
PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments

FULL-SPREADING PLATELETS INDUCED BY THE
RECOMBINANT RHODOSTOMIN ARE VIA BINDING
TO INTEGRINS AND CORRELATED WITH FAK
PHOSPHORYLATION
HSIN-HOU CHANG and SZECHENG J. LO*Institute of Microbiology and Immunology, National Yang-Ming University,
Taipei 11221, Taiwan, ROC
(Received 10 November 1997; accepted 9 February 1998)
H.-H. Chang and S. J. Lo. Full-spreading platelets induced by the recombi-nant rhodostomin are via binding to integrins and correlated with FAKphosphorylation. Toxicon 36, 1087±1099, 1998.ÐWe have previouslyreported that non-activated platelets can be induced by morphologicalchanges from the recombinant fusion protein of GST-rhodostomin [GST-RHO(RGD)], a member of disintegrin with an arginine±glycine±asparticacid (RGD) motif. In this study, we further characterized the factorsinvolved in platelet shape changes induced by rhodostomin. From less tofull-spreading, four cell spreading indexes, p1, p2, s1 and s2, were designatedto the platelet shape based on the scanning electron micrographs. Results ofpeptide competition and antibody blocking con®rmed that interactionbetween the RGD of rhodostomin and the aIIbb3 integrins of platelets wasrequired for induction of a higher percentage of s2 cells. When platelets werepretreated with calphostin C, herbimycin A and cytochalasin B, respectively,the percentage of p1 and p2 cells on rhodostomin-coated plates wasincreased and, concomitantly, the percentage of s1 and s2 cells wasdecreased. Biochemical analyses indicated that the focal adhesion kinase(FAK or pp125FAK) in platelets that adhered to GST-RHO(RGD) was phos-phorylated in contrast to little or no phosphorylation of FAK in cellsadhered to ®brinogen or non-activated cells. Furthermore, the degree ofFAK phosphorylation was consistently correlated with morphologicalchanges in platelets treated with various drugs. Taking all the resultstogether, we suggested that rhodostomin could directly bind to integrins ofplatelets and then trigger signal transduction leading to FAK phosphoryl-ation and actin polymerization and ®nally resulting in platelet full-spreading.# 1998 Elsevier Science Ltd. All rights reserved
Toxicon Vol. 36, No. 8, pp. 1087±1099, 1998# 1998 Elsevier Science Ltd. All rights reserved
Printed in Great Britain0041-0101/98 $19.00+0.00PII: S0041-0101(98)00088-9
*Author to whom correspondence should be addressed.
1087

INTRODUCTION
Snake venoms contain complex mixtures which are generally characterized as neurotox-ins, cardiotoxins, heamorrhagins, coagulants and anticoagulants (Lee, 1972; Jeng et al.,1978; Karlsson, 1979). Many anticoagulants have been puri®ed and identi®ed to be®brinogen-receptor antagonists which interfere with the interaction of ®brinogen andGPIIb/IIIa (integrin aIIbb3) on activated platelets (Teng and Huang, 1991a,b). Theseanticoagulants contain 48 to 84 amino acids (aa) isolated from di�erent snake venomsand all have an arginine±glycine±aspartic acid (RGD) loop and 4 to 7 pairs of disul®debonds. Since they bind to integrin and block the ®brinogen function to result in the in-hibition of platelet aggregation, they have been coined as a family of `disintegrins'(Gould et al., 1990; Huang and Niewiarowski, 1994). The cDNA cloning studiesrevealed that a metalloprotease motif is fused to a disintegrin at the N-terminus(Neeper and Jacobson, 1990; Au et al., 1991; Hite et al., 1994). Recently, similar mol-ecules are also identi®ed in many other animals and renamed as `a disintegrin-like andmetalloprotease' (ADAM in short) or `metalloprotease, disintegrin and cysteine-rich'(MDC in short) proteins (Weskamp and Blobel, 1994; Wolfsberg et al., 1995; Jia et al.,1996; Weskamp et al., 1996), which open a new avenue of cell and developmental bi-ology research.
Rhodostomin from Agkistrodon rhodostoma contains 68 aa with 12 residues ofcysteine and with an RGD sequence at position 49±51 (Gould et al., 1990). It has beenproven to be one of the most potent anticoagulants among the known disintegrins(Teng and Huang, 1991a,b). The IC50 of rhodostomin to inhibit ADP (10 mM)-stimu-lated platelet aggregation is about 3.0�10ÿ8 M (Teng and Huang, 1991a,b). By using asynthetic gene to produce the glutathione S-transferase (GST)-rhodostomin fusion pro-tein, we previously have demonstrated that rhodostomin in a fusion protein form keepsits biological activity and is as potent as the native form in both solution and immobi-lized state (Chang et al., 1993, 1997). To elucidate the mechanism underlying rhodosto-min induction of platelet adhesion and morphological changes, in this study, weemployed various peptides, antibodies and drugs to test the e�ect on platelets adhesionon rhodostomin-coated plates. We were able to demonstrate that phosphorylation offocal adhesion kinase (FAK or pp125FAK) was induced after activation of integrins bythe recombinant rhodostomin and resulted in platelet full-spreading. In contrast, littleor no FAK phosphorylation was found in less spreading platelets if they were pre-treated with inhibitors or directly bound to ®brinogen. Although the present studydemonstrates that a snake venom, a disintegrin in the recombinant fusion protein form,can directly induce the full-spreading of non-activated platelets, the immediate signalraised by rhodostomin and integrin binding remains to be elucidated.
MATERIALS AND METHODS
Chemicals and materialsFibrinogen, thrombin, bovine serum albumin (BSA), Triton X-100 and chemical drugs such as nifedipine,
herbimycin A, calphostin C, cytochalasin B and PMA (phorbol 12-myristate 13-acetate) were purchased fromSigma (St. Louis, MO) as well as apyrase, pepstatin A, leupeptin and sodium orthovanadate. Glutaraldehydefor scanning electron microscopy was from Electron Microscopy Sciences (Washington, PA). IPTG (isopropyl-b-D-thiogalactoside) for induction of fusion protein production was from Biosynth AG (Staad, Switzerland).The glutathione-a�nity column and glutathione for fusion protein puri®cation and protein A-Sepharose beadsfor immunoprecipitation were obtained from Pharmacia (Uppsala, Sweden). An ECL kit for Western blotdetection was from Amersham (Buckinghamshire, U.K.). Puri®ed rhodostomin (product name kistrin) wasfrom Sigma (St. Louis, MO) while peptides of GRGDSP and GRGESP were from Peninsula Lab (Belmont,CA). Antibody against peptide 4559 (Chang et al., 1997) was a gift from Dr S.-H. Chen (National Cheng-
H.-H. CHANG and S. J. LO1088

Kung University, Tainan, Taiwan) and anti-integrin antibodies (AP2 and OPG2) (Pidard et al., 1983; Kunickiet al., 1995) were gifts from Dr T. J. Kunicki (Scripps Research Institute). Other antibodies were purchasedfrom di�erent companies: anti-FAK from Transduction Lab (Lexington, Kentucky), anti-FAK (2A7) forimmunoprecipitation and anti-phosphotyrosine (4G10) from Upstate Biotech (Lake Placid, NY), anti-mouseand anti-rabbit Ig-peroxidase conjugated antibodies from Cappel (West Chester, PA).
Fusion protein preparationEscherichia coli (strain RR1) containing the expression plasmids were grown in 100 ml LB medium sup-
plemented with 50 mg/ml ampicillin by vigorously shaking at 378C until A500 nm reached 0.3±0.5. The synthesisof GST-rhodostomin fusion proteins [GST-RHO(RGD) and GST-RHO(RGE)] (Chang et al., 1997) was theninduced by adding 0.5 mM IPTG to the medium for 1±3 h. Fusion proteins were puri®ed by a glutathione col-umn and eluted by glutathione-containing bu�er as described (Smith and Johnson, 1988). The puri®ed fusionproteins were analyzed by a 0.2% SDS±15% PAGE (Laemmli, 1970).
Platelet preparationWashed platelets were obtained from healthy donors who were not taking any medicines and prepared as
described in the previous study (Chang et al., 1997). Human blood was collected from the forearm vein intotubes containing 1/6 volume of acid-citrate-dextrose (ACD) as anticoagulant. The blood was centrifuged at200g for 15 min at room temperature. The upper platelet rich plasma (PRP) was mixed with 5 mM EDTA andre-centrifuged at 1000g for 12 min. The supernatant was discarded and the platelet pellet was suspended in cal-cium-free Tyrode's bu�er with 0.35% BSA, heparin (50 unit/ml) and apyrase (1 unit/ml). After 20 min incu-bation at 378C, the washed platelet pellet was resuspended in Tyrode's bu�er containing 1 mM calcium andthe cell concentration was adjusted to around 3.5� 108 platelets/ml.
Platelet adhesion study and scanning electron microscopyCoverslips were coated with recombinant fusion proteins or ®brinogen as described previously for the cell
attachment assay (Chang et al., 1993, 1997). Freshly prepared and non-activated blood platelets from healthydonors were incubated on coated-plates for 15 min and then washed with PBS. For some cases, non-activatedplatelets were co-incubated with or pre-treated with di�erent drugs before seeding onto protein-coated plates.Samples were then prepared for scanning electron microscopy as reported before (Lo et al., 1980). In brief,cells were ®xed with glutaraldehyde and subjected to alcohol (50±100%) dehydration and critical drying. Thegold-coated samples were examined under a scanning electron microscope (Joel; JSM-5300) at 15 kV. At least5 di�erent areas were randomly selected and photographed. Classi®cation of platelet morphology was basedon the method described by Jen et al. (1996) with a slight modi®cation (see Section 3).
Analysis of phosphorylation proteins of plateletsPlatelets that adhered to di�erent protein-coated plates were dissolved by the SDS-PAGE sample bu�er.
Proteins were then separated by SDS-PAGE (Laemmli, 1970) and followed by immunoblotting detection.Immunoblotting was performed according to the method of Towbin et al. (1979). Proteins transferred on anitrocellulose membrane were incubated with monoclonal antibody against phospho-tyrosine or FAK. Afterwashing, secondary antibody conjugated with horseradish peroxidase was added and the bound antibody wasdetected by using an ECL kit and following the suggested protocol (Amersham). In some cases of detectionFAK immunoprecipitation was performed using protein A-Sepharose beads bound with anti-FAK beforeECL detection as described by Lipfert et al. (1992) and Shattil et al. (1994).
RESULTS
Morphological classi®cation of platelets adhered to GST-RHO(RGD)-coated coverslipsBecause of the small size of platelets, the detailed morphology, especially the for-
mation of ®lopodia, cannot easily be revealed under a light microscope. We employedscanning electron microscopy to observe platelets that adhered on rhodostomin-coatedplates. We designated the platelets into four di�erent spreading indexes (p1, p2, s1, ands2) according to the previous report (Jen et al., 1996) but with a slight modi®cation, asshown by a higher magni®cation of electron micrographs [Fig. 1(A)±(D)]. P1 representsplatelets with 1 or 2 pseudopodia and non-activated cells [Fig. 1(A)]; p2 is cells withmore than 2 pseudopodia but without any lamellar membrane [Fig. 1(B)]. While s1 rep-
Platelet Spreading Induced by Rhodostomin 1089
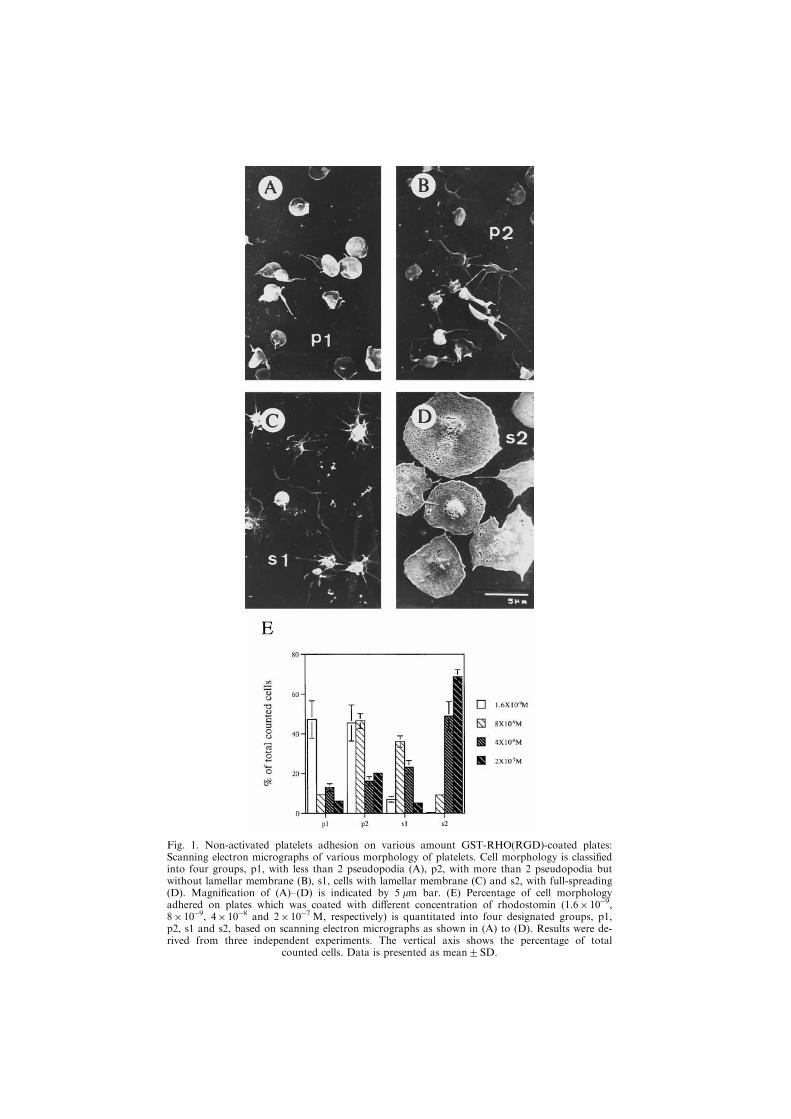
Fig. 1. Non-activated platelets adhesion on various amount GST-RHO(RGD)-coated plates:Scanning electron micrographs of various morphology of platelets. Cell morphology is classi®edinto four groups, p1, with less than 2 pseudopodia (A), p2, with more than 2 pseudopodia butwithout lamellar membrane (B), s1, cells with lamellar membrane (C) and s2, with full-spreading(D). Magni®cation of (A)±(D) is indicated by 5 mm bar. (E) Percentage of cell morphologyadhered on plates which was coated with di�erent concentration of rhodostomin (1.6� 10ÿ9,8� 10ÿ9, 4�10ÿ8 and 2�10ÿ7 M, respectively) is quantitated into four designated groups, p1,p2, s1 and s2, based on scanning electron micrographs as shown in (A) to (D). Results were de-rived from three independent experiments. The vertical axis shows the percentage of total
counted cells. Data is presented as mean2SD.
resents cells with pseudopodia and lamellar membrane [Fig. 1(C)] and s2 is a full-spreading platelet with a smooth edge and a pancake shape [Fig. 1(D)]. To analyze thepercentage of cells in di�erent spreading index under various conditions, we randomlyselected ®ve areas which have at least 100 cells in total. Then based on the above classi-®cation, the percentage of cell spreading index was calculated as a fraction of total cells.For example, platelets adhered to 1.6� 10ÿ9 M GST-RHO(RGD)-coated slips hadabout 48% of cells in p1 shape, 46% of cells in p2, 6% of cells in s1 and less than 1%
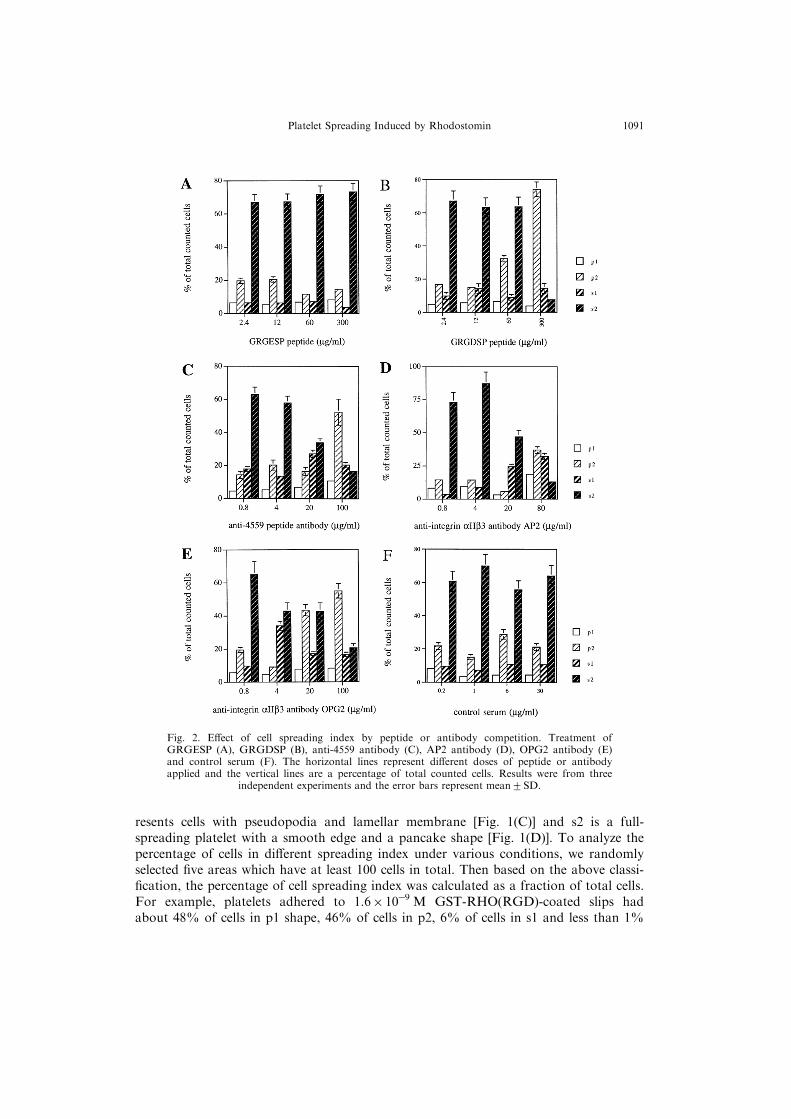
Fig. 2. E�ect of cell spreading index by peptide or antibody competition. Treatment ofGRGESP (A), GRGDSP (B), anti-4559 antibody (C), AP2 antibody (D), OPG2 antibody (E)and control serum (F). The horizontal lines represent di�erent doses of peptide or antibodyapplied and the vertical lines are a percentage of total counted cells. Results were from three
independent experiments and the error bars represent mean2SD.
Platelet Spreading Induced by Rhodostomin 1091

of cells in s2. In contrast, cells attached to 2.0� 10ÿ7 M GST-RHO(RGD)-coated slipshad about 6% of cells in p1 shape, 20% in p2, 4% in s1 and 69% in s2 [Fig. 1(E)].Figure 1(E) shows clearly that the higher percentage of s2 cells adhered on the higherconcentration of recombinant rhodostomin-coated plates.
Adhesion of platelets to GST-RHO(RGD)-coated slips is via RGD and integrin inter-action
In order to verify the higher percentage of fully spreading platelets (s2) on rhodosto-min-coated slips is via the interaction of RGD-motif of substrate and GPIIb/IIIa(aIIbb3) of platelets, we carried out peptide competition and antibody blocking assay.Figure 2(A) shows that platelets treated with various amounts (2.4 mg/ml and with 5-fold increasing up to 300 mg/ml) of hexapeptide GRGESP had little percentage changeof cells among p1, p2, s1 and s2 on the GST-RHO(RGD)-coated slips. In contrast, in-cubation with the hexapeptide GRGDSP in a higher concentration (300 mg/ml) reduceds2 cells from 65 to 10% and simultaneously increased p2 cells from 30 to 75%[Fig. 2(B)]. These results supported that induction of a higher percentage of s2 plateletis indeed via the RGD motif of GST-RHO(RGD).
In antibody blocking studies, three di�erent antibodies, anti-4559 peptide antibody(against a peptide covering 45 to 59 aa of rhodostomin), AP2 and OPG2 (both againstaIIbb3) and a control serum (for a negative control), were used in various amounts(0.8 mg/ml and with 5-fold increasing up to 100 mg/ml), which were co-incubated withseeding platelets on the GST-RHO(RGD)-coated slips. Although the changing patternof the s2 cell number ¯uctuated in the treatment of three antibodies, anti-4559, AP2and OPG2, respectively, there was a common trend showing a decrease of s2 cell per-centages in a higher amount of antibody treatment [Fig. 2(C)±(E)]. No dramatic changeof cell percentages of p1, p2, s1 and s2 in the control serum treatment was observed[Fig. 2(F)]. These results further supported that induction of platelet attachment on theGST-RHO(RGD) slips is via the interaction of RGD and integrin, since the anti-4559antibody is against the peptide of 4559 which covers the RGD motif of rhodostominand AP2 and OPG2 are against aIIbb3.
Protein kinase C (PKC) participates in the signal pathway of inducing platelet full-spreading
Many lines of evidence indicate that PKC activation is one of a signal cascade afterintegrin activation (Shattil et al., 1994; Haimovich et al., 1996). We took advantage ofthe system established in this study that platelets on the ®brinogen-coated slips appearin major populations of p1 and p2 and those on GST-RHO(RGD)-coated slips are ins1 and s2. If PKC participating in platelet full-spreading is true, cells treated with cal-phostin C, a potent PKC inhibitor, will reduce the percentage of s2 and s1 cells on theGST-RHO(RGD)-coated slips; on the other hand, cells treated with PMA (phorbol 12-myristate 13-acetate), a PKC activator, will promote the percentage of s2 and s1 cellson the ®brinogen-coated slips. As shown by scanning electron micrographs, cells treatedwith PMA indeed appeared to have a higher percentage of s2 and s1 cells as comparedto untreated cells on ®brinogen-coated slips [Fig. 3(A) vs Fig. 3(B)]. On the other hand,cells pre-treated with calphostin C for 2 h appeared in a higher percentage of p1 cellson the GST-RHO(RGD)-coated slips [Fig. 3(C) vs Fig. 3(D)], indicating that inhibitionof PKC resulted in a lower percentage of fully spreaded platelets. Figure 3(E) shows
H.-H. CHANG and S. J. LO1092

Fig. 3. E�ect of protein kinase C activity on platelet morphology. Platelets on ®brinogen-coatedplates with (B) or without (A) PMA treatment. Platelets on GST-RHO(RGD)-coated plates with(D) or without (C) pre-treatment of calphostin C for 2 h. Inhibition of PKC activity by variousamount of calphostin C resulted in changing percentage of s2 cells (E). In panel (E), the verticalaxis is the percentage of total counted cells. Results were from three independent experiments
and the error bars represent mean2SD.
Platelet Spreading Induced by Rhodostomin 1093

that pre-treated calphostin C in a dose above 1 mg/ml blocked platelet full-spreading onthe GST-RHO(RGD)-coated slips e�ciently.
Phosphorylation of FAK in platelets can be induced by GST-RHO(RGD) and is corre-lated with its morphology
In order to characterize whether FAK is phosphorylated in platelets that adhered toGST-RHO(RGD) as those activated by thrombin, we performed experiments to analyzeFAK phosphorylation in platelets with di�erent conditions. The ECL Western blottingresults [Fig. 4(A)] showed that platelets that adhered to GST-RHO(RGD)-coated slipsand those treated with thrombin had a phosphorylated band at the molecular weightsimilar to the FAK (lanes 3 and 1) but not in non-activated platelets (lane 2). However,the amount of phosphorylated FAK was greatly reduced in platelets that adhered toGST-RHO(RGD)-coated slips if pre-treated with calphostin C, nifedipine, herbimycinA (a protein kinase inhibitor) or cytochalasin B (an actin polymerization inhibitor)[Fig. 5(A), lanes 4±7, respectively]. Platelets in these conditions appeared in a majorityof p1 and p2 shapes [see Fig. 3(E), Fig. 4(C) and (D), respectively]. Platelets thatadhered to ®brinogen-coated plates, which appear in a higher percentage of p1 and p2shape, also had little or no phosphorylated FAK (lane 8). Cells that adhered to ®brino-gen-coated plates but treated with PMA (TPA), or ionomycin, showed a phosphory-lated band of FAK (lanes 9±10). Figure 4(B) shows those phosphorylation bandspresent in Fig. 4(A) were indeed FAK by reprobing the same blotting membrane withanti-FAK antibody. Therefore, these biochemical data were consistent to the morpho-logical data suggesting that the higher percentage of s1 and s2 cells has a higher amountof phosphorylated FAK while p1 and p2 cells have little or no phosphorylated FAK.
DISCUSSION
Combining the present study with previous reports, a summary of the factors andprocesses which are possibly involved in recombinant rhodostomin induced platelet full-spreading is depicted in Fig. 5. The signaling cascade of full-spreading of the plateletcan be divided into the following steps: (1) interaction of RGD-containing ligand withintegrin causes cell adhesion, (2) elevation of intracellular Ca+2, (3) activation of pro-tein kinase C which in turn phosphorylates downstream substrates, (4) after FAK phos-phorylation, it activates other proteins and/or causes its redistribution into cytoskeletalfraction and (5) polymerization of actin occurs and results in platelet full-spreading.These processes are very similar to the G-protein mediated signal transduction in manymodel systems (Chen et al., 1996), however, linkages between integrin activation andcalcium elevation remain to be determined.
In general, non-activated platelets possess integrin aIIbb3 in an inactive state, whichcan not be bound by ®brinogen in blood stream (Phillips et al., 1991). After thrombinor ADP stimulation, integrin aIIbb3 of platelets is activated through an inside out signaland becomes vulnerable for ®brinogen binding. Because of its structure, rhodostomincan interact with both non-activated and activated integrins (Dennis et al., 1990; Adleret al., 1991; Teng and Huang, 1991a,b). In this study, we demonstrated that non-acti-vated platelet adhesion and spreading on rhodostomin are two independent events; ad-hesion does not necessarily lead to full-spreading but it is a prerequisite step for full-spreading. Apparently, full-spreading of platelets requires further signals, which are
H.-H. CHANG and S. J. LO1094

Fig. 4. Degree of FAK phosphorylation in various treatment of platelets. (A) Detection of anti-phospho-tyrosine and (B) detection of the presence of FAK. The position of FAK is marked byan arrow at the right side and protein size markers are on the left side. Conditions of platelettreatment are indicated on the top of the gel. The concentration of each chemical used is: throm-bin (1 NIH U/ml), calphostin C (1 mg/ml), nifedipine (150 mM), herbimycin (5 mg/ml), cytochala-sin B (10 mg/ml), ®brinogen (F) (20 mg/ml), TPA (0.3 mM) and ionomycin (2 mg/ml). Lanes 3±7are platelets on GST-RHO(RGD)-coated plates but with di�erent treatment while lanes 8±10are cells on ®brinogen (F)-coated plates. Percentage of cell morphology on GST-RHO(RGD)-coated plates after treatment of herbimycin (C) and cytochalasin B (D). In (C) and (D) panels,the vertical lines are percentage of total counted cells. Results were from three independent ex-
periments and the error bars represent mean2SD.
Platelet Spreading Induced by Rhodostomin 1095

absent in those cells that adhered to a low dose of ®brinogen- or GST-rhodostomin-coated plates.
It is well known that Ca2+ is a key factor involved in ®brinogen-induced spreadingof platelets (Pelletier et al., 1992; Winokur and Hartwig, 1995; Jen et al., 1996). In thisstudy, treatment of ionomycin induced cell spreading and a higher phosphorylation ofFAK on ®brinogen-coated plates [Fig. 4(A), lane 10] indicates that extracellular Ca2+
in¯ux could play the role. This observation is consistent with the previous reports(Yamaguchi et al., 1987; Rybak and Renzulli, 1989; Pelletier et al., 1992; Jen et al.,1996), suggesting that Ca2+ in¯ux occurs in intact platelets and stably transfected cellsand the intracellular Ca2+ elevation is correlated with the extent of platelet morphologychange. It is still a controversy that Ca2+ in¯ux occurring in platelets is via a voltage-gated calcium channel (Rink and Sage, 1990). Treatment of o-conotoxin MVII A andMVII C did not reduce the percentage of s2 cells on rhodostomin-coated plate,suggesting that N- and P/Q-type of Ca2+ channels are not responsible for platelet full-spreading (data not shown). However, a higher concentration of nifedipine(1.5� 10ÿ4 M) could inhibit platelet full-spreading induced by rhodostomin (data notshown) as well as reduce FAK phosphorylation [Fig. 4(A), lane 5]. This result is consist-ent with the observation of frog muscle stretch which is enhanced by Ca2+ in¯uxthrough L-type channels after activation of integrins (Chen and Grinnell, 1995). Sinceprevious studies suggested that no L-type Ca2+ channels are present on platelet mem-branes (Rink and Sage, 1990) and the e�ective dosage of nefedipine used in this study isrelative high, the involvement of L-type Ca2+ channels in integrin-mediated plateletspreading remains questionable.
Protein kinase C (PKC) is generally activated following the production of diacylgy-cerol (DG) which is derived from phospholipid degradation (Nishizuka, 1992).
Fig. 5. A hypothetical scheme of signal transduction through rhodostomin and integrin inter-action leading to platelet full-spreading. Brie¯y, integrins are activated by immobilized rhodosto-min which leads to the elevation of intracellular calcium concentration either from outside ofcell or from internal calcium pool and turns on PKC activity. The downstream events, including
phosphorylation of FAK, ®nally lead to actin polymerization and cell shape change.
H.-H. CHANG and S. J. LO1096

However, Ca2+ and phospholipid are also required in certain circumstances (Asaoka etal., 1992). Since PMA-treated platelets appeared in a higher percentage of s1 and s2 ascompared with non-activated cells which appeared in p1 and p2 cell shape when theyadhere to ®brinogen plates [Fig. 3(A) vs (B)], platelets on rhodostomin plates appearingin s1 and s2 shape are likely via the activation of PKC. Such an assumption is furthersupported by results that calphostin C treated platelets greatly reduced the percentageof s1 and s2 shapes [Fig. 3(E)].
Consistent with the observation that PKC regulates phosphorylation of FAK(Haimovich et al., 1996), biochemical results obtained from this study also indicatedthat the amount of phosphorylated FAK is higher in full-spreading cells than that inless-spreading cells [Fig. 4(A)]. We thus could suggest that the degree of FAK phos-phorylation is highly correlated with the morphology of platelets. In addition, thisstudy provided evidence that rhodostomin can activate FAK phosphorylation throughbinding to integrins [Fig. 4(A) lane 3]. Furthermore, actin polymerization participates insignal transduction to FAK phosphorylation since cells treated with cytochalasin B lostthe capability of full-spreading [Fig. 4(D)] and had lower phosphorylation of FAK[Fig. 4(A), lane 8]. In conjunction with the report of Jen et al. (1996) indicating thatcytochalasin B inhibited the elevation of Ca2+, we suggest that actin may play a dualrole in platelet full-spreading. In the early stage, formation of F-actin is required forCa2+ elevation and in the latter stage, actin polymerization is required for platelet full-spreading (Bertagnolli and Beckerle, 1993; Fox et al., 1993).
Actin-based cytoskeleton participating in cell shape change is a well-known phenom-enon. It is becoming clear that cell shape changes based on the actin cytoskeleton arerelated to activation of di�erent small G-proteins in nucleated cells as well as in anu-cleated platelets (Ridley and Hall, 1992; Ridley et al., 1992; Nobes and Hall, 1995;Dedhar and Hannigan, 1996). Although there is a lack of linkage of FAK phosphoryl-ation and small G-protein activation in this study, it has been demonstrated that therecombinant GST-rhodostomin fusion protein alone is able to induce FAK phosphoryl-ation and full-spreading in platelets, which provides an ideal system for the analysis ofmolecules in induction of platelet full-spreading.
AcknowledgementsÐWe thank Dr S.-H. Chen and Dr T. J. Kunicki for providing us antibodies and to DrMary J. Anderson (a Fulbright professor) for English editing of the manuscript. Special thanks are also due tothe EM facility of the Department of Anatomy for the wonderful technical help. This study was supported bygrants (NSC84-2331-B010-097-M15 and NSC85-2331-B010-002) from the National Science Council, Republicof China. S. J. L. is an awardee from the National Science Council and the Medical Research andAdvancement Foundation in Memory of Dr Chi-Shuen Tsou.
REFERENCES
Adler, M., Lazarus, R. A., Dennis, M. S. and Wagner, G. (1991) Solution structure of kistrin, a potent plate-let aggregation inhibitor and GP IIb±IIIa antagonist. Science 253, 445±448.
Asaoka, Y., Nakamura, S.-I., Yoshida, K. and Nishizuka, Y. (1992) Protein kinase C, calcium and phospholi-pid degradation. Trends Biochem. Sci. 17, 414±417.
Au, L. C., Huang, Y. B., Huang, T.-F., Teh, G. W., Lin, H. H. and Choo, K. B. (1991) A common precursorfor a putative hemorrhagic protein and rhodostomin, a platelet aggregation inhibitor of the venom ofCalloselasma rhodosotma: molecular cloning and sequence analysis. Biochem. Biophys. Res. Commun. 181,585±593.
Bertagnolli, M. E. and Beckerle, M. C. (1993) Evidence for selective association of a subpopulation of GPIIb-IIIa with the actin cytoskeletons of thrombin-activated platelets. J. Cell. Biol. 121, 1329±1342.
Chang, H. H., Hu, S. T., Huang, T.-F., Chen, S. H., Lee, Y.-H. W. and Lo, S. J. (1993) Rhodosotmin, anRGD-containing peptide expressed from a synthetic gene in Escherichia coli, facilitates the attachment ofhuman hepatoma cells. Biochem. Biophys. Res. Commun. 190, 242±249.
Platelet Spreading Induced by Rhodostomin 1097

Chang, H. H., Tsai, W. J. and Lo, S. J. (1997) Glutathione S-transferase-rhodostomin fusion protein inhibitsplatelet aggregation and induces platelet shape change. Toxicon 35, 195±204.
Chen, B.-M. and Grinnell, A. D. (1995) Integrins and modulation of transmitter release from motor nerveterminals by stretch. Science 269, 1578±1580.
Chen, M.-Y., Insall, R. H. and Devreotes, P. N. (1996) Signaling through chemoattractant receptors inDictyostelium. Trends Genet. 12, 52±57.
Dedhar, S. and Hannigan, G. E. (1996) Integrin cytoplasmic interactions and bidirectional transmembrane sig-nalling. Curr. Opin. Cell Biol. 8, 657±669.
Dennis, M. S., Henzel, W. J., Pitti, R. M., Lipari, M. T., Napier, M. A., Deisher, T. A., Bunting, S. andLazarus, R. A. (1990) Platelet glycoprotein IIb±IIIa protein antagonists from snake venoms: evidence for afamily of plate-aggregation inhibitors. Proc. Natl. Acad. Sci. U.S.A. 87, 2471±2475.
Fox, J. E. B., Lipfert, L., Clark, E. A., Reynold, C. C., Austin, C. D. and Brugge, J. S. (1993) On the role ofthe platelet membrane skeleton in mediating signal transduction. J. Biol. Chem. 268, 25973±25984.
Gould, R. J., Poloko�, M. A., Friedman, P. A., Huang, T.-F., Holt, J. C., Cook, J. J. and Niewiarowski, S.(1990) Disintegrins: a family of integrin inhibitory proteins from viper venoms. Proc. Soc. Exp. Biol. Med.195, 168±171.
Haimovich, B., Kaneshiki, N. and Ji, P. (1996) Protein kinase C regulates tyrosine phosphorylation ofpp125FAK in platelets adherent to ®brinogen. Blood 87, 152±159.
Hite, L. A., Jia, L. G., Bjarnason, J. B. and Fox, J. W. (1994) cDNA sequences for four snake venom metallo-proteinases: structure, classi®cation and their relationship to mammalian reproductive proteins. Arch.Biochem. Biophys. 308, 182±191.
Huang, T.-F. and Niewiarowski, S. (1994) Disintegrins: the naturally-occurring antagonists of platelet ®brino-gen receptor. J. Toxicol. Toxin Rev. 13, 253±273.
Jen, C. J., Chen, H.-i., Lai, K.-c. and Usami, S. (1996) Changes in cytosolic calcium concentrations and cellmorphology in single platelets adhered to ®brinogen-coated surface under ¯ow. Blood 87, 3775±3782.
Jeng, T. W., Hendon, R. A. and Fraenkel-Conrat, H. (1978) Search for relationship among the hemolytic,phosphorolytic and neurotoxic activities of snake venoms. Proc. Natl. Acad. Sci. U.S.A. 75, 600±604.
Jia, L. G., Shimokawa, K., Bjarnason, J. B. and Fox, J. W. (1996) Snake venom metalloproteinases: structure,function and relationship to the ADAMs family of proteins. Toxicon 34, 1269±1276.
Karlsson, E. (1979) Chemistry of protein toxins in snake toxins. Handb. Exp. Pharmacol. 52, 159±212.Kunicki, T. J., Ely, K. R., Kunicki, T. C., Tomilama, Y. and Amris, D. S. (1995) The exchange of Arg±Gly±Asp (RGD) and Arg±Tyr±ASp (RYD) binding sequences in integrin alpha IIb beta 3 does not alter integrinrecognition. J. Biol. Chem. 270, 16660±16665.
Laemmli, U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4.Nature 227, 680±685.
Lee, C. Y. (1972) Chemistry and pharmacology of polypeptide toxins in snake venoms. Ann. Rev. Pharmacol.12, 265±286.
Lipfert, L., Haimovich, B., Schaller, M. D., Cobb, B. S., Parsons, J. T. and Brugge, J. S. (1992) Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J. Cell Biol.119, 905±912.
Lo, S. J., Taylor, J. D. and Tchen, T. T. (1980) ACTH-induced internalization of plasma membrane inxanthophores of the gold®sh, Carassius auratus L.. Biochem. Biophys. Res. Commun. 86, 748±754.
Neeper, M. P. and Jacobson, M. A. (1990) Sequence of a cDNA encoding the platelet aggregation inhibitortrigramin. Nucl. Acids Res. 18, 4255±4265.
Nishizuka, Y. (1992) Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C.Science 258, 607±614.
Nobes, C. D. and Hall, A. (1995) Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focalcomplexes associated with actin stress ®bers, lamellipodia and ®lopodia. Cell 81, 53±62.
Pelletier, A. J., Bodary, S. C. and Levinson, A. D. (1992) Signal transduction by the platelet integrin aIIbb3:induction of calcium oscillations required for protein-tyrosine phosphorylation and ligand-induced spreadingof stably transfected cells. Mol. Biol. Cell 3, 989±998.
Phillips, D. R., Charo, I. F. and Scarborough, R. M. (1991) GPIIb±IIIa: the responsive integrin. Cell 65, 359±362.
Pidard, D., Montgomery, R. R., Bennett, J. S. and Kunicki, T. J. (1983) Interaction of Ap2, a monoclonalantibody speci®c for the human platelet glycoprotein IIb±IIIa complex, with intact platelets. J. Biol. Chem.258, 12582±12586.
Ridley, A. J. and Hall, A. (1992) The small GTP-binding protein rho regulates the assembly of focal adhesionsand actin stress ®bers in response to growth factors. Cell 70, 389±399.
Ridley, A. J., Paterson, H. F., Johnston, C. L., Diekmann, D. and Hall, A. (1992) The small GTP-bindingprotein rac regulates growth factor-induced membrane ru�ing. Cell 70, 401±410.
Rink, T. J. and Sage, S. O. (1990) Calcium signaling in human platelets. Annu. Rev. Physiol. 52, 431±449.Rybak, M. E. M. and Renzulli, L. A. (1989) Ligand inhibition of the platelet glycoprotein IIb±IIIa complexfunction as a calcium channel in liposomes. J. Biol. Chem. 264, 14617±14620.
H.-H. CHANG and S. J. LO1098

Shattil, S. J., Haimovich, B., Cunningham, M., Lipfert, L., Parsons, J. T., Ginsberg, M. H. and Brugge, J. S.(1994) Tyrosine phosphorylation of pp125FAK in platelets requires coordinated signaling through integrinand agonist receptor. J. Biol. Chem. 269, 14738±14745.
Smith, D. B. and Johnson, K. S. (1988) Single-step puri®cation of polypeptides expressed in Escherichia coli asfusion with glutathione S-transferase. Gene 67, 31±40.
Teng, C.-M. and Huang, T.-F. (1991a) Inventory of exogenous inhibitors of platelet aggregation. Thromb.Haemostas. 65, 624±626.
Teng, C.-M. and Huang, T.-F. (1991b) Snake venom constituents that a�ect platelet function. Platelets 2, 77±87.
Towbin, H., Stachelin, T. and Gordon, J. (1979) Electrophoretic transfer of proteins from polyacrylamide gelsto nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350±4354.
Weskamp, G. and Blobel, C. P. (1994) A family of cellular proteins related to snake venom disintegrin. Proc.Natl. Acad. Sci. U.S.A. 91, 2748±2751.
Weskamp, G., Kratzschmar, J., Reid, M. S. and Blobel, C. P. (1996) MDC9, a widely expressed cellular disin-tegrin containing cytoplasmic SH3 ligand domains. J. Cell Biol. 132, 717±726.
Winokur, R. and Hartwig, J. H. (1995) Mechanism of shape change in chilled human platelets. Blood 85,1796±1804.
Wolfsberg, T. G., Primako�, P., Myles, D. G. and White, J. M. (1995) ADAM, a novel family of membraneproteins containing a disintegrin and metalloprotease domain: multipotential functions in cell±cell and cell±matrix interactions. J. Cell Biol. 131, 275±278.
Yamaguchi, A., Yamamoto, N., Kitagawa, H., Tanoue, K. and Yamazaki, H. (1987) Ca+2 in¯ux mediatedthrough the GPIIb/IIIa complex during platelet activation. FEBS Lett. 225, 228±232.
Platelet Spreading Induced by Rhodostomin 1099
Copyright © 2022 FDOKUMEN




















