
Haematology & The Patient with Chronic Kidney Disease - edtna
194
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Haematology & The Patient with Chronic Kidney Disease - edtna



Haematology & the Patient with Chronic Kidney Disease
An Introductory Guide
This handbook is an initiative of EDTNA/ERCA & ANSA A limited edition will be available in English

All rights are reserved by the author and publisher, including the rights of reprinting, reproduction in any form and translation. No part of this book may be reproduced, stored in a retrieval system or transmitted, in any form or by means, electronic, mechanical, photocopying, recording, or otherwise, without the prior written permission of the publisher.
First edition: September 2009
European Dialysis and Transplant Nurses Association/ European Renal Care Association (EDTNA/ERCA)Pilatusstrase 35, Postfach 3052, 6002 Luzern, Switzerlandwww.edtnaerca.org
ISBN: 978-84-613-4051-4
D.L.: M-34989-2009
Layout, Binding and Printing: Imprenta Tomás HermanosRío Manzanares, 42-44 · E28970 Humanes de MadridMadrid - Spainwww.tomashermanos.com

5
Acknowledgements

6
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
Acknowledgements
This book is a joint initiative of the EDTNA/ERCA and the Anaemia Nurse Specialist Association (ANSA).The EDTNA/ERCA and ANSA would like to thank all the authors of each chapter and the editors for their considerable amount of work in order to complete this book. The support and help given to the editors by the reviewers has also been greatly appreciated and must be acknowledged.
EditorsLesley Bennett, RN, RM, BA, MSc Anaemia, Anaemia Consultant EDTNA/ERCA. Past President & Founder member of ANSA.Oxford Radcliffe Hospitals NHS Trust, Churchill Hospital, Oxford. UK
Susan Pickard, RN ANSA Executive Board MemberRenal Unit Basildon and Thurrock University Hospital Trust, Essex. UK
ReviewersDr. Sarah Gangoli, BSc (Hons) MB BS (Hons) MRCP, Department of Haematology, Belfast City Hospital. N Ireland. UK
Dr. Clare MacEwen, BA (Hons), BMBch (Oxon), MRCP, Oxford Radcliffe Hospitals NHS Trust, Churchill Hospital, Oxford. UK
Alessandra Zampieron, RN, BSN, MSN, DDSI, School of Nursing, Padua University. ITALY

7
Acknowledgements
Sponsor
The printing of the English version of this book has been sponsored by an education grant from Roche Products Ltd (UK) and Syner-Med Ltd (UK) in collaboration with Vifor Pharma International.


9
Table of
Contents

10
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
Preface .............................................................................................................. 15
Lesley Bennett, RN, RM, BA, MSc Anaemia
Oxford Radcliffe Hospitals NHS Trust, Churchill Hospital, Oxford, UK
1. Bleeding in the Uraemic Patient .......................................... 21
Susan Pickard, RN
Renal Unit Basildon and Thurrock University Hospital Trust Essex,
UK
2. Pure Red Cell Aplasia and the Patient with CKD ............................................................. 41
Alison Roche, RN, BSc (Hons) MA
Five Oaks Solutions, Offershaw, UK
3. Vitamin B12 De� ciency and Pernicious Anaemia ............................................................... 57
Karen Jenkins, RN, PG Dip HE, MSc Nursing
Kent Kidney Care Centre, East Kent Hospitals University NHS
Foundation Trust, Canterbury, UK

11
Table of Contents
4. Thalassaemia .............................................................................................. 71
Tai Mooi Ho Wong, RN, RM, DUENephrology Unit, Hospital del Mar, Barcelona, Catalonia, Spain
Anastasia Liossatou, RN, BN, Dip Ed, MSc Nursing Haemodialysis Unit, General Hospital of Kefalonia, Argostoli, Kefalonia, Greece
5. Sickle Cell Disease ............................................................................ 101
Alison Clare Chavanel, B.A.Hons (French and Spanish) In� rmière Diplômée d’Etat (France). Hemodialysis Unit, Centre Hospitalier de Valenciennes, France
6. Making the Connections: Lymphoma, the Kidneys and Transplantation ..... 121
Caroline McCaughey, RGN, BSc (Hons) Specialist Practice (Cancer), PGCE, MSc Nursing, Practice Educator, Queen’s University Belfast and Belfast City Hospital, (Haematology and Oncology), Belfast, N. Ireland
Fiona Murphy, RGN, RNT, Dip Res, Dip Prof Std, BSc (Hons) Renal Nurs, BSc (Hons), Health Stud, PGDip CHScieEduc, PGDip Adv Nurs Scie, MA, MSc, PhD student. Lecturer, Renal Educational
Facilitator, Trinity College, Dublin, Ireland

12
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
7. Myeloma and Renal Failure .................................................... 151
Alison Thornley, RN MSc Autonomous PracticeOxford Radcliffe Hospitals NHS Trust, Churchill Hospital, Oxford, UK
8. Myelodysplasia – The Bare Facts ................................... 175
Sharon Benton, RGN, Dip Ed, PGCE, BSc in Health Studies Duchess of Cornwall Renal Unit, Truro, UK
Cathy Johnson, RGN, BSc (Hons)Derby Hospitals NHS Foundation Trust, Derby, UK

13
Table of Contents


15
Preface

16
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
Preface
Healthcare Professionals (HCP) working within the � eld of Nephrology are used to seeing anaemia in their patient population. This is because renal anaemia, de� ned as a haemoglobin less than 11g/dl, is typical once the estimated glomerular � ltration rate (eGFR) is less than 30ml/min/1.73m² 1 and is almost universal in End Stage Renal Disease (ESRD).
Although the anaemia in patients with Chronic Kidney Disease (CKD) may develop in a response to a wide variety of causes,2
the Anaemia of Chronic Kidney Disease (ACKD) is due primarily to a lack of erythropoietin (EPO). This is because it is this hormone that is involved in the normal physiology of erythropoiesis.
In normal health, the effective circulating red blood cell mass is controlled by specialised interstitial cells that are located in the cortex of the kidney. These cells are very sensitive to small changes in tissue oxygenation.3 If tissue oxygenation decreases in the body, it is these cells that sense the hypoxia and produce erythropoietin.4 On production, erythropoietin binds to erythropoietin receptors that are located on the surface of early red blood cell progenitors, erythroid colony-forming units (CFU-Es) in order to prevent the cells from a pre-programmed cell death (apoptosis). The result is that the CFU-Es will survive and divide thereby increasing the production of reticulocytes, restoring normal circulating red blood cell mass and correcting tissue hypoxia. The entire process is controlled by a negative feedback loop because once the body’s requirement for erythropoietin is reduced, homeostasis results and the body’s requirement for erythropoietin is returned to normal.5

17
Preface
As most of the body’s erythropoietin, is produced from the kidneys, it follows that the majority of people with CKD will eventually become anaemic. However, people with CKD are not immune from having other haematological co-morbid conditions and as such may need to have not only the resulting anaemia appropriately managed but require extra care management programmes to be incorporated into their treatment plans.
The incidence of CKD increases with age,6 and as there are haematological problems associated with aging, for example, Myelodysplasia, people with both conditions may be seen in the CKD population.
Some ethnic populations are known to have a higher prevalence of CKD, these include people from South Asia due to diabetes 7 and Afro-Carribean’s and Africans.8 As some haematological conditions such as Sickle Cell and Thalasaemia are genetically linked and are prevalent in these populations, renal HCP’s may have clients with both conditions. With multi-ethnic populations in many renal units now becoming the norm, location of a unit does not exempt these problems from being seen globally.
The use of immunosuppressive therapy in the transplant population brings with it other associated problems, in particular, the risk of Post Transplant Lymphoproliferative Disorders. In the post transplant period, lymphoma is the second most frequently occurring cancer.9
Healthcare professionals have a responsibility to understand co-morbid conditions in their patient population and provide effective evidence-based care. It is therefore hoped that this introductory guide to haematology for those working within nephrology will be a useful tool and encourage the appropriate management of people with both CKD and haematology problems.

18
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
References1. Erslev AJ, Besarab A. Erythropoietin in the pathogenesis and treatment
of the anemia of chronic renal failure. Kidney International. 1997; 51: 622-630
2. Liossatou A, Jenkins K. Management of Anaemia in Chronic Kidney Disease. In: Mahon A, Jenkins K (eds) Chronic Kidney Disease (Stages 1-3). A Guide to Clinical Practice. Luzern, EDTNA/ERCA 2007 86-103
3. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. American Journal of Kidney Disease. 2001; 38: 415-425
4. Metzen E, Ratcliffe PJ. HIF hydroxylation and cellular oxygen sensing. Biology & Chemistry 2004; 385: 223-230
5. Bennett L. Management of Anaemia in Chronic Kidney Disease, Stages 4 & 5. In: Jenkins K, Mahon A (eds) Chronic Kidney Disease (Stages 4-5). A Guide to Clinical Practice. Luzern, EDTNA/ERCA 2008
6. Rodriguez-Puyol D. Aging Kidney. Kidney International 1998; 54:2247-2265
7. Lightstone L. Preventing Kidney Disease: the ethnic challenge. The National Kidney Research Fund: Peterborough. 2001
8. United States Renal Data System. Annual Data Report:incidence and prevalence of ESRD (2003). American Journal Kidney Disease 2003; 42 (5): S37-41
9. LaCasce, A.S. Post transplant lymphoproliferative disorders. The Oncologist, 2006; 11, pp. 674-680.

19
Preface


Bleeding in the
Uraemic Patient
21

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
22
Introduction Bleeding in a patient with uraemia is a recognised consequence of end stage renal disease and is believed to be multifactoral and complex. This is because the impaired haemostasis and homeostasis of laminar blood � ow in a uraemic patient is related to defective platelet adhesion, secretion or aggregation, anaemia and vascular abnormalities. The unwanted effects from haemodialysis cause further complications.1,2 Nowadays the majority of renal patients are older with co-morbidities such as hypertension and cardiovascular disease, these alone predispose to a higher risk of bleeding. In addition drugs used to treat these conditions such as Aspirin and anti-coagulant therapy will increase the risk by affecting platelet aggregation and coagulation.2
Physiology of normal haemostasisNormal Haemostasis is achieved by three mechanisms responsible for reducing blood loss.3 The physiology of these is as follows:
Learning outcomes
To understand:• The normal mechanisms of haemostasis• Discuss the pathophysiology of platelet dysfunction in
uraemia• Understand presentation and investigations• Be aware of evidence- based treatments

Bleeding in the Uraemic Patient
23Vascular spasm• of the vessel wall occurring immediately following damage to help reduce blood loss. The spasm is a result of the damage to the smooth muscle, activated platelets releasing substances and re� exes from pain receptors
Platelet plug formation• - Platelets, although small contain a vast array of clotting factors such as ade-nosine diphosphate (ADP), adenosine triphosphate (ATP), Ca2+ and serotonin. Enzymes that produce thromboxane A2 (TxA2) an inducer of platelet aggregation, lyosomes, mitochondria and glycogen play a part in this process.The � rst step in plug formation occurs when platelets come into contact with the damaged endothelial surface and adhere to collagen � bres. This is known as platelet adhesion. Production of a protein von Willebrand Factor (vWF) from the endothelial cells forms a bridge between collagen and platelets. On the platelet membrane a Glycoprotein GP1b/IX binds to the vWF capturing platelets. This adhesion results in the platelets becoming activated, resulting in activation of a further platelet receptor protein GPIIb/IIIa that binds to � brinogen.There then follows a platelet release reaction, ADP along with TxA2 are released and bind to receptors on the surface of other platelets resulting in their activation. Once activated the platelets express � brinogen receptors that bind to the plasma protein � brinogen. The � brinogen then forms bridges between the receptors of other platelets. This platelet aggregation results in formation of a platelet plug. At the same timeTxA2 acts as a vasoconstrictor along with serotonin to sustain the contraction of the blood vessel and decrease the blood � ow through the damaged vessel 3
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
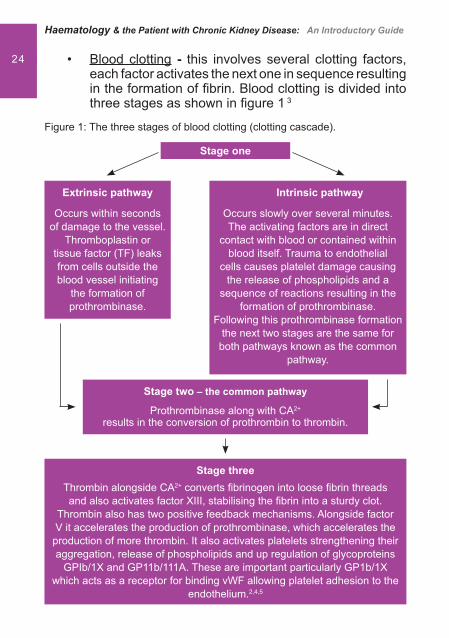
24 Blood clotting• - this involves several clotting factors, each factor activates the next one in sequence resulting in the formation of � brin. Blood clotting is divided into three stages as shown in � gure 1 3
Figure 1: The three stages of blood clotting (clotting cascade).
Intrinsic pathway
Occurs slowly over several minutes. The activating factors are in direct
contact with blood or contained within blood itself. Trauma to endothelial
cells causes platelet damage causing the release of phospholipids and a
sequence of reactions resulting in the formation of prothrombinase.
Following this prothrombinase formation the next two stages are the same for
both pathways known as the common pathway.
Extrinsic pathway
Occurs within seconds of damage to the vessel.
Thromboplastin or tissue factor (TF) leaks from cells outside the blood vessel initiating
the formation of prothrombinase.
Stage two – the common pathway
Prothrombinase along with CA2+ results in the conversion of prothrombin to thrombin.
Stage threeThrombin alongside CA2+ converts � brinogen into loose � brin threads and also activates factor XIII, stabilising the � brin into a sturdy clot.
Thrombin also has two positive feedback mechanisms. Alongside factor V it accelerates the production of prothrombinase, which accelerates the production of more thrombin. It also activates platelets strengthening their aggregation, release of phospholipids and up regulation of glycoproteins
GPIb/1X and GP11b/111A. These are important particularly GP1b/1X which acts as a receptor for binding vWF allowing platelet adhesion to the
endothelium.2,4,5
Stage one

Bleeding in the Uraemic Patient
25Physiology of normal homeostasisUnder normal homeostatic conditions there is a balance between clot formation and bleeding. There are a number of mechanisms responsible that include the following:
Normal endothelial cells release prostacyclin (PGI• 2) a vasodilator prostaglandin, opposing the action of TxA2, inhibiting platelet adhesion and release.3 They also release nitric oxide (NO) causing relaxation of smooth muscle cell and vasodilation.3 NO inhibits platelet to platelet interaction and affects platelet to vessel wall interaction 5
This interaction increases the formation of cyclic • guanosine monophosphate (cGMP), that acts as a cellular messenger causing relaxation of vascular smooth muscle leading to vasodilation and increased blood � ow 2,4,5 Furthermore a second cellular messenger that • modulates intraplatelet function Cyclic Adenosine Monophosphate (cAMP) is responsible for inhibiting platelet activation by counteracting the actions that result from increased cytosolic levels of calcium. By doing so, it inhibits the release of granules that would lead to activation of additional platelets and the coagulation cascade
Laminar blood � ow also in� uences homeostasis. Under the in� uence of a normal haematocrit red blood cells tend to � ow down the centre of the vascular lumen displacing the platelets towards the endothelial lining allowing for them to react quickly to any damage to the vessel.2,4 Prostacyclin and nitric oxide also regulate blood � ow due to their vasodilatory effects.
Pathophysiology of platelet dysfunctionUnderstanding the multifactorial mechanisms of impaired haemostasis and homeostasis helps to explain the increased
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
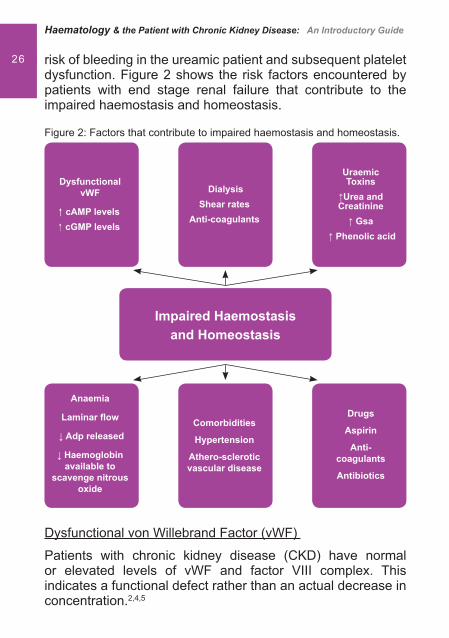
26 risk of bleeding in the ureamic patient and subsequent platelet dysfunction. Figure 2 shows the risk factors encountered by patients with end stage renal failure that contribute to the impaired haemostasis and homeostasis.
Figure 2: Factors that contribute to impaired haemostasis and homeostasis.
Dysfunctional von Willebrand Factor (vWF)
Patients with chronic kidney disease (CKD) have normal or elevated levels of vWF and factor VIII complex. This indicates a functional defect rather than an actual decrease in concentration.2,4,5
Dysfunctional vWF
�� cAMP levels� cGMP levels
Anaemia
Laminar � ow
� Adp released
� Haemoglobin available to
scavenge nitrous oxide
DialysisShear rates
Anti-coagulants
Comorbidities
Hypertension
Athero-scleroticvascular disease
UraemicToxins
�Urea andCreatinine
� Gsa � Phenolic acid
Drugs
Aspirin
Anti-coagulants
Antibiotics
Impaired Haemostasis and Homeostasis

Bleeding in the Uraemic Patient
27In uraemic platelet dysfunction there is thought to be either a decreased interaction between vWF and GP1b/1X, binding af� nity for GP1b/1X receptors or a reduced expression of these receptors on platelets.2,5 This reduced interaction impairs PIP2 (a component of cell membrane) breakdown resulting in a decrease in the production of thromboxane A2.1
Increased Cyclic Adenosine Monophosphate (cAMP)
Patients with a prolonged bleeding time due to CKD have a higher level of PGI2. PGI2 modulates the production of cAMP. A higher level can disrupt PIP2 breakdown resulting in a reduction in TxA2 and ADP.2,4,5
Increased Cyclic Guanosine Monophosphate (cGMP)
Increased levels of this substance due to an increase in nitrous oxide levels have been found in uraemic patients. This is because uraemic plasma is a potent inducer of nitrous oxide formation.6 The increase in these levels reduces TxA2 and ADP levels.
Uraemic toxins
Patients with CKD retain approximately 92 retention solutes or ureamic toxins. These include:
Urea and creatinine• Guanidinosuccinic acid (GSA)• Phenolic acids and methylguanidine•
It is the accumulation of these toxins that interferes with certain biological and biochemical functions.4 For example L-arginine believed to induce NO synthesis is moved as a result of excess urea from the urea cycle, stimulating further GSA. It also transfers an amadine group to aspartic acid, forming GSA, which along with phenolic acid inhibits ADP platelet aggregation.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
28 Dialysis
Haemodialysis itself may contribute to bleeding. This is because the arti� cial surfaces of the dialyser and dialysis tubing cause platelet activation-inducing degranulation and the loss of glycoprotein receptors.1,5
The use of anticoagulants in patients actively bleeding on haemodialysis may therefore need to be reviewed. The use of minimal Heparin, avoidance of anticoagulation or using regional citrate may be advocated.1,2
Anaemia
The majority of patients with CKD will become anaemic due to a decrease in the production of Erythropoietin (EPO) by the kidneys. In normal health EPO increases erythropoiesis and increases the number of circulating red blood cells. Red blood cells displace the platelets closer to the vascular endothelium, which causes a decrease in the response time of the platelets to vascular damage, thereby reducing bleeding time. In the anaemic state the reduction of red blood cells causes the platelets to travel in a mid-stream position i.e. further away from the endothelium. This results in reduced platelet activation when damage occurs.
Red cells also release ADP a potent stimulator of platelet response.5 Other actions of EPO include increasing the number of more metabolically active reticulated platelets, enhancing platelet aggregation and interaction between platelets and the sub endothelium.
Haemoglobin also acts as a scavenger of nitrous oxide. Therefore if haemoglobin levels are normal, NO levels are decreased. This results in a reduced stimulation of guanylyl cyclise and a reduction in the production of cGMP.4

Bleeding in the Uraemic Patient
29Drugs
In CKD some drugs accumulate due to a reduction in renal clearance. If these are administered platelet function may be affected. Examples of these drugs are antibiotics, such as those in the �-lactam group that can interfere with the ADP receptor. Cepholosporins can alter both platelet function and blood coagulation. Aspirin when given at a dose of 100mg daily has been shown to alter the bleeding time.5
Signs of bleeding in uraemia 1,7
Patients usually present with:Purpura• Epistaxis or gingival bleeding• Ecchymoses• Excessive bleeding from venepuncture sites• There may also be evidence of gastrointestinal, • intracranial bleeding or haematuria
InvestigationsDiagnosis is often based on the clinical signs and symptoms but can be evaluated by assessing the bleeding time or by undertaking platelet function studies. These are as follows: 8
Bleeding time• – Two incisions of a � xed depth are made into the skin of the forearm. Using circular � lter paper the blood is mopped up taking care not to disturb any clot formation. Normal range of bleeding time is 7 minutes with > 9 minutes regarded as abnormal.It should be noted that the bleeding time in females is longer than males. The test should not be carried out if the platelet count is <100 x 10 9/L and Aspirin should be withheld at least 7 days beforehand as it will interfere with the result

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
30 Platelet function studies• – blood collected by venepuncture is tested for the following:
Platelet count – (normal range 150 – 400 x 10 9/L), bruising and petechial haemorrhages often occurring when the platelet count drops to <20 x 10 9/LMorphology – a high platelet volume (MPV > 6.5) indicates the platelets are biochemically more activeAggregation – performed on fresh sample. This measures the time taken for � owing blood to occlude a collagen and ADP or collagen and epinephrine coated ring, assessing platelet dependant primary haemostasis 7 Platelet release – not generally available in routine laboratories, but measures granule proteins �- thromboglobulin � -TG and heparin neutralising activity (HNA) which are markers of platelet hyperactivity
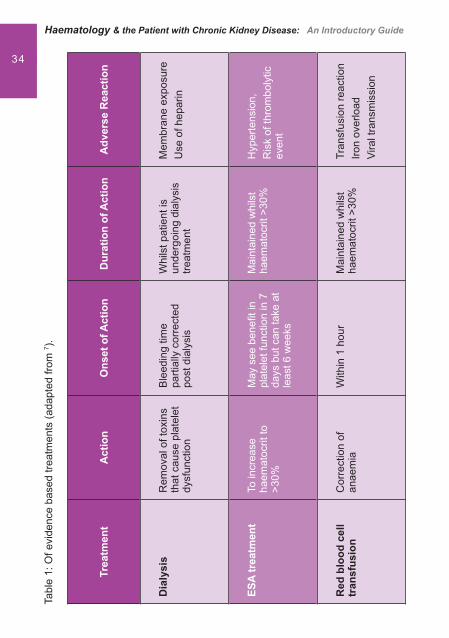
Evidence based treatments
Treatments target the factors that have a role in platelet dysfunction. They can have either an acute or delayed effect and may be used alone or in combination.
Dialysis
Dialysis has been found to only partially correct bleeding tendencies in patients with end stage renal disease.7 The impact in prevention and treatment of uraemic bleeding is still debatable. Studies carried out in dialysis as a prevention or treatment of uraemic bleeding have been of limited use but have shown that haemodialysis may be inferior to peritoneal dialysis. These studies are all at least 25 years old and therefore may not apply to today’s dialysis technology.4

Bleeding in the Uraemic Patient
31Haemodialysis or peritoneal dialysis can help to reverse uraemic bleeding in some patients with end stage renal disease by removing ureamic retention solutes such as urea, creatinine, phenol, phenolic acid and guanidinosuccinic acid (GSA) that contribute to impaired platelet dysfunction. However levels of these toxins do not seem to correlate with either the bleeding time or platelet adhesion.5
High-Flux dialysis dialysis membranes will more effectively remove larger retention solutes such as urea and guanidi-nosuccinic acid. It has also been found that there is a larger volume of distribution of these toxins than urea and that longer or more frequent dialysis is required for their removal.4
The type of dialysis used can have an impact, peritoneal dialysis being more effective in correcting platelet dysfunction by the greater clearance of urea and guanidinosuccinic acid and the absence of platelet activation by the arti� cial surfaces.7
Correction of anaemia with Erythropoietin stimulating agent (ESA)
An Erythropoesis Stimulating Agent (ESA) is often used to correct anaemia in renal disease but can also help in ureamic bleeding in several ways. It is thought that there is a direct effect on platelet function and at the vascular level.1 Treatment with an ESA can be either preventitive or used during acute bleeding in conjunction with other treatments. The aim is to increase the haematocrit to above 30%, which will decrease the bleeding time to a near normal level and allow the platelets to be distributed towards the endothelium. Correction of anaemia can take up to 2 – 3 months but there can be some bene� t on platelet function within 7 days due to the increase in reticulated platelets.4 Dosing regimes may vary and any treatment should be given in accordance with local guidelines.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
32 Blood transfusionsRed blood cell transfusions to increase the haematocrit to 30% can reverse uraemic bleeding by decreasing the bleeding time. Transfusion of platelets alone does not improve bleeding as the same circulating factors that impair native platelets will impair the function of the transfused platelets.7
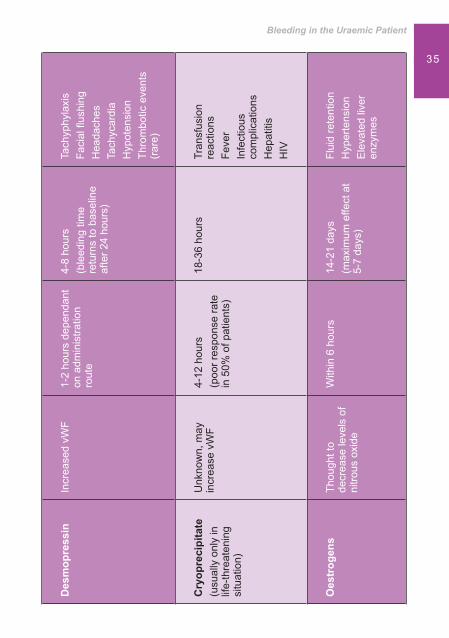
Desmopressin (DDAVP) Desmopressin (DDAVP) is an analogue of argine vasopressin and mainly used to treat Diabetes Insipidus, mild type 1 von Willebrand’s disease and haemophilia A.7 Although not licen-sed in some countries for this purpose it is also commonly used in uraemic bleeding as a � rst line treatment. The action of DDAVP is not fully understood but is thought to exert a haemostatic effect by releasing factor V111 from storage si-tes, increasing the concentration of factor V111 and reducing the effect of dysfunctional vWF.4
DDAVP has a rapid action and bleeding times will improve within an hour.5 Although its short activity means that the bleeding time will return to baseline within 24 hours. It can be used in patients who require immediate surgery or a biopsy and avoids exposure to blood- born pathogens.4
Administration should not be repeated due to the risk of depletion of stores of von Willebrand Factor causing tachyphylaxis. It is administered intravenously or sub-cutaneously at a dose of 0.3u/kg over 30 minutes. It may also shorten bleeding times in 2 hours when administered intra-nasally at a dose of 2 microgrammes/kg.7 Side effects include headache, � ushing, tachycardia, hypotension and rarely thrombotic events.5
CryoprecipitateCryoprecipitate is a blood product that is rich in factor V111, vWF and � brinogen and is used in a variety of bleeding

Bleeding in the Uraemic Patient
33situations, but is usually only administered in life-threatening situations.5
It is thought to increase the amount of clotting factors in plasma and has a bene� cial effect on bleeding time within 4-12 hours in the majority of patients although not all patients will respond.7 It has the potential risk of infectious complications, post-transfusion hepatitis, HIV, fever and allergic reaction.4
Conjugated OestrogensConjugated Oestrogen controls bleeding in both males and females in chronic kidney disease. It is normally used as hormone replacement therapy but when used in uraemic bleeding is thought to decrease the production of L-arginine, a pre-cursor of Nitrous Oxide (NO). Levels of NO are higher in uraemia and by decreasing these levels there is less guanylyl cyclase stimulated and less cGMP production, leading to an increase in TxA2 production and ADP.4
Oestrogens can be administered intravenously at a dose of 0.6mg/kg over 30-40 minutes daily for 4-5 days and have a long action of 14-21 days, with the maximum effect being at 5-7 days.5 The long acting effect is particularly useful in patients with gastrointestinal bleeding or in patients about to undergo surgery.1 They may also be administered orally or transdermally, however most studies have concentrated on intravenous administration and are therefore the preferred route.4 Side effects include � uid retention, hypertension and elevation of liver enzymes.
Tranexamic acidStudies have shown that Tranexamic acid (an anti- � brinolytic drug) may be of use in uraemic bleeding due to an increase in � brinolytic activation markers, resulting in improved or normalisation of bleeding times.9
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
34Ta
ble
1: O
f evi
denc
e ba
sed
treat
men
ts (a
dapt
ed fr
om 7 ).
Trea
tmen
tA
ctio
nO
nset
of A
ctio
nD
urat
ion
of A
ctio
nA
dver
se R
eact
ion
Dia
lysi
sR
emov
al o
f tox
ins
that
cau
se p
late
let
dysf
unct
ion
Ble
edin
g tim
e pa
rtial
ly c
orre
cted
po
st d
ialy
sis
Whi
lst p
atie
nt is
un
derg
oing
dia
lysi
s tre
atm
ent
Mem
bran
e ex
posu
reU
se o
f hep
arin
ESA
trea
tmen
tTo
incr
ease
ha
emat
ocrit
to
>30%
May
see
ben
e� t
in
plat
elet
func
tion
in 7
da
ys b
ut c
an ta
ke a
t le
ast 6
wee
ks
Mai
ntai
ned
whi
lst
haem
atoc
rit >
30%
Hyp
erte
nsio
n,R
isk
of th
rom
boly
tic
even
t
Red
blo
od c
ell
tran
sfus
ion
Cor
rect
ion
of
anae
mia
With
in 1
hou
rM
aint
aine
d w
hils
t ha
emat
ocrit
>30
%Tr
ansf
usio
n re
actio
nIro
n ov
erlo
adVi
ral t
rans
mis
sion
Bleeding in the Uraemic Patient
35D
esm
opre
ssin
Incr
ease
d vW
F1-
2 ho
urs
depe
ndan
t on
adm
inis
tratio
n ro
ute
4-8
hour
s(b
leed
ing
time
retu
rns
to b
asel
ine
afte
r 24
hour
s)
Tach
yphy
laxi
sFa
cial
� us
hing
Hea
dach
esTa
chyc
ardi
aH
ypot
ensi
onTh
rom
botic
eve
nts
(rar
e)
Cry
opre
cipi
tate
(usu
ally
onl
y in
lif
e-th
reat
enin
g si
tuat
ion)
Unk
now
n, m
ay
incr
ease
vW
F4-
12 h
ours
(poo
r res
pons
e ra
te
in 5
0% o
f pat
ient
s)
18-3
6 ho
urs
Tran
sfus
ion
reac
tions
Feve
rIn
fect
ious
co
mpl
icat
ions
Hep
atiti
sH
IV
Oes
trog
ens
Thou
ght t
o de
crea
se le
vels
of
nitro
us o
xide
With
in 6
hou
rs14
-21
days
(max
imum
effe
ct a
t 5-
7 da
ys)
Flui
d re
tent
ion
Hyp
erte
nsio
nE
leva
ted
liver
en
zym
es

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
36 Nursing intervention
The risk of bleeding in patients with end stage renal failure is of clinical concern and nurses working in the renal � eld should be vigilant and able to identify those patients at risk to allow for early intervention.
Regular monitoring of blood results will highlight a • sudden drop in haemoglobin indicating blood loss and require further investigationAny prolonged bleeding from a � stula site should be • reported and reviewedAny prolonged bleeding should be corrected prior to • any invasive surgery or renal biopsy
Conclusion
It is acknowledged that patients with end stage renal disease and uraemia develop bleeding complications. Diagnosis is often based on clinical symptoms but investigation of platelet function studies may be undertaken. Patients may present with epistaxis, purpura, excessive bleeding from venepuncture sites or evidence of gastrointestinal or intracranial bleeding.
The pathophysiology is multifactoral but believed to be due to platelet dysfunction and impairment of normal haemostatic and homeostatic mechanisms. Uraemia and circulating toxins play a major factor, other contributing factors include anaemia, co-morbidity, drugs used for anticoagulation and dialysis itself. Normal haemostatic and homeostatic mechanisms are affected by uraemia resulting in platelet adhesion, secretion and aggregation dysfunction. Clotting mechanisms are altered and changes to laminar blood � ow occur.
Dialysis has some bene� ts but other evidence-based treatments may be required. These include the use of Desmopressin (DDAVP), Cryoprecipitate, conjugated oestrogens and the

Bleeding in the Uraemic Patient
37correction of anaemia with an Erythropoiesis Stimulating Agent (ESA).
The risk of bleeding in patients with end stage renal failure is of clinical concern and any prolonged bleeding should be corrected prior to a renal biopsy or invasive surgery. Patients at risk should be identi� ed to allow for early intervention.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
38 References1. Escolar G, Diaz-Ricart M, Cases A. Uremic Platelet Dysfunction: Past
and Present. Current Hematology Reports 2005; 4:359-367.2. Sohal A S, Gangji A S, Crowther M A, Treleaven D. Uremic bleeding:
Pathophysiology and clinical risk factors. Thrombosis Research 2006; 118: 417-412.
3. Tortora G, Derrickson B. Principles Of Anatomy And Physiology. 11th edition. New Jersey: Von Hoffman Press. 2006. p681-684.
4. Hedges S J, Dehoney S B, Hooper J S, Amanzadeh J, Busti A J. Evidence-based Treatment Recommendations for Uremic Bleeding. National Clinical Practice Nephrology 2007; 3(3): 138-153. http://www.medscape.com/viewarticle/553187(accessed 30/12/08).
5. Kaw D, Malhotra D. Platelet Dysfunction and End-stage Renal Disease. Seminars in Dialysis 2006; 19: 317-322.
6. Noris M, Benigni A, Boccardo P, Aiello S, Gaspari F, Todeschini M et al. Enhanced nitric oxide synthesis in uremia: Implications for platelet dysfunction and dialysis hypotension. Kidney International 1993; 44:445-450.
7. Gangji A S, Sohal A S, Treleaven D, Crowther M A. Bleeding in patients with renal insuf� ciency: A practical guide to clinical management. Thrombosis Research 2006; 118: 423-428.
8. Provan D, Krentz A, editors. Oxford handbook of clinical and laboratory investigation. 2nd ed. Oxford: Oxford University Press, 2002. p209.
9. Downey P, Tagle R, Pereira J, Mezzano D. Tranexamic acid and uremic bleeding: evidence-based treatment recommendations. National Clinical Practice Nephrology 2007; 3. http//www.nature.com/ncneph/journal/v3/n6/full/ncpneph0528.html (accessed 10/2/09).

Bleeding in the Uraemic Patient
39


Pure Red Cell
Aplasia and the
Patient with
CKD
41

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
42 Learning outcomes
• To develop an understanding of the causes of Pure Red Cell Aplasia (PRCA)
• To understand current recommendations for the investigation of a suspected case of PRCA
• To have knowledge of the treatment options available• To understand the measures to be taken to support a
patient through the clinical course of PRCA
Introduction
PRCA (Pure Red Cell Aplasia) is a rare haematological condition where the bone marrow fails to produce red blood cells or their precursor cells resulting in a profound anaemia that usually renders a patient transfusion dependent.
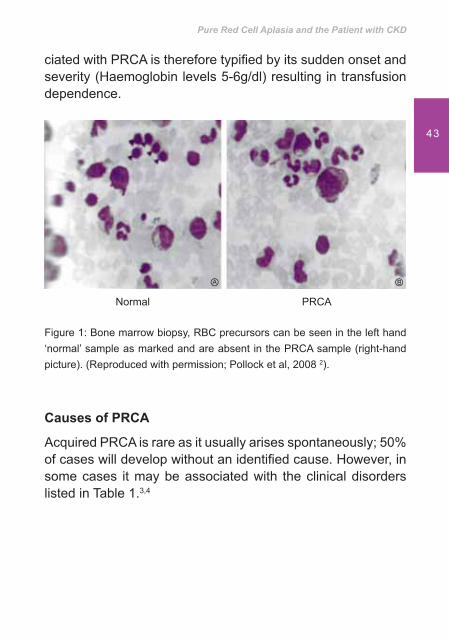
Within the bone marrow, while megakaryocytes (platelet precursors) and white blood cell (WBC) precursors are usually present at normal levels it is the absence of red blood cell (RBC) precursors 1 that is characteristic of PRCA, Figure 1.
Without the development of RBC precursors, erythrocyte production will be diminished. The resulting anaemia asso-
Pure Red Cell Aplasia and the Patient with CKD
43
ciated with PRCA is therefore typi� ed by its sudden onset and severity (Haemoglobin levels 5-6g/dl) resulting in transfusion dependence.
Figure 1: Bone marrow biopsy, RBC precursors can be seen in the left hand ‘normal’ sample as marked and are absent in the PRCA sample (right-hand picture). (Reproduced with permission; Pollock et al, 2008 2).
Causes of PRCA
Acquired PRCA is rare as it usually arises spontaneously; 50% of cases will develop without an identi� ed cause. However, in some cases it may be associated with the clinical disorders listed in Table 1.3,4
Normal PRCA
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
44
Table 1: Known causes of PRCA (after Eckardt et al, 2003 4).
Lymphoproliferative disorders•
Chronic Lymphocytic Leukaemia•
Bone marrow/stem cell transplantation•
Infections•
Parvovirus B19•
Epstein Barr Virus•
Viral Hepatitis•
HTLV Mumps•
Systemic autoimmune disease•
Systemic Lupus Erythematosus•
Rheumatoid Arthritis•
Drugs•
Antiepileptic medicines (e.g. Phenytoin, Carbamazepine, • Sodium Valproate)
Azathioprine•
Chloramphenicol•
Sulfonamides•
Isoniazid•
Procainamide•
Recombinant Human Erythropoietin (rHuEPO)•
Mycophenelate Mofetil (Cellcept• ®)
Thymoma ~ 5% cases •
Unknown Causes ~ 50% cases•

Pure Red Cell Aplasia and the Patient with CKD
45
Incidence of PRCAThe incidence of PRCA is rare in all populations and particularly rare in patients with chronic kidney disease. (CKD). Whilst patients with CKD are not excluded from developing PRCA from any cause the development of PRCA in this patient population has been more commonly associated with drug therapy. In particular with an erythropoietin stimulating agent (ESA) and more recently with Cellcept® (Mycophenelate Mofetil, Hoffman La Roche).
Mycophenelate Mofetil (Cellcept ) and PRCACellCept® is an immunosuppressive agent indicated in combi-nation with ciclosporin and corticosteroids for the prophylaxis of acute transplant rejection in adults receiving allogeneic re-nal, cardiac or hepatic transplants, and in children and adoles-cents (2-18 years) receiving renal transplants.
In June 2009, the manufacturer of Cellcept® issued a ‘Dear Healthcare Professional’ letter notifying clinicians that 41 cases of PRCA have been reported worldwide to date in association with CellCept®.5 Whilst these cases included patients recei-ving other medicines such as alemtuzumab, tacrolimus, azathioprine and co-trimoxazole, which could have contributed to the development of PRCA, alone or in combination, the letter warned that a causal association between CellCept® and PRCA is considered a reasonable possibility.
Patients receiving this drug with a severe anaemia of sudden onset should therefore have PRCA considered as a potential diagnosis.
ESA-associated PRCAKnown as antibody-mediated PRCA, this is due to the presence of anti-erythropoietin (EPO) antibodies and is usually only seen in patients receiving ESA therapy. Antibody-mediated PRCA is now recognised as a rare complication of ESA therapy and cases of this have been described with epoetin

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
46
alfa (Eprex®), epoetin beta (Neorecormon®) and Darbepoetin alfa (Aranesp®).6,7,8
Between 1998 and 2002, there was a rapid increase in the number of cases of antibody mediated PRCA.9 Predominantly associated with Epoetin alfa (Eprex®) formulations, this is thought to have been attributed at least in part to a change in formulation and a subsequent interaction between polysorbate 80, a stabiliser routinely used with injectable proteins and leachates from the rubber stoppers in the syringe.10 Following the addition of Te� on® coating to the rubber stoppers, the incidence of PRCA associated with Eprex® formulations has since fallen back to very low levels. Although new cases do still appear sporadically11, the incidence of this adverse effect peaked in 2002.
It is considered to be very rare for patients to develop antibody-mediated PRCA caused by circulating anti-erythropoietin antibodies as a consequence of ESA therapy in the treatment of anaemia of chronic kidney disease (CKD).9 It is therefore, generally accepted that the bene� ts of treating the anaemia of chronic kidney disease with ESA far out-weighs the very low risk of developing pure red cell aplasia.
Altered erythropoietin physiology
The pathogenesis of PRCA in most drug-induced cases is largely unknown, although the mechanism is believed to be related to direct effects on red cell precursors or the induction of autoimmunity.2
In ESA-associated PRCA, neutralising anti-erythropoietin antibodies develop in patients undergoing treatment with an ESA that essentially destroy all available erythropoietin. These antibodies not only neutralise all currently available ESAs, but also the patient’s own endogenous erythropoietin, consequently stopping any signi� cant erythropoietic activity

Pure Red Cell Aplasia and the Patient with CKD
47
within the bone marrow.12 This leads to a form of anaemia more severe than that occurring before the start of ESA therapy.
Identifying � rst signs/diagnosis
The development of ESA-associated PRCA is sporadic and unpredictable with few identi� ed pre-disposing factors. Patients with this condition experience a rapid decrease in haemoglobin rendering them transfusion dependent. It is associated with a profoundly low reticulocyte count despite often rapidly escalating doses of ESA.
This presenting combination of falling haemoglobin and rising ESA dose is often termed ‘loss of effect’ due to the inability to sustain haemoblobin levels despite escalating ESA dose. There are of course other causes of this ‘loss of effect’ phenomenon, for example occult bleeding, haemolysis, infection or myelo-suppression and these should be excluded as a matter of course. Con� mation of the diagnosis of ESA-associated PRCA therefore requires a systematic approach, beginning with simple measurements such as blood cell counts, as most cases of ESA hyporesponsiveness are attributable to other causes.2
The diagnosis of classical ESA induced PRCA is made by a number of clinical features, including
severe transfusion-dependent anaemia • reticulocytopenia• low or absent erythroblasts in the bone marrow• circulating anti-erythropoietin antibodies•
Clinical signs of antibody-mediated PRCA (ESA-induced PRCA) are listed in Table 2.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
48
Table 2: Clinical features of PRCA (reproduced with permission, after Pollock et al 2).
Severe ESA resistance (>300iu/kg/week or equivalent)•
Rapid fall in haemoglobin•
Transfusion dependence (often on a 1-2 weekly basis)•
Profoundly low reticulocyte count (usually <10 x 109/l)•
Usually normal white cell and platelet counts•
High serum ferritin and transferrin saturation levels•
Bone marrow showing virtual absence of red cell precursors (aplasia)•
Develops at least 2 months after commencing • ESA agent (median approx 10 months)
Presence of antibodies directed against erythropoietin•
Usually associated with subcutaneous administration of ESA•
More recently some cases have been reported in the renal medical literature describing bone marrow � ndings of red cell hypoplasia rather than aplasia; this may represent an earlier presentation of the same condition.13
Treatment of PRCA
The goals of treatment are to restore endogenous red cell production, to maintain Hb at an adequate level, and to treat underlying disorders.1 The mainstay of treatment for PRCA is two-fold, to treat the associated symptoms, most common-ly the profound anaemia, with regular blood transfusions and, to identify the probable causative factors and remove or resolve them.
In the cases of PRCA induced by other drugs (unrelated to anti-erythropoietin antibody production), the PRCA generally resolves within 1-2 weeks after the causative agent is withdrawn.2

Pure Red Cell Aplasia and the Patient with CKD
49
ESA-associated PRCA- treatment recommendationsThe European Best Practice guidelines for the treatment of renal anaemia provide detailed recommendations on how the multi-professional team should proceed in the event of a suspected ESA-associated PRCA case.14 A suggested clinical algorithm has been published within this report, which is still valid, and summarises treatment as:
stopping the ESA agent • implementing an immunosuppressive regimen to • reduce or abolish erythropoietin antibody production
Patients should not be switched to another ESA since the antibodies cross react with all currently available agents.11 Where diagnosis of PRCA is con� rmed by the presence of anti-erythropoietin antibodies, immunosuppressive drugs should be considered in an attempt to suppress antibody formation. Several agents that have been used clinically in these cases include cyclosporine, prednisolone, cyclophosphamide, myco-phenolate and rituximab, all with variable results. 15
Anti-erythropoietin antibody-mediated PRCA is only rarely self-limiting, and treatment has been problematic.16 Treatment with immunosuppressive agents has cured some cases of the disease15 but re-exposure to an erythropoiesis stimulating agent can reincite antibody formation.17
Hematide® is a synthetic peptide (amino acid chain) currently in clinical development (phase 3 clinical trials) for use as an ESA and has been demonstrated under clinical trial conditions to stimulate erythropoiesis, raise haemoglobin and eliminate transfusion requirements in a small number of patients who have con� rmed ESA-mediated PRCA.18 This investigational product may provide hope in the treatment of patients who develop ESA induced PRCA in the future. However, there is currently limited clinical experience with Hematide® particularly with respect to long-term safety and tolerability, and it is not as yet commercially available.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
50
CellCept and PRCA - treatment recommendationsWhere Cellcept® is suspected of being the causative agent of PRCA the recommendation is to reduce the dose in the � rst instance and observe the clinical response.5 Changes to CellCept® therapy should only be undertaken under appropriate supervision in transplant recipients in order to minimise the risk of graft rejection. The rationale for this guidance is due to the potential catastrophic effect of stopping immunosuppressive agents in a patient with a functioning organ transplant.
Nurse’s roleThe role of the nursing team in the management of patients who develop PRCA lies in the ongoing surveillance and support of the patient. The timely evaluation of blood results to identify either a suspected case of ‘loss of effect’ that requires systematic investigation, identifying the need for blood transfusion and/or evaluating the clinical response to treatment can assist in both the diagnosis and evaluating the appropriateness of treatment.
Ideally, a patient speci� c protocol should be established which dictates Haemoglobin (Hb) thresholds for the administration of blood transfusions that maintain Hb levels at a level whereby patients can maintain an acceptable quality of life and are minimally symptomatic of their anaemia.
Accurate feedback and evaluation of co-morbid factors which may impact on Hb results (i.e; intercurrent infection/blood loss via dialyser lines/transfusion reaction/iron overload) is also vital to enable ongoing monitoring of the syndrome in terms of transfusion requirements or syndrome resolution.
Patient Education The nursing team has a major role to play in the provision of support, information and education of patients diagnosed with

Pure Red Cell Aplasia and the Patient with CKD
51
PRCA. Prior to a patient being diagnosed with PRCA it is likely that they will have undergone many investigations to exclude other causes of an unexplained anaemia. These investigations may include:
Blood tests• Haemolysis screening• Screening for infection• Gastro-intestinal examination (OGD or colonoscopy) • to exclude GI bleedBone marrow aspiration/biopsy•
Many of these investigations are invasive but necessary as PRCA is often diagnosed by a process of the elimination of other comorbidity. It is therefore essential that patients be provided with information about the rationale of ongoing investigations to enable them to make informed decisions about treatment.
The education needs of patients with PRCA irrespective of the causative factors are likely to include:
Rationale behind necessary investigations• Bene� ts and side effects of treatment regimens• Frequency of clinical review and evaluation of treat-• mentSymptoms of chronic anaemia• Life style modi� cation to minimise symptoms of anae-• miaThresholds for transfusion and potential for transfusion • reactionInteraction of any treatments for PRCA on usual medi-• cation regimen

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
52
ConclusionsPRCA has been highlighted to the nephrology community due to its occurrence as a serious adverse event of erythropoietin stimulating agent (ESA) therapy 9 and also more recently as a potential but rare side-effect of Cellcept® (Mycophenolate Mofetil).5
PRCA is however a rare phenomenon in patients with chronic kidney disease, and in these patients is most usually although not exclusively associated with the development of anti-ESA antibodies due to the use of ESAs. The incidence of ESA-induced PRCA peaked in 2002 predominantly associated with Eprex® and Erypo® (distributed outside of the USA) and with these cases it is generally accepted that it is thought to be due to a change in formulation of these products. However, cases have been reported with most commercially available ESAs, and new cases still occur sporadically.
Currently, there is no universally accepted commercially available treatment for ESA-induced PRCA, although the cessation of ESA treatment and use of immunosuppression is recommended.15 Most patients with this condition will require regular blood transfusions and thus the usual management guidelines for transfusing blood products apply. Treatment is not universally successful and some patients have remained transfusion dependent inde� nitely. Some hope for future successful treatments for ESA-associated PRCA has been provided by the results of a pilot study using Hematide®,18 a drug currently under clinical development which stimulates erythropoiesis in the presence of anti-erythropoietin antibo-dies.

Pure Red Cell Aplasia and the Patient with CKD
53
References1. Schick, P. Pure Red Cell Aplasia. http://emedicine.medscape.com/
article/205695-overview accessed 24/5/2009.2. Pollock, C, Johnson, DW, Horl, W, et al. Pure red-cell aplasia induced
by erythropoiesis-stimulating agents. Clin J Am Soc Nephrol 2008; 3:193-199.
3. Casadevall N. Antibodies against rHuEPO: native and recombinant. Nephrol Dial Transplant 2002; 17 [Suppl 5]: 42–47.
4. Eckardt K-U, Casadevall N. Pure red-cell aplasia due to anti-erythropoietin antibodies. Nephrol Dial Transplant 2003; 18:865–869.
5. F. Hoffman La Roche. Direct Healthcare Professional Communication on the association of CellCept® (mycophenolate mofetil) with pure red cell aplasia http://www.cbg-meb.nl/NR/rdonlyres/60C14C57-66BA-4C79-B8C3-A9D290E5B09F/0/DHPCCellCeptEng.pdf accessed 16 June 2009.
6. Bennett CL, Luminari S, Nissonson, AR et al. Pure red cell aplasia and epoetin therapy. N Eng J Med 2004; 351:1403-1408.
7. Tolman C, Duja S, Richardson D et al. Four cases of pure red cell aplasia secondary to epoetin beta, with strong temporal relationships. Nephrol Dial Transplant 2004; 19:2133-2136.
8. Jacob A, Sandhu K, Nicholas J et al. Antibody-mediated pure red cell aplasia in a dialysis patient receiving darbepoetin alfa as the sole erythropoietic agent. Nephrol Dial Transplant 2006; 21:2963-2965.
9. Casadevall N, Nataf J, Viron B et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Eng J Med 2002; 346:469-475.
10. Boven K, Stryker S, Knight J et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kid Int 2005; 67:2346-2353.
11. Macdougall IC. Epoetin-induced pure red cell aplasia: diagnosis and treatment. Curr Opin Nephrol Hypertens 2007; 16:585-588.
12. Thorpe R, Swanson SJ. Current methods for detecting antibodies against erythropoietin and other recombinant proteins. Clin Diagn Lab Immun 2005; 12:28-39.
13. Casadevall N, Rossert J, Swanson S. Anti-erythropoietin (EPO) antibodies (Abs) not associated with pure red cell aplasia (PRCA), in patients treated with erythropoiesis-stimulating agents (ESAs) [poster]. American Society of Nephrology Renal Week; 2004.
14. European Best Practice Guidelines Work Group. Section IV: Failure to respond to treatment. Nephrol Dial Transplant 2004; 19:ii32-ii36.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
54
15. Verhelst D, Rossert J, Casadevall N et al. Treatment of erythropoietin-induced pure red cell aplasia: a retrospective study. Lancet 2004; 363:1768-1771.
16. Bennett CL, Cournoyer D, Carson KR. Long-term outcome of indi-viduals with pure red cell aplasia and antierythropoietin antibodies in patients treated with recombinant epoetin: A follow-up report from the Research on Adverse Drug Events and Reports (RADAR) project. Blood 2005; 106: 3343-3347.
17. Andrade J, Taylor PA, Love JM, Levin A. Successful reintroduction of a different erythropoiesis-stimulating agent after pure red cell aplasia: relapse after successful therapy with prednisone. Nephrol Dial Transplant 2005; 20: 2548-51.
18. Macdougall IC, Rossert J, Casadevall N, Stead RB, Duliege AM, Eckardt, KU. Treatment of Anti-Erythropoietin Antibody-Mediated Pure Red Cell Aplasia with a Novel Synthetic Peptide-based Erythropoietin Receptor Agonist. Lancet 2009; (in press).

Pure Red Cell Aplasia and the Patient with CKD
55


Vitamin B12
Defi ciency
and Pernicious
Anaemia
57

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
58
Learning Outcomes• To gain knowledge and understanding of
pernicious anaemia• To be aware of the causes of pernicious anaemia• To understand the tests used to diagnose
pernicious anaemia• To recognise the signs and symptoms of vitamin
B12 de� ciency• To understand the treatments available
IntroductionLack of vitamin B12 can be a contributory factor to anaemia of chronic kidney disease (CKD). Vitamin B12 and folate levels should be measured when screening for causes of anaemia in this population. Those with established renal disease and receiving haemodialysis may require vitamin B12 and folate supplementation as both are water soluble vitamins and are often lost during dialysis, therefore reducing levels.1 Lack of vitamin B12 can lead to pernicious anaemia.
What is pernicious anaemia?Pernicious anaemia is an autoimmune condition usually associated with the development of auto-antibodies against parietal cells or intrinsic factor in the lining of the stomach. Parietal cells make intrinsic factor which helps the body absorb vitamin B12 in the small intestine. In some people, the body’s

Vitamin B12 De� ciency and Pernicious Anaemia
59
immune system may attack and destroy the parietal cells. As a result of this immune system attack, the stomach lining shrinks, and the parietal cells in the lining of the stomach disappear and the stomach stops producing intrinsic factor. An infection such as Helicobacter pylori may initiate a form of autoimmune gastritis which presents as iron de� ciency in the young and as pernicious anaemia in the elderly. If an autoimmune gastritis occurs it leads to loss of intrinsic factor production and consequently a reduction in vitamin B12 absorption and vitamin B12 de� ciency develops.2
A positive response to treatment of the infection means that intrinsic factor production usually returns. Loss of intrinsic factor can also be due to removal of the stomach lining in various kinds of stomach surgery. There is also a rare inherited disorder in which children are born without the ability to produce intrinsic factor. A congenital lack of or abnormality of intrinsic factor will usually present at approximately 2 years of age when stores of B12 derived from the mother in utero have been used up.3
Who is at risk of pernicious anaemia?The incidence of pernicious anaemia increases over the age of 40 years and is more common in women than men.4 The disease is found in all races, but is more prevalent in northern Europe, and Africans, often occurring in families.3 50% of patients are shown to have an antibody to intrinsic factor which inhibits the intrinsic factor binding to B12. Intrinsic factor antibodies are speci� c to pernicious anaemia but occur in the serum of only half of patients.4 Table 1 shows the risk factors associated with pernicious anaemia.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
60
Table 1: Risk Factors.
Family history of pernicious anemia
Diabetes or a thyroid problem
An intestinal disorder that keeps the body from absorbing vitamin B12
Blue eyes
Early graying
Blood group A
Myxoedema
Hashimoto’s disease
Thyrotoxicosis
Hypoparathyroidism
Carcinoma of the stomach
Diagnosing pernicious anaemiaA lack of vitamin B12 means that blood cells do not divide normally. Pernicious anaemia is characterised by the presence of large, immature, nucleated cells (megaloblasts), the fore-runners of red blood cells, (Red blood cells, when mature, have no nucleus), and hypersegmented neutrophils. The com-bination of this and macrocytosis on a blood � lm con� rms diagnosis of the disease which is a type of megaloblastic anaemia.
Most megaloblastic anaemias result from a lack of either vitamin B12 or folate, and it is essential to establish which de� ciency is present and the underlying cause.

Vitamin B12 De� ciency and Pernicious Anaemia
61
Vitamin B12 de� ciencyPeople can develop pernicious anaemia if they don’t get enough vitamin B12 in the foods that they eat. It takes at least two years to develop because this is the time it takes to use up the vitamin B12 already stored in the body (i.e. 1-2�g/day). Some people who are strict vegetarians can develop pernicious anaemia, especially if they do not eat meat, poultry, � sh, eggs, or dairy products, which are the best food sources of vitamin B12. Breastfed infants of strict vegetarian mothers can develop anaemia in a short time because they don’t have enough vitamin B12 stored in their bodies. Some people develop pernicious anaemia because of a poor diet due to conditions such as alcoholism or ageing.
As more elderly people are being identi� ed with Chronic Kidney Disease (CKD) due to estimated glomerular � ltration rate (eGFR) testing it is important to remember that they are susceptible to developing vitamin B12 de� ciency irrespective of having CKD or being on dialysis as they may have a poor dietary intake.5
In summary pernicious anaemia is a condition in which the body does not make enough red blood cells which can be due to a lack of vitamin B12 in the body.
Folate de� ciency Folate is necessary for the production and maintenance of new red blood cells. It is especially important during periods of rapid cell growth such as during pregnancy and infancy. It is needed for DNA synthesis and DNA replication. Since folate de� ciency limits cell division, erythropoiesis (red cell production) is hindered and leads to megaloblastic anaemia. The most common cause of folate (Folic acid) de� ciency is nutritional due to poor diet and/or alcoholism. Although folate is plentiful in liver, greens and yeast, it is easily destroyed by heat during cooking. Body stores are small (5 to 10 mg)

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
62
and individuals who have a folate de� cient diet can develop megaloblastosis within 4 to 5 months. Folate de� ciency is also common in the elderly. In one study the age-speci� c prevalence of folic acid de� ciency was approximately 5 and 10 percent in those age 65 to 74 versus those �75 years of age, respectively.5 Folate is a water soluble vitamin and is removed during haemodialysis. Folic acid supplementation is recommended for those who receive haemodialysis at 5mg per day.6 Testing for folate de� ciency should also be part of routine screening for renal anaemia in all patients with CKD. Any de� ciencies should be treated with folic acid.
PathophysiologyB12 and folic acid are required for DNA synthesis. B12 is also required for neurological functioning. B12 binds with intrinsic factor which is produced by gastric parietal cells and then absorbed in the terminal ileum.Table 2 (Adapted from Lecture notes on Haematology seventh edition 2004)7 shows vitamin B12 Nutritional Absorption in adults.
Table 2: Vitamin B12 absorption.
Dietary sources Only foods of animal origin
Average daily intake
Minimum daily requirement (adults)
Body stores
Requirements for absorption
Site of absorption
Time to develop de� ciency in the absence of absorption
5�g
1-3�g
3-5�g
Intrinsic factor
Terminal Ileum
Anaemia in 2-10yrs

Vitamin B12 De� ciency and Pernicious Anaemia
63
Vitamin B12 Level TestsThe level of vitamin B12 in the bloodstream may be normal or borderline even when the total amount of vitamin B12 in the body is low. Normal values of vitamin B12 are 160-925ng/L or 120-680pmol/L (local laboratories may vary). Other tests can be used to con� rm the diagnosis. Table 3 (adapted from Lecture notes on Haematology 2004)7 shows investigations that may be used to diagnose pernicious anaemia.Table 3: Investigations.
Test Results
Dietary assessment
Haemoglobin
White cell count
Platelets
Mean cell volume
Blood � lm
Serum B12
Red cell folate
Serum methylomalonic acid
Homocysteine levels
Reticulocyte count
Serum Iron Binding capacity
Barium meal and follow through
Assay of intrinsic factor in gastric juice
Bone Marrow aspirate
No intake of animal protein
Low (<6.0g/dL)
Low
Low
Raised (>110fL)
Oval macrocytes
Low
Normal or low
Raised
Raised
Low
Low
Show lesions of small intestine
Very low or absent
Megaloblasts

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
64
Signs and symptoms Table 4 shows the signs and symptoms of vitamin B12 de� ciency.Table 4: Signs and Symptoms.
Macrocytic, megaloblastic anaemia (Hb < 6.0g/dL)
Tiredness
Weakness
Dyspnoea
Neurological symptoms i.e. tingling in hands and feet
Sore tongue (glossitis)
Stomatitis
Gastrointestinal disturbances e.g. anorexia, nausea, vomiting, dyspepsia, constipation diarrhoea
Pallor
Weight loss
Hyperpigmentation of the hands
If left untreated, this disease can cause serious problems for the heart and nerves. The reasons for this are as follows:
Heart
In people with anaemia, the heart has to work harder to pump blood to get enough oxygen to the body’s organs and tissues. This stress on the heart can cause heart murmurs, fast or irregular heartbeats, an enlarged heart, or even heart failure. A lack of vitamin B12 or folic acid (folate) can cause extra problems for the heart because it raises the level in the body of a chemical called homocysteine. Raised homocysteine levels

Vitamin B12 De� ciency and Pernicious Anaemia
65
are associated with an increased incidence of myocardial infarction, peripheral and cerebral vascular disease. Homocysteine levels are higher in men than premenopausal women, old age, impaired renal function, heavy smokers and those who have excessive alcohol consumption.
Nerves
A lack of vitamin B12 can damage nerve cells and cause problems such as tingling and numbness in hands and feet and problems with walking and balance. A vitamin B12 de� ciency can cause changes in taste, smell, and vision. Severe B12 de� ciency can cause progressive neuropathy which affects the peripheral sensory nerves. It is symmetrical and affects lower limbs more commonly than the upper limbs. Symptoms include tingling in the feet, dif� culty in walking and sometimes falling over in the dark. It can also cause mental changes, including memory loss and confusion.
How can pernicious anaemia be treated?
Pernicious anaemia is usually treated with vitamin B12 injections. Hydroxocobalamin is given intramuscularly with an initial course of 1000�g x 6 injections over 2-3 weeks followed by 1000�g every 3 months for life.8
High dose vitamin B12 tablets may also be used if the patient has the ability to absorb B12. Vitamin B12 can also be given in a gel or spray for the nose.
Treatment for the underlying causes of vitamin B12 de� ciency also needs to be considered. For example antibiotics to treat stomach infections or surgery to treat intestinal problems will improve the absorption of B12. If the low vitamin B12 level is due to a poor diet, an improvement in B12 intake will improve levels.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
66
Response to treatment An improvement in symptoms should be seen within 24-48 hours. The haemoglobin should rise by 2-3g/dL each fortnight. White cell and platelet counts become normal within 7-10 days and the marrow is normoblastic in about 48 hours i.e. cells return to normal size and shape. Pernicious anaemia is fully treatable and complications are reversible.
Prophylactic therapyVitamin B12 is given to patients who have total gastrectomy or ileal resection.
How can vitamin B12 de� ciency be prevented?People who are strict vegetarians or who have a poor diet for a long time can develop this condition. Eating foods high in vitamin B12 and folic acid can help prevent low vitamin B12
levels. As the major role of B12 is to serve as a co-factor in the production of folic acid, eating foods high in folic acid can help to prevent the consequences of B12 de� ciency. Some of these foods are:
Eggs, meat, poultry, or shell� sh •
Milk, orange juice, or oranges •
Forti� ed cereals, wheat germ, rice, or barley •
Romaine lettuce, spinach, and other green leafy • vegetables
Sprouts, broccoli, asparagus •
Vitamin B12 also can be found in multivitamins and in B-complex vitamin supplements.

Vitamin B12 De� ciency and Pernicious Anaemia
67
Vitamin B12 de� ciency and Chronic Kidney DiseaseAs previously mentioned vitamin B12 is a water soluble vitamin and may be removed during dialysis when larger membranes are used (due to the size of the molecule). Dialysis induced vitamin B12 de� ciency may occur in patients who undergo high � ux dialysis when a higher percentage of middle molecules are removed. De� ciency of water soluble vitamins in haemodialysis patients has been mainly attributed to either insuf� cient intake, excessive losses in dialysate, or impaired vitamin metabolism.1 Vitamin B12 is not usually removed during peritoneal dialysis.
Patients undergoing maintenance dialysis should have vitamin B12 levels checked at least annually.
ConclusionIn conclusion, pernicious anaemia is a treatable condition. Vitamin B12 de� ciency is common in patients with CKD as vitamin B12 can be lost during dialysis. Patients with CKD should be screened annually for vitamin B12 de� ciency. Dietary assessment is also important.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
68
References
1. Descombes E, Hanck A B, Fellay G. Water soluble vitamins in chronic hemodialysis patients and need for supplementation. Kidney International 1993; 43: 1319–1328.
2. Hershko C, Ronson A, Souroujon M, Maschler I, Heyd J, Patz J. Variable haematological presentation of autoimmune gastritis: age related progression from iron de� ciency to cobalamin depletion. Blood 2006; 106 (4):1673-9.
3. Hoffbrand A V, Moss P A H, Pettit J E. Essential Haematology. 5th ed. Oxford: Blackwell Publishing Ltd, 2006.
4. Provan D, C R J Singer, T Baglin, J Lilleyman. Oxford Handbook of Clinical Haematology. 2nd ed. Oxford: Oxford University Press UK, 2004.
5. Clarke R, Grimley Evans J, Schneede J, Nexo E, Bates C, Fletcher A. Vitamin B12 and folate de� ciency in later life. Age and Ageing 2002; 33(1): 43-41.
6. Schaefer R M, Teschner M, Kosh M. Folate Metabolism in Renal Failure. Nephrology Dialysis and Transplantation 2002; 17: 24-27.
7. Hughes-Jones NC, SN Wickramasinghe, C Hatton. Lecture Notes on Haematology. 7th ed. Oxford. Blackwell Publishing, 2004.
8. British National Formulary (2009) www.bnf.org

Vitamin B12 De� ciency and Pernicious Anaemia
69


Thalassaemia
71

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
72
Learning outcomes• To gain basic knowledge about globin synthesis and
pathophysiology of thalassaemias• To know and understand the diagnostic investigations,
treatment and nursing care for this inherited blood disorder
• To realise the importance of educating patients about adherence to treatment
• To recognise why psychological support care is important to patients and families
• To be able to care adequately for patients with thalassaemia in the renal settings
IntroductionFirst described by Dr. Cooley in 1925,1 Thalassaemia forms part of a group of inherited genetic blood disorders called haemoglobinopathies. Thalassaemia is due to defective synthesis in polypeptide chains of globin, resulting in de� -cient haemoglobin production (quantitative disorder). Anae-mia is therefore often prevalent, although the severity of this will depend upon the subtype of the thalassaemia.2
This disorder was presumably restricted to people from the Mediterranean and certain Asian countries. However, its prevalence has been detected in many parts of the world, especially in the Middle East, Pakistan, India, Southeast Asia, and countries in Northern Africa.3,4 This is because thalassaemia has propagated worldwide due to migration of people and inter-marriage between different ethnic groups. Thus, it is now becoming a global health issue.4 In the UK the

Thalassaemia
73
condition is most common among people of Cypriot, Pakistani, Bangladeshi and Southeast Asian origins.5
Thalassaemia originated in people from areas where malaria was once endemic. Malaria is an infectious disease caused by the anopheles mosquito bite, through which the malaria parasite enters the human’s blood system and attacks the red blood cells (RBC). Overtime, genetic changes in the red cells of these people occurred so that the malaria parasite could not survive and multiply. These series of genetic mutations that originally evolved as a defence mechanism to allow a survival advantage also resulted in these people with mutated forms of haemoglobin (Hb) to develop thalassaemia minor or thalassaemia trait.2,3
Globin synthesis: brief overview 2,6,7
The oxygen transport capability of the RBC depends on haemoglobin (Hb). When there is de� cient Hb in the RBC, body organs are unable to function properly due to lack of oxygen. Haemoglobin synthesis occurs in the developing red cell predominately in mitrochondria.
Haemoglobin is a tetramer protein composed of 2 matching globin chains bound to the heme molecule. There are four principal globins, named after Greek alphabets: alpha (�) and beta (�), gamma () and delta (). Others include epsilon (�) and zeta (�).
In the adult, two � and two � globin chains (�2�2), each with its individual heme molecule, combine to form the dominant and functional haemoglobin (haemoglobin A: HbA).
The minor haemoglobin A2 is composed of two � and two � globin chains (�2�2).
In the foetus, two � and two � globin chains (�2�2) combine to form haemoglobin F: (Hb F). (Table 1)

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
74
Table 1: Differences between foetus and adult haemoglobins.Foetus Adult
There is a different globin chain called gamma (�). Two � chains
and two � chains combine to form haemoglobin F (HbF), which is the primary haemoglobin in the
developing foetus
After birth, the � globin chain then pairs with the � chain
Haemoglobin is a heterogeneous mixture of proteins, consisting
of 98% of the major component haemoglobin A (HbA), 2% of the minor component haemoglobin A2 (HbA2) and traces of foetal
haemoglobin (HbF)
The pairing of alpha chain and non-alpha chain is achieved by a very precisely controlled globin chain production. For instance, if the production is a haemoglobin dimer (2 chains), oxygen will not be delivered ef� ciently. It is necessary that two dimers combine to form a haemoglobin tetramer, which is the functional haemoglobin. (Table 2)
Table 2: Difference between haemoglobin dimer and haemoglobin tetramer.
1 � globin chain + 1 � globin chain = 2 chains
1 pair of haemoglobin (dimer) (Inef� cient oxygen delivery)
2 � globin chains + 2 � globin chains = 4 chains
2 pairs of haemoglobin (tetramer)
(Functional Hb)
Any alteration or mutation in the chains will cause abnormalities in the production, shape, and size of the RBC. These condi-tions will cause varying degrees of anaemia, ranging from asymptomatic to being incompatible with life.
Every individual normally receives a linked pair of � globin genes from each parent. In the case of both parents having thalassaemia trait, the chances of it being inherited is illustrated in Diagram 1. If one parent has thalassaemia trait and the

Thalassaemia
75
other parent has unaffected globin genes, the chances of it being inherited is illustrated in Diagram 2.
Diagram 1: Inheriting thalassaemia Diagram 2: Inheriting thalassaemia
Genes that regulate the synthesis and structure of different globins are arranged into 2 separate clusters. The � and globin-chain genes are encoded on chromosome 16, while the �, �, � and � globin chains are encoded on chromosome 11. In addition, multiple individual genes are expressed at each site. The � complex is called the � globin locus, and the � complex is called the � globin locus. The expression of � and � genes is closely balanced by a complex mechanism.
Normal red cell function depends upon normal red cell structure and shape and functional haemoglobin. Thalassaemia refers to a distinct set of disorders of this balance.
Pathophysiology2,6,7
There are several structural haemoglobin variants, this chapter addresses the two most common: � thalassaemia and � thalassaemia.
Alpha(�) thalassaemia (Most prevalent in Southeast Asia).� thalassaemia arises due to defective �-globin genes, two inherited from each parent leading to genotypic and phenotypic heterogenicity. (Table 3)

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
76
Table 3: Severity of � thalassemia in relation to the number of affected � globin gene.
Mutation of one � gene
Mutation of two � genes
Mutation of three � genes
Mutation of the four � genes
(severe)
The effect is slight,
without clinical symptoms. A person with
this disorder is referred to as “silent carrier”, having slightly reduced mean
corpuscular volume (MCV)
and mean corpuscular
haemoglobin (MCH)
There may be symptoms of mild anaemia
(microcytic hypochromic
anaemia), producing a condition
known as the � thalassaemia
trait.
Note: If both parents with � thalassaemia trait have a
child, there is a 25% chance that their child will inherit the severe form of � thalassaemia
This causes Haemoglobin
H disease. Symptoms
include moderate chronic
haemolytic anaemia and
splenomegaly.
Note: Blood transfusion may be necessary when acute haemolytic
crisis occurs as a response to oxidant drugs and infections
The effect will cause the severe form of
� thalassaemia, known as
� thalassaemia major.
This will result in the production of � tetramers,
with severe anaemia that
is incompatible with life. The
affected foetus typically dies in utero or shortly after birth as a
result of hydrops fetalis
Beta (�) thalassaemia (Most prevalent in the Mediterranean region, parts of Africa, Asia, India and the South Paci� c).
This involves mutations of the � globin gene, resulting in de� cient or absent production of � globin, one of the constituents of the adult haemoglobin molecule (HbA). The relative excess of � globin chains precipitates in erythroblasts (immature red blood cells) resulting in ineffective haematopoiesis. The

Thalassaemia
77
consequence of this is iron overload and haemolysis in the spleen resulting in anaemia. The anaemia is associated with a stimulation of erythropoietin production and resultant marrow hyperplasia that causes the characteristic facies in affected children.
As each individual has two � globin genes, the disorder can be moderate or severe, depending on whether one or both genes are affected. (Table 4)
Table 4: Severity of � thalassaemia in relation to the number of affected � globin gene.
Mutation of one � gene Mutation of both � genes (severe)
This produces the moderate form with mild microcytic anaemia known as � thalassaemia
intermediate (BTI)
Although the production of Hb is reduced, the amount is enough to maintain growth
and development. However, at later stages of childhood or in adulthood, growth may fail or complications may develop,
then regular transfusions may be indicated
This results in the severe form with microcytic, hypochromic anaemia
called � thalassaemia major (BTM) or Cooley’s anaemia
Blood transfusions will be needed for the rest of the life
If not treated adequately, most patients will have hepatomegaly,
splenomegaly, and a short lifespan
The clinical manifestations due to thalassaemia-induced anaemia and the associated complications are summarised in Table 5. (next page)
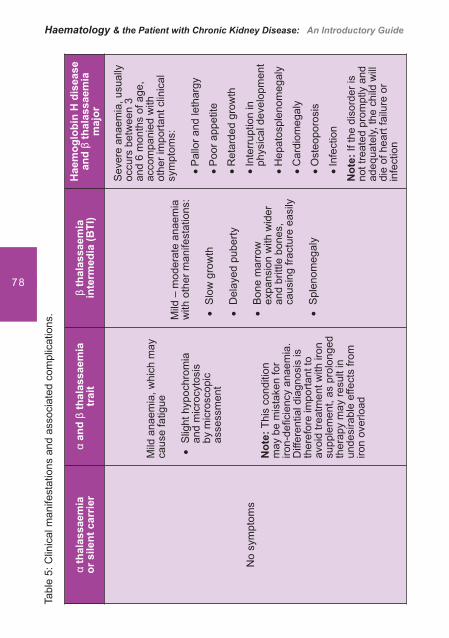
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
78
Tabl
e 5:
Clin
ical
man
ifest
atio
ns a
nd a
ssoc
iate
d co
mpl
icat
ions
.
� th
alas
saem
iaor
sile
nt c
arrie
r�
and
� th
alas
saem
ia
trai
t�
thal
assa
emia
in
term
edia
(BTI
)H
aem
oglo
bin
H d
isea
se
and
� th
alas
saem
ia
maj
or
No
sym
ptom
s
Mild
ana
emia
, whi
ch m
ay
caus
e fa
tigue
Slig
ht h
ypoc
hrom
ia
��an
d m
icro
cyto
sis
by m
icro
scop
ic
asse
ssm
ent
Not
e: T
his
cond
ition
m
ay b
e m
ista
ken
for
iron-
de� c
ienc
y an
aem
ia.
Diff
eren
tial d
iagn
osis
is
ther
efor
e im
porta
nt to
av
oid
treat
men
t with
iron
su
pple
men
t, as
pro
long
ed
ther
apy
may
resu
lt in
un
desi
rabl
e ef
fect
s fro
m
iron
over
load
Mild
– m
oder
ate
anae
mia
w
ith o
ther
man
ifest
atio
ns:
Slo
w g
row
th
��
Del
ayed
pub
erty
��
Bon
e m
arro
w
��ex
pans
ion
with
wid
er
and
britt
le b
ones
, ca
usin
g fra
ctur
e ea
sily
Spl
enom
egal
y��
Sev
ere
anae
mia
, usu
ally
oc
curs
bet
wee
n 3
and
6 m
onth
s of
age
, ac
com
pani
ed w
ith
othe
r im
porta
nt c
linic
al
sym
ptom
s:
Pal
lor a
nd le
thar
gy��
Poo
r app
etite
��
Ret
arde
d gr
owth
��
Inte
rrup
tion
in
��ph
ysic
al d
evel
opm
ent
Hep
atos
plen
omeg
aly
��
Car
diom
egal
y��
Ost
eopo
rosi
s��
Infe
ctio
n��
Not
e: If
the
diso
rder
is
not t
reat
ed p
rom
ptly
and
ad
equa
tely,
the
child
will
di
e of
hea
rt fa
ilure
or
infe
ctio
n

Thalassaemia
79
Diagnostic Investigations Thalassemias should be identi� ed through systematic antenatal screening or diagnosed in early childhood. All at risk pregnant women should be routinely screened for thalassaemia trait. If positive, their partner should also be offered the test, as there is a 25% chance that the offspring will inherit the disease if both parents have the thalassaemia trait.
If thalassaemia is con� rmed, information and counselling is provided to help parents decide whether they wish to continue with the pregnancy or choose to terminate the pregnancy. Babies born affected due to missed identi� cation of risk during pregnancy may be identi� ed through a newborn screening programme.7
In homozygous � thalassaemia and haemoglobin H disease, the clinical and haematological � ndings usually are characte-ristic, resulting in a less complex diagnosis process. In a suspected case, the diagnosis should be established as soon as possible by adopting the recommended diagnostic investigations as shown in Table 6.2,7,8
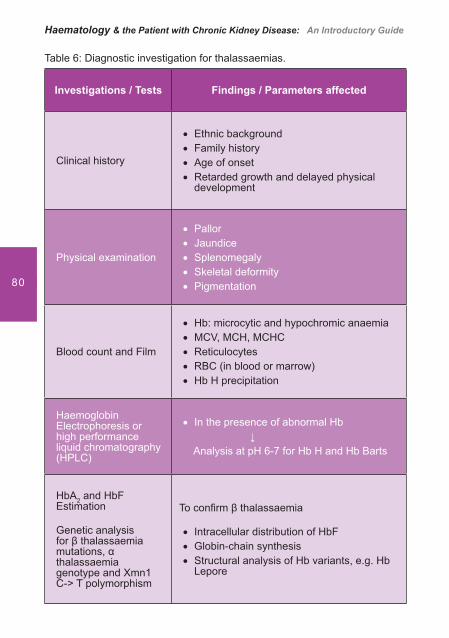
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
80
Table 6: Diagnostic investigation for thalassaemias.
Investigations / Tests Findings / Parameters affected
Clinical history
Ethnic background��Family history��Age of onset��Retarded growth and delayed physical ��development
Physical examination
Pallor��Jaundice ��Splenomegaly��Skeletal deformity��Pigmentation ��
Blood count and Film
Hb: microcytic and hypochromic anaemia ��MCV, MCH, MCHC ��Reticulocytes ��RBC (in blood or marrow) ��Hb H precipitation ��
Haemoglobin Electrophoresis or high performance liquid chromatography (HPLC)
In the presence of abnormal Hb�� Analysis at pH 6-7 for Hb H and Hb Barts
HbA2 and HbF Estimation
Genetic analysis for � thalassaemia mutations, � thalassaemia genotype and Xmn1 C-> T polymorphism
To con� rm � thalassaemia
Intracellular distribution of HbF��Globin-chain synthesis��Structural analysis of Hb variants, e.g. Hb ��Lepore
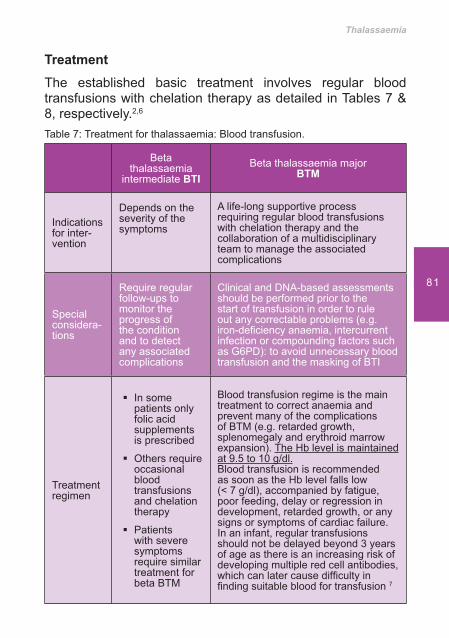
Thalassaemia
81
Treatment The established basic treatment involves regular blood transfusions with chelation therapy as detailed in Tables 7 & 8, respectively.2,6
Table 7: Treatment for thalassaemia: Blood transfusion.
Beta thalassaemia
intermediate BTIBeta thalassaemia major
BTM
Indications for inter-vention
Depends on the severity of the symptoms
A life-long supportive process requiring regular blood transfusions with chelation therapy and the collaboration of a multidisciplinary team to manage the associated complications
Special considera-tions
Require regular follow-ups to monitor the progress of the condition and to detect any associated complications
Clinical and DNA-based assessments should be performed prior to the start of transfusion in order to rule out any correctable problems (e.g. iron-de� ciency anaemia, intercurrent infection or compounding factors such as G6PD): to avoid unnecessary blood transfusion and the masking of BTI
Treatment regimen
In some ��patients only folic acid supplements is prescribed
Others require ��occasional blood transfusions and chelation therapy
Patients ��with severe symptoms require similar treatment for beta BTM
Blood transfusion regime is the main treatment to correct anaemia and prevent many of the complications of BTM (e.g. retarded growth, splenomegaly and erythroid marrow expansion). The Hb level is maintained at 9.5 to 10 g/dl.Blood transfusion is recommended as soon as the Hb level falls low (< 7 g/dl), accompanied by fatigue, poor feeding, delay or regression in development, retarded growth, or any signs or symptoms of cardiac failure. In an infant, regular transfusions should not be delayed beyond 3 years of age as there is an increasing risk of developing multiple red cell antibodies, which can later cause dif� culty in � nding suitable blood for transfusion 7
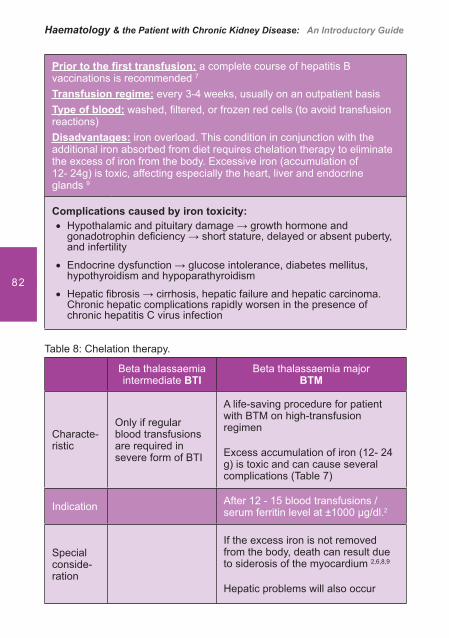
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
82
Prior to the � rst transfusion: a complete course of hepatitis B vaccinations is recommended 7
Transfusion regime: every 3-4 weeks, usually on an outpatient basisType of blood: washed, � ltered, or frozen red cells (to avoid transfusion reactions)Disadvantages: iron overload. This condition in conjunction with the additional iron absorbed from diet requires chelation therapy to eliminate the excess of iron from the body. Excessive iron (accumulation of 12- 24g) is toxic, affecting especially the heart, liver and endocrine glands 9
Complications caused by iron toxicity:Hypothalamic and pituitary damage � growth hormone and ��gonadotrophin de� ciency � short stature, delayed or absent puberty, and infertility
Endocrine dysfunction � glucose intolerance, diabetes mellitus, ��hypothyroidism and hypoparathyroidism
Hepatic � brosis � cirrhosis, hepatic failure and hepatic carcinoma. ��Chronic hepatic complications rapidly worsen in the presence of chronic hepatitis C virus infection
Table 8: Chelation therapy.
Beta thalassaemia intermediate BTI
Beta thalassaemia majorBTM
Characte-ristic
Only if regular blood transfusions are required in severe form of BTI
A life-saving procedure for patient with BTM on high-transfusion regimen
Excess accumulation of iron (12- 24 g) is toxic and can cause several complications (Table 7)
Indication After 12 - 15 blood transfusions / serum ferritin level at ±1000 �g/dl.2
Special conside-ration
If the excess iron is not removed from the body, death can result due to siderosis of the myocardium 2,6,8,9
Hepatic problems will also occur

Thalassaemia
83
Regular and systematic assessment of iron stores.7
• Serum ferritin levels: serially at least every three months. (Note: increased levels occur in intercurrent acute infections, chronic in� ammatory conditions and chronic viral hepatitis)
• Liver R2 or T2 MRI: annually, and additionally with signi� cant therapy changes
• Cardiac T2 MRI: 3 monthly if T2 <10 ms and any sign of cardiac failure, half yearly if T2 <10 ms, annually if T2 = 10-20ms, every 2 years if T2 >20 ms
Magnetic resonance imaging (MRI) assessment should initiate from 8 years of age
Goal of therapy: to attain and maintain annual average serum ferritin at 1000±500�g/dl, liver iron 3-7mg/g dry weight, and cardiac T2 >20 ms.
Chelating drugs bind the iron molecules in the body and then release them through urine or stools. In the UK, there are three chelating drugs currently licensed to treat iron overload in thalassaemia:
Desferrioxamine (DFO, Desferal• ®)Deferiprone (DFP, Ferriprox• ®)Deferasirox (DFX, Exjade• ®)
Table 9 summarises the current indications for use and side effects of the above chelating drugs.2,7
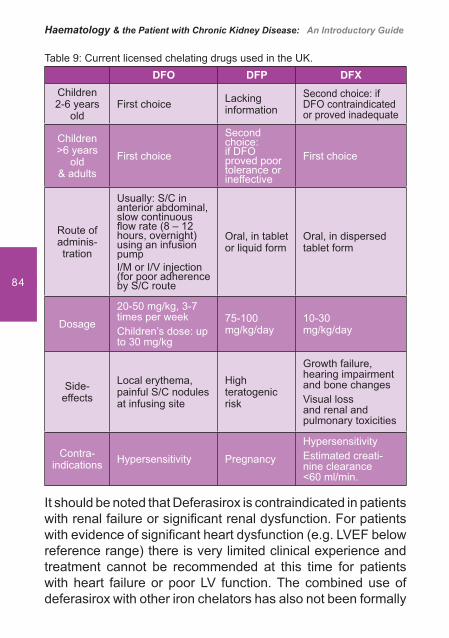
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
84
Table 9: Current licensed chelating drugs used in the UK.DFO DFP DFX
Children2-6 years
oldFirst choice Lacking
informationSecond choice: if DFO contraindicated or proved ina dequate
Children>6 years
old & adults
First choice
Second choice: if DFO proved poor tolerance or ineffective
First choice
Route of adminis-tration
Usually: S/C in anterior abdominal, slow continuous � ow rate (8 – 12 hours, overnight) using an infusion pumpI/M or I/V injection (for poor adherence by S/C route
Oral, in tablet or liquid form
Oral, in dispersed tablet form
Dosage
20-50 mg/kg, 3-7 times per weekChildren’s dose: up to 30 mg/kg
75-100 mg/kg/day
10-30 mg/kg/day
Side-effects
Local erythema, painful S/C nodules at infusing site
High teratogenic risk
Growth failure, hearing impairment and bone changesVisual loss and renal and pulmonary toxicities
Contra-indications Hypersen sitivity Pregnancy
HypersensitivityEstimated creati-nine clearance <60 ml/min.
It should be noted that Deferasirox is contraindicated in patients with renal failure or signi� cant renal dysfunction. For patients with evidence of signi� cant heart dysfunction (e.g. LVEF below reference range) there is very limited clinical experience and treatment cannot be recommended at this time for patients with heart failure or poor LV function. The combined use of deferasirox with other iron chelators has also not been formally

Thalassaemia
85
assessed and therefore cannot be recommended at this time. The drug should not be used in pregnant women.6
The established alternative treatment option to blood trans-fusion and chelation therapy is bone marrow transplantation (BMT), which can offer a cure as discussed in Table 10.7
Table 10: Alternative treatment option for BTM: Bone marrow transplantation.Beta thalassaemia major (BTM)
Characteristic
A procedure which consists of transplanting bone marrow donated by a person without thalassaemia, preferably by another sibling with the same genetic type of human lymphocyte antigen (HLA)
Special consideration Less risk if transplantation is done between 18 months and 3 years of age.10
The risks involve:Graft-versus-host disease (GVHD), a condition in which donor derived - T cells start attacking several parts of the host body. The most commonly affected are the eyes (irritation), skin (rash), gastrointestinal system (nausea, weight loss and jaundice)Increased chance of strokes, seizures and tumours-
The survival and success rates for a bone marrow transplant depend on a series of risk factors.2
Previous poor control of serum ferritin levels- Hepatomegaly- Cirrhosis -
Note: Umbilical cord cells from unaffected younger sibling can serve as a source of stem cells for transplantation.11
Managing Treatment OptionsBlood Transfusion Care12
Patients normally receive 3 units of blood every 3-4 weeks. This may cause huge lifestyle disruptions, with important implications for school or employment. The nurse’s role is to educate patients, parents, teachers as well as employers regarding this special condition, the patients’ needs and the importance of regular treatment. The nurse should also ensure the safe administration of blood and be aware of the related

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
86
transfusion reactions. As venous access can be problematic with prolonged venepuncture cannulation veins can become damaged. The insertion of the needles should therefore be avoided near important blood vessels or nerves, to minimise the risk of damage and/or bleeding. Many patients can require long term access devices that may negatively affect body image and act as constant reminder of the condition. In this instance, the nurse must remain sensitive towards these feelings.
Chelation Therapy Care6 Administering iron chelation subcutaneously (S/C)Iron chelation may impact on patient lifestyle as well as body image. When administered subcutaneously, localised reactions are likely to occur at any site over time and therefore it is important to use different parts of the body and rotate injection sites (see Diagram 3). The abdomen is often the best site, but some patients prefer to use their upper arm or thigh. Over time, the long-term use of DFO may cause lumps to form around the injection site. If pain does occur, it may be reduced by applying topical anaesthetic creams such as Emla 30-60 minutes before starting DFO treatment. Swelling may be reduced by applying a warm compress on the affected area after DFO has been administered. For localised erythema, pain or itching the patient can be prescribed topical heparin cream.Diagram 3: Rotation of Chelation infusion sites.6
Rotation of infusion sites

Thalassaemia
87
Administering Intravenous (IV) Iron chelationContinuous 24-hour intravenous infusionThis method can be life saving in patients suffering severe iron loading and associated cardiac complications as it removes large quantities of iron more quickly than other methods. However, continuous IV infusion also carries signi� cant risks, particularly of infection or thrombosis caused by the in-dwelling line that provides access to the vein. This method should therefore only be used in exceptional cases, where patients exhibit:
Severe iron overload - i.e. ferritin values persistently • > 2500Ìg/l and/or liver iron concentration of > 15Ìg/g/dry weight of liver, established by a liver biopsy
Heart complications resulting from iron overload•
Female patients planning a pregnancy who have high • serum ferritin levels and/or high liver iron concentration (LIC)
Patient’s requiring intensive removal of iron, irrespective • of ferritin or LIC levels, for example before a bone marrow transplant or in patients with chronic active hepatitis C
Intravenous 8-12 hour infusionIntravenous 8-12 hour infusion of DFO, as opposed to continuous 24-hour IV infusion, is another alternative to SC administration, and may be used in cases of serious, localised, problems with the SC method. The dose (40-50mg/kg/day), duration (8-12 hours) and frequency (more than 5 days a week) of infusion are generally the same as in SC infusion.
This method is not as effective as the 24-hour continuous intravenous infusion in cases of severe iron loading and associated cardiac complications. Importantly, the IV use of DFO should be introduced with caution and only where necessary as over the longer-term, the method may damage veins that are essential for blood transfusion.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
88
Adherence to treatment 7 The main problem with iron chelation therapy has been adherence to the regular SC infusions of desferrioxamine. The infusions are time consuming to set up, and require introduction of a subcutaneous needle on each occasion followed by continued attachment to an infuser device over 10-12 hours. Survival has not improved to the extent hoped 30 years ago when desferrioxamine became available and this is probably due to the struggles thalassaemic patients experience in managing regular self-administered infusions.
However, at least in part, due to more tolerable and effective regimens using oral iron chelators, there is growing evidence that life expectancy across whole populations has improved over the last 7 - 8 years. In a large, well organised thalassaemia Centre, physical and psychological problems with adherence can be addressed methodically, and excellent survival can be expected in younger patients.
It is not just the technical care that affects the patient’s life experience, but also the ways in which that care is delivered. The development of patient-centred services is paramount and clinics should take particular care to minimise disruption to education, employment and family life, allowing them to live as fully and normally as possible. Such care can materially improve survival by enhancing adherence to dif� cult treatment regimens.
Patients and carers should be supported in adhering to medical regime and self-management regarding chelation therapy using a multidisciplinary team approach that also includes clinical psychologists and play therapists. Adherence should be monitored regularly, and problems carefully identi� ed and addressed. Patients should be encouraged to use a treatment diary to assist with monitoring.
Information should be given to parents/patients on the options for treatment including the relative bene� ts or disadvantages

Thalassaemia
89
of each option. This should include information on potential consequences of non-adherence to agreed medical treatment. Decisions about treatment regimes (e.g. frequency/days off) should include the patients and all changes in treatment should be discussed with them, and the reasons for changes made clear. Parents/patients should be involved in monitoring their progress (e.g. ferritin levels) and understanding may be enhanced if results are entered into a hand-held record. They should have the opportunity to discuss dif� culties with adherence in con� dence with a psychologist.
Bone Marrow Transplant (BMT) 7
The aim of care in BMT is to ensure that children and families can make properly informed choices about BMT as a treatment option. The care would include:
For all families who have a child with a serious • thalassaemia syndrome to be offered the opportunity to discuss bone marrow transplant as a treatment option at an early stage, usually around 12 - 18 months of age
Discussions to be with a transplant team with speci� c • experience in transplanting for thalassaemia
Counselling for transplanted individuals, when trying • to conceive and partner testing for haemoglobin disorder
Nursing Management Strategy Nurse’s multidimensional roleNursing patients with thalassaemia requires a wide range of skills and knowledge. The role of the nurse is pivotal as the nurse/patient relationship differs to that of the doctor/patient. Due to the often lengthy and regular periods of time spent with the patient, the nurse may be the � rst one to pick up on speci� c problems that a patient may be experiencing.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
90
Nurses should promote a situation where patients can enjoy a near-normal lifestyle and experience regular physical and emotional development from childhood to adulthood, including parenthood, by trying to reduce the degree to which the disease interferes with the patient’s personal and social life. From a practical point of view, nurses and other thalassaemia professionals should:
Manage treatment and monitoring schedules so as • to minimise any unnecessary impact on normal daily activityBe aware of the particular psychological aspects of • health care for this chronic condition
Psychological support care to patient and family6
Thalassaemia has important psychological implications. The way in which the family and the patient come to terms with the disease and its treatment will have a critical effect on the patient’s survival and quality of life. Without an understanding and acceptance of the disease the dif� culties of lifelong transfusion and chelation therapy will not be faced, leading to an increased risk of disease complications and poorer survival. A key role for health care professionals is to help patients and families to face up to the dif� cult demands of treatment, while maintaining a positive role. Adherence to treatment is a basic goal, but a general acceptance by the patient of his/her own condition constitutes the key to normal development from childhood to adulthood. The success of the management of thalassaemia is based, to a greater extent on the establishment of a therapeutic alliance between caring staff and the patient throughout the course of the disease.
The psychology of inherited chronic diseaseEvery genetic disease, regardless of its aetiology, implies a sense of guilt that may interfere with the primary parent-infant

Thalassaemia
91
relationship. Moreover, the treatment is emotionally deman-ding, as therapies require repeated invasive procedures and hospital visits. Chronicity is a powerful source of emotional problems that intensify at each signi� cant developmental stage of the patient’s life. Patients can feel that they are different, limited or isolated. Their state of mind can shift rapidly from depression to anger and vice versa. Health workers must be prepared to accept this shift and to help patients deal with these feelings, � nding a way to their own ‘normalisation’ that implies different individual styles in adult life.
Psychology care goals
In terms of the psychological care of the patient, the aims of healthcare professionals (HCP) are given in Table 11.
Table 11: Aims of psychological care.Provide information that promotes understanding of the illness��Help patient and parents to talk and to express feelings about the ��illnessHelp the patient to accept the illness and to take care of him/herself��Maintain realistic hopes��Facilitate a ‘normal’ lifestyle and encourage self-esteem��Support the full development of an adult life��
Putting these goals into practice requires certain attributes of health professionals. (Table 12)
Table 12: Attributes of HCP.
Open-minded about psychological aspects of having and treating ��inherited disease
Trained in normal psychosocial development from childhood to ��adulthood
Sensitised to the special issues of this chronic hereditary disease��
Available to accompany and support the patient throughout his/her ��life path

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
92
Appropriate psychological support therefore not only requires motivated and able clinicians, but also presupposes an organisational structure that allows for the successful delivery of optimal and comprehensive care.
Communication: healthcare professionals with patientsThe establishment of good communication between health professionals, patients and their families is obligatory if care is to be delivered within all its dimensions. Table 13 lists the skills required for effective communication.Table 13: Effective Communication.
Listen�� – to be interested in the patient’s emotional and real experiences
Accept�� – to respect the patient’s point of view and be sensitive to the timing of personal communication
Share�� – to be consistently close to the patient’s positive and negative feelings
Understand �� – at an emotional and not simply an intellectual level
Maintain boundaries�� – to give help and relief, but keeping in mind his/her role as a health professional
Patient Education
Nurses should educate patients to self-administer their drugs. The avoidance of unhealthy life styles, including smoking, lack of physical exercise and excess alcohol consumption should be promoted. Cardiac function should be monitored as an impaired myocardial function may require speci� c cardiac treatment, but also calls attention to the immediate need for much stricter adherence to chelation protocol or the initiation of a more intensive chelation programme, in order to prevent an inexorable progression to severe cardiac failure.
In general, a transparent discussion of the following issues may help the patient to gain insight into the associated risks.

Thalassaemia
93
Dietary intakePatients with thalassaemia do not have speci� c dietary requirements. During growth, a normal energy intake with normal fat and sugar content is recommended. During adolescence and adult life, a diet low in highly re� ned carbohydrates (sugar, soft drinks, and snacks) may be useful in preventing or delaying the onset of impaired glucose tolerance or diabetes.
IronIncreased iron absorption from the intestinal tract is characteristic of thalassaemia. The amount depends on the degree of erythropoiesis, the haemoglobin level and other potential independent factors. Drinking a glass of black tea with meals reduces iron absorption from food, particularly in thalassaemia intermedia. However, there is no evidence that iron-poor diets are useful in thalassaemia major; only foods very rich in iron should be avoided.
Calcium Many factors in thalassaemia promote calcium depletion. A diet containing adequate calcium is always recommended.
Vitamin EVitamin E requirement is high in thalassaemia. A regular intake of vegetable oils as part of a balanced diet may be recommended.
Zinc de� ciencyZinc de� ciency may occur during chelation, depending on the chelator, dose and duration. Zinc supplementation requires close monitoring.
AlcoholPatients with thalassaemia should be discouraged from consuming alcohol, as it can facilitate the oxidative damage of

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
94
iron and aggravates the effect of HBV (Hepatitis B virus) and HCV (Hepatitis C virus) on liver tissue.
SmokingCigarette smoking may directly affect bone remodelling which is associated with osteoporosis and is related to adverse effects on the general health.
Drug abuseIn many countries, drug abuse is common among adolescents and young adults. For an individual with a chronic disease, drug abuse can be a serious threat to an already challenging condition, upsetting the delicate balance of factors affecting physical and mental health. For young people with thalassaemia, feelings of dependence, difference and anxiety can push patients to seek ‘normality’ through an abuse habit.
Recreational activitiesPhysical activity in general must always be encouraged in patients with a chronic disease. Patients with thalassaemia should have a quality of life and range of life experiences as much like those of others as possible. There is no reason to prevent patients from engaging in physical activity to the limits of what they are capable and interested in doing.
Relation with Kidney DiseaseThalassaemia is a systemic disease in which the renal involvement has not yet been thoroughly studied. Renal tubular function abnor malities are described in beta thalassaemia major13,14,15 beta thalassaemia minor,16,17,18 alphathalassaemia19 as well as in beta thalassemia/Hb E disease.20 Renal failure is a terminal event in thalassaemia major and is usually secondary to heart failure and/or hepatic failure. Acute renal failure following desferrioxamine over dose or haemolysis has been reported.21

Thalassaemia
95
Renal Tubular AbnormalitiesPeople with beta thalassaemia minor are usually asymptomatic.
A variety of renal tubular abnormalities have occasionally been described in patients with beta thalassaemia minor as listed in Table 14.16
Table 14 16
Renal Tubular Abnormalities
Hypercal ciuria
Hypomagnesaemia with renal magnesium wasting
Decreased tubular absorption of phos phorus
Hypouricemia with renal uric acid was ting
Renal glycosuria and tubular proteinuria
Nephrocalcinosis
Often detected as an incidental � nding, Nephrocalcinosis is de� ned as a generalized increase in the calcium content of the kidney. De pending on the location of calci� cation, neph-rocalcinosis can be classi� ed as cortical and medullary. Cortical nephrocalcinosis is rare. Medullary nephrocalcinosis is the typical pat tern seen in 98% of human nephrocalcinosis. The more common causes are hyperparathyroidism, distal tubular acidosis, idiopathic hypercalciuria and hyperoxaluria.20
HaemolysisIt has been postulated that low-grade haemolysis, shortened red cell life span, tubular iron deposition, oxidative lipid peroxidation and toxins derived from erythrocytes might cause renal tubular damage in adult patients with beta-thalassaemia minor.13,16 In addition, increased iron turn over from low grade haemolysis of microcytic erythrocytes evident with increased LDH (de� ne LDH) levels may be another factor.13

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
96
Kidney StonesThalassaemic patients are susceptible to kidney stones, as a result of ineffective erythropoiesis and peripheral haemolysis, which can lead to hydronephrosis and kidney failure. The cause is associated with hypertrophic stones that block the renal tubules and even the calyces. The kidneys are frequently enlarged in thalassaemia, due to the presence of extramedullary haematopoiesis. Less well understood is the tendency for the renal tubules to be dilated. Due to increased concentrations of bile pigments the urine is frequently dark. Large amounts of urate, uric acid and oxalate are also observed.6
Epidemiology of thalassaemia & Chronic Kidney Disease (CKD)
Potentially, there mavbe more thalassaemia patients with CKD in the future. Thalassaemia does not appear to directly affect the kidneys, unlike sickle cell anaemia where kidney damage is common, so renal disease has not been a major problem in thalassaemia, but this will probably change in the future, for several reasons:
Chronic kidney disease is common. In the UK for • example, improved screening by General Practitioners has identi� ed CKD in approximately 10% of the UK population. Any increase in the CKD population could potentially see an increase in those with thalassaemia as a co-morbidityDiabetes and hypertension are becoming more • common as the overall body weight of individuals is increasing. In particular, diabetes represents one of the commonest causes of renal failure and the need for dialysis. People from South East Asia are more prone to both diabetes and thalassaemia and this could impact on the numbers seen with both conditionsThalassaemia patients are living longer and CKD is • primarily a disease of the elderly

Thalassaemia
97
Finally and extremely important, with the explosion in medical therapies, nephrotoxicity has to be taken into consideration.22
ConclusionIn conclusion, thalassaemia is an inherited genetic blood disorder due to defective globin synthesis. Thalassaemia should be identi� ed through systematic antenatal screening or diagnosed in early childhood. Anaemia can be prevalent but the severity will depend upon the subtype of thala-ssaemia. The basic treatment depends on the severity of the disorder and involves regular blood transfusions with chelation therapy to correct iron toxicity. The alternative treatment is bone marrow transplantation.
The nurse’s role is to educate patients, parents, teachers as well as employers regarding thalassaemia, the patients’ needs, and the importance of regular treatment. Psychological support care is important to the patient and the family. The nurse must also ensure the safe administration of blood and promote adherence to chelation treatment.
Renal tubular function abnor malities are described in beta thalassaemia major, beta thalassaemia minor, alpha thala-ssaemia as well as in beta- thalassemia/Hb E disease. Acute renal failure following desferrioxamine over dose or haemolysis has also been reported. In the future there could potentially be more thalassaemia patients with CKD, it is therefore important for nurses in nephrology to understand the complexities of this condition if effective evidence based care is to be provided.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
98
References1. Cooley TB, Lee P. Series of cases of splenomegaly in children with
anemia and peculiar bone change. Trans Am Pediatr Soc 1925; 37:29.
2. Weatherall DJ. Disorders of globin synthesis: the thalassemias. In: Williams Hematology. 7th ed. New York, NY: McGraw-Hill Medical Publishing Division; 2006:633-666.
3. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: An increasing global health problem. Bull World Health Organ. Vol 79 (8) Genebra, 2001.
4. Management of Haemoglobin Disorders. Report of Joint WHO-TIF Meeting, Cyprus. November 2007. (Available from http://www.who.int/genomics/WHO-TIF_genetics_� nal.pdf)
5. Modell B et al. A national register for surveillance of inherited disorders: beta thalassaemia in the United Kingdom. Bulletin of the WHO, 2001; 79(11): 1006-1013.(Available from http://www.who.int/bulletin/archives/79(11) 1006.pdf)
6. Guidelines for the Clinical Management of Thalassaemia. Thalassaemia International Federation. 2nd Revised Edition-November 2008, Cyprus Available at http://www.thalassaemia.org.cy/pdf/Guidelines_2nd_revised_edition_EN.pdf)
7. Standards for the Clinical Care of Children and Adults with Thalassaemia in the UK - 2nd edition 2008 (Available from http://www.ukts.org/pdfs/awareness/ukts-standards-2008.pdf)
8. Kiss TL, Ali MA, Levine M, Lafferty JD. An algorithm to aid in the investigation of thalassemia trait in multicultural populations. Arch Pathol Lab Med. 2000 Sep; 124(9): 1320-3.
9. Gabutti V, Borgna-Pignatti C. Clinical manifestations and therapy of transfusional haemosiderosis. Bailieres Clin Haematol. 1994 Dec; 7(4): 919-40.
10. Lawson SE et al. Bone marrow transplantation for beta thalassaemia major: the UK experience in two paediatric centres. Br J Haematol 2003 Jan; 120(2): 289-95.
11. Locatelli F, et al. Related umbilical cord blood transplantation in patients with thalassaemia and sickle cell disease. Blood 2003 Mar 15; 101(6): 2137-43. Epub 2002 Nov 7.
12. Nursing Adults: The Practice of Caring by Chris Brooker and Maggie Nicol. Mosby, 2003
13. Koliakos G, Papachristou F, Koussi A, et al. Urine biochemical markers of early renal dysfunction are associated with iron overload in beta thalassaemia. Clin Lab Haematol 2003;25(2):105-9.

Thalassaemia
99
14. Michelakakis H, Dimitriou E, Georgakis H, et al. Iron overload and urinary lysosomal enzyme levels in beta thalassaemia major. Eur J Pediatr 1997;156(8):602-4.
15. Aldudak B, Karabay Bayazit A, Noyan A, et al. Renal function in pediatric patients with beta thalassaemia major. Pediatr Nephrol 2000; 15(1-2):109-12.
16. Cetin T, Oktenli C, Ozgurtas T, et al. Renal tubular dysfunction in beta thalassemia minor. Am J Kidney Dis 2003 2; 42(6): 1164-8.
17. Oktenli C, Bulucu F. Renal tubular dysfunction in a patient with beta thalassemia minor. Nephron 2002; 92(1): 222-3.
18. Kalman S, Atay AA, Sakallioglu O, et al. Renal tubular function in children with beta thalassemia minor. Nephrology (Carlton) 2005; 10(5): 427-9.
19. Sumboonnanonda A, Malasit P, Tanphaichitr VS, Ongajyooth S, Petrarat S, Vongjirad A. Renal tubular dysfunction in alpha thalasse-mia. Pediatr Nephrol 2003; 18(3): 257-60.
20. Prabahar MR, Jain M, Chandrasekaran V, Indhumathi E, Soundararajan P. Renal tubular dysfunction with nephrocalcinosis in a patient with beta thalassemia minor. Saudi J Kidney Dis Transpl 2008; 19:964-8.Available from: http://www.sjkdt.org/text.asp?2008/19/6/964/43473
21. Ali D, Mehran K, Moghaddam AG. Comparative Evaluation of Renal Findings in Beta Thalassaemia Major and Intermedia. Saudi J Kidney Dis Transpl [serial online] 2008 [cited 2008 Oct 22]; 19:206-9.
Available from: http://www.sjkdt.org/text.asp?2008/19/2/206/3903122. Monitoring Kidney Function In Relation To Thalassaemia - A
Nephrologist’s View. By Dr Sunil Bhandari Consultant Physician Nephrologist Hull Royal In� rmary, Honorary Senior Lecturer University of Hull, Director of Clinical Studies Hull York Medical School. Available at: http://www.ukts.org/pdfs/complications/KidneyFunction.pdf


Sickle Cell
Disease
101

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
102
Learning outcomes• To gain knowledge and understanding of Sickle cell
disease• To understand some of the complications associated
with Sickle cell disease and it’s multi-disciplinary approach
• To address the treatment options that are offered to patients with this disease
Introduction
Sickle cell disease (SCD), or Sickle cell anaemia, (SCA) is an inherited blood disorder affecting the haemoglobin of the red blood cell. Haemoglobins are tetrameric molecules with 2 �-like and 2 �-like globin polypeptide chains. In sickle cell disease both � globin gene allele carries a mutation.1 This allows valine to be substituted for glutamic acid as the sixth amino acid of the beta globin chain and produces a haemoglobin tetramer that is poorly soluble when deoxygenated –haemoglobin S (HbS).2 In times of low oxygen tension, the abnormal haemoglobin S (HbS) forms strands within the red blood cell causing the cells to be sickle-shaped, stiff and sticky. They therefore do not move easily through the blood vessel and occlude the blood � ow as shown in Figure 1.3
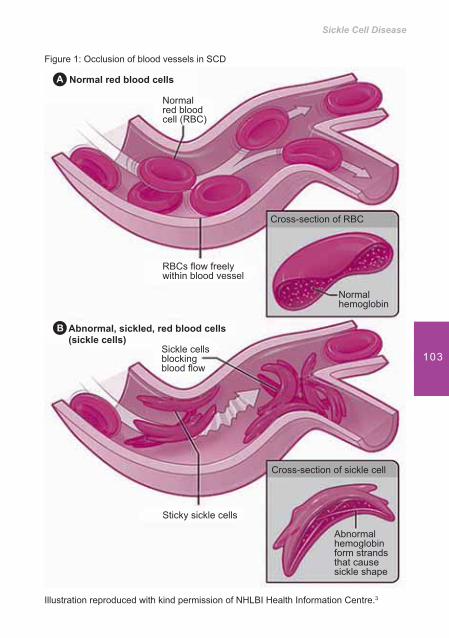
Sickle Cell Disease
103
Figure 1: Occlusion of blood vessels in SCD
Normal red blood cells
Normal red blood cell (RBC)
RBCs � ow freely within blood vessel
Cross-section of RBC
Normal hemoglobin
Cross-section of sickle cell
Abnormal hemoglobin form strands that cause sickle shape
Sticky sickle cells
Sickle cells blocking blood � ow
Abnormal, sickled, red blood cells (sickle cells)
Illustration reproduced with kind permission of NHLBI Health Information Centre.3

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
104
It is cycles of oxygenation and deoxygenation of the red blood cells (RBC) that results in the repeated sickling and unsickling of these cells. As a result, damage to the RBC not only causes an increased rate of red-cell turnover but results in the continuous formation of sub-populations of very high density irreversibly sickled RBC’s that have a reduced survival time.1 Vaso-occlusive phenomena and hemolysis are therefore the clinical hallmarks of Sickle cell disease.
EpidemiologySickle cell disease displays a marked ethnic variation, with those from Africa, Asia, Middle East and Mediterranean populations showing the highest prevalence. The highest incidence of Sickle cell disease is in those of African descent with around 0.8% of newborns carrying the homozygous form and approximately 8% carrying the mutated gene.4
It is thought that the heterozygous state confers a survival advantage from malaria. This is because those with one of the two alleles of the Sickle cell disease are resistant to malaria, as the malaria plasmodium is blocked by the ‘sickling’ of the cells.
Sickle cell disease affects nearly 30 million people worldwide.4 Early diagnosis is crucial and most people with prompt treatment live into their mid-forties.5 The disease has no cure, but treatments are available for symptoms and complications. Bone marrow transplants may offer a cure in a small number of cases.
CharacteristicsThe clinical manifestations of Sickle cell disease vary depen-ding on the genotype. The homozygote form, also known as Sickle cell anaemia is HbSS. The disorder is most severe in these patients as haemoglobin S makes up 75-95% of the

Sickle Cell Disease
105
haemoglobin present. These patients are typically anaemic and will often have recurrent painful vaso-occlusive episodes.6 The clinical signs and symptoms will typically develop at an early age.
Other rarer forms are: Sickle-haemoglobin C disease (HbSC) a double heterozygote for HbS and HbC, characterized by moderate clinical severity; Sickle beta plus thalassaemia (HbS/B+) that has mild to moderate severity and is variable amongst different ethnicities and Sickle-beta zero Thalassaemia (HbS/B°), which is clinically indistinguishable from Sickle cell anaemia, containing a double heterozygote for HbS and B° Thalassamia.4
Sickle Cell Trait
Sickle cell trait (AS), the carrier state, is a heterozygote form, whereby the HbS is present in 35-45% of the haemoglobin. Usually there is an absence of anaemia as the normal allele of haemoglobin A is able to produce over 50% of normal haemoglobin. People with sickle cell trait are generally asymp-tomatic.
Inheritance
Sickle cell disease is inherited from parents. If one parent has sickle cell anaemia (SCA) and the other is normal all the children will have sickle cell trait.7
If one parent has SCA (SS or HbSS) and the other has sickle cell trait (AS) there is a 50% chance of having a child with Sickle cell disease (SS) and a 50% chance of having a child with sickle-cell trait (AS).
If both parents have sickle-cell trait (AS), there is a 25% chance of having a child with Sickle cell disease (SS). This is known as homozygous recessive or “co-dominant”.8
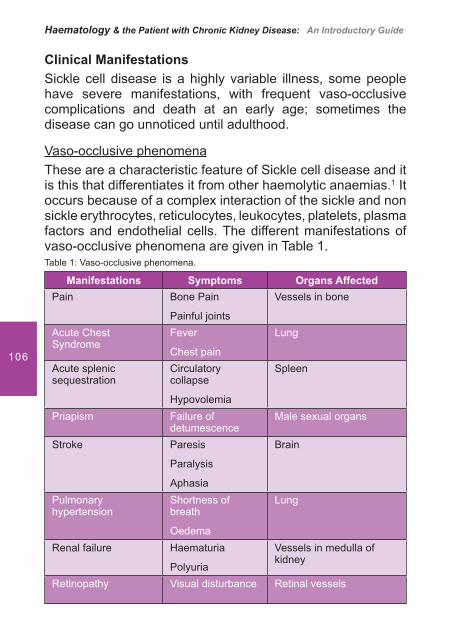
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
106
Clinical ManifestationsSickle cell disease is a highly variable illness, some people have severe manifestations, with frequent vaso-occlusive complications and death at an early age; sometimes the disease can go unnoticed until adulthood.
Vaso-occlusive phenomena These are a characteristic feature of Sickle cell disease and it is this that differentiates it from other haemolytic anaemias.1 It occurs because of a complex interaction of the sickle and non sickle erythrocytes, reticulocytes, leukocytes, platelets, plasma factors and endothelial cells. The different manifestations of vaso-occlusive phenomena are given in Table 1.Table 1: Vaso-occlusive phenomena.
Manifestations Symptoms Organs AffectedPain Bone Pain
Painful joints
Vessels in bone
Acute Chest Syndrome
Fever
Chest pain
Lung
Acute splenic sequestration
Circulatory collapse
Hypovolemia
Spleen
Priapism Failure of detumescence
Male sexual organs
Stroke Paresis
Paralysis
Aphasia
Brain
Pulmonary hypertension
Shortness of breath
Oedema
Lung
Renal failure Haematuria
Polyuria
Vessels in medulla of kidney
Retinopathy Visual disturbance Retinal vessels

Sickle Cell Disease
107
Acute painful crises can be associated with speci� c triggers such as cold, dehydration, intercurrent infection or emotional stress. The most common crises affect the abdomen, bones, joints and soft tissue. Bilateral painful and swollen hands, even feet (dactylitis) can be present. Priapism can also affect male patients.
Over time, chronic complications can occur. These include leg ulcers. chronic renal disease, hyposplenism and consequently severe and recurrent infection.
The most frequent cause of morbidity in Sickle cell disease is stroke and mortality associated with acute chest syndrome and splenic sequestration crises.
HaemolysisHaemolysis occurs due to a decreased red cell survival of 10 -20 days. As a result there is increased red cell production and elevated numbers of immature red blood cell (nucleated red cells and reticulocytes) in peripheral blood and subsequent anaemia as the bone marrow struggles to meet the demands of brisk haemolysis.
AnaemiaDue to haemolysis, the anaemia of Sickle cell disease is usually chronic but reasonably compensated with an appropriate reticulocytosis. However, other factors can contribute to this anaemia. Inappropriately low serum erythropoietin concen-trations may result from renal insuf� ciency. Folate can be a problem due to an increased utilization of folate.9 Whilst iron overload may result from regular blood transfusions, iron de� ciency may occur due to an enhanced urinary loss. Iron de� ciency is present in approximately 20% of patients with Sickle cell disease.10
An acute severe anaemia may cause an acute fall in the haemoglobin level due to splenic sequestrastion crisis (caused by vaso-occlusion within the cells and splenic pooling of red

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
108
cells); aplastic crisis (characterised by the transient arrest of erythropoiesis due to infection and viruses); or hyperhemolytic crisis (a result of sudden exacerbation of anaemia due to haemolysis).11
Complications of Sickle cell disease
Painful CrisisPain is the result of the obstruction of the microcirculation by ‘sickled’ red blood cells. It is the most signi� cant clinical feature of Sickle cell disease and can vary in severity, intensity and frequency.
It is often the � rst cause for emergency and hospitalisation for affected patients. On admission, an initial pain review should be done without delay and should include the patient’s own assessment using adapted pain scales. Re-evaluation should be undertaken as often as necessary until the pain is controlled. Pain is treated by analgesics, with milder crises being managed with Non-Steroidal Anti-in� ammatory Drugs (NSAIDs) if there is no renal insuf� ciency and more severe episodes requiring intravenous opioids until the crisis has passed. If the pain remains intractable an exchange blood transfusion may be indicated.
PriapismPriapism is due to a painful failure of detumescence, clinically described as “scattered episodes or a stuttering pattern”.4 It usually occurs at night and progresses over a short period, with repeated episodes of clustering. Often extremely dif� cult to live with, priapism can lead to impotence and psychological problems.
Leg UlcersThe cause of leg ulcers in Sickle cell disease is unclear. Hypoxia and infarction of the skin caused by the sickling of

Sickle Cell Disease
109
cells, as well as trauma, infection, severe anaemia and warmer temperatures all predispose to ulcer formation.12 Ulcers can affect 10 to 20 percent of patients, from the age of 10 to 50 years. It particularly affects those with the homozygous haemoglobin S genotype (SCD-SS). Ulcers are rare in individuals with SCD-SC +- thalassemia and in those under 10 years of age.
Chronic Renal failure (CRF)Vaso-occlusion provokes papillary necrosis in the kidneys, causing an inability to concentrate urine (hyposthenuria). This can start early in life, with symptoms of enuresis associated with nocturia. Characteristics of the renal medulla are hypoxia, acidosis, hypertonicity. These are all conditions which ignite HbS polymerization and red cell sickling. The factors that appear to predict renal failure in Sickle cell disease patients are hypertension, proteinuria, increasingly severe anaemia and hematuria.12
Not all patients with Sickle cell disease are on dialysis. The incidence rate is reported to be between 4–20%, but this can reach up to 33% among those above 40 years of age. The presence of CRF signi� cantly shortens the survival of patients so it is important that strategies that may prevent chronic kidney disease (CKD) are taken into account and included in the care program.13
InfectionSevere and recurrent infections can be a major cause for concern in patients with Sickle cell disease.
Vaso- occlusion resulting in splenic infarctions and consequent autosplenectomy can lead to severe life-threatening infec-tions. Morbidity and mortality are high, as lower immune defences increase susceptibility to infectious agents such as Haemophilus in� uenzae, Streptococcus pneumoniae, Mycoplasma pneumoniae, Salmonella typhimurium, Staphy-lococcus aureus, and Escherichia coli.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
110
Pneumococcal sepsis can be a major cause for concern for infants and the Parvovirus B19 infection can cause aplasia. Education of parents, with neonatal screening and antibiotic prophylaxis has contributed enormously to reducing morbidity and mortality related to infection.4
Stroke Stroke affects 30% of children and 11% of patients by the age of twenty and is one of the most devastating complications of Sickle cell anaemia.4 It is usually ischemic in children and hemorrhagic in adults. The Tran cranial Doppler (TCD), is an important tool for predicting the risk of stroke as it enables the identi� cation of stroke months to years beforehand.4
Acute chest syndrome (ACS)This is the most common reason for early mortality, as it is associated with pulmonary fat embolism and infection.
ACS often develops after acute infection, painful episodes, rib or bone marrow or pulmonary infarction. It occurs in about 40% of patients with Sickle cell anaemia, and is four times higher in adults than in children.4 Renal failure may result from ACS as well as thrombocytopenic purpura.1
Treatment generally includes the use of antibiotics (macrolide or quinolone) and oxygen therapy for hypoxia. If symptoms persist, with increased use of supplementary oxygen, a blood transfusion can be indicated.
Repeated episodes of acute chest syndrome increase the likelihood of subsequent pulmonary hypertension but early transfusion may prevent this.14
Acute splenic sequestrationAcute abdominal pain can be a manifestation of acute splenic sequestration. Mainly seen in the early years of life it can result in circulatory collapse and death from anaemia and hypovolemic shock in less than 3 hours. De� ned by an enlargement of the

Sickle Cell Disease
111
spleen and a decrease in Hb concentration levels, prompt therapy, including splenectomy (often recommended after a second episode) and early parental action have greatly decreased morbidity and mortality.4
Pulmonary hypertensionThis is a complication of chronic vaso-occlusive events. It is known to affect 20-40% of adult patients with Sickle cell disease and is again associated with a high mortality rate. Older age, renal insuf� ciency, cardiovascular illness, high haemolytic markers and systolic hypertension are associated risk factors.4
DiagnosisEarly diagnosis is essential for health maintenance and surveillance.15 Good pre-natal care includes the sampling of the amniotic � uid and blood tests being taken as soon as possible. Children with Sickle cell disease should undergo close surveillance by a paediatrician and a haematologist to help parents identify early painful crises.
Haemoglobin levels in Sickle cell disease are below normal. In HbSS the Hb ranges between 6–8 g/dL, and is associated with a high reticulocyte count. In other forms of Sickle cell disease, Hb levels tend to be higher.
InvestigationsThe following investigations may be undertaken on blood samples to provide the diagnosis:
Sodium metabisulfate can be used on a blood � lm as it • can induce the sickling of the red blood cellsSodium dithionite, when mixed with normal haemoglobin • gives a clear solution but when mixed with haemoglobin S (HbS) appears turpid

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
112
Haemoglobin electrophoresis, using a form of gel electro-• phoresis, detects abnormal haemoglobin by measuring the speed of various types of haemoglobin
High performance liquid chromatography (HPLC) can be • used to con� rm the diagnosis16
Treatments
As with all patients with chronic disease, patients with Sickle cell disease are best managed in a programme of comprehensive multi-disciplinary care. The care team should be multi-professional and comprise Doctors, Nurses, and Social service co-ordination. The Care program should include not only out patient and inpatient care, but have a day hospital concept, and a pain service. Patient education should include how crises are caused: the prevention of cold, dehydration, infection and the importance of medication and adherence to vaccinations. Close auto-surveillance is necessary to enable prompt action. Readily available data surrounding each patient, and coordination with other medical subspecialties is crucial to support continuation of care.17 This is important as Sickle cell disease should be considered as a “complex, multi-system illness rather than a simple disorder involving deranged red blood cells”. 17
Prophylactic treatmentsPatients will be prescribed 1mg of Folic acid daily for life to deter aplastic crises. The prevention of infection is essential so Penicillin, as prophylaxis will be administered from birth (neonatal screening) to age 5. Most sickle cell patients will be functionally asplenic beyond childhood, so special care and surveillance will be necessary as infection can precipitate vaso occlusive crises and ACS.4 Immunization is essential to prevent childhood illnesses so vaccinations should be kept up to date.

Sickle Cell Disease
113
Blood TransfusionsApproximately 50% of patients with Sickle cell disease will require exchange transfusions to prevent severe organ failure or death. It is also used in the prevention of stroke and it’s recurrence as well as in acute chest syndrome. Transfusion can help to improve the survival and recovery of organ function.14
Methods of transfusion can involve simple transfusion (whereby there is less than 7ml packed RBC/kg to avoid hyperviscosity), partial exchange transfusion or erythrocytapheresis, the latter being bene� cial to sickle cell patients.14
Normal blood cells in the blood transfusion decrease the percentage of haemoglobin S in the patient’s circulation thereby improving microvascular perfusion and correcting the low oxygen-carrying capacity caused by severe anaemia.14 Following exchange transfusion the circulating blood should contain at least 50% HbA.
It should be noted that as most patients with Sickle cell disease are asymptomatic from their anaemia the trigger for blood transfusion for symptomatic relief may be much lower than in patients without Sickle cell disease.
Due to intermittent transfusions, many sickle cell patients can become iron overloaded.14 Iron toxicity can appear after 10-30 units of red cells depending on the patient’s weight.1 Signs and symptoms are painful joints and back problems. As these can be confused with vaso- occlusive crises careful diagnosis is necessary.18 It is important to monitor and treat the iron overload. The magnitude of iron accumulation can be achieved by liver biopsy but this is invasive and has associated risks. MRI and CT scans are not clinically proven and ferritin levels can be unreliable as they can be altered by liver disease, in� ammation and vitamin C reserves. Chelation therapy (Desferrioxamine) should begin when liver iron exceeds 7mg/gm per dry weight.14 Constant surveillance, education and support of the patient are imperative.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
114
Management of anaemia in patients with CKD and SCDPatients with both chronic kidney disease and Sickle cell disease represent a dif� cult cohort of patients to treat in respect of their anaemia. This is because even with high dose Erythropoietin Stimulating Agents (ESA) they are unlikely to achieve a Hb above 8g/dl. The main aim of these patients is therefore to reduce the frequency of blood transfusions rather than correct their anaemia.19 Whilst safety in Sickle cell disease is unclear and controversial there have been studies to support its use.20
Current guidelines suggest titrating the dose of ESA to prevent the level of HbS from becoming greater than 30% to reduce the risk of exacerbating a sickling crisis. Reducing haemolysis with the use of hydroxyurea may also assist in the management of patients with CKD.19
HydroxyureaThis is a drug, taken daily, and is used to reduce the frequen-cy of severe pain, acute chest syndrome, priapism21 and the need for blood transfusions in adult patients with Sickle cell disease.7 Hydroxyurea interferes with the sickle polymeriza-tion process by increasing the production of fetal haemoglobin (HbF).22
Phosphodiesterase-5 (PDE5) inhibitorsPhosphodiesterase-5 (PDE5) inhibitors Sildena� l, Tada� l and Vardena� l, are presumed to enhance production of nitric oxide (NO). Priapism in Sickle cell disease might be a manifestation of reduced NO availibility23 and the PDE5 leads to the re-laxation of the smooth muscles in the corpora cavernosa and penile arteries.
Sildena� l therapy is also used in the treatment of pulmonary hypertension as it reduces the pulmonary artery systolic pre-ssure and increases the 6-minute walk distance.24

Sickle Cell Disease
115
Other therapiesTetrahydrobiopterin (6R-BH4) is used to “improve endothelial dysfunction” which helps to correct the inability of blood vessels to dilate correctly in patients with Sickle cell disease. Tetrahydrobiopterin is an enzyme used in the production of Nitric Oxide (NO) and is essential in the protection of the cardio-vascular system. In Sickle cell disease levels of this enzyme is decreased through in� ammation and high production of superoxide (a highly reactive form of oxygen). Restoring levels of the enzyme have proved bene� cial in nearly 67 per cent of study participants and the treatment appears, so far, to be well tolerated and safe.25
Butyrate and Arginine stimulate haemoglobin production. Studies have shown an increase in healthy haemoglobin and a reduction of symptoms of Sickle cell anaemia.26 In December 2002, researchers found that Arginine had an ‘unexpected side effect’ in the treatment of leg ulcers.27
Poloxamer 188 (Flocor®) is an experimental agent that reduces the length of painful episodes in Sickle cell anaemia by increasing blood � ow to the tiny blood vessels surrounding the painful area, especially in cases of acute chest syndrome.26
Sulphasalazine is a drug that is capable of reducing the number of “sticky” molecules on the red blood cells in Sickle cell anaemia.26
Bone Marrow TransplantsBone Marrow Transplants can be a suitable treatment espe-cially in the case of children. However, there is great dif� culty in � nding suitable donors as patients with Sickle cell disease must have an HLA-identical sibling donor and the procedure is not without risk. The SCD bone marrow is at � rst conditioned by radiation or chemotherapy and then the healthy donor bone marrow is transplanted. The process requires a lengthy

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
116
hospital stay and long term medication as post-transplant immunosuppression is required to prevent Graft versus host disease – a potentially fatal complication.
ConclusionSickle cell disease or Sickle cell anaemia affects a substantial part of the world population (about 30 million) and is a major public health concern because of the associated morbidity and mortality. It is therefore important to understand the manifestations and complications of the disease as well as the treatments available.
For those affected by Sickle cell disease, the complications and treatments can be exhausting and sometimes confusing to live with. By knowing more about the disease, nurses can have a signi� cant role to inform and educate, whether as part of a Care Programme team or on every day duty, by caring and participating in the diagnosis of the complications, or by using the different treatments to reduce the effects of the symptoms. This is fundamental in helping the patient to manage the illness and attain a better quality of life.

Sickle Cell Disease
117
References1. Galacteros F. Sickle cell disease: a short guide to management. In:
Beaumont C, Beris P, Beuzard Y, Brugnara C (Editors) Disorders of Iron homeostasis, erythrocytes, erythropoiesis. Marseille, France: European School of Hematology, 2006; 276-310.
2. Bunn HF. Pathogenesis and treatment of Sickle cell disease. N Eng J Med 1977; 337: 762.
3. Diagram by courtesy of National Heart Lung and Blood Institute, U.S Department of Health and Human services. NHLBI Health Information Center [email protected] [cited Jan.2009]; Available from http://www.nhlbi.nih.gov/health/dci/Diseases/Sca/SCA_WhatIs.html
4. Taher A, Inati A, Kazzi ZN, Dec. 2008. Anemia, Sickle Cell. eMedicine Specialities. Updated Dec.5 2008 [cited 20 Jan 2009]; http://emedicine.medscape.com/article/778971-overview
5. Pearson H. Sickle Cell anaemia and severe infections due to encapsulated bacteria. J. Infect Dis; 136: Suppl S25-30 PMID.
6. Platt OS, Thorington BD, Brambilla DJ et al. Pain in Sickle cell disease: Rates and risk factors. N Eng J Med 1991; 325:11.
7. Sickle Cell Disease Association of America. 2005. [cited Jan.2009] Available from: http://www.sicklecelldisease.org/about_scd/index.phtml
8. “The Open Door Web Site: IB Biology: Genetics: Co-dominance” The Open Door Web Site: Home Page. 9 Sep 2007. 30 Dec.2008 http://www.saburchill.com/IBbiology/chapters03/004.html
9. Lopez R, Shimizu N, Cooperman JM. Recurrent folic acid de� ciency in Sickle cell disease. Am J Dis Child 1971; 122: 48.
10. Vichinsky E, Kleman K, Embury S, Lubin B. The diagnosis of iron de� ciency anemia in Sickle cell disease. Blood 1981; 58: 963.
11. Vichinsky E, Schrier S, Landaw S. Overview of the clinical manifestations of Sickle cell disease. {cited Jan 2009}, http://uptodateonline.com/online/content/topic.do?topicKey=red_cell/24936&view
12. The Sickle Cell Information Center, Atlanta Georgia, U.S.A: Renal Abnormalities in Sickle Cell Disease. Chapter 22 Management of Sickle Cell Disease: Leg ulcers [cited 20/03/2009]. Chapter 19 Renal Abnormalities.http://www.scinfo.org/nihnewchap19.htm
13. Saxena AK, Panhotra BR, Al-Arabi Al-Ghamdi AM. End stage sickle Cell Nephropathy: Determinants of Reduced Survival of Patients on Long term Hemodialysis. Saudi J Kidney Dis Transpl (serial online) 2004; 15:174-5[cited Mar.2009]. Available from http//www.sjkdt.org/article.asp?issn=1319-2442;year=2004;volume=15;issue=2;spag.
14. Vichinsky E. Transfusion therapy in Sickle Cell Disease. Jan 2001. [cited Mar.2009]; http://sickle.bwh.harvard.edu/transfusion.html

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
118
15. Fahner James, (reviewer). Sickle Cell Disease. Kids Health for educational purposes only. Reviewed June 2007 [cited Jan.2009]; Available from: http://kidshealth.org/parent/medical/heart/sickle_cell_anemia.html
16. Clarke GM, Higgins TN. “Laboratory investigation of hemoglobinopathies and thaslassemias: review and update” Clin Chem 200; 46 (8pt 2) 1284-90. http://www.clinchem.org/cgi/content/full/46/8/1284.
17. Benjamin LJ, Swinson GI, Nagel RL Sickle Cell Anemia day hospital: an approach for the management of uncomplicated painful crises. Blood 2000; 95:1130-1136. revised April 19, 2002. Available from: http://sickle.bwh.harvard.edu/comp_care.html
18. V Dimov. Iron Overload due to blood transfusions in sickle cell disease. 2004 [cited Mar.2009] Available from: http://clinicalcases.org/2004/06/iron-overload-due-to-blood-transfusions.html
19. European Renal Association. Revised European Best Practice Guidelines For The Management Of Anaemia In Patients With Chronic Renal Failure. Nephrology, Dialysis, Transplantation 2004; 19 (2) 1-44.
20. Aglieco F, Thomas B, Samson W, Kaplan A. Safe and Effective Administration of Epoetin Alfa in Anemia of Sickle Cell Disease and Kidney Failure. [cited Mar.2009]; Available from: www.kidney.org/news/meetings/abstracts/reviews/embed/Aglieco_safe.pdf
21. Saad ST et al. Follow-up of Sickle Cell Disease: patients with priapism treated hydroxyurea. Hematology and Hemotherapy Am J Hematol 2004; Sep; 77 (1):45-9.
22. Amrolia PJ, Almeida A, Halsey C et al. Therapeutic challenges in childhood sickle cell disease Part 1: current and future treatment options. Br J Haematol 2003; 102:725.
23. Nolan VG, Wyszynski DF, Farrer LA, Steinberg MH. Hemolysis-associated priapism in Sickle Cell Disease. Blood 2005; 106:3264-7.
24. Machado RF et al. Sildena� l therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haemotol 2005 Aug; 130(3): 445-53.
25. Hsu L, Ataga K I, Gordeuk V R, Swerdlow P S, Kutlar A, Smith W, et al. Tetrahydrobiopterin (6R-BH4) Novel therapy for Endolethial Dysfunction in Sickle Cell disease. Presented at ASH (American Society of Hematology) San Francisco CA Dec.6-9 2008, updated Jan.2009 [cited Mar.2009] Available from: http://ash.confex.com/ash/2008/webprogram/Paper15867.html
26. Kugler, M. Treatments for Sickle Cell Anemia, Promising therapies being investigated, Dec.2009 [Butyrate,arginine, Armandola,E A (2002) Management of Sickle Cell Anemia: New approaches. 7th Congress of the European Hematology Association.] [Poloxamer 188, Marlowe, K F, & Chicella, M F (2002). Treatment of Sickle Cell

Sickle Cell Disease
119
pain. Pharmacotherapy 22(4),pp.484-491]. [Sulphasalazine, Solovey, A A., Solovey, A N., Harkness, J., & Hebbel, R P; (2001). Modulation of endothelial cell activation in sickle cell disease: A pilot study. Blood 97, pp.1937-1941]; Available from: http://rarediseases.about.com/cs/sicklecell/a/110602.htm
27. Salodorf Macneil J. Arginine Butyrate Heals Sickle Cell Leg Ulcers. 44th Annual Meeting of the American Society of Hematology: Abstract 26. Presented Dec.8, 2002.


Making the
Connections:
Lymphoma,
the Kidneys and
Transplantation
121

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
122
Learning outcomes• To gain knowledge and understanding of lymphoma
and its association with renal dysfunction• To address the care that renal nurses can provide in
the management of these patients
IntroductionThe lymphomas are a diverse collection of malignant diseases originating in the lymphatic system (see � gure 1). The lymphatic system contains white blood cells called lymphocytes, which are the cells that become cancerous in lymphoma. Lymphoma can manifest in organs which contain lymph tissue, (such as the liver and spleen), and in lymph nodes causing them to enlarge. Within Ireland, lymphoma ranks � fth among cancer cases diagnosed and eighth in cancer related deaths.¹ Although incidence rates continue to rise; there has been an improvement in 5 year survival rates since the 1990’s.2
Figure 1: The Lymphatic System.
Illustration reproduced with kind permission of Richard Henry, Queen’s University Belfast.

Making the Connections: Lymphoma, the Kidneys and Transplantation
123
The disease is characterised by abnormal proliferation of lymphocytes. With the exclusion of skin lymphomas, appro-ximately 85% of lymphomas are derived from B lymphocytes and approximately 15% from T lymphocytes.3,4 Lymphocytes are produced from blood stem cells in the bone marrow (Figure 2) before being released to circulate in the blood and lymph systems. They are part of the body’s immune system and are involved in making antibodies and � ghting infection; therefore lymphoma can affect a person’s immunity and make them more prone to infection.3
Figure 2: Blood Cells.
Illustration reproduced with kind permission of Richard Henry, Queen’s University Belfast.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
124
Classi� cation
The lymphomas can be divided into two broad groups, Hodgkin’s lymphoma (HL) and Non Hodgkin’s lymphoma (NHL). HL is characterised by the presence of unusual B lymphocytes termed Reed – Sternberg cells, while NHL comprises all other lymphomas, arising from B and T lymphocytes. The World Health Organisation (WHO) classi� cation (Table 1) identi� es over 30 subtypes of lymphoma.5 The NHL’s are more varied than HL and are grouped according to their rate of growth, ranging from indolent (low-grade and slow growing) to highly aggressive (high grade and rapidly proliferating).
Indolent Lymphoma
The most common subtype of indolent lymphoma is follicular lymphoma. It is typically widespread throughout the body on diagnosis, but as it is slow growing causes few symptoms and may not initially require treatment until symptoms develop. It is commonly seen in the elderly, and although regarded as incurable, can initially be quite responsive to treatment, but inevitably relapses and becomes more resistant. Average survival times with this condition are 8-10 years.3,6
Aggressive Lymphoma
Diffuse large B cell lymphoma is the most common type of aggressive lymphoma. Aggressive lymphomas are inclined to present in younger patients (less than 60 years) and must be treated immediately as they are rapidly growing and would otherwise be fatal. Most patients have advanced disease on diagnosis; however it is commonly very responsive to treatment and potentially curable.3 Around 40 – 75% of patient with aggressive lymphoma will achieve complete remission.4

Making the Connections: Lymphoma, the Kidneys and Transplantation
125
Non Hodgkin’s Lymphoma B-cell neoplasms
Precursor B-cell neoplasmsPrecursor B-lymphoblastic leukaemia/lymphoma ��
Mature (peripheral) B-cell neoplasmsB-cell chronic lymphocytic leukaemia /small lymphocytic ��lymphomaB-cell prolymphocytic leukaemia��Lymphoplasmacytic lymphoma��Splenic marginal zone B-cell lymphoma ��Hairy-cell leukaemia��Plasma cell myeloma /plasmacytoma��Extra nodal marginal zone B-cell lymphoma of MALT type��Mantle-cell lymphoma��Follicular lymphoma��Nodal marginal zone B-cell lymphoma ��Diffuse large B-cell lymphoma��Burkitt lymphoma��
T - cell neoplasms
Precursor T-cell neoplasmsPrecursor T-lymphoblastic iymphoma/leukemia ��Blastoid NK cell lymphoma��
Mature (peripheral) T-cell neoplasmsT-cell prolymphocytic leukaemia��T-cell granular lymphocytic leukaemia��Aggressive NK-cell leukaemia��Adult T-cell lymphoma/leukaemia (HTLV1+)��Extra nodal NK/T-cell Iymphoma, nasal type��Enteropathy-type T-cell lymphoma��Hepatosplenic y6 T-cell lymphoma��Subcutaneous panniculitis-likeT-cell Lymphoma��
Table 1: R.E.A.L. / WHO Classi� cation of Lymphomas.5

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
126
Clinical Features
Symptoms will depend on the type of lymphoma, its location(s) and the extent of the disease. Commonly in HL a localised, painless swelling of a lymph node (lymphadenopathy) is the only presenting feature. However in more widespread disease there can be lymphadenopathy throughout the body, and the systemic symptoms of fever, night sweats and weight loss, also known as “B” symptoms. “B” symptoms are more common in HL. NHL is more likely to involve not only lymph nodes, but also the sites outside of the nodes (extra nodal), including the liver, spleen and central nervous system (CNS).3 Enlarged lymph nodes can compress nerves resulting in pain, or blood vessels causing obstruction. The latter can manifest as Superior Veno-caval obstruction, an indication for emergency treatment. There may be a cough if there is mediastinal lymphadenopathy or lymphoedema if lymph drainage is obstructed in the nodes of the groin (inguinal) or armpit (axillary). If lymphoma is present within the gastrointestinal (GI) tract, symptoms of abdominal pain, anorexia, vomiting, bleeding, diarrhoea, constipation or ileus may additionally be present and obstruction is possible.
Mature (peripheral) T-cell neoplasmsMycosis fungoides/Sezary syndrome��Anaplastic large-cell Lymphoma, primary cutaneous type��Peripheral T-cell Lymphoma, unspeci� ed��Angioimmunoblastic T-cell Lymphoma��Anaplastic large-cell Lymphoma, primary systemic type��
Hodgkin’s LymphomaLymphocyte predominance, nodular ± diffuse areas��Classical Hodgkin’s disease��
Nodular sclerosisMixed cellularityLymphocyte depletionLymphocyte-rich classical Hodgkin’s disease

Making the Connections: Lymphoma, the Kidneys and Transplantation
127
DiagnosisBiopsyDiagnosis depends on obtaining a biopsy of the effected lymph node or extra nodal mass to determine the histology (the microscopic anatomy of cells) and classi� cation of lymphoma. The surgical removal of the entire node is preferable to a � ne needle biopsy and a bone marrow biopsy should also be obtained for staging purposes.7 Blood and biopsy specimens are prepared and examined by immunophenotyping and molecular studies. Immunophenotyping involves a test called � ow cytometry, which can determine if the lymphoma is of a B or T cell origin and identify special characteristics known as Cluster of Differentiation (CD) markers on the cells surfaces to aid the accuracy of diagnosis.6 Molecular testing, including polymerase chain reaction (PCR) and � uorescence in situ hybridisation (FISH) are used to detect abnormalities within the chromosomes of the cells. This investigational method is useful to obtain both diagnostic and prognostic information.4
Blood TestsBlood tests, obtained for haematology and biochemistry, may include complete blood counts to ascertain if the bone marrow is involved, baseline liver and kidney function in addition to C reactive protein (CRP) and serum lactate dehydrogenase (LDH), as they are useful in monitoring disease progress.8
Protein electrophoresis can be used to determine levels of antibodies and albumin, and blood samples are sent for virology, speci� cally Epstein-Barr virus (EBV) and Human Immunode� ciency virus (HIV) due to their association with the development of lymphoma.8
Imaging TechniquesImaging techniques for example chest X ray, ultrasound, computerised tomography (CT), positron emission tomography (PET) and magnetic resonance imaging (MRI) scans will establish the sites and extent of the disease.
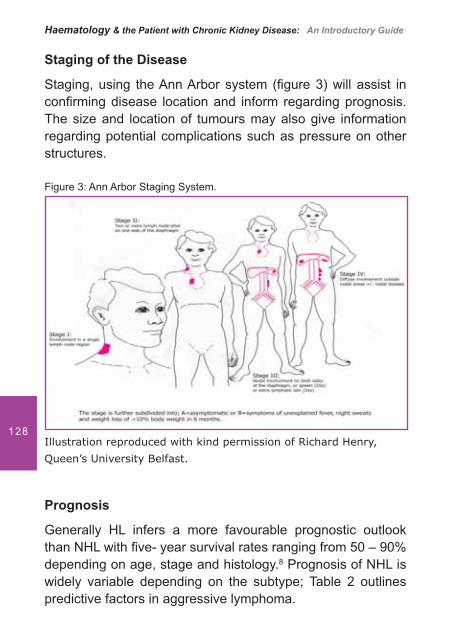
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
128
Staging of the Disease
Staging, using the Ann Arbor system (� gure 3) will assist in con� rming disease location and inform regarding prognosis. The size and location of tumours may also give information regarding potential complications such as pressure on other structures.
Figure 3: Ann Arbor Staging System.
Illustration reproduced with kind permission of Richard Henry,
Queen’s University Belfast.
Prognosis
Generally HL infers a more favourable prognostic outlook than NHL with � ve- year survival rates ranging from 50 – 90% depending on age, stage and histology.8 Prognosis of NHL is widely variable depending on the subtype; Table 2 outlines predictive factors in aggressive lymphoma.
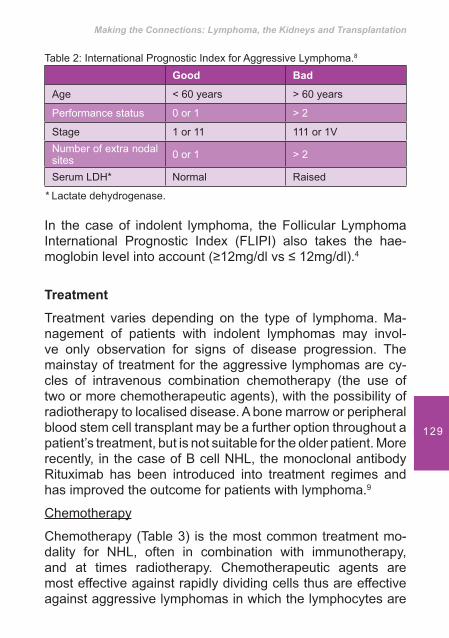
Making the Connections: Lymphoma, the Kidneys and Transplantation
129
Table 2: International Prognostic Index for Aggressive Lymphoma.8
Good Bad
Age < 60 years > 60 years
Performance status 0 or 1 > 2
Stage 1 or 11 111 or 1VNumber of extra nodal sites 0 or 1 > 2
Serum LDH* Normal Raised
* Lactate dehydrogenase.
In the case of indolent lymphoma, the Follicular Lymphoma International Prognostic Index (FLIPI) also takes the hae-moglobin level into account (�12mg/dl vs � 12mg/dl).4
Treatment
Treatment varies depending on the type of lymphoma. Ma-nagement of patients with indolent lymphomas may invol-ve only observation for signs of disease progression. The mainstay of treatment for the aggressive lymphomas are cy-cles of intravenous combination chemotherapy (the use of two or more chemotherapeutic agents), with the possibility of radiotherapy to localised disease. A bone marrow or peripheral blood stem cell transplant may be a further option throughout a patient’s treatment, but is not suitable for the older patient. More recently, in the case of B cell NHL, the monoclonal antibody Rituximab has been introduced into treatment regimes and has improved the outcome for patients with lymphoma.9
Chemotherapy
Chemotherapy (Table 3) is the most common treatment mo-dality for NHL, often in combination with immunotherapy, and at times radiotherapy. Chemotherapeutic agents are most effective against rapidly dividing cells thus are effective against aggressive lymphomas in which the lymphocytes are
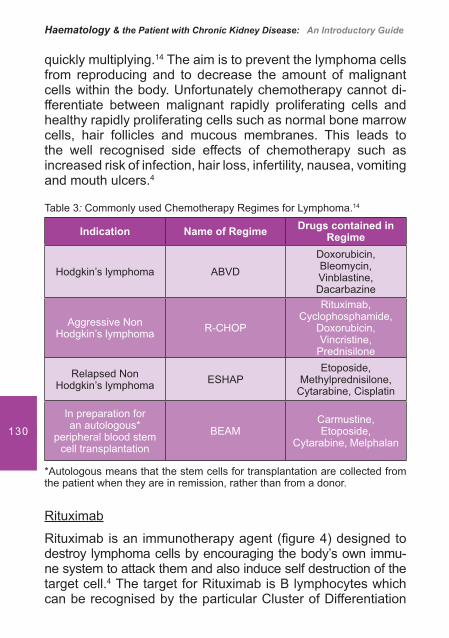
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
130
quickly multiplying.14 The aim is to prevent the lymphoma cells from reproducing and to decrease the amount of malignant cells within the body. Unfortunately chemotherapy cannot di-fferentiate between malignant rapidly proliferating cells and healthy rapidly proliferating cells such as normal bone marrow cells, hair follicles and mucous membranes. This leads to the well recognised side effects of chemotherapy such as increased risk of infection, hair loss, infertility, nausea, vomiting and mouth ulcers.4
Table 3: Commonly used Chemotherapy Regimes for Lymphoma.14
Indication Name of Regime Drugs contained in Regime
Hodgkin’s lymphoma ABVDDoxorubicin, Bleomycin, Vinblastine, Dacarbazine
Aggressive Non Hodgkin’s lymphoma R-CHOP
Rituximab, Cyclophosphamide,
Doxorubicin, Vincristine,
Prednisilone
Relapsed Non Hodgkin’s lymphoma ESHAP
Etoposide, Methylprednisilone,
Cytarabine, Cisplatin
In preparation for an autologous*
peripheral blood stem cell transplantation
BEAMCarmustine, Etoposide,
Cytarabine, Melphalan
*Autologous means that the stem cells for transplantation are collected from the patient when they are in remission, rather than from a donor.
Rituximab
Rituximab is an immunotherapy agent (� gure 4) designed to destroy lymphoma cells by encouraging the body’s own immu-ne system to attack them and also induce self destruction of the target cell.4 The target for Rituximab is B lymphocytes which can be recognised by the particular Cluster of Differentiation

Making the Connections: Lymphoma, the Kidneys and Transplantation
131
marker on their surface called CD20; it is licensed for the treatment of both indolent and aggressive NHL.10,11,12,13 Figure 4: Rituximab binds onto mature B (which expresses CD20 on their cell surface) and encourages other components of the immune system, such as Natural Killer (NK) cells, to destroy them.
Illustration reproduced with kind permission of Richard Henry, Queen’s University Belfast.
Treatment and its effect on Renal Function
Chemotherapy
The effects of chemotherapy on renal function can range from mild and reversible to acute kidney injury (Table 4). Many of the chemotherapy drugs or their metabolites are excreted by the kidneys; therefore assessment of renal function prior to and during chemotherapy cycles is required. Depending on the cytotoxic drugs for administration, this may involve obtaining

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
132
bloods for urea and electrolytes, or a baseline 24 hour urine collection to calculate the glomerular � ltration rate (GFR).
Cisplatin is a chemotherapy drug used in the treatment of lymphoma which is closely associated with nephrotoxicity and can cause signi� cant impairment of the GFR and lead to electrolyte disturbances.15,16 In particular, hyponatraemia, hypocalcaemia, hypocalcaemia and hypomagnesaemia may require correction by supplementation.4 Precautions should be taken to minimise the renal toxicity of this drug. Interventions commonly practiced include aggressive pre and post hy-dration, diuretics prior to administration of the drug, and con-current intravenous Mannitol, in conjunction with accurate � uid balance measurement.14,16
Table 4: Drugs used for the Treatment of lymphoma, which may cause Renal /Bladder Toxicity.14,17
Drug Associated Complications
Cisplatin
Dose limiting nephrotoxicity
Reduced GFR
Electrolyte disturbances
Carmustine Can be associated with delayed renal failure
Melphalan Dose limiting nephrotoxicity
Vincristine Hyponatraemia, nephropathy secondary to induction of tumour lysis syndrome (TLS) (see section on TLS below)
Cyclophosphamide Haemorrhagic cystitis

Making the Connections: Lymphoma, the Kidneys and Transplantation
133
A complication exclusive to the cytotoxic drugs Cyclo phos-phamide and Ifosphamide is haemorrhagic cystitis. It is caused by irritation to the bladder mucosa and can be combated by prophylactic use of the drug Mesna, adequate hydration, frequent voiding of the bladder, and monitoring for haematu-ria.14,16
Complications
Tumour Lysis SyndromeTumour lysis syndrome (TLS) is a potentially life threatening complication that is associated with rapidly proliferative, bulky disease that is treatment sensitive.18 Thus individuals with lymphoma are at risk, particularly if they have a high white cell count and LDH (a marker of proliferation) prior to the commencement of cytotoxic therapy, including chemotherapy, immunotherapy and steroid therapy. Individuals with Burkitt’s lymphoma are at particularly high risk. More rarely TLS can also be initiated by radiotherapy. Further risk factors include patients with underlying dehydration or renal insuf� ciency.19
Pathophysiology
Tumour lysis syndrome is characterised by metabolic distur-bances caused by the massive and rapid release of intracellu-lar products (nucleic acids, potassium and phosphorus) from malignant cells into the circulation, which the kidneys are unable to ef� ciently excrete. This leads to hyperuricaemia, hyperkalaemia, hyperphosphataemia and hypocalcaemia. Hypocalcaemia occurs as serum calcium binds to the increased amounts of phosphorus in the bloodstream.19,20 The crystallisation of uric acid and calcium phosphate in the renal tubules can further impair renal function, and lead to acute kidney injury.19
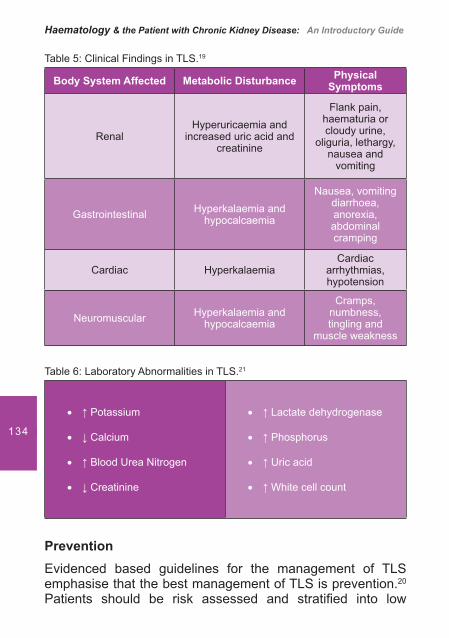
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
134
Table 5: Clinical Findings in TLS.19
Body System Affected Metabolic Disturbance Physical Symptoms
RenalHyperuricaemia and
increased uric acid and creatinine
Flank pain, haematuria or cloudy urine,
oliguria, lethargy, nausea and
vomiting
Gastrointestinal Hyperkalaemia and hypocalcaemia
Nausea, vomiting diarrhoea, anorexia, abdominal cramping
Cardiac Hyperkalaemia Cardiac
arrhythmias, hypotension
Neuromuscular Hyperkalaemia and hypocalcaemia
Cramps, numbness, tingling and
muscle weakness
Table 6: Laboratory Abnormalities in TLS.21
Potassium��
Calcium��
Blood Urea Nitrogen��
Creatinine ��
Lactate dehydrogenase��
Phosphorus��
Uric acid��
White cell count ��
PreventionEvidenced based guidelines for the management of TLS emphasise that the best management of TLS is prevention.20
Patients should be risk assessed and strati� ed into low

Making the Connections: Lymphoma, the Kidneys and Transplantation
135
intermediate and high risk groups and treated accordingly. Whilst patients with indolent lymphoma may be low risk, those with aggressive lymphomas will be intermediate or high risk, depending on their white cell count and LDH.
TreatmentHydration
Aggressive hydration (at least 3 litres/ 24 hours) and main-taining an adequate diuresis are key to the prevention and management of TLS as it promotes the excretion of uric acid and phosphate, and improved renal blood � ow and glomerular � ltration.20 Alkalinisation of the urine is also recommended, by the addition of sodium bicarbonate to the � uid regimen. Uric acid is more soluble in an alkaline environment thus itsexcretion is aided if the urinary PH is maintained at �7.3,20,21
Accurate and frequent monitoring of � uid and electrolyte balance is necessary, including urinary catheterisation to monitor output. The patient with pre existing renal impairment may be more at risk of TLS syndrome and thus they should be jointly managed by a haematologist and renal physician.22
Oral Allopurinol and Intravenous Rasburicase Oral allopurinol is used to decrease the formation of uric acid and thus decrease the incidence of renal tubule obstruction due to uric acid crystal formation. However it has some limitations, as it is ineffective at reducing levels of uric acid formed prior to chemotherapy, is associated with hypersensitivity reactions and takes several days to reduce uric acid levels. Therefore in high- risk patients and to prevent unacceptable delays in chemotherapy treatment, an alternative agent, Rasburicase may be administered intravenously. Whilst Allopurinol is used in the prevention of hyperuricaemia, Rasburicase can not only prevent but also treat acute hyperuricaemia.23 Dose reduction of Allopurinol should be instigated in patients with pre-existing moderate or severe renal insuf� ciency.22,23

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
136
Management of Electrolyte Imbalances
The potassium binder sodium polystyrene sulphonate may also be administered in mild to moderate TLS. However in severe cases, rises in potassium levels may be controlled by administration of Calcium gluconate and dextrose and insulin infusion, in conjunction with ECG monitoring. Aluminium Hydroxide can be prescribed for hyperphosphataemia, although it should be used with caution in severe renal impairment.Haemodialysis or haemo� ltration may be considered.18,20,23
Bone Marrow Suppression
Lymphoma has the potential to in� ltrate the bone marrow, resulting in suppression of the production of healthy blood cells. More commonly however the treatment of lymphoma with chemotherapy temporarily damages the maturing cells within the bone marrow, as they are rapidly dividing cells and therefore are susceptible to the effects of chemotherapy. Thus the individual being treated for lymphoma is more prone to anaemia, infection and bleeding. Prevention of major blood loss, for example by maintaining the platelet count through transfusion of donor platelets, in line with current guidelines
may minimise the impact of hypovolaemia on the individual’s renal function.24
AnaemiaTreatment Induced
The anaemia associated with the lymphomas may have several contributing factors. Most commonly it is treatment-induced anaemia that is transitory in nature and resolves once the bone marrow function is no longer suppressed by cytotoxic agents.25 Thus it is most frequently corrected by blood transfusion, as response to erythropoietin stimulating agents (ESAs) is optimal after 4-6 weeks and the bene� ts may not be experienced in a timeframe to warrant the cost.25

Making the Connections: Lymphoma, the Kidneys and Transplantation
137
Nevertheless with lymphoma there is typically the requirement of repetitive and intensive chemotherapy to induce and main-tain remission and thus anaemia can become a debilitating symptom of both the disease and treatment.26 Certain che-motherapy regimens are associated with more profound anaemia, particularly cisplatin containing schedules.27 Also, during CHOP chemotherapy treatment, a study has reported that 79% of patients developed mild to moderate anaemia and 49% moderate to severe anaemia (Table 7).25
Table 7: National Cancer Institute Toxicity Grading for Anaemia.26
Grade of Anaemia Hb Value
Mild – grade 1 10 - 12g/dL (women) 10 - 14 g/dL (men)
Moderate – grade 2 8.0 – 9.9g/dL
Severe – grade 3 6.5 – 7.9 g/dL
Life threatening – grade 4 <6.5 g/dL
Disease in� ltration of the Bone Marrow
In the event of bone marrow disease in� ltration (stage four disease) causing restraint of red cell production, chemotherapy is often the treatment of choice to eradicate the lymphomatous cells and subsequently re-establish normal haematopoiesis.
In� ammatory Cytokines
As with other malignancies, lymphoma can predispose to anaemia of chronic disease, even in the absence of bone marrow in� ltration. This is due to the production of cytokines (chemical messengers), which can effect erythropoietin pro-duction, growth and development of red cells and cause defective movement of iron into the blood.25,26

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
138
Role of ESA’s and Iron
Due to the negative effect of anaemia on quality of life, intervention with ESAs is advocated by some organisations when the haemoglobin falls below 10-11g/dL, depending on clinical symptoms.25,26 Whilst administration of ESAs can correct anaemia, concomitant use of iron therapy may be necessary to keep pace with the demands of the red cell production (erythropoeisis) stimulated by the erythropoietin therapy. Oral iron is commonly used to treat iron de� ciency; however IV iron may be necessary for those who are intolerant or unresponsive to oral iron.26
Haematinic De� ciencies speci� c to Lymphoma
Iron, vitamin B12 and folate de� ciencies are of signi� cance in lymphoma as the gastrointestinal (GI) tract is a potential site for disease occurrence and therefore occult blood loss or malabsorption can lead to these de� ciencies. Nutritional de� cits may be exacerbated by anorexia associated with disease and treatment related factors. It is therefore important to correct these to prevent recurring anaemia.27
Gastrointestinal Lymphomas
GI lymphomas commonly arise from mucosal associated lymphoid tissue (MALT lymphomas). These are generally indolent, B cell malignancies, but can transform to a more aggressive lymphoma. Whilst MALT lymphomas can effect variable locations of the GI tract, gastric involvement is highly correlated with Helicobacter pylori infection.28 Other risk factors for the development of gastrointestinal lymphoma are Coeliac disease (gluten sensitive enteropathy) and immuno-suppression after solid organ transplantation (See section on post transplant lymphoproliferative disorders).29

Making the Connections: Lymphoma, the Kidneys and Transplantation
139
In Coeliac disease, ingestion of gluten (wheat, rye and barley), triggers atrophy of the villi in the small intestine, resulting in an immune reaction by T lymphocytes, causing damage to the intestinal epithelium; thus malabsorption of vitamins and nutrients occurs, leading to malnutrition. Undiagnosed Coeliac disease or non adherence to a gluten free diet is associated with increased risk of T cell GI lymphoma.29,30,31
Infection
Chemotherapy causes destruction of a type of white blood cell called the neutrophil, which is one of the main white blood cells involved in � ghting bacterial infection.17 The neutropaenic patient therefore often requires the support of intravenous antibiotics which frequently contain an aminoglycoside such as Gentamycin, Vancomycin or Amikacin. As aminoglycoside antibiotics are nephrotoxic, it is imperative that blood levels of these drugs are monitored, and doses adjusted if required, minimising preventable kidney damage.18
Neutropaenic Sepsis
Sepsis is a systemic response to severe infection in the body and is a common cause of morbidity and possible mortality in the cancer patient.18 Septicaemia results in a widespread in� ammatory response to the infection or the toxins produced by the infecting organism.32 As sepsis develops, it is cha-racterised by excessive dilation of blood vessels resulting in hypotension and subsequent decreased organ perfusion. Typically septic shock is diagnosed when � uid replacement fails to increase blood pressure to acceptable levels indicating inadequate perfusion of major organ systems, including the kidneys, with progressive risk to their function.33 Metabolic acidosis can occur also as tissues are unable to receive oxygen and nutrients because of impaired blood � ow.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
140
The abnormal in� ammatory response also causes genera-lised tissue damage, which can initiate bleeding and clotting abnormalities including a serious condition known as disse-minated intravascular coagulation (DIC). This may lead to the development of clots in the small blood vessels leading to major organs and can cause organ failure.18 Thus the kidneys are extremely vulnerable in the event of neutropaenic sepsis (Table 8). The haemorrhagic element in DIC can manifest by oozing from cannula or central line sites and mucosal bleeding; therefore this condition may present dif� culties with line insertion. The coagulation screen is characterised by prolonged Activated Partial Thromboplastin Time (APTT) and Prothrombin Time (PT) and low � brinogen levels, in conjunction with thrombocytopaenia.
Table 8: Management of Sepsis.18,32,34
Management of Neutropaenic Sepsis involves:
Identi� cation of source of infection��
Prompt initiation of broad spectrum intravenous antibiotics ��
Monitoring of vital signs ��
Blood volume replacement- IV � uids��
Maintenance of blood pressure, cardiac output and renal perfusion ��using vasoactive and inotropic agents (for example Dopamine and Doputamine)
Close monitoring of � uid balance, including urinary catheterisation��
Oxygen therapy, with the potential for respiratory assistance��
Correction of electrolyte and metabolic imbalances��
Steroid therapy to decrease in� ammatory response��
Blood product replacement��
Administration of haematopoietic growth factors such as Granulocyte ��Colony Stimulating Factor (G-CSF) to enhance the production and maturation of neutrophils

Making the Connections: Lymphoma, the Kidneys and Transplantation
141
Acute Renal DysfunctionPrimary renal involvement is very rare; however renal fai-lure can occasionally result from secondary in� ltration of kidney tissue.35,36 Furthermore tumour invasion and lympha-denopathy can result in obstruction of the ureters resulting in hydronephrosis.17 Obstruction may be relieved by surgical placement of stents.
The acute side effects of chemotherapy including nausea, vomiting, mouth ulceration, anorexia and diarrhoea may also exacerbate renal impairment as they may contribute to dehydration and electrolyte imbalances (see Table 9, next page).
For patients who require allogeneic bone marrow or peripheral blood stem cell transplantation for aggressive lymphoma, renal function may be increasingly compromised. Allogeneic refers to a transplant whereby the stem cells are obtained from a donor rather than the patient themselves. In addition to the use of more dose intense chemotherapy in preparation of transplant, these individuals may additionally receive total body irradiation with the potential of radiation nephropathy. In addition they will require immunsuppression (increasing the risk of infection and sepsis), using agents such as Cyclosporin, which are nephrotoxic and are correlated with the development of hypertension.37,38
Renal Transplantation and Lymphoma Post transplant malignancy is a signi� cant limitation in renal transplantation and incurs considerable morbidity and mor-tality.40 It is estimated that the risk of developing a malig-nancy is 3-5 times higher in the renal transplant popula-tion than the general public.41,42,43 This risk is highest in the � rst year post transplantation; however there is increased incidence throughout the entire post transplant period.44 With the exception of skin cancers, lymphomas are the most frequently occurring cancer.45,46 The diverse types of

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
142
lymphoma that can present in the renal transplant patient are collectively known as Post Transplant Lymphoprolife-rative Disorders (PTLD’s). These are commonly B cell NHL’s, which are characterised by a more aggressive clini-cal course and a poorer outcome than the lymphomas not associated with solid organ transplantation.44,47
Table 9: Summary of Factors associated with Acute Renal Insuf� ciency in the Lymphoma Patient.17,39
Decreased Perfusion
Obstructive Complications Acute Failure
Dehydration Tumour invasion of kidneys, ureters or
bladderLymphomatous
in� ltration
Bleeding Lymphadenopathy Tumour lysis syndrome
Decreased Perfusion
Obstructive Complications Acute Failure
Gastrointestinal losses Blood clotting Nephrotoxic
chemotherapy
Diuretic therapy
Nephrotoxic drugs: Immunosuppression,
antibiotics, antifungals, antivirals, radiographic contrast,
non steroidal anti in� ammatory drugs
Decreased � uid intake Radiation nephritis
Severe sepsis
Reasons for the Development of PTLDDecreased ImmunsurveillancePTLD is associated with the immunosuppressive thera-py required post transplantation. The length of exposure

Making the Connections: Lymphoma, the Kidneys and Transplantation
143
to immunosuppressive therapy, its intensity and the type of drug(s) used are all correlated with the risk of malignancy development.43,48 More profound immunosuppression (higher doses and a combination of agents) results in an increased risk of cancer and more aggressive disease progression. This is because immunosuppression impairs the ability of healthy immune cells, called cytotoxic T lymphocytes, to “police” the body (immunsurveillance) for abnormal or malignant cells and destroy them, thus allowing cancer cells to multiply undetected.48
Viral InfectionViruses from the herpes family, primarily the Epstein Barr virus (EBV), are a signi� cant cofactor contributing to PTLD. EBV is potentially cancer – inducing (oncogenic) as it has the ability to infect and induce malignant changes in B lymphocytes; furthermore it promotes the growth of these cancerous lymphocytes and prevents cell death (apoptosis). EBV infection is found in 100% of PTLD’s that develop early post transplant.49 EBV can be transmitted either from the donor organ, or reactivate in the recipient as a consequence of their impaired immunity secondary to immunosuppression.41
Immunosuppressive AgentsThere are possible direct cancer-causing (carcinogenic) pro-perties of some immunosuppressive drugs. However emer-ging evidence suggests that whilst Calcineurin Inhibitors such as Ciclosporin and Tacrolimus increase the risk of malignancy, Mammalian Target of Rapamycin (mTOR) Inhibitors for example Sirolimus and Everolimus may have a protective effect against malignancy.40,48 Pre transplant conditioning with lymphocyte depleting antibodies such as Muromonab CD3 (OKT3) are also directly implicated in PTLD development.48 (Table 10).

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
144
Table 10: Risk Factors for the development of Post Renal Transplant Lymphoproliferative Disorders.44,48
Immunosuppressive Therapy��
Antibody induction therapy
High dose immunosuppression
Combination of immunosuppressive agents
Duration of immunosuppression
Viral Infection��
Epstein Barr Virus (EBV). Especially if recipient EBV –ve and donor EBV +ve
Cytomegalovirus (CMV)
Human herpes virus (HHV-8)
Conventional / other Risk Factors��
Genetic factors
Pre transplant malignancy
Smoking
Younger age at transplantation
Management of PTLDImmunosuppression
The reduction or withdrawal of immunosuppression can re-sult in complete tumour regression in early disease; how-ever the graft should be closely monitored for signs of rejection. The management of patients who fail to respond to immunosuppression reduction remains controversial. The recommended options include CHOP chemotherapy and the monoclonal antibody, Rituximab.49 Antiviral therapy such as gancyclovir,41,45 and the monitoring of the EBV viral load by polymerase chain reaction (PCR) to identify those at risk

Making the Connections: Lymphoma, the Kidneys and Transplantation
145
also remains controversial.50 Survival rates range from 25 – 60%.45
The nursing management of the post renal transplant patient should therefore incorporate identi� cation of patients at increased risk and monitoring of immunosuppressive drug levels to ensure they are maintained within the recommended therapeutic range. Patients should also be advised regarding healthy lifestyle choices to minimise their malignancy risk. The nurse should remain vigilant for any signs of PTLD developing such as fever and lymphadenopathy.
ConclusionThe lymphomas can be divided into two broad groups, Hodgkin’s lymphoma (HL) and Non Hodgkin’s lymphoma (NHL) with the symptoms depending upon the type of lymphoma, its location(s) and the extent of the disease. The treatment also varies depending upon the type of lymphoma. There is much interface with lymphoma and renal dysfunction from acute kidney injury through to renal transplantation. It is vital that renal nurses understand this strong correlation and be able to manage the care of these patients. Continuous education is vital to ensure that nurses and other members of the health care team recognise the manifestations of lymphomas and understand how these malignant diseases are so strongly associated with the kidneys and transplantation. There must also be a strong collaborative approach with both the renal and haematological teams to ensure that patients are managed in a holistic manner.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
146
References1. Campo J, Comber H, Gavin A.T. All Ireland Cancer Statistics 1998-
2000. Northern Ireland Cancer Registry / National Cancer Registry Ireland. 2004.
2. Comber, H. and Walsh, P.M. Patterns of care and survival of cancer patients in Ireland 1994-2004. National Cancer Registry Ireland. 2008.
3. Grundy M, Andrews G. Chapter 6. The Lymphomas. In: Grundy M, editor. Nursing in Haematological Oncology. London: Balliere Tindall Elsevier, 2006.
4. Wardley C, Ni Chonghaile M, Goode V. Margulies A. Molassiotis A. Naffziger C. Quinn B. Vaessen G. and Williams K. Non-Hodgkin’s Lymphoma: A self-learning guide for nurses. Nurse Health Liaison Team. UK: Hoffman-La Roche Ltd, 2006.
5. Provan D. Singer C R J. Baglin T, Lilleyman J. Oxford Handbook of Clinical Haematology. 2nd edition. Oxford University Press, 2006
6. Howard M R, Hamilton P J. Haematology. 3rd edition. Philadelphia: Churchill Livingstone Elsevier, 2008
7. British Committee for Standards in Haematology Guidelines for the use of platelet transfusions. British Journal of Haematology 2003; 122(1): 10-23.
8. Hoffbrand A V, Pettit J E. Moss P A H. Essential Haematology. 4th Edition. Oxford: Blackwell Science Ltd, 2005.
9. Castagna L, Magagnoli M, Demarco M, Santoro A. Lymphomas. Update on Cancer Therapeutics 2 2007; 101-110.
10. National Institute for Clinical Excellence. Rituximab for aggressive Non-Hodgkin’s Lymphoma. London: NICE. 2003.
11. National Institute for Health and Clinical Excellence. Rituximab for the treatment of follicular Lymphoma. London: NICE. 2006.
12. National Institute for Health and Clinical Excellence. Rituximab for the treatment of relapsed or refractory stage 111 or 1V follicular Non-Hodgkin’s Lymphoma. London: NICE. 2008a
13. National Institute for Health and Clinical Excellence. Epoetin alfa epoetin beta and darbepoetin alfa for cancer treatment-induced anaemia. London: NICE. 2008b
14. Dougherty L. Bailey C. Chapter 11. Chemotherapy. In: Corner J. and Bailey C. editors. Cancer Nursing. Care in context. Oxford: Blackwell Science Ltd, 2004.
15. Benoehr P. Krueth P. Bokemeyer C. Grenz A. Osswald H, Hartmann J T. Nephroprotection by Theophylline in patients with Cisplatin chemotherapy: A randomised single-blinded placebo-controlled trial. Journal of the American Society of Nephrology 2005; 16:452-458.

Making the Connections: Lymphoma, the Kidneys and Transplantation
147
16. Coward M, Coley H M. Chapter 14. Chemotherapy. In: Kearney A, Richardson A. editors. Nursing Patients with Cancer. Principles and Practice. London: Elsevier Churchill Livingstone. 2006.
17. Dolan S. Chapter 24. Renal effects and tumour lysis. In Brighton D, Wood M. editors. The Royal Marsden Hospital Handbook of Cancer Chemotherapy. London: Elsevier Churchill Livingstone. 2005.
18. Dolan S. Chapter 35. Intensive nursing care of the patient with cancer. In: Kearney A, Richardson A. editors. Nursing Patients with Cancer. Principles and Practice. London: Elsevier Churchill Livingstone. 2006.
19. McGraw B. At an increased risk: Tumor lysis syndrome. Clinical Journal of Oncology Nursing 2008; 12 (4): 563-565.
20. Coif� er B. Altman A. Ching-Hon P. Younes A, Cairo M.S. Guidelines for the management of pediatric and adult tumor lysis syndrome: An evidence-based review. Journal of Clinical Oncology 2008; 26 (16): 2767-2778.
21. Colen F N. Oncologic emergencies: Superior vena cava syndrome tumor lysis syndrome and spinal cord compression. Journal of Emergency Nursing 2008; 34 (6): 535-537.
22. Krishnan K. Hammad A. (2009) Tumour Lysis Syndrome. http://emedicine.medscape.com/article/282171-print. (internet) Accessed 11/05/09.
23. British National Formulary (BNF) (2008) BNF No.56 September. London: BMJ Publishing.
24. British Committee for Standards in Haematology (2003). Guidelines for the use of platelet transfusions. British Journal of Haematology 2003; 122(1): 10-23.
25. Birgegard G. Managing anemia in lymphoma and multiple myeloma. Therapeutics and Clinical Risk Management 2008 4(2): 527-539.
26. Henry D H. Guidelines and recommendations for the management of anaemia in patients with lymphoid malignancies. Drugs 67 2007; (2): 175-194.
27. McClelland D B L. editor. Handbook of transfusion medicine. 4th ed London: TSO. 2007
28. Cohen S M, Petryk M, Varma M, Kozuch P S, Ames E.D, Grossbard M L, Non-Hodgkin’s Lymphoma of Mucosa-Associated Lymphoid Tissue. The Oncologist 2006; 11:1100-1117.
29. Ghai S, Pattison J, Ghai S, O’Malley M E, Khallili K, Stephens M. Primary gastrointestinal lymphoma: spectrum of imaging � ndings with pathologic correlation. Radiographics 2007; 27: 1371-1388.
30. Mendoza N. Coeliac disease: an overview of the diagnosis treatment and management. British Nutrition Foundation Nutritional Bulletin 2005; 30: 231-236.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
148
31. Zarkadas M, Case S. Celiac disease and the gluten-free diet. Topics in Clinical Nutrition 2005; 20 (2): 127-138.
32. Myers J S. Chapter 24. Complications of cancer and cancer treatment. In: Langhorne M, Fulton J S, S E Otto. Oncology Nursing. 5th ed. Missouri: Mosby Elsevier. 2007
33. Sharma S, Mink S. Septic Shock. 2006. Accessed from http://www.emedicine.com/MED/topic2101.htm.
34. Dellinger R P, Levy M M, Carlet J M, Bion J, Parker M M, Jaeschke R et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Critical Care Medicine 2008; 36 (1): 297-327.
35. Ahmad A H, MacLennan T, Listinsky C. (2005) Primary renal lymphoma: a rare neoplasm that may present as a primary renal mass. The Journal of Urology 2005; 173: 239.
36. Cupisti A, Riccioni R, Carulli G, Paoletti S, Tognetti A, Meola M et al. Bilateral primary renal lymphoma treated by surgery and radiotherapy. Case report. Nephrology Dialysis and Transplantation 2004; 19: 1629-1633.
37. Hingorani S. Chronic kidney disease in long- term survivors of hematopoietic call transplantation: Epidemiology pathogenesis and treatment. Journal of the American Society of Nephrology 2006; 17: 1995-2005.
38. Apperley E, Carreras E, Gluckman E, Gratwohl A, Masszi T. editors. The EBMT Handbook. 5th ed. Haematopoietic Stem Cell Transplantation. Paris: European School of Haematology. 2008
39. Humphreys B D, Soiffer R J, Magee C C. Renal failure associated with cancer and its treatment. An update. Journal of the American Society of Nephrology 2005; 16:151-161.
40. Kapoor A. Malignancy in kidney transplant recipients. Drugs 68 2008; Suppl 1; 11-19.
41. Morath C, Mueller M, Goldschmidt H, Schwenger V, Opelz G, Zeier M. Malignancy in renal transplantation. Journal of the American Society of Nephrology 2004; 15: 1582-1588.
42. Sweny P. Infection and cancer following renal transplantation. Saudi Journal of Kidney Diseases and Transplantation 2006; 17 (2): 189-199.
43. Dantal J, Pohanka E. Malignancies in renal transplant: an unmet medical need. Nephrology Dialysis and Transplantation 2007: 22 (suppl 1): i4-i10.
44. Pascual J. Post transplant lymphoproliferative disorder – the potential of proliferation signal inhibitors. Nephrology Dialysis and Transplantation 2007; 22 (Suppl 1): i27-i35.
45. LaCasce A.S. Post transplant lymphoproliferative disorders. The Oncologist 2006;11: 674-680.

Making the Connections: Lymphoma, the Kidneys and Transplantation
149
46. Cowlrick I, Delventhal H, Kaipainen K.,Krcmar C, Petan J, Schieibner S. Three year follow-up of malignancies in tacrolimus treated renal recipients – an analysis of European multicentre studies. Clinical Transplantation 2008; 22: 372-377.
47. Hoshida Y, Aozasa K. Malignancies in organ transplantation recipients. Pathology International 2004; 54: 649-658.
48. Gulierrez-Dalmau A, Campistol J M. Immunosuppressive therapy and malignancy in organ transplant recipients. A systematic review. Drugs 67 2007; (8): 1167-1198.
49. Carbone A, Gloghini A, Dotti G. EBV-Associated Lymphoproliferative Disorders: Classi� cation and Treatment. The Oncologist 2008; 13: 577-585.
50. Tsai D E, Aqui N, Tomaszewski J E, Olthoff K M, Ahya V N, Kotloff R M et al. Serum protein electrophoresis abnormalities in adult solid organ transplant patients with post transplant lymphoproliferative disorder Clinical Transplantation 2005; 19: 644-652.


Myeloma and
Renal Failure
151

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
152
Learning Outcomes
• To understand the pathophysiology of Myeloma
• To understand the process of diagnosis
• To understand special considerations in the care of renal patients with myeloma
• To have a basic understanding of treatments
Pathophysiology of Myeloma
Myeloma is a cancer of the bone marrow, speci� cally of the plasma cells. In normal marrow function, plasma cells develop from specialised B lymphocytes as shown in Figure 1. In a normal bone marrow the role of the plasma cells is to produce a variety of anti-bodies in response to the range of infections that the body may be exposed to. In myeloma, the plasma cells overproduce a single anti-body or immunoglobulin and this is often detected in the blood or urine. As a consequence there may be a limitation to the other anti-bodies the bone marrow can produce, and this will result in a reduced ability to � ght infection.
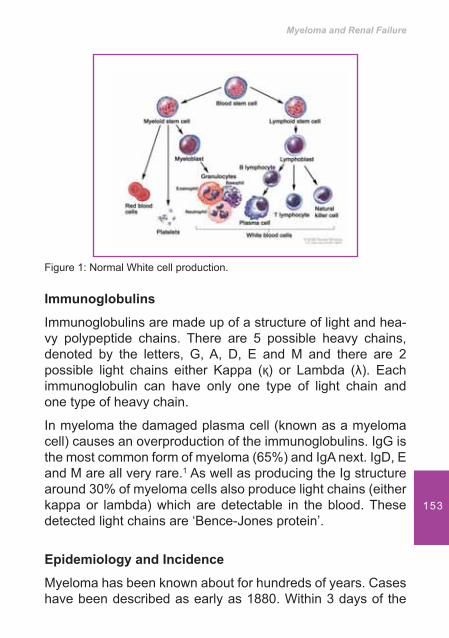
Myeloma and Renal Failure
153
Figure 1: Normal White cell production.
Immunoglobulins
Immunoglobulins are made up of a structure of light and hea-vy polypeptide chains. There are 5 possible heavy chains, denoted by the letters, G, A, D, E and M and there are 2 possible light chains either Kappa (�) or Lambda (�). Each immunoglobulin can have only one type of light chain and one type of heavy chain.
In myeloma the damaged plasma cell (known as a myeloma cell) causes an overproduction of the immunoglobulins. IgG is the most common form of myeloma (65%) and IgA next. IgD, E and M are all very rare.1 As well as producing the Ig structure around 30% of myeloma cells also produce light chains (either kappa or lambda) which are detectable in the blood. These detected light chains are ‘Bence-Jones protein’.
Epidemiology and Incidence
Myeloma has been known about for hundreds of years. Cases have been described as early as 1880. Within 3 days of the

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
154
Hiroshima blast of 1945 the risk of dying from myeloma had risen to 60%.
Myeloma has an incidence of about 50 per million population in the UK and Scandinavian countries. There are in the region of 3970 new cases diagnosed each year in the UK.2 The median age at diagnosis is 65-70 years and 2% of cases are diagnosed in the under 40’s.3 The age range of patients diagnosed with myeloma has implications for the different types of treatments that are discussed later.
Myeloma has a higher incidence in Afro-Caribbean ethnic groups, but little else is known about its epidemiology.4
Presenting Symptoms
Patients with myeloma present with a variety of symptoms and these are shown in table 1 together with their causes.Table 1: Signs and Symptoms of myeloma.
Symptom Cause
Pain, fractures Damage to bone from plasma cells(often back/pelvic pain)
Confusion, red eyes, constipation Hypercalcaemia as a result of damaged bone
Recurrent or persistent infectionFailure of white cell production.Patients may present with neutropaenia or leucopaenia
Fatigue Anaemia typically normochromic
Breathlessness, swollen ankles
May be as a result of renal impairment.Nephrotic syndrome as a result of cardiac failure or amyloidosis
Nausea, loss of appetiteHigh paraprotein levels cause renal tubular damage which may require renal replacement therapy
Headache, blurred visionRaised viscosity as result of excess paraprotein in the circulating blood volume

Myeloma and Renal Failure
155
DiagnosisNew guidelines for the classi� cation of myeloma were released in 2002.5
A diagnosis of myeloma is dependent on the amount of protein in the urine or blood, the number of plasma cells found in the bone marrow and any other organ related myeloma damage (lytic lesions on X-ray, anaemia, raised calcium levels or renal impairment). The diagnosis is usually con� rmed by the demonstration of a paraprotein in the serum or urine and lytic lesions on x-ray together with over 10% of plasma cells in the bone marrow.5
The diagnosis of myeloma is therefore made using a varied approach including
Blood tests• Urine Test• Bone Marrow Aspirate and Trephine• X Ray•
Blood Tests
Some tests are screening tests, and others are speci� c to a diagnosis of myeloma. Table 2 lists the blood tests that may be used and how they maybe altered in myeloma.
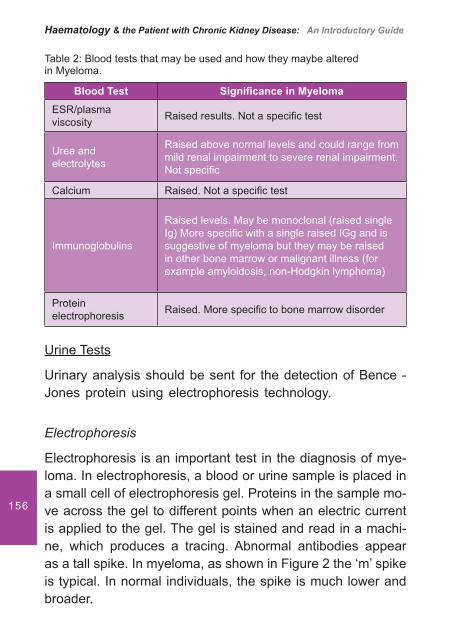
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
156
Table 2: Blood tests that may be used and how they maybe altered in Myeloma.
Blood Test Signi� cance in MyelomaESR/plasma viscosity Raised results. Not a speci� c test
Urea and electrolytes
Raised above normal levels and could range from mild renal impairment to severe renal impairment. Not speci� c
Calcium Raised. Not a speci� c test
Immunoglobulins
Raised levels. May be monoclonal (raised single Ig) More speci� c with a single raised IGg and is suggestive of myeloma but they may be raised in other bone marrow or malignant illness (for example amyloidosis, non-Hodgkin lymphoma)
Protein electrophoresis Raised. More speci� c to bone marrow disorder
Urine Tests
Urinary analysis should be sent for the detection of Bence - Jones protein using electrophoresis technology.
Electrophoresis
Electrophoresis is an important test in the diagnosis of mye-loma. In electrophoresis, a blood or urine sample is placed in a small cell of electrophoresis gel. Proteins in the sample mo-ve across the gel to different points when an electric current is applied to the gel. The gel is stained and read in a machi-ne, which produces a tracing. Abnormal antibodies appear as a tall spike. In myeloma, as shown in Figure 2 the ‘m’ spike is typical. In normal individuals, the spike is much lower and broader.

Myeloma and Renal Failure
157
Figure 2: Diagram showing electrophoresis.
Bone Marrow Aspirate and Trephine
A bone marrow aspirate from the hip or the sternum will be taken to look at the percentage of plasma cells in the bone marrow. A trephine will offer more accurate information about the in� ltration of the myeloma to the bone marrow. The in� ltration of the bone marrow of more than 10% plasma cells contributes to a diagnosis of myeloma.2 A high level of proliferation in the bone marrow is associated with a poorer prognosis.6
X-Ray
As myeloma cells invade the bone marrow X-rays maybe taken of the cervical, thoracic and lumbar spine, skull, chest, pelvis, humeri and femora. These may show lytic lesions (holes) as shown in � gure 3.
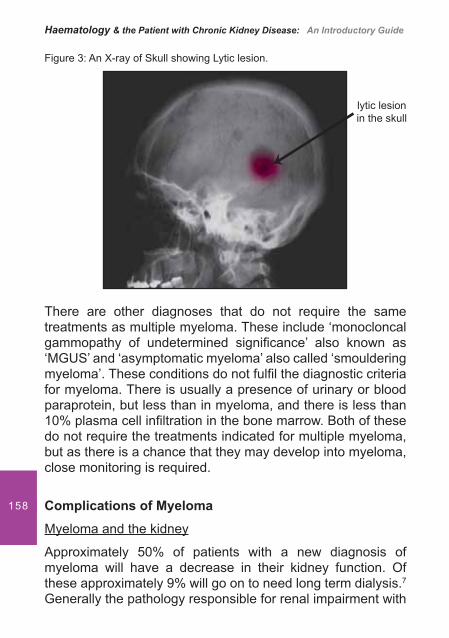
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
158
Figure 3: An X-ray of Skull showing Lytic lesion.
There are other diagnoses that do not require the same treatments as multiple myeloma. These include ‘monocloncal gammopathy of undetermined signi� cance’ also known as ‘MGUS’ and ‘asymptomatic myeloma’ also called ‘smouldering myeloma’. These conditions do not ful� l the diagnostic criteria for myeloma. There is usually a presence of urinary or blood paraprotein, but less than in myeloma, and there is less than 10% plasma cell in� ltration in the bone marrow. Both of these do not require the treatments indicated for multiple myeloma, but as there is a chance that they may develop into myeloma, close monitoring is required.
Complications of Myeloma
Myeloma and the kidney
Approximately 50% of patients with a new diagnosis of myeloma will have a decrease in their kidney function. Of these approximately 9% will go on to need long term dialysis.7 Generally the pathology responsible for renal impairment with
lytic lesion in the skull

Myeloma and Renal Failure
159
myeloma is cast nephropathy. This is because a high light chain secretion rate causes a blockage of the renal tubules as the light chains are � ltered through the glomeruli.8 However, many patients can produce large numbers of light chains and still not reach end stage renal failure.Kidney failure can be further exacerbated by:
Hypercalcaemia as a result of myeloma bone effects. • High calcium levels cause vasoconstriction and a possible decrease in glomerular � ltration rate.Non-steroidal use (commonly prescribed for treatment • of pain, often apparently innocent back pain). Non steroidal medications affect the blood supply to the kidney through vasoconstriction and the altering of the renal blood � ow.Dehydration- causing an increased concentration of • light chains.Radio-contrast- that may be given in the investigation • phase.
Not all patients will go on to require lifelong renal replacement. In respect of the recovery of renal function studies do vary. Some say that minimal recovery is possible whilst others predict a recovery rate of up to 83%.9,10 Recovery of renal function does seem to be related to an increased survival rate (2 years versus 5 months).
Recommended preventative measures include the use of adequate hydration (3 litres per day), and calcium lowering biphosphinate medications.
In elderly patients other causes of renal impairment will also need to be excluded for example, renal vascular disease and obstructive uropathy.
Anaemia and myelomaAnaemia is a feature of myeloma and several causes of this have been described. They include;

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
160
Decreased level of erythropoietin (epo) production• Insuf� cient response to the EPO hormone• Bone marrow replacement by tumour cells • Cytokine mediated suppressive effect on erythro-• poiesis. This is described as the cytokine anaemia of chronic disease. Cytokines (such as tumour necrosis factor and interferon and interleukin) affect anaemia by shortening red cell survival, blunting erythropoietin production and reducing availability of ironMedication used to treat myeloma as they are often • myelosuppresents and reduce bone marrow function
It appears that 50% of patients newly diagnosed with myeloma will present with anaemia and the remainder will develop it during the course of their illness.11 The degree of anaemia will be greater if renal failure is a feature of the disease process.
Use of Erythropoetin Stimulating Agents (ESA’s)
There is good evidence that erythropoietin stimulating agents (ESA) are effective in the treatment of anaemia in myeloma, both with and without renal failure.12,13,14 It is important to pre-vent anaemia occurring as studies have reported that patients who are given ESA’s to prevent anaemia have an improved quality of life. During a course of chemotherapy fatigue is the most commonly reported symptom (75% of patients report this) and often the effect of even mild anaemia (de� ned as Hb of 8-10g/dl) is under-estimated by care teams. Research shows that as few as 23% of oncologists would treat mild anaemia15 yet typically between 50 and 70% of patients will respond to ESA’s.16
Studies have also shown that ESA used pre high dose chemotherapy or pre bone marrow transplant has a bene� cial effect and patients need fewer blood transfusions.17 The timing for the initiation of ESA maybe important as during

Myeloma and Renal Failure
161
chemotherapy the bone marrow is at its most suppressed and hence the effect of the ESA may be less. Evidence shows the positive effects of starting ESA’s four weeks before chemotherapy, with 87% patients achieving a normal haemoglobin (Hb) in 12 weeks.18,19 A cost bene� t analysis of ESA versus blood transfusion would have to be analysed when deciding on local protocols.20
It is also recognised that patients with renal failure may need higher doses of ESA’s, but no speci� c formula is offered in the literature.21
Some controversial evidence has been published about ESA use in other cancer patients that suggests poor survival.22 This may be due to exacerbation of the hypercoaguable state of cancer and venous embolism seems to be implicated but this has not been speci� cally related to the treatment of myeloma.
The European Organisation for research and treatment of Cancer published guidelines in 2007 that recommended the use of ESA’s and a target Hb of 12.0 g/dl. These guidelines are summarised by Aapro and recommend that in cancer therapy ESA’s can a) decrease transfusion needs; (b) sus-tain Hb levels, which is not the case with intermittent trans-fusions; (c) increase quality of life; and (d) ESAs should be used within the guidelines.23
Use of iron therapyThere seems to be little comment in the literature discussing the use of iron in management of anaemia of renal failure and myeloma. On this basis, local protocols of maintaining adequate iron levels remain important in the management of anaemia.
Myeloma and BonesMyeloma cells divide and invade the bone marrow and can attack various sites (hence why myeloma is sometimes called

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
162
multiple myeloma). This can be the skull, spine, hips, ribs or shoulders. Rapid growth of myeloma cells inhibits normal bone-forming cells and damages bone. Osteoclasts normally break down old or worn out bone and work with bone-forming cells to repair bone. In myeloma there is production of substances that activate the osteoclasts resulting in increased activity, causing bone loss and loss of bone repair. Fractures may therefore be an aspect of this disease.
Treatments of Myeloma
When deciding the most appropriate treatment of myeloma several factors should be considered. The ultimate aim of treatment is to control the disease, maximise the quality of life and prolong survival.
In considering therapies the relative risk versus the impact of the therapy should be considered. This risk is in� uenced by age and co-morbidities. It is therefore important that the side effects of the therapy do not out-weigh any potential bene� ts and that strategies are available for dealing with relapse in the younger patient.
There is a range of therapies available, some very new (Thalidomide and Bortezomib) as well as very traditional. The role of the renal nurse is to have some understanding of these therapies in order to support the patient in their time receiving dialysis.
Recent therapies:
Thalidomide and its analogues (e.g. Lenolidamide)
This group of drugs are commonly used in early disease. Their role in maintenance therapy is being evaluated and there is some evidence of it being a successful rescue therapy after � rst chemotherapy.

Myeloma and Renal Failure
163
The optimum dose of the drug varies between studies but seems to range from 200-800 mg. The aim of the treatment seems to be, rather than having an optimal effective dose to have a dose that the patient can tolerate taking for as long as possible. This is because the length of therapy seems signi� cant.24 It is thought that the effectiveness of Thalidomide in myeloma is as a result of it’s anti-angiogenesis (ability to inhibit the growth of new blood cells), its inhibition of tumour cell growth in the bone marrow (speci� cally stromal cells), the alteration of cytokines involved in the growth of myeloma and the stimulation of T-cells from the immune system that help the body attack and destroy tumour cells.25
Newer studies also seem to demonstrate the positive bene� t of this group of drugs being given in combination with steroids to provide higher response rates and lower relative side effects.26 Studies have shown this group of drugs to have a response rate of 30% 27 but when added to dexmethasone this increases to 60%.
The side effects of these drugs are given in Table 3.Table 3: Side effect of Thalidomide type drugs.28
Side Effect Incidence and ActionsNeuropathy 30%- reduce dose or discontinue therapy
Somnolence 60%- give drug at night
Constipation 50%- dietary advice or laxatives
Thrombo-embolism 5-25%-consider anticoagulation in patients with previous emobolism
Rashes 15%
Bortezomib (Velcade)
Velcade is another one of the newer therapies. It has only recently been approved in the USA and UK as � rst line therapy.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
164
In the UK it was approved by the National Institute of Clinical Excellence in 2007.29
Velcade is a proteasome inhibitor. Proteasome are a complex of enzymes found within cells that play a role in the regulation of cell function and growth. Proteasomes break down and clear out proteins. There is some evidence that cancer cells appear to be particularly dependent on proteasomes to grow and survive so if proteasome activity is blocked the cancer cells will undergo programmed cell death (apoptosis).
Velcade is used in newly diagnosed myeloma in combination with traditional treatments (Melphalan and Dexmethasone). It is an intravenous therapy that is given usually twice a week for a period of six to eight weeks. Maintenance therapy may be once a week for a further 4 weeks. Side effects may limit therapy and are described in Table 4.
Table 4: Side Effects of Velcade.30
Side Effect Incidence and Actions
Neuropathy 50%- reduce dose or discontinue therapy
Haematological disturbance
49% Low platelet counts commonly- reduce or withhold dose
GI side effects- nausea and diarrhoea 10-33%. Adjust � uid intake
Pyrexia 29%
Peripheral oedema 20%
Velcade has the highest response rate for newly diagnosed therapies.
Traditional therapies for myelomaA range of therapies that may be considered lower risk can be used for treatment and may delay the progression of the

Myeloma and Renal Failure
165
disease. The choice of therapy lies in the balance of risk to the patient. Steroids (usually Dexmethasone) are shown to increase response rates in many types of cancer. Steroids may be used alone or in combination with a drug called Melphalan.
Melphalan is an alkylating agent that works by disrupting the cancer cells DNA and preventing it dividing. Melphalan is available as an oral and intravenous therapy although side effects are more pronounced with IV therapy. The side effects are similar to those experienced with many chemotherapy treatments and include anaemia, fatigue, nausea, diarrhoea and hair loss. Melphalan is a higher risk treatment for patients with renal failure. It needs dose adjustment and can cause severe bone marrow depression. This means that it is rarely used for dialysis patients.31
A more commonly used treatment in patients with renal failure may be Vincristine, Adriamycin and Dexmethasone (VAD), a combination chemotherapy but this is being somewhat superseded by Thalidomide agents.
Intensive Treatment for myelomaIntensive treatment for myeloma is rarely available for patients with renal failure, but not totally contra-indicated. With this treatment the bone marrow is destroyed by high dose chemotherapy and then a transplant of stem cells into the bone marrow takes place. These may be the patients own stem cells (autologous transplant) or those of a matched donor (allogenic transplants). Whilst the side effects related to this therapy can be high, if successful there can be long periods of remission.
Outcome of treatmentsWhichever treatment is used it is important to be able to quantify the success of the treatment. Table 5 therefore provides details
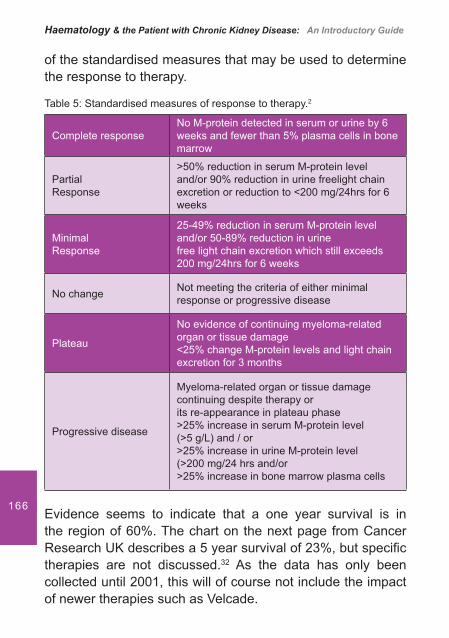
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
166
of the standardised measures that may be used to determine the response to therapy.
Table 5: Standardised measures of response to therapy.2
Complete responseNo M-protein detected in serum or urine by 6 weeks and fewer than 5% plasma cells in bone marrow
PartialResponse
>50% reduction in serum M-protein level and/or 90% reduction in urine freelight chain excretion or reduction to <200 mg/24hrs for 6 weeks
MinimalResponse
25-49% reduction in serum M-protein level and/or 50-89% reduction in urinefree light chain excretion which still exceeds 200 mg/24hrs for 6 weeks
No change Not meeting the criteria of either minimal response or progressive disease
Plateau
No evidence of continuing myeloma-related organ or tissue damage<25% change M-protein levels and light chain excretion for 3 months
Progressive disease
Myeloma-related organ or tissue damage continuing despite therapy orits re-appearance in plateau phase>25% increase in serum M-protein level (>5 g/L) and / or>25% increase in urine M-protein level (>200 mg/24 hrs and/or>25% increase in bone marrow plasma cells
Evidence seems to indicate that a one year survival is in the region of 60%. The chart on the next page from Cancer Research UK describes a 5 year survival of 23%, but speci� c therapies are not discussed.32 As the data has only been collected until 2001, this will of course not include the impact of newer therapies such as Velcade.
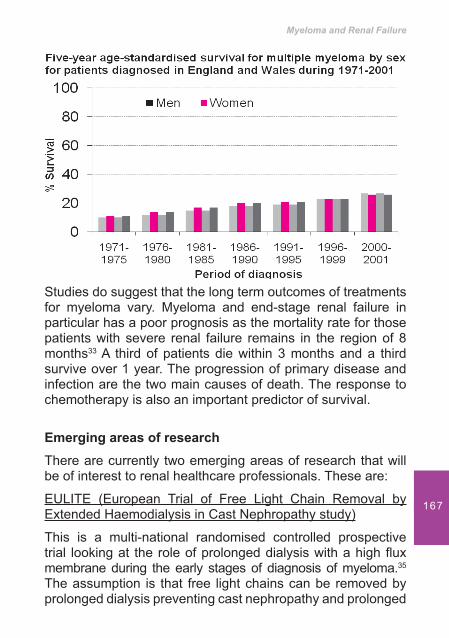
Myeloma and Renal Failure
167
Studies do suggest that the long term outcomes of treatments for myeloma vary. Myeloma and end-stage renal failure in particular has a poor prognosis as the mortality rate for those patients with severe renal failure remains in the region of 8 months33 A third of patients die within 3 months and a third survive over 1 year. The progression of primary disease and infection are the two main causes of death. The response to chemotherapy is also an important predictor of survival.
Emerging areas of researchThere are currently two emerging areas of research that will be of interest to renal healthcare professionals. These are:
EULITE (European Trial of Free Light Chain Removal by Extended Haemodialysis in Cast Nephropathy study)
This is a multi-national randomised controlled prospective trial looking at the role of prolonged dialysis with a high � ux membrane during the early stages of diagnosis of myeloma.35 The assumption is that free light chains can be removed by prolonged dialysis preventing cast nephropathy and prolonged

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
168
renal failure. The study uses a leaky dialyser in order to clear immunoglobulin whilst the � rst cycles of chemotherapy are given. The regime is intense for patients and involves alternate day dialysis for up to 10 hours. This regime continues for up to 22 days.
Gene therapy
Gene therapy is being explored for myeloma and there are studies that are exploring the use of radiation therapy to eliminate myeloma.
Nursing care of patients with MyelomaEducation
Myeloma is not yet a curable disease and may well follow a relapsing and remitting pathway. Symptoms and problems range from bone pain, immunological complications and anaemia to renal failure. Juggling chemotherapy and dialysis is an enormous challenge, especially alongside the physical and psycho-social impact of the disease. The nurse’s role as a key consistent contact in the patients care is therefore vital to provide education, communication and support.
To ensure nurses are equipped to do this resources are vital. An example of such a resource is Myeloma UK that have set up a training course called ‘Magic’. This provides nurses with up to date information about treatments and support, with information on areas from active treatment to palliative care. The course can be accessed via the intranet (www.myeloma.org.uk).
Symptom control
Pain
A systematic approach to analgesia must form part of the integrated care plan of the myeloma patient. This is because

Myeloma and Renal Failure
169
pain arising from the skeletal complications of myeloma is the commonest presenting symptom and results from pathologies such as vertebral compression/collapse from osteoporosis. It will arise in up to 80% of patients during the course of their disease.36 The pain may be generalised or local.
Chemotherapy is also a key component of pain management in myeloma as it is aimed at the underlying pathological process. Pain improves with the response to chemotherapy.37
An active approach to pain management is required that encompasses systemic analgesia and the use of local measures. Strategies for managing pain are therefore;
Systemic Analgesia. These may include non-opoid • preparations, such as Paracetamol, weak opioids such as Co-codamol or Codeine or strong opioids such as Morphine. In renal failure, synthetic opioids such as Oxycodeine and Fentanyl are useful. Non- steroidal analgesics should be avoided because of the potential effect on renal function and risk of gastric irritationRadiotherapy. Local radiotherapy is effective for pain • relief for skeletal disease and may also palliate soft tissue diseaseVertebroplasty. This involves the percutaneous injec-• tion, under local anaesthetic and light sedation, and using imaging, of biomaterial into the vertebral body (several vertebrae can be treated simultaneously). The injection allows local pain relief and bone strengthening but will not restore vertebral height
HypercalcaemiaHypercalcaemia, symptomatic or asymptomatic, occurs in up to 30% of myeloma patients and typically occurs in the presence of active disease. Prompt recognition and treatment minimises renal damage. Management requires rehydration with close monitoring of � uid balance and renal function combined with

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
170
intravenous bisphosphonate therapy. Lower calcium can also be achieved by altering dialysate � uid.
Infection
The risk of infection is increased in myeloma at diagnosis or during the course of disease due to the disease and its therapy. During chemotherapy, variable degrees of neutropaenia may occur. Prolonged and high-dose steroids also compromise defences against fungal and viral infection.
ConclusionsThe needs of the patient with myeloma are great and extend from active disease management, optimal renal management as well as psychological support and symptom control. The renal nurse needs a good understanding of the facets of this disease if they are to provide high quality, effective care.

Myeloma and Renal Failure
171
Referenceshttp://www.myeloma.org.uk1. UK Myeloma Forum and Nordic Myeloma Study Group (2005). 2. Guidelines on the diagnosis and management of multiple myeloma (accessed on British Committee for standards in Haematology (BCSH) website. http//wwwbcshguidelines.com/page411.Jemal A, Siegal R, Ward E, Hao Y, Xu J, Murray T, Thun M. Cancer 3. Statistics. Cancer Journal Clinics 2008; 58(2): 71-96.Riedal D, Pottern L. The Epidemiology of Multiple Myeloma. 4. Haematology Oncology Clinics of North America 1992; 6(2): 225-47.Greipp P. Advances in the diagnosis and management of Myeloma. 5. Seminars Haematology 1993; 29:24-45.Greipp P, Miguel J, Durie B, Crowley J. International Staging System 6. for multiple Myeloma. Journal of Clinical Oncology 2005; 23:3412-3420.Goldschmidt H, Lannert H, Bommer J, Ho A. multiple Myeloma and 7. Renal Failure. Nephrology Dialysis and Transplantation 2000; 15: 301-304.Ronco P, Aucouturier P, Mougenot B. Kidney involvement in plasma 8. cell dyscrasias. Davison A, Cameron J, Grunfeld JP, Ponticelli C, Ritz E, Winearls C, Van Ypersele C. (eds). Oxford Textbook of Clinical Nephrology. (3rd ed) 2005 p 714. Oxford University Press.Winearls C. Nephrology Forum. Acute Myeloma Kidney. 9. Kidney International 1995; 1347-1361Misani R. Management of Myeloma Kidney. 10. American Journal Of Kidney Disease 1987; 10: 28-33.Littlewood T, Collins G. Granulocyte and Erythropoietic stimulating 11. proteins after high-dose chemotherapy for myeloma. Bone marrow transplantation 2007: 40:1147-1155.Ludwig H, Rai K, Blade J, Dammacco F, Degos L, Itri L et al. 12. Management of disease- related anaemia in patients with multiple myeloma or chronic lymphocytic leukaemia:epoetin treatment recommendations. Haematology Journal 2002; 3:121-130.Barlogie B, Beck T. Recombinant human erythropoietin and the 13. anaemia of multiple myeloma. Stem Cells 1993; 11:88-94.Littlewood T, Bajetta E, Nortier J, Vercammen E, Rapoport B. Effects 14. of epoetin alfa on hematologic parameters and quality of life in cancer patients receiving non-platinum chemotherapy: results of a randomised, double-blind, placebo controlled trial. Journal of Clinical Oncology 2001; 19:2865-74. Littlewood T. Tired of being tired- why we should treat cancer related 15. anaemia. Hospital Pharmacist 2004; 11(8): 337 338.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
172
Birgegard G. Managing anaemia in lymphoma and multiple myeloma. 16. Therapeutic Clinical Risk Management 2008; April; 4(2): 527-539.Martino M, Oliva E, Console G, Stelitano C, Fujo M, Messina G et 17. al. Administration of recombinant human erythropoietin alpha before autologous stem cell transplantation reduces transfusion requirement in multiple myeloma patients. Support Care Cancer 2005; 13:182-87.Baron F, Frere P, Fillet G, Beguin Y. Recombinant human erythropoietin 18. therapy is very effective after autologous peripheral blood stem cell transplant when started soon after engraftment. Clinical Cancer Research 2003; 9:5566-5572.Dammacco F, Luccarelli G, Prete M, Sivestris F. The role of 19. recombinant human erythropoietin alpha, chronic anaemia in the treatment of multiple myeloma. Rev Clin Hematol Suppl 2002.Sraus D, Testa M, Sarokhan B et al. Quality of life and health bene� ts 20. of early treatment of mild anaemia: a randomised trial of epoetin alfa in patients receiving chemotherapy for hematologic malignancies. Cancer 2006; 107:1909- 1917.http://www.kidney.org/professionals/kdoqi/guidelines - updates21. Bohlius J, Wilson J, Seiden� eld J, Piper M, Schwarzer G, Sandercock 22. et al. Recombinant human erythropoietins and cancer patients: Updated Meta-analysis of 57 studies including 9353 patients. Journal of the National Cancer Institute 2006; 98(10): 708-714.Aapro. Update on EORTC Guidelines and Anaemia Management with 23. Erythropoiesis Stimulating Agents 2007; The Oncologist; 13 (3): 33-36.Abdalla S, Mahmud S. Thalidomide in relapsed or refractory multiple 24. myeloma:how much and for how long? Leukaemia and Lymphoma 2003; 44:989-991.http://www.multiplemyeloma.org/treatments. Multiple myeloma re-25. search foundation.http://www.multiplemyeloma.org/treatments. Multiple myeloma re-26. search foundation. Velcade.Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P et 27. al. Anti tumour activity of Thalidomide in refractory multiple myeloma. New England Journal of Medicine 1999; 341:1565-1571.UK Myeloma Forum and the Nordic Myeloma Study Group: Guidelines 28. on the diagnosis and Management of Multiple Myeloma 2005. British Journal of Haematology 2006; p23.National Institute for Clinical Excellence (2007). Guidance for the use 29. of Velcade is a win-win situation for multiple myeloma patients and the NHS.

Myeloma and Renal Failure
173
San Miguel J, Schlag R, Nuriet K, Khuageva et al. Botezomib plus 30. Melphalan and Prednisolone for initial treatment of multiple myeloma. New England Journal of Medicine 2008; 359: 906-917.Goldschmidt H, Lannert H, Bommmer J, Ho A. Multiple myeloma 31. and renal failure. Nephrology Dialysis and Transplantation 2000; 15:301-304.http://info.cancerresearchuk.org/cancerstats/types/multiplemyeloma/32. survival.Irish A, Winearls C, Littlewood T. Presentation and survival of patients 33. with severe renal failure and myeloma.Quarterly Journal of Medicine 1997; 90(12): 773-780.Ronco P, Aucouturier P, Mougenot B. Kidney involvement in plasma 34. cell dyscrasias. In Davison A, Cameron J, Grun� eld J P, Ponticelli C, Ritz E, Winearls C et al. editors. Oxford Textbook of Clinical Nephrology. 3rd ed. Oxford University Press. 2005; p217.http://www.freelite.co.uk35. Kyle R. Multiple myeloma: Review of 869 cases. Mayo Clinic 36. Proceedings 1975; 50:29-40.Mcloskey E, MacLennan I et al. A randomised trial of the effect of 37. Clodronate on skeletal morbidity in multiple myeloma. British Journal of Haematology 1998; 100:317-325.


Myelodysplasia
The Bare Facts
175

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
176
Learning outcomes• To understand the physiology and pathophysiology of
myelodysplasia (MDS)• Be aware of the different classi� cations and sub types
of MDS• Identify how MDS impacts on quality of life• Know what investigations are needed to identify MDS• Discuss the treatments available to treat MDS• Be aware of the nephrology nurse’s role in relation to
the patient with MDS
IntroductionMyelodysplasia (MDS) is an abnormality of the bone marrow that inhibits its ability to produce healthy blood cells. All myeloid lineages are affected by MDS although MDS primarily affects red cell production. There are different sub-types of MDS and they are classi� ed according to peripheral blood and bone marrow � ndings. The World Health Organisation de� nes MDS as; “A group of clonal haematopoietic stem cell disorders characterized by cytopenias, dysplasia in one or more of the major myeloid cell lines, ineffective haematopoiesis and an increased risk of development of acute myeloid leukaemia”.1
Sideroblastic anaemia which is abbreviated to RARS (refractory anaemia with ring sideroblasts) usually occurs as part of myelodysplastic syndrome and can occasionally evolve into haematological malignancies. This is where the developing red cells show an internal ring of iron granules. These cells are called sideroblasts.2 Sideroblasts occur because the body has iron available but cannot incorporate

Myelodysplasia – The Bare Facts
177
it into haemoglobin. There is therefore a failure to completely form haem molecules. This leads to deposits of iron in the mito-chondria within the cell that form a ring around the nucleus of the developing red blood cell.2 Microscopically, sideroblasts are seen as nucleated erythrocytes with granules of iron in their cytoplasm.3 This type of MDS is generally associated with a better prognosis as only 1-2% of patients go on to develop acute myelogenous leukaemia (AML).4
Medically known as MDS, myelodysplasia typically develops slowly and is more prevalent in people over 65 years of age, though a ‘high risk’ (see classi� cation of MDS’ section) MDS will progress to AML quite quickly in some cases. The median age at diagnosis is between 60 and 75 years, few patients are less than 50, and it is rare in children. Males are slightly more commonly affected than females.
Physiology / Normal HaemopoiesisWithin the normal bone marrow the process for stem cell production is that cells will either differentiate into myeloid or lymphoid precursors.
Lymphoid precursors become lymphocytes. Lymphocytes are white blood cells that are responsible for the body’s immune defences. Therefore lymphocytes develop from a common lymphoid progenitor cell and mediate the functions of the adaptive immune system. There are two main types of lymphocytes: B cells and T cells. The principal functions of B cells are to make antibodies against antigens, perform the role of Antigen Presenting Cells (APCs) and eventually develop into memory B cells after activation by antigen interaction. B cells are an essential component of the adaptive immune system. T cells play a central role in cell-mediated immunity. They can be distinguished from other lymphocyte types, such as B cells, by the presence of a special receptor on their cell surface called T cell receptors (TCR). The abbreviation T,

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
178
in T cell, stands for thymus, since this is the principal organ responsible for the T cell’s maturation.
Myeloid precursors develop into red cells or platelets. Red cells contain the haemoglobin that transports oxygen around the body. Platelets are an irregular, disc-shaped element in the blood that assists in blood clotting.
When stem cells are evolving through their different stages they are called blasts. The earliest recognizable myeloid cell in the bone marrow is the myeloid blast. Blasts can be measured as a percentage and should not exceed 5% in normal bone marrow. If the blast percentage is greater than 5% it is suggestive of MDS as excessive numbers of blasts are observed in MDS (5-15% of non erythroid bone marrow cells). In AML >20% of non erythroid marrow cells are seen.
Pathophysiology of MDSThe pathophysiology of MDS involves many individual bio-logical pathways and accounts for the wide spectrum of symptoms, the variability in the severity of the illness and the potential for progression. The presence of detectable cytogenetic abnormalities is 40-70% of patients with primary MDS and 80% with secondary MDS.5 Causes of secondary MDS have been related to chemotherapy and radiotherapy (see: Causes and Categorisation of MDS.) The underlying pathophysiology of MDS is not entirely understood. The development of cytopenias is attributed to an enhanced aptosis (programmed cell death) of the cells despite a characteristically hyperplastic bone marrow.6 A hyperplastic bone marrow has a proliferation of cells beyond that which is ordinarily seen.
EpidemiologyThe prevalence of MDS is unknown, in part due to under diagnosis and the lack of no registry collecting data. In the

Myelodysplasia – The Bare Facts
179
United States it has been suggested that there are between 10 and 20000 new cases each year, with the incidence of MDS rising as the population ages.
Aetiology (causes) and Categorisation of MDSIn MDS the majority of cases (60–70%) will have no de� ned aetiology. These patients will be diagnosed with primary or de novo myelodysplastic syndrome. In a small number of peo-ple, a myelodysplastic syndrome occurs following an expo-sure to benzene, occupational chemicals, radiation treatment and chemotherapy.7 There have also been associations with tobacco, alcohol, viral infections and autoimmune disorders. This is known as therapy-related or secondary myelodysplas-tic syndrome. There are also congenital disorders associated with MDS such as Fanconi anaemia and dyskeratosis con-genital.
The pathogenesis of MDS has been divided into 5 categories as shown in Table 1.5
Classi� cation of MDSClassi� cation systems are used to organise MDS into subtypes. This aims to aid prognosis and help with clinical decision making in regards to treatment. Initially classi� ed by the French American British (FAB) system (table 2)6, MDS sub types have recently been re-classi� ed by the World Health
Table 1: The Five Categories of Myelodysplasia.Cellular damage related to toxic environmental exposure and cellular aging
Genetic alterations due to cytogenetic and epigenetic abnormalities
Changes in bone marrow microenvironment
Dysregulation of the immune system
Altered cell cycle regulation and blocked differentiation
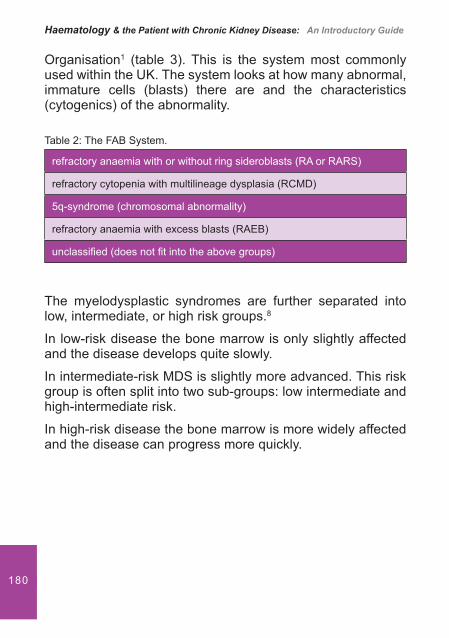
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
180
Organisation1 (table 3). This is the system most commonly used within the UK. The system looks at how many abnormal, immature cells (blasts) there are and the characteristics (cytogenics) of the abnormality.
The myelodysplastic syndromes are further separated into low, intermediate, or high risk groups.8
In low-risk disease the bone marrow is only slightly affected and the disease develops quite slowly.
In intermediate-risk MDS is slightly more advanced. This risk group is often split into two sub-groups: low intermediate and high-intermediate risk.
In high-risk disease the bone marrow is more widely affected and the disease can progress more quickly.
Table 2: The FAB System.
refractory anaemia with or without ring sideroblasts (RA or RARS)
refractory cytopenia with multilineage dysplasia (RCMD)
5q-syndrome (chromosomal abnormality)
refractory anaemia with excess blasts (RAEB)
unclassi� ed (does not � t into the above groups)
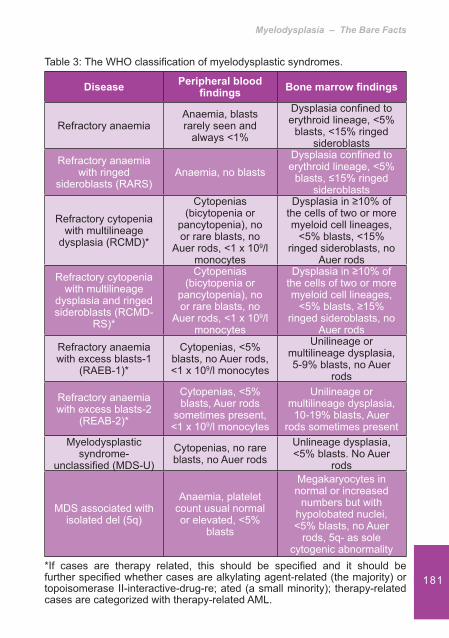
Myelodysplasia – The Bare Facts
181
Table 3: The WHO classi� cation of myelodysplastic syndromes.
Disease Peripheral blood � ndings Bone marrow � ndings
Refractory anaemiaAnaemia, blasts rarely seen and
always <1%
Dysplasia con� ned to erythroid lineage, <5%
blasts, <15% ringed sideroblasts
Refractory anaemia with ringed
sideroblasts (RARS)Anaemia, no blasts
Dysplasia con� ned to erythroid lineage, <5%
blasts, �15% ringed sideroblasts
Refractory cytopenia with multilineage
dysplasia (RCMD)*
Cytopenias (bicytopenia or
pancytopenia), no or rare blasts, no
Auer rods, <1 x 109/l monocytes
Dysplasia in �10% of the cells of two or more myeloid cell lineages,
<5% blasts, <15% ringed sideroblasts, no
Auer rodsRefractory cytopenia
with multilineage dysplasia and ringed sideroblasts (RCMD-
RS)*
Cytopenias (bicytopenia or
pancytopenia), no or rare blasts, no
Auer rods, <1 x 109/l monocytes
Dysplasia in �10% of the cells of two or more myeloid cell lineages,
<5% blasts, �15% ringed sideroblasts, no
Auer rodsRefractory anaemia with excess blasts-1
(RAEB-1)*
Cytopenias, <5% blasts, no Auer rods, <1 x 109/l monocytes
Unilineage or multilineage dysplasia, 5-9% blasts, no Auer
rods
Refractory anaemia with excess blasts-2
(REAB-2)*
Cytopenias, <5% blasts, Auer rods
sometimes present, <1 x 109/l monocytes
Unilineage or multilineage dysplasia, 10-19% blasts, Auer
rods sometimes presentMyelodysplastic
syndrome-unclassi� ed (MDS-U)
Cytopenias, no rare blasts, no Auer rods
Unlineage dysplasia, <5% blasts. No Auer
rods
MDS associated with isolated del (5q)
Anaemia, platelet count usual normal or elevated, <5%
blasts
Megakaryocytes in normal or increased
numbers but with hypolobated nuclei, <5% blasts, no Auer
rods, 5q- as sole cytogenic abnormality
*If cases are therapy related, this should be speci� ed and it should be further speci� ed whether cases are alkylating agent-related (the majority) or topoisomerase II-interactive-drug-re; ated (a small minority); therapy-related cases are categorized with therapy-related AML.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
182
Signs and symptoms of MDSPatients with MDS are often asymptomatic and may be referred for further investigation due to the � nding of one or more cytopenia on routine blood testing. Other patients may present with a gradual onset of symptoms attributable to anaemia, thrombocytopenia or neutropenia.9 As the signs and symptoms of MDS are non- speci� c those patients that are symptomatic at presentation tend to describe a gradual onset. Indeed the main signs of myelodysplasia syndromes include anaemia, repeated infections and bruising or bleeding. Anaemia can result in other symptoms that include fatigue and shortness of breath. Thrombocytopenia can lead to pinpoint-sized red spots just beneath the skin caused by the bleeding (petechiae).10 Systemic symptoms such as fever and weight loss are uncommon and generally present as late manifestations of the disease or its attendant complications. A small number of people will have an enlarged spleen.11 The spleen is part of the body’s defence system against infection. Infection remains the principal cause of death in patients with MDS. Although fungal, viral, and bacterial infections can occur they are rare in the absence of concurrent administration of immunosuppressive agents.12
Quality of Life (QOL)The QOL for people with MDS has been studied. Within Steenma et al’s study13 patients were described as typical of MDS patients in terms of demographics, blood counts, and disease subtype. The study found that the patients reported high levels of excessive fatigue and recorded poor scores on QOL assessments such as the Functional Assessment of Cancer Therapy-Anaemia (FACT-An) and the Brief Fatigue Inventory (BFI). It was also noted that the patients’ debilitating fatigue correlated with a low haemoglobin level and that fatigue was associated with signi� cant impairment of both health-related QOL and the ability to work or participate in desired activities.

Myelodysplasia – The Bare Facts
183
Investigations to determine the presence of MDSAlthough a sub-set of patients can have dysplastic features that are solely con� ned to the erythroid cell line, others will have abnormalities in all of them. For these patients there will also be neutrophil and platelet abnormalities.
Investigations to diagnose MDS are therefore:Full blood count (FBC) to measure the number of all • blood cells. The FBC result will show a low white cell count if leucopoenia is present, a low red blood cell count if anaemia is present and a low platelet count if thrombocytopenia is presentMicroscopic review of the red blood cells because if • the red blood cells have basophilic stippling (minute indentations on the cells) this is highly suggestive of MDSReview of the white blood cells to see if the white blood • cells are morphological (the nucleus is misshapen) as this may indicate MDSA bone marrow biopsy. Bone marrow may be obtained • from the posterior pelvic bones. This involves taking a sample of bone marrow from the back of the pelvis or occasionally the sternum.4 The bone marrow sample is taken under a local anaesthetic and a tiny sample of the marrow is aspirated into a syringe. Sometimes a small core of marrow is also needed (a trephine biopsy). A trephine biopsy is usually taken from the back of the hipbone. The bone marrow aspirate and trephines are examined for cytology and histology respectively to see if it contains any abnormal cellsCytogenetic tests. The tests on the blood and bone • marrow sample will include a chromosome analysis to look for any particular changes in the chromosomes. These tests, known as cytogenetic tests can identify particular cytogenic abnormalities associated with high, intermediate and low risk MDS, and in the case of 5q-, can guide therapy

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
184
Biochemistry blood analysis. Secondary to ineffective • erythropoesis a mild increase in bilirubin and a mild increase in lactate dehydrogenase (LDH) may be present 14
Measurement of haptoglobins. Haptoglobin is a • protein and in blood plasma haptoglobin binds free haemoglobin (Hb) released from erythrocytes. The haptoglobin-hemoglobin complex is removed by the reticuloendothelial system (mostly the spleen). In clinical settings the haptoglobin assay is used to screen for and monitor hemolytic anemia. In MDS the haptoglobin level may decrease due to ineffective erythropoesis 14
Treatment options for MDSTreatment options for MDS are either supportive care or treatments to cure the disease.
Supportive CareIn MDS there are a number of supportive care options that are used to control symptoms, prevent complications and improve quality of life. They include the following:
Red Blood Cell TransfusionsThe mainstay for the management of myelodysplastic syndromes (MDS) is supportive therapy with red blood cell (RBC) transfusions to improve the patient’s quality of life. This is because RBC transfusions enable adequate tissue oxygenation and increase haemoglobin levels, improve fatigue, and improve the physical and intellectual activity of patients. Up to 90% of patients with MDS will receive RBC transfusions during the course of their disease, and many will become chronically dependent on transfusions to manage their anaemia. Blood transfusions are problematic in that they can lead to an accumulation of excess iron that, in turn, can lead to iron overload if multiples of blood transfusions are given. It

Myelodysplasia – The Bare Facts
185
is therefore recommended that the iron burden of transfused patients be monitored regularly and that iron chelation therapy be considered to maintain serum ferritin levels of <1000 ng/mL. While increased ferritin is associated with a decreased survival rate, it remains unclear if a reduction in the ferritin level leads to improved survival.5
It should be noted that even after allogeneic stem cell transplantation RBC transfusion might be required.15
Platelet transfusionsPlatelet transfusions are needed when the individual has a low platelet count/thrombocytopenia.
AntibioticsAs MDS affects lymphocytes production leading to neutropenia there is an increased susceptibility to infections. Antibiotics are needed to treat existing and new infections. Depending on the organism grown a variety of antibiotics maybe used. For patients who are neutropenic, extra precautions should be advised. This includes the avoidance of crowds and of anyone with symptoms of transmissible infection. Strict hand washing and good general hygiene are also of importance.
Hematopoietic Growth Factors Research has shown that it may be possible to boost the number of healthy red and white cells by administering growth factors. Growth factors such as recombinant human granulocyte colony-stimulating factor (G-CSF), recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) and ESA’s (erythropoietin stimulating agents) may be used in MDS. There is evidence that using a combination of therapies may be more effective than ESA alone.16,17 However, whilst the drug Granulocyte colony- stimulating factor (G-CSF), usually given subcutaneously can boost the number of white cells it may not be effective for everyone who has myelodysplasia.18

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
186
Epigenetic Modi� cationA number of drugs that target epigenetic changes have been reported.19,20
Erythropoietin Stimulating Agents (ESA’s)As a result of the pathophysiology of MDS all patients will be anaemic. In addition to this the anaemia will be exacerbated if the patient is receiving chemotherapy. This is because anaemia is a common negative side effect induced by chemotherapy10 as patients will have innately low erythropoietin (EPO) levels caused by cytotoxic suppressive effects on erythroid progenitor cells or through renal tubular effects on erythropoietin production.21 In such cases the EPO assay is measured to determine the endogenous EPO level. If this is low the patient can be treated ESA’s. Table 4 shows a validated decision model that uses the baseline EPO assay and previous blood transfusion requirements to predict the patient response to ESA.17
ESAs improve symptoms of anaemia, increase haemoglobin levels and decrease transfusion requirements in patients with low risk MDS.8 The recommended ESA dose requirement for individuals who have MDS is 40,000iu to 60,000iu one to three times each week of epoetin alfa/beta or 150�g to 300 �g/wk of darbepoetin alfa. All patients should receive ESAs for at least eight weeks before a decision is made regarding effectiveness.15
Table 4: Predictor for Patient Response to ESA’s.
Predictive Model for Response to ESA’s in MDS
Patient Criteria Probability of Response
Transfusion need <2 units/month and serum EPO <500 74%
Only one of the above criteria met 23%
Neither criteria met 7%

Myelodysplasia – The Bare Facts
187
Immune TherapyImmunomodulatory agents such as Cyclosporine and Lenalidomide have been used in the treatment of MDS. 21,22 The reason why immunosuppressant drugs are used is that they can suppress the immune system and may help to increase the number of blood cells produced in the bone marrow. They may also help to control the progression of MDS. The immunosuppressant drugs that are most commonly used are cyclosporine or anti-thymocyte globulin (ATG).23
ChemotherapyChemotherapy is the use of cytotoxic drugs. Administered either as tablets, capsules or intravenously it is given to help control the disease and any symptoms. In MDS the treatment may involve just one type of chemotherapy or a combination of drugs.
Treatments that may cure MDSBone marrow transplantationCurrently the only cure for MDS, stem cell transplants are generally only used for younger patients, as the treatment is very intensive and the risks involved with a transplant increase as you get older. The treatment usually involves high doses of chemotherapy or radiotherapy being given to destroy the unhealthy bone marrow. Healthy stem cells are then given intravenously into the blood stream whereupon they make their way into the bone marrow and start to produce red and white cells and platelets again.
MDS and Chronic Kidney Disease (CKD)Whilst patients with CKD may present with MDS, very little literature exists relating the kidney to MDS. One paper reports on a case of glomerulonephritis and MDS suspected to be as a result of angiogenesis stimulated through the over production of cytokines through an unknown mechanism.24 Other case reports have linked MDS to nephropathy and glomerulonephritis.25
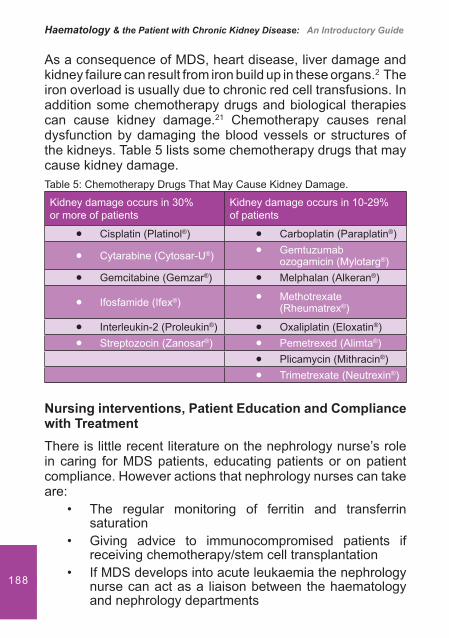
Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
188
As a consequence of MDS, heart disease, liver damage and kidney failure can result from iron build up in these organs.2 The iron overload is usually due to chronic red cell transfusions. In addition some chemotherapy drugs and biological therapies can cause kidney damage.21 Chemotherapy causes renal dysfunction by damaging the blood vessels or structures of the kidneys. Table 5 lists some chemotherapy drugs that may cause kidney damage.
Nursing interventions, Patient Education and Compliance with TreatmentThere is little recent literature on the nephrology nurse’s role in caring for MDS patients, educating patients or on patient compliance. However actions that nephrology nurses can take are:
The regular monitoring of ferritin and transferrin • saturationGiving advice to immunocompromised patients if • receiving chemotherapy/stem cell transplantationIf MDS develops into acute leukaemia the nephrology • nurse can act as a liaison between the haematology and nephrology departments
Table 5: Chemotherapy Drugs That May Cause Kidney Damage.
Kidney damage occurs in 30% or more of patients
Kidney damage occurs in 10-29% of patients
Cisplatin (Platinol�� ®) Carboplatin (Paraplatin�� ®)
Cytarabine (Cytosar-U�� ®) Gemtuzumab ��ozogamicin (Mylotarg®)
Gemcitabine (Gemzar�� ®) Melphalan (Alkeran�� ®)
Ifosfamide (Ifex�� ®) Methotrexate ��(Rheumatrex®)
Interleukin-2 (Proleukin�� ®) Oxaliplatin (Eloxatin�� ®)Streptozocin (Zanosar�� ®) Pemetrexed (Alimta�� ®)
Plicamycin (Mithracin�� ®) Trimetrexate (Neutrexin�� ®)

Myelodysplasia – The Bare Facts
189
The implementation and the use of hand held patient • plans could provide a useful tool to bene� t the communication between different departmentsNurses should also act as a resource for patients by • providing patient education
In addition to this the nephrology nurse must be aware of the issues associated with thrombocytopenia in the patient with kidney disease. In particular the presence of thrombocytopenia will have an impact on the use of anticoagulant therapies used in haemodialysis treatments. Staff should also be educated on the risks of both blood and platelet transfusion.
The use of desferrioxamine brings with it extra challenges for the nurse and this has been discussed within the chapter on Thalasaemia.
Conclusion
Myelodysplasia is a disorder of the bone marrow and occurs with increasing age. Most patients will develop anaemia. Leukopenia and thrombocytopenia are also common. In most cases no cause for the disease developing can be identi� ed. Treatment is determined by the classi� cation of the myelodysplagia syndrome though most cases of MDS are treated with supportive therapies. The median survival age is only three years26 though those patients with RA, RARS and MDS associated with del (5q) are in the favourable risk group. Most patient’s die from either infection or haemorrhage and one third of patients will go on to develop acute myeloid leukaemia.26 It is likely that at some point in your nursing role you will come into contact with a patient who has MDS. By gaining knowledge and having a better understanding of MDS you will be able to provide a more holistic approach to their care.

Haematology & the Patient with Chronic Kidney Disease: An Introductory Guide
190
References1. Classi� cation of Tumours of Haematopoietic and Lymphoid Tissues.
Published by the World Health Organisation. October 2008. 4th edition. Chapter 3, p160.
2. Oliver G, Grundy M, Bratt-Wyton, R. Nursing in Haemotological Oncology. 2000. Saunders (W.B) Co Ltd. ISBN 978-0702023231. p 21-30.
3. Papadakis, Maxine A, Tierney, Lawrence M, McPhee, Stephen J. (2005). Sideroblastic Anemia. Current Medical Diagnosis & Treatment. 2006. McGraw-Hill Medical. ISBN 0-07-145410-1. p 427.
4. Longmore, Wilkinson, Rajagopalan. Oxford Handbook of Clinical Medicine. 6th Edition. Oxford University Press. 2004. Section 625.
5. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classi� cation of the myelodysplastic syndromes. British Journal of Haematology. 1982; 51 (2): 189-199.
6. Myelodyspastic Syndrome.The Leukaemia and Lymphoma society. White Plains, New York. 2001. p 6.
7. Fontenay-Roupie M, Bouscary D, Guesnu M et al. Ineffective erythropoiesis in myelodysplastic syndromes: correlation with Fas expression but not with lack of erythropoietin receptor signal transduction. British Journal of Haematology. August 1999; 106/2: 464-73.
8. Warlick, Smith. Myelodysplastic syndromes: review of pathophysiology and current novel treatment approaches. Current Cancer Drug Targets. September 2007; 7/6:541-58.
9. Komrokji RS, Bennett JM. The clinical implications of the World Health Organization’s classi� cation of myelodysplastic syndromes. Current Hematology Reports. May 2005; 4/3: 175-81.
10. Hoffbrand, Pettit, Moss. Essential Haematology. Blackwell Scienti� c Publications; 2006. Chapter 14. p182 -187.
11. The Myelodyspastic Syndromes. www.cancerbackup.org.uk. Retrieved 24/04/2009. Health professionals section, search ‘myelodysplastic syndromes’.
12. Meyers C.A., Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer. 2005; 104/4: 788-93.
13. Steensma DP, Heptinstall KV, Johnson VM et al. Common troublesome symptoms and their impact on quality of life in patients with myelodysplastic syndromes (MDS): results of a large internet-based survey. Leukemia Research. May 2008; 32/5:691-8.

Myelodysplasia – The Bare Facts
191
14. Matthes T, Beris P. Sideroblastic anaemias. In: Beaumont C, Beris P, Beuzard Y, Brugnara C (eds). Disorders of iron homeostasis, erythrocytes, erythropoiesis. 2006, chapter 21 European School of Haematology, France.
15. Platzbecker U, Bornhauser M, Germing U et al. Red blood cell transfusion dependence and outcome after allogeneic peripheral blood stem cell transplantation in patients with de novo myelodysplastic syndrome. Biology of Blood & Marrow Transplantation. 2008; 14/11:1217-25.
16. Moyo V, Lefebvre P, Duh et al. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Annals of Hematology. 2008; 87/7: 527-36.
17. Park S, Grabar S, Kelaidi C et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. January 2008; 111/2: 574-82.
18. Koppel A, Schiller G. Myelodysplastic syndrome: an update on diagnosis and therapy. Current Oncology Reports. 2008; 10/5: 372-8.
19. Wijermans PW, Lubbert M, Verhoef G et al. An epigenetic approach to the treatment of advanced MDS; the experience with the DNA
20. Negrin RS, Haeuber DH, Nagler A, et al, 1989: Treatment of myelodysplastic syndromes with recombinant human granulocyte colony-stimulating factor. A phase I-II trial. Ann Intern Med 1989; 110:976-984.
21. Wijermans PW, Lubbert M, Verhoef G et al. An epigenetic approach to the treatment of advanced MDS; the experience with the DNA demethylating agent 5-aza-2’-deoxycytidine (decitabine) in 177 patients. Annals of Hematology. 2005; 84 Suppl 1: 9-17.
22. Chen S, Jiang B, Da W et al. Treatment of myelodysplastic syndrome with cyclosporin A. International Journal of Hematology. 2007; 85/1; 11-7.
23. Galili N, Raza A. Immunomodulatory drugs in myelodysplastic syndromes. Expert Opinion on Investigational Drugs. 2006; 15/7: 805-13.
24. Doukkali O, Tarrass F, Medkouri G et al. Extramembranous glomerulonephritis and myelodysplastic syndrome. Nephrologie. 2004; 25/2: 59-61.
25. Malcovati L, Della Porta MG, Cazzola M. 2006: Predicting survival and leukemia evolution in patients with myelodysplastic syndrome. Haematologica. 2006 91:1588-1590.
26. Kintzel PE. Anticancer drug-induced kidney disorders. Drug Saf. 2001 Jan; 24(1): 19-38.























