
In search of a treatment for HIV--current therapies and the role of non-nucleoside reverse...
31
In Search of a treatment for HIV – Current Therapies and the Role of Non -nucleoside Reverse Transcriptase Inhibitors(NNRTIs) Journal: Chemical Society Reviews Manuscript ID: CS-TRV-02-2012-035058 Article Type: Tutorial Review Date Submitted by the Author: 29-Feb-2012 Complete List of Authors: Reynolds, Chevonne; University of the Witwatersrand, de Koning, Charles; University of the Witwatersrand, Pelly, Stephen; Stellenbosch University, van Otterlo, Willem; Stellenbosch University, Bode, Moira; University of the Witwatersrand, Chemical Society Reviews
Transcript of In search of a treatment for HIV--current therapies and the role of non-nucleoside reverse...

In Search of a treatment for HIV – Current Therapies and
the Role of Non -nucleoside Reverse Transcriptase
Inhibitors(NNRTIs)
Journal: Chemical Society Reviews
Manuscript ID: CS-TRV-02-2012-035058
Article Type: Tutorial Review
Date Submitted by the Author: 29-Feb-2012
Complete List of Authors: Reynolds, Chevonne; University of the Witwatersrand,
de Koning, Charles; University of the Witwatersrand, Pelly, Stephen; Stellenbosch University, van Otterlo, Willem; Stellenbosch University, Bode, Moira; University of the Witwatersrand,
Chemical Society Reviews

Scope of the Journal Chemical Society Reviews (Chem Soc Rev) publishes accessible, succinct and reader-friendly articles on topics of current interest in the chemical sciences. The promotion of international and multidisciplinary awareness and co-operation is particularly encouraged. Article types Tutorial Reviews Tutorial reviews should be written to be of relevance both to the general research chemist who is new to the field, as well as the expert, and must be accessible to advanced undergraduates and beyond. They should provide an essential introduction to the field, serving as a springboard to further reading, and should have particular appeal to younger researchers seeking new fields to explore. Authors are required to write succinctly and select references carefully in order to give an enticing flavour of the topic rather than a comprehensive treatise. The implications of recent developments for the wider scientific community should be emphasised. Tutorial Reviews must be: Short: 10 – 12000 word equivalents: the entire manuscript (double-spaced text and artwork) should not exceed 30 - 35 pages of A4 or American Quarto (10 -12 journal pages) Lightly referenced: A maximum of 50 - 60 citations is strongly recommended. Multiple referencing (the use of a, b, etc.) is strongly discouraged. Jargon free: All specialist terms and symbols should be defined. Authors should not presume knowledge beyond undergraduate level. Fundamental ideas should be simply explained. For most articles a broad outlook is required: authors should credit the major contributors within the citation limit. The 'and references therein' tag may be useful to direct the reader to further reading without having to list whole series of papers from one research group. Critical Reviews Critical Reviews should provide a deeper understanding of the topic in hand, yet retain their accessibility through an introduction written for the general reader. They should give a critical discussion of the existing state of knowledge of the subject matter, and while not exhaustive in coverage give a balanced assessment of the current primary literature, normally concentrating on the previous 5-10 years. The implications of recent developments for the wider scientific community should be emphasised and authors should aim to stimulate progress in the field. Critical Reviews must be: Carefully referenced: 250–300 citations are recommended. References should be selected to give a balanced view of the field but do not need to be exhaustive. Jargon free: All specialist terms and symbols should be defined. Fundamental ideas should be simply explained Articles should be of a suitable length to give an in depth discussion of the field but concise writing is encouraged and manuscripts should normally be between 20 and 30 journal pages in length (20-30000 word equivalents, no more than 90 A4/American quarto pages in length (double-spaced text and artwork). A good introduction is of prime importance and should include a historical perspective and set the topic for discussion in the context of current chemical research. The introduction must be accessible to the general chemist. The body of the review should provide an in-depth, critical (yet fair) discussion of the current field and aim to stimulate further research. A section of concluding remarks should include comment on applications or future prospects for the field.
Page 1 of 30 Chemical Society Reviews

Dear Reviewers,
Please find below our reasoning and justification for writing this review.
Article Title: In Search of a treatment for HIV – Current Therapies and the Role of
Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
Current importance of the field
There are currently 34 million people world-wide living with HIV/AIDS, with the
largest affected population (5.6 million) living in South Africa. A number of therapies
now exist that improve the life expectancy and quality of life of sufferers, but the
search continues for better drugs in existing and new therapeutic classes to combat
the problem of drug resistance. The non-nucleoside reverse transcriptase inhibitors
(NNRTIs) are an important class of compounds used in most first-line HAART (highly
active antiretroviral therapy) regimens. The advantage of this therapeutic class is
that the drugs generally have an excellent selectivity index and are thus relatively
safe. However, they have traditionally suffered from the disadvantage of having a
low barrier to the development of drug resistance, some becoming ineffective in the
presence of only one amino acid mutation. Newer drugs in this class have
completely altered this reality, and show good potency even in the presence of two
or more amino acid mutations. Research is ongoing to find new antiretroviral agents
with improved potency and pharmacokinetic properties.
Communities of readers that will find the article of interest
This review tutorial should be of value to research chemists interested in recent HIV
advances, but who do not necessarily understand the biology of how the current
regimen of HIV drugs (NNRTIs in particular) interact with their protein targets. The
biological aspects of the tutorial, including the discussion on mutations, are written
by chemists and are therefore described in terms readily understandable to
chemists.
In addition, the article is written in language that can be understood by an educated
lay person and contains interesting historical information about HIV and AIDS. As the
authors of the tutorial review are working in the scientific community of South Africa
Page 2 of 30Chemical Society Reviews

there is a South African flavour to the article including, for example, statistics and
current South African HIV treatment regimens.
Page 3 of 30 Chemical Society Reviews

1
In Search of a treatment for HIV – Current Therapies and the Role of Non-nucleoside
Reverse Transcriptase Inhibitors (NNRTIs)
Chevonne Reynolds,1 Charles B. de Koning,
1 Stephen C. Pelly,
2 Willem A. L. van Otterlo
1,2 and
Moira L. Bode1*
1Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3,
PO WITS, 2050, South Africa
2Department of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1,
Matieland, 7602, South Africa
ABSTRACT
The Human Immunodeficiency Virus (HIV) causes AIDS (acquired immune deficiency syndrome),
a disease in which the immune system progressively deteriorates, making sufferers vulnerable to all
manner of opportunistic infections. Currently, world-wide there are estimated to be 34 million
people living with HIV, with the vast majority of these living in sub-Saharan Africa. Therefore, an
important research focus is development of new drugs that can be used in the treatment of
HIV/AIDS. This review gives an overview of the disease and addresses the drugs currently used for
treatment, with specific emphasis on new developments within the class of non-nucleoside reverse
transcriptase inhibitors (NNRTIs).
Page 4 of 30Chemical Society Reviews

2
INTRODUCTION
Acquired Immune Deficiency Syndrome or AIDS is a disease caused by the Human
Immunodeficiency Virus (HIV) and is responsible for more than 20 million deaths globally.1,2
With
an estimated 34 million people living with HIV in 2010,3 and no viable cure, we are facing one of
the worst pandemics in human history. Although HIV is a devastating disease for sufferers, it has
further reaching implications in terms of economic, social and political stability.1 South Africa is
estimated to have 5.6 million people currently living with HIV, more than any other country in the
world.3 In the worst-affected province of Kwa-Zulu Natal (KZN), 26.4% of the working age
population is thought to be HIV positive.4 The predictions for KZN show that at the current
infection rate, two-fifths of the adult population will die of AIDS related illnesses by 2025. By way
of understanding the serious impact of the disease on this region, Thurlow et al.4 predicted that the
collective effects of HIV over all economic sectors means that the KZN economy will be 43%
smaller by 2025. Combating the AIDS epidemic is thus of paramount importance in order to secure
economic stability, as well as to address the devastating social decline which results.1
Brief History of Human Immunodeficiency Virus (HIV)
Along with many discoveries in science, the discovery of the HI virus as the cause of AIDS, was
one touched by serendipity. HIV was first isolated in 1983 by Luc Montagnier2 and after work
carried out in the laboratory of Robert Gallo, was generally accepted as the causative agent of AIDS
by 1984.5 Had it not been for research into cancer-causing viruses in the same time period as the
first identified AIDS patients, the knowledge and tools for the discovery of the HI virus may not
have existed.2 Many factors including fungi, chemicals and autoimmune diseases were thought to
be the cause of AIDS, but it was the earlier discovery in 1970 by Temin and Baltimore of the
enzyme reverse transcriptase, present in all retroviruses, that ultimately paved the way for the
discovery that a retrovirus was the cause of AIDS.2 Gallo isolated the first human retrovirus, human
T-cell leukaemia virus type 1 (HTLV-1) in 1979, followed by HTLV-2 in 1982 from patients with
T-cell leukaemia.5 Animal studies revealed that HLTV could cause an AIDS-like syndrome and this
result, together with the similar epidemiology of HTLV, pointed scientists in the right direction
towards a new retrovirus, eventually named HIV, as the cause of AIDS.6
From 1983-1985 rapid advancements, including genome sequencing, understanding of
pathogenesis, blood tests and the development of AZT, led to the expectation that AIDS would be
quickly combated.6 However, by the early 1990s it was clear that if a suitable vaccine was not
Page 5 of 30 Chemical Society Reviews

3
developed HIV would probably become a permanent infection in our species. The use of multiple
drug routines over the last 15 years has significantly extended the period between infection with the
virus and the onset of AIDS symptoms, but they do not represent a cure for the disease.6 Until a
vaccine is discovered the production of more potent HIV drugs which can significantly inhibit viral
replication and reduce viral load is vital to slowing the mortality rate of this relentless disease.
Mode of Infection of HIV
As a retrovirus, HIV has a single strand RNA containing its genomic material. The viral enzyme
reverse transcriptase enables the virus to utilise this single strand RNA genome as a template for the
production of an intermediate single DNA strand, and ultimately double-stranded DNA.2,7
There are
two major types of HIV, viz. HIV-1 and HIV-2 which are thought to have originated from two
different primate species.6 HIV-1, which is the more virulent type, is more easily transmitted and
accounts for the vast majority of global HIV infections.8 Hence for the purposes of this tutorial
review all information on the pathogenesis of HIV will pertain to HIV-1. Heterosexual transmission
by unprotected sexual intercourse with an infected person accounts for 80% of all HIV-1 positive
people.9 Other modes of infection include direct exposure to infected body fluids (blood, semen
etc.), either by accident or intravenously, and direct mother to child transmission.1
Once the HI virus has entered the body of the host, a viral envelope protein facilitates entry of the
virus into a host cell. This protein consists of two parts, one protruding outwards from the virus
particle called the docking glycoprotein (gp120) and the second part called the transmembrane
glycoprotein (gp41) that anchors the protein in the viral envelope. The docking glycoprotein
(gp120) attaches to the CD4 receptor protein of a helper T cell, its primary target (Figure 1).7,10
Although these cells are considered primary targets,11
many other cell types that carry the CD4
receptor are susceptible to infection, including macrophages, microglial, dendritic and Langerhans
cells, endothelial cells and gastrointestinal epithelial cells.12
Initial interaction between gp120 and
CD4 receptors is followed by binding of gp120 to a host cell co-receptor (CCR5 or CXCR4).
Viruses making use of the CCR5 co-receptor for entry are called CCR5- or R5-tropic and those
making use of the CXCR4 co-receptor are called CXCR4- or X4-tropic. Some viruses are able to
use either of the two co-receptors and these are dual-tropic viruses.10
This co-receptor binding
results in a conformational change in the gp120 protein that allows the transmembrane gp41 to
anchor into the host cell membrane, resulting in fusion of the viral envelope and the host cell
membrane. This allows the viral core or capsid containing the nucleic acid and enzymes to enter the
host cell cytoplasm.7,10
Once in the cytoplasm, host enzymes remove the capsid thereby releasing
Page 6 of 30Chemical Society Reviews

4
the viral RNA and viral enzymes into the cell. The viral enzyme reverse transcriptase (RT) now
catalyses the synthesis of a complementary DNA strand to the genomic RNA to give a DNA/RNA
hybrid.10
This enzyme is both an RNA- and DNA-dependent DNA polymerase, which means that it
can grow a complementary DNA strand from an RNA template, as well as from a DNA template.
After formation of the hybrid strand, the RNA strand is destroyed by another domain of RT, known
as RNase H.13
Reverse transcriptase catalyses the synthesis of double stranded (ds) DNA from the
complementary ssDNA.7 This dsDNA migrates to the host cell’s nucleus under direction of the viral
enzyme integrase, which then catalyses integration of the viral DNA into the host’s chromosome.10
Once incorporated into the chromosomal DNA of the host cell, the viral DNA, now known as the
provirus, can use host enzymes to transcribe viral genomic RNA and mRNA. The viral mRNA then
leaves the nucleus and initiates the synthesis of viral proteins on host ribosomes. Translation of
viral mRNA gives rise to non-functional polyproteins (Gag and GagPol) that must be cleaved by the
viral enzyme protease into functional proteins.14
The viral genomic RNA and viral polyproteins are
then assembled by host enzymes into new virus particles which exit the cell using the host plasma
membrane to make the viral envelope. These are immature virus particles and they mature and
become infective virions shortly after budding, when viral polyproteins are cleaved by protease into
functional proteins.14
Courtesy of Northwest AETC, University of Washington
Figure 1. Schematic of HIV mode of infection and replication
Page 7 of 30 Chemical Society Reviews

5
Immediately following primary infection, the virus replicates rapidly and high levels of virus
particles are present in the blood.15
These particles spread throughout the body, seeding in many
different organs, particularly lymphoid organs such as the thymus, spleen and lymph nodes. In
adition, the number of CD4+ T cells in the bloodstream decreases by 20-40% because CD4+ cells
activated by infection are highly susceptible to the virus. There are a number of ways in which these
cells are destroyed by the virus: they can be killed directly when large amounts of virus are
produced and bud off; they may be killed by apoptosis (programmed cell death); and uninfected
cells may die if HIV particles bind to their cell surface, making them vulnerable to destruction by
killer T cells.16
About 70% of HIV-infected individuals report flu-like symptoms 2-4 weeks after
exposure to the virus. It has been shown that acutely infected individuals, in the stage immediately
following primary infection, are most infective and able to transmit the virus to others.15
This is a
result of the high viral load during this phase of the infection.
Following the acute stage of the infection starts the phase known as clinical latency. The immune
system fights the virus with CD8+ T cells (killer cells) and antibodies. Levels of HIV in the blood
are dramatically reduced and CD4+ T cell levels rise again, for some people back to normal levels.
The infected individual is usually symptom-free during this stage. However, the virus continues to
replicate in lymphoid organs and slow destruction of the immune system continues. Cells such as
monocytes and macrophages appear relatively resistant to killing by the virus but they become
reservoirs of infection as the HIV provirus remains integrated in their DNA. Even some infected
CD4+ cells survive and return to a resting state, contributing to the viral reservoir. One of the areas
in particular that harbours dormant cells is the digestive mucosa.16
Towards the end of the clinical
latency period, CD4+ cell levels decline and viral load, measured as HIV RNA copies/ml plasma,
rises.
The progression from HIV infection to AIDS varies considerably between individuals. A small
proportion of individuals develop AIDS and die within months or a few years of primary infection,
while 5% of infected individuals show no sign of the disease even after 12 years. The average
length of the clinical latency period in otherwise healthy individuals is 8-10 years and in the
absence of treatment, the average length of time between HIV infection and death is 10 years.17
There are some indicators with respect to progression of the disease from infection to AIDS; people
with high viral load are more likely to progress to AIDS faster. In addition, the viral set point (level
of HIV in the blood after the first few months of infection) also influences AIDS progression, with
a higher set point resulting in faster progression to AIDS. The age of the individual at the time of
Page 8 of 30Chemical Society Reviews

6
infection is also a factor, with those infected at a younger age (15-24) progressing slower to AIDS
than those infected at an older age (45-54).18
An HIV-infected person is diagnosed with AIDS when
they have had one or more opportunistic infections such as pneumonia or tuberculosis, and have
less than 200 CD4+ T cells per cubic millimetre of blood. This means the immune system has
deteriorated to the point that it is unable to fight opportunistic infections caused by bacteria, fungi
and parasites.17
In South Africa, the prevalence of tuberculosis (TB) is increasing steadily, largely as
a result of HIV. Currently, 70% of TB patients are HIV-positive and the problem of drug-resistant
TB is of great concern, with 7386 cases of multidrug-resistant TB and 741 cases of extensively
drug-resistant TB being confirmed in South Africa in 2010.19
IN SEARCH OF A TREATMENT FOR HIV/AIDS
Science has had significant success in prolonging the life span of HIV positive people and
improving their quality of life through the development of antiretroviral drugs.1 However, since
there is as yet no cure for HIV/AIDS research into new treatments and possible prophylaxis are of
crucial importance, particularly to the heavily burdened sub-Saharan African countries.
AZT: The proposed wonder drug
Azidothymidine or AZT 1 (Figure 2) was the first drug approved for the treatment of HIV/AIDS.6
AZT was first synthesised in 1964 by Jerome P. Horwitz for use as an anti-cancer agent.20
He
wanted to design a novel drug which would prevent the growth of cancer cells by preventing cell
replication.21
A group of dideoxythymidine compounds were synthesised that could mimic DNA
nucleosides and act as chain terminators in DNA synthesis, thus preventing the growth of cancer
cells. AZT was among the drugs synthesised and screened, but showed no activity and so was
quickly forgotten. In 1974 AZT was revisited by Wolfram Ostertag at the Max Planck Institute for
Experimental Medicine, who tested it against Friend Leukaemia Virus (FLV), a retrovirus, and
found it to successfully inhibit the virus in murine cell culture.22
Burroughs Wellcome, a
pharmaceutical company in the United States, then bought AZT from Ostertag and conducted
intensive animal tests, but ultimately decided not to develop the drug further as it did not seem to
have applications for human use. At the time it was not known that retroviruses were able to infect
humans. AZT was thus shelved as Burroughs Wellcome compound BW A509U and did not make
an appearance again until 1984 when pharmaceutical companies began screening their compound
libraries in the hopes of finding a drug which could be used to treat the newly emerging disease,
AIDS.21
Page 9 of 30 Chemical Society Reviews

7
On 29 October 1984 Janet Rideout selected compound BW A509U for testing against retroviruses
FLV and Harvey Sarcoma Virus (HaSV). AZT showed good activity against these two retroviruses
and was sent for testing against live HIV by Samuel Broder and Hiroaki Matsuya at the US
National Cancer Institute in Bethesda, Maryland. Their positive findings were confirmed at Duke
University by Dani Bolognesi who showed the drug to be active against HIV in vivo. These three
institutions worked together and published joint findings on the efficacy of AZT.23
On 3 July 1985 a
furniture salesman from Massachusetts named Joseph Rafuse became the first person to be given
AZT for the treatment of HIV and after six weeks of administration of the drug his T-cell count had
increased significantly.21
The discovery of AZT and initial positive results of the clinical trial led
many researchers to the expectation that AIDS would quickly be eradicated; however, the rapid
mutation rate of the HI virus and its subsequent resistance to AZT meant this was only the start of
what subsequently became a world-wide research effort. The search for compounds active against
HIV has resulted in FDA-approval of drugs for HIV treatment in many different classes, including
those active against RT, protease, integrase and those that prevent viral entry or fusion.
Figure 2. Structure of AZT
Current Treatment Regimens
At some point between clinical latency and progression to AIDS, antiretroviral therapy is
introduced. Based on the rapid development of resistance to drugs used as monotherapy, first
observed for AZT, a combination of three drugs called highly active antiretroviral therapy
(HAART) or triple therapy is now typically used. HAART is extremely effective at lowering viral
load and increasing CD4+ T cell levels. There has been some debate as to the ideal point for
introduction of HAART, and in 2010 the World Health Organisation (WHO) changed their
recommendation from commencement at a CD4+ T cell count of 200 cells/mm3
or below to
commencement at 350 cells/mm3 as this results in considerably better health outcomes. However, in
poorer-resourced countries such as South Africa, government-funded antiretrovirals are still only
made available at a CD4+ T cell count of 200 cells/mm3 or below, unless there is a tuberculosis co-
infection or pregnancy.
Page 10 of 30Chemical Society Reviews

8
The rapid development of drug resistance is a result of HIV reverse transcription being extremely
inaccurate, with a coding error rate of 1 per 1700 nucleotides incorporated.24
In addition, there is
evidence that the in vivo mutation rate increases under the pressure of drugs such as AZT.25
These
rapid changes in the HIV genome allow drug resistant mutant forms of HIV to emerge that are no
longer inhibited by the drug therapies in use. This has seriously hindered efforts to block viral
replication and to treat HIV infection effectively.26
Use of HAART has proven successful in
slowing down the appearance of resistant virus and this therapy is able to radically increase the
survival of HIV-infected individuals. The first-line treatment regimen now employed worldwide
generally consists of two nucleoside reverse transcriptase inhibitors (NRTIs), plus either a non-
nucleoside reverse transcriptase inhibitor (NNRTI) or a protease inhibitor (PI).27
Due to the essential role that reverse transcriptase plays in viral replication it is a major target for
antiretroviral drugs.28
There are two classes of reverse transcriptase (RT) inhibitors, the nucleoside
(NRTIs) and non-nucleoside (NNRTIs) reverse transcriptase inhibitors, each affecting the RT
enzyme at a different location. NRTIs are analogues of deoxyribonucleosides which lack the
hydroxyl group on the 3' carbon of the deoxyribose sugar. In order for these compounds to have
antiviral activity they must be metabolically converted by host cellular kinases to the triphosphate
form, which allows them to be mistaken by RT for a natural nucleotide.28,29
These phosphorylated
NRTIs can now function as chain-terminators in DNA synthesis. Azidothymidine 1 (AZT or
Retrovir®
) is an example of an NRTI that mimics the nucleoside thymidine, but the 3'-OH group is
replaced by a 3'-azide (N3) group, thus stopping further inclusion of nucleotides and terminating
DNA chain extension.29
A total of eight NRTIs have been approved for clinical use, although a few
of them are no longer in common use. South Africa’s first-line regimen includes the NRTIs
tenofovir 2 (TDF or Viread®
, strictly speaking a nucleotide inhibitor or NtRTI as it contains a
phosphonate) and lamivudine 3 (3TC or Epivir®
) or emtricitabine 4 (Emtriva®
) (Figure 3).28,30
32
N
N N
N
NH2
OP
O
HO
HO
N
N
NH2
O
S
O
HO
4
N
N
NH2
O
S
O
HO
F
Figure 3. Some commercially available NRTIs
NNRTIs on the other hand are small molecules that are chemically distinct from nucleosides and
are not dependent on host cell metabolism to be converted into an active form. NNRTIs are a group
Page 11 of 30 Chemical Society Reviews

9
of diverse hydrophobic molecules which inhibit the HIV-1 RT catalytic activity through interaction
with an allosteric site of the enzyme.28,30
The binding of a non-competitive inhibitor in this
allosteric site effects a change in conformation of the substrate-binding site which substantially
reduces the rate of incorporation of nucleotides, thereby halting DNA synthesis.31
Five NNRTIs
have been approved by the FDA for clinical use including nevirapine 5 (Viramune®
), delavirdine 6
(Rescriptor®
), efavirenz 7 (Sustiva®
, Stocrin®
), etravirine 8 (Intelence®
) and, most recently in May
2011, rilpivirine 9 (Edurant®
) (Figure 4), and several more have entered into clinical trials and
development.31
Nevirapine 5 and delavirdine 6 are considered “first-generation” NNRTIs that are
sensitive to the development of drug resistance, even with single amino acid mutations in RT.32
Efavirenz 7, a “second-generation” NNRTI maintains antiviral activity against several common
NNRTI mutants and is currently approved as first-line regimen treatment in South Africa (replaced
by nevirapine for pregnant women). The most recently approved NNRTIs, etravirine 8 and
rilpivirine 9, are believed to require at least three amino acid mutations in the NNRTI region before
clinically significant resistance is observed.33
This higher barrier to development of resistance is
considered possible because of the relatively large number of conformations that these molecules
can adopt, which allows multiple binding modes within the allosteric site of RT.31
This is believed
to be a result of the structures being less rigid, with considerable torsional flexibility, when
compared to earlier NNRTIs. These properties allow amino acid changes within the allosteric site to
be well tolerated. A more detailed discussion of the NNRTIs follows in a later section.
Figure 4. Commercially available NNRTIs
The next class of therapeutic agents most commonly used in HAART are the HIV protease
inhibitors. HIV protease plays a crucial role in the viral life cycle as it is essential for generating
mature infectious virus particles by cleavage of peptide bonds of large proteins and polypeptides
into smaller proteins.14,34
There are currently nine protease inhibitors approved for clinical use
including the older drugs saquinavir 10 (Invirase®
), indinavir 11 (Crixivan®
) and lopinavir 12
(Figure 5). Interestingly, although ritonavir 13 is a protease inhibitor in its own right, it is used to
boost the activity of other protease inhibitors by inhibiting the activity of Cytochrome P34A, which
Page 12 of 30Chemical Society Reviews

10
rapidly metabolises most of the protease inhibitors. In South Africa the lopinavir/ritonavir
combination (Kaletra®
) is commonly used as a second-line regimen treatment. The other older
protease inhibitors are largely falling into disuse and being replaced by newer drugs such as
tipranavir 14 (Aptivus®
) and darunavir 15 (Prezista®
) (Figure 5).35
The protease inhibitors act as
competitive peptidomimetic inhibitors by imitating the natural substrate of the HIV protease
enzyme.14
These mimics contain a non-scissile hydroxyethylene core instead of the amide link of
the natural substrate. The fact that the protease inhibitor cannot be cleaved by the protease enzyme
results in blockage of the enzyme site and prevents maturation of the viral particles.14
An exception
within the approved protease inhibitor class is tipranavir 14, which is a non-peptidomimetic
protease inhibitor.
Figure 5. Some commercially available PIs
More Recent Developments and Strategies for Treating HIV
The first drug developed to fall outside of the traditional RT and PI classes was enfuvirtide
(Fuzeon®
), which is a fusion inhibitor.36
This is a synthetic 36 amino acid peptide that inhibits the
fusion of viral and host cell membranes. Enfuvirtide is not widely used as it requires administration
by means of subcutaneous injection, and is extremely expensive. This drug finds use in salvage
therapy, where two or more regimens have failed and the patient exhibits evidence of ongoing viral
replication, even while undergoing HAART. The first small-molecule entry inhibitor, maraviroc 16
Page 13 of 30 Chemical Society Reviews

11
(Celsentri®
, Selzentry®
, Figure 6), was only approved by the FDA in 2008. This drug is a CCR5 co-
receptor antagonist and acts by preventing the virus from attaching to the CCR5 co-receptor,
preventing viral entry into the host cell. Maraviroc is generally used for treatment-experienced
patients, ie. those for whom other regimens have already been used and failed.37
Obviously, patients
need to be tested prior to being prescribed maraviroc to determine if they are infected with CCR5-
tropic virus, as the drug is ineffective in individuals infected with CXCR4-tropic or dual-tropic
virus. It has been shown that 50-60% of treatment-experienced patients tested are infected with
CCR5-tropic virus.38
HIV integrase is one of the three viral enzymes essential for viral replication (along with RT and
protease); this makes integrase an excellent drug target.39,40
HIV integrase is responsible for the
insertion of pro-viral DNA into the host-cell’s genome39,41
and a class of compounds known as
integrase inhibitors have been developed that inhibit this activity. Raltegravir (Isentress®
) 17
(Figure 6) was approved by the FDA on 12 October 2007 for use in antiretroviral therapy in patients
with extreme drug resistance and was the first HIV integrase inhibitor to be used as an antiretroviral
drug.39
The integration of viral DNA into the human genome is a complex process starting with the
binding of integrase to the viral DNA to form a stable pre-integration complex.39,40
This pre-
integration complex then migrates from the cytoplasm of the host cell to the nucleus where the viral
DNA is transferred and integrated into the host DNA. Raltegravir functions by blocking the active
site in the pre-integrated enzyme complex, thus preventing the viral DNA strand from being
incorporated into the host cell’s genome. A structural feature common to most compounds showing
activity against integrase is a di-keto acid, beta-keto acid or beta-hydroxy ketone capable of
chelating metal ions. Thus, the mechanism of inhibition appears to be through chelation of divalent
cations such as Mn2+
or Mg2+
within the enzyme active site. HIV integrase inhibitors have just
recently been included in the antiretroviral regimen for treatment of individuals with multi-drug
resistant HIV and have shown great success in boosting immune recovery in these patients.
However, this drug class is once again not impervious to mutations in the HI virus which render the
drugs much less effective.42
An application to the FDA for approval of a new integrase inhibitor,
elvitegravir 18 (Figure 6), is expected in the first half of 2012.
Page 14 of 30Chemical Society Reviews

12
Figure 6. Maraviroc 16, Raltegravir 17 and Elvitegravir 18, members of newer classes of ARVs
Prevention of infection: vaccines and microbicides
It has been widely established that a safe, effective and affordable HIV vaccine is needed if we wish
to successfully combat the spread of AIDS.43
The first HIV vaccine was developed in 1987 and
since then over 35 different vaccines have been tested in phase I and II clinical trials. A few HIV
vaccines have reached phase III trials, but researchers believe we are still many years away from a
successful HIV vaccine due to the seemingly insurmountable number of challenges scientists face
when dealing with this disease.43,44
When one considers the biology of HIV it becomes apparent
what the complexities associated with an HIV vaccine are and why development of an effective
vaccine is such a challenge.45
Perhaps the biggest hurdle which must be overcome is the ability of
HIV to persist in the host’s immune cells by crippling the immune system and avoiding an adaptive
immune response. These problems are further complicated by the rapid mutation of the HI virus,
which aids in evading the immune response and seriously hampers progress in developing an HIV
vaccine.45
A recent advancement in the field of HIV vaccine development was reported by Jin and
co-workers46
who submitted a novel polypeptide vaccine of HIV T helper epitopes (EP-1043) and a
DNA vaccine of HIV CTL epitopes for phase I clinical trials. The group demonstrated that 64% of
their test subjects (non HIV-infected adults) had a positive CD4+ T response after two vaccinations.
Whether the vaccine could be used to prevent HIV infection or simply to slow disease progression
was not established, but it is hoped that upon further study EP-1043, or a modified version of it,
could be used as a novel HIV vaccine. A recent Phase III trial conducted in Thailand was the first
Page 15 of 30 Chemical Society Reviews

13
human HIV vaccine trial to show a decrease in HIV infection after immunisation, albeit a modest
30% reduction.44,47
While immunologists and scientists work to develop a successful vaccine and more effective drugs
to treat HIV infected patients, the most effective solution for stopping the spread of HIV is that of
education. Educating the public on the facts of how HIV is transmitted and empowering women in
rural communities is the best way of slowing the spread of HIV/AIDS in a South African context.
One such means of empowerment for women is developing a method for preventing HIV infection
that is within their own control. This is the rationale behind the development of microbicides as
prophylactic agents. Microbicides are agents that can be applied topically prior to sexual intercourse
in order to prevent sexually transmitted HIV infections.48
A recent double-blind controlled trial in
South Africa comparing the use of tenofovir gel versus placebo as a microbicidal agent promisingly
showed a reduction in HIV acquisition of 39% in the tenofovir arm of the trial.49
THE USE OF NNRTIS AS DRUGS TARGETING RT
With the advent of HAART therapy the use of NNRTIs has been firmly established in the treatment
of HIV infection, but due to drug resistance many of the earlier NNRTIs are no longer as potent.26
Thus the synthesis and development of novel NNRTIs with increased activity against resistant
strains has become the focus of many research groups in the fight against HIV/AIDS.50
The RT Enzyme and Mode of Action of NNRTIs
HIV-1 RT is a heterodimer consisting of p66 and p51 subunits (Figure 7), where the p66 subunit is
the larger of the two and contains both NRTI and NNRTI binding sites.26,51
The p66 subunit
consists of RNA- and DNA-dependent DNA polymerase and ribonuclease H (RNase H) domains,
both of which are vital for the process of converting single-stranded RNA into double-stranded
DNA.26,28
The p51 subunit, on the other hand, plays a structural rather than catalytic role, although
the protein sequence is identical to that of p66 except that it excludes the RNase H domain.13
Page 16 of 30Chemical Society Reviews

14
Figure 7. HIV-1 RT heterodimer showing Nevirapine bound in the allosteric site (superimposition
of PDB structures 1VRT and 2HMI). This figure was prepared using Accelrys Discovery Studio.
NNRTIs interact with the HIV-1 RT by binding to a site on the p66 subunit called the NNRTI
binding pocket, which is an allosteric site situated 10 Ǻ from the RT polymerase and 60 Ǻ from the
RNase H active site.26,28
In contrast to NRTIs, which are competitive inhibitors and actively
compete for the active site of the polymerase, NNRTIs are non-competitive inhibitors. When
unliganded and NNRTI-bound RT crystal structures are compared, it is evident that the NNRTI
binding pocket is only created by the binding of the inhibitor itself.26
The NNRTI binding site is
surrounded by hydrophobic amino acids and thus π‒π interactions between the inhibitor and the
amino acid side chains tend to be the dominant interactions in this site. Inhibitor binding is
characterised by the movement of the side chains of the key amino acids Tyr181 and Tyr188 from a
‘down’ position to an ‘up’ position. The importance of these residues for binding of the inhibitor is
shown by how favourably the virus selects for the mutated enzyme where these two residues are
aliphatic so that favourable π-stacking interactions with aromatic inhibitors is avoided.26
It is thus
Page 17 of 30 Chemical Society Reviews

15
obvious that design of new NNRTIs should seek to avoid this key interaction in the hopes of
significantly reducing the rate at which HIV becomes drug resistant.
In order to design new and effective NNRTIs it is important to understand the mode of inhibition
and the specific binding actions of the ligands in the enzyme. A number of different mechanisms
have been proposed for NNRTI inhibition of RT. The simplest explanation is that the binding of the
NNRTI in the allosteric site of the enzyme prevents the domain movements which are imperative to
the catalytic cycle.26
Another possibility is that on binding of the NNRTI in the allosteric site there
is a significant and consistent movement of the β chains which contain the important amino acids
Asp110, Asp185 and Asp186 (catalytic triad).26
It is assumed that when these catalytically essential
amino acid residues are shifted in the active site by the change in conformation of the enzyme,
catalysis is halted. It is generally accepted that binding of the NNRTI in the allosteric site brings
about a change in conformation of the active site, reducing the rate of incorporation of nucleotides,
thereby slowing down or halting viral DNA synthesis.31
The recent determination of the crystal
structure of an RT-DNA-nevirapine complex has shed more light on the inhibition mechanism of
NNRTIs.52
This study showed that binding of nevirapine had a dramatic effect on the RT-DNA
conformation, with the β-sheet containing the catalytic triad moving away from the β-sheet
containing the “primer grip” that positions the primer correctly for polymerase activity. The result
of this movement is improper alignment of the different elements required for nucleotide
incorporation.52
Current NNRTIs and the Mutations Which Confer Resistance
There are unfortunately a wide range of drug resistance mutations described in the literature for
NNRTIs and this highlights the problem with many of the NNRTIs available for use in HAART.
Understanding the mechanisms by which these mutations confer resistance is therefore imperative
in designing novel NNRTIs which may be more effective in treating HIV infections.26,30
First
generation NNRTIs such as nevirapine and delavirdine lose potency in the presence of single point
mutations, whilst the second generation compound efavirenz maintains potency in the presence of a
variety of single point mutations.51
As might be expected, most of the NNRTI resistance mutations
observed are for amino acids immediately surrounding the allosteric binding pocket. This is in stark
contrast to the NRTIs, where mutations occur at positions distal to the catalytic site.51
The
Tyr181/Tyr188 mutations which have already been discussed, greatly reduce the efficacy of the
NNRTI nevirapine (see Fig. 7 expansion).26,33
Typically the mutations are Tyr181Cys (Y181C) or
Tyr188Cys/Leu (Y188C/L), where the aromatic tyrosine side-chain is replaced by an aliphatic
group. This results in the loss of favourable aromatic stacking between the pyridine ring of
Page 18 of 30Chemical Society Reviews

16
nevirapine and the aromatic side chains of the Tyr181/Tyr188 residues. The Y181C mutation, not
surprisingly, only confers a small loss in potency with the NNRTI efavirenz as the cyclopropyl
group already has less effective contacts with the Tyr181 residue (Figure 8). Instead, the dominant
binding interaction for efavirenz is a hydrogen bonding interaction with the main chain carbonyl
oxygen of Lys101,53
an interaction that is maintained in the binding of the newer inhibitors
rilpivirine and etravirine.54
These newer inhibitors maintain excellent potency against the Y181C
and Y188C/L mutants.
The most widely reported clinically relevant resistance mutation is K103N (Figure 8), the mutation
of the lysine residue at 103 to an asparagine residue.26
Structural studies indicate that this mutation
allows favourable H-bonding interactions within the site, possibly between the Tyr188 hydroxyl
group and the amide group of Asn103, in the absence of an inhibitor. This acts to stabilise the
closed NNRTI binding site,55
creating an energy barrier to inhibitor binding. This mutation reduces
the activity of many FDA approved NNRTIs (nevirapine, delavirdine and efavirenz).56
The binding
of nevirapine is weakened 40-fold in the presence of the K103N mutation, and that for efavirenz is
weakened 6-fold.53
Interestingly, efavirenz appears able to bind to the K103N mutant with Tyr181
in both an “up” and a “down” position51
(see Figure 8). The newer NNRTIs etravirine and
rilpivirine are virtually impervious to this mutation, maintaining almost full potency.54
In addition,
their binding mode in the wild-type and K103N mutant appears unchanged. The fact that these new
NNRTIs maintain potency against K103N mutants means that the increased stability of the
unliganded form may only be a partial explanation for the cross-resistance this mutant displays
towards the first and second generation NNRTIs.
Page 19 of 30 Chemical Society Reviews

17
Figure 8. Efavirenz in the active site of wild-type (PDB 1FK9) and K103N mutant (PDB 1FKO)
with Tyr181 occupying an “up” or “down” position. This figure was prepared using Accelrys
Discovery Studio.
Other mutations commonly observed for amino acids surrounding the binding site are Leu100Ile
(L100I), Val106Ala (V106A) and Val108Ile (V108I). Conversion of leucine and valine into
isoleucine is likely to have consequences of a steric nature, where unfavourable steric interactions
arise with the inhibitor. Contrastingly, conversion of valine into alanine would result in a loss of the
potential for hydrophobic interactions.33
A further mutation which confers moderate resistance to a number of NNRTIs is mutation at
positions 101 and 138.26
Lys101 is found at the surface of the NNRTI pocket and interacts with
Glu138 (of the p51 subunit) by formation of a salt bridge. In the case of nevirapine, there is no
direct contact between Lys101 and the inhibitor and therefore it is difficult to explain why mutation
at this position leads to an 8 fold reduction in potency. However, there appear to be indirect contacts
between Lys101 and nevirapine via three water molecules. A Lys101Glu (L101E) mutation inverts
the charge at this position, converting the positively charged amino side-chain into the negatively
charged glutamic acid side-chain. Now, instead of a salt bridge between 101 and 138, there is
repulsion of like charges and the amino acids move away from each other and the NNRTI binding
pocket, leading to reduced affinity of nevirapine for the binding site. In the case of efavirenz, there
is direct interaction between the inhibitor and Lys101, explaining selection of the L101E mutant in
the clinical use of efavirenz.51
The high barrier to the development of drug-resistance exhibited by the most recently FDA-
approved NNRTIs etravirine and rilpivirine is believed to be the result of torsional flexibility that
allows the compounds to bind effectively to mutants by changing their bound conformations in the
allosteric site.31,32,54
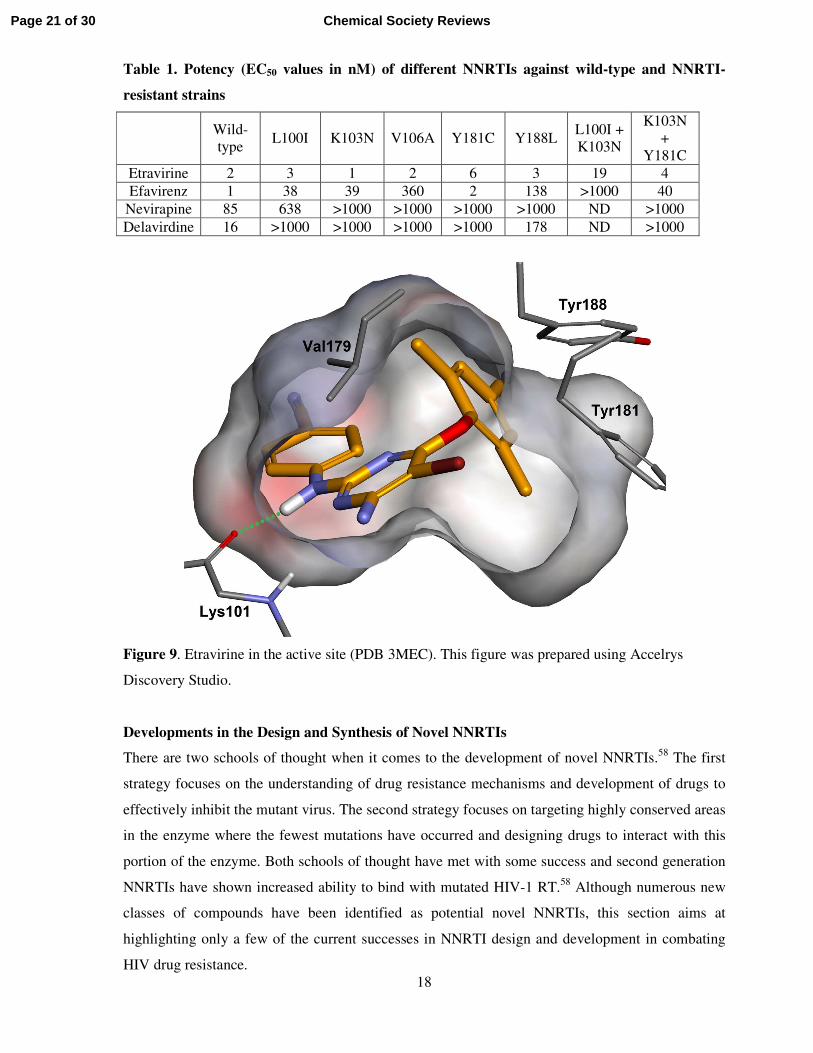
Etravirine and rilpvirine both maintain a hydrogen bond to Lys101, as shown
for etravirine in Figure 9. The in vitro activity of etravirine against wild-type and mutant strains is
shown in Table 1 and compared with the activities of the earlier NNRTIs nevirapine, delavirdine
and efavirenz.57
Page 20 of 30Chemical Society Reviews
18
Table 1. Potency (EC50 values in nM) of different NNRTIs against wild-type and NNRTI-
resistant strains
Wild-
type L100I K103N V106A Y181C Y188L
L100I +
K103N
K103N
+
Y181C
Etravirine 2 3 1 2 6 3 19 4
Efavirenz 1 38 39 360 2 138 >1000 40
Nevirapine 85 638 >1000 >1000 >1000 >1000 ND >1000
Delavirdine 16 >1000 >1000 >1000 >1000 178 ND >1000
Figure 9. Etravirine in the active site (PDB 3MEC). This figure was prepared using Accelrys
Discovery Studio.
Developments in the Design and Synthesis of Novel NNRTIs
There are two schools of thought when it comes to the development of novel NNRTIs.58
The first
strategy focuses on the understanding of drug resistance mechanisms and development of drugs to
effectively inhibit the mutant virus. The second strategy focuses on targeting highly conserved areas
in the enzyme where the fewest mutations have occurred and designing drugs to interact with this
portion of the enzyme. Both schools of thought have met with some success and second generation
NNRTIs have shown increased ability to bind with mutated HIV-1 RT.58
Although numerous new
classes of compounds have been identified as potential novel NNRTIs, this section aims at
highlighting only a few of the current successes in NNRTI design and development in combating
HIV drug resistance.
Page 21 of 30 Chemical Society Reviews

19
One such case is the synthesis and development of novel second generation diaryl ethers as lead
compounds.59
Tucker et al. discovered compound 19 via high throughput screening (HTS), but the
disappointing pharmacokinetic results forced the group to re-think their starting position. Using
elements of compound 19 and combining these with elements from an already known NNRTI 20
via molecular modelling, they were able to identify a novel diphenyl ether substructure 21 (Figure
10) which showed good docking results within the NNRTI binding site. The molecular modelling
study showed that the amide carbonyl was imperative in maintaining a hydrogen bond with the
backbone NH of Lysine 103, which is considered crucial in maintaining good potency against
mutants at this position, such as K103N.59
The modelling study also indicated that a di-meta
substitution pattern on the A ring, as seen in compound 20, and introduction of a 2- or 3- substituent
on the B ring would help optimise interactions with the binding site and help to improve the
potency of this series of compounds. The model also indicated that the sulfonamide was situated at
the enzyme/water interface and was thus fully solvent exposed, indicating the potential for various
polar substitutions in this area.
Figure 10. Diaryl ethers as lead compounds
The group synthesised a variety of compounds based on the modelling study with an indazole
compound 22 (Figure 10) showing the best NNRTI activity.59
The key carbonyl H-bonding
interaction is replaced by the indazole moiety which is capable of forming two H-bonds with amino
acid residue K103, one with the backbone NH and one with the backbone carbonyl, thus increasing
its potency. The di-meta substitution on the A aryl-ring once again increased the potency of the
compound and this is thought to be due to the improved π-π stacking of the A-aryl ring and the
Page 22 of 30Chemical Society Reviews

20
Tyr188 residue. The chlorine atom on the B aryl-ring fits very well into a small lipophilic region
behind the Val179 residue and once again increases binding potency. Unfortunately this compound
showed low bioavailability and poor solubility, but subsequent synthetic efforts led to compound 23
(MK-4965),60
which, together with high potency, showed excellent pharmacokinetic properties and
good bioavailability. These compounds, like etravirine, are also expected to display torsional
flexibility, an advantage against mutant viral strains. Further modifications are being made to
improve the properties of these biaryl ethers, based on the structure of lead compound 23.61-63
In the quest for novel NNRTIs HTS identified sulfanyltriazoles (24)64
and sulfanyltetrazoles (25)65-
67 (Figure 11) as potent inhibitors of both wild-type and resistant viral strains. One of the problems
associated with these compounds was poor metabolic stability, and in an attempt to address this, the
sulphur atom was replaced by oxygen or carbon for the sulfanyltetrazole series (25). This led to a
dramatic reduction in antiviral potency and did not improve the metabolic properties.66
Based on
these findings, the sulphur was retained and a series of compounds was prepared in order to
improve potency and pharmacokinetic properties. Compounds such as 26 were found to show
excellent activity against both wild-type and double mutant K103N/Y181C viral strains.66
Figure 11. Triazole and tetrazole thioacetanilide based NNRTIs
Molecular modelling of this compound in the allosteric site showed a number of key interactions.
Most notable was a hydrogen bond between the inhibitor amide carbonyl and the backbone NH of
K103. In addition, there was a perpendicular CH-π interaction between the 4-substituent of the
phenyl-tetrazole and the indole side-chain of Trp229, which is a highly conserved residue. The
para-substituent of the anilide ring was found to be directed towards the solvent interface,
increasing the likelihood of hydrophilic substituents being acceptable in this position. Interestingly,
substitution at this position was found to increase potency in both the sulfanyltetrazole as well as
Page 23 of 30 Chemical Society Reviews

21
the sulfanyltriazole series of compounds.67
No specific interactions were observed between RT and
the thio-tetrazole ring and the tetrazole ring was found to be orientated perpendicular to the plane of
the attached 4-tert-butyl-2-chlorophenyl ring. This was also found to be true for compound 27
which exhibited nanomolar potency against both wild-type and K103N/Y181C double mutant
strains, as well as an acceptable pharmacokinetic profile.67
These examples highlight the importance of processes such as HTS, SAR studies and molecular
modelling in finding potent and selective NNRTIs. A recent example showing the successful
application of a computational approach, particularly the use of free-energy perturbation
calculations, to the design of novel NNRTIs was published by Jorgensen et al.68
Their efforts led to
a compound showing potent activity against both wild-type and Y181C variants. There is ongoing
interest in the design and development of novel comounds in this important antiretroviral class, with
a particular emphasis on finding compounds with potent activity against mutant strains.69-74
Figure 12. Lersivirine 28 and Capravirine 29
A notable success in achieving this aim is found in the example of lersivirine 28 (Figure 12),75
which was recently tested in a successful Phase IIb clinical trial. The starting point for the design of
lersivirine was capravirine 29 (Figure 12),76,77
a compound for which development was
discontinued after disappointing Phase IIb trial results. Lersivirine has a unique resistance profile
compared to other NNRTIs because it binds to RT in a novel way (Figure 13). There is an edge-to-
face π interaction between the dicyanophenyl ring and residue Trp229. In addition, the Tyr181
residue, which usually moves to the “up” position on NNRTI binding, remains in the “down”
position on binding lersivirine.78
Reports indicate that this promising candidate drug inhibits 60% of
viruses that carry key RT mutations.75
Activity was maintained against 14 of 15 single point
mutations most commonly observed in NNRTI-experienced patients, as well as against two double
mutants (Y181C/Y188C and V106A/Y181C). This will make it a valuable addition to the current
selection of NNRTIs available for clinical treatment, should planned Phase III trials prove
successful.
Page 24 of 30Chemical Society Reviews

22
Figure 13. A comparison of the binding modes of efavirenz (PDB 1FK9) and lersivirine (PDB
2WON) in the NNRTI allosteric site. This figure was prepared using Accelrys Discovery Studio.
CONCLUSION
The development of new drugs for HIV treatment over the last two decades has resulted in 25
different drugs now being commercially available, as well as several drugs that are formulated as
combinations. These drugs fall into 6 different classes, depending on their mode of action. NNRTIs
are firmly established as valuable contributors to HIV/AIDS treatment regimens; initially as first-
line treatments but increasingly for treatment-experienced populations. Of particular importance
going forward is the development of drugs with a high barrier to resistance; where more than one
mutation is required before there is any loss in drug potency. In countries such as South Africa,
where antiretroviral coverage has thus far been low and the bulk of HIV-positive people are still
treatment naive, resistance to these drugs has not yet been of widespread clinical significance.
However, now that the country is in the process of scaling up antiretroviral coverage, drug
resistance will play an increasingly significant role both within the treatment-naive and treatment-
experienced groups; and new drugs will play a crucial role in treating the 5.6 million HIV/AIDS
sufferers.
Page 25 of 30 Chemical Society Reviews

23
ACKNOWLEDGEMENTS
The following funders are gratefully acknowledged: CSIR Thematic Fund, National Research
Foundation (NRF, GUN 2053652 and IRDP of the NRF for financial support provided by the
Research Niche Areas programme), the University of the Witwatersrand (Science Faculty Research
Council) and the Mellon Postgraduate Mentoring Programme (sponsored by the Andrew W. Mellon
Foundation).
REFERENCES
1. R. C. Gallo and L. Montagnier, Science, 2002, 298, 1730-1731.
2. R. C. Gallo and L. Montagnier, N. Eng. J. Med. 2003, 349, 2283-2285.
3. “UNAIDS World AIDS Day Report 2011”, Joint United Nations Programme on
HIV/AIDS (UNAIDS), Geneva, ISBN: 978-92-9173-904-2.
4. J. Thurlow, J. Gow and G. George, J. Int. AIDS Soc. 2009, 12, 18-31.
5. R. C. Gallo, Science, 2002, 298, 1728-1730.
6. R. C. Gallo, Retrovirology, 2006, 3, 72-78.
7. E. M. Campbell and T. J. Hope, Trends Microbiol. 2008, 16, 580-587.
8. M. Ndour, P. S. Sow, A. M. Coll-Seck, S. Badiane, C. T. Ndour, N. Diakhaté, B. Diop,
M. Faye, M. Soumaré, G. Diouf and R. Colebunders, Trop. Med. Int. Health, 2000, 5,
687-691.
9. R. J. Shattock and J. P. Moore, Nat. Rev. Microbiol. 2003, 1, 25-34.
10. E. O. Freed, Somat. Cell Mol. Genet., 2001, 26, 13-33.
11. A. T. Haase, Nature, 2010, 464, 217-223.
12. F. Groot, S. Welsch and Q. J. Sattentau, Blood, 2008, 111, 4660-4663.
13. N. Sluis-Cremer, D. Arion and M. A. Parniak, CMLS, Cell. Mol. Life Sci., 2000, 57,
1408-1422.
14. A. M. J. Wensing, N. M. van Maarseveen and M. Nijhuis, Antiviral Res., 2010 85, 59-
74.
15. T. D. Hollingsworth, R. M. Anderson, C. Fraser, J. Infect. Dis., 2008, 198, 687-693.
16. V. Dahl, L. Josefsson, S. Palmer, Antiviral Res., 2010, 85, 286-294.
17. S. Jaffer, A. D. Grant, J. Whitworth, P. G. Smith and H. Whittle, Bull. WHO, 2004, 82,
462-469.
Page 26 of 30Chemical Society Reviews

24
18. Collaborative Group on AIDS Incubation and HIV Survival including the CASCADE
EU Concerted Action, Lancet, 2000, 355, 1131-1137.
19. South African Government National Strategic Plan on HIV, STIs and TB: 2012-2016,
accessed 27 February 2012, http://www.doh.gov.za/docs/stratdocs/2011/hiv_nsp.pdf.
20. J. P. Horwitz, J. Chua, M. Noel and V. Nucleoside, J. Org. Chem., 1964, 29, 2076-2078.
21. M. Yarchoan, “The Story of AZT: Partnership and Conflict”, accessed 11 November
2011, www.scribd.com/doc/1049/The-History-of-AZT.
22. W. Ostertag, G. Roesler, C. J. Krieg, J. Kind, T. Cole, T. Crozier, G. Gaedicke, G.
Steinheider, N. Kluge and S. Dube, Proc. Nat. Acad. Sci. USA, 1974, 71, 4980-4985.
23. H. Mitsuya, K. J. Weinhold, P. A. Furman, M. H. St. Clair, S. N. Lehrman, R. C. Gallo,
D. Bolognesi, D. W. Barry and S. Broder, Proc. Nat. Acad. Sci. USA, 1985, 82, 7096-
7100.
24. J. D. Roberts, K. Bebenek and T. A. Kunkel, Science, 1988, 242, 1171-1173.
25. L. M. Mansky and L. C. Bernard, J. Virol., 2000, 74, 9532-9539.
26. J. Ren and D. K. Stammers, Virus Res., 2008, 134, 157-170.
27. F. Maggiolo, J. Antimicrob. Chemother., 2009, 64, 910-928.
28. N. Sluis-Cremer and G. Tachedjian, Virus Res., 2008, 134, 147-156.
29. G. Wang, N. Boyle, F. Chen, V. Rajappan, P. Fagan, J. L. Brooks, T. Hurd, J. M. Leeds,
V. K. Rajwanshi, Y. Jin, M. Prhavc, T. W. Bruice and P. D. Cook, J. Med. Chem., 2004
47, 6902-6913.
30. E. De Clercq, Antiviral Res., 1998, 38, 153-179.
31. D. Jochmans Virus Res., 2008, 134, 171-185.
32. R. Pauwels Curr. Opin. Pharmacol., 2004, 4, 437-446.
33. A. C. Anderson, ACS Chem. Biol., 2012, 7, 278-288.
34. K. D. Phillips, J. Assoc. Nurses, AIDS Care, 1996, 7, 57-71.
35. W. D. F. Venter, R. Osih, S. Andrews and F. Conradie, S. Afr. J. HIV Med., 2008, 9, 44-
49.
36. J. A. Esté and A. Telenti, Lancet, 2007, 370, 81-88.
37. R. M. Gulick, J. Lalezari, J. Goodrich, N. Clumeck, E. DeJesus, A. Horban, J. Nadler, B.
Clotet, A. Karlsson, M. Wohlfeiler, J. B. Montana, M. McHale, J. Sullivan, C. Ridgway,
S. Felstead, M. W. Dunne, E. van der Ryst and H. Mayer. N. Engl. J. Med., 2008, 359,
1429-1441.
38. J. Edmunds-Ogbuokiri, HIV Clinician, 2009, 21, 11-14.
39. J. Cocohoba and B. J. Clin. Ther., 2008, 30, 1747-1765.
Page 27 of 30 Chemical Society Reviews

25
40. S. Lee-Huang, P. L. Huang, D. Zhang, J. W. Lee, J. Bao, Y. Sun, Y.-T. Chang, J. Zhang
and P. L. Huang, Biochem. Biophys. Res. Commun., 2007, 354, 879-884.
41. V. R. de Soultrait, P.-Y. Lozach, R. Altmeyer, L. Tarrago-Litvak, S. Litvak, M. L.
Andréola, J. Mol. Biol., 2002, 324, 195-203.
42. P. A. Cane, J. Antimicrob. Chemother., 2009, 64(s 1), i37-i40.
43. M. P. Girard, S. K. Osmanov and M. P. Kieny, Vaccine, 2006, 24, 4062-4081.
44. H. W. Virgin and B. D. Walker, Nature, 2010, 464, 224-231.
45. P. Mooij and J. L. Heeney, Vaccine, 2001, 20, 304-321.
46. X. Jin, M. J. Newman, S. De-Rosa, C. Cooper, E. Thomas, M. Keefer, J. Fuchs, W.
Blattner, B. D. Livingston, D. M. McKinney, E. Noonan, A. deCamp, O. D. Defawe, M.
Wecker, Vaccine, 2009, 27, 7080-7086.
47. S. Rerks-Ngarm, P. Pitisuttithum, S. Nitayaphan, J. Kaewkungwal, J. Chiu, R. Paris, N.
Premsri, C. Namwat, M. de Souza, E. Adams, M. Benenson, S. Gurunathan, J. Tartaglia,
J. G. McNeil, D. P. Francis, D. Stablein, D. L. Birx, S. Chunsuttiwat, C.
Khamboonruang, P. Thongcharoen, M. L. Robb, N. L. Michael, P. Kunasol and J. H.
Kim, N. Engl. J. Med., 2009, 361, 2209-2220.
48. J. Balzarini and L. Van Damme, Lancet, 2007, 369, 787-797.
49. Q. Abdool Karim, S. S. Abdool Karim, J. A. Frohlich, A. C. Grobler, C. Baxter, L. E.
Mansoor, A. B. M. Kharsany, S. Sibeko, K. P. Mlisana, Z. Omar, T. N. Gengiah, S.
Maarschalk, N. Arulappan, M. Mlotshwa, L. Morris and D. Taylor, Science, 2010, 329,
1168-1174.
50. M.-P., De Béthune, Antiviral Res., 2010, 85, 75-90.
51. J. Ren, C. E. Nichols, A. Stamp, P. P. Chamberlain, R. Ferris, K. L. Weaver, S. A. Short
and D. K. Stammers, FEBS Journal, 2006, 273, 3850-3860.
52. K. Das, S. E. Martinez, J. D. Bauman and E. Arnold, Nat. Struct. Mol. Biol., 2012, 19,
253-259.
53. J. Ren, J. Milton, K. L. Weaver, S. A. Short, D. I. Stuart, D. K. Stammers, Structure,
2000, 8, 1089-1094.
54. E. B. Lansdon, K. M. Brendza, M. Hung, R. Wang, S. Mukund, D. Jin, G. Birkus, N.
Kutty and X. Liu, J. Med. Chem., 2010, 53, 4295-4299.
55. R. A. Domaoal and L. M. Demeter, Int. J. Biochem. Cell Biol., 2004, 36, 1735-1751.
56. M. Udier-Blagovic, E. K. Watkins, J. Tirado-Rives and W. L. Jorgensen, Bioorg. Med.
Chem. Lett., 2003, 13, 3337-3340.
57. K. Das, A. D. Clark, Jr., P. J. Lewi, J. Heeres, M. R. de Jonge, L. M. H. Koymans, H. M.
Vinkers, F. Daeyaert, D. W. Ludovici, M. J. Kukla, B. De Corte, R. W. Kavash, C. Y.
Page 28 of 30Chemical Society Reviews

26
Ho, Hong Ye, M. A. Lichtenstein, K. Andries, R. Pauwels, M.-P. de Béthune, P. L.
Boyer, P. Clark, S. H. Hughes, P. A. J. Janssen and E. Arnold, J. Med. Chem., 2004, 47,
2550-2560.
58. D. G. Prajapati, R. Ramajayam, M. R. Yadav and R. Giridhar, Bioorg. Med. Chem.,
2009, 17, 5744-5762.
59. T. J. Tucker, S. Saggar, J. T. Sisko, R. M. Tynebor, T. M. Williams, P. J. Felock, J. A.
Flynn, M.-T. Lai, Y. Liang, G. McGaughey, M. Liu, M. Miller, G. Moyer, V. Munshi,
R. Perlow-Poehnelt, S. Prasad, R. Sanchez, M. Torrent, J. P. Vacca, B.-L. Wan and Y.
Yan, Bioorg. Med. Chem. Lett., 2008, 18, 2959-2966.
60. T. J. Tucker, J. T. Sisko, R. M. Tynebor, T. M. Williams, P. J. Felock, J. A. Flynn, M.-T.
Lai, Y. Liang, G. McGaughey, M. Liu, M. Miller, G. Moyer, V. Munshi, R. Perlow-
Poehnelt, S. Prasad, J. C. Reid, R. Sanchez, M. Torrent, J. P. Vacca, B.-L. Wan and Y.
Yan, J. Med. Chem., 2008, 51, 6503-6511.
61. D.-S. Su, J. J. Lim, E. Tinney, B.-L. Wan, M. B. Young, K. D. Anderson, D. Rudd, V.
Munshi, C. Bahnck, P. J. Felock, M. Lu, M.-T. Lai, S. Touch, G. Moyer, D. J.
DiStefano, J. A. Flynn, Y. Liang, R. Sanchez, R. Perlow-Poehnelt, M. Miller, J. P.
Vacca, T. M. Williams and N. J. Anthony, J. Med. Chem., 2009, 52, 7163-7169.
62. D.-S. Su, J. J. Lim, E. Tinney, T. J. Tucker, S. Saggar, J. T. Sisko, B.-L. Wan, M. B.
Young, K. D. Anderson, D. Rudd, V. Munshi, C. Bahnck, P. J. Felock, M. Lu, M.-T.
Lai, S. Touch, G. Moyer, D. J. DiStefano, J. A. Flynn, Y. Liang, R. Sanchez, R. Perlow-
Poehnelt, M. Miller, J. P. Vacca, T. M. Williams and N. J. Anthony, Bioorg. Med.
Chem. Lett., 2010, 20, 4328-4332.
63. R. Gomez, S. Jolly, T. Williams, T. Tucker, R. Tynebor, J. Vacca, G. McGaughey, M.-
T. Lai, P. Felock, V. Munshi, D. DeStefano, S. Touch, M. Miller, Y. Yan, R. Sanchez,
Y. Liang, B. Paton, B.-L. Wan and N. Anthony, Bioorg. Med. Chem. Lett., 2011, 21,
7344-7350.
64. Z. Wang, B. Wu, K. L. Kuhen, B. Bursulaya, T. N. Nguyen, D. G. Nguyen and Y. He,
Bioorg. Med. Chem. Lett., 2006, 16, 4174-4177.
65. E. Muraglia, O. D. Kinzel, R. Laufer, M. D. Miller, G. Moyer, V. Munshi, F. Orvieto,
M. C. Palumbi, G. Pescatore, M. Rowley, P. D. Williams and V. Summa, Bioorg. Med.
Chem Lett., 2006, 16, 2748-2752.
66. J. A. O’Meara, A. Jakalian, S. LaPlante, P. R. Bonneau, R. Coulombe, A.-M. Faucher, I.
Guse, S. Landry, J. Racine, B. Simoneau, B. Thavonekham and C. Yoakim, Bioorg.
Med. Chem. Lett., 2007, 17, 3362-3366.
Page 29 of 30 Chemical Society Reviews

27
67. A. Gagnon, M. H. Amad, P. R. Bonneau, R. Coulombe, P. L. DeRoy, L. Doyon, J.
Duan, M. Garneau, I. Guse, A. Jakalian, E. Jolicoeur, S. Landry, E. Malenfant, B.
Simoneau and C. Yoakim, Bioorg. Med. Chem. Lett., 2007, 17, 4437-4441.
68. W. L. Jorgensen, M. Bollini, V. V. Thakur, R. A. Domaoal, K. A. Spasov, K. S.
Anderson, J. Am. Chem. Soc., 2011, 133, 15686-15696.
69. M. L. Mitchell, J. C. Son, I. Y. Lee, C.-K. Lee, H. S. Kim, H. Guo, J. Wang, J. Hayes,
M. Wang, A. Paul, E. B. Lansdon, J. M. Chen, G. Eisenberg, R. Geleziunas, L. Xu and
C. U. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 1585-1588.
70. A.-M. Monforte, P. Logoteta, L. De Luca, N. Iraci, S. Ferro, G. Maga, E. De Clercq, C.
Pannecouque and A. Chimirri, Bioorg. Med. Chem., 2010, 18, 1702-1710.
71. Z.-S. Zeng, Q.-Q. He, Y.-H. Liang, X.-Q. Feng, F.-E. Chen, E. De Clercq, J. Balzarini
and C. Pannecouque, Bioorg. Med. Chem., 2010, 18, 5039-5047.
72. D. J. Kertesz, C. Brotherton-Pleiss, M. Yang, Z. Wang, X. Lin, Z. Qiu, D. R. Hirchfeld,
S. Gleason, T. Mirzadegan, P. W. Dunten, S. F. Harris, A. G. Villaseňor, J. Q. Hang, G.
M. Heilek and K. Klumpp, Bioorg. Med. Chem. Lett., 2010, 20, 4215-4218.
73. M. Venkatraj, K. K. Ariën, J. Heeres, B. Dirié, J. Joossens, S. Van Goethem, P. Van der
Veken, J. Michiels, C. M. L. Vande Velde, G. Vanham, P. J. Lewi and K. Augustyns,
Bioorg. Med. Chem., 2011, 19, 5924-5934.
74. M. S. Novikov, O. N. Ivanova, A. V. Ivanov, A. A. Ozerov, V. T. Valuev-Elliston, K.
Temburnikar, G. V. Gurskaya, S. N. Kochetkov, C. Pannecouque, J. Balzarini and K. L.
Seley-Radtke, Bioorg. Med. Chem., 2011, 19, 5794-5802.
75. R. Corbau, J. Mori, C. Phillips, L. Fishburn, A. Martin, C. Mowbray, W. Panton, C.
Smith-Burchnell, A. Thornberry, H. Ringrose, T. Knöchel, S. Irving, M. Westby, A.
Wood and M. Perros, Antimicrob. Agents Chemother., 2010, 54, 4451-4463.
76. C. E. Mowbray, C. Burt, R. Corbau, M. Perros, I. Tran, P. A. Stupple, R. Webster and A.
Wood, Bioorg. Med. Chem. Lett., 2009, 19, 5599-5602.
77. C. E. Mowbray, C. Burt, R. Corbau, S. Gayton, M. Hawes, M. Perros, I. Tran, D. A.
Price, F. J. Quinton, M. D. Selby, P. A. Stupple, R. Webster and A. Wood,. Bioorg.
Med. Chem. Lett., 2009, 19, 5857-5860.
78. L. H. Jones, G. Allan, R. Corbau, D. S. Middleton, C. E. Mowbray, S. D. Newman, C.
Phillips, R. Webster and M. Westby, Chem. Biol. Drug Des., 2011, 77, 393-397.
Page 30 of 30Chemical Society Reviews


![5 H-pyrrolo[1,2- b][1,2,5]benzothiadiazepines (PBTDs): A novel class of non-nucleoside reverse transcriptase inhibitors](https://static.fdokumen.com/doc/165x107/63144331fc260b71020f7b4c/5-h-pyrrolo12-b125benzothiadiazepines-pbtds-a-novel-class-of-non-nucleoside.jpg)

















