
GUIA CLINICA ENFERMEDAD DREPANOCITICA SVH 2013 1
242
2013 GUÍA DE PRÁCTICA CLÍNICA EN ENFERMEDAD DREPANOCÍTICA
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of GUIA CLINICA ENFERMEDAD DREPANOCITICA SVH 2013 1

2013
GUÍA DE PRÁCTICA CLÍNICA EN ENFERMEDAD DREPANOCÍTICA

1
2013
GUÍA DE PRÁCTICA CLÍNICA EN ENFERMEDAD DREPANOCÍTICA

2
Corrección de estilo: Mariló MorenoDiagramación y Diseño: El Ejemplar C.A.Coordinación general: Dra. Olimpia Pérez-BándezCoordinación editorial: Dr. J. Ildefonzo Arocha RodulfoImpresión: XXXXDepósito legal: If25220136101708ISBN: 978-980-7612-00-5Todos los derechos reservados, Asociación Venezolana de Drepanocitosis y Talasemias.

3
A todas las personas que presentan Drepanocitosis y a sus familiares, fuente de inspira-ción de la Guía de práctica clínica en enfermedad drepanocítica.
A la memoria de nuestro amado e inolvidable maestro Dr. Tulio Arends W, padre de la investigación de las hemoglobinas anormales en Venezuela. Henry Adams dijo: ”El maestro deja una huella para la eternidad; nunca puede decir cuando se detiene su influencia” .
Dedicatoria

4
Agradecemos a todas las personas y organizaciones que de manera directa e indirecta hicieron posible concretar este proyecto, mencionarlos a todos en tan pocas líneas, resultaría imposible.
La producción de “Guía de práctica clínica en enfermedad drepanocítica” no hubiese sido posible sin el apoyo de Laboratorios Novartis, que de manera constante han contri-buido grandemente con la adquisición y actualización de conocimientos por parte de los hematólogos venezolanos, hecho que influye directamente en la calidad de la atención brindada a los pacientes.
Agradecimiento especial merecen la licenciada Kelly López y el veterinario Javier Pirra-glia quienes se mantuvieron atentos y vigilantes de cada detalle durante el desarrollo de las múltiples reuniones realizadas.
No encontramos palabras para agradecer la labor realizada por la licenciada Denise Bustos quien, por su excelente formación profesional, paciencia y diplomacia, supo sor-tear todo tipo de escoyos hasta llevar la nave a puerto seguro. Denise muchas gracias.
Agradecemos al Dr. José Idelfonzo Arocha Rodulfo por haber ejercido la coordinación editorial y encargarse del aspecto legal.
A Mariló Moreno por la corrección de estilo.
Gracias al Sr. Douglas Limonchy por su paciencia ante tanto “quita aquí y coloca allá”.
A todos nuestros colegas interesados en el tema, que nos alentaron al preguntar insis-tentemente ¿Cuándo estará lista?
Hasta escribir una cuartilla consume parte del valioso tiempo de familiares y amigos, por ese motivo nuestro agradecimiento a ellos por su comprensión.
Por último, gracias a La Vida por brindarnos la oportunidad de compartir, aprender y aportar un muy pequeño grano de arena para mejorar las expectativas de vida de los pacientes con Drepanocitosis.
Agradecimientos

5

6
.01
.02
.04
.03
1.1. Epidemiología de la enfermedad drepanocítica. 1.2. Fisiopatología.
............................. 20.................................................. 25
...................................................................................... 8
.................................................................................... 10
.................................................................................... 12
................................................ 30
......................................... 32................................. 34
2.1. Pesquisa neonatal a nivel nacional.2.2. Diagnóstico posterior al período neonatal.2.3. Asesoramiento genético.
4.1. Dolor.4.2. Infección.4.3. Anemia aguda.4.4. Aplasia transitoria de serie roja.4.5. Ictus cerebral.4.6. Complicaciones oculares.4.7. Complicaciones pulmonares.4.8. Secuestro esplénico.4.9. Priapismo.4.10. Sindrome de embolización sistémica.4.11. Dolor abdominal.
3.1. En el niño.3.2. En el adolescente.3.3. En el adulto joven.3.4. En el adulto mayor de 40 años.3.5. Papel de la enfermera.3.6. Educación del paciente y de los familiares.3.7. Manejo psico-social.3.8. Cuidados odontológicos.
......................................................... 38 ............................................ 46
............................................ 47....................... 52
.................................... 53
........................................... 55........................................ 60
................................. 62
................................................................. 64 ........................................................... 79
................................................. 84....................... 86
................................................... 88............................... 96
....................... 101..................................... 109
...................................................... 113........ 122
......................................... 125
Epidemiología y Fisiopatología.
Integrantes del grupo.Prólogo.Presentación y generalidades.
Diagnóstico y asesoramiento genético.
Tratamiento de las complicaciones agudas.
Medidas generales.
Contenido

7
.055.1. Cardiovasculares.5.2. Hepatobiliares.5.3. Renales.5.4. Osteomusculares.5.5.Úlceras en miembros inferiores.
Tratamiento de las complicaciones crónicas.
........................................... 130............................................... 134
.......................................................... 138.......................................... 147
................... 154
.06
.07
.08
6.1. Rasgo drepanocítico. 6.2. Embarazo. 6.3. Anestesia y cirugía.6.4. Transfusión. 6.5. Sobrecarga de hierro y quelación.6.6. Trasplante en enfermedad drepanocítica.
7.1. Inducción de hemoglobina fetal.7.2. Otras estrategias farmacológicas.
8.1. Estrategias futuras en el diagnóstico de la enfermedad drepanocítica.8.2. Estrategias futuras en el tratamiento de la enfermedad drepanocítica.
...................................... 160....................................................... 162
........................................ 171.................................................... 177
................ 194
................................................. 201
................... 208................ 217
..................................................... 226
..................................................... 227
............................................................................................................ 229...................................... 236
....................................................................... 238
Tópicos especiales.
Estrategias farmacológicas en el tratamiento de la enfermedad drepanocítica.
Estrategias futuras en el diagnóstico y trata-miento de la enfermedad drepanocítica.
Apéndices:Hoja de control anual.Hoja de control de exanguinotransfusion y/o eritrocitaféresis.Historia inmunohematólogica y serológica.

8
Integrantes
José Ildefonzo Arocha R.
Jaime Bracho.
Adrián Cárdenas
Richar Figueredo
Leslie González
José Vicente Gil
Amel Guánchez
Marcos Hernández
Clementina Landolfi
María E. López.
Esther Graciela León de González
Internista. Cardiólogo
Hematólogo. Médico Internista. Hospital Dr. Rafael Zamora ArveloValle de La Pascua. Estado Guárico. Venezuela. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Hematólogo. PediatraJefe del servicio de Hematología Hospital Pastor Oropeza Barquisimeto. Estado Lara. Venezuela.Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias
Hematólogo. Jefe del Servicio de Hematología Hospital Dr. Egor Nucete. San Carlos. Estado Cojedes. Venezuela.Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Hematólogo. Internista. Adjunto del Servicio de Hematología Hospital Universitario de Caracas. Caracas. Venezuela.
Pediatra. Neumonólogo Infantil. Policlínica Metropolitana. Caracas. Venezuela.
Hematólogo. Internista.Profesor. Universidad de Oriente. Núcleo Anzoátegui. Puerto La Cruz. Estado Anzoátegui. Venezuela.
Hematólogo. Pediatra. Jefe de la Unidad de Trasplante. Ciudad Hospitalaria Enrique Tejera. Valencia. Estado Carabobo. Venezuela. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Hematólogo Pediatra Policlínica Metropolitana. Caracas. VenezuelaPresidente de la Sociedad Venezolana de Hematología. 2011-2013.
Neurólogo Pediatra Policlínica Metropolitana.Medico adjunto del Insti-tuto Venezolano de Desarrollo Integral del Niño (INVEDIN)Profesora invitada del Post-Grado de Neurología Pediátrica del Hospital Universitario de Caracas.
Hematólogo. Medico consultor Banco Municipal de Sangre del Distrito Capital. Caracas. Venezuela. Docente de Postgrado de He-matología. Universidad Central de Venezuela. Sede Banco Municipal de Sangre del Distrito Capital. Caracas. Venezuela. Directora del Banco de Sangre Instituto Diagnóstico de San Bernardino. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.

9
Nirka Lourdes Marcano Arévalo
Luz Marina Navarrete Grau.
Olimpia C. Pérez-Bández
Francisco Ramírez Osío
Evelio Rubio
María Rubio
Maritza Suárez Darauche
Yraida Tersek
Betty Urdaneta de Ramos
Dalia Velásquez de Lara
Francia Yépez de Velásquez
Urólogo. Jefe del Departamento de Urología. Hospital Militar Dr. Arvelo Caracas. Venezuela.
Internista. Nefrólogo.
Hematólogo. Bioquímica. Docente de Postgrado de Hematología. Universidad Central de Venezuela. Sede Banco Municipal de Sangre del Distrito Capital. Caracas. Venezuela. Fundadora de la Asociación Venezolana de Drepanocitosis y Talasemias.Coordinadora de la Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias
Hematólogo. Pediatra. Unidad de Trasplante de Médula Ósea. Hospital de Clínicas Caracas. Caracas. Venezuela. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Neurólogo pediatra. Adjunto del Servicio de Neurología Hospital. Dr. Domingo. Luciani El Llanito. Estado Miranda. Venezuela.
Hematólogo. Jefe del Servicio de Hematología. Maternidad Dr. Armando Castillo Plaza. Maracaibo. Estado Zulia. Centro Médico. Paraíso. Maracaibo. Estado Zulia. Venezuela.Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Hematólogo. Pediatra. Servicio de Hematología. Hospital de Niños J. M. de los Ríos. Caracas. Venezuela. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.
Hematólogo. Pediatra. Jefe del Servicio de Hematología PediátricaHospital Niño Jesús San Felipe. Estado Yaracuy. Venezuela.Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemia.
Hematólogo. Pediatra. Coordinadora del servicio de Hematología y Banco de Sangre Hospital de Especialidades Pediátricas.Maracaibo. Estado Zulia. Venezuela.
Hematólogo. Internista. Jefa de Servicio de Hematología. Hospital Universitario de Caracas. Caracas. Venezuela. Directora del Postgrado de Hematología. Universidad Central de Venezuela. Sede Hospital Universitario de Caracas. Caracas. Venezuela.
Hematólogo. Internista. Servicio de Hematología. Maternidad Concepción Palacios. Caracas. Venezuela. Comisión Médica de la Asociación Venezolana de Drepanocitosis y Talasemias.

10
Prólogo
Sindrome drepanocítico, es el término que engloba un conjunto de trastornos heredi-tarios caracterizados por la presencia del gen de la hemoglobina S, la mas frecuente de las hemoglobinas anormales, comprende el rasgo drepanocítico y la enfermedad drepanocítica, a su vez esta última representada por la anemia drepanocítica (Hb SS) y la asociación de Hb S con otras hemoglobinas anormales o con los diversos tipos de talasemias.
La primera descripción del drepanocito (del griego drépanon, hoz y kytos, célula) o cé-lula falciforme, data de 1910 cuando James Herrick y Ernest Irons describen la presen-cia de hematíes elongados, en forma de hoz, en la sangre de un estudiante procedente de Granada que consultaba por dolor abdominal y articular, fatiga, anemia y úlcera en miembro inferior.
Desde entonces, el fenómeno falciforme y sus consecuencias clínicas han sido objeto de numerosas investigaciones con repercusión en el campo de la biología, la medicina y en el estudio de poblaciones humanas. Su expresión clínica más severa, la anemia drepanocítica, fue el modelo que Linus Pauling utilizó en 1949 para desarrollar el con-cepto de enfermedad molecular, confirmado por Vernon Ingram en 1956 al demostrar que era el resultado de la sustitución de un único aminoácido en la posición sexta de la cadena ß de la globina.
En Venezuela, el estudio de las hemoglobinopatías está vinculado a la labor pione-ra del Doctor Tulio Arends quien junto a sus colaboradores establece pautas para el diagnóstico de laboratorio y determina su frecuencia en la población venezolana. De este modo se confirma que la hemoglobina S, en su forma heterocigota, muestra una frecuencia variable que puede alcanzar el 19% en poblaciones formadas por descen-dientes africanos.
La OMS considera que la anemia drepanocítica tiene importante repercusión de salud pública y es un hecho conocido que la morbilidad, mortalidad y calidad de vida del pa-ciente dependen en mayor medida de la precocidad del diagnóstico y de los cuidados médicos que reciba en todas las etapas de la vida.
Es en este contexto, siento especial complacencia en escribir estas palabras introduc-torias con las que presento a ustedes la guía de práctica clínica sobre la enfermedad drepanocítica, preparada con el objetivo de mejorar el conocimiento sobre la enferme-dad, sus complicaciones, prevención y tratamiento. Contiene información actualizada y con un enfoque práctico sobre la fisiopatología, diagnóstico, asesoramiento genético, complicaciones agudas y crónicas, tratamiento general, integral y en situaciones es-peciales, sin olvidar la importancia de los aspectos psicosociales y la educación del paciente y su familia.

11
Esta publicación fue elaborada por un grupo de hematólogos, miembros de la Socie-dad Venezolana de Hematología e integrantes de la comisión médica de la Asociación Venezolana de Drepanocitosis y Talasemias (AVDT), con la colaboración de profesiona-les especialistas en áreas diferentes a la hematología y experiencia en el tratamien-to de las complicaciones propias de la enfermedad. Ha sido coordinada por Olimpia Pérez-Bández hematólogo de gran trayectoria y con amplia experiencia en el manejo integral del paciente con enfermedad drepanocítica, y fundadora de la AVDT.
Con la certeza de la utilidad de su contenido solo queda agradecer a los autores por su desinteresado esfuerzo.
Arlette V. Ruíz de Sáez

12
Presentación y generalidades
El propósito de elaborar la Guía de práctica clínica sobre la enfermedad drepanocítica es brindar una herramienta de consulta a los especialistas, residentes, enfermeras y otros profesionales de la salud vinculados con esta patología en un intento de unificar conceptos, criterios y conductas sobre esta condición clínica.Esta guía está conformada por cinco capítulos: diagnóstico y asesoramiento genético, medidas generales en niños, adolescentes y adultos, tratamiento de complicaciones agudas, tratamiento de complicaciones crónicas y tópicos especiales.Revisamos publicaciones sobre consensos, revisiones de evidencias relacionadas con distintos tópicos de la enfermedad drepanocítica y guías utilizadas por grandes centros dedicados al tratamiento integral de pacientes con esta enfermedad. Encontramos que no existen evidencias suficientes en muchos de los esquemas de tratamiento utilizados así como en los agentes terapéuticos empleados en las diversas complicaciones, sien-do el problema mayor cuando se trata de adultos. Para dar inicio a este proyecto, que forma parte de uno mas grande orientado al logro de la atención integral del paciente con enfermedad drepanocítica, hemos elaborado la guía tomando muchos puntos de los ya conocidos y adaptando otros a las evidencias publicadas, a las condiciones socioculturales y grado de instrucción de nuestros pa-cientes y a la disponibilidad de medicamentos por parte de las instituciones de salud pública.La Guía de práctica clínica sobre la enfermedad drepanocítica será sometida a revisión en el año 2018, para realizar las modificaciones pertinentes en base a las evidencias existentes para ese momento y a la experiencia de los hematólogos que conformarán el Grupo Cooperativo Venezolano de Hemoglobinopatías.
Definiciones
Síndrome drepanocítico. Genotipos.Para mejor comprensión hemos seguido la nomenclatura aceptada internacionalmen-te, estableciendo que el síndrome drepanocítico incluye el rasgo drepanocítico (Hb AS) y la enfermedad drepanocítica, la cual presenta varios tipos (Tabla 1).
Tabla 1. Tipos más frecuentes de enfermedad drepanocítica1.
Genotipos Nombre completo Abreviatura
βS/βC
βS/β+ talasemia
Enfermedad drepanocítica -SC
Enfermedad drepanocítica- Sβ+ talasemia
ED-SC
ED-S β+ tal
βS/βS
βS/β0 talasemia
Enfermedad drepanocítica-SS
Enfermedad drepanocítica- Sβ0 talasemia
ED-SS
ED-S β0 tal
Olimpia C. Pérez-Bández / Médico Hematólogo
13
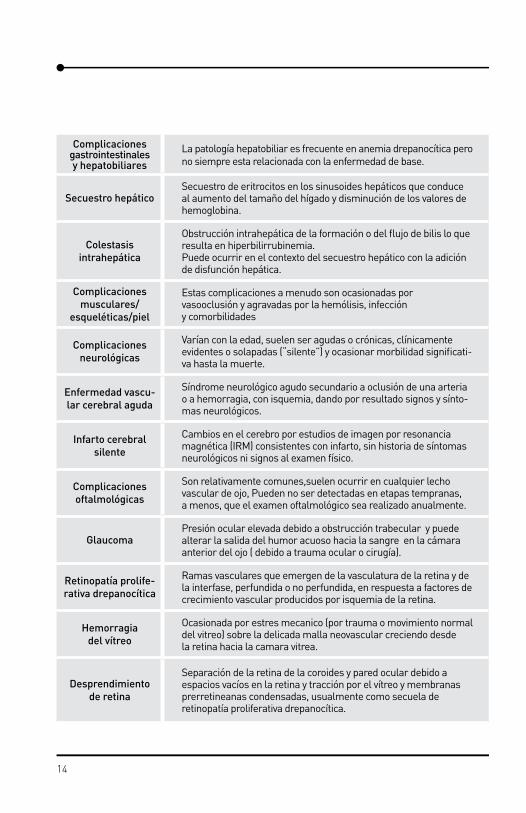
FenotiposEn ausencia de una nomenclatura de aceptación internacional y de los criterios diag-nósticos para las diversas complicaciones de la enfermedad drepanocítica que hace confundir a pacientes, familiares, público en general y proveedores de salud y que ha obstaculizado los esfuerzos clínicos para colectar datos y comparar métodos y hallaz-gos, en 1910 se publicó el resultado de un consenso de las manifestaciones fenotípicas de la enfermedad drepanocítica, con el propósito de facilitar la investigación al esta-blecer una base común que permita la comparación de resultados (Tabla 2).
Tabla 2. Definición de las manifestaciones más comunes de la enfermedad drepanocítica2.
Manifestaciones Definición
Exacerbaciónde la anemia
Disminución aguda de los niveles de hemoglobina, por lo menos 2g/dL del valor basal.
Hiperhemólisis Esta complicación es a menudo ocasionada por vasooclusión y agra-vadas por la hemólisis, infección y comorbilidades
Secuestro esplénico agudo
Atrapamiento rápido por parte del bazo de los elementos formes de la sangre, lo que ocasiona disminución de los valores de hemoglobina, por lo general, asociada con trombocitopenia absoluta o relativa (<150.000 x 106/L) e hipovolemia.
Crisis aplásicaSupresión transitoria, total o parcial, de la eritropoyesis, caracterizada por disminución de los valores de hemoglobina y reticulocitopenia absoluta (<50.000 x 106/L).
Complicaciones cardíacas
Disfunción cardíaca con cambios fisiológicos y morfológicos, entre otros, adelgazamiento del septum interventricular, aumento de la masa muscular, llenado diastólico anormal y disfunción diastólica del ventrículo izquierdo.
Cardiomiopatía
Enfermedad del corazón que afecta la conducción ventricular o altera su función. Alto gasto cardiaco crónico, asociada o no con hipertrofia ventricular y desarrollo de cardiomiopatía hipertrófica. La sobrecarga de hierro puede ocasionar cardiomiopatía dilatada o siderosis cardíaca.
Cardiomegalia Dilatación del corazón que involucra los ventrículos, aurículas o ambos.
Insuficiencia cardiaca
congestiva
Disfunción del ventrículo izquierdo con síntomas/signos de congestión pulmonar y/o sistémica, a menudo asociado a edema periférico. Puede ser aguda o crónica.
Alteraciones en el crecimiento
y desarrollo
Falla del potencial crecimiento lineal, medido por un promedio-Z, mayor o igual a dos desviaciones estándar por debajo del promedio de los valores referenciales para la población
14
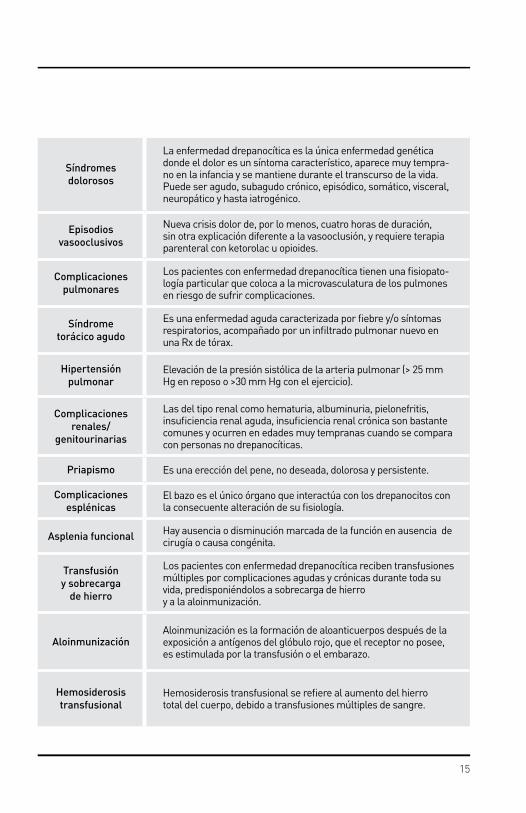
Complicaciones gastrointestinales y hepatobiliares
La patología hepatobiliar es frecuente en anemia drepanocítica pero no siempre esta relacionada con la enfermedad de base.
Secuestro hepáticoSecuestro de eritrocitos en los sinusoides hepáticos que conduce al aumento del tamaño del hígado y disminución de los valores de hemoglobina.
Colestasis intrahepática
Obstrucción intrahepática de la formación o del flujo de bilis lo que resulta en hiperbilirrubinemia. Puede ocurrir en el contexto del secuestro hepático con la adición de disfunción hepática.
Complicaciones musculares/
esqueléticas/piel
Estas complicaciones a menudo son ocasionadas por vasooclusión y agravadas por la hemólisis, infección y comorbilidades
Complicaciones neurológicas
Varían con la edad, suelen ser agudas o crónicas, clínicamente evidentes o solapadas (“silente”) y ocasionar morbilidad significati-va hasta la muerte.
Enfermedad vascu-lar cerebral aguda
Síndrome neurológico agudo secundario a oclusión de una arteria o a hemorragia, con isquemia, dando por resultado signos y sínto-mas neurológicos.
Infarto cerebral silente
Cambios en el cerebro por estudios de imagen por resonancia magnética (IRM) consistentes con infarto, sin historia de síntomas neurológicos ni signos al examen físico.
Complicaciones oftalmológicas
Son relativamente comunes,suelen ocurrir en cualquier lecho vascular de ojo, Pueden no ser detectadas en etapas tempranas,a menos, que el examen oftalmológico sea realizado anualmente.
GlaucomaPresión ocular elevada debido a obstrucción trabecular y puede alterar la salida del humor acuoso hacia la sangre en la cámara anterior del ojo ( debido a trauma ocular o cirugía).
Retinopatía prolife-rativa drepanocítica
Ramas vasculares que emergen de la vasculatura de la retina y de la interfase, perfundida o no perfundida, en respuesta a factores de crecimiento vascular producidos por isquemia de la retina.
Hemorragia del vítreo
Ocasionada por estres mecanico (por trauma o movimiento normaldel vitreo) sobre la delicada malla neovascular creciendo desdela retina hacia la camara vitrea.
Desprendimiento de retina
Separación de la retina de la coroides y pared ocular debido a espacios vacíos en la retina y tracción por el vítreo y membranas prerretineanas condensadas, usualmente como secuela de retinopatía proliferativa drepanocítica.
15
Síndromes dolorosos
La enfermedad drepanocítica es la única enfermedad genética donde el dolor es un síntoma característico, aparece muy tempra-no en la infancia y se mantiene durante el transcurso de la vida. Puede ser agudo, subagudo crónico, episódico, somático, visceral, neuropático y hasta iatrogénico.
Episodios vasooclusivos
Nueva crisis dolor de, por lo menos, cuatro horas de duración, sin otra explicación diferente a la vasooclusión, y requiere terapia parenteral con ketorolac u opioides.
Complicaciones pulmonares
Los pacientes con enfermedad drepanocítica tienen una fisiopato-logía particular que coloca a la microvasculatura de los pulmones en riesgo de sufrir complicaciones.
Síndrome torácico agudo
Es una enfermedad aguda caracterizada por fiebre y/o síntomas respiratorios, acompañado por un infiltrado pulmonar nuevo en una Rx de tórax.
Hipertensión pulmonar
Elevación de la presión sistólica de la arteria pulmonar (> 25 mm Hg en reposo o >30 mm Hg con el ejercicio).
Complicaciones renales/
genitourinarias
Las del tipo renal como hematuria, albuminuria, pielonefritis, insuficiencia renal aguda, insuficiencia renal crónica son bastante comunes y ocurren en edades muy tempranas cuando se compara con personas no drepanocíticas.
Priapismo Es una erección del pene, no deseada, dolorosa y persistente.
Complicaciones esplénicas
El bazo es el único órgano que interactúa con los drepanocitos con la consecuente alteración de su fisiología.
Asplenia funcional Hay ausencia o disminución marcada de la función en ausencia de cirugía o causa congénita.
Transfusión y sobrecarga
de hierro
Los pacientes con enfermedad drepanocítica reciben transfusiones múltiples por complicaciones agudas y crónicas durante toda su vida, predisponiéndolos a sobrecarga de hierro y a la aloinmunización.
AloinmunizaciónAloinmunización es la formación de aloanticuerpos después de la exposición a antígenos del glóbulo rojo, que el receptor no posee, es estimulada por la transfusión o el embarazo.
Hemosiderosis transfusional
Hemosiderosis transfusional se refiere al aumento del hierro total del cuerpo, debido a transfusiones múltiples de sangre.

16
Las manifestaciones clínicas incluyen tres grupos de síntomas y signos:• Anemia hemolítica y sus complicaciones (Tabla 3).• Síndromes dolorosos drepanocíticos y manifestaciones relacionadas (Tabla 4). • Complicaciones de órganos mayores y sus secuelas (Tabla 5).
Exacerbación de la anemia
Hiperhemólisis
Secuestro agudo esplénico
Crisis aplásica
Complicaciones relacionadas a la transfusión
Síndrome de hiperviscosidad
Hemólisis inmune
Hemosiderosis transfusional
Neurológicas
Enfermedad vásculo cerebral aguda
Aneurisma
Enfermedad vásculo cerebral aguda hemorrágica
Enfermedad vásculo cerebral aguda isquémica o infarto
Moyamoya
Infarto cerebral silente
Ataque isquémico transitorio
Velocidad alta de flujo medida por eco doppler transcraneal
Convulsiones
Episodios vasooclusivos
Falla aguda multiorgánica
Síndromes dolorosos iatrogénicos
Neuropático
Tabla 3. Anemia hemolítica y sus complicaciones
Tabla 4. Síndromes dolorosos drepanocíticos y manifestaciones relacionadas2
Tabla 5. Complicaciones de órganos mayores y sus secuelas5

17
Oftalmológicas
Lesiones sol de rayos negros
Signo de coma conjuntival
Glaucoma
Hifema
Retinopatía proliferativa drepanocítica
Hemorragia del vítreo
Desprendimiento de retina
Hemorragia en parche
Cardíacas
Cardiomegalia
Cardiomiopatía
Insuficiencia cardíaca congestiva
Prolapso de la válvula mitral
Hipertensión arterial
Pulmonares
Sindrome torácico agudo
Hipertensión pulmonar
Gastrointestinales y hepatobiliares
Colecistitis
Colelitiasis/barro biliar
Secuestro hepático
Colestasis intrahepática
Hepatitis viral
Renales/genitourinarias
Insuficiencia renal aguda
Insuficiencia renal crónica
Hematuria
Priapismo
Proteinuria y sindrome nefrótico
Pielonefritis

18
Esplénicas
Infarto agudo esplénico
Asplenia funcional
Hiperesplenismo
Secuestro agudo esplénico
Musculares/esqueléticas/piel
Necrosis avascular
Dactilitis (sindrome mano/pie)
Úlceras en pienas
Miositis/mionecrosis/fascitis
Osteomielitis
Osteopenia/Osteoporosis
Alteraciones en el crecimiento y desarrollo
Retardo en el crecimiento
Hipertensión arterial
Haplotipos.El término polimorfismo del ADN, se refiere a secuencias de nucleótidos diferentes a la normal que no alteran la función del gen. En el bloque génico β se han descrito 5 zo-nas de polimorfismo cercanas al gen βS, que dan origen a 5 haplotipos cuyos nombres corresponden al sitio geográfico donde predominan, así tenemos: Senegal (SEN), Be-nín (BEN), República Centroafricana (CAR), Camerún (Cam) y Arabia-Indio. El haplotipo CAR está asociado a formas severas de la enfermedad, en tanto que el SEN se asocia a formas menos severas.
Según Arends A y colaboradores, en Venezuela los haplotipos mas frecuentes en ED-SS son BEN y CAR, seguidos de SEN y Cam y un alto porcentaje de heterocigocidad entre los haplotipos3.
Referencias:1. Statement of the Council of Regional Networks for Genetics Services (CORN). J Pediatr 2000; (4 Su-ppl):S1-46.2. Ballas SK, Lieff S , Benjamin LJ ,Dampier CD , Heeney M, Hoppe C et al .Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol 2010; 85:6–13.3.Arends A,Chacín M,Bravo-Urquiola M,Montilla S,Guevara JM, Velásquez D,García G,Alvarez M, Castillo O.Hemoglobinopatías en Venezuela. Asociación Interciencia.2007. Investigaciones HighBeam. <HYPER-LINK “http://www.highbeam.com/”

19

20
Epidemiología y fisiopatología.01 Richar Figueredo. Médico hematólogo
Yraida Tersek. Médico hematólogo
1.1 Epidemiología de la enfermedad drepanocítica
Las hemoglobinopatías, alteraciones hereditarias de la estructura o síntesis de la he-moglobina, son las enfermedades monogénicas mas frecuentes en el mundo. La Organi-zación Mundial de la Salud (OMS), estima que por lo menos el 5,2% de la población global y más del 7% de las mujeres embarazadas, son portadores de una variante significativa de la hemoglobina. La Hb S representa el 40% de ellas pero es responsable del 80% de los enfermos, debido a la alta prevalencia de portadores localizada en algunos países: cerca del 85% de las personas con enfermedad drepanocítica y aproximadamente el 70% de todos los nacimientos afectados, ocurren en África. Se estima que cada año nacen en el mundo mas de 300.000 niños con hemoglobinopatías (83% con enfermedad drepano-cítica y 17% con talasemias).1,2 Cerca de 1,1% de las parejas en todo el mundo tienen el riesgo de hijos con un trastorno de la hemoglobina y 2,7 por 1.000 son afectados. Los que nacen en los países desarrollados, sobreviven con la enfermedad pero los que nacen en los poco desarrollados, mueren antes de los 5 años.1
Muchos de los países de poco o mediano desarrollo, poseen limitaciones extremas para la atención y control de los afectados por la enfermedad drepanocítica, factores que influyen sobre su esperanza y calidad de vida. La enfermedad drepanocítica ha sido ignorada en gran proporción tanto por los gobiernos de los países donde la fre-cuencia es alta como por los organismos internacionales.3
En el 2006, en su resolución WHA57.13 sobre la genómica y la salud mundial, la Asam-blea General de la OMS, instó a los estados miembros a que movilicen recursos para la acción4 y considera necesaria la creación de grupos de trabajo de expertos regionales, nuevas alianzas a nivel nacional, regional y mundial y una labor de sensibilización a alto nivel para asegurar que los gobiernos de los países más afectados y los organismos de ayuda internacional estén plenamente concientizados de la magnitud del problema y presten mayor atención a las hemoglobinopatías e insta a los estados miembros, entre otras cosas, a que diseñen, apliquen y refuercen de modo sistemático, equitativo y eficaz programas nacionales exhaustivos e integrados para la prevención y el manejo de las hemoglobinopatías, en los que se incluyan la vigilancia, la difusión de informa-ción, la concientización y la detección, y que estén adaptados a sus contextos socioe-conómicos y culturales específicos, todo ello con el objetivo de reducir la incidencia, la morbilidad y la mortalidad de estas enfermedades genéticas.
Distribución y frecuencia de la enfermedad drepanocítica.La distribución de la Hb S y por tanto la enfermedad drepanocítica, es amplia a través de la mayor parte de África subsahariana, con focos en el Mediterráneo y Medio Orien-te y relativamente frecuente en ciertas regiones de la India. Las grandes migraciones

21
humanas han distribuido este gen a través del mundo. Originalmente las alteraciones hereditarias de la hemoglobina se encontraban en el 60% de 229 países, afectando potencialmente al 75% de los nacimientos, pero hoy son comunes en 71% de los países y afectan al 89% de los nacimientos.1
En Venezuela la distribución de la Hb S estuvo limitada durante mucho tiempo a la zona norte costera no detectándose en estados andinos5. En poblaciones que se mantuvie-ron relativamente aisladas la prevalencia del gen es alta, así tenemos Tapipa (estado Miranda) con una prevalencia del gen de 19,2%6 y en isla Toas (estado Zulia) una pre-valencia de 15%.7,8
Estudios mas recientes demuestran la extensión del gen a todos los estados del país9 y no se dispone de data relacionada al número de personas afectadas por la enfermedad ni del número de niños nacidos con la misma. Extrapolando cifras obtenidas del Reino Unido, Estados Unidos, Canadá y Australia, se estima que en Venezuela, basado en una población de 25.017.387, existen 6.622 personas con anemia drepanocítica (enfer-medad-SS), y que cada año nacen 735 niños con la enfermedad.1,10 El cálculo para la extrapolación es automatizado, no toma en cuenta ninguna diferencia genética, étnica, cultural, social, otras, entre los países a los cuales fueron extrapolados. La extrapo-lación puede ser altamente imprecisa para los países en desarrollo como el nuestro y para los países no desarrollados y sólo da una idea de lo que pudiera estar sucediendo en una determinada área en relación a la enfermedad3.
Causas que explican lo común de la enfermedad drepanocítica y la tendencia de hacerse mas frecuente.Hay muchas razones que pudieran explicar el por qué las hemoglobinopatías son comunes, sin embargo es indudable que la principal es la selección natural. Existen fuertes evidencias de que la Hb S, Hb C y α talasemias son altamente frecuentes de-bido a la protección que confieren al heterocigoto contra la infección por plasmodium falciparum.11,12
El mecanismo por el cual los heterocigotos para estas variantes son resistentes a la in-fección, no es bien conocido. Indudablemente ocurre una variación en la interacción del parásito con la célula anormal, pero existen otros factores como la respuesta inmune. También hay evidencias de interacciones epistáticas entre las variantes hemoglobíni-cas y la protección contra malaria, por ejemplo mientras los heterocigotos para Hb S o α + talasemia obtienen un grado fuerte de protección, los que heredan ambos genes pierden por completo dicha protección13. Otro factor importante es el maridaje entre consanguíneos, muy común en regiones de alta frecuencia de estos desórdenes14.
No hay que dejar a un lado el hecho de que en algunos países la tasa de mortalidad neonatal e infantil ha disminuido debido a que mejoraron las condiciones sociales y de
Epidemiología y fisiopatología

22
salud pública, por tanto niños que antes morían ahora viven lo suficiente como para ser diagnosticados, controlados y tratados.3
Los factores demográficos también son importantes, aun cuando la malaria fuera completamente erradicada, tardaría muchas generaciones para que las hemoglobino-patías declinen; por otra parte la población tiende a aumentar en algunos países donde la prevalencia de la enfermedad es grande. Predicciones recientes sugieren que en los próximos años la enfermedad drepanocítica constituirá un problema de salud a nivel global.3
Mortalidad y expectativas de vida.A excepción de Jamaica, existen pocos estudios relacionados con la evolución natural de la enfermedad y la data de muerte es escasa. En Estados Unidos cerca del 50% de los pacientes con anemia drepanocítica sobreviven hasta la quinta década, y la edad de muerte es de 42 años para los hombres y de 48 años para las mujeres; para la enfermedad-SC es de 60 y 68 respectivamente15. En África subsahariana, pocos niños llegan a los 5 años16. En Venezuela no hay datos relacionados con esperanza de vida y mortalidad.
Control poblacional.Aun cuando las manifestaciones de la enfermedad y de sus complicaciones han sido definidas claramente, persisten muchas interrogantes en relación a qué hacer para controlar la enfermedad sobre todo en los países pobres.
Si bien el despistaje neonatal en talasemias tiene un valor limitado, está demostrado que es extremadamente importante en el control de la enfermedad drepanocítica si se asocia con el asesoramiento genético, de allí que la OMS ha recomendado el desarrollo global de estos servicios4,17.
Los programas de detección necesitan ser apoyados por la educación de la población y por estructuras que la reglamenten, de modo que las personas puedan tomar deci-siones con conocimiento de causa y se les asegure una protección contra la discrimi-nación a consecuencia de los resultados de las pruebas. En países desarrollados esta estrategia se lleva a cabo en tres etapas1:• Primera. Información retrospectiva a los padres de niños enfermos, sobre el 25% de probabilidades en cada embarazo, de tener otro niño enfermo. Esta medida puede disminuir significativamente la prevalencia de nacimientos de niños afectados.• Segunda. Diagnóstico prenatal. Tiene poco efecto sobre la prevalencia debido a la influencia de factores económicos, médicos, sociales, éticos, religiosos y legales.• Tercera. Información y detección prospectiva para la población general. La selección de esta estrategia varía con las actitudes sociales, costos y oportunidades dentro del sistema de salud. Puede ofrecerse a los jóvenes o antes de decidir formar una pareja.
.01

23
La demanda pública aumenta cuando el examen no se propone como una opción de diagnóstico prenatal18. El despistaje a nivel de población no es la única opción útil, también justifica el costo ofrecerlo a familias donde la consanguinidad es común19, o donde la prevalencia de portadores es baja20.
La data relacionada con la enfermedad drepanocítica, indica que en esta enfermedad, es muy bajo el uso del dictamen prenatal y que existe un aumento de niños afectados con diagnóstico neonatal21,22.
El diagnóstico al momento de nacer, seguido del uso de penicilina profiláctica y va-cunación contra los patógenos mas frecuentes, contribuyen de manera importante en la disminución de la mortalidad por infección. Por otra parte existen evidencias considerables de la eficacia del uso de hidroxicarbamida (hidroxiurea) tanto en niños como en adultos. La enfermedad cerebro vascular aguda se puede reducir mediante la pesquisa regular y transfusión crónica y un enfoque mejor definido en el manejo de las diferentes complicaciones23,24.
Uno de los mayores problemas prácticos en el manejo y control de la enfermedad dre-panocítica, es su extraordinaria heterogeneidad fenotípica. Aun cuando se han identi-ficado numerosos modificadores genéticos, falta mucho por aprender y todavía no es posible predecir el curso de la enfermedad en base del genotipo del paciente25.
La OMS en su Asamblea 158, del año 2006, considera improbable que los países en desarrollo organicen programas de alcance nacional desde el principio, pues tienen otras prioridades y disponen de infraestructuras insuficientes para prestar el servicio4.Por consiguiente, el objetivo inicial consistirá en establecer uno o más centros de refe-rencia con capacidad para desarrollar métodos apropiados de prevención y tratamien-to. A medida que aumente la demanda, esos servicios pueden ser transferidos a otros centros del país.
Para que la atención prestada sea apropiada, es indispensable que exista una estrecha relación de trabajo entre los dispensadores de atención primaria y esos centros. El personal de la institución debe facilitar la articulación de programas nacionales efica-ces integrados en los servicios de salud nacionales, elaborar directrices y materiales educativos, y coordinar y cooperar con las asociaciones nacionales de padres y pacien-tes. También considera que es urgente la creación de grupos de trabajo de expertos regionales en el manejo de hemoglobinopatías, para que coordinen las actividades de dichos centros.
Costo económico.Indudablemente que el costo económico es un factor importante a considerar cuan-do se plantea el control y manejo de la enfermedad en los países en desarrollo. El
Epidemiología y fisiopatología

24
ministerio de salud requiere conocer el número de pacientes y la carga clínica de la enfermedad, al mismo tiempo necesita realizar un análisis costo/beneficio de formas específicas cómo prevenirla y tratarla. La ausencia de data relacionada con mortali-dad e historia natural de la enfermedad hace extremadamente difícil estos cálculos, sin embargo parece que la detección y el diagnóstico prenatal sería rentable siempre y cuando se dirija solamente a afrodescendientes y no a la población general3 ; sin embargo esto es válido en países donde existe poco mestizaje y no en países como Venezuela donde el mestizaje es alto.
El programa “Carga general de enfermedades” de la OMS está analizando los datos, para tratar de estimar la carga general de los trastornos de la hemoglobina en térmi-nos de DALYs (disability adjusted life years), el cual se considera un parámetro mas preciso para la determinación de costos3, razón por la cual es importante comenzar a trabajar con esta finalidad1.
Debido a la diversidad y heterogeneidad de la distribución de las hemoglobinopatías, las políticas deben adaptarse a las condiciones de cada país en particular. Las hemo- globinopatías pueden utilizarse como indicadores de salud de una población.
Referencias 1. Modell, B. & Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization. 2008;86: 480-487. Diponible en http://www.who.int/bulletin/volu-mes/86/6/06-036673/en/2. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, Weatherall DJ, Hay SI. Global distri-bution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 2010;1: 104.3. Weatherall, DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol 2011; 154: 736–744 4. Sickle cell anaemia. Agenda item 11.4. In: 59th World Health Assembly, 27 May 2006. WHA 59.20. Available from: http://www.who.int/gb/ebwha/pdf_files/WHA59-REC1/e/WHA59_2006_REC1-en.pdf [p. 26; accessed on 6 February 2008].5. Arends T. Epidemiología de las variantes hemoglobínicas en Venezuela. Gaceta Médica de Caracas 1984;92:189-224. 6. Arends T, Gallango ML, Muller A, González-Marrero M, Pérez-Bández O. Tapipa: A negroid Venezuelan isolate. En Meyer RJ, Otten CM, Ab-del-Hameed F (Eds.) Evolutionary Models and Studies in Human Diversi-ty. Mounton. La Haya, Holanda. 1978, pp. 201-214. 7. Núñez-Montiel A, Arteaga-Pérez R, Montilla RLG, Ferrer A. Estudios hematológicos sobre la población de la isla de Toas (Estado Zulia). Acta Cient Venez 1962;13:94-97.8. Pineda de del Villar l, BorjaS. La hemoglobina S en la isla de Toas. ¿Un problema genético de salud pública? Inv Clin 1986;27:5-14.9. Arends A, Chacín M, Bravo -Urquiola M, Montilla S, Guevara JM et al. Hemoglobinopatías en Venezuela. Interciencia 2007;32:516-521.
.01

25
10. Statistics by Country for Sickle Cell Anemia. Disponible en http://www.rightdiagnosis.com/s/sickle_cell_anemia/stats-country.htm11. Weatherall DJ. Genetic variation and susceptibility to infection: the red cell and malaria. Brit J Haematol 2008;141:276–286.12. Williams TN. Red blood cell defects and malaria. Mol Biochem Parasitol 2006;149:121–127.13. Williams TN, Mwangi TW, Wambua S, Peto TE, Weatherall DJ, Gupta S et al. Negative epistasis between the malaria-protective effects of alpha+-thalassemia and the sickle cell trait. Nat Genet 2005; 37: 1253–1257.14. Bittles AH, Mason WM, Greene J, Rao NA. Reproductive behavior and health in consanguineous marria-ges. Science 1991; 252:789–794.15. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639-44. 16. Molineaux L, Grammiccia G. The Garki Project. World Health Organization, Geneva. 1980.17. Thalassaemia and other haemoglobinopathies. Agenda item 5.2. 6. In: 59th World Health Assembly, 27 May 2006. EB118.R1. Available from: http://www.who.int/gb/ebwha/pdf_files/EBSS-EB118-2006-REC1/engli-sh/Res/res-eb118_2006_rec1-en.pdf [accessed on 6 February 2008].18. Samavat A, Modell B. Iranian national thalassaemia screening programme. BMJ 2004. 329:1134-7. 19. Ahmed S, Saleem M, Modell B, Petrou M. Screening extended families for genetic haemoglobin disorders in Pakistan. N Engl J Med 2002; 347:1162-68. 20. Martins MC, Olim G, Melo J, Magalhaes HA, Rodrigues MO. Hereditary anaemias in Portugal: epidemio-logy, public health significance and control. J Med Genet 1993;30:235-39. 21. Modell B, Petrou M, Layton M, Varnavides L, Slater C, Ward RHT et al. Audit of prenatal diagnosis for haemoglobin disorders in the United Kingdom: the first 20 years. BMJ 1997; 315:779-84. 22. The United Nations Demographic Yearbook. New York: United Nations; 2003. Available from: http://uns-tats.un.org/unsd/demographic/products/dyb/ [accessed on 6 February 2008.23. Serjeant GR, Serjeant BE. Sickle Cell Disease. Oxford University Press, Oxford. 2001.24. Steinberg MH, Ohene-Frempong K, Heeney MM. Clinical and pathophysiological aspects of sickle cell anaemia. In: Disorders of Hemoglobin (eds by MH Steinberg, BG Forget, DR Higgs, DJ Weatherall), 2009. pp. 437–496. Cambridge University Press, Cambridge.25. Steinberg MH, Nagel RL. Genetic modulation of sickle cell disease and thalassemia. In: Disorders of Hemoglobin (eds by MH Steinberg, BG Forget, DR Higgs, DJ. Weatherall, 2009: pp. 638–657. Cambridge University Press, Cambridge.
1.2. Fisiopatología de la enfermedad drepanocítica.
La enfermedad drepanocítica es una condición caracterizada por la presencia de Hb S, que resulta de una mutación puntual (GAG → GTG) en el exón 1 del gen β de la globina, dando por resultado la sustitución de ácido glutámico por valina en la posición 6 de la cadena β de la globina1. Aunque es una mutación sencilla, el gen de la Hb S es un gen pleitrópico, ocasiona diversidad de expresión fenotípica, que se traduce en las múltiples complicacio-nes de la enfermedad drepanocítica en general y de la anemia drepanocítica en particular.
Epidemiología y fisiopatología

26
Polimerización de la hemoglobina S.La polimerización es un complejo de reacciones en cadena, que se han estudiado ex-tensivamente in vitro pero que aún sigue sin entenderse completamente. En el estado desoxigenado la valina se encuentra encerrada en un bolsillo hidrofóbico sobre una cadena β globina adyacente juntando entre sí, las moléculas de hemoglobina2, trans-formándolas en un núcleo crítico, donde otras moléculas estabilizan el polímero, cre-ce en longitud formando fibras helicoidales que se entrecruzan por la intervención de mas moléculas, en un proceso autocatalítico3. Los polímeros existen en equilibrio dinámico con la hemoglobina soluble y en una cantidad suficiente para formar un gel insoluble que aumenta la viscosidad del citoplasma y ocasiona daño a la membrana del eritrocito, disminuyendo la flexibilidad de la misma y dando origen a la forma de hoz o drepanocito, término del cual se deriva el nombre de la enfermedad4. La rigidez es un biomarcador potencialmente útil, sin embargo no se investiga de rutina debido a que los métodos consumen mucho tiempo y requiere de sangre fresca y equipo es-pecializado5.
Existe un tiempo medible entre la nucleación y la polimerización, el cual es influen-ciado por diversos factores locales, entre ellos: tensión de oxígeno, pH, temperatura, niveles de 2,3-DPG, y de monóxido de carbono. La concentración de Hb S ejerce un fuerte efecto sobre la polimerización6.
En su tránsito por la micro circulación, el eritrocito discoideo se transforma en dre-panocito, este hecho provoca retardo en su paso a nivel de las arteriolas terminales, lecho capilar y vénulas post capilares. Es en este sitio donde la saturación de oxígeno es menor y el eritrocito necesita deformarse para pasar por un vaso de muy pequeño diámetro, el drepanocito causa obstrucción debido a las interacciones con el endotelio y otras células.
La hemoglobina fetal (Hb F) interfiere con la polimerización de la Hb S mediante dos mecanismos, primero sí la concentración de hemoglobina total es constante, un au-mento de la Hb F intracelular trae como consecuencia menor concentración de Hb S. Segundo los tetrámeros de Hb F pueden disociarse en dímeros y conjugarse con dí-meros de Hb S dando origen a híbridos (α2 βSγ), que no participan en la polimerización7.
Dado que esta ocurre a escala molecular no es sorprendente que no existan biomar-cadores que midan directamente la tasa de polimerización en el eritrocito y tampoco se ha desarrollado la tecnología para llevar a cabo tal medición5.
La vasooclusión es el fenómeno más importante en la enfermedad drepanocítica, ocu-rre tanto en los vasos pequeños como en los grandes. Anteriormente este fenómeno se atribuía a un efecto netamente mecánico provocado por los drepanocitos, hoy a la luz de nuevas investigaciones, se sabe que además de la adherencia de los eritrocitos
.01

27
a las células endoteliales, también se adhieren leucocitos y plaquetas, además hay inflamación, hipercoagulabilidad, disfunción endotelial, alteración en el metabolismo del óxido nítrico y daño por isquemia-reperfusión8-21.
Aumento de la adhesión de glóbulos rojos. La membrana del eritrocito normal es asimétrica y las enzimas se encargan de mantenerla. En el drepanocito se pierde la asimetría de la membrana provocan-do la exposición de fosfatidil serina y fosfatidil etanolamina, lo cual puede ser me-dible por citometría de flujo. Fosfatidil serina y fosfatidil etanolamina, activan el complemento y aumentan la adhesividad del eritrocito5. Otras proteínas expre-sadas de manera anormal en los reticulocitos de pacientes con enfermedad dre-panocítica, son CD36 y α4β1 integrinas; CD36 permite la adhesión del eritrocito al endotelio vascular por la unión a la trombospondina, por su parte α4β1 se une a fi-bronectina11 y a la molécula 1 de adhesión (VCAM-1) sobre el endotelio vascular. El factor iniciador del fenómeno de vasooclusión es la adherencia de los glóbulos ro-jos a las células endoteliales, a favor de este hecho está la relación existente entre la severidad de la anemia drepanocítica y la extensión de la adhesividad de los drepano-citos8-13.
Aumento de la adhesión de leucocitos.En la enfermedad drepanocítica es típico el mayor número de leucocitos, secundario a hiperesplenismo e inflamación. Se ha correlacionado tal incremento con la frecuencia de episodios dolorosos22, aumento de enfermedad vascular cerebral hemorrágica23 y muerte temprana24. Los neutrófilos juegan un papel protagónico atrapando drepano-citos, por tanto en la vasooclusión, los drepanocitos parecen ser más adherentes a los neutrófilos que los glóbulos rojos normales. Los drepanocitos también inducen la actividad oxidativa de los neutrófilos, lo que parece ser importante en el daño tisular inducido por los neutrófilos durante los episodios vasooclusivos14.
Hipercoagulabilidad.En estado basal los pacientes con enfermedad drepanocítica exhiben en el plasma niveles altos de marcadores de generación de trombina, disminución de los inhibido-res naturales de la coagulación, actividad reducida de la fibrinólisis y aumento de la expresión del factor tisular, las plaquetas y otros elementos celulares también están activados, lo que pudiera disponer a tromboembolismo15, 16. Se considera un estado de hipercoagulabilidad debido al aumento de generación de trombina y fibrina y de la actividad pro coagulante del factor tisular17-19.
También se ha demostrado la presencia de marcadores de activación de la coagula-ción (dímeros D, F1+2 y complejo trombina-antitrombina)19, además de la exposición de fosfatidil serina y fosfatidil etanol amina por parte de la membrana eritrocitaria, plaquetas, eritrocitos, leucocitos y células endoteliales26.
Epidemiología y fisiopatología

28
A pesar de lo anterior no está claro que el estado de hipercoagulabilidad contribuya a la vasooclusión, no existen estudios que muestren de manera convincente una relación exacta entre los biomarcadores de la coagulación y la frecuencia de complicaciones vasooclusivas mas importantes como dolor agudo y síndrome torácico agudo. Igual-mente no hay publicaciones que demuestren el beneficio de anticoagulantes o terapia antiplaquetaria ya que los protocolos clínicos no se han cumplido adecuadamente5.
Daño por reperfusión y óxido nítrico (ON).Un aspecto importante de la vasculopatía drepanocítica, es la alteración de la regula-ción vasomotora del tono del endotelio, trombosis e inflamación8. La oclusión vascu-lar intermitente trae como consecuencia daño por reperfusión, el cual está asociado con acumulación de granulocitos y producción de especies reactivas de oxígeno, el ON tiende a contrarrestar las reacciones de estrés oxidativo, lo que ocasiona, reducción de la biodisponibilidad de ON además de contribuir a la disfunción vascular en la en-fermedad drepanocítica9.
Papel de los mediadores inflamatorios.Existe un consenso emergente en relación a que un estado proinflamatorio contribuye a las complicaciones vasooclusivas que ocurren en la enfermedad drepanocítica. El daño tisular como consecuencia de la vasooclusión provoca la liberación de numero-sos mediadores inflamatorios que inician la transmisión del estímulo y la percepción del dolor. Las citoquinas plasmáticas y factores relacionados representan un área de investigación floreciente relacionado a la patogénesis de la enfermedad drepanocítica. Se ha responsabilizado a citoquinas derivadas de plaquetas, leucocitos y células endo-teliales en el desarrollo de diversas secuelas de la enfermedad 21, 26.
Es importante tener en cuenta los diversos aspectos de la fisiopatología de la enferme-dad drepanocítica, con el propósito de racionalizar las medidas terapéuticas existentes y las que seguramente se implementaran en un futuro cercano.
Referencias 1. Ingram, VM. Gene mutations in human hemoglobin: the chemical difference between normal and sickle hemoglobin. Nature 1957; 180:326 – 328. 2. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997; 337:762-769.3. Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J Mol Biol 1985;183:611-631. 4. Ferrone F, Nagel RR. Polymer structure and polymerization of deoxyhemoglobin S. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2001:577-610.5. Rees DC, Gibson J. Biomarkers in sickle cell disease. Br J Haematol 2012;156:433-45.6. Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood 1987; 70:1245-1266.7. Poillon WN, Kim BC, Rodgers GP, Noguchi CT, Schechter AN. Sparing effect of hemoglobin F and hemoglobin A2 on
.01

29
the polymerization of hemoglobin S at physiologic ligand saturations. Proc Natl Acad Sci USA 1993; 90:5039-5043.8. Aslan M, Freeman BA. Redox-dependent impairment of vascular function in sickle cell disease. Free Radic Biol Med 2007; 43:1469-1483.9. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: Inflammation and a chronic vasculopathy. Microcirculation 2004;11:129–151.10. Mehta SR, Afenyi-Annan A, Byrns PJ, Lottenberg R. Opportunities to improve outcomes in sickle cell disease. Am Fam Physician 2006;74:303-310.11. Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation 2009; 16:97–111.12. Styles LA, Lubin B, Vichinsky E, Lawrence S, Hua M, Test S, Kuypers F. Decrease of very late activation antigen-4 and CD36 on reticulocytes in sickle cell patients treated with hydroxyurea. Blood 1997; 89:2554–2559.13. Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med 1980; 302:992-995.14. Grigg AP. Granulocyte colony-stimulating factor induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta thalassemia. Blood 2001; 97:3998-3999.15. Ataga KI, Key NS. Hypercoagulability in sickle cell disease new approaches to an old problem. Hematolo-gy Am Soc Hematol Educ Program 2007:91-96.16. Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, et al Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol 2000;111:474-481.17. Mousa S, Al Momen A, Al Sayegh, Al Jaouni S, Nasrullah Z, Al Saeed H, et al. Management of Painful Vaso-Occlusive Crisis of Sickle-Cell Anemia: Consensus Opinion. Clinical and Applied Thrombosis/Hemosta-sis. 2010; 16 (4):365-376.18. Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med 2001; 137:398–407.19. Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA et al. Whole blood tissue factor pro-coagulant activity is elevated in patients with sickle cell disease. Blood 1998; 91:4216-4223.20. Ataga KI, Moore CG, Hillery CA, , Jones S, Whinna HC, Strayhorn D et al.Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica 2008; 93:20-26 21. Etienne-Julan M, Belloy MS, Decastel M,Dougaparsad S, Ravion S, Hardy-Dessources MD.Childhood sickle cell crises: clinical severity, inflammatory markers and the role of interleukin-8. Haematologica 2004;89:863-864.22. Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, Kinney TR. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–16.23. Ohene-Frempong K, Weiner S J, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow C H, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998;91:288–294.24. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–1644.25. Shet AS, Aras O, Gupta K, Hass MJ, Rausch J, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue -positive microparticles derived from cells and monocytes. Blood 2003; 102:2678–2683.26. Ballas SK. Current issues in sickle cell pain and its management. Hematology Am Soc Hematol Educ Program 2007:97-105.
Epidemiología y fisiopatología

30
Diagnóstico y Asesoramiento Genético.02
2.1. Pesquisa neonatal de drepanocitosis.Olimpia C. Pérez-Bández / Médico hematólogo. El despistaje neonatal conduce al diagnóstico temprano de la enfermedad drepanocí-tica, que unido a la educación de los padres y al cuidado integral del paciente a cargo de profesionales expertos, reduce considerablemente la morbimortalidad y aumenta la esperanza de vida de estos pacientes1, 2; además también permite descartar niños con otras hemoglobinopatías, a los portadores, y en algunos casos niños con sindromes α talasemia3.
Para que la pesquisa neonatal de hemoglobinopatías a nivel nacional sea eficiente, se requiere de un programa gubernamental. En los actuales momentos se realiza sólo en algunos centros que están dentro del radio de acción del proyecto de pesquisa de en-fermedades heredo metabólicas, llevado por el Instituto de Estudios Avanzados (IDEA). El estudio de hemoglobinas se lleva a cabo en el laboratorio de hemoglobinas anor-males, que funciona en el Instituto Anatómico de la Universidad Central de Venezuela.
MétodosLa mayoría de los programas de pesquisa neonatal, utilizan la técnica de enfoque isoeléctrico de un eluído de manchas de sangre seca3, 4, pocos emplean cromatografía líquida a alta presión, conocida por sus siglas en inglés HPLC y electroforesis en ace-tato de celulosa como método de detección inicial4.
La sensibilidad y especificidad de ambos métodos es excelente, sin embargo algunos casos de drepanocitosis pueden quedar sin identificar debido a3:
• Prematuridad extrema.• Transfusión antes de la toma de muestra.• Error en la identificación de la muestra.• Errores humanos en el laboratorio.• Imposibilidad de encontrar al niño después de egresar
de la institución hospitalaria.
La pesquisa neonatal permite identificar a la Hb H, la cual es reportada en porcentaje con respecto al total de la hemoglobina presente. En el recién nacido normal la he-moglobina fetal (Hb F) está en mayor proporción que la hemoglobina A (Hb A), por eso la mayoría de recién nacidos presentan Hb FA.
En el recién nacido, podemos identificar los cuatro genotipos mas comunes descritos en la abla 2.1 Los recién nacidos con hemoglobinopatías, igual como sucede en los recién nacidos normales, presentan predominantemente hemoglobina fetal (Hb F).

31
Aquellos con enfermedad drepanocítica mostrarán:• Hb S en ausencia de Hb A (Hb FS).• Hb S con otra variante.
· Hb FSC. · Hb FSD Punjab
· Hb FSA, donde la de Hb S está en mayor proporción que la A, en este caso se trata de Enfermedad–Sβ+talasemia.
Tabla 2.1. Diagnóstico de los sindromes drepanocíticos.Separación de hemoglobina (electroforesis de hemoglobina, enfoque isoeléctrico o cromatografía líquida a alta presión5.
1. β0 talasemia. Talasemia donde hay ausencia de producción de cadenas β.2. β+ talasemia. Talasemia donde hay reducción pero no ausencia de producción de cadenas β.3. Las hemoglobinas se reportan en orden mayor producción a menor producción. Ej. FS = F>S; FSA= F>S>A.4. En ocasiones la cantidad de Hb A es tan pequeña que no se puede medir.5. En casos raros, la anemia drepanocítica (Hb SS) puede cursar con Hb F tan elevada que justifica hacer el diagnóstico diferencial con Hb SS asociada a persistencia hereditaria de Hb F (HbSS-PHHF). Se debe realizar coloración de extendidos de sangre, para observar la distribución intracelular de Hb F.NB. Cuando se utiliza la electroforesis como método de separación de hemoglobinas, la Hb A2 no se puede medir ya que no se separa de la Hb C.
Cuando en la pesquisa neonatal se encuentra solamente Hb FS, se plantean los si-guientes diagnósticos:
• Anemia Drepanocítica (Hb SS).• Enfermedad-S β0 talasemia.• Enfermedad-S δβ talasemia.• Anemia Drepanocítica (Hb SS) con persistencia de hemoglobina fetal. • Muchos niños con S-β+ talasemia presentan Hb SF en la pesquisa, esto sucede-
Tipos desíndromes
EDAD
Neonatal 6 semanas> 5 años
Hb A (%) Hb S (%) Hb F (%) Hb A2 (%) Hb C (%)
Anemia drepanocítica (Hb SS) FS3 FS 0 75-95 2-255 <3,5 0
Enfermedad drepanocítica-Sβ0 talasemia1 FS FS 0 80-92 2-15 3,5-7 0
Enfermedad drepanocítica-SC FSC FSC 0 45-50 1-5 ND 45-50
Enfermedad drepanocítica-Sβ+ talasemia2
FSA o FS4 FSA 5-30 65-90 2-10 3,5-6 0
Rasgo drepanocítico FA S FAS 50 - 60 35-45 <2 <3,5 0
Normal FA FA o AF 95 -98 0 <2 <3,5 0

32
cuando la cantidad de Hb A es tan pequeña que no puede ser detectada3,5. Los niños que resulten positivos para Hb S en el momento del despistaje, deben in-cluirse en el programa de atención integral, iniciar penicilina profiláctica antes de los dos meses y educar a los padres en relación a la enfermedad6. A los dos meses de edad, se debe practicar el estudio confirmatorio a aquellos niños que en el despistaje resultaron con hemoglobinas anormales. El estudio puede realizarse mediante elec-troforesis de hemoglobina (acetato de celulosa y agar citrato), electroforesis capilar, enfoque iso eléctrico o cromatografía líquida a alta presión. En los casos donde se plantea el diagnóstico de talasemia parece ser útil el estudio de los padres o de ADN6.
Referencias1. Vichinsky E, Hurts D, Earles A, Kleeman K, Lubin. B Newborn screening for sickle cell disease: effect on mortality. Pediatrics 1988; 81:749-55.2. Eckman JR Dent, Bender D, Henson MA, Myers C Follow-up of infants detected by neonatal screening in Georgia, Louisiana, and Mississippi (abstr). Proceeding of the 14th National Screening symposium. Associa-tion of Public Health Laboratories. Washington DC, 1999.3. National Institutes of Health, National Heart, Lung and Blood Institute. Neonatal Screening. Chap 2 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethes-da.2002; 7-14.4. Pass KA, Lane PA, Fernhoff PM, Hinton CF, Panny SR, Parks JS et al. U.S. new-born screening system gui-deline II: follow-up of children, diagnosis, management, and evaluation. Statement of the Council of Regional Networks for Genetics Services. J pediatrics 2000; 137 (Supll):S1-46.5. Lane PA: Sickle cell disease. Pediatr Clin North Am 1996; 43:639-64. Modificada por Sickle Cell Disease Critical Elements of Care. 2006. Center for Children with Special Health Needs Children’s Hospital and Re-gional Medical Center, Seattle, WA6. American Academy of Pediatrics. Section of Hemathology/Oncology. Committee of Genetics. Health super-vision for children with sickle cell disease. Pediatrics. 2002;109 (3):526-535.
2.2. Diagnóstico posterior al período neonatal.Betty Urdaneta de Ramos / Médico hematólogo.
El estudio confirmatorio de los recién nacidos que presentaron una banda de hemoglo-bina S en las pruebas de pesquisa y de pacientes mayores que consultan por síntomas relacionados con enfermedad drepanocítica, requiere la separación de la hemoglobina S mediante alguno de los siguientes métodos1,2:
• Uso combinado de electroforesis a pH alcalino y electroforesis a pH ácido. • Enfoque isoeléctrico.• Cromatografía líquida de alta presión. • Electroforesis micro capilar.
.02

33
Personas que requieren estudio para descartar la presencia de hemoglobina S2. • Persona de cualquier edad, con antecedentes familiar de sindrome drepanocítico,
clínica de anemia hemolítica, y cualquiera de las siguientes manifestaciones: · Anemia + dolor. · Anemia + meningitis + sepsis + neumococo. · Anemia + enfermedad vásculo cerebral aguda. · Anemia + sindrome torácico agudo. · Anemia + priapismo. · Anemia + úlceras en las piernas. · Anemia + hiperesplenismo agudo. · Anemia + pérdida brusca de la visión.
• Familiares de una persona con sindrome drepanocítico. · Padres de una persona con enfermedad drepanocítica. · Hijos(as) de hombre o mujer con enfermedad drepanocítica. · Hermanos(as) de una persona con diagnóstico de enfermedad drepanocítica. · Pareja de un hombre o mujer con enfermedad drepanocítica. · Pareja de un hombre o mujer portador (a) de rasgo drepanocítico.
Procedimiento para la toma de muestra3.• Sangre venosa, 3-5 mL de sangre con anticoagulante (EDTA o heparina). Se debe
conservar y transportar a una temperatura de 4 o C.• Tomar la muestra antes de transfundir o después de tres meses de practicada la
última transfusión.• Identificar la muestra con nombres y apellidos.• Indicar fecha de la toma de muestra.• Enviar al laboratorio en las primeras 24 o 48 horas.
En caso de sospechar la existencia de hemoglobina inestable lo ideal es que la mues-tra se tome en el laboratorio donde va a ser procesada, ya que el estudio ha de reali-zarse de inmediato. En caso de electroforesis convencional, analizar la hemoglobina obtenida por hemolisado rápido y la de una solución de hemoglobina.
• La muestra debe acompañarse de una hoja de referencia que incluya los siguientes datos: · Nombres y apellidos del paciente. · Fecha de nacimiento (edad). · Lugar de nacimiento. · Lugar de nacimiento de ambos padres. · Posible diagnóstico. · Fecha de la última transfusión.
• El estudio de ambos padres contribuye al diagnóstico.
Diagnóstico y Asesoramiento Genético

34
Referencias 1. Pass KA, Lane PA, Fernhoff PM, Hinton CF, Panny SR, Parks JS, et al. U.S. new-born screening system gui-deline II: follow-up of children, diagnosis, management, and evaluation. Statement of the Council of Regional Networks for Genetics Services. J pediatrics 2000; 137 (Supll):S1-46.2. Cantalejo López MA. Protocolo de anemia de células falciformes o drepanocitosis. Bol S Vasco Nav Pediatr 2005; 38: 20-38.3. Manual de procedimientos del Laboratorio de Hemoglobinas Anormales. Banco Municipal de Sangre del Distrito Metropolitano.2006.
2.3. Asesoramiento GenéticoClementina Landolfi / Médico hematólogo
Debido a que la enfermedad drepanocítica es una condición hereditaria, el ase-soramiento genético constituye un elemento importante en la atención inte-gral del paciente, el cual no sólo incluye lo relacionado al patrón de heren-cia, sino también la discusión sobre el origen y significado de los diversos genes de la hemoglobina y las opciones futuras en cuanto a la reproducción. Cuando un paciente tiene claro todos los aspectos concernientes a su enfermedad, tiene la oportunidad de tomar decisiones mas acertadas en relación con el tratamiento médico y planificar mejor la familia1.
ObjetivosEducar e instruir sobre:
• Enfermedad drepanocítica.• Rasgo drepanocítico.• Necesidad de tomar decisiones en relación a la sexualidad y concepción.
Personas que deben realizar el asesoramiento genético2
• Médico especialista con entrenamiento en asesoramiento para hemoglobinopatías.
Quiénes deben recibir asesoramiento genético1
• Padres y familiares de niños con rasgo o niño (a) con enfermedad drepanocítica (Hb SS, ED-SC, ED-S β0 tal, ED- Sβ+ tal, ED-SD, asociación de Hb S con hemoglo-binas anormales diferentes a la Hb C y Hb D).
• Mujer u hombre con rasgo o enfermedad drepanocítica (Hb SS, ED-SC, ED-S β0 tal, ED- Sβ+ tal, ED-SD, asociación de Hb S con otras hemoglobinas anormales diferentes a la Hb C y Hb D.
Momento de realizar el asesoramiento1
• Al realizarse el diagnóstico.
.02

35
• Durante la adolescencia y antes de cada embarazo, al paciente con enfermedad drepanocítica o a la persona portadora de rasgo drepanocítico.
• En cualquier momento cuando el paciente o el familiar lo solicite y cuando el mé-dico lo considere pertinente.
Puntos que deben discutirse durante el asesoramiento1-3
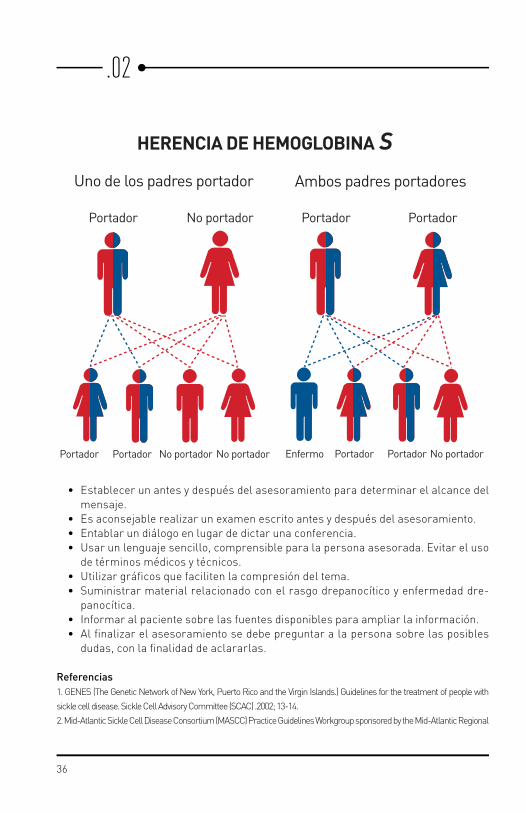
• Informar sobre la finalidad del asesoramiento.• Función de la hemoglobina normal.• Bases genéticas de la Hb S.• Diferencias entre rasgo drepanocítico y enfermedad drepanocítica.• Porcentaje de probabilidades en cada embarazo, de tener: niños sanos, con rasgo
drepanocítico, o con enfermedad drepanocítica (Figura 2.1).• Opciones de planificación familiar.• Significado médico de la Hb S y otras hemoglobinas anormales asociadas, si fuese
el caso.• Tipos de enfermedad drepanocítica.• Manifestaciones clínicas de la enfermedad drepanocítica.• Variabilidad entre una persona a otra, de las manifestaciones clínicas de la enfer-
medad drepanocítica. • Complicaciones que pudieran surgir en el curso de la enfermedad.• Imposibilidad de predecir el curso de la enfermedad.• Expectativas de vida de una persona con enfermedad drepanocítica.• Opciones de tratamiento.• Importancia de:
· Control estricto con el hematólogo. · Asistir a consulta de niño sano con el pediatra. · Acudir a las consultas con los especialistas que sugiera el hematólogo. · Practicar todas las exploraciones solicitadas por el hematólogo. · Adherirse al tratamiento indicado por los especialistas. · Cumplir las medidas para prevenir complicaciones.
• Necesidad de estudio de todo el grupo familiar.• Diferencia entre rasgo drepanocítico y enfermedad drepanocítica.• Manifestaciones clínicas que pudiera presentar una persona con rasgo drepano-
cítico.• Importancia de pertenecer a una asociación de pacientes con enfermedad dre-
panocítica.• ¿Cómo contactar a la(s) asociación(es) de pacientes con enfermedad drepanocíti-
ca, existente(s) en el país o a la Asociación Venezolana de Drepanocitosis y Tala-semias (AVDT)?
Instrucciones para el asesoramiento3
• Elaborar un protocolo de entrevista con el propósito de no dejar puntos sin tratar.
Diagnóstico y Asesoramiento Genético
36
HERENCIA DE HEMOGLOBINA SUno de los padres portador Ambos padres portadores
Portador Enfermo No portadorNo portador No portador
No portador
PortadorPortador Portador
PortadorPortadorPortador
• Establecer un antes y después del asesoramiento para determinar el alcance del mensaje.
• Es aconsejable realizar un examen escrito antes y después del asesoramiento.• Entablar un diálogo en lugar de dictar una conferencia.• Usar un lenguaje sencillo, comprensible para la persona asesorada. Evitar el uso
de términos médicos y técnicos.• Utilizar gráficos que faciliten la compresión del tema. • Suministrar material relacionado con el rasgo drepanocítico y enfermedad dre-
panocítica.• Informar al paciente sobre las fuentes disponibles para ampliar la información.• Al finalizar el asesoramiento se debe preguntar a la persona sobre las posibles
dudas, con la finalidad de aclararlas.
Referencias 1. GENES (The Genetic Network of New York, Puerto Rico and the Virgin Islands.) Guidelines for the treatment of people with sickle cell disease. Sickle Cell Advisory Committee (SCAC) .2002; 13-14.2. Mid-Atlantic Sickle Cell Disease Consortium (MASCC) Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional
.02

37
Diagnóstico y Asesoramiento Genético
Human Genetics Network (MARHGN). Diagnosis, guidelines for comprehensive care, and protocols for management of acuteand chronic complications.2001; 7-8.3. National Institutes of Health, National Heart, Lung and Blood Institute. Child and Adolescent Health Care Maintenance. Chap 4 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda.2002; 19-22

38
Medidas Generales.033.1. En el niñoMaritza Suárez, Adrián Cárdenas / Médicos hematólogos
El manejo adecuado de la enfermedad drepanocítica durante la infancia, repercute enormemente en las condiciones físicas y calidad de vida del adulto y el mantenimiento del estado de salud de estos niños ha de incluir unas series de medidas para prevenir complicaciones propias o específicas de su enfermedad y disminuir el impacto que ocasiona el diagnóstico en el paciente y su familia. El pediatra juega papel importante en el diagnóstico de la enfermedad, ya que es el profesional que entra en contacto por primera vez con el paciente y sus familiares, sospechando o haciendo el diagnóstico y orientando a los familiares acerca de la enfermedad y de la necesidad de seguir sus controles con el hematólogo pediatra y por equipo multidisciplinario integrado, ade-más, por: pediatra, psicólogo, neurólogo, nutrólogo, odontólogo, trabajador social, en-fermera especializada, además de nefrólogo, cardiólogo, oftalmólogo y neumonólogo1.
Dieta, nutrición y suplemento de vitaminasA menos que el niño reciba lactancia materna, debe recibir fórmula fortificada con hie-rro durante el primer año. La dieta será la óptima en todas las edades. Se recomienda el uso de multivitamínicos que contienen hierro, hasta los 2 años de edad y aún mas tarde si la dieta es errática o no balanceada. La rutina con ácido fólico (1mg/d) no es esencial para todos los pacientes, pero es necesaria si existe hemólisis importante y si se sospecha que el niño no ingiere alimentos ricos en esta vitamina2. En niños con en-fermedad drepanocítica SC y Sβ+talasemia es controversial3. Si se considera necesa-rio indicar ácido fólico se puede hacer siguiendo las dosis presentadas en la Tabla 3.1.
Tabla 3.1. Requerimientos de ácido fólico según la edad4.
Profilaxia con penicilina La medida mas importante en el manejo rutinario de los niños con enfermedad drepa-nocítica SS y Sβ0 talasemia es la profilaxis con penicilina oral dos veces diarias, para prevenir la infección por neumococo1,2,4. La rutina de penicilina profiláctica en niños con enfermedad drepanocítica SC y Sβ+ talasemia es controversial3. El tratamiento profiláctico, puede detenerse a los 5 años5.
• Penicilina profiláctica1,2,4. · 2 meses a 3 años.
Edad Dosis
Desde los 12 meses hasta los 5 años 5mg una vez a la semana
Desde los 5 años hasta la adolescencia 5mg dos veces a la semana
Durante la adolescencia 5mg diarios

39
• Penicilina VK, 125 mg vía oral, dos veces al día. · 3años-5 años.
• Penicilina VK, 250 mg vía oral dos veces al día. · Una alternativa de la penicilina oral es una inyección de 1.2 millones de unida-
des de penicilina G benzatínica cada 3 semanas1. · Otra alternativa es la amoxicilina a razón de 20mg/kg/d5. · Para pacientes alérgicos a la penicilina, emplear etil succinato de eritromicina
(20mg/Kg) dividido en dos dosis diarias1,2.
InmunizaciónAdemás de las de rutina, los niños con enfermedad drepanocítica, deben recibir inmu-nización contra neumococos, hepatitis B y Haemophilus influenzae1.• Streptococcus pneumoniae. • Vacunas
· Heptavalente (PCV7, siglas en inglés). Contiene polisacáridos capsulares de los serotipos 4, 6B, 9V, 14, 18C, 19F y 23F, conjugados con el transportador proteico CRM197, variante no tóxica de la toxina diftérica. En Venezuela, la cobertura que brinda la PCV7 representa el 61,4% de los casos identificados6.
· Veintitrésvalente (PPSV23, siglas en inglés). Se recomienda a partir de los 2 años de edad, incluye los siguientes 23 serotipos de polisacáridos purificados: 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F y 33F. La PPSV23 ha reportado una eficacia promedio del 57% en niños mayores de 6 años de edad. Sus anticuerpos se mantienen en concentraciones protectoras durante un máximo de 5 años, con disminución a niveles previos a la vacunación a los 10 años, la cual será más acelerado en niños inmunocom-prometidos, dentro de los cuales se incluyen a los pacientes pediátricos con anemia falciforme7,8.
· Trecevalente (PCV13, siglas en inglés). Incluye los serotipos 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F y 23F conjugados con CRM197.
· Decavalente (PCV10, siglas en inglés). Vacuna conjugada antineumocóccica de polisacáridos (adsorbida), comprende los serotipos 1, 4, 5, 6B, 7F, 9V, 14, 18C, 19F y 23F. Esta vacuna tiene 3 proteínas transportadoras que son: proteína D (derivada de Haemophilus influenzae no tipificable), toxoide tetánico a la cual se fija el serotipo 18C y toxoide diftérico a la cual se fija el serotipo 19F.
En la tabla 3.2 se presenta el esquema de inmunización con las diferentes vacunas antineumococos.• Haemophilus influenzae.
· Vacunación de rutina.• Neisseria meningitides.
· No se justifica vacunación a menos que haya exposición.• Infecciones por virus de la influenza.

40
· Vacunación anual.• Hepatitis B.
· Niños. Vacunación de rutina. · .Adultos. Cuando sean seronegativos.
Tabla 3.2. Esquema de vacunación contra neumococos9.
Fuente: Primer Consenso Venezolano de Enfermedad Neumocóccica. Archivos Venezolanos de Puericultura Y Pediatría Abril, 2009; volumen 72 Suplemento1
Medicación crónica.El uso de ácido fólico y penicilina profiláctica fue ya discutido. Los otros medicamentos que pudieran recibir los pacientes con enfermedad drepanocítica, son hidroxiurea y quelante de hierro, los cuales serán discutidos en los capítulos correspondientes, igual que las transfusiones crónicas.
Tanto el hematólogo como la enfermera especializada, han de estar atentos a la ad-herencia al tratamiento por parte del paciente e insistir en cada consulta, sobre la importancia de su cumplimiento.
Practicat en todos los pacientes una evaluación médica integral periódica con el pro-pósito de documentar manifestaciones previas de la enfermedad, datos basales del examen físico y de valores de laboratorio, evaluar el crecimiento, detectar síntomas y signos de daño crónico y elaborar planes individuales2. En cada visita el especialista debe enfatizar la enseñanza a los familiares de como palpar el bazo, de la importancia de las inmunizaciones, de la adecuada alimentación, de la profilaxis con penicilina, de los signos y síntomas de alerta, que les permitan tomar la conducta adecuada. Ver guía de educación de pacientes y familiares.
Frecuencia de las consultas con el hematólogo.• Depende de la edad. En los primeros dos años de vida los pacientes tienen mayor
Esquema de administración (edad)
1ra. dosis 2da. dosis 3ra. dosis Refuerzo 1ra. dosisRevacunación (lapso
después de última dosis)2 meses 4 meses 6 meses 12-15
meses ≥ 24 meses
PCV7 x x x x
PCV13 x x x x
PCV10 x x x x
PPSV23 No indicada en niños menores de 24 meses x
5 añosEn niños <10 años de edad
con drepanocitosis conside-rar 3-5 años
.03

41
riesgo de sepsis y de hacer otras complicaciones como secuestro esplénico, crisis dolorosa en pies y manos, motivo por el cual se recomiendan controles cada 2-3 meses. Después de 2 años de edad la frecuencia de las consultas dependerá de las necesidades del paciente y de sus familiares, pero debe ser al menos cada 6 meses1 (Tabla 3.3).
• El hematólogo también debe evaluar al paciente en la sala de emergencia y al salir de una hospitalización10.
Tabla 3.3. Esquema recomendado para la consulta con el hematólogo1,3
*National Institutes of Health, National Heart, Lung and Blood Institute1. **American Academy of Pediatrics2.
Estudios que deben solicitarse una vez realizado el diagnóstico de enfermedad drepanocítica11.
• Hematología (hemoglobina, hematocrito, reticulocitos, índices hematimétricos, leucocitos, hemograma de Schilling y plaquetas).
• Determinación en suero de: · Creatinina y BUN. · Electrolitos. · Bilirrubina total y fraccionada, ALT, AST, proteínas totales, albúmina, globuli-
nas, LDH. · Hierro sérico, capacidad total de fijar hierro (TIBC siglas de total iron binding
capacity), saturación de transferrina. · Ferritina a partir de los 2 años.
• Uroanálisis.• Proteinuria en 24 horas (si es mayor de 8 años).• Serología para: hepatitis B (antígeno de superficie y anti core), hepatitis C y virus
de la inmunodeficiencia humana.• Grupo sanguíneo e inmunofenotipo (al menos para Rh, Duffy y Kell). Lo ideal es hacerlo antes de
recibir la primera transfusión. Si ha sido transfundido, debe tener mínimo tres meses sin recibir transfusión.
• Ultrasonido abdominal.• Eco-doppler transcraneal (si tiene entre 2-16 años de edad) (Ver sección de ictus cerebral agudo).• RMN craneal a partir de los 4 años de edad y hasta los 10 años, con el propósito de investigar
infartos silentes.
Edad Frecuencia de las citas
<2 años* Cada 2 o 3 meses
2 años - 5 años** Cada 6 meses (ED-SS y ED- Sβo talasemia)
Mínimo cada 12 meses (ED-SC y ED- Sβ+ talasemia)
Medidas Generales

42
• Espirometría. Si tiene antecedentes de sindrome torácico agudo.• Estudio de trombofilia, de ser posible.• Electroforesis de hemoglobina de padres, hermanos y pareja (si es un adulto)• Realizar estudios de HLA al paciente y hermanos, en posibles candidatos a trasplante de
médula ósea (TMO).
Seguimiento del paciente• En cada visita de control del paciente el hematólogo realizará lo siguiente9,11:
· Anamnesis minuciosa: crisis dolorosas, ictericia, coluria, nicturia, priapismo, cansancio, fatiga, escolaridad. Revisar si cumple el tratamiento prescrito: ácido fólico, penicilina oral; si ha realizado los exámenes solicitados, si ha recibido el esquema de vacunación acorde a la edad más las vacuna antineumococo y anti Haemophilus influenzae. Si ha estado hospitalizado en otro centro hospitalario o sufrido algún proceso infeccioso que no ameritó hospitalización.
· Examen físico completo, que incluya peso y talla. · Estudios de laboratorio (Tabla 3.4). · Informar a los familiares y pacientes acerca de las medidas preventivas de sa-
lud. Ver guía de educación. · Otros estudios están descritos en las tablas 3.5 y 3.6. · Los niños y sus familiares recibirán orientación en relación a las actividades
recreativas y viajes. • Es aconsejable que los pacientes realicen ejercicio regularmente en base a la auto
limitación. • En la edad escolar participar en la educación física, pero con el debido permiso
para descansar e ingerir líquidos después del ejercicio. • El niño ha de conocer los riesgos a los cuales se expone si realiza ejercicios fuer-
tes. • Es recomendable la asistencia a campamentos, si éstos son especiales para niños
con drepanocitosis, sería mucho mejor1.• Los viajes en aviones presurizados usualmente no representan problemas, sin
embargo el niño debe abrigarse, ingerir líquidos en abundancia y moverse con frecuencia cuando sea posible. Los viajes por encima de los 4.500 m en vehículos no presurizados, representan un riesgo de complicaciones vasooclusivas1.
• Por lo general los viajes en carro, autobús, o tren no se asocian con aumento en el riesgo de complicaciones, aun cuando se aconseja detenerse para descansar y refrescarse.
• Se sugiere al paciente y a sus familiares que consulten a su hematólogo antes emprender un viaje, sobre la conveniencia de llevar consigo un informe donde se indica el diagnóstico, valores hematológicos basales, tratamiento actual, nombre y teléfono de su médico. · Es recomendable que el médico suministre a los familiares del niño, el número
de teléfono donde puede ser localizado en caso de una emergencia1.
.03

43
Tabla 3.4. Evaluación integral rutinaria en niños y adolescentes con enfermedad drepanocítica1,10
Mínimo 1 vez al año. Según criterio clínico. Una sola vez. Documentar en información basal.
Infancia Niñez temprana Niñez tardía
Adoles-cencia
Neo-natal
2 meses
4 meses
6 meses
9 meses
12 meses
15 meses
18 meses
2-5 años
5-13 años
13-21 años
Educación x x x x x x x x x x x
Consejo genético x
Evaluación psicosocial x x x x x x x x x x x
Evaluación hematólogo
Historia x x x x x x x x x x x
Examen físico x x x x x x x x x x x
Laboratorio
Hematología incluya índices y reticulocitos
x x x x x x x x x x x
Hb fetal x x 2 años
Pruebas hepáticas: ALT, bilirrubina
x x x x
Pruebas renales: uroanálisis, creatinina,
x x x x
Patrón de hierro x x x x x x
Otras evaluaciones
Oximetría de pulso x x x x x x x x x x x
Fenotipo antígenos eritrocita-rios
Medidas Generales
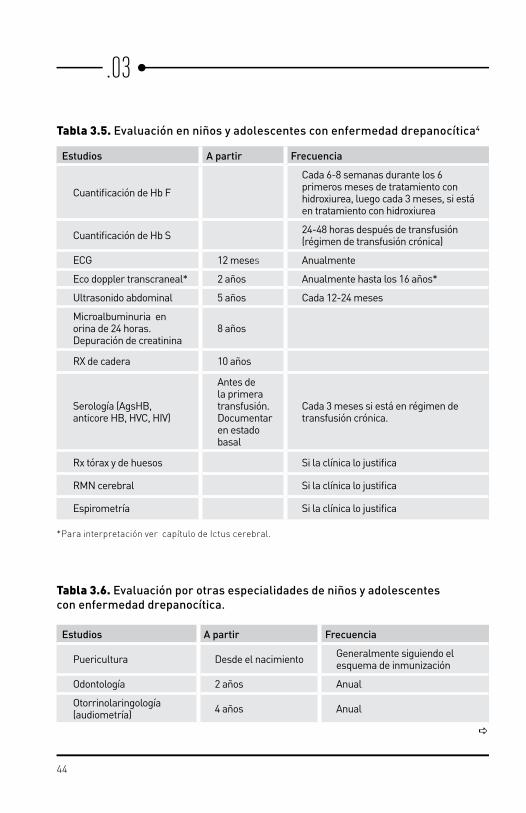
44
Tabla 3.5. Evaluación en niños y adolescentes con enfermedad drepanocítica4
*Para interpretación ver capítulo de Ictus cerebral.
Tabla 3.6. Evaluación por otras especialidades de niños y adolescentes con enfermedad drepanocítica.
Estudios A partir Frecuencia
Cuantificación de Hb F
Cada 6-8 semanas durante los 6 primeros meses de tratamiento con hidroxiurea, luego cada 3 meses, si está en tratamiento con hidroxiurea
Cuantificación de Hb S 24-48 horas después de transfusión (régimen de transfusión crónica)
ECG 12 meses Anualmente
Eco doppler transcraneal* 2 años Anualmente hasta los 16 años*
Ultrasonido abdominal 5 años Cada 12-24 meses
Microalbuminuria en orina de 24 horas.Depuración de creatinina
8 años
RX de cadera 10 años
Serología (AgsHB, anticore HB, HVC, HIV)
Antes de la primera transfusión. Documentar en estado basal
Cada 3 meses si está en régimen de transfusión crónica.
Rx tórax y de huesos Si la clínica lo justifica
RMN cerebral Si la clínica lo justifica
Espirometría Si la clínica lo justifica
Estudios A partir Frecuencia
Puericultura Desde el nacimiento Generalmente siguiendo el esquema de inmunización
Odontología 2 años Anual
Otorrinolaringología (audiometría) 4 años Anual
.03
a

45
Referencias1. National Institutes of Health, National Heart, Lung and Blood Institute. Child Health Care Mainte-nance. Chap 5 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda. 2002; 25-34. 2. GENES (The Genetic Network of New York, Puerto Rico and the Virgin Islands.) Guidelines for the treatment of people with sickle cell disease. Sickle Cell Advisory Committee (SCAC) 2002; 23-25.3. American Academy of Pediatrics. Section of Hemathology/Oncology. Committee of Genetics. Health supervision for children with sickle cell disease. Pediatrics 2002;109:526-535.4. Cervera A. Salud general infantil en Guía de manejo de las enfermedades falciformes en Asociación Española de Hematología. Grupo de Eritropatología. 2009; 3-11.5. Cober MP, Phelps SJ. Penicillin Prophylaxis in Children with Sickle Cell Disease. J Pediatr Pharmacol Ther 2010;15:152–1596. Gabastou JM, Agudelo CI, Brandileoine MC, Castañeda E, Silva de Lemos AP, DiFabio Jl y col. Carac-terización de aislamientos invasivos de S pneumoniae, H influenzae y N meningitidis en América Latina y el Caribe: SIREVA II 2000-20005. Rev Panam Salud Publica-2008 (1)1-15.7. Recommendation of the Advisory Committee on Immunization Practices – Prevention of Pneumococ-cal Disease, Morbidity and Mortality Weekly Report. 46(RR-8):1997; April 4: 1-25.8. Butler JC, Breiman RF, Campbell JF, Lipman HB, Broome CV, Facklam RR. Pneumococcal polysac-charide vaccine efficacy. An evaluation of current recommendations. JAMA 1993;270: 1826-31.9. Primer Consenso Venezolano de Enfermedad Neumocóccica. Archivos Venezolanos de Puericultura Y Pediatría Abril, 2009; volumen 72 Suplemento 1.10. Mid-Atlantic Sickle Cell Disease Consortium (MASCC) Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional Human Genetics Network (MARHGN). Sickle cell disease in children and adolescents: diagnosis, guidelines for comprehensive care, and protocols for management of acute and chronic complications.2001; 1-76.11. Cantalejo López MA. Protocolo de anemia de células falciformes o drepanocitosis. Bol S Vasco Nav Pediatr. 2005; 38: 20-38.
Estudios A partir Frecuencia
Oftalmología (especialista en retina) 8 años Anual
Nefrología Si hay microalbuminuria
NeumonologíaSi hay antecedentes de síndrome torácico agudo
Cardiología
En caso de fatiga, síncope o dolor torá-cico, hipoxia basal, sospecha de hiper-tensión pulmonar
Medidas Generales

46
3.2. En el adolescenteYraida Tersek / Médico hematólogo
La transición de la niñez a la adolescencia, es un período sumamente difícil para los niños con enfermedades crónicas y aquellos con enfermedad drepanocítica no esca-pan de esta realidad. Por otra parte, los adolescentes frecuentemente se sienten frus-trados y presentan dificultad para expresar sus sentimientos; se preocupan por su estatura, desarrollo sexual, manejo del dolor y hasta por la muerte, lo cual lo expresan como rebeldía, depresión, y negativa a asistir a la consulta, seguir un tratamiento y a escuchar los consejos del médico1. A los problemas inherentes a la adolescencia, se suman la existencia de una enfermedad crónica y discapacitante, la transición de niño a adolescente y de adolescente a adulto y por último el cambio de médico y de institución hospitalaria. La transición de adolescencia a adulto debe ser un proceso progresivo, por tal razón merece la creación de un programa o esquema de atención adecuado que le permita al paciente entender su condición y asumir la responsabilidad de su enfermedad.
El programa de atención general al adolescente incluye los siguientes aspectos2:• Educación. Ver guía relacionada con educación del paciente.• Asesoramiento genético.• Evaluación psicosocial. Ver guía relacionada con evaluación
psicosocial del paciente.• Evaluación médica.
Asesoramiento genético• Revisar con el paciente, la genética de la enfermedad drepanocítica.• Hablar sobre las oportunidades de tener niños enfermos,
si la pareja es portadora.• Resaltar la necesidad de descartar hemoglobinopatías en el novio o novia.
Evaluación médica.En la historia y examen físico, cubrir los puntos de la infancia tardía, con énfasis en:
• Eventos vaso-oclusivos.• Examen músculo-esquelético con consideración específica en posible necrosis de
la cabeza del fémur.• Síntomas de colelitiasis.• Asistencia a la escuela y rendimiento, tomando nota de cualquier cambio negativo
en la funcionalidad que haga pensar en enfermedad vascular cerebral.• Hallazgos de enfermedad respiratoria crónica.• Crecimiento y desarrollo.• Palidez de la piel o ictericia. • Examen neurológico completo.
.03

47
El período de transición debe abarcar los siguientes aspectos3:• Referencia a un hematólogo experimentado en el tratamiento de adultos con en-
fermedad drepanocítica.• Redacción de un informe completo de la historia del paciente, que incluya resul-
tados de: · Electrocardiograma y ecocardiograma. · Oftalmología. · Audiometría. · Función pulmonar. · Eco doppler transcraneal. · Otros de interés.
• Promoción de auto cuidado y auto responsabilidad.
Referencias1. National Institutes of Health, National Heart, Lung and Blood Institute. Child and Adolescent Health Care Maintenance. Chap 5 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 35-39.2. Mid-Atlantic Sickle Cell Disease Consortium (MASCC). Mid-Atlantic Regional Human Genetics Network (MARHGN). Sickle cell disease in children and adolescents, in Diagnosis, guidelines for comprehensive care, and protocols for management of acute and chronic complications : 2001; 9-193. GENES (The Genetic Network of New York, Puerto Rico and the Virgin Islands.) Guidelines for the treatment of people with sickle cell disease. Sickle Cell Advisory Committee (SCAC) .2002; 23-25.
3.3. En el adulto jovenDalia Velásquez de Lara / Médico hematólogo.
En los últimos 30 años el pronóstico de la enfermedad drepanocítica, ha mejorado significativamente. Platt y colaboradores1 señalaron que en el caso de anemia drepa-nocítica (Hb SS) las expectativas de vida eran de 42 años para la mujer y 48 años para los hombres, en tanto que para la enfermedad por asociación de Hb S con Hb C, era de 60 años y 68 años respectivamente. En la actualidad hay pacientes que viven hasta la séptima y octava década. En países con programas de salud para pacientes con enfer-medad drepanocítica, más del 90% de los pacientes viven más de 20 años y existe un número significativo mayores de 50 años1. El riesgo de muerte temprana en los adultos está asociado a complicaciones tales como sindrome torácico agudo, anemia aguda, insuficiencia renal crónica y enfermedad pulmonar crónica2.
El aumento de la esperanza de vida ofrece oportunidades de mejorar la calidad de la misma, pero igualmente retos para mantener la salud. Durante las etapas tempranas de la edad adulta se nota un incremento de los episodios dolorosos, la prevalencia de retinopatía también aumenta con la edad, igualmente las probabilidades de insuficien-
Medidas Generales

48
cia renal crónica, enfermedad pulmonar crónica. Las úlceras en las piernas, necrosis de la cabeza del fémur y húmero ocasionan dolor crónico y discapacidad que requieren ajuste social y vocacional. Por otra parte es necesario recordar que los pacientes con enfermedad drepanocítica no están exentos de padecer: hipertensión arterial, diabe-tes, cáncer, asma, artritis, ateroesclerosis y otras enfermedades crónicas propias de las personas adultas2. El hematólogo que trata pacientes adultos, debe tener expe-riencia en el manejo de problemas hematológicos, dolor, úlceras en los miembros, enfermedades renales, medicina del adolescente, psiquiatría, medicina preventiva y abordaje individual, familiar y comunitario de los problemas que plantea la enferme-dad. Esta concepción holística y desarrollada por un solo profesional (y su equipo) es lo que necesitan los pacientes con enfermedad drepanocítica3.
El éxito de las medidas a tomar en el adulto con enfermedad drepanocítica, exige el cumplimiento de cuatro aspectos claves que son4:
• Combinar medidas preventivas y curativas.• Entender bien los mecanismos fisiopatológicos para poder establecer las estra-
tegias adecuadas.• Reforzar los procedimientos de pesquisa de complicaciones.• Al momento que surjan las complicaciones, aplicar un enfoque sistemático em-
pleando maneras más simples.
Las evidencias han demostrado que los adultos con enfermedad drepanocítica deben ser sometidos rutinariamente a pesquisa de enfermedades, igual que el resto de la po-blación, (altamente recomendado), a saber: hipertensión arterial, trastornos lipídicos, cáncer colorectal, cáncer de mama, depresión, prevención primaria de eventos car-diovasculares5. Por ejemplo, los pacientes con enfermedad drepanocítica presentan cifras de presión más bajas que las personas no drepanocíticas de su misma edad, por eso aumentos discretos pudieran significar una complicación renal u otro trastorno6.
Los cuidados del adulto incluyen reconocimiento y tratamiento oportunos de las com-plicaciones, evaluación de la condición social, asesoría y apoyo psicológico y la conti-nuación de la educación del paciente.
Lo anterior es posible solo cuando ellos cumplen adecuadamente con las citas esta-blecidas y exista una excelente relación médico paciente.
Existen muy pocos protocolos o guías dedicadas a los pacientes adultos con enfer-medad drepanocítica, de allí que adoptaremos las medidas sugeridas por el National Institute of Health (NIH)2 de los EE.UU.
En la primera visita, realizar la historia clínica que incluya lo siguiente.Información general:
.03

49
• Nombres y apellidos.• Fecha de nacimiento.• Número telefónico.• Nombre y número telefónico de familiar de contacto.
Elaborar perfil social y sicológico.• Nivel de instrucción (éxito en los estudios).• Historia ocupacional, pasatiempos.• Fuentes de ingresos.• Adhesión a los tratamientos.• Modo de enfrentar la enfermedad (depresión, estrés).• Apoyo y participación de la familia en el cuidado del paciente y modo de enfrentar
complicaciones previas.
Hábitos• Tabaquismo.• Alcohol.• Uso y abuso de drogas ilegales.• Historia sexual incluido el control de natalidad y técnicas de sexo seguro.
Historia de la enfermedad drepanocítica relacionada con complicaciones y otros problemas médicos desde el nacimiento.Características de las crisis dolorosas:
• Frecuencia.• Duración.• Tratamiento habitual en el hogar.• Tratamiento usual en emergencia.• Promedio del número y duración de las hospitalizaciones.
Complicaciones mayores:• Enfermedad vascular cerebral.• Hepática.• Renal.• Oculares (retina).
Tratamientos en el pasado:• Cirugía.• Transfusiones.
· Número. · Reacciones adversas. · Alo anticuerpos.
• Historia de inmunizaciones.
Medidas Generales

50
• Medicación y alergias.• Historia familiar.
Datos objetivos• Examen físico completo.• Hematología (hemoglobina, hematocrito, reticulocitos, índices hematimétricos,
leucocitos, hemograma de Schilling y plaquetas).
Determinación en suero de: · Creatinina y BUN. · Electrolitos. · Bilirrubina total y fraccionada, ALT, AST, proteínas totales, albúmina,
globulinas, LDH. · Hierro sérico, TIBC, saturación de transferrina, ferritina.
(Si ha recibido transfusiones).
• Uroanálisis incluyendo proteinuria en orina de 24 horas.• Serología para: hepatitis B (antígeno de superficie y anti core), hepatitis C y virus
de la inmunodeficiencia humana.• Grupo sanguíneo e inmunofenotipo (al menos para Rh, Duffy y Kell). Si ha sido
transfundido, debe tener mínimo tres meses sin recibir transfusión.• Rx de tórax y otros estudios radiológicos, dependiendo de los hallazgos de la his-
toria y el examen físico. • Ecosonograma abdominal si hay manifestaciones de colelitiasis.• Electrocardiograma en los pacientes mayores o en aquellos con hallazgos de sín-
tomas cardiacos.• Evaluación por oftalmología, incluido el estudio de retina.• Espirometría y evaluación por neumonología si hay antecedentes de tórax agudo.
Seguimiento• Cada 6-12 meses.
· Consulta con el hematólogo y estudios paraclínicos.• Este período puede ser mas corto si el paciente presenta alguna complicación o
está recibiendo hidroxiurea, quelante de hierro u otro medicamento que amerite control mas frecuente.
• En cada visita:• Examen físico completo.• Presión arterial. Los pacientes con enfermedad drepanocítica poseen cifras más
bajas que las personas normales, por lo que es necesario estar atentos a cual-quier aumento que pudiera significar un problema renal o de otra índole.
• Consignar los hallazgos nuevos o relevantes.• Se solicitarán los siguientes estudios ( se realizaran con mas frecuencia si el pa-
.03

51
ciente presenta alguna complicación o está recibiendo hidroxiurea, quelante de hierro u otro medicamento). · Hematología que incluya: hemoglobina, hematocrito, reticulocitos, índices
hematimétricos, leucocitos y fórmula leucocitaria y plaquetas. · Saturación de oxígeno transcutánea. · Electrolitos. · Uroanálisis. · Depuración de creatinina, proteinuria en orina de 24 horas. · Bilirrubina total, directa e indirecta, ALT. · Hierro, TIBC, Saturación de transferrina y ferritina. · Serología para HVC. AgsHB, Anti core, HIV. Si el paciente recibió transfusión
antes de la cita. Cada tres meses si está en régimen de transfusión crónica.
• Cada año han de practicarse las siguientes evaluaciones: · Espirometría (Si hay antecedentes de sindrome torácico agudo). Ver guía de
complicaciones agudas. · Ultrasonido abdominal (si hay antecedentes de colelitiasis). · Otorrinolaringológica: audiometría. · Cardiológica: ECG o ecocardiograma para descartar hipertensión pulmonar.
Ver guía de complicaciones cardíacas. · Nefrológica: si hay hipertensión, micro albuminuria, o hematuria. Ver guía de
complicaciones renales. · Odontológicas. Ver guía de medidas generales.
• Cada 2 años. · Oftalmológica: evaluación de retina. Ver guía de complicaciones oculares · Inmunizaciones5. · Neumococo: Antineumococo con polisacárido neumococo 23 valente (Pneumo-
vax). Refuerzo cada 5 años. Tabla 3.5 · Haemophilis influenzae tipo b. Cada año. · Meningococos (si no la ha recibido). · Hepatitis B, si no la ha recibido. Previo estudio serológico si ha sido transfundi-
do. Ver guía de transfusión.
Medicamentos.• Tratamiento para el dolor.
· El tratamiento en un hospital de día o su equivalente, reduce efectivamente los días de hospitalización5. Las evidencias son sólidas en relación a la hidratación con soluciones hipotónicas, vía endovenosa; el mantenimiento debe limitarse a dar 1,5 veces del volumen requerido, para evitar la sobrehidratación5.
· Dolores moderados e intensos deben tratarse con morfina. No administrar meperidina.
Medidas Generales

52
· Tratamiento farmacológico del dolor y otras complicaciones. Ver guías · correspondientes. ·
• Hidroxiurea. · Reduce la frecuencia de crisis de dolor, sindrome torácico agudo, transfusio-
nes y hospitalizaciones.• Captopril reduce la albuminuria en pacientes normotensos.
Referencias 1. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 3430:1639-1644.2. National Institutes of Health, National Heart, Lung and Blood Institute. Adult Health Care Maintenance. Chap 7 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 41-46.3. Abreu MA. Cuidados de la salud del adulto con enfermedad falciforme. Educación sanitaria. Papel de la atención primaria en Salud general infantil en Guía de manejo de las enfermedades falciformes. Asociación Española de Hematología. Grupo de Eritropatología 2009;17-26.4. Bertolucci P, Galactéros F. Clinical management of adult sickle-cell disease. Current Opinion. Disponible en http://www.co-hematology.com.5. Lottenberg R, Hassell KL. An Evidence-Based Approach to the Treatment of Adults with Sickle Cell Disea-se. Hematology 2005; 58-65.6. Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G et al. Natural history of blood pressu-re in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med 1997;102:171-177.7. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV et al. Effect of hydroxyurea on the fre-quency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317-1322.
3.4. En el adulto mayor de 40 añosJaime Bracho / Médico hematólogo
Actualmente, la esperanza de vida de los pacientes con drepanocitosis se está ele-vando posiblemente debido al diagnóstico precoz, la educación del paciente y al tra-tamiento. Muchas publicaciones reportan grupos con más de 20 años de edad1. Es un fenómeno relativamente frecuente ver pacientes con drepanocitosis viviendo más de 40 años, lo que podría explicarse porque existen sujetos con ventajas fisiológicas a los cuales la medicina moderna les brinda la plataforma para lograr una larga sobrevida. Este grupo de pacientes con más de 40 años de edad es considerado por algunos auto-res como el grupo de edad avanzada 2-4.
Hay un reporte publicado de 5 pacientes con más de 70 años de edad5, sin embargo no
.03

53
se conoce cuáles características puedan ser ventajosas para tal longevidad6. Losada publicó en el 20067, las características clínicas y los resultados de laboratorio en 40 pacientes con drepanocitosis y edades mayores de 40 años radicados en Trinidad y Tobago. La edad promedio fue de 48,82 (40-66) años: 25 (62,5 %) tenían entre 40 y 49 años de edad; 11 (27,5 %) entre 50-59 años; y 4 (10 %) 60 años ó más. Trece (32,5 %) eran del sexo masculino y 27 (67,5 %) del femenino; 33 (82,5 %) presentaron fenotipo SS y 7 (17,5 %) el SC. Las características del cuadro clínico y los resultados de las pruebas de laboratorio mostraron poca severidad de la enfermedad, constituyendo un subgrupo con características fisiopatológicas distintivas.
Obviamente, las complicaciones crónicas serán más frecuentes en los pacientes de ead, por lo cual las medidas estarán enfocadas en esos aspectos como se discute en la sección correspondiente de esta guía.
Un importante aspecto a considerar son los cambios fisiológicos de la menopausia en las pacientes drepanocíticas de este grupo etario especial.
Referencias1. Platt OS, Brambilla DJ, Rosse WF. Mortality in sickle cell disease. N Eng J Med 1994; 330:1639-1644.2. Morris J, Dunn D, Beckford M, Grandison Y, Mason K, Higgs D, et al. The haematology of homozygous SCD after the age of 40 years. Br J Haematol 1991; 77:382-385.3. Hayes RJ, Beckford M, Grandison Y, Mason K, Serjeant BE, Serjeant GR. The haematology of the steady state homozygous SCD: Frecuency, distributions, variations with age and sex, longitudinal observations. Br J Haematol 1985; 59:369-382.4. Shurafa MS, Prasad AS, Ruccknagel DL, Kan YW. Long survival in sickle cell anemia. Am J Hematol 1982; 12:357-655. Steinberg MH, Jackson MS, Ballas SK, Brunson CY, Bookchin R. Sickle cell anemia in septuagenarians. Blood 1995; 86:3997-3998.6. McKerrell TD, Cohen HW, Billet HH. The older sickle cell patient. Am J Hematol 2004; 76:101-106.7. Losada R, Bravo I, Charles K, Capildeo K, Agramonte O, Silva J. Pacientes con drepanocitosis y edad avan-zada en Trinidad y Tobago. Rev Cubana Hematol Inmunol Hemoter 2006; 22(2). Versión on line.
3.5. Papel de la (el) enfermera(o).María Rubio / Médico hematólogo.
La atención del paciente con enfermedad drepanocítica, requiere la participación de un equipo multidisciplinario integrado por: hematólogo, enfermera(o), enfermera(o) auxiliar, trabajador social y psicólogo. Cada servicio de hematología, debe contar por lo menos con una enfermera(o) con experiencia en el manejo de estos pacientes.
Estos profesionales deben poseer conocimiento de la enfermedad, su papel abarca
Medidas Generales

54
desde cuidados preventivos y primarios, manejo del dolor agudo, terapia transfusional y quelante (en caso de usar deferoxamina), educación del paciente y familiares hasta el entrenamiento de sus colegas.
Su actuación es fundamental en la atención integral del paciente, mediante su parti-cipación en:
• Programas educativos dirigidos al paciente y a sus familiares.• Reconocimiento de signos y síntomas de complicación aguda.• Coordinación oportuna de las citas con otras especialidades. • Desarrollo y orientación emocional del paciente así como en la enseñanza rela-
cionada con los derechos y deberes de los mismos.
Responsabilidades1,2.
Observación/Historia/Estado mental• Color de la piel: Detectar si hay palidez, especialmente en niños pequeños. Deter-
mine si apareció bruscamente y si está acompañada de debilidad, letargo, desma-yo u otro comportamiento inapropiado. Precisar si hay ictericia.
• Respiración: Determinar si hay dificultad para respirar, profundidad y calidad de la respiración. Practicar oximetría de pulso. Apreciar si hay dolor en el pecho, disnea o tos.
• Posición del paciente, signos y síntomas de dolor, incluyendo expresión facial, mueca; si está mintiendo; si usa férula.
• Evaluar el estado mental, con o sin dolor. Si hay depresión. Una observación mi-nuciosa puede permitir descubrir intenciones suicidas.
• Determinar los signos vitales: Temperatura, presión arterial, pulso, frecuencia respiratoria. Fiebre, taquicardia e hipotensión pueden ser indicios de sepsis.
• Examinar el estado de hidratación.• Trate de obtener la historia actual, si hubo exposición a una enfermedad viral o
bacteriana.• Si es un paciente masculino, indague sobre priapismo.• Fijarse en la forma de caminar y determine si hay cojera, consulte si hay dolor.• Alertar al médico sobre cualquier anormalidad que haya detectado.
Intervención específica en el tratamiento del paciente en el centro de atención1, 2.• Suministrar los medicamentos prescritos por el médico.• Administrar rápidamente el medicamento para el dolor.• Proveer el antibiótico prescrito por el médico.• Cumplir las órdenes relacionadas con estudios que haya que practicar al paciente.• Reevaluar el estado del paciente cada 15-30 minutos. Determinar si hay cambios.• Revisar el estado de la infusión o transfusión.• Notificar al médico sobre cualquier cambio.
.03

55
Intervención en la consulta1, 2
• Control de signos vitales.• Pesar y determinar la talla.• Tomar la historia nutricional. Enfatizar la importancia de una adecuada ingesta
calórica y la ingestión de abundante cantidad de líquido.• Recordar al paciente que debe recibir todas las inmunizaciones acorde con la
edad.• Averigüar si el paciente cumple el tratamiento indicado.• Recordar al paciente que debe cuidarse las piernas y evitar traumatismos• Revisar los conocimientos del paciente acerca de la enfermedad. • Recordar al paciente y a familiares que la enfermedad se transmite
genéticamente. • Reforzar la educación sobre la naturaleza de la enfermedad, el reconocimiento de
los síntomas, especialmente la temperatura y palpar el bazo (en niños), la impor-tancia de asistir prontamente al centro hospitalario en caso de fiebre, tratamiento del dolor en el hogar, evitar situaciones que pudieran desencadenar una crisis.
• Fomentar la participación en grupos de apoyo (Asociación Venezolana de Drepa-nocitosis y Talasemias)
• Insistir en la necesidad de cumplir las citas con el hematólogo y otros especia-listas.
Referencias1. Guidelines for the Treatment of People with Sickle Cell Disease. SCAC (the Sickle Cell Advisory Committee) of GENES (The Genetic Network of New York, Puerto Rico and the Virgin) .2002: 19-20.2. Brent Sickle Cell & Talassaemia Center. Management of People with Sickle Cell Disease http://www.sic-kle-thal.nwlh.nhs.uk/ ForHealthcareProfessionals/ Management PeopleWithSickleCellDisease.aspx.
3.6. Educación del paciente y de los familiaresMaritza Suárez, Olimpia C. Pérez-Bández / Médicos hematólogos. Educación de los familiares de niños con enfermedad drepanocítica.La educación de los pacientes y padres de niños con enfermedad drepanocítica, junto con la implementación de medidas profilácticas juega un papel preponderante en la prevención y diagnóstico temprano de las complicaciones de la enfermedad. El tema está muy bien tratado en Management of Sickle Cell Disease del NIH1-3.Los hematólogos pediatras deben instruir a los padres, madres y representantes acer-ca de la enfermedad de sus hijos, a saber:
• Importancia de las evaluaciones periódicas con el hematólogo y otros especialistas
• Necesidad de la profilaxia con penicilina (si aplica). Acudir urgentemente a un cen-tro hospitalario, si hay temperatura mayor de 38,5°C.
Medidas Generales

56
• Trascendencia de la adherencia al tratamiento (penicilina, ácido fólico, transfu-sión crónica, hidroxiurea, quelante de hierro, otros.)
• Signos y manejo apropiado de la crisis, dactilitis y otras manifestaciones de dolor; sindrome torácico agudo.
• Enseñar a palpar el bazo. Acudir urgentemente a un centro hospitalario si hay crecimiento del bazo, dolor en hipocondrio izquierdo, palidez y otros síntomas que sugieran incremento de la anemia.
• Manifestaciones del sistema nervioso central (SNC). Signos o síntomas que hagan pensar en ictus cerebral), ataque de isquemia transitoria (AIT). Importancia del eco doppler transcraneal. Acudir urgentemente a un centro hospitalario si hay manifestaciones neurológicas.
• Es aconsejable que los pacientes realicen ejercicio regularmente en base de la autolimitación.
• Enuresis y su relación con la enfermedad drepanocítica.• Pueden participar en la educación física, pero se les debe permitir descansar e
ingerir líquidos después del ejercicio. • El niño debe conocer los riesgos a los cuales se expone si realiza ejercicios fuer-
tes. • Es recomendable la asistencia a campamentos, si éstos son especiales para niños
con drepanocitosis sería mucho mejor.• Los viajes en aviones presurizados usualmente no representan problemas, sin
embargo el niño debe abrigarse, ingerir líquidos en abundancia y moverse con frecuencia cuando sea posible.
• Los viajes por encima de los 4.200 metros en vehículos no presurizados, puede provocar complicaciones vasooclusivas.
• Por lo general los viajes en carro, autobús, o tren no se asocian con aumento en el riesgo de complicaciones, aun cuando se aconseja detenerse para descansar y refrescarse.
• Importancia de las evaluaciones periódicas con el hematólogo y otros especia-listas.
• Se aconseja al paciente y a sus familiares que consulten a su hematólogo antes emprender un viaje, quien le informará si debe llevar consigo un informe donde se indica el diagnóstico, valores hematológicos basales, tratamiento actual, nombre y teléfono de su médico.
• Considerar modalidades de tratamiento en el hogar, dependiendo del grado de instrucción y compromiso de la familia.
• Es conveniente que el médico le de a los familiares del niño, el número de teléfono donde puede ser localizado en caso de una emergencia.
• Los padres deben recibir asesoramiento genético.
Educación del adolescente• Conversar con el paciente.
.03

57
· Sobre la naturaleza de la enfermedad, utilizando un lenguaje acorde con la edad.
· Insistir en la importancia de la adherencia al tratamiento (ácido fólico, transfu-sión crónica, hidroxiurea, quelante de hierro, otros).
· Revisar la genética. Incluyendo la necesidad de practicar detección de hemog-lobinopatías a la futura pareja.
· Revisar los temas relacionados con el impacto de la enfermedad en la adolescencia.
· Insistir en la necesidad de la evaluación periódica con el hematólogo y otros especialistas como forma de prevenir complicaciones y tratarlas de manera precoz si aparecieran.
· Evaluar el rendimiento escolar. · Discutir la participación en las actividades deportivas, la necesidad de ingerir
abundantes líquidos y descansar, aprender a manejar el estrés. · Prevenir y cuidar las úlceras de las piernas. · Revisar el manejo del dolor en el hogar, principios del manejo del mismo y
cuando acudir al centro de emergencia. · Revisar los síntomas de litiasis vesicular. · Revisar los síntomas de necrosis avascular de la cabeza del fémur. · Discutir sobre sexualidad y forma de control de la natalidad, de una manera
apropiada con la edad del paciente. · Hablar sobre los métodos existentes para evitar el embarazo: barreras, admi-
nistración intramuscular de medroxiprogesterona y dosis baja de estrógenos por vía oral.
· En caso de paciente femenina, conversar sobre los riesgos de un embarazo asociado a enfermedad drepanocítica.
· Recordar que si están tomando hidroxiurea (varones y hembras) no deben concebir.
· En caso de pacientes masculinos, hablar sobre priapismo. · Proporcionar asesoramiento sobre educación, educación superior y planifica-
ción profesional. · Abordar la inquietud sobre la transición a la atención médica para el adulto y
facilitar la transferencia cuando sea apropiado y deseado. · Asesorar al paciente y a los padres sobre los efectos adversos del cigarrillo,
alcohol y drogas ilícitas. · Considerar modalidades de tratamiento en el hogar. · Es conveniente que el médico suministre a los familiares del niño, el número
de teléfono donde puede ser localizado en caso de una emergencia. · Los padres deben recibir asesoramiento genético.
Educación del paciente adultoLa primera visita incluye la información al paciente y a los familiares con el propósito
Medidas Generales

58
de que conozcan y entiendan la enfermedad. La actividad educativa se debe enfocar hacia el conocimiento de las complicaciones tales como:
• Infección.• Litiasis vesicular.• Necrosis aséptica.• Sindrome torácico agudo.• Ulceras en las piernas.• Priapismo.
Se debe instruir al paciente en relación a que debe acudir al médico en caso de:• Temperatura de 38°C persistente. • Dolor en tórax, tos y respiración entrecortada.• Síntomas de anemia aguda como: disnea, mareos y debilidad• Dolor abdominal con nauseas o vómitos.• Infección respiratoria con tos productiva.• Síntomas de infección urinaria.• Dolor inusual de cabeza.
La educación del paciente adulto también incluye recomendaciones relacionadas con:
• Protección del tercio inferior de las piernas.• Práctica de sexo seguro.• Importancia a la adhesión al tratamiento (ácido fólico, transfusión crónica, hi-
droxiurea, quelante de hierro, otros. • El riesgo específico del uso del alcohol, marihuana, cocaína, cigarrillos, crack y
otras drogas ilícitas.• Mantener una nutrición balanceada.• Disminución de peso como medida para minimizar el riesgo de necrosis aséptica
de la cabeza del fémur y aparición de diabetes.• Ingestión de ácido fólico, aun cuando se desconoce la dosis necesaria para nor-
malizar la homocisteína. • Ausencia de evidencias científicas que justifiquen el suministro de otras vitaminas.
En relación al dolor• El paciente ha de aprender a manejar el dolor.• Reconocer el sitio de origen e intensidad.• Conocer que medidas y medicamentos utilizar para manejar en el hogar, el dolor
leve y moderado. • Saber cómo actuar en un momento determinado. • La presencia de dolor crónico debido a úlceras en las piernas, necrosis aséptica
u otra complicación, requiere una instrucción especial de modo que el paciente entienda que los objetivos de la terapia del dolor crónico son diferentes a los del
.03

59
control del dolor durante una crisis aguda. • Considerar modalidades de tratamiento en el hogar, dependiendo del grado de
instrucción y compromiso de la familia.
Actividades y ejercicios físicos.• Los adultos jóvenes pueden realizar actividades siempre y cuando eviten estrés,
cansancio, deshidratación y temperaturas extremas.• El paciente debe practicar la moderación y el autocontrol del nivel de esfuerzo.• Es preferible el ejercicio individual a los deportes en equipo.
Vuelos en vehículos aéreos.• Los viajes en aviones presurizados no representan problema y durante el vuelo
necesita ingerir suficiente cantidad de líquido.• En viajes largos, dentro de las posibilidades, levantarse, caminar y movilizarse. • Los pacientes pueden presentar problemas si viajan en aviones no presurizados,
ascienden a montañas por encima de 4.200 m, o vuelan en parapente, globos, ícaros, y otros.
• Existen evidencias anecdóticas que pacientes con función esplénica, ejemplo en-fermedad por asociación de Hb S- Hb C, pueden presentar mayor riesgo durante el vuelo aéreo.
• En caso de antecedentes de haber tenido problemas en viajes anteriores, o la exis-tencia de un problema médico que pudiera incrementar el riesgo de complicacio-nes, es recomendable llevar consigo un informe médico, y solicitar a la aerolínea la provisión de oxígeno suplementario.
La paciente y su pareja necesitan ser instruidas en relación a:• Riesgo durante el embarazo.• Riesgo potencial de aborto espontáneo y parto prematuro.• Riesgo de tener un mortinato.• Uso de métodos anticonceptivos.• Informar que no existe evidencias de que una paciente con enfermedad drepano-
cítica, que toma anticonceptivos, tenga bajas mas problemas de trombosis que una mujer sana.
• Riesgo de infecciones con el uso de dispositivos intrauterinos.
Vocación y profesión.• Estimular al paciente para que culmine su educación y formación profesional.• Alentarlo para que estudie y trabaje en función de su vocación.• Descartar aquellos trabajos o profesiones que impliquen ejercicio físico extremo,
largas horas de trabajo, guardias nocturnas, exposición a temperaturas extremas y/o a condiciones de hipoxia.
• Instruir al paciente para que adquiera excelentes hábitos de estudio y la práctica
Medidas Generales

60
de actividades que conduzcan a la reducción de la frecuencia de complicaciones.• Es posible que sea necesaria la intervención del hematólogo, para que el paciente
pueda conservar su empleo. • Los pacientes con enfermedad cerebral severa, y necrosis avascular, suelen pre-
sentar discapacidad, en estos casos el médico debe proporcionar todos los datos necesarios que permitan al paciente obtener beneficios por parte del Estado o de alguna institución u organización privada.
• Es conveniente que el médico suministre el número de teléfono donde puede ser localizado en caso de una emergencia.
• Los padres deben recibir asesoramiento genético.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Child Health Care Maintenance. Chap 5 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 25-39.2. National Institutes of Health, National Heart, Lung and Blood Institute. Adolescent Health Care and Tran-sition Maintenance. Chap 6 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 35-39.3. National Institutes of Health, National Heart, Lung and Blood Institute. Adult Health. Chap 7 in The Mana-gement and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 41-46.
3.7. Manejo psico-socialAmel Guánchez / Médico hematólogo
Al evaluar la gravedad de la enfermedad drepanocítica, no se debe subestimar su impacto emocional y social. La repercusión sicológica sobre el individuo es debido a tres factores importantes: manejo de síntomas y discapacidades, mantener relaciones adecuadas con el personal de salud y el control de las consecuencias emocionales y sociales de la enfemedad1.
Si bien no existe consenso en relación a indicadores de severidad de la enfermedad drepa-nocítica, el dolor se ha tomado como tal por ser el síntoma mas frecuente y el que causa mayor impacto en la vida del paciente. Para la familia, no hay nada más desgarrador que ver a sus hijos soportar el dolor extremo y que amenaza la vida. El paciente sufre no sólo el dolor en sí, sino también la tensión emocional de episodios impredecibles, miedo a la muerte, y el tiempo perdido; el aislamiento social en la escuela y el trabajo, el rendimiento escolar es bajo cuando se compara con niños sanos de la misma edad, incluso en niños con una baja frecuencia de hospitalización. Otro factor importante que afecta el desenvolvi-miento social, escolar y laboral, es la ocurrencia de úlceras en las piernas, debido a que el
.03

61
dolor y la presencia de un vendaje rezumante, hace que los pacientes se queden en casa2.Por lo general, el personal que atiende a los pacientes en los servicios de emergen-cia carece de la experiencia necesaria y de protocolos adecuados para el tratamiento del dolor; el desacuerdo entre médico-paciente y enfermera-paciente compromete las buenas relaciones interpersonales y el sentido de colaboración. Los desacuerdos ge-neralmente surgen en relación a cuán agresivamente debe ser tratado el dolor ya que con frecuencia el personal de salud considera que el paciente exagera y/o sospecha de adicción a narcóticos3.
Las frecuentes complicaciones sicológicas en los pacientes con enfermedad drepa-nocítica abarcan un amplio rango que va desde estrategias inadecuadas para afrontar la enfermedad, disminución de la calidad de vida, limitación de actividad física y de roles hasta alteraciones neurocognitivas. Las mismas se deben, principalmente, al impacto del dolor y otros síntomas sobre su vida diaria y de las actitudes sociales hacia ellos. Los problemas sicológicos más frecuentes son: ansiedad, depresión, aislamien-to social, agresividad, pocas relaciones interpersonales y bajo rendimiento escolar. Algunos estudios describen altos niveles de ansiedad en los padres, sobreprotección y excesivo sentimiento de responsabilidad y culpa1.
La alteración de la función neurocognitiva es una complicación no visible de la enfer-medad drepanocítica, no detectable por estudios de imagen, más bien es definida por neurosiquiatría y técnicas que estudian el comportamiento. Está asociada a la anemia y a la edad. A diferencia de las manifestaciones neurológicas, no está relacionada a vasooclusión ni a hemólisis intravascular. En adultos se han descrito trastornos en la función cognitiva global, memoria, pruebas de velocidad y de la función ejecutora que empeoran con la edad. En niños con infarto silente antiguo, está informada la altera-ción en las funciones cognitivas, de modo que no está claro si se debe a la anemia o a los infartos4.
Los pacientes con enfermedad drepanocítica y los cuidadores enfrentan a menudo grandes obstáculos en la búsqueda de apoyo psicológico. Generalmente los pacientes no reciben atención de apoyo, incluso de base, que podrían ayudar a reducir la ansiedad y la intensidad del dolor que se produce cuando se presenta un episodio vasooclusivo.Muchos pacientes con enfermedad drepanocítica, viven en comunidades que por lo ge-neral carecen de los servicios que pudieran satisfacer sus necesidades, y los profesio-nales que trabajan en sus centros médicos, a menudo están sobrecargados de trabajo. En los actuales momentos las evidencias que existen en relación a la eficacia de la psicoterapia en enfermedad drepanocítica son limitadas, lo que hace necesario la rea-lización de estudios aleatorios y controlados5.
En ausencia de una cura universal, se recomienda incorporar el tratamiento psicoló-gico a los protocolos de tratamiento de los pacientes con enfermedad drepanocítica y
Medidas Generales

62
ofrecerlo como un elemento de ayuda a mejorar la calidad de vida.Las siguientes son algunas medidas que pudieran ser útiles en el tratamiento de esta enfermedad:
• Reducción de estrés. · Métodos de relajación. · Técnicas de respiración. · Meditación.
• Terapia Cognitivo-Conductual. · Habilidades de afrontamiento adoptando una actitud positiva frente al dolor. · Habilidades de adaptación. Se refieren a la capacidad del paciente para res-
ponder a los síntomas, como dolor.• Asociaciones de apoyo. Las asociaciones de pacientes y de profesionales de apoyo
siguen ofreciendo las mejores fuentes de ayuda y son menos costosas.• Otros factores importantes son aquellos que ayudan a mantener actitudes posi-
tivas como: · La espiritualidad. · El humor. · Tener metas importantes de la vida (por ejemplo, tener un hogar
o una carrera).
Referencias 1. Anie KA. Psychological complications in sickle cell disease Br J Haematol 2005; 129:723–729.2.Alleyne SI, Wint E, Serjeant GR. Social effects of leg ulceration in sickle cell anemia. Southern Med J 1977;70:213-214.3. Barbarin O. The Social and Cultural Context of Coping With Sickle Cell Disease: I. A Review of Biomedical and Psychosocial Issues. J Black Psychology 1999;25: 277-293.4. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Mana-gement. Scientific World Journal 2012;2012:949535. 5. Anie KA, Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database of Systematic Reviews 2012, Issue 2. Art. No.: CD001916. DOI: 10.1002/14651858.CD001916.pub2.
3.8. Cuidados odontológicosBetty Urdaneta de Ramos / Médico hematólogo.
En la enfermedad drepanocítica es primordial el cuidado dental ya que las caries por pequeñas que sean representan un punto de partida de infección. Es necesario que exista una estrecha comunicación entre el hematólogo y el odontólogo que conduzca a una atención óptima del paciente. Por lo tanto se recomienda:
• Un sistema de citas que minimize la inasistencia a la escuela y al trabajo.• Visitas anuales al odontólogo, comenzando después de la erupción del primer
.03

63
diente (6-12 meses de edad).• Algunos niños (basado en la evaluación del dentista) necesitan ser vistos con ma-
yor frecuencia (cada 3-4 meses).• Implementar y constatr un régimen adecuado de higiene oral y dieta. • Tratar intensivamente toda infección dental con un agente antibiótico adecuado.• En todo procedimiento invasivo se recomienda la profilaxis antimicrobiana con:
· Amoxicilina 50mg/kg, máximo 2,0 g VO o ampicilina 50mg/kg, máximo 2g IM o EV una hora antes del procedimiento.
· En caso de alergia a la penicilina, usar clindamicina 20mg/kg, máximo 600mg por vía oral o endovenosa, o azitromicina o claritromicina 15mg/kg por vía oral una hora antes del procedimiento.
· Otros medicamentos alternativos: Cefalexina 50mg/kg (máximo 2,0 g) vía oral y cefazolina 25mg/kg (máximo 1,0 g) IM o EV.
• Preferir la restauración a la extracción.• Si se necesitan extracciones, es preferible la anestesia local.
· No hay evidencia que apoye el uso de un anestésico local sin vasoconstrictor. • En niños inquietos o que no cooperen, implementar la sedación, la cual será lle-
vada a cabo por un anestesiólogo. · Cuando se utiliza óxido nitroso para la sedación por inhalación, evitar:
• La difusión.• La hipoxia mediante la administración de 100% de oxígeno al menos durante 5
minutos después de finalizar el procedimiento. · Estos niños deben estar bien hidratados.
En relación a la anestesia general: · Como alternativa solamente en los casos donde la sedación no tiene éxito y/o
en procedimientos extensos y complicados. · Sólo realizarla en una institución que cuente con hematólogo. · En caso de disfunción cardiovascular, neurológica o pulmonar preexistentes,
transfundir para llevar los valores de hemoglobina a 10-11g/dL (pero no supe-rior a 12g/dL).
· Seguir los cuidados contemplados en la guía de anestesia y cirugía.• El control del dolor se hará según lo recomendado en la guía de dolor.
Referencias1. Sakhalkar VS , Dunbar L, Davidson L, Management Guidelines Pediatric Sickle Cell Disease Program .Uni-versity of Florida. http://residency.pediatrics.med.ufl.edu/files/2012/03/pediatric_sickle_cell_program.pd
Medidas Generales

64
Tratamiento de las complicaciones agudas .04
4.1. Dolor agudoJaime Bracho, Adrián Cárdenas / Médicos hematólogos.
El dolor es la principal característica clínica de la enfermedad drepanocítica, constitu-yendo la primera causa de hospitalización, se estima que hasta un 16% de los pacientes egresados de hospitalización por esta causa sean readmitidos por el mismo motivo1.El dolor es básicamente de tipo nociceptivo, aunque con menor frecuencia de tipo neu-ropático, con o sin evidencia de lesión de tejido neuronal central o periférico. Las lla-madas “crisis dolorosas agudas” de la enfermedad drepanocítica comprenden cuatro fases bien delimitadas, a saber: transducción, transmisión, modulación y percepción, acopladas con cambios específicos en ciertos marcadores de la enfermedad. Un abor-daje diagnóstico y terapéutico inadecuado del dolor pudiera, en ciertos casos, generar un síndrome de dolor crónico de manejo más difícil2,3.
La percepción de la experiencia desagradable de crisis dolorosa en pacientes drepa-nocíticos se ve afectada no solo por factores biológicos intrínsecos y extrínsecos a los eritrocitos sino por factores psicológicos, sociales, culturales y espirituales por lo que resulta mandatorio el tratamiento integral e individualizado acorde a las necesidades y particularidades de cada paciente3. Se recomienda efectuar evaluación anual, del aspecto psicosocial del paciente con la finalidad de conocer el comportamiento del dolor; la evaluación se practicará a intervalos menores si el dolor se presenta con mucha frecuencia4.
Como en otros aspectos de la enfermedad, la educación y entrenamiento del paciente drepanocítico y de su entorno familiar redundará en un manejo adecuado en el hogar, del dolor leve a moderado mediante del uso de medidas de soporte y analgesia vía oral. La frecuencia de la crisis del dolor varía con cada individuo y depende hasta cierto pun-to del fenotipo de la hemoglobina, condición física, enfermedad concurrente y variables sicológicas o sociales.
Factores que determinan la severidad y frecuencia de las crisis de dolor5.• Genéticos
· Niveles de hemoglobina fetal. · Coherencia de talasemias (alfa o beta) disminuye frecuencia de las crisis. · Coherencia de otras hemoglobinas anormales (Hb C). Disminuye frecuencia. · Haplotipos beta. Senegal presenta menos crisis que el Benín o CAR. · Genes epistáticos modificadores. · Género. Las mujeres son hospitalizadas menos que los hombres.
• Celulares · Hay menos crisis cuando:
· La deformabilidad de eritrocitos es menor.

65
· Hay aumento de células densas en estado basal. · Existe anemia severa. · La viscosidad de la sangre es menor.
· Crisis mas frecuentes: · Cifras altas de hemoglobina.
• Ambientales (factores controlables) · Hipoxia nocturna. · Apnea del sueño. · Deficiencia de vitamina A.
Factores que determinan la oclusión de los vasos, en pacientes con enfermedad drepanocítica.
• Factores intrínsecos del eritrocito2. · Polimerización de la hemoglobina S. · Reología del drepanocito.
· Deshidratación celular. · Deformabilidad y fragilidad mecánica del eritrocito. · Células densas.
• Factores extrínsecos al eritrocito. · Viscosidad de la sangre completa. · Inherentes a los leucocitos. · Endoteliales.
· Adhesión de los drepanocitos al endotelio. · Hiperplasia de la íntima.
· Hemostáticos. · Vasculares.
De acuerdo a su duración el dolor se clasifica en agudo y crónico.
Características del dolor agudo1,5:• Es la primera causa de consulta a la emergencia.• Inicio brusco.• Impredecible.• La mayoría de las veces no posee desencadenante.• La intensidad en la escala visual del dolor generalmente es >6.• Dura horas o pocos días.• Ser persistente o recurrente y puede migrar.
Características del dolor crónico5:• La duración es mayor, usualmente 3 a 6 meses. • Debilitante. • Debe diferenciarse de las crisis de dolor agudo recurrente.

66
Condiciones clínicas causantes de dolor en pacientes drepanocíticos:
• Dolor agudo: Síndrome mano-pie, crisis vasooclusiva, síndrome torácico agudo, colecistitis, priapismo, sindrome de cuadrante superior derecho, secuestro es-plénico4.
• Dolor crónico: artritis, artropatía, necrosis aséptica (avascular) de huesos, úlce-ras en piernas, aplastamiento vertebral4.
• Dolor neuropático: La causa es el daño y disfunción de los nervios. No se conoce bien su etiopatogenia en la enfermedad drepanocítica5.
• “Brusco y desgarrador”: Es súbito e incidental, precipitado por movimientos y du-ración de minutos o pocas horas5.
El dolor óseo como resultado de la hipoxia relativa existente en los sinusoides de la médula ósea y los múltiples infartos isquémicos de las trabéculas óseas, puede pre-sentarse en cualquier hueso, pero es más frecuentes en la columna vertebral, pelvis y huesos largos (húmero, tibia y fémur, sobre todo en su segmento distal).
La dactilitis o síndrome “mano-pie” es un fenómeno vasooclusivo limitado que se pro-duce en las manos y los pies de los lactantes menores de 1 año. Afecta a una o más extremidades al mismo tiempo.
El dolor leve y moderado generalmente se trata en el hogar y responde bien al trata-miento con acetaminofén y antiinflamatorios no esteroideos (AINE), mientras que el más intenso generalmente responde a una de las combinaciones de AINE con opiáceos orales6 o por vía parenteral.
Se recomienda que el paciente acuda al centro asistencial más cercano a su domicilio en busca de atención médica en casos de persistencia o agravamiento del dolor a pesar de medidas iniciales y analgesia oral o en presencia de cefalea acompañada o no de signos neurológicos como: parálisis o paresia, dolor torácico asociado o no a síntomas respiratorios, priapismo, signos de flogosis articular, vómitos recurrentes con o sin dolor abdominal.
El tratamiento del dolor en el hospital es problemático. De manera constante los pa-cientes se quejan por no ser tratados de manera adecuada y oportuna cuando acuden a las salas de emergencia, debido a las siguientes causas:
• Por lo general el personal de salud que trabaja en estas áreas desconoce la na-turaleza del dolor agudo en la enfermedad drepanocítica.
• Consideran que el paciente exagera con la finalidad de lograr el suministro de opiáceos.
• Los opiáceos se administran solo o combinado con AINE, en base de resolver una contingencia, por no existir guías de tratamiento.
.04

67
Existen pocos estudios grandes, aleatorios y controlados que pudieran servir de guía en relación a la dosificación de opiáceos. La titulación de la dosis adecuada para elimi-nar el dolor resulta efectiva en los hospitales de día, especializados, pero esta práctica es difícil de implementar en los servicios de emergencia tradicionales5.
En relación a los líquidos, la mayoría de las guías de tratamiento del dolor contem-plan su administración oral o endovenosa, con el objetivo de detener o disminuir la intensidad y duración del dolor. Lottenberg y colaboradores7 consideran que existen evidencias fuertes en cuanto a que la hidratación con soluciones hipotónicas, vía en-dovenosa es necesaria; en tanto que Okomo8 y colaboradores señalan que no existen estudios aleatorios y controlados que demuestren cuál es la mejor vía de suministro, tipo de solución y cantidad necesaria para el tratamiento del dolor agudo en pacientes con enfermedad drepanocítica.
Normas generales del tratamiento del dolor agudo intenso.• Disponer de un protocolo de tratamiento para estos casos. • Notificar al hematólogo tratante la llegada de un paciente con enfermedad drepa-
nocítica al servicio de emergencia.• Los casos complicados, serán tratados por un equipo multidisciplinario.• A su egreso del servicio de emergencia, el paciente debe contar con los analgési-
cos necesarios para evitar la readmisión.• El paciente que reingresa en un lapso no mayor de 48 horas, requiere de una
evaluación minuciosa.
Recomendamos el esquema del National Institutes of Health, National Heart, Lung and Blood Institute4, que a nivel mundial es seguido por la mayoría de las instituciones que manejan pacientes con enfermedad drepanocítica.
Los pasos a seguir son los siguientes:• Evaluación clínica para determinar causa del dolor.• Evaluar de una manera rápida la intensidad del dolor, utilizando una herramienta
sencilla para tal fin (Figura 4.1).Tomar una vía venosa e iniciar una hidratación vigorosa con soluciones hipotónicas como dextrosal 0,45 + 20 mEq NaCl, este último ajustado a los resultados de la química sanguínea. El total de líquidos utilizados no debe exceder a 1,5 veces del volumen de mantenimiento. (incluyendo el volumen de infusión de drogas).
Durante la evaluación es importante establecer una comunicación efectiva y fluida con el paciente y su familia, para hacer una estimación lo más objetiva y real posible de la gravedad del dolor, a través del uso de escalas de medición del dolor, tanto en niños como en adultos, así como erradicar del equipo de salud, pacientes y familiares, los tabúes relacionados a la posibilidad de adicción a los medicamentos opioides.
Tratamiento de las Complicaciones Agudas

68
Grado Escala de Dolor
Sin dolor Dolor moderado Peor dolor posible0 1 2 3 4 5 6 7 8 9 10
La evaluación del dolor incluye:• Anamnesis y exploración física exhaustivas, para determinar la causa y los posi-
bles factores precipitantes. Lo más importante es lo que el propio paciente refiere en relación al dolor (Figura 4.2).
• Categorizar la intensidad para valorar de forma más objetiva la respuesta al tra-tamiento. Para esto se pueden usar escalas validadas de evaluación de intensidad del dolor de acuerdo al grupo etario, tales como Escala del Dolor de Wong-Baker (Figura 4.1) y escalas analógicas visuales.
• Determinación de las características: aparición, localización, intensidad, conco-mitantes, irradiación, atenuantes y causas que lo aumentan, duración y extensión
• Conocimiento de la historia, a saber: frecuencia de dolor agudo en años previos, número de visitas a la sala de emergencia el año pasado, frecuencia y duración de las hospitalizaciones, otros tipos de dolor, historia de medicación para el dolor y medicación actual (Figura 4.3).
• Determinación de: presión arterial, frecuencia respiratoria y cardíaca, tempera-tura, movilidad de articulaciones, morbilidad asociada, hematología con reticulo-citos y plaquetas, oximetría de pulso, uroanálisis y Rx de tórax.
• Si existe morbilidad asociada, incluir en la evaluación: proteína C reactiva, quími-ca sanguínea, hemocultivo, urocultivo, y coprocultivo. En caso de lactantes meno-res de un año y signos de meningitis, efectuar punción lumbar.
• Evaluar la respuesta al tratamiento y posibles efectos secundarios cada 15-30 minutos para ver si se requieren dosis adicionales de medicamento o drogas ad-yuvantes (Figura 4.4).
• Después de 2- 8 horas decidir hospitalización de acuerdo a la respuesta del dolor y evolución del paciente (Figura 4.5).
.04
Figura 4.1.

69
Figura 4.2. Conducta a través de la evaluación clínica.
Determinar tipo yseveridad del dolor
Si No
¿Dolor típico de enfermedad drepanocitica?
Seleccione tratamiento basado en severidad e historia del tratamiento
Determinar causa, evaluar signos y síntomas considerar infección,
comorbilidad y factores precipitantes
Evaluar dolor, tratar de acuerdo a la etiología
Reevaluar de acuerdo a respuesta a terapia y eventos adversos
Si es necesariomodificar tratamiento
Reevaluar frecuentemente
Tratamiento de las Complicaciones Agudas

70
Figura 4.3. Características del dolor.
National Institutes of Health, National Heart, Lung and Blood Institute. Pain. Chap 10 in The Manage-ment and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda.2002; 59-74.
Características del dolor (Inicio, duración, frecuencia)
Evaluación integral
Factores psicosociales y demográficos
Impacto del dolor en su vida diaria
Síntomas relacionados
Determinar causa probable
Relacionado con ED No relacionado con ED
Tratamiento basado en historia Determinar etiología
Tratamiento basadoen evaluación integral
CrónicoAgudo
Momentáneo Persistente
Características, localización,
intensidad(auto-reporte)
.04

71
Figura 4.4. Evaluación del dolor a los 15-20 minutos de iniciado el tratamiento.
National Institutes of Health, National Heart, Lung and Blood Institute. Pain. Chap 10 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda.2002.
Determinar tipo de dolor, iniciación,inflamación, duración, frecuencia
Determine síntomas relacionados(infección, complicaciones, otras
comorbilidades y agentes precipitantes)
Dolor severo
Llegadaal hospital
15-20minutos
Determine la causa
Examine factoresfísicos pertinentes
Causa relacionada a enfermedaddrepanocítica
¿Dolor tipico? No
Determine caracteristicas, locación, intensidad y cualidades
en base de su observación
¿Habitualemtne no responde a AINEy requiere opiáceos?
Seleccione medicacíon aplique dosis inicial en base de evalución e historia de tratamiento
Precise el dolor trate y conduzca el plan de trabajo para
determinar etiología
No
Sí
Sí
Comenzar con la siguiente dosis: adultos/niños <50kg
morfina: 0,1-05mg/kghydromorfina: 0,015-0,020mg/kg
adultos/niños >50kgmorfina: 5-10mg
hydromorfona 1,5mg
Obtenga la historia de tratamiento, medicación ambulatoria, dolor agudo,
hospitalización por dolor, RX, medicación en las últimas 24H
Resuma, determine un perfil y seleccione tratamiento basado en caracteriticas,
historia de tratamiento anterior y hallazgos físicos
Administre por vía EV(si es facil el acceso) o vía subcutánea
(si acceso endovenoso es difícil)
Tratamiento de las Complicaciones Agudas
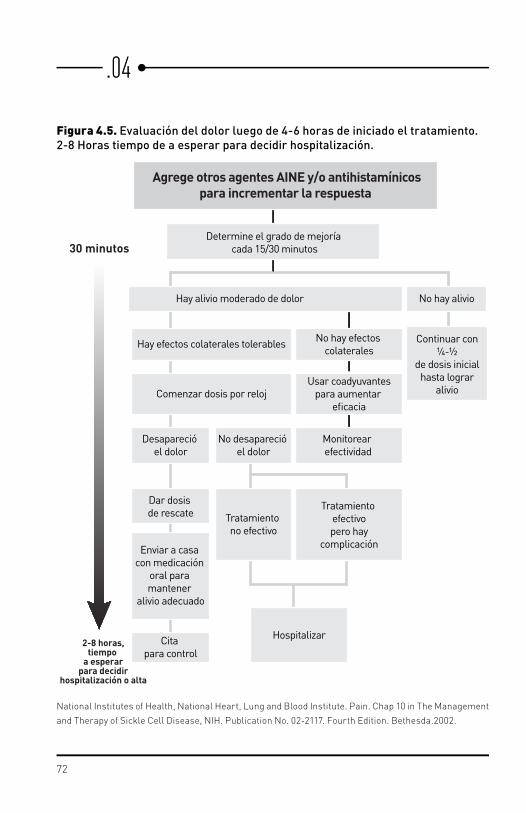
72
Figura 4.5. Evaluación del dolor luego de 4-6 horas de iniciado el tratamiento.2-8 Horas tiempo de a esperar para decidir hospitalización.
National Institutes of Health, National Heart, Lung and Blood Institute. Pain. Chap 10 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda.2002.
Agrege otros agentes AINE y/o antihistamínicospara incrementar la respuesta
No hay alivio
Continuar con¼-½
de dosis inicialhasta lograr
alivio
Hay alivio moderado de dolor
No hay efectos colaterales
Usar coadyuvantespara aumentar
eficacia
Monitorear efectividad
Hay efectos colaterales tolerables
Comenzar dosis por reloj
Desapareció el dolor
No desapareció el dolor
Dar dosis de rescate Tratamiento
no efectivo
Tratamiento efectivopero hay
complicación
Hospitalizar
Enviar a casa con medicación
oral para mantener
alivio adecuado
Cita para control
Determine el grado de mejoría cada 15/30 minutos30 minutos
2-8 horas,tiempo
a esperarpara decidir
hospitalización o alta
.04.04

73
Pautas generales del manejo farmacológico del dolor en enfermedad drepanocítica
• El manejo racional y eficaz del dolor incluye una evaluación profunda, además la individualización de una terapia que combine elementos no farmacológicos y farmacológicos.
• El tratamiento del dolor agudo debe ser rápido y agresivo con el fin de disminuir su intensidad y permitir al paciente recobrar su capacidad funcional en el menor tiempo posible.
• El dolor severo es considerado una emergencia médica, y el tratamiento oportuno y suficiente ha de ser proporcionado hasta que el dolor llegue a ser tolerable.
• El dolor varía latitudinalmente entre un paciente y otro y longitudinalmente en el mismo paciente.
• Adaptar las medidas de analgesia a cada paciente. • Los AINE o acetaminofén son ideales para manejar el dolor de leve a moderada
intensidad, a menos que estén contraindicados. • Si el dolor persiste, agregar una droga opioide al esquema de tratamiento.• El tipo de opioide usado se decide en base a las características y la duración
del dolor. • El tratamiento con opioides durante períodos prolongados conduce a la tolerancia
y dependencia física y no debe confundirse con dependencia sicológica (adicción).• Indicar dosis equivalentes de opioides orales para su uso en el hogar en los casos
de dolor crónico y crisis recurrentes frecuentes.• El uso de medidas sicológicas, físicas y ambientales incrementan el efecto de la
analgesia; por ejemplo, el calor local, la relajación, la distracción, la música, el masaje, la vibración, la práctica de la oración en pacientes que profesan deter-minadas religiones, los ejercicios terapéuticos, el frote con cremas de mentol, la acupresión o digito presión, la acupuntura, y la estimulación nerviosa eléctrica transcutánea (TENS). Al momento no hay estudios clínicos controlados publica-dos sobre la eficacia de estas modalidades terapéuticas del dolor en la enferme-dad drepanocítica, pareciéndose limitar su utilidad a episodios de dolor leve no complicado5.
• El no tratar agresivamente una crisis de dolor agudo, eventualmente, conduce al sindrome de dolor crónico intratable.
• Administrar bicarbonato a una dosis de 80mEq/L, en caso de acidosis, (pH <7,15-7,20 cuando no mejora con el tratamiento de la causa subyacente).
• Mascarilla con O2 sólo si hay hipoxia. • Si la crisis dolorosa es muy severa y se prolonga por más de 10 días a pesar de tra-
tamiento convencional en condición hospitalizado, realizar transfusión simple de concentrado de hematíes o exanguinotransfusión parcial. Ver guía de transfusión.
• En casos graves, asociar metilprednisolona a 15mg/Kg/día, vía endovenoso por 2 días, máximo un gramo por dosis.
Tratamiento de las Complicaciones Agudas

74
Manejo farmacológico del dolor agudo Incluye tres tipos de compuestos: No opiáceos, opiáceos y adyuvantes:
Tabla 4.1. Medicamentos empleados en el tratamiento del dolor agudo.
Se utilizan en el tratamiento del dolor leve y moderado• Acetaminofén (Paracetamol). Se cree que el paracetamol aumenta el umbral al
dolor inhibiendo las ciclooxigenasas en el sistema nervioso central (COX 3).• Inhibidores no selectivos de COX. El efecto analgésico parece depender de la inhi-
bición de la síntesis de las prostaglandinas, las cuales sensibilizan los receptores del dolor a la estimulación mecánica o a otros mediadores químicos (Ej. bradiqui-nina, histamina), además de prevenir la síntesis de prostaglandinas involucradas en el dolor. Presentan efectos analgésicos principalmente periféricos. Además su mecanismo de acción antiinflamatorio contribuye al efecto analgésico.
• Posología y efectos adversos descritos en la tabla 4.2
MorfinaAgonista de los receptores opiáceos µ. Mecanismo de acción: disminuye el AMP-cíclico intracelular inhibiendo la adenilato ciclasa, una enzima que modula la liberación de neurotransmisores nociceptivos como la sustancia P, el GABA o la dopamina. Desde del punto de vista clínico, la estimulación de los receptores µ producen analgesia, euforia, depresión circulatoria, disminución del peristaltismo, miosis y dependencia.
No opiáceos Analgésicos opiáceos Adyuvantes
Paracetamol o acetaminofén Codeína Antihistamínicos
Inhibidores no selectivos de COX Hidrocodona /acetaminofén Antidepresivos
Ácido acetilsalicílico Hidrocodona/ibuprofeno Anticonvulsivantes
Salicilados no acetilados. Oxicodona Benzodiacepinas
Ibuprofeno Oxicodona/codeína Fenotiazinas
Naproxen Morfina Antieméticos
Ketorolac Meperidina Agonistas α 2 adrenérgico
Tramadol Hidromorfona
Tramadol / Paracetamol Levorfanol
Tramadol /acetaminofén Oxymorfona
Metadona
Fentanyl
.04

75
Tabla 4.2. Posología y efectos adversos de los analgésicos no opiáceos.
Posología: Tratamiento del dolor agudo o crónico moderado o intenso descrito en la tabla 4.2.Niños >6 meses: las dosis iniciales no deben ser superiores a los 15mg/dosis. Dosis inicial de 0,1mg/kg EV, SC o IM cada 3 o 4 horas. Dosis de mantenimiento: 0,03 a 0,15 mg/kg/hora.Infantes <6 meses y neonatos: se recomienda una dosis inicial de 0,03-0,05mg/kg EV, SC o IM cada 3 o 4 horas. Alternativamente, puede administrarse una infusión intrave-nosa continua a razón de 0,01mg/kg/hora.Si se administran dosis >30mg/día de morfina, la interrupción del tratamiento ha de hacerse lentamente para evitar los signos y síntomas de abstinencia de opiáceos. Se recomienda disminuir las dosis en un 50% durante uno o dos días, al 25% durante
Analgésico/AINES Niños <12 años Adultos y niños >12 años Efectos adversos
Acetaminofén
Vía oral o rectal 10-15mg/kg cada 4-6 horas. No administrar más de cinco dosis en 24 horas.
Vía oral o rectal 325-650mg cada 4-6 horas.Alternativamente, 1.000mg, 2-4 veces al día. No deben sobrepa-sarse más de 1g dosis única o más de 4g al día.
Toxicidad hepática, necrosis tubular renal y nefropatía en analgésica crónica.
Inhibidores de la ciclooxigenasa
Ibuprofeno
20mg/kg/día cada 6 horas. Dosis máx. 3200 mg/día. Niños < de 30 kg. No exceder los 500mg/día.
400mg cada 4 a 6 hs/ día. Dosis máx. 3200mg/día.
TrombocitopeniaAnemia aplásicaAgranulocitosis Pancitopenia.HipersensibilidadNefropatía Hepato-toxicidadRabdomiolisis
Diclofenac Oral y rectal: 0.5 a 3mg/kg
Oral: 75-100mg/d Parenteral: 75mg cada 12 - 24 horas.
Ketoprofeno
0.5 mg/kg/6-8 horas. Dosis máxima: 2mg/kg/día.
Oral 50mg cada 12 horas. Forma LP: 200mg al día. Parenteral: 100mg cada 12-24 horas
Tratamiento de las Complicaciones Agudas

76
otros dos días hasta alcanzar dosis diarias de 15 a 30mg. Después, interrumpir el tra-tamiento.
TramadolAnalgésico de acción central, agonista puro no selectivo de los receptores opioides µ, δ y κ, con mayor afinidad por los µ.Posología: Ajustar según intensidad del dolor y respuesta. Administrar el tiempo es-trictamente requerido. Posología descrita en la tabla 4.3.Niños <12 años: no recomendado; sólo puede usarse vía parenteral a una dosis unitaria de 1-1,5mg/kg.
Fentanilo Mecanismo de acción: Agonista opiáceo, produce analgesia y sedación por interacción con el receptor opioide µ, principalmente en SNC.Vía de administración: transdérmico (parches). Se utiliza para control del dolor crónico que requiera analgesia con opioides. Posología: Tabla 4.3. En caso de pacientes que no estén recibiendo opiáceos, la dosis máxima inicial es 25 mcg/h; si está en tratamiento previo con opioides será de 25, 50, 75 ó 100mcg/h; de-pendiendo de la necesidad analgésica durante las 24 horas previas, si ésta es ≤135 mg/24 h de morfina oral, administrar 25mcg/h fentanilo; por cada 90 mg adicionales de morfina oral aumentar en 25 mcg/h la dosis de fentanilo, máximo hasta 300 mc-g/h; retirar gradualmente analgésico previo. Mantenimiento: ajustar cada 3 días en incrementos de 12 ó 25mcg/h; se puede usar más de un parche para dosis >100mcg/h. Cambiar el parche cada 72h. Suspender gradualmente el tratamiento
OxicodonaMecanismo de acción: Agonista de los receptores opioides del cerebro y de la médula espinal. El efecto terapéutico es principalmente analgésico, ansiolítico y sedante. Posología: Principios generales: Ingerir enteros los comprimidos de liberación prolon-gada. La terapia será revisada regularmente y ajustada en base a los informes sobre el dolor del paciente y los efectos secundarios.
Comienzo de la terapia: Pacientes sin recibir opiáceos: Dosis inicial 10 mg cada 12 horas. Pacientes bajo terapia opiácea: Determinar dosis diaria total (24 horas) del otro opiáceo.Individualización de la dosis: El ajuste de dosis puede realizarse cada 1 a 2 días. Lo más adecuado es aumentar la dosis cada 12 horas y no la frecuencia. Como orien-tación, indicar un incremento de 10mg a 20mg cada 12 horas, la dosis diaria total de oxicodona usualmente puede ser aumentada en un 25% a 50% de la dosis corriente en cada incremento.
Analgesia suplementaria: La mayoría de los pacientes que están sometidos a terapia
.04
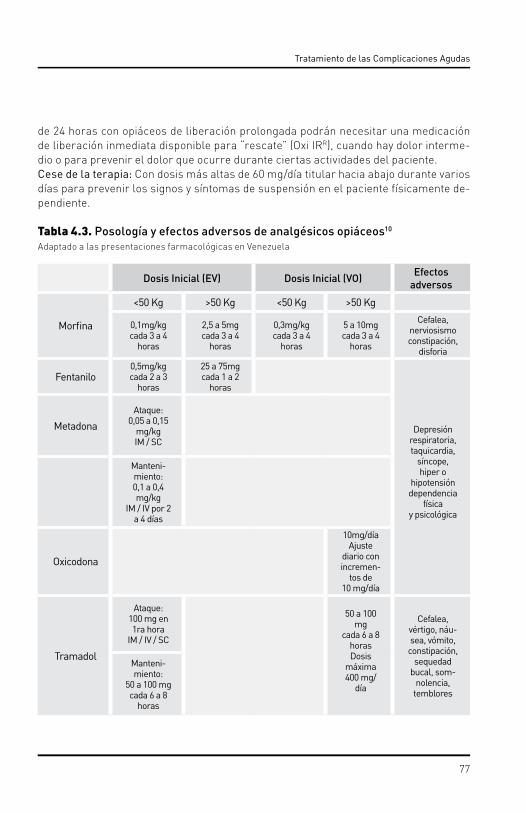
77
de 24 horas con opiáceos de liberación prolongada podrán necesitar una medicación de liberación inmediata disponible para “rescate” (Oxi IRR), cuando hay dolor interme-dio o para prevenir el dolor que ocurre durante ciertas actividades del paciente. Cese de la terapia: Con dosis más altas de 60 mg/día titular hacia abajo durante varios días para prevenir los signos y síntomas de suspensión en el paciente físicamente de-pendiente.
Tabla 4.3. Posología y efectos adversos de analgésicos opiáceos10
Adaptado a las presentaciones farmacológicas en Venezuela
Dosis Inicial (EV) Dosis Inicial (VO) Efectos adversos
Morfina
<50 Kg >50 Kg <50 Kg >50 Kg
0,1mg/kgcada 3 a 4
horas
2,5 a 5mgcada 3 a 4
horas
0,3mg/kgcada 3 a 4
horas
5 a 10mgcada 3 a 4
horas
Cefalea, nerviosismoconstipación,
disforia
Fentanilo0,5mg/kgcada 2 a 3
horas
25 a 75mgcada 1 a 2
horas
Depresión respiratoria,taquicardia,
síncope,hiper o
hipotensióndependencia
físicay psicológica
MetadonaAtaque:
0,05 a 0,15 mg/kgIM / SC
Manteni-miento:0,1 a 0,4 mg/kg
IM / IV por 2 a 4 días
Oxicodona
10mg/díaAjuste
diario conincremen-
tos de10 mg/día
Tramadol
Ataque:100 mg en 1ra hora
IM / IV / SC
50 a 100 mg
cada 6 a 8 horasDosis
máxima400 mg/
día
Cefalea, vértigo, náu-sea, vómito, constipación,
sequedad bucal, som-
nolencia,temblores
Manteni-miento:
50 a 100 mgcada 6 a 8
horas
Tratamiento de las Complicaciones Agudas

78
Combinaciones de analgésicos
Acetaminofén + codeína: • Presentación: Tabletas de acetaminofén 500mg + codeína: 25mg.• Posología: 2 tabletas cada 6 horas.
Tramadol + acetaminofén:Presentación: Tabletas de tramadol 37,5mg + acetaminofén: 325mg.Posología: 2 tabletas cada 8 horas. Se pueden indicar dosis adicionales según sea necesario, sin exceder de 8 comprimidos (equivalente a 300 mg de tramadol y 2600mg de paracetamol) al día.
Referencias 1. Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: Frequency, etiology, and prognostic significance. Am J Hematol. 2005; 79:17-25.2. Ballas K. Current Issues in Sickle Cell Pain and Its Management. Hematology 2007; 97-105.3. Schnog JJ, Lard LR, Rojer RA, Van der Dijs FP, Muskiet FA, Duits AJ. New concepts in assessing sickle cell disease severity. Am J Hematol 1998; 58:61-66.4. National Institutes of Health, National Heart, Lung and Blood Institute. Pain. Chap 10 in The Manage-ment and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. Fourth Edition. Bethesda.2002; 59-74. 5. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. ScientificWorldJournal. 2012;2012:949535. doi: 10.1100/2012/949535. 6. Dampier C, Ely E, Brodecki D, O’Neal P. Home management of pain in sickle cell disease: a daily diary study in children and adolescents. J Pediatr Hematol Oncol 2002;24(8):643-47.7. Lottenberg R, Hassell KL. An evidence-based approach to the treatment of adults with sickle cell disea-se. Hematology Am Soc Hematol Educ Program 2005:58-65.8. Okomo U, Meremikwu MM. Replacing fluids to treat acute episodes of pain in people with sickle cell disease. The Cochrane Library, 2008 Issue 3. Chichester, UK: John Wiley & Sons. Disponible en: http://summaries.cochrane.org/CD005406/replacing-fluids-to-treat-acute-episodes-of-pain-in-people-with-sickle-cell-disease9. Remacha S AF. Dolor y crisis vasooclusivas en Guía de manejo de las enfermedades falciformes. Aso-ciación Española de Hematología. Grupo de Eritropatología.2009; 51 – 57.10. Lobo C, Neves V, Silva R. Crises dolorosas na doenca falciforme. Rev Bras Hematol Hemoter 2007;29(3):247-258.
.04

79
4.2. Infección Adrián Cárdenas / Médico hematólogo.
Las infecciones bacterianas son frecuentes en todos los pacientes con enfermedad drepanocítica y constituyen la causa más importante de mortalidad en niños. Los epi-sodios vasooclusivos recurrentes a nivel del bazo, conducen a infartos y consecuen-temente a la auto esplenectomía, predisponiendo a infecciones severas por gérmenes capsulados (Haemophilus influenzae, Streptococcus pneumoniae).
Por otra parte, niveles bajos de IgM inducen dificultad en la opsonización y en trastor-nos en la vía alterna del complemento e incrementan la sensibilidad a infecciones por Mycoplasma pneumoniae, Salmonella typhimurium, Staphylococcus aureus, y Escherichia coli. Las infecciones mas comunes incluyen neumonía, bronquitis, colecistitis, pielone-fritis, cistitis, osteomielitis, meningitis y sepsis1.
Además de las inmunizaciones de rutina, existen otras medidas preventivas descritas a continuación.
Medidas preventivas en niños. Ver guía de medidas generales.
Medidas preventivas en adultos.
Virtualmente todos los adultos con anemia drepanocítica son asplénicos desde el pun-to de vista funcional, pero el sistema inmune ha madurado y permite la formación de anticuerpos específicos contra polisacáridos. Debido a que no son susceptibles como los niños, la incidencia de sepsis es baja.
En pacientes con enfermedad drepanocítica se recomienda la vacunación contra Strep-tococcus pneumoniae. El manejo de un paciente con fiebre, varía según el grado de tem-peratura y de los signos y síntomas asociados. Hasta el momento no existen evidencias que avalen la superioridad de un antibiótico con respecto a otro.
Su administración se hace de manera empírica y todas las guías recomiendan iniciar con una cefalosporina y cambiar o añadir otros, de acuerdo a la evolución y el germen aislado en cada caso en particular. Sugerimos el esquema recomendado por el Centro de Investigación y Tratamiento de Drepanocitosis de Colorado, revisado y actualiza-do por hematólogos de Arizona, Colorado, Georgia, Missouri, New México, Tennessee, Texas, y Utah2.
Consulta: Hematólogo, Internista, Infectólogo.
La conducta varía de acuerdo a la temperatura (Figura 4.6).
Tratamiento de las Complicaciones Agudas

80
Figura 4.6. Conducta ante un paciente febril con enfermedad drepanocítica.
Hipertemia
Examen físico irrelevante
temperatura <38ºC
Hematologia, hemocultivo, uroanálisis, urocultivo,
RX tórax, oximetría
Lab: negRX: normal
Enviar a casacontrol a las24-48 horas
Temperatura >3,58ºC y <39,5ºC con o sin síntomas
Hospitalizar
Examen físico negativo
para infección
Uroanáalisis, urocultivo,
hematología, hemocultivo,
RX tórax, antibióticos STAT.
Ej. ceftriaxona
Ver laboratoriohozpitalizaru observar1-2 horas
Buen nivelintelectual enviar
a casa controla las 24 horas
Febril"luce enfermo"
o cultivos positivosa las 24 horashospitalizar
Afebril cultivosnegavitos dar
2da dosisCeftriaxone
antibióticos vo
Controla intervalos
determinadossi reaparece fiebre
hospitalizar
Temperatura igual o>39,5ºC
Hospitalizar
Examen físico positivo para
infeccion respiratoria y SAT 02
.04

81
Si la temperatura es menor a 38°, el examen físico irrelevante y los análisis de labora-torio dentro de los parámetros considerados normales, el paciente se envía a casa con control dentro de 24 a 48 horas.
Si la temperatura es igual o mayor de 38°C: · Pasar inmediatamente al paciente a la sala de examen.
Realizar historia breve y examen físico con énfasis en:• Signos vitales.• Determinar grado de palidez.• Evidencias de sepsis (frialdad, disminución de perfusión periférica), o infección
localizada. • Estado cardiopulmonar.• Tamaño del bazo (en relación al estado basal).• Examen neurológico.
STAT. Tomar una vía con el propósito de extraer muestra para:• Hematología (que incluya recuento diferencial, reticulocitos y plaquetas).• Hemocultivo.• Ordenar infusión inmediata de antibióticos: NO ESPERAR RESULTADOS DE ES-
TUDIOS SOLICITADOS PARA INICIARLO. LOS PACIENTES CON SEPTICEMIA EX-PIRAN EN POCAS HORAS. · Ceftriaxone (50-100 mg/kg, la dosis sencilla no debe sobrepasar los 2,0 g).
Evaluar cada 2 horas. · Los pacientes conocidos o con sospecha de ser alérgicos a las cefalosporinas,
recibirán clindamicina, a razón de 10-15mg/kg. La dosis sencilla no debe pasar de 600mg.
· Si la enfermedad es severa o se sospecha infección del sistema nervioso cen-tral, añadir vancomicina (10-15mg/kg EV).
• Grupo sanguíneo, Rh, y pruebas cruzadas (si hay palidez extrema, signos respira-torios o neurológicos o aumento del bazo).
• Considerar transfusión de concentrado eritrocitario, leucorreducido e inmunofe-notípicamente compatible para antígenos Rh, Duffy y Kell. Ver guía correspon-diente a transfusión. · Solicitar uroanálisis y urocultivo y otros cultivos dependiendo de la clínica. · Indicar Rx de tórax. · Practicar saturación de oxígeno. · Realizar punción lumbar en niños con facies tóxica y en aquellos con signos de
meningitis. · Administrar acetaminofén a razón de 15mg/kg/VO, si no lo ha recibido en las
últimas 4 horas y/o ibuprofeno, 10mg/kg/VO. No suministrar ibuprofeno en caso de gastritis, úlcera, coagulopatía, o enfermedad renal.
Tratamiento de las Complicaciones Agudas

82
Hospitalizar si existen uno o mas de los siguientes criterios:• Temperatura >40°.• Niños <1 año con diagnóstico de ED-SS o ED-Sβ0 talasemia.• Historia previa de sepsis. • Leucocitos >30 x 109/L o < de 5 x 109/L.• Plaquetas < de 100 x109/L.• Hemoglobina < de 5g/dL.• Signos de toxicidad sistémica.• Pacientes que recibieron clyndamicina o vancomicina.• Evidencia de otro episodio agudo: crisis de dolor, secuestro esplénico, aplasia eri-
troide, sindrome de tórax agudo, ictus cerebral agudo, priapismo.• También deben hospitalizarse con la finalidad de recibir antibióticos aquellos ni-
ños no tóxicos con temperatura inferior a 40º pero con cualquiera de los siguien-tes elementos:
• Infiltrado pulmonar en Rx de tórax.• Saturación de oxígeno anormal.
· Los niños y adultos con facies tóxica y aquellos con temperatura superior a 40° deben tratarse de inmediato con antibióticos vía intravenosa, sin esperar resultados de Rx o de laboratorio. Hospitalizar de una vez.
· Si el paciente ha ingresado por el servicio de Emergencia, llamar al hematólo-go tratante o a hematólogo con experiencia en enfermedad drepanocítica.
· La bacteriemia documentada será tratada parenteralmente durante 7 días y los niños con meningitis recibirán tratamiento por lo menos durante 14 días.
· Si hay evidencia de enfermedad respiratoria, suministrar oxígeno mediante mascarilla facial.
Tratar ambulatoriamente:• Niños no tóxicos y temperatura inferior a 40ºC, quienes nunca han tenido sepsis,
presentan Rx de tórax y saturación de oxígeno normal, leucocitos, plaquetas y he-moglobina en valores basales. Ellos pueden observarse en el hogar, después de haber recibido una dosis de antibiótico de larga duración que cubra Streptococcus pneumoniae y Haemophilus influenzae (Ej. Ceftriaxone, 75mg/kg); si:
• Han permanecido clínicamente estables por 3 horas.• Los padres están debidamente entrenados, el paciente tiene adherencia al tra-
tamiento con penicilina profiláctica, cumplen con las citas a cabalidad y tienen acceso rápido al hospital.
• Evaluación a las 24 horas.
Manejo del paciente hospitalizado• Controlar.
· Signos vitales cada 2 horas hasta que el paciente se estabilice. · Considerar monitor cardio-respiratorio y unidad de cuidados intensivos si
.04

83
existe inestabilidad cardio-respiratoria. · Control de líquidos (parenteral y oral). Peso diario. · Oximetría de pulso.
• Infección severa.• Signos y síntomas respiratorios.• Diagnóstico (si no se obtuvo previamente)• Hematología (que incluya recuento diferencial, reticulocitos y plaquetas. Diaria-
mente hasta mejorar. Comparar con valores basales.• RX de tórax, si hay taquipnea, tos, dolor torácico o abdominal o existe cualquier
síntoma respiratorio subyacente.• Hemocultivo, uroanálisis. Considerar urocultivo u otro cultivo de acuerdo a la clínica.• Evaluar función hepática , renal y pruebas para descartar coagulación intravas-
cular diseminada, en caso de dolor muy intenso o signos encefalopatía (daño multi orgánico).
• Solicitar ultrasonido, amilasa y lipasa en caso de dolor abdominal intenso (cole-cistitis, colelitiasis, pancreatitis).
Grupo y Rh. Considerar transfusión:• Si la Hb está 1-2gr por debajo de los valores basales.• Si hay signos evidentes de sindrome torácico agudo.• En caso de transfusión, hacerlo con concentrado globular, leucodepletado, inmu-
nofenotípicamente compatible, al menos para Rh, Duffy y Kell.• Requerir consulta con ortopedia, con aspiración para cultivo en caso de sospecha
de osteomielitis o artritis séptica.
Líquidos. • El incremento de líquidos es necesario si hay deshidratación o pérdidas no visibles
por fiebre sostenida. • Evitar la sobrehidratación por riesgo de precipitar sindrome de tórax agudo.• Los líquidos por vía oral + los suministrados por vía endovenosa no deben superar
a 1 ½ veces la necesidad de mantenimiento.
Medicamentos.• Administrar acetaminofén a razón de 15mg/kg/VO, cada 4 horas (dosis máxima
diaria 75mg/kg) y/o ibuprofeno, 10 mg/kg/VO, cada 6-8 horas. No suministrar ibu-profeno en caso de gastritis, úlcera, coagulopatía, o enfermedad renal.
• Ceftriaxone: 50mg/kg, EV c/8 h.• Los pacientes conocidos o con sospecha de ser alérgicos a las cefalosporinas,
recibirán clindamicina, a razón de 10-15mg/kg, EV, c/8h.• Si la enfermedad es severa o se sospecha infección en el sistema nervioso cen-
tral, añadir vancomicina (10-15 mg/kg, EV).• Suspender penicilina profiláctica mientras dure el tratamiento con antibióticos de
Tratamiento de las Complicaciones Agudas

84
amplio espectro. • Oxígeno nasal por cánula o máscara, para mantener la oximetría de pulso >92%
Evitar el exceso de O2 por riesgo de disminuir la producción de reticulocitos y oca-sionar anemia.
• Considerar transfusión cuando la Hb esté 1-2gr por debajo de los valores basales.
Criterios para el alta:• Afebril durante >24 horas con cultivos negativos >24 o 48 horas • Capacidad de ingerir líquidos y por tanto medicamentos.• Resolución de cualquier signo de infección pulmonar y ser capaz de mantener una
oxigenación adecuada, respirando el aire ambiental.• No evidencias de anemia (aplasia eritroide o secuestro). Hemoglobina y hemato-
crito estables. • Adherencia a las citas y tratamiento.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Infection. Chap 11 in The Ma-nagement and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 75-79.2. Lane PA, Buchanan GR, Hutter JJ, Robert F. Austin RF, Britton HA, Rogers ZR et al. Sickle cell disease care consortium. Management of febrile illness, in Sickle cell disease in children and adolescents: Diag-nosis, guidelines for comprehensive care, and care Paths and protocols for management of acute and chronic complications. 2001. Disponible en: http://scinfo.org/care-paths-and-protocols-children-ado-lescents/care-path-outpatient-evaluation-and-management-of-febrile-illness.
4.3. Anemia aguda Jaime Bracho / Médico hematólogo.
El factor determinante de la anemia en la enfermedad drepanocítica es la tasa de he-mólisis. La anemia crónica es propia de la anemia drepanocítica (Hb SS) y de la Sβ0-Ta-lasemia. En la enfermedad SC la anemia es moderada y en ocasiones hasta mínima. El nivel de hemoglobina es una característica clínica individual la cual permanece más o menos estable sin requerir terapia transfusional. Cualquier cambio descendente del nivel basal puede ser clínicamente significativo y amerita ser investigado1. El efecto de la anemia crónica es parcialmente compensado por un aumento del gasto cardíaco en reposo. El empeoramiento de la anemia es común e implica el riesgo de un impacto pronóstico inmediato2. Esta es la razón por la cual, cada paciente necesita ser carac-terizado en su fenotipo eritrocitario en las primeras consultas, para que en caso ne-cesario, reciba la terapia transfusional adecuada e individualizada. El comienzo clínico es por lo general brusco con debilidad, astenia, palidez, disnea, palpitaciones, dolor abdominal y mayor ictericia que la usual. Los hallazgos clínicos incluyen taquicardia,
.04

85
taquipnea, palidez, hepato o esplenomegalia súbita, ritmo de galope y/o insuficiencia cardíaca congestiva (cor anémico). Es importante comparar los valores de hemo- globina, hematocrito, reticulocitos y bilirrubina con cifras previas consideradas como basales para cada paciente en particular. En general, un descenso de 25% a 30% de los valores basales, requerirá alguna intervención individualizada, siempre en el contexto de una situación clínica específica. Además de la simple explicación de la insuficiente ingesta de folatos, la agudización de la anemia crónica resulta por destrucción ace-lerada de eritrocitos (hiperhemólisis) y secuestro esplénico ó hepático3,4; sangrado, generalmente gastrointestinal, urinario o ginecológico o como consecuencia de una falla de la eritropoyesis, mayormente secundaria a infección por parvovirus B19 o a ex-tensa necrosis medular5,6. Causas menos frecuentes son las hemólisis autoinmunes y las reacciones hemolíticas tardías transfusionales. Comorbilidades como inflamación crónica, hipotiroidismo, déficit de vitamina B12, insuficiencia renal e hiperesplenismo crónico situan el nivel de hemoglobina más bajo aún a largo plazo. (Tabla 4.4)
Tabla 4.4. Causas de aumento de la anemia en la enfermedad drepanocítica.
Referencias1. Serjeant GR, Grandison Y, Lowrie Y, Mason K, Phillips J, Serjeant BE, Vaidya S. The development of haematologi-cal changes in homozygous sickle cell disease: a cohort study from birth to 6 years. Br J Haematol 1981; 48: 533-43.2. Brown AK, Sleeper LA, Miller ST, Pegelow CH, Gill FM, Waclawiw MA. Reference values and hematologic chan-ges from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med 1994; 48:796-804.3. Emond AM, Collis R, Darvill D, Higgs DR, Maude GH, Serjeant GR. Acute splenic sequestration in homozygous sickle cell disease: natural history and management. J Pediatr 1985;107:201-206.4. Gover R, Wethers DL. Management of acute splenic sequestration crisis in sickle cell disease. J Assoc Acad Minor Phys 1990; 1: 67-70.5. Cole TB, Smith SJ, Buchanan GR. Hematologic alterations during acute infection in children with sickle cell disea-se. Pediatric Infect Dis J 1987; 6(5):454-57.6. Serjeant GR, Serjeant BE, Thomas PW et al. Human parvovirus infection in homozygous sickle cell disease. Lan-cet 1993; 341: 1237-1240.
Anemia sintomática súbita Anemia crónica mas severa
Crisis de hiperhemólisis Inflamación crónica severa
Secuestro esplénico/hepático Hipotiroidismo
Aplasia eritroide por eritrovirus B19 Deficiencia de vitamina B12
Necrosis medular extensa Insuficiencia renal
Hemólisis autoinmune Hiperesplenismo crónico
Reacción hemolítica post transfusional
Sangrado agudo urinario, gastrointestinal o ginecológico
Tratamiento de las Complicaciones Agudas

86
4.4. Aplasia transitoria de serie roja.Betty Ramos de Urdaneta / Médico hematólogo.
La aplasia eritroide es una complicación grave en pacientes con enfermedad drepa-nocítica. El número de reticulocitos desciende considerablemente (<1%). Si cursa con aumento del tamaño del bazo, pensar en secuestro esplénico. Entre 70 y 100% de los casos son originados por la infección aguda por el eritrovirus B-19 (B19V). El eritro-virus también ha estado vinculado a otras complicaciones agudas de la enfermedad drepanocítica que pueden ocurrir durante la crisis de aplasia, como: dolor, necrosis medular, sindrome torácico agudo y enfermedad vascular cerebral aguda Este virus causa la quinta enfermedad, un trastorno de la infancia, usualmente benigna, asociada a fiebre, malestar general y erupción cutánea leve.
El virus infecta células progenitoras de la serie roja en médula ósea, mediada lo que ocasiona disminución de la división celular transitoria1. En personas sanas se observa disminución discreta del hematocrito, pero en pacientes con enfermedad drepanocíti-ca, quienes ya tienen una sobrevida eritrocitaria acortada, se produce descenso rápido de los valores de hemoglobina. La condición es auto limitada, la médula ósea se recu-pera en 7-10 día con reticulocitosis1.
No existen estudios grandes, aleatorios y controlados relacionados con el manejo clí-nico de esta condición. En un estudio realizado en Jamaica, el 87% de los pacientes requirieron transfusión1.
Se sugiere el esquema recomendado por el Centro de Investigación y Tratamiento de Drepanocitosis de Colorado, recientemente revisado y actualizado por hematólogos de Arizona, Colorado, Georgia, Missouri, New México, Tennessee, Texas, and Utah2.
Signos y síntomas• Generalmente precedida de fiebre.• No hay rash.• Presencia de fatiga.• Debilidad evidente.• En el laboratorio.
· Anemia. · Reticulocitopenia (usualmente <1%).
Conducta• Consulta con el hematólogo.• Hospitalizar:
· Si hay evidencias de compromiso cardio respiratorio. · Si no se dispone del componente adecuado para transfundir en forma ambulatoria.
.04

87
· Dificultad para el control ambulatorio.• Control de signos vitales cada 2 horas hasta estabilizar, luego cada 4 horas una
vez hospitalizado el paciente.• Indicar monitoreo cardio respiratorio y oximetría de pulso continua.• Solicitar ingreso en UCI si hay compromiso cardiovascular.• Control de ingestión y eliminación de líquidos. Pesar diariamente.
Diagnóstico• Hematología incluyendo reticulocitos y plaquetas. Después del contaje inicial de
reticulocitos, repetir cada 12-24 horas. • Determinación de grupo y Rh. Considerar realizar inmunofenotipo Rh, Duffy y Kell. • Si hay fiebre, practicar
· Uroanálisis y urocultivo. · Hemocultivo. · Otros cultivos según la clínica.
• Si además de fiebre existen signos de infección respiratoria, solicitar RX de tórax.• Estudios para parvovirus: Títulos de Ig G e Ig M.
Cuidados generales• Administración de líquidos. Vía oral + EV, para cubrir mantenimiento. En caso de
fiebre persistente, pudiera ser necesario el suministro de mayor cantidad de líqui-dos. El exceso ocasionaría sobrecarga.
• Tratamiento. · Bajar rápidamente la fiebre, mediante el suministro de acetaminofén a razón de 15
mg/kg/VO, si no lo ha recibido en las últimas 4 horas y/o ibuprofeno, 10mg/kg/VO. No dar ibuprofeno en caso de gastritis, úlcera, coagulopatía, o enfermedad renal.
· Indicar transfusión en caso de anemia sintomática y/o Hb < 5 g/dL sin evidencias de recuperación (reticulocitopenia persistente con o sin disminución progresiva de la hemoglobina). En ocasiones es necesario repetir transfusión. Suministrar concen-trado globular, leucorreducido e inmunofenotípicamente compatible al menos para Rh, Duffy y Kell. Ver Guía de transfusión.
· Administrar inmunoglobulina en caso de anemia sintomática y/o Hb <5g/dL sin evi-dencias de recuperación, a la dosis de 400 mg/kg/día/EV durante 5 días.
· Si la anemia compromete la vida, suministrar oxígeno 100%. · En caso de temperatura >38,5°, suministrar antibióticos siguiendo lo indicado en la
guía para infección y fiebre. · Continuar penicilina profiláctica si el paciente no recibe antibióticos; en caso con-
trario suspender hasta finalizar el tratamiento con antibióticos de amplio espectro. · Continuar ácido fólico.
• Criterios para dar el alta. · Cifras estables de hemoglobina · Afebril durante 24 horas.
Tratamiento de las Complicaciones Agudas

88
· Ingestión adecuada de líquidos y de medicamentos.
Control después del egreso• Control hematológico, incluyendo reticulocitos, una vez por semana, hasta que la
hemoglobina y reticulocitos lleguen a niveles basales para el paciente.• Repetir determinación de títulos de Ig G e Ig M para parvovirus, si fueron negativos
inicialmente.• Tomar muestras para determinación de títulos de Ig G e Ig M para parvovirus, a los
familiares. También hacer esto al momento de diagnosticar al paciente.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Transient red cell aplasia. Chap 12 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Be-thesda.2002; 81-82.2. Lane PA, Buchanan GR, Hutter JJ, Robert F. Austin RF, Britton HA, Rogers ZR et al. Sickle cell di-sease care consortium. Aplastic crisis, in Sickle cell disease in children and adolescents: Diagnosis, guidelines for comprehensive care, and care Paths and protocols for management of acute and chronic complications. 2001. Disponible en http://scinfo.org/care-paths-and-protocols-children-adolescents/care-path-aplastic-crisis.
4.5. Ictus cerebral y enfermedad del sistema nervioso central (SNC)María E. López - Evelio Rubio / Médicos especialistas en Neurología Infantil. El ictus cerebral y la isquemia cerebral crónica constituyen unas de las complicacio-nes más devastadoras de la enfermedad drepanocítica1. Los eventos vásculocerebra-les, tales como el accidente isquémico transitorio, infartos cerebrales, hemorragias intracraneales y alteraciones silentes de la microvasculatura cerebral que conllevan a disfunción neuropsicológica son evidenciadas en una alta proporción de los pacientes, siendo la complicación de mayor impacto en esta enfermedad y la principal causa de muerte en la población pediátrica y adulta. La tasa de presentación llega a ser 300 ve-ces mayor al comparar pacientes drepanocíticos con la población general. La ocurren-cia de eventos vasculares se relaciona con variables genéticas y epigenéticas. El Es-tudio Cooperativo de Enfermedad Drepanocítica (ECED) demostró que la prevalencia e incidencia de ictus en pacientes con ED-SS es cuatro veces mayor que en pacientes con ED-SC2. Debido a esta diferencia en el riesgo las recomendaciones para evalua-ción y prevención primaria de ictus se aplican a pacientes con ED-SS asintomáticos neurológicamente y no a pacientes con ED-SC y las correspondientes para tratamiento y prevención secundaria en pacientes sintomáticos se aplican a todos los pacientes con enfermedad drepanocítica.
Los niños con enfermedad drepanocítica pueden tener una variedad de anomalías ana-
.04

89
tómicas y fisiológicas que afectan el SNC, aun cuando ellos parezcan neurológicamen-te “normales” y tales anomalías estar asociadas con deterioro de la función cognitiva con efectos sobre el aprendizaje y la conducta e incrementar el riesgo para daño clí-nico y subclínico al SNC en el futuro. Ictus cerebral agudo y enfermedad de sistema nervioso central en niños con enfermedad drepanocítica.
Los infartos cerebrales se asocian principalmente con la estenosis u oclusión de las arterias cerebrales de gran calibre, como la arteria cerebral media y la carótida inter-na1. Los resultados del ECED demuestran que el 11% de los niños con ED-SS presentan ictus cerebral antes de los 20 años y el 24% de los adultos antes de los 45 años3.
El riesgo es mayor en niños en edades comprendidas entre 5 y 8 años, en las cuales es frecuente la aplasia de serie eritroide secundaria a infección por eritrovirus, cuya recuperación cursa con reticulocitosis, trombocitosis y leucocitosis, factores que pre-disponen a esta complicación.
La alta incidencia de ictus cerebral en niños, adolescentes y adultos jóvenes se ha relacionado con la mayor velocidad de flujo sanguíneo cerebral en las estructuras vas-culares de gran calibre en estas edades.
Adicionalmente:• El retardo y oclusión del flujo sanguíneo en vasos distales de menor calibre por
eritrocitos rígidos conlleva a las alteraciones en la microcirculación cerebral.• La ocurrencia de otros factores intercurrentes como la anemia crónica compro-
mete la reserva funcional cerebro vascular. • El daño de la íntima vascular agrava la adherencia de las células falciformes y
genera mayor daño endotelial. • La hipertrofia de la musculatura lisa perivascular y el desarrollo de vasos de mo-
ya-moya que consisten en la presencia de pequeñas arterias reunidas en forma de red vascular que producen en los estudios de arteriografía una imagen de “bo-canada de humo”.
• La hipercoagulabilidad sanguínea. • La existencia de una mayor susceptibilidad de trombosis cerebral relacionada con
el HLA, son todas variables de importancia en la patogénesis del daño cerebral en la drepanocitosis4-6.
Manifestaciones clínicasLa disfunción cerebral ocurre cuando el suplemento de oxígeno al cerebro cae a un nivel crítico en relación a las necesidades. Los síntomas de isquemia incluyen déficit neuromotor lateralizado tipo hemiplejia o hemiparesia, con o sin afasia; trastornos vi-suales y del lenguaje; convulsiones (especialmente focales) y alteración de la actividad mental y el estado de alerta, estupor o coma. Cualquier signo o síntoma neurológico
Tratamiento de las Complicaciones Agudas

90
diferente de cefalea leve, aun cuando sea transitorio, amerita consulta médica urgen-te. Hay evidencias que las demandas de oxigeno son más altas en niños que en adultos por lo que el niño con drepanocitosis que, además tiene anemia significativa, esté a un riesgo particular. En este orden de ideas se establece que la presencia de síntomas neurológicos con una duración mínima de 24 horas, como déficits focales motores, trastornos del lenguaje, alteraciones de la marcha y/o cambios del sensorio definen clínicamente la presencia de un accidente vásculocerebral7. Aun cuando los pacientes no fallecen agudamente por un infarto cerebral, estos se acompañan de un incremento significativo de la morbilidad.
El ictus cerebral suele acompañarse de manifestaciones clínicas en un 11% de los pa-cientes en la primera década de la vida, con una mayor incidencia entre los dos y cinco años de edad o también presentarse como infartos cerebrales silenciosos en un 17 a 22% de los niños afectados.
Los infartos cerebrales recurrentes pueden ocurrir en las dos terceras partes de pa-cientes en un período de dos años posterior al evento inicial8. Este riesgo se incremen-ta en aquellos pacientes que han sufrido un primer evento antes de los 20 años.
Factores de riesgoEntre los factores de riesgo de ictus cerebral, los principales son:
• Ocurrencia de accidentes isquémicos transitorios (AIT). Sin embargo no son fácil-mente detectados en la población pediátrica.
• Episodios de dolor torácico agudo: Se ha identificado una asociación temporal en la que estos parecen preceder en un lapso de una a dos semanas a los eventos vasculares, pudiendo considerarse como un evento premonitorio2.
• Anemia severa.• Incremento del flujo sanguíneo cerebral y de su velocidad que representan un
riesgo de generar daño vascular.• Elevación de las cifras de presión arterial sistólica.
Evaluación del riesgo de ictus cerebral• Eco doppler transcraneal (EDT) El EDT mide la velocidad media del flujo sanguí-
neo en los vasos intracraneales de gran calibre. La velocidad del flujo está inver-samente relacionada con el diámetro de la luz arterial. Un incremento focal de la velocidad del flujo es indicativo de estenosis arterial, mientras que si el incremen-to se registra de manera bilateral es un indicador de enfermedad vascular arterial bilateral. Permite seguir un control de los cambios de la circulación cerebral en condiciones normales y patológicas. Es un procedimiento no invasivo, rápido y sin efectos colaterales, útil en la detección temprana de aquellos pacientes con riesgo elevado de padecer ictus cerebral.
• Por ser un procedimiento con alta sensibilidad para la detección de alteraciones
.04

91
del flujo sanguíneo cerebral, el EDT se ha establecido como herramienta para despistaje precoz en niños drepanocíticos en situación de riesgo. Se recomienda su utilización desde los dos años de edad cronológica, con nuevas determinacio-nes a intervalos variables de acuerdo a resultados del estudio precedente10.
Interpretación del EDT y sugerencias de acuerdo a resultados9:• Normal: velocidad de flujo en la arteria cerebral media menor de 170cm/seg. Se
recomienda realización de EDT anualmente hasta que el paciente cumpla 16 años.• Condicional: velocidad de flujo en la arteria cerebral media entre 170 y 199cm/seg.
Repetir el EDT cada seis meses o antes a criterio del médico tratante.• Anormal: velocidad de flujo en la arteria cerebral media mayor o igual a
200cm/seg. Se sugiere iniciar régimen transfusional crónico y EDT mensual hasta que se normalice la velocidad.
• Tomografía Axial Computarizada (TAC), Resonancia Magnética Nuclear (RMN), Angiorresonancia: Indicadas en pacientes con síntomas y signos neurológicos, incluyendo anomalías en las funciones cognitivas y el lenguaje.
• Saturación de oxígeno (SpO2): su monitorización se utiliza para identificar a los niños con desaturación de la Hb que están en riesgo de sufrir un ictus cerebral. En estos pacientes la aplicación de intervenciones terapéuticas destinadas a mejorar la saturación de la hemoglobina, disminuyen el riesgo de infarto cerebral.
ConductaNo existen estrategias basadas en la evidencia en relación al tratamiento del ictus cerebral en ED. Se sugiere el esquema recomendado por Lane Peter y colaboradores11
(Figura 4.7).
Tratamiento de las Complicaciones Agudas

92
Figura 4.7. Enfermedad cerebral aguda.
Ictus Cerebral
Signos de focalización
Hematología tipeje y pruebas cruzadas química, acceso EV.
manejo en UCI
Cefalea, obnubilación,meningismo, convulsiones
Resonancia magnética, TACconsulta con neurólogocontrol con hematólogo
Infarto Hemorragia Normal
Punción lumbar normal
Punción lumbar anormal
Ver guía de:infección, sepsis
meningitisevaluaciónneurológica
Resonancia magnética
angiografía/resonanciaeco doppler transcraneal
si no fué hecha en inicio
Obeservaciónconstante
y cuidadosapor neurólogo
Anormalconsiderartransfusión
crónica
Intervención continua
del hematólogo
Programatransfusión
crónicamantener Hb S <30%
Exaguinotransfusión
Angiografíaevaluación por neurocirujanointervencion
médica/quirúrgica
Ver guía deanestesiay cirugía
.04

93
Plan de trabajo• Historia clínica:
· Enfermedad actual. · Antecedentes de eventos neurológicos, traumas o cualquier otro pertinente. · Estudios anteriores: EDT, TAC, IRM, angiorresonancia. · Examen neurológico.
Laboratorio · Tipeaje sanguíneo e inmunofenotipo de grupos sanguíneos. · Hematologia completa y reticulocitos. · Pruebas generales de coagulación. · Pruebas hepáticas y renales. · Electrolitos. · Cultivos (sangre, orina, otros) en caso de fiebre.
Estudios de imágenes · RMN, angiorresonancia. Si al momento no están disponibles realizar TAC SIN
CONTRASTE, para descartar lesiones susceptibles de tratamiento quirúrgico (hematoma subdural, hemorragia parenquimatosa, aneurisma, absceso). Pue-de ser normal hasta 2-4 días después. La RMN es más sensible para identificar las lesiones isquémicas que la TAC por lo que si no se hace inicialmente debe realizarse en algún momento para establecer con propiedad el diagnostico de la lesión cerebral.
El estudio por imágenes no debe retrasar el inicio del tratamiento con transfusiones.
Tratamiento• Hidratación parenteral: 1 vez el mantenimiento.• Terapia transfusional: el objetivo es disminuir los niveles de Hb S a 30% o menos,
manteniendo la Hb entre 9 y 10g/dL. Se logra mediante transfusiones simples, exanguinotransfusión parcial o eritraferesis. Ver guía de transfusión.
• Control de resultados de la terapia transfusional mediante la cuantificación de Hb S.
• Suministrar O2 por cánula nasal o mascara facial para mantener la oximetría de pulso igual a, o por encima de, 92%.
• En presencia de fiebre (>38ºC) iniciar tratamiento con cefotaxima 50mg/kg, EV, c/8 horas. En caso de alergia a las cefalosporinas, indicar clindamicina 10mg/kg, EV, c/6 horas. Si hay evidencias o sospecha de infección de SNC o proceso infec-cioso grave, asociar vancomicina 10-15mg/kg, EV, c/8 horas.
• Suspender profilaxis con penicilina (si es el caso), mientras el paciente reciba an-tibioticoterapia de amplio espectro.
Tratamiento de las Complicaciones Agudas

94
• Si existe hemorragia por aneurisma sangrante, puede estar indicada la cirugía. En adultos con accidentes hemorrágicos está indicado la nimodipina, antagonista del calcio que contrarresta el vasoespasmo, a dosis de 60mg VO c/4 horas por 21 días. Esta opción no está aprobada en niños.
• En caso de asociación con crisis dolorosa, síndrome torácico agudo, secuestro esplénico, crisis aplásica, priapismo, consultar las guías correspondientes.
Control ambulatorio.• Iniciar programa de transfusión crónica cada 3 o 4 semanas para mantener Hb S
igual o menor a 30%. La tasa de recurrencia en pacientes que no reciben terapia transfusional se eleva hasta 70%, a menudo con deterioro neurológico adicional y muerte en alrededor del 25%, mientras que los pacientes tratados con transfusio-nes y que logran mantener niveles de Hb S en 30% o menos, la tasa de recurrencia registrada es del 10%.
• La terapia transfusional es reconocida como la primera opción de tratamiento para la prevención del ictus cerebral en niños de alto riesgo y para disminuir la tasa de recurrencia en quienes ya han sufrido un evento. Se recomienda de mane-ra indefinida en los pacientes pertenecientes a estas categorías.
• Evaluación de los niveles de hierro y/o sobrecarga de hierro con determinación de T2* cardiaco (recíproco del parámetro R2* de relajación ventricular, medido por RMN, se expresa en mili segundos) o en su defecto hierro sérico, TIBC, saturación de transferrina y ferritina antes del inicio del régimen transfusional y posterior-mente cada 3 a 4 meses.
• Iniciar tratamiento con quelantes de hierro en caso de sobrecarga de este ele-mento. Ver guía de tratamiento de sobrecarga de hierro post transfusional.
• Si por alguna razón no se cumpliera la terapia transfusional crónica, se puede iniciar tratamiento con hidroxiurea aun cuando todavía no se ha determinado su utilidad en la prevención primaria de ictus cerebral agudo. Ver guía relacionada con tratamiento con hidroxiurea.
• Si existe donante compatible considerar trasplante con células progenitoras he-matopoyéticas.
• De ser posible y si aplica, practicar estudios de trombofilia.
Ictus y enfermedad cerebral en adultos con enfermedad drepanocíticaEn pacientes adultos, el ictus cerebral más frecuente es de origen trombótico. A pesar del riesgo de sangrado no existe una justificación clara para excluir a estos pacientes del uso de activador tisular del plasminogeno (t-PA) recombinante.
• Indicaciones del t-PA: · Pacientes mayores de 18 años de edad. · Ictus cerebral agudo isquémico con déficit neurológico significativo. · Pacientes sin evidencias de hemorragia en los estudios de imágenes. · La terapia con t-PA debe iniciarse dentro de las tres primeras horas después
.04

95
de ocurrido el evento. · En las primeras 24 horas después de la administración de t-PA no administrar
heparina ni antiagregantes plaquetarios. · En caso de producirse alguna hemorragia indicar tratamiento con crioprecipi-
tados, plasma fresco y/o plaquetas.
Contraindicaciones del t-PA• Ictus cerebral de más de 3 horas de evolución.• Presión arterial sistólica de mayor de 185 mm Hg y/o diastólica mayor de 110mm Hg.• Rápida progresión de los síntomas neurológicos.• Convulsiones como síntoma inicial.• Uso de heparina en las últimas 48 horas.• INR >1,7 o una diferencia del Tiempo de Protrombina mayor de 3 segundos en
relación al control.• Glicemia menor de 50mg/dL o mayor de 400mg/dL.• PTTa prolongado.• Contaje plaquetario <100.000/µL• Historia de ictus cerebral en los 3 últimos meses.• Referencia de cirugía mayor en los 14 días precedentes.• Precedente de hemorragia intracraneal.• Antecedentes de sangrado gastrointestinal o genitourinario en los 21 días precedentes:
· Si no se usa t-PA, indicar aspirina: 325mg/día. · La transfusión crónica tiene las mismas indicaciones que en los pacientes
pediátricos.
Referencias 1.- Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurology 2006;5:501-512.2. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW et al. and The Cooperative Study of Sickle Cell Disease. Cerebrovascular accidents in Sickle Cell Diseasae: Cerebrovascular acci-dents in sickle cell disease: rates and risk factors. Blood 1998; 91:288-94.3. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, hagar W, Darbari DS, Malik P. Beyond the definitions of the phenotypic complications of Sickle Cell Disease: An update on ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi: 10.1100/2012/949535 in http://www.tswj.com/2012/949535/4. Wood KC, Granger DN. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin Exp Pharmacol Physiol 2007; 34: 926-932.5. DobsonSR, Holden KR, Nietert PJ, Cure JH. Moyamoya Syndrome in childhood in sickle cell disease: A predictive factor for recurrent cerebrovascular event. Blood 2002; 99:3144-50.6. Styles LA, Hoppe C, Klitz W et al. evidence for HLA-related susceptibility for stroke in children with sickle cell disease. Blood 2000; 95:3562-67.7. Ohene-Frempong K, Weiner SJ, Sleeper LA et al. Cerebrovascular accidents in sickle cell disease rates
Tratamiento de las Complicaciones Agudas

96
and risk factors. Blood. 1998;91:288-94. 8. Balkaran B, Char G, Morris JS et al. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr 1992;120:360-669. Adams RJ, Mckie VC, Carl EM et al. Long-term stroke risk in children with sickle cell disease screened with transcraneal Doppler. Ann Neurol 1997; 42: 699-704.10. Sloan MA, Alexandrov AV, Tegeler CH, Spencer MP, Caplan LR, Feldmann E et al; Therapeutics and Te-chnology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004;62:1468-81. 11. Lane PA, Buchanan GR, Hutter JJ et al. Sickle cell disease care consortium. Acute stroke or neurolo-gic event in Diagnosis, guidelines for comprehensive care, and care paths and protocols for management of acute and chronic complications. 2001. Disponible en http://scinfo.org/care-paths-and-protocols-chil-dren-adolescents/care-path-acute-strke-or-neurologic-event
4.6. Complicaciones ocularesAmel Guánchez / Médico hematólogo.
Las manifestaciones oculares de la enfermedad drepanocítica, consecuencias de la vasooclusión, son el resultado de la afectación de la conjuntiva, segmento anterior, retina, coroides y nervio óptico con riesgo potencial de ceguera. Tales complicaciones pueden pasar desapercibidas, a menos que se practique un examen formal por un oftalmólogo1.
Indicación de consulta urgente con oftalmología para evaluación exhaustiva: • Trauma ocular en cualquier persona con sindrome drepanocítico, incluyendo el
rasgo drepanocítico.• Todo paciente con sindrome drepanocítico que presente cambio agudo en la visión.
Cuadros clínicosSindrome de compresión orbitaria.Es muy raro. El infarto del esfenoides puede ocasionar hematoma subperióstico y re-acción inflamatoria aguda2.
• Síntomas · Proptosis · Dolor periorbital · Limitación de movimientos · Neuropatía compresiva del nervio óptico.
• Diagnóstico. Imagen mediante resonancia magnética.• Tratamiento.
· Corticoesteroides. · Antibióticos si coexiste infección. · Quirúrgico, si falla el tratamiento médico.
.04

97
ConjuntivaDilataciones vasculares transitorias en formas saculares o de “salchicha”, rojas os-curas con segmentos vasculares en forma de coma, mas comunes en la conjuntiva bulbar inferior. Las comas son mas frecuentes en la ED-SS que en la ED-SC e infre-cuentes en los pacientes que presentan niveles elevados de hemoglobina fetal. El signo de coma es un excelente indicador para el diagnóstico de enfermedad drepanocítica, pero está ausente en los portadores de Hb S2.
• Etiopatogenia. Proliferación endotelial, agregación de eritrocitos en la porción distal de los capilares y dilatación y adelgazamiento en el segmento proxima2.
Cámara anterior La presencia de sangre en la cámara anterior del ojo constituye una verdadera emer-gencia aún para las personas portadoras del rasgo drepanocítico y el ojo debe ser sometido rápidamente a examen, que incluya medida de la presión intraocular (menor de 25mm Hg)2.
• Tratamiento. · La paracentesis generalmente es suficiente. · La oxigenoterapia transcorneal puede reducir la presión intraocular en glauco-
ma secundario a hifema inducido por drepanocitos.
Segmento posterior• Evaluación2:
· Medida precisa de la agudeza visual. · Reactividad pupilar. · Evaluación cuidadosa de las estructuras de la cámara anterior mediante bio-
microscopio con lámpara de hendidura. · Examen de las estructuras posteriores y retina, previa dilatación de la pupila.
En el estudio de retina incluir angiografía con fluoresceína.• Anormalidades de la cámara posterior2:
· Cambios en el disco óptico.
Puntos intravasculares de color rojo oscuro (oclusión intravascular), son transitorios, no ocasionan manifestaciones clínicas. Son mas comunes en ED-SS pero también pueden verse en pacientes con ED-SC y ED-S talasemia.
· Vasooclusión de los vasos posteriores de la retina y de la mácula. · Oclusión de la arteria central o de sus colaterales. · La vasooclusión a nivel peripapilar o arteriolar macular son raras. · También es infrecuente la oclusión de la vena central de la retina. · Los cambios crónicos de la mácula (maculopatía drepanocítica) ocurren por
oclusión vascular crónica; se manifiesta como microaneurismas y se observan como puntos en forma de horquilla o bucles.
· Zona foveal avascular anormal.
Tratamiento de las Complicaciones Agudas

98
· Oclusión vascular coroidal, extremadamente rara. Se han descrito muy pocos casos.
Cambios retinianos no proliferativos:• Tortuosidad vascular. Probablemente por anastomosis arteriovenosas desde la
periferia de la retina. Presente en muchos pacientes con ED-SS y ED-SC.• Hemorragias color salmón. Se deben a hemorragias intrarretinianas superficia-
les. Vistas a mitad de la periferia de la retina, adyacente a una arteriola. • Cavidades. Son espacios causados por la desaparición de hemorragia intrarreti-
niana. La retinopatía no proliferativa característica de la enfermedad drepanocí-tica se observa como manchas iridiscentes y cuerpos brillantes, refringentes en el interior de la cavidad.
• Sol de rayos negros. Corresponden a cicatrices coriorretinianas, ubicadas gene-ralmente en la zona ecuatorial del fondo del ojo. Son el resultado del acúmulo de pigmentos alrededor de un vaso. No causan síntoma visual.
Cambios retinianos proliferativos. Corresponden a la retinopatía proliferativa drepa-nocítica y representan el cambio ocular mas severo de la enfermedad. Por ser una patología progresiva, lo ideal es tratar el tejido neo vascularizado antes de que ocurra la hemorragia en el vítreo. Es mas frecuente en pacientes ED-SC, pero también se observan en pacientes con ED-S talasemia, ED-SS, Hb AS y Hb AC. Existen 5 estadios o categorías.
• Oclusión arteriolar periférica.• Anastomosis arteriovenular.• Proliferación neo vascular.• Hemorragia del vítreo.• Desprendimiento de retina.
Tratamiento3,4
• Se dirige hacia la prevención de pérdida de visión por hemorragia vítrea, despren-dimiento de retina, y las membranas epirretinianas.
• Puede indicarse tratamiento tópico. Evitar el uso de inhibidores de la anhidrasa carbónica ya que favorecen la formación de drepanocitos empeorando la circula-ción del líquido acuoso.
• Dada la alta tasa de regresión espontánea y la falta de progresión de la neovas-cularización en algunos pacientes, las indicaciones para su tratamiento no son siempre claras.
• La intervención terapéutica, a carga de un especialista en retina, generalmente se recomienda en: · Enfermedad bilateral proliferativa. · Hemorragias espontáneas. · Grandes zonas neo vascularizadas elevadas.
.04

99
· Casos en los que casi se ha pedido la visión de un ojo, debido a la retinopatía proliferativa.
• Idealmente debe iniciarse en el estadio III, antes de que exista hemorragia del vítreo y/o desprendimiento de retina.
• Técnicas tales como la diatermia, la crioterapia y la fotocoagulación con láser pro-ducen involución de las lesiones de neovascularización De todos estos métodos, la fotocoagulación con láser tiene menos efectos secundarios.
• Para los estadios avanzados, existen técnicas intensivas de microcirugía vitro-retinal, donde se incluyen la vitrectomía con o sin la colocación de un cerclaje escleral.
• A pesar de microcirugía vítreo-retiniana moderna puede mejorar la visión en muchos pacientes con retinopatía falciforme avanzada, cabe destacar que la cirugía conlleva un riesgo significativo de complicaciones intra y postoperatorias, incluyendo isque-mia ocular severa, hemorragias recurrentes y la presión ocular elevada.
• La oclusión de la arteria central de la retina, un evento que ocasiona pérdida permanente de la visión, demanda intervención urgente dentro de los primeros minutos u horas después de iniciarse los síntomas. El tratamiento consiste en híperoxigenación combinada con rápida reducción de la presión del ojo utilizando técnicas médicas y quirúrgicas.
• Deben considerarse riesgos/beneficios de la exanguino transfusión prequirúrgi-ca, en el contexto del paciente individual.
• Emplear anestesia local y no usar simpaticomiméticos.• Minimizar el uso de simpaticomiméticos tópicos o locales.• Oxígeno durante 48 horas del postoperatorio.• Control periódico de la presión intraocular.
Recomendaciones1. (Figura.4.8)• Los pacientes con enfermedad drepanocítica necesitan ser evaluados cada año
por un especialista en retina.• Comenzando en la infancia, a los 10 años, y continuar durante la adultez. • El estudio requiere dilatación previa de la retina.• Todo paciente con sindrome drepanocítico que consulte por cambios bruscos de la
visión, debe referirse de inmediato al especialista en retina.
Tratamiento de las Complicaciones Agudas
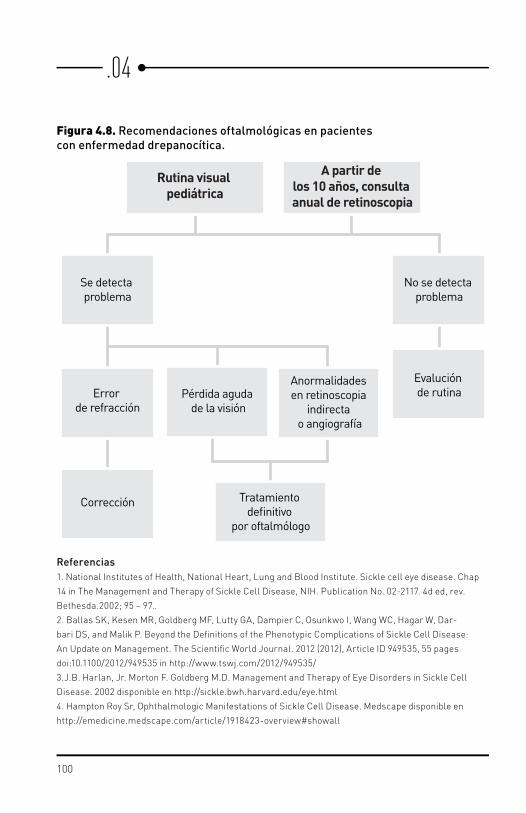
100
Figura 4.8. Recomendaciones oftalmológicas en pacientes con enfermedad drepanocítica.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Sickle cell eye disease. Chap 14 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 95 – 97.. 2. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Dar-bari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/3.J.B. Harlan, Jr. Morton F. Goldberg M.D. Management and Therapy of Eye Disorders in Sickle Cell Disease. 2002 disponible en http://sickle.bwh.harvard.edu/eye.html4. Hampton Roy Sr, Ophthalmologic Manifestations of Sickle Cell Disease. Medscape disponible en http://emedicine.medscape.com/article/1918423-overview#showall
Rutina visual pediátrica
A partir de los 10 años, consulta anual de retinoscopia
Se detecta problema
No se detecta problema
Evalución de rutina
Tratamiento definitivo
por oftalmólogo
Corrección
Error de refracción
Pérdida aguda de la visión
Anormalidades en retinoscopia
indirecta o angiografía
.04

101
4.7. Complicaciones pulmonaresJosé Vicente Gil / Médico neumonólogo pediatra.
Las complicaciones pulmonares en drepanocitosis pueden ser agudas y crónicas y se resumen en la tabla 4.5.
Tabla 4.5. Complicaciones pulmonares en enfermedad drepanocítica1,2
Esta sección se ocupa únicamente del síndrome torácico agudo y de la crisis de asma por ser las más frecuentes y con mayor riesgo.
Sindrome torácico agudo (STA)La llegada de un paciente con enfermedad drepanocítica a la sala de urgencia, con síntomas respiratorios (tos o taquipnea, expectoración, disnea o dolor torácico, sibi-lancias) requiere su inmediata asistencia y el obligatorio descarte del STA definido por la presencia de un infiltrado pulmonar nuevo, que afecte al menos un segmento pul-monar completo (sin atelectasia). En niños la bronquiolitis por virus sincisial o exacer-bación del asma se considera como un STA1.
El STA es un término genérico para las complicaciones pulmonares agudas por cau-sas heterogéneas3. Tal diversidad puede incluir cuadros relativamente banales que presentan una regresión espontánea y otros que evolucionan a un síndrome de dis-tress respiratorio con necesidad de ventilación mecánica4. Cerca del 50% de los STA, no tienen una causa definida5.
Causas de sindrome torácico agudo6
• Relacionadas a la Hb S. · Consecuencia directa:
· Infarto pulmonar por falciformación “in situ”. · Etiología desconocida. · Hipoventilación secundaria a:
· Infarto esterno costal.
Complicaciones agudas Complicaciones crónicas
Sindrome torácico agudo (STA) Enfermedad pulmonar crónica del drepanocítico
Neumonía Apnea obstructiva del sueño
Embolismo graso Alteraciones de la función pulmonar
Infarto por trombosis in situ Hipertensión pulmonar crónica HPC
Infarto de costillas y esternón
Edema agudo de pulmón
Tratamiento de las Complicaciones Agudas

102
· Administración de narcóticos. · Atelectasia post operatoria.
· Infarto por embolismo. · Necrosis de la médula ósea/graso. · Drepanocitos formados a distancia (sinusoides hepáticos).
· Edema pulmonar secundario a sobrecarga de líquido.
Consecuencia indirecta. Infección.• Bacteriana.• Viral.• Micótica.• Protozoarios.• No relacionada a la Hb S.
· Tromboembolismo venoso. · Infecciones oportunistas relacionadas a VIH. · Obstrucción bronquial secundaria a cuerpo extraño o neoplasma. · Otra (aspiración y/o trauma).
El 50% de los pacientes con enfermedad drepanocítica han presentado al menos un episodio de STA y la mortalidad oscila entre 10% y 12%. Es responsable del 50% de todas las muertes en enfermedad drepanocítica. La incidencia del STA es mayor en el genotipo βS/βS y mas bajo en βS/β+ talasemia. Es la segunda causa de hospitalización y la más frecuente de muerte tanto en niños como en adultos y la complicación más común en cirugía y anestesia. Los niños presentan mayor incidencia que los adultos pero menor mortalidad7.
En la figura 4.9 se indican los pasos a seguir frente a un paciente con manifestaciones de tórax agudo.
.04

103
Síndrome Torácido Agudo
Dolor en tórax Taquipnea, distress respiratorio RALES
Hospitalizar, consulta con hematólogo, internista, neumonólogo e infectólogo
Sintomas respiratorios ausentes o leves
Analgesia oral, liquidos orales,
oximetria de pulso
Taquipneadistress respiratorio
RALES
Aumento de síntomasrespiratorios sindrome
de distress respiratorio agudo
Considerar UCI
Hospitalización cosulta con hematólogo
Síntomas respiratorios: moderados o severos
Rx tórax, contaje leucocitos, oximetría de pulso
Disminución de la saturación de oxígeno (oximetría de
pulso) y/o dolor persistente
Analgesia (narcóticos con precaución), hidratación (1-1,5 del mantenimien-
to), no sobrehidratar, oximetría de pulso, terapia respiratoria.
Rx de tórax, gases arteriales, oxígeno,
hidratación (pérdidas más mantenimiento), antibióticos
(incluir para gérmenes atípicos).
Considerar transfusión o eritracitaferesis por
persistencia o empeoramiento de anemia, dificultad
respiratoria o hipoxemia .
Tratamiento de las Complicaciones Agudas
Figura 4.9. Evaluación del paciente con sindrome torácico.El paciente con STA, debe ser atendido de inmediato, cumpliendo los siguientes pasos8:

104
Historia • Investigar síntomas como: fiebre, tos, dificultad para respirar y dolor en el pecho.• Documentar síntomas asociados incluyendo dolor, especialmente dolor en los
huesos y extremidades ya que el tórax agudo está asociado con embolismo graso.• Investigar dolor abdominal ya que este síntoma puede estar asociado con neumo-
nía basal.• Revisar historia de requerimiento de oxígeno. Tratar de determinar la línea basal
de saturación de oxígeno del paciente.• Interrogar sobre antecedentes de asma.• Indagar sobre medicación actual, incluyendo inhaladores utilizados en asma.• Documentar alergia a drogas.• Investigar sobre exposición reciente a infecciones.• Revisar la historia médica enfocándose en problemas relacionados con la enfer-
medad drepanocítica, especialmente complicaciones pulmonares. Preguntar si el paciente ha tenido STA, neumonía, y hospitalizaciones previas por problemas respiratorios.
• Revisar la última evaluación integral, con el propósito de determinar niveles ba-sales del paciente.
Conducta.• Hospitalizar.• Consulta con el hematólogo.• Consulta con el neumonólogo.• Control de signos vitales cada 2-4 horas.• Oximetría de pulso continua.• Control de líquidos ingeridos y eliminados. Peso diario.• Hematología incluyendo plaquetas y reticulocitos.• RX de tórax, repetir si empeoran las manifestaciones clínicas.• Tipeaje y pruebas cruzadas en pacientes con descenso de valores de Hb >2g/dL en
relación a sus valores basales.• Hemocultivo si hay fiebre o historia febril reciente.• Gases arteriales en caso de enfermedad grave.• Bioquímica renal y hepática.
Líquidos, nutrición y cuidados generales.• Conservar la volemia normal. Líquidos orales e intravenosos 1-1½ veces el volu-
men de mantenimiento. Rresulta inapropiado administrar mas líquido y sólo se justifica si el paciente está deshidratado por pérdidas insensibles (fiebre persis-tente). Administrar dextrosa al 5% o dextrosal 0,45%.
• Incentivar la espirometría. Hacer 10 respiraciones profundas cada 2 horas mien-tras esté despierto.
• Fomentar la deambulación.
.04

105
Tratamiento• Suministrar oxígeno para mantener los valores de oximetría de pulso >92% o en
niveles basales del paciente si eran >92%.• Indicar acetaminofén a razón de 15mg/kg/VO, cada 4 horas o cuando la tempera-
tura sea mayor de 38°C (dosis máxima 75mg/kg/día).• Ibuprofeno, 10mg/kg/VO. No administrar ibuprofeno en caso de gastritis, úlcera,
coagulopatía, enfermedad renal o estar en tratamiento con ketorolac y por no más de 72 horas.
• Morfina. · Adultos y niños <50Kg 0,1-0,15 mg/kg cada 2-4 horas, EV. · Niños y adultos >50Kg 0,5-1,0 mg cada 2-4 horas, EV.
Si hay dolor severo, dar 1/3-1/2 de la dosis total de morfina en infusión continua y 1/2-2/3 en forma de bolo. En ocasiones se puede requerir la dosis total de morfina en infusión, más bolos de 0,1mg/kg/hora, pero siempre con cautela.
• Otros medicamentos alternativos, con la finalidad de disminuir la dosis de mor-fina. · Hidromorfona: 0,015-0,020 mg/kg cada 3-4 horas en adultos y niños <50kg;
1,5mg cada 3-4 horas en niños y adultos >50Kg. · Ketorolac.
· Adultos. · Dosis única.
· Endovenosa (disuelto) 30mg (0,5mg/kg). · Intramuscular: 60mg (1mg/kg).
· Dosis múltiple (Máximo 5 días). · Endovenosa (disuelto): 30 mg cada 6 horas (dosis máxima 120mg/día). · Intramuscular: 30 mg cada 6 horas (dosis máxima 120mg/día). · Vía oral: 10-20 mg cada 4-6 horas (dosis máxima 40 mg/día).
· Niños mayores de 3 años, ancianos, insuficiencia renal. · Dosis única.
· Endovenosa (disuelto): 0,25mg/kg; · Intramuscular. 0,5 mg/kg.
· Dosis múltiple (Máximo 5 días). · Endovenosa (disuelto) 0,25mg/kg cada 6 horas (dosis máxima 60mg/día. · Intramuscular: 0,25mg/kg cada 6 horas (dosis máxima 60mg/día. · Vía oral 10mg cada 4-6 horas (dosis máxima 40mg/día).
• Cefotaxime 50mg/kg, EV cada 8h. Si se conoce o sospecha alergia a las cefalospo-rinas, sustituir por la clindamicina 10mg/kg, EV, cada 6 horas.
• En niños, el uso de la clindamicina tiene mayor riesgo de reacciones adversas importantes como la rectocolitis pseudomembranosa (entre otras) por lo que se sugiere usar otros antibióticos.
• Azitromicina 10mg/kg vía oral la primera dosis, luego 5mg/kg o sustituir por eri-
Tratamiento de las Complicaciones Agudas

106
tromicina a razón de 10mg/kg cada 6 horas o por otro macrólido.• En un paciente drepanocítico con STA al cual se quiere proteger contra una in-
fección por neumococo (resistente o no), utilizar teicoplanina a razón de 5mg/kg/dosis las primeras 3 dosis y después 5mg/kg/dosis cada 24h.
• Considerar seriamente añadir vancomicina 10-15mg/kg, cada 8 horas, por vía en-dovenosa, si la enfermedad es severa o existe derrame pleural.
• Furosemida a razón de 0,5-1mg/kg vía endovenosa, si hay sobrecarga hídrica.• Considerar la posibilidad de usar broncodilatadores, especialmente si el paciente
tiene antecedentes de enfermedad respiratoria reactiva o si al examen físico pre-senta sibilancias9.
• Estimar la necesidad del uso de ventilación a presión si el paciente hace poco esfuerzo para respirar o presenta ventilación reducida.
• Valorar la necesidad de transfusión. Ver guía de transfusión. · Transfusión simple para enfermedad moderada, sobre todo si el descenso de
los valores de Hb son >1g/dL en relación a los valores basales del paciente. No transfundir para lograr valores de Hb >10g/dL y de Hto >30.
· Exanguino transfusión parcial o eritrocitaferesis para lograr valores de Hb >10g/dL, Hto >30 y Hb S o Hb S+ C (en caso de ED-SC) a <30% , para la enfer-medad severa y rápidamente progresiva.
• Solicitar ingreso a la unidad de cuidados intensivos en caso de enfermedad severa que progrese rápidamente.
• Remover catéter femoral o central una vez realizada la eritrocitaferesis para evi-tar trombosis.
No existen evidencias que avalen la eficacia de un antibiótico con respecto a otro10, la mayoría de las guías recomiendan el uso de una cefalosporina con un macrólido11.
Criterios para el alta• Mejoría de los síntomas pulmonares y evidencias documentadas de que el paciente res-
pira espontáneamente sin dificultad. • Paciente afebril por >24 horas, cultivos negativos durante >24-48 horas (si es aplicable).• Hemoglobina/hematocrito estables.• El paciente está en condiciones de ingerir líquidos y cumplir tratamiento vía oral • Alivio considerable del dolor y capacidad de manejarlo con medicamentos orales, en
el hogar.• Coordinación adecuada con el servicio de Hematología, tomando en cuenta la necesi-
dad de transfusión crónica y tratamiento con hidroxiurea.• Coordinación adecuada con el propósito de realizar evaluación de función respiratoria.
Prevención• Hidroxiurea, puede disminuir en 50% la frecuencia del STA. Ver guía de tratamien-
to con hidroxiurea.
.04

107
• El régimen de transfusión crónica puede prevenir la repetición de otro evento. Ver guía de transfusión.
AsmaRecientemente han aumentado las evidencias en relación a que el asma predispone a ciertas complicaciones de la enfermedad drepanocítica incluyendo crisis de dolor, sin-drome torácico agudo12, hipertensión pulmonar, enfermedad vascular cerebral aguda5 y muerte13. Cerca de las dos terceras partes de los pacientes drepanocíticos tienen enfermedad obstructiva pulmonar ya sea por la misma enfermedad drepanocítica o porque tienen asma asociada2,14. En la infancia el asma se clasifica en fenotipos: el 70% de los niños menores de 2 años que tiene sibilantes deja de hacerlo alrededor de los 10 años y la mayoría (40%) antes de los 4 años (sibilantes transitorios tempranos); el 30% restante son casi siempre pacientes con asma desencadenada por virus y no están ligadas a un aumento de IgE, solo el 30% del asma en la infancia es de origen alérgico y será el que se mantendrá durante toda la vida.
Debido a que el asma y la enfermedad drepanocítica, son entidades diferentes pero que pueden coexistir, es recomendable descartar el diagnóstico de asma en pacientes con episodios múltiples de dolor o sindrome torácico agudo15. Es necesario precisar diagnóstico diferencial entre los sibilantes de asma que pudieran responder a esteroi-des orales o inhalados, de sibilantes relacionados con la enfermedad drepanocítica, neumonía o anormalidades de la función pulmonar preexistentes con la finalidad de instaurar un tratamiento adecuado.
Diagnóstico (niños y adultos)15,16
• La evaluación neumonológica de un niño drepanocítico sin síntomas pulmonares debe iniciarse a los 2 años de edad.
• Si hay historia de asma, referir a neumonólogo de inmediato.• Función pulmonar por estudios de:
· Espirometría (pre y post broncodilatadores). · Volúmenes pulmonares por pletismografía. · Resistencia de vías aéreas (Rint) que no amerita colaboración y se puede reali-
zar en niños muy pequeños. · Oscilometría de impulso (IOS) que se puede practicar en niños pre escolares
que colaboren.
Edad de inicio • Niños.
· Comenzar a los 6 años. · Repetir:
· Cada 5 años en aquellos sin manifestaciones de asma o debidas a enferme-dad drepanocítica.
Tratamiento de las Complicaciones Agudas

108
· Cada 2-3 años en niños con asma o manifestaciones debidas a enfermedad drepanocítica.
• Adultos. · Iniciar cuando consulte por primera vez.
· Repetir cada 2-3 años si la primera es anormal. · Si hay restricción u obstrucción, referir a neumonólogo.
Tratamiento• Los pacientes con enfermedad drepanocítica y asma, deben ser tratados igual que
los pacientes sin enfermedad drepanocítica.• Metilprednisolona (IV) 1-2mg/kg/día cada 12 horas o hidrocortisona (EV)
10-20mg/kg/dia cada 6 a 8 horas.• En los últimos años han aparecido alternativas para menores de 5 años como son
los antileucotrienos (VO) que se pueden utilizar como monoterapia en casos leves y como ahorradores de esteroides en terapia combinada en asma moderada a severa14.
• Consulta con neumonólogo · Asma leve: por lo menos anualmente. · Asma moderada-severa por lo menos cada 6 meses.
Referencias 1. Minter KR, Gladwin MT. Pulmonary complications of sickle cell anemia. A need of increased recogni-tion, treatment and research. Am J Resp Crit Care Med 2001; 164: 2016-2019.2. Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest 2010; 138:973-83.3. Vichinsky EP, Neumayr LD, Earles NA, Williams R, Lennette ET, Dean D et al. et al. National Acute Chest Syndrome Study Group. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000;342:1855-65. 4. Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B . Acute chest syndrome in sickle cell disease: clinical presentation and course. Blood 1997; 89:1787-92.5. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/.6. Mid-Atlantic Sickle Cell Disease Consortium (MASCC). Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional Human Genetics Network (MARHGN). Diagnosis, guidelines for comprehensive care, and protocols for management of acute and chronic complications.2001; 20-28.7. National Institutes of Health, National Heart, Lung and Blood Institute. Acute Chest syndrome and other pulmonary complications. Chap 16 in The Management and Therapy of Sickle Cell Disease, NIH. Publica-tion No. 02-2117. 4d ed, rev. Bethesda.2002; 103-109.8. Lane PA, Buchanan GR, Hutter JJ, Robert F. Austin RF, Britton HA, Rogers ZR et al. Sickle cell disease care consortium. Acute chest syndrome in Sickle cell disease in children and adolescents: Diagnosis, guidelines for comprehensive care, and care Paths and protocols for management of acute and chro-
.04

109
nic complications. 2001. Disponible en . Disponible en http://scinfo.org/care-paths-and-protocols-chil-dren-adolescents/care-path-acute-chest-syndrome9. Knight-Madden JM, Hambleton IR. Inhaled bronchodilators for acute chest syndrome in people with sickle cell disease. Cochrane Database of Systematic Reviews 2012, Issue 7. Art. No.: CD003733. DOI: 10.1002/14651858.CD003733.pub210. Martí-Carvajal AJ, Conterno LO, Knight-Madden JM. Antibiotics for treating acute chest syndrome in people with sickle cell disease. Cochrane Database of Systematic Reviews 2013, Issue 1. Art. No.: CD006110. DOI: 10.1002/14651858.CD006110.pub3. 11. Miller S. How I treat acute chest syndrome in children with sickle cell disease. Blood 2011.;117:5297-5305. 12. Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with acute chest syndrome and pain in children with sickle cell anemia. Blood 2006; 108:2923-2927.13. Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with increased mortality in indi-viduals with sickle cell anemia. Haematologica 2007; 92:1115-1118.14. Knigth-Madden JM, Forrester TS, Lewis NA, Greenough A. Asthma in children with sickle cell disease and its association with acute chest syndrome. Thorax 2005; 60:206-10.15. Field JJ, DeBaun MR. Asthma and sickle cell disease: two distinct diseases or part of the same pro-cess? Hematology Am Soc Hematol Educ Program 2009; 45-53.16. Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma (GINA) 2012. Available from: http://www.ginasthma.org/.
4.8. Secuestro esplénicoMaritza Suárez / Médico hematólogo.
Definición.
El Estudio Cooperativo de Enfermedad Drepanocítica lo define como un evento agudo, asociado con disminución de la hemoglobina o del hematocrito en, lo menos, 20% por debajo de los valores basales del paciente con aumento del tamaño del bazo palpable de, al menos, 2 cm por encima del tamaño basal1. A menudo se acompaña de tromboci-topenia moderada, reticulocitosis mayor o igual al valor basal. Si hay reticulocitopenia debe pensarse en crisis aplásica asociada. El secuestro esplénico es una situación de urgencia ya que pone en riesgo la vida del paciente2.
Edad de aparición: La mayoría sucede entre los 3 meses y 5 años, pero se han descrito casos tan temprano como a las 5 semanas de vida y en adultos2.
El secuestro esplénico ocasiona un incremento de la anemia en pacientes con enfer-medad drepanocítica lo cual es causa significativa de morbilidad y mortalidad. Esta complicación se observa en niños con ED-SS, ED-SC y ED S-β talasemia. La tasa de recurrencia es de 50% en aquellos que sobreviven el primer episodio3.
Tratamiento de las Complicaciones Agudas

110
Síntomas• Palidez.• Letargia.• Hipotensión.• Aumento del tamaño del bazo.
Enfermedad actual y antecedentes• Aparición y duración de síntomas abdominales como dolor y aumento del tamaño
del bazo.• Signos asociados como palidez, fiebre o letargo.• Transfusiones recientes.• Tamaño previo del bazo.• Valores hematológicos basales incluyendo reticulocitos y plaquetas.• Episodios previos de secuestro.
Examen físico inicial• Signos vitales y saturación de oxígeno.• Grado de palidez.• Estado cardiopulmonar.• Estado neurológico. • Documentar palpación abdominal, tamaño del hígado y del bazo.• Determinar si hay o no shock hipovolémico.
Estudios iniciales• Hematología completa y reticulocitos.• Rx de tórax, si hay fiebre y signos o síntomas de enfermedad respiratoria. • Si la temperatura es >38°. Ver guía de infección y sepsis.• Ecosonograma abdominal.
Conducta. Medidas generales• El paciente ser atendido y tratado con urgencia debido al potencial riesgo de
muerte (Figura 4.10). • Hospitalizar.• Establecer acceso venoso.• Indicar tipeaje y transfusión con concentrado globular.• Considerar el ingreso a unidad de cuidados intensivos, si existe compromiso car-
diovascular.• Control de signos vitales cada 2 horas hasta que el paciente se estabilice, luego
cada 4 horas.• Monitoreo cardiopulmonar.• Balance hídrico.• Oximetría de pulso continua.
.04

111
• De ser necesario, repetir la transfusión para mantener la estabilidad hemodinámica.• Al recuperarse el tamaño del bazo, es posible que haya autotransfusión, ocasio-
nando valores de Hb mayores que lo esperado. El objetivo es que la Hb no supere 8g/dL.
• En casos severos con insuficiencia cardíaca, se sugiere practicar exanguino transfusión.
• Administrar oxígeno 100% en pacientes con Hb extremadamente baja e inestabi-lidad cardiaca hasta que los valores de Hb asciendan y el paciente se estabilice.
• En caso de dolor moderado a severo (ver guía del dolor), administrar acetami-nofén a razón de 15mg/kg/VO, cada 4 horas (Dosis máxima diaria 75mg/Kg) y/o ibuprofeno, 10mg/kg/VO, cada 6-8 horas. No dar ibuprofeno en caso de gastritis, úlcera, coagulopatía, o enfermedad renal.
• En pacientes adultos en quienes el secuestro esplénico generalmente es de inten-sidad moderada, la conducta puede ser de observación.
• No existen evidencias suficientes que indiquen que la esplenectomía aumenta la sobrevida y disminuye la morbilidad en pacientes que han sufrido secuestro es-plénico4. Sin embargo en caso de planificar esplenectomía electiva es necesaria la vacunación antineumococo, antimeningococo.
• Los niños que hacen un secuestro esplénico tienen más probabilidades de repetir el evento, lo que hace necesario planificar la esplenectomía electiva2.
• Reeducar a los padres y otros cuidadores acerca de medir el bazo y la necesidad de buscar atención médica inmediata si observa aumento brusco o rápido del ta-maño del mismo, palidez y dolor abdominal.
Tratamiento de las Complicaciones Agudas

112
Figura 4.10. Conducta ante la sospecha de problema esplénico.
Hematologíareticulocitos
Esplenomegaliainicio desconocido
Crónica, grande o recurrentePocos o mínimos
cambios de hemoglobina basal
Desaparición de síntomas.Control. Alta
Seguimiento hematología reticulocitos plaquetas
Evaluación cuidadosa
Régimen de transfusión crónica.
Esplenectomía.
Educación padres y pacientes
Hospitalización vigilancia estricta transfundir.(Guía de transfusión)
Dolor en cuadrante superior izquierdo, o torácico inferior, esplenomegalia, palidez de:
labios, lecho ungueal, piel, lengua aumento del abdomen y/o malestar abdominal
Esplenomegalia súbita(Guía de secuestro agudo)
Esplenomegalia importan-te con o sin síntomas palidez súbita, dolor
abdominal, deblidiad, shock
Vigilancia estricta, tipeaje, pruebas cruzadas, control seriado de hemoglobina
y reticulocitos
.04

113
Referencias 1. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/.2. Mid-Atlantic Sickle Cell Disease Consortium (MASCC). Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional Human Genetics Network (MARHGN). Evaluation and Management of Acute Splenic Sequestration, In Children with Sickle Cell Disease, in Sickle Cell Disease in Children and Ado-lescents: Diagnosis, Guidelines for Comprehensive Care, and Protocols for Management Of Acute And Chronic Complications.2001; 66 – 67.3. National Institutes of Health, National Heart, Lung and Blood Institute. Splenic Sequestration. Chap 18 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethes-da.2002; 119 – 122.4. Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database of Systematic Reviews 2002, Issue 4. Art. No.: CD003425. DOI: 10.1002/14651858.CD003425.
4.9. PriapismoNirka Lourdes Marcano Arévalo / Médico urólogo. Dalia Velásquez de Lara / Médico hematólogo.
El priapismo, definido como una erección sostenida, no relacionada a estimulación sexual, dolorosa y no deseada, es una complicación bien reconocida de la drepanoci-tosis1,2. La edad promedio a la cual ocurre es a los 12 años, y para la segunda década el 89% de los hombres con drepanocitosis han experimentado uno o más episodios de priapismo1. En la literatura no existen estudios controlados de esta complicación de la enfermedad drepanocítica y la mayoría de los reportes son observacionales3.
FisiopatologíaEl mecanismo fisiopatológico no está muy claro, parece ser debido a una combinación de hipoxia y alteración del flujo venoso del pene. La disminución del flujo venoso a tra-vés del pene durante la erección normal aumenta la extracción de oxígeno, como re-sultado de la hipoxia hay falciformación con la consecuente congestión de los cuerpos cavernosos, incremento de la viscosidad, disminución de la salida de sangre venosa y mayor hipoxia. La sangre extraída de los cuerpos cavernosos durante un priapismo, es oscura y con bajos niveles de glucosa y PO2.
3
Factores precipitantes y factores de riesgoEl 75% de los priapismos suceden entre la media noche y la 6 de la mañana. La acidosis resultante de la deshidratación y la hipoventilación durante el sueño, pueden ser fac-tores precipitantes. Otros factores desencadenantes descritos son: el acto sexual5, la
Tratamiento de las Complicaciones Agudas

114
masturbación6, la ingestión de alcohol7, infección de la próstata o de la vejiga, trauma reciente, medicamentos con efectos adversos autonómicos8; sin embargo, la mayoría de los casos no tiene etiología precisa.
Entre los factores de riesgo están la leucocitosis, niveles bajos de hemoglobina fetal y hemólisis severa. Estudios recientes han ligado a la hemólisis severa con úlcera en las piernas, priapismo e hipertensión pulmonar9.
Aspectos psicosocialesPrecozmente desde la niñez, los pacientes del sexo masculino deben conocer que el priapismo es un aspecto de la drepanocitosis y que no es un evento que cause ver-guenza. Los niños y hombres jóvenes, al igual que sus familiares, necesitan saber que han de estar preparados para buscar atención médica tan pronto como comience un episodio de priapismo ya que si no es tratado puede ocasionar impotencia en el futuro. Los hombres necesitan saber que una vejiga llena puede disparar el priapismo, lo cual obliga a orinar regularmente. Además evitar actividad sexual prolongada, por ser un factor desencadenante. Si tienen más de un episodio, se pueden prescribir medica-mentos que prevengan las recurrencias.
ClasificaciónEl priapismo ha sido clasificado de diversas formas3:
• Desde el punto de vista fisiopatológico. · Isquémico (bajo flujo): Trastorno del flujo venoso y/o estasis. · Recurrente: Erecciones dolorosas recurrentes con períodos intermitentes de
detumescencia. · No isquémico (de “alto flujo”) es un trastorno de afluencia de la circulación
arterial.
En la enfermedad drepanocítica el tipo de priapismo que ocurre corresponde al pria-pismo de “bajo flujo” o priapismo isquémico.
• Por su duración: · <4 horas de duración.
· Aislados e infrecuentes. · No requiere intervención médica.
· >4 horas · Tan prolongado como 12 horas. · Riesgo de impotencia si no se atiende inmediatamente10. · A menudo requiere hospitalización y tratamiento:
· Médico. · Quirúrgico.
• Intermitente · Pronóstico bueno.
.04

115
· Doloroso. · De corta duración. · Repetitivo · Detumescencia a las pocas horas del primer episodio. · Rara vez requiere de intervención médica.
• Desde el punto de vista anatómico · Bicorporal
· Es frecuente en niños. · Involucra ambos cuerpos cavernosos. · Patrón intermitente.
· Tricorporal · Es común en hombres de más edad. · Involucra ambos cuerpos cavernosos y el cuerpo esponjoso · Se asocia con ictus cerebral, enfermedad pulmonar crónica, insuficiencia
renal crónica, úlceras en piernas11.
Tratamiento• Objetivos
· Tratar el dolor · Evitar la impotencia
Priapismo <de 2-4 horas de duración (puede ser inicio de priapismo prolongado)• <2 horas3
· Generalmente se trata en el hogar. · Control del dolor con analgésicos de uso habitual en el hogar. · Hidratación vía oral. · Estimular micción. · Ejercicio moderado. · Baño tibio. · No usar hielo, bolsas de hielo o enemas de agua fría. · Benzodiacepinas.
• >2 horas: El paciente debe acudir a la Emergencia donde se seguirá el protocolo Interconsulta urgente con urólogo con experiencia en el tratamiento de enferme-dad drepanocítica.
Conducta inicial • Pasar de inmediato, al paciente a la sala de examen o consultorio.• Realizar historia con énfasis en:
· Inicio y duración de la erección. · Intensidad del dolor. · Factores desencadenantes.
Tratamiento de las Complicaciones Agudas

116
· Trauma. · Infección. · Uso de drogas
· Alcohol · Cocaína.
· Medicamentos · Psicotrópicos. · Sildenafil. · Testosterona.
· Síntomas asociados · Fiebre. · Disuria. · Deshidratación. · Dolor en otro sitio.
· Historia de episodios anteriores · Tratamiento y efectividad del mismo. · Apnea del sueño.
· Practicar examen físico que incluya · Signos vitales. · Estado de hidratación. · Grado de palidez. · Estado cardiopulmonar. · Examen del pene y del periné.
· Laboratorio, solicitar lo siguiente: · Hematología completa y reticulocitos. · Uroanálisis y urocultivo (si hay manifestaciones de infección). · Examen toxicológico (si hay indicación). · Hemocultivo (si hay fiebre. Ver guía de infección y sepsis). · Tipeaje y pruebas cruzadas si hay: Palidez extrema.
Signos o síntomas respiratorios, neurológicos y de secuestro esplénico.• En caso de transfusión ordenar concentrado globular, leuco-reducido e inmu-
nofenotípicamente compatible al menos para antígenos del Rh, Duffy y Kell. Ver guía de transfusión.
• Hidratar por vía endovenosa a razón de 10cc/Kg a pasar en una hora de solución 0,30% Y luego continuar con mantenimiento.
• Analgesia: En caso de dolor severo, administrar morfina: · Adultos y niños <50kg: 0,1-0,15mg/Kg cada 2-4 horas, EV. · Adultos y niños >50kg: 5-10mg cada 2-4 horas, EV.
Siguiendo el esquema si hay dolor severo: 1/3-1/2 de la dosis total de morfina en in-fusión continua y 1/2-2/3 en forma de bolo. En ocasiones se puede requerir la dosis
.04

117
total de morfina en forma de infusión, más bolos de 0,1mg/kg/hora, pero siempre con cautela. Ver guía del dolor.
• Oximetría de pulso continua.• Oxígeno por cánula o máscara facial para mantener la saturación de O2 en >92%.• Si no hay desentumecimiento en la primera hora de iniciado el tratamiento, se re-
quiere practicar aspiración e irrigación. Este procedimiento debe ser practicado tan pronto como sea posible por un urólogo: · Generalmente no requiere sedación. · Limpiar el pene con betadine. · Aplicar 0,5mL de lidocaína al 1%, en la parte lateral del pene; primero infiltrar
la piel y luego albugínea del pene. · Insertar una aguja 19G en el cuerpo cavernoso del pene y por medio de una
jeringa de 10mL con llave de tres vías, extraer tanta sangre como sea posible. En caso de niños se utiliza una aguja 23G.
· Con otra jeringa de 10mL, inyectar intracavernosamente, 1-2mL de solución diluida de fenilefrina, preparada de la forma siguiente: añadir 1mL de fenilefri-na (10mg/mL) a 99mL de solución salina normal para una concentración final de 100µg/mL.
· Se repite la inyección a intervalos de 2 minutos hasta lograr la detumescencia, sin sobrepasar un total de 10mL de solución inyectada.
· En caso de no haber fenilefrina, utilizar una solución de epinefrina al 1:1.000.000 (es decir, 1mL de 1:1000 epinefrina diluida en 1 litro de solución salina normal).
· Si la detumescencia no se logra con la fenilefrina o epinefrina, irrigar el cuerpo cavernoso con solución salina normal con o sin heparina utilizando la tercera vía.
· Una vez terminado el procedimiento, extraer la aguja y ejercer presión firme durante 5 minutos (por reloj), para evitar la formación de hematoma.
· Si el pene permanece desentumecido durante >1 hora, puede darse el alta y control posterior con el hematólogo y urólogo.
· Si el priapismo recurre, repetir aspiración e irrigación 3-4 veces si es necesario. · Si el procedimiento falla, hospitalizar al paciente para manejo intrahospitalario.
Los agentes simpaticomiméticos incluyen epinefrina, norepinefrina y fenilefrina. Aun-que estos fármacos portan el riesgo de efectos sistémicos tales como hipertensión, taquicardia, palpitaciones y arritmias cardiacas, la fenilefrina es el agente preferido para uso en el tratamiento de priapismo dada su selectividad por los receptores alfa adrenérgicos y sus limitados efectos cardiovasculares.
Independientemente de cuál sea el agente empleado, se recomienda el monitoreo no invasivo de los pacientes durante y después de su administración. Si hay priapismo con más de 4 horas de duración, realizar conducta inicial. En la figura 4.11 se resumen los procedimientos a cumplir según el caso en particular.
Tratamiento de las Complicaciones Agudas
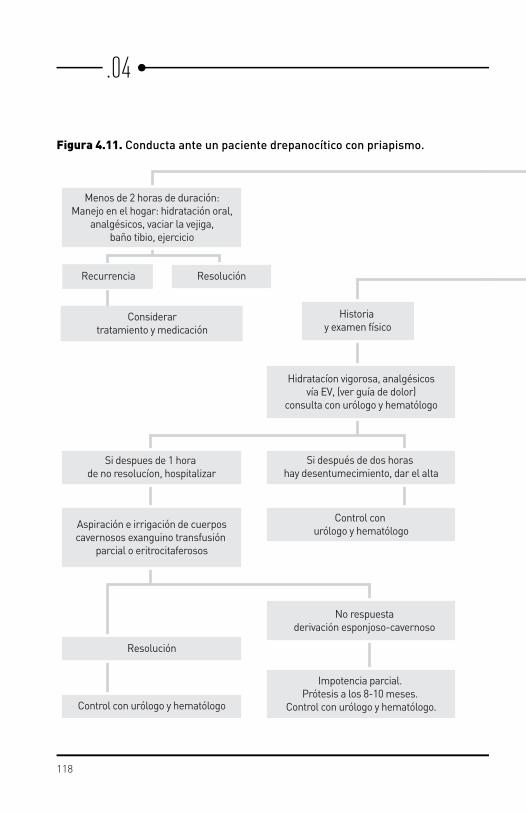
118
Figura 4.11. Conducta ante un paciente drepanocítico con priapismo.
Priapismo
Recurrencia
Considerartratamiento y medicación
Más de 4 horas de duración
Hidratación vigorosa, analgésicos vía EV (ver guía de dolor)
consulta con urólogo y hematólogo
Aspiración e irrigaciónde cuerpos cavernosos
Historia y examen físico Consulta urgente con:
hematólogo y urólogo
Hidratacíon vigorosa, analgésicosvía EV, (ver guía de dolor)
consulta con urólogo y hematólogo
Si despues de 1 hora de no resolucíon, hospitalizar
Control con urólogo y hematólogo
Si después de dos horas hay desentumecimiento, dar el alta
Aspiración e irrigación de cuerposcavernosos exanguino transfusión
parcial o eritrocitaferosos
Control con urólogo y hematólogo
No respuestaderivación esponjoso-cavernoso
Impotencia parcial. Prótesis a los 8-10 meses.
Control con urólogo y hematólogo.
Menos de 2 horas de duración:Manejo en el hogar: hidratación oral,
analgésicos, vaciar la vejiga,baño tibio, ejercicio
Más de 2 horas y menos de 4 horas de duración.
Consulta a emergencia del hospital
Si a las 24 horas no hay respuesta, practicar derivación
esponjoso-cavernoso.
Si a las 12 horas no hay mejoríapracticar exanguino transfusión
parcial o eritracitafaresis
Resolución
Resolución
.04

119
Priapismo
Recurrencia
Considerartratamiento y medicación
Más de 4 horas de duración
Hidratación vigorosa, analgésicos vía EV (ver guía de dolor)
consulta con urólogo y hematólogo
Aspiración e irrigaciónde cuerpos cavernosos
Historia y examen físico Consulta urgente con:
hematólogo y urólogo
Hidratacíon vigorosa, analgésicosvía EV, (ver guía de dolor)
consulta con urólogo y hematólogo
Si despues de 1 hora de no resolucíon, hospitalizar
Control con urólogo y hematólogo
Si después de dos horas hay desentumecimiento, dar el alta
Aspiración e irrigación de cuerposcavernosos exanguino transfusión
parcial o eritrocitaferosos
Control con urólogo y hematólogo
No respuestaderivación esponjoso-cavernoso
Impotencia parcial. Prótesis a los 8-10 meses.
Control con urólogo y hematólogo.
Menos de 2 horas de duración:Manejo en el hogar: hidratación oral,
analgésicos, vaciar la vejiga,baño tibio, ejercicio
Más de 2 horas y menos de 4 horas de duración.
Consulta a emergencia del hospital
Si a las 24 horas no hay respuesta, practicar derivación
esponjoso-cavernoso.
Si a las 12 horas no hay mejoríapracticar exanguino transfusión
parcial o eritracitafaresis
Resolución
Resolución
Tratamiento de las Complicaciones Agudas

120
Cuidados generales• Tomar una vía venosa e iniciar hidratación con soluciones hipotónicas, dextrosal
0,45 a razón de 10mL/Kg, a pasar en una hora, luego continuar con líquidos vía oral y parenteral (Dextrosal 0,45% + 20mEq KCl, este último ajustado a los resul-tados de la química sanguínea). El total de líquidos utilizados no debe exceder a 1,5 veces el volumen de mantenimiento (incluyendo el volumen de infusión de drogas).
• Incentivar la espirometría. Hacer 10 respiraciones profunda cada 2 horas mien-tras esté despierto.
• Administrar O2 por cánula nasal o máscara facial necesaria para mantener la oximetría de pulso >92% o en cifras basales del paciente, si es >92%. .
• Fomentar la deambulación.
Tratamiento• Aplicar las medidas generales como en el priapismo ≥ dos horas de duración.• Analgesia
· Dolor severo · Morfina.
· Adultos y niños <50 kg: 0,1-0,15mg/kg cada 2-4 horas, EV. ó 0,05-0,1mg/kg/h en infusión continua. · Adultos y niños >50kg: 5-10 mg cada 2-4 horas, EV.
· Otros medicamentos alternativos, con la finalidad de disminuir la dosis de morfina. · Hidromorfona.
· Adultos y niños <50 kg: 0,015-0,020mg/kg cada 3-4 horas. · Niños y adultos >50kg: 1,5mg cada 3-4 horas.
· Dolor moderado · Acetaminofén + codeína: 1mg/kg, vía oral, cada 4 horas. · Ibuprofeno, 10mg/kg/vía oral, cada 6-8 horas. No indicarlo en caso de
gastritis, úlcera, coagulopatía, enfermedad renal, o estar en tratamiento con ketorolac, ni por mas de 72 horas.
· Evalúe el dolor al menos dos veces al día. Si hay mejoría del dolor, disminuya la dosis, no aumente el tiempo entre dosis.
· Exanguino transfusión parcial o eritrocitaferesis, con la finalidad de llevar la Hb a 10g/dL y los niveles de Hb S a <30%.
· Considerar la transfusión simple como alternativa a la exanguino transfusión parcial, si la Hb es igual o inferior a 6g/dL (No transfundir en bolo para llevar la Hb a 10g/dL y hematocrito a 30).
· El médico, la enfermera, el paciente y los familiares deben estar alerta ante la aparición de cefalea intensa o cualquier signo o síntoma neurológicos, ya en el lapso de 1-10 días del inicio del priapismo, puede aparecer enfermedad vascu-lar cerebral isquémica, el riesgo es mayor después de la transfusión.
· La respuesta clínica a la exanguino transfusión es variable, y los efectos colate-
.04

121
rales van desde cefalea o convulsiones hasta obnubilación requiriendo soporte ventilatorio. La asociación de drepanocitosis, priapismo, exanguino transfusión y eventos neurológicos recibe el nombre de síndrome de Aspen.
· Si el priapismo no responde a las medidas generales, al drenaje e irrigación y pasa de 24 horas, evaluar la posibilidad de una derivación esponjoso-cavernoso. (Derivación de Winter). Con un tru-cut (aguja utilizada para biopsia), se crea una derivación entre el glande y la porción distal del cuerpo cavernoso, que permite que la sangre fluya desde el cuerpo cavernoso distendido al cuerpo esponjoso indemne.
No existen evidencias suficientes relacionadas con los riesgos y beneficios de los di-versos tratamientos del priapismo en paciente con enfermedad drepanocítica. Se re-quiere realizar estudios aleatorios y controlados para tener evidencias suficientes.
Complicaciones del priapismo Sangrado por los orificios en el pene, como parte de los procedimientos de aspiración y derivación.
• Infección.• Necrosis de piel.• Daño o estenosis de la uretra.• Fístulas. • Impotencia.
Criterios para el alta• Resolución del priapismo (desentumecimiento y desaparición del edema).
Tarda 3-4 semanas.• Ausencias de otras complicaciones.
Medidas preventivas• Considerar:
· Uso de seudoefedrina todas las noches. Concentración: 7,5mg/mL. · Niños menores de 10 años 30mg/día/VO. · Mayores de 10 años: 60mg/día/VO.
· Régimen de transfusión crónica durante 6-12 meses, en caso de priapismo recurrente (>1 en un mes o >4 en un año).
• Si la impotencia persiste por 12 meses o más, el paciente considerar la implanta-ción de una prótesis peneana semirrígida.
Referencias 1. Mantadakis E, Cavender JD, Rogers ZR, Ewalt DH, Buchanan GR. Prevalence of priapism in children
Tratamiento de las Complicaciones Agudas

122
and adolescents with sickle cell anemia. Am J Pediatr Hematol Oncol 1999;21:518-22.2. Miller ST, Rao SP, Dunn EK, Glassberg KI. Priapism in children with sickle cell disease. J Urol 1995; 154:844-8477.3. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/.4. Olujohungbe B, Adeyoju A, Yardumian A, Akinyanju O, Morris J , Westerdale N et al. A prospective diary study of stuttering priapism in adolescents and young men with sickle cell anemia: report of an interna-tional randomized control trial–the priapism in sickle cell study. J Androl 2011; 32 (4): 375–382.5. Krauss L, Fitzpatrick T. The treatment of priapism by penile aspiration under controlled hypotension. J Urol 1961; 85: 595–598. 6. Karayalcin G, Imran M, Rosner F. Priapism in sickle cell disease: report of five cases. Am J Med Sci 1972: 264:289–293. 7. Conrad ME, Perrine GM, Barton JC, Durant JR. Provoked priapism in sickle cell anemia. Am J Hematol 1980;9:121–122. 8. Galloway SJ and Harwood-Muss AL. Sickle-cell anemia. A review. J Emer Med 1988;6:213–226. 9. Kato GJ, Gladwin MT and Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical sub phenotypes. Blood Rev 2007;21:37–47.10. National Institutes of Health, National Heart, Lung and Blood Institute. Priapism. Chap 20 in The Ma-nagement and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 95 – 97.11. Chinegwundoh FI, Anie KA. Treatments for priapism in boys and men with sickle cell disease. Cochrane Database of Systematic Reviews 2004, Issue 4. Art. No.: CD004198. DOI: 10.1002/14651858.CD004198.pub2.
4.10. Sindrome de embolización sistémicaDalia Velásquez de Lara / Médico hematólogo.
El infarto óseo y necrosis es una de las complicaciones de la enfermedad drepanocíti-ca. Cuando el infarto es masivo con necrosis de la médula ósea existe un alto riesgo de embolismo graso hacia los vasos pulmonares.
Factores de riesgo1
• Genotipo βS/βC.• Embarazo.• Tratamiento con corticoesteroides.
Síntomas y signos2
• El embolismo se inicia lentamente y se completa en 24-48 horas.• Generalmente precedido por una crisis dolorosa.• Fiebre presente o ausente.
.04

123
• Pulmones. · Disfunción respiratoria presente en el 75% de los casos:
· Taquipnea. · Disnea. · Cianosis. · Hipoxemia (puede detectarse antes de la instalación de los síntomas).
• Cerebro. · Disfunción cerebral observada en 86% de los casos:
· Confusión. · Rigidez. · Coma. · Edema cerebral contribuye al deterioro de las condiciones general.
• Piel. · Rash petequial, no detectable en conjuntivas, tórax, axilas y cuello; aparece
dentro de las 24-36 horas y desaparece dentro de la primera semana en el 20%-50% de los pacientes.
• Riñón. · Lipiduria. · Oliguria. · Anuria.
• Hígado. · Ictericia.
• Retina. · Exudados. · Edema. · Hemorragia. · Glóbulos de grasa intravasculares.
• Laboratorio. · Anemia progresiva. · Eritroblastemia. · Trombocitopenia. · Coagulación vascular diseminada (CID).
• Diagnóstico2
· Se basa en los criterios de Gurd y Wilson (Tabla 4.5). · Se requiere la presencia de, al menos, 1 criterio mayor y 4 menores.
Tratamiento de las Complicaciones Agudas

124
Tabla 4.6. Criterios de Gurd y Wilson para el diagnóstico de embolismo graso2
• Estudios2
· Gases arteriales. · Hematología completa y reticulocitos. · VSG. · En el examen citológico de orina, sangre y esputo aparición de glóbulos de
grasa. Su ausencia no excluye el embolismo graso. · La Rx de tórax seriado revela de infiltrado difuso. · La TAC de tórax puede ser normal. · RNM de cerebro, normal en algunos casos, pero en pocos se observan imáge-
nes hiperdensas no confluentes. · El lavado bronquial demuestra gotas de grasa en los macrófagos, estos tam-
bién pueden estar presente en pacientes con sepsis y en hiperlipidemia.
• Tratamiento3
· Administrar oxígeno por cánula o máscara facial para mantener la saturación de O2 en >92%.
· Solicitar transfusión o exanguinotransfusión lo más pronto posible. Los pocos pacientes con enfermedad drepanocítica que han sobrevivido a eventos de este tipo han recibido transfusión. Ver guía de transfusión.
· Mantener el volumen intravascular. · Indicar ventilación mecánica. · Ningún medicamento ha sido efectivo.
Criterios Mayores Criterios Menores
Rash petequial Taquicardia
Insuficiencia respiratoria Fiebre
Manifestaciones cerebrales Cambios en retina
Ictericia
Signos renales
Trombocitopenia
Anemia
VSG elevada
Glóbulos de grasa
.04

125
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Acute chest syndrome and other pulmonary complications. Chap 16 in The Management and Therapy of Sickle Cell Disease, NIH. Publica-tion No. 02-2117. 4d ed, rev. Bethesda.2002; 103 –109.2. Nissar Shaikh. Emergency management of fat embolism syndrome. Emerg Trauma Shock. 2009;2:29-33.Disponible en http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2700578/3. Mid-Atlantic Sickle Cell Disease Consortium (MASCC). Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional Human Genetics Network (MARHGN). Diagnosis, guidelines for comprehensive care, and protocols for management of acute and chronic complications.2001; 20-28.
4.11. Dolor abdominalJaime Bracho / Médico hematólogo.
La crisis de dolor abdominal amerita inmediata asistencia. En la mayoría de las ocasio-nes es auto limitada y se resuelve espontáneamente, siendo un componente frecuente de los episodios vasooclusivos, sin embargo es necesaria la evaluación para descartar otras causas (hepáticas, biliares, intestinales, pancreáticas, vertebrales, urológicas, ginecológicas o pulmonares), considerando como prioritarias las quirúrgicas1.
Sindrome de cuadrante superior derecho (SCSD)Esta condición se define como el dolor abdominal que se localiza con predominio en el hipocondrio derecho y se asocia a ictericia, náuseas y vómitos, febrícula y hepato-megalia dolorosa con elevación de las transaminasas e hiperbilirrubiemia. Puede ser debido a complicaciones de la propia enfermedad por vaso-oclusión intra-hepática, colelitiasis (cólico biliar o colecistitis), hepatitis viral (infecciosa o post-transfusional) o hepatotoxicidad inducida por drogas. No obstante, la distinción entre ellas a veces es difícil e inclusive pueden concurrir en un mismo paciente1 (Figura 4.12).
Tratamiento de las Complicaciones Agudas
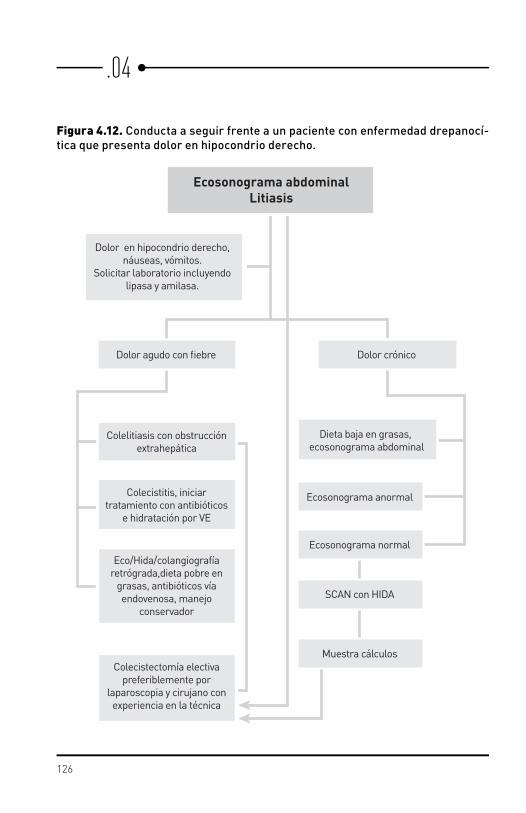
126
Figura 4.12. Conducta a seguir frente a un paciente con enfermedad drepanocí-tica que presenta dolor en hipocondrio derecho.
.04
Dolor en hipocondrio derecho,náuseas, vómitos.
Solicitar laboratorio incluyendolipasa y amilasa.
Ecosonograma abdominalLitiasis
Dolor agudo con fiebre Dolor crónico
Ecosonograma anormal
Ecosonograma normal
SCAN con HIDA
Muestra cálculos
Dieta baja en grasas, ecosonograma abdominal
Colelitiasis con obstrucción extrahepática
Colecistitis, iniciar tratamiento con antibióticos
e hidratación por VE
Eco/Hida/colangiografía retrógrada,dieta pobre en
grasas, antibióticos vía endovenosa, manejo
conservador
Colecistectomía electiva preferiblemente por
laparoscopia y cirujano con experiencia en la técnica

127
Las complicaciones derivadas de la propia enfermedad presentan una base fisiopato-lógica común. Desde un punto de vista clínico predominan algunas de las manifesta-ciones que definen este síndrome en cada entidad:
• La crisis aguda hepática es un síndrome generalmente auto limitado que cursa con2: · Dolor en el hipocondrio derecho · Elevación de los valores de ALT (generalmente menos de 300UI/L) que dismi-
nuye a los pocos días. · Hepatomegalia
• Secuestro hepático agudo: Es una complicación vasooclusiva, rara vez diagnosti-cada que pone en riesgo la vida cuando sucede en niños. Puede formar parte del síndrome de falla multiorgánica3. · Manifestaciones clínicas:
· Aumento rápido del tamaño del hígado en relación al tamaño basal. · Dolor en hipocondrio derecho. · Hígado de consistencia blando.
· Laboratorio · Disminución de los valores de hemoglobina y hematocrito y aumento
de reticulocitos. · Bilirrubina puede ser tan alta como 24mg/dL, a predominio de la conjugada. · Fosfatasa alcalina normal o tan alta como 650UI/L.
· Imágenes · Ecosonograma y TAC: hepatomegalia.
• Colestasis intrahepática: Constituye la forma más grave de estas com¬plicacio-nes. El hígado es afectado por cambios vasooclusivos o isquemia frecuente y daño por reperfusión, sobrecarga de hierro secundaria a transfusiones, disfunción del endotelio vascular, estado de hipercoagulabilidad y destaca la ictericia por hiper-bilirrubiemia acentuada. Se acompaña o no de insuficiencia renal aguda, coagu-lación intravascular diseminada, acidosis láctica y encefalopatía3. Se distinguen dos categorías: · Benigna
· Ictericia y prurito. · Aumento importante de bilirrubina incluso por encima de 50mg/dL, a expen-
sas de la bilirrubina conjugada (directa). · Aumento discreto de:
· Fosfatasa alcalina. · ALT y AST.
· No hay alteración en la síntesis hepática. · Pruebas de coagulación (PT,PTT y Fibrinógeno) normales. · Histología hepática: Colestasis intrasinusal debido a drepanocitos e hiperpla-
sia de células de Kupffer con fagocitosis de drepanocitos.
Tratamiento de las Complicaciones Agudas

128
· Progresiva · Ausencia de cirrosis. · Bilirrubina extremadamente alta. · Fosfatasa alcalina muy elevada. · Elevación variable de ALT y AST. · Insuficiencia renal. · Trombocitopenia. · Pruebas de Coagulación alteradas: PT y PTT prolongados y fibrinógeno bajo. · Histología hepática: Colestasis intrasinusal debido a drepanocitos. · Hiperplasia de células de Kupffer con fagocitosis de drepanocitos.
• Conducta · Constituye una verdadera emergencia. Los pasos a seguir son los siguientes:
· Hospitalización y evaluación del paciente para determinar causa e intensidad del dolor.
· Garantizar un acceso venoso. · Registro de signos vitales.
· Exámenes complementarios iniciales a solicitar: · Hematología completa y reticulocitos. · Bilirrubina total, conjugada y no conjugada, ALT, AST · Amilasa, lipasa. · Creatinina. · Electrolitos séricos. · Gasometría arterial. · Cultivos de sangre y orina si hay fiebre. · Serología para hepatitis viral. · Rx simple de abdomen (de pie). · Ecosonograma abdominal. · Rx Tórax AP y lateral, en los casos con síntomas respiratorios asociados. · Evaluación por cirugía.
• Manejo terapéutico inicial · Hidratación EV. · Dieta absoluta. · Oxigenoterapia. · Analgesia sin dosis altas de opiáceos. · Antibióticos EV (si hay sospecha de infección).
El tratamiento definitivo dependerá del cuadro clínico. La rapidez de aparición de los síntomas y de la corrección de la ALT orienta la terapia. Los síntomas que comienzan bruscamente a menudo son típicos de un proceso autolimitado. Síntomas que inician de manera insidiosa durante días o semanas pueden deberse a una condición más severa como hepatitis viral o hepatitis autoinmune, infarto hepático, o disfunción de la
.04

129
vesícula biliar; la elevación de la bilirrubina por encima de 30mg/dL representa insufi-ciencia hepática aguda o colestasis intrahepática.
Si la condición es típica de enfermedad drepanocítica e inflamación, la ALT disminuye en pocos días. La elevación severa y persistente de la ALT puede relacionarse con infarto hepático.
• Crisis hepática aguda. · Generalmente basta con terapia de soporte con hidratación y analgesia. Ver
sección de tratamiento del dolor.
• Secuestro hepático. · Tratar de revertir rápidamente el volumen sanguíneo y masa eritrocitaria e in-
tentar revertir la falciformación mediante transfusión de concentrado globular, expansores del plasma o exanguino transfusión. Ver sección de transfusión.
· Oxigenación con inspiraciones incentivadas.
• Colestasis intrahepática. · Recomendaciones:
· Benigna. · Observación, por lo general la colestasis se resuelve de manera espontánea
en 2-8 semanas.
· Progresiva. · Terapia transfusional precoz: mediante transfusión simple, exanguinotrans fusión o eritrocitaferesis según sea el caso (Ver guía de transfusión). · Plasmaferesis. · Medidas de soporte como plasma fresco congelado, fibrinógeno en caso de coagulopatía. · Hemodiálisis en caso de insuficiencia renal.
Referencias1. González F FA. Dolor abdominal. Síndrome del cuadrante abdominal superior. Secuestro esplénico en Guía de manejo de las enfermedades falciformes. Grupo de eritropatología. Asociación Española de Hematología y Hemoterapia.2000; 59-62.2. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/3. National Institutes of Health, National Heart, Lung and Blood Institute. Gall Bladder and Liver. Chap 17 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Be-thesda.2002; 111-117.
Tratamiento de las Complicaciones Agudas

130
Tratamiento de las complicaciones crónicas.05
5.1. Cardiovasculares
Complicaciones cardiovasculares de la drepanocitosisDr. J. Ildefonzo Arocha Rodulfo / Médico cardiólogo.
Siendo los eritrocitos las células predominantes en la sangre, sus características bio-mecánicas determinan principalmente las propiedades reológicas y hemodinámicas sanguíneas, tanto en condiciones normales como patológicas. En la enfermedad dre-panocítica, los eritrocitos mecánicamente frágiles y deformados contribuyen al de-terioro del flujo sanguíneo y de otros aspectos fisiopatológicos de la enfermedad. La mayor causa subyacente de tales alteraciones es la polimerización de la hemoglobina S y el comportamiento del eritrocito falciforme bajo condiciones de desoxigenación1.La recurrencia periódica de crisis vasooclusivas dolorosas es la característica que define a la enfermedad drepanocítica y entre las múltiples patologías asociadas a esta enfermedad, se ha considerado a la interacción del eritrocito falciformado con el endo-telio como un mecanismo potencial desencadenante de tales eventos2.
La oclusión vascular de los pequeños y grandes vasos puede conducir al daño crónico de múltiples órganos incluyendo el cerebro, pulmones, hueso, riñón, hígado, bazo y retina. Sin embargo, poco se conoce del impacto de la enfermedad drepanocítica sobre la función cardíaca cuyas manifestaciones incluyen la disfunción ventricular derecha e izquierda, sistólica y diastólica, incremento en el gasto cardíaco, cardiomegalia e isquemia miocárdica. El daño cardíaco progresivo proveniente de la sobrecarga de hierro ocurre en los pacientes que requieren rutinariamente transfusión de sangre2. Los mecanismos fisiopatológicos radican fundamentalmente en la presencia de diver-sos cambios que se suceden a nivel celular con las siguientes características3-6:
• Las modificaciones impuestas al eritrocito por la existencia de la HbS y su proce-so de polimerización, la desoxigenación y consecuente polimerización de la HbS trae como consecuencia profundas alteraciones en la estructura y función de la membrana 2 y la deformación del hematíe que adopta diferentes formas según la velocidad en que la desoxigenación se produce.
• Reacciones de adhesión. Desde hace muchos años se conoce que los glóbulos rojos SS se adhieren al endotelio in vitro, y que esta acción se correlaciona con la severidad de la enfermedad. Las interacciones adhesivas son complejas e involu-cran a una gran cantidad de ligandos, receptores e interacciones no específicas que varían dependiendo de la edad y densidad de la célula, las alteraciones del endotelio, factores en el plasma y la velocidad del flujo circulatorio. La adhesión ocurre entre los hematíes y las células endoteliales, entre los hematíes y la matriz

131
subendotelial que se expone por lesión de las células endoteliales o por retracción inducida por trombina, y en determinadas condiciones, los hematíes entre sí.
• Alteraciones en la coagulación, incluyendo la activación de las plaquetas. Aunque los estudios de las anormalidades de la coagulación no son concluyentes, es muy posible que, sobre todo durante las crisis vasculares, los factores procoagulan-tes, anticoagulantes y las plaquetas desempeñen un papel en la oclusión vas-cular. Esto es particularmente importante en las interacciones que tienen lugar en la superficie del endotelio, donde el factor Von Willebrand, las plaquetas y la trombospondina aumentan la adhesividad. La oclusión vascular es favorecida por la generación de trombina in vivo. El endotelio activado y la fosfatidilserina en la superficie de los hematíes condicionan un estado trombofílico.
• Disfunción endotelial. El endotelio participa activamente en el proceso de la oclu-sión vascular, presenta alteraciones histológicas en casi todos los órganos y en los sitios en que existe hiperplasia de la íntima se observa la formación de trom-bos. Además, el endotelio se encuentra bajo la influencia de un conjunto de estí-mulos que lo conducen a un estado de activación crónica, con un número alto de células endoteliales que expresan las moléculas de adhesión VCAM-1, ICAM-1, E selectina y P selectina.
• Anemia hemolítica. Se debe a profundas alteraciones de la membrana del gló-bulo rojo y es fundamentalmente extravascular por eritrofagocitosis, pero tam-bién intravascular. Durante mucho tiempo se consideró que la anemia hemolítica desempeñaba un papel secundario en el cuadro clínico de la drepanocitosis en relación con los fenómenos vasooclusivos. Normalmente el óxido nítrico se une con la guanilato ciclasa soluble que transforma la guanosina-trifosfato en gua-nosina monofosfato cíclica. Estas enzimas, que participan en el metabolismo de los nucleótidos, producen relajación del músculo liso y vasodilatación. Cuando la Hb plasmática liberada por la hemólisis intravascular consume óxido nítrico, el balance normal entre vasoconstricción y vasodilatación es desviado hacia la vasoconstricción.
Hipertensión arterial pulmonar (HAP)La HAP es una de las causas más importantes de morbilidad y mortalidad en los pa-cientes adultos con enfermedad drepanocítica. Sin embargo existen numerosas lagu-nas en el conocimiento de las variables de predicción, modalidades seguras para su pesquisa, fisiopatología y tratamiento.
La HAP es definida cuando la presión arterial media (PAM) en la arteria pulmonar, medida por cateterismo derecho, es ≥25mm Hg, pero la naturaleza invasiva de este procedimiento lo hace prohibitivo para ser utilizado como herramienta de pesquisa. Por otro lado, no existe una técnica no invasiva para la cuantificación fiel de la PAM. La velocidad del jet de regurgitación tricuspídea, determinada por ecocardiografía Do-ppler con histología virtual, es utilizada para estimar la presión arterial sistólica/pico

132
en arteria pulmonar con una baja sensibilidad (33%) pero con una alta especificidad (100%) para identificar pacientes con HAP y valor predictivo negativo de 84,7%7. No obstante, una velocidad elevada del jet está vinculada con mal pronóstico, sugiriendo que la patología endotelial/cardíaca asociada conduce a un incremento en la PAM y anuncia una peor evolución.
El pronóstico de la HAP en la enfermedad drepanocítica es serio por las siguientes razones8,9:
• Representa un riesgo de mortalidad del 40 al 50% en un lapso de dos a tres años.• No todos los pacientes con enfermedad drepanocítica desarrollan HAP y los bio-
marcadores que los identifican no están bien definidos, por lo que se pierde un tiempo precioso en una posible intervención terapéutica temprana.
En razón de la anterior, se requiere de métodos diagnósticos no invasivos y biomarca-dores confiables para la detección precoz de la HAP en los pacientes susceptibles. De allí que algunos autores recomiendan el tamizaje de rutina en niños con asma y enfer-medad drepanocítica por medio de la ecocardiografía para medir la velocidad del jet de regurgitación tricuspídea6. En este aspecto, la reciente introducción del ultrasonido intravascular de la arteria pulmonar es una contribución importante ya que permite el análisis de las propiedades elásticas y rigidez arterial10.
Fisiopatología de la HAP. Es de origen multifactorial e incluye hemólisis, deterioro en la biodisponibilidad de óxido nítrico, hipoxemia crónica e incremento de la endotelina-1, un potente vasoconstrictor pulmonar11. Las alteraciones hemodinámicas comprenden la elevación de la PAM, hipertensión venosa pulmonar y aumento de la presión con-dicionada por el estado hiperquinético asociado con hipertrofia y disfunción diastólica del ventrículo derecho y/o izquierdo12-15.
Tratamiento. Las opciones terapéuticas de la HAP en los pacientes con enfermedad drepanocítica son extremadamente limitadas. Se han utilizado diversos agentes en-tre los cuales se cuentan los análogos de la prostacilcina (treprostinil), bloqueadores del receptor de endotelina-1 (bosentan, ambrisentan, macitentan), inhibidores de la fosfodiesterasa, bloqueadores de los canales de calcio, agentes inotrópicos y anticoa-gulantes, sin que los resultados hayan mostrado un marcado efecto sobre la super-vivencia16. Entre los nuevos fármacos promisorios se encuentran los inhibidores de la rho-quinasa y los estimuladores de la guanilato ciclasa soluble (receptor intracelular del óxido nítrico) que han demostrado efectos beneficiosos en estudios experimentales y clínicos, pero no son selectivos en sus acciones sobre el lecho vascular pulmonar17.
Referencias1.- Voskaridou E, Christoulas D, Terpos E. Sickle-cell disease and the heart: review of the current litera-ture. Br J Haematol 2012;157:664-73.
.05

133
2.- Wood JC. Cardiac iron across different transfusion-dependent diseases. Blood Rev 2008;22(Suppl 2): S14–S21.3.- Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng 2010;12:345-67.4.- Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation 2009;16:97-111.5.- Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation 2011;123:1227-32.6.- Hassell KL. Pulmonary hypertension, tricuspid regurgitant velocity screening, and the nitric oxide pathway. Hematology Am Soc Hematol Educ Program 2011;2011:419-26.7.- Hammerstingl C, Schueler R, Bors L, Momcilovic D, Pabst S, Nickenig G, Skowasch D. Diagnostic value of echocardiography in the diagnosis of pulmonary hypertension. PLoS One 2012;7(6):e38519.8.- Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disalease. N Engl J Med. 2004 Feb 26;350(9):886-95.9.- Castro O, Gladwin MT. Pulmonary hypertension in sickle cell disease: mechanisms, diagnosis, and management. Hematol Oncol Clin North Am. 2005;19(5):881-96, vii.10.- Lau EM, Iyer N, Ilsar R, Bailey BP, Adams MR, Celermajer DS. Abnormal pulmonary artery stiffness in pulmonary arterial hypertension: in vivo study with intravascular ultrasound. PLoS One 2012;7(3):e33331.11.- Machado RF, Gladwin MT. Pulmonary hypertension in hemolytic disorders: pulmonary vascular di-sease: the global perspective. Chest 2010 Jun;137(6 Suppl):30S-38S.12.- Johnson MC, Kirkham FJ, Redline S, Rosen CL, Yan Y et al. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood. 2010 Jul 8;116(1):16-21.13.- Klings ES. Pulmonary hypertension of sickle cell disease: more than just another lung disease. Am J Hematol. 2008 Jan;83(1):4-5.14.- Klings ES, Anton Bland D, Rosenman D, Princeton S, Odhiambo A, Li G et al. Pulmonary arterial hypertension and left-sided heart disease in sickle cell disease: clinical characteristics and association with soluble adhesion molecule expression. Am J Hematol. 2008 Jul;83(7):547-53.15.- Anthi A, Machado RF, Jison ML, Taveira-Dasilva AM, Rubin LJ, Hunter L et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med 2007;175:1272-79.16.- Murthy SN, Nossaman BD, Kadowitz PJ. New approaches to the treatment of pulmonary hyperten-sion: from bench to bedside. Cardiol Rev 2010;18:76-84.17.- Nossaman B, Pankey E, Kadowitz P. Stimulators and activators of soluble guanylate cyclase: review and potential therapeutic indications. Crit Care Res Pract 2012;2012:290805.
Tratamiento de las Complicaciones Crónicas

134
5.2. HepatobiliaresMaría Rubio / Médico hematólogo.
En la enfermedad drepanocítica, las causas de afectación de las vías biliares son múltiples1:• Inherentes a la enfermedad.
· Isquemia. · Hiperbilirrubinemia.
· Barro biliar · Cálculos de bilirrubina
• Secundarias al tratamiento · Transfusionales: Hepatitis viral, sobrecarga de hierro y medicamentos
Hiperbilirrubinemia.La hemólisis crónica cursa con niveles de bilirrubina total que no exceden a los 6mg/dL, a expensas de bilirrubina no conjugada o indirecta (BI) hasta en un 90%. Hay correlación directa entre los niveles de bilirrubina no conjugada y los niveles de lactato deshidrogenasa (LDH) y en cierto grado con los de as-partato amino transferasa (AST). El aumento de los valores de alanino amino-transferasa (ALT) y de fosfatasa alcalina (FA) son indicativos de daño hepáti-co, sobre todo si aparecen en el curso de una complicación aguda2 (Figura 5.1). Existen publicaciones donde se describe un aumento marcado de bilirrubina no con-jugada, asociada con defecto genético de la UDP-glucoronil transferasa (sindrome de Gilbert)2.
Barro biliarEs un material viscoso detectable por imágenes no acústicas, compuesto de bilirrubi-nato de calcio, cristales de colesterol, bilis viscosa, moco y proteínas. Según la historia natural de la enfermedad en el lapso de 2, 1 años, el 65% de los pacientes hacen cál-culos en la vesícula. El ceftriaxone favorece la formación de barro biliar. Por lo general carece de importancia clínica2.
• Recomendaciones · Ultrasonido cada 12-24 meses. · Colecistectomía si ocurre colestasis.
ColelitiasisPuede aparecer tan temprano como los 2-4 años de edad (12% en algunos estudios). La prevalencia se incrementa con la edad, hasta en 30 % a 50% a la edad de 20 años. Es más frecuente en ED-SS y en enfermedad- Sβ0 talasemia y mucho menos en en-fermedad-SC y enfermedad-S β+ talasemia. La coexistencia de α talasemia parece disminuir su frecuencia debido a que la hemólisis es menor. La dieta y factores genéti-cos parecen influir en su aparición. Las cefalosporinas de tercera generación pueden cristalizar en la vesícula biliar y favorecer la aparición de litiasis2.
.05

135
Figura 5.1. Conducta ante un paciente con enfermedad drepanocítica e hiperbili-rrubinemia por encima de valores basales.
Tratamiento de las Complicaciones Crónicas
A expensas de directa
Hematocrito estable
Hiperbilirrubinemia
Observacion Consulta con hematólogo
Enzimas hepáticas anormales
Asintomático.Continuar control
rutinario
Ecosonograma STAT considerar TAC⁄ HIDA⁄ colangio-retrógada
Consulta con hematólogogastroenterólogo, STAT
Colecistectomía eletiva en 4-6 semanas, preferible laparoscopia
Llenura postprandial nauseas vómitos
Ultrasonido abdominal consulta con
gastroenterólogo
Dolor agudofiebre, nauseas, vómitos
Hospitalizar, hidratartratar el dolor, dar antibióticos
Posible hepatitis cirrosis o hemocromatosis

136
El paciente puede permanecer asintomático durante muchos años y luego presentar síntomas agudos o crónicos (Figura 4.12).
• Tratamiento2
· Síntomas agudos. Ver guías de dolor y dolor en cuadrante superior derecho. · Colecistectomía.
· Pacientes sintomáticos pero sin colecistitis. · Colecistectomía electiva, mediante laparoscopia, previa preparación ade cuada. La colecistectomía aporta los siguientes beneficios:
· Reduce el riesgo de complicaciones por colelitiasis. · Elimina la enfermedad vesicular como diagnóstico etiológico de dolor en cuadrante superior derecho.
· El procedimiento debe ser realizado por un cirujano con amplia experiencia en la técnica.
· Se recomienda practicar colangiopancreatografía retrógrada previo a la colecistectomía por laparoscopia ya que la colangiografía intraoperatoria da un 25% de falsos positivos.
• Ver guía de anestesia y cirugía.
Hemosiderosis. Ver guía de tratamiento de sobrecarga de hierro
Hepatitis viral.Hay un aumento de la prevalencia de las hepatitis virales en comparación con la pobla-ción general, relacionado con las transfusiones múltiples.
• Hepatitis B2. · Relacionada con la endemicidad del virus y con transfusiones: · Se sugiere inmunización temprana. · Su cronicidad es inversamente proporcional a la edad.
• Indicaciones de tratamiento. · HBsAg positivo por más de 6 meses. · ADN VHB positivo. · Elevación persistente de ALT o biopsia con evidencias de hepatitis crónica.
• El manejo de la hepatitis crónica y de la cirrosis es competencia del hepatólogo.• Hepatitis C2
· Relacionada con transfusiones. · Al igual que en la población general, el 65% desarrolla hepatitis crónica. · Es subclínica: Solo 25% de los pacientes presentan ALT y AST dos veces por
encima de lo normal. · Puede evolucionar hacia cirrosis. · Manifestaciones extra hepáticas que se confunden y complican el manejo de la
enfermedad drepanocítica: · Vasculitis leucocitoclástica. · Crioglobulinemia.
.05

137
· Púrpura trombocitopénica inmune. · Artralgias. · Glomerulonefritis. · Neuropatía periférica.
• Criterios para tratamiento · RCP para ARN virus C, positiva. · Elevación persistente de AST/ALT o biopsia con evidencias de hepatitis crónica.
• El manejo de la hepatitis crónica y de la cirrosis es competencia del hepatólogo.
Hepatitis autoinmuneSe acompaña de rash, úlceras en piel y manifestaciones articulares. La etiología, na-turaleza, curso y el tratamiento en pacientes con enfermedad drepanocítica no está clara2. Si los síntomas persisten y los anticuerpos antimúsculo liso están presentes, se recomienda la referencia del paciente al hepatólogo.
Síndromes hepato renal, hepato pulmonar y porto pulmonar.Pueden complicar la enfermedad hepática de pacientes con enfermedad drepanocítica2.
REFERENCIAS 1. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/2. National Institutes of Health, National Heart, Lung and Blood Institute. Gall bladder and liver. Chap 17 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethes-da.2002; 111-117.
Tratamiento de las Complicaciones Crónicas

138
5.3. RenalesDra. Luz Marina Navarrete Grau / Médico nefrólogo.
La enfermedad drepanocítica (ED) es la hemoglobinopatía estructural más frecuente en el mundo. La afectación renal clínicamente significativa ocurre más comunmente en los individuos homocigotos que en los heterocigotos o con hemoglobinopatías mix-tas, con la excepción del carcinoma medular renal, que ocurre en mayor número en los portadores del rasgo drepanocítico1.
Las manifestaciones renales de la ED abarcan desde anormalidades funcionales hasta alteraciones anatómicas, que aparecen a lo largo de todo la nefrona2,3. La afectación renal en la ED comienza en la infancia, siendo su manifestación más común la hema-turia macroscópica asintomática, microinfartos, necrosis papilar renal y los defectos de la función tubular, todos ellos desencadenados por fenómenos vasooclusivos oca-sionados por la hipoxia1,4 (Tabla 5.1).
El medio ambiente de la médula se caracteriza por ser hipóxico, isquémico (dismi-nución de flujo plasmático medular), ácido e hipertónico. Las condiciones descritas promueven la polimerización de la hemoglobina y la falciformación2,5.
Los estudios de microradioangiografía en pacientes con ED evidencian y demuestran una disminución significativa del número de vasas rectas, anormales dilataciones y obliteración de los capilares2. La obstrucción por hematíes falciformes de las vasas rectas provoca microtrombos, infarto y formación de vasos colaterales. Esto conlleva a la reducción del número de vasas rectas funcionantes y a la pérdida de la arquitectu-ra normal de la médula renal, lo cual interfiere con el mecanismo de contracorriente, necesario para la concentración adecuada de la orina y el balance hidro-electrolíticolo y se traduce en una incapacida para la concentración urinaria (Figura 5.2)
.05

139
Tabla 5.1. Manifestaciones renales de la enfermedad falciforme1,2
Tipos de manifestación renal Comentarios
Alteraciones funcionales de la nefrona distal•Hipostenuria.•Acidosistubularrenaldistal
No debidas a la anemia Síntoma mas frecuente
Función tubular proximal supranormal•Reabsorciónaumentadadesodio•Reabsorciónaumentadadefosfatos•Secreciónaumentadadeuratos•Reabsorciónaumentadade β2-microglobulina
Sin significación patológica.No requiere tratamiento adicional.Precaución con retenedores de K+ y AINE
Hematuria La manifestación más frecuente, 10% bilateral. 80% procede de riñón izquierdo
Necrosis papilar renalAparece en un 30-40% de los homocigotos.Coexiste frecuentemente con infección urinaria
Glomerulopatía drepanocítica Su prevalencia aumenta con la edad
Albuminuria, proteinuria, insuficiencia renal
La estimación de FG mediante fórmulas para cribado de ERC en la ED no es recomendable hasta que FG <35-40mL/min/1,73m2
Son útiles los inhibidores del sistema renina angiotensina (SRA)
Insuficiencia renal aguda Múltiples etiologías
Hipertensión arterialPrevalencia menor que en la población generalTratar en rango de prehipertensión
Carcinoma medular renal Detección obligada en hematuria macroscópica
Tratamiento de las Complicaciones Crónicas
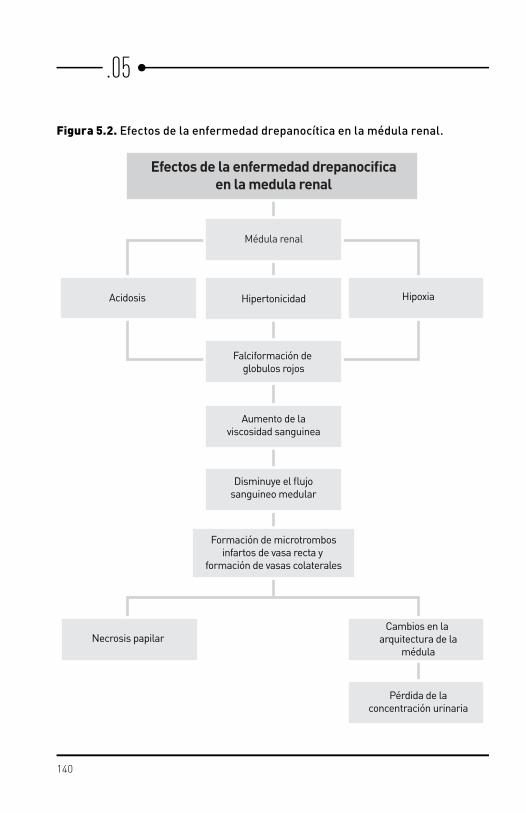
140
Figura 5.2. Efectos de la enfermedad drepanocítica en la médula renal.
Médula renal
Hipoxia
Aumento de laviscosidad sanguinea
Hipertonicidad
Falciformación de globulos rojos
Efectos de la enfermedad drepanocificaen la medula renal
Acidosis
Disminuye el flujosanguineo medular
Formación de microtrombosinfartos de vasa recta y
formación de vasas colaterales
Necrosis papilarCambios en la
arquitectura de lamédula
Pérdida de laconcentración urinaria
.05

141
Defectos tubulares funcionales• Alteraciones de la nefrona distal.
· Hipostenuria: Es el defecto tubular que se presenta más precozmente en la ED, así como el más frecuente, en pacientes homocigotos (SS). La concentración de orina máxima que puede alcanzar estos pacientes es de 400-450mOsm/kg en estado de deprivación de agua entre 8-10 horas1,3. Se expresa en la niñez con enuresis, poliuria, nicturia en este caso estaríamos ante una diabetes insípida nefrogénica irreversible6. La prevalencia de enuresis en los niños con ED va del 20 al 69%, mientras que la nicturia llega al 68% en los mismos7,8; la poliuria conduce a estados de deshidratación. Los defectos de concentración puede ser reversibles o aminorados en pacientes de 10-15 años de edad con transfusión de concentrados globulares1.
· Acidosis tubular distal: Acompaña a la hipostenuria, la hiperpotasemia es rara a no ser que se afecten los mecanismos compensatorios renales. Disminuye la la acidez titulable, de amonio e hidrogeniones1,3. Se pondría de manifiesto tras sobrecarga con cloruro amónico que conseguiría una acidificación urinaria disminuida a pH 5,8 frente a 5,1 en sujetos normales a pesar de una excreción de amonio normal. La hiperpotasemia puede aparecer con la progresión de la insuficiencia renal.
Función tubular proximal supranormalExiste alteración en la función tubular manifestada con reabsorción aumentada de sodio y una disminución de su excreción urinaria por lo que la respuesta a los diu-réticos del asa está disminuida o abolida. Incremento en la reabsorción de fosfatos y de β2-microglobulina e incremento en la secreción de ácido úrico y de creatinina. Por esta razón la depuración de creatinina está significativamente sobrestimada; se han referido diferencias de hasta un 30% en la estimación de FG.3
El mecanismo etiopatogénico, propuesto por De Jones y colaboradores para estas al-teraciones9, sería la liberación de prostaglandinas estimulada por la isquemia en la médula renal. Se ha observado un efecto inhibidor en la reabsorción proximal de sodio por la indometacina mayor en pacientes con ED que en pacientes sin ED. La inhibición de las prostaglandinas restablece la normalidad de reabsorción proximal de sodio y restaura la respuesta natriurética de los diuréticos de asa.
Tratamiento• La mayoría de los defectos tubulares no requieren de medidas terapéuticas adi-
cionales al tratamiento sintomático.• Ingerir abundantes líquidos, para compensar la pérdida de los mismos debido a
la hipostenuria.• La depleción de líquidos que potencialmente ocurre durante las crisis, se trata
con soluciones hipotónicas vía intravenosa.
Tratamiento de las Complicaciones Crónicas

142
Medidas preventivas • Evitar fármacos favorecedores de hiperpotasemia como los betabloqueantes,
inhibidores de la ciclooxigenasa 2, o inhibidores del sistema renina-angiotensi-na-aldosterona (SRAA) y evitar nefrotóxicos, especialmente en aquellos pacientes con hipostenuria.
Hematuria y necrosis papilar• Hematuria
· Es un hallazgo común en la ED. · Es frecuente en el rasgo drepanocítico (Hb AS). · Puede ser microscópica y más frecuentemente macroscópica y autolimitada. · Habitualmente es unilateral, por lo general debido a la longitud de la vena
renal izquierda y su localización anatómica que por compresión entre la aorta y la arteria mesentérica superior estaría sometida a una mayor presión venosa con hipoxia relativa en la médula renal que favorece la falciformación1.
· El sangramiento ocurre por polimerización del eritrocito dentro de la médula renal3.
· La hematuria macroscópica continuada o persistente en un paciente con EF o con rasgo drepanocítico (HbAS) suele representar una crisis falciforme renal1.
· El paciente que acude a urgencias puede tener dolor en el flanco, dolor abdo-minal, vómitos y fiebre que se asocian a infección o a necrosis papilar extensa9.
· El sedimento de orina puede mostrar dismorfia eritrocitaria con eritrocitos fal-ciformes. El hallazgo de eritrocitos, leucocitos y papilas desprendidas sugiere necrosis papilar renal1.
• Tratamiento · Como la hematuria es autolimitada, generalmente, el tratamiento es conser-
vador: Reposo en cama. Mantener alto el flujo de orina, mediante la hidratación oral y control del hematocrito.
· Si es necesario transfundir o si persiste más de una semana, se deberá hidra-tar al paciente con líquidos hipotónicos (4L/l,73 m2 por día). · Administrar bicarbonato, alcalinizar la orina, potencialmente útil para au-
mentar la afinidad de la Hb por el O2 y reducir la falciformación, además de disminuir la toxicidad tubular de la hemoglobinuria, aunque no ha demostra-do su eficacia como medida terapéutica. Se recomienda NaHCO3 2 a 3 g, QID
· Diuréticos de asa (furosemida: 40mg IV /BID)1,6,7. · Acetazolamida 250mg/QID6. · Cuando estas medidas fallan, la desmopresina (DDAVP) endovenosa ha de-
mostrado disminuir el sangrado al aumentar las concentraciones del Factor VIII y el factor Von-Willebrand, aunque hay pocos estudios que confirmen su utilidad a largo plazo10,11.
· Se ha utilizado el ácido épsilon amino capróico (AEAC) por sus propiedades antifibrinolíticas10.
.05

143
· Si el sangrado es prolongado, la embolización segmentaria es una alternati-va a tener en cuenta7.
· Dejar la nefrectomía unilateral como última opción.
Necrosis Papilar Renal (NPR)• La médula y papila renal son particularmente vulnerables a la necrosis isquémica
por su ambiente hipertónico y el carácter peculiar de la perfusión sanguínea12. • La NPR ocurre en un 15 a 50% de pacientes tanto con ED o con rasgo falciforme.
Estos eventos isquémicos generalmente son subagudos, siendo la NPR más co-munmente diagnosticada en pacientes asintomáticos o durante la evaluación de una hematuria3,6,7.
• Es más frecuente entre los 30-40 años6. • Para el diagnóstico de NPR es muy importante la sospecha clínica. Puede haber
cólico renal, dolor intenso, fiebre, sepsis y cuadro de retención aguda de orina11. • La ecografía renal es la técnica de elección para su detección y diagnóstico dife-
rencial con la litiasis o el carcinoma medular renal. El hallazgo más precoz de la NPR es una ecogenicidad aumentada de las pirámides medulares (la zona más interna medular), lo que en ausencia de hipercalciuria en un paciente con ED y hematuria sugiere NPR. Posteriormente aparece calcificación en las pirámides medulares1.
• Evita la urografía IV por el riesgo elevado de nefrotoxicidad por contrastes yodados.• La TAC helicoidal es más sensible que la ecografía para detectar precozmente
NPR. Utilizar contrastes No-iónicos.• Tratamiento de la NPR.
· Es esencial la terapia antibiótica agresiva para el tratamiento de infecciones y la derivación precoz de la vía urinaria en caso de obstrucción por papilas desprendidas.
· En caso necesario, lavados manuales y sondaje vesical de tres vías para lavado vesical continuo.
· Evitar AINE.
Glomerulopatía falciformeLos pacientes con ED tienen riesgo de desarrollar proteinuria e insuficiencia renal que progresa a enfermedad renal crónica terminal (ERCT)1,3.
La albuminuria marcador inicial de daño renal, su prevalencia aumenta con la edad, entre los 3 y 20 años se ha descrito albuminuria en un 21,3% a un 28%13 de los casos, de los cuales un 10,5% progresan a proteinuria en un tiempo de seguimiento de 20 meses que en la mayoría de los casos (72%) conduce con el paso de los años a insuficiencia renal, aunque estas series son retrospectivas.
Guasch y colaboradores4 determinaron la prevalencia de albuminuria en pacientes
Tratamiento de las Complicaciones Crónicas

144
adultos con ED encontrando 68% de excreción urinaria de albúmina (EUA): 26% protei-nuria, 42% de albuminuria, solo 32 % de los adultos con ED eran normoalbuminúricos. No había diferencias en relación al género. En cuanto a la edad se demostró que la prevalencia aumentaba de 61%, entre los 18-30 años, a 79% en mayores de 40 años. La insuficiencia renal crónica se presentó en 21%, en pacientes con ED (SS), la progresión a ERCT fue del 29%.
Becton y colaboradores14 en un estudio transversal, encontraron que la prehiperten-sión, la hipertensión y la anemia se asocian de manera independiente a la albuminuria. La proteinuria se acompaña de mayor anemia, hemólisis y reticulocitosis y también se ha relacionado con la incidencia de crisis dolorosas, colelitiasis, síndrome torácico agudo e ictus1,3. La proteinuria puede estar en el rango nefrótico (síndrome nefrótico).Day y colaboradores15 encontraron una fuerte asociación entre los marcadores de he-mólisis (reticulocitos, hemoglobina, LDH, bilirrubina) y la albuminuria de pacientes con ED (SS).
Los mejores predictores de fallo renal son la hiperfiltración, proteinuria, hipertensión, anemia grave y hematuria.
La prevalencia de hipertensión arterial (HTA) en la ERC de la ED es notablemente me-nor que en otras causas de ERC.
• Anatomía Patológica: Existen 4 tipos de glomerulopatías descritas en la ED: · Glomeruloesclerosis segmentaria y focal (GESF), la más frecuente se presenta
en 39% de los pacientes. · Glomerulonefritis membranoproliferativa (GNMP). · Glomerulopatía específica de la ED (GEF). · Microangiopatía trombótica (MAT) la cual se acompaña de fibrosis intersticial.
Los glomérulos se encuentran hipertrofiados con capilares distendidos por los eritrocitos falciformes, lo que se describe como GEF.
· Hallazgo casi universal son los depósitos de hemosiderina en las células tubulares.
• Diagnóstico: Todo paciente con ED ha de ser considerado en riesgo de desarrollar ERC. Por lo cual se recomienda la evaluación periódica de la relación albumina/creatinina (RAC) en la primera orina de la mañana. Si es positiva, confirmar con segunda muestra. Los criterios de positividad para albuminuria (RACu >30 mg/g) y proteinuria (RACu >300 mg/g) son los vigentes para enfermedad renal crónica (ERC). Siempre estimar la tasa de filtración glomerular, a través de las fórmulas recomendadas actualmente: la MDRM-4, la Cockroft-Gault y la CKD-EPI. Debe plantearse biopsia renal en casos de síndrome nefrótico de brusca instauración o enfermedad renal rápidamente progresiva.
.05

145
Tratamiento. · No existe terapéutica específica para disminuir o prevenir la progresión de la
nefropatía por enfermedad drepanocítica hacia ERC terminal. · La evidencia señala que la disminución de la proteinuria puede resultar bene-
ficioso. · Los inhibidores de la enzima convertidora de la angiotensina (IECA) están indi-
cados aún en ausencia de hipertensión. · Los bloqueadores de receptores de la angiotensina también pueden ser útiles. · La eritropoyetina no resulta muy útil en pacientes con enfermedad drepanocíti-
ca e insuficiencia renal terminal. · Si la eritropoyetina es inefectiva, se debe transfundir.
• Los pacientes con ERC estadio 5 necesitarán tratamiento sustitutivo renal: · Hemodiálisis · Diálisis peritoneal. · Trasplante de riñón2. · Complicaciones post-trasplante renal · Eventos vasooclusivos frecuentes. · Infarto renal · Reaparición de la nefropatía por enfermedad drepanocítica en el riñón tras-
plantado.
• Falla renal aguda1,3,7
· El fallo renal agudo usualmente está relacionado a factores prerrenales por depleción de volumen pero también a infección concomitante (sepsis) o por rabdomiolisis por esfuerzos físicos intensos, acidosis, hipoxia o anestesia y menos frecuentemente con trombosis de la vena renal o a hemolisis intravas-cular. Otras son las causas obstructivas como coágulos o menos frecuente-mente por necrosis papilar.
· Se caracteriza por la aparición brusca de disfunción severa de al menos dos órganos mayores (riñones, pulmones, hígado), durante un episodio de dolor agudo en pacientes con ED.
· La insuficiencia renal también puede acompañar a la rabdomiolisis.
• Carcinoma medular renal1, 7
· Esta neoplasia es rara, de muy mal pronóstico y descrito casi exclusivamente en la población negra con rasgo falciforme y menos frecuente en la ED. Predo-mina en el género masculino. Representa aproximadamente el 2% de todos los tumores renales primarios entre los 10 a 20 años de edad.
· Los síntomas más frecuentes en el momento del diagnóstico son dolor en el flanco o dolor abdominal, hematuria macroscópica con o sin coágulos o sínto-mas sistémicos asociados a enfermedad metastásica. El tumor se localiza en
Tratamiento de las Complicaciones Crónicas

146
el lado derecho en un 75% de los casos. Debe considerarse este diagnóstico en todo paciente joven con rasgo falciforme y hematuria.
· La sobrevida tras la cirugía varía entre 1 a 5 meses.
• Infecciones urinarias1,7
· Los pacientes con ED tienen la inmunidad humoral afectada por los infartos esplénicos, lo que les predispone a infecciones por bacterias encapsuladas, entre ellas las infecciones urinarias (ITU). En un estudio se mostró que la prevalencia de bacteriuria asintomática en pacientes con ED es, aproximada-mente, de un 10,9%.
· Se ha demostrado un incremento en la incidencia de pielonefritis durante el embarazo en pacientes con la enfermedad.
Referencias 1. López Revuelta K, Ricard Andrés MP. Afectación renal en la enfermedad falciforme. Nefrología 2011;31(5):591-601.2. Phuong-Thu TP, Phuong-Chi TP, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney International 2000;57:1-8.3. Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol 2000; 63:205–211. 4. Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglo-binopathies: Prevalence and clinical correlations of progressive renal failure. J Am Soc Nephrol 2006; 17:2228-35.5.National Institutes of Health, National Heart, Lung and Blood Institute. Renal abnormalities in sickle cell disease. Chap 19 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda. 2002; 123 -1276. Zadeii G, Lohr J. Renal papillary necrosis in a patient with sickle cell trait. J Am Soc Nephrol 1997; 8: 1034-1040 7. García A, Samper AO, Rojas C, Gascón LG, Sanjuan JB, Villavicencio H. Manifestaciones genitourinarias de la enfermedad de células falciformes. Arch Esp Urol 2011;64: 597-604.8. Field JJ, Austin PF, An P, Yan Y, DeBaun MR. Enuresis is a common and persistent problem among children and young adults with sickle cell anemia. Urology 2008;72:81-849. De Jong PE, Statius van Eps LW. Sickle cell nephropathy: new insights into its pathophysiology. Kidney Int 1985; 27:711-7.10. Kiryluk K, Jadoon A, Gupta M, Radhakrishnan J. Sickle cell trait and gross hematuria. Kidney Int 2007;71:706-10.11. Molitierno JA, Jr., Carson CC, 3rd. Urologic manifestations of hematologic disease sickle cell, leuke-mia, and thromboembolic disease. Urol Clin North Am 2003;30:49-6112. Jung DC, Kim SH, Jung SI, Hwang SI. Renal papillary necrosis: review and comparison of findings at multi-detector row CT and intravenous urography. Radiographics 2006; 26:1827-36.13. McKie KT, Hanevold CD, Hernandez C, Waller JL, Ortiz L, McKie KM. Prevalence, prevention, and treatment of microalbuminuria and proteinuria in children with sickle cell disease. J Pediatr Hematol Oncol 2007; 29:140-4.
.05

147
14. Becton LJ, Kalpatthi RV, Rackoff E, Disco D, Orak JK, Jackson SM, et al. Prevalence and clinical corre-lates of microalbuminuria in children with sickle cell disease. Pediatr Nephrol 2010;25:1505-11.15. Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 2012; 97:201-205.
5.4. OsteomuscularesRichar Figueredo / Médico hematólogo.
Las principales causas de complicaciones osteomusculares1 son:• Hiperplasia de la médula ósea, lo cual puede provocar distorsión y alteraciones
en el crecimiento óseo, especialmente en el cráneo, vértebras y huesos largos.• Episodios vasooclusivos que conducen a infartos en las metáfisis y diáfisis óseas
y necrosis del hueso yuxta-articular.• Infección bacteriana hematógena que provoca osteomielitis y artritis séptica.
Hiperplasia de la médula ósea2.La hiperplasia de la serie eritroide secundaria a la hemólisis crónica, conduce a las siguientes alteraciones óseas:
• Las trabéculas de los huesos del cráneo tienen apariencia de “borde de cepillo”.• Aumento del espacio entre el diploe interno y diploe externo da origen a protrusión
frontal.• En el maxilar superior ocasiona protrusión de los incisivos y del paladar.• Protrusión acetabular de una o ambas caderas.• Osteopenia y osteoporosis.• Causas
· Hipogonadismo (retardo en la pubertad y/o vinculado a sobrecarga de hierro). · Retardo constitucional en el crecimiento y maduración asociado a deficiencia
de la hormona del crecimiento, hipotiroidismo y micronutrientes · Expansión de la médula ósea. · Infartos óseos recurrentes. · Inflamación crónica. · Deficiencia de vitamina D. · Sedentarismo. · Sobrecarga de hierro (inflamación crónica, inhibición funcional de los osteo-
blastos. • Es una complicación frecuente en niños y adultos.• La prevalencia de la osteopenia oscila entre el 30 y 80% con predilección por co-
lumna lumbar. El aplastamiento de las vértebras conduce a cifosis.• Es necesaria la evaluación músculoesqueletica minuciosa con el propósito de de-
tectar fracturas en sitios de riesgo (cadera, hombros y columna). Las fracturas de huesos largos y de la columna son subdiagnosticados en parte por presencia de
Tratamiento de las Complicaciones Crónicas

148
dolor por otras causas.• Es asintomática, pero cuando da síntomas es porque hay fractura que se expresa
por dolor, deformidad y cifosis.• No existen guías de consenso de tratamiento de la osteoporosis secundaria a la
enfermedad drepanocítica.• La prevención de la osteoporosis comienza en la niñez, con el uso adecuado de:
· Vitamina D y calcio. · Ejercicio. · Detección y tratamiento temprano del hipogonadismo, hormona de crecimiento
y de la sobrecarga de hierro. · En relación al uso de bifosfonatos, no hay evidencias suficientes que justifiquen
su uso en el tratamiento de la osteoporosis en la enfermedad drepanocítica2.
Episodios vasooclusivos.• Dactilitis o “síndrome mano-pie”2.
· Generalmente es la primera manifestación osteomuscular. · Aparece en niños muy pequeños teniendo su máxima incidencia entre los 6-12
meses. · Prevalencia antes de los 2 años es de 45%. · Aparece frecuentemente en la temporada de frío y está asociada a bajos nive-
les de hemoglobina fetal y alta reticulocitosis. · Es considerado un factor de riesgo para alta morbilidad. · Tratamiento AINE vía oral e hidratación vigorosa, compresas calientes (Ver la
guía del dolor).• Infartos en diáfisis y metáfisis1.
· Los huesos y articulaciones son los sitios más frecuentes de episodios vasoo-clusivos. Para su tratamiento ver la guía del dolor.
· Con frecuencia se acompañan de signos de inflamación. · En ocasiones es difícil distinguir entre infarto óseo y osteomielitis. · Para su tratamiento ver guía del dolor.
• Miositis/Mionecrosis/Fascitis2
· Aparecen en personas con larga historia de episodios vasooclusivos y porcen-tajes bajos de hemoglobina fetal.
· Más frecuente en músculos proximales. · Comienza como miositis que progresa hacia mionecrosis y eventual miofibro-
sis que a largo término ocasiona contractura muscular, induración y atrofia muscular.
· Síntomas: · Dolor agudo, localizado en un pequeño grupo muscular, referido como “dife-
rente” al dolor típico de las crisis de dolor. · Edema.
· Puede asociarse infección por Staphylococcus aureus, Streptococcus pneumo-
.05

149
niae, con extensión progresiva a huesos y articulaciones que ocasiona daño orgánico, sepsis y falla multiorgánica.
· Diagnóstico · El aumento de LDH y CPK son sugestivos aunque no siempre ocurren. · El aumento en la Proteína C reactiva es indicativo para iniciar antibioterapia. · Imagen por resonancia magnética y la biopsia son confirmatorias.
· Tratamiento. · Inicial. · Es igual para las tres entidades: · Reposo en cama. · Inmovilización de la parte afectada durante un período corto. · Hidratación por vía endovenosa. · AINE. · Analgésicos opiáceos para el dolor intenso (Ver guía de tratamiento del
dolor). · Osteonecrosis2.
· El diagnóstico temprano es fundamental (Figuras 5.3 y 5.4). · Ocurre antes de los 35 años de edad. Con una prevalencia de:
· 26% en niños con ED-SS (edad promedio: 9,8 años). · 48,6% en adultos con ED-SS (edad promedio: 26,7 años).
· Factores de riesgo: · Vasooclusión recurrente. · Género masculino. · Cifras altas de hemoglobina. · Niveles bajos de hemoglobina fetal. · Deficiencia de vitamina D.
· Coexistencia de rasgo de α talasemia. · Ocurre en la zona yuxtaarticular del hueso. · Se debe a trombosis de los vasos endarteriales. · Hay destrucción de la articulación adyacente. · Las zonas más afectadas son las cabezas del fémur, húmero y vértebras. · Tratamiento (Figuras 5.3 y 5.4).
· Se requiere un equipo multidisciplinario conformado por ortopedista, hema-tólogo, fisioterapeuta y nutricionista.
· Preferible acudir a un centro donde haya especialistas con experiencia en enfermedad drepanocítica.
· Puntos básicos: · Aliviar las molestias. · Evitar la progresión. · Incrementar la función de la articulación afectada.
· Tratar la deficiencia de vitamina D, zinc y proteínas. · Fisioterapia.
Tratamiento de las Complicaciones Crónicas

150
· Si el tratamiento conservador no surte efecto y/o hay progresión a la destruc-ción de la articulación, fracturas o dolor discapacitante, se requiere tratamien-to quirúrgico: · Descompresión del core (no es superior a la fisioterapia intensiva)2,3. · Artroplastia. · Remplazo completo de la articulación:
· La falla de la prótesis es frecuente en pacientes con enfermedad drepano cítica. Entre 31-63% de ellos pierden el efecto de la prótesis antes de los 10 años en comparación con el 10% de los no drepanocíticos. · La longevidad de las prótesis ha aumentado.
· Existen nuevas técnicas quirúrgicas. · Desarrollo de los implantes de cerámica y polietileno.
· El uso de hidroxiurea y el régimen de transfusión crónica no es efectivo. · Aparentemente, la hidroxiurea empeora la necrosis avascular. · Tratamiento perioperatorio
· Hidratación. · Transfusión. · Ver guía de anestesia y cirugía.
· Terapias nuevas prometedoras. · Células progenitoras con o sin osteotomía. Utiliza injerto de hueso vascula-
rizado y/o inyección de un volumen reducido de médula ósea autóloga dentro de la cabeza femoral siguiendo a la descompresión del core.
· Útil en etapas 2-3 de la necrosis avascular.
Figura 5.3. Conducta en la necrosis avascular de cabeza de fémur o húmero en su estadio inicial.
Rx normal o cambios tempranos
Imagen de resonancia magnética
Normal Anormal
Referir a ortopedistaObservación
Dolor severolimitación de movimientos
Manejo médico, AINE, fisioterapia Considerar descompresión de core
.05

151
Figura 5.4. Conducta en la necrosis avascular de cabeza de fémur o húmero en su estadio tardío.
OsteomielitisLa osteomielitis se caracteriza por un proceso inflamatorio, acompañado de destruc-ción ósea. Las formas aguda y crónica están particularmente asociadas a la necrosis avascular e infartos óseos. La prevalencia varía entre el 12% y el 17%. Es menos fre-cuente en el haplotipo Bantú. Es más común en menores de 20 años, y se presenta más usualmente en la infancia asociándose con el riesgo de complicaciones físicas y psicológicas. Los huesos más afectados son el fémur (26%), tibia (23%) y húmero (21%). La desvitalización del tejido óseo por las crisis vasooclusivas con aparición de zonas isquémicas, la saturación de los macrófagos con productos derivados de la hemólisis crónica y la disfunción esplénica predisponen a la infección ósea.
Los gérmenes asociados con estos procesos son la salmonella, en alrededor de un 70%, especialmente serotipos no típicos (Salmonella typhimurium, Salmonella enteriti-dis, Salmonella choleraesuis y Salmonella paratyphi B), el resto corresponde a Staphyilo-cocus aureus y bacilos entéricos Gram negativos4,5.
• Síntomas · Fiebre. · Dolor agudo persistente que va aumentando en intensidad.
Manejo médico con analgésicos (AINE y acetaminofén con codeína),
fisioterapia
Siempre es necesaria consulta con ortopedista
con experiencia en drepanocitosis.
Incapacidad:cirugía a veces garantizada
en lesiones femorales
Considerar:reemplazo de cadera
fijación o reconstruciónde cadera
Tratamiento de las Complicaciones Crónicas

152
· Inflamación. · Enrojecimiento
• Diagnóstico2 (Figura 5.5)
Figura 5.5. Conducta ante la sospecha de osteomielitis en un paciente con enfermedad drepanocítica.
Examen físico: disminución de motilidad, reblandecimiento, edema.
Leucocitos, conteo diferencial, proteína C reactiva, mielocultivo, consulta con hematólogo.
Hospitalizar.Consultar con ortopedista e infectología
iniciar antibióticos.
Historia de fiebre dolor local
Dar medicación para el dolor y observar.
Si no mejora, considerar scan óseacon leucocitos marcados,
resonancia magnética con galodinium.
RX, biopsia o aspirado,cultivo, ultrasonido, scan óseo
con leucocitos marcados.
Cultivos y otros, negativos,paciente mejor,
suspender antibióticos.
Cultivos positivos, estudios radiográficosindican osteomielitis,conducta quirúrguica.
Absceso: drenaje quirúrgico antibiótico EV.No absceso: antibióticos EV.
Apreciar respuestas
Adecuada: Antibióticos VO, dosis mínima necesaria.
Inadecuada: repetir scan óseo, cosiderar drenaje quirúrgico,
repetir cultivo, cambiar terapia.
Leucocitos >25.000 Proteína C reactiva elevada, mielocultivo positivo.
leucocitos y proteína C reactiva normales, mielocultivo negativo.
.05

153
Tratamiento.
• El principio fundamental de la terapia inicial es tomar en cuenta los gérmenes causales de osteomielitis en el paciente con drepanocitosis. Antes de disponer de los resultados de cultivos: · Terapia Inicial
· Cloxacilina, vía endovenosa. · Adulto: 2gr c/4h. · Niños: 150-200 mg/kg/dia, c/4h.
· Cefotaxime, vía endovenosa. · Adulto: 2g c/8h. · Niños: 100-150mg/kg/día, c/8h.
· Si hay alergia a los betalactámicos, utilizar. · Clindamicina: 40mg/kg/día, c/8h.
Al recibir los resultados de cultivos, indicar la antibióticoterapia específica.Si no hay respuesta a la terapia específica o si hay secuestro óseo o abscesos subpe-riostios, realizar drenaje quirúrgico. Si ha ocurrido una evolución tórpida, la terapia EV no debe ser menor de 3 semanas.
• Duración del tratamiento. · El tratamiento EV inicial es de, por lo menos, 2 semanas. · El paso a la vía oral dependerá de la mejoría de los parámetros clínicos y de
laboratorio. Si no ha habido drenaje quirúrgico el tratamiento por vía oral ha de prolongarse por 2 a 3 semanas.
• Dependiendo de la evolución, el tratamiento puede prolongarse hasta 2 a 3 meses. · En cuanto al tratamiento por vía oral:
· Si se usó cloxacilina EV se pasa a cloxacilina VO. · Si se usó cefotaxime se pasa a cefixima.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Bones and joints. Chap 21 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 133–137.2. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/3. Martí-Carvajal AJ, Solà I, Agreda-Pérez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane Database of Systematic Reviews 2012, Issue 5. Art. No.: CD004344. DOI: 10.1002/14651858.CD004344.pub4
Tratamiento de las Complicaciones Crónicas

154
4. Atkins BL, Price EH, Tillyer L, Novelli V, Evans J. Salmonella osteomyelitis in sickle cell disease chil-dren in the east end of London. J Infect 1997;34:133-38. 5. Burnett MW, Bass JW, Cook BA. Etiology of osteomyelitis complicating sickle cell disease. Pediatrics 1998;101:296–297.
5.5. Úlceras en miembros inferioresRichar Figueredo / Médico hematólogo.
Las úlceras en miembros inferiores son relativamente comunes, y discapacitantes, la prevalencia es variable siendo baja antes de los 10 años. Son más frecuentes en ED-SS y menos en ED-SC y en ED-Sβ talasemia. Su distribución geográfica es variable1.
Patogénesis: es compleja, entre los factores predisponentes se cuentan1:• Obstrucción mecánica por células densas.• Comunicación arterio-venosa.• Infección bacteriana.• Control autonómico anormal con vasoconstricción excesiva.• Trombosis microvascular.• Disminución de la biodisponibilidad de óxido nítrico y consecuente daño endotelial.
TraumatismoAparecen en los sitios donde hay menos tejido graso, piel delgada y disminución del flujo venoso (maléolos), menos frecuente en la región tibial anterior, dorso del pie y tendón de Aquiles.
• Factores de riesgo2,3: · Traumatismo. · Infección. · Anemia severa. · Hemólisis intensa (LDH elevada, reticulocitosis, cifras baja de hemoglobina y
de hemoglobina fetal). · Condición socioeconómica. · Incompetencia venosa. · Genéticos. HLA3B y Cw44.
Se han reportado en mayor proporción en los portadores del haplotipo CAR-β globina. Algunos estudios demuestran que son más ocurrentes en hombres que en mujeres y su aparición aumenta con la edad1. En la enfermedad drepanocítica se ha propuesto un sub fenotipo hemolítico que cursa clínicamente con hemólisis severa, aumento de la incidencia de úlceras en miembros inferiores, hipertensión pulmonar y priapismo, estos dos últimos son marcadores de la gravedad de la enfermedad5.
.05

155
Los índices de severidad se basan en: tamaño, profundidad y duración. No existe un consenso para la clasificación según la duración, sin embargo se acepta que una úlce-ra aguda cicatriza en menos de un mes, la úlcera crónica dura mas de 6 meses, no son raras las ulceras de varios años de evolución. A menudo las úlceras duelen con fre-cuencia, son intratables y tardan meses o años en cicatrizar. El dolor suele ser agudo, penetrante, severo e insoportable. La mayoría de los pacientes requieren analgésicos, en ocasiones opiáceos vía oral o parenteral para aliviar el dolor6.
Prevención7
Obviar punción de la venas de los miembros inferiores a menos que sea estrictamente indispensable.
• Usar repelente contra insectos con el propósito de evitar picaduras.• Usar cremas emolientes para prevenir resequedad de la piel.• Rehabilitación ocupacional con la finalidad del combatir el estasis venoso.• Descansar en cama durante unos minutos.• No estar de pie durante mucho tiempo.• Instruir a los jóvenes en el uso de vendajes no elásticos para proteger la parte
inferior de la pierna, los maléolos y el dorso del pie.• Evitar situaciones que pudieran ocasionar trauma en la parte inferior de la pierna,
los maléolos y el dorso del pie.
Una vez que la úlcera se ha curado, se debe instruir al paciente sobre los siguientes aspectos:
• Impedir traumatismos en la zona afectada.• Lubricar el área con cremas para la piel.• Usar medias elásticas y mantener la pierna en alto para evitar el edema y por
tanto la recurrencia de la úlcera.
Manejo7 (Figura 5.6)• Datos subjetivos.
· Documentar · Fecha de inicio de la úlcera. · Historia. · Recurrencia. · Edema maleolar. · Trauma. · Dolor. · Inflamación. · Fiebre. · Determinar tipo y resultado de tratamientos anteriores.
Tratamiento de las Complicaciones Crónicas

156
Datos objetivos• Registrar signos vitales, especialmente temperatura.• Evaluar en las extremidades.
· Tamaño de la úlcera. · Profundidad. · Presencia y características de exudado. · Presencia de tejido de granulación. · Edema o eritema circundante. · Si la úlcera es simple, medirla en dos direcciones perpendiculares. · Si la úlcera es compleja, dibujarla y medir todas las direcciones.
• Ganglios linfáticos · Determine:
· Presencia y tamaño de los mismos. · Inflamación de ganglios inguinales y femorales. · Si hay o no linfangitis.
Laboratorio• Solicitar hematología completa y reticulocitos.• Cultivo si hay evidencia de infección de la úlcera o celulitis.• Rx en los siguientes casos: la úlcera parece estar infectada, hay leucocitosis con
desviación a la izquierda o el paciente presenta fiebre.
Adicionales• Gammagrama óseo con galio para descartar osteomielitis, si la úlcera es profun-
da, hay síntomas sistémicos, dolor intenso.• Hemocultivo en paciente con fiebre, neutrofilia, celulitis, linfangitis
u osteomielitis.
Criterios de hospitalización• Osteomielitis o linfadenitis para tratamiento con antibióticos por vía endovenosa.• El tratamiento de úlceras grandes, necróticas y purulentas se facilita con el repo-
so en cama y debridación frecuente.
Terapia específica• Se inicia con medidas simples y se escala en intensidad si no hay respuesta al
tratamiento. · Úlceras agudas (Menos de 6 meses).
· Reposo con la pierna levantada durante 7-10 días. · Sulfato de zinc a razón de 200mg/d/VO. Favorece la cicatrización de las
úlceras. · Higiene máxima.
.05

157
Cultivo radiografía/scan/IRM
si está indicado
Consulta con hematólogo,infectólogo, dermatólogo, cirujano.
Osteomielitis o celulitis con fiebre No osteomielitis ni celulitis
Tratamiento enel hogar
Hospitalizarver guía de osteomielitis
Examen local: Temperatura, localización, profundidad,apariencia, tamaño, compromiso de piel linfadenopatía
Mejoría No mejoría
Continuartratamientohasta curar
Consulta con:hematólogo
cirujano
Considerar TTO alternativo bota de unna, transfusión, oxigeno terapia,
factores de crecimiento, injerto de piel
Tratamiento de las Complicaciones Crónicas
Figura 5.6. Conducta ante un paciente con enfermedad drepanocítica y úlceras maleolares.

158
Debridamiento con: · Cura húmeda hasta dejar secar, 4 veces al día. Usar gasa húmeda con solución
salina, una vez seca retirar. Resistir la tentación de humedecer para evitar dolor.
· Utilizar cura con soluciones salinas hipertónicas en caso de que haya mucho exudado.
· Aplicar Duoderm R, con lo cual: · Su desprendimiento es menos doloroso. · Provoca debridamiento de tejido muerto y alrededor de la úlcera, de modo
que ésta aumentará de tamaño antes de comenzar la curación. · Informar al paciente de lo anterior para que no pierda la confianza en el
tratamiento. · Cambiar semanalmente o antes si el exudado comienza a salir. · La aparición de líquido o gel amarillo es buen signo · Se retira el Duoderm, se irriga el fondo con salina por el menor tiempo posi-
ble y se coloca nuevo Duoderm.
• Utilizar bota de Unna. · Útil en caso de
· Buena granulación. · Edema.
· La úlcera requiere estar relativamente limpia. · Procedimiento.
· Aplicar una venda impregnada de óxido de zinc, desde el tobillo hasta por debajo de la rodilla.
· Envolver con una venda elástica de 4 pulgadas la cual debe quedar suave, cómoda. Disminuir la tensión hacia la parte proximal. La venda se volverá más apretada en la medida que vaya secando.
· Se cambia una vez a la semana o cada 3-4 días si la úlcera no estaba com-pletamente limpia.
· Es conveniente continuar con el vendaje durante varias semanas después de la curación de la úlcera.
• Tratamiento quirúrgico si existe tejido que no cura. · Si hay infección, indicar antibióticos vía sistémica. Los antibióticos tópicos
parecen no ser efectivos. · Si no hay curación o la úlcera es muy grande, referir para evaluación y posible
injerto de piel. En general, los resultados del injerto no son muy halagadores, con alta incidencia de recaída y pérdida del injerto.
Úlceras crónicas (Más de 6 meses).• Considerar programa de transfusión crónica (cada 4 semanas) para llevar el he-
matocrito a 30- 35 % y los niveles Hb A a 70%. Ver guía de transfusión. Parece ser
.05

159
beneficioso en algunos pacientes. · Si la úlcera cura, interrumpir progresivamente. · Si en 6 meses no hay curación, suspender de una vez.
• Antibióticos vía oral o parenteral, se reserva para úlceras complicadas con infec-ción sistémica u osteomielitis. Estudios controlados demuestran la utilidad del uso de antibióticos tópicos.
• Sulfato de zinc a razón de 200mg/TID/VO. • Si no hay curación, o la úlcera es muy grande, referir para evaluación y posible
injerto de piel, cuyos resultado se pueden favorecer mediante el uso de: · Transfusión crónica durante un período breve. · Antiagregantes plaquetarios. · Anticoagulantes.
• Los reportes de oxígeno hiperbárico, butirato de arginina, aplicaciones tópicas de hierbas y factor de crecimiento tópico, resultan anecdóticos.
Referencias 1. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, and Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Ma-nagement. The Scientific World Journal. 2012 (2012), Article ID 949535, 55 pages doi:10.1100/2012/949535 in http://www.tswj.com/2012/949535/2. Koshy Rentsuah, A. Koranda, Kraus AP, Johnson R, Bellvue R et al. Leg ulcers in patients with sickle cell disease. Blood 1989;74:1403–1408. 3. Cumming V, King L, Fraser R , Serjeant G, Reid M. Venous incompetence, poverty and lactate dehy-drogenase in Jamaica are important predictors of leg ulceration in sickle cell anaemia. Br J Haematol 2008;142:119-25.4. Eckman JR. Leg ulcers in sickle cell disease. Hematol Oncol Clin North Am 1996;10:1333–1344.5. Kato G J, McGowan V, Machado RF, Little JA, Taylor J. Lactate dehydrogenase as a biomarker of he-molysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006; 107:2279–2285.6. Minniti C P Eckman J, Sebastiani P, Steinberg M H, ,Samir K, Ballas S. Leg ulcers in Sickle Cell Disease. Am J Hematol 2010;10:831–833.7. Sickle Cell Information Center. Specifics problems: leg ulcers. Edit Eckman J and Platt A disponible en http://scinfo.org/problem-oriented-clinical-guidelines/specific-problems-leg-ulcers
Tratamiento de las Complicaciones Crónicas

160
6.1. Rasgo DrepanociticoYraida Tersek / Médico hematólogo.
Rasgo drepanocítico es el término utilizado para describir la presencia del estado he-terocigoto de la Hb S (Hb AS). En el mundo existen alrededor de 300 millones de per-sonas con rasgo drepanocítico.
Hasta hace pocos años se consideraba que las personas con rasgo drepanocítico, en condiciones fisiológicas no presentaban eventos vasooclusivos y tenían expectativas de vida normal, sin embargo evidencias recientes demuestran que el rasgo drepanocítico no debe considerarse como una entidad completamente benigna ni como una enfer-medad real, sino como una condición de riesgo resultante de la interacción de factores genéticos y ambientales1.
En casos extremos las siguientes condiciones pueden representar riesgo para perso-nas con rasgo drepanocítico2:
• Baja de oxígeno. · Entrenamiento militar intenso. · Deportes de competencia.
• Deshidratación.• Ascenso a alturas elevadas.
· Vuelos en aeronaves no presurizadas. · Montañismo.
En la tabla 6.1, se presentan las complicaciones que se han descrito en personas con rasgo drepanocítico.
Hematuria importante• Puede ser ocasionada por ejercicio intenso aun cuando suele presentarse de ma-
nera espontánea3.• Manejo conservador.
· Evaluación por urólogo con la finalidad de descartar neoplasma o litiasis renal. · Evitar ejercicios intensos. · Reposo en cama. · Indicar hidratación agresiva con solución de NaCl 0.45 %. · Utilizar bicarbonato de sodio a razón de 650mg-1.200 mg diarios, vía oral. · Si el sangrado persiste, indicar un antifibrinolítico tipo ácido epsilón aminoca-
proico (AEAC), a razón de 3g, vía oral, 3-4 veces diarias. Estudios controlados. evidencian mejoría en resolución de la hematuria en 2-3 días con esta droga.
.06 Tópicos especiales
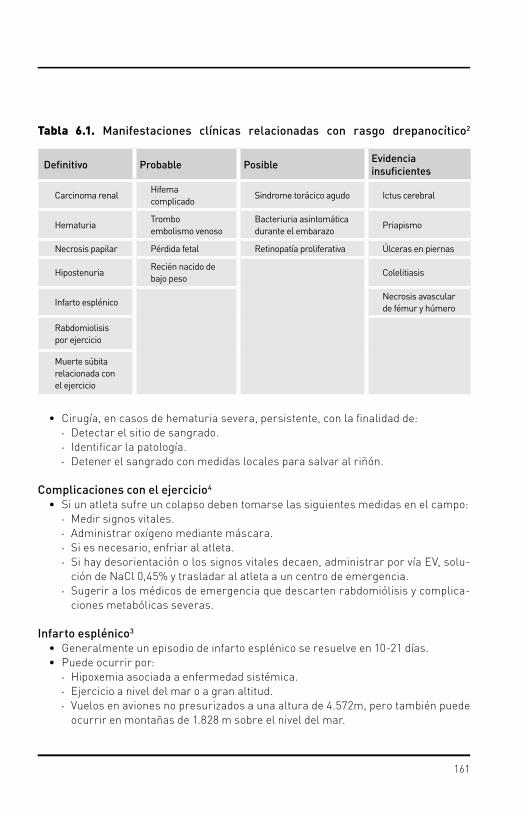
161
Tabla 6.1. Manifestaciones clínicas relacionadas con rasgo drepanocítico2
• Cirugía, en casos de hematuria severa, persistente, con la finalidad de: · Detectar el sitio de sangrado. · Identificar la patología. · Detener el sangrado con medidas locales para salvar al riñón.
Complicaciones con el ejercicio4
• Si un atleta sufre un colapso deben tomarse las siguientes medidas en el campo: · Medir signos vitales. · Administrar oxígeno mediante máscara. · Si es necesario, enfriar al atleta. · Si hay desorientación o los signos vitales decaen, administrar por vía EV, solu-
ción de NaCl 0,45% y trasladar al atleta a un centro de emergencia. · Sugerir a los médicos de emergencia que descarten rabdomiólisis y complica-
ciones metabólicas severas.
Infarto esplénico3
• Generalmente un episodio de infarto esplénico se resuelve en 10-21 días. • Puede ocurrir por:
· Hipoxemia asociada a enfermedad sistémica. · Ejercicio a nivel del mar o a gran altitud. · Vuelos en aviones no presurizados a una altura de 4.572m, pero también puede
ocurrir en montañas de 1.828 m sobre el nivel del mar.
Definitivo Probable Posible Evidencia insuficientes
Carcinoma renalHifema complicado
Sindrome torácico agudo Ictus cerebral
HematuriaTrombo embolismo venoso
Bacteriuria asintomática durante el embarazo
Priapismo
Necrosis papilar Pérdida fetal Retinopatía proliferativa Úlceras en piernas
HipostenuriaRecién nacido de bajo peso
Colelitiasis
Infarto esplénicoNecrosis avascular de fémur y húmero
Rabdomiolisis por ejercicio
Muerte súbita relacionada con el ejercicio

162
Cirugía3. • No existe riesgo aumentado para la anestesia.• No hay limitaciones en la selección del tipo de anestesia.• Educación y asesoramiento genético de la persona con rasgo drepanocítico o de
los padres de un niño con rasgo drepanocítico. · Las personas deben instruirse en relación a la herencia del rasgo drepanocítico,
a la necesidad de estudiar a la pareja y sobre las eventuales complicaciones, según el siguiente esquema: · Las personas que presentan rasgo drepanocítico, tienen dos tipos de hemog-
lobina en sus glóbulos, hemoglobina A que es la normal y hemoglobina S. · El glóbulo rojo posee suficiente hemoglobina A (hemoglobina normal) para
cumplir sus funciones. · La persona portadora de rasgo drepanocítico lo heredó de uno de los padres. · La persona con rasgo tiene probabilidades de transmitirlo a sus hijos. · La persona que nace con rasgo drepanocítico, siempre lo tendrá. · El rasgo drepanocítico es una condición de riesgo en condiciones extrema de
hipoxia, deshidratación y ejercicio extremo. · Por lo general no requiere tratamiento. · El rasgo no se contagia como una gripe. · Si usted o su niño es portador de un rasgo drepanocítico necesita comuni-
cárselo a cada médico en la oportunidad debida. Esta información es de gran ayuda al interpretar los síntomas o signos que presenta en un momento de-terminado.
Referencias 1. Key NS and Derebail VK. Sickle-Cell Trait: Novel Clinical Significance. Hematology.2010; 1:48 – 422. Disponible en:http://asheducationbook.hematologylibrary.org/content/2010/1/418.full.pdf+html.2. Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med 2009; 122:507–512.3. National Institutes of Health, National Heart, Lung and Blood Institute. Sickle cell trait. Chap 3 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Bethesda.2002; 15-18. 4. The National Athletic Trainers’ Association (Nata). Releases “Sickle Cell Trait And The Athlete” Consen-sus Statement. Dallas 2007. Disponible en : http://www.nata.org/NR062107.
6.2. Embarazo Francia Yépez - María Rubio / Médicos hematólogos.
En décadas pasadas, los índices de mortalidad materna y perinatal eran altos en muje-res con enfermedad drepanocítica. Hendrickse y colaboradores1 en 1970 reportan una
.06

163
mortalidad materna de 11,5%. Frente a este hecho se aconsejaba a las mujeres con enfermedad drepanocítica, no embarazarse, la esterilización primaria o post parto y el aborto. La esperanza de vida ha mejorado, en el 2008 Villers y colaboradores2 con-siguen una mortalidad materna de 72,4/100.000 partos. Por otra parte, siguen sien-do frecuentes las complicaciones maternas y fetales con aumento de la incidencia de mortalidad perinatal3-5, parto prematuro3-6, retardo en el crecimiento fetal3-7, episodios de dolor severo durante el embarazo5,8-10.
Algunos estudios describen aborto espontáneo11, hospitalización durante el embarazo7, muerte materna2, parto por cesárea2,7, infección, eventos trombo embolicos12, sangra-do durante el embarazo2. Diversos autores reportan aumento del riesgo de eclampsia e hipertensión inducida por el parto2,5-7, pero otros no4,8,11. En la enfermedad drepano-cítica-SC se reportan menos complicaciones, pero existe un aumento en la incidencia de episodios de dolor severo durante el embarazo13.
Medidas para el momento en que la paciente decide salir embarazada14. • Es necesario:
· Informar a la paciente acerca de como el embarazo afecta a la enfermedad dre-panocítica y los efectos de ésta sobre el embarazo.
· Investigar si existe daño orgánico asociado con la enfermedad drepanocítica. · Indagar el estado serológico relacionado con enfermedades virales. · Interrogar sobre el estado de inmunizaciones. · Si está recibiendo hidroxiurea, interrumpirla por lo menos tres meses antes de
la concepción. · Suspender IECA y bloqueadores del receptor de angiotensina, (si los recibe).
Información relevante para la paciente que ha decidido concebir. • Papel de la deshidratación, frío y calor extremo, ejercicios y estrés en la frecuen-
cia de las crisis de dolor.• Informar que los vómitos durante el embarazo pueden ocasionar deshidratación
y desencadenar crisis.• Existe un mayor incremento en el riesgo de:
· Anemia. · Episodios de dolor. · Síndrome torácico agudo. · Infecciones, sobre todo a nivel del tracto genito urinario. · Probabilidades de tener un niño con bajo peso, lo cual eleva las probabilidades de:
· Sufrimiento fetal. · Inducción del parto. · Cesárea.
• Probabilidades de tener un niño portador o con enfermedad drepanocítica (si el padre es portador de hemoglobinopatía o talasemia).
Tópicos Especiales

164
Investigación de enfermedad crónica • Es necesario practicar:
· Toma de presión arterial y uroanálisis para detectar hipertensión arterial y/o proteinuria.
· Creatinina. · Pruebas hepáticas. · Urocultivo, debido a la alta frecuencia de infecciones urinarias. De ser positivo,
indicar la antibioterapia correspondiente y realizar cultivos mensualmente15.
· Despistaje de anticuerpos contra los glóbulos rojos: · La presencia de anticuerpos puede indicar aumento de riesgo de enfermedad
hemolítica en el recién nacido.
· Despistaje de sobrecarga de hierro: · Especialmente en mujeres que hayan recibido más de 10 unidades de concen-
trado globular. · Dependiendo de los niveles de ferritina, se indicará o no hierro vía oral.
· Ecocardiograma para descartar hipertensión pulmonar: · Su presencia en la embarazada está asociada con mayor mortalidad. · Una velocidad de regurgitación tricuspídea de más de 2,5m/seg, está asociada
a alto riesgo de hipertensión pulmonar.
· Estudio de retina · Determinar el estado serológico relacionado con enfermedades virales.
· Si la embarazada no está inmunizada contra Hepatitis B, administrar la vacu-na correspondiente.
· Determinar el estado de inmunización asociado con enfermedades infecto-contagiosas.
· Inmunizar contra H. influenzae tipo b y la vacuna conjugada contra el meningo-coco C, si no lo está, además contra H1N115.
Suplemento de vitaminas • Ácido fólico.
· Aumentarlo de 1mg/día a 5 mg/día durante la etapa preconcepción y durante el embarazo para16: · Prevenir eventuales defectos del tubo neural. · Compensar la demanda durante el embarazo.
El desarrollo de la atención multidisciplinaria parece estar asociado con una mejoría de las complicaciones maternas y fetales. El control del embarazo debe iniciarse lo más temprano posible por un equipo que incluya un hematólogo con experiencia en
.06

165
enfermedad drepanocítica y embarazo y un obstetra especializado en embarazo de alto riesgo (Figura 6.1).
Figura 6.1. Conducta a seguir en caso de una mujer con enfermedad drepanocí-tica y embarazo
Control Prenatal La frecuencia de las consultas se presenta en la tabla 6.2.
Primera consulta • Ofrecer información, asesoramiento y apoyo en relación a las medidas generales
para mantener un estado saludable.• Informar y asesorar sobre la necesidad de practicar estudio de hemoglobina a la
pareja, si no se lo han hecho.• Realizar historia exhaustiva para determinar extensión de la enfermedad drepa-
nocítica y de sus complicaciones.• Revisar los medicamentos y suspender hidroxiurea, IECA y bloqueadores del re-
ceptor de angiotensina, si los estuviera recibiendo.• Solicitar hematología que incluya reticulocitos y plaquetas.• Indicar ecocardiograma y estudio de retina, si no los hubiese practicado el año
Consulta de alto riesgo obstétrico con obstetra con experiencia en drepanocitosis en colaboración con hematólogo
Consultar poranemia
y tranfusión.Ver guía
de transfusión
Aconsejar partovaginal si es
necesario cesárea.Ver guía de
anestesia y cirugía
Informe con datos basales para obstetra
Consultapor episodios agudos.
Ver guíacorrespondientes
Asesoramiento genético
Tópicos Especiales

166
anterior o en el período antes de la concepción.• Documentar presión arterial y saturación de oxígeno basales.• Solicitar examen de orina (chorro medio).• Establecer la basal de función renal, relación proteína en orina/creatinina • Consignar la basal de función hepática• Determinar niveles de hierro sérico, TIBC, saturación de transferrina y ferritina• Ordenar fenotipo extendido de glóbulos rojos (si no se ha realizado)• Indicar ácido fólico (5mg/día) y penicilina profiláctica si no los estuviera recibiendo.
Tabla 6.2. Frecuencia de las consultas con el hematólogo durante el embarazo complicado con enfermedad drepanocítica.
Segunda consulta• Conversar acerca de cómo la enfermedad drepanocítica afecta al embarazo.• Revisar resultados de estudios solicitados en la primera consulta.• Para estudios complementarios, ver tabla 6.3.
Transfusión Las indicaciones de transfusión se observan en la tabla 6.4. Para detalles relacionados con la técnica de transfusión revisar guía relacionado con transfusión.
Tratamiento del dolor El dolor es la primera complicación durante el embarazo, cerca de 27-50% de las mu-jeres embarazadas presentan eventos dolorosos5,8-10. No existen estudios aleatorios y controlados relacionados con el tratamiento del dolor durante el embarazo, de modo que se seguirán las indicaciones realizadas en la guía del manejo del dolor.
Los opiáceos no se han asociado con teratogenia pero pueden disminuir transitoria-mente los movimientos fetales y la frecuencia del ritmo cardiaco fetal.
Si la mujer ha recibido opiáceos en la etapa tardía del embarazo, observar al recién nacido con el propósito de detectar signos de abstinencia de opiáceos.
El uso de AINE está limitado entre las 12 y 28 semanas de gestación.
Edad gestacional Frecuencia
< 28 semanas Cada 3 semanas
28 semanas - 32 semanas Cada 2 semanas
> 32 semanas Cada semana
.06

167
Tabla 6.3. Estudios complementarios durante el embarazo complicado con enfermedad drepanocítica14.
Edad gestacional Estudios complementarios
7 semanas - 9 semanas UltrasonidoConfirmar viabilidad en vista del riesgo de aborto espontáneo
12 semanasConsiderar aspirina a dosis bajasSolicitar marcadores ecosonográficos y serológicos para malformaciones
16 semanas Repetir uroanálisis, (chorro medio)
20 semanas Ultrasonido. Repetir uro análisis (chorro medio)
24 semanas
Ultrasonido de 2º y 3º nivelUltrasonido tridimensionalControl de crecimiento fetal Medida de volumen de líquido amnióticoMarcadores para cromosomopatíasRepetir uroanálisis, (chorro medio)
26 semanas Repetir uroanálisis (chorro medio)
28 semanasUltrasonido control de crecimiento fetalMedida de volumen de líquido amnióticoRepetir uroanálisis, (chorro medio)
30 semanas Repetir uroanálisis, (chorro medio)
32 semanasUltrasonido control de crecimiento fetal Medida de volumen de líquido amnióticoRepetir uroanálisis, (chorro medio)
34 semanas Repetir uroanálisis, (chorro medio)
36 semanas
Ultrasonido control de crecimiento fetal Medida de volumen de líquido amnióticoEco Doppler. Permite visión exacta de la circulación materno fetal y signos pronóstico de preclampsia, monitoreo no estresante para ver movimientos fetales, perfil biofísico para movimientos fetales, respiración y tono muscularRepetir uroanálisis, (chorro medio)Ofrecer información y asesoramiento sobre:•Tiempo,modoymanejodelparto•Analgesia,anestesia•Cuidadosdelniñodespuésdelnacimiento
38 semanas - 40 semanas
Control de rutinaTratar de llevar el embarazo hasta la 40 semana
Tópicos Especiales

168
Tabla 6.4. Indicaciones de transfusión en embarazo complicado por enfermedad drepanocítica14
Indicaciones de hospitalización • Dolor que no cede con analgésicos usados en el hogar.• Dolor atípico.• Dolor en el tórax. • Fiebre. • Dificultad para respirar.
Otras complicaciones agudas deben tratarse según lo indicado en las guías co-rrespondientes.
Cuidados durante el trabajo de parto14
El parto ha de tener lugar en una institución hospitalaria capaz de manejar tanto las complicaciones de la enfermedad drepanocítica como embarazo de alto riesgo.
Las mujeres con enfermedad drepanocítica y un feto con crecimiento normal, no tienen contraindicación para ofrecerles un parto electivo a través de inducción del parto o por cesárea si está indicado, después de la 38 semana de gestación.
La enfermedad drepanocítica no es contraindicación para el parto vaginal, aún des-pués de una cesárea anterior.
Las pruebas cruzadas necesitan realizarse con anticipación por si hubiese un anti-cuerpo que demorará la entrega de sangre en el momento oportuno.
Indicaciónes Comentarios
Mujeres con antecedentes de compli-cación médica, obstétrica o fetal
Transfusión crónica o exanguinotransfusión parcial dependiendo de indicación clínica. A ser decidida conjuntamente con el obstetra
Mujeres sometidas a régimen de transfusión crónica, antes del embarazo por prevención primaria o secundaria de ictus cerebral agudo o prevención de complicación severa
Debe continuar durante el embarazo
Embarazo múltiple Considerar transfusión crónica por el alto riesgo de complicaciones
Anemia aguda Transfusión simple
Sindrome torácico agudo o ictus cerebral agudo Exanguinotransfusión parcial
.06

169
En casos de mujeres con prótesis a nivel de cadera, estudiar previamente la posición para el momento del parto.
Condiciones óptimas durante el trabajo de parto• En cuanto se inicie el trabajo de parto, informar al equipo multidisciplinario (resi-
dente, obstetra, hematólogo, anestesiólogo y neonatólogo)• La mujer debe mantenerse abrigada para evitar el enfriamiento y recibir abundan-
te líquido; si no tolera la vía oral, administrar soluciones hipotónicas (Dextrosal 0,45%) EV.
• Practicar control permanente de la frecuencia cardiaca ya que el feto pudiera presentar sufrimiento fetal que amerita una acción inmediata.
• Determinación de oxígeno mediante oxímetro de pulso.• Administrar oxígeno por mascarilla si la saturación de oxígeno es igual o menor
de 94%.• Instaurar heparina de bajo peso molecular a dosis profilácticas, mantener hasta 7
días después de dar el alta en caso de parto vaginal y durante 6 semanas en caso de cesárea.
• El trabajo prolongado de parto (más de 12 horas) incrementa el riego de crisis de dolor secundario a deshidratación. · Si la mujer está bien hidratada y el trabajo progresa, supervisar cuidadosamen-
te. · Si el trabajo no progresa, plantear cesárea.
• Si la temperatura supera 37,5°C, se requiere investigar la causa.
Analgesia y anestesia durante el trabajo de parto14 • No usar petidina. Se puede usar otros opiáceos. • En caso de cesárea se recomienda anestesia regional.• En lo posible evitar anestesia general. • La indicación de analgesia peridural durante el trabajo de parto es igual que en
mujeres normales, y está determinada por: · Nivel de dolor experimentado. · A elección por la paciente. · Ausencia de contraindicaciones.
Cuidados postparto14 • El mismo nivel de cuidados y vigilancia, ya que durante el puerperio continua el
riesgo de eventos vasooclusivos, más aún si se administró anestesia general17: · Mantener:
· Saturación de oxígeno superior a 94% hasta el alta. · Hidratación adecuada hasta el egreso. · Heparina de bajo peso molecular hasta 7 días después del parto vaginal y du-
rante 6 semanas si el parto fue por cesárea.
Tópicos Especiales

170
· Deambulación precoz. · Manejar crisis de dolor de igual manera que en mujeres no embarazadas. · Los AINE son usados durante el puerperio y pueden continuar usándose durante
la lactancia. · Recomendar lactancia materna.
Referencias 1. Hendrickse JP, Watson-Williams EJ, Luzzatto L, Ajabor LN. 1. Pregnancy in homozygous sickle-cell anaemia. J Obstet Gynaecol Br Commonw. 1972; 79(5):396-409. 2. Villers MS, Jamison MG, De Castro LM, James AH. Morbidity associated with sickle cell disease in pregnancy. Am J Obstet Gynecol 2008; 199:125.e1-5 4. 3. Sun PM, Wilburn W, Raynor BD, Jamieson D. Sickle cell disease in pregnancy: twenty years of experien-ce at Grady Memorial Hospital, Atlanta, Georgia. Am J Obstet Gynecol 2001; 184: 1127-30. 4. Afolabi BB, Iwuala NC, Iwuala IC, Ogedengbe OK. Morbidity and mortality in sickle cell pregnancies in Lago, Nigeria: a case control study. J Obstet Gynaecol 2009;29:104–106. 5. Al Jama FE, Gasem T, Burshaid S, Rahman J, Al Suleiman SA, Rahman MS. Pregnancy outcome in patients with homozygous sickle cell disease in a university hospital, Eastern Saudi Arabia. Arch Gynecol Obstet 2009;280:793–797. 6. Smith JA, Espeland M, Bellevue R, Bonds D, Brown AK, Koshy M. Pregnancy in sickle cell disease: experience of the Cooperative Study of Sickle Cell Disease. Obstet Gynecol 1996; 87:199–204. 7. Chakravarty EF, Khanna D, Chung L. Pregnancy outcomes in systemic sclerosis, primary pulmonary hypertension, and sickle cell disease. Obstet Gynecol 2008;111:927–934. 8. Rajab KE, Issa AA, Mohammed AM, Ajami AA. Sickle cell disease and pregnancy in Bahrain. Int J Gy-naecol Obstet 2006; 93:171–175. 9. Powars DR, Sandhu M, Niland-Weiss J, Johnson C, Bruce S, Manning PR. Pregnancy in sickle cell disease. Obstet Gynecol 1986;67:217–28. 10. Howard RJ, Tuck SM, Pearson TC. Pregnancy in sickle cell disease in the UK: results of a multicentre survey of the effect of prophylactic blood transfusion on maternal and fetal outcome. Br J Obstet Gynaecol 1995;102;947–51. 11. Serjeant GR, Loy LL, Crowther M, Hambleton IR, Thame M. Outcome of pregnancy in homozygous sickle cell disease. Obstet Gynecol 2004;103:1278–85. 12. el-Shafei AM, Sandhu AK, Dhaliwal JK. Maternal mortality in Bahrain with special reference to sickle cell disease. Aust N Z J Obstet Gynaecol 1988;28:41–44. 13. Serjeant GR, Hambleton I, Thame M. Fecundity and pregnancy outcome in a cohort with sickle cell–haemoglobin C disease followed from birth. BJOG 2005; 112:1308–1314. 14. Royal College of Obstetrician and Gynaecologists. London. 2011. Management of Sickle Cell Disease in Pregnancy.Green-top Guideline No. 61 15. Vichinsky EP. Pregnancy in sickle cell disease. Disponible en http://www.uptodate.com/contents/pregnancy-in-sickle-cell-disease 16. Charache S, Niebyl JR. Pregnancy in sickle cell disease. Clin Haematol 1985;14:729–46. 17. Camous J, N’da A, Etienne-Julan M, Stéphan F. Anesthetic management of pregnant women with sic-kle cell disease –effect on postnatal sickling complications. Can J Anaesth 2008;55:276–83.
.06

171
6.3. Anestesia y Cirugía Graciela León / Médico hematólogo.
Los pacientes con enfermedad drepanocítica ameritan de cirugía por complicaciones como necrosis aséptica y colelitiasis o por condiciones no relacionadas con la enfer-medad. El riesgo de morbilidad y mortalidad es mayor que en la población general, debido a1:
• Anemia. • Tendencia a la vasooclusión. • Presencia de daño orgánico. • Riesgo de hipoxia. • Efectos de la asplenia.
El riesgo en pacientes con ED-SS (anemia drepanocítica) y en ED-Sβ0 talasemia, es mayor que en pacientes con ED-SC y ED-Sβ+ talasemia, siendo el síndrome toráci-co agudo la complicación mas frecuente1. Los estudios en grandes series datan de aproximadamente 20 años; desde entonces se ha incrementado el conocimiento de la fisiopatología de la enfermedad drepanocítica. La existencia de nuevos anestésicos y técnicas quirúrgicas novedosas hace necesario realizar estudios prospectivos que permitan conocer el estado actual de la morbilidad y mortalidad de la enfermedad drepanocítica vinculada a complicaciones por anestesia y cirugía. La anestesia general está asociada con un riesgo significativo de complicaciones postoperatorias, especialmente síndrome torácico agudo. Con el propósito de mini-mizar los riesgos, la cirugía debe llevarse a cabo en una institución hospitalaria que cuente con profesionales con experiencia en el manejo de enfermedad drepanocítica y existir una estrecha comunicación entre el hematólogo, cirujano, anestesiólogo y ban-co de sangre (figura 6.2)2.
Evaluación preoperatoria Documentar el estado de salud del paciente y prestar atención a aquellos que son predictores de complicaciones:
• Tipo de intervención quirúrgica y riesgo: bajo, moderado o alto.• Edad, está asociado con la progresión de la enfermedad drepanocítica.• El número de hospitalizaciones es un marcador de gravedad en la enfermedad
drepanocítica.
Identificar si hay daño orgánico y enfermedad coexistente, ya que aumentaría el riesgo de complicaciones perioperatoria. Los pacientes con alto riesgo, especialmente de síndrome torácico agudo y eventos vasooclusivos son aquellos con historia de:
• Enfermedad pulmonar. • Asma. • Síndrome torácico agudo.
Tópicos Especiales
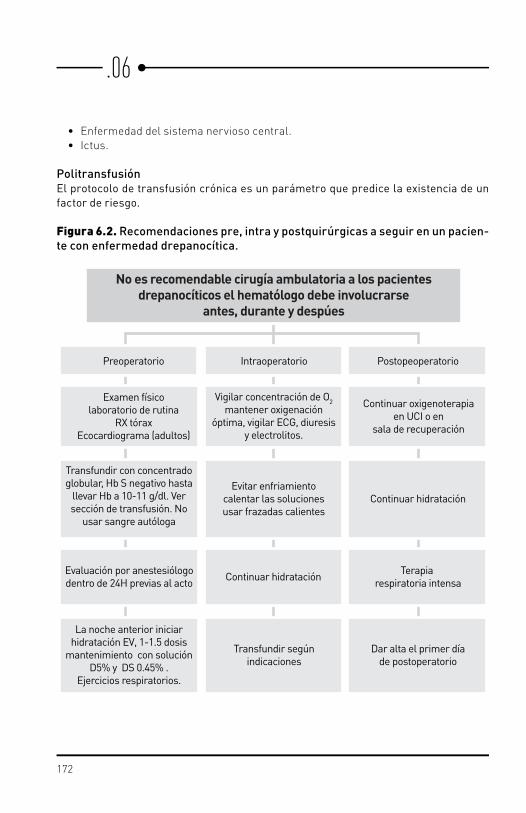
172
• Enfermedad del sistema nervioso central.• Ictus.
PolitransfusiónEl protocolo de transfusión crónica es un parámetro que predice la existencia de un factor de riesgo.
Figura 6.2. Recomendaciones pre, intra y postquirúrgicas a seguir en un pacien-te con enfermedad drepanocítica.
No es recomendable cirugía ambulatoria a los pacientesdrepanocíticos el hematólogo debe involucrarse
antes, durante y despúes
Continuar oxigenoterapiaen UCI o en
sala de recuperación
Examen físicolaboratorio de rutina
RX tóraxEcocardiograma (adultos)
Preoperatorio Intraoperatorio Postopeoperatorio
Evaluación por anestesiólogodentro de 24H previas al acto
Transfundir segúnindicaciones
Dar alta el primer díade postoperatorio
Terapia respiratoria intensa
Continuar hidratación
Continuar hidratación
Evitar enfriamientocalentar las solucionesusar frazadas calientes
Transfundir con concentrado globular, Hb S negativo hasta
llevar Hb a 10-11 g/dl. Ver sección de transfusión. No
usar sangre autóloga
Vigilar concentración de O2 mantener oxigenación
óptima, vigilar ECG, diuresis y electrolitos.
La noche anterior iniciar hidratación EV, 1-1.5 dosis
mantenimiento con solución D5% y DS 0.45% .
Ejercicios respiratorios.
.06

173
Revisión de la historia de transfusión con énfasis en: • Reacciones transfusionales previas.• Presencia de anticuerpo(s) contra los glóbulos rojos.
Investigar signos de actividad actual de la enfermedad drepanocítica• Vasooclusión. • Fiebre. • Deshidratación.
· El embarazo aumenta el riesgo materno.
Solicitar pruebas de laboratorio • Generales.
· Hemograma completo que incluya plaquetas y reticulocitos. · Pruebas generales de coagulación. · Urea y creatinina (suelen estar por debajo de los valores normales, en ausencia
de proteinuria). · Uroanálisis. · Oximetría de pulso. · Para pacientes con antecedentes de síndrome torácico agudo, asma u otra en-
fermedad pulmonar, considerar radiología simple de tórax y función pulmonar con análisis de respuesta a broncodilatadores.
· Ultrasonido abdominal. · Electrocardiograma y eco cardiograma, especialmente en pacientes con ante-
cedentes de hipertensión pulmonar y sobrecarga de hierro por transfusiones repetidas.
· Tipeaje completo extendido e investigación de anticuerpos irregulares. Y prue-bas cruzadas. Ver guía de transfusión.
Complementarias • Función pulmonar y gasometría arterial (si hay datos de disfunción pulmonar gra-
ve o disnea paroxística).• Pruebas de función hepática (y serología viral).
En la indicación del tratamiento perioperatorio, se sugieren los siguientes fundamentos3:• En pacientes con enfermedad drepanocítica.
· La deshidratación conduce a complicaciones. Los niños son propensos a des-hidratación por incapacidad de concentrar la orina. La sobrehidratación puede desencadenar problemas pulmonares en pacientes con alto riesgo.
· La hipoxia ocasiona falciformación. Por otra parte, la hiperoxia prolongada es innecesaria y conllevar a aumento del riesgo de atelectasia y a depresión de la eritropoyesis.
· La hipotermia origina escalofríos y estasis periférico que ocasiona hipoxia y fal-
Tópicos Especiales

174
ciformación. · La infección puede desencadenar crisis de dolor y sindrome torácico agudo. · Procurar la movilización precoz. · Algunos pacientes presentan riesgo elevado de complicaciones y se benefician
de la transfusión simple o de la exanguino transfusión. Sin embargo la transfu-sión implica algunos riesgos como aloinmunización, transmisión de enfermeda-des e hiperviscosidad que desencadena crisis vasooclusivas.
· Existen pocas publicaciones contradictorias en relación al uso de torniquetes. Esto no es una contraindicación absoluta sobre todo para aquellos fenotipos que cursan con una enfermedad moderada o presentan niveles altos de hemoglo-bina fetal.
Transfusión preoperatoria• Ver guía de transfusión.• Considerar electroforesis de hemoglobina y cuantificación de Hb S postransfusión.
72 horas antes de la operación4 • Realizar hematología que incluya reticulocitos. • Si el paciente recibió transfusión simple, asegurarse de que la hemoglobina sea
>10 g/dL.
Todos los pacientes deben ser evaluados por el anestesiólogo el día anterior a la cirugía.
12 horas antes de la cirugía4 • Iniciar hidratación con solución de NaCl 0,45% y dextrosa al 5% (Dextrosal 0,45%).
Administrar 1-1½ veces el requerimiento de mantenimiento.• Control estricto de peso.• Control estricto de líquidos ingeridos y eliminados.• Familiarizar al paciente con la espirometría intensa o forzada.
Cuidados intra operatorios4 • Hacer todo lo posible por evitar:
· Hipoxia. · Hipercapnia. · Hiperventilación. · Enfriamiento.
• Controlar a cada paciente, al menos, con un ECG y determinación del oxígeno ins-pirado, mediante oximetría de pulso o determinación de gases sanguíneos.
• Al momento de iniciar la anestesia general y durante la intervención el paciente debe estar normotérmico y bien hidratado.
• Está contraindicada la técnica de recuperación de sangre para autotransfusión.• Oxigenación: el anestesiólogo mantendrá la oxigenación utilizada en personas
.06

175
normales y la aumentará en presencia de disfunción orgánica.• Hidratación: La tendencia actual hacia el uso de un menor volumen de líquido en
cirugía mayor, sugiere que la administración del volumen adecuado en el momen-to oportuno es probablemente la mejor forma de manejar a los pacientes.
Postoperatorio4 • Administrar oxígeno hasta que el efecto de la anestesia haya desaparecido. En
pacientes donde la cirugía entorpece la respiración, prolongar el suministro de oxígeno. Continuar con oximetría de pulso durante 18h-24h, para estar seguros de que el suministro de oxígeno es el adecuado para mantener la saturación >95%.
• Continuar la hidratación por vía EV, pasar a vía oral en cuanto el paciente esté en condiciones de ingerir líquidos.
• Evitar la hidratación excesiva que pudiera desencadenar síndrome torácico agu-do. No exceder 1-1 ½ veces el requerimiento de mantenimiento (líquidos intrave-nosos + líquidos vía oral + medicación).
• Si hay signos de sobrehidratación, contemplar el uso de diuréticos tipo furosemida.• Aumentar el volumen de hidratación si el paciente tiene signos de deshidratación
por pérdidas insensibles. • Para minimizar las complicaciones pulmonares se sugiere espirometría forzada.
Durante el día, si el paciente está despierto, con el espirómetro ha de realizar, mínimo, 10 inspiraciones cada 2 horas. Durante la noche y durante el día si el paciente está descansando, despertarlo cada 4 horas para realizar los ejercicios respiratorios. En niños se recomienda hacer burbujas.
• Si requiere analgésicos, usarlos según lo indicado en la guía de tratamiento del dolor. · Evitar el uso de narcóticos debido a que provocan hipoventilación. · Individualizar la dosis adecuada que elimine el dolor y facilite la deambulación,
sin ocasionar hipoventilación.• Hasta que el paciente esté estable, practicar diariamente hematología incluyendo
conteo diferencial (hemograma de Schilling), plaquetas y reticulocitos.• Se recomienda un ablandador de heces o laxante suave para evitar estreñimiento. • Continuar el uso de:
· Ácido fólico. · Penicilina profiláctica (si es aplicable). · Si surge una complicación: fiebre, síndrome torácico agudo, crisis vasooclusi-
va, aplasia eritroide, secuestro esplénico, enfermedad cerebro vascular aguda, priapismo), ver la guía correspondiente para el tratamiento de las mismas.
Criterios para el alta4 • Se recomienda permanecer en el hospital al menos 24 horas después de la inter-
vención.• Oxigenación adecuada sin el uso de suplemento con oxígeno.
Tópicos Especiales

176
• Afebril durante más de 24 horas y sin evidencias de infección.• Ingestión adecuada de líquidos, incluyendo medicamentos vía oral.• Control adecuado del dolor con analgésicos vía oral.• No dejar hospitalizado al paciente de manera innecesaria.
Tratamiento ambulatorio y seguimiento4 • Asegurarse que el paciente tenga acceso a los analgésicos indicados.• Coordinar la cita de control con el cirujano.• Concertar la cita con el hematólogo en 2-3 semanas.• Indicaciones en caso de aparición de dolor agudo.• Sugerencia para iniciar actividades normales acordes con la edad.
Recomendaciones finales4 • Lograr que el equipo multidisciplinario esté consciente que el paciente con diag-
nóstico de enfermedad drepanocítica requiere atención especial.• Los pacientes con ED-SS y ED-Sβ0 talasemia deberán recibir transfusión simple
de glóbulos rojos para lograr niveles de Hb de 10g/dL antes de cualquier procedi-miento invasivo de bajo riesgo.
• En pacientes con ED-SC, puede ser necesaria la realización de exanguino transfu-sión parcial para evitar complicaciones relacionadas con hiperviscosidad.
• El riesgo de aloinmunización postransfusional debe ser minimizado mediante la selección de concentrados de glóbulos rojos al menos con igual fenotipo Rh y Kell.
• Independientemente del fenotipo, en los pacientes con enfermedad drepanocítica está indicada atención especial con monitorización intraoperatoria de: · Presión arterial. · Balance hídrico. · Niveles de hemoglobina. · Niveles de oxigenación (saturación arterial y venosa). · Consumo de oxígeno. · Perfusión. · Tensión tisular o coeficiente de extracción.
• La monitorización intraoperatoria de: presión arterial, ritmo cardiaco y oxigena-ción deberá llevarse de manera continua en todos los procedimientos quirúrgicos.
Referencias 1. National Institutes of Health, National Heart, Lung and Blood Institute. Anesthesia and Surgery. Chap 24 in The Management and Therapy of Sickle Cell Disease, NIH. Publication No. 02-2117. 4d ed, rev. Be-thesda.2002; 149 - 151. 2. Lane PA, Buchanan GR, Hutter JJ, Austin RF, Britton H A, Rogers ZA. Eckman JE et al. General Anes-thesia and Surgery in Sickle cell disease in children and adolescents: Diagnosis, guidelines for compre-hensive care, and care paths and protocols for management of acute and chronic complications. Consor-tium Annual Meeting of the Sickle Cell Disease Care .2001:25-26.
.06

177
3. Marchant WA, Walker I. Anaesthetic management of the child with sickle cell disease. Pediatr Anesth 2003:13:473-489. 4. Mid-Atlantic Sickle Cell Disease Consortium (MASCC). Practice Guidelines Workgroup sponsored by the Mid-Atlantic Regional Human Genetics Network (MARHGN). Management of Children with Sickle Cell Disease Undergoing General Anesthesia and Surgery: Diagnosis, Guidelines for Comprehensive Care, and Protocols For Management Of Acute And Chronic Complications.2001; 42-46.
6.4. Transfusión Graciela León / Médico hematólogo.
La transfusión juega un papel importante en el manejo integral del paciente con en-fermedad drepanocítica, sin embargo debe utilizarse con precaución, no solo por las complicaciones postransfusionales del paciente politransfundido, sino para prevenir elevación excesiva del hematocrito (Hto), que pudiera incrementar la viscosidad san-guínea a niveles peligrosos. Objetivos de la transfusión1-2:
• Mejorar la oxigenación tisular al aumentar las cifras de Hb/Hto. • Sustituir la Hb S por Hb A con el propósito de:
· Evitar o reducir la vasooclusión. · Mejorar la microcirculación. · Incrementar la saturación de oxígeno.
• Reducir la hiperviscosidad por dilución de la Hb S.• Reducir la eritropoyesis al minimizar la producción de eritrocitos autólogos.• Retardar la aparición de lesión vascular al disminuir la hemólisis y la inflamación.
Características de los eritrocitos a transfundir: • Hb S negativo. Los eritrocitos de personas portadoras de Hb S tienen dificultad
para atravesar los filtros de leucorreducción, limitando la recuperación de los mismos3.
• Solicitar leucorreducidos con el propósito de disminuir las reacciones febriles y prevenir la alosensibilización a leucocitos y plaquetas (muy importante en pacien-tes candidatos a trasplante de médula ósea (TMO)4.
• Preferiblemente lavados, en casos de reacciones alérgicas severas5.• De menos de 2 semanas de extraídos5. Para los recambios sanguíneos, menos de
5-7 días de la extracción.• De igual fenotipo Rh y Kell para evitar sensibilización6.
Tipos de transfusiónLa terapia transfusional puede efectuarse a través de transfusiones simples (TS) o
Tópicos Especiales

178
mediante procedimientos de recambio sanguíneo, realizando una exanguino trans-fusión parcial (ETp) manual o automatizada o por procedimientos de eritrocitaféresis (EAF). Esto dependerá del objetivo de la transfusión.
A) Transfusión ocasional, aguda o intermitente
Se utiliza la TS para mejorar la oxigenación tisular al corregir la anemia aguda o la hipovolemia 7-9 (Hb <5gr/dL):
• Caída del 20% del valor basal durante una afección aguda.• Reducción de 2 g/dL del valor basal siempre que este sea <7g/dL.
El uso de eritrocitos es preferible a la sangre total, excepto cuando se requiere expan-dir la volemia.
La cantidad de eritrocitos a transfundir en una TS se puede calcular según la siguiente fórmula:8
Volumen a transfundir (mL) = (Hbdeseada – Hbinicial) x Kg x K Donde K=3 si son eritrocitos; K=4 si es sangre plasma reducido; K=6 si es sangre total.
La TS está indicada en pacientes anémicos con descompensación fisiológica aguda secundaria a descenso importante de hemoglobina y que presenten2, 7-8, 10,11:
• Sobrecarga del trabajo cardíaco.• Insuficiencia cardíaca.• Disnea.• Hipotensión postural.• Astenia intensa.• Angina. • Disfunción cerebral. • Preparación pre-quirúrgica para mantener la Hb S en menos del 30% y el Hto en
30%. Puede utilizarse ETp o EAF, dependiendo de la urgencia y tipo de cirugía12-13 .
Causas del descenso importante de hemoglobina2,7
• Secuestro esplénico.• Secuestro hepático.• Aplasia transitoria de serie roja (ej. infección por parvovirus B19).• Exacerbación de la hemólisis por diversas causas.• Hemorragia.
B) Se utilizan ETp o EAF en problemas agudos cuando se desea recambiar en corto tiempo (horas a 1-2 días) la Hb S por Hb A. Mejora el transporte de oxígeno y disminuye las complicaciones al prevenir los fenómenos vasooclusivos, evita la hiperviscosidad y
.06

179
reduce la sobrecarga de hierro en los regímenes crónicos14-16.La ETp o EAF esta indicada en2, 7,8:
• Síndrome torácico agudo rápidamente progresivo.• Falla multiorgánica.• Shock séptico.• Ictus (isquémicos o hemorrágicos), incluyendo los transitorios.• Previo a estudios angiográficos con contraste hipertónico.• Oclusión de arteria de la retina.• Profilaxia preoperatoria, sobre todo en cirugías largas o que requieran el uso de
torniquete.• Priapismo que no mejora en 12 horas (controversial).• Secuestro hepático (en ausencia de colapso circulatorio).• Colestasis intrahepática severa (es preferible la ETp a la EAF para reducir la hi-
perbilirrubinemia).• Excepcionalmente en crisis dolorosas severas que no ceden al tratamiento con-
vencional.• Cuando se requiere aumentar los niveles de Hb sin ocasionar incremento de so-
brecarga de volumen, en pacientes con volemia plasmática expandida en res-puesta al estado de anemia crónica y que presentan disfunción cardíaca o renal.
• En úlceras rebeldes (discutible).• Cirugía oftálmica (discutible).
Esquemas de exanguinotransfusión parcial manual: The National Institute of Health (NIH)17 señala que el volumen que se debe recambiar en las ETp se tiene que calcular en base al peso del paciente, al hematocrito inicial, y al hematocrito y concentración de Hb A que se desean alcanzar con el procedimiento:
En adultos pueden realizarse extracciones y transfusiones de 500mL por vez, repitien-do de 6 a 8 veces. Para algunos clínicos, la ETp realizada con ST es más efectiva que con eritrocitos solos.
El cálculo en niños, utilizando ST cuyo hematocrito se encuentra en 38 a 40%, es a razón de 50 a 60mL/Kg y los volúmenes a recambiar deben ser pequeños.
Al final del procedimiento la hemoglobina no debe exceder de 10 a 12g/dL para evitar la hiperviscosidad y la Hb A debe estar entre 60-70%. Hay autores18,19 que aconsejan realizar una sangría previa antes de iniciar el procedimiento, extrayendo una fracción del volumen del paciente y reemplazándola con solución salina o albúmina. Esto acor-ta el tiempo del procedimiento y ahorra volumen a transfundir. En adultos se sugiere extraer hasta un 15% de la volemia y en niños hasta un 10%. El presangrado es útil cualquiera que sea el hematocrito inicial. No se debe hacer sangría previa en la enfer-medad vascular cerebral aguda9.
Tópicos Especiales

180
Para el cálculo del hematocrito (Hto) después del presangrado se utiliza la siguiente fórmula: Sangre extraída (mL) Hto inicial x [1 ] Kg de peso x 80 Swerdlow18 modificó el esquema del NIH calculando el volumen a exanguinar en 1,5 veces la volemia globular. Calcular la volemia a 70 ml x Kg en pacientes con más de 20 Kg de peso y a 85 ml x Kg en los de menos de 20Kg y la volemia globular = volemia x Hto. Considera que una unidad (ud) estándar tiene 200 mL de eritrocitos aproximadamente (Hto ~ 40% x 500mL).
El procedimiento en adultos se haría de la siguiente manera: 1. Extraer 500mL de ST e infundir 500 mL de solución salina 0,9%.2. Extraer 500mL de ST e infundir dos unidades de eritrocitos.3. Repetir 1 y 2 hasta que el volumen de los eritrocitos administrados sea igual a lo calculado (puede repetirse tres o cuatro veces en adultos).
Si el nivel de hemoglobina del paciente está cercano a 10g/dL, el uso de este protocolo puede generar valores más altos después del equilibrio de fluidos post ETp.
El volumen de eritrocitos extraídos en dos sangrías de 500mL de ST es menor que el volumen de eritrocitos administrado con las dos unidades de eritrocitos, ya que la he-moglobina del paciente es menor a lo normal. Considerar extraer 500mL de ST al final o transfundir una unidad de eritrocitos en vez de dos unidades en el segundo ciclo (y en el cuarto si es necesario).
En pacientes con hemoglobina baja, es poco probable que con este esquema se exceda de 10 g/dL al final del proceso. En niños, los volúmenes a extraer y transfundir se calculan en 5 a 10mL por Kg y el volumen total de eritrocitos a recambiar en 1 a 1,25 veces la volemia globular.1. Por un brazo extraer 5-10mL/kg peso de sangre total y por el otro brazo infundir 5-10 mL de solución salina 0,9%2. Seguidamente, por un brazo extraer 5-10mL/kg de sangre total y por el otro brazo infundir 5-10mL/kg peso de glóbulos rojos3. Repetir 1 y 2 hasta que el volumen de los glóbulos rojos administrados sea igual a lo calculado.
Piomelli y colaboradores19 diseñaron una guía orientadora en cuanto al volumen de ST, o de ST o sangre reconstituida (SR) y eritrocitos (procedimiento mixto) a utilizar para el recambio, dependiendo del hematocrito inicial y proyectando el hematocrito final con una concentración de HbS igual al 30%. (Tabla 6.5)
.06
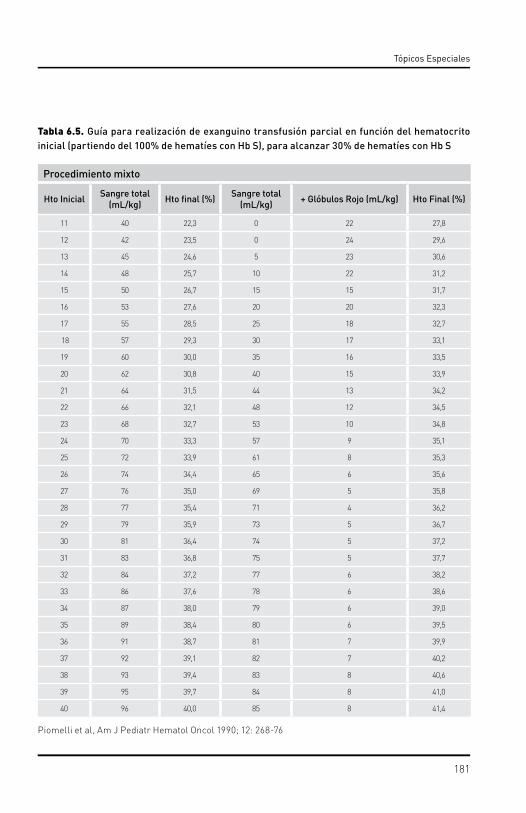
181
Tabla 6.5. Guía para realización de exanguino transfusión parcial en función del hematocrito inicial (partiendo del 100% de hematíes con Hb S), para alcanzar 30% de hematíes con Hb S
Piomelli et al, Am J Pediatr Hematol Oncol 1990; 12: 268-76
Tópicos Especiales
Procedimiento mixto
Hto Inicial Sangre total(mL/kg) Hto final (%) Sangre total
(mL/kg) + Glóbulos Rojo (mL/kg) Hto Final (%)
11 40 22,3 0 22 27,8
12 42 23,5 0 24 29,6
13 45 24,6 5 23 30,6
14 48 25,7 10 22 31,2
15 50 26,7 15 15 31,7
16 53 27,6 20 20 32,3
17 55 28,5 25 18 32,7
18 57 29,3 30 17 33,1
19 60 30,0 35 16 33,5
20 62 30,8 40 15 33,9
21 64 31,5 44 13 34,2
22 66 32,1 48 12 34,5
23 68 32,7 53 10 34,8
24 70 33,3 57 9 35,1
25 72 33,9 61 8 35,3
26 74 34,4 65 6 35,6
27 76 35,0 69 5 35,8
28 77 35,4 71 4 36,2
29 79 35,9 73 5 36,7
30 81 36,4 74 5 37,2
31 83 36,8 75 5 37,7
32 84 37,2 77 6 38,2
33 86 37,6 78 6 38,6
34 87 38,0 79 6 39,0
35 89 38,4 80 6 39,5
36 91 38,7 81 7 39,9
37 92 39,1 82 7 40,2
38 93 39,4 83 8 40,6
39 95 39,7 84 8 41,0
40 96 40,0 85 8 41,4

182
The Georgia Comprehensive Sickle Cell Center recomienda20
Con hematocrito hasta 19%. • Por un brazo extraer 30mL/kg peso de sangre total e igualmente transfundir por
el otro brazo igual cantidad de glóbulos rojos.• Luego extraer 40mL/kg peso de sangre total y paralelamente transfundir sangre
total a razón de 40mL/Kg peso. Con hematocrito entre 20-30%
• Por un brazo extraer 10mL/kg peso de sangre total y simultáneamente transfun-dir por el otro brazo igual cantidad de glóbulos rojos.
• Luego extraer 70mL/kg de sangre total y simultáneamente transfundir sangre total a razón de 70mL /kg peso.
Existen otros esquemas de ETp, entre ellos, para un individuo de 50Kg y Hto pre pro-cedimiento en 25%21. Un método sencillo podría ser:
· Tener preparada una unidad de glóbulos rojos (SOS). · Por un brazo, remover 500mL de sangre total. · Por el otro brazo administrar 300mL de solución salina 0,9%. · Extraer 500mL de sangre total. · Transfundir 5 unidades de glóbulos rojos, en un lapso de 10 horas.
Otro esquema• Primer día: Por un brazo remover 500-700mL de sangre total y por el otro brazo
transfundir 2 unidades de glóbulos rojos.• Segundo día: Igual al primero.• Tercer día: Transfundir 2 unidades de glóbulos rojos.• Se logra alcanzar nivel de Hb A entre 60-70%.
Recambios con equipos automatizados Con estos equipos se realiza exanguinotransfusión pero se utilizan mayor-mente para EAF. Con la EAF el recambio de eritrocitos es casi total y se evi-ta la sobrecarga de volumen porque el balance de fluidos puede ser con-trolado muy de cerca, aproximándose a las condiciones de euvolemia22. Las máquinas de aféresis calculan el volumen de eritrocitos requerido para el procedi-miento en base al peso, talla, hematocrito previo, hematocrito de las uds de eritrocitos, volumen de líquido a recambiar y el hematocrito final deseado17. Hay que tener una estimación previa para hacer la solicitud de las unidades de eritrocitos requeridas al banco de sangre10,11.
• Cálculo del número de unidades de eritrocitos para la EAF: Volemia de eritrocitos (VE) = Volemia x Hto.
• Cantidad aproximada de eritrocitos por unidad (CAEU)= Hto de la unidad de eritro-
.06

183
citos x volumen de la unidad de eritrocitos/Número de unidades necesarias para recambiar un volumen= VE/CAEU.
• Después del procedimiento realizar control de hematocrito y de electroforesis de hemoglobina.
Con una ETp se reduce la HbS a aproximadamente una tercera parte (33%). Con la EAF se podrían hacer dos recambios de volemia eritrocítica con lo que la Hb S debe llegar al 10%, y luego se podría transfundir para aumentar la hemoglobina a más de 10g/dL.
La EAF tiene como desventajas su elevado costo, alto consumo de eritrocitos, uso de catéteres venosos centrales, poca disponibilidad de las máquinas a nivel hospitalario e imposibilidad de utilizarla en niños pequeños o con peso menor de 20-25kg.
Transfusión crónica Los regímenes de transfusión crónica se emplean en pacientes con complicaciones crónicas o recaídas frecuentes. Deben mantenerse por largo tiempo según la evolu-ción del problema, pudiéndose observar recaídas al suspender dichos programas. Los pacientes con daño crónico severo y descompensación manifiesta, recibirán transfu-siones durante toda su vida2,10,23.
Indicaciones2, 5, 7, 9, 15, 17, 22,24-30: • Prevención de ictus cerebral.• Prevención de la recurrencia de ictus.• Secuestro esplénico recurrente.• Síndrome torácico recurrente.• Hipertensión pulmonar y otras causas de hipoxia crónica.• Embarazo complicado.• Fallo orgánico crónico.• Insuficiencia renal progresiva.• Insuficiencia cardíaca.• Dolor recurrente discapacitante con más de tres hospitalizaciones al año.• Priapismo recurrente.• Úlceras maleolares crónicas de evolución tórpida.• Curva pondoestatural plana cuando otras causas se han descartado.
La TS se utiliza en los programas de transfusión crónica. Una vez que la concentración de Hb A logra un 60-70% (lo ideal es alcanzarla con un recambio sanguíneo) y el hema-tocrito se encuentra ≤30%, se deben mantener dichos valores durante el tiempo que sea necesario según la causa que indujo a la inclusión en este tipo de programa. Las transfusiones se realizarán cada 3 a 4 semanas, a dosis de 10 a 20mL por kg de peso. La eficacia de este tipo de régimen será monitoreada periódicamente con electrofo-resis de hemoglobina y cuantificación de los valores de hemoglobina y hematocrito. El
Tópicos Especiales

184
hematocrito post transfusión podrá ser mayor a 30%, mientras mayor sea el nivel de Hb A alcanzado en el paciente. Sin embargo, las cifras hematológicas no deben sobre-pasar los 11,5-12g/dL de hemoglobina o 35-36% de hematocrito2,7-10.
Ha de vigilarse la hiperviscosidad, la sobrecarga de fluidos y de hierro postransfu-sional y considerar el uso de quelantes (deferoxamina, deferasirox)31 para reducir o prevenir la sobrecarga de hierro y ciertas medidas farmacológicas para minimizar las transfusiones como la administración de hidroxiurea32,33, la cual incrementa los nive-les de Hb F, el hematocrito y disminuye los síntomas.
A diferencia de la TS crónica, cuando se utilizan recambios sanguíneos, la frecuencia es cada 3 a 6 meses según las experiencias reportadas16. Después de realizada la pri-mera ETp, los recambios subsiguientes serán de sólo un volumen, aunque siempre hay que hacer ajustes según la necesidad y los niveles de Hb A alcanzados. En los niños hay que vigilar el incremento del tamaño del bazo que a su vez puede elevar los reque-rimientos transfusionales. Si esto ocurre se deberá plantear la esplenectomía lo más pronto posible.
Efectos adversos de la transfusión y procedimientos de aféresis • No inmunológicas5,24,34:
· Hiperviscosidad. · Sobrecarga de hierro. · Sobrecarga de volumen. · Transmisión de infecciones.
• Inmunológicas4-7, 35,36
· Aloinmunización · 5% - 36% se sensibilizan. · Los sistemas antigénicos involucrados con más frecuencia son el sistema Rh,
Kell, MNS, Duffy y Kidd. · En los pacientes con enfermedad drepanocítica es frecuente el haplotipo R0
(Dce), el cual es raro en la población caucásica; de allí que la transfusión sea un desencadenante de la sensibilización contra los antígenos C y E, habitual-mente presentes en el resto de la población.
· Pueden carecer de los antígenos del sistema Duffy (Fy(a-b-) y con frecuencia producen anti-Fya.
· Después de ocurrida la sensibilización, muchos de estos anticuerpos (inde-pendientemente de la especificidad) descienden sus títulos precozmente man-teniéndose muy bajos o imperceptibles. Este comportamiento ocasiona que en el momento de preparar una nueva transfusión, un paciente sensibilizado pudiera dar el rastreo de anticuerpos (pantallas) negativo y la prueba cruzada ser aparentemente compatible, con la consecuente generación de una reac-ción hemolítica post-transfusional.
.06

185
· Es importante que el paciente sensibilizado conserve un informe expedido por el banco de sangre en el que se señale su fenotipo eritrocitario completo, la especificidad de sus anticuerpos y se sugiera las características de los eri-trocitos a administrar, sean o no detectables los anticuerpos en las pruebas pre-transfusionales.
· Aloinmunización contra antígenos del sistema HLA. · Se estima que entre el 70-85% de los pacientes politransfundidos que han recibido 50 o más transfusiones están aloimunizados para el sistema HLA y/o contra antígenos plaquetarios.
• Reacción hemolítica post transfusional. • Síndrome hiperhemolítico. • Reacciones o complicaciones durante los procedimientos de aféresis (EAF):
· Descompensación hemodinámica: · Hipotensión. · Taquicardia. · Hipovolemia.
· Calambres y parestesias por el citrato. · Hipotermia por el uso de componentes fríos. · Obstrucción de accesos venosos y hemólisis.
Hoy día, estos efectos son mucho mejor controlados y las complicaciones severas son muy raras.
Recomendaciones pretransfusionales • Información dirigida al paciente y a los familiares: Antes de transfundir por pri-
mera vez y previo al iniciar un esquema de transfusión crónica es fundamental conversar con el paciente y/o sus familiares con el fin de: · Explicar la trascendenia de la transfusión crónica en la recurrencia de una com-
plicación aguda. · Informar que la transfusión tendrá lugar cada 3-4 semanas. · Señalar la importancia de mantener el régimen de transfusión crónica por el
tiempo indicado. · Educar sobre el riesgo de la transfusión. · Informar sobre los posibles efectos adversos de la transfusión. · Obtener el consentimiento informado, antes de iniciar el programa de transfusión. · Instruir acerca de la necesidad de acudir a la institución por lo menos 24 horas
antes de la transfusión, con la finalidad de tomar muestra para hematología que incluya reticulocitos y para las pruebas pre transfusionales.
· El médico debe indicar en la historia, la justificación para el régimen de trans-fusión crónica.
· Evaluación diagnóstica. · Antes del inicio de la terapia transfusional, es recomendable hacer estudio in-
Tópicos Especiales

186
munohematolológico que contemple fenotipo eritrocitario completo. En caso de que no fuera posible, al menos para los sistemas Rh y Kell.
· Antes de iniciar el régimen de transfusión crónica, solicitar los siguientes es-tudios: · Ferritina, hierro sérico, TIBC y saturación de transferrina. · Serología para Hepatitis A, B y C. · Estado de inmunización para Hepatitis A y B. · Serología para VIH. · Enzimas hepáticas. · Considerar estudio audiológico y oftalmológico. · Electro y ecocardiograma.
· Antes de cada transfusión, indicar:
· Hematología con conteo de reticulocitos. · Peso, signos vitales y saturación de oxígeno. · Pruebas pretransfusionales que incluyan rastreo de anticuerpos. En caso de
ser positivo, identificar especificidad(es). · Seleccionar eritrocitos compatibles, preferiblemente de igual fenotipo Rh y
Kell. · Considerar electroforesis de Hb para obtener el estado inicial de Hb S.
· A realizar durante el régimen de transfusión crónica: · Evaluación física cada 3 meses. · Uroanálisis y enzimas hepáticas cada 6 meses. · Serología de hepatitis B, C y VIH cada tres meses. · Estudio tiroideo, glicemia y otros estudios endocrinos según esquema general
para pacientes con enfermedad drepanocítica. · Considerar electro y ecocardiograma anualmente y antes de iniciar quelación.
Ver guía de sobrecarga de hierro.
Las tablas 6.6 y 6.7 y figura 6.3 muestran un resumen del manejo transfusional en enfermedad drepanocítica
.06

187
Tabla 6.6. Manejo transfusional en complicaciones agudas de la enfermedad drepanocítica
Complicación aguda Tipo de transfusión
Secuestro esplénico TS rápida pero fraccionada para evitar rebote.ETp en caso de distress cardio respiratorio.
Aplasia transitoria de células rojas. TS cuando la Hb desciende 2g/dL por debajo de los niveles basales.
Incremento de hemólisis TS cuando la Hb está en 5g/dL y el Hto en 15%.
Sindrome torácico agudo TS o ETp / EAF. Depende de evolución clínica.
Falla multiorgánica aguda ETp / EAF.En caso de anemia severa, iniciar con TS
Ictus cerebralSe sugiere ETp/EAF al instalarse el cuadro agudo y para prevenir recurrencias en forma crónica.También se puede emplear TS.
Shock séptico ETp/EAF
Preparación para anestesia generalTS o ETp / EAF en caso de cirugía mayor. No hay diferencias significativas entre el tipo de abordaje transfusional y las complicaciones post quirúrgicas.
Angiografía cerebral / uso de medios de contraste ETp / EAF en caso de utilizar contrastes hipertónicos
Oclusión de arteria central de la retina ETp / EAF
Cirugía oftálmica Como cualquier cirugía mayor, aunque es discutible
Priapismo que no responda a otras medidas ETp / EAF
TS : Transfusión simple. ETp: Exanguinotransfusión parcial. EAF: Eritracitaféresis
Tópicos Especiales
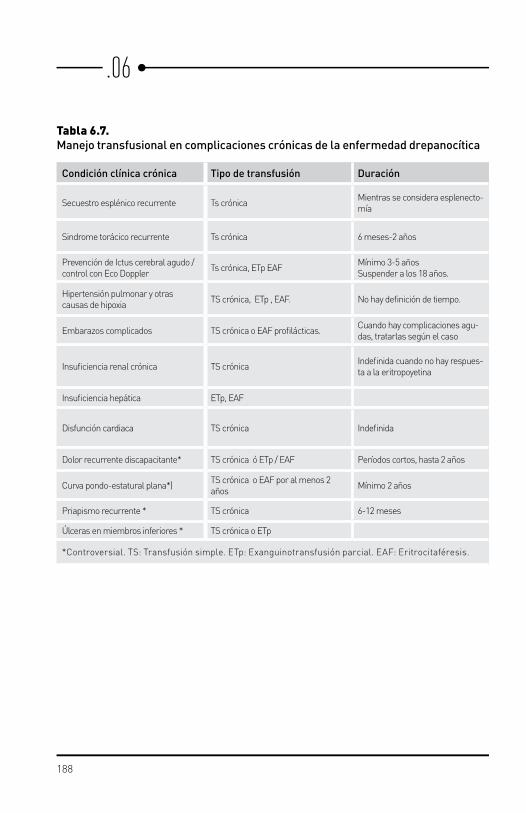
188
Tabla 6.7. Manejo transfusional en complicaciones crónicas de la enfermedad drepanocítica
Condición clínica crónica Tipo de transfusión Duración
Secuestro esplénico recurrente Ts crónica Mientras se considera esplenecto-mía
Sindrome torácico recurrente Ts crónica 6 meses-2 años
Prevención de Ictus cerebral agudo / control con Eco Doppler Ts crónica, ETp EAF Mínimo 3-5 años
Suspender a los 18 años.
Hipertensión pulmonar y otras causas de hipoxia TS crónica, ETp , EAF. No hay definición de tiempo.
Embarazos complicados TS crónica o EAF profilácticas. Cuando hay complicaciones agu-das, tratarlas según el caso
Insuficiencia renal crónica TS crónica Indefinida cuando no hay respues-ta a la eritropoyetina
Insuficiencia hepática ETp, EAF
Disfunción cardiaca TS crónica Indefinida
Dolor recurrente discapacitante* TS crónica ó ETp / EAF Períodos cortos, hasta 2 años
Curva pondo-estatural plana*) TS crónica o EAF por al menos 2 años Mínimo 2 años
Priapismo recurrente * TS crónica 6-12 meses
Úlceras en miembros inferiores * TS crónica o ETp
*Controversial. TS: Transfusión simple. ETp: Exanguinotransfusión parcial. EAF: Eritrocitaféresis.
.06

189
Tópicos Especiales
Figura 6.3. Terapia transfusional (páginas 190 y 191)
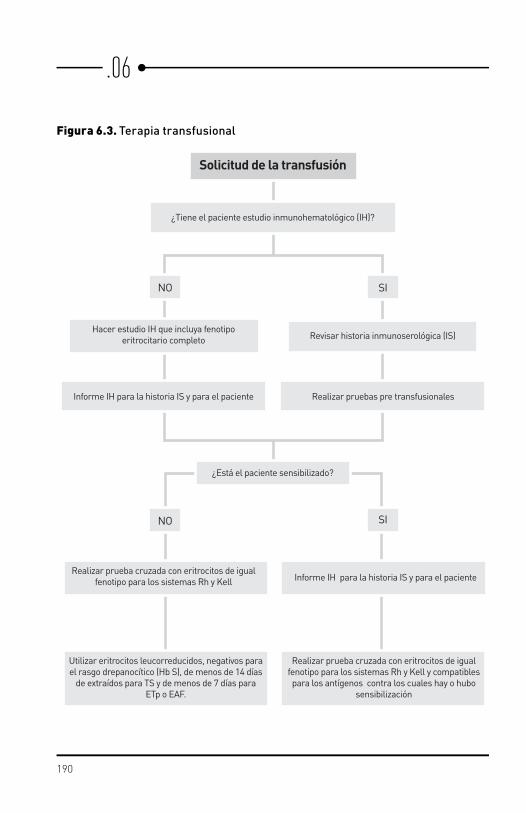
190
Figura 6.3. Terapia transfusional
.06
Solicitud de la transfusión
¿Tiene el paciente estudio inmunohematológico (IH)?
NO SI
NO SI
Hacer estudio IH que incluya fenotipoeritrocitario completo
Realizar prueba cruzada con eritrocitos de igual fenotipo para los sistemas Rh y Kell
Utilizar eritrocitos leucorreducidos, negativos para el rasgo drepanocítico (Hb S), de menos de 14 días
de extraídos para TS y de menos de 7 días para ETp o EAF.
Realizar prueba cruzada con eritrocitos de igual fenotipo para los sistemas Rh y Kell y compatibles para los antígenos contra los cuales hay o hubo
sensibilización
Informe IH para la historia IS y para el paciente
Revisar historia inmunoserológica (IS)
Realizar pruebas pre transfusionales
¿Está el paciente sensibilizado?
Informe IH para la historia IS y para el paciente
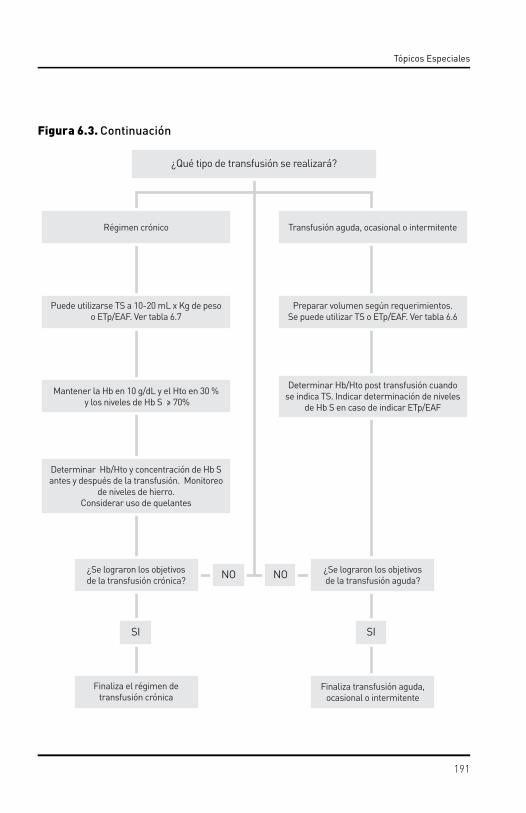
191
Figura 6.3. Continuación
Tópicos Especiales
¿Qué tipo de transfusión se realizará?
SI SI
NONO
Régimen crónico Transfusión aguda, ocasional o intermitente
Puede utilizarse TS a 10-20 mL x Kg de peso o ETp/EAF. Ver tabla 6.7
Preparar volumen según requerimientos. Se puede utilizar TS o ETp/EAF. Ver tabla 6.6
Mantener la Hb en 10 g/dL y el Hto en 30 % y los niveles de Hb S ≥ 70%
Determinar Hb/Hto y concentración de Hb S antes y después de la transfusión. Monitoreo
de niveles de hierro. Considerar uso de quelantes
¿Se lograron los objetivos de la transfusión crónica?
Finaliza el régimen de transfusión crónica
¿Se lograron los objetivos de la transfusión aguda?
Finaliza transfusión aguda, ocasional o intermitente
Determinar Hb/Hto post transfusión cuando se indica TS. Indicar determinación de niveles
de Hb S en caso de indicar ETp/EAF

192
Referencias 1. Rosse WF, Telen M, Ware RE. The pathophysiology of sickle cell disease. In: Transfusion in Sickle Cell Disease. Bethesda - Maryland, AABB Press; 1998. p. 31-42. 2. Josephson CD, Su LL, Hillyer KL, Hillyer CD. Transfusion in the patient with sickle cell disease: a critical review of the literature and transfusion guidelines. Trans Med Rew 2007;21:118-133. 3. Schuetz AN, Hillyer KL, Roback JD, Hillyer CD. Leukoreduction filtration of blood with sickle cell trait. Transfus Med Rev 2004;18:168-176. 4. Friedman DF, Lukas MB, Jawad A, Larson PJ, Ohene-Frempong K, Manno CS.. Alloimmunization to platelets in heavily transfused patients with sickle cell disease. Blood 1996;88:3216-3222. 5. Sharon B. Transfusion Therapy in Congenital Hemolytic Anemias. In: Mintz P. Transfusion Therapy: Clinical principles and practice. 3 ed. Bethesda, Maryland, AABB Press; 2011. p. 87-126. 6. Win N. Hyperhemolysis syndrome in sickle cell disease. Expert Rev Hematol 2009;2:111-115. 7. Telen MJ. Principles and problems of transfusion in sickle cell disease. Semin Hematol 2001;38:315-323. 8. Cantalejo MA. Protocolo de anemia de células falciformes o drepanocitosis. Sociedad Española de Hematología Pediátrica .DREP-2002-SEHP. 9. Cantalejo MA. Protocolo de anemia de células falciformes o drepanocitosis. Bol S Vasco-Nav Pediatr 2005;38 (1):20-38. 10. Eckman JR. Techniques for blood administration in sickle cell patients. Semin Hematol 2001; 38 (1 Suppl 1):23-29. 11. Vichinsky EP. Current issues with blood transfusions in sickle cell disease. Semin Hematol 2001;38 (1 Suppl 1):14-22. 12. Koshy M, Weiner SJ, Miller ST, Sleeper LA, Vichinsky EP, Brown AK, et al. Surgery and anesthesia in sickle cell disease. Cooperative Study Sickle Cell Disease. Blood 1995;86:3676-3684. 13. Hirst C, Williamson L. Preoperative blood transfusions for sickle cell disease. Cochrane Database of Systematic Reviews. 2012 Issue 3 (DOI: 10.1002/14651858). 14. Ware RE, Russell W, Zimmerman S, Sylvestre P, Mortier N, Davis J, Treem W, Schultz W. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxiurea and phlebotomy. J Pediatr 2004;145:346-352. 15. Quirolo K. How do I transfuse patients with sickle cell disease? Transfusion 2010;50:1881-1886. 16. Masera N, Tevecchia L, Pozzi L, Riva F, Vimercati Ch, Calabria M. Periodic erythroexchange is an effective strategy for high risk pediatric patients with sickle cell disease. Transfusion and Apheresis Science 2007;37:241-247. 17. National Heart, Lung, and Blood Institute. Transfusion, Iron Overload, and Chelation. The Manage-ment of Sickle Cell Disease. Bethesda, MD: US Department of Health and Human Services, National Institutes of Health, NIH Publication No. 02-2117. 4 ed. June 2002. p. 153-160. Disponible en: http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf . Acceso el 15 de enero de 2012. 18. Swerdlow PS. Red cell Exchange in sickle cell disease. Haematology Am Soc Hematol Educ Program 2006;48-53. 19. Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated argini-ne metabolism, hemolysis-associated pulmonary hypertension, and mortality sickle cell disease. JAMA 2005;294:81-90. 20. Gee Beatrice. Transfusion Therapy. In Eckman J, Platt A. The Sickle Cell Information Center. The
.06

193
Tópicos Especiales
Georgia Comprehensive Sicle Cell Center at Grady Health System. www.scinfo.org/transfus.htm. Acceso mayo 2007.21. Rosse WF, Telen M, Ware RE. The pathophysiology of sickle cell disease. In: Transfusion support for patients with sickle cell disease. Bethesda - Maryland, AABB Press; 1998. p. 1-13. 22. Hagar W, Vichinsky E. Advances in clinical research in sickle cell disease Br J Hematol 2008;141:346-356. 23. Boga C, Kozanoglu I, Ozdogu H, Sozer O, Sezqin N, Bakar C. Alterations of circulating endothelial cells after apheresis in patients with sickle cell disease: a potential clue for restoration of pathophysiology. Transfus Apher Sci 2010;43:273-279. 24. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998;91(1):288-294. 25. Hulbert ML, Scothorn DJ, Panepinto JA, Scott P, Buchanan G, Sarnaik S. Exchange blood transfusión compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: A retrospective cohort study of 137 children with sickle cell anemia. J Pediatr 2006;149:710-712. 26. Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials 1998;19:110-129. 27. Adams RJ, Brambilla D. STOP 2 Trial investigators: discontinuing prophylactic transfusions to prevent stroke in sickle cell disease. N Engl J Med 2005;353:2769-2778. 28. Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886-895. 29. Kosby M. Sickle cell disease and pregnancy. Blood Rev 1995;9:157- 164. 30. Seoud MA, Cantwell C, Nobles G, Levy DL. Outcome of pregnancies complicated by sickle cell and sickle-C hemoglobinopathies. Am J Perinatol 1994;11:187-91. 31. Vichinsky E, Onyekwere O, Porter J, Swerdlow P, Eckman J, Lane P, et al. A randomized comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol 2007;136:501-508. 32. Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxiurea: effects on hemog-lobin F production inpatients with sickle cell anemia. Blood 1992;79:2555-2565. 33. Vichinsky EP. New therapies in sickle cell disease. Lancet 2002;360:629-631. 34. Schmalzer EA, Lee JO, Brown AK, Usami S, Chien S. Viscosity of mixtures of sickle and normal red cells at varying hematocrit levels. Implications for transfusion. Transfusion 1987;27:228-233. 35. Olujohungbe A, Hembleton I, Steohens L, Serjeant B, Serjeant G. Red cell antibodies in patients with homozygous sickle cell disease: a comparison of patients in Jamaica and United Kingdom. Br J Hematol 2001;113:661-665. 36. Osby M, Shulman IA. Phenotype matching of donor red blood cell units for nonalloimmunized sickle cell. A survey of 1182 North American laboratories. Arch Pathol Lab Med 2005;129:190-93.

194
6.5. Sobrecarga de hierro y quelaciónOlimpia C. Pérez- Bández / Médico hematólogo.
Los nuevos conocimientos sobre el metabolismo del hierro y la fisiopatología de la en-fermedad drepanocítica (ED), permiten entender mejor lo que sucede en la sobrecarga de hierro en pacientes con esta enfermedad. Por una parte el metabolismo del hierro es controlado por proteínas tales como: hepcidina, ferroportina, factor 1 inducible por hipoxia, y factor 15 de crecimiento y diferenciación, las cuales están relacionadas con el daño orgánico; por otra parte en la enfermedad drepanocítica el componente infla-matorio juega un papel preponderante en la fisiopatología de la enfermedad. El hierro no unido a transferrina y el depósito en miocardio es mas bajo en ED que en talasemia. Cabe destacar que algunos pacientes con ED están sometidos a régimen crónico de transfusión que ocasiona sobrecarga de hierro1.
Walter y colaboradores1 sugieren que el metabolismo y movilización de hierro es di-ferente en la enfermedad drepanocítica, cuando se compara con las talasemias. Los altos niveles de citoquinas inflamatorias presentes en la primera, aumentan el hierro en los macrófagos/células retículo endoteliales y su retención en las células renales, esto hace que los órganos afectados en ED sean diferentes a los de talasemia, donde corazón y glándulas endocrinas son los más afectados. Con respecto a la hepcidina, está baja en pacientes pediátricos no transfundidos en tanto que está alta en adultos que han recibido transfusión, sugiriendo que la carga de hierro y la inflamación, junto con la hipoxia secundaria a anemia, son responsables de su aumento. Un hecho inte-resante es que la pérdida de hierro es mayor en la ED que en talasemia, quizás debido al depósito en riñón o escape por la orina. Se precisan más estudios para conocer con exactitud los aspectos del metabolismo del hierro en esta afección.
La transfusión es una de las tres alternativas efectivas de tratamiento utilizadas en los actuales momentos y específicamente en la ED-SS; las otras dos son la hidro-carbamida mejor conocida como hidroxiurea y el trasplante de células progenitoras hematopoyéticas (TCPH).
El estudio Stroke Prevention Trial en Enfermedad Drepanocítica (STOP 1), demostró que la transfusión crónica (para mantener los niveles de Hb S <30%), resultó ser efec-tiva en disminuir la recurrencia de enfermedad vascular cerebral aguda en niños2,3; posteriormente se comprobó que la transfusión disminuyó en 92% el riesgo del primer evento de enfermedad vascular cerebral aguda en niños con alto riesgo, quienes pre-sentaban velocidad alta de flujo en la carótida interna o arteria cerebral media, deter-minada por eco doppler transcraneal. El estudio STOP 2 aleatorio, reveló que el flujo se normalizó a los 30 meses de la transfusión crónica, pero hubo que detenerlo por la reaparición del aumento en la velocidad del flujo arterial y ocurrencia de enfermedad vascular cerebral aguda en el grupo de niños que no recibía transfusión4. Además de
.06

195
Tópicos Especiales
prevenir la enfermedad vascular cerebral aguda, la transfusión crónica disminuyó la aparición del sindrome torácico agudo y de eventos de dolor severo5.
Métodos para cuantificar hierro. • Hierro hepático. Existen tres métodos que se basan en la cuantificación del hierro
hepático como indicador de los depósitos del cuerpo6:A) Determinación directa de hierro en tejido hepático (requiere biopsia del órgano)
· Valores normales : 0,4-2,2mg/g de tejido seco · Es exacto. · Es invasivo. · De costo alto. · No está disponible en Venezuela. · La terapia debe iniciarse cuando el hierro alcance niveles de 3mg/g de tejido
seco.
B) Resonancia magnética. Permite estimar el hierro tisular. Ha reempla-zado la biopsia hepática como método de determinación del hierro en di-cho órgano en pacientes con sobrecarga de hierro secundaria a transfusión crónica. El impacto mayor ha sido su uso en la determinación de hierro extrahe-pático, ejemplo el hierro cardiaco a través del (T2*) y páncreas7. El T2* se expresa en milisegundos. Su valor es inversamente proporcional al hierro en el miocardio. Valores normales >20 ms. En resumen, es un método:
· Exacto. · No invasivo. · Costoso. · Requiere calibración y validación para otros órganos diferentes a hígado, cora-
zón y páncreas (específicamente riñón y gonadas). · Actualmente, es más útil para talasemias que para enfermedad drepanocítica. · Está disponible en muchas instituciones en Venezuela.
C) SQUID (Superconducting Quantum Interference Device). · Método desarrollado en 1967 por Bauman y Harris8. Mide la fuerza y dirección
de la respuesta magnética ocasionada en el tejido por la aplicación de un cam-po magnético constante9. En el mundo existen pocos equipos y están dedicados a la investigación.
• Ferritina sérica. Es un parámetro: · No invasivo. · Relativamente económico. · Disponible en Venezuela.
Para que este parámetro sea confiable se recomienda su determinación seriada, al menos tres veces en estado basal, acompañarla de hierro sérico, TIBC y saturación

196
de transferrina. Los valores de ferritina sérica superiores a 1.000ug/L y saturación de transferrina >50%, en estado estacionario son indicativos de sobrecarga de hierro.
Los estudios STOP1 y STOP2 también permitieron determinar que• Valores de ferritina >1500ng/mL se correlacionan con sobrecarga post transfu-
sional baja y concentración baja de hierro hepático.• Valores de ferritina >3.000ng/mL se correlacionan muy bien con valores de hierro
hepático >10mg/g. • Valores intermedios no tienen una correlación lineal con el hierro hepático10. • En enfermedad drepanocítica, los valores aislados de ferritina >1.000ng/mL no
deben utilizarse como parámetro de sobrecarga de hierro, como se hace en tala-semia. Una mejor variable de estimación no invasiva es la carga de hierro trans-fundido, ya que:
· En niños, una carga de hierro transfundido de >100mg/Kg se correlaciona bastante bien con niveles altos de hierro hepático y es indicación para iniciar quelación.
· En adultos, no existen estudios al respecto pero 30 U de concentrado de gló-bulos rojos equivale a una carga de hierro de 94mg/kg6.
· En una enfermedad tan fuertemente inflamatoria como es la enfermedad dre-panocítica, la ferritina no es un parámetro fiable de los depósitos de hierro por tratarse de un reactante de fase aguda.
Quelantes. Existen tres quelantes clínicamente efectivos6,11,12:• Deferoxamina (Desferal R)
· Eficaz. · Seguro. · Vía de administración, subcutánea o EV. · Vida media corta por lo que requiere ser administrado durante 8-12 horas. · Buena acción sobre hierro cardiaco y hepático. · Se elimina por la orina. · Poca adherencia por parte del paciente. · Reacciones locales. · Disponible en el país para aquellos casos que no responden al deferasirox.
• Deferiprona · Eficaz. · Seguro. · Vía de administración oral, tres veces al día. · Parece ser superior a la deferoxamina en relación al hierro cardiaco. · Se elimina por las heces. · Buena adherencia por parte del paciente. · No disponible en Venezuela.
.06

197
Tópicos Especiales
• Deferasirox (Exjade R) · Eficaz. · Seguro. · Vía de administración oral, una vez al día. · Acción similar a la deferoxamina. · Actúa sobre hierro no unido a transferrina y sobre hierro plasmático lábil. · Buena acción sobre hierro cardiaco y hepático. · Se elimina por las heces. · Buena adherencia por parte del paciente. · Disponible en el IVSS, a nivel nacional, para todos los pacientes que requieran
tratamiento por sobrecarga de hierro.
Consideraciones• La sobrecarga de hierro está relacionada con el número de transfusiones.• El resultado de la terapia quelante depende de la cantidad de hierro transfundido.• La severidad de la sobrecarga de hierro y la continuidad en las transfusiones han
de tomarse en cuenta para definir las dosis terapéuticas de quelante.• La toxicidad es inversamente proporcional a la sobrecarga. No administrar que-
lante si la ferritina es <500ng/mL.• El objetivo no es eliminar la sobrecarga, sino mantener niveles de hierro que no
ocasionen daño orgánico.
Criterios para iniciar tratamiento quelanteBasados en datos disponibles6 (extrapolados de otras enfermedades), en pacientes con enfermedad drepanocítica la terapia quelante se establece cuando:
• Pacientes adultos hayan recibido 20-30 unidades de concentrado globular.• Pacientes pediátricos cuando la carga de hierro transfusional sea de >100mg/kg
y/o la concentración de hierro hepático exceda de 7-9mg/g.
Cuando la ferritina sea >3000 ng/mL es muy probable que la concentración de hierro hepático esté alto.
Otro criterio puede ser el siguiente:Cuando en estado estacionario la cifra de ferritina sea ≥1000ng/mL con saturación de transferrina >50% y/o el hierro hepático ≥3,2mg/g o si la cantidad de sangre transfun-dida supera los 120mL de eritrocitos/kg o el paciente ha recibido más de una transfu-sión mensual por año1.
Deferoxamina13.• Antes de iniciar tratamiento se recomienda investigar valores basales de:
· Ferritina, hierro sérico, TIBC y % saturación de transferrina. · Audiometría.

198
· Visión. · Función cardiaca. · Función hepática. · Función renal. · Se recomienda evaluación por crecimiento y desarrollo (niños).
• Dosis de inicio. · Niños a partir de 2 años. 20mg/kg/día, vía subcutánea, mediante bomba de infu-
sión, usando un scalp con aguja 25G, durante 10 o 12 horas. Adaptar el número de días a la semana, a los niveles de ferritina.
· Mayores de 10 años. 30-50mg/kg/día, vía subcutánea, mediante bomba de infu-sión, durante 10 ó 12 horas (5-7 días/semana).
· Cada 500 mg de deferoxamina se disuelve en mínimo 2mL de agua libre de pre-servativo. El DesferalR se almacena a temperatura ambiente. Una vez reconsti-tuido no debe guardarse por más de una semana.
· Administrarse en los sitios donde haya más grasa y rotar el sitio de aplicación cada día.
• Efectos adversos: Induración y eritema en el sitio de aplicación, reacciones alér-
gicas, efecto vagal si se administra muy rápido, neumonitis intersticial, tinitus, disminución de la audición, insuficiencia renal, displasia ósea, osteopenia y os-teoporosis cuando se inicia antes de los tres años.
• Controles · Cada 3-6 meses:
· Peso, talla. · Ferritina, ALT. · Investigar aceptación y adherencia por parte del paciente y familiares.
· Anualmente: · Oftalmología. Antes si aparecen síntomas. · Audiología. Antes si hay tinitus o disminución de la audición. · Considerar ECG y ecocardiograma. · Considerar proteinuria en orina de 24 horas. · Rx columna y metafiseal. · Otros según lo indicado en la sección de evaluación de niños, adolescentes y
adultos.
Deferasirox14
• Antes de iniciar tratamiento: · Ferritina, hierro sérico, TIBC y % saturación de transferrina. · ALT. · Creatinina.
.06

199
Tópicos Especiales
• Consideraciones generales · Presentación en tabletas dispersables · Administrar una vez al día con el estómago vacío, 30 minutos antes de las comi-
das preferiblemente a la misma hora todos los días. · Disolver en agua o jugo de naranja (100-200mL), agitar hasta obtener suspen-
sión fina. · No masticar las tabletas ni tragarlas enteras. · NO COMBINAR CON OTROS QUELANTES, hasta que se demuestre la seguridad
de tal asociación.
• Dosis de inicio · Niños a partir de 2 años: 20mg/kg/día. · Mayores de 10 años. 20-30mg/kg peso día en una sola toma. Con aumento gra-
dual si fuese necesario hasta un máximo de 40mg/kg/día.
• Efectos adversos12
· En general es tolerado. · Trastornos gastrointestinales durante los 10 primeros días.
· Nauseas en el 14,6% de los pacientes. · Diarrea en el 10,8% de los pacientes.
· Proteinuria y aumento de creatinina (33% sobre valor basal), en el 36%, se nor-maliza al suspender el medicamento. No existen evidencias que los pacientes con enfermedad drepanocítica presenten aumento de la proteinuria por el uso de deferasirox1.
· Rash cutáneo en el 7% de los pacientes. · Aumento de enzimas hepáticas en el 2% de los casos.
• Exámenes de control · Cada 15 días los dos primeros meses
· Creatinina. · Depuración de creatinina.
• Posteriormente cada mes · ALT y AST. · Creatinina. · Proteinuria.
• Cada tres meses · Ferritina y saturación de transferrina.
• Deferiprona1. Antes de iniciar el tratamiento, establecer valores basales de · Leucocitos y neutrófilos. · Ferritina, hierro sérico, TIBC y % saturación de transferrina. · ALT. · Creatinina.

200
• Consideraciones generales · Fue el primer quelante de uso oral. · Parece ser superior a la deferoxamina en la remoción del hierro cardiaco. · Los estudios publicados en enfermedad drepanocítica son muy limitados.
• Dosis · 75-100mg/kg/día, dividido en tres dosis diarias.
• Efectos adversos11
· En general es bien tolerado. · Trastornos gastrointestinales. · Dolores articulares. · Neutropenia. · Agranulocitosis en el 5% de los casos. · Elevación de enzimas hepáticas.
• Controles · Cada 15 días
· Leucocitos y neutrófilos. · Cada 3 meses
· Ferritina, hierro sérico, TIBC y % saturación de transferrina. · ALT, AST. · Creatinina.
Observación. Se requieren de estudios clínicos aleatorios y controlados adicionales para obtener mayor número de evidencias sobre seguridad, eficacia y relación costo beneficio del uso de quelantes en pacientes con enfermedad drepanocítica.
Referencias 1. Walter PB, Harmatz P, Vichinsky E. Iron Metabolism and Iron Chelation in Sickle Cell Disease. Acta Haematol 2009;122:174–183.2. Russell MO, Goldberg HI, Hodson A. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood 1984;63:162–169.3. Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatrics 1995;126:896–899.4. Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med 2005;353:2769–2778.5. Miller ST, Wright E, Abboud M, Berman B, Files B, Scher CD, Styles L, Adams RJ et al; STOP Investi-gators. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatrics 2001;139:785–89.6. Raghupathy R, Manwani D, Little J. Iron overload in sickle cell Disease. Adv Hematol 2010;2010:272940. doi: 10.1155/2010/272940.
.06

201
Tópicos Especiales
7. Wood JC. Impact of Iron Assessment by MRI. Strategies for optimal management in thalassemia—now and in the future. Hematology Am Soc Hematol Educ Program 2011;2011:443-50.8. Bauman JH, Harris JW. Estimation of hepatic iron stores by in-vivo measurements of magnetic suscep-tibility. J Lab Clin Med 1967;70:246-257. 9. Brittenham GM, Farrel DE, Harris W, Feldman ES, Danish EH, Muir WA et al. Magnetic susceptibility measurement of human iron stores. N Engl J Med 1982;307:1671-1675.10. Adamkiewicz TV, Abboud MR, Paley C, Olivieri N, Kirby-Allen M, Vichinsky E et al. Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood 2009;114:4632–4638.11. Kwiatkowski JL. Real-World Use of Iron Chelators. Hematology Am Soc Hematol Educ Program. 2011;2011:451-8. 12. Vichinsky E, Onyekwere O, Porter J, Swerdlow P , Eckman J, Lane P et al. A randomized comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol 2007;136:501–50813. Gee.B.Specific Problems: Iron Overload and Chelation in Therapy in Sickle Cell Information Center. Ed Eckmand and Platt A .2010; 1-14. Disponible en http://scinfo.org/problem-oriented-clinical-guidelines/specific-problems-iron-overload-and-chelation-therapy14. Data on file Novartis Pharma AG, Basel, Switzerland.2006.
6.6. Trasplante en enfermedad drepanocíticaFrancisco Ramírez Osío - Marcos Hernández J. / Médicos Hematólogos
La ED-SS, ED-Sβ0 talasemia y la ED-SC pueden ser enfermedades graves hasta en un 20% de los casos, llevando a disminución de la esperanza y calidad de vida por afectación crónica multiorgánica dado el daño endotelial y hemolisis sostenida. Las infecciones, el síndrome torácico agudo recurrente y la enfermedad vascular cerebral aguda son causas de mortalidad; igualmente a largo plazo, la hipertensión pulmonar, insuficiencia cardiaca e insuficiencia renal, retinopatía proliferativa y otros trastornos vasculares, son causas importantes de morbilidad.
El abordaje precoz a través del despistaje neonatal, la profilaxis de infecciones median-te las inmunizaciones y suministro de penicilina, la disponibilidad de mejores métodos diagnósticos incluyendo el eco doppler transcraneal, el tratamiento con hidroxiurea y transfusión crónica, la prevención de la sobrecarga de hierro con el uso de quelantes de hierro, la educación del paciente y de sus familiares y el mejor conocimiento de la enfermedad drepanocítica por parte del personal de salud, han mejorado grandemen-te el manejo de la misma, igual que la calidad y expectativas de vida de los pacientes.
ObjetivosRestablecer la hematopoyesis normal, detener y recuperar las lesiones vasculares y evi-tar la hemolisis controlando y mejorando con el tiempo el daño multiorgánico crónico. La

202
curación de la enfermedad con trasplante con células progenitoras hematopoyéticas (TCPH) en pacientes bien seleccionados, tomando en cuenta los criterios de inclusión y exclusión, usando como donante un hermano HLA idéntico, portador o no de rasgo drepanocítico, ha sido bien demostrada, reportándose una sobrevida, mayor de 85%, que justifica plenamente la indicación de este procedimiento a pesar del 15% de riesgo que incluye enfermedad injerto contra huésped, rechazo y otras complicaciones rela-cionadas al trasplante1.
Se han utilizado otros tipos de trasplantes para ampliar la obtención de donantes usan-do células de cordón umbilical y donantes voluntarios de los registros internacionales. También se investiga con regímenes menos intensos para disminuir la toxicidad y lo-grar un quimerismo mixto conveniente, principalmente en adultos, todos estos repor-tes con experiencia limitada2.
Experiencia mundial con TCPH en enfermedad drepanocíticaEn 1980 en el Hospital St. Jude se realizó el primer trasplante en enfermedad drepa-nocítica en una niña de 8 años que también tenía leucemia2. En 1988, Vermylen reporta la primera serie de pacientes trasplantados3.
Durante lo siguientes años, grupos europeos y americanos han demostrado una so-brevida global de 88% a 93% y sobrevida libre de enfermedad de 80% a 84%, en pa-cientes seleccionados y recibiendo el trasplante de un hermano HLA idéntico, usando regímenes mieloablativos con busulfán y ciclofosfamida, añadiendo además globuli-na antitimocito para disminuir el rechazo tardío. Actualmente, a nivel mundial se han trasplantado más de 400 pacientes4-7. Estas experiencias hacen que se plantee la indi-cación del trasplante temprano, con mayor aplicación en niños muy sintomáticos y her-mano compatible no afectado, para evitar la aparición de complicaciones relacionadas con la enfermedad.
Experiencia en VenezuelaHasta la fecha se han realizado cuatro trasplantes, el primero en Valencia en el año 2000 y tres mas en Caracas, siguiendo todos las recomendaciones internacionales6, 8 (en cuanto a los criterios de inclusión y exclusión, evaluaciones pre trasplante y los regímenes de acondicionamientos (ver mas adelante). El acondicionamiento consistió en busulfán vía endovenosa, 16 mg-kg los días -10 a -7, ciclofosfamida 200mg los días -5 a -2, globulina antitimocito equina 90mg-kg los días -6 a -4, con la finalidad de evitar rechazo tardío; metilprednisolona 1mg-kg desde el día -7, para evitar enfermedad de suero. Previamente se practicó exanguino transfusión para lograr nivel de hemog-lobina S menor a 30%, luego terapia transfusional para mantener nivel de hemoglo-bina mayor de 9g/dL y plaquetas por aféresis para mantener su cifra por encima de 50 x 109/L. Para evitar convulsiones, se indica clonazepan antes de comenzar busulfán y se mantiene hasta suspender ciclosporina. Se practica inmunoprofilaxis con ciclos-
.06

203
Tópicos Especiales
porina desde el día -1 (pre-trasplante) hasta 8 a 12 meses postrasplante, metotrexate 4 dosis, los días +1, +3, +6,+11 postrasplante (Figura 6.4).
Figura 6.4. Régimen de acondicionamiento en enfermedad drepanocítica para TCPH alogénico familiar6,8
Busulfán 1mg/kg/dosis cada 6h dias -10 a -7
-10 -9 -8 -7
Ciclofosfamida 50 mg/kg días -5 a -2
-5 -4 -3 -2
Globulina antitimocito conejo 5 mg/kg días -6 a -3 equina 30 mg-kg 3 días
-6 -5 -4 -3
MetrotexateD+1 (15 mg/m2) y días +3, 6, y 11 (10 mg/m2)
+1 +3 +6 +11
Ciclosporina día -1 hasta 8 a 12 meses
INFUSIÓN CPH

204
En la actualidad todos los pacientes están vivos, sin enfermedad drepanocítica, ni en-fermedad injerto contra huésped con más de 2 años de seguimiento.
Criterios de inclusión para trasplante• Edad menor de 16 años.• Tipo de enfermedad drepanocítica: ED-SS, ED-SC y ED-Sβ°.
Antecedentes de• Enfermedad vascular cerebral aguda • Eco doppler transcraneal reportado como de alto flujo de manera persistente.• Síndrome torácico agudo recurrente.• Más de 3 crisis vasooclusivas que requieran hospitalización, en un año.• Priapismo recurrente.• Enfermedad pulmonar o fibrosis I y II.• Nefropatía. Filtración glomerular con compromiso 30-50% de lo normal.• Osteonecrosis múltiple.• Aloinmunización con más de 2 anticuerpos. • Sin respuesta a hidroxiurea.
Criterios de exclusión de trasplante• Karnosky menor de 70%.• Enfermedad hepática significativa, con fibrosis.• Enfermedad pulmonar con hipertensión o fibrosis III y IV.• Nefropatía con filtración glomerular menor a 30%.• Enfermedad neurológica severa.
Otras fuentes de células progenitoras hematopoyéticas (CPH) y regímenes para trasplante.Trasplante utilizando células de cordón umbilical familiar9
• Recuperación de neutrófilos día 23 (12-60), plaquetas día 39 (19-22). • Enfermedad injerto contra huésped (EICH):
· Aguda grado II 4/38 (10%). · Crónica limitada 2/36.
• Probabilidad de EICH crónica: 6%.• 90% de sobrevida.• Se puede justificar la recolección de sangre deñ cordón umbilical y placenta de
madres con hijos que padecen enfermedad drepanocítica.
Trasplante de cordón umbilical no familiar2
• Poca experiencia reportada, por dificultad de encontrar cordón compatible.• Alta incidencia de EICH, la compatibilidad HLA 6-6 es difícil de obtener.• El porcentaje de rechazo del injerto es mayor y ocurre al suspender la inmunosupresión.
.06

205
Tópicos Especiales
• Mayor riesgo de infección.
Regímenes de intensidad reducida en enfermedad drepanocítica2.• Menor mortalidad relacionada al trasplante.• 7 pacientes: 6 ED-SS, 1 Talasemia.• Régimen de acondicionamiento: fludarabina, irradiación corporal total (ICT) con
ATG o aletuzumab.• Investigación en adultos para lograr quimerismo estable con formación de he-
moglobina A9. • Inmunoprofilaxis: tacrolimus/micofenolato (MMF) /sirolimus o CsA / MMF.• De 7 pacientes analizados 6 hicieron buen injerto de las células del donante, evi-
denciado por 25% - 85% de quimerismo.• No hubo complicaciones de enfermedad drepanocítica, mientras se mantuvo qui-
merismo con células de donante.• Al finalizar la inmunosupresión hay alto riesgo de perder injerto.
Estudios recomendados al paciente y a familiares para selección de donantes. Paciente
• Informe clínico detallado: señalando evolución y criterios de inclusión.• Histocompatibilidad.• Serológicos: con menos de 30 días de practicados
· VIH. · CMV. · HTLV I-II. · Herpes simple. · VDRL. · Chaga.s · Títulos de toxoplasma.
• Hematológicos · Hematología incluyendo reticulocitos y plaquetas. · Pruebas generales de coagulación. · Investigación de hemoglobinopatías.
• Banco de sangre · Grupo sanguíneo: ABO y RH. · Investigación de anticuerpos irregulares. · Selección de donantes para aféresis.
• Perfil bioquímico.• Coproanálisis.• Uroanálisis.
Evaluación por siguientes especialidades• Cardiología

206
· Electrocardiograma. · Ecocardiograma.
Neumonología.• Función pulmonar.• Radiología.
Otorrinolaringología.• TAC de senos nasales. • Paranasales.
Odontología• Tratamientos de caries e infecciones.
Psiquiatría.
Dermatología.
Endocrinología• Edad ósea (niños). • Cuantificación de hormonas.
Orientación en consulta de fertilidad.
Neurología. Eco Doppler trascraneal.
Donante• Histocompatibilidad.• Serológicos
· VIH. · Hepatitis B y C. · CMV. · HTLV I-II. · Herpes simple. · VDRL. · Chagas. · Títulos de toxoplasma.
Hematológicos• Hematología incluyendo reticulocitos y plaquetas. • Pruebas generales de coagulación.
.06

207
Tópicos Especiales
· Hemoglobinopatías.
Banco de sangre • Sistema ABO y RH. • Anticuerpos irregulares.
Perfil bioquímico.• Evaluación por cardiología
· Electrocardiograma · Ecocardiograma · Radiografía de tórax.
Conclusiones• El TCPH alogénico familiar ofrece cura para la enfermedad drepanocítica.• Detiene el progreso de daño multiorgánico.• Algunas alteraciones severas pueden no ser reversibles.• El TCPH seleccionado es asociado a una morbilidad y mortalidad razonable me-
nor de 20%. En niños con enfermedad drepanocítica severa, siempre investigar la posibilidad de un hermano HLA idéntico, portador o no de Hb S.
• Para otros tipos de trasplante, debe esperarse mayor evidencia para su uso.
Referencias 1- Angelucci E, Baronciani D. Haemoglobinopathies HSCT for children and adolescents, chapter 20.9, The EBMT Handbook, 6th edition 2012.2- Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic Hematopoietic stem cell transplantation for sickle cell disease: the time is now. Blood 2011; 118: 1197-12073- Bernaudin F, Souillet G, Vannier JP, Michel G, Lutz P et al for the SFGM. Sickle cell disease (SCD) and BMT: report of the French experience concerning 26 children transplanted for severe SCD. Bone Marrow Transplant 1997; 19(suppl 2):112-115.4- Vermylen C, Cornu G, Ferster A, Brichard B , Ninane J, Ferrant A, et al. Haematopoietic stem cell trans-plantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998; 22:1-6.5- Walters MC, Storb R, Patience M, Leisenring W, Taylor T, Sanders JE, et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood 2000; 95:1918-1924.6- Bernaudin F, Socié G, Kuentz M, Chevret S, Duval M, Bertrand Y. Long-term results of related myeloa-blative stem-cell transplantation to cure sickle cell disease. Blood 2007; 110: 2749-2756.7- Pieroni F, Barros GMN, Voltarelli JC, Simoes BP. Hematopoietic stem cell transplantation in sickle cell anemia. Rev Bras Hematol Hemoter 2007;29: 327-330.8- Gluckman E, Bernaudin F. Hematopoietic stem cell transplantation, in patients with sickle cell disease. Disorders of iron homeostasis, erythorcytes, erythropoiesis. ESH European School of Haematology. The Handbook, 2006 Editions, Chapter 14, page 325.9- Locatelli F, Rocha V, Reed W, Bernaudin F, Ertem M, Grafakos S.Related umbilical cord blood trans-plantation in patients with thalassemia and sickle cell disease. Blood.2003;101:2137-2143.10- Walters MC, Patience M, Leisering W, Taylor T, Sanders JE et al. Bone marrow transplantation for sickle cell disease. N Engl J Med 1996; 335:369-376.

208
7.1. Inducción de hemoglobina fetal Olimpia C. Pérez-Bández - Leslie González / Médicos hematólogos.
Años atrás el descubrimiento de la base molecular de la enfermedad drepanocítica1, representó un punto clave para la medicina molecular; hoy, las herramientas de la biología celular y molecular han aportado un cúmulo de conocimientos que permiten entender mejor la fisiopatología de la enfermedad drepanocítica y establecer nuevas modalidades terapéuticas. La vasooclusión, evento cardinal de la enfermedad dre-panocítica, es consecuencia de la combinación de anormalidades en la estructura y función de la hemoglobina, integridad de la membrana del eritrocito, densidad de los mismos, activación endotelial, tono microvascular, mediadores inflamatorios, factores de coagulación; por tanto, las estrategias farmacológicas se enfocan en modificar o corregir tales anormalidades:
a. Inducción de la producción de hemoglobina fetal.b. Prevención de la deshidratación celular.c. Vasodilatadores.d. Contrarrestar la inflamación y proteger al endotelio.e. Anticoagulantes y agentes antiplaquetarios.f. Otras drogas.
a.- Inducción de la producción de hemoglobina fetal (Hb F). Esta forma de hemoglobina es un potente inhibidor de la polimerización de la deoxihemoglobina S, debido a que ni la Hb F (α2γ2) ni el tetrámero híbrido (α2γβS son incorporados a la fase de polímeros2. La historia natural de la enfermedad drepanocítica, reportada por el Cooperative Study of Sickle Cell Disease (CSSCD), de Estados Unidos, demuestra una correlación inversa entre concentración de Hb F y frecuencia de crisis de dolor, sindrome torácico agudo y mortalidad3-5. El objetivo de los tratamientos con agentes inductores de Hb F, es lograr una concentración de Hb F dentro de cada eritrocito, capaz de inhibir o retardar la po-limerización de la deoxihemoglobina S.
Los agentes inductores de Hb F actúan mediante diferentes mecanismos6.• Inhibición de la ribunucleótido reductasa.• Hipometilación del ADN.• Inhibición de la histona deacetilada.
Inhibición de la ribonucleótido reductasa.• Hidrocarbamida o hidroxiurea.
En la actualidad es la única droga aprobada por US Food and Drug Administration (FDA),
Estrategias farmacológicas en el tratamiento de la enfemedad drepanocítica..07

209
para el tratamiento en adultos con anemia drepanocítica severa (1998).
La hidroxiurea fue sintetizada en 1869, en Alemania, por Dresler. Hace cerca de 50 años fue desarrollada como droga antineoplásica7 y en la enfermedad drepanocítica fue uti-lizada por primera vez en 1984. Charache y colaboradores8 demostraron su eficacia en reducir la morbilidad en adultos con ED-SS. Los estudios iniciales mostraron aumento de eritrocitos que contenían hemoglobina fetal. A mediados de los años 90, más de 300 pacientes con enfermedad drepanocítica que tuvieron más de tres episodios de dolor severo en el curso de un año, se incluyeron en un estudio aleatorio (hidroxiurea o placebo), el cual fue detenido prematuramente porque se observó claramente que la hidroxiurea disminuyó el número e intensidad de los episodios de dolor severo9.
Mecanismo de acción. La hidroxiurea pertenece a la clase de compuestos denomina-dos ácidos hidroxámicos, compuestos con capacidad de unir metales. El efecto citotó-xico primario es debido a su capacidad de inhibir la ribonucleótido reductasa, al unir los dos hierro de la reductasa e inhibir el radical tyrosil que es un sitio crítico10. Este efecto citotóxico disminuye la producción de células que contienen cantidades altas de Hb S (las cuales tienen una gran capacidad de división) y favorece la producción de células con alta concentración de Hb F que surgen de progenitores que se dividen menos. Esta droga también desciende el número de leucocitos y plaquetas reduciendo potencialmente el papel que juegan estas células en la producción del daño vascular11. Otro efecto importante es que el metabolismo de la hidroxiurea conduce a la produc-ción de óxido nítrico el cual estimula a la guanilato ciclasa aumentando la síntesis de hemoglobina fetal y por otra parte, compensa la menor producción de este gas como consecuencia de la hemólisis intravascular12. La hidroxiurea disminuye la adhesivi-dad de los eritrocitos portadores de Hb S reduciendo la expresión de receptores de adhesión como CD36+ y VCAM-1, la expresión de fosfatidil serina en la membrana de eritrocitos y plaquetas, y la adhesión a proteínas de la matriz extracelular como lami-nina y trombospondina. Sin embargo, los mecanismos moleculares por los cuales la hidroxiurea ejerce estos efectos no han sido esclarecidos13.
En relación a la hidroxiurea el consenso realizado por el National Institutes of Health de los Estados Unidos en el año 2008, concluyó14:
• La eficacia de la hidroxiurea, definida como el efecto terapéutico en un grupo con-trolado, varía con el genotipo (ED-SS, ED-Sβ0talasemia, ED-Sβ+talasemia, ED-SC, ED-SD, ED-SOarab), con el haplotipo y con factores demográficos (edad y sexo).
• La evidencia es fuerte en adultos pero es débil en niños, ya que el único estudio aleatorio realizado en éstos tiene un diseño deficiente, pocos casos y corta dura-ción.
• La data de la efectividad de la hidroxiurea, definida como el efecto terapéutico en la práctica diaria, es limitada; pero la experiencia de tantos clínicos, sugiere que la droga es altamente efectiva en la práctica generalizada pero está subutilizada;

210
solamente un pequeño número de pacientes que pueden beneficiarse de la hi-droxiurea lo están haciendo.
• El efecto de la hidroxiurea a corto y largo plazo. · Corto plazo (definido como los primeros 6 meses).
· Leucopenia. · Trombocitopenia. · Anemia. · Descenso en el número de reticulocitos. · Disminución reversible de la producción de esperma. No existen reportes de
malformaciones en hijos(as) de hombres que reciben hidroxiurea. · Sequedad de la piel (corto y largo plazo). · Pigmentación de las uñas. · Menor riesgo de úlceras en las piernas determinado por un estudio aleatorio.
· Largo plazo (definido por más de 6 meses). Los efectos potenciales son: · Defectos congénitos en hijos de padres (hombres y mujeres) que reciben hidroxiu-
rea. · Alteraciones en crecimiento de niños que reciben la droga. · Cáncer. · Los efectos a largo plazo pueden ser permanentes e irreversibles, pero hasta
ahora no se han descrito. · Los efectos sobre el feto aun permanecen por determinar, sin embargo no
parece haber aumento de la aparición de malformaciones en niños de madres que tomaron hidroxiurea durante el embarazo.
· Niños entre 5 y 15 años que han recibido y continúan con hidroxiurea, muestran un crecimiento igual a aquellos niños con enfermedad drepanocítica sin haberla recibido.
· Se han descrito muy pocos casos de leucemia y otros tipo de cáncer en pacien-tes con enfermedad drepanocítica tratados con hidroxiurea, pero no parecen ser más comunes que en la población general; por otra parte no hay diferencia entre pacientes con enfermedad drepanocítica que la reciben o no.
Deben recibir hidroxiurea9 Adultos, adolescentes y niños (los dos últimos, previo consentimiento de los padres), con los siguientes genotipos:
• Anemia drepanocítica (Hb SS).• ED-S β0 talasemia. • Que hayan presentado los siguientes eventos en los años anteriores:
· Tres o más crisis de dolor que requirieron hospitalización. · Sindrome torácico agudo. · Enfermedad cerebro vascular aguda. · Necrosis de fémur o húmero. · Anemia severa sintomática.
.07

211
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
• No administrar hidroxiurea en las siguientes condiciones9
· Embarazo. · Alergia a la droga. · Trombocitopenia. · Neutropenia.
• Realizar los siguientes estudios previos al tratamiento9
· Hematología. · Índices hematimétricos. · Determinación del porcentaje de Hb F. · Pruebas hepáticas. · Creatinina. · Uroanálisis. · Prueba de embarazo.
La disfunción renal no es contraindicación para el tratamiento con hidroxiurea, pero si influye en la dosis.Esquema de tratamiento9
• Iniciar con 15 mg/kg/día. Una sola dosis. Durante 6-8 semanas. · Si la depuración de creatinina es <60 mL/minuto (1mL por segundo), iniciar con
7,5mg/kg/día.• Después de comenzar el tratamiento, a las dos semanas practicar hematología
que incluya plaquetas y reticulocitos. Para este momento debe haber: · Disminución del número de leucocitos. · Disminución de plaquetas. · Aumento del volumen corpuscular medio. · Si estos cambios no han sucedido, es necesario revisar:
· Dosis. · Marca de hidroxiurea que el paciente está usando. · Esquema de dosis y la adherencia al tratamiento.
• Practicar hematología cada dos semanas con la finalidad de ajustar la dosis y establecer la dosis óptima para el paciente. (Tabla 7.1)
• El objetivo es administrar la dosis más alta sin que haya signos de mielotoxicidad.• Si no hay signos de toxicidad, aumentar 5 mg/kg/día cada 6-8 semanas, hasta
alcanzar la dosis necesaria para lograr el efecto deseado.• Dosis máxima: 35 mg/kg/día.

212
Tabla 7.1 Esquema de estudios para clínicos, sugeridos en adultos con enfermedad drepanocítica.
Efectos deseados9
• Menos dolor.• Aumento de Hb F hasta 15%-20%.• Incremento de cifras de hemoglobina (en caso de anemia severa).• Sensación de bienestar.• Mielotoxicidad aceptable.
· Granulocitos >2,0 x 109/L. · Plaquetas >80 x 109/L.
• Suspender el tratamiento, si no se obtiene respuesta después de 6 meses de
Frecuencia Estudios
Cada 2-6 meses
• Hematología que incluya: hemoglobina, hematocrito, reticuloci-tos, índices hematimétricos, leucocitos y fórmula leucocitaria y plaquetas.
• Uroanálisis.• Saturación de oxígeno, transcutánea.
Cada 3 meses
• Cuantificación de Hb F1
• Cuantificación de Hb S2 • Serología HVB3, HVC3, HIV3
• Hierro sérico4, TIBC4, saturación de transferrina4, ferritina4 y T2*4
• ALT4, y AST4 • Glicemia4
• BUN4, Creatinina4
• Hormonas tiroideas4 • Estudio de paratiroides4
Cada año
• ALT, AST, bilirrubina total y fraccionada. HDL• Glicemia• Función renal (creatinina, BUN, proteínas en orina de 24 horas)• Ultrasonido abdominal• Espirometría. Si tiene antecedentes de sindrome de tórax agudo
1. Durante el primer año de tratamiento con hidroxiurea.2. 24-48 horas después de la transfusión sí el paciente está en régimen de transfusión crónica.3. Antes de la transfusión sí el paciente está en régimen de transfusión crónica.4. Si el paciente recibe tratamiento con deferasirox.
.07

213
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
recibir el medicamento.
Realizar control de:9
• Cuenta de leucocitos cada 2 semanas. • Hb F cada 3-4 meses.• Pruebas hepáticas y renales mensualmente los primeros 4 meses, con el propó-
sito de detectar si hay idiosincrasia a la hidroxiurea. • Si a los 6 meses no se aprecia mejoría, suspender.• Generalmente los pacientes respondedores, lo hacen en un período menor de 6
meses.• Después de lograr dosis ideal, practicar hematología mensualmente durante 4
meses, luego cada 3 meses durante un año y posteriormente cada 3-6 meses los años siguientes.
• Por lo general a los 6 meses la Hb F se ha duplicado, los niveles de Hb aumentan 1g/dL, hay disminución de valores absolutos de reticulocitos, bilirrubina y deshi-drogenasa láctica15.
Toxicidad6
• Neutrófilos <2 x 109/L.• Plaquetas <80 x 109/L.• Reticulocitos absolutos <80 x 109/L, si hemoglobina es <9,0 g/dL.• Creatinina sérica >1,0mg/dL o 50% por encima del valor basal.• Incremento en ALT del 100%.
Si alguno de los valores hematológicos disminuye a niveles considerados como índice de mielotoxicidad
• Suspender el tratamiento mínimo durante una semana o hasta que pase la toxi-cidad.
• Luego reiniciar hidroxiurea a la misma dosis o disminuir 2,5-5mg/kg en relación a la dosis que recibía el paciente.
• Después de 12 semanas de reiniciada la hidroxiurea, si la toxicidad no reaparece, dejar igual dosis o aumentar 2,5-5mg/kg según la conducta que se haya tomada al momento de reiniciarla.
• Si reaparece toxicidad, suspende r de nuevo hasta que desaparezca y restablecer a una dosis menor.
Precauciones9
• Se debe indicar método anticonceptivo en hombre y mujeres, ya que la hidroxiurea es teratogénica en ratas y se desconoce su efecto durante el embarazo humano.
• Daño renal o hepático, iniciar con la mitad de la dosis.• Valores iniciales de Hb <5,5g/dL, no contraindican el tratamiento.

214
Efectos colaterales9
• Eritema, ocasionalmente.• Ulceras maleolares.• Híperpigmentación o atrofia de piel y uñas, muy rara vez.• Cambios malignos en piel (infrecuente).
Fallas en el aumento de Hb F o de VCM9
• Incapacidad biológica para responder al tratamiento · Reserva medular disminuida antes de iniciar el tratamiento. · Factores genéticos.
• No adherencia al mismo (la mayoría de los casos) · Efectos adversos no deseables. · Dificultad en el control.
Si hay respuesta clínica dejar igual dosis.Si no hay respuesta clínica, aumentar la dosis diaria a 2.000 o 2.500 mg (35 mg/Kg/d) y seguir muy de cerca. Los pacientes que responden a la hidroxiurea, la pueden recibir indefinidamente a menos que haya efectos adversos9. Los pacientes que fueron enro-lados en el primer protocolo con hidroxiurea, tienen más de 25 años bajo tratamiento sin presentar efectos adversos.
Uso de hidroxiurea en niños. Su empleo no ha sido aprobado por US Food and Drug Ad-ministration (FDA, sin embargo se han publicado pocos estudios cuyos resultados ya fueron comentados. Durante el Hydroxyurea Safety and Organ Toxicity (HUSOFT trial)16, se realizó la primera preparación de hidroxiurea oral en un jarabe saborizado a una concentración de 100 mg/cc con reposiciones cada 4 semanas durante el tiempo del estudio. Esta solución fue preparada basada en la receta del St. Jude Children Hospi-tal para el tratamiento de neuroblastoma encontrándose una estabilidad de 1 mes a temperatura ambiente y de 3 meses en refrigeración. Para el estudio BABY-HUG trial17 se tomó la misma receta publicada en marzo de 2004 por Mathew M. Heeney18, expo-niendo el uso de hidroxiurea como formulación oral, bajo preparación extemporánea a una concentración de 100mg/mL a partir de su presentación original de cápsula de 500mg usando agua estéril y Syrpalta® (vehículo saborizado que consiste en: sucrosa, agua estéril, saborizante, glicerina, benzoato de sodio y cloruro de benzalconium) de-mostrando ser químicamente estable. En el hospital Universitario de Caracas se utiliza desde el año 2007 con buenos resultados19.
Otros fármacos5-azacitidinaFue el primer fármaco utilizado en ratones con efecto inductor de la producción de hemoglobina fetal, sin embargo, los efectos cancerígenos secundarios limitaron su desarrollo clínico. El mecanismo de acción es la hipometilación del ADN que forma el
.07

215
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
gen responsable de la síntesis de las cadenas γ de la hemoglobina fetal20,21.
Decitabina La 5-aza-2’-deoxicitidina es un análogo de la 5-azacitidina, con similar efecto inductor de la produc-ción de hemoglobina F, pero mejor perfil de seguridad porque a diferencia de la 5-azacitidina, no se incorpora al ARN de la célula sino al ADN por lo que no afecta la síntesis de proteínas. Después de integrarse al ADN se une covalentemente a la DNA metiltransferasa (DNMT) provocando disminución de la enzima y consecuente hipo-metilación del ADN22. Se han realizado pocos estudios de corta duración que incluyen pacientes con enfermedad drepanocítica que no han respondido a la hidroxiurea, los resultados han sido satisfactorios. El único efecto colateral observado fue leucopenia de discreta a moderada de pocos días de duración. Sin duda, la decitabina ofrece una esperanza en el tratamiento de los pacientes con enfermedad drepanocítica que no re-accionan a la hidroxiurea, pero son necesarios más estudios para determinar la dosis efectiva del medicamento con menos citotoxicidad23.
Butiratos Son ácidos grasos de cadena corta que inhiben la deacetilación de las histonas, pro-moviendo la expresión del gen de las cadenas gamma e incremento de la hemoglo-bina fetal. En los primeros estudios el butirato fue administrado en infusión continua durante 2-3 semanas y los resultados no fueron concluyentes; sin embargo, estudios recientes han demostrado que la administración en pulsos tiene mejores efectos por lo que son más esperanzadores.
Las investigaciones también sugieren que hay una actividad sinérgica sin resistencia cruzada con la hidroxiurea; el pretratamiento con hidroxiurea puede seleccionar una población de precursores eritroides cuyos genes de cadenas γ se encuentran activos, facilitando la respuesta al butirato. Los butiratos no parecen ser citotóxicos y su uso sigue siendo experimental. Lo difícil de su administración, necesidad de grandes dosis por vía venosa central y su vida media corta, hace poco práctico su uso24.
Eritropoyetina recombinanteEs otro fármaco que también produce aumento de hemoglobina fetal. El aumento de la viscosidad y dolor óseo se han descrito como efectos colaterales. Es necesario esperar el resultado de estudios sobre su efecto trombótico25.
Tricostatina A Usada en ratones transgénico con drepanocitosis severa. Además de inhibir el factor tisular (FT), provoca aumento de hemoglobina fetal e impide de manera importante la expresión de moléculas de adhesión celular vascular (VCAMP) en las venas pulmona-res. Reduce también la estasis vascular, sugiriendo que puede tener un mecanismo multimodal26.

216
PolimolamidaEs un inmunomodulador derivado de la talidomida, probado en ratones drepanocíticos transgénicos, aumentó la producción de hemoglobina fetal y disminuyó en un 50% la inflamación y necrosis hepática, sin modificar el número de leucocitos26. Se requiere de estudios clínicos para investigar eficacia y seguridad en humanos.
Referencias 1. Ingram, VM. Gene mutations in human hemoglobin: the chemical difference between normal and sickle hemoglobin. Nature 1957; 180:326-328. 2. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997; 337:762-769. 3. Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse, WF, Vichinsky E, Kinney TR. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11-16.4. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH et al. Mortality in sickle cell disea-se. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639-1644.5. Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette Pet et al. The acute chest syndro-me in sickle cell disease: incidence and risk factors. The cooperative study of sickle cell disease. Blood 1994;84:643-649.6. Fathallah H. Atweh GF. Induction of Fetal Hemoglobin in the Treatment of Sickle Cell Disease. Hema-tology 2006; 2006 (1): 58 – 62.7. Cancercare.org .Ontario: Cancer Care Ontario Formulary. Disponible en: http://www.cancercare.on.ca/pdfdrugs/HYDROXYU.pdf.8. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB and, Eckert SV. Multicenter Study of Hydroxyu-rea in Sickle Cell Anemia. Effect of Hydroxyurea on the frequency of painful crisis in sickle cell anemia. N Engl J Med 1995;332:1317 – 22.9. Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglo-bin production in sickle cell anemia. J Clin Invest 1984;74:652-56.10. Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol 1992;19: Suppl 9: 1-10. 11. Platt OS. Hydroxyurea for the treatment in sickle cell anemia. N Engl J Med 2008; 358:1362 – 69.12. Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest 2003; 111:231-239.13. Johnson C, Telen M. Adhesion molecules and hydroxyurea in the pathophysiology of sickle cell disea-se. Haematologica 2008; 93:481-486.14. Brawley OW, Cornelius LJ, Edwards LR, Gamble VN, Green BL, Inturrisi C et al. National Institutes of Health Consensus Development Conference Statement: Hydroxyurea Treatment for Sickle Cell Disease. Ann Inter Med 2008;148:932–38.15. Kinney TR, Helms RW, O’Branski EE, Ohene-Frempong K, Wang W, Daeschner C et al. Safety of hy-droxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Blood 1999; 94:1550-554.16. Hankins JS, Ware RS, Rogers ZR, Wynn WL, Lane PA, Scott JP, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood 2005; 106:2269-274.17. Wang W, Helms RW, Lynn HS, Redding-Lallinger R, Gee B, Ohene-Frempong K et al. Effect of Hy-
.07

217
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
droxyurea in children with sickle cell anemia: Results of the HUG-KIDS study. J Pediatr 2002;140:225-29.18. Heeney MM. Chemical and functional analysis of hydroxyurea oral solutions. J Pediatr Hematol Oncol 2004;26:179-18.19. González LD. Hidroxiurea: Evaluación Clínico Terapéutica en niños drepanocíticos de un centro asis-tencial universitario. 2009. UCV-Facultad de Medicina-Comisión de Postgrado.20. Ley TJ, DeSimone J, Noguchi CT, Turner PH, Schechter AN, Heller P and Nienhuis AW. 5-Azacytidine increases gamma-globin synthesis and reduces the proportion of dense cells in patients with sickle cell anemia. Blood 1983; 62:370-80.21. Carr BI, Rahbar S, Asmeron Y, Riggs A, Winberg CD. Carcinogenicity and haemoglobin synthesis in-duction by cytidine analogues. Br J Cancer 1988;57:395-402.22. Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythro-leukemia cell differentiation by 5-azacytidine and 5-aza- 2’-deoxycytidine. J Biol Chem 1982;257:2041-48.23. Atweh GF, DeSimone J, Saunthararajah Y, Fathallah H, Weinber RS, Nagel RL, Fabry ME, Adams RJ. Hemoglobinopathies. Hematology Am Soc Hematol Educ Program 2003;14-3924. Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest 2007;117:850–58.25. Bennett CL, Silver S.M, Djulbegovic B, Samaras AT, Blau CA, Gleason, KJ et al. Venous thromboem-bolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. JAMA 2008; 299:914–24.26. Ataga KI. Novel therapies in sickle cell disease. Hematology Am Soc Hematol Educ Program 2009;2009:54-61. Disponible en http://asheducationbook.hematologylibrary.org/content/2009/1/54.full.pdf+html
7.2. Otras estrategias farmacológicas Clementina Landolfi / Médico hematólogo.
Además de las drogas dirigidas a la inducción de la producción de hemoglobina fetal, existen otras tantas que aún están en diversas etapas de investigación, con la finalidad de disminuir, bloquear o estimular, según sea el caso, los diferentes procesos bioquí-micos moleculares que se encuentran alterados en la enfermedad drepanocítica.
Prevención de la deshidratación celular La polimerización de la Hb S está relacionada de manera exponencial a la concentra-ción intracelular de hemoglobina, por tanto el estado de hidratación es crítico para la polimerización. Existen varias vías celulares que regulan el paso de iones y agua: los canales Gardos (canales de K+, losdependientes de Ca++), los canales de transporte K y Cl y bomba de Na+. La inhibición de cualquiera de estos canales previene la deshidra-tación celular y produce un beneficio clínico1.
• El magnesio, puede reducir la actividad de cotransporte Cl/K con la consecuente disminución de2,3: · Concentración de hemoglobina corpuscular media (CHCM).

218
· Densidad de los eritrocitos. · Número de reticulocitos. Su uso oral en adultos (0,6mEq/kg/día) parece ser
beneficioso en prevenir las crisis vasooclusivas; también el sulfato de magne-sio vía endovenosa, reduce la estancia hospitalaria debido a ellas.
El efecto colateral mas frecuente es dolor abdominal y diarrea, sin embargo es tole-rado.
La fase I de un estudio en niños con ED-SS, combinando pidolato de magnesio con hidroxiurea, demostró reducción significativa de la actividad del cotransporte K-Cl, siendo el dolor abdominal y diarrea los efectos adversos más reportados4.
• El sulfato de zinc también puede prevenir la deshidratación celular.
Clotrimazol5
• Es un potente bloqueador de los canales Gardos.• Previene la deshidratación.• Induce disminución de:• CHCM.• Densidad de los eritrocitos.• Número de reticulocitos.• Marcadores bioquímicos de hemólisis (deshidrogenasa láctica, DHL, bilirrubina
indirecta)• No previene los episodios dolorosos.
Efectos colaterales• Nauseas.• Vómitos.• Aumento de los niveles de enzimas hepáticas.
Dipiridamol6
• Inhibe el flujo de cationes inducido por la desoxigenación. • Poco tóxico. • Potencialmente útil en enfermedad drepanocítica.
VasodilatadoresLa vasooclusión es el síntoma cardinal de la enfermedad drepanocítica. El óxido nítrico (ON) es el vasodilatador natural más potente que se forma a partir de la L-arginina por acción de la ON sintetasa. El ON media la relajación vascular vía formación de guano-sina monofosfato cíclico (cGMP), la cual señala el secuestro intracelular de Ca++ y la vasodilatación7.
La hemólisis intravascular que sucede en la enfermedad drepanocítica, inactiva rápi-damente al ON vía destrucción por la hemoglobina libre y por reacción entre las formas
.07

219
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
férrica y ferrosa del hierro, formándose nitrosil hemoglobina que consume al ON8. La hemólisis intravascular también genera arginasa, lo que hace que disminuya la arginina que es el precursor del ON. En la enfermedad drepanocítica, la hipertensión pulmonar, priapismo, úlceras en las piernas y la enfermedad vascular cerebral aguda se asocian a la hemólisis inducida por vasculopatía y la hemólisis con disminución de ON parece jugar un papel importante en su desarollo8,9.
TratamientoON inhalado. Existen publicaciones donde este procedimiento ha resultado beneficioso en casos de sindrome torácico agudo complicado con insuficiencia respiratoria, hi-poxia e hipertensión pulmonar que no responde al tratamiento convencional. También parece que el uso de ON durante los episodios de dolor, disminuye la necesidad de morfina; sin embargo, un estudio grande, multicéntrico, controlado, demostró que no hubo beneficio en adultos con episodios de dolor1,10.
Tetrahidrobiopterina (BH4). Es un cofactor importante en la producción del ON y su deficiencia es responsable de disfunción endotelial en pacientes con enfermedad dre-panocítica11. L-arginina. El tratamiento de 10 pacientes con hipertensión pulmonar, me-diante la administración de L-arginina (0,1g/kg tres veces/día por VO) redujo la presión sistólica de la arteria pulmonar medida antes y después de cinco días de tratamiento (de 63,9 ± 13 mm de Hg a 54,2 ± 12mm de Hg); sin embargo, los resultados preliminares de un estudio multicéntrico en Fase II con arginina oral no demostró aumento en sus niveles después de 3 meses de tratamiento12.
Sildenafilo• Inhibe la 5 fosfodiesterasa, por tanto aumenta el efecto biológico del óxido nítrico
sobre la guanilato ciclasa. • Reduce la activación de las plaquetas13.• Aumenta niveles de Hemoglobina F14.• Puede ser de utilidad clínica en pacientes con hipertensión pulmonar. Este fárma-
co está aprobado para su uso en pacientes con hipertensión pulmonar idiopática. • No ha demostrado beneficio en los estudios clínicos en pacientes con enfermedad
drepanocítica en la prevención de eventos dolorosos15,16.
Contrarrestar la inflamación y proteger al endotelio
EstatinasIndependientemente de su efecto de bajar los niveles de colesterol, se ha demostrado que puede prevenir el daño vascular por varios mecanismos incluyendo la regulación del ON endotelial. Dado que el metabolismo del ON está alterado en la enfermedad drepanocítica, el uso de estos fármacos parece ser beneficioso. Es necesario esperar los resultados de los estudios clínicos1,6.

220
Esteroides • Disminuye la duración de los episodios dolorosos severos, pero aumenta las pro-
babilidades de readmisión17.• Atenúa la severidad del sindrome torácico agudo en niños y adolescentes18.• Aumenta el riesgo de hemorragia cerebral en niños con enfermedad drepanocítica19.• Se requieren mas estudios para determinar el beneficio en pacientes con enfer-
medad drepanocítica.
Niacina• Los pacientes con enfermedad drepanocítica, presentan niveles bajos de apolipo-
proteína A-I (apoA-I).• La niacina está aprobada para uso clínico en pacientes con aterosclerosis y nive-
les bajos de apoA-I y HDL-C).
Sulfasalazina (Inhibidor del factor nuclear kappa B).1,20,21
• En humanos reduce la expresión de: · VCAMP. · ICAMP (Molécula de adhesión intercelular). · Selectina.
• En ratones transgénicos: · Disminuye la adhesión de leucocitos. · Mejora el flujo microvascular.
Antiagregantes plaquetarios y anticoagulantes.• Heparina de bajo peso molecular, tinzaparina (Innohep R) (Dupont.Wilmington,-
DE). Un estudio aleatorio, doble ciego y controlado con placebo, en pacientes con episodios de dolor dio por resultado22: · Reducción del promedio de duración del episodio doloroso. · Disminución de días de hospitalización en los pacientes con dolor severo. · Dosis recomendada
· Dolor severo y en embarazadas con alto riesgo de trombosis23: · 175UI/kg/d durante 2-7 días. · Profiláctica 75UI/kg/día.
· No existen estudios relacionados con su uso en las crisis de dolor durante el embarazo.
• Eptifibatida (Inhibidor de la glicoproteína IIb/IIIa). Utilizado en un estudio piloto con pacientes con ED-SS, demostró ser útil al disminuir: · La agregación plaquetaria. · Los niveles de mediador inflamatorio, ligando soluble CD24.
No obstante, no existen suficientes evidencias de su uso en enfermedad drepanocítica.• Aspirina. Excelente antiinflamatorio y antiagregante. Tampoco posee suficientes
.07

221
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
evidencias que avalen su uso en la enfermedad drepanocítica. Otros agentes
• Polaxamer purificado 188. Es un copolímero no iónico, surfactante, que aumenta el flujo vascular al disminuir la viscosidad, la agregación eritrocitaria y la fricción entre los eritrocitos y la pared del vaso. Ha sido utilizado en el tratamiento de eventos de dolor severo con buenos resultados. A la espera de resultados de es-tudios en fase III (http://www.clinicaltrials.gov # NCT00004408)6.
• Nix-0699 (NiprisanR, NicosanR, HemoxinR). Es un producto vegetal que contiene 4 plantas diferentes: Piper guineense, Pterocarpus osun, Eugenia caryophyllum, y Sorghum bicolor. Aún cuando se desconoce su principio activo y su mecanismo de acción, los estudios in vitro han demostrado que inhiben la formación de drepano-citos y provocan desviación a la izquierda de la curva de disociación de oxígeno de la Hb S. Un estudio doble ciego, controlado con placebo, que incluyó 98 pacientes, reveló que Nix-0699 es seguro y además disminuyó la frecuencia de episodios do-lorosos25. En el 2005 fue declarada como droga huérfana por la European Medicine Evaluation Agency y en el 2006 por la FDA. En el año 2006 fue aprobada en Nigeria donde se encuentra disponible en el mercado6.
• Inmunoglobulina EV. En ratones transgénicos26: · Disminuye la adhesión de leucocitos al endotelio. · Disminuye el número de eritrocitos interactuando con leucocitos. · Aumenta el flujo microvascular.
• Es necesario esperar resultados de estudios clínicos1 (http://www.clinicaltrials.gov #NCT00644865.
• Bonsentan. Es un antagonista del receptor de endotelina · En ratones transgénicos previene:
· Congestión micro vascular en riñones y pulmones. · Inflamación sistémica. · Formación de eritrocitos densos. · Infiltración de neutrófilos activados dentro de un tejido siguiendo a la hipoxia/
reoxigenación. · Previno la muerte después de hipoxia severa27. · Los estudios clínicos en pacientes con enfermedad drepanocítica fueron
suspendidos al provoca aumento de la anemia y hepatopatía16.
Hace más de 35 años, De Vita y colaboradores28 demostraron en la enfermedad de Hodgkin que el uso de múltiples drogas con diferentes mecanismos de acción y sin superposición de efectos adversos, curaban los estadios avanzados de la enfermedad, estableciendo un nuevo paradigma en el uso de la quimioterapia. Ese concepto se ha extendido, así hoy se usan combinaciones de antibióticos, antihipertensivos y otros.
En anemia drepanocítica, por ejemplo pudiera indicarse hidroxiurea más decitabina o un ácido graso de cadena corta para lograr la máxima concentración de hemoglobina

222
fetal; otra factibilidad es la combinación de drogas que aumenten la biodisponibilidad de ON, reduzcan la densidad de los eritrocitos y su interacción con las células endote-liales más un antiinflamatorio. Si un esquema de dos drogas demuestra ser superior a la monoterapia, se podría añadir una tercera y así sucesivamente29. Aun no existe ningún estudio en este sentido pero es el futuro a mediano plazo en la terapia de la enfermedad drepanocítica.
En la tabla 7.2 se enumera una lista de drogas, el aspecto fisiopatológico sobre el cual actúa y su mecanismo de acción.
Tabla 7.2. Mecanismo fisiopatológico sobre el cual actúan ciertas drogas28
Fisiopatología Droga Mecanismo de acción
Polimerización de la Hb S
Hidroxiurea Aumento de inducción de Hb F: eritropoyesis de estrés
Decitabina Aumento de inducción de Hb F por hipome-tilación
Valproato Favorece la inducción de Hb F por desacetila-ción de histona
Daño de la membrana del eritrocito
Glutamato, vitamina C Antioxidante
Deferiprona Quelación del hierro de membrana
Hemólisis y otras indicaciones Terapia transfusional Reemplazo de eritrocitos con hemoglobina S
Sobrecarga de hierro
Deferasirox, Deferiprona Quelación oral
Deferoxamina Quelación IV
Aferesis, exanguino transfusión Reducción de carga de hierro
Deshidratación del eritrocito
Clotrimazol, ICA-17403 Inhibición de los canales de Gardos
Pidolato de Mg Inhibición de los canales K:CI
Dipiridamol Inhibición del flujo catiónico
.07

223
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
Referencias 1. Ataga KI. Novel therapies in sickle cell disease. Hematology 2009; 2009:54-61. 2. De Fransceschi L, Beuzard Y, Jouault H, Brugnara C. Modulation of erythrocyte potassium chloride co-transport, potassium content, and density by dietary magnesium intake in transgenic SAD mouse. Blood 1996; 88:2738-44. 3. De Fransceschi L, Bachir D, Galacteros F, Tchernia G, Cynober T, Neuberg D, Beuzard Y, Brugnara С. Oral magnesium pidolate: effects of long-term administration in patients with sickle cell disease. Br J Haematol 2000; 108:284-289. 4. Molavi MA, QasemyZadeh M, Nazemi A, Goodarzi R, Moghtadaie M, Hosaini S , Molavi K. Asian J. Med.
Fisiopatología Droga Mecanismo de acción
Adhesión del eritrocito al endotelio
Polaxamer 188 (Flocor) Surfactante
Sulfasalazina Inhibición del factor κ nuclear
Anticuerpo monoclonal integrina αVβ3 Bloqueo de trombospondina
Sulfato dextran, sulfato condroitin Inhibición de unión de glicolípidos
Tono vascular
Inhalación de NO Vasodilatación
Arginina Sustrato de ON
Hidroxiurea Aumenta ON
InflamaciónHidroxiurea Reducción de neutrófilos
¿Esteroides? Anti inflamatorio
Hipercoagulabilidad
Aspirina, ticlopidina Inhibición plaquetaria
Heparina, enoxaparina Anticoagulación
Ácidos grasos omega 3 Reducción de la actividad del factor tisular
Levostatin Disminución de la expresión del factor tisular
Activador del plasminóge-no tisular Trombolítico
Defecto genético Terapia génica Reemplazo genético
Trasplante de células progenitoras Reemplazo de médula ósea
Tabla 7.2. Continuación

224
Pharm. Res 2013; 3 : 32-34. 5. Brugnara C, Gee B, Armsby CC, Kurth S, Sakamoto M, Rifai N, Alper SL, Platt, OS. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest 1996; 97:1227–34. 6. Joiner CH, Jiang M, Claussen WJ, Roszell NJ, Yasin Z, Franco RS. Dipyridamole inhibits sickling-indu-ced cation fluxes in sickle red blood cells. Blood 2001; 97:3976-83. 7. Hankins J, Aygun B. Pharmacotherapy in sickle cell disease – state of the art and future prospects. Br J Haematol 2009; 145:296-308. 8. Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002; 8:1383–89. 9. Kato G.J, McGowan V, Machado RF, Little J.A, Taylor J, Morris CR et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hyper-tension, and death in patients with sickle cell disease. Blood 2006; 107:2279–85. 10. Atz AM, Wessel DL. Inhaled nitric oxide in sickle cell disease with acute chest syndrome. Anesthesio-logy 1997; 87:988-990. 11. De Franscechi L, Platt OS, Malpeli G, Janin A, Scarpa A, Leboeuf Ch ,et al. Protective effects of phos-phodiesterase-4 (PDE4) inhibition in the early phase of pulmonary arterial hypertension in transgenic sickle cell mice. FASEB J 2008; 22:1849-1860. 12. Styles L, Kuypers F, Kesler K, Reiss U, Lebeau P, Nagel R, Fabry M. Arginine therapy does not benefit children with sickle cell anemia – results of the CSCC clinical trial consortium multi-institutional study Blood 2007;110:2252. 13. Villagra J, Shiva S, Hunter LA, Machado RA, Gladwin MT and Kato GJ. Platelet activation in patients with sickle cell disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007; 110:2166-72. 14. Little JA, Hauser KP, Martyr SE, Harris A, Maric I, Morris CA et al. Hematologic, biochemical, and cardiopulmonary effects of Larginine supplementation or phosphodiesterase 5 inhibition in patients with sickle cell disease who are on hydroxyurea therapy. Eur J Haematol 2009; 82:315-21. 15. Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol 2005;130:445-53. 16. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hagar W, Darbari DS, Malik P. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. The Scientific World Journal 2012; 2012:949535. doi: 10.1100/2012/949535. 17. Griffin TC, McIntire D, Buchanan GR. High-dose intravenous methylprednisolone therapy for pain in children and adolescents with sickle cell disease. N Engl J Med 1994; 330:733-58. 18. Bernini JC, Rogers ZR, Sandler ES, Reisch JS, Quinn ChT, Buchanan GR .Beneficial effect of intra-venous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood 1998; 92:3082–89. 19. Strouse JJ, Takemoto CM, Keefer JR, Kato GJ, Casella JF. Corticosteroids and increased risk of readmission after acute chest syndrome in children with sickle cell disease. Pediatr Blood Cancer 2008;50:1006-12. 20. Soloveyt AA, Solovey AN, Harkness J, Hebbel RP. Modulation of endothelial cell activation in sickle cell disease: a pilot study. Blood 2001; 97:1937-41. 21. Kaul DK, Liu XD, Choong S, Belche JD, Vercellotti GM, Hebbel RP. Anti-inflammatory therapy ame-
.07

225
Estrategias Farmacológicas en el Tratamiento de la Enfermedad Drepanocítica
liorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle cell mice. Am J Physiol Heart Circ Physiol 2004; 287:H293-301. 22. Qari MH, Aljaouni SK, Alardawi MS, Fatani H Alsayes F M, Zografos P ,Alsaigh M, Alalfi A et al. Re-duction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost 2007; 98:392-96. 23. Mousa S, Al Momen A, Al Sayegh, Al Jaouni S, Nasrullah Z, Al Saeed H et al. Management of Painful Vaso-Occlusive Crisis of Sickle-Cell Anemia: Consensus Opinion. Clin Appl Thromb Hemost 2010; 16:365-76. 24. Lee SP, Ataga KI, Zayed M, Manganello JM, Orringer EP, Phillips DR, and Parisee LV et al. Phase I study of eptifibatide in patients with sickle cell anaemia. Br J Haematol 2007; 139: 612-20. 25. Wambebe C, Khamofu H, Momoh JA, Ekpeyong M, Audu BS, Njoku OS et al. Double-blind, place-bo-controlled, randomised cross-over clinical trial of NIPRISAN in patients with sickle cell disorder. Phy-tomedicine 2001;8:252–61. 26. Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vaso-occlusive crises in mice through rapid inhibition of neutrophil adhesion. Blood 2008; 111:915-23. 27. Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H et al. Endothelin receptor anta-gonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle cell disease. J Clin Invest 2008; 118:1924-33. 28. DeVita VT Jr, Serpick AA, and Carbone PP. Combination chemotherapy in the treatment of advanced Hodgkin ’s disease. Ann Intern Med 1970; 73:881–895. 29. Steinberg MH. Clinical trials in sickle cell disease: adopting the combination chemotherapy paradigm. Am J Hematol 2008; 83:1–3.

226
8.1. Estrategias futuras en el diagnóstico. Clementina Landolfi / Hematólogo pediatra.
El diagnóstico de la enfermedad drepanocítica, generalmente es fácil y sencillo, rara vez significa un problema, si bien las células en forma de hoz no siempre se observan en el frotis de sangre periférica, basta con la corrida electroforética a dos pH para identificar a la Hb S.
La electroforesis permite determinar algunos pero no todos los genotipos de la enfer-medad drepanocítica, por ejemplo cuando en la electroforesis se encuentra un porcen-taje de Hb A inferior al de Hb S, es posible establecer que se trata de ED-Sβ+talasemia; sin embargo, la ED-Sβ0 talasemia es mas problemática de diferenciar de la ED-SS, debido a que en ambas hay ausencia de Hb A; si bien la presencia de Hb A2 elevada y el estudio de ambos padres puede ser de gran utilidad en el diagnóstico diferencial , no siempre es así y en esos casos se impone el estudio por biología molecular para un diagnóstico definitivo.
La mayor dificultad es el diagnóstico prenatal en etapas tempranas del embarazo, ac-tualmente es posible realizar el diagnóstico mediante el análisis del ADN obtenido por biopsia de vellosidades coriónicas1, posiblemente en un futuro el diagnóstico de Hb S de las pocas células fetales circulantes en la sangre materna2 estará al alcance de todos.
Se conoce que una mutación única en la cadena β de la globina es responsable de la fisiopatología tan compleja de la enfermedad drepanocítica; igualmente, las manifes-taciones clínicas son extremadamente heterogéneas debido a factores múltiples como la interacción del gen βS con el gen β talasemia, niveles de hemoglobina fetal (Hb F) y coherencia de α talasemia, las cuales modulan la expresión clínica.
En la medida que los conocimientos relacionados con la fisiopatología han aumentado, también se ha elevado el número de potenciales genes epistáticos. Se han encontrado genes ubicados fuera del bloque génico β que modulan la expresión de los genes γ responsables de la producción de hemoglobina fetal, como el cMYB (114)3.
Se conoce el efecto potencial de la metilentetrahidrofolato reductasa y la patogénesis de la necrosis avascular, el factor V R485K y la trombosis venosa, y el polimorfismo de UDP glucoronil transferasa-1 y los niveles de bilirrubina sérica.
La investigación basada en métodos moleculares está evolucionando de tal forma, que en un futuro será posible identificar el gen que aumenta el riesgo de una complicación determinada en un paciente en particular utilizando las pocas células fetales que se pueden obtener de la circulación materna.
Estrategias futuras en el diagnóstico Y tratamiento de la enfermedad drepanocítica.08

227
8.2. Estrategias futuras en el tratamiento No existe tratamiento curativo para la enfermedad drepanocítica diferente al trasplan-te alogénico de células progenitoras hematopoyéticas; sin embargo esta opción no está al alcance de todos por la ausencia de donante HLA compatible.
En la actualidad se encuentra en investigación una serie de estrategias genéticas, con el propósito de tratar la anemia y disminuir la formación de drepanocitos. Ellas incluyen:
• Transferir un gen de globina regulado a células progenitoras hematopoyéticas autólogas.
• Reparar o eliminar los ARNm de hemoglobinas mutantes.• Corregir la mutación en células progenitoras hematopoyéticas.
El objetivo común es elevar los niveles de Hb A o Hb F en las células progenitoras eritroides en cantidad suficiente para prevenir la formación de drepanocitos, poste-riormente trasplantar las células progenitoras modificadas genéticamente, con un mínimo de toxicidad a corto plazo (causado por el régimen de acondicionamiento) y a largo plazo (generado por la genotoxicidad).
Aún cuando la terapia génica está en fase experimental, tiene el potencial de dar origen a un tratamiento curativo de la enfermedad drepanocítica. Usando modelos de ratones “humanizados” se ha logrado la corrección de la enfermedad drepanocítica mediante terapia génica en células progenitoras pluripotenciales inducidas (iPS) que se pueden diferenciar hacia la serie eritroide6.
En el 2009, Ye y colaboradores7 lograron reprogramar fibroblastos obtenidos por biop-sia de piel de un paciente con B0 talasemia y homocigoto para la deleción 41/42 4-bp (CTTT). Los fibroblastos fueron inducidos a formar iPS y éstas se diferenciaron hacia células hematopoyéticas que sintetizaron hemoglobina fetal. Igual procedimiento pue-de realizarse con células de pacientes con enfermedad drepanocítica.
Las células tomadas para el diagnóstico prenatal pueden ser transformadas en célu-las iPS para ser usadas en el período perinatal. El tratamiento en fases tempranas tie-ne la ventaja de requerir un número menor de células que el tratamiento de un adulto, además de prevenir el daño orgánico7.
Referencias 1. Orkin SH, Little PF, Kazazian HH Jr. and Boehm C D. Improved detection of the sickle mutation by DNA analysis: application to prenatal diagnosis. N Engl J Med 1982;307:32–36.2. Cheung M.C, Goldberg JD, Kan YW. Prenatal diagnosis of sickle cell anaemia and thalassaemia by analysis of fetal cells in maternal blood. Nat. Genet 1996;14:264–68. 3. Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin

228
Invest 2007; 117:850–58. 4. Kutlar A, Kutlar F, Turker I, Tural C. The methylene tetrahydrofolate reductasa (C677T) mutation as a potential risk factor for avascular necrosis in sickle cell disease. Hemoglobin 2001; 25:213–17.5. Helley D, Besmond C, Ducrocq R da Silva F, Guillin MC, Bezeaud A, Elion J et al. Polymorphism in exon 10 of the human coagulation factor V gene in a population at risk for sickle cell disease. Hum Genet 1997; 100:245–48.6. Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP et al. Treatment of sickle cell ane-mia mouse model with iPS cells generated from autologous skin. Science 2007; 318:1920-23.7. Ye L, Chang JC, Lin C, Sun X, Yu J, Kan YW. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci USA 2009;106:9826-30.
.08

229
Hoja de control anual
Objetivos:1. Proporcionar un documento de fácil acceso e interpretación que permita conocer en pocos minutos: a. Estado de la enfermedad del paciente. b. Estudios realizados y resultados de los mismos. c. Estudios que faltan por realizar. d. Tratamientos que recibe.
2. Facilitar los datos necesarios para la elaboración del resumen de eventos a final de año.
3. Brindar de manera rápida los datos necesarios para el levantamiento de los censos de morbilidad y mortalidad.

230
Apendice 1

231
Control anual de pacientes con enfermedad drepanocíticaED-SS, ED-SC, ED-Sβ0 ,ED-Sβ+ Tal, ED-SD, otras asociaciones (Llenar en enero)
Fecha: / /Hospital: Ciudad: Estado:Hematólogo responsable:Teléfono fijo:Tlf. movil: e-mail:Fax:
Datos del pacienteApellidos: Nombres:Edad: Domicilio actual:
Ciudad: Municipio: Estado:C.I.: Nº Historia:Tlf. Móvil: Tlf. Fijo:e-mail:
Familiar A Quien Llamar En Caso De EmergenciaApellidos: Nombres:Teléfono: Peso paciente: kg (percentil): Talla: cm (percentil):
Datos de análisis de laboratorio del dia del reporte: Hb(g/dl) Hto(%) Eritrocitos (x1012/L) υcm (fl) Reticulocitos (x 109/L)Plaquetas (109/L) Leucocitos(x109/L) Neutrófilos (x109/L)Aclaramiento creatinina (ml/min) Bilirrubina total/indirecta (mg/dl) /GOT/GPT/GGT (U/L) / / Ldh (U/L): Hierro Sérico (Υg/dl)% se saturación Ferritina (ng/ml)
Ultimo eco-doppler transcraneal (niños entre 2 años y 16 años) Fecha: / /
Normal No realizado Patologico: valores lado derecho: Izquierdo:

232
En regimen de transfusión crónica SiNo ¿Desde cuando? / /
Ultima RMN craneal:Fecha: / /
Patológica: SiNoDescriba:
Examen neuropsicologico: Fecha / /
Resultado: Normal Anormal:
Ecocardiograma:Fecha: / / Normal Anormal:
Ultrasonido abdominal: Normal Anormal:
Otras evaluaciones especializadas:
*Describir alteraciones:
No realizado Normal Alterado* Fecha
Oftalmologia
O.R.L.
Neumonologia
Nefrologia
Traumatología

233
Recibe hidroxiurea
SINO ¿Desde cuando? / /
Complicaciones crónicas presentes hasta el año 20
(Reporte inicial y luego cada 5 años)
Complicaciones agudas en el año 20___ de Enero a Diciembre
ENE FEB MAR ABR MAY JUN JUL AGO SEP OCT NOV DIC
Hospitalización
Dolor severo
Infección
Aplasia transitoria serie roja
ECVA
Isquemia transitoria aguda (< 24 h de instalación)
Ocular
Sindrome torácico agudo
Secuestro esplénico
Priapismo
Sindrome de cuadrante superior derecho (colelitiasis,
secuestro hepático)
Sindrome de embolización sistémica
Transfusión de CG. ¿Cuántas?
¿Exanguinotransfusión?
*Describir alteraciones:

234
Retraso pondoestatural Oftalmológicas Auditivas Neurológicas Cardiovasculares Pulmonares Hepatobiliares Renales Osteomusculares Úlceras maleolares Transfusionales (infecciosas, aloinmunización, sobrecarga de hierro)
¿Recibe quelantes?SINO¿Cuál? ¿Desde cuándo? / /Situación actual:Vivo: Muerto:Edad: Fecha de la muerte: / /Causa de la muerte:

235
Apendice 2

236
Hoja de control de exanguinotransfusion y/o eritrocitaféresisDatos de identificacion del paciente
Nombre y apellido: Edad: Sexo:
Diagnóstico: CI: Nº historia:
Procedimiento: Peso: Talla:
Hora de inicio: Hora de finalización: Volemia: Volumen total de GR (VTGR):
Hospital/Servicio: Sala/Hab: Cama:
Valores pre procedimiento
Fecha: % HbS: % HbA: % HbA2: % HbF:
Fecha: Hb: Hto: Leuco: Plaq:
Fecha: Fe sérico: TIBC: Sat de transf: Ferritina:
Manejo de volúmenes
Equipo utilizado: Anticoagulante:
Volumen extraído: Volumen de ST administrado:
Volumen de GR administrado: Volumen de sol. salina administrado:
Reacciones adversas durante el procedimiento:
Valores post procedimiento
Fecha: % HbS: % HbA: % HbA2: % HbF:
Hora: Hb: Hto: Leuco: Plaq:
Observaciones
Médico responsable:
Hemoterapista responsable:

237
Apendice 3
238
Historia inmunohematólogica y serológica.Pacientes drepanocíticos y talasémicos.
Datos de identificacion del paciente
Nombre y apellido: Edad: Sexo:
Diagnóstico: CI: Nº historia:
Hospital/Servicio: Sala/Hab: Cama:
Antecedentes
% Hb A: %Hb S: %Hb A2: %Hb C: % HbF:
Tipeaje ABO/Rh Antecedente de transfusión: SI__ NO__ Antecedente de embarazo: SI__ NO__
Fecha de 1ra transfusión: Nº de transfusiones recibidas: Nº de embarazos:
Nº de transfusiones sin fenotipo recibidas: ¿Tiene antecedentes de reacciones a la transfusión? SI___ NO___
Señale el tipo de reacciones adversas a la transfusión presentadas:1.:2.:3.:
Acs irregulares detectados:
Fenotipo eritrocitario
Fenotipo (X) C: c: E: e: K: K: Fya: Fyb: Jka: Jkb: M: N:
Control inmunohematológico
Fecha Especificidad de Ac(s) Título (s) Coombs Directo PE Título/Score
Control de marcadores serologicos
Fecha AgsHB Anti-cHB Anti-VHC Agp24-Anti-VIH 1/2 Anti-Tcruzi (Chagas) Anti-HTLV Sífilis
Observaciones:
S: s: P1: Lea: Leb: Lua: Dia: Otros:
Observaciones:
Fecha: / /

239
Observaciones:
Recomendaciones:

240
Observaciones:
Recomendaciones:

Publicación cortesía de Novartis de Venezuela S.A.




















