
Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms...
12
Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments Mirella L. Meyer-Ficca, 1 Ralph G. Meyer, 1 Donna L. Coyle, Elaine L. Jacobson, and Myron K. Jacobson * Department of Pharmacology and Toxicology, College of Pharmacy, Arizona Cancer Center, University of Arizona, Tucson, AZ 85724, USA Received 12 February 2004, revised version received form 22 March 2004 Available online 27 April 2004 Abstract Poly(ADP-ribose) glycohydrolase (PARG) is the only protein known to catalyze hydrolysis of ADP-ribose (ADPR) polymers to free ADP-ribose. While numerous genes encode different poly(ADP-ribose) polymerases (PARPs) that all synthesize ADP-ribose polymer, only a single gene coding for PARG has been detected in mammalian cells. Here, we describe two splice variants of human PARG mRNA, which lead to expression of PARG isoforms of 102 kDa (hPARG102) and 99 kDa (hPARG99) in addition to the full-length PARG protein (hPARG111). These splice variants differ from hPARG111 by the lack of exon 1 (hPARG102) or exons 1 and 2 (hPARG99). They are generated by the utilization of ambiguous splice donor sites in the PARG gene 5V untranslated region. The hPARG111 isoform localizes to the nucleus, whereas hPARG102 and hPARG99 are cytoplasmic proteins. The nuclear targeting of hPARG111 is due to a nuclear localization signal (NLS) in exon 1 that was mapped to the amino acids (aa) 10 CTKRPRW 16 . Immunocytochemistry, immunoblotting, and PARG enzyme activity measurements show that the cytoplasmic isoforms of PARG account for most of the PARG activity in cells in the absence and presence of genotoxic stress. The predominantly cytoplasmic location of cellular PARG is intriguing as most known cellular PARPs have a nuclear localization. D 2004 Elsevier Inc. All rights reserved. Keywords: PARG; Poly(ADP-ribosyl)ation; PARP; Splice variants; Nuclear localization signal Introduction The synthesis and rapid turnover of ADP-ribose (ADPR) polymers are immediate responses of cells to DNA damage [1–3]. This unique biopolymer is synthe- sized by members of the poly(ADP-ribose) polymerase (PARP) family as a posttranslational protein modification and consists of up to 200 ADP-ribose residues covalently bound to acceptor proteins [4–8]. PARP-1 is the major activity that catalyzes polymer synthesis in response to DNA strand breaks, and polymer levels can increase more than 100-fold in minutes [6,9]. Once synthesized, ADPR polymers are rapidly degraded by the action of poly(ADP- ribose) glycohydrolase (PARG) [6,10]. Nuclear ADPR polymer metabolism modulates cellular responses to gen- otoxic stress including stimulation of DNA repair [11], and thus it is an important factor for maintenance of genomic integrity [6,12–14]. In addition to PARP-1, the first discovered and most abundant PARP, a family of proteins located throughout the cell, has been shown to have PARP activity [15]. Most known PARPs are located in the nucleus. PARP-1 [16] and PARP-2 [17] are located throughout the nucleus, tankyrase-1 is associated with telomeres [18], PARP-2 with centromeres in mitotic chro- mosomes [19], and several PARPs are associated with centrosomes [20,21]. Extranuclear PARPs include VPARP, which is associated with vaults, large ribonucleoprotein complexes with unknown function [22], and tankyrase-2 which interacts with proteins located in the Golgi complex [23] and endosomes [24]. In contrast to many genes that encode PARPs, thus far, only a single gene has been found to code for PARG in 0014-4827/$ - see front matter D 2004 Elsevier Inc. All rights reserved. doi:10.1016/j.yexcr.2004.03.050 * Corresponding author. College of Pharmacy, University of Arizona, Room 3985 Arizona Cancer Center, 1515 North Campbell Avenue, Tucson, AZ 85724. Fax: +1-520-626-8657. E-mail address: [email protected] (M.K. Jacobson). 1 The first two authors contributed equally to this work. www.elsevier.com/locate/yexcr Experimental Cell Research 297 (2004) 521 – 532
Transcript of Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms...

www.elsevier.com/locate/yexcr
Experimental Cell Research 297 (2004) 521–532
Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice
variants yielding isoforms that localize to different cell compartments
Mirella L. Meyer-Ficca,1 Ralph G. Meyer,1 Donna L. Coyle,Elaine L. Jacobson, and Myron K. Jacobson*
Department of Pharmacology and Toxicology, College of Pharmacy, Arizona Cancer Center, University of Arizona, Tucson, AZ 85724, USA
Received 12 February 2004, revised version received form 22 March 2004
Available online 27 April 2004
Abstract
Poly(ADP-ribose) glycohydrolase (PARG) is the only protein known to catalyze hydrolysis of ADP-ribose (ADPR) polymers to free
ADP-ribose. While numerous genes encode different poly(ADP-ribose) polymerases (PARPs) that all synthesize ADP-ribose polymer, only a
single gene coding for PARG has been detected in mammalian cells. Here, we describe two splice variants of human PARG mRNA, which
lead to expression of PARG isoforms of 102 kDa (hPARG102) and 99 kDa (hPARG99) in addition to the full-length PARG protein
(hPARG111). These splice variants differ from hPARG111 by the lack of exon 1 (hPARG102) or exons 1 and 2 (hPARG99). They are
generated by the utilization of ambiguous splice donor sites in the PARG gene 5V untranslated region. The hPARG111 isoform localizes to the
nucleus, whereas hPARG102 and hPARG99 are cytoplasmic proteins. The nuclear targeting of hPARG111 is due to a nuclear localization
signal (NLS) in exon 1 that was mapped to the amino acids (aa) 10CTKRPRW16. Immunocytochemistry, immunoblotting, and PARG enzyme
activity measurements show that the cytoplasmic isoforms of PARG account for most of the PARG activity in cells in the absence and
presence of genotoxic stress. The predominantly cytoplasmic location of cellular PARG is intriguing as most known cellular PARPs have a
nuclear localization.
D 2004 Elsevier Inc. All rights reserved.
Keywords: PARG; Poly(ADP-ribosyl)ation; PARP; Splice variants; Nuclear localization signal
Introduction ribose) glycohydrolase (PARG) [6,10]. Nuclear ADPR
The synthesis and rapid turnover of ADP-ribose
(ADPR) polymers are immediate responses of cells to
DNA damage [1–3]. This unique biopolymer is synthe-
sized by members of the poly(ADP-ribose) polymerase
(PARP) family as a posttranslational protein modification
and consists of up to 200 ADP-ribose residues covalently
bound to acceptor proteins [4–8]. PARP-1 is the major
activity that catalyzes polymer synthesis in response to
DNA strand breaks, and polymer levels can increase more
than 100-fold in minutes [6,9]. Once synthesized, ADPR
polymers are rapidly degraded by the action of poly(ADP-
0014-4827/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.yexcr.2004.03.050
* Corresponding author. College of Pharmacy, University of Arizona,
Room 3985 Arizona Cancer Center, 1515 North Campbell Avenue, Tucson,
AZ 85724. Fax: +1-520-626-8657.
E-mail address: [email protected] (M.K. Jacobson).1 The first two authors contributed equally to this work.
polymer metabolism modulates cellular responses to gen-
otoxic stress including stimulation of DNA repair [11], and
thus it is an important factor for maintenance of genomic
integrity [6,12–14]. In addition to PARP-1, the first
discovered and most abundant PARP, a family of proteins
located throughout the cell, has been shown to have PARP
activity [15]. Most known PARPs are located in the
nucleus. PARP-1 [16] and PARP-2 [17] are located
throughout the nucleus, tankyrase-1 is associated with
telomeres [18], PARP-2 with centromeres in mitotic chro-
mosomes [19], and several PARPs are associated with
centrosomes [20,21]. Extranuclear PARPs include VPARP,
which is associated with vaults, large ribonucleoprotein
complexes with unknown function [22], and tankyrase-2
which interacts with proteins located in the Golgi complex
[23] and endosomes [24].
In contrast to many genes that encode PARPs, thus far,
only a single gene has been found to code for PARG in

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532522
mammals, suggesting that products of this single gene are
needed to complete ADP-ribose polymer cycles. Although
the cDNAs coding for the human [25,26], bovine [27],
murine [28], and rat PARG [29] have been cloned, the
subcellular localization of PARG and the mechanisms by
which PARG is targeted to different subcellular compart-
ments are poorly understood. There are reports of putative
nuclear localization signal (NLS) [27] and nuclear export
signals (NES) [29] in PARG. A nuclear localization of an
overexpressed PARG-GFP fusion protein [25] has been
observed, while other studies have indicated a cytosolic
location of overexpressed PARG [30,31]. Here, we dem-
onstrate three splice variants of human PARG, which are
expressed from a single gene and give rise to PARG
isoforms targeted to the nuclear and cytoplasmic compart-
ments of the cell. Our results demonstrate that alternative
splicing provides a mechanism by which a single PARG
gene can generate a family of different PARG proteins that
can complete ADP-ribose polymer cycles in multiple
cellular locations. Our results show that, in contrast to
the known PARPs, most of the cellular PARG activity is
present in the cytoplasmic rather than nuclear compartment
of the cell.
Materials and methods
Cloning of human PARG cDNA by 5VRACE and
construction of expression vectors
Human total RNA was isolated from cultured CF3
normal skin fibroblasts as described earlier [26]. Briefly,
CF3 cells were cultured in DMEM (Sigma, St. Louis, MO)
with 10% calf serum (Hyclone) under standard cell culture
conditions (5% CO2, 37jC). Cells were removed from the
dish by treatment with trypsin, and total RNA was isolated
from 106 cells using a kit (Qiagen). Additionally, com-
mercially available total human skin RNA (Stratagene) was
reversely transcribed and processed to ensure reproduction
of the results from a different RNA source. One micro-
gram of total RNA each was subjected to reverse tran-
scription using a RACE kit (Clontech) which utilizes
SMARTk cDNA technology and Powerscriptk moloney
murine leukemia virus (MoMuLV) reverse transcriptase
from Clontech. After first-strand synthesis, PCR amplifi-
cation was directly performed using a proofreading DNA
polymerase mix (Expand, Roche) in a ‘‘touch-down PCR’’
protocol which omits a separate annealing step in favor
of higher amplification specificity. A universal primer
mix (UPM, Clontech) was used for forward priming
in the 5VRACE reaction in combination with a
specific reverse primer that binds in exon 3 (hPARG-
exon3 rev; 5 V-CCGTAGTTCTGCTTTGCATTTG-
CAAGCTTTACAGTTGT-3V) or exon 6 (hPARG-exon6-
rev: 5V-GTGCAGTCTGAATGAGCTCCCACCGGCTC-3V)to amplify the 5V untranslated region of the PARG gene.
Full-length cDNA clones were generated by using the
UPM primer in combination with a primer hPARG-
exon18rev which specifically binds at the end of the
coding region in exon 18 (5V-GGACCGGTCCTCAGGT-CCCTGTCCTTTGCCCTGAATG-3V) including the trans-
lation stop codon. As an alternative to UPM, either a
forward primer which binds in the distal portion of the
PARG 5V-UTR (hPARG-UTRfor: 5V-CGGAATTCGG-
GAAAGTGAACGAATCCCGAATCAAAGCGGCGC-3V)or a forward primer specific for exon 1 of PARG (hPARG-
exon1fo r : 5 V-GCGAATTCCCAGCATGAATGC-
GGGCCCCGGC-3V) was used with primer hPARG-exon6-
rev or with hPARG-exon18rev. PCR products were
purified by agarose gel electrophoresis, and the bands
were excised. Purified fragments were cloned into
pcDNA3.1/V5-His-TOPO using a topoisomerase TA clon-
ing kit (Invitrogen). After transformation of TOP10 bacte-
ria (Invitrogen), resulting pcDNA-PARG clones were
analyzed by complete sequencing of the inserts (both
strands). A representative DNA fragment generated with
primers UPM and hPARGrev1 coding for the human
PARG 5V end and N-terminal untranslated region isolated
from reversely translated CF3 RNA was subcloned into
pcDNA-hPARG [26] using restriction enzymes AflII and
BstX1. The resulting plasmid pcDNA-hPARG102 was
sequenced to ensure ORF integrity and used for transfec-
tion studies. By cloning a complete 5VRACE product
encoding hPARG99 amplified with primers UPM and
hPARG-exon18rev into pcDNA3.1 using the TOPO TA
kit, pcDNA-hPARG99 was generated.
Computational sequence analyses
Sequence alignments were performed by comparing
PARG cDNA to the NCBI database using the BLAST
[32] program which is accessible via the World Wide Web
(http://www.ncbi.nlm.nih.gov/BLAST). Cloned cDNA 5Vends that were identified to contain PARG sequences were
further analyzed for the presence of the 5VUTR and for the
correct sequence of the predicted splicing products of the
gene. The human EST database was searched for examples
of 102-kDa and 99-kDa hPARG splice variant clones using
the same program. The PARG protein sequence or frag-
ments of it were analyzed using the computer program
PSORTII (http://psort.nibb.ac.jp).
In vitro expression and immunoblot analyses of human
PARG
Coupled in vitro transcription and translation analyses
were carried out using a rabbit reticulocyte-based TNT
expression kit (Promega) according to the protocol supplied
by the manufacturer. As templates, 2 Ag of each pcDNA-
hPARG111, pcDNA-hPARG102 or pcDNA-hPARG99, lin-
earized with ScaI, or pT7luc control DNA (Promega), was
used per reaction. Samples of the positive control reactions

Fig. 1. Specific PCR amplification of PARG cDNA products. Using primers
in the 5VUTR (UTRfor) upstream of the newly identified splice donor sites
and at the end of the ORF in exon 18 (Exon18rev, lane a), two bands (i and
ii) were produced corresponding to hPARG111 (band i, with an expected
size of 3193 bp) and hPARG102 (band ii, 2761–2794 bp) respectively. The
latter included hPARG99 (band ii, 2694 or 2698 bp, depending on the
splice donor sites used in the individual cDNA). When a primer specific for
exon 1 (Exon1for) was used instead of UTRfor (lane b), only one band with
the expected size of 2954 bp (band iii) encoding the hPARG111 ORF was
obtained. Similarly, using the UTRfor primer in combination with a reverse
primer located in exon 6 (Exon6rev, lane c), two bands, representing
hPARG111 (band iv, 1880 bp) and hPARG102 (band v, 1448–1481 bp), as
well as hPARG99 (band v, 1385 or 1381 bp) were observed. If primer
Exon1for was used instead of UTRfor (lane d), a single band marked the
expected 1641-bp fragment of the hPARG111 ORF (band vi). All bands (i –
vi) were reproducibly obtained, cloned, and sequenced from CF3 fibroblast
RNA and from commercially available human skin total RNA (Stratagene).
M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532 523
were subjected to luciferase activity assays to ensure func-
tionality of the in vitro transcription and translation proce-
dure. Total cell extracts of HeLa cells (ATCC) were
prepared according to standard procedures [33]. Western
blot analyses of in vitro translated protein and total
cell lysates were performed using a standard SDS–PAGE
method with 8% polyacrylamide gels. After electrotransfer
to PVDF membranes (Millipore), PARG protein was
detected using a polyclonal rabbit antiserum raised against
peptide KNSCQDSEADEETSP in the N-terminal region of
PARG and a secondary goat anti rabbit antibody coupled to
horseradish peroxidase (Jackson Laboratories) with an en-
hanced chemoluminescence (ECL) reaction.
Construction of a GFL reporter gene fusion with human
PARG exon 1
A DNA fragment coding for human PARG exon 1 was
generated by PCR using Expand PCR polymerase (Roche)
and oligonucleotide primers designated PGEXFOR (5VCGACTCACTATAGGGAGACCCAAGCTGGCTAGC G
3V, containing an additionalNheI recognition site, underlined)and PGEXREV (5V CCTTTAGTGTCCATGG AACTGG-
TAATAGTCTTTTGTTGTTTGAAAACAAGC 3V which
has an additional NcoI site; underlined). The PCR product
was subcloned into plasmid pGFL [34] using restriction sites
NcoI and NheI, yielding plasmid pEx1-GFL. In frame fusion
of the ORF of the PCR product to the reporter gene was
verified by sequencing. The NheI/BglII fragment of pEx1-
GFL which contained the exon 1 reporter gene fusion was
subcloned into vector pcDNA3.1 (precut with NheI and
BamHI) leading to the expression plasmid pCMV-Ex1-GFL.
Site-directed mutagenesis
To define the amino acids (aa) that are essential for NLS
function, a PCR-based site-directed mutagenesis was per-
formed using the Quikchange Site-directed Mutagenesis Kit
(Stratagene) according to the manufacturer’s instruction. The
two candidate NLS sequences that had been identified by the
PSORTII program in exon 1 were altered sequentially with
two specific primer pairs using plasmid pEx1-GFL as a
template. For mutation of putative NLS1, primers NLS1MF
(5V-GCTGTGAACCCTGCACCGCCGGCCCCCGCTGG-GGCGCCGC-3V) and NLS1MR (5V-GCGGCGCCCCAGC-GGGGGCCGGCGGTGCAGGGTTCACAGC-3V) were
used. For alteration of putative NLS2, primers NLS2MF
(5V-GTCCTTGGGGTCGAGGACGGCGCCCTGCCTGC-TGGGAAAGC-3V and NLS2MR 5VGCTTTCCCAGCAGG-CAGGGCGCCGTCCTCGACCCCAAGGAC-3V) were
used.
Fusion of the putative bipartite NLS region to GFL
Sequence analyses published previously had indicated
the presence of a putative bipartite NLS in the central
region of the protein, between amino acids 422 and 439
of human PARG [27]. To determine if this peptide
sequence also mediates nuclear localization, a PCR frag-
ment coding for this region was amplified using primer
pair NLSfor EcoRI (5 VGTGAATTCATGAGAC-
ATGAGACTGCCCAAAGCAGAGGAC3V, additional
EcoRI site underlined) and NLSrevNcoI (5VCTG-
CCATGGCCTTCCTTTCTGTTCTTTGATGTTTGGTTT-
CCCA3V; additional NcoI site underlined). The PCR
product was digested with EcoRI and NcoI and subcloned
into plasmid pGFL, fusing the PARG peptide ORF in
frame to the 5V terminus of the GFL reporter gene. The
complete open reading frame consisting the PARG pep-
tide and the GFL reporter was then placed into plasmid
pcDNA3.1 using the restriction endonucleases NheI,
BamHI and BglII. The resulting expression plasmid
pCMV-bpNLS-GFL was used for transfection and over-
expression of the fusion protein.
Culture, transfection, and treatment of cells
HeLa cells and the human embryonal kidney cell line
HEK293 were obtained from ATCC and cultured in
DMEM (Sigma) supplemented with 10% fetal bovine
serum (Hyclone). For overexpression of human PARG
M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532524
protein or reporter protein, HEK293 cells were seeded at
a density of 5 � 105 cells/well into six-well plates which
contained one sterile coverslip per well. After 24 h, cells
were transfected with 1 Ag plasmid per well using
FuGene6 transfection reagent (Roche) according to the
manufacturer’s protocol. The treatment with N-methyl-N V-nitro-N-nitrosoguanidine (MNNG), an alkylating agent
that induces base excision repair and PARP-1 and
PARP-2 activation, was performed on HEK293 cells in
T75 flasks in culture medium with a final concentration
of 500 AM. After 15 min of treatment, cells were
harvested and assayed as described below.
Microscopic preparations and immunofluorescence staining
One day after transfection, cells were washed with
PBS twice, then fixed for 10 min at room temperature
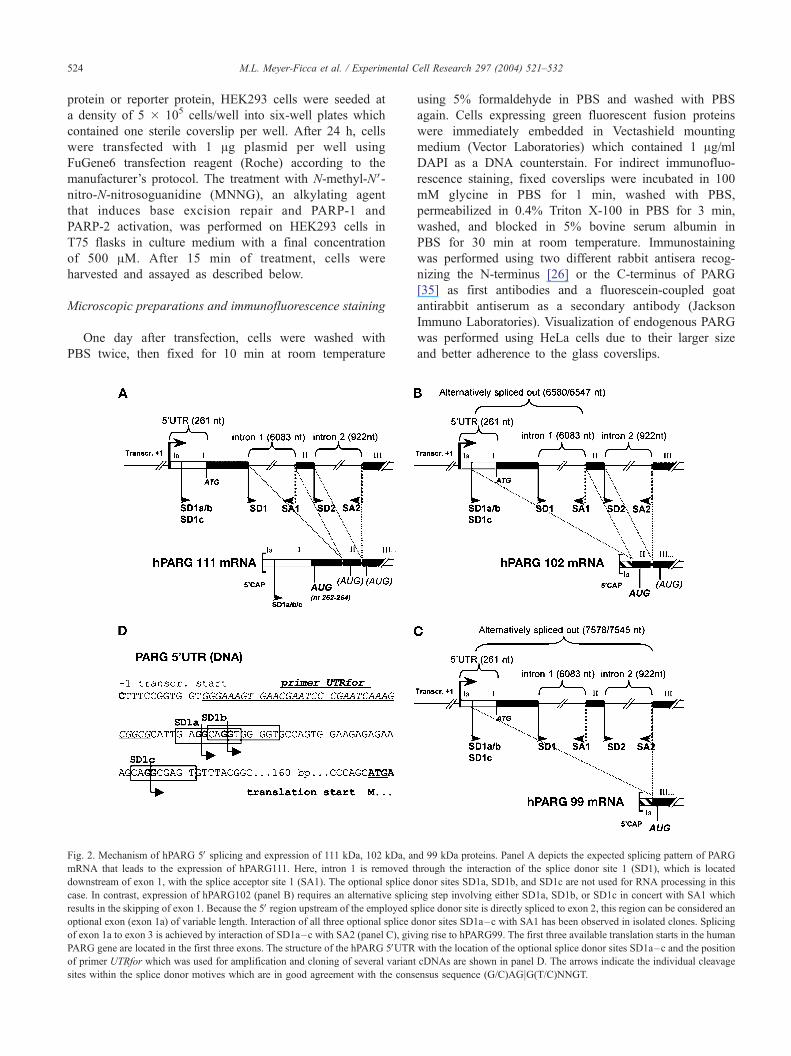
Fig. 2. Mechanism of hPARG 5V splicing and expression of 111 kDa, 102 kDa, an
mRNA that leads to the expression of hPARG111. Here, intron 1 is removed t
downstream of exon 1, with the splice acceptor site 1 (SA1). The optional splice d
case. In contrast, expression of hPARG102 (panel B) requires an alternative splic
results in the skipping of exon 1. Because the 5V region upstream of the employed s
optional exon (exon 1a) of variable length. Interaction of all three optional splice d
of exon 1a to exon 3 is achieved by interaction of SD1a–c with SA2 (panel C), giv
PARG gene are located in the first three exons. The structure of the hPARG 5VUTRof primer UTRfor which was used for amplification and cloning of several variant
sites within the splice donor motives which are in good agreement with the cons
using 5% formaldehyde in PBS and washed with PBS
again. Cells expressing green fluorescent fusion proteins
were immediately embedded in Vectashield mounting
medium (Vector Laboratories) which contained 1 Ag/ml
DAPI as a DNA counterstain. For indirect immunofluo-
rescence staining, fixed coverslips were incubated in 100
mM glycine in PBS for 1 min, washed with PBS,
permeabilized in 0.4% Triton X-100 in PBS for 3 min,
washed, and blocked in 5% bovine serum albumin in
PBS for 30 min at room temperature. Immunostaining
was performed using two different rabbit antisera recog-
nizing the N-terminus [26] or the C-terminus of PARG
[35] as first antibodies and a fluorescein-coupled goat
antirabbit antiserum as a secondary antibody (Jackson
Immuno Laboratories). Visualization of endogenous PARG
was performed using HeLa cells due to their larger size
and better adherence to the glass coverslips.
d 99 kDa proteins. Panel A depicts the expected splicing pattern of PARG
hrough the interaction of the splice donor site 1 (SD1), which is located
onor sites SD1a, SD1b, and SD1c are not used for RNA processing in this
ing step involving either SD1a, SD1b, or SD1c in concert with SA1 which
plice donor site is directly spliced to exon 2, this region can be considered an
onor sites SD1a–c with SA1 has been observed in isolated clones. Splicing
ing rise to hPARG99. The first three available translation starts in the human
with the location of the optional splice donor sites SD1a–c and the position
cDNAs are shown in panel D. The arrows indicate the individual cleavage
ensus sequence (G/C)AGjG(T/C)NNGT.

Table 1
Characteristics of human PARG cDNA variants cloned from total RNA
derived from human fibroblasts (CF3), skin (Stratagene), or placentaa
(P, Clontech)
PARG
variant
Prot. size
kDa (aa)
Length
of cDNA
[bp]
Cloned from
(Pa/CF3/skin)
EST evid.
(GenBank)
GenBank
acc. #
hPARG111 111.1
(976)
3193 Pa,
CF3,
skin
BQ220449.1
BQ232736.1
BG714165.1
and others
AY258587
AF005043
hPARG102 102.3
(894)
2761
(SD1a)
CF3,
skin
BG719380 AY575848
2765
(SD1b)
2794
(SD1c)
hPARG99 99.3
(867)
2698
(SD1b)
CF3 AY575849
The accession numbers of the cloned cDNAs and EST clones with
similarity to them are indicated.a See reference [26].
M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532 525
Determination of PARG activity in nuclear and cytosolic
fractions
Nuclear and cytosolic fractions were isolated from
HEK293 and HeLa cells using the NucleiPURE Prep Nuclei
Isolation Kit (Sigma, St. Louis, MO) according to the
manufacturer’s instructions. The protein content of each
fraction was determined using the BCA Protein Assay
(Pierce Biotechnology, Inc., Rockford, IL). PARG activity
was measured in each fraction following sonication (3 � 10
s; on ice) as described by Menard and Poirier [36].
Fig. 3. Coupled T7RNA polymerase-dependent in vitro transcription/
translation of hPARG expression plasmids encoding the 111-kDa (a), 102-
kDa (b), and 99-kDa (c) forms of hPARG. In this experiment, expression of
the 111-kDa variant always showed three bands (arrows), presumably due
to usage of two additional downstream translation start codons in exon 2
(M83) and 3 (M110). Accordingly, the hPARG102 cDNA is translated in two
forms, corresponding to the 102-kDa and the 99-kDa protein variants. The
hPARG99 plasmid led to the formation of one band only (c). A firefly
luciferase plasmid (pT7-luc, Promega) was used as a control (d).
Results
Human PARG is expressed as multiple splice variants
To search for possible splice variants of PARG mRNA,
two separate strategies were used. First, a 5’RACE-PCR
amplification of hPARG cDNA was completed using a
universal primer mix in combination with a specific reverse
primer in exons 6 or 18 to search for splice variants with
differences in the 5’ region of PARG mRNA. This system
resulted in the generation of PCR products of at least two
distinct sizes in both CF3 and total skin RNA. Second, based
on sequence analyses of the PCR products, a forward primer
located in the 5’UTR of the PARG gene (UTRfor) was
designed for subsequent PCR experiments. Fig. 1 shows
examples using the UTRfor primer where agarose gel
electrophoresis revealed that the size of the major PCR
product derived from CF3 fibroblasts (Fig. 1, bands i and
iv) corresponded to the 111-kDa PARG mRNA described
previously [26]. However, bands of reduced intensity (Fig. 1,
bands ii and v) were also observed. Control PCR reactions
with a primer specific for exon 1 and reverse primers in exon
6 (Fig. 1, band vi) or exon 18 (Fig. 1, band iii) yielded
only a single band containing the sequences predicted for
hPARG111. Sequence analyses of the cloned fragments
isolated from band ii showed that it contained two alterna-
tively spliced mRNA species of PARG that encode proteins
of calculated molecular masses of 102 kDa and 99 kDa that
were designated hPARG102 and hPARG99, respectively.
The newly identified hPARG splice variants do not contain
exon 1
Recently, we have reported the structure of the human
PARG gene [26], (NCBI GenBank Accession Number
AY258587) with its intron/exon organization and its
complete 5V upstream sequence (GenBank Accession
Number AY258588). Comparing the cDNA sequences
of the hPARG102 and hPARG99 splice variants to this
published cDNA revealed that hPARG102 does not con-
tain exon 1 and that hPARG99 does not contain either
exon 1 or exon 2, although both splice variants share the
first 51 or 55 nucleotides of the 5’UTR of the full-length
PARG cDNA, depending upon which splice donor site is
used as described below. In addition, a second hPARG102
splice variant was detected, which shares the first 74 bp
of the 5VUTR of hPARG111. Three putative splice donor
sites were identified in the 5VUTR of the PARG gene,
each of which is consistent with a consensus splice donor
sequence (G/C)AGjG(T/C)NNGT. These splice donor sites
have been designated SD1a (GAGjGCAGGT), SD1b,

M.L. Meyer-Ficca et al. / Experimental C526
which partially overlaps with SD1a (CAGjGTGGGGT,with the overlapping sequences given in italics) and
SD1c (CAGjGCGAGT) (Fig. 2, panel D). The structures
of hPARG102 and hPARG99 are consistent with the
absence of exon 1 or both exons 1 and 2, respectively,
due to these alternative splice donor sites. A schematic
model of the splicing process leading to the different
splice variants is shown in Fig. 2, panels A, B, and C for
hPARG111, hPARG102, and hPARG99, respectively. Ad-
ditional information concerning these splice variants and
EST evidence obtained through the NCBI BLAST pro-
gram supporting the occurrence of these splice variants is
presented in Table 1.
Fig. 4. hPARG111 is a nuclear protein; hPARG102 and hPARG99 are localized
HEK293 cells overexpressing different PARG isoforms (plasmids pcDNA-hPAR
antibodies recognizing the PARG N-terminal portion (a, c, e) and the PARG C-term
m). PARG fluorescence signals in cells overexpressing hPARG111 were observed i
seen in the cytoplasm (c, d, i, k; e, f, l, m). Human PARG111 protein corresponds
portion coded for by exon1 and hPARG99 lacks the region coded for by exons
putative PARG protein domains [26,46].
In vitro transcription and translation of human PARG cDNA
variants yielded proteins of different sizes
In vitro transcription and translation of the hPARG111
cDNA yielded three distinct bands of protein in an immuno-
blot analysis (Fig. 3, lane a) as observed previously [26]. The
largest product is expected to represent the 976-amino acid,
111-kDa isoform of PARG, and the two smaller bands most
likely represent alternative translation start codon usage that
results in smaller products. In vitro expression of hPARG102
yielded two detectable bands (Fig. 3, lane b), and expression
of hPARG99 resulted in detection of only a single band (Fig.
3, lane c). In contrast to hPARG111, hPARG102 does not
ell Research 297 (2004) 521–532
in the cytoplasm. Immunofluorescence staining of transiently transfected
G111, pcDNA-hPARG102, and pcDNA-hPARG99) was performed using
inal portion (g, i, l), and DNAwas counterstained using DAPI (b, d, f, h, k,
n the nucleus (a, b, g, h), while both hPARG102 and hPARG99 signals were
to the full-length cDNA, while splice isoform hPARG102 lacks the protein
1 and 2 (see scheme below images). Bold letters A, B, C, D designate the

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532 527
contain the 72-amino acid segment encoded by exon 1, and
omission of exon 1 results in a transcript in which an ORF is
dependent on a translation initiation codon (ATG) which is
located further downstream in the cDNA. Translation is
predicted to start in exon 2 at amino acid 83 (M83) as the
first available methionine encoded in frame, giving rise to a
(893 aa) 102-kDa protein. At this site, the coding sequence
TGGATGG does not represent a perfect Kozak consensus
sequence (ANNATGG, [37], with the underlined nucleotides
being preferably A or G). The next available translation start
is located in exon 3 at M109 (TCCATGA) or, more likely,
M110 (ATGATGA), which would give rise to a protein of 99
kDa (hPARG99). Therefore, the observed double band of
hPARG102 likely corresponds to the 102-kDa and the 99-
kDa forms, with the latter being the predominant product
Fig. 5. PARG exon 1 contains a nuclear localization signal. Transiently transfecte
region to a GFL reporter protein. The fusion protein (plasmid pCMV-Ex1-GFL) w
EGFPnuc, Clontech; (c, d), while the unfused reporter protein (pRSV-GFL) was
contains two motives with putative NLS function (NLS #1 and NLS #2). Site-dire
n), while mutagenesis of NLS #2 had no effect on nuclear localization (i, k, n). A
been described of having homology to the PARP-1 bipartite nuclear localization s
panel o).
under the conditions of the experiment. Translation of
hPARG99 resulted in a single detectable product, which
was expected as M109 or M110 are the first available
translation initiation sites in this splice variant. In conclu-
sion, the three products of hPARG111 expression would
correspond to translation of the 111-kDa, 102-kDa, and 99-
kDa ORFs; the two products of hPARG102 expression
would correspond to the 102-kDa and 99-kDa ORFs; and
hPARG99 expression would correspond to the 99-kDa ORF.
Overexpressed hPARG111 localizes predominantly to the
nucleus, while hPARG102 and hPARG99 are cytoplasmic
To determine if the three PARG splice variants encode
proteins targeted to different subcellular compartments,
d HEK293 cells overexpress a fusion protein of the PARG exon 1 encoded
as targeted to the nucleus (a, b) as observed with a positive control (pEF1-
preferentially cytoplasmic (e, f). The peptide sequence encoded by exon 1
cted mutagenesis of NLS#1 completely abolished nuclear localization (g, h,
fusion protein of the GFL reporter with a PARG region that has previously
ignal [27] was only observed to be cytoplasmic (l, m; pCMV-bpNLS-GFL,
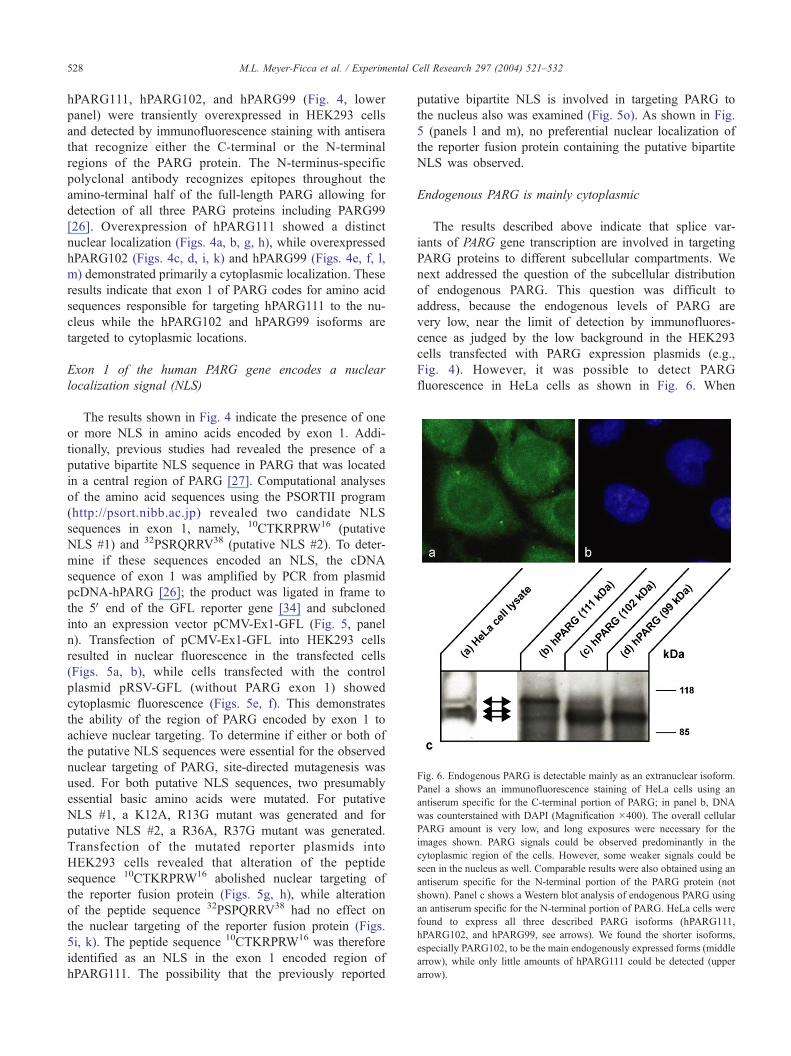
Fig. 6. Endogenous PARG is detectable mainly as an extranuclear isoform.
Panel a shows an immunofluorescence staining of HeLa cells using an
antiserum specific for the C-terminal portion of PARG; in panel b, DNA
was counterstained with DAPI (Magnification �400). The overall cellular
PARG amount is very low, and long exposures were necessary for the
images shown. PARG signals could be observed predominantly in the
cytoplasmic region of the cells. However, some weaker signals could be
seen in the nucleus as well. Comparable results were also obtained using an
antiserum specific for the N-terminal portion of the PARG protein (not
shown). Panel c shows a Western blot analysis of endogenous PARG using
an antiserum specific for the N-terminal portion of PARG. HeLa cells were
found to express all three described PARG isoforms (hPARG111,
hPARG102, and hPARG99, see arrows). We found the shorter isoforms,
especially PARG102, to be the main endogenously expressed forms (middle
arrow), while only little amounts of hPARG111 could be detected (upper
arrow).
M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532528
hPARG111, hPARG102, and hPARG99 (Fig. 4, lower
panel) were transiently overexpressed in HEK293 cells
and detected by immunofluorescence staining with antisera
that recognize either the C-terminal or the N-terminal
regions of the PARG protein. The N-terminus-specific
polyclonal antibody recognizes epitopes throughout the
amino-terminal half of the full-length PARG allowing for
detection of all three PARG proteins including PARG99
[26]. Overexpression of hPARG111 showed a distinct
nuclear localization (Figs. 4a, b, g, h), while overexpressed
hPARG102 (Figs. 4c, d, i, k) and hPARG99 (Figs. 4e, f, l,
m) demonstrated primarily a cytoplasmic localization. These
results indicate that exon 1 of PARG codes for amino acid
sequences responsible for targeting hPARG111 to the nu-
cleus while the hPARG102 and hPARG99 isoforms are
targeted to cytoplasmic locations.
Exon 1 of the human PARG gene encodes a nuclear
localization signal (NLS)
The results shown in Fig. 4 indicate the presence of one
or more NLS in amino acids encoded by exon 1. Addi-
tionally, previous studies had revealed the presence of a
putative bipartite NLS sequence in PARG that was located
in a central region of PARG [27]. Computational analyses
of the amino acid sequences using the PSORTII program
(http://psort.nibb.ac.jp) revealed two candidate NLS
sequences in exon 1, namely, 10CTKRPRW16 (putative
NLS #1) and 32PSRQRRV38 (putative NLS #2). To deter-
mine if these sequences encoded an NLS, the cDNA
sequence of exon 1 was amplified by PCR from plasmid
pcDNA-hPARG [26]; the product was ligated in frame to
the 5V end of the GFL reporter gene [34] and subcloned
into an expression vector pCMV-Ex1-GFL (Fig. 5, panel
n). Transfection of pCMV-Ex1-GFL into HEK293 cells
resulted in nuclear fluorescence in the transfected cells
(Figs. 5a, b), while cells transfected with the control
plasmid pRSV-GFL (without PARG exon 1) showed
cytoplasmic fluorescence (Figs. 5e, f). This demonstrates
the ability of the region of PARG encoded by exon 1 to
achieve nuclear targeting. To determine if either or both of
the putative NLS sequences were essential for the observed
nuclear targeting of PARG, site-directed mutagenesis was
used. For both putative NLS sequences, two presumably
essential basic amino acids were mutated. For putative
NLS #1, a K12A, R13G mutant was generated and for
putative NLS #2, a R36A, R37G mutant was generated.
Transfection of the mutated reporter plasmids into
HEK293 cells revealed that alteration of the peptide
sequence 10CTKRPRW16 abolished nuclear targeting of
the reporter fusion protein (Figs. 5g, h), while alteration
of the peptide sequence 32PSPQRRV38 had no effect on
the nuclear targeting of the reporter fusion protein (Figs.
5i, k). The peptide sequence 10CTKRPRW16 was therefore
identified as an NLS in the exon 1 encoded region of
hPARG111. The possibility that the previously reported
putative bipartite NLS is involved in targeting PARG to
the nucleus also was examined (Fig. 5o). As shown in Fig.
5 (panels l and m), no preferential nuclear localization of
the reporter fusion protein containing the putative bipartite
NLS was observed.
Endogenous PARG is mainly cytoplasmic
The results described above indicate that splice var-
iants of PARG gene transcription are involved in targeting
PARG proteins to different subcellular compartments. We
next addressed the question of the subcellular distribution
of endogenous PARG. This question was difficult to
address, because the endogenous levels of PARG are
very low, near the limit of detection by immunofluores-
cence as judged by the low background in the HEK293
cells transfected with PARG expression plasmids (e.g.,
Fig. 4). However, it was possible to detect PARG
fluorescence in HeLa cells as shown in Fig. 6. When

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532 529
the PARG C-terminus-specific antiserum was used, which
should detect all PARG variants equally well, endogenous
PARG was observed throughout the cell, although cyto-
plasmic fluorescence was more intense than nuclear
fluorescence. This result predicts that the cytoplasmic
isoforms of PARG should be more prevalent than nuclear
isoforms. This prediction was confirmed by a Western
blot analysis of HeLa cell lysates. Fig. 6, panel C,
compares the detection of PARG isoforms derived from
in vitro translation of hPARG111 (lane b), hPARG 102
(lane c), and hPARG 99 (lane d) with endogenous PARG
(lane a). Note that the detection of the multiple bands of
hPARG111 and hPARG102 is not as well resolved as
shown in Fig. 3 as the immunoblot had to be over-
exposed to detect the endogenous PARG. To further
examine the subcellular distribution of PARG, HEK293
and HeLa cell extracts were fractionated into nuclear and
cytosolic fractions and assayed for PARG activity. The
purity of the isolated fractions was verified by immuno-
blotting using an antihistone antibody (results not shown).
Table 2 shows that, although the specific activity of
PARG was higher in HeLa cells than in HEK293 cells,
the majority of total PARG activity was associated with
the cytosolic fraction for both cell types. In untreated cells,
the nuclear fraction accounted for only 19% and 8% of
total PARG activity for HEK293 and HeLa cells, respec-
tively. For HEK293 cells, the distribution of PARG activity
in cells treated for 15 min with 500 AM MNNG was
compared to untreated cells. The MNNG treatment resulted
in a decrease in activity in the nuclear fraction and an
increase in the cytosolic fraction. Taken together, the
results of Fig. 6 and Table 1 provide a consistent picture,
indicating that the majority of PARG has a cytoplasmic
location due to the predominance of the PARG102 and
PARG 99 isoforms.
Table 2
PARG activity in nuclear and cytoplasmic fractions of HEK293 and HeLa
cells
Cell line and condition Poly(ADP-ribose) glycohydrolase activity
Specific activity Total activity
Nuclear Cytosolic Nuclear Cytosolic
pmol/
min/
mg
pmol/
min/
mg
Total
units
%
Total
Total
units
%
Total
HEK293
untreated
366
(F38)
111
(F4)
553
(F57)
19 2330
(F84)
80
HEK293
MNNG
treated
248
(F15)
145
(F13)
393
(F24)
10 3402
(F305)
90
HeLa
untreated
550
(F70)
512
(F30)
780
(F99)
8 8850
(F520)
92
Nuclear and cytosolic fractions were isolated and assayed for PARG
activity as described in Materials and methods. PARG activities shown
represent the mean and SEM of duplicate analyses. Values shown are from
a representative experiment.
Discussion
We have observed that the gene encoding human PARG
is transcribed and processed into splice variants that encode
at least three different protein isoforms with different
molecular weight, namely, 111 kDa, 102 kDa and 99 kDa.
While the 111-kDa PARG is located in the nucleus, the other
two proteins appear to be cytoplasmic. Our results demon-
strate that the amino acids encoded by exon 1, specifically
residues 10CTKRPRW16, represent an NLS for targeting
PARG to the nucleus (Figs. 4 and 5). This sequence is
highly conserved among all known PARG proteins of
mammalian species, and it is in good agreement with the
consensus sequence proposed for monopartite nuclear lo-
calization signals (K/R) RX (K/R) [38]. Subcellular target-
ing of a protein is a result of the influence of signaling
sequences present within the protein and potentially the
interactions with other proteins. Although we have shown
the presence of an active NLS in the N-terminal region of
PARG encoded by exon 1, we cannot exclude that this
region could also be capable of mediating nuclear targeting
by other mechanisms, such as the mediation of protein–
protein interactions between hPARG111 and other NLS-
containing proteins.
Previously, we have reported that PARG contains an
evolutionary conserved region (amino acids 414–450)
showing close analogy to the bipartite NLS in PARP-1
[27]. However, this amino acid sequence does not target a
reporter protein to the nucleus (Figs. 5l, m), and thus it does
not appear to code for a NLS. This region of PARG
probably serves as a hinge region between the putative
regulatory domain A (Fig. 4, lower panel) and the catalytic
domain (B, C, D). This view is supported by the observation
that isolated PARG protein is highly sensitive to protease
cleavage in this region [27,30]. However, our results indi-
cate that nuclear hPARG111 is not cleaved to a great extent
as antibodies directed against both amino terminal (Figs. 4a,
b) and carboxyl terminal regions (Figs. 4g, h) of PARG
indicate a primarily nuclear localization for hPARG111,
although some faint cytoplasmic localization is observed.
A putative nuclear export signal (NES) has been described
in amino acids 126–134 of PARG [29], which might
explain the preferential cytoplasmic localization of the
PARG isoforms lacking exon 1. However, the significance
of this NES is still not entirely clear, as PARG fragments of
74 kDa and 85 kDa, lacking the NES, have also been shown
to reside in the cytoplasm [39]. The presence of other not
yet identified NES sequences or an interaction between
PARG and another NES-containing protein therefore still
needs to be investigated.
Our results provide an explanation of previous studies
that have observed different subcellular localizations of
PARG. In other reports of bovine PARG overexpression
[30,31] that resulted in a perinuclear localization of the
protein, a cDNA that lacked the first 74 amino acids present
in PARG111 was used and thus did not contain the NLS

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532530
identified in the study reported here. A recent report
suggested that the previously expressed bovine PARG
protein was indeed an alternatively spliced PARG isoform
as the cDNA used in that study lacked PARG exon 1 [39].
This isoform, therefore, corresponds to the human
PARG102 isoform described here. Another recent study
has reported that fusion of GFP to the amino terminus of
a full-length human PARG cDNA targets GFP to the
nucleus during interphase and to the cytoplasm during
mitosis [25]. This result is consistent with our finding that
the NLS of PARG is present in the GFP-PARG fusion
protein used in that study.
By virtue of the lack of amino acids encoded by exon 1,
hPARG102 and hPARG99 are targeted to a cytoplasmic
location within the cell. Further studies will be needed to
determine the significance of two different extranuclear iso-
forms of PARG. While our studies demonstrate that alterna-
tive splicing of PARG transcripts can achieve both nuclear
and cytoplasmic targeting, results presented here also suggest
another possibility for achieving different PARG isoforms.
As shown in Figs. 3 and 6c (lane b), in vitro transcription and
translation of hPARG111 resulted in the presence of several
PARG isoforms. They likely arise via alternative sites of
translation initiation, because their size corresponds to the
sizes of the splice variants, which use the respective start
codons, but could theoretically also be proteolytic cleavage
products. However, our results suggest that alternative splic-
ing rather than alternative translation initiation of hPARG111
is the primary mechanism for generation of PARG isoforms
in vivo. This conclusion is based on the observation that
overexpression of hPARG111 from a cDNA construct in
transfected cells (Figs. 4a, b) led to an increase in nuclear
PARG with only minor expression of cytoplasmic PARG.
Our results indicating that the cytoplasmic isoforms of
PARG predominate within the cell (Fig. 6, Table 1) are
somewhat surprising as most PARPs identified to date are
nuclear enzymes. A similar finding describing the majority
of PARG in the soluble fractions of HeLa S3 cells [39],
which correspond to a cytoplasmic localization, is also in
line with our report. It is not clear at present whether the
presence of most PARG in the cytoplasmic cell compart-
ment(s) indicates the presence of as yet undetected PARPs
located outside of the nucleus or as yet undiscovered roles
for PARG. Hanai et al. [40] have shown that a cytoplasmic
accumulation of poly(ADP-ribose) observed in Drosophila
melanogaster PARG loss-of-function mutants resulted in
severe neurodegeneration. Therefore, the presence of PARG
in the cytoplasm might be essential for proper DNA damage
or cell death signaling.
A PARG nuclear-shuttling mechanism has been proposed
[25,41,42] by which cytoplasmic PARG can translocate to
the nucleus under conditions of genotoxic stress, although
our results shown in Table 1 do not support such a
mechanism as the PARG activity in isolated nuclear frac-
tions decreased in MNNG-treated cells compared to un-
treated cells. However, our experiments could not rule out
a mechanism by which PARG would be lost from the
nucleus in MNNG-treated cells.
It has been proposed that 35–60% of all human genes
show alternative splicing [43], leading to more than one
gene product. The mechanism of alternative splicing lead-
ing to expression of proteins targeted to different locations
within the cell has been observed for other proteins. This
mechanism has been described for the uracil-DNA glyco-
sylases UNG1 (mitochondrial) and UNG2 (nuclear), which
rely on alternative splicing in the 5Vregion of the UNG
gene to generate isoforms targeted to the mitochondrial
and nuclear compartments of the cell [44]. These variants
have an additional noncoding exon 1a, as also found in the
PARG gene (see Fig. 2), but in addition, there is also a
second transcription start that is created by another up-
stream promoter. Intriguingly, the uracil-DNA glycosylases
are important participants in the base excision repair
pathway, in which PARP-1 and PARG are also involved
[11]. Another pair of enzymes, the dUTPases DUT-N
(nuclear) and DUT-M (mitochondrial), is spliced alterna-
tively at their 5V end to form two different isoforms [45] by
a mechanism that closely resembles the one reported here
for PARG.
Taken together, the discovery of the PARG protein family
reported here resolves some of the dilemmas raised by the
presence of a PARP enzyme family with more than seven
members that localize in many different cellular compart-
ments, while only a single gene encodes poly(ADP-ribose)
glycohydrolase. On the other hand, the presence of the
majority of PARG activity in the cytoplasmic compartment
of the cell raises interesting new questions concerning the
function of PARG.
Acknowledgments
This work was supported by research grants from the
NIH (CA-43894) and Niadyne, Inc. MKJ and ELJ are
principals in Niadyne Inc., whose sponsored research is
managed in accordance with University of Arizona conflict-
of-interest policies. RGM was supported by NIH training
grant CA-09213. DNA sequence analyses were performed
by the University of Arizona Genetic Analysis and
Technology Core Service Facility.
References
[1] S. Shall, G. de Murcia, Poly(ADP-ribose) polymerase-1: what have we
learned from the deficient mousemodel?Mutat. Res. 460 (2000) 1–15.
[2] Z. Herceg, Z.Q. Wang, Functions of poly(ADP-ribose) polymerase
(PARP) in DNA repair, genomic integrity and cell death, Mutat. Res.
477 (2001) 97–110.
[3] S. Smith, The world according to PARP, Trends Biochem. Sci. 26
(2001) 174–179.
[4] K. Ueda, O. Hayaishi, ADP-ribosylation, Annu. Rev. Biochem. 54
(1985) 73–100.

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532 531
[5] F.R. Althaus, C. Richter, ADP-ribosylation of proteins. Enzymology
and biological significance, Mol. Biol. Biochem. Biophys. 37 (1987)
1–237.
[6] D. D’Amours, S. Desnoyers, I. D’Silva, G.G. Poirier, Poly(ADP-
ribosyl)ation reactions in the regulation of nuclear functions, Bio-
chem. J. 342 (Pt. 2) (1999) 249–268.
[7] G. de Murcia, J. Menissier de Murcia, Poly(ADP-ribose) polymer-
ase: a molecular nick-sensor, Trends Biochem. Sci. 19 (1994)
172–176.
[8] M.K. Jacobson, E.L. Jacobson, Discovering new ADP-ribose poly-
mer cycles: protecting the genome and more, Trends Biochem.
Sci. 24 (1999) 415–417.
[9] A.A. Pieper, A. Verma, J. Zhang, S.H. Snyder, Poly (ADP-ribose)
polymerase, nitric oxide and cell death, Trends Pharmacol. Sci. 20
(1999) 171–181.
[10] J.C. Ame, E.L. Jacobson, M.K. Jacobson, ADP-ribose polymer me-
tabolism, in: G. de Murcia, S. Shall (Eds.), From DNA Damage and
Stress Signalling to Cell Death: Poly ADP-Ribosylation Reactions,
2000, pp. 1–34.
[11] A. Burkle, Poly(ADP-ribosyl)ation: a posttranslational protein modi-
fication linked with genome protection and mammalian longevity,
Biogerontology 1 (2000) 41–46.
[12] L. Van Gool, R. Meyer, E. Tobiasch, C. Cziepluch, J.C. Jauniaux,
A. Mincheva, P. Lichter, G.G. Poirier, A. Burkle, J.H. Kupper,
Overexpression of human poly(ADP-ribose) polymerase in trans-
fected hamster cells leads to increased poly(ADP-ribosyl)ation and
cellular sensitization to gamma irradiation, Eur. J. Biochem. 244
(1997) 15–20.
[13] R. Meyer, M. Muller, S. Beneke, J.H. Kupper, A. Burkle, Negative
regulation of alkylation-induced sister-chromatid exchange by po-
ly(ADP-ribose) polymerase-1 activity, Int. J. Cancer 88 (2000)
351–355.
[14] A. Burkle, PARP-1: a regulator of genomic stability linked with
mammalian longevity, Chembiochemistry 2 (2001) 725–728.
[15] J.P. Gagne, R.G. Shah, G.G. Poirier, Analysis of ADP-ribose
polymer sizes in intact cells, Mol. Cell. Biochem. 224 (2001)
183–185.
[16] K. Ikai, K. Ueda, Immunohistochemical demonstration of poly(ade-
nosine diphosphate-ribose) synthetase in bovine tissues, J. Histochem.
Cytochem. 31 (1983) 1261–1264.
[17] J.C. Ame, V. Rolli, V. Schreiber, C. Niedergang, F. Apiou, P. Decker,
S. Muller, T. Hoger, J. Menissier-de Murcia, G. de Murcia, PARP-2, a
novel mammalian DNA damage-dependent poly(ADP-ribose) poly-
merase, J. Biol. Chem. 274 (1999) 17860–17868.
[18] S. Smith, I. Giriat, A. Schmitt, T. de Lange, Tankyrase, a poly
(ADP-ribose) polymerase at human telomeres, Science 282 (1998)
1484–1487.
[19] A. Saxena, L.H. Wong, P. Kalitsis, E. Earle, L.G. Shaffer, K.H. Choo,
Poly(ADP-ribose) polymerase 2 localizes to mammalian active cen-
tromeres and interacts with PARP-1, Cenpa, Cenpb and Bub3, but not
Cenpc, Hum. Mol. Genet. 11 (2002) 2319–2329.
[20] M. Kanai, W.M. Tong, E. Sugihara, Z.Q. Wang, K. Fukasawa, M.
Miwa, Involvement of poly(ADP-ribose) polymerase 1 and poly
(ADP-ribosyl)ation in regulation of centrosome function, Mol. Cell.
Biol. 23 (2003) 2451–2462.
[21] A. Augustin, C. Spenlehauer, H. Dumond, J. Menissier-De Murcia,
M. Piel, A.C. Schmit, F. Apiou, J.L. Vonesch, M. Kock, M. Bornens,
G. De Murcia, PARP-3 localizes preferentially to the daughter centri-
ole and interferes with the G1/S cell cycle progression, J. Cell Sci. 116
(2003) 1551–1562.
[22] V.A. Kickhoefer, A.C. Siva, N.L. Kedersha, E.M. Inman, C.
Ruland, M. Streuli, L.H. Rome, The 193-kD vault protein, VPARP,
is a novel poly(ADP-ribose) polymerase, J. Cell Biol. 146 (1999)
917–928.
[23] N.W. Chi, H.F. Lodish, Tankyrase is a Golgi-associated mitogen-
activated protein kinase substrate that interacts with IRAP in
GLUT4 vesicles, J. Biol. Chem. 275 (2000) 38437–38444.
[24] R.J. Lyons, R. Deane, D.K. Lynch, Z.S. Ye, G.M. Sanderson, H.J.
Eyre, G.R. Sutherland, R.J. Daly, Identification of a novel human
tankyrase through its interaction with the adaptor protein Grb14,
J. Biol. Chem. 276 (2001) 17172–17180.
[25] S. Ohashi, M. Kanai, S. Hanai, F. Uchiumi, H. Maruta, S. Tanuma, M.
Miwa, Subcellular localization of poly(ADP-ribose) glycohydrolase
in mammalian cells, Biochem. Biophys. Res. Commun. 307 (2003)
915–921.
[26] R.G. Meyer, M.L. Meyer-Ficca, E.L. Jacobson, M.K. Jacobson, Hu-
man poly(ADP-ribose) glycohydrolase (PARG) gene and the common
promoter sequence it shares with inner mitochondrial membrane
translocase 23 (TIM23), Gene 314 (2003) 181–190.
[27] W. Lin, J.C. Ame, N. Aboul-Ela, E.L. Jacobson, M.K. Jacobson, Iso-
lation and characterization of the cDNA encoding bovine poly(ADP-
ribose) glycohydrolase, J. Biol. Chem. 272 (1997) 11895–11901.
[28] J.C. Ame, F. Apiou, E.L. Jacobson, M.K. Jacobson, Assignment of
the poly(ADP-ribose) glycohydrolase gene (PARG) to human chro-
mosome 10q11.23 and mouse chromosome 14B by in situ hybridiza-
tion, Cytogenet. Cell Genet. 85 (1999) 269–270.
[29] T. Shimokawa, M. Masutani, S. Nagasawa, T. Nozaki, N. Ikota, Y.
Aoki, H. Nakagama, T. Sugimura, Isolation and cloning of rat po-
ly(ADP-ribose) glycohydrolase: presence of a potential nuclear ex-
port signal conserved in mammalian orthologs, J. Biochem. (Tokyo)
126 (1999) 748–755.
[30] E. Winstall, E.B. Affar, R. Shah, S. Bourassa, A.I. Scovassi, G.G.
Poirier, Poly(ADP-ribose) glycohydrolase is present and active in
mammalian cells as a 110-kDa protein, Exp. Cell Res. 246 (1999)
395–398.
[31] E. Winstall, E.B. Affar, R. Shah, S. Bourassa, I.A. Scovassi, G.G.
Poirier, Preferential perinuclear localization of poly(ADP-ribose) gly-
cohydrolase, Exp. Cell Res. 251 (1999) 372–378.
[32] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W.
Miller, D.J. Lipman, Gapped BLAST and PSI-BLAST: a new gen-
eration of protein database search programs, Nucleic Acids Res. 25
(1997) 3389–3402.
[33] F.M. Ausubel, R. Brent, R.E. Kingston, D.D. Moore, J.G. Seid-
man, J.A. Smith, K. Struhl, Current Protocols in Molecular Biology,
Wiley-Interscience, New York, 1998.
[34] R.G. Meyer, J.H. Kupper, R. Kandolf, H.P. Rodemann, Early growth
response-1 gene (Egr-1) promoter induction by ionizing radiation in
U87 malignant glioma cells in vitro, Eur. J. Biochem. 269 (2002)
337–346.
[35] J.C. Ame, E.L. Jacobson, M.K. Jacobson, Molecular heterogeneity
and regulation of poly(ADP-ribose) glycohydrolase, Mol. Cell. Bio-
chem. 193 (1999) 75–81.
[36] L. Menard, G.G. Poirier, Rapid assay of poly(ADP-ribose) glycohy-
drolase, Biochem. Cell Biol. 65 (1987) 668–673.
[37] M. Kozak, Compilation and analysis of sequences upstream from the
translational start site in eukaryotic mRNAs, Nucleic Acids Res. 12
(1984) 857–872.
[38] M.R. Hodel, A.H. Corbett, A.E. Hodel, Dissection of a nuclear local-
ization signal, J. Biol. Chem. 276 (2001) 1317–1325.
[39] M.E. Bonicalzi, M. Vodenicharov, M. Coulombe, J.P. Gagne, G.G.
Poirier, Alteration of poly(ADP-ribose) glycohydrolase nucleocyto-
plasmic shuttling characteristics upon cleavage by apoptotic pro-
teases, Biol. Cell 95 (2003) 635–644.
[40] S. Hanai, M. Kanai, S. Ohashi, K. Okamoto, M. Yamada, H. Takaha-
shi, M. Miwa, Loss of poly(ADP-ribose) glycohydrolase causes pro-
gressive neurodegeneration in Drosophila melanogaster, Proc. Natl.
Acad. Sci. U. S. A. 101 (2004) 82–86.
[41] E.B. Affar, M. Germain, E. Winstall, M. Vodenicharov, R.G. Shah,
G.S. Salvesen, G.G. Poirier, Caspase-3-mediated processing of poly
(ADP-ribose) glycohydrolase during apoptosis, J. Biol. Chem. 276
(2001) 2935–2942.
[42] L. Davidovic, M. Vodenicharov, E.B. Affar, G.G. Poirier, Importance
of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-
ribose) metabolism, Exp. Cell Res. 268 (2001) 7–13.

M.L. Meyer-Ficca et al. / Experimental Cell Research 297 (2004) 521–532532
[43] B. Modrek, C. Lee, A genomic view of alternative splicing, Nat.
Genet. 30 (2002) 13–19.
[44] H. Nilsen, M. Otterlei, T. Haug, K. Solum, T.A. Nagelhus, F. Skor-
pen, H.E. Krokan, Nuclear and mitochondrial uracil-DNA glycosy-
lases are generated by alternative splicing and transcription from
different positions in the UNG gene, Nucleic Acids Res. 25
(1997) 750–755.
[45] R.D. Ladner, S.J. Caradonna, The human dUTPase gene encodes both
nuclear and mitochondrial isoforms. Differential expression of the iso-
forms and characterization of a cDNA encoding the mitochondrial
species, J. Biol. Chem. 272 (1997) 19072–19080.
[46] M.A. Oliveira, D. Koh, C.N. Patel, M.K. Jacobson, Structure-
based characterization of a novel anticancer target poly(ADP-ribose)
glycohydrolase (PARG): evidence for the presence of an ADP-ribo-
syl-transferase (ADPRT) fold, Proc. Am. Assoc. Cancer Res. 42
(2001) 832.




















