil samples. In order to eliminate this possibility, nylon mesh will be used instead of metal and these nylon sieves will not be used again for later samples. 1.8 Materials 1.8.1 Parafilm M 1.8.2 Large Ziploc Storage Bags 1.8.3 1000ml Wide Mouth Nalgene Sample Jars, with lids 1.8.4 500ml Wide Mouth Nalgene Sample Jars, with lids 1.8.5 Nylon Mesh opening size <250µm (McMaster-Carr #9318T16) 1.8.6 Nylon Mesh opening size <75µm (McMaster-Carr #9318T22) 1.8.7 Nylon Mesh opening size <2000µm (McMaster-Carr #9318T26) 1.8.8 Heat Sealable Plastic Bags (20.5 cm opening) (McMaster-Carr # 2062T44) 1.8.9 Paper Towels or Kimwipes 1.8.10 laboratory tape 1.8.11 Sharpi magic marker 1.8.12 plastic o-ring 11.5cm in diameter, Dash # 155 (McMaster-Carr # 9452K176) 1.8.13 Scissors 1.8.14 plastic spatulas 1.8.15 decontaminating detergent (Picoclean) 1.8.16 PACE Multi Arm Evac II Fume Extraction System with HEPA/Gas Filtration
Cartridge 1.8.17 Grade #1 Filter Paper 3.2cm diameter (Whatman # 1001-032, VWR# 28450-021) 1.8.18 Respirator with HEPA filter cartridge 1.8.19 Timer 1.8.20 appropriate radiation survey instruments 1.8.21 TORBAL* Flexible Glove Cabinet with Frame (Vertex# GC-2 VWR# 32980-
000) 1.8.22 personal safety equipment: elbow-length gloves, Tyvek laboratory coat, eye
protection 1.8.23 Sieve Shaker, CSC Scientific - Catalogue Number 18480 1.8.24 H-Shaped Anchor 1.8.2 Equipment 1.8.2.1 appropriate radiation survey instruments 1.8.2.2 glove box 1.8.2.3 H-Shaped Anchor 1.8.2.4 HEPA filter unit 1.8.2.5 personal safety equipment: gloves, laboratory coat, eye protection 1.8.2.6 Sieve Shaker, CSC Scientific - Catalogue Number 18480 1.9 Safety 1.9.1 All experiments will be carried out in compliance with safety recommendations
provided by the Radiation Safety Committee at Rutgers University, which is

159
authorized by the Nuclear Regulatory Commission and the State of New Jersey (see reference 1.20.2.)
1.9.2 Samples will be stored at room temperature in closed containers. 1.9.3 Soil, which can be released as a dispersible dust or aerosol, represents a potential
inhalation hazard. The work being performed creates fine particles, which must be contained to avoid exposure to the worker and others in the vicinity.
1.9.4 Soil drying is accomplished with minimum disturbance to the media and any
outflow is filtered where possible. 1.9.5 To contain the samples during sieving and sample transfers requires working in a
closed, HEPA-filtered chamber or glovebox. The entire assembly is placed in a fume hood.
1.9.6 Soil materials shall be contained within and transferred between sealed vessels. The
sieving takes place while shaking within a sealed assembly. Opening the assembly is done in a filtered environment, after settling has take place. Dust creation is minimized by not allowing the soil to dry completely. Static electricity is avoided by not using plastic bags as the primary containment. Double containment is used, such as a bottle within a bag, or sealing an assembly with plastic wrap within a filtered chamber.
1.9.7 Samples must be in closed containers when removed from the filtered enclosure or
fume hood. 1.9.8 The potential for radiation exposure from soils will be determined prior to starting
work. The quantities approved for use will generally be limited by the exposure potential such that exceeding regulatory limits will not be possible, or determined to be extremely unlikely.
1.9.9 Radiation dosimetry badges will be worn if necessary. 1.9.10 All samples are labeled with the sample identifier code and radiation hazard class.
A chain of custody form is kept with each container. (See reference 1.) 1.9.11 Protective eyeglasses, dust mask, long-sleeved laboratory coat, long pants, and
two pairs of disposable vinyl gloves will be worn when working with radioactive samples. Gloves are to be surveyed periodically during work, and when removed. Hands must be washed after gloves are removed.
1.9.12 Laboratory coats, safety glasses and other safety wear must remain in room 149.
Do not bring the items into the counting room (room 145.) 1.9.13 All items shall be surveyed for contamination before exiting the laboratory.

160
1.9.14 Bench surfaces shall be covered with disposable bench paper. 1.9.15 Survey the work area and the laboratory for radioactive contamination at the end
of each day. 1.9.16 Wastes are contained in closed containers for pick-up by REHS and disposed per
agreement with the Savannah River Site. (See references 1 and 2) 1.10 Sample Collection, Handling, and Preservation Soil samples will be collected by the Department of Energy and will be stored at Rutgers Environmental Health and Safety Department’s Environmental Services Building. Samples are stored in 1-liter polyethylene bottles, wrapped in a polyethylene bag, and placed in styrofoam coolers/shipping boxes. Samples will be requested by the researcher by completing a sample request form and samples will only be transported by REHS. A record of sample flow will be recorded with a chain of custody form that will be assigned to each soil sample (See reference 1.) 1.11 Quality Control Nylon mesh will be used with the appropriate opening size. Nylon mesh is preferred over wire mesh to eliminate the possibility of metal leaching from a metal sieve. To eliminate cross contamination, the sieve mesh will not be reused rather disposed after a soil sample has been sieved. Samples will be dried to the same extent, and sieved for an equal amount of time (per the timer located on the sieving device). All samples will be sieved at the sieve shaker control rate of 1-3 (per the shaker control on the front of the sieve shaker), and once chosen this sieve rate will remain constant for all soil samples. Samples will be labeled with a batch number, the date of sieving, and the sample identification number in order to eliminate possible identification confusion. Finally, any balances used will be checked with a test weight prior to use. 1.12 Calibration Balances will be calibrated on an annual basis. This calibration will be checked by a test weight before use. The sieve shaker timer will be used during every sieving procedure to ensure equal sieving time. 1.13 Calculations None 1.14 Procedure Read this entire document before beginning any work in this section.

161
Note: When working with radioactive soils, keep materials and equipment away from the
edge of the laboratory bench at all times. 1.14.1 Soil must be dried to constant weight before sieving (See Soil Drying Procedure
SOP#001). 1.14.2 Set up the sieving apparatus according to the desired particle size. The situation
of each part of the sieve apparatus (from top to bottom) will follow this outline; soil drying jar, modified lid, desired particle size (250µm) Nylon mesh, modified central jar, modified bottom lid, and the soil collection jar.
1.14.3 Before assembly of the sieve apparatus put down an extra piece of bench paper,
which may be discarded after sieving. Note: Sieve apparatus will be assembled starting at the bottom collection jar to the top
soil drying jar. 1.14.4 Modify the lid of a 500ml Nalgenetm sample jar by cutting a hole into the top of
the lid just inside the white central indentation. 1.14.5 Secure this lid onto the jar, making the soil collection unit. 1.14.6 Modify the lid of this jar by cutting a hole into the top of the lid just inside the
white central indentation. 1.14.7 Cut out the bottom of the central jar to make the central sieve cylinder. 1.14.8 Drill a hole into the side of the central sieve cylinder, and secure a screw and a
nut into the hole surrounded by two Nylon washers. 1.14.9 Prepare the central unit by cutting a 6” x 6” piece of Nylon mesh from the desired
mesh size(250µm). 1.14.10 Place the Nylon mesh over the mouth of the central sieve cylinder, and secure it
with the modified lid of the jar. Make sure the sieve is tightly in place before continuing with the assembly of the sieve.
1.14.11 Once the sieve is in place, connect a conducting wire to the screw on the inside of
the central sieve cylinder so that it runs along the nylon mesh. 1.14.12 Place the central sieve unit on top of the soil collection unit. Secure this fitting
by placing an o-ring around the bottom of the central sieving unit, which will fit just inside of the white indentation on the soil collection unit.
1.14.13 Cut strips of Parafilm double the length of a Nalgenetm jar circumference.

162
1.14.14 Secure these two units with Parafilm strips and secure the Parafilm with
laboratory tape. 1.14.15 Work a heat sealable bag up over the soil collection unit and the central unit
leaving enough slack to enclose the upper soil drying jar as well. 1.14.16 Gather and twist the soil collection side end of the heat sealable bag and secure
with laboratory tape. 1.14.17 Invert the central and soil collection units together and place them on top of the
soil drying jar. 1.14.18 Secure the entire bottom unit to the soil drying jar with strips of Parafilm and
secure the Parafilm with strips of laboratory tape. 1.14.19 Pull the heat sealable bag up to enclose the entire sieving apparatus. 1.14.20 Gather and twist the top end of the heat sealable bag, and secure with laboratory
tape. Note: The grounding wire will exit the top of the heat sealable bag, and can be grounded
to the HEPA vacuum. 1.14.21 Turn on the HEPA vacuum for 1 minute before continuing work. 1.14.22 Wipe outside of the heat sealable bag with a moistened paper towel. 1.14.23 Do a swipe of the outside of the heat sealable bag and count with the appropriate
survey equipment. 1.14.24 Invert the entire enclosed apparatus and place onto the sieve shaker. 1.14.25 Place the h-shaped sieve anchor on top of the sieving apparatus, inserting the
top of the apparatus into the slots on the h-shaped sieve anchor. 1.14.26 Place the two fasteners on either of the sieve anchor in the holes on the bottom
of either side of the sieve shaker. 1.14.27 Secure this whole apparatus by tightly screwing down the knobs on either side of
the horizontal bar on the top of the sieve anchor. 1.14.28 One can assure that the screws are horizontal by making sure that the top two
screws, and the top bar, are horizontal with respect to the sieve lid. If the two top screws and the horizontal bar are not horizontal, the sieve may vibrate.

163
1.14.29 When the sieve is secure, and the top bar is parallel with the sieve, turn the sieve on.
1.14.30 The sieve will vibrate a little, but if it is too noisy the top screws need to be
tightened further. As one tightens the sieve screws, make sure to check the position of the horizontal bar and the top two screws with respect to the sieve lid keeping everything parallel.
1.14.30 Sieve soil for approximately fifteen minutes, at a shaking level no greater than 3.
The sieving time may change but should remain constant for each succeeding soil sample.
Note: The sieving apparatus should not run unattended, the technician must be present
during the whole sieving process. 1.14.31 Once the sieving time has expired, turn off sieve and wait for the vibration to
come to a full stop. 1.14.32 Unscrew and remove clamp. 1.14.34 Remove sieve apparatus from the sieve shaker. 1.14.35 Let sieving apparatus rest for one hour to allow dust to settle. 1.14.36 To remove the soil collection jar, carefully slide the heat sealable bag up the soil
collection jar until it can be removed. 1.14.37 Remove tape and Parafilm from around the bottom soil collection jar. 1.14.38 Invert top and central jar as a unit and set down. 1.14.39 Remove the nylon mesh from the central unit, and shake any remaining soil from
the central cylinder into the heat sealable bag. Place the central cylinder in a plastic bag until it can be rinsed for future use.
1.14.40 Carefully unscrew the lid of the soil collection jar. 1.14.41Place the lid into the heat sealable bag containing the soil drying jar. 1.14.42 Cap the soil collection jar. 1.14.43 Turn on the HEPA vacuum for 1 minute. 1.14.44 Now, dispose of the heat sealable bag (Nylon mesh and modified lid) into the
solid waste container.

164
1.14.45 Allow the sample container to sit so that dust can settle, then wipe around the outside of the jar with a moistened paper towel.
1.14.46 Take a swipe of the outside of the soil sample jar including the inner accessible
part of the lid. 1.14.47 Enclose the sample bottles with a large Ziploctm bag. 1.14.47 Measure activity of the soil sample, and mark the reading on the front of the wide
mouth Nalgenetm sample bottle. Specific counting methods will include a total gamma count of the bottle, and two prepared scintillation vials for gross alpha and beta.
1.14.49 Label sample bottle with sample identification number, activity of the sample,
date of sieving, and batch number. 1.14.50 Complete previous steps for all desired particle sizes. 1.15 Storage and containment 1.15.1 All samples will be stored in sealed polypropylene or polyethylene containers. 1.15.2 Polypropylene jars will then be placed inside Ziploctm storage bags as a secondary containment measure. 1.15.3 Samples will be stored in the dark, under conditions of reasonable humidity and room temperature. 1.16 Cleaning or disposal of glassware and equipment 1.16.1 All weighing papers, spatulas, soil sieve and catch basin that exhibit radioactivity
when measured with a radiation meter will be disposed of in a container marked “radioactive solid waste”.
1.16.2 Dispose of radioactive soils, which do not pass through the sieve in the container
for radioactive solid wastes. 1.16.3 Remove all other solid radioactive wastes to a container for radioactive wastes. 1.16.4 Rinse all non-contaminated equipment with DI water and acid washed materials
with 10% reagent grade nitric acid. 1.16.5 Remove the liquid radioactive wastes to a container for radioactive wastes.

165
1.16.6 After operations are completed, check all work surfaces, non-waste materials, and the regular trash, with radiation probes to assure that no radioactivity is present outside of permitted storage and waste containers.
1.16.7 Check for contamination on all equipment and containers and record the survey
results before removal from the radioactive facility. Check the activity of the normal trash as well, to ensure that no radioactive waste has been placed there accidentally.
1.16.8 After all operations are completed, perform swipes on all work areas and count in
a scintillation counter. 1.17 Waste Minimization By enclosing the sieving apparatus in a small, filtered environment, we will minimize the spread of contamination and, therefore, the amount of cleanup. In addition, it may be possible to use the waste soil for other experiments that do not need smaller, sieved fractions. 1.18 Waste Disposal Wastes generated by these procedures will include dry solid waste such as gloves, paper towels, dust-containment apparatus and unused soil. Liquid waste will include any aqueous waste generated for the purpose of dust minimization/collection and decontamination procedures. All waste from these procedures will be collected in appropriate containers for transport to the Savannah River Site for disposal. 1.19 Accident Instructions 1.19.1 Prior to beginning work, discuss with the OJT trainers how to respond to accidents. Accidents that might result in an unplanned external radiation exposure or in any internal exposure (inhalation or ingestion) require immediate REHS notification. Review the instructions on pages 24, 25, 29 and 30 of reference 1.20.2. 1.19.2 Small, localized spills (with no personal contamination) will be handled by the technician. Use a spray bottle with water to prevent the spilled material from drying out and becoming airborne. Use moist paper towels to cleanse the spill area with a blotting action to prevent the spread of radioactive material. Use decontaminating detergent to clean surfaces. Check for persistent contamination following cleaning. Contact REHS immediately (445-2550, or 5-2550 from laboratory phones) if the area cannot be decontaminated. Dispose of the contaminated cleaning materials in the radioactive waste. Report the lost material to the Sample Manager. 1.19.3 If there is personal contamination, remove contaminated clothing. Use a spray bottle with water to prevent the spilled material from drying out and becoming airborne, and cover with damp paper towels. Wash contaminated skin with mild soap and water.

166
Do not use abrasives. Limit contact with others to avoid spreading contamination into clean areas. Exit the immediate area of the spill, but do not leave the laboratory area until advised by REHS or Rutgers Police. 1.19.4 Contact REHS for assistance in cleaning large spills. 1.19.5 In the event of dispersal of radioactive material outside of the ventilated enclosure,
exit the laboratory immediately and contact REHS. Restrict access to area. 1.20 References 1.20.1 EOHSI/CRESP Radiation Safety Global Procedures Manual. 1.20.2 Rutgers University Radiation Safety Guide, 7th edition, 1989. 1.21 Attachments/Enclosures 1.21.1 REHS Radioisotope Disposal Form, DY 6/93 AF/disp. 1.21.2 Figure 1, Soil Shaker 1.21.3 Chain-of-Custody form 1.21.4 Waste Log

167
3. CRESP SOP-003 “Bioaccessibility Assay” 1.0 Cover Page Author(s): Kristie Ellickson Date Robert Meeker Date Scott Petlick Date Mike Pollock Date
Carl Schopfer Principal Investigator(s): David Kosson, Ph.D. Date Paul Lioy, Ph.D. Date Robert Tata, Ph.D. Date Authoree: David Kosson, Ph.D. Date EOHSI Radiation Safety Committee Chair: Arthur Upton, M.D. Date

168
2.0 Experimental Bioaccessibility is the amount of a contaminant that becomes dissolved in the gastrointestinal system, whereas bioavailability is the amount of a contaminant that actually crosses the intestinal wall to become an internal dose. The bioaccessibility assay is a method for the estimation of potential dose to a contaminant by mimicking the human gastrointestinal system. The specific exposure scenario of interest is inadvertent or purposeful ingestion of a contaminated soil. Two current methods involved in the assessment of potential dose by incidental soil ingestion are animal bioavailability models and microwave acid digestion procedures. The microwave digestion procedure, may not provide a reliable estimate of bioaccessibility or bioavailability as biological fluids (e.g. intestinal fluid) may solubilize only a small percentage of the total elements present in a soil. Animal models are more reliable and closer estimates, but are highly expensive and time consuming. Animal models, therefore, would be impractical as the main vehicle for determining the bioavailable fraction with the high number of DOE hazardous waste sites in the United States at present. The bioaccessibility procedure involves the introduction of a soil sample to physiologically based extraction fluids, in a sequential manner from saliva to gastric to intestinal fluid. This in vitro extraction then will be compared to assimilation values of an in vivo rat model, as a comparison of bioaccessibility and bioavailability. 2.1 Hypothesis Our hypotheses are: 1) the more closely the synthetic, in vitro, extractant mimics the extraction properties of the human digestive biological fluids, the more accurate will be the estimate of an internal dose; 2) performances can be evaluated by in vivo studies with a rat model and quantitative examination of a mass balance, calculation and dose estimates from model simulations for the in vitro and in vivo system; and 3) the concentration of the elements, present in the bioaccessible fraction obtained with a synthetic extraction system, will be a better indicator of contaminant ingestion from a contaminated soil because it represents the portion of the mass which can yield exposure, uptake and the internal dose to an individual. 2.2 Objectives Several surface soils will be processed through a bioaccessibility assay as a means to approximate the potential dose one may receive due to incidental ingestion of that soil. Gamma spectroscopy and liquid scintillation of the total soil and the extractant in each synthetic biofluid will measure radioactivity. If further alpha analysis is desired, isolation methods (wet chemical or electroplating) will be used and counted with alpha spectrometry. Experiments will include a quality control procedure including laboratory blanks, method blanks, lab controls, and method controls. The following elements and total counts analysis are of interest: cesium, uranium, gross alpha, gross beta, and all gamma emitters; however others may be incorporated into the design as the work progresses.

169
2.3 Experimental Design The dependent variable of interest is the percent bioaccessible, and the research question is to understand how various factors affect this value. Independent factors of interest are biological fluid components and their quantities, soil characteristics, specific elements and isotopes. It is important to understand both how closely the human gastrointestinal fluid can be practically mimicked, as well as the factors, which most affect bioaccessibility. For each unique sample, four replications will be run. The order of experimentation and analysis of these samples will be randomized by the assignment of a number to each sample with the key located only in the laboratory notebook. Sample order will be randomized both in the execution of the procedure and the instrumental analysis. Specific factors will be tested for significant influence on bioaccessibility using SPSS software. 2.4 Method Summary Soil preparation will involve the drying and sieving of the soil into one of the following particle sizes of <2000µm, <250µm, and <75µm (see PIP-001 “soil drying” and PIP-002 “preparation of soil sub-fractions”). The sample volumes that are to be analyzed are approximately 10ml fluid samples. Twelve of these samples may be processed through the bioaccessibility procedure in parallel, where four identical samples will be run as replicates. A 0.05g - 0.5g soil sample is weighed into a polyethylene Nalgene bottle. Approximately 208 milliliters of synthetic biological fluid is added sequentially to the bottle. An aliquot of the supernatant is taken from each step. This fluid sample is centrifuged, filtered to <0.45µm, and diluted to a suitable concentration. The remaining soil will be collected using vacuum filtration with a HEPA trap attached to the vacuum apparatus. The original soil sample will be counted before bioaccessibility analysis to provide a direct measurement of the total contaminant level. Gamma spectrometry and liquid scintillation will be used for all activity analyses. 2.5 Procedure Quality Objectives 2.5.1 Results of these procedures need to have adequate precision and accuracy to
distinguish differences between the affects of varying gastrointestinal fluid components, soil characteristics and other dependent variables. Method detection limits will be measured upon receiving soil samples in order to estimate the number of samples needed for sufficient statistical power.
2.5.2 Method control, laboratory control, method blanks and laboratory blanks will be run
in parallel to all experiments in order to detect matrix effects. Three replicates of each sample will also be run.
3.0 Radionuclides 3.1 Radionuclides present in the soils to be processed are from core samples of seepage
basins used to dispose of radioactive wastes over several decades. Other samples to

170
be processed may include surface soil samples from areas thought to be radioactively contaminated. Also, background samples will be processed. (Background soils contain no known radionuclide contaminants other than that which may be present naturally or from global deposition/contribution.)
3.2 The EOHSI Radiation Safety committee will review radionuclide characterization
data prior to releasing each sample. All samples, including background samples, are characterized for radionuclides prior to use.
3.3 The principal investigator, prior to authorizing work with any sample, shall consider
the possibility of the presence of other toxic contaminants, such as metals or organics. 3.4 Each principal investigator supervising persons using this procedure shall review the
known or suspected contents of each sample, the sample quantity and its physical form and may consult with members of the EOHSI Radiation Safety committee if desired.
3.5 The principal investigator shall discuss with the technician any hazards in the sample. 3.6 Technicians processing soils shall observe any quantity limits imposed by the EOHSI
Radiation Safety Committee and shall follow instructions regarding custodianship of samples and contaminant inventory.
4.0 List of Procedures 4.1 This protocol includes one standard operating procedure, SOP-003, ”Bioaccessibility
Assay.” 4.2 This protocol requires the procedures SOP-001 “Soil Drying” and SOP-002 “Soil
Sieving” for soil preparation. 4.1 Format The procedure SOP-003 follows a formal written procedure format. 4.1.1 Purpose The bioaccessibility is used to approximate the potential dose one would receive by the inadvertent or purposeful ingestion of contaminated soil. Of specific interest is the potential dose to radionuclides present in soil. The values resulting from this work will be compared to dose estimations obtained from computer models and animal models. 4.1.2 Scope This procedure applies to experiments requiring uniform particle sizes from soils and sediments that are declared to be or which may be radioactive, or have been determined to require handling appropriate for radioactive hazards.

171
Approximately 200 samples will be processed by this method. Approximately 40 of these will be samples containing substantial amounts of radioactive isotopes. One hundred fifty four of these samples will contain background or low-levels of radioactive contaminants. 4.1.3 Definitions 4.1.3.1 Glove Bag - a completely sealed, disposable, plastic containment apparatus with
an internal frame used to avoid release of dust, aerosols, or other contaminants to the laboratory.
4.1.3.2 HEPA filter box - an apparatus containing two suction hoses which will be fitted
into the wall of the glove box and uses a blower or fan to suction away any airborne contaminants from the glove box. The box contains a HEPA filter that traps soil particles.
4.1.4 Responsibilities 4.1.4.1 All technicians performing work under this protocol are responsible for following
all instructions as written. 4.1.4.2 The principal investigator supervises persons implementing this protocol. 4.1.4.3 The authoree, when not the principal investigator, approves the written protocol
and may provide instructions to the technician as appropriate. 4.1.4.4 EOHSI Radiation Safety Committee members may provide advice on protocols
and materials safety as requested. 4.1.4.5 Scott Petlick acts as Sample Manager. 4.1.4.6 The following people can provide on-the-job training (OJT) on this protocol. Kristie Ellickson – bioaccessibility assay (module OJT-002) Carl Schopfer - health physics surveys and instrumentation (module OJT-001) 4.1.5 Prerequisites All soil samples must be air-dried and sieved prior to this procedure. Worker must read entire Standard Operating Procedure before starting any laboratory work. 4.1.6 Interferences As small-particle dispersion is a concern with the weighing and transfer of soil, these actions will be conducted in a glove bag with a HEPA vacuum attached. Other

172
interferences of concern are the cross contamination of soil samples. In order to eliminate this possibility, sample bottles will not be reused. 4.1.7 Materials 4.1.7.1 250 ml Nalgene wide mouth plastic bottles (Fisher# 02-923-7) 4.1.7.2 Teflon coated spatula for weighing (Fisher# 21-401-50B) 4.1.7.3 Weigh paper 4” X 4” (Fisher# 09-898-12B) 4.1.7.4 Deionized water 4.1.7.5 NaCl (<2 ppm heavy metals) 4.1.7.6 NaHCO3 (<2 ppm heavy metals) 4.1.7.7 Pepsin, purified powder (Fisher# p53-100) 4.1.7.8 High purity HCl for trace metal analysis (Fisher# A508-500) 4.1.7.9 Glass volumetric flask, 1L 4.1.7.10 Glass volumetric flask, 100ml 4.1.7.11 0.1 - 2.5 ml Rainin automatic pipette 4.1.7.12 Wheaton 0.5 - 5.0 ml pipette 4.1.7.13 2-10ml adjustable pipette 4.1.7.14 Pipette tips for 2-10ml pipette (UMDNJ stockroom# 175680115P) 4.1.7.15 100ml reusable glass pipette and bulb 4.1.7.16 Rainin microliter pipette tips, RC-2500 (VWR# 53550-452) 4.1.7.17 Oxford 914 pipette tips (VWR# 53503-826) 4.1.7.18 Nalgene bottles, 30 ml (UMDNJ stockroom# 175680022P) 4.1.7.19 Parafilm M 4.1.7.20 Teflon-coated tweezers 4.1.7.21 Polystyrene screw top centrifuge tubes, Falcon 15ml conical (Fisher# 05-
527-90) 4.1.7.22 Appropriate Survey Instrument 4.1.7.23 Personal Safety Equipment: laboratory gloves, laboratory coat with long
sleeves, and eye protection. 4.1.7.24 Protective barrier for working with high activity levels (lead, or acrylic) 4.1.7.25 Glove bag with frame (Torbal* VWR# 32980-000) 4.1.7.26 Nalgene Analytical Test Filter Funnels (funnels are polypropylene and
filters are cellulose nitrate) (VWR# 28198-865) 4.1.7.27 Polypropylene propylene flask with angular tubulation (Fisher# 29417-
047) 4.1.7.28 Silicone No. 8 Stoppers 4.1.7.29 100 x 15mm Standard Petri Dishes 4.1.7.30 Retort Stand 4.1.7.31 Fixed Angle Clamps 4.1.7.32 Personal safety equipment: elbow length gloves, laboratory coat, eye
protection 4.1.7.33 Tygon Tubing 3603 4.1.8 Equipment

173
4.1.8.1 HEPA filter trap for the vacuum apparatus (Fisher #09-743-28), or Desiccant Vacuguard (Fisher #09-746B)
4.1.8.2 Sieve Shaker, CSC Scientific - Catalogue Number 18480 4.1.8.3 IEC Centra CL2 Centrifuge (capacity - 50ml samples) (Fisher #05-101-7) 4.1.8.4 Magni Whirl Constant Temperature Water Bath (Blue M) 4.1.8.5 Analytical microbalance 4.1.9 Safety 4.1.9.1 All experiments will be carried out in compliance with safety recommendations
provided by the Radiation Safety Committee at Rutgers University, which is authorized by the Nuclear Regulatory Commission and the State of New Jersey (see reference 1.20.2.)
4.1.9.2 Samples will be stored at room temperature. 4.1.9.3 Soil, which can be released as a dispersible dust or aerosol, represents a potential
inhalation hazard. The work being performed creates fine particles, which must be contained to avoid exposure to the worker and others in the vicinity.
4.1.9.5 To contain the samples during weighing and sample transfers requires working in
a closed, HEPA-filtered chamber or glovebox. The entire assembly is placed in a fume hood.
4.1.9.6 Samples must be in closed containers when removed from the filtered enclosure
or fume hood. 4.1.9.7 The potential for radiation exposure from soils will be determined prior to starting
work. The quantities approved for use will generally be limited by the exposure potential such that exceeding regulatory limits will not be possible, or determined to be extremely unlikely.
4.1.9.8 Radiation dosimetry badges will be worn if necessary. 4.1.9.9 All samples are labeled with the sample identifier code and radiation hazard class.
A chain of custody form is kept with each container. (See reference 1.) 4.1.9.10 Protective eyeglasses, dust mask, long-sleeved laboratory coat, long pants, and
two pairs of disposable vinyl gloves will be worn when working with radioactive samples. Gloves are to be surveyed periodically during work, and when removed. Hands must be washed after gloves are removed.
4.1.9.11 Laboratory coats, safety glasses and other safety wear must remain in room 149.
Do not bring the items into the counting room (room 145.) 4.1.9.12 All items shall be surveyed for contamination before exiting the laboratory.

174
4.1.9.13 Bench surfaces shall be covered with disposable bench paper. 4.1.9.14 Survey the work area and the laboratory for radioactive contamination at the end
of each day. 4.1.9.15 Wastes are contained in closed containers for pick-up by REHS and disposed per
agreement with the Savannah River Site. (See references 2 and 3.) 4.1.10 Sample Collection, Handling, and Preservation Soil samples will be collected by the Department of Energy and will be stored at Rutgers Environmental Health and Safety Department’s Environmental Services Building. Samples are stored in 1-liter polyethylene bottles, wrapped in a polyethylene bag, and placed in styrofoam coolers/shipping boxes. Samples will be requested by the researcher by completing a sample request form and samples will only be transported by REHS. A record of sample flow will be recorded with a chain of custody form that will be assigned to each soil sample (See reference 1.) 4.1.11 Quality Control A NIST standard reference material will be used to maintain appropriate quality control. This material will be run in parallel to all experiments. Every experiment will also involve a method blank, and a method control, wherein a mixed gamma standard and Uranium and Thorium spike will be used to follow method recoveries and detect possible matrix interferences. Samples will be labeled with a batch number, the date of sieving, and the sample identification number in order to eliminate possible identification confusion. The pH and redox potential of each synthetic gastrointestinal fluid will be measured before the start of each experiment. Finally, any balances used will be checked with a test weight prior to use. 4.1.12 Calibration Balances will be calibrated on an annual basis. This calibration will be checked by a test weight before use. The sieve shaker timer will be used during every sieving procedure to ensure equal sieving time. The pH meter will be calibrated before each using using buffers of pH 4, 7, and 11. 4.1.13 Calculations The bioaccessibility of an element is calculated using the mass balance equation, which is outlined below. Mass refers to the mass can apply to the mass of a metal, radionuclide, or other analytes.
MB = MGJ + MIJ where MB = mass bioaccessible

175
MGJ = mass soluble in gastric fluid MIJ = mass soluble in intestinal fluid percent bioaccessible = (MGJ + MIJ)/MT where MT = total mass in soil
The mass in intestinal fluid cannot be measured directly, as the resulting fluid is a mixture of saliva, gastric, and intestinal fluids. The analysis of the intestinal fluid mixture without soil present, and the subsequent analysis of this same mixture with soil present measure this fraction. In this way the change in metal concentration from the addition of soil can be analyzed, which allows an indirect measurement of the influence of synthetic intestinal fluid on metal dissolution from soils. The mass soluble in the intestinal mixture with soil will be subtracted from the mass soluble in the intestinal mixture alone as an indirect measurement of the mass soluble in the intestinal mixture. The relationship is described below;
MIJ = Mijs - Mij where Mij = mass soluble in intestinal fluid, Mijs = mass soluble from intestinal mixture with soil present, and Mij = mass soluble from intestinal mixture with no soil present
The mass total can be measured by an EPA 3051 microwave digestion, or by the summation of the bioaccessible components including the concentration found in soil which is recovered at the end of the procedure. Another way of estimating bioaccessibility is the measurement of solubility ar one point in the system. For example, if the site of absorption is known well(ie. Upper jejunal for Hg) the solubility may be measured at the conditions of that section of intestine. In this way, the additon of the final Mijs and recovered soil mass will result in the total metal mass, and Mijs/MT are the bioaccessible fraction. 4.1.14 Procedure Preparation of Artificial Fluids
Artificial Saliva 1.14.1 Weigh 0.8g of CaCl2 •4H2O, 1.0g urea, 0.6g Na2HPO4, 0.4g KCl, 0.4g NaCl, 4g
mucin individually onto weigh paper. 1.14.2 Add the chemicals to a 1L volumetric flask, and dilute to volume. Artificial Gastric Fluid 1.14.3 Weigh 2.0g NaCl onto weigh paper. 1.14.4 Measure 7ml HCl into a graduated cylinder (or enough HCl to result in a pH=2
fluid). 1.14.5 Add these chemicals to a 1L volumetric flask and dilute up to 250ml volume. 1.14.6 Add 3.2 g pepsin to the flask directly before the assay is run, and then the
components are diluted up to a final volume of 1L. 1.14.7 Adjust the pH to 2 using HCl. Artificial Synthetic Intestinal Fluid 1.14.8 Add 16.8g NaHCO3 to a 1L volumetric flask and dilute up to volume. Soil Preparation 1.14.9 Dry and Sieve the soil according to The Drying and Sieving of Soil procedures
(SOP-001, and SOP-002).

176
Bioaccessibility Assay 1.14.10 Add 8 ml of synthetic saliva to the 250 ml Nalgene sample bottles. 1.14.11 Add 100 ml of synthetic gastric fluid to the Nalgene sample bottles. 1.14.12 Place the balance in the glove bag. 1.14.13 Place a folded piece of weigh paper in a petri dish, and place the petri dish on the
scale. 1.14.14 Using a Teflon coated spatula for transfer, weigh 0.05 - 0.5g of soil onto weigh
paper in the glove bag (which is located under a fume hood), and empty into the 250ml Nalgene wide mouth sample bottle containing synthetic saliva and gastric fluid.
1.14.15 Before removing sample bottles from the glove box, wipe the outside with a moist paper towel and secure rim with parafilm.
1.14.16 Check bottles for outside contamination by taking a wipe with small filter paper and counting the paper with Geiger counter or appropriate probe.
1.14.17 Place the samples in a water bath at 37°C and shake at approximately 125 cycles per minute for two hours.
1.14.18 Wipe the balance and soil sample bottle with a moist paper towel before removing from the glove bag.
1.14.19 Acid (1% nitric acid) rinse the Teflon coated spatula and the rinse with DI-water and dry, before removing from glove bag.
1.14.20 After processing in the water bath, remove sample bottles from the water bath and place into the glove bag.
1.14.21 Pipette a 10 ml aliquot from the supernatant of the samples and dispense into a 15ml conical centrifuge tube.
1.14.22 Replace lids onto the sample bottles and secured bottles with parafilm. 1.14.23 Centrifuge the 250ml sample bottles for 10 minutes until the soil has settled to
the bottom of the bottles. 1.14.24 Decant the liquid portion of the saliva and gastric fluid sample so the intestinal
fluid may be added to the saliva and gastric fluid alone (without soil). 1.14.25 Weigh each bottle, and an empty 250ml bottle to obtain total liquid remaining in
the bottle containing soil. 1.14.26 Secure the lid onto the soil-containing bottle with parafilm and store in a glove
box. 1.14.27 Add 100 ml of synthetic intestinal fluid to the Nalgene sample bottles containing
saliva, gastric, and intestinal fluid (without soil). Note: This may be done before hand to decrease time between sample processing. If so
pour the gastric fluid directly into bottles containing intestinal fluid. 1.14.28 Place the sample bottles once again into the water bath at 37°C for two hours to
shake at approximately 125 cycles per minute. 1.14.29 After the two hour processing period, remove sample bottles and place them in
the glove bag. 1.14.30 Pipette a 10 ml aliquot from the supernatant of each sample into a 15ml conical
centrifuge tube.

177
1.14.31 Add the saliva, gastric, and intestinal fluid to the bottles containing soil (making sure to transfer between the correct sample numbers)
1.14.32 Before placing the sample bottles back into the water bath, wipe down the outside with a moist paper towel.
1.14.33 Process the samples in the shaker bath at 125 cycles per minute for two hours at 37°C.
1.14.34 After processing, pipette a 10ml aliquot from the supernatant of the samples and dispense into a 15ml Conical screw top test tube.
Bioaccessibility Fluid Sample Preparation 1.14.35 Make sure the lids for the centrifuge tubes from the saliva-gastric, saliva-gastric-
intestinal, and saliva-gastric-intestinal-soil samples, are secured before centrifugation.
1.14.36 After lids are secured, wrap the lids with parafilm. 1.14.37 Wipe the centrifuge tubes with a moist paper towel before removing from the
glove bag. 1.14.23 Centrifuge 15ml Falcon tubes for 20 minutes at 3400 rpm. 1.14.38 After centrifugation and within a glove bag, press the samples through a
Whatman 0.45 syringe filter into a 30ml Nalgene sample bottle. 1.14.39 Between samples, rinse out the glass syringes with 1% nitric acid, and then DI
water. 1.14.40 Secure the lids on the sample bottles, and then wrap parafilm around the bottle
lid. Soil Collection Procedure 1.14.41 The remaining soil will be collected onto a cellulose nitrate filter using a vacuum
filtration apparatus (with HEPA filter trap or Desiccant Vacuguard attached, and within a glove bag).
1.14.42 Place a piece of Tygon R-3603 into a Nalgene Analytical Test Filter Funnel (250ml capacity).
1.14.43 Parafilm the filter funnel to a glass erlenmeyer flask (1000ml capacity), and attach a vacuum tube.
1.14.44 The vacuum apparatus “train” will be set up as follows 1. House vacuum, 2. Vacuum hose, 3. HEPA or Desiccant Vacuguard, 4. Vacuum hose, 5. Erlenmeyer Flask #2, 6. Vacuum hose, 7. Erlenmeyer Flask #1.
1.14.45 Clamp the entire apparatus to a retort stand using fixed angle clamps. 1.14.46 Secure the vacuum hose to the Erlenmeyer flask tube(#2) and to the house
vacuum, then turn the vacuum on. 1.14.47 Pour the remaining contents from the Nalgene wide mouth bottles into the
collection beaker, and allow filtering until the soil appears dry. 1.14.48 Rinse the filter with DI water to collect the total amount of soil remaining on the
rim of the collection beaker. 1.14.49 Remove the collection beaker from the connector and cellulose nitrate filter, and
place the filters flat into a 100 x 15mm petri dish. 1.14.50 Secure the petri dish with laboratory tape or parafilm. 1.14.51 Allow the filters to dry to constant weight in a vacuum dessicator. 4.1.15 Storage and containment

178
4.1.15.1 All samples will be stored in sealed polypropylene sample bottles or petri dishes. 4.1.15.2 Place polypropylene jars and the petri dishes inside a plastic bin with a cover as
a secondary containment measure. 4.1.15.3 Store samples in the dark, under conditions of reasonable humidity and room
temperature. 4.1.16 Cleanup or disposal of glassware and equipment 1.16.1 All weighing papers, spatulas, and polypropylene filtration equipment that exhibit
radioactivity when measured with a radiation meter will be disposed of in a container marked “radioactive solid waste”.
1.16.2 Remove all other solid radioactive wastes to a container for radioactive wastes. 1.16.3 The acid rinse waste will be kept separate from all deionized water rinse and
disposed separately. 1.16.4 Rinse all non-contaminated equipment with a 1% reagent grade nitric acid rinse
solution. Equipment will be rinsed into a vacuum filtration apparatus equipped with a Buchner funnel and a filter, where the liquid will empty into an Erlenmeyer flask. For safety precautions two Erlenmeyers will be placed in line to avoid contamination of the vacuum pump.
1.16.5 The acid wash will be followed by a deionized water rinse as per step 1.16.4. This water rinse should be emptied into a different Erlenmeyer flask than the rinse from step 1.16.4.
1.16.6 Remove the liquid (non-acidic) radioactive wastes to a container for radioactive wastes.
1.16.7 Remove the acidic liquid radioactive wastes to a separate waste container. 1.16.8 After operations are completed, check all work surfaces, non-waste materials, and
the regular trash, with radiation probes to assure that no radioactivity is present outside of permitted storage and waste containers.
1.16.9 Check for contamination on all equipment and containers and record the survey results before removal from the radioactive facility. Check the activity of the normal trash as well, to ensure that no radioactive waste has been placed there accidentally.
1.16.10 After all operations are completed, perform swipes on all work areas and count in a scintillation counter.
1.16.11 Before removing equipment and supplies from the glove bag, wipe these down with a moist paper towel.
4.1.17 Waste Minimization 1.17.1 The conservative estimates of solid wastes (given in Table 1), are based upon the
weights of samples, glassware, and materials that actually come in contact with the soil samples. The maximum estimates are approximate summations of the weights of all samples, glassware, and materials used in each procedure. As many of the wastes are very dilute, some samples may be decontaminated after use, and can be reused. In this case the following tables may represent overestimates of contaminant levels.

179
Table 1. Conservative and Maximum Estimates of Radioactive Solid Wastes per Soil Sample.
Procedure Conservative estimates of per sample waste (g)
Maximum estimate of per sample waste (g)
Bioaccessibility 130 1575* Maximum values for the generation of liquid radioactive waste have been estimated. These estimates have been made with the assumption that all liquid wastes will show signs of radioactivity. The synthetic biological fluids will dilute the radioactivity, thereby minimizing the total activity of the resulting waste. Table 2. Maximum Estimates for the generation of Liquid Radioactive Wastes.
Procedure Maximum estimate of per sample Waste (milliliters)
Bioaccessibility Assay 250ml** * refers to the actual sample, dilutions, and the cleaning of the microwave vessels. ** refers to the actual sample, dilutions, and the cleaning of the microwave vessels and
the filtering apparatus. 4.1.18 Waste Disposal 4.1.18.1 Wastes generated by these procedures will include dry solid waste such as
gloves, paper towels, dust-containment apparatus and unused soil. Liquid waste will include any aqueous waste generated for the purpose of dust minimization/collection and decontamination procedures. All waste from these procedures will be collected in appropriate containers for transport to the Savannah River Site for disposal.
4.1.19 Accident Instructions 4.1.19.1 Prior to beginning work, discuss with the OJT trainers how to respond to
accidents. Accidents that might result in an unplanned external radiation exposure or in any internal exposure (inhalation or ingestion) require immediate REHS notification. Review the instructions on pages 24, 25, 29 and 30 of reference 1.20.2.
4.1.19.2 Small, localized spills (with no personal contamination) will be handled by the
technician. Use a spray bottle with water to prevent the spilled material from drying out and becoming airborne. Use moist paper towels to cleanse the spill area with a blotting action to prevent the spread of radioactive material. Use decontaminating detergent to clean surfaces. Check for persistent contamination following cleaning. Contact REHS immediately (445-2550, or 5-2550 from laboratory phones) if the area cannot be decontaminated. Dispose of the contaminated cleaning materials in the radioactive waste. Report the lost material to the Sample Manager.

180
4.1.19.3 If there is personal contamination, remove contaminated clothing. Use a spray
bottle with water to prevent the spilled material from drying out and becoming airborne, and cover with damp paper towels. Wash contaminated skin with mild soap and water. Do not use abrasives. Limit contact with others to avoid spreading contamination into clean areas. Exit the immediate area of the spill, but do not leave the laboratory area until advised by REHS or Rutgers Police.
4.1.19.4 Contact REHS for assistance in cleaning large spills. 4.1.19.5 In the event of dispersal of radioactive material outside of the ventilated
enclosure, exit the laboratory immediately and contact REHS. Restrict access to area.
4.20 References 4.20.1 EOHSI/CRESP Radiation Safety Global Procedures Manual. 4.20.2 Rutgers University Radiation Safety Guide, 7th edition, 1989. 4.20.3 Hamel, S.C., Buckley, B., Lioy, P.J., “Bioaccessibility of Metals in Soils for
Different Liquid to Solid Ratios in Synthetic Gastric Fluid” Environmental Science and Technology Vol. 32. Pps. 358-362. 1998.
4.21 Attachments/Enclosures 1.21.1 REHS Radioisotope Disposal Form, DY 6/93 AF/disp. 1.21.2 Chain-of-Custody form 1.21.3 Waste Log 4.2.2 References and standardized procedures. 3-PIP-BIO, Bioaccessibility Assay is an approved “plug-in” procedure for use by CRESP personnel. PIP-001 “Soil Drying” and PIP-002 “Preparation of Soil Sub-Fractions” are needed to carry out this procedure. Kingston, H.M., Test for Evaluating Solid Waste, Physical methods, 3rd Edition; U.S. Environmental Protection Agency, Office of Solid Waste and Emergency Response. U.S Government Printing Office: Washington D.C. SW- 846-3051, (1986). 5.0 Training Task-specific training, laboratory and radiation safety training are required before this procedure is used to process soil samples. All training modules must be completed successfully before work may begin.

181
5.0.1 On-the-Job Training (SOP-003, Bioaccessibility Assay) is required. 5.0.2 The technician shall demonstrate to the OJT trainer familiarity and understanding of
all written procedures and manuals. 5.0.3 The technician shall obtain adequate training on the use of all equipment and an
understanding of expected outcomes. 5.0.4 The technician shall demonstrate the ability to successfully operate the equipment
under normal conditions with typical samples, and to recognize abnormal situations. 5.0.5 The technician shall demonstrate proficiency by obtaining the desired product or
results while performing the procedure under the direction of the OJT trainer. 5.0.6 Safety training includes at a minimum, the following components: Rutgers or UMDNJ Laboratory Safety Training Right-to-Know Training Radiation Safety Training On-the-Job Training - Health Physics Surveys and Instrumentation 6.0 Facility 6.0.1 All work using radioactive materials will be performed in room #149 in the
Environmental and Occupational Health Sciences Institute of the University of Medicine and Dentistry of New Jersey and Rutgers University in Piscataway, New Jersey.
6.0.2 Preparation of the synthetic fluids, which does not involve radioactive materials, will be performed in room # 360 of EOHSI (see 6.1).
6.1 Location Work conducted under this procedure will be performed in room 149, EOHSI. 6.2 Major Equipment Fume hood. 6.3 Time Factors, Staffing, and Resources No critical issues. 7.0 Other Reviews A copy of this procedure has been provided to REHS.

182
4. CRESP SOP-004 “Acid Digestion of Soil or House Dust for Liquid Scintillation Counting” 1.0 Cover Page Author(s): Kristie Ellickson Date Robert Meeker Date Scott Petlick Date Mike Pollock Date
Carl Schopfer Principal Investigator(s): David Kosson, Ph.D. Date Paul Lioy, Ph.D. Date Robert Tata, Ph.D. Date Authoree: David Kosson, Ph.D. Date EOHSI Radiation Safety Committee Chair: Arthur Upton, M.D. Date

183
2.0 List of Procedures This protocol includes one standard operating procedure, SOP-004, “Acid Digestion of Soil or House Dusts for Liquid Scintillation Counting.” 2.1 PURPOSE As soil will attenuate radioactive emissions, heavy metal or organic availability, these contaminants need to be released from the soil for accurate analysis. Depending on soil type and the age of contaminant, a nitric acid digestion can completely release some metals from the soil matrix into the liquid phase. A possible further step in the removal of radionuclides from a soil matrix is the complete dissolution of the soil matrix. This is carried out with the incorporation of hydrofluoric acid and hydrogen peroxide, providing the most accurate assessment of the non-volatile contaminants in the soil matrix. 2.2 SCOPE The resultant acid solution from this procedure can be used for any gross alpha or beta analysis. Analysis of specific radionuclides may require further chemical or electroplating techniques if interferences exist. When applicable, heavy metal analysis by such methods as ICP/MS or GFAA is also possible for long-lived isotopes or heavy metals. 2.3 DEFINITIONS or TERMINOLOGY 2.3.1 Environmental and Occupational Health Sciences Institute Hydrofluoric Acid
Policy (EOHSI HF policy) – a document, housed in the EOHSI, outlining specific daily and emergency procedures for dealing with HF. This document also contains health effect information and references.
2.3.2 Liquid Scintillation Counter - A type of particle or radiation counter that makes
use of a flash of light(scintillation) emitted by an excited atom falling back to its ground state after having been excited by a passing photon or particle. The liquid scintillation medium is used in connection with a photomultiplier tube, which produces a pulse of current for each scintillation.
2.3.3 Scintillator - A phosphor, a substance capable of luminescence, that releases its
energy after a short delay of 10-10 to 10-4 seconds. 2.3.4 Suspension - A mixture in which small liquid or solid particles are incorporated
into a liquid or a gas for a prolonged period of time relative to larger particles. 2.3.5 Leach - Extraction of soluble components of a solid mixture by percolating a
solvent through it. 2.3.6 Digestion - The process of adsorbing and incorporating materials into a medium.
A digestion is more aggressive than a leaching technique. 2.3.7 Digestate – The solution resulting from an acidic digest of a soil.

184
2.3.8 Matrix - The components of tissues, solution or coarser grained rocks into which a
material is embedded. It is generally considered the material that surrounds an analyte.
2.3.9 Sample Processing Container - A 30ml capacity Teflon container with a screw top
lid. The top of the container has been equipped with a Teflon tube fitting, and approximately 1 foot of Teflon tubing. This allows refluxing of the acid, so this method does not require the addition of acid during the digestion process.
2.4 RESPONSIBILITIES Carl Schopfer - leaching techniques, health physics surveys and instrumentation. Kristie Ellickson - leaching techniques and a portion of instrumentation work. Vito Ilacqua – Perform extractions of house dusts from the Tom’s River Cancer Cluster project, which are unrelated to CRESP work. Mr. Ilacqua will not be processing radioactive samples from CRESP. 2.5 PREREQUISITES Soil samples must be dried before use in these procedures. (see CRESP procedure entitled “Soil Drying”) Worker must read the entire protocol before starting any laboratory work. All workers involved with these procedures must have the proper training (outlined in the safety section). 2.6 INTERFERENCES Any particulate material will interfere with the counting results. Certain solvents may not be compatible with the liquid scintillation cocktail. Hydrofluoric acid must never be stored in glass containers. 2.7.0 STANDARDS, REAGENTS and MATERIALS 2.7.1 Trace Metal Nitric Acid, Fisher A509-500 2.7.2 Trace Metal Hydrofluoric Acid, Fisher A513-500 2.7.3 Hydrogen Peroxide, Fisher 50% Catalogue # H341-500 2.7.4 The appropriate mixed or single isotope standard for efficiency calculations 2.7.5 Deionized Water 2.7.6 Filter Paper - Whatman No. 41 or equivalent 2.7.7 Whatman GD/X 13 or Whatman Puradisc syringe filters 2.7.8 Funnel 2.7.9 Pipette or Volumetric measuring device 2.7.10 Teflon screw top sample jars, Savillex Co. Part #0201 2.7.11 Teflon tubing, McMaster-Carr 5239K14 2.7.12 Tube Fitting, McMaster-Carr 52195K56 2.7.13 Tap, McMaster-Carr 8322A62

185
2.7.14 HF Use Kit: • Calcium gluconate tubes • Heavy duty polyethylene bags for disposal of contaminated cleaning supplies. • Laminated Caution/Danger sign for HF use • 2 neoprene gloves (outer gloves), nitrile gloves (inner gloves) • Orange hazardous waste labels from REHS • HF Spill kit including: 3 Calcium Gluconate Gel tubes, 5 heavy duty polyethylene
bags, laminated caution/danger sign, 2 pair nitrile gloves (inner), hazardous waste labels, copy of EOHSI HF guideline, neoprene gloves(outer), absorbent cotton sheets, plastic tongs.
• Rubber Apron • Face Shield • Calcium carbonate decontamination solution • Laminated wallet sized card outlining HF medical procedures and safety. • Plastic tongs • Tyvek overalls and booties • Brightly colored stickers to place on samples containing (neutralized or not) HF. 2.8.0 EQUIPMENT or APPARATUS 2.8.1 Hot plate 2.8.2 Thermometer 2.8.3 Analytical Balance 2.8.4 Laboratory standing clamp 2.9 SAFETY General • All samples are labeled with the sample identifier code, radiation hazard class and a
chain of custody form. The labels shall indicate the presence of hydrofluoric acid. • Bench surfaces shall be covered with disposable bench paper. • Secondary containment is required for both radioactivity-contaminated samples and
samples containing HF and workstations where HF is used. Radioactivity Related • All experiments will be carried out in compliance with safety recommendations
provided by the Radiation Safety Committee at Rutgers University, which is authorized by the Nuclear Regulatory Commission and the State of New Jersey. Anyone conducting work with radionuclides must attend yearly radioactive training courses given by REHS.
• Soil represents a potential inhalation hazard, because it can be dispersed as a dust or
aerosol. The work must be performed with the proper containment to avoid exposure

186
to the worker and other in the vicinity. Dry soil will be handled in a glove bag within the fume hood. Liquid samples will be handled inside the fume hood.
• Radiation dosimetry badges will be worn if necessary. • The potential for radiation exposure from soils will be determined prior to starting
work. The quantities approved for use will generally be limited by the exposure potential such that exceeding regulatory limits will not be possible, or determined to be extremely unlikely.
♦ All safety equipment, including gloves, laboratory coat, face shield and safety glasses
must be kept in room 149. Do not bring these items into the counting room.(room 145)
♦ All items shall be surveyed for contamination before exiting the laboratory ♦ Survey the work area and the laboratory for radioactive contamination at the end of
each day. ♦ Wastes are contained in closed containers for pick-up by REHS and disposed per
agreement with the Savannah River Site. Hydrofluoric Acid Related All hydrofluoric acid use will be done only if the Environmental and Occupational
Health Sciences Institute Hydrofluoric Acid Policy is followed, the worker is trained for work and had obtained prior approval.
Nitric and Hydrofluoric Acids are highly caustic agents. The worker must wear
elbow-length neoprene gloves (outer) (as per REHS glove chart), nitrile gloves(inner), a face shield, hard shoes, lab coat and long durable pants during the entire experiment. Hands must be washed after gloves are removed.
After working with HF the worker shall carry home with them: 1. Calcium Gluconate
Gel, 2. A laminated card outlining HF safety procedures (provided by Scott Petlick at EOHSI).
A HF kit shall be in place at the workstation before beginning any HF involved work.
This kit shall contain: HF spill kit, 3 Calcium Gluconate Gel tubes, 5 heavy-duty polyethylene bags, laminated Caution/Danger sign, 2 pairs of nitrile gloves (inner), 2 pairs neoprene gloves (outer), hazardous waste labels, plastic tongs, absorbent cotton sheet, copy of the EOHSI HF guideline. There should be one of these kits in the laboratory where work will be performed and in the Employee Health Clinic at EOHSI.

187
The buddy system must be used while working with hydrofluoric acid. When laboratory work is to be performed, make sure someone else knows when and where you will be working. As your work proceeds have them check on your progress personally every 30 minutes.
While working with hydrofluoric acid, post a sign on the laboratory door that states:
“Hydrofluoric Acid in Use.” Work with hydrofluoric acid can be performed only during normal business hours of
the EOHSI (Monday-Friday, 8:30 to 16:30). The technician’s/student’s primary supervisor or primary investigator shall be informed when HF work is in progress.
All HF work must be performed in the fume hood, with the sash at operating height.
Protective equipment will be donned before hydrofluoric acid is removed from the
storage area. Persons who have been working with HF during the day should carry a calcium
gluconate tube with them for the following 24 hours. In case a burn should become apparent at any time, apply the gel and seek medical attention immediately. If the worker must seek medical attention, they must bring the HF treatment card and the calcium gluconate card with them to the medical attendant.
2.10 COLLECTION, HANDLING, and PRESERVATION 2.10.1 Wet soil samples, once delivered from REHS will be stored in a locked cabinet. 2.10.2 Dried soil samples will be stored in a vacuum dessicator until the time of use. 2.10.3 Reagent or sample bottles containing hydrofluoric acid will be labeled upon
delivery (or manufacture) with a printed label reading: “Training Required for the use of this chemical. See EOHSI procedure #”.
2.10.4 Reagent or sample bottles must be securely capped when not actively transferring
materials. 2.11 QUALITY CONTROL The samples will be run in triplicate for proper replication. A spiked solution will be run as a method control. Method blanks will include the solution without the soil or spike present. Laboratory blanks will include the scintillation cocktail only. If necessary, a laboratory control will be run using a spiked liquid scintillation cocktail. The method should detect no less than 50 pCi per gram of dry soil. 2.12 CALIBRATION

188
All balances will be calibrated on a biannual basis, and checked with a known weight and bubble level, before use. 2.13 CALCULATIONS 1pCi = 0.037Bq The HF solutions will be neutralized with boric acid before further analysis or processing. The quantities of boric acid to HF are based on the chemical equation below: H3BO3 + 4HF HBF4 + H2O 1) 1.5ml HF is used per sample. 2) (1.5ml HF) * (1.13g/mlHF) * (20.01g/mole)-1 = 0.085moles
If we can neutralize 4moles of HF with 1 mole of H3BO3, we need a total of 0.021moles(or 1.31g) of H3BO3 (61.81g/mole).
3) So, we can add 100ml of a 4% boric acid solution to each prepared sample-this triples the amount of boric acid necessary to neutralize the HF.
3.0.0 PROCEDURE 3.0.1 Dry the soil in the sample processing container according to CRESP SOP-001. 3.0.2 If necessary, sieve the soil according to CRESP SOP-002 (Preparation of Soil
Sub-fractions) 3.0.3 Weigh 1gram of soil into the sample-processing container. Note: All work involving HF shall be performed in the fume hood. Double gloves shall
be worn when using HF. The outer neoprene gloves shall remain in the fume hood after use.
3.0.4 Once the samples have been dried to constant weight and weighed into the sample processing container (figure 1), add 9ml of a 1:1 HNO3 mixture (50% DI water) to each 1 gram soil sample.
3.0.5 Place a beaker of water on the hot plate and heat water to 95°C, and place the sample processing container in the beaker.
3.0.6 Secure the sample in the water bath with a laboratory stand clamp. 3.0.7 Reflux the acid solution in the sample processing container at 95°C ± 5°C for two
hours without letting the sample boil. Note: Maintain the acid levels in the bottle, do not let evaporate to dryness. 3.0.8 After the two hour time period, cool sample to room temperature. 3.0.9 Once the sample is cooled, add 0.5ml 30% hydrogen peroxide. 3.0.10 Note: Add the hydrogen peroxide very slowly so that there is no vigorous
effervescence. 3.0.11 Add hydrogen peroxide slowly until there is no more effervescence. You may
need to heat the solution in order for the effervescence to occur, however do not add hydrogen peroxide to hot digestate. Do not add more than 10 ml total of hydrogen peroxide total.
3.0.12 Cool digestate to room temperature in the fume hood.

189
3.0.13 Once cool, add 1.5ml of 50% hydrofluoric acid. 3.0.14 Heat digestate to 95°C ± 5°C for up to one hour. Condensation can be tested by
holding a pH strip at the top of the Teflon tube. 3.0.15 Complexation Reaction (to be used for all samples). 1 Fill a 125ml wide mouth polyethylene bottle with 100ml of a 4% boric acid solution. 2 Pour entire digestate into the sample bottle with the boric acid solution. 3 Mark the outside of the wide mouth sample bottle with a brightly colored sticker-to
advise others that the sample contained HF. 4 Secure the top of the wide mouth sample bottle with Parafilm, and place in a
refrigerator for 72 hours. 5 Double contain the vial in a larger wide mouth Nalgene jar, if storage outside the
refrigerator is required. 3.0.16 Counting Preparation 1 Add 18ml scintillation cocktail to scintillation vials. The Packard Ultima Gold A/B
liquid scintillation cocktail can be used, or Packard Ultima Gold LLT liquid scintillation cocktail if low background is necessary.
2 Add 2ml of the acid mixture to a syringe filter apparatus-with a 0.45µm polypropylene syringe filter in place.
3 Note: Use of either Whatman GD/X 13 or Whatman Puradisc syringe filters will work for all the leaching procedures. The first syringe filter type is more expensive, but requires less force by the technician.
4 Filter the sample into a liquid scintillation vial containing scintillation cocktail. 5 Count samples using the liquid scintillation counting procedure. 6 Gamma Spectra: These samples can be diluted and counted directly using the gamma
counter. In this case the samples would be diluted with water and placed into a plastic bottle with the appropriate geometry standards. Samples must be parafilmed and placed in a plastic zip-loc bag before placing onto the gamma counter detector.
7 To measure specific Sr-90, Ra-226 or Ra-228 activities the samples may be filtered using the appropriate 3M Empore Rad disk. The counting procedures for these disks are available from the 3M Company.
4.1 STORAGE and CONTAINMENT Dried soil samples will be contained in a vacuum dessicator.
Wet soils samples will be contained in a locked cabinet in room 149 in the EOHSI
building. All samples containing HF, or liquid samples with radioactivity, will be double
contained before storage. The double containment should be durable plastic (compatible with HF) and able to hold the entire volume of the samples.
4.2 WASTE MINIMIZATION Kim wipes will be used rather than paper towels as a way to reduce the mass of solid
waste.

190
Teflon sample processing containers will be washed and reused by refluxing with
10ml 50% reagent grade nitric acid. 4.3 WASTE DISPOSAL The residual acid will be collected into a common container, neutralized with sodium
bicarbonate or calcium carbonate and then disposed of as liquid radioactive waste. All solid wastes including gloves, sample containers, stir bars, etc. will be collected into a common solid radioactive waste container and disposed of as solid waste. All wastes will be collected in their appropriate containers for transport to Savannah River Site for disposal.
Segregate solid radioactive wastes from solid wastes that have HF contamination.
REHS will pickup HF containing waste as hazardous waste. The acidic liquid wastes
will be neutralized with sodium bicarbonate or calcium carbonate to reduce the level of hazard. Liquid waste should be placed in compatible bottles with a tight fitting cap.
Contaminated gloves, vials, towels and other disposable objects should be placed in
sturdy polyethylene bags. Hazardous waste labels should be filled in and applied to the containers or bags.
4.4 ACCIDENT INSTRUCTIONS 4.4.1. Radioactive Sample Considerations • Prior to beginning work, discuss with the OJT (On the Job Trainers) how to respond
to accidents. Accidents that might result in an unplanned external radiation exposure or in any internal exposure (inhalation or ingestion) require immediate REHS notification. Review the instructions on pages 24, 25, 29 and 30 of the EOHSI/CRESP Radiation Safety Global Procedures Manual.
• Small localized spills (with no personal contamination and assuming no HF has been
added to the sample at this time) will be handled by the technician. Use a spray bottle with water to prevent the spilled material from drying out and becoming airborne. Use moist paper towels to cleanse the spill area with a blotting action to prevent the spread of radioactive material. Use decontaminating detergent to clean surfaces (Sparkleen 1 or Picoclean). Check for persistent contamination following cleaning. Contact REHS immediately (445-2550, or 5-2550 from laboratory phones) if the area cannot be decontaminated. Dispose of the contaminated cleaning materials in the radioactive mixed waste. Report the lost material to the Sample Manager.
• If there is a spill of radionuclide contaminated soils, the worker shall use a spray
bottle with water to prevent the spilled material from drying out and becoming airborne, and cover with a damp paper towels.

191
• If spill is on glove, remove outer glove immediately, but leave it in the hood. Check
for contamination of the inner glove. Remove the inner glove and check for skin contamination. Dispose of contaminated gloves as hazardous material.
4.4.2 Hydrofluoric Acid Spill Considerations ♦ In general, HF will be neutralized with a basic compound on contaminated objects.
The rationale for this action is that the fluoride ion (F-) so formed, though toxic, has much less potential to cross the body barriers than HF does.
♦ If HF (1% or more) is spilled in any amount outside of a chemical hood, evacuate the
area close the doors, and post the area with a sign to prevent others from entering. The cleaning procedure may require the use of a respirator and the judgement is not to be made while breathing HF. If the ventilation system is not working, a 50-m3 room may reach OSHA PEL with the evaporation of only 125 mg of HF (approx. 0.25 mL of 50% solution)! A few milliliters of diluted 20% acid may be cleaned up by trained personnel at EOHSI. For larger spills or more concentrated acid call REHS. If the spill is on disposable objects, place them in a polyethylene bag without cleaning them up. Non-disposable objects should be washed in the hood with calcium carbonate solution before being washed with water.
♦ Spills on the workbench in the hood are best neutralized by slowly covering them
with dry sodium or calcium carbonate, bicarbonate or hydroxide, as available. The worker shall keep one of these powders or salts in the hood while working. To facilitate the process of picking up the salts you may carefully add some water to form slurry. This procedure is safe for dilute solutions (50% or less) and up to 20-30 mL. Dispose of the resulting salts as hazardous waste.
♦ For larger spills, up to 100 mL of dilute acid, in the hood use the spill kit included in
the HF spill kit. A new kit should be ordered afterwards as necessary. In the case of such spills of concentrated acid (50% or more) or for larger amounts of dilute solutions, contain the spill as best as you can, turn over the warning signal, close the hood and call REHS. These spills require trained personnel. (EOHSI HF Policy) All wastes from clean up of HF are regulated hazardous waste; if also radioactive it is considered a mixed waste.
♦ For localized well-defined personal contamination, neutralize clothing with Calcium
Gluconate Gel, remove clothing, rinse affected area on skin with water for 1 minute and then treat skin with Calcium Gluconate Gel. To treat the contaminated skin with Calcium Gluconate Gel: wash contaminated skin with mild soap and water and then apply gel liberally. Reapply for 10-15 minutes while seeking medical attention. Do not use abrasives when washing skin. Limit contact with others to avoid spreading contamination into clean areas. If there is a hazardous condition the worker shall exit the immediate area of the spill, but do not leave the laboratory area until advised by REHS or Rutgers Police.

192
♦ For non-localized spill, remove contaminated clothing, rinse skin with water for 1 minute and then treat contaminated skin with the Calcium Gluconate Gel.
♦ To check for workbench contamination, take a pH strip and wipe it across the area. A
low reading (pH of 1-4) could mean that there is still some residual contamination in the area of the spill. You may conclude that there is no contamination in the area if the pH is above 6.
♦ Contact REHS for assistance in cleaning large spills. ♦ In the event of dispersal of radioactive material outside of the ventilated enclosure,
exit the laboratory immediately and contact REHS. Restrict access to area. 5.0 REFERENCES EOHSI/CRESP Radiation Safety Global Procedures Manual. Rutgers University Radiation Safety Guide, 7th edition, 1989. Kingston, H.M. Test for Evaluating Solid Waste, Physical Methods, 3rd Edition; US Environmental Protection Agency, Office of Solid Waste and Emergency Response. US Government Printing Office: Washington DC SW-846-3051, (1986). Environmental and Occupational Health Sciences Institute Hydrofluoric Acid Policy 6.0 ATTACHMENTS/ENCLOSURES 1. Environmental and Occupational Health Sciences Institute Hydrofluoric Acid Policy 2. Figure 1: Schematic of the Sample Processing Container

193
5. Data for Bioaccessible Radionuclides (137Cs & 90Sr) 5.1. Cesium-137
Sample Weight (g)
Concentration (Bq/g)
Bioaccessibility (%)
Recovery (%)
1Berm1 Gas 1 0.2 59.1640 10.06 Gas 2 0.2 51.8844 8.82 Gas 3 0.2 65.0989 11.06 Soil 1 0.2 158.5107 37.00 Soil 2 0.2 249.0262 51.14 Soil 3 0.2 211.0695 46.94 Int 1 0.2 77.8546 13.23 Int 2 0.2 77.1764 13.12 Int 3 0.2 81.1098 13.79 Soil 1 0.2 176.3620 43.21 Soil 2 0.2 143.2791 37.47 Soil 3 0.2 232.7617 53.35
1Berm2 Gas 1 0.2079 11.8649 15.74 Gas 2 0.2054 11.0739 14.69 Gas 3 0.2029 10.1463 13.46 Soil 1 0.2079 30.9733 56.83 Soil 2 0.2054 25.6339 48.69 Soil 3 0.2029 17.5425 36.73 Int 1 0.2037 10.5871 14.04 Int 2 0.2145 10.8129 14.34 Int 3 0.2086 10.5985 14.06 Soil 1 0.2037 14.1021 32.75 Soil 2 0.2145 16.7659 36.58 Soil 3 0.2086 16.0365 35.33
1Berm3 Gas 1 0.515 1.2479 14.95 Gas 2 0.540 0.9942 11.91 Gas 3 0.509 0.8393 10.05 Soil 1 0.515 2.2369 41.74 Soil 2 0.540 2.3839 40.46 Soil 3 0.509 1.2985 25.60 Int 1 0.516 1.3646 16.34 Int 2 0.535 1.2691 15.20 Int 3 0.519 1.4438 17.29 Soil 1 0.516 3.9356 63.48 Soil 2 0.535 3.1447 52.86 Soil 3 0.5191 2.3046 44.89

194
Sample Weight (g)
Concentration (Bq/g)
Bioaccessibility (%)
Recovery (%)
2Berm1 Gas 1 1.5860 0.4975 7.42 Gas 2 1.7904 0.6240 9.30 Soil 1 0.5694 4.1662 71.42 Soil 2 0.6277 4.7327 79.87 Soil 3 0.5601 4.2370 72.48 Int 1 1.7180 0.9396 14.01 Int 2 2.3465 0.6461 9.63 Soil 1 0.5841 4.5533 77.52 Soil 2 0.5295 4.1415 71.38 Soil 3 0.6107 4.1892 72.09
2Berm2 Gas 1 1.6241 0.0760 9.96 Gas 2 0.5232 0.0367 4.81 Gas 3 1.9148 0.0945 12.38 Soil 1 0.6325 0.4904 76.63 Soil 2 0.5557 0.3588 59.40 Soil 3 0.7266 0.5239 81.03 Int 1 1.5944 0.0757 9.92 Int 2 1.5944 0.0503 6.59 Soil 1 0.5145 0.5382 77.12 Soil 2 0.5736 0.5425 77.67
2Berm3 Gas 1 1.00 0.0273 6.33 Soil 1 0.198 0.3916 99.08 Soil 2 0.250 0.2881 74.60 Soil 3 0.218 0.3387 86.58 Int 1 1.7180 0.0590 14.01 Soil 1 0.2282 0.2912 82.84 Soil 2 0.2278 0.0019 14.41 Soil 3 0.2282 0.3721 101.98
3Berm1 Gas 1 0.4831 1.1771 21.31 Gas 2 1.5524 1.3617 24.66 Gas 3 1.5808 1.1149 20.19 Soil 1 0.5609 2.2092 60.19 Soil 2 0.5181 3.1143 76.58 Soil 3 0.5018 3.3111 80.14 Int 1 1.6460 1.7696 32.04 Soil 1 0.5335 2.7392 80.78 Soil 2 0.5712 3.6664 97.57 Soil 3 0.5494 3.4777 94.15

195
Sample Weight (g)
Concentration (Bq/g)
Bioaccessibility (%)
Recovery (%)
4Berm1 Gas 1 1.6403 0.0394 5.44 Gas 2 1.6403 0.1003 13.86 Soil 1 0.5512 0.4421 74.93 Soil 2 0.5395 0.3148 57.35 Soil 3 0.5496 0.3493 62.12 Int 1 1.7020 0.0918 12.69 Soil 1 0.5031 0.6055 96.33 Soil 2 0.5508 0.6149 97.63 Soil 3 0.6485 0.5751 92.13
4Berm2 Gas 1 0.5197 0.2253 12.90 Gas 2 1.5642 0.1559 8.93 Gas 3 2.0611 0.1537 8.80 Soil 1 0.6636 1.1684 75.70 Soil 2 0.6817 1.0396 68.32 Soil 3 0.7158 0.7576 52.18 Int 1 1.6973 0.2121 12.15 Int 2 1.6973 0.3131 17.93 Soil 1 0.6038 0.9173 64.66 Soil 2 0.5782 0.7918 57.48 Soil 3 0.5153 0.7943 57.62

196
5.2. Strontium-90 Sample Weight
(g) Concentration
(Bq/g) Bioaccessibility
(%) 1Berm1
Gas 1 0.20 32.8461 95.91% Gas 2 0.20 30.0162 87.64% Gas 3 0.20 35.5194 103.71% Int 1 0.20 21.1046 61.62% Int 2 0.20 18.3956 53.71% Int 3 0.20 20.9096 61.05%
1Berm2 Gas 1 0.2079 17.2776 106.05% Gas 2 0.2054 14.7164 90.33% Gas 3 0.2029 16.1460 99.10% Int 1 0.2037 9.4196 57.82% Int 2 0.2145 10.2064 62.64% Int 3 0.2086 10.2929 63.18%
1Berm3 Gas 1 0.5150 2.4549 79.20% Gas 2 0.5360 2.5767 83.13% Gas 3 0.5090 2.2485 72.54% Int 1 0.5160 1.1680 37.68% Int 2 0.5350 1.4825 47.83% Int 3 0.5190 1.6836 54.32%
2Berm2 Gas 1 0.4952 4.2015 94.36% Gas 2 0.5230 4.0806 91.65% Gas 3 0.6059 4.1403 92.99% Int 1 0.5145 1.3850 31.11% Int 2 0.5063 1.0362 23.27% Int 3 0.5736 1.6087 36.13%
2Berm3 Gas 1 0.5319 2.7746 88.94% Gas 2 0.5207 2.9531 94.66% Gas 3 0.5060 2.7719 88.85% Int 1 0.4862 1.5388 49.33% Int 2 0.4612 1.6771 53.76% Int 3 0.3256 1.7809 57.08%
2Berm4 Gas 1 0.5139 0.9848 91.96% Gas 2 0.5024 1.0081 94.14% Gas 3 0.5255 1.0145 94.74% Int 1 0.4503 0.7269 67.87% Int 2 0.4780 0.7455 69.61% Int 3 0.4494 0.8089 75.54%

197
Sample Weight (g)
Concentration (Bq/g)
Bioaccessibility (%)
3Berm1 Gas 1 0.4831 0.2348 33.18% Gas 2 0.5640 0.2190 30.94% Gas 3 0.5053 0.2139 30.22% Int 1 0.5335 0.1508 21.31% Int 2 0.5712 0.1092 15.43% Int 3 0.5494 0.1186 16.75%
3Berm2 Gas 1 0.5364 0.4642 72.53% Gas 2 0.5713 0.4857 75.90% Gas 3 0.5844 0.4402 68.79% Int 1 0.5608 0.3883 60.67% Int 2 0.5826 0.3557 55.58% Int 3 0.5002 0.3486 54.48%
4Berm1 Gas 1 0.5395 0.0695 47.96% Gas 2 0.5496 0.0795 54.83% Gas 3 0.5512 0.0668 46.08% Int 1 0.5031 0.0815 56.22% Int 2 0.5508 0.0624 43.07% Int 3 0.6485 0.0648 44.70%
4Berm2 Gas 1 0.5197 0.1376 105.49% Gas 2 0.5195 0.1081 82.86% Gas 3 0.5250 0.1128 86.46% Int 1 1.6973 0.0632 48.47% Int 2 1.6973 0.0572 43.87%

198
6. Data for Bioaccessible Lead 6.1. Three Particle Size Fractions
Extracts Recaptured Type Sample Weight (g) (ng/g) (mL) (ng/g) (mL)
Bioaccessibility(%)
Home 001 Gas 1 0.1334 2032 52.85 299.9 45 73.40 Gas 2 0.1342 2212 52.91 261.2 45 79.52 Gas 3 0.1351 2192 52.03 267.5 45 76.97 Int 1 0.1380 97.2 102.73 2252 45 6.60 Int 2 0.1377 93.0 103.08 2015 45 6.35
<75µm
Int 3 0.1321 98.0 103.48 2146 45 7.00 Gas 1 0.1387 1846 52.23 408.9 45 72.44 Gas 2 0.1366 1765 52.04 410.3 45 70.07 Gas 3 0.1348 1948 52.03 387.4 45 78.35 Int 1 0.1327 121.5 102.97 2850 45 9.82 Int 2 0.1394 134.0 103.42 2205 45 10.36
<150µm
Int 3 0.1383 129.8 103.31 2477 45 10.10 Gas 1 0.1318 1684 52.50 553 45 73.21 Gas 2 0.1280 1646 51.47 520.3 45 72.07 Gas 3 0.1298 1727 51.89 341.3 45 75.35 Int 1 0.1304 129.5 101.80 2258 45 11.03 Int 2 0.1363 137.0 102.71 2275 45 11.27
<250µm
Int 3 0.1376 154.5 103.42 2295 45 12.67 Home 002
Gas 1 0.1364 889 52.80 216.0 45 57.74 Gas 2 0.1377 1246 52.90 157.0 45 80.31 Gas 3 0.1397 1352 52.23 194.3 45 84.81 Int 1 0.1377 53.2 103.11 1783 45 6.68 Int 2 0.1322 52.1 104.16 1944 45 6.89
<75µm
Int 3 0.1350 49.9 103.37 2203 45 6.41 Gas 1 0.1322 1312 52.32 274.9 45 74.20 Gas 2 0.1336 1226 52.06 231.4 45 68.27 Gas 3 0.1344 1274 52.56 246.4 45 71.20 Int 1 0.1370 77.8 102.75 2457 45 8.34 Int 2 0.1360 75.8 103.83 2282 45 8.27
<150µm
Int 3 0.1335 69.0 103.51 2082 45 7.65 Gas 1 0.1304 1503 52.41 287.9 45 78.98 Gas 2 0.1320 1253 53.03 209.7 45 65.81 Gas 3 0.1318 1363 51.82 226.9 45 70.06 Int 1 0.1303 54.5 103.42 1942 45 5.66 Int 2 0.1344 61.0 103.59 1876 45 6.15
<250µm
Int 3 0.1354 68.4 101.01 2348 45 6.67

199
Extracts Recaptured Type Sample Weight
(g) (ng/g) (mL) (ng/g) (mL) Bioaccessibility
(%) Home 004
Gas 1 0.1422 379.7 52.61 60.5 45 64.92 Gas 2 0.1321 386.4 53.22 34.2 45 71.94 Gas 3 0.1349 403.3 53.04 33.3 45 73.28 Int 1 0.1360 23.0 103.61 419.9 45 8.10 Int 2 0.1347 26.1 104.32 403.2 45 9.34
<75µm
Int 3 0.1383 23.5 103.16 518 45 8.10 Gas 1 0.1355 402.0 53.29 54.2 45 68.38 Gas 2 0.1447 421.3 53.47 66.5 45 67.33 Gas 3 0.1409 436.9 52.51 59.7 45 70.42 Int 1 0.1383 30.8 102.69 532 45 9.89 Int 2 0.1365 27.2 103.25 493.5 45 8.90
<150µm
Int 3 0.1382 27.5 103.04 495 45 8.87 Gas 1 0.1345 377 52.36 69.3 45 61.15 Gas 2 0.1431 585 53.40 90.5 45 90.95 Gas 3 0.1398 485 52.70 64.4 45 76.17 Int 1 0.1293 37.8 103.68 377 45 12.63 Int 2 0.1301 54.0 104.68 616 45 18.10
<250µm
Int 3 0.1324 31.1 104.41 322 45 10.22 Home 005
Gas 1 0.1395 2055 52.46 194.0 45 68.67 Gas 2 0.1312 1898 53.34 104.0 45 68.57 Gas 3 0.1376 2049 52.49 368.0 45 69.46 Int 1 0.1329 262.1 104.35 2723 45 18.29 Int 2 0.1347 248.7 103.55 2613 45 16.99
<75µm
Int 3 0.1356 265.6 103.61 2827 45 18.03 Gas 1 0.1255 2107 54.33 160.0 45 78.17 Gas 2 0.1238 2178 52.71 213.0 45 79.47 Gas 3 0.1276 2398 52.63 298.0 45 84.76 Int 1 0.1331 301.0 104.08 3102 45 20.17 Int 2 0.1346 388.7 102.84 3722 45 25.45
<150µm
Int 3 0.1345 347.5 104.54 3491 45 23.15 Gas 1 0.1367 1964 51.77 178.0 45 73.36 Gas 2 0.1366 2115 53.10 162.0 45 81.09 Gas 3 0.1384 2191 53.37 139.0 45 83.34 Int 1 0.1330 315.0 104.94 2766 45 24.52 Int 2 0.1377 286.3 104.23 3057 45 21.38
<250µm
Int 3 0.1375 267.3 104.13 3094 45 19.97

200
Extracts Recaptured Type Sample Weight (g) (ng/g) (mL) (ng/g) (mL)
Bioaccessibility(%)
Home 006 Gas 1 0.1394 639.0 52.67 77.8 45 67.51 Gas 2 0.1394 681.0 52.62 72.6 45 71.88 Gas 3 0.1310 668 52.20 73.8 45 74.43 Int 1 0.1335 39.0 102.93 911 45 8.41 Int 2 0.1383 35.0 102.34 977 45 7.24
<75µm
Int 3 0.1385 30.3 101.82 1032 45 6.23 Gas 1 0.1357 557 52.68 79.5 45 62.59 Gas 2 0.1332 613 52.70 117.6 45 70.20 Gas 3 0.1389 602 52.26 110.3 45 65.60 Int 1 0.1365 50.9 103.02 738 45 11.12 Int 2 0.1383 64.4 103.52 707 45 13.95
<150µm
Int 3 0.1380 55.6 104.37 822 45 12.17 Gas 1 0.1328 525.6 52.88 122.8 45 67.72 Gas 2 0.1315 522.4 53.26 86.0 45 68.46 Gas 3 0.1339 481 53.21 170.7 45 61.85 Int 1 0.1374 63.1 103.96 740 45 15.45 Int 2 0.1371 56.4 104.16 825 45 13.87
<250µm
Int 3 0.1322 50.8 102.85 658 45 12.79

201
6.2. Particle Size < 75 µm Fractions Type Sample Weight
(g) Volume
(mL) Concentration
(ng/g) Bioaccessibility
(%) Home 007
Gas 1 0.1361 52.21 2487 72.70 Gas 2 0.1367 51.56 2583 74.24 Gas 3 0.1341 52.19 2598 77.04 Int 1 0.1315 102.75 100.6 5.99 Int 2 0.1333 103.94 98.5 5.85
<75µm
Int 3 0.1315 104.48 101.7 6.16 Home 009
Gas 1 0.1335 52.31 1380 76.22 Gas 2 0.1322 53.23 1372 77.87 Gas 3 0.1348 52.93 1398 77.38 Int 1 0.1350 103.35 104.6 11.29 Int 2 0.1325 104.25 99.5 11.03
<75µm
Int 3 0.1390 103.61 104.9 11.02 Home 010
Gas 1 0.1394 51.72 686 52.30 Gas 2 0.1317 52.81 659 54.30 Gas 3 0.1317 53.45 618 51.54 Int 1 0.1350 106.9 201.0 32.70 Int 2 0.1358 104.77 197.3 31.28
<75µm
Int 3 0.1371 104.62 205.4 32.21 Home 013
Gas 1 0.1318 52.30 1023 55.48 Gas 2 0.1309 52.97 988 54.64 Gas 3 0.1341 52.71 1067 57.32 Int 1 0.1314 106.42 45.5 5.04 Int 2 0.1340 102.65 47.1 4.93
<75µm
Int 3 0.1321 104.50 42.8 4.63 Home 014
Gas 1 0.1342 52.32 310 57.91 Gas 2 0.1325 52.51 307 58.30 Gas 3 0.1370 53.28 295 54.88 Int 1 0.1329 103.95 24.7 9.26 Int 2 0.1334 103.77 23.2 8.65
<75µm
Int 3 0.1387 103.83 27.8 9.97 Home 016
Gas 1 0.1309 52.77 1430 70.28 Gas 2 0.1357 53.25 1580 75.59 Gas 3 0.1384 52.4 1560 72.01 Int 1 0.1308 104.17 57.8 5.61 Int 2 0.1323 104.03 59.2 5.68
<75µm
Int 3 0.1386 105.06 64.6 5.97

202
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility(%)
Home 021 Gas 1 0.1317 52.35 1043 61.30 Gas 2 0.1315 52.02 1038 60.71 Gas 3 0.1294 52.34 962 57.53 Int 1 0.1282 104.08 102.4 12.29 Int 2 0.1291 104.74 108.0 12.96
<75µm
Int 3 0.1348 102.96 102.4 11.56 Home 023
Gas 1 0.1337 53.93 2445 55.72 Gas 2 0.1336 52.72 2548 56.81 Gas 3 0.1369 53.07 2409 52.76 Int 1 0.1303 103.35 147.0 6.59 Int 2 0.1334 103.84 141.6 6.23
Int 3 0.1347 104.35 144.2 6.31 Home 027
Gas 1 0.1316 53.07 1032 51.16 Gas 2 0.1306 53.27 1048 52.55 Gas 3 0.1350 52.99 1106 53.37 Int 1 0.1297 104.53 307.9 30.51 Int 2 0.1298 103.73 299.1 29.39
<75µm
Int 3 0.1330 104.31 273.4 26.36 Home 029
Gas 1 0.1332 52.32 738 53.57 Gas 2 0.1369 53.34 744 53.57 Gas 3 0.1305 52.24 731 54.08 Int 1 0.1376 104.8 127.8 17.99 Int 2 0.1397 103.03 122.0 16.63
<75µm
Int 3 0.1261 104.46 120.4 18.43

203
7. Data for Bioaccessible Mercury 7.1. Tuna Steak
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
1st Tuna Steak Test Gas Liq 1 2.9800 56.34 19.5 54.34 Gas Liq 2 2.9200 55.99 15.0 42.39 Gas Liq 3 2.9400 56.85 18.0 51.30 Gas Sol 1 2.9800 40 8.4 16.62 Gas Sol 2 2.9200 40 12.0 24.23 Gas Sol 3 2.9400 40 8.6 17.24 Int Liq 1 2.9000 107.45 6.7 36.70 Int Liq 2 2.9400 106.97 7.0 37.54 Int Liq 3 2.9400 106.82 8.12 43.48 Int Sol 1 2.9000 40 21.0 42.69 Int Sol 2 2.9400 40 30.0 60.16
Raw Steak
Int Sol 3 2.9400 40 36.0 72.19 Gas Liq 1 2.9600 56.74 30.0 56.38 Gas Liq 2 2.9400 56.21 33.0 61.86 Gas Liq 3 2.8800 57.26 31.5 61.40 Gas Sol 1 2.9600 40 15.0 19.87 Gas Sol 2 2.9400 40 15.0 20.01 Gas Sol 3 2.8800 40 9.1 12.39 Int Liq 1 2.9000 107.37 3.9 14.23 Int Liq 2 2.9600 106.51 6.2 21.73 Int Liq 3 2.9500 105.85 8.6 30.25 Int Sol 1 2.9000 40 54.0 73.02 Int Sol 2 2.9600 40 49.0 64.92
Cooked Steak
Int Sol 3 2.9500 40 49.0 65.14 2nd Tuna Steak Test
Gas Liq 1 2.9400 56.06 23.8 69.39 Gas Liq 2 2.9700 56.22 21.0 60.78 Gas Liq 3 2.9600 54.45 26.6 74.82 Gas Sol 1 2.9400 40 3.9 8.11 Gas Sol 2 2.9700 40 3.6 7.41 Gas Sol 3 2.9600 40 5.0 10.33 Int Liq 1 3.0200 102.77 6.2 32.05 Int Liq 2 2.9700 102.08 6.2 32.37 Int Liq 3 2.9500 104.16 6.9 37.04 Int Sol 1 3.0200 40 18.0 36.45 Int Sol 2 2.9700 40 20.0 41.19
Raw Steak
Int Sol 3 2.9500 40 20.0 41.47

204
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
Gas Liq 1 2.9300 55.19 33.6 61.45 Gas Liq 2 2.9200 55.11 35.0 64.13 Gas Liq 3 3.0200 55.48 39.2 69.92 Gas Sol 1 2.9300 40 11.0 14.58 Gas Sol 2 2.9200 40 11.0 14.63 Gas Sol 3 3.0200 40 12.0 15.43 Int Liq 1 2.9300 103.52 14.3 48.98 Int Liq 2 2.9200 101.18 14.0 47.10 Int Liq 3 3.0000 101.99 15.4 50.83 Int Sol 1 2.9300 40 23.0 30.48 Int Sol 2 2.9200 40 26.0 34.58
Cooked Steak
Int Sol 3 3.0000 40 27.0 34.95 3rd Tuna Steak Test
Gas Liq 1 2.9400 55.25 18.2 49.89 Gas Liq 2 2.9500 56.30 16.8 46.77 Gas Liq 3 2.9800 55.50 18.2 49.45 Gas Sol 1 2.9400 30 11.0 16.37 Gas Sol 2 2.9500 30 10.0 14.84 Gas Sol 3 2.9800 30 14.0 20.56 Int Liq 1 2.9800 102.68 9.4 47.15 Int Liq 2 2.9700 103.55 6.0 30.62 Int Liq 3 2.9500 103.44 4.6 23.63 Int Sol 1 2.9800 30 26.1 38.33 Int Sol 2 2.9700 30 24.4 35.95
Raw Steak
Int Sol 3 2.9500 30 26.1 38.72 Gas Liq 1 2.9500 53.61 19.2 35.68 Gas Liq 2 2.9800 56.60 26.6 51.66 Gas Liq 3 2.9700 55.77 31.2 59.90 Gas Sol 1 2.9500 30 47.0 48.87 Gas Sol 2 2.9800 30 15.0 15.44 Gas Sol 3 2.9700 30 13.0 13.43 Int Liq 1 2.9200 102.51 11.1 39.70 Int Liq 2 2.9800 102.11 14.0 49.05 Int Liq 3 2.9400 102.60 13.2 47.10 Int Sol 1 2.9200 30 33.0 34.67 Int Sol 2 2.9800 30 35.0 36.03
Cooked Steak
Int Sol 3 2.9400 30 35.0 36.52

205
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
4th Tuna Steak Test Gas Liq 1 2.9300 55.92 25.2 68.71 Gas Liq 2 2.9300 55.60 28.0 75.90 Gas Liq 3 2.9300 56.23 30.8 84.44 Gas Sol 1 2.9300 20 17.0 16.58 Gas Sol 2 2.9300 20 12.0 11.70 Gas Sol 3 2.9300 25 11.0 13.41 Int Liq 1 2.9600 101.86 7.0 34.41 Int Liq 2 2.9400 103.04 7.8 39.25 Int Liq 3 2.9200 102.31 11.8 58.86 Int Sol 1 2.9600 20 27.0 26.06 Int Sol 2 2.9400 20 31.0 30.13
Raw Steak
Int Sol 3 2.9200 20 29.0 28.38 Gas Liq 1 2.9400 55.40 40.6 68.92 Gas Liq 2 2.9500 58.08 40.6 72.01 Gas Liq 3 2.9800 56.44 42.0 71.66 Gas Sol 1 2.9400 20 23.0 14.10 Gas Sol 2 2.9500 20 23.0 14.05 Gas Sol 3 2.9800 20 25.0 15.12 Int Liq 1 2.9600 102.71 16.3 51.02 Int Liq 2 2.9600 101.35 18.2 56.14 Int Liq 3 2.9600 103.48 21.0 66.14 Int Sol 1 2.9600 20 49.0 29.83 Int Sol 2 2.9600 20 57.0 34.70
Cooked Steak
Int Sol 3 2.9600 20 51.0 31.04 5th Tuna Steak Test
Gas Liq 1 2.9500 55.25 23.8 61.57 Gas Liq 2 2.9500 56.44 29.4 77.69 Gas Liq 3 2.9600 56.36 26.4 69.43 Gas Sol 1 2.9500 25 13.0 15.22 Gas Sol 2 2.9500 25 11.0 12.88 Gas Sol 3 2.9600 25 10.0 11.67 Int Liq 1 2.9200 101.66 10.8 51.84 Int Liq 2 2.9400 102.92 8.8 42.65 Int Liq 3 2.9600 102.80 12.7 61.11 Int Sol 1 2.9200 25 22.0 26.02 Int Sol 2 2.9400 25 26.0 30.54
Raw Steak
Int Sol 3 2.9600 25 21.0 24.50

206
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
Gas Liq 1 2.9600 54.49 34.8 51.87 Gas Liq 2 2.9300 55.96 43.4 67.12 Gas Liq 3 2.9300 55.68 43.4 66.78 Gas Sol 1 2.9600 25 48.0 32.83 Gas Sol 2 2.9300 25 33.0 22.80 Gas Sol 3 2.9300 25 36.0 24.87 Int Liq 1 2.9500 102.42 6.2 17.32 Int Liq 2 2.9200 102.97 7.1 20.22 Int Liq 3 2.9400 102.86 10.6 30.14 Int Sol 1 2.9500 25 96.0 65.88 Int Sol 2 2.9200 25 91.0 63.09
Cooked Steak
Int Sol 3 2.9400 30 79.0 65.27

207
7.2. Canned Tuna Type Sample Weight
(g) Volume
(mL) Concentration
(ng/g) Bioaccessibility / Recovery (%)
1st Canned Tuna Test Gas Liq 1 2.9500 55.01 1.5 55.23 Gas Liq 2 2.9200 55.38 1.5 56.17 Gas Liq 3 2.9700 56.52 1.3 46.11 Gas Sol 1 2.9500 35 1.0 22.82 Gas Sol 2 2.9200 35 1.1 25.36 Gas Sol 3 2.9700 35 0.8 18.13 Int Liq 1 2.9600 102.17 0.14 9.29 Int Liq 2 2.9200 101.33 0.14 9.34 Int Liq 3 2.9700 102.16 0.14 9.2 Int Sol 1 2.9600 35 2.7 61.40 Int Sol 2 2.9200 35 2.9 66.85
Yellow Can
Int Sol 3 2.9700 35 2.9 65.72 Gas Liq 1 2.9600 56.01 7.0 57.42 Gas Liq 2 2.9300 56.06 8.4 69.68 Gas Liq 3 2.9300 56.28 8.4 69.95 Gas Sol 1 2.9600 35 3.7 18.97 Gas Sol 2 2.9300 35 5.3 27.45 Gas Sol 3 2.9300 35 4.5 23.30 Int Liq 1 2.9500 103.79 0.14 2.14 Int Liq 2 2.9200 101.50 0.14 2.11 Int Liq 3 2.9400 103.24 0.14 2.13 Int Sol 1 2.9500 35 16.0 82.30 Int Sol 2 2.9200 35 17.0 88.34
White Can
Int Sol 3 2.9400 35 15.0 77.42 2nd Canned Tuna Test
Gas Liq 1 2.9287 56.43 1.1 41.50 Gas Liq 2 2.9300 56.69 1.5 57.30 Gas Liq 3 2.9109 56.33 1.4 52.10 Gas Sol 1 2.9287 20 2.2 28.89 Gas Sol 2 2.9300 20 2.4 31.50 Gas Sol 3 2.9109 20 2.0 26.43 Int Liq 1 2.9147 102.28 0.1 9.45 Int Liq 2 2.9143 103.15 0.1 9.53 Int Liq 3 2.9225 103.69 0.1 9.55 Int Sol 1 2.9147 20 5.6 73.90 Int Sol 2 2.9143 20 5.7 75.23
Yellow Can
Int Sol 3 2.9225 20 5.1 67.12

208
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
Gas Liq 1 2.9826 55.80 6.4 52.95 Gas Liq 2 2.9281 55.03 7.0 56.25 Gas Liq 3 2.9830 55.76 6.6 53.37 Gas Sol 1 2.9826 20 10.0 29.47 Gas Sol 2 2.9281 20 9.2 26.87 Gas Sol 3 2.9830 20 9.4 27.34 Int Liq 1 2.9911 103.75 0.14 2.15 Int Liq 2 2.9799 104.01 0.14 2.16 Int Liq 3 2.9887 103.65 0.14 2.13 Int Sol 1 2.9911 19 30.0 84.29 Int Sol 2 2.9799 19 31.0 87.35
White Can
Int Sol 3 2.9887 19 31.0 86.51 3rd Canned Tuna Test
Gas Liq 1 2.9354 55.09 2.0 70.74 Gas Liq 2 2.9358 56.21 1.7 61.86 Gas Liq 3 2.9185 56.34 1.8 67.57 Gas Sol 1 2.9354 20 2.1 27.52 Gas Sol 2 2.9358 20 2.1 27.51 Gas Sol 3 2.9185 20 2.0 26.36 Int Liq 1 2.9373 102.43 0.14 9.39 Int Liq 2 2.9118 102.21 0.14 9.45 Int Liq 3 2.9097 104.41 0.14 9.66 Int Sol 1 2.9373 20 4.7 61.54 Int Sol 2 2.9118 20 5.3 70.01
Yellow Can
Int Sol 3 2.9097 20 5.4 71.38 Gas Liq 1 2.9826 55.35 8.5 68.71 Gas Liq 2 2.9281 55.86 7.0 57.89 Gas Liq 3 2.9830 54.85 8.1 64.73 Gas Sol 1 2.9826 20 8.6 25.00 Gas Sol 2 2.9281 20 7.5 22.21 Gas Sol 3 2.9830 20 9.4 27.32 Int Liq 1 2.9911 103.18 0.14 2.09 Int Liq 2 2.9799 102.84 0.14 2.09 Int Liq 3 2.9887 102.98 0.14 2.09 Int Sol 1 2.9911 19 31.0 85.37 Int Sol 2 2.9799 19 32.0 88.45
White Can
Int Sol 3 2.9887 19 32.0 88.19

209
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
4th Canned Tuna Test Gas Liq 1 2.9587 54.47 1.0 34.70 Gas Liq 2 2.9537 55.35 0.8 30.27 Gas Liq 3 2.9526 54.49 1.3 44.72 Gas Sol 1 2.9587 20 2.5 32.50 Gas Sol 2 2.9537 20 2.1 27.35 Gas Sol 3 2.9526 20 2.2 28.66 Int Liq 1 2.9606 102.17 0.14 9.29 Int Liq 2 2.9688 101.08 0.14 9.17 Int Liq 3 2.9422 102.30 0.14 9.36 Int Sol 1 2.9147 20 5.6 73.90 Int Sol 2 2.9143 20 5.7 75.23
Yellow Can
Int Sol 3 2.9225 20 5.1 67.12 Gas Liq 1 2.9479 54.66 5.6 45.02 Gas Liq 2 2.9618 55.67 6.2 50.20 Gas Liq 3 2.9341 55.18 5.7 46.80 Gas Sol 1 2.9479 20 8.2 24.12 Gas Sol 2 2.9618 20 8.0 23.42 Gas Sol 3 2.9341 20 7.5 22.16 Int Liq 1 2.9772 102.88 0.14 2.10 Int Liq 2 2.9651 101.71 0.14 2.08 Int Liq 3 2.9453 102.93 0.14 2.12 Int Sol 1 2.9772 20 28.0 81.54 Int Sol 2 2.9651 20 27.0 78.95
White Can
Int Sol 3 2.9453 20 26.0 76.54 5th Canned Tuna Test
Gas Liq 1 2.9126 56.70 1.1 41.93 Gas Liq 2 2.9016 56.71 1.1 42.10 Gas Liq 3 2.9046 55.92 0.8 31.10 Gas Sol 1 2.9126 20 1.6 21.13 Gas Sol 2 2.9016 20 1.8 23.86 Gas Sol 3 2.9046 20 1.9 25.16 Int Liq 1 2.9178 102.83 0.14 9.49 Int Liq 2 2.9440 103.21 0.14 9.44 Int Liq 3 2.9347 103.80 0.14 9.52 Int Sol 1 2.9178 20 4.7 61.95 Int Sol 2 2.9440 20 5.0 65.32
Yellow Can
Int Sol 3 2.9347 20 4.8 62.91

210
Type Sample Weight (g)
Volume (mL)
Concentration (ng/g)
Bioaccessibility / Recovery (%)
Gas Liq 1 2.9395 55.26 6.2 50.20 Gas Liq 2 2.9431 54.62 6.7 54.07 Gas Liq 3 2.9264 55.29 6.9 56.19 Gas Sol 1 2.9395 20 8.7 25.66 Gas Sol 2 2.9431 20 8.6 25.34 Gas Sol 3 2.9264 20 8.2 24.30 Int Liq 1 2.9255 101.90 0.14 2.11 Int Liq 2 2.9425 103.63 0.14 2.14 Int Liq 3 2.9464 102.85 0.14 2.12 Int Sol 1 2.9255 20 29.0 85.95 Int Sol 2 2.9425 20 28.0 82.51
White Can
Int Sol 3 2.9464 20 28.0 82.40

211
References 1. Aberg, B., Ekman, L., Falk, R., Greitz, U., Persson, G., and Snihs, O. S.
Metabolism of Methyl Mercury (203Hg) Compounds in Man. Archives of Environmental Health, Vol. 19, pp 478-484, 1969.
2. Akagi, H., Malm, O., Branches, F. J. P., Kinjo, Y., Kashima, Y., Guimaraes, J. R.
D., Oliveira, R. B., Haraguchi, K., Pfeiffer, W. C., Takizawa, Y, and Kato, H. Human Exposure to Mercury due to Gold Mining in the Tapajos River Basin, Amazon, Brazil: Speciation of Mercury in Human Hair, Blood and Urine. Water, Air, and Soil Pollution, Vol. 80, pp 85-94, 1995.
3. Amano. H., and Onuma, Y. Depth Profile of Long Lived Radionuclides in
Chernobyl Soils Sampled around 10 Years after the Accident, Journal of Radioanalytical and Nuclear Chemistry, Vol. 255, No. 1, pp 217-222, 2003.
4. Ambrecht, V., Jensen, J., Eden, S., and Stockbrugger, R. Assessment of Oro-
caecal Transit Times by Means of a Hydrogen (H2) Breath Test as Compared with Radiological Control Method. Scandinavian Journal of Gastroenterology, Vol. 21, pp 669-677, 1986.
5. Anspaugh, L. R., Shinn, J. H., Phelps, P. L., and Kennedy, N. C. Resuspension
and Redistribution of Plutonium in Soils, Health Physics, Vol. 29, pp 571-582, 1975.
6. Annals of the ICRP Publication 56, Age-dependent Doses to Members of the
Public from Intake of Radionuclides: Part 1. Vol. 20, No. 2, 1989. 7. Annals of the ICRP Publication 71, Age-dependent Doses to Members of the
Public from Intake of Radionuclides: Part 4 Inhalation Dose Coefficients, Vol. 25, No. 3/4, 1995.
8. Barltrop D. and Meek, F. Effect of Particle Size on the Lead Absorption form the
Gut. Archives of Environmental Health, July/August, 34(4) pp 280-285, 1979. 9. Berglund, F., Berlin, M., Birke, G., Cederlof, R., Von Euler, U., Friberg, L.,
Holmsteadt, B., and Johnson, E. Methylmercury in Fish: a Toxicological-Epidemiologic Evaluation of Risks (Report from an Expert Group). Nordisk Hygenisk Tidskrift. Suppl. 4, Stockholm, 1971.
10. Berlin, M. Interactions Between Selenium and Inorganic Mercury. Environmental
Health Perspectives, Vol. 25, pp 67-69, 1978. 11. Bornmann, G., Henke, G., Alfes, H., and Mollmann, H. Intestinal Absorption of
Metallic Mercury. Archives of Toxicology, Vol. 26, No. 3, pp 203-209, 1970.

212
12. Brady, N.C., and Weil, P.R. Elements of the Nature and Properties of Soils 11th Edition. Prentice Hall, Upper Saddle River, New Jersey, 1995.
13. Burger, J. Fishing and Risk along the Savannah River: Possible Intervention.
Journal of Toxicology and Environmental Health, Part A, Vol. 55, pp 405-419, 1998.
14. Burger, J., Stephens, W.L., Boring, C.S., Kuklinski, M., Gibbons, J.W., and
Gochfeld, M. Factors in Exposure Assessment: Ethnic and Socioeconomic Differences in Fishing and Consumption of Fish Caught along the Savannah River. Risk Analysis, Vol. 19, No. 3, pp 427~437, 1999.
15. Burger, J. Gender Differences in Meal Patterns: Role of Self-caught Fish and
Wild Game in Meat and Fish Diets. Environmental Research, Part A, Vol. 83, pp 140-149, 2000.
16. Burger, J., Gaines K. F., Boring C. S., Stephens, W. L., Snodgrass, J. J., and
Gochfeld, M. Mercury and Selenium in Fish from the Savannah River: Species, Tropic Level, and Locational Differences. Environmental Research, Part A, Vol. 87, pp 108-118, 2001.
17. Burger, J. and Gochfeld, M. Mercury in Canned Tuna: White versus Light and
Temporal Variation, Environmental Research, Vol. 96, pp 239-249, 2004. 18. Burger, J., Stern, A. H., and Gochfeld, M. Mercury in Commercial Fish:
Optimizing Individual Choices to Reduce Risk, Environmental Health Perspectives, Vol. 113, No. 3, pp 1-6, 2005.
19. Burson, Z. G. and Profio, A. E. Structure Shielding in Reactor Accidents, Health
Physics, Vol. 33, pp 287-299, 1977. 20. Charbonneau, S. M., Munro, I. C., Nera, E. A., Armstrong, F. A., Willes, R. F.,
Bryce, F., and Nelson, R. F. Chronic Toxicity of Methylmercury in the Adult Cat, Interim Report. Toxicology, Vol. 5, No. 3, pp 337-349, 1976.
21. Clapp, T. C., Umbreit, T. M., Meeker, R. J., Kosson, D. S., Gray, D., and Gallo,
M. A. Bioavailability of Lead and Chromium from Encapsulated Pigment Materials. Bulletin of Environmental Contamination and Toxicology, Vol. 46, No. 2, pp 271-275, 1991.
22. Clarkson, T. W. Environmental Contaminants in Food Chain, American Journal
of Clinical Nutrition, Vol. 61, No. 3, pp 682-686, 1995. 23. Clayton, C.A., Pellizzari, E.D., Whitmore, R.W., Perritt, R. L., and Quackenboss,
J. J. National Human Exposure Assessment Survey (NHEXAS): Distribution and Associations of Lead, Arsenic, and Volatile Organic Compounds in EPA Region

213
5. Journal of Exposure Analysis and Environmental Epidemiology, Vol. 9, No. 5, pp 381-392, 1999.
24. Crystal Ball 2000 User Manual. Decisoneering Denver, CO. 25. Cuvin-alalar, L. A. and Furness, R. W. Mercury and Selenium Interaction: A
Review. Ecotoxicology and Environmental Safety, Vol. 21, pp 348-364, 1991. 26. Davis, A., Drexler, J. W., Ruby, M. V., and Nicholson, A. Micromineralogy of
Mine Wastes in Relation to Lead Bioavailability, Butte, Montana. Environmental Science and Technology, Vol. 27, No. 7, pp 1415-1425, 1993.
27. Davis, A., Ruby, M. V., Goad P., Eberle, S., and Chryssoulis, S. Mass Balance on
Surface-Bound, Mineralogic, and Total Lead Concentrations as Related to Industrial Aggregate Bioaccessibility. Environmental Science and Technology, Vol. 31, No. 1, pp 31-37, 1997.
28. Driver, J. H., Konz, J. J., and Whitmyre, G. K. Soil Adherence to Human Skin,
Bulletin of Environmental Contamination and Toxicology, Vol. 43, pp 814-820, 1989.
29. Duggan, M. J. and Inskip, M. J. Childhood Exposure to Lead in Surface Dust,
Public Health Review, No. 13, pp 1-54, 1985. 30. Edwards, R. D. and Lioy, P. J. The EL Sampler: A Press Sampler for the
Quantitative Estimation of Dermal Exposure to Pesticides in Household Dust. Journal of Exposure Analysis and Environmental Epidemiology, Vol. 9, pp 521-529, 1999.
31. Ellickson, K. M., Meeker, R. J., Gallo, M. A., Buckley, B. T., and Lioy, P. J. Oral
Bioavailability of Lead and Arsenic from a NIST Standard References Soil Material. Archives of Environmental Contamination and Toxicology, Vol. 40, pp 128-135, 2001.
32. Ellickson, K.M. The Bioaccessibility of Selected Radionuclides and Heavy
Metals: An Investigation of Bioaccessibility, Bioavailability, and Natural Soil Characteristics. Ph. D. Dissertation, Rutgers and UMDNJ, May 2001.
33. Ellickson, K.M., Schopfer, C.J. and Lioy, P.J. The Bioaccessibility of Low Level
Radionuclides from Two Savannah River Site Soils. Health Physics, Vol. 83, No. 4, pp 476-484, October 2002.
34. Freeman, G. B., Johnson, J. M., Killinger, J. M., Liao, S. C., Feder, P. I., Davis, A.
O., Ruby, M. V., Chaney, R. L., Lovre, S. C., and Bergstrom, P. D. Relative Bioavailability of Lead from Mining Waste Soil in Rats. Fundamental and Applied Toxicology, Vol. 19, pp 388-398, 1992.

214
35. Freeman, G. B., Dill, J. A., Johnson, P. J., Parham, F., and Matthews, H. B.
Comparative Absorption of Lead from Contaminated Soil and Salts by Weanling Fisher 344 Rats. Fundamental and Applied Toxicology, Vol. 33, pp 109-119, 1996.
36. Fusayama, T., Katayori, T., and Nomoto, S. Corrosion of Gold and Amalgam
Placed in Contact with Each Other. Journal of Dental Research, Vol. 42, pp 1183-1197, 1963.
37. Ganther, H.E., Goudie, C., Sunde, M.L., Kopecky, M. J., Wagner, P., Oh, S., and
Hoekstra, W. G. Selenium Relation to Decreased Toxicity of Methylmercury Added to Diets Containing Tuna. Science, Vol. 175, pp 1122-1124, 1972.
38. Gargas, M. L., Norton, R. L., Harris, M. A., Paustenbach, D. J., and Finley, B. L.
Urinary Excretion of Chromium Following Ingestion of Chromite-ore Processing Residues in Humans: Implications for Biomonitoring. Risk Analysis, Vol. 14, No. 6, pp 1019-1024, 1994.
39. Gibaldi, M. Biopharmaceutics and Clinical Pharmacokinetics. 3rd Edition. Lea
and Febiger, Philadelphia, 1984. 40. Goheen, S. C. Method RP515, Rapid Determination of Radiostrontium Using
Empore™ Sr Rad Disks, DOE Methods for Evaluating Environmental and Waste Management Samples. Columbus, OH, Batelle Press, 1997.
41. Gallegos, G. Surveillance Monitoring of Soils for Radioactivity: Lawrence
Livermore National Laboratory 1976 to 1992. Health Physics, Vol. 69, No. 4, pp 487-493, October 1995.
42. Groen, K., Vaessen, H. A. M. G., Liest, J. J. G., De Boer, J. L. M., Van Ooik, T.,
Timmerman, A., and Vlug, R. F. Bioavailability of Inorganic Arsenic from Bog Ore-containing Soil in the Dog. Environmental Health Perspectives, Vol. 102, No. 2, pp 182-184, 1994.
43. Hamel, S. C., Buckley, B., and Lioy, P. J. Bioaccessibility of Metals in Soils for
Different Liquid to Solid Ratios in Synthetic Gastric Fluid. Environmental Science and Technology, Vol. 32, No. 3, pp 358-362, 1998.
44. Hamel, S. C. The Estimation of Bioaccessibility of Heavy Metals in Soils Using
Artificial Biofluids. Ph. D. Dissertation, Rutgers and UMDNJ, May 1998. 45. Hamel, S.C., Ellickson, K.M., and Lioy, P.J. The Estimation of the
Bioaccessibility of Heavy Metals in Soils Using Artificial Biofluids by Two Novel Methods: Mass-balance and Soil Recapture. Science of Total Environment, Vol. 243/244, pp 273-283, 1999.

215
46. Hatch, M. C. Centers Needed to Study Women’s Environmental Health.
Environmental Health Perspectives, Vol. 108, No. 1, A10-A11, January 2000. 47. Hubal, E.A.C., Sheldon, L.S., Burke, J.M., McCurdy, T. R., Berry, M. R., Rigas,
M. L., Zartarian, V. G., and Freeman, N. C. G. Children’s Exposure Assessment: A Review of Factors Influencing Children’s Exposure and the Data Available to Characterize and Assess that Exposure. Environmental Health Perspectives, Vol. 108, No. 6, pp 475-486, 2000.
48. Institute of Medicine (IOM). Seafood Safety. National Academy Press,
Washington D.C., 1991. 49. Institute of Medicine (IOM). Gender Differences in Susceptibility to
Environmental Factors: A Priority Assessment. National Academy Press, Washington D.C., 1998.
50. Jacobson, J.L., Humphrey, H.E.B., Jacobson, S.W., Schantz, S. L., Mullin, M. D.,
and Welch, R. Determinants of Polychlorinated Biphenyls (PCBs), Polybrominated Biphenyls (PBBs), and Dichlorodiphenyl Trichloroethane (DDT) Levels in the Sera of Young Children. American Journal of Public Health, Vol. 79, No. 10, pp 1410-1404, 1989.
51. Jacobson, J.L., Jacobson, S.W., and Humphrey, J.B. Effects of in Utero Exposure
to Polychlorinated Biphenyls and Related Contaminants on Cognitive Functioning in Young Children. Journal of Pediatrics, Vol. 116, No. 1, pp 38-45, 1990.
52. Jacobson, J.L., and Jacobson, S.W. Intellectual Impairment in Children Exposed
to Polychlorinated Biphenyls in Utero. New England Journal of Medicine, Vol. 335, No. 11, pp 783-789, 1996.
53. Kimura, Y., Nagata, Y., Bryant, C. W., and Buddington, R. K. Nondigestible
Oligosaccharides Do Not Increase Accumulation of Lipid Soluble Environmental Contaminants by Mice. Journal of Nutrition, Vol. 132, pp 80-87, 2002.
54. Landi, M. T., Consonni, D., Patterson, D. G. Jr., Needham, L. L., Lucier, G.,
Brambilla, P., Cazzaniga, M. A., Mocarelli, P., Pesatori, A. C., Bertazzi, P. A., Caporaso, N. E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin Plasma Levels in Seveso 20 Years after the Accident. Environmental Health Perspectives. Vol. 106, No. 5, pp 273-277, 1998.
55. Lanphear, B. P., Weitzman, M., Winter, N. L., Eberly, S., Yakir, B., Tanner, M.,
Emond, M., and Matte, T. D. Lead-Contaminated House Dust and Urban Children’s Blood Lead Levels. American Journal of Public Health, Vol. 86, No. 10, pp 1416-1421, 1996.

216
56. Lewis, R. G., Fortune, C. R., Willis, R. D., Camann, D. E., and Antley, J. T. Distribution of Pesticides and Polycyclic Aromatic Hydrocarbons in House Dust as a Function of Particle Size. Environmental Health Perspectives, Vol. 107, No. 9, pp 721-726, 1999.
57. Lioy, P. J. Assessing total human exposure to contaminants. Environmental
Science and Technology, Vol. 24, No. 7, pp 938-945, 1990. 58. Lioy, P. J., Freeman, N. C. G., and Millette, J. R. Dust: A Metric for Use in
Residential and Building Exposure Assessment and Source Characterization. Environmental Health Perspectives, Vol. 110, No. 10, pp 969-983, 2002.
59. Maddaloni, M., LoIacono, N., Manton W., Blum, C., Drexler, J., and Graziano, J.
Bioavailability of Soilborne Lead in Adults, by Stable Isotope Dilution. Environmental Health Perspectives, Vol. 106. Suppl. 6, pp 1589-1594, 1998.
60. Malagelada, J. R., Longstreth, G. F., Summerskill, W.H. J., and Go, V. L. W.
Measurement of Gastric Functions During Digestion of Ordinary Solid Meals in Man. Gastroenterology, Vol. 70, No. 2, pp 203-210, 1976.
61. Malek, M. A., Hinton, T. G., and Webb, S. B. A comparison of 90Sr and 137Cs
Uptake in Plants via Three Pathways for Two Chernobyl-Contaminated Sites. Journal of Environmental Radioactivity, Vol. 58, pp 129-141, 2002.
62. Malmstrom, M., Sundquist, J., Johansson, S. E. Neighborhood Environment and
Self-reported Health Status: A Multilevel Analysis. American Journal of Public Health. Vol. 89, No. 8, pp 1181-1186, 1999.
63. Marieb, E. N. Essentials of Human Anatomy and Physiology, 5th Edition.
Benjamin/Cummings Publishing Company, 1997. 64. Mason, R. P., Lawson, N. M., Lawrence, A. L., Leaner, J. L., Lee, J. G., and Sheu,
G. R. Mercury in the Chesapeake Bay. Marine Chemistry. Vol. 65, pp 77-96, 1999.
65. Miettinen, J.K. Absorption and Elimination of Dietary (Hg2+) and Methylmercury
in Man. Mercury, Mercurial and Mercaptans, Chapter 13, Springfield, IL. Thomas, 1973.
66. Miller D.D., Schricker B.R., Rasmussen R.R., and Van Campen, D. An In Vitro
Method for Estimation of Iron Availability from Meals. The American Journal of Clinical Nutrition, Vol. 34, pp 2248-2256, 1981.
67. National Academy of Sciences. Risk Assessment in the Federal Government:
Managing the Process, National Research Council, National Academy Press, Washington D.C., 1983.

217
68. National Academy of Sciences. Toxicological Effects of Methylmercury,
Committee on the Toxicological Effects of Methylmercury, Board on Environmental Studies and Toxicology, National Research Council. National Academy Press, Washington D.C., 2000.
69. National Council on Radiation Protection and Measurements. NCRP Reports 93:
Ionizing Radiation Exposure of the Population of the United States. Bethesda, Maryland, 1987.
70. National Council on Radiation Protection and Measurements. NCRP Reports 115:
Risk Estimates for Radiation Protection. Bethesda, Maryland, 1993. 71. National Council on Radiation Protection and Measurements. NCRP Reports 116:
Limitation of Exposure to Ionizing Radiation. Bethesda, Maryland, 1993. 72. Navarro, M., Lopez, M. C., and Lopez, H. Microwave Dissolution for the
Determination of Hg in Fish by Cold Vapor Atomic Absorption Spectrometry. Analytica Chimica Acta, Vol. 257, No. 1, pp 155-158, 1992.
73. Olden, K. and Newbold, R. R. Women’s Health and the Environment in the 21st
Century. Environmental Health Perspectives, Vol. 108, Suppl. 5, pp 767-768, October 2000.
74. Oliver, D. P., McLaughlin, M. J., Naidu, R., Smith, L. H., Maynard, E. J., and
Calder, I. C. Measuring Pb Bioavailability from Household Dusts Using an In Vitro Model. Environmental Science and Technology, Vol. 33, No. 24, pp 4434-4439, 1999.
75. Oomen A.G., Hack A., Minekus M., Zeijdner, E., Cornelis, C., Schoeters, G.,
Verstrete, W., Van De Wille, T., Wragg, J., Romperberg, C. J. M., Sips, A. J. A. M., and Van Wilnen, J. H. Comparison of five In Vitro Digestion Models to Study the Bioaccessibility of Soil Contaminants, Environmental Science and Technology, Vol. 36, No. 15, pp 3226-3334, 2002.
76. Oomen, A. G., Rompelberg, J. M., Bruil, M. A., Dobble, C. J. G., Pereboom, D. P.
K. H., and Sips, A. J. A. M. Development of an In Vitro Digestion Model for Estimating the Bioaccessibility of Soil Contaminants. Archives of Environmental Contamination and Toxicology, Vol. 44, pp 281-287, 2003.
77. Paustenbach, D. J. The Practice of Exposure Assessment: A state-of-the-art
Review. Journal of Toxicology and Environmental Health, Part B, Vol. 3, pp 179-291, 2000.

218
78. Petoussi, N., Jacob, P., Zankl, M., and Saito, K. Organ Doses for Fetuses, Babies, children, and Adults from Environmental Gamma Rays. Radiation Protection Dosimetry, Vol. 37, pp 31-41, 1991.
79. Piotrowski, J.K., Szymanska, J.A., Skrypinska-Gawrysiak, M, Kotelo, J., and
Sporny, S. Intestinal Absorption of Mercury in Rat. Pharmacological Toxicology. Vol. 70, pp 53-55, 1992.
80. Que Hee, S. S., Peace, B., Clark, C. S., Boyle, J. R., Bornshein, R. L., and
Hammond, P. B. Evolution of Efficient Methods to Sample Lead Sources, such as House Dust and Hand Dust, in the Homes of Children. Environmental Research, Vol. 38, pp 77-95, 1985.
81. Rahola, T., Hattula, T., Korolainen, A., and Miettinen, J. K. Elimination of Free
and Protein-bound Ionic Mercury 203Hg2+ in Man. Annals of Clinical Research. Vol. 5, No. 4, pp 214-219, 1973.
82. Rhoads, G. G., Ettinger, A. S., Weisel, C. P., Buckley, T. J., Goldman, K. D.,
Adgate, J. L., and Lioy, P. J. The Effect of Dust Lead Control on Blood Lead in Toddlers: A Randomized Trial. Pediatrics, Vol. 103, pp 551-555, 1999.
83. Rodes, C. E., Newsome, J. R., Vanderpool, R. W., Antley, J. T., and Lewis, R. G.
Experimental Methodologies and Preliminary Transfer Factor Data for Estimation of Dermal Exposures to Particles. Journal of Exposure and Analysis and Environmental Epidemiology. Vol. 11, No. 2, pp 123-139, 2001.
84. Rodriguez, R. R., Basta, N. T., Casteel, S. W., and Pace, L. W. An In Vitro
Gastrointestinal Method to Estimate Bioavailable Arsenic in Contaminated Soils and Solid Media. Environmental Science and Technology, Vol. 33, No. 4, pp 642-649, 1999.
85. Rowland, I. R., Mallett, A. K., Flynn, J., and Hargreves, R. J. The Effect of
Various Dietary Fibers on Tissue Concentration and Chemical Form of Mercury after Methylmercury Exposure in Mice. Archives Toxicology, Vol. 59, No. 2, pp 94-98, 1986.
86. Ruby, M.V., Davis A., Kempton J. H., Drexler, J. W., and Bergstrom, P. D. Lead
Bioavailability: Dissolution Kinetics under Simulated Gastric Conditions. Environmental Science and Technology Vol. 26, pp1242-1248, 1992.
87. Ruby, M.V., Davis A., Link T. E., Schoof, R., Chaney, R. L., freeman, G. B., and
Bergstrom, P. Development of an In Vitro Screening Test to Evaluate the In Vivo Bioaccessibility of Ingested Mine-waste Lead. Environmental Science and Technology, Vol. 27, pp 2870-2877, 1993.

219
88. Ruby, M.V., Davis, A., Schoof, R., Eberle, S., and Sellstone, C. M. Estimation of Lead and Arsenic Bioavailability Using a Physiologically Based Extraction Test. Environmental Science and Technology, Vol. 30, No. 2, pp 422-430, 1996.
89. Ruby, M.V., Schoof, R., Brattin, W., Goldade, M., Post, G., Harnois, M., Mosby,
D. E., Casteel, S. W., Berti, W., Carpenter, M., Edwards, D., Cragin, D., and Chappell, W. Critical Review: Advances in Evaluating the Oral Bioavailability of Inorganics in Soil for Use in Human Health Risk Assessment. Environmental Science and Technology, Vol. 33, pp 3697-3705, 1999.
90. Savannah River Site. Environmental Report for 2000. Westinghouse Savannah
River Company, Aiken, South Carolina, 2000. 91. Savannah River Site. Environmental Data for 2000. Westinghouse Savannah
River Company, Aiken, South Carolina, 2000. 92. Satoh, H., Yasuda, N., and Shimai, S. Development of Reflexes in Neonatal Mice
Prenatally Exposed to Methylmercury and Selenite. Toxicology Letters, Vol. 25, pp 199-203, 1985.
93. Sheppard, S. C., Eveden, W. G., and Schwartz, W. J. Ingested Soil:
Bioavailability of Sorbed Lead, Cadmium, Cesium, Iodine, and Mercury. Journal of Environmental Quality, Vol. 24, pp 498-505, 1995.
94. Simon, S.L. Soil Ingestion by Humans: A Review of History, Data and Ecology
with Applications to Risk Assessment of Radioactively Contaminated Soils. Health Physics, Vol. 74, No. 6, pp 647-672, 1998.
95. Sparks, P., and Shepherd, R. Public Perceptions of the Potential Hazards
Associated with Food Production and Food Consumption: an Empirical Study. Risk Analysis, Vol. 14, pp 799-806, 1994.
96. Storelli, M. M., Stuffler, R. G., and Marcotrigiano, G. O. Total and
Methylmercury Residues in Tuna-fish from the Mediterranean Sea, Food Additives and Contaminants, Vol. 19, No. 8, pp 715-720, 2002.
97. Suzuki, T., Hongo T., Yoshnaga J., Imai, H., Nakazawa, M. Matsuo, M., and
Akagi, H. The Hair-organ Relationship in Mercury Concentration in Contemporary Japanese. Archives of Environmental Health, Vol. 48, No. 4, pp 221-229, 1993.
98. Thornton I., Davies D.J., Watt J.M., and Quinn, M. J. Lead Exposure in Young
Children from Dust and Soil in the United Kingdom. Environmental Health Perspectives, Vol. 89, pp 55-90, 1990.

220
99. U.S. Department of Housing and Urban Development. Guidelines for the Evaluation and Control of Lead-based Paint Hazard in Housing. HUD-1539-LBP, Washington D.C.: U.S. HUD, 1995.
100. U.S. Department of Health and Human Services, Agency for Toxic Substance and
Disease Registry. Toxicological Profile for Mercury. Atlanta, Georgia: U.S. DHHS-ATSDR, 1999.
101. U.S. Department of Health and Human Services, Agency for Toxic Substance and
Disease Registry. Toxicological Profile for Lead. Atlanta, Georgia: U.S. DHHS-ATSDR, 1999.
102. U.S. Department of Health and Human Services, Agency for Toxic Substance and
Disease Registry. Toxicological Profile for Ionizing Radiation. Atlanta, Georgia: U.S. DHHS-ATSDR, 1999.
103. U.S. Department of Health and Human Services, Agency for Toxic Substance and
Disease Registry. Toxicological Profile for Cesium. Atlanta, Georgia: U.S. DHHS-ATSDR, 2004.
104. U.S. Department of Health and Human Services, Agency for Toxic Substance and
Disease Registry. Toxicological Profile for Strontium. Atlanta, Georgia: U.S. DHHS-ATSDR, 2004.
105. U.S. Environmental Protection Agency. Limiting Values of Radionuclide Intake
and Air Concentration and Dose Conversion Factors for Inhalation, Submersion, and Ingestion. Federal Guidance Report, No. 11, September 1988.
106. U.S. Environmental Protection Agency. Guidelines for Exposure Assessment.
EPA/600/Z-92/001, U.S. EPA, May 1992. 107. U.S. Environmental Protection Agency. External Exposure to Radionuclides in
Air, Water, and Soil. Federal Guidance Report, No. 12, September 1993. 108. U.S. Environmental Protection Agency. Method 7471A, Revision 1: Mercury in
Solid or Semisolid Waste (Manual Cold-vapor Technique). U.S. EPA, September 1994.
109. U.S. Environmental Protection Agency. Method 1631, Revision 0: Microwave
Assisted Acid Digestion of Sediments, Sludges, Soils, and Oils. U.S. EPA, September 1994.
110. U.S. Environmental Protection Agency. Sampling House Dust for Lead -Basic
Concepts and Literature Review. EPA747R-95-007, Washington D.C.: U.S. EPA, 1995.

221
111. U.S. Environmental Protection Agency. Mercury Study Report to Congress Vol. 1-7. U.S. EPA, December 1997.
112. U.S. Environmental Protection Agency. Exposure Factors Handbook. Washington
D.C.: U.S. EPA, 1999. 113. U.S. Environmental Protection Agency. Protect Your Family from Lead in Your
Home. EPA747-K-99-001, Washington D.C.: U.S. EPA, 2001. 114. U.S. Environmental Protection Agency. User’s Guide for the Integrated Exposure
Uptake Biokintetic Model for Lead in Children (IEUBK) Windows® version. EPA 540-K-01-005, Washington D.C.: U.S. EPA, 2002.
115. U.S. Environmental Protection Agency. EPA Facts About Cesium-137. U.S. EPA,
July 2002. 116. U.S. Environmental Protection Agency. EPA Facts About Strontium-90. U.S.
EPA, July 2002. 117. U.S. Environmental Protection Agency. Method 1631, Revision E: Mercury in
Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry. U.S. EPA, August 2002.
118. U.S. Environmental Protection Agency. Children’s Environmental Exposures.
U.S. EPA, March 2003. 119. U.S. Food and Drug Administration. Mercury in Fish: Cause for Concern? U.S.
FDA, March 2001. 120. U.S. Pharmacopoeia. The United States Pharmacopoeia 22 Inc. U.S.
Pharmacopoeia Convention, Twinbroke Parkway, Rockville, Maryland, 1990. 121. Vander, A. J., Sherman, J. H., and Luciano, D. S. Human Physiology: The
Mechanism of Body Function, 7th Ed. McGraw Hill, New York, 1997. 122. Watras C.J., and Bloom N.S. Mercury and Methylmercury in Individual
Zooplankton for Bioaccessibility. Limnology Oceanography, Vol. 37, pp 1313-1318, 1992.
123. Wilson C. G., Parr G. D., Kennerley J. W., Taylor M. J., Davis, S. S., Hardy, J. G.,
and Rees, J. A. Pharmacokinetics and In Vivo Scintigraphic Monitoring of a Sustained Release Acetylsalicylic Acid Formulation. International Journal of Pharmacy, Vol. 18, pp 118, 1984.

222
124. Yannai, S., and Sachs, K.M. Absorption and Accumulation of Cadmium, Lead and Mercury from Foods by Rats. Food and Chemical Toxicology. Vol. 31, pp 351-355, 1993.
125. Yiin, L., Rhoades, G. G., Rich, D. Q., Zhang, J., Bai, Z., Adgate, J. L., Ashley, P.
J., and Lioy, P. J. Comparison of Techniques to Reduce Residential Lead Dust on Carpet and Upholstery: The New Jersey Assessment of Cleaning Techniques Trial. Environmental Health Perspectives, Vol. 110, No., 12, pp 1233-1237, 2002.

223
Curriculum Vitae
Chang Ho Yu
1997 B.S. Degree, Environmental Engineering, Youngnam University, Kyungsan, KOREA (R.O.K.)
2000 M.S. Degree, Environmental Engineering, Kyungpook National
University, Taegu, KOREA (R.O.K.) 2000-present Graduate Student, Exposure Assessment Option, Environmental Sciences,
Rutgers-The State University of New Jersey, New Brunswick/Piscataway, New Jersey & UMDNJ-University of Medicine and Dentistry of New Jersey, Piscataway, New Jersey
2000-2002 Research Assistant, CRESP (Consortium for Risk Evaluation with Stake-
holders Participation) Project. 2003-2004 Part-time Lecturer, Environmental Sciences, Rutgers-The State University
of New Jersey, “Principles of Air Pollution” (3 semesters) 2003-2005 Research Assistant, Community Help to Reduce Environmental Lead
Exposure Project. 2003-2005 Project Coordinator, ECSC (Evaluation of Dry Steam Cleaning in
Reducing Contaminants in Carpets) Project. 2001 Jo, W. and Yu, C. H. Public Bus and Taxicab Drivers’ Exposure to
Aromatic Work-time Volatile Organic Compounds. Environmental Research, Section (A)86, pp 66-72, 2001.
2005 Yu, C. H., Yiin, L., and Lioy, P. J. The Bioaccessibility of Lead (Pb) from
Vacuumed House Dust on Carpet in Urban Residences. Risk Analysis, In Press.
Copyright © 2022 FDOKUMEN




















