
A nonlinear, recurrence-based approach to traffic classification
Upload
independentCategory
view
0download
0
Genes Associated With Progression and Recurrenceof Hepatocellular Carcinoma in Hepatitis C Patients
Waiting and Undergoing Liver Transplantation:Preliminary Results
Valeria R. Mas,1,2 Robert A. Fisher,1,5 Kellie J. Archer,3,4 Kenneth C. Yanek,1 Bridgette Williams,1
Catherine I. Dumur,2 and Daniel G. Maluf 1
Background. Liver transplantation (LT) represents a curative treatment for small hepatocellular carcinoma (HCC).Potentially curable higher-stage HCC patients are denied LT due to the lack of cancer markers that predict progressionand recurrence.Methods. Thirty-eight candidates for LT with hepatitis C virus (HCV) cirrhosis and HCC were studied. Gene expres-sion (Gexp) analysis of tumor samples was performed using microarrays.Results. Twenty patients underwent transplantation, 13 progressed while waiting for transplantation, 4 are aliveawaiting transplantation, and 1 died without progression while waiting for LT. Differences in GExp among patientswho underwent LT or did not progress (n�25) versus those whose disease progressed while waiting for LT (n�13) wereassessed. Thus, 54 probe sets (Pset) were significantly differentially expressed. Among LT patients, 10 Psets weredifferentially expressed between LT patients with the same explanted stage that recurred (n�5) versus LT patients whodid not recur (n�5). Ninety-eight Psets were significantly associated with survival at the ��0.005 level.Conclusions. Here, we have identified genes associated with HCC progression in HCV-HCC patients awaiting LTtransplantation. A limited number of genes were related to overall survival and cancer-free survival after LT. Incorpo-ration of these molecular markers could help to improve organ allocation for HCV-HCC patients.
Keywords: Liver transplantation, Hepatocellular carcinoma, Gene expression analysis.
(Transplantation 2007;83: 973–981)
Hepatocellular carcinoma (HCC) is the most commonprimary solid tumor of the liver (1–3). Although much is
known about both the cellular changes that lead to HCC andthe etiological agents responsible for the majority of HCC, themolecular pathogenesis of HCC is not well understood.
The poor prognosis of HCC is largely the result of ahigh rate of recurrence after surgery or of intrahepatic metas-tases (4, 5). Liver transplantation (LT) represents the besttreatment option for patients with early stages of HCC (4 –7),offering the advantage of radical tumor removal. However,until organ availability improves, transplantation for HCChas been offered to patients whose survival is predicted to besimilar to that in patients transplanted for non-HCC disease(5, 6). Despite the use of living donor liver allograft and an
allocation point system to benefit HCC patients, a notewor-thy proportion of patients are excluded from LT because oftumor progression (6 –9). Moreover, there is not an agree-ment in the selection criteria that could simultaneouslymaximize the number of viable HCC candidates for trans-plantation and reject the smallest number of those who couldbenefit (10).
The current clinical results, using LT in HCC patientsobtained using the established criteria for patient selection,lack specificity (10 –12). As a consequence, the necessity ofdeveloping tools to refine HCC patient selection is vital toexpanding utility of this therapy.
HCC heterogeneity might be a consequence of the dif-ferent molecular defects that can induce similar tumor phe-notypes. The prognostic variability likely reflects a molecularheterogeneity that has not been appreciated from methodstraditionally used to characterize HCC (13). Improving theclassification of HCC patients into groups with homogeneousprognosis, as well as a more comprehensive understanding ofthe underlying biology of HCC development at the molecularlevel, would improve patient selection for the currently avail-able treatment modalities.
The analysis of gene expression profiles using microar-rays offers the possibility for identifying new approaches fordiagnosing and stratifying patients with regard to risk of pooroutcome and adds new diagnostic and prognostic informa-tion not provided by standard clinical and laboratory exam-inations (14 –18). This approach has resulted in successfulmolecular classification of several human malignant tumorswith respect to their stage, prognostic outcome, or responseto therapy (19, 20).
This project was partially supported by the Commonwealth Foundation forCancer Research as a pilot project of the Massey Cancer Center.
1 Division of Transplantation, Department of Surgery, Virginia Common-wealth University, Richmond, VA.
2 Department of Pathology, Virginia Commonwealth University, Richmond,VA.
3 Department of Biostastistics, Virginia Commonwealth University, Richmond,VA.
4 Center for the Study of Biological Complexity, Virginia CommonwealthUniversity, Richmond, VA.
5 Address correspondence to: Robert A. Fisher, M.D., FACS, Division ofTransplant, Department of Surgery, West Hospital, 9th Floor, Room 313,1200 East Broad Street, PO Box 980057, Richmond VA 23298-0057.
E-mail: [email protected] 5 January, 2007.Accepted 5 January, 2007.Copyright © 2007 by Lippincott Williams & WilkinsISSN 0041-1337/07/8307-973DOI: 10.1097/01.tp.0000258643.05294.0b
Transplantation • Volume 83, Number 7, April 15, 2007 973

In this preliminary study, we compared gene expres-sion profiles by identifying genes implicated in tumor pro-gression in hepatitis C virus (HCV)-HCC patients awaitingtransplantation. In addition, we analyzed differences in geneexpression for distinguishing HCC patients with a potentialfor developing recurrence after LT.
PATIENTS AND METHODSFrom December 1997 to February 2004, 163 patients
diagnosed with cirrhosis and HCC were evaluated at theHume-Lee Transplant Center, Medical College of VirginiaHospitals. The Institutional Review Board at Virginia Com-monwealth University approved the study protocol. Afterstaging, based on serial liver magnetic resonance imaging(MRI), chest computed tomography (CT), bone scan, alpha-fetoprotein (AFP), and a physical examination, the patientswere listed for LT according to the United Network for OrganSharing (UNOS) criteria. Restaging occurred every 3 monthswhile awaiting transplantation. This study was restricted tothe HCV-HCC patients. Thirty-eight HCV patients with di-agnosis of HCC (HCV-HCC) were recruited in the studysince they were enlisted for LT (Table 1). Three ablation tech-niques previously characterized (6) were utilized as primarytreatment for ablating HCC, including: transcatheter hepaticarterial chemoembolization (TACE); transcatheter hepaticarterial chemoinfusion (TACI); and radiofrequency micro-wave ablation (RFA). Histopathological classification wasperformed according to the Edmondson grading system.Clinical stages were determined according to the AmericanTumor Study Group modified tumor-node-metastasis (TNM)classification mandated by UNOS (21). Tumor tissue sampleswere collected from biopsies (n�18) and explanted livers(n�20) from 38 HCV-HCC patients. Tumor samples withmore than 85% of tumor cell content were used for the mi-croarray studies.
In addition, six liver samples were obtained from ex-planted liver donors chosen for isolated hepatocyte prepara-tion for transplantation. Liver function and histopathologyfor these liver donors were shown to be normal. All six pa-tients were seronegative for HCV antibody (Ab). These sam-ples were used to perform class comparisons for filtering thedata.
RNA Isolation, cDNA Synthesis, and In VitroTranscription for Labeled cRNA Probe
The sample preparation protocol follows the Af-fymetrix GeneChip Expression Analysis Manual (SantaClara, CA). Total RNA was extracted from tumor tissue sam-ples using TRIzol (Life Technologies, Rockville, MD). Integ-rity of RNA was checked using Agilent 2100 Bioanalyzer.Briefly, total RNA was reverse-transcribed using T7-polydTprimer and converted into double-stranded cDNA (One-Cycle Target Labeling and Control Reagents, Affymetrix,Santa Clara, CA), with templates being used for an in vitrotranscription reaction to yield biotin-labeled antisense cRNA.The labeled cRNA was chemically fragmented and made intothe hybridization cocktail according to the AffymetrixGeneChip protocol, which was then hybridized to HG-U133A and U133A 2.0 GeneChips. The array image was gen-erated by the high-resolution GeneChip Scanner 3000 by
Affymetrix (Affymetrix, Santa Clara, CA). The data was ana-lyzed using Affymetrix Microarray Suite Software, Version5.0 and Bioconductor packages (22) available in the R pro-gramming environment (23).
Data AnalysisProbe level data from the two different sets of
GeneChips (11 HG-U133A and 27 HG-U133Av2) were com-bined by probe sequence, using the matchprobes Bioconduc-tor package, then probe set expression summaries wereestimated using the robust-multiarray average (RMA) (24,25) method. Quality assessment was performed by assessingthe average background, scaling factor, RNA degradationplots, percent present calls, and 3�:5� ratios of glyceraldehyde-3-phosphatase dehydrogenase (GAPDH) and �-actin. Prior toconducting statistical analyses, all control probe sets were re-moved, leaving 22,215 probe sets for statistical analysis.
TABLE 1. Characteristics of patients and tumors
Characteristic n
n 38
Sex
Female 5
Male 33
Age, mean years�SD (range) 55.1�7.8 (41–75)
Race
White 24
Black 7
Hispanic 5
Other 2
Number of tumors
One 19
Two 12
Three 6
Four� 1
Vascular invasion
Microvascular 36
Macrovascular 2
Lobar distribution
Right 22
Left 4
Bilobar 12
Pretransplant stage
T1N0M0 1
T2N0M0 17
T3N0M0 19
T4N0M0 0
Explant stage
T1N0M0 1
T2N0M0 9
T3N0M0 5
T4N0M0 5
Cirrhosis
Yes 38
No 0
974 Transplantation • Volume 83, Number 7, April 15, 2007

Procedure for Filtering GenesThe six normal liver samples were used to reduce the
number of genes for subsequent analyses. Specifically, the sig-nificance analysis of microarray method (SAM) (26) was usedto perform class comparisons of the 6 normal versus 38 can-cerous tissues by probe set. All probe sets identified as signif-icantly differentially expressed at a false discovery rate of 10%(using ��1.0) were retained (n�13,741 probe sets) for sub-sequent analyses involving HCV-HCC samples.
ClusteringTo determine whether the 38 HCV-HCC samples clus-
tered into homogeneous groups, partitioning around me-diods (PAM) (27) was used where the number of clusters wasallowed to range from k�2, . . ., 7. The average silhouettewidth, which provides an estimate of strength of the cluster-ing results, was used to determine the most appropriate valueof k. Specifically, for each GeneChip i, the silhouette valuess(i) were calculated as follows: let A denote the assigned clus-ter (assuming A is not a singleton) and a(i) represent theaverage dissimilarity of i to all other objects in A. When con-sidering cluster C different from A, let d(i, C) represent theaverage dissimilarity of i to all objects in C. After computing d(i, C) for all clusters C�A, let b(i)�minC�A d(i, C). That is,cluster B would be the second best cluster assignments forobject i. The silhouette value s(i)�b(i)�a(i)/max{a(i), b(i)}measures how well object i matches the cluster assignment athand, with the property �1�s(i)�1. The average silhouettewidth for a partitioning of i�1, . . ., N objects into k clusters,s̄(k), was used for selecting the best value of k, by choosingthat k for which s̄(k) is a maximum. Multidimensional scalingwas used to display the resulting k clusters.
Class ComparisonsClass comparisons were performed using Welch’s t test
and an adjusted �-level to control for multiple hypothesistesting. Survival analytic methods were also used to accountfor the time-dependent and censored data values associatedwith time to progression, recurrence, and death. Patient sur-vival was defined to be the number of months from date ofdiagnosis until the date of death or last follow-up, censoringfor patients still alive. Among patients transplanted, the prob-ability of survival was estimated using the Kaplan-Meiermethod for explant stage. The groups were compared usingthe log-rank test.
In addition, the time-dependent covariate “transplant”was modeled using Cox proportional hazards model to ac-count for potential differences in survival among patientstransplanted versus not-transplanted. An analysis restrictedto transplanted patients sought to identify genes significantlyassociated with cancer-free survival. Cancer-free survival wasdefined as the time from transplant to recurrence or death,censoring for patients still alive.
N-fold Cross ValidationN-fold cross-validation was used to provide an unbi-
ased estimate of generalization error when classifying patientsas progressed while waiting for transplant versus did notprogress. The analysis started with all probe sets that wereidentified as significantly differentially expressed when com-
pared the normal to the HCV-HCC tissue samples. One at atime each patient’s sample was left out and Welch’s t test wasused for identifying genes significantly differentially ex-pressed comparing the two groups (� level�0.005). After-ward, k-nearest neighbors (knn) (28) was used to construct aclassifier using all but the left out sample as the training set.The left-out sample was then used as a test set. N-fold cross-validation was also used in conjunction with the support vec-tor machine (SVM) method (29). For both knn and SVM, thegeneralization error was estimated as the percent of misclas-sified test samples.
Validation of Microarray ResultsWe carried out a quantitative reverse transcriptase real-
time polymerase chain reaction (PCR) for OAS1, GPC3, andEGFR mRNAs from the same RNAs samples that were sub-jected to microarray study. OAS1 was differentially expressedin HCC patients with recurrence, and GPC3 and EGFR weredifferentially expressed in the tumor samples when comparedto normal tissues. Total RNA was subjected to reverse tran-scription using TaqMan Reverse Transcription Reagents(Applied Biosystems, Foster City, CA) according to the man-ufacturer’s protocol. Real-time PCR reactions then were car-ried out in a 25-�L reaction mixture in an ABI Prism 7700Sequence Detection System, using TaqMan PreDevelopedAssay Reagents for Gene Expression (Applied Biosystems).Data was analyzed according to the comparative cycle thresh-old (Ct) method and was normalized by GAPDH expressionin each sample. Pearson’s correlation coefficient (r) was cal-culated to examine the relation between microarray and real-time PCR results. P values �0.05 were considered significant.
RESULTS
Patients and SamplesCharacteristics of the 38 HCV-HCC patients and tu-
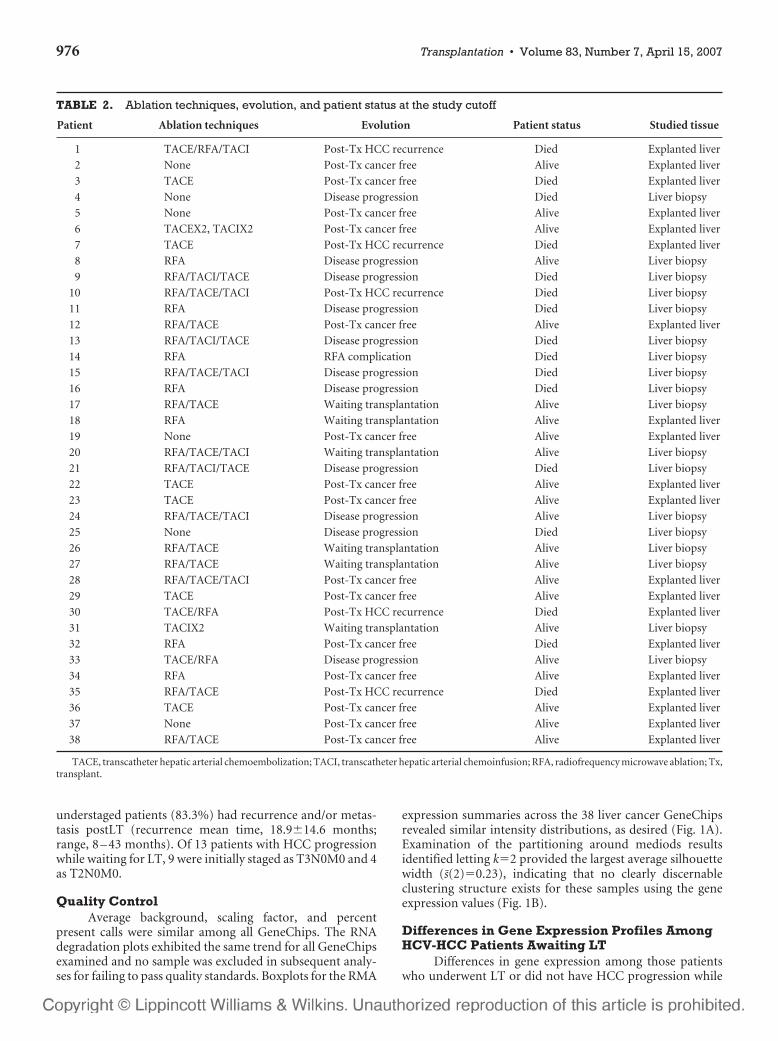
mors are presented in Table 1. AFP levels at diagnosis were55.9�114.3 ng/dL (range 2.0 –538.0 ng/dL). No associationwas found between AFP levels at diagnosis and pretransplantHCV-HCC stage (P�0.40). Thirty-two patients received ab-lation therapy (1.9�1.4 procedures/patient; Table 2). Sixteenof the 32 ablated patients were followed by LT. Of the 38studied patients, 20 (52.6%) underwent LT, 13 (34.2%) HCCprogressed while waiting for transplantation, 4 (10.5%) arealive awaiting transplantation, and 1 (2.6%) died withoutprogression due to clinical complications while waiting fortransplantation (mean follow-up time among patients stillalive, 26.4�21.7 months; range, 24 – 88 months). HCC pro-gression was defined as an increase or advance in the TNMstage (compared with diagnostic HCC stage) and determinedby radiological studies. For patients who underwent LT(n�20) or who progressed while waiting transplantation(n�13), there was no significant difference between time totransplant and time to progression (9.1�10.4 vs. 9.5�11.3months, P�0.93).
The pretransplant HCC stage for the studied patients atthe on-study time included: 2 T1N0M0, 17 T2N0M0, and 19T3N0M0. However, when pretransplant liver MRIs werecompared to the findings of the explanted livers (for those 20patients undergoing LT), 40.0% of the patients (n�8) werefound to be under- (n�6) or overstaged (n�2). Five of the six
© 2007 Lippincott Williams & Wilkins 975Mas et al.
understaged patients (83.3%) had recurrence and/or metas-tasis postLT (recurrence mean time, 18.9�14.6 months;range, 8 – 43 months). Of 13 patients with HCC progressionwhile waiting for LT, 9 were initially staged as T3N0M0 and 4as T2N0M0.
Quality ControlAverage background, scaling factor, and percent
present calls were similar among all GeneChips. The RNAdegradation plots exhibited the same trend for all GeneChipsexamined and no sample was excluded in subsequent analy-ses for failing to pass quality standards. Boxplots for the RMA
expression summaries across the 38 liver cancer GeneChipsrevealed similar intensity distributions, as desired (Fig. 1A).Examination of the partitioning around mediods resultsidentified letting k�2 provided the largest average silhouettewidth (s̄(2)�0.23), indicating that no clearly discernableclustering structure exists for these samples using the geneexpression values (Fig. 1B).
Differences in Gene Expression Profiles AmongHCV-HCC Patients Awaiting LT
Differences in gene expression among those patientswho underwent LT or did not have HCC progression while
TABLE 2. Ablation techniques, evolution, and patient status at the study cutoff
Patient Ablation techniques Evolution Patient status Studied tissue
1 TACE/RFA/TACI Post-Tx HCC recurrence Died Explanted liver
2 None Post-Tx cancer free Alive Explanted liver
3 TACE Post-Tx cancer free Died Explanted liver
4 None Disease progression Died Liver biopsy
5 None Post-Tx cancer free Alive Explanted liver
6 TACEX2, TACIX2 Post-Tx cancer free Alive Explanted liver
7 TACE Post-Tx HCC recurrence Died Explanted liver
8 RFA Disease progression Alive Liver biopsy
9 RFA/TACI/TACE Disease progression Died Liver biopsy
10 RFA/TACE/TACI Post-Tx HCC recurrence Died Liver biopsy
11 RFA Disease progression Died Liver biopsy
12 RFA/TACE Post-Tx cancer free Alive Explanted liver
13 RFA/TACI/TACE Disease progression Died Liver biopsy
14 RFA RFA complication Died Liver biopsy
15 RFA/TACE/TACI Disease progression Died Liver biopsy
16 RFA Disease progression Died Liver biopsy
17 RFA/TACE Waiting transplantation Alive Liver biopsy
18 RFA Waiting transplantation Alive Explanted liver
19 None Post-Tx cancer free Alive Explanted liver
20 RFA/TACE/TACI Waiting transplantation Alive Liver biopsy
21 RFA/TACI/TACE Disease progression Died Liver biopsy
22 TACE Post-Tx cancer free Alive Explanted liver
23 TACE Post-Tx cancer free Alive Explanted liver
24 RFA/TACE/TACI Disease progression Alive Liver biopsy
25 None Disease progression Died Liver biopsy
26 RFA/TACE Waiting transplantation Alive Liver biopsy
27 RFA/TACE Waiting transplantation Alive Liver biopsy
28 RFA/TACE/TACI Post-Tx cancer free Alive Explanted liver
29 TACE Post-Tx cancer free Alive Explanted liver
30 TACE/RFA Post-Tx HCC recurrence Died Explanted liver
31 TACIX2 Waiting transplantation Alive Liver biopsy
32 RFA Post-Tx cancer free Died Explanted liver
33 TACE/RFA Disease progression Alive Liver biopsy
34 RFA Post-Tx cancer free Alive Explanted liver
35 RFA/TACE Post-Tx HCC recurrence Died Explanted liver
36 TACE Post-Tx cancer free Alive Explanted liver
37 None Post-Tx cancer free Alive Explanted liver
38 RFA/TACE Post-Tx cancer free Alive Explanted liver
TACE, transcatheter hepatic arterial chemoembolization; TACI, transcatheter hepatic arterial chemoinfusion; RFA, radiofrequency microwave ablation; Tx,transplant.
976 Transplantation • Volume 83, Number 7, April 15, 2007

awaiting LT (n�25) versus those whose disease progressedwhile waiting for LT (n�13) were assessed using Welch’s ttest (��0.005). Fifty-four probe sets were significantly differ-entially expressed (Table 3) (Fig. 2).
Genes encoding ribosomal proteins (i.e., RPL27,RPL21, RPS7) were relatively highly expressed in patientswith HCC progression during the waiting time, an expressionpattern characteristically observed in growing cells (18).HDAC1 (histone deacetylase 1) was also overexpressed in theprogression group. The protein encoded by this gene is acomponent of the histone deacetylase complex. It has beendemonstrated that p53 activity is upregulated in part by post-translational acetylation (30). Together with metastasis-
associated protein-2, it deacetylates p53 and modulates itseffect on cell growth and apoptosis.
MT1M (metallothionein 1M) expression was down-regulated in those patients with HCC progression whileawaiting transplantation. The expression and induction ofthe metallothioneins have been associated with protectionagainst DNA damage, oxidative stress, and apoptosis (31).
SOCS-1 (suppressor of cytokine signaling-1) is a nega-tive regulatory of Janus kinase and signal transducer andactivation of transcription pathway and its expression wasunder regulated in the progression group. Recently (32), itwas demonstrated that SOCS-1 gene was silenced frequentlyby methylation in HCC. However, its role in HCC progres-sion remains unclear.
The expression of PTPRB (protein tyrosine phospha-tase, receptor type, B) was also downregulated. The proteinencoded by this gene is a member of the protein tyrosinephosphatase (PTP) family. PTPs are known to be signalingmolecules that regulate a variety of cellular processes includ-ing cell growth, differentiation, mitotic cycle, and oncogenictransformation (33). As a final point, genes related to apopto-sis induction were downregulated in the progression group(i.e., GADD45B, ZBTB16). Specifically, GADD45B is a mem-ber of a group of genes whose transcript levels are increasedfollowing stressful growth arrest conditions and treatmentwith DNA-damaging agents. The function of these genes, ortheir protein products, is involved in the regulation of growthand apoptosis. These genes are regulated by different mecha-nisms, but they are often coordinately expressed and canfunction cooperatively in inhibiting cell growth.
N-fold Cross-validationTo estimate generalization error for a classifier derived
using the limited number of samples we had available fortraining, we used N-fold cross-validation (CV). Specifically,in classifying a patient as either progressed while waiting fortransplant or did not progress, we first began with the 13,471probe sets differentially expressed between normal and HCC
FIGURE 1. (A) Boxplots for the RMA expression summaries across the 38 liver cancer samples. The plot revealed similarintensity distributions, as desired. (B) The average silhouette width, which provides an estimate of strength of the clusteringresults, was used to determine the most appropriate value of k. Examination of the partitioning around mediods resultsidentified letting k�2 provided the largest average silhouette width (s̄(2)�0.23). The observed average silhouette widthestimates were very small for all values of k(s̄(k)�0.25 indicates no discernable structure), indicating that no clearlydiscernable structure exists in these data.
TABLE 3. Relevant genes related to HCC progression
ProcessGene
symbolLocuslink
Foldchange
Ubiquitin-dependent proteincatabolism
USP22 23326 3.22
Negative regulation ofprogression through cellcycle
GMNN 51053 3.77
Protein biosynthesis RPL27 6155 3.26
RPL21 6144 3.27
RPL0 6175 3.52
RPL30 6156 3.59
Antiapoptosis HDAC1 3065 3.51
Oxidative stress and apoptosis MT1M 4499 �3.68
GADD45B 4626 �3.27
ZBTB16 7704 �4.17
Regulation of cell cycle SOCS1 8651 �4.51
Regulation of transcription DSCR1 1827 �3.25
TCEB3 6924 �3.21
ETS2 2114 �3.13
© 2007 Lippincott Williams & Wilkins 977Mas et al.

tissue samples, identified from the SAM analysis. One at atime, each patient’s gene expression vector was left out andthe remaining patients’ gene expression values were used toselect differentially expressed genes comparing those patientswho underwent or did not have HCC progression whileawaiting LT (n�25) versus those whose disease progressedwhile waiting for LT (n�13) at � level�0.005. A k-nearestneighbors classifier was derived using the n-1 observation,which was subsequently used to classify the left-out sample.The same N-fold CV procedure was applied for support vec-tor machines (SVM). From the N-fold procedure, the averagenumber of significant probe sets was 56 (range 35–121). Thegeneralization error estimated using N-fold cross-validationwas 39.5% for k-nearest neighbors and 34.2% for SVM.
Differentially Expressed Genes Among LTPatients With Advanced HCC Stages With andWithout Recurrence
Among LT patients with an explanted stage of ad-vanced HCV-HCC, 10 probe sets were differentially expressedcomparing those that had recurrence (n�5) versus those thathad no recurrence (n�5; Supplemental Table 1, http://www.vcohealth.org/transplant/text-research.htm). The most note-worthy changes in gene expression affected the transcrip-tional network regulated by interferons (IFNs), specificallyIFN-�/�-inducible genes (STAT1, OAS1, MX1). All thesegenes are involved in the response to virus. In addition, theantiapoptotic gene FAIM3 was overexpressed in thesepatients. USP18 was overexpressed in patients with HCC re-currence and is a member of the deubiquitinating proteasefamily of enzymes, which removes ubiquitin adducts from abroad range of protein substrates. The under expressed genesin this group of patients included TFPI, HIST1H4E, andNRG1.
Genes Related to SurvivalNumber of tumors, lobar distribution, and Model for
End-Stage Liver Disease score were not significant predictors
of survival in the study group (P�0.20, P�0.50, and P�0.94,respectively). Age, race, and AFP levels at diagnosis were alsonot significant predictors of survival (P�0.10, P�0.67, andP�0.52, respectively). However, there were 98 probe sets thatwere significantly associated with survival after adjusting forthe time-dependent covariate “transplant” at the ��0.005level (Supplemental Table 2). To investigate the biologicalfunctions associated with the survival genes, we analyzed theGO category for the survival gene list.
UBE2D3 was one of the significant genes related tosurvival. The modification of proteins with ubiquitin is animportant cellular mechanism for targeting abnormal orshort-lived proteins for degradation. Ubiquitination involvesat least three classes of enzymes: ubiquitin-activating en-zymes (E1s), ubiquitin-conjugating enzymes (E2s), andubiquitin-protein ligases (E3s). This gene encodes a memberof the E2 ubiquitin-conjugating enzyme family. This enzymefunctions in the ubiquitination of the tumor-suppressor pro-tein p53, which is induced by an E3 ubiquitin-protein ligase.Genes related to ubiquitination have been recently related toHCC prognosis (18). Moreover, the proteasome is an ubiq-uitous enzyme complex that plays a critical role in the degra-dation of many proteins involved in cell cycle regulation,apoptosis, and angiogenesis. Because these pathways are fun-damental for cell survival and proliferation, particularly incancer cells, the inhibition of proteasome is an attractive po-tential anticancer therapy (34).
Another gene present in the survival list was DLC1. Thisgene is a candidate tumor suppressor gene for human livercancer, as well as for prostate, lung, colorectal, and breastcancers (35). The protein encoded by CDC14B gene, alsopresent in the survival gene list, is a member of the dual spec-ificity protein tyrosine phosphatase family. This protein hasbeen shown to interact with and dephosphorylates tumorsuppressor protein p53, and is thought to regulate the func-tion of p53. The other gene related to survival was CTTN. Itsaberrant regulation contributes to tumor cell invasion andmetastasis.
FIGURE 2. Two-dimensional hierarchical clustering results for differentially expressed genes between patients whounderwent or did not have HCC progression while awaiting LT (n�25) vs. those whose disease progressed while waiting forLT (n�13). This set was created using Pearson (centered) correlation cluster analysis, using the BRB-ArrayTools v3.0.2package. Each vertical column represents an independent experiment. The fold changes in mRNA levels in the samples arerepresented by green and red squares, showing decreased and increased levels between the RNA samples, respectively.The color scale indicates the magnitude of fold changes.
978 Transplantation • Volume 83, Number 7, April 15, 2007

In addition, the survivor gene list was enriched withgenes related to regulation of apoptosis (i.e., CFLAR,NGFRAP1). Of the 98 probe sets present in the gene survivallist, sixteen genes were annotated with the cellular compo-nent of the integral membrane proteins.
There were six probe sets that were significantly associ-ated with survival after adjusting for the time-dependent co-variate “HCC-progression” at the ��0.005 level. The probesets included CDH6 (cadherin 6), UROD (uroporphyrinogendecarboxylase), UGT2B28 (UDP glucuronosyltransferase 2family), CCDC76 (coiled-coil domain containing 76), RET(ret proto-oncogene), and YKT6 (YKT6 v-SNARE homolog(S. cerevisiae). CDH6 gene encodes a type II classical cadherinfrom the cadherin superfamily. Cadherins mediate cell-cellbinding. Strong transcriptional expression of this gene hasbeen observed in hepatocellular and renal carcinoma celllines, suggesting a possible role in metastasis and invasion(36, 37).
Finally, an analysis restricted to transplanted patientssought to identify genes significantly associated with cancer-free survival. Only one probe gene (USP14, ubiquitin specificpeptidase 14 (tRNA-guanine transglycosylase)) was signifi-cantly associated with cancer-free survival at the ��0.005level. The cancer-free survival gene list included eight probesets when a less conservative alpha-level (��0.01) was used(Table 4).
Gene Expression Quantitation Using Real-TimePCR
Expression levels of the OAS1, GPC3, and EGFRmRNAs were further confirmed using a quantitative reverse-transcriptase, real-time PCR. The results from the microarraywere reproduced by reverse-transcriptase, real-time PCR(r�0.78, P�0.001; ��0.82, P�0.001; and r�0.87, P�0.01respectively).
DISCUSSIONHCC is a complex disease resulting from multiple mu-
tations that induce dramatic changes in expression patternsof genes and proteins that function in networks controllingcritical cellular events (38 – 40). Characterization of the com-plex molecular networks that drive HCC development hasbeen facilitated by novel high-throughput technologies, in-
cluding DNA microarrays (13–17). This new technology hasbeen used successfully to predict clinical outcome and sur-vival as well as to classify different types of cancer (18, 19).
Although the first-line surgical therapy for HCC is liverresection, the concomitant cirrhosis most often leaves LT,and not liver resection, as the only potentially curative option.As a consequence of the growing shortage of organs availablefor transplantation plus the lack of predictive power of cur-rently used staging systems, the study of predictive markers ofrecurrence-free survival is critically needed.
The goal of all staging systems is to separate patientsinto groups with homogeneous prognosis, which then formthe bases for the selection of the most appropriate treatments.Much work has been devoted to establishing prognostic mod-els for HCC by using clinical information and pathologicalclassification in order to provide information at diagnosis onboth survival and treatment options (41, 42). However, manyissues still remain unresolved. The selection of candidates foreither resection or LT is continuously evolving and a redefi-nition of tumor criteria is required to further improve patientselection (43, 44). Molecular tumor profiling may offer a bet-ter estimation of risk of recurrence, and, consequently, im-proved selection of patients.
Several recent studies have employed the gene expres-sion profiling to address the issue of HCC recurrence follow-ing resection and intrahepatic metastasis (45, 46). Iizuka et al.(45) investigated gene expression profiles in tissue specimensfrom patients with HCC using high-density oligonucleotidemicroarrays. This training set was used to construct a predic-tive system, consisting of 12 genes. The predictive perfor-mance of the system was then compared on 27 newly enrolledpatients. The system correctly predicted early intrahepatic re-currence or nonrecurrence in 93% of the samples. Kurokawaet al. (46) addressed the issue of intrahepatic recurrence byanalyzing gene expression using a PCR-based array platformin 100 HCC patients. The authors selected 92 genes that dem-onstrated distinct expression patterns differing significantlybetween recurrence and recurrence-free cases.
In the present study, we analyzed genes involved indisease progression during the waiting time using geneexpression profiles in HCV-HCC patients awaiting LT. Werestricted the study to the HCV-HCC patients because differ-ences in HCC profiles have been associated with different
TABLE 4. Description of probe sets (AffyID) significantly related to cancer-free survival
Locus link Gene symbol Gene name Chromosome Map Hazard
23175 LPIN1 Lipin 1 2 2p25.1 276
771 CA12 Carbonic anhydrase XII 15 15q22 23.8
164 AP1G1 Adaptor-related protein complex 1, gamma 1 subunit 16 16q23 4.9
9097 USP14 Ubiquitin specific peptidase 14 (tRNA-guaninetransglycosylase)
18 18p11.32 33.7
4613 MYCN V-myc myelocytomatosis viral related oncogene,neuroblastoma derived (avian)
2 2p24.1 4.39E-08
27348 TOR1B Torsin family 1, member B (torsin B) 9 9q34 114
57460 PPM1H Protein phosphatase 1H (PP2C domain containing) 12 12q14.1-q14.2 6.8
25959 ANKRD25 Ankyrin repeat domain 25 19 19p13.2 167.7
The last column is the estimated hazard for a one unit change in gene expression Cox PH model. For all genes reported in the table, the hazard indicates thatthe risk of death is higher with an increase in gene expression.
© 2007 Lippincott Williams & Wilkins 979Mas et al.

HCC etiologies (14). Moreover, HCV-HCC patients repre-sent the more important number of patients in our livertransplant waiting list. We observed a set of 54 genes differ-entially expressed. In the HCC progression group, the over-expressed genes suggested a higher cell division and cellactivity in these tumors. To design a predictive scoring systemwith the number of samples we had available for training, weadopted a cross-validation approach. The system correctlyclassified up to 70% of the samples. The predictive power ofthe resulting HCC progression gene expression profile is cur-rently being validated in an independent HCV-HCC dataset.
From the patients that underwent LT five presentedrecurrence posttransplantation. The analysis of the genes dif-ferentially expressed resulting from the comparison of HCV-HCC patients undergoing LT with HCC recurrence vs. thepatients with the same explanted HCC stages without recur-rence showed over expression of genes associated with im-mune response and specific response to virus, supporting theimportance for studying the biology of HCC disease progressionsegregated to the underlying etiology of the liver cirrhosis.
Survival prediction of HCC patients is more challeng-ing than other cancers. This is, in the specific case of HCV, theconsequence of the underlying liver disease including chronichepatitis and cirrhosis. In a recent gene expression analysis ofhuman HCC, Lee et al. (18) identified two distinctive sub-classes that were highly associated with the survival of thepatients. A limited number of genes were identified as predic-tors of survival of the HCC patients.
In the present preliminary report, we also identifiedgenes related to overall survival and cancer-free survival.Ninety-six probe sets were associated with overall survival.From the analysis of the survival list genes, we observed tu-mor suppressor genes and genes involved in the regulation oftranscription and cell cycle. However, only eight genes werefound significantly associated with cancer-free survival whenthe analysis was restricted to transplanted patients. Furtherbiological studies on these genes are required to gain key in-sights into our findings.
Other than predicting HCC progression, the multigeneexpression profile extracted from this microarray study mayprovide further insights into the complex molecular processinvolved in the HCV-HCC and recurrence after LT. The anal-ysis of the differentially expressed genes among patients withadvanced stages of HCC with and without recurrence after LTshowed the overexpression of interferon inducible genes aswell genes involved in apoptosis regulation. It indicates thatthe study of the underlying cirrhosis might be critical for pre-dicting outcomes after LT. All the studied HCV-associatedtumors were associated with cirrhosis. Thus, it is apparentthat C-type HCC is closely related to chronic inflammation(47), suggesting that the immune response-related genesidentified here serve as molecular targets for chemopreven-tion and treatment of C-type HCC (14).
Our preliminary report includes very selected HCV-HCC patient candidates for LT. Although a longer follow-uptime and sample size might be necessary for analysis, ouranalyzed cases were sufficient for our focus on HCC progres-sion in the waiting list and early recurrence posttransplanta-tion. Our results showed that the incorporation of thesemolecular markers could help to improve organ allocationfor patients with HCC. A prospective multicenter study, be-
ing conducted by our group and nested in the Adult to AdultLiving Donor Liver Transplantation Cohort Study, will pro-vide a larger data set of patients for a stronger evaluation ofthese preliminary results.
REFERENCES1. Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer
burden: Globocan 2000. Int J Cancer 2001; 94: 153.2. El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma
in the United States. N Engl J Med 1999; 340: 745.3. Davila JA, Morgan RO, Shaib Y, et al. Hepatitis C infection and the
increasing incidence of hepatocellular carcinoma: a population-basedstudy. Gastroenterology 2004; 127: 1372.
4. Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplan-tation for hepatocellular carcinoma. Semin Liver Dis 2005; 25: 181.
5. Llovet JM. Updated treatment approach to hepatocellular carcinoma.J Gastroenterol 2005; 40: 225.
6. Fisher RA, Maluf D, Cotterell AH, et al. Non-resective ablation therapyfor hepatocellular carcinoma: effectiveness measured by intention-to-treat and dropout from liver transplant waiting list. Clin Transplant2004; 18: 502.
7. Schwartz M. Liver transplantation for hepatocellular carcinoma. Gas-troenterology 2004; 127 (Suppl 1): S268.
8. Maluf D, Fisher RA, Maroney T, et al. Non-Resective Ablation andLiver Transplantation in Patients with Cirrhosis and HepatocellularCarcinoma (HCC): Safety and Efficacy. Am J Transplant 2003; 3: 312.
9. Figueras J, Ibanez L, Ramos E, et al. Selection criteria for liver trans-plantation in early-stage hepatocellular carcinoma with cirrhosis: re-sults of a multicenter study. Liver Transpl 2001; 7: 877.
10. Marin-Hargreaves G, Azoulay D, Bismuth H. Hepatocellular carcino-ma: surgical indications and results. Crit Rev Oncol Hematol 2003;47: 13.
11. Olthoff KM. Surgical options for hepatocellular carcinoma: resectionand transplantation. Liver Transpl Surg 1998; 4: S98.
12. Herrero JI, Sangro B, Quiroga J, et al. Influence of tumor characteristicson the outcome of liver transplantation among patients with liver cir-rhosis and hepatocellular carcinoma. Liver Transpl 2001; 7: 631.
13. Thorgeirsson SS, Lee JS, Grisham JW. Molecular prognostication ofliver cancer: end of the beginning. J Hepatol 2006; 44: 798.
14. Iizuka N, Oka M, Yamada-Okabe H, et al. Comparison of gene expres-sion profiles between hepatitis B virus- and hepatitis C virus-infectedhepatocellular carcinoma by oligonucleotide microarray data on thebasis of a supervised learning method. Cancer Res 2002; 62: 3939.
15. Okabe H, Satoh S, Kato T, et al. Genome-wide analysis of gene expres-sion in human hepatocellular carcinomas using cDNA microarray:identification of genes involved in viral carcinogenesis and tumor pro-gression. Cancer Res 2001; 61: 2129.
16. Shirota Y, Kaneko S, Honda M, et al. Identification of differentiallyexpressed genes in hepatocellular carcinoma with cDNA microarrays.Hepatology 2001; 33: 832.
17. Mas VR, Maluf DG, Stravitz R, et al. Hepatocellular carcinoma inHCV-infected patients awaiting liver transplantation: genes involvedin tumor progression. Liver Transpl 2004; 10: 607.
18. Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival inhepatocellular carcinoma by gene expression profiling. Hepatology2004; 40: 667.
19. Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse largeB-cell lymphoma identified by gene expression profiling. Nature 2000;403: 503.
20. van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signa-ture as a predictor of survival in breast cancer. N Engl J Med 2002; 19:1999.
21. American Liver Tumor Study Group. A randomized prospective multi-institutional trial of orthotopic liver transplantation or partial hepaticresection with or without adjuvant chemotherapy for hepatocellularcarcinoma. Investigators Booklet and protocol 1998. United Networkfor Organ Sharing. Policy 1998; 3.6.4.4.
22. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open softwaredevelopment for computational biology and bioinformatics. GenomeBiology 2004; 5: R80.81-R80.16.
23. R Development Core Team. R: A language and environment for statis-tical computing. R Foundation for Statistical Computing, Vienna,Austria, 2005.
980 Transplantation • Volume 83, Number 7, April 15, 2007

24. Irizarry RA, Bolstad BM, Collin F, et al. Summaries of AffymetrixGeneChip probe level data. Nucleic Acids Research 2003; 31: e15.
25. Irizarry RA, Hobbs B, Collin F, et al. Exploration, Normalization, andSummaries of High Density Oligonucleotide Array Probe Level Data.Biostatistics 2003; 4: 249.
26. Tusher V, Tibshirani R, Chu G. Significance analysis of microarraysapplied to the ionizing radiation response. Proc Natl Acad Sci 2001; 98:5116.
27. Kaufman L, Rousseeuw PJ: Finding Groups in Data: An Introductionto Cluster Analysis. New York, John Wiley & Sons; 1990.
28. Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimationmethods for DNA microarrays. Bioinformatics 2001; 17: 520.
29. Furey TS, Cristianini N, Duffy N, et al. Support vector machine classi-fication and validation of cancer tissue samples using microarray ex-pression data. Bioinformatics 2000; 16: 906.
30. Juan LJ, Shia WJ, Chen MH, et al. Histone deacetylases specificallydown-regulate p53-dependent gene activation. J Biol Chem 2000; 275:20436.
31. Weinlich G, Eisendle K, Hassler E, et al. Metallothionein-overexpressionas a highly significant prognostic factor in melanoma: a prospective studyon 1270 patients. Br J Cancer 2006; 94: 835.
32. Miyoshi H, Fujie H, Shintani Y, et al. Hepatitis C virus core proteinexerts an inhibitory effect on suppressor of cytokine signaling(SOCS)-1 gene expression. J Hepato 2005; 43: 757.
33. Freiss G, Vignon F. Protein tyrosine phosphatases and breast cancer.Crit Rev Oncol Hematol 2004; 52: 9.
34. Lugwig H, Khayat D, Giacome G, Facon T. Proteasome inhibition andits clinical prospects in the treatment of hematologic and solid malig-nancies. Cancer 2005; 104: 1794.
35. Yuan BZ, Durkin ME, Popescu NC. Promoter hypermethylation ofDLC-1, a candidate tumor suppressor gene, in several common humancancers. Cancer Genet Cytogenet 2003; 140: 113.
36. Iso Y, Sawada T, Okada T, Kubota K. Loss of E-cadherin mRNA andgain of osteopontin mRNA are useful markers for detecting early re-
currence of HCV-related hepatocellular carcinoma. J Surg Oncol 2005;92: 304.
37. Shimazui T, Yoshikawa K, Uemura H, et al. The level of cadherin-6mRNA in peripheral blood is associated with the site of metastasis andwith the subsequent occurrence of metastases in renal cell carcinoma.Cancer 2004; 101: 963.
38. Guan XY, Fang Y, Sham JS, et al. Recurrent chromosome alterations inhepatocellular carcinoma detected by comparative genomic hybridiza-tion. Genes Chromosomes Cancer 2000; 29: 110.
39. Nishida N, Nishimura T, Ito T, et al. Chromosomal instability andhuman hepatocarcinogenesis. Histol Histopathol 2003; 18: 897.
40. Buendia MA. Genetic alterations in hepatoblastoma and hepatocellularcarcinoma: common and distinctive aspects. Med Pediatr Oncol 2002;39: 530.
41. Chevret S, Trinchet JC, Mathieu D, et al. A new prognostic classifica-tion for predicting survival in patients with hepatocellular carcinoma.Groupe d’Etude et de Traitement du Carcinome Hepatocellulaire.J Hepatol 1999; 31: 133.
42. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet2003; 362: 1907.
43. Figueras J, Jaurrieta E, Valls C, et al. Resection or transplantation forhepatocellular carcinoma in cirrhotic patients: outcome based on indi-cated treatment strategy. J Am Coll Surg 2000; 190: 580.
44. Marsh JW, Finkelstein SD, Demetris AJ, et al. Genotyping of hepato-cellular carcinoma in liver transplant recipients adds predictive powerfor determining recurrence-free survival. Liver Transpl 2003; 9: 664.
45. Iizuka N, Oka M, Yamada-Okabe H, et al. Oligonucleotide microarrayfor prediction of early intrahepatic recurrence of hepatocellular carci-noma after curative resection. Lancet 2003; 361: 923.
46. Kurokawa Y, Matoba R, Takemasa I, et al. Molecular-based predictionof early recurrence in hepatocellular carcinoma. J Hepatol 2004; 41 284.
47. Shimotohno K. Hepatitis C virus and its pathogenesis. Semin CancerBiol 2000; 10: 233.
© 2007 Lippincott Williams & Wilkins 981Mas et al.
Copyright © 2022 FDOKUMEN




















