
Androgen receptor-modulatory microRNAs provide insight into ...

This article was downloaded by: [Professor Teodorico C. Ramalho]On: 17 August 2012, At: 07:52Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Journal of Biomolecular Structure and DynamicsPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/tbsd20
First principles calculations of thermodynamicsand kinetic parameters and molecular dynamicssimulations of acetylcholinesterase reactivators: canmouse data provide new insights into humans?Karina S. Matos a , Elaine F.F. da Cunha a , Arlan da Silva Gonçalves b , Alan Wilter c , KamilKuča d e , Tanos C.C. França f & Teodorico C. Ramalho a
a Department of Chemistry, Federal University of Lavras, Campus Universitário, 37200-000,Lavras, MG, Brazilb Chemistry Department, Federal Institute of Education Science and Technology, Estrada daTartaruga, S/N, 29215-090, Bairro Muquiçaba, Guarapari/ES, Brazilc Universal Protein Resource (UniProt), EMBL-EBI, European Bioinformatics Institute,Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UKd Faculty of Military Health Sciences UO, Department of Toxicology, University of Defence,Hradec Králové, Czech Republice University Hospital Hradec Králové, Sokolská 581, 50005, Hradec Králové, Czech Republicf Laboratory of Molecular Modeling Applied to the Chemical and Biological Defense (LMCBD),Military Institute of Engineering, Rio de Janeiro, 22290-270, Brazil
Version of record first published: 25 Jun 2012
To cite this article: Karina S. Matos, Elaine F.F. da Cunha, Arlan da Silva Gonçalves, Alan Wilter, Kamil Kuča, Tanos C.C.França & Teodorico C. Ramalho (2012): First principles calculations of thermodynamics and kinetic parameters and moleculardynamics simulations of acetylcholinesterase reactivators: can mouse data provide new insights into humans?, Journal ofBiomolecular Structure and Dynamics, 30:5, 546-558
To link to this article: http://dx.doi.org/10.1080/07391102.2012.687521
PLEASE SCROLL DOWN FOR ARTICLE
Full terms and conditions of use: http://www.tandfonline.com/page/terms-and-conditions
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form toanyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae, and drug doses shouldbe independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims,proceedings, demand, or costs or damages whatsoever or howsoever caused arising directly or indirectly inconnection with or arising out of the use of this material.

First principles calculations of thermodynamics and kinetic parameters and moleculardynamics simulations of acetylcholinesterase reactivators: can mouse data provide newinsights into humans?
Karina S. Matosa, Elaine F.F. da Cunhaa, Arlan da Silva Gonçalvesb, Alan Wilterc, Kamil Kučad,e, Tanos C.C. Françaf
and Teodorico C. Ramalhoa*aDepartment of Chemistry, Federal University of Lavras, Campus Universitário, 37200-000 Lavras, MG, Brazil; bChemistryDepartment, Federal Institute of Education Science and Technology, Estrada da Tartaruga, S/N, 29215-090 Bairro Muquiçaba,Guarapari/ES, Brazil; cUniversal Protein Resource (UniProt), EMBL-EBI, European Bioinformatics Institute, Wellcome TrustGenome Campus, Hinxton, Cambridge CB10 1SD, UK; dFaculty of Military Health Sciences UO, Department of Toxicology,University of Defence, Hradec Králové, Czech Republic; eUniversity Hospital Hradec Králové, Sokolská 581, 50005 HradecKrálové, Czech Republic; fLaboratory of Molecular Modeling Applied to the Chemical and Biological Defense (LMCBD),Military Institute of Engineering, Rio de Janeiro 22290-270, Brazil
Communicated by Ramaswamy H. Sarma
(Received 5 September 2011; final version received 12 March 2012)
We have applied a theoretical methodology, previously developed to evaluate the association and kinetic reactivationconstants of oximes, comparing theoretical data obtained for human acetylcholinesterase (HsAChE) with in vitro resultsfrom Mus musculus AChE (MmAChE) previously reported in the literature. Our results, further checked by additionalmolecular dynamics simulations steps, showed a good correlation between the theoretical and experimental data, support-ing the methodology as appropriate for prediction of thermodynamic and kinetic parameters and corroborated MmAChEas a suitable model for studies with HsAChE.
Keywords: acetylcholinesterase; QM/MM; chemical mechanism of reactivation; neurotoxic agents
1. Introduction
The enzyme acetylcholinesterase (AChE) promotes thetermination of impulse transmission at cholinergic syn-apses by rapid hydrolysis of the neurotransmitter acetyl-choline (ACh). This is the principal biological role ofAChE (Barnard, 1974). The powerful acute toxicity oforganophosphorus poisons is primarily due to the factthat they are potent irreversible AChE inhibitors, forminga covalent bond with a serine residue at the active site(Quinn, 1987). The neurotoxics are chemical warfareagents that act as inhibitors of AChE, interrupting thehydrolysis of ACh, and can lead to an irreversible inhibi-tion of this enzyme (aging) thus triggering the choliner-gic syndrome (Black & Harrison, 1996; Kryger et al.,2000; Somani, Solana, & Dube, 1992). A nucleophile,like an oxime, whose hydroxyl group is able to removethe nerve agent from the active site and reactivate AChE,can be used to avoid the inhibition, as shown in Figure 1.This reactivation reaction, illustrated in Equation (1),
involves, first, the association of the oxime to the inhib-ited enzyme (EIOx) and then the reactivation of theenzyme by the leaving of the oxime complexed to theneurotoxic agent (IOx) (Kassa, Kuca, Bartosova, & Kun-esova, 2007; Ramalho et al., 2010).
EIþ Ox$KR EIOx!kR Eþ IOx (1)
where KR and kr are the dissociation constants, whichrepresent the affinity of oximes for the inhibited AChE,and the rate constant for the decomposition of the stableenzyme-inhibitor-reactivator complex, respectively (Kassaet al., 2007; Ramalho et al., 2010).
Many structurally different oximes are reported toperform the reactivation of AChE inhibited by severaldifferent neurotoxic agents, but one structure able to actefficiently against all the existing neurotoxic agents hasnot yet been reported (Kassa et al., 2007; Ramalho et al.,2010). Furthermore, oximes that are efficient against one
*Corresponding author. Email: [email protected]
Journal of Biomolecular Structure and DynamicsVol. 30, No. 5, 2012, 546–558
ISSN 0739-1102 print/ISSN 1538-0254 onlineCopyright � 2012 Taylor & Francishttp://dx.doi.org/10.1080/07391102.2012.687521http://www.tandfonline.com
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

specific agent can be completely ineffective with another(Black & Harrison, 1996; Kassa et al., 2007; Ramalhoet al., 2010; Sidell, Takafuji, & Franz, 1997; Somaniet al., 1992). This probably happens because theirmechanisms of action are not well elucidated so far(Ekström et al., 2006) and some relevant factors like theadequate orientation of the phosphoryl bond inside theactive site, the suitable oxime’s charge, the most ade-quate angles for attacking the phosphylated serine, theinfluence of the oxime’s isomery, and, also, the chemicalenvironment of the oxime group inside AChE remainsunknown despite been a recurrent issue in literature(Ashani et al., 1995; Bernard et al., 2000; Ekström,Astot, & Pang, 2007; Ekström et al., 2006; Gonçalves,França, Figueroa-Villar, & Pascutti, 2010, 2011; Gonçal-ves, França, & Wilter, 2006; Guimarães et al., 2011;Matos et al., 2011; Nemukhim, Lushchekina, Bochenk-ova, Golubeva, & Varfolomeev, 2008; Steven et al.,2011; Wong et al., 2000; Worek et al., 2007).
The computer-aided drug design has been a promisingstrategy for identifying potential lead compounds andmolecular structural features that are related to biologicalactivity. Structure-based investigations using first princi-ples, docking and molecular dynamics (MD) simulationshave been widely used to study ligand and receptor inter-action and have been applied in the research against bothchemical and biological warfare agents (Akten, Cansu, &Doruker, 2009; Andrianov, 2009; Borkar, Ghosh, & Bhat-tacharyya, 2010; Cambria, Di Marino, Falconi, Garava-glia, & Cambria, 2010; Chen, 2009, 2010; Chen et al.,2009; Cordomi & Perez, 2009; da Cunha, Barbosa, Oli-veira, & Ramalho, 2010; Hage-Melim, da Silva, Semigh-ini, Taft, & Sampaio, 2009; Huang, Lee, Yu, Chen, Hsu,et al., 2010; Huang, Lee, Yu, Chen, Tsai et al., 2010; Jin,Lee & Kim, 2010; Kahlon, Roy, & Sharma, 2010; Koshy,Parthiban, & Sowdhamini, 2010; Likhatskaya et al.,2005; Meynier, Guerlesquin & Roche, 2009; Mohan,Perry, Poulose, Nair, & Anilkumar, 2009; Mehrnejad &
Zarei, 2010; Bairagya, Mukhopadhyay, & Sekar, 2009;Ramalho et al., 2009; Roy & Thakur, 2010; Sille & Rem-ko, 2009; Sharadadevi & Nagaraj, 2010; Sharma, Sonav-ane, & Joshi, 2010; Tao, Rao, & Liu, 2010; Timofeyevaet al., 2009; Varughese, Chalovich, & Li, 2010; Zhang,2009; Zhong & Xie, 2009).
Several molecular modeling studies available in litera-ture suggest important features on the oximes structuresthat could be very useful to guide experimental researchon this issue (Ashani et al., 1995; Bernard et al., 2000;Ekström et al., 2006, 2007; Gonçalves et al., 2010;Gonçalves et al., 2011; Gonçalves, França, Wilter, & Fig-ueroa-Villar, 2006; Guimarães et al., 2011; Matos et al.,2011; Nemukhim et al., 2008; Steven et al., 2011; Wonget al., 2000; Worek et al., 2007). However, it is worthmentioning that, at both experimental and theoretical lev-els, these works have been most performed with modelsystems like Mus musculus AChE (MmAChE) instead ofthe human enzyme due to the intrinsic difficultiesinvolved in conducting this kind of study with humans.So, the usual strategy to evaluate the biological activityof oximes is to use MmAChE for the experimental tests.Despite the success of this methodology and high homol-ogy in the active site between MmAChE and HsAChEenzymes, some criticism occur due to the extrapolation ofreactive data among species. In this context, it raises thequestion if the usual model is adequate to represent thebehavior of the AChE reactivators in humans.
In this line, we have explored some molecularaspects of the reactivation process (Ekström et al., 2006;Kassa et al., 2007; Matos et al., 2011; Ramalho et al.,2010; Sidell et al., 1997) employing docking studies anddensity functional theory (DFT) calculations for evalua-tion of KR and kr of oximes in MmAChE inhibited bytabun, and by cyclosarin, using Molegro® VirtualDocker 2006 (MVD) (Thomsen & Christensen, 2006),Spartan08® (Hehre, Deppmeier, & Klunzinger, 1999),and Gaussian03® (Frisch et al., 2004) softwares. Our
Figure 1. Inhibition, desinhibition, and aging of AChE. X is the leaving group.
Acetyl cholinesterase reactivators 547
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

results corroborated this methodology as suitable for theprediction of kinetic and thermodynamic parameters foroximes that might be helpful for the design and selectionof new and more effective oximes. Herein, we haveapplied the same methodology developed previously toevaluate the kinetic and thermodynamics parameters of thereactivation process in a set of oximes, now for HumanAChE (HsAChE) inhibited by the nerve agent cyclosarin(GF). This time our results were additionally checked andcorroborated by additional steps of MD simulations of thebest poses of the oximes inside the HsAChE/GF com-plexes obtained from the docking studies.
2. Methodology
2.1. Ligands dataset and docking energy calculations
The in vitro data of KR and kr for the oximes studied inthis work (Figure 2) regarding MmAChE inhibited byGF, were reported by Kassa et al. (2007). Crystallo-graphic coordinates of HsAChE (PDB code: 3LII) (Dvir,Silman, Harel, Rosenberry, & Sussman, 2010) weretaken from the Brookhaven Protein Data Bank (PDB)(Guex & Peitsch, 1997) and had the crystallographicwater molecules removed in the Swiss-PdbViewer(SPDBViewer) software (Guex & Peitsch, 1997) for thebest performance of the calculations, due to the existenceof hydrophobic areas distinct from the binding site insideAChE (Ekström et al., 2009). Crystallographic coordi-nates of the nerve agent sarin (GB) and the oxime HI-6complexed inside MmAChE (PDB code: 2WHP)(Ekström et al., 2009) were used to model GF and HI-6inside HsAChE, using SPDBViewer (Guex & Peitsch,1997). The GB structure inside the crystallographicstructure of MmAChE was completed through the inser-tion of the group –CH2CH2CH2– using the softwareSpartan08® (Hehre et al., 1999) (MMFF94 force field),in order to get the structure of GF. The FASTA sequenceof HsAChE and MmAChE were copied from the PDB
(Guex & Peitsch, 1997) to Expasy (http://au.expasy.org/)in order to evaluate the sequential identity between theenzymes. To verify the similarity between the residues ofthe active sites of HsAChE and MmAChE and to deter-mine the degree of identity between them, we alignedHsAChE and MmAChE primary sequences using theSPDBViewer program (Guex & Peitsch, 1997). The 3Dstructures of each oxime were built in the Spartan08®(Hehre et al., 1999) software based on the bioactive con-formation of HI-6 inside this structure, and subsequently,the overall geometry optimizations and partial atomiccharge distribution calculations were performed withGaussian03® (Frisch et al., 2004) using the AM1 semi-empirical molecular orbital method. These quantummechanics (QM) electrostatic charges were incorporatedin the AMBER force field (Cornell et al., 1995; Maseras& Morokuma, 1995) in order to evaluate coulomb inter-actions in the docking procedure.
In the same way as before (Ramalho et al., 2010), thecompounds were docked into the HsAChE binding siteusing MVD® (Thomsen & Christensen, 2006), a programfor predicting the most likely conformation of how aligand will bind to a macromolecule. The MolDock scor-ing function (MolDock Score) used by MVD® is derivedfrom the piecewise linear potential and further extendedin generic evolutionary method for molecular DOCK(GEMDOCK) with a new hydrogen-bonding term andnew charge schemes. The docking search algorithm usedin MVD® (Thomsen & Christensen, 2006) is based oninteractive optimization techniques inspired by the Dar-winian evolution theory (evolutionary algorithms [EA]).Binding sites were restricted within spheres with radiusfrom 5 to 15Å, according to the ligand size, centered atthe HI-6 binding site in the protein complex and enclos-ing all the active site residues. Ligand molecules and asubset region composed of all amino acid residues (sidechain) having at least one atom within 12Å of the centerof HI-6, were considered flexible during the docking sim-ulation. Due to the stochastic nature of the ligand–protein
Figure 2. Structures of the oximes studied.
548 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

docking search algorithm, about 10 runs were conductedand 30 docking solutions were retained for each ligand.The best superimposing poses related to HI-6, were cho-sen for the analysis performed in this work.
2.2. DFT studies
QM/molecular mechanics (MM) techniques were per-formed with the selected structures in the docking proce-dure, to determine the preferred route for the reactivationprocess, combining docking techniques, and DFT calcu-lations at the QM/MM interface for the enzymatic mech-anism. The QM/MM approach seeks to partition thetarget system under study into QM and MM regions.The hydrogen is employed as the link atom to saturatethe free valency, so that the electronic effects resultingfrom QM/MM partitioning are minimized. The currentresearch utilized the ONIOM methodology (Jorgensen,Chandrasekhar, Madura, Impey, & Klein, 1983). Allclassical MM calculations have been performed with theAMBER all-atom force field (Cornell et al., 1995; Mas-eras & Morokuma, 1995). The QM calculations werecarried out in the Spartan08® (Hehre et al., 1999) andGaussian03® (Frisch et al., 2004) packages. The QMregions were cut out from the docking results in theSPDBViewer software (Dvir et al., 2010) and consistedof residues, neighboring peptide bonds, link atoms, crys-tallographic water molecules, ligand, and GF inside asphere with radius of 15 , centered at each oxime. Allthe transition states, intermediates, and precursorsinvolved were calculated. Each conformer was fully opti-mized at the DFT level with B3LYP/6-311G(d) (Rama-lho, Caetano, & Cunha, 2009; Ramalho & Taft, 2005).Furthermore, after each optimization, a force constantcalculation was made in order to verify whether the opti-mized structures were indeed local minima (no imaginaryfrequencies) or transition states (one imaginary fre-quency).
2.3. MD simulations
Before performing the MD simulations, it was necessaryto parameterize the ligands so they could be recognizedby the force field OPLASS (Jorgensen, Maxwell, & Tira-do-Rives, 1996) from the program GROMACS 4.5(Hess, Kutzner, van der Spoel, & Lindahl, 2008). Toobtain the parameters and topologies for the referredcompounds, we used AnteChamber PYthon Parser inter-facE (ACPYPE) (Sousa da Silva & Vranken, 2012). It isa tool based on Python programming language to useANTECHAMBER (currently bundled in AmberToolsversion 1.4) (Wang, Wang, Kollman, & Case, 2006) togenerate parameters and topologies for chemical com-pounds and to interface with other python applicationslike CCPN tools (Vranken et al., 2005) or ARIA2 (Rie-ping et al., 2007). ACPYPE is currently able to generateoutput files for the following MM softwares: CNS/
XPLOR (Brunger, 2007; Schwieters, Kuszewski, &Clore, 2006), GROMACS (Hess et al., 2008),CHARMM, and AMBER (Cornell et al., 1995; Jorgen-sen et al., 1983). The atomic partial charges of theligands were calculated by the semi-empirical quantumchemistry program SQM (Walker, Crowley, & Case,2008) (via ANTECHAMBER), according to AM1-BCCparameters (parameterized to reproduce HF/6-31G_RESPcharges). SQM was modified to include six decimals ofprecision instead of the default three.
The HsAChE/GF/oxime complexes were simulatedusing the GROMACS 4.5 package (Hess et al., 2008), incubic boxes of approximately 750 nm3 containingaround 21,000 water molecules. These systems wereminimized using the force field OPLSAA (Jorgensenet al., 1996). The minimization algorithms used weresteepest descent with position restrained (PR) of theligands and convergence criterion of100.00 kcal mol�1 Å�1, followed by steepest descentwithout PR, conjugate gradients and finally, quasi-New-ton–Raphson until an energy of 1.00 kcal mol�1 Å�1.The minimized complexes were then submitted to MDsimulations in two steps. Initially, we performed 500 psof MD at 300K with PR for the entire system, exceptthe water molecules, in order to insure a balance of thesolvent molecules around the residues of the enzyme.Subsequently, there were performed 10,000 ps of MD at300K without any restriction, using 2 fs of integrationtime and a cutoff of 10Å for long-distance interactions.A total of 500 conformations were obtained during eachsimulation. In this step, the lists of pairs (pairlists) wereupdated every 20 steps, all Arg and Lys residues wereassigned with positive charges and the residues Glu andAsp were assigned with negative charges.
To analyze the structures generated after the optimi-zation and MD steps, we used the VMD (Humphrey,Dalke, & Schulte, 1996) program. Plots of variation oftotal energy and root-mean-square deviation (RMSD)along the MD simulation were generated with the Micro-soft Excel®. Qualitative spatial RMSD pictures weregenerated in the MolMol (Koradi, Billeter, & Wüthrich,1996) and the figures of the frames of MD simulationswere generated in the PyMOL program (Warren, 2002).
3. Results and discussion
3.1. Docking results
The sequential identity between the HsAChE andMmAChE was evaluated and the value observed was of88.5%. It was also verified that the amino acids residuesin the active site of both enzymes are 100% conserved(see alignment in Figure 3). This means that, based onsequence similarity, comparisons of alpha-carbon coordi-nates, as well as number and type of amino acid residuespresent in the active site of the HsAChE and MmAChE
Acetyl cholinesterase reactivators 549
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

enzymes were equivalent. This suggests that experimen-tal results obtained with MmAChE could be extrapolatedto HsAChE.
After docked inside the HsAChE active site, the oxi-mes (Figure 2) had all their reasonable binding orienta-tions for the HsAChE reactivation investigated,according to a search of the conformational space fordifferent ligand orientations performed by MVD®(Thomsen & Christensen, 2006). As mentioned before,the water molecules were removed to perform the calcu-lations due to the hydrophobic site for the alkoxy leavinggroup of the substrate, including residues Trp86, Tyr337,and Phe338, which are among the key elements main-taining the functional architecture of the active center,contributing to the stabilization of the Michaelis–Mentencomplexes (Ordentlich et al., 1996). The low-energyinteraction modes were, then, chosen for further minimi-zations (Ramalho et al., 2010). From the molecular dock-ing simulations performed between each studiedcompound and the enzyme, the binding modes with the
lowest docked energies were selected (Ramalho et al.,2010). It should be kept in mind, however, that the con-formational search methods should be typically validatedagainst standard benchmark datasets, because dockingscenarios may typically fall in some systems. In this con-text, an alternative approach is to compare generatedconformations by docking calculations against experi-mentally derived X-ray structures. Thus, it is possible todetect whether experimentally determined bioactive con-formations are present in various generated conforma-tional ensembles. In the current case, we have used thecrystal structure of HI-6 complexed inside MmAChE todo this procedure.
Our final results suggest that the correlation betweentheoretical and experimental data is satisfactory, with a R2
value equal to 0.96. Wherever possible the derived dataon the bioactive conformation from experimental resultsshould be verified, considering that the identification of alow-energy conformation is an important step in theunderstanding of the relationship between structure and
Figure 3. Sequence alignment of the primary sequences of HsAChE and MmAChE. Nonmatching residues are shown in red and theactive site residues are shown in blue.
550 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

biological activity of a molecule (da Cunha, Ramalho,Mancini, Fonseca, & Oliveira, 2010; Jorgensen, 1991).
As shown in Table 1, the oximes with the mostpromising in vitro data were HI-6 and K005, which cor-roborates the theoretical data, since their interactionenergy values were the lowest found. The favorable ori-entation of the HI-6 inside the HsAChE active siteinvolves nine hydrogen bonds with GF and the residuesTyr124, Arg296, and Tyr337, while K005 involves 11hydrogen bonds with GF and the residues Tyr72,Asp74, Ans87, Tyr124, and Ser125 (Figure 4). Table 2
presents the predicted dissociation free energies of theoximes with HsAChE and the experimental valuesreported by Kassa et al. (2007), in the same way asdemonstrated before for MmAChE (Matos et al., 2011;Ramalho et al., 2010), thus confirming our theoreticalfindings that point out that a more quantitative explana-tion to structure–activity relationship of the inhibitorymechanism for these inhibitors can be based on thebinding free energies and their correlation with KR ofthe inhibited enzyme–reactivator complexes (Ramalhoet al., 2010).
Table 1. Energy values for HsAChE reactivators in the active site and dissociation constants of the inhibited MmAChE-reactivatorcomplex.
Ligand pKRa (μM) ΔEb (kcalmol�1) ΔEc (kcal mol�1) Residue Strength bond (kcalmol�1) Distance (Å)
K005 5.30 �143.48 �140.67 Asp74 �0.31 2.99Tyr72 �2.50 2.61Ser125 �1.70 3.26Ser125 �2.50 2.66Asn87 �1.69 3.26Tyr124 �2.50 3.04Tyr124 �2.18 3.16Tyr124 �2.46 3.11Tyr124 �2.50 3.00GF �1.39 3.32GF �0.60 3.46
HI-6 4.92 �142.98 �140.77 Tyr124 �2.22 2.56Tyr124 �1.39 3.32Tyr124 �1.70 2.50Tyr124 �1.13 3.26Tyr337 �2.28 3.14Tyr337 �2.50 2.99Arg296 �2.50 2.73GF �2.50 2.80GF �2.13 2.85
HS-6 4.35 �142.67 �133.62 Tyr124 �2.50 2.73Tyr124 2.83 1.97Tyr124 �2.33 3.13Tyr124 �2.50 2.61Arg296 �2.50 3.09Ser293 �2.50 3.01Tyr341 �0.79 3.44GF �2.50 3.04
HLö-7 3.00 �141.91 �129.93 Tyr124 �1.81 2.83Tyr124 �2.30 3.14GF �2.50 2.95GF �2.50 2.92
BI-6 1.00 �140.21 �121.29 Tyr72 0.73 2.22Gln71 �2.34 3.13Ser125 2.22 2.04Asp74 �1.64 2.83Asn87 �2.49 2.59Tyr124 �2.17 3.16Tyr124 �2.50 2.97Tyr124 �2.50 3.08GF �0.13 2.31
aMmAChE experimental data.bTheoretical data to MmAChE (Matos et al., 2011).cTheoretical data to HsAChE. The percentage of homological identity between the enzymes is 88.5%.
Acetyl cholinesterase reactivators 551
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

3.2. Mechanistic studies
The understanding of the reactivation process mechanismof oximes is an important step to the design of new andmore selective reactivation agents. Also the dynamiceffects on both, the reaction mechanism and ligand ori-entation should be kept in mind. The mechanism goesthrough an addition–elimination pathway (Ramalhoet al., 2010). This QM/MM study is a first step in under-standing the interaction between the complex HsAChE/GF and reactivators in a theoretical way. From Table 2,the kinetic parameters and ΔΔE# predicted by theoreticalcalculations of the oximes and the experimental valuesreported by Kassa et al. (2007) are observed. However,in our present study, we have compared the ΔE valuesbetween the transition state and the initial system foreach oxime. In this way, we have obtained the tendencyof reactivity of oximes, avoiding thus the direct compu-tation of absolute energy values. The correlation amongthese values is better expressed in Figure 5 and demon-strates that the kinetic parameters and the experimentalvalues are in reasonable agreement, as demonstratedpreviously (Ramalho et al., 2010). After the optimization
of the selected conformers, a force constant calculationwas carried out to assure that the structures reported inTable 2 are all transition states. Thus, we have used thesame computational procedure previously employed suc-cessfully for tabun-inhibited (Somani et al., 1992) andcyclosarin-inhibited (Matos et al., 2011) MmAChE andon similar systems (Cunha et al., 2010; Jorgensen,1991). Again an extra stabilization of the transition statewas observed through long-range interactions, such ascation–π, π–π staking or electrostatic interactions, thistime with the residues Gln71, Tyr72, Asp74, Ans87,Tyr124, Ser125, Ser293, Phe295, Arg296, Tyr337, andTyr341.
We believe that, here, the stabilization of the transi-tion state is favored by hydrogen bonding with the clos-est amino acids residues: Tyr72, Asp74, Ans87, Tyr124,Arg296, Tyr337, and GF which orient the oxime towardsthe adequate TS geometry. In that conformation, theoxime interacts more strongly with GF. Furthermore, thelong-range interactions among the oximes and the otheramino acids could also take place, such as the cation–πinteractions with Tyr72, Tyr124, Arg296, Tyr337, andTyr341; electrostatic interactions with Asp74, Arg296,
Figure 4. Hydrogen bonds formed between K005 andresidues of the active site of HsAChE.
Table 2. Relative activation energies of the transition states and kinetic parameters.
Ligand kra (min�1) ΔΔE#b,c (kcal mol�1) ΔΔE#b,d (kcal mol�1) Frequency (cm�1)
HI-6 0.350 220.85 0.6781 i260.20BI-6 0.150 170.43 0.4623 i198.66HS-6 0.156 131.12 0.4044 i199.02K005 0.010 31.96 0.2817 i239.81HLö-7 0.008 0.00 0.00 i192.53
aRate constants of reactivation (Kassa et al., 2007).bΔΔE# =ELIGi � EHLö-7.cTheoretical data to MmAChE (Matos et al., 2011).dTheoretical Data to HsAChE.
Figure 5. Plot of �log kr ×ΔΔE# for the oximes studied.
1 =HI-6, 2 =BI-6, 3 =HS-6, 4 =K005, and 5 =HLö-7.
552 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012
and a π–π staking interaction among the oxime’s pyridi-nium ring and Tyr124 which occurs in the transitionstate geometry. Thus, our theoretical data point out thatthe oximes K005, HI-6, and Hlö-7 could be promisingHsAChE/GF reactivators. K005 and HI-6 are consideredamong the best reactivators of HsAChE/GF and showgood KR results (Table 1), and a high affinity for theintact enzyme (like Hlö-7) showing, consequently, a highreactivity with the enzyme (Table 2). However, theexcess of those oximes can also inhibit the reactivatedenzyme (Kassa et al., 2007; Ramalho et al., 2010).
3.3. MD simulations
After the docking and mechanistic studies, the best posesof the oximes in Figure 2 inside HsAChE/GF were sub-mitted to 10,000 ps of MD simulations. The goal of thesesimulations was to observe the dynamic behavior of theoximes inside the complexes in order to check the dock-ing and QM/MM results, obtaining additional informa-tion for supporting our methodology.
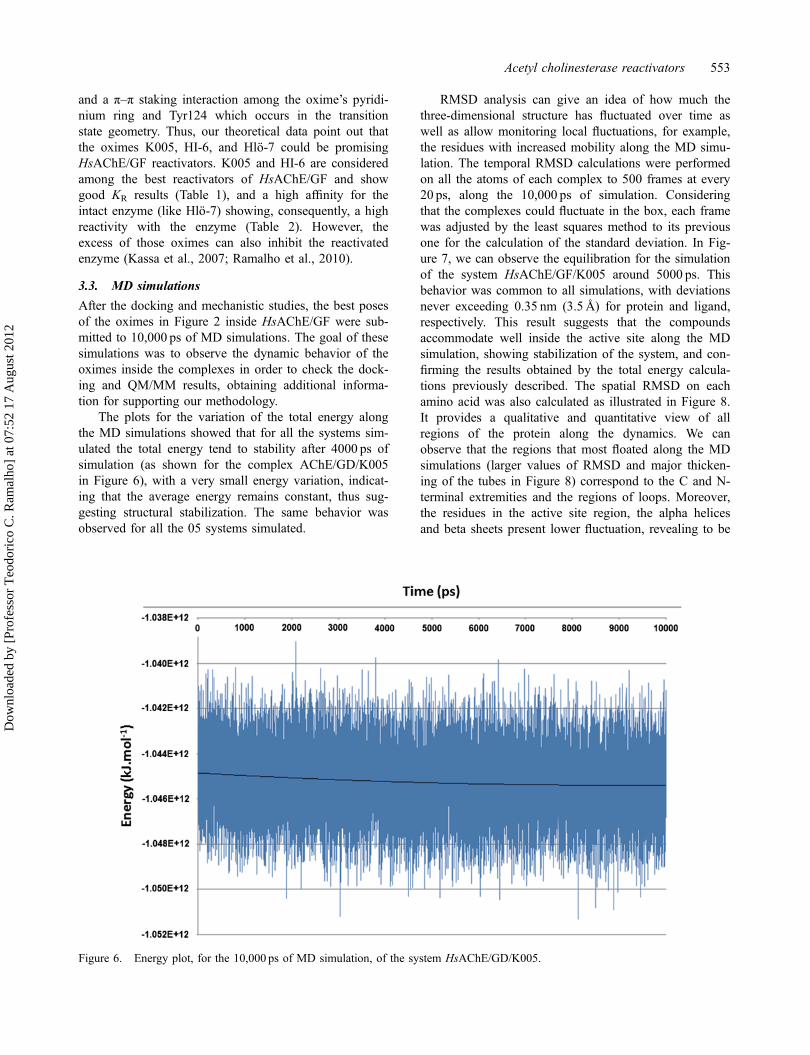
The plots for the variation of the total energy alongthe MD simulations showed that for all the systems sim-ulated the total energy tend to stability after 4000 ps ofsimulation (as shown for the complex AChE/GD/K005in Figure 6), with a very small energy variation, indicat-ing that the average energy remains constant, thus sug-gesting structural stabilization. The same behavior wasobserved for all the 05 systems simulated.
RMSD analysis can give an idea of how much thethree-dimensional structure has fluctuated over time aswell as allow monitoring local fluctuations, for example,the residues with increased mobility along the MD simu-lation. The temporal RMSD calculations were performedon all the atoms of each complex to 500 frames at every20 ps, along the 10,000 ps of simulation. Consideringthat the complexes could fluctuate in the box, each framewas adjusted by the least squares method to its previousone for the calculation of the standard deviation. In Fig-ure 7, we can observe the equilibration for the simulationof the system HsAChE/GF/K005 around 5000 ps. Thisbehavior was common to all simulations, with deviationsnever exceeding 0.35 nm (3.5Å) for protein and ligand,respectively. This result suggests that the compoundsaccommodate well inside the active site along the MDsimulation, showing stabilization of the system, and con-firming the results obtained by the total energy calcula-tions previously described. The spatial RMSD on eachamino acid was also calculated as illustrated in Figure 8.It provides a qualitative and quantitative view of allregions of the protein along the dynamics. We canobserve that the regions that most floated along the MDsimulations (larger values of RMSD and major thicken-ing of the tubes in Figure 8) correspond to the C and N-terminal extremities and the regions of loops. Moreover,the residues in the active site region, the alpha helicesand beta sheets present lower fluctuation, revealing to be
Figure 6. Energy plot, for the 10,000 ps of MD simulation, of the system HsAChE/GD/K005.
Acetyl cholinesterase reactivators 553
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012
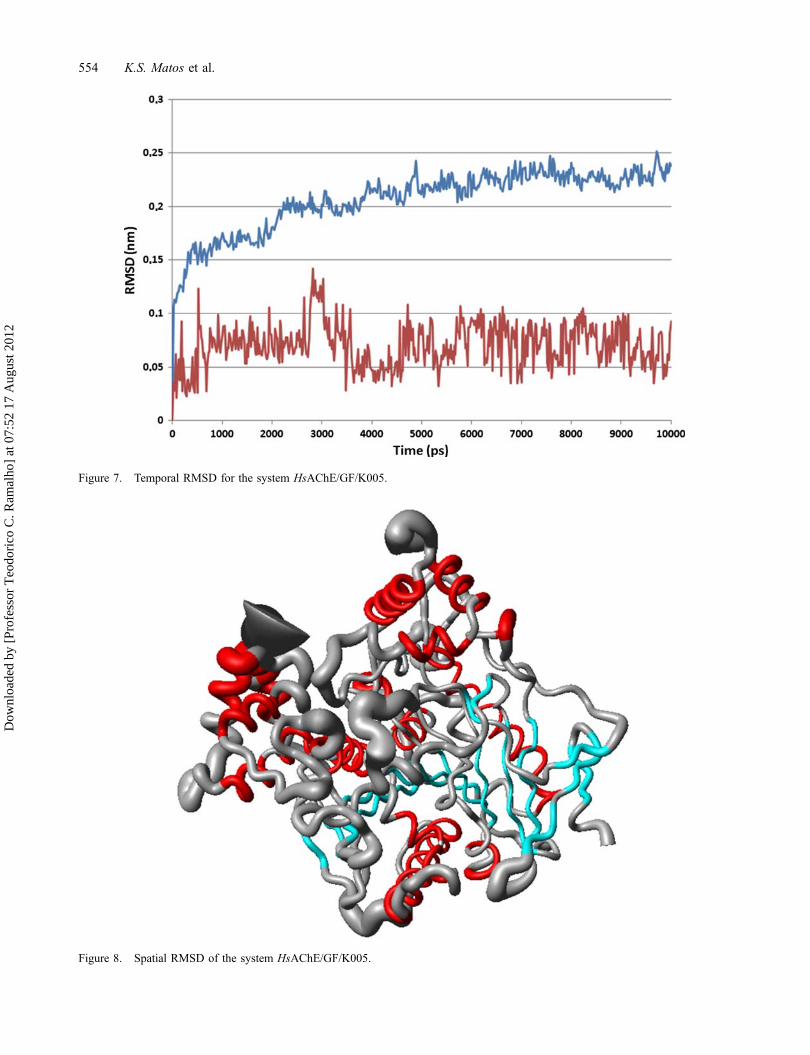
Figure 7. Temporal RMSD for the system HsAChE/GF/K005.
Figure 8. Spatial RMSD of the system HsAChE/GF/K005.
554 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

the more stable regions, as expected. This behavior wascommon to all the 05 systems simulated.
The illustrations in Figure 9 show the succession offrames for each oxime studied here inside the HsAChE/GF active site along the 10,000 ps of MD simulation.Analyzing these illustrations, we can access the dynamicbehavior of the oximes, GF, and the nearby residuesalong the 10,000 ps simulated. For all cases, we observedthat the oximes remained well behaved inside the activesites with its oxime group establishing H-bonds with thephosphate group of GF. Besides, all the residues pre-dicted in the docking studies were observed along thesimulations to be close enough to the oximes to establishinteractions. We believe that these results corroborate ourdocking and mechanistic results.
4. Conclusion
Our results again showed good agreement between thetheoretical dissociation free energies of the oximes andexperimental data, corroborating the methodology used
for the prediction of kinetic and thermodynamic parame-ters that might be helpful for the design of newHsAChE reactivators. This findings, suggest that thebinding process of oximes in HsAChE is favorablethrough the residue Tyr124. We observed again that thenumber of hydrogen bonds with Tyr124 is a key featureto determine the oxime binding mode. Regarding thereaction pathway, we noticed that Tyr124 is also respon-sible for the transition state stabilization. It is importantto mention that all oximes interacted with the phosphategroup of GF. This interaction can be due to interactionsof the oximes with Tyr124, which is located at theactive site close to GF, stabilizing the oxime, andenhancing the reaction. Finally, the results obtained here,besides corroborating deeply with our previous study(Matos et al., 2011; Somani et al., 1992), were checkedby 10,000 MD simulations that showed the same inter-actions former predicted in the docking studies, reinforc-ing the idea that the calculated kinetic parameters mightbe helpful for the design and selection of new and moreeffective oximes.
Figure 9. Frames of (A) K005, (B) HI-6, (C) HS-6, (D) HLo7, and (E) BI-6 inside the active site of HsAChE along the MDsimulations.
Acetyl cholinesterase reactivators 555
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

Acknowledgments
The authors are grateful to Fundação de Amparo a Pesquisa doEstado de Minas Gerais (FAPEMIG) (TCR and EFFC),Coordenação de Aperfeiçoamento de Pessoal de Nível Superior(CAPES) (KSM), Fundação de Amparo ao Ensino e Pesquisa doEstado do Rio de Janeiro (FAPERJ) (TCCF) and ConselhoNacional de Desenvolvimento Científico e Tecnológico (CNPq)(TCR and EFFC) for financial support. We also would like tothank the Centro Nacional de Processamento de Alto Desempenhoem São Paulo (CENAPAD-SP) for the computational facilities.This study was also supported by Project No. FVZ0000604 (KK).
ReferencesAkten, E.D., Cansu, S., & Doruker, P. (2009). A docking study
using atomistic conformers generated via elastic networkmodel for cyclosporin a/cyclophilin a complex. Journal ofBiomolecular Structure & Dynamics, 27, 13–25.
Andrianov, A.M. (2009). Immunophilins and HIV-1 V3 loopfor structure-based anti-AIDS drug design. Journal of Bio-molecular Structure & Dynamics, 26, 445–454.
Ashani, Y., Radic, Z., Tsigelny, I., Vellom, D.C., Pickering, N.A., Quinn, D.M., … Taylor, P. (1995). Amino acid residuescontrolling reactivation of organophosphonyl conjugates ofacetylcholinesterase by mono- and bisquaternary oximes.Journal of Biological Chemistry, 11, 6370–6380.
Bairagya, H.R., Mukhopadhyay, B.P., & Sekar, K. (2009). Aninsight to the dynamics of conserved water molecular triadin IMPDH II (Human): Recognition of cofactor and sub-strate to catalytic Arg 322. Journal of Biomolecular Struc-ture & Dynamics, 27, 149–158.
Barnard, E.A. (1974). Neuromuscular transmission – enzymaticdestruction of acetylcholine. In J.I. Hubbard (Ed.), Theperipheral nervous system (pp. 30–50). New York, NY:Plenum Press.
Bernard, P.P., Kireev, D.B., Pintore, M., Chrétien, J.R., Fortier,P.L., & Froment, D. (2000). A CoMFA study of enantio-meric organophosphorus inhibitors of acetylcholinesteras.Journal of Molecular Modeling, 6, 618–629.
Black, R.M., & Harrison, J.M. (1996). The chemistry of orga-nophosphorus chemical warfare agents. In F.R. Hartley(Ed.), The chemistry of organophosphorus compounds (Vol.4, pp. 120–240). Chichester: Wiley.
Borkar, A., Ghosh, I., & Bhattacharyya, D. (2010). Structureand dynamics of double helical DNA in torsion anglehyperspace: A molecular mechanics approach. Journal ofBiomolecular Structure & Dynamics, 27, 695–712.
Brooks, B.R., Brooks C.L. III, MacKerell Jr., A.D., Nilsson,L., Petrella, R.J., Roux, B.
Brunger, A.T. (2007). Version 1.2 of the crystallography andNMR system. Nature Protocols, 2, 2728–2733.
Cambria, M.T., Di Marino, D., Falconi, M., Garavaglia, S., &Cambria, A. (2010). Docking simulation and competitiveexperiments validate the interaction between the 2,5-xylidine Inhibitor and Rigidoporus lignosus laccase.Journal of Biomolecular Structure & Dynamics, 27, 501–509.
Chen, C.Y.C. (2009). Weighted equation and rules – a novelconcept for evaluating protein–ligand interaction. Journalof Biomolecular Structure & Dynamics, 27, 271–282.
Chen, C.Y. (2010). Virtual screening and drug design forPdDe-5 receptor from traditional Chinese medicine data-base. Journal of Biomolecular Structure & Dynamics, 27,627–640.
Chen, C.Y., Chang, Y.H., Bau, D.T., Huang, H.J., Tsai, F.J., Tsai,C.H., & Chen, C.Y.C. (2009). Ligand-based dual target drugdesign for H1N1: Swine flu – a preliminary first study. Jour-nal of Biomolecular Structure & Dynamics, 27, 171–178.
Cordomi, A., & Perez, J.J. (2009). Structural rearrangements ofrhodopsin subunits in a dimer complex: A moleculardynamics simulation study. Journal of Biomolecular Struc-ture & Dynamics, 27, 127–147.
Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., … Kollman, P.A. (1995). A secondgeneration force field for the simulation of proteins, nucleicacids, and organic molecules. Journal of the AmericanChemical Society, 117, 5179–5197.
da Cunha, E.F.F., Barbosa, E.F., Oliveira, A.A., & Ramalho, T.C. (2010). Molecular modeling of mycobacterium tubercu-losis DNA gyrase and its molecular docking study withgatifloxacin inhibitors. Journal of Biomolecular Structure& Dynamics, 27, 619–625.
da Cunha, E.F.F., Ramalho, T.C., Mancini, D.T., Fonseca, E.M.B., & Oliveira, A.A. (2010). New approaches to thedevelopment of anti-protozoan drug candidates: A reviewof patents. Journal of the Brazilian Chemical Society, 21,1787–1806.
Dvir, H., Silman, I., Harel, M., Rosenberry, T.L., & Sussman,J.L. (2010). Acetylcholinesterase: From 3D structure tofunction. Chemico-Biological Interactions, 187, 10–22.
Ekström, F.J., Astot, C., & Pang, Y.P. (2007). Novel nerve-agent antidote design based on crystallographic and massspectrometric analyses of tabun-conjugated acetylcholines-terase in complex with antidotes. Clinical Pharmacologyand Therapeutics, 82(3), 282–293.
Ekström, F., Hörnberg, A., Artursson, E., Hammarström, L.,Schneider, G., & Pang, Y. (2009). Structure of HI-6NSarin-Acetylcholinesterase determined by X-ray crystallographyand molecular dynamics simulation: Reactivator mechanismand design. PLoS ONE, 4, e5957.
Ekström, F.J., Yuan-Ping, P., Boman, M., Artursson, E., Akfur,C., & Börjegren, S. (2006). Crystal structures of acetylcho-linesterase in complex with HI-6, Ortho-7 and obidoxime:Structural basis for differences in the ability to reactivatetabun conjugates. Biochemical Pharmacology, 72, 597–607.
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E.,Robb, M.A., Cheeseman, J.R., … Pople, J.A. (2004).Gaussian, Inc. Wallingford, CT.
Gonçalves, A.S., França, T.C.C., Figueroa-Villar, J.D., & Pas-cutti, P.G. (2010). Conformational analysis of toxogonine,TMB-4 and HI-6 using PM6 and RM1 methods. Journal ofthe Brazilian Chemical Society, 21, 179–184.
Gonçalves, A.S., França, T.C.C., Figueroa-Villar, J.D., &Pascutti, P.G. (2011). Molecular dynamics simulations andQM/MM studies of the reactivation by 2-PAM of tabuninhibited human acethylcolinesterase. Journal of the Brazil-ian Chemical Society, 22, 155–165.
Gonçalves, A.S., França, T.C.C., Wilter, A., & Figueroa-Villar,J.D. (2006). Molecular dynamics of the interaction of prali-doxime and deazapralidoxime with acetylcholinesteraseinhibited by the neurotoxic agent tabun. Journal of the Bra-zilian Chemical Society, 17, 968–975.
556 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

Guex, N., & Peitsch, M.C. (1997). Peitsch, swiss-model andSwiss-PdbViewer: An environment for comparative proteinmodeling. Electrophoresis, 18, 2714–2723.
Guimarães, A.P., França, T.C.C., Ramalho, T.C., Rennó, M.N.,da Cunha, E.F.F., Matos, K.S., Mancini, D.T., & Kuča, K.(2011). Docking studies and effects of syn-anti isomery ofoximes derived from pyridine imidazol bicycled systems aspotential human acetylcholinesterase reactivators. Journalof Applied Biomedicine, 9, 163–171.
Hage-Melim, L.I.D., da Silva, C.H.T.D., Semighini, E.P., Taft,C.A., & Sampaio, S.V. (2009). Computer-aided drug designof novel PLA2 inhibitor candidates for treatment of snake-bite. Journal of Biomolecular Structure & Dynamics, 27,27–35.
Hehre, W.J., Deppmeier, B.J., & Klunzinger, P.E. (1999). PCSPARTAN Pro. Irvine, CA: Wavefunction.
Hess, B., Kutzner, C., van der Spoel, D., & Lindahl, E. (2008).GROMACS 4: Algorithms for highly efficient, load-bal-anced, and scalable molecular simulation. Journal of Chem-ical Theory and Computation, 4, 435–447.
Huang, H.J., Lee, K.J., Yu, H.W., Chen, C.Y., Hsu, C.H., Chen,H.Y., Tsai, F.J., & Chen, C.Y.C. (2010). Structure-basedand ligand-based drug design for HER 2 receptor. Journalof Biomolecular Structure & Dynamics, 28, 23–37.
Huang, H.J., Lee, K.J., Yu, H.W., Chen, H.Y., Tsai, F.J., &Chen, C.Y. (2010). A novel strategy for designing theselective PPAR agonist by the “sum of activity” model.Journal of Biomolecular Structure & Dynamics, 28, 187–200.
Humphrey, W., Dalke, A., & Schulte, K. (1996). VMD – visualmolecular dynamics. Journal of Molecular Graphics, 14,33–38.
Jin, B., Lee, H.M., & Kim, S.K. (2010). Conformational analy-sis of genotoxic benzo[a]pyrene-7,8-dione-duplex DNAadducts using a molecular dynamics method. Journal ofBiomolecular Structure & Dynamics, 27, 457–464.
Jorgensen, W.L. (1991). Rusting of the lock and key model forprotein-ligand binding. Science, 254, 954–955.
Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W.,& Klein, M.L. (1983). Comparison of simple potentialfunctions for simulating liquid water. Journal of Computa-tional Physics, 79, 926–935.
Jorgensen, W.L., Maxwell, D.S., & Tirado-Rives, J. (1996).Development and testing of the OPLS all-atom force fieldon conformational energetics and properties of organic liq-uids. Journal of the American Chemical Society, 118(45),11225–11236.
Kahlon, A.K., Roy, S., & Sharma, A. (2010). Molecular dock-ing studies to map the binding site of squalene synthaseinhibitors on dehydrosqualene synthase of Staphylococcusaureus. Journal of Biomolecular Structure & Dynamics,28, 201–210.
Kassa, J., Kuca, K., Bartosova, L., & Kunesova, G. (2007).The development of new structural analogues of oximes forthe antidotal treatment of poisoning by nerve agents andthe comparison of their reactivating and therapeutic efficacywith currently available oximes. Current Organic Chemis-try, 11, 267–283.
Koradi, R., Billeter, M., & Wüthrich, K. (1996). MOLMOL: Aprogram for display and analysis of macromolecular struc-tures. Journal of Molecular Graphics, 14, 51–55.
Koshy, C., Parthiban, M., & Sowdhamini, R. (2010). 100 nsmolecular dynamics simulations to study intramolecularconformational changes in bax. Journal of BiomolecularStructure & Dynamics, 28, 71–83.
Kryger, G., Harel, M., Giles, K., Toker, L., Velan, B., Lazar,A., … Sussman, J.L. (2000). Structures of recombinantnative and E202Q mutant human acetylcholinesterase com-plexed with the snake-venom toxin fasciculin-II. Acta Crys-tallographica. Section D, Biological Crystallography, 56,1385–1394.
Likhatskaya, G.N., Solov’eva, T.F., Novikova, O.D., Issaeva,M.P., Gusev, K.V., Kryzhko, I.B., … Nurminski, E.A.(2005). Homology models of the Yersinia pseudotuberculo-sis and Yersinia pestis general porins and comparative anal-ysis of their functional and antigenic regions. Journal ofBiomolecular Structure & Dynamics, 23, 113–232.
Maseras, F., & Morokuma, K. (1995). IMOMM: A new inte-grated ab initio + molecular mechanics geometry optimiza-tion scheme of equilibrium structures and transition states.Journal of Computational Chemistry, 16, 1170–1179.
Matos, K.S., Mancini, D.T., da Cunha, E.F.F., Kuča, K., Fran-ça, T.C.C., & Ramalho, T.C. (2011). Molecular aspects ofthe reactivation process of acetylcholinesterase inhibited bycyclosarin. Journal of the Brazilian Chemical Society, 22,1999–2004.
Mehrnejad, F., & Zarei, M. (2010). Molecular dynamics simu-lation study of the interaction of piscidin 1 with DPPCbilayers: Structure–activity relationship. Journal of Biomo-lecular Structure & Dynamics, 27, 551–559.
Meynier, C., Guerlesquin, F., & Roche, P. (2009). Computationalstudies of human galectin-1: Role of conserved tryptophanresidue in stacking interaction with carbohydrate ligands.Journal of Biomolecular Structure & Dynamics, 27, 49–57.
Mohan, S., Perry, J.J.P., Poulose, N., Nair, B.G., & Anilkumar,G. (2009). Homology modeling of GLUT4, an insulin reg-ulated facilitated glucose transporter and docking studieswith ATP and its inhibitors. Journal of Biomolecular Struc-ture & Dynamics, 26, 455–464.
Nemukhim, A.V., Lushchekina, S.V., Bochenkova, A.V.,Golubeva, A.A., & Varfolomeev, S.D. (2008). Characteriza-tion of a complete cycle of acetylcholinesterase catalysis byab initio QM/MM modelin. Journal of Molecular Model-ing, 14, 409–416.
Ordentlich, A., Barak, D., Kronman, C., Ariel, N., Segall, Y.,Velan, B., & Shafferman, A. (1996). The architecture ofhuman acetylcholinesterase active center probed by interac-tions with selected organophosphate inhibitors. Journal ofBiological Chemistry, 271(20), 11953–11962.
Quinn, D.M. (1987). Acetylcholinesterase: Enzyme structure,reaction dynamics, and virtual transition states. ChemicalReviews, 87, 955–975.
Ramalho, T.C., Caetano, M.S., Cunha, E.F.F., Souza, T.C.S., &Rocha, M.V.J. (2009). Construction and assessment of reac-tion models of class I EPSP synthase: Molecular dockingand density functional theoretical calculations. Journal ofBiomolecular Structure & Dynamics, 27, 195–207.
Ramalho, T.C., França, T.C.C., Rennó, M.N., Guimarães, A.P.,Cunha, E.F.F., & Kuča, K. (2010). Development of newacetylcholinesterase reactivators: Molecular modeling versusin vitro data. Chemico-Biological Interactions, 185, 73–77.
Ramalho, T.C., & Taft, C.A. (2005). Thermal and solventeffects on the NMR and UV parameters of some bioreduc-tive drugs. Journal of Chemical Physics, 123(5), 054319.
Rieping, W., Habeck, M., Bardiaux, B., Bernard, A., Malliavin,T.E., & Nilges, M. (2007). ARIA2: Automated NOEassignment and data integration in NMR structure calcula-tion. Bioinformatics, 23, 381–382.
Roy, S., & Thakur, A.R. (2010). 20ns Molecular dynamics sim-ulation of the antennapedia homeodomain-DnaA complex:
Acetyl cholinesterase reactivators 557
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012

Water interaction and DnaA structure analysis. Journal ofBiomolecular Structure & Dynamics, 27, 443–455.
Schwieters, C.D., Kuszewski, J.J., & Clore, G.M. (2006).Using Xplor–NIH for NMR molecular structure determina-tion. Progress in Nuclear Magnetic Resonance Spectros-copy, 48, 47–62.
Sharadadevi, A., & Nagaraj, R. (2010). A molecular dynamicsstudy of human defensins HBD-1 and HNP-3 in water.Journal of Biomolecular Structure & Dynamics, 27, 541–550.
Sidell, F.R., Takafuji, E.T., & Franz, D.R. (Eds.). (1997). Medi-cal aspects of chemical and biological warfare-textbook ofmilitary medicine. Washington, DC: Office of the SurgeonGeneral, US Army.
Sharma, S., Sonavane, U.B., & Joshi, R.R. (2010). Moleculardynamics simulations of cyclohexyl modified peptidenucleic acids (PNA). Journal of Biomolecular Structure &Dynamics, 27, 663–676.
Sille, J., & Remko, M. (2009). Computational study of the sulf-onylated amino acid hydroxamates binding to the zinc ionwithin the active site of carbonic anhydrase. Journal ofBiomolecular Structure & Dynamics, 26, 431–444.
Somani, S.M., Solana, R.P., & Dube, S.N. (1992). Toxicologyof nerve agents. In S.M. Somani (Ed.), Chemical warfareagents (pp. 67–123). San Diego, CA: Academic Press.
Sousa da Silva, A.W., & Vranken, W.F. (BMC Research Notes,in press). ACPYPE – AnteChamber python parser interface.http://www.ccpn.ac.uk/software/ACPYPE-folder.
Steven, S.Z., Peterson, M.W., Hamza, A., Zhan, C.G., Cerasoli,D.M., & Chang, W.E. (2011). Computational characteriza-tion of how the VX nerve agent binds human serum parao-xonase 1. Journal of Molecular Modeling, 17, 97–109.
Tao, Y., Rao, Z.H., & Liu, S.Q. (2010). Insight derived frommolecular dynamics simulation into substrate-inducedchanges in protein motions of proteinase K. Journal of Bio-molecular Structure & Dynamics, 28, 143–158.
Thomsen, R., & Christensen, M.H. (2006). MolDock: A newtechnique for high-accuracy molecular docking. Journal ofMedicinal Chemistry, 49, 3315–3321.
Timofeyeva, N.A., Koval, V.V., Knorre, D.G., Zharkov, D.O.,Saparbaev, M.K., … Fedorova, O.S. (2009). Conforma-tional dynamics of human AP endonuclease in base exci-sion and nucleotide incision repair pathways. Journal ofBiomolecular Structure & Dynamics, 26, 637–652.
Varughese, J.F., Chalovich, J.M., & Li, Y. (2010). Moleculardynamics studies on troponin (TnI-TnT-TnC) complexes:Insight into the regulation of muscle contraction. Journal ofBiomolecular Structure & Dynamics, 28, 159–174.
Vranken, W.F., Boucher, W., Stevens, T.J., Fogh, R.H., Pajon,A., Llinas, M., … Laue, E.D. (2005). The CCPN datamodel for NMR spectroscopy: Development of a softwarepipeline. Proteins: Structure, Function, and Bioinformatics,59, 687–696.
Walker, R.C., Crowley, M.F., & Case, D.A. (2008). The imple-mentation of a fast and accurate QM/MM potential methodin amber. Journal of Computational Physics, 29, 1019–1031.
Wang, J., Wang, W., Kollman, P.A., & Case, D.A. (2006).Automatic atom type and bond type perception in molecu-lar mechanical calculations. Journal of Molecular Graphicsand Modelling, 25, 247–260.
Warren, D. (2002). The PyMOL molecular graphics system.San Carlos, CA: DeLano Scientific.
Wong, L., Radic, Z., Brüggemann, R.J.M., Hosea, N., Berman,H.A., & Taylor, P. (2000). Mechanism of oxime reactiva-tion of acetylcholinesterase analyzed by chirality and muta-genesis. Biochemistry, 39, 5750–5757.
Worek, F., Aurbek, N., Koller, M., Becker, C., Eyer, P., & Thi-ermann, H. (2007). Kinetic analysis of reactivation andaging of human acetylcholinesterase inhibited by differentphosphoramidates. Biochemical Pharmacology, 73(11),1807–1817.
Zhang, J.P. (2009). Studies on the structural stability of rabbitprion probed by molecular dynamics simulations. Journal ofBiomolecular Structure & Dynamics, 27, 159–162.
Zhong, L.H., & Xie, J.M. (2009). Investigation of the effect ofglycosylation on human prion protein by molecular dynam-ics. Journal of Biomolecular Structure & Dynamics, 26,525–533.
558 K.S. Matos et al.
Dow
nloa
ded
by [
Prof
esso
r T
eodo
rico
C. R
amal
ho]
at 0
7:52
17
Aug
ust 2
012
Copyright © 2022 FDOKUMEN




















