
Improved expansion of human bone marrow-derived mesenchymal stem cells in microcarrier-based...
16
Improved expansion of human bone marrow-derived mesenchymal stem cells in microcarrier-based suspension culture Yifan Yuan 1 , Michael S. Kallos 1 , Christopher Hunter 2 and Arindom Sen 1 * 1 Pharmaceutical Production Research Facility (PPRF), Schulich School of Engineering, University of Calgary, Calgary, Alberta, Canada 2 Mechanical and Manufacturing Engineering, Schulich School of Engineering, University of Calgary, Calgary, Alberta, Canada Abstract Human bone marrow-derived mesenchymal stem cells (hBM-MSCs) have potential clinical utility in the treatment of a multitude of ailments and diseases, due to their relative ease of isolation from patients and their capacity to form many cell types. However, hBM-MSCs are sparse, and can only be isolated in very small quantities, thereby hindering the development of clinical therapies. The use of microcarrier-based stirred suspension bioreactors to expand stem cell populations offers an approach to overcome this problem. Starting with standard culture protocols commonly reported in the literature, we have successfully developed new protocols that allow for improved expansion of hBM-MSCs in stirred suspension bioreactors using CultiSpher-S microcarriers. Cell attachment was facilitated by using intermittent bioreactor agitation, removing fetal bovine serum, modifying the stirring speed and manipulating the medium pH. By manipulating these parameters, we enhanced the cell attachment efficiency in the first 8 h post-inoculation from 18% (standard protocol) to 72% (improved protocol). Following microcarrier attachment, agitation rate was found to impact cell growth kinetics, whereas feeding had no significant effect. By serially subculturing hBM-MSCs using the new suspension bioreactor protocols, we managed to obtain cell fold increases of 10 3 within 30 days, which was superior to the 200-fold increase obtained using the standard protocol. The cells were found to retain their defining characteristics after several passages in suspension. This new bioprocess represents a more efficient approach for generating large numbers of hBM-MSCs in culture, which in turn should facilitate the development of new stem cell-based therapies. Copyright © 2012 John Wiley & Sons, Ltd. Received 13 May 2011; Revised 29 January 2012; Accepted 28 February 2012 Supporting information may be found in the online version of this article. Keywords human mesenchymal stem cells; suspension culture; spinner flask; microcarrier; cell expan- sion; bioreactor; scale-up 1. Introduction Human bone marrow-derived mesenchymal stem cells (hBM-MSCs) have been shown to differentiate to connective-tissue cell phenotypes such as bone, cartilage and fat (Pittenger et al., 1999). Studies suggest that these cells may have clinical utility in the allogeneic treatment of prevalent medical conditions, such as intervertebral disc disorders, arthritis, osteoporosis, cardiac disease and cancer, due to their multilineage differentiation potential and immunoprivileged nature (Allon et al., 2010; Tyndall and van Laar, 2010; Clines, 2010; Wen et al., 2011; Dwyer et al., 2010). The generation of new therapies using these cells is currently hindered, in part, by the limited quantities in which they can be isolated from tissue (Pittenger et al., 1999; Dominici et al., 2006). Thus, the development of robust and effective production methods to rapidly expand these cell populations to clinical quantities will *Correspondence to: A. Sen, Pharmaceutical Production Research Facility (PPRF), Schulich School of Engineering, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: [email protected] Copyright © 2012 John Wiley & Sons, Ltd. JOURNAL OF TISSUE ENGINEERING AND REGENERATIVE MEDICINE RESEARCH ARTICLE J Tissue Eng Regen Med (2012) Published online in Wiley Online Library (wileyonlinelibrary.com) DOI: 10.1002/term.1515
Transcript of Improved expansion of human bone marrow-derived mesenchymal stem cells in microcarrier-based...

Improved expansion of human bone marrow-derivedmesenchymal stem cells in microcarrier-basedsuspension cultureYifan Yuan1, Michael S. Kallos1, Christopher Hunter2 and Arindom Sen1*1Pharmaceutical Production Research Facility (PPRF), Schulich School of Engineering, University of Calgary, Calgary, Alberta, Canada2Mechanical and Manufacturing Engineering, Schulich School of Engineering, University of Calgary, Calgary, Alberta, Canada
Abstract
Human bone marrow-derived mesenchymal stem cells (hBM-MSCs) have potential clinical utility inthe treatment of a multitude of ailments and diseases, due to their relative ease of isolation frompatients and their capacity to form many cell types. However, hBM-MSCs are sparse, and can onlybe isolated in very small quantities, thereby hindering the development of clinical therapies. Theuse of microcarrier-based stirred suspension bioreactors to expand stem cell populations offers anapproach to overcome this problem. Starting with standard culture protocols commonly reportedin the literature, we have successfully developed new protocols that allow for improved expansionof hBM-MSCs in stirred suspension bioreactors using CultiSpher-S microcarriers. Cell attachmentwas facilitated by using intermittent bioreactor agitation, removing fetal bovine serum, modifyingthe stirring speed and manipulating the medium pH. By manipulating these parameters, we enhancedthe cell attachment efficiency in the first 8 h post-inoculation from 18% (standard protocol) to 72%(improved protocol). Following microcarrier attachment, agitation rate was found to impact cellgrowth kinetics, whereas feeding had no significant effect. By serially subculturing hBM-MSCs usingthe new suspension bioreactor protocols, we managed to obtain cell fold increases of 103 within30 days, which was superior to the 200-fold increase obtained using the standard protocol. Thecells were found to retain their defining characteristics after several passages in suspension. Thisnew bioprocess represents a more efficient approach for generating large numbers of hBM-MSCsin culture, which in turn should facilitate the development of new stem cell-based therapies.Copyright © 2012 John Wiley & Sons, Ltd.
Received 13 May 2011; Revised 29 January 2012; Accepted 28 February 2012
Supporting information may be found in the online version of this article.
Keywords human mesenchymal stem cells; suspension culture; spinner flask; microcarrier; cell expan-sion; bioreactor; scale-up
1. Introduction
Human bone marrow-derived mesenchymal stem cells(hBM-MSCs) have been shown to differentiate toconnective-tissue cell phenotypes such as bone, cartilageand fat (Pittenger et al., 1999). Studies suggest that these
cells may have clinical utility in the allogeneic treatmentof prevalent medical conditions, such as intervertebraldisc disorders, arthritis, osteoporosis, cardiac disease andcancer, due to their multilineage differentiation potentialand immunoprivileged nature (Allon et al., 2010; Tyndalland van Laar, 2010; Clines, 2010; Wen et al., 2011; Dwyeret al., 2010). The generation of new therapies using thesecells is currently hindered, in part, by the limited quantitiesin which they can be isolated from tissue (Pittenger et al.,1999; Dominici et al., 2006). Thus, the development ofrobust and effective production methods to rapidlyexpand these cell populations to clinical quantities will
*Correspondence to: A. Sen, Pharmaceutical ProductionResearch Facility (PPRF), Schulich School of Engineering,University of Calgary, 2500 University Drive NW, Calgary,Alberta T2N 1N4, Canada. E-mail: [email protected]
Copyright © 2012 John Wiley & Sons, Ltd.
JOURNAL OF TISSUE ENGINEERING AND REGENERATIVE MEDICINE RESEARCH ARTICLEJ Tissue Eng Regen Med (2012)Published online in Wiley Online Library (wileyonlinelibrary.com) DOI: 10.1002/term.1515

facilitate the development of new therapies. hBM-MSCshave traditionally been grown in static tissue cultureflasks (T-flasks), because these vessels are simple to use,effective for cell expansion and provide a plastic surfaceonto which these anchorage-dependent hBM-MSCs canattach in order to survive and proliferate (Dominiciet al., 2006). However, scaling-up cell production by simplyincreasing the number of T-flasks is not an effective means ofcell expansion; it can be very labour-intensive, is not amena-ble to control of many culture parameters and is subject tovariability between culture vessels. Bioreactors are scalableculture vessels in which the environment can be controlledso that important parameters, e.g. pH, oxygen tensionand nutrient levels, can be maintained at optimum levelsthroughout the expansion process.Moreover, another advan-tage over static T-flask cultures is that the mechanical envi-ronment, e.g. shear stresses, hydrodynamic pressures andmedium flow patterns, which is known to impact cell charac-teristics, can be manipulated (Huang et al., 2006). We havepreviously shown that suspension culture bioreactors canbe used to expand embryonic stem cells (Cormier et al.,2006), neural stem cells (Baghbaderani et al., 2008, 2010;Mukhida et al., 2008; McLeod et al., 2006) and cancer stemcells (Youn et al., 2006).
In order to expand anchorage-dependent hBM-MSCpopulations in suspension culture, it is necessary tosupply a surface for cell attachment. Microcarriers aresmall spherical beads (< 200 mm in diameter), whichcan be maintained in a suspended state within a stirred bio-reactor and provide a large overall surface area onto whichcells can attach. There are several types of microcarriersthat are commercially available, each with differentcharacteristics. The choice of microcarrier for a particularapplication is an important consideration, as it has beenreported that different cell types prefer onemicrocarrier typeover another. For example, several adult cell types have beenshown to prefer Cytodex 1 microcarriers (Frauenschuh et al.,2007; Malda et al., 2003; Schop et al., 2008, 2010; Ng et al.,1996),whilemouse embryonic stem cells have been reportedto grow well on Cytodex 3 microcarriers (Abranches et al.,2007; Alfred et al., 2011) and human embryonic stem cellsshowed enhanced attachment and growth on Hillex andCultiSpher microcarriers (Phillips et al., 2008). Cytodex 1and Cytodex 3 microcarriers have been shown to supporthuman MSC growth in suspension culture (Schop et al.,2010). However, these microcarriers are microporous, andthus only allow for cell growth on their external surfacearea. CultiSpher-S microcarriers, on the other hand, aremacroporous and, if the internal surface area is taken intoconsideration, provide a much larger surface area per beadfor cell attachment than an equivalent sized microporousmicrocarrier. Another potential advantage of growing cellswithin a porous microcarrier is that they are sheltered fromshear stresses present in a stirred bioreactor (Nilsson et al.,1986). Shear stress has been shown to promote cell differ-entiation (Huang et al., 2006), and protecting stem cellsfrom this stimulus may serve to encourage proliferation ofthese cells without promoting differentiation. Moreover,these particular microcarriers are produced from a highly
crosslinked gelatin matrix, which can easily be fullydissolved in 0.05% trypsin–EDTA, thereby facilitatingthe recovery of viable cells from microcarriers. For thesereasons, CultiSpher-S microcarriers may be a good choiceof microcarrier for the growth of hBM-MSCs. Eibes andcolleagues (2010) have shown that hBM-MSCs can indeedsurvive and grow within CultiSpher-S microcarriers. How-ever, they did not evaluate how inoculation and growthconditions affect the eventual overall cell yield. In thepresent study, we have evaluated whether manipulationof key variables can improve overall yields of hBM-MSCson CultiSpher-S microcarriers.
The expansion of adherent cells in suspension culturecan be divided into two distinct phases. The first phaseis called the inoculation phase and is a period of timeduring which cells adhere to microcarriers and spread out.The second phase is called the proliferation phase, duringwhich attached cells actively divide on the microcarriers.The attachment efficiency of anchorage-dependent cells tomicrocarriers during the inoculation phase has been foundto have a significant effect on subsequent cell yield duringthe proliferation phase (Forestell et al., 1992). Starting withsuspension culture protocols that have been reported tosupport cell attachment to microcarriers and expansion ofcells on microcarriers (Schop et al., 2008, 2010; Forestellet al., 1992), we investigated the impact of several para-meters on the overall attachment efficiency and subsequentexpansion of hBM-MSCs on CultiSpher-S microcarriers.Inoculation phase variables that were investigated includestirring regimen (intermittent vs continuous agitation),agitation rate, serum level, cell inoculation (cell:bead ratio)and pH level. We also tested different agitation rates andthe effect of feeding during the proliferation phase.We havedetermined values for the tested parameters which allowfor significantly greater cell yields than those previouslyreported in the literature for the culture of hBM-MSCs onCultiSpher-S microcarriers.
2. Methods and materials
2.1. Static culture cultivation of hBM-MSCs
Cells from two donors were used in this study. Human bonemarrow-derived mononuclear cells were obtained commer-cially (Lonza Inc., Switzerland, Cat. Nos.1423C and 2422B)and cultured at an inoculation density of 5000 cells/cm2 in75 cm2 Nunc tissue culture flasks (T-75), with a mediumcomposed of 10% fetal bovine serum (FBS; Invitrogen,Burlington, Canada, Cat. No.12483) in Dulbecco’s modifiedEagle’s medium (DMEM; Invitrogen, Burlington, Canada,Cat. No. 31600034). This medium will be referred to as10% FBS/DMEM. Cells were subcultured every 5 days. Onday 5 post-inoculation, cells were detached from thesurface of T-flasks (Nunc, Rochester, NY, Cat. No. 156499)using 4.0 ml 0.05% trypsin–EDTA (Invitrogen, Cat. No.25300–120) for each T-75 flask. Cells were enumeratedon a haemocytometer (VWR, Mississauga, Canada, Cat.
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

No.1483), using 0.1% trypan blue (Sigma,Oakville, Canada,Cat. No. T8154) exclusion. hBM-MSCs were inoculatedinto new T-flasks containing fresh 10% FBS/DMEMmedium at a density of 5000 cells/cm2, and the T-flaskswere placed in a humidified incubator (37�C, 5% CO2 inair). All institutional biosafety guidelines were adheredto for this study.
2.2. Microcarrier preparation
A 0.125 g sample of dry CultiSpher-S (Sigma, Cat. No.M9043-10G) microcarriers (equivalent to 0.1 millionCultiSpher-S beads) was hydrated for 3 h in a Pyrex flask,using 50 ml 1� PBS (Invitrogen, Cat. No. 14200–075).The flasks were coated with Sigmacote (Sigma, Cat. No.SL2-100ml) to minimize microcarrier attachment to theinner walls of the flasks. The hydrated microcarriers wereautoclaved and stored at 4�C for later use. Autoclavedmicrocarriers were washed twice with sterile 1� PBS,followed once by inoculation medium before being addedto the spinner flasks. The medium used was dependentupon the experiment being conducted.
2.3. Spinner flask culture
Spinner flask bioreactors (125 ml; NDS Technologies, NJ,USA) were coated with the silicone product Sigmacoteand autoclaved. Washed microcarriers (0.1 million/spinnerflask) were aseptically added to each spinner flask, alongwith 58ml of inoculation medium. Whereas the normalworking volume of these reactors is 125 ml, this lowmedium volume was used during the inoculation phaseto increase the probability of a collision between cells andmicrocarriers. Unless otherwise specified, the mediumused for all experiments was 10% FBS/DMEM. In the eventthat the medium was modified during a particularexperiment (e.g. to evaluate the impact of FBS levelon cell attachment), the modification will be clearlydescribed when presenting the results of that experi-ment. A suspension of single hBM-MSCs (cell numbervaried with experiment) was then carefully added toeach spinner flask using a sterile pipette. Each inoculatedspinner flask was placed on a magnetic stir plate in ahumidified incubator (37�C, 5% CO2) to allow the cellsto attach. The appropriate agitation rate during theinoculation phase was a parameter that was investigatedas part of our studies (described in Section 3.1.1). Duringthis phase, the goal was to maximize cell attachment.The appropriate duration of the inoculation phase wasalso determined (see Section 3.2). At the end of theinoculation phase, sterile medium was added to theflask to a final working volume of 125 ml (10% FBS).During the proliferation phase, which followed theinoculation phase, the effect of agitation rate was againinvestigated. During this phase, the goal was to maxi-mize cell proliferation.
2.4. Spinner flask sampling
The contents of each spinner flask were periodicallysampled by removing two representative 2.0 ml samples,using a sterile pipette. This medium was not replaced asthis would cause the bead concentration to vary andnutrient levels to fluctuate. Each 2.0 ml sample was pouredinto a 70 mm cell strainer (BD Falcon, Mississauga, Canada,Cat. No. 352350) to separate the liquid and non-attachedcell fraction (which passed through the filter into a 50 mlconical tube) from the microcarriers. The microcarriers re-tained in the cell strainer were rinsed twice with 1� PBS andthen placed in a well-plate, where they were digestedover the course of 8 min using 2.0 ml 0.05% trypsin–EDTA in order to recover the attached cells. The cell sus-pension was then centrifuged, the supernatant removedand the cells rinsed in 1� PBS. We have previouslydetermined that cells liberated in this manner throughtransient exposure to trypsin do not exhibit any detrimen-tal effects (data not shown). Enumeration of cells derivedfrom CultiSpher-S microcarriers was carried out by 0.1%trypan blue exclusion, using a haemocytometer. Allexperiments involving spinner flasks were conducted induplicate. Two samples were taken from each of the twospinner flasks at designated time points, and each samplewas counted in duplicate.
2.5. Microcarrier cultivation of hBM-MSCs:serial passaging
Every 6 days, the spinner flasks were taken from theincubator and placed in a biosafety cabinet, where themicrocarriers were allowed to settle for 1–2 min. Super-natant (the upper 100 ml of medium in the vessel) wasremoved carefully from each spinner flask by pipette. Theremaining 25 ml medium (containing the settled microcar-riers) was then collected and passed through a 70 mm cellstrainer into a 50 ml conical tube. The microcarriers onthe cell strainer were washed with 1� PBS and thenplaced in a different 50 ml conical tube. hBM-MSCsexpanded on the microcarriers were recovered by using20.0 ml 0.05% trypsin–EDTA to digest the beads for8 min. The cell number and viability were microscopicallydetermined on a haemocytometer, using trypan blueexclusion, and cells were inoculated as a single-cellsuspension into a new spinner flask containing 125 ml freshmedium and 0.1 million new CultiSpher-S microcarriers (cellinoculation level depended on the specific experiment).
2.6. Measurement of glucose, glutamine, lacticacid and ammonia
The glucose, glutamine, lactic acid and ammonia concen-trations of fresh and spent culture medium were measuredusing a Nova BioProfile 100 Analyser (Nova Biomedical,Ontario, Canada). This instrument was operated andcalibrated according to the manufacturer’s instructions.
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

Prior to analysis, medium samples were centrifuged at600� g for 5 min to remove any cells and the superna-tant was stored at �20�C. After thawing, samples ofthawed medium (0.7 ml) were analysed. All feedingregimen experiments requiring nutrient and waste productanalyses were conducted in duplicate, and each replicatewas analysed twice.
2.7. Medium replacement
Medium replacement was carried out by removing aspinner flask from the incubator, placing it in a biosafetycabinet, and allowing the microcarriers to settle to thebottom of that flask for a period of one minute. An appro-priate amount of medium was removed (see Results forexact volumes, as the amount removed varied), using a pi-pette from the upper volume of the flask contents (so asnot to remove any microcarriers), and replaced by anequal volume of pre-warmed fresh medium. The spinnerflask was then placed back into the incubator.
2.8. Cell characterization – flow cytometryanalysis
The cell surface markers on hBM-MSCs grown in static orsuspension culture were analysed using a FACSCaliber(Becton-Dickinson, Mississauga, Canada, Cat. No.E02300006). The FACSCaliber was calibrated using cali-brite beads (BD Biosciences, Mississauga, Canada, Cat.No. 340486). For each analysis, 5�105 single cells wereplaced into a 15 ml centrifuge tube, washed three timeswith 10.0 ml of 1� PBS (centrifuged at 300� g for 5 minbetweenwashes) and then resuspended in 10.0ml blockingsolution (3% FBS in 1� PBS). After being incubated in thedark on ice for 30 min, the sample was centrifuged at300� g for 5 min. For each marker being tested, 0.1 mlcells in blocking solution (3% FBS/PBS) was placed intoseparate 15 ml centrifuge tubes. Next, 5 ml CD105, CD90,CD73, CD34 antibodies or isotype control (IgG1 and IgG1k)antibodies (Table 1) was added to each tube. The sampleswere incubated in the dark on ice for 30 min. After beingwashed three times with 1� PBS, the samples were ana-lysed. The data were analysed by FACSComp software(BD Biosciences).
2.9. Characterization – multilineagedifferentiation
2.9.1. Osteoblastic differentiation
Osteoblastic differentiation of hBM-MSCs was induced insix-well plates. After harvesting cells from spinner flasks,each well was inoculated with 3.0�104 cells (3.1�103
cells/cm2) in 2.5 ml culture medium. Cells were allowedto attach to the tissue culture surface for 24 h at 37�Cand 5% CO2. After 24 h, the culture medium was replacedwith osteoblastic induction medium (OIM), whichconsisted of culture medium supplemented with 10 mM
b-glycerophosphate (Calbiochem, NJ, USA, Cat. No.35675), 0.1 mM dexamethasone (Sigma, Cat. No. D2915)and 0.05 mM ascorbate-2-phosphate (Sigma, Cat. No.A4544). The samples were cultured for 13 days in OIM.Control samples were cultured in 10% FBS/DMEM. Acomplete medium change was performed twice weekly forall wells.
Osteoblastic differentiation was identified throughstaining with alizarin red, which identifies regions ofcalcium deposition. Osteoblastic induction medium wasdiscarded from each well and the cells rinsed twice with2.0ml/well of 1� PBS. The cells were then fixed in a fumehood at room temperature, using 2.0ml/well of 10% buff-ered formalin (Fisher Scientific, Ottawa, Canada, Cat. No.SF100-4). After 1 h, the formalin was discarded and thecells were washed twice with 2.0ml of double-distilled H2O(ddH2O). Alizarin red solution (2.0ml/well) was then addedto the cells. Alizarin red solution was prepared by adding1.0g alizarin red powder (Sigma, Cat. No. A5533-25G) to100 ml ddH2O. The pH was adjusted to 4.1–4.3 usingammonium hydroxide(Fisher Scientific, Ottawa, Canada,Cat. No. A669). The solution was filtered using a 0.2 mmfilter and stored at room temperature until ready to use.After being incubated in alizarin red at room temperaturefor 20 min, the stain was discarded and the cells werewashed with 2.0ml of ddH2O. The washes were repeateduntil all excess stain had been removed. The cells were storedin 2.0ml of ddH2O and analysed under the microscope forevidence of calcium deposition.
2.9.2. Adipogenic differentiation
Adipogenesis was induced in six-well plates. After harvestingcells from spinner flasks, each well was inoculated with2.0�105 cells/well (2.1�104cells/cm2) in 2.0ml of culturemedium. The cells were allowed to reach confluence, atwhich time the medium was replaced with 2.0ml of adipo-genic induction medium (AIM), which consisted of cul-ture medium with 0.5 mM isobutylmethylxanthine(Sigma, Cat. No. I5879), 0.5 mM dexamethasone and 0.1mM indomethacin (Sigma, Cat. No. I7373). The cells werecultured in AIM for 19 days at 37�C and 5% CO2. Controlsamples were cultured in 10% FBS/DMEM. A complete me-dium change was performed twice weekly for all wells.
Adipogenic differentiation was identified by stainingwith oil red O (Sigma, Cat. No. O0625-25G) which stains
Table 1. Antibodies used for immunofluorescence staining ofhBM-MSCs in flow cytometry analysis
Antibody Manufacturer Cat. No.
Mouse anti-human CD90-IgG1-FITC SeroTec MCA90FMouse anti-human CD105-IgG1-FITC SeroTec MCA1557FMouse anti-human CD34-IgG1k-PE BD Biosciences 550822Mouse anti-human CD73-IgG1k-PE BD Biosciences 550257Mouse IgG1 negative control-FITC SeroTec MCA928FMouse IgG1k isotype-PE BD Biosciences 555749
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

intracellular lipids. Upon completion of adipogenic dif-ferentiation, the medium in each well was removed andthe cells rinsed with 2.0 ml of 1� PBS. Next, 2.0ml of10% buffered formalin was added in a fume hood andthe cells were incubated at room temperature for 1 h.The formalin was discarded and the cells were washed with2.0 ml of ddH2O. Next, 2.0 ml of 60% isopropanol wasadded and the samples were incubated at room temperaturefor 2–5 min, after which the isopropanol was discarded.Then 2.0 ml of oil red O working solution was added andthe sample was incubated at room temperature. After5 min, the oil red O was discarded and the samples werewashed with 2.0ml of ddH2O. The washes were repeateduntil all excess stain had been removed. The cells werestored in 2.0ml of ddH2O and analysed under the micro-scope for evidence of intracellular lipids.
2.10. Cell labelling
In order to facilitate the visualization of cells on microcar-riers, the cells were stained with the lipophilic dye DiI(Invitrogen, Cat. No. V22888). Briefly, DiI stain wasadded to a hBM-MSC suspension (106 cells/ml) at a levelof 7 ml/ml, the cell suspension was gently mixed with apipette to distribute the dye and the cells were incubatedfor 30 min at 37�C and 5% CO2. The samples were thencentrifuged (200� g for 5 min), the supernatant removedand the cells gently resuspended in 5.0 ml warm (37�C)medium. This washing procedure was repeated twicemore to remove any residual dye.
2.11. Statistical analysis
The values shown graphically in figures are mean� rangeof data. A one-way analysis of variance (ANOVA) wasused to examine various conditions for cell expansion.Statistical analysis was performed using GraphPad Prism.A significance level of p< 0.05 was selected.
3. Results
3.1. Microcarrier cultivation of hBM-MSCs –inoculation phase
3.1.1. Intermittent agitation vs continuous agitation
We first investigated the effect of continuous agitation(60 rpm) or intermittent agitation (repeated cycles of3 min at 60 rpm followed by 27 min without stirring)during the first 24 h of bioreactor culture. After preparingthe spinner flasks, hBM-MSCs harvested from tissueculture flasks were inoculated into each spinner flask at acell:bead ratio of 9:1 with 0.001 dry g/ml prepared micro-carriers (refers to final concentration in 125 ml of medium)and incubated at 37�C and 5% CO2.. Since CultiSpher-Smicrocarriers have a surface area of 15 000 cm2/g, this
translated to a cell inoculation density of 480 cells/cm2.Spinner flasks were placed on a magnetic stir plate for24 h with intermittent agitation or continuous agitation.As shown in Figure 1A, over the course of 24 h an aver-age cell attachment efficiency of 55% was achieved usingintermittent agitation compared to only 34% for continu-ous agitation. The attachment efficiency was defined asthe percentage of inoculated cells that attached to themicrocarriers. It was clear to see that there weresignificantly fewer unattached cells in suspension withintermittent agitation than with continuous agitation(Figure 1B, C). It should be noted that under bothstirring protocols, the viability of the attached cellsexceeded 98%. We also investigated the effect of chang-ing the agitation rate from 60 rpm to 100 rpm, and foundno difference in the final attachment efficiency after24 h (Figure 1D). However, at 100 rpm, the cellsappeared to reach maximum cell attachment efficiency(37% at 4 h) faster than at 60 rpm (24% at 4 h). Moreover,at this higher agitation rate, the microcarriers had lessof a tendency to aggregate together. Therefore, anagitation rate of 100 rpm was used for all subsequentexperiments.
3.1.2. Fetal bovine serum concentration
The impact of FBS concentration during the inoculationphase was evaluated. hBM-MSCs were harvested fromstatic tissue culture vessels and inoculated at a densityof 480 cells/cm2 into spinner flasks containing microcar-riers (0.001 g/ml). Each spinner flask contained DMEMsupplemented with 0%, 5% or 10% FBS. The spinnerflasks were placed on magnetic stir plates (intermittentagitation at 100 rpm) and incubated at 37�C and 5%CO2 for 24 h. It was found that the absence of serum(0% FBS) significantly increased attachment efficiencycompared to 5%and10%FBSover thefirst 12h (Figure 1E).This effect was especially pronounced over the first4 h post-inoculation, during which the attachmentefficiency was 63% for 0% FBS vs 37% for 5% and10% FBS. The viability of the cells attached to micro-carriers was above 98% in all cases throughout theexperiment.
3.1.3. Cell inoculation (cell:bead ratio)
The impact of initial cell:bead ratio on cell attachmentefficiency was evaluated. hBM-MSCs were harvested fromstatic tissue culture vessels and inoculated into eachspinner flask with 0.001 g/ml beads in 125 ml serum-freeDMEM. Cell:bead ratios of 5:1, 9:1 and 60:1 (267, 480and 3200 cells/cm2, respectively) were evaluated induplicate. The spinner flasks were placed on stir plates(intermittent agitation) and incubated at 37�C and 5%CO2 for 12 h. A shorter attachment period was evaluatedin this case as higher attachment efficiencies were notedup to 12 h in our previous experiment (Figure 1E). Asshown in Figure 1F, there was no difference in cellattachment efficiency between the different cell:bead
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term
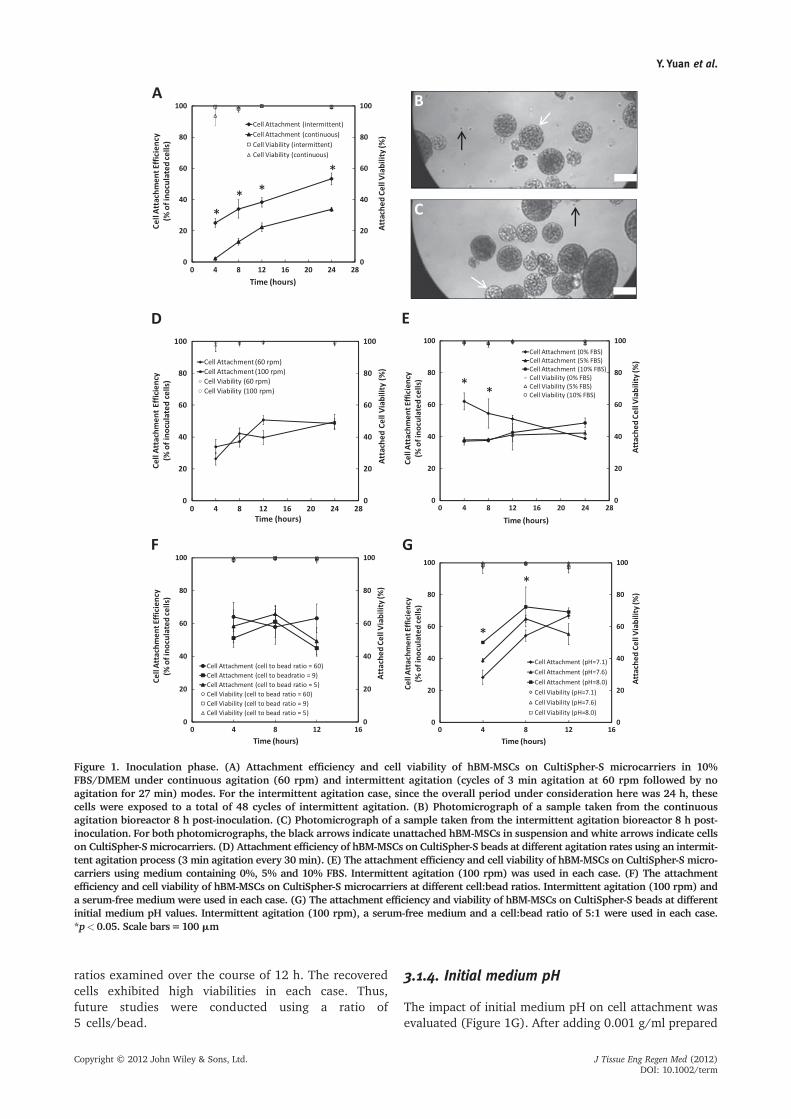
ratios examined over the course of 12 h. The recoveredcells exhibited high viabilities in each case. Thus,future studies were conducted using a ratio of5 cells/bead.
3.1.4. Initial medium pH
The impact of initial medium pH on cell attachment wasevaluated (Figure 1G). After adding 0.001 g/ml prepared
Figure 1. Inoculation phase. (A) Attachment efficiency and cell viability of hBM-MSCs on CultiSpher-S microcarriers in 10%FBS/DMEM under continuous agitation (60 rpm) and intermittent agitation (cycles of 3 min agitation at 60 rpm followed by noagitation for 27 min) modes. For the intermittent agitation case, since the overall period under consideration here was 24 h, thesecells were exposed to a total of 48 cycles of intermittent agitation. (B) Photomicrograph of a sample taken from the continuousagitation bioreactor 8 h post-inoculation. (C) Photomicrograph of a sample taken from the intermittent agitation bioreactor 8 h post-inoculation. For both photomicrographs, the black arrows indicate unattached hBM-MSCs in suspension and white arrows indicate cellson CultiSpher-S microcarriers. (D) Attachment efficiency of hBM-MSCs on CultiSpher-S beads at different agitation rates using an intermit-tent agitation process (3 min agitation every 30 min). (E) The attachment efficiency and cell viability of hBM-MSCs on CultiSpher-S micro-carriers using medium containing 0%, 5% and 10% FBS. Intermittent agitation (100 rpm) was used in each case. (F) The attachmentefficiency and cell viability of hBM-MSCs on CultiSpher-S microcarriers at different cell:bead ratios. Intermittent agitation (100 rpm) anda serum-free medium were used in each case. (G) The attachment efficiency and viability of hBM-MSCs on CultiSpher-S beads at differentinitial medium pH values. Intermittent agitation (100 rpm), a serum-free medium and a cell:bead ratio of 5:1 were used in each case.*p<0.05. Scale bars=100 mm
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term
microcarriers into each spinner flask, hBM-MSCs wereharvested from static tissue culture vessels, and the cellswere inoculated into each spinner flask at a cell:bead ratioof 5:1. The serum-free medium added to a particularspinner flask had the initial pH adjusted to 7.1, 7.6 or 8.0by adding varying ratios of 1.0 M stock solution of 4-(2-hydroxyethyl)-1-piperazine-ethanesulphonic acid (HEPES;Sigma-Aldrich, Cat. No. H4034, Lot No. 095K5410) andsodium bicarbonate (Sigma-Aldrich, Cat. No. S5761, LotNo. 035K0074). The osmolality change due to this additionof these buffers was negligible (data not shown). Themedia were allowed to equilibrate for 2–4 h in the incuba-tor before cells were added and each pH was evaluated induplicate. The spinner flasks were placed on magnetic stirplates (intermittent agitation) and incubated at 37�C and5% CO2. Over the first 8 h post-inoculation, a higherattachment efficiency was found at pH 8.0 (72% at 8 h)compared to media with lower pH values of 7.1 and 7.6(54% and 61% at 8 h, respectively). The cell viability atthe different pH levels was always maintained above 98%over 12 h. Thus, an initial medium pH of 8.0 was used inall subsequent studies.
3.2. Inoculation phase: new conditions vsoriginal conditions
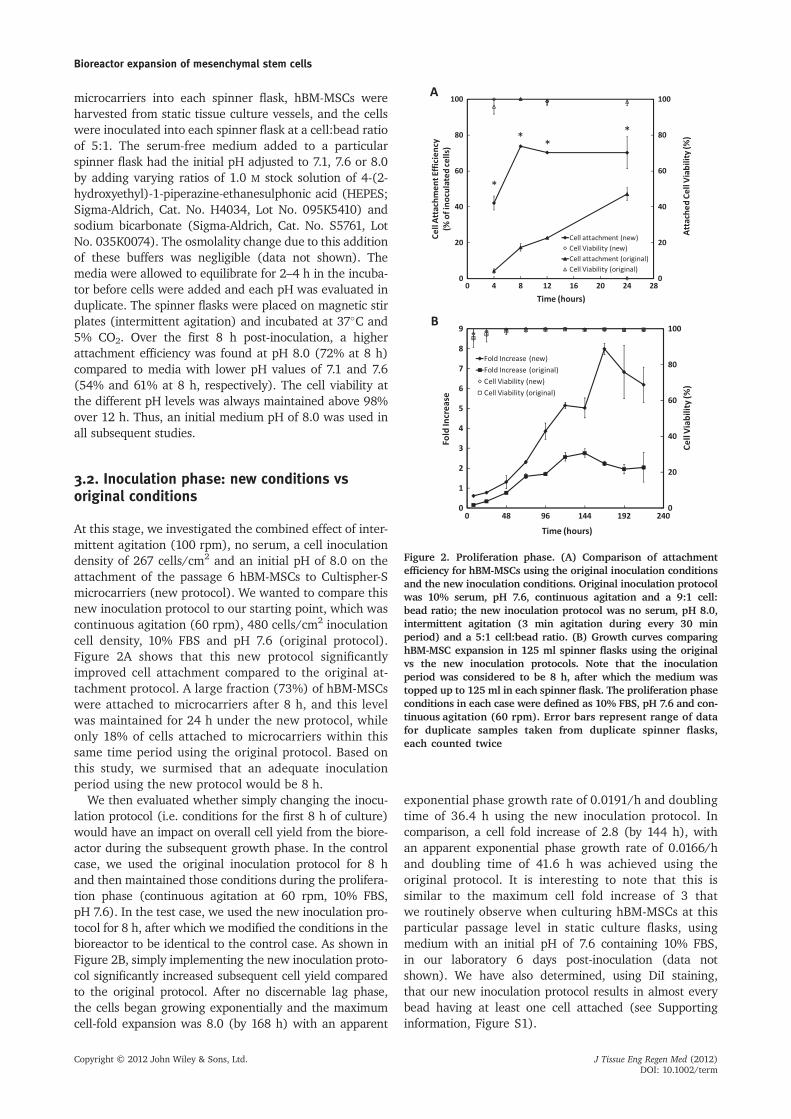
At this stage, we investigated the combined effect of inter-mittent agitation (100 rpm), no serum, a cell inoculationdensity of 267 cells/cm2 and an initial pH of 8.0 on theattachment of the passage 6 hBM-MSCs to Cultispher-Smicrocarriers (new protocol). We wanted to compare thisnew inoculation protocol to our starting point, which wascontinuous agitation (60 rpm), 480 cells/cm2 inoculationcell density, 10% FBS and pH 7.6 (original protocol).Figure 2A shows that this new protocol significantlyimproved cell attachment compared to the original at-tachment protocol. A large fraction (73%) of hBM-MSCswere attached to microcarriers after 8 h, and this levelwas maintained for 24 h under the new protocol, whileonly 18% of cells attached to microcarriers within thissame time period using the original protocol. Based onthis study, we surmised that an adequate inoculationperiod using the new protocol would be 8 h.
We then evaluated whether simply changing the inocu-lation protocol (i.e. conditions for the first 8 h of culture)would have an impact on overall cell yield from the biore-actor during the subsequent growth phase. In the controlcase, we used the original inoculation protocol for 8 hand then maintained those conditions during the prolifera-tion phase (continuous agitation at 60 rpm, 10% FBS,pH 7.6). In the test case, we used the new inoculation pro-tocol for 8 h, after which we modified the conditions in thebioreactor to be identical to the control case. As shown inFigure 2B, simply implementing the new inoculation proto-col significantly increased subsequent cell yield comparedto the original protocol. After no discernable lag phase,the cells began growing exponentially and the maximumcell-fold expansion was 8.0 (by 168 h) with an apparent
exponential phase growth rate of 0.0191/h and doublingtime of 36.4 h using the new inoculation protocol. Incomparison, a cell fold increase of 2.8 (by 144 h), withan apparent exponential phase growth rate of 0.0166/hand doubling time of 41.6 h was achieved using theoriginal protocol. It is interesting to note that this issimilar to the maximum cell fold increase of 3 thatwe routinely observe when culturing hBM-MSCs at thisparticular passage level in static culture flasks, usingmedium with an initial pH of 7.6 containing 10% FBS,in our laboratory 6 days post-inoculation (data notshown). We have also determined, using DiI staining,that our new inoculation protocol results in almost everybead having at least one cell attached (see Supportinginformation, Figure S1).
Figure 2. Proliferation phase. (A) Comparison of attachmentefficiency for hBM-MSCs using the original inoculation conditionsand the new inoculation conditions. Original inoculation protocolwas 10% serum, pH 7.6, continuous agitation and a 9:1 cell:bead ratio; the new inoculation protocol was no serum, pH 8.0,intermittent agitation (3 min agitation during every 30 minperiod) and a 5:1 cell:bead ratio. (B) Growth curves comparinghBM-MSC expansion in 125 ml spinner flasks using the originalvs the new inoculation protocols. Note that the inoculationperiod was considered to be 8 h, after which the medium wastopped up to 125 ml in each spinner flask. The proliferation phaseconditions in each case were defined as 10% FBS, pH 7.6 and con-tinuous agitation (60 rpm). Error bars represent range of datafor duplicate samples taken from duplicate spinner flasks,each counted twice
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term
3.3. Microcarrier cultivation of hBM-MSCs –
proliferation phase
Once proper conditions for cell attachment were identi-fied (intermittent agitation at 100 rpm, no serum, a cellinoculation density of 267 cells/cm2 and an initial pH of8.0), they were used during the inoculation phase for allsubsequent studies that focused on the proliferation phase(defined as the culture period after the 8 h inoculationperiod). Following the inoculation phase, the volume ofmedium in the spinner flask was always topped up to125 ml. The initial pH at the beginning of the proliferationphase was always 7.6, and medium used during this phasecontained 10% serum in all cases.
3.3.1. Agitation rate
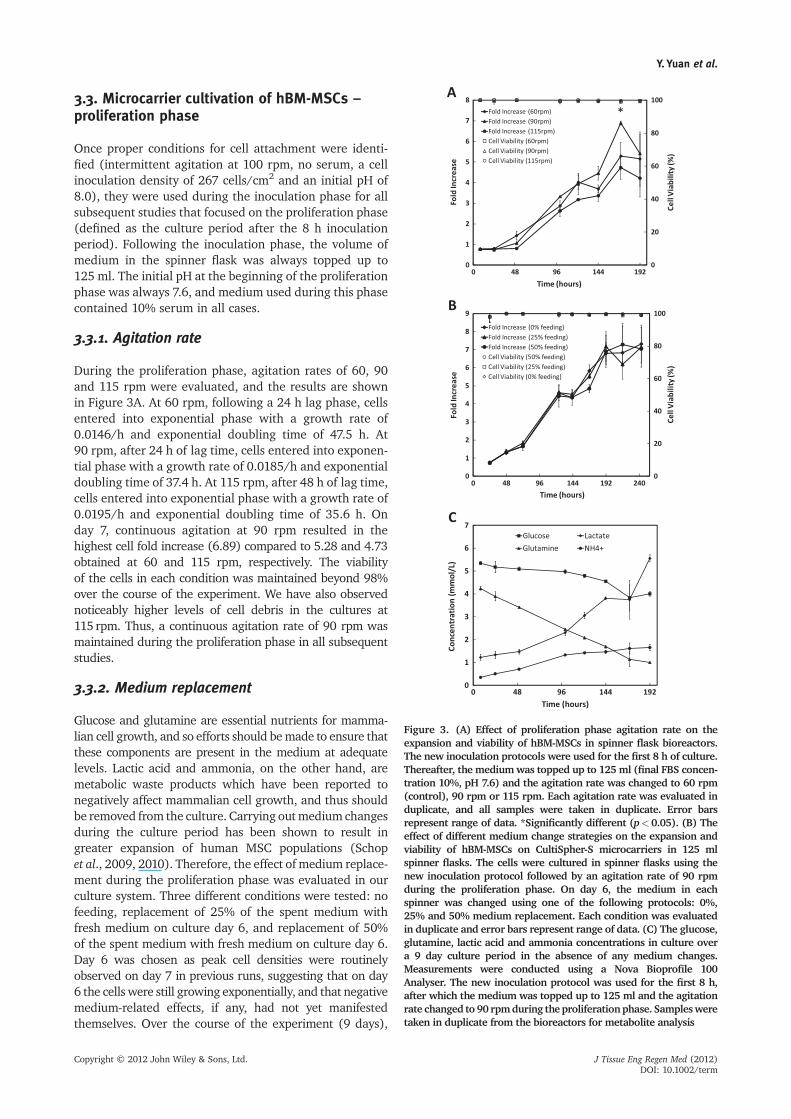
During the proliferation phase, agitation rates of 60, 90and 115 rpm were evaluated, and the results are shownin Figure 3A. At 60 rpm, following a 24 h lag phase, cellsentered into exponential phase with a growth rate of0.0146/h and exponential doubling time of 47.5 h. At90 rpm, after 24 h of lag time, cells entered into exponen-tial phase with a growth rate of 0.0185/h and exponentialdoubling time of 37.4 h. At 115 rpm, after 48 h of lag time,cells entered into exponential phase with a growth rate of0.0195/h and exponential doubling time of 35.6 h. Onday 7, continuous agitation at 90 rpm resulted in thehighest cell fold increase (6.89) compared to 5.28 and 4.73obtained at 60 and 115 rpm, respectively. The viabilityof the cells in each condition was maintained beyond 98%over the course of the experiment. We have also observednoticeably higher levels of cell debris in the cultures at115 rpm. Thus, a continuous agitation rate of 90 rpm wasmaintained during the proliferation phase in all subsequentstudies.
3.3.2. Medium replacement
Glucose and glutamine are essential nutrients for mamma-lian cell growth, and so efforts should bemade to ensure thatthese components are present in the medium at adequatelevels. Lactic acid and ammonia, on the other hand, aremetabolic waste products which have been reported tonegatively affect mammalian cell growth, and thus shouldbe removed from the culture. Carrying outmedium changesduring the culture period has been shown to result ingreater expansion of human MSC populations (Schopet al., 2009, 2010). Therefore, the effect of medium replace-ment during the proliferation phase was evaluated in ourculture system. Three different conditions were tested: nofeeding, replacement of 25% of the spent medium withfresh medium on culture day 6, and replacement of 50%of the spent medium with fresh medium on culture day 6.Day 6 was chosen as peak cell densities were routinelyobserved on day 7 in previous runs, suggesting that on day6 the cells were still growing exponentially, and that negativemedium-related effects, if any, had not yet manifestedthemselves. Over the course of the experiment (9 days),
Figure 3. (A) Effect of proliferation phase agitation rate on theexpansion and viability of hBM-MSCs in spinner flask bioreactors.The new inoculation protocols were used for the first 8 h of culture.Thereafter, the mediumwas topped up to 125 ml (final FBS concen-tration 10%, pH 7.6) and the agitation rate was changed to 60 rpm(control), 90 rpm or 115 rpm. Each agitation rate was evaluated induplicate, and all samples were taken in duplicate. Error barsrepresent range of data. *Significantly different (p<0.05). (B) Theeffect of different medium change strategies on the expansion andviability of hBM-MSCs on CultiSpher-S microcarriers in 125 mlspinner flasks. The cells were cultured in spinner flasks using thenew inoculation protocol followed by an agitation rate of 90 rpmduring the proliferation phase. On day 6, the medium in eachspinner was changed using one of the following protocols: 0%,25% and 50% medium replacement. Each condition was evaluatedin duplicate and error bars represent range of data. (C) The glucose,glutamine, lactic acid and ammonia concentrations in culture overa 9 day culture period in the absence of any medium changes.Measurements were conducted using a Nova Bioprofile 100Analyser. The new inoculation protocol was used for the first 8 h,after which the medium was topped up to 125 ml and the agitationrate changed to 90 rpmduring theproliferationphase. Samplesweretaken in duplicate from the bioreactors for metabolite analysis
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

there was no difference in the cell growth kinetics for thedifferent feeding regimens (Figure 3B). The concentrationsof the nutrients and wastes were tracked for the bioreactorswith 0% medium change. The glucose concentrationdecreased from 5.34 mM (8 h) to 4.00 mM (192 h), whilethe concentration of glutamine decreased from 4.11 mM to0.99 mM over the same time period. The concentration oflactate increased from 1.22 mM to 5.55 mM and NH4
+ con-centration increased from 0.35mM to 1.65 mM (Figure 3C).Both of these levels have previously been shown to benon-toxic to hBM-MSCs (Schop et al., 2009).
3.4. Serial subculture – new inoculation andproliferation conditions
The new inoculation phase conditions (intermittentagitation, 267 cells/cm2 inoculation cell density, pH 8.0and 0% FBS) and new proliferation phase conditions(agitation rate of 90 rpm) were combined (Table 2) toevaluate how they compared to the original cultureconditions during the serial passages of hBM-MSCs in spin-ner flask bioreactors. As shown in Figure 4, the cumulativefold increase in cell number using the new protocol wassignificantly higher than the increase obtained over thesame time period using the original culture conditions forhBM-MSCs isolated from two separate donors. Cumulativefold increase refers to the fold increase in cell number thatwould have been obtained over the course of multiplepassages if all cells were serially subcultured and nonewere discarded. It was calculated by multiplying togetherthe measured cell fold increase measured at each passagelevel. For donor 1, the cell fold increase was approximately1098 over five passages using the new conditions vs 176using the original conditions (Figure 4A). Similarly, fordonor 2, the cumulative cell fold increase was approxi-mately 934 under the new conditions vs 162 usingthe original conditions (Figure 4D). The fold increaseduring every passage under the new culture conditionswas higher than under the original conditions (Figure 4B,E) and the average doubling times in each passage using thenew protocols were shorter than observed under the originalprotocols (Figure 4C, F).
3.5. Characterization of multilineagedifferentiation potential
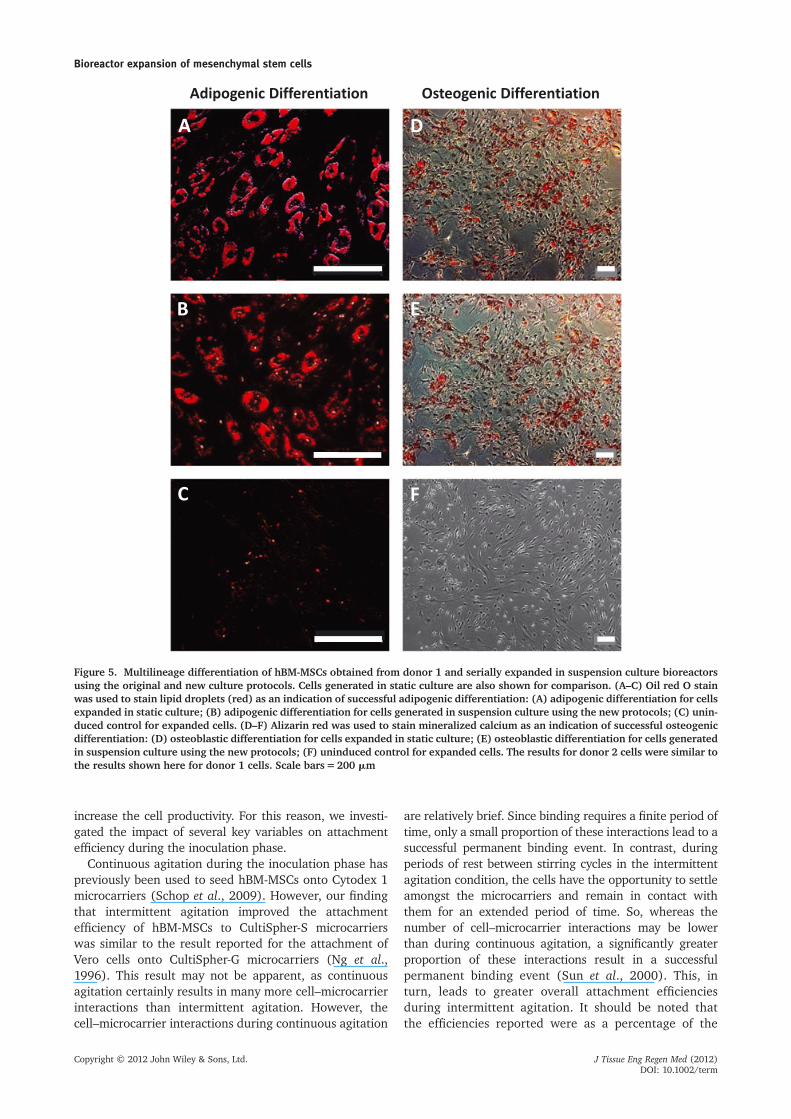
The differentiation potential of hBM-MSCs seriallyexpanded in spinner flask bioreactors using the newculture conditions was evaluated for cells isolated fromtwo donors (donor 1 for five passages, donor 2 for fourpassages). The cells were harvested after passaging andinoculated into six-well plates. Since we wanted toensure that expansion in suspension culture did notadversely affect the multilineage differentiation capacityof the hBM-MSCs, cells grown under static conditions intissue culture flasks were also inoculated into six-wellplates to serve as a control. Donor 1 cells expandedunder static or suspension conditions were at passagelevel 11 at the time of these differentiation assays,whereas the cells from donor 2 were at passage level 8.Adipogenic and osteogenic differentiation was performedfor all of these cells. After 19 days of exposure to adipo-genic induction medium, the cells expanded in staticculture as well as in spinner flasks were observed tocontain intracellular lipid droplets, which stained positivelywith oil red O (Figure 5A, B). There was no red stain foundin uninduced groups, which were treated with 10%FBS/DMEM instead of adipogenic induction medium(Figure 5C).
For osteoblastic differentiation, after 13 days oftreatment with osteoblastic differentiation medium,the cells expanded in both static and suspensioncultures were shown to stain positive for alizarin red,suggesting that some of these cells maintained anability to deposit calcium, thereby indicating an osteogeniccapacity (Figure 5D, E). Uninduced groups treatedwith 10% FBS DMEM and not exposed to osteoblasticinduction medium did not stain positive for alizarin red(Figure 5F).
3.6. Characterization of surface markers
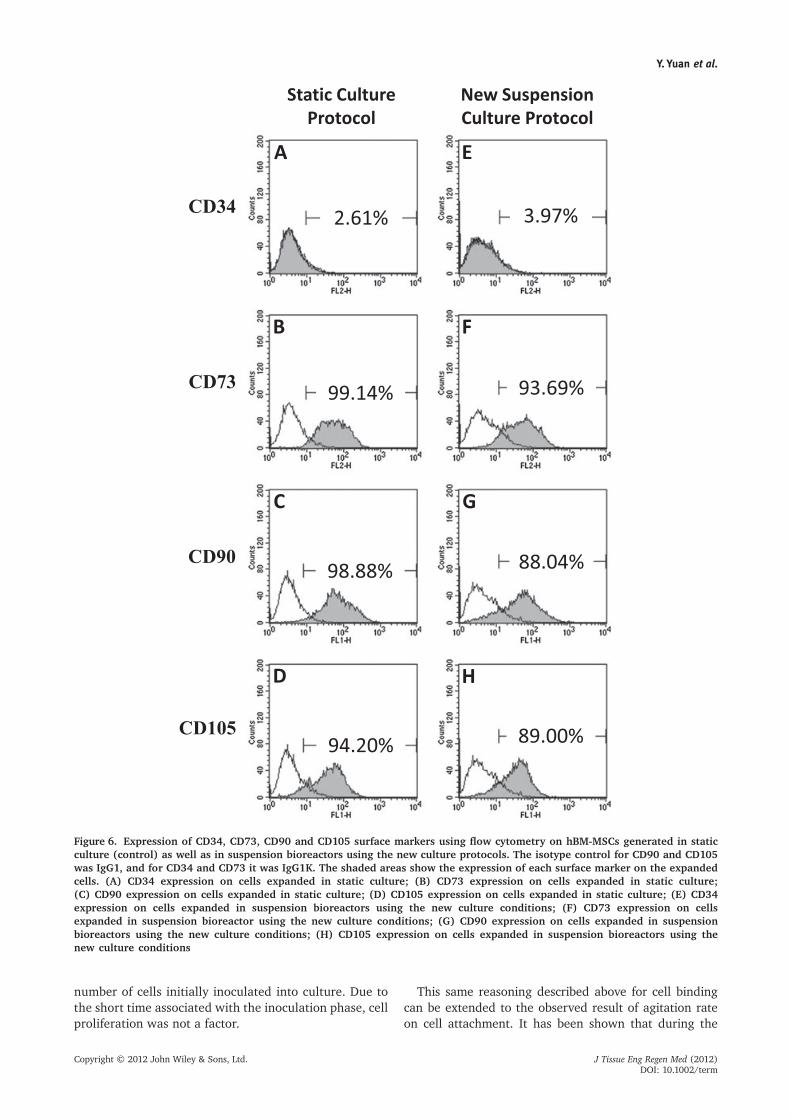
hBM-MSCs serially expanded in spinner flasks (newsuspension culture protocol) were harvested after fourpassages to evaluate surface marker expression. Cells
Table 2. A summary of the new culture conditions developed for the expansion of hBM-MSCs in suspension culture usingCultiSpher-S microcarriers.
Parameter
New culture conditions
New inoculation conditions (0–8 h)New proliferation conditions
(from 8 h until harvest)
Cell : bead ratio 5:1 5:1Agitation rate 100 rpm 90 rpmAgitation type Intermittent (repeated cycles of 3 min of agitation
followed by 27 min without agitation)Continuous
Serum (FBS) level 0% 10%pH 8.0 7.6
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

grown in static culture were used as controls. We foundthat the post-expansion hBM-MSC populations fromsuspension culture were positive for CD90 (88.04%),CD105 (89.00%) and CD73 (93.69%) but negativefor CD34 (3.97%). These results are similar to thoseobtained for the cells expanded in static culture, whichwere positive for CD90 (98.88%), CD105 (94.20%)and CD73 (99.14) but negative for CD34 (2.61%)(Figure 6).
4. DiscussionThe importance of the inoculation phase on overall cellyield from a microcarrier-based suspension bioreactor wasdemonstrated by Forestell and colleagues (1992). Theyshowed that simply manipulating serum concentration,pH level, cell inoculation (cell:bead ratio) and initial agita-tion rate could lead to> 90% attachment efficiencies ofMRC-5 cells on Cytodex 1 microcarriers, and significantly
Figure 4. Serial passaging of hBM-MSCs in suspension bioreactors using the original and new culture protocols. Cells from asingle source were inoculated into duplicate bioreactors at the same time and ultimately harvested at the same time, so the cultureperiod across all vessels was identical during each passage. (A) Cumulative fold increase of donor 1 hBM-MSCs over five passages,using the new and original culture conditions. (B) Cell fold increase per passage for donor 1 hBM-MSCs serially subcultured forfive passages, using the new and original culture protocols. (C) Average doubling time per passage for donor 1 hBM-MSCs seriallysubcultured for five passages, using the new and original culture protocols. (D) Cumulative cell fold increase for donor 2 hBM-MSCsover five passages, using the new and original culture protocols. (E) Cell fold increase for donor 2 hBM-MSCs serially subculturedfor five passages, using the new and original culture protocols. (F) Average doubling time per passage for donor 2 hBM-MSCsserially subcultured for five passages, using the new and original culture conditions. All the conditions were evaluated in duplicate;*p<0.05
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term
increase the cell productivity. For this reason, we investi-gated the impact of several key variables on attachmentefficiency during the inoculation phase.
Continuous agitation during the inoculation phase haspreviously been used to seed hBM-MSCs onto Cytodex 1microcarriers (Schop et al., 2009). However, our findingthat intermittent agitation improved the attachmentefficiency of hBM-MSCs to CultiSpher-S microcarrierswas similar to the result reported for the attachment ofVero cells onto CultiSpher-G microcarriers (Ng et al.,1996). This result may not be apparent, as continuousagitation certainly results in many more cell–microcarrierinteractions than intermittent agitation. However, thecell–microcarrier interactions during continuous agitation
are relatively brief. Since binding requires a finite period oftime, only a small proportion of these interactions lead to asuccessful permanent binding event. In contrast, duringperiods of rest between stirring cycles in the intermittentagitation condition, the cells have the opportunity to settleamongst the microcarriers and remain in contact withthem for an extended period of time. So, whereas thenumber of cell–microcarrier interactions may be lowerthan during continuous agitation, a significantly greaterproportion of these interactions result in a successfulpermanent binding event (Sun et al., 2000). This, inturn, leads to greater overall attachment efficienciesduring intermittent agitation. It should be noted thatthe efficiencies reported were as a percentage of the
Figure 5. Multilineage differentiation of hBM-MSCs obtained from donor 1 and serially expanded in suspension culture bioreactorsusing the original and new culture protocols. Cells generated in static culture are also shown for comparison. (A–C) Oil red O stainwas used to stain lipid droplets (red) as an indication of successful adipogenic differentiation: (A) adipogenic differentiation for cellsexpanded in static culture; (B) adipogenic differentiation for cells generated in suspension culture using the new protocols; (C) unin-duced control for expanded cells. (D–F) Alizarin red was used to stain mineralized calcium as an indication of successful osteogenicdifferentiation: (D) osteoblastic differentiation for cells expanded in static culture; (E) osteoblastic differentiation for cells generatedin suspension culture using the new protocols; (F) uninduced control for expanded cells. The results for donor 2 cells were similar tothe results shown here for donor 1 cells. Scale bars=200 mm
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term
number of cells initially inoculated into culture. Due tothe short time associated with the inoculation phase, cellproliferation was not a factor.
This same reasoning described above for cell bindingcan be extended to the observed result of agitation rateon cell attachment. It has been shown that during the
Figure 6. Expression of CD34, CD73, CD90 and CD105 surface markers using flow cytometry on hBM-MSCs generated in staticculture (control) as well as in suspension bioreactors using the new culture protocols. The isotype control for CD90 and CD105was IgG1, and for CD34 and CD73 it was IgG1K. The shaded areas show the expression of each surface marker on the expandedcells. (A) CD34 expression on cells expanded in static culture; (B) CD73 expression on cells expanded in static culture;(C) CD90 expression on cells expanded in static culture; (D) CD105 expression on cells expanded in static culture; (E) CD34expression on cells expanded in suspension bioreactors using the new culture conditions; (F) CD73 expression on cellsexpanded in suspension bioreactor using the new culture conditions; (G) CD90 expression on cells expanded in suspensionbioreactors using the new culture conditions; (H) CD105 expression on cells expanded in suspension bioreactors using thenew culture conditions
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

inoculation phase, agitation can exert two contradictoryeffects. At higher rates, the number of interaction eventsincreases, which should lead to greater attachmentefficiency. Moreover, the average energy associated witheach interaction should also be greater, since both thecells and microcarriers have more kinetic energy atimpact, and this should facilitate the formation of apermanent bond. However, the increased shear effects athigher agitation rates would promote the separation of acell from a microcarrier. Moreover, the time associatedwith each interaction event would be shorter, therebydecreasing the probability of a permanent attachmentforming. We evaluated the impact of agitation rate(60–100 rpm) during intermittent agitation and foundno difference in overall cell attachment efficiency(Figure 1D). However, the rate at which the maximumattachment efficiency was reached was faster at 100 rpm.These results suggest that a greater proportion of interac-tions during a 3 min agitation phase went on to makesuccessful permanent bonds during the ensuing 27 minrest period until a maximum binding efficiency wasreached. It should also be noted that the probability ofbinding decreases over time during the inoculation phase,because (a) a portion of the microcarrier surface is coveredby cells during previous binding events, leaving less roomfor new binding events, and (b) the number of cells remain-ing in suspension decreases with time. From our study, itappears that intermittent agitation is superior to continuousagitation for the binding of cells to microcarriers. It hasbeen suggested in the literature that the introduction ofintermittent agitation in bioreactors may adversely affectcell adhesion and compromise cell viability as the cellsand microcarriers settle in a region that may not be suffi-ciently oxygenated (Takagi et al., 1999). However, in ourstudies, the viability remained> 98% in all cases andintermittent agitation was superior to continuous agita-tion, indicating that oxygen deficiency was not a factor.
Based on current literature, there is no consensus onthe need for serum to be present for the attachment ofcells to microcarriers. Serum contains fibronectin, whichneeds to be adsorbed onto the culture surface in orderfor some cells, such as rat liver cells and baby hamsterkidney cells, to attach and spread (Forestell et al., 1992;Grinnell et al., 1977). However, other cell types, such asdiploid fibroblasts, can secrete fibronectin themselves,thereby obviating the requirement for serum during cellattachment (Grinnell, 1978). In contrast, Himes and Hu(1987) reported that the presence of serum had a nega-tive effect on FS-4 cell attachment. Our findings suggestthat hBM-MSCs can attach with high efficiency to Culti-Spher-S microcarriers without serum being present. Also,in contrast to a previous study (Forestell et al., 1992),which indicated that the absence of serum in theinoculation phase would negatively impact the subse-quent growth phase by extending the lag phase, wefound that subsequent cell division of hBM-MSCs in sus-pension bioreactors was not negatively impacted duringthe proliferation phase by 8 h exposure to 0% FBS duringthe inoculation phase. Recently, several groups have
investigated serum-free media for the proliferation ofhBM-MSCs in static culture (Meuleman et al., 2006; Junget al., 2010, 2012). However, the current study is the firstto examine serum-free medium for human mesenchymalstem cell attachment on gelatin microcarriers in suspensionbioreactors.
Ideally, at the end of the inoculation period, eachmicrocarrier should have a single attached cell (i.e. acell:bead ratio of 1). However, in practice, cell attachmentis a random process that has been shown to follow aPoisson distribution, meaning that the number of cellsper microcarrier at the end of the inoculation phase variesover a range. To minimize the number of empty microcar-riers, cell:bead ratios greater than unity are typically used.However, very high cell:bead ratios tend to be avoided, asthis approach leads to a large number of cells that remainunattached and die, thereby wasting inoculum. Thus, it isimportant to determine a cell:bead ratio which minimizesboth the number of lost cells and empty microcarriers. Inour study, we examined the impact of using cell:beadratios of 5:1, 9:1 and 60:1. Surprisingly, there were nosignificant differences in measured attachment efficien-cies between these three cases. Using a membrane cellmarker, we were able to visually confirm that 8 h afterinoculation, almost every microcarrier had at least onecell attached in all cases, including those cultures whichwere inoculated at the lowest rate of five cells/bead (seeSupporting Information, Figure S1). Thus, we chose acell:bead ratio of 5:1 for future studies, as a relativelysmall number of cells could be used to inoculate the vastmajority of microcarriers with at least one cell on each.This ratio is in line with the minimum that has beenreported for other cell types (Hu et al., 1985a, 1985b).Given that the average cell attachment efficiency at thiscell:bead ratio was approximately 50% after 12 h, weachieved a final average cell attachment rate of 2.5cells/bead during that time period. Studies in which cellattachment to microcarriers occurred in the absence ofserum (i.e. no biochemical attachment factors) suggestthat electrostatic interactions play a role in cell binding(Himes and Hu, 1987). Since medium pH can impact thecharge density on the microcarrier surface, it is likely thatpH can also impact initial attachment for certain cell–microcarrier combinations. Forestell et al. (1992) per-formed a study on mammalian cell attachment to Cytodex1 microcarriers at different pH levels and found that cellattachment increased as the pH decreased (between pH8.2 and 6.7). In contrast, Ng et al. (1996) found that theattachment of Vero cells on CultiSpher-G microcarrierswas not significantly impacted by pH 6.8–7.8. Resultsfrom our study indicate a small but significant positivecorrelation between increasing pH and hBM-MSC attach-ment efficiency over the first 8 h after inoculation.Since FBS had been eliminated from the inoculationphase for this study, it is likely that the medium pH wasimpacting both the charge density on the CultiSpher-Sbeads (which are made from gelatin) and the attach-ment molecules on the cell surface. It has beenreported that binding between gelatin and fibronectin,
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

which plays important role in cell adhesion, is depen-dent on pH (Vuento et al., 1982).
The studies described to this point were carried outsequentially by testing single parameters deemed to beimportant for cell attachment (stirring regimen, agitationrate, cell:bead ratio, serum concentration and pH level).In each case, the most promising result from one studywas carried into the next experiment. Whereas a factorialapproach was not utilized and our new protocol may not beoptimized, our approach did lead to the development of anew and superior inoculation protocol for the attachmentof hBM-MSCs to CultiSpher-S microcarriers (Figure 2A).It is interesting to note the rapid rate at which attachmenttook place using our new protocol. An attachmentefficiency approaching 75% was achieved within an 8 hperiod, whereas it was approximately 45% after 24 h usingthe original protocol. Reducing the length of the inocula-tion phase is a bioprocess improvement, as it significantlyshortens the amount of time needed to generate agiven number of cells in a bioreactor. Having a shorterinoculation phase may also be beneficial for permanentcell binding, since exposure to serum-free condi-tions facilitates initial cell attachment but appears to bedetrimental for permanent binding (Figure 1E).Interestingly, this effect was not seen in Figure 2A, whenthe pH and cell:bead ratio were also changed. It may beworthwhile to perform further investigations into alter-native methods for achieving permanent cell binding inthe absence of serum.
As indicated earlier, serum may facilitate the spreadingand permanent binding of the cells to these microcarriers.Thus, having a short 8 h inoculation phase followed by theimmediate addition of serum to start the growth phasemay serve to convert many of those initial bindings intopermanent interactions. Note that cell spreading was notstudied here because of the macroporous nature of themicrocarrier.
The importance of the inoculation phase on ultimatecell yield from a bioreactor is exemplified by the resultsshown in Figure 2B. It is clear from these results thatsimply changing from the original inoculation protocolto the new inoculation protocol increased the maximumcell fold increase from 3 to 8 while maintaining extremelyhigh viabilities. This is a very significant and positive result.
We next turned our attention to the growth phase todetermine whether modifying certain culture parameterscould improve cell productivity. To our knowledge, noone had investigated the effect of agitation rate on hBM-MSC expansion using CultiSpher-S microcarriers. Wefound that an agitation rate of 90 rpm could supportslightly higher cell fold-increase values compared to60 rpm. Higher agitation rates are known to improveculture homogeneity and improve mass transfer to cellson a microcarrier surface. Moreover, a higher agitationrate may have promoted convective flow into the macro-porous microcarriers, thereby providing improved accessto oxygen and nutrients, as well as removal of metabolicwaste products. However, increasing the agitation ratefurther to 115 rpm was found to be detrimental compared
to 90 rpm. The observation of more single cells in suspen-sion suggests that the shear in the bioreactor at 115 rpmwas sufficient to remove a proportion of cells from themicrocarriers. Moreover, the noticeably larger amount ofcell debris means that a significant percentage of thedetached cells were destroyed. The destroyed cells wouldnot have been included in trypan blue exclusion assays,which would explain why the cell viabilities at 115 rpmremained high (Figure 3A).
It is standard practice to feed MSCs which are beinggrown in static culture vessels. It is known that the concen-tration of key medium components such as glucose andglutamine, and metabolic by-products such as lactate andammonia, can affect the growth of hBM-MSCs in staticculture (Schop et al., 2009). Glucose and glutamine areused for the generation of cellular energy and should bekept above limiting values. Lactate and NH4
+ are the pro-ducts of metabolism and are known to inhibit mammaliancell growth at high concentrations. Replacing old mediumwith fresh medium removes wastes from culture andsupplies new nutrients. We regularly feed our hBM-MSCsin static culture, and have found that not feeding the cellsresults in lower cell yields (data not shown). Schop and hercolleagues (2010) evaluated different feeding regimen forMSC expansion on Cytodex 3microcarriers and determinedthat medium replacement every 3 days during a 9 dayculture period was sufficient to maintain continuous prolif-eration (Schop et al., 2010). As such, we were surprisedwhen we found that medium replacement on day 6 insuspension culture did not promote cell expansion insuspension culture relative to no medium replacement.However, it should be noted that Schop and colleaguesstarted with 4 million cells seeded into 50 ml medium,whereas we inoculated a total of 500 000 cells into 125 mlmedium (growth phase). Our higher medium volume:cellratio would have provided significantly more nutrients ona per cell basis compared to Schop and colleagues,possibly explaining why a medium change was not neces-sary during the culture period evaluated. Subsequentanalysis of the growth medium (Figure 3C) showed thatglucose and glutamine had not been depleted, and thatlactate and ammonia were well below levels known tobe toxic to mammalian cells. Future studies utilizing ourinoculation and expansion protocols should evaluatewhether medium replacement later in the culture periodcan extend the proliferation phase, assuming that themicrocarriers are not confluent.
We found that our new protocols could successfullysupport the serial expansion of hBM-MSCs isolated fromtwo independent donors, and achieve cell fold increasesthat were significantly higher than those achieved usingthe original protocols (Figure 4). Specifically, 5�105 mes-enchymal stem cells could be expanded to approximately6�108 cells within a 33 day period using the new protocol,whereas the original protocol would yield only 8.4�107
cells over that same time period. Immunocytochemicalstaining following induction showed that the expansionmethods did not compromise the capacity of the cellpopulations to differentiate down osteogenic and adipogenic
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

lineages (Figure 5). Moreover, the serially expanded cellpopulations from each donor generally maintained theirMSC surface marker expression profile after expansion inbioreactors, although there was a slight decrease in theexpression levels of positive markers relative to cells fromstatic culture. Together these results suggest that expansionin suspension culture using our new protocols did notnegatively impact the defining characteristics of thesehBM-MSC populations, and is a reasonable approach forthe generation of large numbers of these cells.
This study illustrates that control of culture parametersduring the inoculation phase can significantly impact theultimate productivity of a bioreactor and that our newprotocols, in conjunction with CultiSpher-S microcarriers,can be used to efficiently and reproducibly generate largenumbers of hBM-MSCs in suspension bioreactors. Theability to rapidly expand stem cell populations undercontrolled conditions will facilitate the development ofnew therapies, as such bioprocesses, will be able to gener-ate the many billions of cells required not only to carry outbasic research in a laboratory, but also to enable clinicaltrials. Once a treatment has been developed, large numb-ers of cells will again be required to implement thesetherapies clinically (Bernardo et al., 2011; Penfornis andPochampally, 2011). For example, Chen et al. (2004)indicated that 8–10 billion cells may be required to treata single patient with acute myocardial infarction. Therepair of bone and joint defects would also require manymillions, or billions, of MSCs, with the exact number ofcells dependent on the size of the defects being treated(Dennis et al., 2007). An important consideration is thatthere is evidence to suggest that MSCs are immunoprivi-leged, suggesting that cells from one person could be usedallogeneically without the need for immunosuppression(Dwyer et al., 2010). If this is the case, then it would bebeneficial to isolate an MSC population from one person,and expand it under controlled conditions to generatesufficient numbers of cells to treat multiple patients. Thecells could be cryogenically preserved in a cell bank andused in an off-the-shelf manner when needed. Assumingthat cell immunogenicity is not impacted by cultivation insuspension culture, the development of controlled biopro-cesses to efficiently and reproducibly expand stem cells, such
as the one described here, is an important step towards theeventual clinical implementation of MSC-based therapies.
5. Conclusions
We have shown that manipulation of parameters duringthe inoculation phase can significantly impact the ulti-mate productivity of a bioreactor. Moreover, we haveidentified improved inoculation and growth protocols thatcan support the rapid expansion of hBM-MSC populationswithout compromising the defining characteristics of thecells. These protocols form the foundation of a bioprocessthat can be used to generate the large numbers of cellsrequired to develop stem cell-based therapies for clinicalapplications.
Acknowledgements
AS, MSK and CJH were funded by the Natural Sciences andEngineering Research Council of Canada (NSERC) and theCanadian Institutes of Health Research (CIHR) as part of theCollaborative Health Research Projects (CHRP) programme.AS and MSK were also funded by NSERC through the DiscoveryGrants programme. Additionally, AS was funded by AlbertaIngenuity through the New Faculty Grants programme.
Supporting information on theinternet
The following supporting information may be found inthe online version of this article:Figure S1. (A) Brightfield photomicrograph of DiI-stained
hBM-MSCs that were used to inoculate a suspension culturespinner flask bioreactor. (B) Same image as (A) underfluorescence, showing that DiI-stained cells appear red.(C) Image of CultiSpher-S microcarriers sampled from asuspension culture spinner flask 8 h after it was inoculatedwith DiI-stained hBM-MSCs (cell:bead ratio = 5). This is arepresentative image showing that each microcarrier hadat least one cell attached.
References
Abranches E, Bekman E, Henrique D et al.2007; Expansion of mouse embryonic stemcells on microcarriers. Biotechnol Bioeng96(6): 1211–1221.
Alfred R, Radford J, Fan J et al. 2011;Efficient suspension bioreactor expansionof murine embryonic stem cells on micro-carriers in serum-free medium. BiotechnolProg 27(3): 811–823.
Allon AA, Aurouer N, Yoo BB. 2010; Struc-tured coculture of stem cells and disc cellsprevent disc degeneration in a rat model.Spine J 10(12): 1089–1097.
Baghbaderani BA, Mukhida K, Sen A et al.2010; Bioreactor expansion of humanneural precursor cells in serum-free media
retains neurogenic potential. BiotechnolBioeng 105(4): 823–833.
Baghbaderani BA, Behie LA, Sen A et al.2008; Expansion of human neural pre-cursor cells in large-scale bioreactorsfor the treatment of neurodegenerativedisorders. Biotechnol Prog 24(4):859–870.
Bernardo ME, Cometa AM, Pagliara D et al.2011; Ex vivo expansion of mesenchymalstromal cells. Best Pract Res Clin Haematol24(1): 73–81.
Chen SL, Fang WW, Ye F et al. 2004; Effect onleft ventricular function of intracoronarytransplantation of autologous bone mar-row mesenchymal stem cell in patients
with acute myocardial infarction. Am JCardiol 94(1): 92–95.
Clines GA. 2010; Prospects for osteopro-genitor stem cells in fracture repair andosteoporosis. Curr Opin Organ Transpl15(1): 73–78.
Cormier JT, zur Nieden NI, Rancourt DE etal. 2006; Expansion of undifferentiatedmurine embryonic stem cells as aggregatesin suspension culture bioreactors. TissueEng 12(11): 3233–3245.
Dennis JE, Esterly K, Awadallah A et al. 2007;Clinical-scale expansion of a mixed popula-tion of bone-marrow-derived stem and pro-genitor cells for potential use in bone-tissueregeneration. Stem Cells 10: 2575–2582.
Bioreactor expansion of mesenchymal stem cells
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term

Dominici M, Le Blanc K, Mueller I et al. 2006;Minimal criteria for defining multipotentmesenchymal stromal cells. The Interna-tional Society for Cellular Therapy positionstatement. Cytotherapy 8(4): 315–317.
Dwyer RM, Khan S, Barry FP et al.2010; Advances in mesenchymal stemcell-mediated gene therapy for cancer.Stem Cell Res Ther 1(3): 25.
Eibes G, dos Santos F, Andrade PZ et al.2010; Maximizing the ex vivo expansionof human mesenchymal stem cells using amicrocarrier-based stirred culture system.J Biotechnol 146(4): 194–197.
Forestell SP, Kalogerakis N, Behie LA et al.1992; Development of the optimal inocula-tion conditions for microcarrier cultures.Biotechnol Bioeng 39(3): 305–313.
Frauenschuh S, Reichmann E, Ibold Y et al.2007; A microcarrier-based cultivationsystem for expansion of primary mesen-chymal stem cells. Biotechnol Prog 23(1):187–193.
Grinnell F. 1978; Cellular adhesiveness andextracellular substrata. Int Rev Cytol 53:65–144.
Grinnell F, Hays DG, Minter D. 1977; Celladhesion and spreading factor: partialpurification and properties. Exp Cell Res110(1): 175–190.
Himes VB, Hu WS. 1987; Attachment andgrowth ofmammalian cells onmicrocarrierswith different ion exchange capacities.Biotechnol Bioeng 29: 1155–1163.
Hu WS, Meier J, Wang DIC. 1985a; A mecha-nistic analysis of the inoculum require-ment for the cultivation of mammaliancells on microcarriers. Biotechnol Bioeng27(5): 585–595.
Hu WS, Giard DJ, Wang DIC. 1985b; Serialpropagation ofmammalian cells onmicrocar-riers. Biotechnol Bioeng 27(10): 1466–1476.
Huang CY, Hagar KL, Frost LE et al. 2006;Effects of cyclic compressive loading onchondrogenesis of rabbit bone-marrow
derived mesenchymal stem cells. StemCells 22(3): 313–323.
Jung S, Sen A, Rosenberg L et al. 2010; Iden-tification of growth and attachment factorsfor the serum-free isolation and expansionof human mesenchymal stromal cells.Cytotherapy 12(5): 637–657.
Jung S, Sen A, Rosenber L et al. 2012;Human mesenchymal stem cell culture:rapid and efficient isolation and expansionin a defined serum-free medium. J TissueEng Regen Med 6(5): 391–403.
Malda J, van Blitterswijk CA, Grojec M et al.2003; Expansion of bovine chondrocytesonmicrocarriers enhances redifferentiation.Tissue Eng 9(5): 939–948.
McLeod M, Hong M, Sen A et al. 2006; Trans-plantation of bioreactor-produced neuralstem cells into the rodent brain. CellTranspl 15(8–9): 689–697.
Meuleman N, Tondreau T, Delforge A et al.2006; Human marrow mesenchymal stemcell culture: serum-free medium allowsbetter expansion than classical a-MEMmedium. Eur J Haematol 76(4): 309–316.
Mukhida K, Baghbaderani BA, Hong M et al.2008; Survival, differentiation and mig-ration of bioreactor-expanded humanneural precursor cells in a rat model ofParkinson’s disease. Neurosurg Focus24(3–4): E8.
Nilsson K, Buzsaky F, Mosbach F. 1986;Growth of anchorage dependent cells onmacroporous microcarriers. Bio/Technology4: 989–990.
Ng YC, Berry JM, Butler M. 1996; Optimi-zation of physical parameters for cellattachment and growth on macroporousmicrocarriers. Biotechnol Bioeng 50(6):627–635.
Penfornis P, Pochampally R. 2011; Isolationand expansion of mesenchymal stemcells/multipotential stromal cells fromhuman bone marrow. Methods Mol Biol698: 11–21.
Phillips BW, Horne R, Lay TS et al. 2008;Attachment and growth of human embry-onic stem cells on microcarriers. J Biotechnol138(1–2): 24–32.
PittengerMF, Mackay AM, Beck SC et al. 1999;Multilineage potential of adult human mes-enchymal stem cells. Science 284(5411):143–147.
Schop D, Janssen FW, Borgart E et al. 2008;Expansion of mesenchymal stem cellsusing a microcarrier-based cultivationsystem: growth and metabolism. J TissueEng Regen Med 2(2–3): 126–135.
Schop D, Janssen FW, van Rijn LD et al.2009; Growth, metabolism, and growthinhibitors of mesenchymal stem cells.Tissue Eng Part A 15(8): 1877–1886.
Schop D, van Dijkhuizen-Radersma R,Borgart E et al. 2010; Expansion of humanmesenchymal stromal cells on microcar-riers: growth and metabolism. J TissueEng Regen Med 4(2): 131–140.
Sun X, Zhang Y, TanW et al. 2000; Attachmentkinetics of Vero cells onto CT-3 microcar-riers. J Biosci Bioeng 90(1): 32–36.
Takagi M, Sasaki T, Yoshida T. 1999; Spatialdevelopment of the cultivation of a bonemarrow stromal cell line in porous car-riers. Cytotechnology 31(3): 225–231.
Tyndall A, van Laar JM. 2010; Stem cells inthe treatment of inflammatory arthritis.Best Pract Res Clin Rheumatol 24(4):565–574.
Vuento M, Salonen E, Osterlund K et al.1982; Essential charged amino acids inthe binding of fibronectin to gelatin.Biochem J 201(1): 1–8.
Wen Z, Zheng S, Zhou C et al. 2011; Repairmechanisms of bone marrow mesenchy-mal stem cells in myocardial infarction.J Cell Mol Med 15(5): 1032–1043.
Youn BS, Sen A, Kurpios N et al. 2006; Scale-up of breast cancer stem cell aggregatecultures to suspension bioreactors. Biotech-nol Prog 22(3): 801–810.
Y. Yuan et al.
Copyright © 2012 John Wiley & Sons, Ltd. J Tissue Eng Regen Med (2012)DOI: 10.1002/term




















