
Genetic diversity in the VP1 gene of foot-and-mouth disease virus serotype Asia 1
18
Arch Virol (2002) 147: 85–102 Genetic diversity in the VP1 gene of foot-and-mouth disease virus serotype Asia 1 C. B. Gurumurthy,A. Sanyal, R. Venkataramanan, C. Tosh, M. George, and D. Hemadri Project Directorate on Foot-and-Mouth Disease, Indian Veterinary Research Institute, Mukteswar-Kumaon, Nainital, India Accepted August 1, 2001 Summary. Complete nucleotide sequence of the 1D (VP1-encoding) gene of 61 foot-and-mouth disease (FMD) serotype Asia 1 virus isolates recovered from different outbreaks in India between 1985 and 1999 including two vaccine strains currently used were determined. The sequences were compared with each other and those from other Asian countries. On the basis of phylogenetic analysis the viruses could be grouped into four genotypes (genotypes I–IV). All the 61 isolates from India belong to a single genotype (genotype-II) which is further subdivided into three lineages (B1, B2 and B3) under the same genotype. The viruses of the lineage B1 and B3 were found to be more prevalent before 1996 while the viruses of lineage B2 appeared to be new variants responsible for most of the recent outbreaks. Most of the isolates of lineage B1 lack one amino acid in the VP1 protein (position 44) whereas most of the isolates of lineage B2 and B3 contain it which indicates the possibility of these lineages having evolved independently. The rate of evolution of FMDV Asia 1 virus was also estimated and found to be 2.7 × 10 −2 synonymous substitutions per nucleotide per year. Introduction Foot-and-mouth disease virus (FMDV), a member of the genus aphthovirus, family Picornaviridae, is the causative agent of one of the most economically important diseases of domestic livestock. The virus exists as seven immunologi- cally distinct serotypes (i.e., O, A, C, Asia 1 and SAT 1–3) with multiple subtypes within a serotype [19]. Disease outbreaks due to serotypes O, A and C are re- ported from Europe, South America, Asia and Africa, whereas, serotypes Asia 1 and SAT1–3 are restricted toAsian countries andAfrica, respectively. The disease is endemic in Asian countries, and in India after serotype O, Asia 1 causes the largest number of outbreaks. The country has a large population (470 million) of
Transcript of Genetic diversity in the VP1 gene of foot-and-mouth disease virus serotype Asia 1

Arch Virol (2002) 147: 85–102
Genetic diversity in the VP1 gene of foot-and-mouthdisease virus serotype Asia 1
C. B. Gurumurthy, A. Sanyal, R. Venkataramanan, C. Tosh,M. George, and D. Hemadri
Project Directorate on Foot-and-Mouth Disease, Indian Veterinary Research Institute,Mukteswar-Kumaon, Nainital, India
Accepted August 1, 2001
Summary. Complete nucleotide sequence of the 1D (VP1-encoding) gene of 61foot-and-mouth disease (FMD) serotype Asia 1 virus isolates recovered fromdifferent outbreaks in India between 1985 and 1999 including two vaccine strainscurrently used were determined. The sequences were compared with each otherand those from other Asian countries. On the basis of phylogenetic analysis theviruses could be grouped into four genotypes (genotypes I–IV).All the 61 isolatesfrom India belong to a single genotype (genotype-II) which is further subdividedinto three lineages (B1, B2 and B3) under the same genotype. The viruses ofthe lineage B1 and B3 were found to be more prevalent before 1996 while theviruses of lineage B2 appeared to be new variants responsible for most of therecent outbreaks.Most of the isolates of lineageB1 lack one amino acid in theVP1protein (position 44) whereas most of the isolates of lineage B2 and B3 containit which indicates the possibility of these lineages having evolved independently.The rate of evolution of FMDV Asia 1 virus was also estimated and found to be2.7 × 10−2 synonymous substitutions per nucleotide per year.
Introduction
Foot-and-mouth disease virus (FMDV), a member of the genus aphthovirus,family Picornaviridae, is the causative agent of one of the most economicallyimportant diseases of domestic livestock. The virus exists as seven immunologi-cally distinct serotypes (i.e., O,A, C,Asia 1 and SAT 1–3) with multiple subtypeswithin a serotype [19]. Disease outbreaks due to serotypes O, A and C are re-ported from Europe, South America, Asia and Africa, whereas, serotypes Asia 1and SAT1–3 are restricted toAsian countries andAfrica, respectively. The diseaseis endemic in Asian countries, and in India after serotype O, Asia 1 causes thelargest number of outbreaks. The country has a large population (470 million) of

86 C. B. Gurumurthy et al.
FMD susceptible livestock comprising of cattle, buffalo, sheep, goats and pigs.There is no regular vaccination, less than 2% of the susceptible population isvaccinated annually. Under these conditions, it is essential that a thorough studyof the epidemiology of the disease be carried out.
FMDV, like other RNA viruses, exhibits high mutation rates during repli-cation [6] and remains, as non-identical but related populations termed viralquasispecies [5] and antigenic variants are selected from such virus pools dueto positive selection. As VP1 is the most immunodominant structural protein ofFMDV and antigenic variants are selected in the presence of antibodies to it, theVP1 gene is the preferred gene for comparison of outbreak strains. A previousstudy of Asia 1 FMDV, involving viruses isolated from Asia between 1954 and1992 [2] based on partial (approximately 165 nucleotides) VP1 gene sequencehad provided evidence for the prevalence of a single genotype of Asia 1 viruses.Five outbreak viruses from India (isolated between 1971 and 1988) were alsoincluded in this study. A more recent report [14] where three outbreak virusesfrom Bangladesh and one each from Thailand and Israel were compared on theother hand, indicated the circulation of three genotypes of Asia 1 viruses.
In this study, we have sequenced and analyzed sixty-one Asia 1 isolates (in-cluding 2 vaccine viruses) recovered from different outbreaks in 14 states of Indiawithin a period of 15 years (1985–1999) to study their genetic divergence andevolutionary rate.
Materials and methods
Virus
A total of 61 serotype Asia 1 FMDV field isolates (including 2 vaccine strains) recoveredfrom different outbreaks were used in the study. The sampling areas and date of isolationare summarized in Table 1. All the viruses were adapted in BHK-21 cells and the clarifiedinfected cell culture supernatants were used for extraction of RNA. Additional sequencesused in the study obtained from GenBank/published earlier were: PAK/1/54 (AJ251478),Shamir/89 [27] and China (AF241566).
RNA extraction, RT-PCR and sequencing
The viral RNA was extracted from 460 �l of the clarified infected cell culture supernatantusing RNeasy kit (Qiagen) as per the recommendations of the supplier. The VP1 gene wasamplified from viral RNA by RT-PCR following the method described earlier [29]. ThePCR products were purified using Wizard preps (Promega) and sequenced using fmol cyclesequencing system (Promega). The primers used for RT-PCR amplification and cycle se-quencing are listed in Table 2. The reaction products were resolved in 6% polyacrylamidedenaturing gels and the bands were developed by silver staining (non-radioisotopic method)(Promega).
Phylogenetic analysis
Molecular sequences (nucleotide and amino acids) were aligned using Clustal W algo-rithm [28], implemented in the OMIGA 2.0 program (Oxford Molecular Ltd, UK) andrefined manually. Phylogenetic trees were generated by neighbour-joining method basedon the nucleotide and deduced amino acid sequences with the PHYLIP package,
Sequence analysis of FMDV Asia 1 87
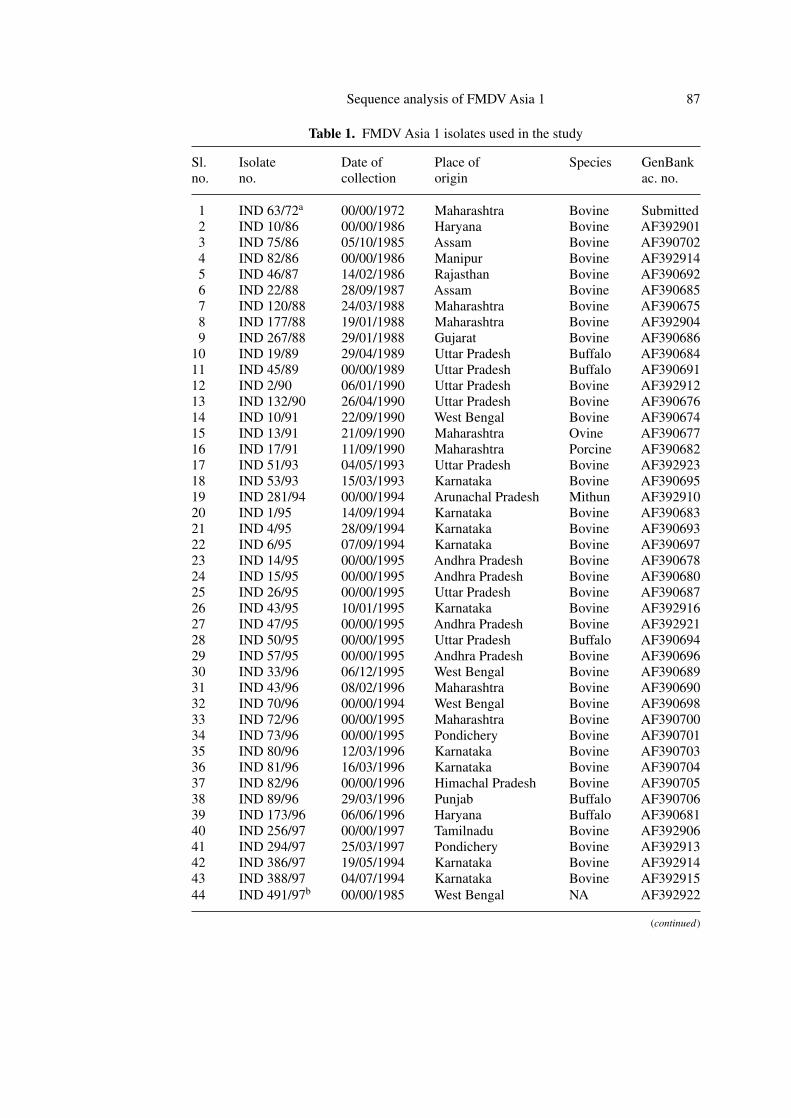
Table 1. FMDV Asia 1 isolates used in the study
Sl. Isolate Date of Place of Species GenBankno. no. collection origin ac. no.
1 IND 63/72a 00/00/1972 Maharashtra Bovine Submitted2 IND 10/86 00/00/1986 Haryana Bovine AF3929013 IND 75/86 05/10/1985 Assam Bovine AF3907024 IND 82/86 00/00/1986 Manipur Bovine AF3929145 IND 46/87 14/02/1986 Rajasthan Bovine AF3906926 IND 22/88 28/09/1987 Assam Bovine AF3906857 IND 120/88 24/03/1988 Maharashtra Bovine AF3906758 IND 177/88 19/01/1988 Maharashtra Bovine AF3929049 IND 267/88 29/01/1988 Gujarat Bovine AF390686
10 IND 19/89 29/04/1989 Uttar Pradesh Buffalo AF39068411 IND 45/89 00/00/1989 Uttar Pradesh Buffalo AF39069112 IND 2/90 06/01/1990 Uttar Pradesh Bovine AF39291213 IND 132/90 26/04/1990 Uttar Pradesh Bovine AF39067614 IND 10/91 22/09/1990 West Bengal Bovine AF39067415 IND 13/91 21/09/1990 Maharashtra Ovine AF39067716 IND 17/91 11/09/1990 Maharashtra Porcine AF39068217 IND 51/93 04/05/1993 Uttar Pradesh Bovine AF39292318 IND 53/93 15/03/1993 Karnataka Bovine AF39069519 IND 281/94 00/00/1994 Arunachal Pradesh Mithun AF39291020 IND 1/95 14/09/1994 Karnataka Bovine AF39068321 IND 4/95 28/09/1994 Karnataka Bovine AF39069322 IND 6/95 07/09/1994 Karnataka Bovine AF39069723 IND 14/95 00/00/1995 Andhra Pradesh Bovine AF39067824 IND 15/95 00/00/1995 Andhra Pradesh Bovine AF39068025 IND 26/95 00/00/1995 Uttar Pradesh Bovine AF39068726 IND 43/95 10/01/1995 Karnataka Bovine AF39291627 IND 47/95 00/00/1995 Andhra Pradesh Bovine AF39292128 IND 50/95 00/00/1995 Uttar Pradesh Buffalo AF39069429 IND 57/95 00/00/1995 Andhra Pradesh Bovine AF39069630 IND 33/96 06/12/1995 West Bengal Bovine AF39068931 IND 43/96 08/02/1996 Maharashtra Bovine AF39069032 IND 70/96 00/00/1994 West Bengal Bovine AF39069833 IND 72/96 00/00/1995 Maharashtra Bovine AF39070034 IND 73/96 00/00/1995 Pondichery Bovine AF39070135 IND 80/96 12/03/1996 Karnataka Bovine AF39070336 IND 81/96 16/03/1996 Karnataka Bovine AF39070437 IND 82/96 00/00/1996 Himachal Pradesh Bovine AF39070538 IND 89/96 29/03/1996 Punjab Buffalo AF39070639 IND 173/96 06/06/1996 Haryana Buffalo AF39068140 IND 256/97 00/00/1997 Tamilnadu Bovine AF39290641 IND 294/97 25/03/1997 Pondichery Bovine AF39291342 IND 386/97 19/05/1994 Karnataka Bovine AF39291443 IND 388/97 04/07/1994 Karnataka Bovine AF39291544 IND 491/97b 00/00/1985 West Bengal NA AF392922
(continued)

88 C. B. Gurumurthy et al.
Table 1 (continued)
Sl. Isolate Date of Place of Species GenBankno. no. collection origin ac. no.
45 IND 270/98 25/03/1998 West Bengal Bovine AF39290746 IND 271/98 16/04/1998 West Bengal Bovine AF39290847 IND 451/98 00/00/1998 Haryana Buffalo AF39291748 IND 452/98 00/00/1998 Haryana Bovine AF39291849 IND 453/98 02/12/1998 Haryana Buffalo AF39291950 IND 454/98 02/12/1998 Haryana Buffalo AF39292051 IND 92/99 00/00/1998 Haryana Bovine AF39292552 IND 102/99 07/01/1999 Haryana Buffalo AF39289753 IND 104/99 07/01/1999 Haryana Buffalo AF39289854 IND 105/99 00/00/1999 Haryana Bovine AF39289955 IND 107/99 31/12/1998 Punjab Bovine AF39290056 IND 108/99 00/00/1999 Punjab Bovine AF39290257 IND 126/99 17/12/1998 Karnataka Bovine AF39290358 IND 134/99 16/01/1999 Haryana Bovine Submitted59 IND 196/99 02/02/1999 Karnataka Bovine AF39290560 IND 277/99 12/03/1999 Karnataka Bovine AF39290961 IND 286/99 00/00/1999 Haryana Bovine AF392911
aIND 63/72 – Vaccine virus of IVRIbWBN 117/85 – Vaccine virus of Indian Immunologicals Ltd.
Table 2. Location and sequence of oligonucleotide primers
Primer Sequence (5′-3′) Location Polarity Use Referencename
NK61 GACATGTCCTCCTGCATCTG 2B, 58–77 Antisense RT-PCR [12]1C505 TACACTGCTTCTGACGTGGC VP3, 505–524 Sense RT-PCR, [12]
SequencingAs9 CGAGACCACAAGTGGCAGG VP3, 522–541 Sense Sequencing This studyAs5 GGACTTTGAGTTTCGACTGC VP3, 618–637 Sense Sequencing This studyMG47 GAGAACTACGGAGGAGAGACTCA VP1, 75–97 Sense Sequencing This studyMG1D180 ATGCAGATCCCCTCACACACGCTG VP1, 180–203 Sense Sequencing This studyMG48 CTCCCCTACACCGCCCCCCA VP1, 348–367 Sense Sequencing This studyP1RS GTCCACCAGTTTGGAGAAGTT 2B, 28–48 Antisense Sequencing This studyNK72 GAAGGGCCCAGGGTTGGACTC 2A/2B, Antisense Sequencing [12]
34–48/1–6
�Fig. 1. Neighbor-joining tree based on complete (633 nucleotides) VP1 encoding gene se-quence showing evolutionary relationship within Asia 1 FMDV. The bootstrap confidencevalues above 50% (out of 1000 bootstrap replicates) are shown near the nodes of the tree.Genotypes, (designated as I–IV) of the viruses were shown on the branches. The scale barbelow the tree indicates genetic distance. O1/Kaufbeuren [8] was used as outgroup and thebranch length of the outgroup has been reduced by 50% to save space. The arrow indicatesthe monophyletic cluster used in the calculation of evolution rate in the virus

Sequence analysis of FMDV Asia 1 89

90 C. B. Gurumurthy et al.
Table 3. Estimation of nucleotide substitution rates among Asia 1 FMDV
Substitution rates (substitution/nt./year)a
Synonymous R2 Nonsynonymous R2
2.7 × 10−2 0.2723 1.1 × 10−3 0.1079(P = 0.001) (P = 0.05)
a36 isolates from 1993 to 1999
version 3.5c [7]. The evolutionary distances were calculated in the programs DNADIST(Kimura 2-parameter model) and PROTDIST (Dayhoff PAM matrix), wherever applicable.The nucleotide sequence based tree diagram was produced in the DRAWGRAM program.The tree topology was statistically evaluated with 1000 replicas of the original data setusing the programs BOOTSTRAP, DNADIST, NEIGHBOUR and CONSENSE, available inthe package.Adendrogrambased onUPGMAmethod for the same sequenceswas constructedusing the “EpiSeq” program (written by N. J. Knowles). Pairwise sequence homologies wereestimated using the DNASTAR program package (DNASTAR Inc, Madison, USA).
Estimation of nucleotide substitution rates
For calculation of substitution rates in VP1 gene sequence, the isolates were selected basedon the relationships in the phylogenetic tree (Fig. 1). Two different substitution rates (synony-mous substitutions per synonymous site and nonsynonymous substitutions per nonsynony-mous site) were calculated using the method of [17], available in DnaSP 3.50 program [21].The substitution rates were calculated by linear regression analysis. The genetic distancesof each isolate from the oldest one in the monophyletic cluster were plotted along the Y-axis, and the time of isolation (years) were plotted along the X-axis. The calculated rates ofsubstitution are shown in Table 3.
GenBank accession numbers
The nucleotide sequences generated in this study have been deposited in the GenBankdatabase under the accession numbers: AF390674–AF390678, AF390680–AF390687,AF390689–AF390698, AF390700–AF390706 and AF392897–AF392925.
Results and discussion
Molecular epidemiology of field isolates is a powerful tool to monitor the cir-culation of viruses. Worldwide, molecular epidemiological data is available forFMDV serotypes O,A and C whereas for serotypeAsia 1, much less informationis available. The present work was undertaken to study the molecular phylogenyand evolution of Asia 1 virus isolates recovered from India.
Sixty-one FMDV Asia 1 isolates collected from 14 states in India between1985 and 1999, including two vaccine strains, IND 63/72 and IND 491/97 wereused in this study. The viral RNA extracted from the BHK-21 cell culture infectedsupernatants was amplified by RT-PCR in the VP1 coding region to obtain PCRproducts of 908/911 base pairs length. The purified PCR products were sequenceddirectly by cycle sequencing using the primers listed in Table 2. For most of the

Sequence analysis of FMDV Asia 1 91
isolates the primers As 9, MG 47 and NK72/MG P1RS were found sufficient indeducing the entire VP1 gene sequence. However, in some instances the otherprimers were needed for clarifying ambiguities.
Genotype identification of FMDV Asia 1 viruses
Several studies on phylogenetic analysis of FMDV have been performed usingeither full or part (as less as 165 nucleotides) ofVP1 gene sequences [2, 3, 10, 11,13, 18, 23, 26, 30]. However, it has been suggested [22] that precise phylogeneticanalysis may require at least 250 nucleotides, if not the complete VP1 gene se-quence. Since a portion ofVP1 sequence at the 5′ terminus was not available in anisolate from China and an Indian isolate was also incomplete in this region, a den-drogram was drawn using the incomplete nucleotide sequence (597 nucleotides)corresponding to the nucleotide position 37 to 633.
We have identified four major genotypes in serotype Asia 1 FMDV based onthe branching pattern of the isolates in the dendrogram (Fig. 2).A 15% nucleotidedivergence between isolates was used as cut off value to define the genotype.Similar criteria has been proposed for identifying genotypes for FMDV serotypeO [24] and Polio virus [20] using partial sequence data. The Asia 1 prototypestrain from Pakistan, PAK/1/54 formed genotype I and all Indian isolates includ-ing the two vaccine viruses (IND 63/72, isolated during 1972 and IND 491/97,isolated during 1985) formed a single genotype, which was named genotype II.One strain each from Israel (Shamir/89) and China (China) formed genotype IIIand IV, respectively. A previous phylogenetic analysis of Asia 1 viruses (whichincluded five Indian viruses, PAK/1/54 and the Israeli isolate, Shamir/89) basedon partial nucleotide sequences (165 nts) [2], clustered them into a single geno-type. However, in the present study, when the analysis was extended to 597 basesand 64 viruses were compared, PAK/1/54 and Shamir/89, each formed a dis-tinct genotype separated from the Indian isolates. Segregation into individualgenotypes was supported by significant bootstrap values. Thus the results of thisstudy show the existence of at least 4 different genotypes circulating in Asiancountries.
Sequence analysis of VP1 gene
To investigate the sequence identity between the Asia 1 isolates, complete VP1nucleotide and deduced amino acid sequences were aligned separately and pair-wise calculations were made. The overall nucleotide identity between the Indianisolates and PAK/1/54 ranged from 76.5 to 83.3%, while that between the Indianand Chinese isolate was 74.3 to 80.8% and that between the Indian and Israeliisolate was 73.7 to 87.2%. However, all the Indian isolates were related to eachother by 86–100%, which show a high degree of genetic heterogeneity within theoutbreak strains studied. This is similar to that observed for Indian type O isolates[10, 18].
Though the amino acid sequence alignment (Fig. 3) shows that substitutionsare scattered along the entire length of the sequence, there are areas where the

92 C. B. Gurumurthy et al.
Fig. 2. Dendrogram showing genetic relationship betweenAsia 1 FMDV. The genotypes areshown in Roman numerals (I–IV) on the branches. ∗, Vaccine strains

Sequence analysis of FMDV Asia 1 93
Fig. 3 (continued)

94 C. B. Gurumurthy et al.
Fig. 3 (continued)

Sequence analysis of FMDV Asia 1 95
Fig. 3 (continued)

96 C. B. Gurumurthy et al.

Sequence analysis of FMDV Asia 1 97
frequency of substitution is higher. Three such regions ie., the G-H loop (position140–160), the B-C loop, (position 40–60), and the C-terminus of VP1 (position210–211) are known to be antigenically significant in other FMD viruses [15].A higher frequency of substitutions is also seen in the E-F loop but the roleof changes at this position in terms of antigenicity remains to be elucidated.One extra amino acid was found at positionVP1 44 (Fig. 3) in more than 79% (51out of the 64 isolates compared) of viruses. The residue observed in Indian isolateswas either A/T, in China isolate it was Q whereas in Israeli isolate (Shamir/89) itwas T. Since information on the antigenic sites/epitopes and their exact locationon theVP1 polypeptide of FMDV serotypeAsia 1 is not available, no significancein terms of antigenicity could be attributed to this insertion.
The integrin receptor-binding motif, RGD (Arginine-Glycine-Aspartic acid)tripeptide, was found to be conserved in the Asia 1 isolates compared, whereasresidues on both sides were highly variable (Fig. 3). Systematic replacementof amino acid residues within the RGD loop of FMDV type C-S8c1 [16] hasshown that apart from the RGD triplet, two conserved leucine residues, locatedat positions +1 and +4 downstream to RGD and to a lesser extent, the residueat position +2 are the only critical and specific determinants within the loopin promoting cell-recognition of the virus. In Asia 1, the situation seems to beslightly different, since the leucine at +1 was not conserved, while the leucine at+4 and an alanine at +2 were totally conserved in all viruses except in GUS 48,where at +4, it was isoleucine.
Phylogenetic analysis
To examine the phylogenetic relationship betweenAsia 1 FMDV, we constructeda neighbor-joining tree from the complete nucleotide sequences of VP1 gene(Fig. 1). The tree showed 3 major branches, termed A, B and C. The majorbranches were supported with more than 70% bootstrap confidence. The branchA and C was represented by one isolate each from Pakistan (PAK/1/54) andChina, respectively. The branch B was further subdivided in to four lineages,designated B1, B2, B3 and B4. The branch B4 was represented by one isolatefrom Israel (Shamir/89), and B1, B2 and B3 comprised of 61 Indian isolates.From the above result it is evident that there are at least three different lineagesof FMDV type Asia 1 in India. Examination of phylogenetic distances basedupon the alignment of complete nucleotide sequence (633 nucleotides) of VP1gene indicated that the overall genetic distances between the 64 Asia 1 isolatescompared varied from 0.0096 to 0.2357 nucleotide substitutions per site. The
�Fig. 3. Alignment of deduced 211 amino acids of Asia 1 FMDV in the VP1 protein. Dotsrepresent sequence identity with the consensus, amino acid changes relative to the consensusare indicated using single letter codes, asterisk represents missing amino acid, gap in thesequence indicates region not sequenced. Cell recognition RGD motif is shown in bold-faced letters. Protein secondary structures (�-helix and �-sheet) corresponding to FMDV O1BFS [1] are indicated on top

98 C. B. Gurumurthy et al.
distance observed in Indian isolates ranged from 0.0096 to 0.1873 nucleotidesubstitutions per site.
The predicted amino acid sequences from all the above isolates and the 6 otherisolates whose amino acid sequences only were available [14, 25] were used toconstruct a phylogenetic tree (Fig. 4). On the basis of the clustering pattern theisolates from Bangladesh (BAN 5/87, BAN 2/96 and BAN 4/96) and India weregrouped together.Although one isolate from the former USSR (GUS 48) groupedwith the isolates from China, they showed about 15% divergence at amino acidlevel. One isolate from Thailand (TAI 5/96) grouped separately from the restof the isolates. Two isolates from Israel (Shamir/89 and ILKK/85) with 98.6%identity at amino acid level [14] were grouped separately from the others in thephylogenetic tree.
Estimation of evolutionary rate
To calculate the rate of evolution in Asia 1 viruses a monophyletic cluster wasselected within the Indian isolates, which spanned a period of 7 years (1993 to1999) (Fig. 1). We have taken a subset of data (forming a monophyletic cluster)from the (Indian) lineage B2 (Fig. 1) for the calculation of rate of evolution inAsia 1 viruses. Study with type A viruses from Europe has shown that someof the outbreaks were originated from the vaccine virus [3]. Further it has beensuggested that outbreak strains with less than 2% nucleotide diversity were epi-demiologically related to each other [30]. Though the lineages B1 and B3 formindividual monophyletic clusters, they were not taken in the calculation as bothincluded the vaccine strains and some strains epidemiologically related to thevaccine virus (Fig. 1). Inclusion of vaccine related strains in the calculation couldlead to erroneous results. In the monophyletic cluster analyzed, IND 53/93 wasfound to be the oldest isolate and hence maybe regarded as the common ancestorfor all the viruses in the cluster. Therefore we estimated the genetic distances ofeach isolate from this virus (IND 53/93) in the cluster. Synonymous and non-synonymous changes were plotted separately. The linear regression analysis ofthe genetic distances has enabled us to estimate the rate of evolution in Asia 1FMDV. The results of the estimation are shown in Table 3. The rate of evolutionof FMDV Asia 1 was estimated to be 2.7 × 10−2 synonymous substitutions pernucleotide per year. This is similar to that observed (0.9 to 7.4 × 10−2) in ani-mals persistently infected with FMDV serotype C [9] and in the case of anotherpicornavirus, enterovirus 71 (1.35× 10−2) [4]. The nonsynonymous substitutionrate was estimated to be 1.1 × 10−3 substitutions per nucleotide per year.
Molecular epidemiology
FMD is endemic in India and affects a vast population of susceptible animals.Though vaccination is the only choice of disease control in India, only∼2% of thesusceptible population are vaccinated annually. Therefore, the disease becomessevere, as there is not enough immune population to restrict the spread of dis-ease. The situation is further complicated due to selection of antigenic variants

Sequence analysis of FMDV Asia 1 99
Fig. 4. Neighbor-joining tree based on deduced amino acid sequence of VP1 protein. Thescale bar below the tree indicates genetic distance

100 C. B. Gurumurthy et al.
in presence of antibodies either from previous vaccination or infection leading toprevalence of multiple lineages as revealed in this study.
Of the three lineages identified in India, lineage B2 seems to be dominant asit is responsible for the majority of the outbreaks. This lineage is responsible formost of the recent outbreaks and distributed at least in nine different states in thenorthern and the southern parts of the country. Further, the prevalence of virusesfrom more than one genetic lineage is seen in the same state/region within aperiod of one year. The above observations are to be examined in the light of thefact that there is no restriction on animal movement in the country, except thoseimposed by natural barriers. When outbreaks occur year round and in all parts ofthe country, unrestricted animal movement could introduce new strains into areaswhere they were not quite prevalent, and these new strains could dominate overstrains that are currently prevalent at that place. The endemicity of the diseaseand the quasispecies nature of the causative virus contribute to the continuousevolution of FMD viruses in the subcontinent and the small percentage of vac-cinated animals also play a minor, nevertheless important role in the emergenceof dominant viral strains in a region. This is evident in lineage B1, where a clearepidemiological relationship could be established between the vaccine virus (IND63/72) and some of the field isolates. This could either be due to inadvertent re-lease of vaccine virus in the field or incomplete inactivation of the virus duringvaccine preparation.
Alignment of amino acid sequences from isolates of Bangladesh with Indianisolates showed a close relationship (92–96.7%) at amino acid level (comparisonat the nucleotide level was not made since sequences were not available for theseviruses). In a recent report [14], though the Indian vaccine strain (IND 63/72) wasshown to be different from the isolates of Bangladesh, our present studywith largenumber of recent field isolates, shows that they are more related to the virusesfrom India. This is not surprising as the two countries share a land border.
Conclusions
Foot-and-mouth disease has been, and still is a major livestock disease, whichis endemic in India and type Asia 1 is the second most prevalent serotype in thecountry. Sequencing and phylogenetic analysis of 64 Asia 1 viruses distributedthem into four different genotypes: the prototypeAsia 1 strain, PAK/1/54, formedgenotype I; genotype II was formed of the 61 viruses from India, while genotypeIII and IVwere represented by one isolate each from Israel (Shamir/89) andChina,respectively. The Indian Asia 1 viruses clustered into three genetic lineages ofwhich viruses of lineage B2 seemed to have evolved in the recent past. A highdegree of genetic heterogeneity was observed among Indian isolates and theirdistribution in the country probably reflects the lack of restriction on animalmovement. The results also provide evidence in support of outbreaks attributableto the vaccine virus. Baseline information on the genetic diversity of Asia 1viruses as is generated in this work would be of great assistance in the continuousepidemiological surveillance that is invaluable to understand the natural history

Sequence analysis of FMDV Asia 1 101
of viruses circulating in an FMD-endemic country like India. Further, such datawould also be useful at the time of launching a control programme.
Acknowledgement
We are thankful to the Indian Council of Agricultural Research and the Director, IndianVeterinary Research Institute for providing necessary facilities to carry out this work.
References
1. Acharya R, Fry E, Stuart D, Fox G, Rowlands D, Brown F (1989) The three-dimensionalstructure of foot-and-mouth disease virus at 2.9Å resolution. Nature 337: 709–716
2. Ansell DM, Samuel AR, Carpenter WC, Knowles NJ (1994) Genetic relationships be-tween Foot-and-mouth disease type Asia 1 viruses. Epidemiol Infect 112: 213–224
3. Beck E, Strohmaier K (1987) Subtyping of European FMD virus strains by nucleotidesequence determination. J Virol 61: 1621–1629
4. Brown BA, Oberste MS, Alexander Jr JP, Kennett ML, Pallansch M (1999) Molecularepidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. JVirol73: 9969–9975
5. Domingo E, Escarmis C, Martinez MA, Martinez-Salas E, Mateu MG (1992) Foot-and-mouth disease virus populations are quasispecies. Curr Top Microbiol Immunol 176:33–47
6. Domingo E, Holland JJ (1997) DNA virus mutations and fitness for survival. Ann RevMicrobiol 51: 151–178
7. Felsenstein J (1993) PHYLIP inference Package, Version 3.5c. Department of Genetics,University of Washington, Seattle, WA, USA
8. Forss S, Strebel K, Beck E, Schaller H (1984) Nucleotide sequence and genome organi-zation of foot-and-mouth disease virus. Nucleic Acids Res 12: 6587–6601
9. Gebauer F, de La Torre JC, Gomes I, MateuMG, Barahona H, Tiraboschi B, Bergmann I,Auge de Mello P, Domingo E (1988) Rapid selection of genetic and antigenic variantsof foot-and-mouth disease virus during persistence in cattle. J Virol 62: 2041–2049
10. Hemadri D, Tosh C, Venkataramanan R, Sanyal A, Samuel AR, Knowles NJ,Kitching RP (2000) Genetic analysis of foot-and-mouth disease virus type O isolatesresponsible for field outbreaks in India between 1993 and 1999. Epidemiol Infect 125:729–736
11. Knowles NJ, Ansell DM, Samuel AR (1998) Molecular comparison of recent foot-and-mouth disease typeA viruses fromWestAfrica with historical and reference virus strains.Rpt Sess Res Gp Stand Tech Comm Eur Comm Control of FMD (FAO), Aldershot, UK,pp 41–48
12. Knowles NJ, Samuel AR (1994) Polymerase chain reaction amplification and cyclesequencing of the 1D (VP1) gene of foot-and-mouth disease viruses. Rpt Sess Res GpStand Tech Comm Eur Comm Control of FMD (FAO), Vienna, Austria, pp 45–53
13. Marquardt O, Haas B (1998)VP1-coding sequences of recent isolates of foot-and-mouthdisease virus types A, O and Asia 1. Virus Genes 16: 185–193
14. Marquardt O, Rahman MM, Freiberg B (2000) Genetic and antigenic variance of foot-and-mouth disease virus type Asia 1. Arch Virol 145: 149–157
15. Mateu MG (1995) Antibody recognition of Picornaviruses and escape from neutraliza-tion: a structural view. Virus Res 38: 1–24
16. Mateu MG,Valero ML,Andreu D, Domingo E (1996) Systematic replacement of aminoacid residues within an Arg-Gly-Asp-containing loop of foot-and-mouth disease virusand effect on cell recognition. J Biol Chem 271: 12814–12819

102 C. B. Gurumurthy et al.: Sequence analysis of FMDV Asia 1
17. Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymousand nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426
18. Pattnaik B, Venkataramanan R, Tosh C, Sanyal A, Hemadri D, Samuel AR, KnowlesNJ, Kitching RP (1998) Genetic heterogeneity of Indian field isolates of foot-and-mouthdisease virus serotype O as revealed by partial sequencing of 1D gene. Virus Res 55:115–127
19. Pereira HG (1977) Subtyping of foot-and-mouth disease virus. Dev Biol Stand 35:167–174
20. Rico-Hesse R, Pallansch MA, Nottay BK, Kew OM (1987) Geographic distribution ofwild polio virus type 1 genotypes. Virol 160: 311–322
21. Rozas J, Rozas R (1999) DnaSP version 3: an integrated program for molecular popula-tion genetics and molecular evolution analysis. Bioinformatics 15: 174–175
22. Saiz JC, Sobrino F, Dopazo J (1993)Molecular epidemiology of foot-and-mouth diseasevirus type O. J Gen Virol 74: 2281–2285
23. Samuel AR, Knowles NJ, Kitching RP, Hafez SM (1997) Molecular analysis of typeO foot-and-mouth disease viruses isolated in Saudi Arabia between 1983 and 1995.Epidemiol Infect 119: 381–389
24. Samuel AR, Knowles NJ (2001) Foot-and-mouth disease type O viruses exhibit genet-ically and geographically distinct evolutionary lineages (topotypes). J Gen Virol 62:609–621
25. Sonsnovtsew SV, Onishchenko AM, Petrov NA, Mamaeva NV, Kalashnikova TI,Perevozchikova NA, Ivaniushchenkov VN, Burdov AN, Vasilenko SK (1989) Primarystructure of the gene for VP1 protein of the foot-and-mouth disease virus of Asia 1serotype. Mol Gen Microbiol Virusol 12: 44–46
26. StramY, Chai D, Fawzy HED, Molad T, Meiri N, Van-Ham M, El-Kilani S, Fahamy F,Moussa AAM, Yadin H (1995) Molecular epidemiology of foot-and-mouth disease inIsrael in 1994 and in other Middle-Eastern countries in the years 1992–1994.ArchVirol140: 1791–1797
27. Stram Y, Laor O, Molad T, Chai D, Moore D, Yadin H, Becker Y (1994) Nucleotidesequence of the P1 region of serotypeAsia 1 foot-and-mouth disease virus. Virus Genes8: 275–278
28. Thomson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity ofprogressivemultiple sequence alignment through sequenceweighting, positions-specificgap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680
29. Tosh C, Hemadri D, Sanyal A, Pattnaik B, Venkataramanan R (1997) One-tube andone-buffer system of RT-PCR amplification of 1D gene of foot-and-mouth disease virusfield isolates. Acta Virol 41: 153–155
30. Vosloo W, Kirkbride E, Bengis RG, Thomson GR (1995) Genomic variation in the SATtypes of FMD viruses prevalent in buffalo (Syncerus caffer) in the Kurger National Parkand other regions of South Africa, 1986–93. Epidemiol Infect 114: 203–218
Author’s address: Dr. R. Venkataramanan, Project Directorate on Foot-and-Mouth Dis-ease, Indian Veterinary Research Institute, Mukteswar-Kumaon, Nainital 263 138, India;e-mail: [email protected]
Received May 7, 2001




















