
Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains
13
Neurobiology of Aging 27 (2006) 54–66 Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains Michele Mishto a,f,∗ , Elena Bellavista a , Aurelia Santoro a,f , Alexandra Stolzing h , Claudia Ligorio d , Benedetta Nacmias c , Liana Spazzafumo b , Martina Chiappelli a , Federico Licastro a , Sandro Sorbi c , Annalisa Pession d , Thomas Ohm e , Tilman Grune g,h , Claudio Franceschi a,b,f a Department of Experimental Pathology, University of Bologna, 40126 Bologna, Italy b Italian National Research Center on Aging, Ancona, Italy c Department of Neurological and Psychiatric Sciences, University of Florence, Italy d Department of Oncological Sciences, Bellaria Hospital, Bologna, Italy e Department of Clinical Cell and Neurobiology, Institute of Anatomy, Charit´ e, Humboldt University, Berlin, Germany f Interdepartmental Center for Studies on Biophysics, Bioinformatics and Biocomplexity ‘L. Galvani’ (CIG), Bologna, Italy g Research Institute of Environmental Medicine, Heinrich Heine University, Duesseldorf, Germany h Neuroscience Research Center, Charit´ e, Humboldt University, Berlin, Germany Received 6 August 2004; received in revised form 23 November 2004; accepted 1 December 2004 Abstract In this study, we investigated the presence and role of immunoproteasome and its LMP2 subunit polymorphism at codon 60 in Alzheimer’s disease (AD). Immunoproteasome was present in brain areas such as hippocampus and cerebellum and localized in neurons, astrocytes and endothelial cells. A higher expression of immunoproteasome was found in brain of AD patients than in brain of non-demented elderly, being its expression in young brain negligible or absent. Furthermore, AD affected regions showed a partial decrease in proteasome trypsin-like activity. The study of LMP2 polymorphism (R/H) showed that it does not influence LMP2 expression (neither the mRNA nor mature protein) in brain tissue. However, control brain areas of AD patients carrying the RR genotype showed an increased proteasome activity in comparison with RH carriers. To test whether this effect of the genotype might be related to AD onset we performed a genetic study, which allowed us to exclude an association of LMP2 codon 60 polymorphism with AD onset, despite its influence on the proteasome activity in human brain. © 2005 Elsevier Inc. All rights reserved. Keywords: Alzheimer’s disease; LMP2 polymorphism; Immunoproteasome; Ageing; Neuroinflammation; Proteasome 1. Introduction Alzheimer’s disease (AD) is a devastating neurodegener- ative disorder of the central nervous system (CNS) occurring most frequently in later stages of adulthood. AD is associated with a specific pattern of pathological changes in brain that result in neurodegeneration and progressive development of dementia. The pathological hallmarks of AD are neuronal loss accompanied by intraneuronal neurofibrillary tangles ∗ Corresponding author. Tel.: +39 051 2094757; fax: +39 051 2094757. E-mail address: [email protected] (M. Mishto). (NFTs) formed of tau-based paired helical filaments (PHFs) and extracellular senile plaques (SPs) of -amyloid [25,41]. Recently, it has been reported that PHFs inhibit proteasome activity and it has been suggested that this inhibition may induce neuronal damage in AD [21]. Furthermore, the ubiquitin-proteasome system is also involved in the control of the physiological maturation of the -amyloid precursor protein (APP) through the modulation of the intracellular concentration of presenilins [7]. Proteasomes are multicatalytic enzyme complexes that are responsible for degradation of short-lived, damaged and antigenic proteins [39]. 26S proteasomes consist of a catalytic 20S core and 0197-4580/$ – see front matter © 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2004.12.004
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains

Neurobiology of Aging 27 (2006) 54–66
Immunoproteasome and LMP2 polymorphism in aged andAlzheimer’s disease brains
Michele Mishtoa,f,∗, Elena Bellavistaa, Aurelia Santoroa,f , Alexandra Stolzingh,Claudia Ligoriod, Benedetta Nacmiasc, Liana Spazzafumob, Martina Chiappellia,
Federico Licastroa, Sandro Sorbic, Annalisa Pessiond, Thomas Ohme,Tilman Gruneg,h, Claudio Franceschia,b,f
a Department of Experimental Pathology, University of Bologna, 40126 Bologna, Italyb Italian National Research Center on Aging, Ancona, Italy
c Department of Neurological and Psychiatric Sciences, University of Florence, Italyd Department of Oncological Sciences, Bellaria Hospital, Bologna, Italy
e Department of Clinical Cell and Neurobiology, Institute of Anatomy, Charite, Humboldt University, Berlin, Germanyf Interdepartmental Center for Studies on Biophysics, Bioinformatics and Biocomplexity ‘L. Galvani’ (CIG), Bologna, Italy
g Research Institute of Environmental Medicine, Heinrich Heine University, Duesseldorf, Germanyh Neuroscience Research Center, Charite, Humboldt University, Berlin, Germany
A
lzheimer’sd strocytes ande erly, beingi trypsin-likea protein)i comparisonw wed us toe n brain.©
K
1
amwrdl
Fs)
ometion,the
esle forteinsand
0d
Received 6 August 2004; received in revised form 23 November 2004; accepted 1 December 2004
bstract
In this study, we investigated the presence and role of immunoproteasome and its LMP2 subunit polymorphism at codon 60 in Aisease (AD). Immunoproteasome was present in brain areas such as hippocampus and cerebellum and localized in neurons, andothelial cells. A higher expression of immunoproteasome was found in brain of AD patients than in brain of non-demented eld
ts expression in young brain negligible or absent. Furthermore, AD affected regions showed a partial decrease in proteasomectivity. The study of LMP2 polymorphism (R/H) showed that it does not influence LMP2 expression (neither the mRNA nor mature
n brain tissue. However, control brain areas of AD patients carrying the RR genotype showed an increased proteasome activity inith RH carriers. To test whether this effect of the genotype might be related to AD onset we performed a genetic study, which alloxclude an association of LMP2 codon 60 polymorphism with AD onset, despite its influence on the proteasome activity in huma2005 Elsevier Inc. All rights reserved.
eywords: Alzheimer’s disease; LMP2 polymorphism; Immunoproteasome; Ageing; Neuroinflammation; Proteasome
. Introduction
Alzheimer’s disease (AD) is a devastating neurodegener-tive disorder of the central nervous system (CNS) occurringost frequently in later stages of adulthood. AD is associatedith a specific pattern of pathological changes in brain that
esult in neurodegeneration and progressive development ofementia. The pathological hallmarks of AD are neuronal
oss accompanied by intraneuronal neurofibrillary tangles
∗ Corresponding author. Tel.: +39 051 2094757; fax: +39 051 2094757.E-mail address: [email protected] (M. Mishto).
(NFTs) formed of tau-based paired helical filaments (PHand extracellular senile plaques (SPs) of�-amyloid[25,41].Recently, it has been reported that PHFs inhibit proteasactivity and it has been suggested that this inhibimay induce neuronal damage in AD[21]. Furthermorethe ubiquitin-proteasome system is also involved incontrol of the physiological maturation of the�-amyloidprecursor protein (�APP) through the modulation of thintracellular concentration of presenilins[7]. Proteasomeare multicatalytic enzyme complexes that are responsibdegradation of short-lived, damaged and antigenic pro[39]. 26S proteasomes consist of a catalytic 20S core
197-4580/$ – see front matter © 2005 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2004.12.004

M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 55
either the 19S or 11S regulatory complex. 20S proteasomesare a four-ring structure with seven different subunits in eachring, arrayed as�7�7�7�7 [46]. In cells exposed to IFN-�or tumor necrosis factor-� (TNF-�), the � subunits withcatalytic activity (�1, �2, �5) of constitutive proteasomesare replaced by other catalytic subunits, respectively, LMP2,MECL-1 and LMP7 that are incorporated into an alternativeproteasome form[26]. This isoform known as immuno-proteasome, has an altered activity of the three catalyticsites[40], which enhance the capacity to generate antigenicepitopes and modify turnover rate of specific proteins such astau[5,16,24]. Constitutive and immunoproteasomes usuallycoexist in cell, but the ratio between the two isoforms variesin base of cell type, tissue, condition of environment, etc.[36]. Recently, immunoproteasomes have been detected inhuman brains[10,37]. Hence, we investigated its expressionin brains from AD patients and age-matched controls.Furthermore, we investigated whether immunoproteasomeexpression was related not only to AD neuroinflammationbut also to the ageing process in CNS. Moreover, evidencesfor chronic immune reaction in AD brains have emerged bygenetic association of AD with polymorphisms of severalcytokines and detection in brains of inflammatory mediators[2,6,31,29]. One of these cytokines, namely TNF-�, hasbeen shown to play a role in neuronal degeneration[48]. Wepreviously reported that the polymorphism at codon 60 of theLa iven 0 (ine orH ffer-e dt oni tivitya
2
2
ini-c 70.3,r stage0 entedy 40–50y from4 10c a-t and6 am-p
2
andc ered
saline, and subsequently lysed by repeated freeze-thawing cy-cles and homogenized using a potter. The insoluble materialwas removed by centrifugation, the supernatant, containingthe proteins, was stored in aliquots at−80◦C and quantifiedusing the Coomassie staining. Polypeptides were separatedby 12% SDS-polyacrylamide gel, stained in a concentratedmethanol/acetic acid/Coomassie Brilliant Blue R250 solutionovernight at 4◦C on shaking and destained in the same solu-tion without the dye at room temperature for 4 h. The intensityof the dye in each single lane was compared with a sampleused as internal standard, loaded in every gel, whose con-centration was known. This strategy was preferred to probehousekeeping proteins (e.g., actin or GAPDH) because age-ing and AD impinge upon their expression in human brains[38].
2.3. Western blot analysis
Extracted proteins (10–15�g) were separated in a 12%polyacrylamide gel and transferred to a nitrocellulose filter(Trans–Blot Transfer Medium, Bio Rad, Hercules, CA). As-pecific sites on the membrane were blocked overnight in a 5%non-fat dry milk/0.01% Tween 20-TBS solution. Proteasomesubunits were detected using the following primary antibody(Affiniti Research, PA): anti-HC3 (1:500), anti-�1 (1:5000),anti-LMP2 and -LMP7 (both 1:2000). The filters were incu-b n-fatd useh wasu ogy,C g toe ware( -m lts. Ino werer bandd dedot HC3a ed
2
sin-l flu-o m),S A( uo-r erei MTD efl 0 nme onlyp ioned
MP2 gene influences the susceptibility to TNF-�-inducedpoptosis[33]. This polymorphism is a non-conservatucleotide base pair change at amino acid position 6xon 3) in LMP2, resulting in two alleles, Arginine (R)istidine (H), which has been already associated with dint autoimmune diseases[19,45,30]. Hence, we investigate
he possible role of LMP2 codon 60 polymorphismmmunoproteasome expression, brain proteasome acs well as on risk and age of onset of AD.
. Materials and methods
.1. Brain tissue samples
Autopsy brain samples from a total number of 10 clally demented AD patients (Braak stage 5–6; mean ageange 49–84 years), 12 non-demented elderly (Braak–1; mean age 70.2, range 50–86 years) and 6 non-demoung subjects (Braak stage 0–1; mean age 42, rangeears) were examined. The post-mortem interval rangedto 41 h in all groups. Ten hippocampus (HPG-AD) and
erebellum (CBL-AD) regions were available from AD pients, 12 hippocampus regions from elderly controls (K)
hippocampus areas from young subjects (Y); all the sles were stored at−80◦C.
.2. Protein quantification
Brain tissue samples from the different areas of ADontrol patients were homogenized in phosphate-buff
ated with the antibody 1 h at room temperature in 5% nory milk/0.01% Tween 20 in TBS. A secondary anti-moorseradish peroxidase-conjugated antibody (Bio Rad)sed followed by ECL detection (Santa-Cruz BiotechnolA). Densitometry analysis of the band correspondinach subunit was performed using QuantityOne SoftBio Rad) and data represented as INT mm2. Each experient was repeated and we considered the mean of resurder to compare samples loaded in different gels, dataeported as ratio between samples and control’s specificensity; sample “CBL-AD 8” was used as control and loan each gel. LMP2 (immunoproteasome) and�1 (constitu-
ive proteasome) contents were reported as ratio LMP2/nd�1/HC3 to avoid loading errors (� and� subunits weretected on the same Western blot).
.4. Proteasome activity
The three activities of the 20S proteasome (chymotrypike, caspase-like, trypsin-like) were measured using therogenic substrates: Suc-Leu-Leu-Val-Tyr-MCA (Bacheuc-Pro-Phe-Arg-MCA (Alexis), Suc-Leu-Leu-Glu-MC
Bachem). Twenty micrograms of the sample material, flogenic substrates (final concentration of 200 nmol/ml) wncubated together for 1 h in a buffer containing 50 mris–HCl (pH 7.8), 20 mM KCl, 5 mM MgAc and 0.5 mMTT. Incubation took place at 37◦C in a 96-well plate. Thuorescence was determined at 380 nm excitation/44mission in an plate-reader. Since proteasome is not therotease which might be able to degrade the above ment

56 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
fluorogenic peptide substrates, we used the selective protea-some inhibitor lactacystin (final concentration of 20 nmol/ml)for testing of the proteasomal share. This share was above85% in the tested brain samples. The proteasome activity re-ported in this manuscript represented the total minus the valuein presence of lactacystin (i.e., specific activity attributed tothe proteasome).
2.5. Immunohistochemistry
Immunostaining was performed on 2�m thick sections,serially cut from the selected blocks (hippocampus and cere-bellum). The following antibodies were employed: mousemonoclonal antibody to human 20S proteasome subunit�1i (Affiniti Reasearch, clone LMP2-13, diluted 1:300),mouse monoclonal antibody to human 20S proteasome sub-unit �5i (Affiniti Reasearch, clone LMP7-1, diluted 1:100)and mouse monoclonal antibody to 20S proteasome sub-unit �1 (Affiniti Reasearch, diluted 1:300). Sections weredeparaffinized, rehydrated, submitted to 40-min treatmentat 98◦C by MS UNMASKER buffer 10× and cooled atroom temperature. Endogenous peroxidase was blockedin 1% H2O2 in distilled H2O. All antibodies were ap-plied at room temperature for 1 h. The processing was per-formed using biotinylated goat anti-polyvalent antibody ands ont,C em( per-o er’sh
2
at− re-d liq-u -n rityo iono so-l n or-g wasr for1f air-d anti-fi Ae in-t re-cc on-t hens t lastt tedw
2.7. RT-PCR
The first strand cDNA was prepared from total RNA us-ing M-MLV Reverse Transcriptase system (Life Technolo-gies). Briefly, 5�g of RNA was primed with 0.5�g oligo(dT)12–18, the mixture was incubated at 65◦C for 5 min with10 mM dNTPs and quick chilled on ice. After addiction of 5×first-strand buffer and 0.1 M DTT, the mixture was incubatedat 37◦C for 2 min with 40 units of RNaseOUT Recombi-nant Ribonuclease Inhibitor. Two hundred units of M-MLVRT were added to the samples and then incubated at 37◦Cfor 50 min. The reaction was inactivated by heating at 70◦Cfor 15 min. Afterwards, samples of cDNA were amplified forLMP2 mRNA by PCR, utilizing as primers (Roche Diagnos-tic, Basel, Switzerland):
LMP2S (Fw; exon 3): 5′-CGAGGCGGTGGTGAACCG-3′LMP2F (Rv; exon 4): 5′-TCTCACCACATTTGCAGCA-GCC-3′.
The reaction was performed using standard PCR methodsas previously described[33], obtaining an amplified fragmentof 185 bp.
2.8. mRNA allelic isoform comparison
To evaluate mRNA expression of LMP2 codon 60 al-l com-p mit-t uss di-goas Ac ento
Af les;a byH ffera oths e gelt pper( andsw ; ther d andc scer-t tionsd ed as platec ,R plesw ev r thee latec ted)
treptavidin peroxidase (LabVision Corporation, FremA, USA). DAKO liquid DAB-substrate chromogen syst
Dako, Carpintera, CA, USA) was used to detect thexidase activity. Sections were counterstained with Mayematoxylin.
.6. Tissue homogenization and nucleic acids isolation
Two hundred micrograms of frozen tissue (stored80◦C) from the different areas of brain samples, wasuced to a powder in a potter with continuous addition ofid nitrogen. Two milliliter of TRIZOL reagent (Life Techologies, Paisley, UK) was added to maintain the integf the RNA, and to disrupt the cell component. Additf chloroform followed by centrifugation separates the
ution into an aqueous phase containing the RNA and aanic phase containing the DNA and the protein. RNAecovered by mixing with isopropyl alcohol, incubating0 min at 4◦C and followed by centrifugation at 12,000× g
or 10 min at 4◦C [8]. The pellet was washed, and thenried. RNA was dissolved in RNase-free water, and qued at the spectrophotometer (λ = 260 nm). We checked RNxtraction quality by the OD ratio 260/280 nm and RNAegrity in MOPS-formaldehyde agarose gel. DNA wasovered by sequential precipitation with ethanol[9]. Afterentrifugation, the pellet was washed with a solution caining 0.1 M sodium citrate in 10% ethanol, twice and tuspended in 75% ethanol, centrifuged and air-dried. Ahe DNA was dissolved in 8 mM NaOH and the pH adjusith Hepes.
elic isoforms, we set up a new approach based onarison of intensity of ethidium bromide fluorescence e
ed by bands from cDNA amplified from heterozygoamples (RH for LMP2 codon 60 polymorphism) andested with the restriction enzymeHhaI (New England Bi-labs, UK) at 37◦C for 6 h (see Section2.10). cDNA wasmplified as previously described (see Section2.7). Re-triction enzymeHhaI was able to digest only the cDNodon 60 Arginine isoform, yielding a detectable fragmf 133 bp.
To calibrate the assay, we initially amplified cDNrom three homozygous (RR in LMP2 codon 60) sampfterwards 50% of the amplified cDNA was digestedhaI enzyme, while the other 50% was left in same bund condition without the restriction enzyme. Then, bamples were loaded in the same lane of 2.5% agaroso simulate an heterozygote. The intensity of the u185 bp; not digested) and lower (133 bp; digested) bas determined using QuantityOne Software (Bio Rad)
atio between the upper and lower bands was calculateonsidered as a “equimolar calibration standard”. To aain that the ratio obtained was not subjected to variaue to the amount of the loaded sample, we performcalar dose experiment using different amounts of temDNA (2.15, 1.85, 1.55, 1.2, 0.9 and 0.6�g). AfterwardsNA of nine heterozygous (RH in LMP2 codon 60) samas amplified, digested withHhaI and fragments werisualized by electrophoresis (2.5% agarose gel). As foquimolar calibration standard, different amount of tempDNA were used. Intensity of upper (185 bp; not diges

M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 57
and lower (133 bp; digested) bands was determined andratio between upper and lower bands was calculated. Theobtained results were compared to the equimolar calibrationstandard. A characteristic of this approach is that it does notrequire as a reference housekeeping gene. In several samples,digestion fragments were sequenced in order to verify thedata obtained by restriction analysis as previously reported[33].
2.9. Patients and controls for genotyping
Patients with AD were consecutively collected amongout patients attending the Department of Neurology at theUniversity of Florence. Clinical assessment of patientswas done according to published guidelines[44] and ADdiagnosis fulfilled Diagnostic and Statistical Manual ofMental Disorders criteria (DSM-IV)[1]. All controls werecarefully assessed with a rigorous diagnostic evaluation inorder to exclude diagnosis of any neurological disorder.DNA was extracted from peripheral blood of controlsand AD patients as previously described[42]. Informedconsent was obtained from patients or legal represen-tatives.
2.10. ApoE and LMP2 genotyping
tan-dq ing.L eth-o ibed[ ncedi is asp
2
theM nda oly-m n wes uset byP (K).H firsts per-fo ndS tw so-f ancew y dis-tA ardyW kov
Chain, according to Gou and Thompson, implemented in Ar-lequin Package.
3. Results
3.1. Immunoproteasomes are expressed in the brain ofAD patients and non-demented elderly controls
In order to study the immunoproteasome and the putativeeffect of LMP2 codon 60 polymorphism in AD, we initiallyverified the presence of immunoproteasomes in the brain, byWestern blot detection of LMP2 and LMP7 subunits, in hip-pocampus (10 affected brain region, HPG-AD) and cerebel-lum (10 non-affected, CBL-AD) of patients affected by AD,as well as in hippocampus of 10 age-matched controls (K).Immunochemical detection of LMP2 (Fig. 1A) and LMP7subunits (data not shown) in protein extracts showed thepresence of immunoproteasomes in AD brains (both HPG-AD and CBL-AD) as well as in age-matched brains. Thepresence of immunoproteasomes was confirmed by analyz-ing LMP2 mRNA by RT-PCR on RNA extracts from brainmaterial (Fig. 1B). Indeed, the presence of LMP2 mRNAin each sample tested (HPG-AD, CBL-AD as well as K)was revealed, despite different post-mortem delay among thea opro-t anda andL withl ckedw nors,t rolsw
F ssed inh -PCR( (A)W ctsw ri non-s CR-a s.
We analyzed the Apo E gene polymorphism using sard PCR methods as previously described[42]. The fre-uencies of ApoE� alleles were estimated by gene countMP2 genotyping was performed using standard PCR mds and DNA enzymatic digestions as previously descr
33]. In several samples, digestion fragments were sequen order to verify the data obtained by restriction analysreviously reported[33].
.11. Statistical analysis
Statistical analysis of data was performed usingann–WhitneyU-test for results from Western blotting activity assays. When we compared LMP2 codon 60 porphism effects on proteasome activity and expressio
tandardized data of controls (Cnt: CBL-AD + K), becahey were coming from two different areas, not affectedHF-tau and�-amyloid as cerebellum and hippocampusence, in those analysis, also data sets from AD weretandardized and then analyzed. Standardization wasormed as following: (xi − xm)/S.D., wherexi is the valuef each brain andxm is the mean of the brain population, a.D. is the standard deviation from the mean.t-Student’s tesas used for valuation of LMP2 codon 60 allelic mRNA i
orm expression. In each data set, homogeneity of varias checked by Levene’s test. Comparisons of frequenc
ributions were assessed by Monte Carlo Pearson’sχ2-test.ll the analyses were implemented in SPSS software. Heinberg equilibrium was tested by Monte Carlo Mar
vailable brain samples. Thus, the presence of immuneasome in hippocampus and cerebellum of AD patientsge-matched controls was verified by analyzing LMP2MP7 subunits. As brain has been considered an organ
ow or null expression of immunoproteasomes, we chehether this expression was influenced by the age of do
aking into account that most of AD patients and contere over 60-year-old.
ig. 1. Immunoproteasome and constitutive proteasomes are expreuman brains. Detection was performed by Western blot (A) and RTB) in HPG-AD (lanes 1), CBL-AD (lanes 2) and K (lanes 3) samples.estern blot analysis of HPG-AD, CBL-AD and K total protein extraith antibodies for� subunit (HC3),�1 subunit and� subunit specific fo
mmunoproteasomes (LMP2). The upper band (not marked) is likely apecific band due to HC3 probing. (B) Electrophoretic analysis of Pmplified LMP2 subunit cDNA from HPG-AD, CBL-AD and K sample

58 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
3.2. Brain expression of proteasome andimmunoproteasome subunits is age-dependent
We compared the expression of immunoproteasome andconstitutive proteasome subunits in hippocampi of elderlyand young control donors and patients affected by AD.LMP2, LMP7, �1 and HC3 expression in protein extractsfrom hippocampi were measured using Western blot analy-sis. These experiments showed that LMP2 and LMP7 werepresent in brain samples from elderly (K) and in very lowamount in brain samples from young hippocampi: only oneout of six young samples showed a low LMP2 expressionwhile the other five samples were negative (Fig. 2A); at vari-ance, LMP7 antibody gave a weak signal in young samples,but only when 30�g of total proteins were loaded in the gelinstead of the usual 10�g (data not shown). Thus, immuno-proteasome seemed to be strongly upregulated during ageingin hippocampus. A comparison between young and elderlysamples showed that also the amount of proteasomes is al-tered during ageing. Indeed, the HC3 subunit was increasedby 54% (Mann–Whitney value = 4;p = 0.006) in elderly sub-jects (Fig. 2B), and�1 subunit detection confirmed this result(data not shown).
3.3. Immunoproteasomes are expressed in neurons,astrocytes and endothelium
entst im-m mpusa derlys ls own,
F mpus.( nd 2a weret erb havea siso iess ungs (HC3)o neyt
respectively, inFig. 3A and B. Intriguingly both anti-LMP2and -LMP7 antibodies revealed that neurons and astrocytesexpressed immunoproteasomes, while oligodendrocytesseemed to be negative (Fig. 3C). On the contrary,�1 subunitprobing was diffused in all cell types (Fig. 3E), testifyingthe ubiquitous presence of constitutive proteasomes. Re-garding the different expression of immunoproteasome andconstitutive proteasomes, it is worthy to note that Purkinjecells were weakly reactive to immunoproteasome specificantibodies (Fig. 3D), while they were strongly positiveto �1 constitutive proteasome subunit (Fig. 3F). We alsoobserved several endothelial cells, in blood vessels, positiveto antibodies toward immunoproteasome and constitutiveproteasome subunits (Fig. 3C–E). Furthermore, theseexperiments described a scenario of immunoproteasomeexpression that, similar in hippocampus and cerebellum, wasbasically conserved both in AD and non-demented elderlysamples (data not shown). Immunohistochemistry stainingwas specific because immunoreactivity was negative whenprimary antibodies were omitted or a non-relevant antibodywas used.
3.4. Immunoproteasome expression is increased in ADaffected regions
To investigate how much AD alters immunoproteasomee andc l asC ADa meda ons(H singW mesa /HC3as l con-t iveo ectedt e ofi ofp es).T a ob-t unit,a -AD,C nif-i wasd sh AD( yv 7;p ese hadtd e inH
We next performed immunohistochemistry experimo investigate the cell type(s) in which the induction of theunoproteasomal subunits occurs. Samples of hippocand cerebellum from AD patients and non-demented elubjects were tested for�1i, �5i and �1 subunits. Typicatructures of hippocampus and cerebellum are sh
ig. 2. Proteasomes are differently expressed upon ageing in hippocaA) Western blot analysis of non-demented elderly (K) in lanes 1 and young samples (Y) in lanes 3 and 4. Total protein brain extracts
reated with antibodies anti-�1 subunit (upper bands) and anti-LMP2 (lowands). Both Y samples are negative to LMP2 immunodetection anddecrease of�1 signal. (B) Optical density from Western blot analy
f K (n = 12) and Y (n = 5) total protein extracts probed with antibodpecific for� subunit (HC3). Values are given as mean + S.E.M. In yoamples, emerged a decrease in optical density of proteasome bandf 35% (p = 0.006). Significance (*) was calculated by Mann–Whit
est.
xpression, we compared the immunoproteasomeonstitutive proteasome content in HPG-AD as welBL-AD and K. Furthermore, we also merged the CBL-nd K groups in the non-affected sample group (renas “Cnt”) and we compared with AD affected brain regiHPG-AD). To these purposes, we measured LMP2,�1 andC3 expression in all protein extracts of brain samples uestern blot analysis; relative levels of immunoproteaso
nd constitutive proteasomes were reported as LMP2nd�1/HC3, respectively. For each sample, LMP2 and�1ubunit levels were measured in order to have an internarol. Indeed, assuming the� subunit (HC3) as a representatf all proteasomes and its constant expression we exp
hat to an increase of LMP2 protein level (representativmmunoproteasomes) should correspond a decrease�1rotein level (representative of constitutive proteasomhis assumption was experimentally verified and the dat
ained indicate that no variation in the amount of HC3 subnd thus of all proteasome content, occurred in HPGBL-AD and K (data not shown). On the contrary, a sig
cant increase in LMP2 subunit expression in HPG-ADetected (Fig. 4A). Indeed, relative�1i subunit content waigher in hippocampus of AD patients compared to CBL-Mann–Whitney value = 11;p = 0.03), K (Mann–Whitnealue = 16;p = 0.04) and Cnt (Mann–Whitney value = 2= 0.02). Accordingly, relative constitutive proteasomxpression showed an inverse trend, where HPG-ADhe lower relative amount of�1/HC3 (Fig. 4B). Suchifference, characterized by a borderline significancPG-AD versus K (Mann–Whitney value = 12;p = 0.07) and

M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 59
Fig. 3. Immunoproteasomes are expressed mainly in astrocytes, neurons and vessels in CNS of AD patients and non-demented elderly subjects. Immunohis-tochemical LMP2 probing of hippocampus (A) and cerebellum (B) of AD patients revealed that astrocytes, endothelial and neuronal cells were positive, onthe contrary of oligodendrocytes. (C) High magnification photo of AD hippocampus stained with anti-LMP2 antibody. Examples of neurons are marked with1, astrocytes with 2 and endothelial cells with 3. (D) Particular of cerebellar granules probed with anti-LMP2 antibody, where Purkinje cells (arrows) werenegative to anti-LMP2 antibody, while they were strongly positive to�1 subunit (F). (E) AD hippocampus stained with anti-�1 antibody. On the contrary ofanti-LMP2 probing, all cell types were positive to�1 subunit, as expected taking into account its ubiquitous expression. A and B scale bar, 150�m; C scalebar, 15�m; D and F scale bar, 40�m; E scale bar, 30�m.
HPG-AD versus Cnt (Mann–Whitney value = 25;p = 0.07)was in agreement with LMP2 results since as assumed, anincrease of relative expression of LMP2 corresponded adecrease of its constitutive homologue�1 subunit in eachsample.
3.5. Trypsin-like proteasome activity is decreased in ADaffected regions
In order to get information not only about the quantitybut also about the proteolytic function of the proteasomes in
Fig. 4. Immunoproteasomes are differently expressed in brain areas affected by AD. Here LMP2/HC3 (A) and�1/HC3 (B) values, obtained through subunitdetection by Western blot in brain samples of HPG-AD (n = 7; hippocampus), K (n = 11; hippocampus), CBL-AD (n = 9; cerebellum) and Cnt (n = 20) arereported. Values are given as mean + S.E.M. (A) Relative expression of LMP2 was significantly increased in hippocampus of AD patients (HPG-AD) comparedto K (* p = 0.04), CBL-AD (** p = 0.03) and Cnt (*** p = 0.02). (B) Relative expression of�1 showed a inverse trend with borderline significance (HPG-AD vs.K: p = 0.07; HPG-AD vs. Cnt:p = 0.07). These results confirmed LMP2/HC3 data. Statistical significance (*, **, ***) was calculated by Mann–Whitney test.

60 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
Fig. 5. Proteasome activity in brain tissues of HPG-AD, K, CBL-AD and Cnt (CBL-AD + K). Trypsin-like (A), chymotrypsin-like (B) and caspase-like (C)activities were measured with specific short fluorogenic peptides in protein extracts of HPG-AD, CBL-AD and K. Cnt is the sum of CBL-AD and K samples.Activity was calculated as specific activity (nmol (mg min)−1). Values of 8 HPG-AD, 10 CBL-AD and 9 K are given as mean + S.E.M. Only HPG-AD andCBL-AD showed a significant difference (*) (Mann–Whitney test) in trypsin-like activity (p = 0.03).
our brain samples, proteasome activity (chymotrypsin-like,caspase-like and trypsin-like) was measured in HPG-AD(n = 8), CBL-AD (n = 10) and K (n = 9) samples. Similar toWestern blot experiments, we compared HPG-AD eitherto Cnt (CBL-AD + K) or to CBL-AD and K, separately.This approach was adopted in order to directly comparenot only different areas of AD subjects (namely HPG-ADversus CBL-AD) as well as hippocampi of AD patients andof non-demented elderly (namely HPG-AD versus K), butalso brain areas non-affected (Cnt) and affected (HPG-AD)by NFTs and SPs, known to impair proteasome function.The results (Fig. 5) showed that a significant decrease intrypsin-like activity was evident comparing in HPG-ADversus CBL-AD (Mann–Whitney value = 16;p = 0.03). Thistendency emerged also comparing HPG-AD versus K, orHPG-AD versus Cnt (Mann–Whitney value = 43;p = 0.08)(Fig. 5A). On the contrary, chymotrypsin-like activity of thethree brain populations seemed to be similar (Fig. 5B). Evenwhen we compared the caspase-like activity, we did notobserve a clear decrease in this activity in affected brain areas(HPG-AD) with respect to non-affected (CBL-AD + K) aswell as to the two populations, i.e., CBL-AD and K separately(Fig. 5C).
3.6. LMP2 polymorphism does not alter the LMP2transcript and protein expression
P2c ome
functionality in brain tissue, and in particular, in the brainareas affected by AD (presence of NFTs and SPs), all brainsamples were genotyped for LMP2 codon 60 polymorphism.Neither in 10 HPG-AD (as well as in 10 CBL-AD) nor in12 K, we identified subjects with an HH genotype. Thus, inthe following analysis and experiments, we compared brainsfrom subjects with RR versus RH genotype and verifiedwhether these LMP2 genotypes might alter the transcriptionof LMP2 mRNA and/or the expression of LMP2 mature pro-tein. To this purpose, we set up a methodology (see Section2.8) that combines the RT-PCR of exons 3 and 4 of LMP2mRNA to a subsequent enzymatic digestion withHhaI, anenzyme able to discriminate between the two-allele variantsat codon 60 of the gene. This technique was applied to set upa calibration method using RNA from three different brainsamples, RR homozygotes at the LMP2 polymorphism,as described in Section2.8. This calibration allowed us toobtain a “relative ratio” of the densitometric values of R(133 bp) and H (185 bp) equimolar bands. We then amplifiedand digested the RNA of 9 (3 HPG-AD, 3 CBL-AD and 3K) heterozygous (RH) brain samples at the codon 60 LMP2polymorphism. Then, we measured the densitometry of the R(133 bp) and H (185 bp) bands and calculated the relative ra-tio (upper/lower bands). For each sample (both in calibrationand in RH heterozygote assays), we tested different cDNAconcentrations in the PCR reaction. In the final analysis, dataw ction2 tionv ozy-
In order to answer the question whether the LModon 60 polymorphism influences the immunoproteas
hich suggested a heteroduplex’s formation (see Se.8) were excluded. We finally compared the calibraalues with the results obtained with the nine RH heter

M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 61
Fig. 6. LMP2 codon 60 R/H polymorphism does not alter its mRNA expression and protein level. (A) Example of electrophoretic analysis of cDNA resultingfrom RT-PCR for exon 3–4 of LMP2 mRNA followed by digestion with enzymeHhaI. Lanes 1 and 2: heterozygous (RH) samples for LMP2 codon 60. Lanes3 and 4: homozygous (RR) samples for calibration. In lanes 1 and 2, the amplified cDNA has been digested withHhaI enzyme; in this case, 185 bp bandcorresponds to H variant and 133 bp to R variant. In lanes 3 and 4, half of RR samples amplified cDNA has been digested withHhaI enzyme (133 bp band), whilethe other half has not been (185 bp). Both digested and not digested amplified cDNA has been loaded in the same lane. With this methodology (calibration), weobtained two bands corresponding to equimolar fragments that have the same sequence of fragments produced in RH cDNA digestion. Afterwards, densitometricquantification of the 185 bp and 133 bp fragments has been performed. For each sample, the upper/lower band ratio has been calculated. The upper/lower bandratio from calibration and from RH samples have been compared and analyzed with Student’st-test. (B) Histograms showing the upper/lower band ratio ofheterozygotes RH (1) versus calibration (2). No difference emerged, meaning that the R and H mRNA variants are equally expressed. (C) Relative expressionof �1 (�1/HC3) and LMP2 (LMP2/HC3) proteins in RR and RH carriers (LMP2 codon 60 polymorphism) are reported. Protein measurement was performedby Western blot assay. Both ratio are the mean of all 27 samples tested (HPG-AD + CBL-AD + K). Values are given as mean + S.E.M. Significant difference ismarked with (*). Value has been analyzed, after standardization, with Mann–Whitney test.
gotes. The analysis, reported inFig. 6B, did not show anydifference in the relative ratio between the two bands in RHheterozygotes and calibration with RR homozygotes. Thisresult allowed us to assume that the LMP2 mRNA expressionin brain tissue is not influenced by its polymorphism atcodon 60.
To confirm this result, we compared LMP2 and�1 relativeprotein expression in brain extracts of 7 HPG-AD and 20 Cnt(9 CBL-AD + 11 K), with Western blot assay. Even in theseexperiments, we checked�1 subunit in order to have an ad-ditional control. To minimize the difference due to differentbrain area populations, we standardized the LMP2/HC3 and�1/HC3 values within each population (CBL-AD, HPG-ADand K), and then standardized values were compared amongRR and RH carriers in the three brain populations. The re-sults showed that LMP2 codon 60 variants did not affect theamount of HC3 subunit, and thus proteasomes content in allbrains (data not shown). On the contrary, a borderline dif-ference (Mann–Whitney value = 34;p = 0.05) between RRand RH carriers emerged as far as LMP2 relative expression.However, the statistical significance of this difference is du-bious because when the�1 relative amount was checked nodecrease of the relative�1 subunit in RR carriers emerged(Fig. 6C). Furthermore, the variance of LMP2/HC3 of allthe samples was not homogeneous (Levene test value = 4.53;p = 0.045), and not in any brain area RR carriers had highera P2
codon 60 polymorphism likely does not alter LMP2 expres-sion.
3.7. LMP2 polymorphism influences the activity ofproteasomes in non-affected brain areas (Cnt) but not inaffected AD areas (HPG-AD)
Once we proved that the R and H variants in LMP2 codon60 did not alter messenger and, likely, protein expressionof �1i subunit, we focused our attention on the possibledirect effect of the polymorphism on proteasome activity.Accordingly, we measured the ex vivo chymotrypsin-like,caspase-like and trypsin-like proteasome activities in brainareas affected (8 HPG-AD samples) and not affected (Cnt:10 CBL-AD and 8 K samples) by NFTs and SPs, taking intoaccount RR and RH genotypes. Since Cnt samples derivedfrom different brain areas (hippocampus and cerebellum),we analyzed the proteasome activity standardizing theresults within each brain populations (namely CBL-AD andK). To this purpose, the standardized values of CBL-AD andK were combined, analyzed for their proteasome activitiesin order to assess the role of RR versus RH genotype inunaffected brain regions. The same analysis was performedin HPG-AD samples in order to assess the role of RR versusRH genotype in affected brain regions. This part of the studyconfirmed the difference between HPG-AD and Cnt (Fig. 7)s ces
mount of relative LMP2 subunit, suggesting that LM howing that LMP2 codon 60 polymorphism influen
62 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
Fig. 7. LMP2 codon 60 polymorphism influences proteasome activity in brain Cnt (CBL-AD + K) tissues but not in HPG-AD. Trypsin-like (A), chymotrypsin-like (B) and caspase-like (C) activities has been measured with specific short fluorogenic peptides in protein extracts of HPG-AD and Cnt. Activity hasbeencalculated as specific activity (nmol/mg/min). Samples has been genotyped for LMP2 codon 60 polymorphism (R/H). We compared proteasome activity ofRR and RH both in 8 HPG-AD and 18 Cnt (10 CBL-AD and 8 K), as well as value of CBL-AD and K samples separately. Values are given as mean + S.E.M. InCnt, RR vs. RH showed an increased activity both in chymotrypsin-like (p = 0.04) and trypsin-like activity (p = 0.01), but not in caspase-like activity. Even inRR samples of control hippocampus, we observed a significant increase in trypsin activity (p = 0.02). On the contrary in HPG-AD did not emerge a significantdifference in proteasome activity of RR vs. RH. Significant differences are marked with. Value has been analyzed, after standardization, with Mann–Whitneytest.
chymotrypsin-like (Mann–Whitney value = 18;p = 0.04)and trypsin-like (Mann–Whitney value = 13;p = 0.01)proteasome activity in brain area not affected by NFTs andSPs (Fig. 7A and B). Indeed, RR subjects showed a 45.4%(chymotrypsin-like) and 40.1% (trypsin-like) increasedactivity in Cnt in comparison with RH subjects. This highactivity of RR samples was present both in CBL-AD and Kbrain samples (Fig. 7A). At variance, caspase-like activitydid not show any significant difference between RR and RHin Cnt brain samples (Fig. 7C). A different scenario emergedwhen the proteasome activities in brain areas affected byNFTs and SPs were analyzed. The samples from RR subjectsdid not show any increase of proteasome activity consideringthe three-proteolitic activities (Fig. 7A–C). These resultsindicate that LMP2 codon 60 polymorphism modulates pro-teasome activity in unaffected but not in affected brain areas,and suggest that this polymorphism likely does not play a rolein the late stages of AD progression, when the most importantmarkers of AD phenotype, i.e., NFTs and SPs, are fullyexpressed within the brain tissue. This interpretation leavesopen the possibility that the LMP2 codon 60 polymorphismmay influence the early phases of AD onset, by differentlyaffecting the proteasome activity in the hippocampus (K).In order to verify this hypothesis, a genetic study on ADand control population was performed, paying particularattention to the distinction between early and late onset AD.
3.8. LMP2 polymorphism has not effect on AD risk andage of onset
We studied a population of 154 patients affected by ADand 406 age-matched controls. All donors were genotypedfor LMP2 codon 60 and ApoE polymorphisms, the lastconsidered the main genetic risk factor for sporadic AD. Asexpected the frequency of ApoE�4 allele in AD patients wasmuch higher (44.6%) in comparison with elderly subjects(14.7%). The results did not show any significant differencebetween groups (Table 1A). Afterwards, the LMP2 codon60 polymorphism distribution in AD patients consideringgender (Table 1B) or ApoE �4 (�4+ and�4−) (Table 1C)was analyzed. The distribution of LMP2 polymorphism didnot change in the various data sets (Table 1E). Finally, theLMP2 codon 60 polymorphism distribution in AD early(EO) versus late (LO) onset (Table 1D) was comparedand again no association of LMP2 genotype with the ADage of onset emerged (Table 1E). For each data set, theHardy–Weinberg equilibrium was verified by Monte CarloMarkov Chain test (p > 0.05).
4. Discussion
In this study, we investigated the expression and the activ-i erly
ty of immunoproteasomes in brains of AD patients, eld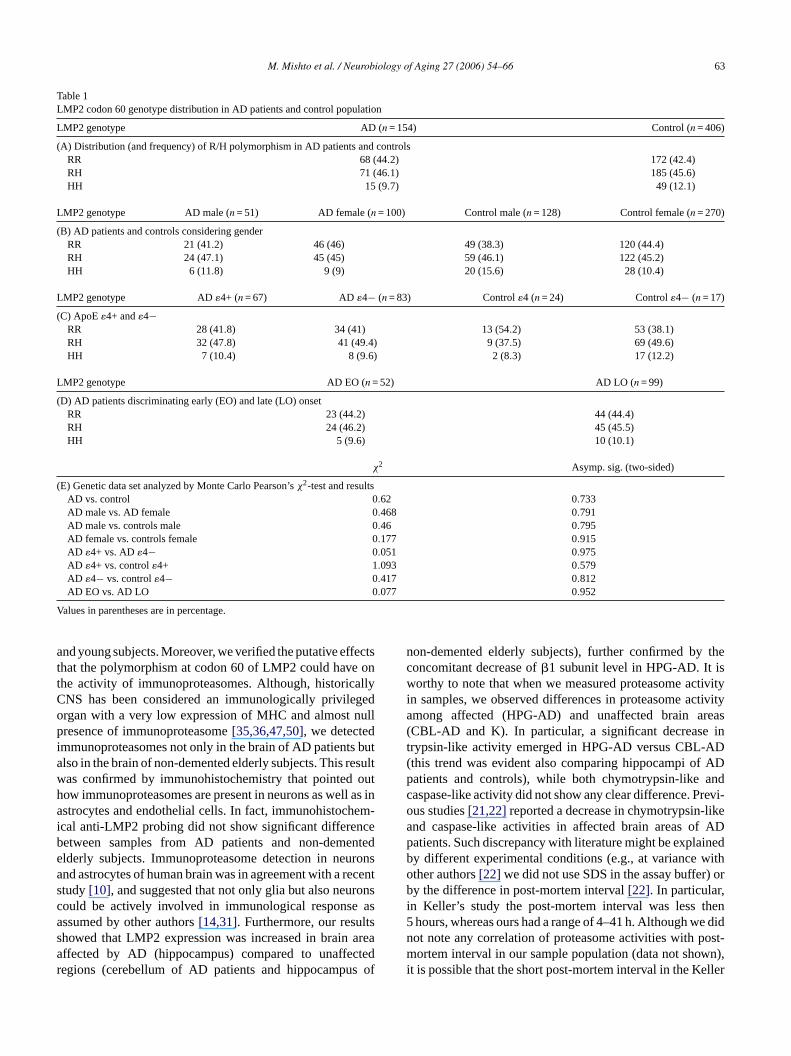
M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 63
Table 1LMP2 codon 60 genotype distribution in AD patients and control population
LMP2 genotype AD (n = 154) Control (n = 406)
(A) Distribution (and frequency) of R/H polymorphism in AD patients and controlsRR 68 (44.2) 172 (42.4)RH 71 (46.1) 185 (45.6)HH 15 (9.7) 49 (12.1)
LMP2 genotype AD male (n = 51) AD female (n = 100) Control male (n = 128) Control female (n = 270)
(B) AD patients and controls considering genderRR 21 (41.2) 46 (46) 49 (38.3) 120 (44.4)RH 24 (47.1) 45 (45) 59 (46.1) 122 (45.2)HH 6 (11.8) 9 (9) 20 (15.6) 28 (10.4)
LMP2 genotype AD�4+ (n = 67) AD �4− (n = 83) Control�4 (n = 24) Control�4− (n = 17)
(C) ApoE�4+ and�4−RR 28 (41.8) 34 (41) 13 (54.2) 53 (38.1)RH 32 (47.8) 41 (49.4) 9 (37.5) 69 (49.6)HH 7 (10.4) 8 (9.6) 2 (8.3) 17 (12.2)
LMP2 genotype AD EO (n = 52) AD LO (n = 99)
(D) AD patients discriminating early (EO) and late (LO) onsetRR 23 (44.2) 44 (44.4)RH 24 (46.2) 45 (45.5)HH 5 (9.6) 10 (10.1)
χ2 Asymp. sig. (two-sided)
(E) Genetic data set analyzed by Monte Carlo Pearson’sχ2-test and resultsAD vs. control 0.62 0.733AD male vs. AD female 0.468 0.791AD male vs. controls male 0.46 0.795AD female vs. controls female 0.177 0.915AD �4+ vs. AD�4− 0.051 0.975AD �4+ vs. control�4+ 1.093 0.579AD �4− vs. control�4− 0.417 0.812AD EO vs. AD LO 0.077 0.952
Values in parentheses are in percentage.
and young subjects. Moreover, we verified the putative effectsthat the polymorphism at codon 60 of LMP2 could have onthe activity of immunoproteasomes. Although, historicallyCNS has been considered an immunologically privilegedorgan with a very low expression of MHC and almost nullpresence of immunoproteasome[35,36,47,50], we detectedimmunoproteasomes not only in the brain of AD patients butalso in the brain of non-demented elderly subjects. This resultwas confirmed by immunohistochemistry that pointed outhow immunoproteasomes are present in neurons as well as inastrocytes and endothelial cells. In fact, immunohistochem-ical anti-LMP2 probing did not show significant differencebetween samples from AD patients and non-dementedelderly subjects. Immunoproteasome detection in neuronsand astrocytes of human brain was in agreement with a recentstudy[10], and suggested that not only glia but also neuronscould be actively involved in immunological response asassumed by other authors[14,31]. Furthermore, our resultsshowed that LMP2 expression was increased in brain areaaffected by AD (hippocampus) compared to unaffectedregions (cerebellum of AD patients and hippocampus of
non-demented elderly subjects), further confirmed by theconcomitant decrease of�1 subunit level in HPG-AD. It isworthy to note that when we measured proteasome activityin samples, we observed differences in proteasome activityamong affected (HPG-AD) and unaffected brain areas(CBL-AD and K). In particular, a significant decrease intrypsin-like activity emerged in HPG-AD versus CBL-AD(this trend was evident also comparing hippocampi of ADpatients and controls), while both chymotrypsin-like andcaspase-like activity did not show any clear difference. Previ-ous studies[21,22]reported a decrease in chymotrypsin-likeand caspase-like activities in affected brain areas of ADpatients. Such discrepancy with literature might be explainedby different experimental conditions (e.g., at variance withother authors[22] we did not use SDS in the assay buffer) orby the difference in post-mortem interval[22]. In particular,in Keller’s study the post-mortem interval was less then5 hours, whereas ours had a range of 4–41 h. Although we didnot note any correlation of proteasome activities with post-mortem interval in our sample population (data not shown),it is possible that the short post-mortem interval in the Keller

64 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
study was a factor in uncovering the reported differencein chymotrypsin-like activity. Furthermore, in our study,it emerged that immunoproteasome, while being highlyexpressed in the brain (hippocampus) of old non-dementedsubjects appears to be present also in the brain (hippocampus)of young subjects but at a much lower level. Thus, all theseobservations are compatible with the following scenario: (i)immunoproteasome expression is increased in brain regionswhere NFTs and SPs are present and stimulate neuroinflam-mation. In particular,�APP induces activation of NF-�Btranscription factor directly involved in LMP2 expression[2]; such pathway could explain the observed immunoprotea-some increase in hippocampus of AD patients. Moreover, thepresence in hippocampus of molecules, such as A� and PHF-tau that inhibit proteasome activity[18,21,27]could explainthe observed decrease in trypsin-like activity. Intriguingly,a similar increase in immunoproteasome content is presentin brains affected by Huntington, as recently reported[10];(ii) immunoproteasomes are strongly induced by the ageingprocess in the brain (or at least in hippocampus) as shown byits faint presence in young donors, indirectly confirming thepossible role of immunoproteasome in immunosenescence[34]. Furthermore, such result agrees with the theory of achronic neuroinflammation in aged brain, as suggested byother study[28], and with the general concept of inflamma-tion ageing, which points out that a chronic inflammatoryp cence[ ntentc zedp ono esf noto ntents gly,a seb rsd ,m tt ationa s ourfi nh e tod a int catet ngesw erepc reaseo ld bed omes fp ion,r uldb rtantm ordert
Furthermore, our results suggest not only a variation ofproteasome amount and activity in brains of AD patientsand aged individuals, but also an effect of LMP2 codon 60polymorphism on proteasome activity. As expected, in ourcase, record HH subjects were not present, owing to their lowfrequency in the general population[33]. Therefore, RR withRH subjects were compared. To this point, it is importantto consider that RH subjects may have, from this pointof view, three immunoproteasome isoforms as in the twoLMP2 subunits in the upper and lower� rings, two Arginine,two Histidine or one Arginine and one Histidine can bepresent. Furthermore, we can speculate that, assuming thesubstitution RH involves a conformational change (Arginineand Histidine have different chemical features), the isoformcarrying both Arginine and Histidine could be asymmetric.Such structural modification could affect the differentimmunoproteasome functions and not only the caspase-likeactivity that is mainly in charge of�1i subunit. At variance,RR subjects do not have the asymmetric isoform, and thusRR and RH genotypes, rather than R and H alleles, must beconsidered. The first point, we considered, was to determinewhether the two alleles of LMP2 could alter the stability orthe expression of LMP2 subunit. To address this topic, weinvestigated the level of mRNA of the 2 LMP2 isoforms,adopting as far as we know, a novel technique that allowedus to compare, in heterozygotes (RH), the relative amounto outa iedp didn edbs allys omeo leasti oesn r thei udedt ino ccur.O ationo don6 andS 45.4a likea ucht andh ore,t thatt re ofi us, itw ntals s wep to stedp ingi at
rocess is characteristic of ageing and of immunosenes15]. The observed increase in immunoproteasome coould also be in part due to the high levels of oxidiroteins in aged brains[13], which induce the expressif �i subunits[12]. Intriguingly, recent study on muscl
rom aged mice[20] reported an age-related increase innly of immunoproteasome, but also of proteasome coimilar to that we observed in human brain. Accordinn interesting review[17], showed that in aged mourains an increase in 20S� subunit occurs (although otheescribed a decrease in proteasome content[23]). Moreovericro-array study[17] reported a decrease in�2 subunit tha
aking into account of the age-dependent neuroinflammnd increased proteasome content indirectly reconcilendings about the reduction of�1 subunit in aged humaippocampi. Despite some inconsistencies, likely duifferent models or experimental conditions, the dat
he literature, including the data here reported, indihat proteasome composition and activity in CNS chaith ageing, likely affecting neural homeostasis whroteasomes play a pivotal role[11,43]. In particular, weould be tempted to speculate that the observed incf proteasome content in aged human hippocampi couue to a progressive impairment of ubiquitin-proteasystem[23] that leads to a increasedde novo expression oroteasomes[32]. In human brain, such feedback regulateported by Meiners et al. in several in vitro models, coe interpret as a attempt of brain to restore a so impoechanism such as ubiquitin-proteasome system, in
o preserve non-replication neurons from death.
f the two R and H variants, respectively. The data rulevariation of LMP2 mRNA levels depending on the studolymorphism, since the ratio between the two isoformsot diverge from the calibration. This result was confirmy the experiments on the expression of the� and� matureubunits by Western blot assay. In fact, no statisticignificant difference in the content of immunoproteasr constitutive proteasomes emerged, confirming at
n brain tissue that LMP2 codon 60 polymorphism dot alter neither the total amount of proteasomes no
mmunoproteasome relative amount. It cannot be exclhat this polymorphism could affect LMP2 expressionther tissue, since a tissue specific regulation could on the contrary, our data suggest that a strong modificf the activity of proteasomes depending on LMP2 co0 polymorphism, in brain area not affected by NFTsPs (namely CBL-AD and K), where RR subjects hadnd 40.1% of increased chymotrypsin-like and trypsin-ctivity, respect to RH subjects. It is worthy to note that s
rend was equally present in cerebellum of AD patientsippocampi of non-demented elderly controls. Furtherm
hese differences are likely underestimated consideringhe proteasome activities we tested came from a mixtummunoproteasome and constitutive proteasomes. Thill be interesting to check these results in an experimeystem, which expresses only immunoproteasomes areviously suggested[34]. Intriguingly, the biological effecf the LMP2 polymorphism did not emerge when we teroteasome activity in hippocampi of AD patients. Tak
nto account previous studies[18,21,27], we can argue th

M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66 65
the inhibitory effect of molecules, such as�-amyloid andPHF-tau, may affect proteasome activity, thus masking the ef-fect of the LMP2 polymorphism. Accordingly, LMP2 codon60 polymorphism could have a restricted window to exert itsinfluence on AD, likely in the phase preceding the overt brainphenotype characterized by the presence of NFTs and SPs. Inorder to check this hypothesis we carried out a genetic studyon AD and age-matched populations, which did not show anyassociation between LMP2 codon 60 polymorphism and AD,neither stratifying them for gender, presence of the ApoE�4allele or age of onset. These results do not allow to excludedefinitively any possible role for LMP2 codon 60 polymor-phism in AD, as suggested by data regarding polymorphismsof biologically relevant molecules such as p53 which indicatethat a polymorphism can still exert a strong functional roleemerging with age[4] despite the fact that its frequency doesnot change with age[3]. In any case, the data here reportedshow that LMP2 codon 60 polymorphism is capable of mod-ulating proteasome activity in brain tissue (both cerebellumand hippocampus). This observation, together with otherdata of literature, such as its influence on the susceptibilityto TNF-�-induced apoptosis in PBMCs[33], its associationswith autoimmune diseases[19,30,45] and the fact that itis conserved in mice[49], strongly suggest an importantbiological role related to immunoproteasome function.
A
jectP Eu-r oft lth( eman lat-t diA Wet icala ially,w ds
R
nualiatric
m-eta-uro-
P,althy
, eteles
cell
[5] Cardozo C, Michaud C. Proteasome-mediated degradation oftau proteins occurs independently of the chymotrypsin-like ac-tivity by a non-processive pathway. Arch Biochem Biophys2002;408(1):103–10.
[6] Caruso C, Franceschi C, Licastro F. Genetics of neurodegenerativedisorders. N Engl J Med 2003;349(2):193–4.
[7] Checler F, da Costa CA, Ancolio K, Chevallier N, Lopez-PerezE, Marambaud P. Role of the proteasome in Alzheimer’s disease.Biochim Biophys Acta 2000;502(1):133–8.
[8] Chomezynski P, Sacchi N. Single-step method of RNA isolation byacid guanidinium thiocyanate–phenol–chloroform extraction. AnalBiochem 1987;162(1):156–9.
[9] Chomezynski P. A reagent for the single step simultaneous isolationof RNA, DNA and protein from cell and tissue samples. Biotech-niques 1993;15:532–6.
[10] Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, et al. Neuronal inductionof the immunoproteasome in huntington’s disease. J Neurosci2003;23(37):11653–61.
[11] Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, CuervoAM, et al. Characterization of chronic low-level proteasome inhibi-tion on neural homeostasis. J Neurochem 2003;86(2):489–97.
[12] Ding Q, Reinacker K, Dimayuga E, Nukala V, Drake J, Butter-field DA, et al. Role of the proteasome in protein oxidation andneural viability following low-level oxidative stress. FEBS Lett2003;546:228–32.
[13] Floyd RA, Hensley K. Oxidative stress in brain aging. Implica-tions for therapeutics of neurodegenerative diseases. Neurobiol Aging2002;23(5):795–807.
[14] Foster JA, Quan N, Stern EL, Kristensson K, Herkenham M. In-duced neuronal expression of class I major histocompatibility com-
oim-
[ nes-no-ine
[ is-codedicat-
[ nd34):
[ WE,pro-
[ lynomeilib-
1.
[ LV,ition
[ byer’s
[ nc-
[ eing
[ dic-Eng
[ urol
cknowledgments
This work was financed in part by a grant from proROTAGE and FUNCTIONAGE, sponsored by the
opean Commission fifth Framework Program “Aginghe Population”, finalized projects from Minister of Hea“Sviluppo e maturazione delle placche senili nel sistervoso di primati come modello per lo studio della ma
ia di Alzheimer” and “Farmacogenetica della malattialzheimer: ottimizzazione delle terapie e dei servizi”).
hank Prof. R. Ferracini for his help in immunistochemssay, Prof. G. Cenacchi for her logistic support. Spece thank Dr. M. Bonafe and Dr. S. Salvioli for their help anupervision of the experiments.
eferences
[1] American Psychiatric Association. Diagnostic and statistical maof mental disorders. 4th ed. Washington, DC: American PsychAssociation; 1994.
[2] Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflamation and Alzheimer’s disease: critical roles for cytokine/A binduced glial activation, NF-kappaB, and apolipoprotein E. Nebiol Aging 2000;21(3):427–32.
[3] Bonafe M, Olivieri F, Mari D, Baggio G, Mattace R, Sansoniet al. p53 variants predisposing to cancer are present in hecentenarians. Am J Hum Genet 1999;64(1):292–5.
[4] Bonafe M, Salvioli S, Barbi C, Trapassi C, Tocco F, Storci Gal. The different apoptotic potential of the p53 codon 72 allincreases with age and modulates in vivo ischaemia-induceddeath. Cell Death Differ 2004;11(9):962–73.
plex mRNA in acute and chronic inflammation models. J Neurmunol 2002;131(1-2):83–91.
15] Franceschi C, Bonafe M, Valensin S. Human immunosecence: the prevailing of innate immunity, the failing of clotypic immunity, and the filling of immunological space. Vacc2000;18(16):1717–20.
16] Fruh K, Gossen M, Wang K, Bujard H, Peterson PA, Yang Y. Dplacement of housekeeping proteasome subunits by MHC-enLMPs: a newly discovered mechanism for modulating the multalytic proteinase complex. EMBO J 1994;13:3236–44.
17] Gray DA, Tsirigotis M, Woulfe J. Ubiquitin, proteasomes, athe aging brain. Sci Aging Knowledge Environ 2003;2003(RE6.
18] Gregori L, Fuchs C, Figueiredo-Pereira ME, Van NostrandGoldgaber D. Amyloid beta-protein inhibits ubiquitin-dependenttein degradation in vitro. J Biol Chem 1995;270(34):19702–8.
19] Heward JM, Allahabadia A, Sheppard MC, Barnett AH, FrankJA, Gough SC. Association of the large multifunctional proteas(LMP2) gene with Graves’ disease is a result of linkage disequrium with the HLA haplotype DRB1*0304-DQB1*02-DQA1*050Clin Endocrinol 1999;51(1):115–8.
20] Husom AD, Peters EA, Kolling EA, Fugere NA, ThompsonFerrington DA. Altered proteasome function and subunit composin aged muscle. Arch Biochem Biophys 2004;421(1):67–76.
21] Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibitionpaired helical filament-tau in brains of patients with Alzheimdisease. J Neurochem 2003;85:115–22.
22] Keller JN, Hanni KB, Markesbery WR. Impaired proteasome fution in Alzheimer’s disease. J Neurochem 2000;75(1):436–9.
23] Keller JN, Gee J, Ding Q. The proteasome in brain aging. AgRes Rev 2002;1(2):279–93.
24] Kesmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Pretion of proteasome cleavage motifs by neural networks. Protein2002;15(4):287–96.
25] Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Ne1985;42(11):1097–105.

66 M. Mishto et al. / Neurobiology of Aging 27 (2006) 54–66
[26] Kloetzel PM. Antigen processing by the proteasome. Nat Rev2001;2:179–87.
[27] Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R,et al. Inhibition of the ubiquitin-proteasome system in Alzheimer’sdisease. Proc Natl Acad Sci USA 2000;97(18):9902–6.
[28] Lee CK, Weindruch R, Prolla TA. Gene-expression profile of theageing brain in mice. Nat Genet 2000;25(3):294–7.
[29] Licastro F, Grimaldi LM, Bonafe M, Martina C, Olivieri F, CavalloneL, et al. Interleukin-6 gene alleles affect the risk of Alzheimer’sdisease and levels of the cytokine in blood and brain. NeurobiolAging 2003;24(7):921–6.
[30] Maksymowych WP, Tao S, Vaile J, Suarez-Almazor M, Ramos-Remus C, Russell AS. LMP2 polymorphism is associated withextraspinal disease in HLA-B27 negative Caucasian and Mexi-can Mestizo patients with ankylosing spondylitis. J Rheumatol2000;27(1):183–9.
[31] McGeer L, McGeer E. Local neuroinflammation and the progressionof Alzheimer’s disease. J Neurovirol 2002;8(6):529–38.
[32] Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM,et al. Inhibition of proteasome activity induces concerted expressionof proteasome genes and de novo formation of mammalian protea-somes. J Biol Chem 2003;278(24):21517–25.
[33] Mishto M, Bonafe M, Salvioli S, Olivieri F, Franceschi C. Agedependent impact of LMP polymorphisms on TNF alpha-inducedapoptosis in human peripheral blood mononuclear cells. Exp Geron-tol 2002;37(2–3):301–8.
[34] Mishto M, Santoro A, Bellavista E, Bonafe M, Monti D,Franceschi C. Immunoproteasomes and immunosenescence. ARR2003;2:419–32.
[35] Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHCclass I genes in neurons. Science 1995;269(5223):549–52.
[ ssue, and–54.
[ doman.
[ manbrainRes
[39] Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, etal. Inhibitors of the proteasome block the degradation of most cellproteins and the generation of peptides presented on MHC class Imolecules. Cell 1994;78:761–71.
[40] Rock KL, Goldberg AL. Degradation of cell proteins and the gen-eration of MHC class I-presented peptides. Annu Rev Immunol1999;17:739–79.
[41] Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron1991;6(4):487–98.
[42] Sorbi S, Nacmias B, Forleo P, Latorraca S, Gobbini I, Bracco L, et al.ApoE allele frequencies in Italian sporadic and familial Alzheimer’sdisease. Neurosci Lett 1994;177:100–2.
[43] Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, DingQ, et al. Proteasome inhibition alters neural mitochondrial home-ostasis and mitochondria turnover. J Biol Chem 2004;279(20):20699–707.
[44] The Dementia Study Group of the Italian Neurological Society,Guidelines for the diagnosis of dementia and Alzheimer’s disease.Neurol Sci 2000;21(4):187–194.
[45] Vinasco J, Fraile A, Nieto A, Beraun Y, Pareja E, Mataran L,et al. Analysis of LMP and TAP polymorphisms by polymerasechain reaction-restriction fragment length polymorphism in rheuma-toid arthritis. Ann Rheum Dis 1998;57(1):33–7.
[46] Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecularmachine designed for controlled proteolysis. Annu Rev Biochem1999;68:1015–68.
[47] Zanelli E, Zhou P, Cao H, Smart MK, David CS. Genomic organi-zation and tissue expression of the mouse proteasome gene Lmp-7.Immunogenetics 1993;38(6):400–7.
[48] Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasser-man AJ, et al. The induction of the TNFalpha death domain
Res
[ eticea-81–
[ andomics
36] Noda C, Tanahashi N, Shimbara N, Hendil KB, Tanaka K. Tidistribution of constitutive proteasomes, immunoproteasomesPA28 in rats. Biochem Biophys Res Commun 2000;277(2):348
37] Piccinini M, Mostert M, Croce S, Baldovino S, Papotti M, RinauMT. Interferon-gamma-inducible subunits are incorporated in hubrain 20S proteasome. J Neuroimmunol 2003;135(1-2):135–40
38] Preece P, Cairns NJ. Quantifying mRNA in post-mortem hubrain: influence of gender, age at death, post-mortem interval,pH, agonal state and inter-lobe mRNA variance. Mol Brain2003;118(1–2):60–71.
signaling pathway in Alzheimer’s disease brain. Neurochem2003;28(2):307–18.
49] Zhou P, Cao H, Smart M, David C. Molecular basis of genpolymorphism in major histocompatibility complex-linked protsome gene (LMP-2). Proc Nat Acad Sci USA 1993;90:264.
50] Zhou P, Zanelli E, Smart M, David C. Genomic organizationtissue expression of mouse proteasome gene Lmp-2. Gen1993;16(3):664–8.




















