
Transcriptional regulation is insufficient to explain substrate ...

doi:10.1006/jmbi.2000.4018 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 301, 1207±1220
How Does a Symmetric Dimer Recognize anAsymmetric Substrate? A Substrate Complex ofHIV-1 Protease
Moses Prabu-Jeyabalan, Ellen Nalivaika and Celia A. Schiffer*
Department of Pharmacologyand Molecular ToxicologyUniversity of MassachusettsMedical School, WorcesterMA 01655, USA
E-mail address of the [email protected]
0022-2836/00/051207±14 $35.00/0
The crystal structure of an actual HIV-1 protease-substrate complex ispresented at 2.0 AÊ resolution (R-value of 19.7 % (Rfree 23.3 %)) between aninactive variant (D25N) of HIV-1 protease and a long substrate peptide,Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser, which covers a full bindingepitope of capsid(CA)-p2, cleavage site. The substrate peptide is asym-metric in both size and charge distribution. To accommodate this asym-metry the two protease monomers adopt different conformations buryinga total of 1038 AÊ 2 of surface area at the protease-substrate interface. Thespeci®city for the CA-p2 substrate peptide is mainly hydrophobic, asmost of the hydrogen bonds are made with the backbone of the peptidesubstrate. Two water molecules bridge the two monomers through theloops Gly49-Gly52 (Gly490-Gly520) and Pro790-Val820 (Pro79-Val82). Whenother complexes are compared, the mobility of these loops is correlatedwith the content of the P1 and P10 sites. Interdependence of the confor-mational changes allows the protease to exhibit its wide range of sub-strate speci®city.
# 2000 Academic Press
Keywords: HIV-1 protease; homodimer; substrate speci®city;crystallography; molecular recognition
*Corresponding authorIntroduction
Molecular recognition between proteins is one ofthe most fundamental events in biology. How doesone protein surface speci®cally recognize another?The question becomes even more fascinating whenone molecular surface can recognize a variety ofother seemingly non-homologous surfaces. Such isthe case when HIV protease recognizes its variouscleavage sites in the gag-pol and gag polyproteins.
For the AIDS virus to mature, HIV proteasemust process the viral polyproteins gag and gag-polspeci®cally at nine non-homologous sites (Chouet al., 1996; Henderson et al., 1988; Kohl et al., 1988;Robins & Plattner, 1993) (Table 1). The proteasehas been a prime target in the massive structure-based drug design effort to combat AIDS(Wlodawer & Erickson, 1993), since it is essentialto viral maturation. As a result of this effort nearly200 crystal structures of complexes are now publi-cally available (Vondrasek et al., 1997). The homo-dimeric HIV protease (Wlodawer et al., 1989),undergoes a large conformational change upon
ing author:
binding substrates or inhibitors. From peptide stu-dies, the enzyme optimally binds at least eight resi-dues prior to the cleavage event. The active site ofthis aspartyl protease is at the dimer interface,with each monomer contributing a single asparticacid residue. Yet, how this symmetric enzymerecognizes its various substrates is still poorlyunderstood.
In this manuscript we begin to elucidate how asymmetric enzyme adjusts to recognize its asym-metric substrates by describing the structure of theprotease bound to a substrate peptide. The particu-lar peptide crystallized corresponds to the sub-strate site that releases the capsid protein from thepolyprotein, CA-p2. The sequence of this substrate,Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser, is notsymmetric around the cleavage site, Leu-Ala, ineither the size of the amino acid residues or theirelectrostatic character. In this particular case, thecatalytic aspartic acid has been changed to anasparagine. This renders the enzyme inactive, andthe substrate is therefore not cleaved. Four regionsin the protein backbone between the monomersrearrange in different manners to bind this sub-strate, burying a total of 1038 AÊ 2 of protease sur-face area. In addition side-chains and water
# 2000 Academic Press

Table 1. The nine recognition sequences that the HIV-1protease cleaves speci®cally in the virus's gag-pol poly-proteins
Peptide sequences Cleavage domain
Cleavage sites in gag:SQNY * PIVQ MA-CAARVL * AEAM CA-p2ATIM * MQRG p2-NCQANF * LGKI NC-p1PGNF * LQSR p1-p6
Cleavage sites in pol:SFNF * PQIT TF-PRTLNF * PISP PR-RTAETF * YVDG RT-RHRKIL * FLDG RH-IN
The substrate used in the current investigation has been high-lighted and the cleavage sites are denoted by an asterisk.
NC, nucleocapsid; MA, matrix; CA, capsid; TF, trans framepeptide; PR, protease; RT, reverse transcriptase; IN, integrase;RH, RNAse H.
1208 HIV-1 Protease-substrate Complex
molecules adopt a variety of positions for optimalbinding. These adjustments function in an interde-pendent manner to accommodate the residues inthe substrate, and are suggestive of the mechanismby which HIV protease achieves substrate speci-®city. The patterns observed here provide adetailed example of protein-protein recognitionand thereby offer a paradigm to study a variety ofbiological systems, where molecules undergo con-formational changes to recognize each other withhigh speci®city.
Results
Structure of the complex
The crystal structure of the HIV-1 proteasemutant D25N complexed with a decameric peptide(P5-P50) corresponding to the CA-p2 cleavage site,has been solved and re®ned to an R value of19.4 % (Rfree 23.9 %) at 2.0 AÊ resolution. In theinitial cycles of re®nement, both the difference den-sity maps, 2Fo ÿ Fc and Fo ÿ Fc, indicated the pos-ition of the central eight residues, P4-P40, of thesubstrate unambiguously. The complex is in thebiologically relevant form with the peptide bondbetween the P1 and P10 sites, straddling the ``cata-lytic'' Asn25s. The cleavage site of the substrate isbetween leucine (P1) and alanine (P10). The peptideadopts an extended conformation (Figure 1(a)), asfound in other complexes of peptidomimetics withHIV-1 protease (Gustchina et al., 1994; Weber et al.,1997), forming a parallel b-sheet with the ¯ap ofthe ®rst monomer and anti-parallel b-sheet withthe ¯ap of the second monomer (Figure 1(b)). Thetransition from the parallel to the anti-parallel b-sheet conformation takes place at P1 (f � ÿ 85,c � 50). Consequently, a kinked geometry, asobserved in another substrate analog proteasecomplex (Swain et al., 1990), enables the carbonyloxygen of LeuP1 to make a hydrogen bond with
Asn250ND2. This complex, therefore, represents abiologically relevant transition state of the pro-tease-substrate complex.
The peptide, Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser, is highly asymmetric in sequence andstructure about the cleavage site. On either side ofthe cleavage site, the side-chains are pointing awayfrom the center of the dimer. One side of the pep-tide (Figure 1(c)) is made up of alternating residuesP3(Arg), P1(Leu), P20(Glu) and P40(Met), which arephysically large and two of them are charged,while the residues on the other side P4(Ala),P2(Val), P10(Ala), and P30(Ala) are small andhydrophobic. The side-chains of P5 and P50 are notshown, as they cannot be modeled unambiguouslyin the electron density (Figure 1(a)). This asymme-try of the substrate side-chains results in distinctadaptations of each monomer, a necessary require-ment for speci®city of the protease.
Enzyme-substrate interactions
The binding interface observed in the crystalstructure between the inactive enzyme and thepeptide substrate is substantial. Upon binding theenzyme, 1038 AÊ 2 of solvent-exposed surface area(Lee & Richards, 1971) is buried on the substrate.Of this area, 732 AÊ 2 (71 %) of the buried surfacearea is hydrophobic. In this extensive bindinginterface, the peptide forms 24 hydrogen bonds(Figure 2); 16 are directly to the protein (Table 2A)and eight are water mediated (Table 2B). Eighteenof the hydrogen bonds involve the backbone atomsof the substrate, and therefore do not account forsubstrate speci®city. Only two side-chains of thesubstrate, ArgP3 and GluP20, form hydrogenbonds with the protein. ArgP3 hydrogen bondsindirectly with the protein via Wat48 to the back-bone carbonyl of Pro810. GluP20 forms the onlydirect hydrogen bond between a substrate side-chain and the protein. This side-chain hydrogenbonds with both the backbone and the side-chainof Asp300 and the backbone of Asp290. The hydro-gen bonding between the two acidic groups, GluP20 and Asp300, indicates that at least one of theside-chains is protonated. This interaction has beensuggested to be speci®c for the CA-p2 cleavage site(Weber et al., 1997) and is the only direct hydrogenbond that can be implicated in the speci®city ofsubstrate recognition. The rest of the speci®citydeterminants of the protease for this substratemust come from shape complementarity.
The shape complementarity (Lawrence &Colman, 1993) between the interacting surfaces ofthe peptide and the protein is very tight, with avalue of 0.64 (0.5 or higher is considered comp-lementary). Such a high level of complementarityis attained by way of the protein changing its con-formation to accommodate the substrate. Watermolecules within the complex play an integral rolein assisting this adaptation. In addition to thewater molecule mediating the interaction withArgP3, ®ve other water molecules mediate inter-
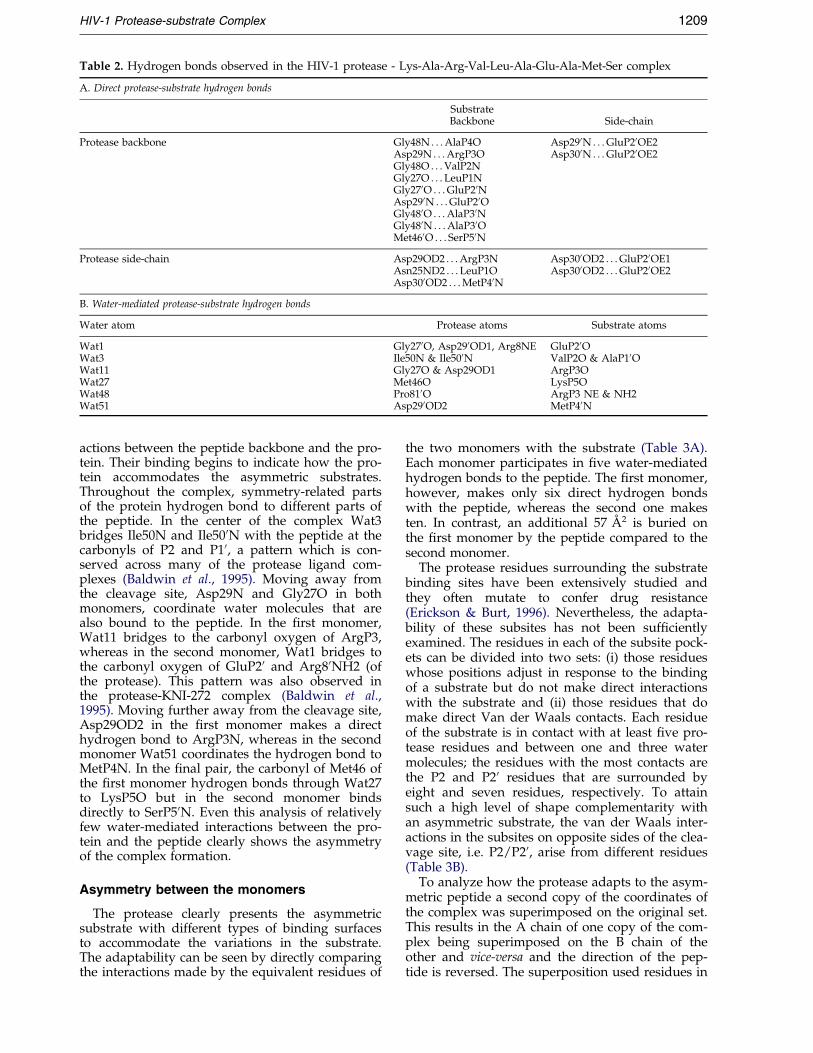
Table 2. Hydrogen bonds observed in the HIV-1 protease - Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser complex
A. Direct protease-substrate hydrogen bonds
SubstrateBackbone Side-chain
Protease backbone Gly48N . . . AlaP4O Asp290N . . . GluP20OE2Asp29N . . . ArgP3O Asp300N . . . GluP20OE2Gly48O . . . ValP2NGly27O . . . LeuP1NGly270O . . . GluP20NAsp290N . . . GluP20OGly480O . . . AlaP30NGly480N . . . AlaP30OMet460O . . . SerP50N
Protease side-chain Asp29OD2 . . . ArgP3N Asp300OD2 . . . GluP20OE1Asn25ND2 . . . LeuP1O Asp300OD2 . . . GluP20OE2Asp300OD2 . . . MetP40N
B. Water-mediated protease-substrate hydrogen bonds
Water atom Protease atoms Substrate atoms
Wat1 Gly270O, Asp290OD1, Arg8NE GluP20OWat3 Ile50N & Ile500N ValP2O & AlaP10OWat11 Gly27O & Asp29OD1 ArgP3OWat27 Met46O LysP5OWat48 Pro810O ArgP3 NE & NH2Wat51 Asp290OD2 MetP40N
HIV-1 Protease-substrate Complex 1209
actions between the peptide backbone and the pro-tein. Their binding begins to indicate how the pro-tein accommodates the asymmetric substrates.Throughout the complex, symmetry-related partsof the protein hydrogen bond to different parts ofthe peptide. In the center of the complex Wat3bridges Ile50N and Ile500N with the peptide at thecarbonyls of P2 and P10, a pattern which is con-served across many of the protease ligand com-plexes (Baldwin et al., 1995). Moving away fromthe cleavage site, Asp29N and Gly27O in bothmonomers, coordinate water molecules that arealso bound to the peptide. In the ®rst monomer,Wat11 bridges to the carbonyl oxygen of ArgP3,whereas in the second monomer, Wat1 bridges tothe carbonyl oxygen of GluP20 and Arg80NH2 (ofthe protease). This pattern was also observed inthe protease-KNI-272 complex (Baldwin et al.,1995). Moving further away from the cleavage site,Asp29OD2 in the ®rst monomer makes a directhydrogen bond to ArgP3N, whereas in the secondmonomer Wat51 coordinates the hydrogen bond toMetP4N. In the ®nal pair, the carbonyl of Met46 ofthe ®rst monomer hydrogen bonds through Wat27to LysP5O but in the second monomer bindsdirectly to SerP50N. Even this analysis of relativelyfew water-mediated interactions between the pro-tein and the peptide clearly shows the asymmetryof the complex formation.
Asymmetry between the monomers
The protease clearly presents the asymmetricsubstrate with different types of binding surfacesto accommodate the variations in the substrate.The adaptability can be seen by directly comparingthe interactions made by the equivalent residues of
the two monomers with the substrate (Table 3A).Each monomer participates in ®ve water-mediatedhydrogen bonds to the peptide. The ®rst monomer,however, makes only six direct hydrogen bondswith the peptide, whereas the second one makesten. In contrast, an additional 57 AÊ 2 is buried onthe ®rst monomer by the peptide compared to thesecond monomer.
The protease residues surrounding the substratebinding sites have been extensively studied andthey often mutate to confer drug resistance(Erickson & Burt, 1996). Nevertheless, the adapta-bility of these subsites has not been suf®cientlyexamined. The residues in each of the subsite pock-ets can be divided into two sets: (i) those residueswhose positions adjust in response to the bindingof a substrate but do not make direct interactionswith the substrate and (ii) those residues that domake direct Van der Waals contacts. Each residueof the substrate is in contact with at least ®ve pro-tease residues and between one and three watermolecules; the residues with the most contacts arethe P2 and P20 residues that are surrounded byeight and seven residues, respectively. To attainsuch a high level of shape complementarity withan asymmetric substrate, the van der Waals inter-actions in the subsites on opposite sides of the clea-vage site, i.e. P2/P20, arise from different residues(Table 3B).
To analyze how the protease adapts to the asym-metric peptide a second copy of the coordinates ofthe complex was superimposed on the original set.This results in the A chain of one copy of the com-plex being superimposed on the B chain of theother and vice-versa and the direction of the pep-tide is reversed. The superposition used residues in

Figure 1 (legend opposite)
1210 HIV-1 Protease-substrate Complex
both monomers from the terminal domain. Fourregions (Figure 3), residues 12-19, 36-46, 49-53, and79-82, representing 28 % of the protein have devi-ations of 0.4 AÊ or greater, some of which may bedue to crystal packing. However, residues Lys45,Met46, Gly49 and Ile50 located in the ¯aps, makeat least one direct van der Waals contact with thesubstrate (Table 3B). The superposition of the
dimer on itself also allows the close examination ofthe local deviations occurring between the sub-strate binding site pairs (i.e. P1 & P10, P2 & P20 . . . ).The set of residues that make direct van der Waalscontacts is only partially overlapping (Table 3B).However, the residues forming the binding pocketsare necessarily the same, as it is a homodimer.Since the a-carbon positions of most of the residues

Figure 1. (a) Stereo view of the electron density corresponding to the simulated annealed Fo ÿ Fc omit map con-toured at 2s level for the CA-p2 substrate (Sack, 1988), clearly showing the unique conformation of the substrate inthe bound state. (b) The substrate peptide (in green) bound to the protease (in magenta and cyan) viewed down thedimer interface. Residues within van der Waals contact of the substrate are displayed on the protease backbone(Nicholls et al., 1991). (c) Stick model of the substrate viewed down the molecular 2-fold of the protease (Ferrin et al.,1988).
HIV-1 Protease-substrate Complex 1211
in this region do not shift, the changes in localinteractions are due to side-chain rotations inthe protease, rearrangement of water and alteredpositioning of the substrate residue in the pocket.In the following sections, the four substrate bind-ing subsite pairs, P1/P10 - P4/P40 (Figure 4) areexamined in detail:
P1/P10 site (Leu/Ala)
In the ®rst subsite, the positions of the a-carbonsof LeuP1 and AlaP10 are 0.9 AÊ apart (Figure 4(a)).The smaller alanine side-chain is located deeper inthe pocket, burying 94 AÊ 2 of surface area com-pared with 142 AÊ 2 on the leucine. The LeuP1CD2also makes van der Waals contact with ArgP3CB.The loops containing the ``catalytic'' residue Asn25extending through Asp29 form an almost invariantbase to the P1/P10 pocket. One side of the pocketis made of the tip of the protease ¯ap, Gly48-Gly49-Ile50-Gly51-Gly52. The central isoleucine,surrounded by two glycine residues on either side,allows the ¯ap the adaptability to adjust to theasymmetry in the substrate sequence. Surprisingly,changes in side-chain rotamers of residues Leu230,Val820, Ile840, and Ile50 surrounding the P10 seemto open up more space for the smaller alanine resi-due in comparison to Leu23, Val82, Ile84, andIle500 surrounding P1 leucine. However, both thealternative rotamers of the side-chains and also arealignment of the entire P1-loop (residues 78-84)are responsible for the change in size of the bind-ing site. The distance between the a-carbons of
Pro810 (the central residue in the P1 loop) andLeuP1 is 0.6 AÊ closer when compared with Pro81and AlaP10. Since the alanine is small, two watermolecules (Wat37 and Wat38) ®ll the empty spacein the pocket, even though it is primarily hydro-phobic. This larger pocket could easily accommo-date a bigger side-chain like phenylalanine that isoften seen in the protease substrates at this site(Wlodawer & Erickson, 1993). The fact that thispocket remains large when the substrate side-chainis an alanine, implies that the shape of the pocketis in¯uenced by other interactions in the complex.
P2/P2 0 site (Val/Glu)
In contrast to the P1/P10 pockets, the structuresof the P2/P20 pockets are quite rigid (Figure 4(b)).The positions occupied by ValP2 and GluP20 whensuperimposed, however, are quite different. Thedistance between their a-carbons is 1.3 AÊ . Bothside-chains make extensive van der Waals contactsand bury a considerable amount of surface area:ValP2, 136 AÊ 2 and GluP20, 147 AÊ 2. There are somekey differences, however, in the residues withwhich they make these contacts (Table 3B). Thevaline makes hydrophobic interactions with Ile840and Val320, and the glutamic acid rotates about thew1 torsion angle to make ionic contacts with Asp29and Asp30. Thus a large variation in environmentsexists within this relatively rigid pocket, allowingvarious side chains to each ®nd their optimalorientation.

Figure 2. The hydrogen bonds between the substrate and the protease. The two monomers and the peptide areshown in cyan, magenta and green, respectively (Ferrin et al., 1988). The water bridge involving Wat48, P3 Arg NEand Pro810 O (not shown) and the hydrophobic side-chains are omitted for clarity.
1212 HIV-1 Protease-substrate Complex
P3/P3 0 site (Arg/Ala)
At ®rst glance, the most striking observationabout the P3/P30 superposition is that the largerArgP3 is located deeper in the pocket than thesmaller AlaP30 (Figure 4(c)). The separation of theira-carbon positions is 1.9 AÊ . This large shift resultsin the hydrogen bonds from the protease to thebackbone of the substrate being mediated bydifferent atoms (Table 2A). The backbone hydro-gen bonds of P3 are primarily with the side-chainand backbone of Asp29, and the backbone hydro-gen bonds of P30 are with the backbone atoms ofGly480. This extensive rearrangement may be facili-tating a key role for the Arg side-chain at P3, forthis side-chain strongly impacts the P1 bindingsite. The side-chain of ArgP3 not only makes a vander Waals contact with the side-chain of LeuP1 asnoted earlier, but ArgP3 may also be responsiblefor the tightening of the P1 pocket. The side-chaincoordinates a water molecule, Wat48, to hydrogenbond with Pro810O (the central residue of the P1loop), drawing the loop closer into the P1 pocket.This accounts for the pocket around the Leu P1being smaller than the pocket around the Ala P10,
where no such analogous interaction as withArgP3 exists.
P4/P40 site (Ala/Met)
The location of the binding pocket for P4/P40runs along the base of the leading strand of theprotease ¯ap. Once again the a-carbon positions ofthese two residues are signi®cantly offset, this timeby 2.0 AÊ , and the larger residue is deeper in thebinding pocket. This time, however, it is on theopposite side of the peptide Met at P40. As the siteis fairly solvent-exposed, the methionine only bur-ies 112 AÊ 2 and the alanine buries 89 AÊ 2. To accom-modate the longer methionine side-chain, Lys450rotates out, offering a hydrophobic slot for the sub-strate side-chain.
Each of the eight substrate subsite pocketsthus adapts to facilitate the binding of a particu-lar substrate side-chain and together they de®nethe manner in which the protease is able torecognize the sequence of a substrate. Althoughthis is the ®rst HIV-1 protease-substrate complexto be analyzed, the features underlying theobserved adaptability in recognizing this sub-

Table 3. Interactions between the substrate peptide and the protease
A. Summary of the interactions for each monomerType of interaction Monomer A Monomer B
Direct hydrogen bonds 6 10Backbone hydrogen bonds 4 5Hydrogen bonds involving at least oneside-chain atom 2 5Water-mediated hydrogen bonds 5 5Surface area buried (hydrophobic) (AÊ 2) 393 (226) 336 (194)
Summary of the interactions between the protein and peptide divided by monomer. Six residues forming Van der Waals interac-tions were found to be common between the monomers. The additional ones are Ile84, Val32 and Pro810.
B. Van der Waals contacts with protease and water
Substrate Contacts Substrate Contacts
LeuP1 Asn25, Gly27, Ile50, Asn250,Gly270
AlaP10 Asn25, Ile84, Gly270, Gly480,Gly490
Wat3, Wat11 Wat3, Wat37ArgP3
ValP2 Gly27, Ala28, Val32, Ile47,Gly48, Gly49, Ile84
GluP20 Ile50
Ile500Gly270, Ala280, Asp290, Asp300,Ile470, Gly480
Wat3 Wat1, Wat37ArgP3 Gly27, Ala28, Asp29, Gly48 AlaP30 Arg8
Pro810 Asp290, Asp300, Ile470, Gly480Wat11, Wat48, Wat88 Wat37, Wat57
LeuP1AlaP4 Asp29, Asp30, Lys45, Ile47,
Gly48MetP40 Asp300, Lys450, Met460, Ile470,
Gln580Wat27 Wat51, Wat65
van der Waals interactions within 4.2 AÊ of the substrate. Amino acid residues found to be common between the subsite pairs arehighlighted.
HIV-1 Protease-substrate Complex 1213
strate are also found in many of the peptidomi-metic complexes in the database (Laco et al.,1997; Weber et al., 1997).
Discussion
Although the crystal structure of HIV-1 proteasehas been solved many times in the presence ofpeptidomimetics and inhibitors, the complexdescribed here is the closest representation of anactual protease-substrate complex that has yet beenanalyzed. Unlike other complexes, the length of thepeptide (ten residues) is long enough to cover theentire binding epitope that has been determined byenzyme kinetics (Rasnick, 1997). This study eluci-dates the recognition of an asymmetric substrateby this homodimeric enzyme.
Some similar patterns have been seen in otherprotease complexes, though the interactions are notidentical. A crystal structure has previously beensolved with a shorter CA-p2 peptidomimic, RVL-r-FEA(Ahx-NH2) (Weber et al., 1997) where a non-homologous phenylalanine has been substitutedfor the wild-type alanine at the P10 position(Table 1) and the cleavage site is a reduced peptidebond. In our complex the wild-type sequence isused and the P1 and P10 are leucine and alanine(Wlodawer & Erickson, 1993). To prevent the pro-tease from cleaving the substrate the catalytic
aspartic acid residues have been replaced by isos-teric asparagine residues. A similar study was car-ried out on an inactive FIV protease bound to asimilar substrate, where P2 through P20 are alsothose of the CA-p2 cleavage site (Laco et al., 1997).However, in that case the crystallization did notdistinguish the direction of the bound substrate,therefore the asymmetry of the protease could notbe analyzed.
There are nearly 200 crystal structures availablein the database (http://www.ncifcrf.gov/CRYS/HIVdb/NEW DATABASE/) (Vondrasek et al.,1997) involving HIV-1 protease. If the ligand wasor resembled a peptide and the complex was deter-mined to at least 2.5 AÊ resolution it was includedin our analysis. This resulted in the inclusion ofonly 19 structures (Table 4). Most of the ligands inthese 19 complexes are relatively short, primarilycovering only the P3-P30 subsites. An averagestructure from the positions of the a-carbons of the20 structures (19 plus ours) was computed asdescribed in Materials and Methods. The meandeviation from this average structure was calcu-lated at each residue and is plotted in Figure 5.There are four regions in each monomer where theprotein structures deviate from each other. Theseare Ile15-Leu19 (Ile150-Leu190), Met35-Lys45(Met350-Lys450), Gly49-Gly52 (Gly490-Gly520) andPro79-Val82 (Pro790-Val820) (Peaks 1/10, 2/20, 3/30

Figure 3. Histogram of the deviations between the monomers in AÊ ; superimposition was performed using the a-carbons of residues 1-9, 86-99 from each monomer.
1214 HIV-1 Protease-substrate Complex
and 4/40, respectively, in Figure 5(a)). Mutationalstudies on HIV-1 protease (Loeb et al., 1989) indi-cate that the activity of the enzyme is compro-mised by mutations that occur in the regions of thethree-dimensional structure that are rigid. How-ever, many of the residues in the four variablepeak regions are associated with drug-resistantmutations (Erickson & Burt, 1996). Peak 1 is thehinge between the terminal domain and the activesite. Peak 2 is the hinge before the ¯ap. Peak 3 isthe tip of the ¯ap containing Ile50 that borders theP1, P2 and P3 substrate subsites (Table 3B). Peak 4is the P1-loop that stabilizes the P1 and P3 sub-strate subsite (Figure 5(b)). The average tempera-ture factors of the residues in peaks 3 and 4 arerelatively low, indicating that the deviations arelikely a direct result of the particular substrate ana-log that is bound. Mediating these two regions andbridging between the monomers in many of thecomplex structures are two water molecules.Above the P1 and P3 substrate sites is a water,Wat7, that bridges Pro790O to Ile50O, and abovethe P10 and P30 substrate site is a water, Wat70,that bridges Pro79O to Gly510N. Both of thesewater molecules are present in nine of the com-
plexes and another seven have one of them. There-fore, these water molecules likely facilitate thecooperative structural adaptation between the sub-strate subsites.
Brie¯y examining the details of the 19 proteasestructures in this database (Table 4), the extent ofthese adaptations can be further elucidated. How-ever, in almost half of these structures the crystalli-zation did not distinguish the direction of thebound substrate, thus the asymmetry of the pro-tease could not be unambiguously analyzed.Nevertheless, some patterns became clear whenthese structures were grouped into three classes(Table 4) from the sequence of their P1 and P10sites: (i) complexes with Leu (or Met) at P1 (sixincluding ours), (ii) complexes with ringed or aro-matic residues at P1 and a small residue at P10 (six)and (iii) complexes with aromatic residues orringed residues at both P1 and P10 (eight). In mostcases P1 and P3 make Van der Waals contact witheach other but P10 and P30 do not, except in thecases where P10 and P30 are very large. In group 2 ,when P1 is an aromatic and P10 is small, the intera-tomic distances between both the a-carbons of P1and P3, and Pro810 are closer to each other, by

Figure 4. Stereo view of the superimposed (a) P1/P10 site, (b) P2/P20 site, (c) P3/P30 site and (d) P4/P40 site. TheHIV-1 protease dimer is placed onto itself by superimposing the a-carbons of residues 1-9 and 86-99 of the monomersupon each other. In one structure, the monomers are distinguished by cyan and magenta, the substrate is green andthe water molecules are red spheres. In the other structure, the monomers are distinguished in white and grey, thesubstrate is yellow and the water molecules are white spheres. Red, blue and black sticks indicate oxygen, nitrogenand sulfur atoms, respectively.
HIV-1 Protease-substrate Complex 1215
approximately 1.0 AÊ , than in either of the othertwo groups . This is the case, regardless of the sizeof the residue at P30. The largest distances are seen
for the third group, where P1 (P10) contacts bothP3 (P30) and Pro810(Pro81). In addition, if either P1or P10 is a ringed or aromatic residue, the water

Table 4. Protease complexes bound to peptidic and peptidomimetic ligands
Structure P5 P4 P3 P2 P1 P10 P20 P30 P40 P50 Res
CA-p2 Lys Ala Arg Val Leu Ala Glu Ala Met Ser 2.0 AÊ
1A8K (Weber et al., 1997) Arg Val Leu Phe Glu Ala (Met) Nle NH2 2.0 AÊ
*3FIV (Laco et al., 1997)a Ace (Trp) Nal Val Leu Ala Glu (Trp) Nam 1.85 AÊ
8HVP (Jaskolski et al., 1991) Val Ser Gln Asn Leu Val Ile Val 2.5 AÊ
5HVP (Fitzgerald et al., 1990) Ace Val Val (Leu) Sta Ala (Leu) Sta 2.0 AÊ
4HVP (Miller et al., 1989) Ace Thr Ile (Met) Nle (Met) Nle Gln Arg NH2 2.3 AÊ
7HVP (Swain et al., 1990) Ace Ser Leu Asn Phe Pro Ile Val Ome 2.4 AÊ
1YTH (Rose et al., 1996) Ace Ser Leu Asn Phe 2.2 AÊ
1YTG (Rose et al., 1996) Pro Ile Val 2.3 AÊ
1CPI (Martin et al., 1999) CH2 Cbg Asn Tyr Pro Ile Val NH2 2.05 AÊ
1HIV (Thanki et al., 1992) (Trp) Noa His (Phe) Cal Val Ile (Phe) Apy 2.0 AÊ
1GNM (Hong et al., 1996) (Phe) Cha Val Gln Arg 2.3 AÊ
1AAQ (Dreyer et al., 1992) Ala Ala Phe Gly Val Val 2.5 AÊ
1HEF (Murthy et al., 1992) Ala Ala Phe Phe Val Val 2.2 AÊ
1HIH (Priestle et al., 1995) Ace Val Phe (Phe) Cha Val Ace 2.2 AÊ
1HOS (Abdel-Meguid et al.,1993)
(Phe) Cbz Val NH-Phe Phe Val (Phe) Cbz 2.3 AÊ
1HVI (Hosur et al., 1994),(Baldwin et al., 1995)
Phe Val Phe Phe Val Phe 1.8 AÊ
1DIF (Silva et al., 1996) (Phe) Nhe Val Phe Phe Val (Phe) Nhe 1.7 AÊ
1HVC (Bhat et al., 1994) Phe(N) Val Phe Phe Val Phe(N) 1.8 AÊ
1HXB (Krohn et al., 1991) (Trp) Qcn Asn (Phe) Hph (Trp) D1q NT3 2.3 AÊ
1MTR (Martin et al., 1999) BOC (Phe) Phm Tyr Ile Gly 1.75 AÊ
The 19 structures from of HIV protease (and one FIV protease structure awhich was not included in the calculation) from the Pro-tein Data Bank (PDB) (Bernstein et al., 1977) complexed with a peptidomimetic and peptidic ligands and in addition to the Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser complex (indicated as CA-p2) used for the analysis. The structures are grouped by the sequenceof P1 and P10. For non-natural amino acids, the name listed in the coordinate ®le was used, with the most similar natural aminoacid listed above it. In all cases except CA-p2, 3FIV, 1YTH and 1YTG the cleavable peptide bond has been replaced by a variety ofstructurally similar constructs. Furthermore, the Table is subdivided into three groups based on the content of the P1 and P10 residuesites.
1216 HIV-1 Protease-substrate Complex
(Wat7 or Wat70), which bridges the P1-loop to the¯ap, is seen less than 50 % of the time. Whereas,when a smaller side-chain is in the P1 or P10 site,the bridging water is present almost 80 % of thetime. This analysis con®rms that patterns of coop-erative structural adaptations exist when HIV pro-tease recognizes ligands. The full extent of thepatterns will only be comprehensively describedonce a series of structures of substrate complexesare determined and compared.
Materials and Methods
Protein purification and crystallization
The mutant protease gene Q7 K; D25N(Babe et al.,1995), a gift from Dr C.S. Craik, University of California,San Francisco, was cloned into pet11a (Novagen). Escher-ichia coli BL21(DE3) cells containing the plasmid weregrown at 37 �C to an A600 � 0.7 and induced with2.0 mM isopropyl-beta-D-thiogalactoside (IPTG). Thecells were harvested after 3.0 hours and the pellet frozenat ÿ80 �C. The protease was puri®ed via a two columnprotocol (Hui et al., 1993; Rose et al., 1995).
50 mg of the 10-mer peptide was purchased fromQuality Controlled Biochemicals (QCB), Inc. at 95 %purity. The ends of the peptide were blocked with neu-tral acetyl and amide groups. The peptides were ®rst dis-solved in DMSO, solubilized and then mixed with theprotein's buffer.
The puri®ed protein was concentrated to 2.5 mg/ml.A database was generated using all the available crystal-lographic data for HIV-1 protease and a table was con-
structed containing the various crystallization conditions.The protein was equilibrated on ice for 30 minutes witha ®ve molar excess of the peptide. The crystals wereobtained in hanging drops under more than one con-dition. The crystals used were grown with a reservoirsolution of 126 mM phosphate buffer (pH 6.2), 63 mMsodium citrate and 37 % ammonium sulfate (Thanki et al.,1992). Each droplet contained 2.5 ml of the protease-pep-tide mixture and 2.5 ml of the reservoir solution. Thebrick-shaped crystals grew in two days time.
Data collection
Intensity data were collected using a R-axis IV imageplate mounted on a Rigaku rotating anode X-ray genera-tor. A total of 200 frames were collected using a 1� oscil-lation. The raw data were reduced using DENZO andScalePack (Otwinowski, 1993) (Table 5).
Crystallographic refinement
Crystallography and NMR System (CNS) (Brungeret al., 1998) was used for carrying out crystallographicre®nement with 10 % of the re¯ections set aside for thecalculation of Rfree (Brunger, 1992). The HIV-1 protease-inhibitor complex (code: 1MTR) (Martin et al., 1999) hadthe closest cell dimensions to the one we observed andwas therefore used as the model to solve HIV-1 protease:Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser. Alanine resi-dues were substituted wherever there were differencesin sequence and the ligand was removed. In the ®rstFo ÿ Fc and 2Fo ÿ Fc maps the peptide could be unam-biguously placed in a unique orientation. Model buildingwas carried out using CHAIN (Sack, 1988). In the ®nal

Figure 5. (a) Histogram representing the mean deviation in the a-carbons between the 19 structures and the aver-age structure. The second monomer starts at residue 101. The four peaks are labeled in each of the monomers. (b)The deviations in the a-carbons are mapped on the three-dimensional ribbon diagram of HIV protease with the lar-gest deviations shown in yellow, and the four regions of the greatest deviations are labeled on one monomer.
HIV-1 Protease-substrate Complex 1217
model 96 water molecules and six acetate ions wereincluded. Eight lysine and one glutamic acid residueswere left as alanine due to ambiguity in determining
their side-chain positions. The stereo chemical par-ameters were checked using PROCHECK (Laskowskiet al., 1993). The Ramachandran map (Ramachandran &

Table 5. Crystallographic and re®nement statistics
Parameter Values
Space group P212121
Cell dimensions (AÊ ) a = 51.615b = 59.043c = 61.351
Oligomeric state DimerResolution (AÊ ) 50-2.0Total number of reflections 49,525Number of unique reflections 12,474Rsymm � �jIÿIj2/�jIj2 (%) 4.8Completeness (%) 94.3Crystallographic R-factor (%) 19.7Rfree (10 % data) (%) 23.3Sigma cutoff NoneRMS deviation Bond = 0.005 AÊ
Angle = 1.3�Ramachandran violations NoneContent of asymmetric unit One dimer:
Residues 207Waters 96Acetates 6
1218 HIV-1 Protease-substrate Complex
Sasisekharan, 1968) showed no violations. The ®nal crys-tallographic R-factor the free R-value are 19.7 % and23.3 %, respectively (Table 5). The average temperaturefactor of all the atoms is 31.9 AÊ 2. The average tempera-ture factor values for backbone atoms, side-chain atoms,substrate atoms and water oxygen atoms are 30.0, 34.0,32.5, 34.5 AÊ 2, respectively.
Comparison of the subsite pairs
The dimer was superimposed onto itself to comparethe differences between the monomers on binding anasymmetric ligand, by superimposing monomer A ontomonomer B of the copy. The a-carbons from residues inthe relatively immobile (Rose et al., 1998) terminal region(1-9 and 86-99) from each monomer were used for thesuperposition. This superposition also resulted in thepeptide subsite pairs being superimposed on each other(i.e. P2 on P20). By doing this the environments sur-rounding peptide units P1 through P4 were able to becompared with the positions of P10 through P40.
Calculation of the average structure
To compare the different HIV-1 protease-inhibitor/substrate analog complexes, an averaged structurescheme was followed. Nineteen HIV-1 protease struc-tures, which contain a peptidomimetic ligand at theiractive sites, were chosen from the Protein Data Bank(PDB) (Bernstein et al., 1977; Vondrasek et al., 1997)(Table 3). Once again the structures were superimposedusing the a-carbon backbone of the relatively immobile(Rose et al., 1998) terminal domain of all these structures(residues 1-9 and 86-99) with the program MIDAS(Ferrin et al., 1988). The ligands were also, when possible,aligned in the same direction. An average structure wascalculated from the a-carbon positions. Deviation of eachresidue from this average structure was also computedand plotted using the program XMGR (copyright 1995,Paul J. Turner).
Protein Data Bank accession code
The coordinates are deposited in the Protein DataBank entry code 1F7A.
Acknowledgments
The authors acknowledge Charles S. Craik for provid-ing the plasmid and Hema Bashyam, Jennifer Foulkes,Nancy King, Jeanette Lavasta, Kai Lin, Wendy Mathews,William Royer and Walter Scott for many helpfulsuggestions. This research was supported by the Ameri-can Cancer Society grant RPG-99-213-01-MBC.
References
Abdel-Meguid, S. S., Zhao, B., Murthy, K. H. M.,Winborne, E., Choi, J.-K., DesJarlais, R. L., Minnich,M. D., Culp, J. S., Debouck, C., Tomaszek, T. A.,Meek, T. D. & Dreyer, G. B. (1993). Inhibition ofhuman immunode®ciency virus-1 protease by a C2-symmetric phosphinate. Synthesis and crystallo-graphic analysis. Biochemistry, 32, 7972-7980.
Babe, L. M., Rose, J. & Craik, C. S. (1995). Trans-domi-nant inhibitory human immunode®ciency virustype 1 protease monomers prevent protease acti-vation and virion maturation. Proc. Natl Acad. Sci.USA, 92, 10069-10073.
Baldwin, E. T., Bhat, T. N., Gulnik, S., Liu, B., Topol,I. A., Kiso, Y., Mimoto, T., Mitsuya, H. & Erickson,J. W. (1995). Structure of HIV-1 protease withKNI-272, a tight-binding transition-state analog con-taining allophyenylnorstatine. Structure, 3, 581-590.
Bernstein, F. C., Koetzle, T. F., Williams, G. J., Meyer,E. F. J., Brice, M. D., Rodgers, J. R., Kennerd, O.,Shimanouchi, T. & Tasumi, M. (1977). The proteindata bank: a computer based archival ®le formacromolecular structures. J. Mol. Biol. 112, 535-542.
Bhat, T. N., Baldwin, E. T., Liu, B., Cheng, Y. S. &Erickson, J. W. (1994). Crystal structure of a teth-ered dimer of HIV-1 proteinase complexed with aninhibitor. Nature Struct. Biol. 1, 552-556.
Brunger, A. T. (1992). The free R value: a novel statisti-cal quantity for assessing the accuracy of crystalstructures. Nature, 355, 472-474.
Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S.,Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J.,Rice, L. M., Simonson, T. & Warren, G. L. (1998).Crystallography & NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905-921.
Chou, K.-C., Tomasselli, A. G., Reardon, I. M. &Heinrikson, R. L. (1996). Predicting human immu-node®ciency virus protease cleavage sites in pro-teins by a discriminant function method. Proteins:Struct. Funct. Genet. 24, 51-72.
Dreyer, G. B., Lambert, D. M., Meek, T. D., Carr, T. J.,Tomaszek, J. T. A., Fernandez, A. V., Bartus, H.,Cacciavillani, E., Hassel, A. M., Minnich, M.,Petteway, S. R. & Metcalf, B. W. (1992). Hydro-xyethylene isostere inhibitors of human immunode-®cency virus-1 protease: structure-activity analysisusing enzyme kinetics, X-ray crystallography, andinfected T-cell assays. Biochemistry, 31, 6646-6659.
Erickson, J. W. & Burt, S. K. (1996). Structural mechan-isms of HIV drug resistance. Annu. Rev. Pharmacol.Toxicol. 36, 545-571.
Ferrin, T. E., Huang, C. C., Jarvis, L. E. & Langridge, R.(1988). The MIDAS display system. J. Mol. Graphics,6, 13-27.

HIV-1 Protease-substrate Complex 1219
Fitzgerald, P. M., McKeever, B. M., Van Middlesworth,J. F., Springer, J. P., Heimbach, J. C., Leu, C. T.,Herber, W. K., Dixon, R. A. & Darke, P. L. (1990).Crystallographic analysis of a complex betweenhuman immunode®ciency virus type 1 protease andacetyl-pepstatin at 2.0 AÊ resolution. J. Biol. Chem.265, 14209-14219.
Gustchina, A., Sansom, C., Prevost, M., Richelle, J.,Wodak, S. Y., Wlodawer, A. & Weber, I. T. (1994).Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 7, 309-317.
Henderson, L. E., Copeland, T. D., Sowder, R. C.,Schultz, A. M. & Oraszlan, S. (1988). Human Retro-viruses, Cancer and AIDS: Approaches to Preventionand Therapy, Liss, New York.
Hong, L., Treharne, A., Hartsuck, J. A., Foundling, S. &Tang, J. (1996). Crystal structures of complexes of apeptidic inhibitor with wild-type and two mutantHIV-1 proteases. Biochemistry, 35, 10627-10633.
Hosur, M. V., Bhat, T. N., Kempf, D. J., Baldwin, E. T.,Liu, B., Gulnik, S., Wideburg, N. E., Norbeck, D. W.,Apelt, K. & Erickson, J. W. (1994). In¯uence ofstereochemistry on activity and binding modes ofC2 symmetry-based diol inhibitors of HIV-1 pro-tease. J. Am. Chem. Soc. 116, 847-855.
Hui, J. O., Tomasselli, A. G., Reardon, I. M., Lull, J. M.,Brunner, D. P., Tomich, C.-S. C. & Heinrikson, R. L.(1993). Large scale puri®cation and refolding ofHIV-1 protease from Escherichia coli inclusionbodies. J. Protein Chem. 12, 323-327.
Jaskolski, M., Tomasselli, A. G., Sawyer, T. K., Staples,D. G., Heinrikson, R. L., Schneider, J., Kent, S. B. H.& Wlodawer, A. (1991). Structure of 2.5 AÊ resol-ution of chemically synthesized human immunode-®ciency virus type 1 protease complexed with ahydroxyethylene-based inhibitor. Biochemistry, 30,1600-1609.
Kohl, N. E., Emini, E. A., Schleif, W. A., Davis, L. J.,Heimbach, J. C., Dixon, R. A., Scolnick, E. M. &Sigal, I. S. (1988). Active human immunode®ciencyvirus protease is required for viral infectivity. Proc.Natl Acad. Sci. USA, 85, 4686-4690.
Krohn, A., Redshaw, S., Ritchie, J. C., Graves, B. J. &Hatada, M. H. (1991). Novel binding mode ofhighly potent HIV-proteinase inhibitors incorporat-ing the (R)-hydroxyethylamine isostere. J. Med.Chem. 34, 3340-3342.
Laco, G. S., Schalk-Hihi, C., Lubkowski, J., Morris, G.,Zdanov, A., Olson, A., Elder, J. H., Wlodawer, A. &Gustchina, A. (1997). Crystal structures of the inac-tive D30N mutant of feline immunode®ciency virusprotease complexed with a substrate and an inhibi-tor. Biochemistry, 36, 10696-10708.
Laskowski, R. A., Mac Arthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK. A program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283-291.
Lawrence, M. G. & Colman, P. M. (1993). Shape comple-mentarity at protein/protein interfaces. J. Mol. Biol.234, 946-950.
Lee, B. & Richards, F. M. (1971). The interpretation ofprotein structures: estimation of static accessibility.J. Mol. Biol. 55, 379-400.
Loeb, D. D., Swanstrom, R., Everitt, L., Manchester, M.,Stamper, S. E. & Hutchison, C. A. D. (1989). Com-plete mutagenesis of the HIV-1 protease. Nature,340, 397-400.
Martin, J. L., Begun, J., Schindeler, A., Wickramasinghe,W. A., Alewood, D., Alewood, P. F., Bergman,
D. A., Brinkworth, R. I., Abbenante, G., March,D. R., Reid, R. C. & Fairlie, D. P. (1999). Molecularrecognition of macrocyclic peptidomimeticinhibitors by HIV-1 protease. Biochemistry, 38, 7978-7988.
Miller, M., Schneider, J., Sathyanarayana, B. K., Toth,M. V., Marshall, G. R., Clawson, L., Selk, L., Kent,S. B. & Wlodawer, A. (1989). Structure of complexof synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 AÊ resolution. Science, 246,1149-1152.
Murthy, K. H. M., Winsborne, E. L., Minnich, M. D.,Culp, J. S. & Debouck, C. (1992). The crystal struc-tures of 2.2 AÊ resolution of hydroxyethylene-basedinhibitors bound to human immunode®ciency virustype 1 protease show that the inhibitors are presentin two distinct orientations. J. Biol. Chem. 267,22770-22778.
Nicholls, A., Sharp, K. & Honig, B. (1991). Protein fold-ing and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281-296.
Otwinowski, Z. (1993). Data Collection and Processing,SerC Daresbury Laboratory, England.
Priestle, J. P., Fassler, A., Rosel, J., Tintelnot-Blomley,M., Strop, P. & Grutter, M. G. (1995). Comparativeanalysis of the X-ray structures of HIV-1 and HIV-2proteases in complex with CGP 53820, a novelpseudosymmetric inhibitor. Structure, 3, 381-389.
Ramachandran, G. N. & Sasisekharan, V. (1968). Confor-mation of polypeptides and proteins. Advan. ProteinChem. 23, 283-438.
Rasnick, D. (1997). Kinetics analysis of consecutive HIVproteolytic cleavages of the Gag-Pol polyprotein.J. Biol. Chem. 272, 6348-6353.
Robins, T. & Plattner, J. (1993). HIV protease inhibitors:their anti-HIV activity and potential role in treat-ment. J. Acquir. Immune De®c. Syndr. 6, 162-170.
Rose, J. R., Babe, L. M. & Craik, C. S. (1995). De®ningthe level of human immunode®ciency virustype 1 (HIV-1) protease activity required for HIV-1particle maturation and infectivity. J. Virol. 69, 2751-2758.
Rose, R. B., Craik, C. S., Douglas, N. L. & Stroud, R. M.(1996). Three dimensional structures of HIV-1 andSIV protease product complexes. Biochemistry, 35,12933-12944.
Rose, R. B., Craik, C. S. & Stroud, R. M. (1998). Domain¯exibility in retroviral proteases: structural impli-cations for drug resistant mutations. Biochemistry,37, 2607-2621.
Sack, J. S. (1988). CHAIN - a crystallographic modelingprogram. J. Mol. Graphics, 6, 224-225.
Silva, A. M., Cachau, R. E., Sham, H. L. & Erickson,J. W. (1996). Inhibition and catalytic mechanism ofHIV-1 aspartic protease. J. Mol. Biol. 255, 321-346.
Swain, A. L., Miller, M. M., Green, J., Rich, D. H.,Schneider, J., Kent, S. B. H. & Wlodawer, A. (1990).X-ray crystallographic structure of a complexbetween a synthetic protease of human immuno-de®ciency virus 1 and a substrate-based hydro-xyethylamine inhibitor. Proc. Natl Acad. Sci. USA,87, 8805-8809.
Thanki, N., Rao, J. K. M., Foundling, S. I., Howe, J.,Moon, J. B., Hui, J. O., Tomasselli, A. G.,Heinrikson, R. L., Thaisrivongs, S. & Wlodawer, A.(1992). Crystal structure of a complex of HIV-1 pro-tease with a dihydroxyethylene-containing inhibitor:

1220 HIV-1 Protease-substrate Complex
comparisons with molecular modeling. Protein Sci.1, 1061-1072.
Vondrasek, J., van Buskirk, C. P. & Wlodawer, A.(1997). Database of three-dimensional structures ofHIV proteinases. Nature Struct. Biol. 4, 8.
Weber, I. T., Wu, J., Adomat, J., Harrison, R. W.,Kimmel, A. R., Wondrak, E. M. & Louis, J. M.(1997). Crystallographic analysis of human immu-node®ciency virus 1 protease with an analog ofthe conserved CA-p2 substrate - interactions withfrequently occurring glutamic acid residue at P20
position of substrates. Eur. J. Biochem. 249, 523-530.
Wlodawer, A. & Erickson, J. W. (1993). Structure-basedinhibitors of HIV-1 protease. Annu. Rev. Biochem.62, 543-585.
Wlodawer, A., Miller, M., Jaskolski, M., Sathyanarayana,B. K., Baldwin, E., Weber, I. T., Selk, L. M.,Clawson, L., Schneider, J. & Kent, S. B. (1989).Conserved folding in retroviral proteases: crystalstructure of a synthetic HIV-1 protease. Science, 245,616-621.
Edited by I. Wilson
(Received 21 March 2000; received in revised form 3 July 2000; accepted 6 July 2000)
Copyright © 2022 FDOKUMEN




















