Properties of different ethyl alcohol based secondary fluids ...
Upload
independentCategory
view
1download
0
B
FI
DA*L
Bno(filfl[cc(t(taRtrnto[waAltpmctrAAc
GASTROENTEROLOGY 2006;130:781–793
ASIC–LIVER, PANCREAS, AND BILIARY TRACT
atty Acid Ethyl Esters Cause Pancreatic Calcium Toxicity vianositol Trisphosphate Receptors and Loss of ATP Synthesis
AVID N. CRIDDLE,* JOHN MURPHY,*,‡ GREGORIO FISTETTO,* STEPHANIE BARROW,*LEXEI V. TEPIKIN,* JOHN P. NEOPTOLEMOS,‡ ROBERT SUTTON,‡ and OLE H. PETERSEN*Medical Research Council Secretory Control Research Group, Physiological Laboratory, and ‡Division of Surgery and Oncology, University of
iverpool, Liverpool, United KingdomEqpmtofaetptopcaaotc
we(a
nity[mrm
See editorial on page 992.
ackground & Aims: Fatty acid ethyl esters are etha-ol metabolites inducing sustained, toxic elevationsf the acinar cytosolic free calcium ion concentration[Ca2�]C) implicated in pancreatitis. We sought to de-ne the mechanisms of this elevation. Methods: Iso-
ated mouse pancreatic acinar cells were loaded withuorescent dyes for confocal microscopy to measureCa2�]C (Fluo 4, Fura Red), endoplasmic reticulumalcium ion concentration ([Ca2�]ER, Mg Fluo 4), mito-hondrial membrane potential (TMRM), ADP:ATP ratioMg Green), and NADH autofluorescence in responseo palmitoleic acid ethyl ester and palmitoleic acid10–100 �mol/L). Whole-cell patch clamp was usedo measure the calcium-activated chloride current andpply ethanol metabolites and/or ATP intracellularly.esults: Intracellular delivery of ester induced oscilla-
ory increases of [Ca2�]C and calcium-activated cur-ents, inhibited acutely by caffeine (20 mmol/L), butot atropine, indicating involvement of inositolrisphosphate receptor channels. The stronger effectf extracellular ester or acid caused depletion ofCa2�]ER, not prevented by caffeine, but associatedith depleted ATP, depleted NADH autofluorescence,nd depolarized mitochondria, suggesting calcium-TPase pump failure because of lack of ATP. Intracel-
ular ATP abolished the sustained rise in [Ca2�]C, al-hough oscillatory signals persisted that wererevented by caffeine. Inhibition of ester hydrolysisarkedly reduced its calcium-releasing effect and
onsequent toxicity. Conclusions: Fatty acid ethyl es-er increases [Ca2�]C through inositol trisphosphateeceptors and, following hydrolysis, through calcium-TPase pump failure from impaired mitochondrialTP production. Lowering cellular fatty acid substrate
oncentrations may reduce cell injury in pancreatitis.thanol consumption is important among the manycauses of acute pancreatitis because ethanol fre-
uently induces severe disease, may precipitate extensiveancreatic necrosis, and, with continued intake, is theost common cause of progression to chronic pancreati-
is.1 The pancreas contains high concentrations of non-xidative synthase enzymes that combine ethanol withatty acids, forming fatty acid ethyl esters.2,3 There arelso high concentrations of hydrolytic esterases that op-rate in the reverse direction,4 permitting oxidative me-abolism of both products. Increasing evidence has im-licated fatty acid ethyl esters, rather than ethanol itself,o be the cause of pancreatic damage.2–8 Administrationf high doses of ethanol for prolonged periods by oral orarenteral routes to various species does not inducehanges characteristic of either acute or chronic pancre-titis.7 This is in marked contrast to the effects of fattycid ethyl esters,3,5,8 or of ethanol with inhibition of itsxidative metabolism,6 or with corn oil,9,10 or with secre-agogues at doses that would otherwise not induce pan-reatitis.7,11–13
In this study, we have investigated the mechanism byhich fatty acid ethyl esters and fatty acids induce
levations of the cytosolic free calcium ion concentration[Ca2�]C) in isolated mouse pancreatic acinar cells usingcombination of whole-cell, patch-clamp current record-
Abbreviations used in this paper: BAPTA, 1,2-bis(O-aminophe-oxy)ethane-N,N,N’,N’-tetraacetic acid; [Ca2�]C, cytosolic free calcium
on concentration; [Ca2�]ER, endoplasmic reticulum calcium concen-ration; CCCP, carbonyl cyanide 3-chlorophenylhydrazone; EGTA, eth-lene glycol-bis (�-aminoethyl ether)-N,N,N’,N’-tetraacetic acid;Mg2�]C, cytosolic free magnesium ion concentration; PMCA, plasma
embrane calcium ATPase pump; SERCA, sarcoplasmic/endoplasmiceticulum calcium ATPase pump; TMRM, tetramethyl rhodamineethyl ester.© 2006 by the American Gastroenterological Association Institute
0016-5085/06/$32.00
doi:10.1053/j.gastro.2005.12.031
ippl[ttrpprId[tSetirpfppwetpeft
caCe2e1pf(oeaadbc
HMw
wGMmcapo[flwd�miauuecctmac
pmtps(rawtmgimtdtptsmfls
782 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
ng and confocal microfluorimetry. We found that bothalmitoleic acid ethyl ester and its hydrolytic productalmitoleic acid uncouple oxidative phosphorylation,eading to a global, sustained, and toxic increase inCa2�]C because of endoplasmic reticulum calcium leak,hrough compromise of calcium pump functions in bothhe endoplasmic reticulum (sarcoplasmic/endoplasmiceticulum calcium ATPase pump; SERCA) and thelasma membrane (plasma membrane calcium ATPaseump; PMCA). The calcium pump activities could beestored by intracellular patch-pipette delivery of ATP.ntracellular delivery of palmitoleic acid ethyl ester in-uced immediate, transient, oscillatory increases ofCa2�]C that were blocked by caffeine, used as an inositolrisphosphate receptor antagonist, but not by atropine.imilar oscillatory increases of [Ca2�]C were seen withxtracellular application of palmitoleic acid ethyl esterogether with intracellular delivery of ATP, demonstrat-ng 2 independent actions of this ethanol metabolite: (1)elease of calcium from the endoplasmic reticulum de-endent on inositol trisphosphate receptors and (2) de-ective clearance of cytosolic calcium by calcium-ATPaseumps compromised by impaired mitochondrial ATProduction. The latter action and its consequent toxicityere found to depend on the hydrolysis of the fatty acid
thyl ester to liberate its fatty acid. These findings iden-ify molecular mechanisms by which ethanol can induceancreatic acinar cell injury, also likely in hyperlipid-mia, and suggest that strategies that reduce intracellularatty acid substrate concentrations may be beneficial inhose at risk of pancreatitis.
Materials and Methods
Cell Preparation and Solutions
Freshly isolated pancreatic acinar cells and acinar celllusters of 2 or 3 cells were prepared from the pancreases ofdult CD1 mice using collagenase (Worthington Biochemicalorporation, Lakewood, NJ) as in our previous work.8,14 Allxperiments were performed at room temperature (23°C–5°C), and all cells were used within 4 hours of isolation. Thextracellular solution contained (mmol/L): 140 NaCl, 4.7 KCl,.13 MgCl2, 1 CaCl2, 10 D-glucose, 10 HEPES (adjusted toH 7.35 using NaOH); in some experiments, CaCl2 (calcium-ree extracellular solution), or CaCl2 together with MgCl2
calcium- and magnesium-free extracellular solution) weremitted; where stated, ethylene glycol-bis (�-aminoethylther)-N,N,N’,N’-tetraacetic acid (EGTA; 1 mmol/L) wasdded to the calcium-free solution (all chemicals, highest gradevailable from Sigma, Gillingham, United Kingdom). Stan-ard, whole-cell, patch-clamp current recordings (as describedelow) were performed using an internal pipette solution of
omposition (mmol/L): 140 KCl; 1.5 MgCl2; 2 MgATP; 10 IEPES; 0.1 EGTA; and pH 7.2. In some experiments,gATP was omitted from the internal pipette solution,hereas, in others, this was raised to 4 mmol/L.
Confocal Imaging
Confocal imaging of cells loaded with fluorescent dyesas performed using a Zeiss LSM510 system (Carl Zeiss JenambH, Jena, Germany or Leica AOBS SP2 system (Leicaicrosystems AG, Wetzlar, Germany) for Mg Green experi-ents to measure the intracellular free ionized magnesium
oncentration, [Mg2�]C, for determination of ATP changes) tossess the effects of ethanol and its nonoxidative metabolitesalmitoleic acid ethyl ester (dissolved in 850 mmol/L ethanol8
r 0.5% vol/vol dimethyl sulfoxide) and palmitoleic acid onCa2�]C (all highest grade from Sigma). Cells were loaded withuorescent dyes as cell-permeable, hydrophobic esters (-AM),hich accumulate following hydrolysis, rendering them hy-rophilic. Fluo 4-AM (2.5 �mol/L) or Fura Red-AM (5mol/L) were loaded for 30 minutes at room temperature toeasure [Ca2�]C. Fura Red loading of cells was used in exper-
ments with caffeine because the fluorescence of this dye isffected less by caffeine. The fluorescence of Fluo 4 was excitedsing an argon 488 nm laser line, with emitted light collectedsing an LP505 filter. The fluorescence of Fura Red was alsoxcited by an argon 488 nm laser line, but emitted lightollected using an LP585 filter. The images collected wereomposed of 256 � 256 pixels, and the optical section selectedo be 5–6 �m. A C-Apochromat �63 objective with a nu-erical aperture of 1.2 was used in all experiments, and image
nalysis carried out using Zeiss confocal LSM510 or Leicaonfocal AOBS SP2 image software.
Mg Fluo 4-AM (5 �mol/L) was loaded into cells in theresence of 0.01% pluronic acid for 20–30 minutes at 37°C toeasure the endoplasmic reticulum free calcium ion concen-
ration ([Ca2�]ER) as described previously.15 Experiments wereerformed using the multitrack configuration of the LSM510ystem, which allowed simultaneous measurement of [Ca2�]ER
excitation 488 nm, emission �510 nm) and NADH autofluo-escence (excitation 351 nm, emission 385–470 nm) to obtainmeasure of mitochondrial metabolism.16 Further experimentsere done to determine changes of intracellular ATP concen-
rations after loading with Mg Green-AM (4 �mol/L) for 30inutes at room temperature. Because ATP has a 10-fold
reater affinity for magnesium ions than ADP, and mostntracellular magnesium is present as MgATP,17,18 this dye
onitors changes in ADP:ATP ratios that may indicate main-enance or depletion of intracellular ATP supplies, the latteremonstrated by an increase in Mg Green fluorescence (exci-ation 476 nm, emission 500–550 nm) as [Mg2�]C rises in theresence of a calcium- and magnesium-free extracellular solu-ion.17,18 Mitochondrial membrane potential changes were as-essed by loading cells with 100 nmol/L tetramethyl rhoda-ine methyl ester (TMRM) for 30 minutes at 37°C to measure
uorescence in the perigranular mitochondrial region as de-cribed previously (excitation 488 nm, emission �550 nm).14
n some experiments, cells were coloaded with TMRM and
F[Larc
ta�h�(d
nfP(wswsb
aO(Sa
etcpr(sotlbp
F�(roa(t(tgcin
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 783
luo 4-AM to measure mitochondrial membrane potential andCa2�]C, respectively, using the multitrack configuration of theSM510 system. Palmitoleic acid (10 or 50 �mol/L) was thenpplied, after which the protonophore carbonyl cyanide 3-chlo-ophenylhydrazone (CCCP; 10 �mol/L) was added to induceomplete mitochondrial depolarization.14
Cell Toxicity Measurement
Isolated acinar cells were incubated for 1 hour at roomemperature with palmitoleic acid ethyl ester. Some cells werelso preincubated with bis-(4-nitrophenyl) phosphate (200mol/L) for 10 minutes, an inhibitor of fatty acid ethyl esterydrolase.4 Cells were then incubated in propidium iodide (1mol/L) for 10 minutes and observed by confocal microscopy
Zeiss LSM510; excitation 363 nm, emission �400 nm) toetect necrotic changes.19
Patch-Clamp Current Recording
The whole-cell configuration of the patch-clamp tech-ique was used to record calcium-activated chloride currentsrom single pancreatic acinar cells as previously described.20–23
atch-pipettes were pulled from borosilicate glass capillariesHarvard Apparatus, Edenbridge, Kent, United Kingdom)ith a resistance of 2–3 M� when filled with an intracellular
olution (containing 140 mmol/L KCl). Whole-cell currentsere sampled at 10 KHz using an EPS8 amplifier and Pulse
oftware (HEKA, Lambrecht, Pfalz, Germany) with the mem-rane voltage clamped at �30 mV.
Chemicals
Fluo 4-AM, Fura Red-AM, Mg Fluo 4-AM, Mg Green,nd TMRM were purchased from Molecular Probes (Eugene,R) and palmitoleic acid ethyl ester from ICN Biomedicals
Cleveland, OH). All other chemicals were purchased fromigma (Gillingham, United Kingdom) of the highest gradevailable.
Results
Intracellular Fatty Acid Ethyl Ester InducesOscillatory [Ca2�]C Increases
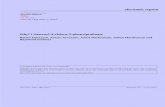
Intracellular application of palmitoleic acid ethylster (100 �mol/L, with 850 mmol/L ethanol to main-ain solubility) via a patch-pipette (whole-cell recordingonfiguration) induced repetitive [Ca2�]C spikes accom-anied by associated calcium-activated chloride ion cur-ents in voltage-clamped single pancreatic acinar cellsFigure 1A; 16 of 18 cells). The [Ca2�]C oscillations wereimilar to those observed following external applicationf physiologic doses of secretagogues.21 In many cases,ransient increases in [Ca2�]C were apparent on estab-ishing the giga-seal between the pipette and acinar cell,efore cell membrane rupture to allow current recording,
resumably because of diffusion of the membrane per- uigure 1. (A) Typical effects of palmitoleic acid ethyl ester (100mol/L POAEE) applied internally to the acinar cell via patch pipette
whole-cell recording configuration) on calcium-activated chloride cur-ent and [Ca2�]C. Inset shows isolated acinar cell doublet with patchn recorded cell. (B) Absence of effect of ethanol (850 mmol/L) alonepplied internally on calcium-activated chloride current and [Ca2�]C.C) Representative recording showing the comparative effects of in-racellular and extracellular application of palmitoleic acid ethyl ester100 �mol/L) on [Ca2�]C, to produce transient and sustained eleva-ions, respectively (n � 8). The Ca2�-sensitive Fluo 4 fluorescence isiven as the fluorescence ratio (F/F0), where F is the fluorescence thathanges with time, increasing with increasing [Ca2�]C, and F0 is thenitial (prestimulation) basal fluorescence. Inset shows isolated aci-ar cell doublet from which recordings were taken, with patch on
pper cell.
mbltp([cen[awoplpcwtgcvcebctm
papvpavceaccaisepetec
te
cpftecoar(pof2lttfvtispt
sataitlse
e(lo�[scel
784 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
eant ester across the isolated part of the plasma mem-rane covered by the patch pipette. Less frequent oscil-atory increases in [Ca2�]C were seen in cells adjacent tohose receiving palmitoleic acid ethyl ester via the patch-ipette, as in doublets (Figure 1C). In contrast, ethanol850 mmol/L) alone produced no current or changes inCa2�]C (Figure 1B, 5 of 8 cells) or only minimal inwardurrents (3 of 8 cells). Previously, we observed thatxternal application of ethanol (850 mmol/L) alone toonpatched cells produced only a small increase ofCa2�]C in most cells tested, in contrast to palmitoleiccid ethyl ester (100 �mol/L, with 850 mmol/L ethanol),hich consistently induced global, sustained elevationsf [Ca2�]C.8 In the current experimental protocol usingatched cells, when the same concentration of palmito-eic acid ethyl ester (100 �mol/L) that was used in theipette solution (applied to a single, small area of theell) was subsequently applied outside the cells (to thehole of the cell surface except the small part covered by
he patch pipette), the oscillations were replaced by alobal, sustained [Ca2�]C elevation (Figure 1C; 8 of 8ells). Thus, intracellular delivery of fatty acid ethyl esteria a patch clamp pipette was less effective than extra-ellular application, almost certainly because of the greatase with which this compound passes the plasma mem-rane. It would appear that most of the ester entering theell via a pipette rapidly escapes to the exterior throughhe plasma membrane adjacent to the pipette attach-ent.To test whether ethanol was necessary for the effects of
almitoleic acid ethyl ester, dimethyl sulfoxide was useds an alternative solvent. Intracellular application ofalmitoleic acid ethyl ester (100 �mol/L with 0.5%ol/vol dimethyl sulfoxide to maintain solubility) via aatch-pipette again induced repetitive [Ca2�]C spikesccompanied by calcium-activated chloride currents inoltage-clamped, single pancreatic acinar cells (7 out of 7ells), similar to those induced by palmitoleic acid ethylster with ethanol as the solvent. Dimethyl sulfoxidelone (0.5% vol/vol) applied intracellularly (5 out of 5ells) or extracellularly (6 out of 6 cells) produced nourrent or changes in [Ca2�]C (5 out of 5 cells). Externalpplication of palmitoleic acid ethyl ester (100 �mol/L)n 0.5% vol/vol dimethyl sulfoxide produced global,ustained elevations in [Ca2�]C (9 out of 13 cells). Thesexperiments demonstrated independence of the effects ofalmitoleic acid ethyl ester from its solvent. Becausethanol and not dimethyl sulfoxide is present for someime after abusive alcohol intake, and the effects ofthanol (850 mmol/L) alone on mouse pancreatic acinar
ells have been documented in detail,8 we used ethanol as the solvent for palmitoleic acid ethyl ester in all otherxperiments.
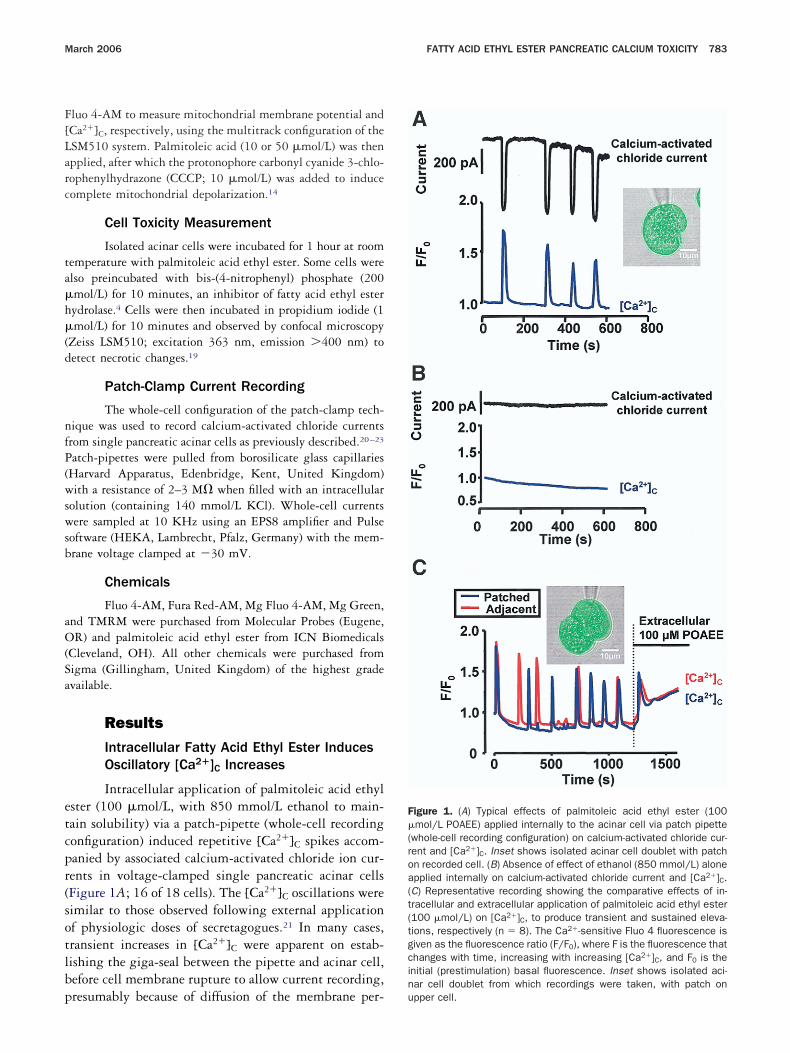
Because the oscillatory increases in [Ca2�]C and asso-iated inward chloride currents induced by intracellularalmitoleic acid ethyl ester appeared similar to thoseollowing acetylcholine administration, the effect of in-racellular palmitoleic acid ethyl ester on [Ca2�]C wasxamined in the presence of atropine (10 �mol/L) andaffeine (20 mmol/L) to determine any role for activationf their respective receptors. The effects of palmitoleiccid ethyl ester were clearly not mediated by muscariniceceptors because atropine failed to inhibit the responses4 out of 4 cells), but caffeine caused an immediate androlonged inhibition of the calcium oscillations (11 outf 11 cells). In several experiments, however, this effectaded in the continuing presence of the agent (FigureA). [Ca2�]C oscillations elicited by external acetylcho-ine application can be mimicked by intracellular inositolrisphosphate infusion and are inhibited by inositolrisphosphate receptor antagonists, including caf-eine.20,23 Although caffeine is better known as an acti-ator of calcium-induced calcium release in musclehrough the ryanodine receptor, it is in fact also annositol trisphosphate receptor antagonist, and the ino-itol trisphosphate receptor antagonist action is com-letely independent of the effect on the ryanodine recep-or.20,23,24
The caffeine experiments (Figure 2) indicate that ino-itol trisphosphate receptors play a role in the palmitoleiccid ethyl ester-elicited [Ca2�]C oscillations, but the facthat the effect is weaker than in the case of the action ofcetylcholine, in which all [Ca2�]C rises are abol-shed,20,25 may indicate that inositol trisphosphate recep-or activation is not the only cause of the [Ca2�]C oscil-ations. This conclusion was reinforced by studies of thetronger effects of extracellular palmitoleic acid ethylster application (Figure 1C).
Extracellular Fatty Acid Ethyl Ester or FattyAcid Induces Sustained [Ca2�]C Increases
Extracellular application of palmitoleic acid ethylster induced global, sustained elevations of [Ca2�]C
Figure 1C) as previously described.8 Similarly, extracel-ular application of palmitoleic acid induced steady risesf [Ca2�]C, which were slower to occur. Atropine (10mol/L) had no effect on the sustained elevations of
Ca2�]C induced by either metabolite, as well as the farmaller elevations induced by ethanol alone (n � 7–12ells, data not shown). Caffeine (20 mmol/L) had noffect on the elevations of [Ca2�]C induced by extracel-ular palmitoleic acid ethyl ester (Figure 2B) or palmi-
oleic acid (Figure 2C).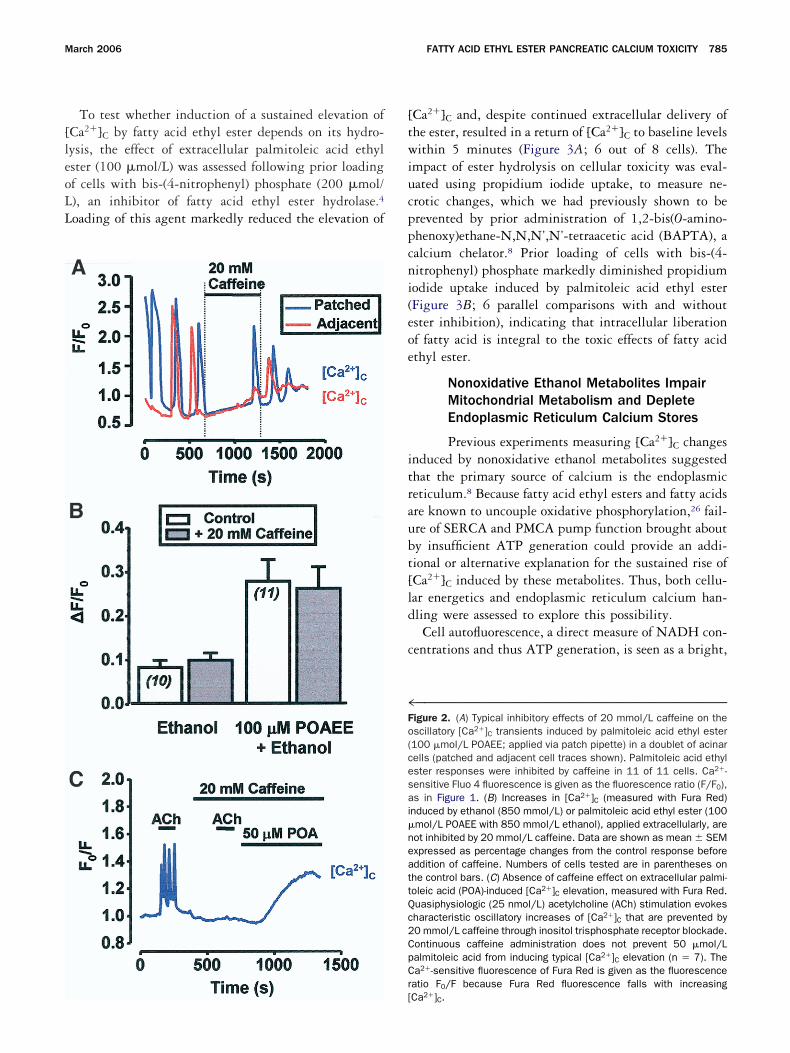
[leoLL
[twiucppcni(eoe
itraubt[ld
c
4Fo(cesai�neattQc2CpCr
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 785
To test whether induction of a sustained elevation ofCa2�]C by fatty acid ethyl ester depends on its hydro-ysis, the effect of extracellular palmitoleic acid ethylster (100 �mol/L) was assessed following prior loadingf cells with bis-(4-nitrophenyl) phosphate (200 �mol/), an inhibitor of fatty acid ethyl ester hydrolase.4
oading of this agent markedly reduced the elevation of
[
Ca2�]C and, despite continued extracellular delivery ofhe ester, resulted in a return of [Ca2�]C to baseline levelsithin 5 minutes (Figure 3A; 6 out of 8 cells). The
mpact of ester hydrolysis on cellular toxicity was eval-ated using propidium iodide uptake, to measure ne-rotic changes, which we had previously shown to berevented by prior administration of 1,2-bis(O-amino-henoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA), aalcium chelator.8 Prior loading of cells with bis-(4-itrophenyl) phosphate markedly diminished propidiumodide uptake induced by palmitoleic acid ethyl esterFigure 3B; 6 parallel comparisons with and withoutster inhibition), indicating that intracellular liberationf fatty acid is integral to the toxic effects of fatty acidthyl ester.
Nonoxidative Ethanol Metabolites ImpairMitochondrial Metabolism and DepleteEndoplasmic Reticulum Calcium Stores
Previous experiments measuring [Ca2�]C changesnduced by nonoxidative ethanol metabolites suggestedhat the primary source of calcium is the endoplasmiceticulum.8 Because fatty acid ethyl esters and fatty acidsre known to uncouple oxidative phosphorylation,26 fail-re of SERCA and PMCA pump function brought abouty insufficient ATP generation could provide an addi-ional or alternative explanation for the sustained rise ofCa2�]C induced by these metabolites. Thus, both cellu-ar energetics and endoplasmic reticulum calcium han-ling were assessed to explore this possibility.Cell autofluorescence, a direct measure of NADH con-
entrations and thus ATP generation, is seen as a bright,
™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™igure 2. (A) Typical inhibitory effects of 20 mmol/L caffeine on thescillatory [Ca2�]C transients induced by palmitoleic acid ethyl ester100 �mol/L POAEE; applied via patch pipette) in a doublet of acinarells (patched and adjacent cell traces shown). Palmitoleic acid ethylster responses were inhibited by caffeine in 11 of 11 cells. Ca2�-ensitive Fluo 4 fluorescence is given as the fluorescence ratio (F/F0),s in Figure 1. (B) Increases in [Ca2�]C (measured with Fura Red)
nduced by ethanol (850 mmol/L) or palmitoleic acid ethyl ester (100mol/L POAEE with 850 mmol/L ethanol), applied extracellularly, areot inhibited by 20 mmol/L caffeine. Data are shown as mean SEMxpressed as percentage changes from the control response beforeddition of caffeine. Numbers of cells tested are in parentheses onhe control bars. (C) Absence of caffeine effect on extracellular palmi-oleic acid (POA)-induced [Ca2�]C elevation, measured with Fura Red.uasiphysiologic (25 nmol/L) acetylcholine (ACh) stimulation evokesharacteristic oscillatory increases of [Ca2�]C that are prevented by0 mmol/L caffeine through inositol trisphosphate receptor blockade.ontinuous caffeine administration does not prevent 50 �mol/Lalmitoleic acid from inducing typical [Ca2�]C elevation (n � 7). Thea2�-sensitive fluorescence of Fura Red is given as the fluorescence
atio F0/F because Fura Red fluorescence falls with increasing
Ca2�]C.
psamdt(slmfawaoa
ptcotllmnfllbMclao[elde�5cddidabA
ll
Fpse6r(l2lt�nteffects of bis-(4-nitrophenyl) phosphate.
786 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
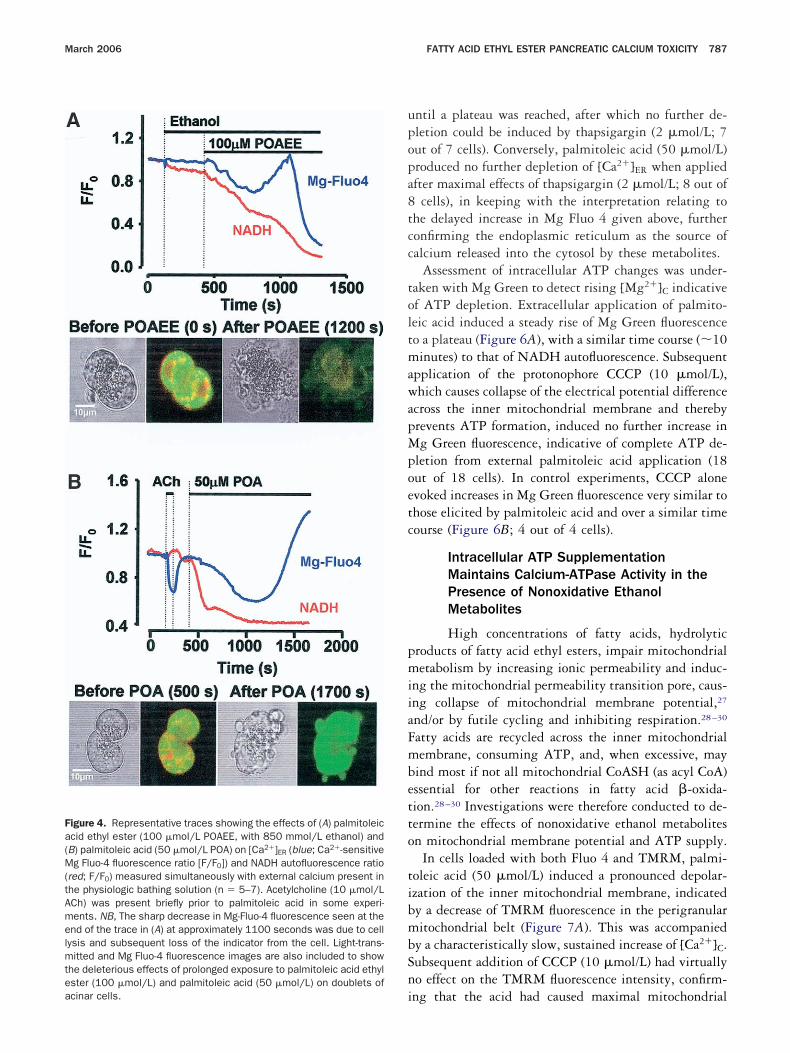
erigranular mitochondrial belt that increases in re-ponse to physiologic doses of acetylcholine,16 indicatingcoordinated increase in oxidative phosphorylation toeet cellular demands. NADH autofluorescence was re-
uced, however, by external application of both palmi-oleic acid ethyl ester (100 �mol/L) and palmitoleic acid50 �mol/L), diminishing to markedly low levels over aimilar time course (10 minutes) with both metabo-ites (Figure 4A and 4B). Simultaneous direct measure-ent of [Ca2�]ER with Mg Fluo 4 demonstrated pro-
ound depletion of the endoplasmic reticulum followingpplication of both metabolites that was more immediateith the fatty acid ethyl ester (Figure 4A and 4B). The
ctions of both metabolites were thus distinct from thosef acetylcholine (10 �mol/L), which increased NADHutofluorescence while decreasing [Ca2�]ER (Figure 4B).
Initially, Mg Fluo 4 fluorescence fell steadily afteralmitoleic acid ethyl ester or palmitoleic acid applica-ion, demonstrating depletion of endoplasmic reticulumalcium stores (Figures 4 and 5). This is consistent withur previous observations, which demonstrated no fur-her increase in [Ca2�]C in response to agents (acetylcho-ine, thapsigargin) that empty the endoplasmic reticu-um store following extracellular administration of eitheretabolite (50–100 �mol/L).8 In the presence of exter-
al calcium, a delayed diffuse increase in Mg Fluo 4uorescence was seen after application of either metabo-ite (Figure 4A and 4B), associated with membrane bleb-ing and eventual cell lysis (Figure 4A and 4B). Becauseg Fluo 4 is present in the cytosol, although at lower
oncentrations than in the endoplasmic reticulum,15 theate diffuse increase in fluorescence is likely to result fromcumulative rise of [Ca2�]C (Figure 4A and 4B) becausef continued calcium entry driven by the reducedCa2�]ER. To test this interpretation directly, similarxperiments were performed in the absence of extracel-ular calcium (calcium-free with 1 mmol/L EGTA). Un-er these conditions, both extracellular palmitoleic acidthyl ester (100 �mol/L) and palmitoleic acid (50mol/L) reduced NADH levels and [Ca2�]ER (FigureA–C), but no delayed increase in Mg Fluo 4 fluores-ence then occurred. These results confirmed that theelayed increase in Mg Fluo 4 fluorescence followingepletion of [Ca2�]ER induced by the alcohol metabolitesn the presence of extracellular calcium (1 mmol/L) wasue to a buildup of [Ca2�]C from continued store-oper-ted calcium entry driven by the low [Ca2�]ER. Com-ined SERCA and PMCA pump failure from diminishedTP production would account for these findings.In separate experiments using calcium-free extracellu-
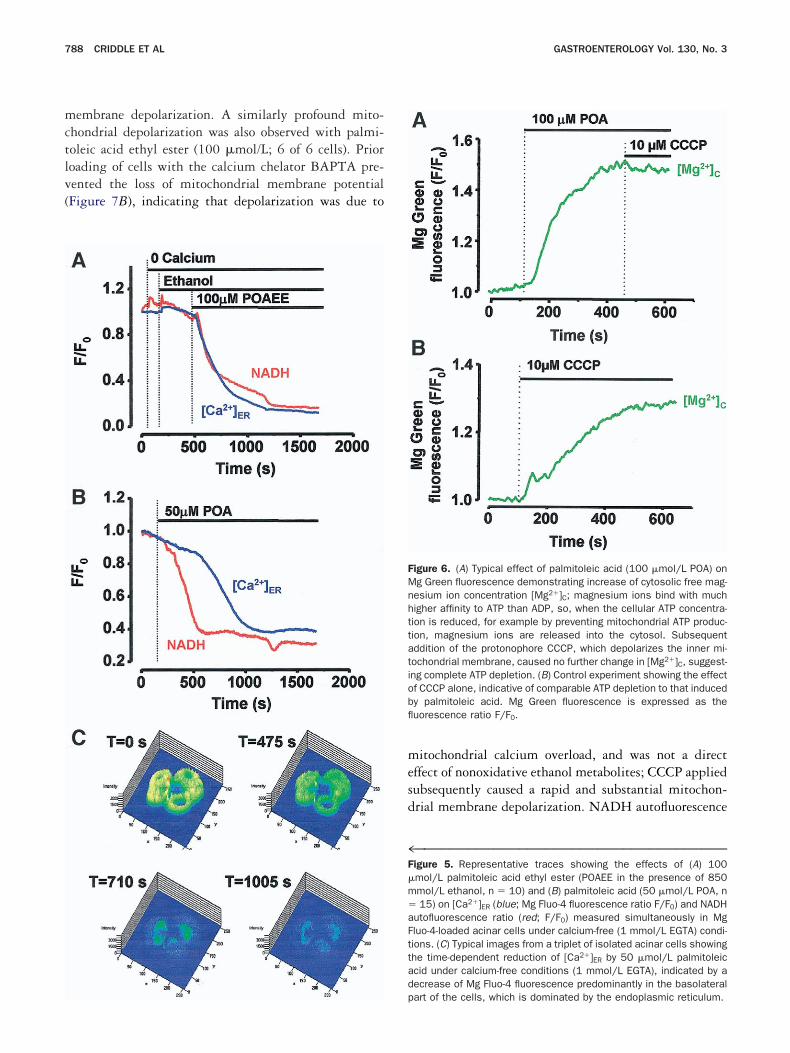
ar solution, reduction of [Ca2�]ER induced by palmito-
igure 3. (A) Typical effect of 200 �mol/L bis-(4-nitrophenyl) phos-hate (BNPP), a fatty acid ethyl ester hydrolase inhibitor, to inhibit theustained elevation of [Ca2�]C (measured with Fluo 4) induced byxternally perfused palmitoleic acid ethyl ester (100 �mol/L POAEE;of 8 cells). (B) Typical light-transmitted and propidium iodide fluo-
escence images of isolated acinar cells after 1-hour incubation withi) 100 �mol/L palmitoleic acid ethyl ester alone, showing morpho-ogic disruption and propidium iodide staining of the nucleus, and (ii)00 �mol/L bis-(4-nitrophenyl) phosphate and 100 �mol/L palmito-
eic acid ethyl ester together. (iii) Propidium iodide fluorescence in-ensity following staining of cells after 1-hour coexposure to 100mol/L palmitoleic acid ethyl ester and 200 �mol/L bis-(4-nitrophe-yl) phosphate, compared with cells exposed to 100 �mol/L palmi-oleic acid ethyl ester alone (n � 6), showing significant protective
eic acid (50 �mol/L) was monitored using Mg Fluo 4
upopa8tcc
toltmawapMpoetc
pmiiaFmbetto
tibmbSni
Fa(M(tAmelmteacinar cells.
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 787
ntil a plateau was reached, after which no further de-letion could be induced by thapsigargin (2 �mol/L; 7ut of 7 cells). Conversely, palmitoleic acid (50 �mol/L)roduced no further depletion of [Ca2�]ER when appliedfter maximal effects of thapsigargin (2 �mol/L; 8 out of
cells), in keeping with the interpretation relating tohe delayed increase in Mg Fluo 4 given above, furtheronfirming the endoplasmic reticulum as the source ofalcium released into the cytosol by these metabolites.
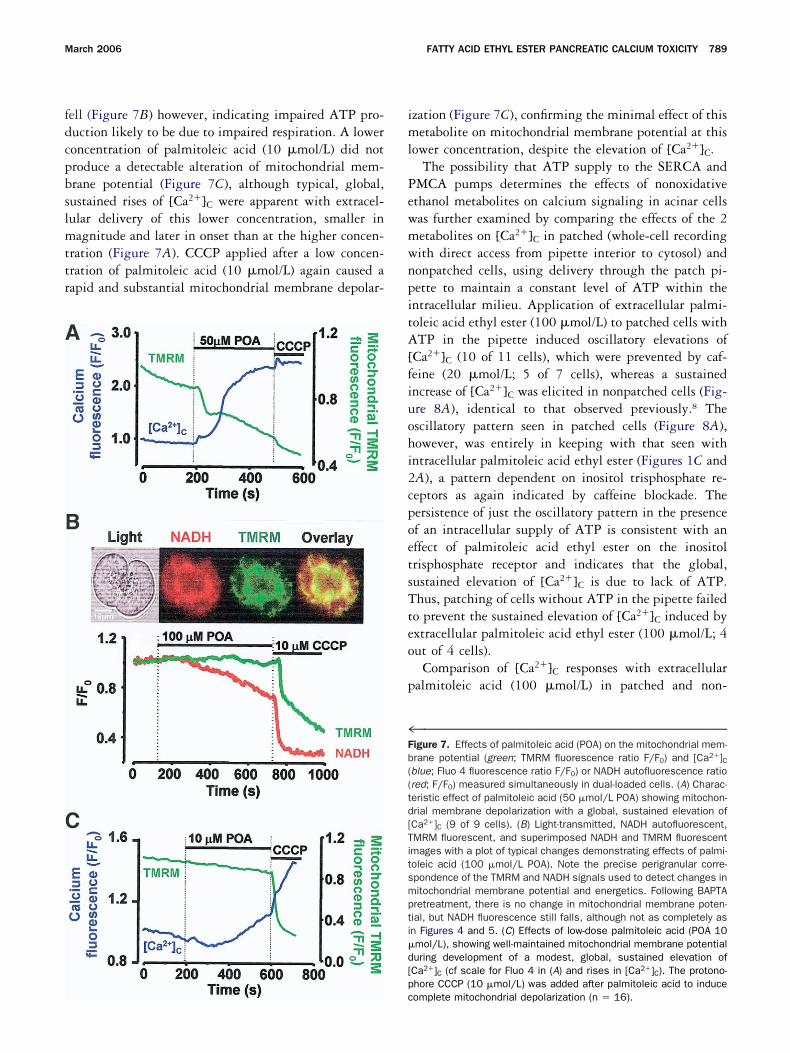
Assessment of intracellular ATP changes was under-aken with Mg Green to detect rising [Mg2�]C indicativef ATP depletion. Extracellular application of palmito-eic acid induced a steady rise of Mg Green fluorescenceo a plateau (Figure 6A), with a similar time course (10inutes) to that of NADH autofluorescence. Subsequent
pplication of the protonophore CCCP (10 �mol/L),hich causes collapse of the electrical potential difference
cross the inner mitochondrial membrane and therebyrevents ATP formation, induced no further increase ing Green fluorescence, indicative of complete ATP de-
letion from external palmitoleic acid application (18ut of 18 cells). In control experiments, CCCP alonevoked increases in Mg Green fluorescence very similar tohose elicited by palmitoleic acid and over a similar timeourse (Figure 6B; 4 out of 4 cells).
Intracellular ATP SupplementationMaintains Calcium-ATPase Activity in thePresence of Nonoxidative EthanolMetabolites
High concentrations of fatty acids, hydrolyticroducts of fatty acid ethyl esters, impair mitochondrialetabolism by increasing ionic permeability and induc-
ng the mitochondrial permeability transition pore, caus-ng collapse of mitochondrial membrane potential,27
nd/or by futile cycling and inhibiting respiration.28–30
atty acids are recycled across the inner mitochondrialembrane, consuming ATP, and, when excessive, may
ind most if not all mitochondrial CoASH (as acyl CoA)ssential for other reactions in fatty acid �-oxida-ion.28–30 Investigations were therefore conducted to de-ermine the effects of nonoxidative ethanol metabolitesn mitochondrial membrane potential and ATP supply.
In cells loaded with both Fluo 4 and TMRM, palmi-oleic acid (50 �mol/L) induced a pronounced depolar-zation of the inner mitochondrial membrane, indicatedy a decrease of TMRM fluorescence in the perigranularitochondrial belt (Figure 7A). This was accompanied
y a characteristically slow, sustained increase of [Ca2�]C.ubsequent addition of CCCP (10 �mol/L) had virtuallyo effect on the TMRM fluorescence intensity, confirm-
igure 4. Representative traces showing the effects of (A) palmitoleiccid ethyl ester (100 �mol/L POAEE, with 850 mmol/L ethanol) andB) palmitoleic acid (50 �mol/L POA) on [Ca2�]ER (blue; Ca2�-sensitiveg Fluo-4 fluorescence ratio [F/F0]) and NADH autofluorescence ratio
red; F/F0) measured simultaneously with external calcium present inhe physiologic bathing solution (n � 5–7). Acetylcholine (10 �mol/LCh) was present briefly prior to palmitoleic acid in some experi-ents. NB, The sharp decrease in Mg-Fluo-4 fluorescence seen at thend of the trace in (A) at approximately 1100 seconds was due to cellysis and subsequent loss of the indicator from the cell. Light-trans-itted and Mg Fluo-4 fluorescence images are also included to show
he deleterious effects of prolonged exposure to palmitoleic acid ethylster (100 �mol/L) and palmitoleic acid (50 �mol/L) on doublets of
ng that the acid had caused maximal mitochondrial
mctlv(
mesd
4F�m�aFttad
FMnhttatiobfl
788 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
embrane depolarization. A similarly profound mito-hondrial depolarization was also observed with palmi-oleic acid ethyl ester (100 �mol/L; 6 of 6 cells). Prioroading of cells with the calcium chelator BAPTA pre-ented the loss of mitochondrial membrane potentialFigure 7B), indicating that depolarization was due to
p
itochondrial calcium overload, and was not a directffect of nonoxidative ethanol metabolites; CCCP appliedubsequently caused a rapid and substantial mitochon-rial membrane depolarization. NADH autofluorescence
™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™igure 5. Representative traces showing the effects of (A) 100mol/L palmitoleic acid ethyl ester (POAEE in the presence of 850mol/L ethanol, n � 10) and (B) palmitoleic acid (50 �mol/L POA, n15) on [Ca2�]ER (blue; Mg Fluo-4 fluorescence ratio F/F0) and NADH
utofluorescence ratio (red; F/F0) measured simultaneously in Mgluo-4-loaded acinar cells under calcium-free (1 mmol/L EGTA) condi-ions. (C) Typical images from a triplet of isolated acinar cells showinghe time-dependent reduction of [Ca2�]ER by 50 �mol/L palmitoleiccid under calcium-free conditions (1 mmol/L EGTA), indicated by aecrease of Mg Fluo-4 fluorescence predominantly in the basolateral
igure 6. (A) Typical effect of palmitoleic acid (100 �mol/L POA) ong Green fluorescence demonstrating increase of cytosolic free mag-esium ion concentration [Mg2�]C; magnesium ions bind with muchigher affinity to ATP than ADP, so, when the cellular ATP concentra-ion is reduced, for example by preventing mitochondrial ATP produc-ion, magnesium ions are released into the cytosol. Subsequentddition of the protonophore CCCP, which depolarizes the inner mi-ochondrial membrane, caused no further change in [Mg2�]C, suggest-ng complete ATP depletion. (B) Control experiment showing the effectf CCCP alone, indicative of comparable ATP depletion to that inducedy palmitoleic acid. Mg Green fluorescence is expressed as theuorescence ratio F/F0.
art of the cells, which is dominated by the endoplasmic reticulum.
fdcpbslmttr
iml
PewmwnpitA[fiuohi2cpoetsTteo
p
4Fb((td[Titsmpti�d[p
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 789
ell (Figure 7B) however, indicating impaired ATP pro-uction likely to be due to impaired respiration. A loweroncentration of palmitoleic acid (10 �mol/L) did notroduce a detectable alteration of mitochondrial mem-rane potential (Figure 7C), although typical, global,ustained rises of [Ca2�]C were apparent with extracel-ular delivery of this lower concentration, smaller inagnitude and later in onset than at the higher concen-
ration (Figure 7A). CCCP applied after a low concen-ration of palmitoleic acid (10 �mol/L) again caused aapid and substantial mitochondrial membrane depolar-
c
zation (Figure 7C), confirming the minimal effect of thisetabolite on mitochondrial membrane potential at this
ower concentration, despite the elevation of [Ca2�]C.The possibility that ATP supply to the SERCA and
MCA pumps determines the effects of nonoxidativethanol metabolites on calcium signaling in acinar cellsas further examined by comparing the effects of the 2etabolites on [Ca2�]C in patched (whole-cell recordingith direct access from pipette interior to cytosol) andonpatched cells, using delivery through the patch pi-ette to maintain a constant level of ATP within thentracellular milieu. Application of extracellular palmi-oleic acid ethyl ester (100 �mol/L) to patched cells withTP in the pipette induced oscillatory elevations of
Ca2�]C (10 of 11 cells), which were prevented by caf-eine (20 �mol/L; 5 of 7 cells), whereas a sustainedncrease of [Ca2�]C was elicited in nonpatched cells (Fig-re 8A), identical to that observed previously.8 Thescillatory pattern seen in patched cells (Figure 8A),owever, was entirely in keeping with that seen withntracellular palmitoleic acid ethyl ester (Figures 1C andA), a pattern dependent on inositol trisphosphate re-eptors as again indicated by caffeine blockade. Theersistence of just the oscillatory pattern in the presencef an intracellular supply of ATP is consistent with anffect of palmitoleic acid ethyl ester on the inositolrisphosphate receptor and indicates that the global,ustained elevation of [Ca2�]C is due to lack of ATP.hus, patching of cells without ATP in the pipette failed
o prevent the sustained elevation of [Ca2�]C induced byxtracellular palmitoleic acid ethyl ester (100 �mol/L; 4ut of 4 cells).
Comparison of [Ca2�]C responses with extracellularalmitoleic acid (100 �mol/L) in patched and non-
™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™igure 7. Effects of palmitoleic acid (POA) on the mitochondrial mem-rane potential (green; TMRM fluorescence ratio F/F0) and [Ca2�]Cblue; Fluo 4 fluorescence ratio F/F0) or NADH autofluorescence ratiored; F/F0) measured simultaneously in dual-loaded cells. (A) Charac-eristic effect of palmitoleic acid (50 �mol/L POA) showing mitochon-rial membrane depolarization with a global, sustained elevation ofCa2�]C (9 of 9 cells). (B) Light-transmitted, NADH autofluorescent,MRM fluorescent, and superimposed NADH and TMRM fluorescentmages with a plot of typical changes demonstrating effects of palmi-oleic acid (100 �mol/L POA). Note the precise perigranular corre-pondence of the TMRM and NADH signals used to detect changes initochondrial membrane potential and energetics. Following BAPTAretreatment, there is no change in mitochondrial membrane poten-ial, but NADH fluorescence still falls, although not as completely asn Figures 4 and 5. (C) Effects of low-dose palmitoleic acid (POA 10mol/L), showing well-maintained mitochondrial membrane potentialuring development of a modest, global, sustained elevation ofCa2�]C (cf scale for Fluo 4 in (A) and rises in [Ca2�]C). The protono-hore CCCP (10 �mol/L) was added after palmitoleic acid to induce
omplete mitochondrial depolarization (n � 16).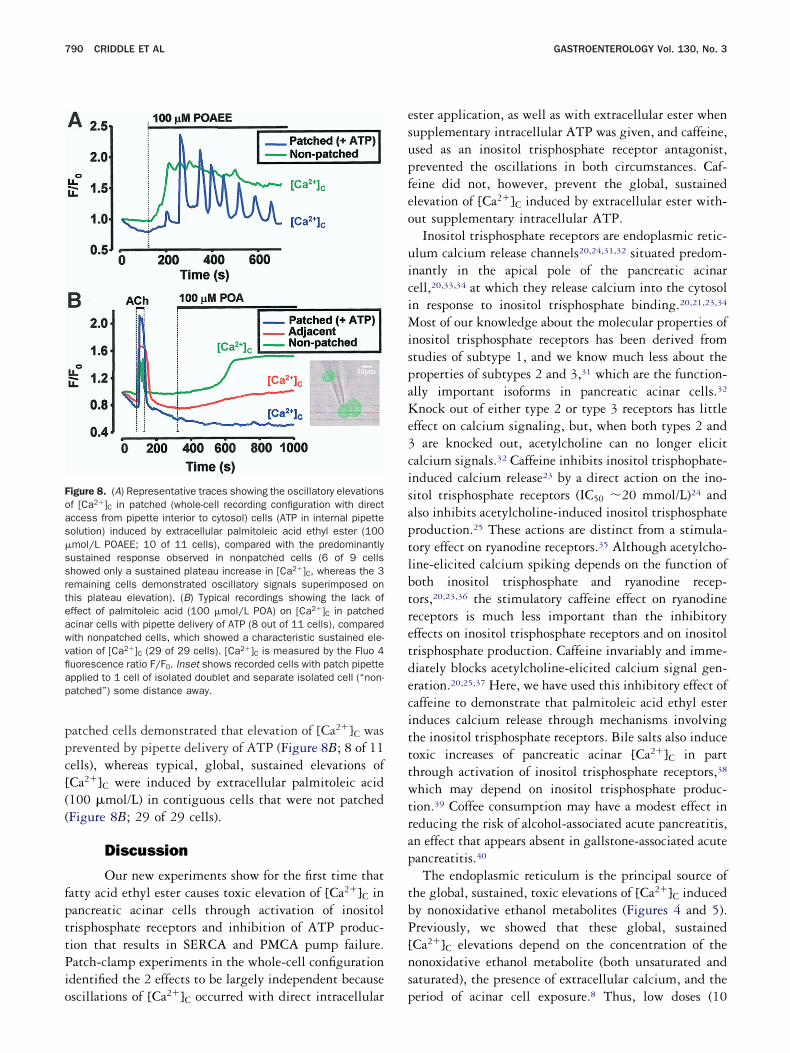
ppc[((
fpttPio
esupfeo
uiciMispaKe3cisaptlbtretdecitttwtrap
tbP[ns
Foas�ssrteawvflap
790 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
atched cells demonstrated that elevation of [Ca2�]C wasrevented by pipette delivery of ATP (Figure 8B; 8 of 11ells), whereas typical, global, sustained elevations ofCa2�]C were induced by extracellular palmitoleic acid100 �mol/L) in contiguous cells that were not patchedFigure 8B; 29 of 29 cells).
Discussion
Our new experiments show for the first time thatatty acid ethyl ester causes toxic elevation of [Ca2�]C inancreatic acinar cells through activation of inositolrisphosphate receptors and inhibition of ATP produc-ion that results in SERCA and PMCA pump failure.atch-clamp experiments in the whole-cell configuration
dentified the 2 effects to be largely independent because
igure 8. (A) Representative traces showing the oscillatory elevationsf [Ca2�]C in patched (whole-cell recording configuration with directccess from pipette interior to cytosol) cells (ATP in internal pipetteolution) induced by extracellular palmitoleic acid ethyl ester (100mol/L POAEE; 10 of 11 cells), compared with the predominantlyustained response observed in nonpatched cells (6 of 9 cellshowed only a sustained plateau increase in [Ca2�]C, whereas the 3emaining cells demonstrated oscillatory signals superimposed onhis plateau elevation). (B) Typical recordings showing the lack offfect of palmitoleic acid (100 �mol/L POA) on [Ca2�]C in patchedcinar cells with pipette delivery of ATP (8 out of 11 cells), comparedith nonpatched cells, which showed a characteristic sustained ele-ation of [Ca2�]C (29 of 29 cells). [Ca2�]C is measured by the Fluo 4uorescence ratio F/F0. Inset shows recorded cells with patch pipettepplied to 1 cell of isolated doublet and separate isolated cell (“non-atched”) some distance away.
scillations of [Ca2�]C occurred with direct intracellular p
ster application, as well as with extracellular ester whenupplementary intracellular ATP was given, and caffeine,sed as an inositol trisphosphate receptor antagonist,revented the oscillations in both circumstances. Caf-eine did not, however, prevent the global, sustainedlevation of [Ca2�]C induced by extracellular ester with-ut supplementary intracellular ATP.
Inositol trisphosphate receptors are endoplasmic retic-lum calcium release channels20,24,31,32 situated predom-nantly in the apical pole of the pancreatic acinarell,20,33,34 at which they release calcium into the cytosoln response to inositol trisphosphate binding.20,21,23,34
ost of our knowledge about the molecular properties ofnositol trisphosphate receptors has been derived fromtudies of subtype 1, and we know much less about theroperties of subtypes 2 and 3,31 which are the function-lly important isoforms in pancreatic acinar cells.32
nock out of either type 2 or type 3 receptors has littleffect on calcium signaling, but, when both types 2 and
are knocked out, acetylcholine can no longer elicitalcium signals.32 Caffeine inhibits inositol trisphophate-nduced calcium release23 by a direct action on the ino-itol trisphosphate receptors (IC50 20 mmol/L)24 andlso inhibits acetylcholine-induced inositol trisphosphateroduction.25 These actions are distinct from a stimula-ory effect on ryanodine receptors.35 Although acetylcho-ine-elicited calcium spiking depends on the function ofoth inositol trisphosphate and ryanodine recep-ors,20,23,36 the stimulatory caffeine effect on ryanodineeceptors is much less important than the inhibitoryffects on inositol trisphosphate receptors and on inositolrisphosphate production. Caffeine invariably and imme-iately blocks acetylcholine-elicited calcium signal gen-ration.20,25,37 Here, we have used this inhibitory effect ofaffeine to demonstrate that palmitoleic acid ethyl esternduces calcium release through mechanisms involvinghe inositol trisphosphate receptors. Bile salts also induceoxic increases of pancreatic acinar [Ca2�]C in parthrough activation of inositol trisphosphate receptors,38
hich may depend on inositol trisphosphate produc-ion.39 Coffee consumption may have a modest effect ineducing the risk of alcohol-associated acute pancreatitis,n effect that appears absent in gallstone-associated acuteancreatitis.40
The endoplasmic reticulum is the principal source ofhe global, sustained, toxic elevations of [Ca2�]C inducedy nonoxidative ethanol metabolites (Figures 4 and 5).reviously, we showed that these global, sustainedCa2�]C elevations depend on the concentration of theonoxidative ethanol metabolite (both unsaturated andaturated), the presence of extracellular calcium, and the
eriod of acinar cell exposure.8 Thus, low doses (10
�alt�lodtlc5cbifipgmtn
aefhocaehecaetufa
pmia(altdplb
ias
buolprdei7ogtN�coad[TimmwacmcawcpMtinEcattCTdcd
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 791
mol/L) were less likely to induce sustained elevations,nd, when such elevations occurred, these were moreikely to reverse on early withdrawal of either the me-abolite or extracellular calcium, whereas high doses (100mol/L) consistently produced elevations that were less
ikely to reverse on withdrawal of either the metaboliter extracellular calcium.8 We now demonstrate that,uring sustained stimulation with these metabolites, inhe absence of external calcium, [Ca2�]ER falls to veryow levels and remains low, indicating almost completealcium depletion of the endoplasmic reticulum (Figure). The sustained, high [Ca2�]C under such conditions8
ould most easily be explained by failure to clear calciumy SERCA and PMCA pumps, most likely because ofnadequate ATP supply. This interpretation was con-rmed by the experimental results showing that neitheralmitoleic acid ethyl ester nor palmitoleic acid inducedlobal sustained elevations of [Ca2�]C in cells supple-ented with intracellular ATP via a patch pipette, al-
hough such elevations developed and persisted in nearbyonpatched cells (Figure 8).The pancreas forms more fatty acid ethyl ester than
ny other human organ,2,3 catalyzed by fatty acid ethylster synthases and acyl-coenzyme A:ethanol O-acyltrans-erase.4 The fatty acid ethyl ester concentrations testedere of 10–100 �mol/L are equivalent to those that mayccur within pancreatic acinar cells following acute al-ohol intoxication, at up to 500 nmol/g or nmol/g/h asmount present (nmol/g) or amount synthesized fromthanol and fatty acids by pancreatic tissue (nmol/g/).2–6 Although the nonoxidative synthase and esterasenzymes that metabolize ethanol are as yet inadequatelyharacterized,4 and any allelic variation unknown, fattycid ethyl ester synthases are up-regulated by chronicthanol consumption.41 Fatty acid ethyl esters bind tohe inner mitochondrial membrane and accumulate andndergo hydrolysis to release fatty acids,26 catalyzed byatty acid ethyl ester hydrolases including carboxylester-se.4
Global elevations of [Ca2�]C induced by a high dose ofalmitoleic acid proved more difficult to reverse by re-oval of extracellular calcium than those induced by an
dentically high dose of its ethyl ester8; also, the fattycid induced a more rapid fall in NADH than the estercf, Figures 4A and 4B). These data are consistent withn effect of fatty acid ethyl esters that is dependent on theiberation and activity of their fatty acids, as indicated byhe marked diminution in the increase of [Ca2�]C in-uced by extracellular palmitoleic acid ethyl ester in theresence of an inhibitor of fatty acid ethyl ester hydro-ase. Hydrolase inhibition also permitted restoration of
asal [Ca2�]C concentrations (Figure 3), suggesting that onhibition of enzymes contributing to the intracellularccumulation of fatty acid substrates could limit theeverity of pancreatitis.
High concentrations of long-chain fatty acids haveeen shown to impair mitochondrial metabolism,27–30
ncoupling oxidative phosphorylation through alterationf ion channel function leading to mitochondrial depo-arization and opening of the permeability transitionore27 and/or through futile cycling and inhibition ofespiration.28–30 In the present experiments, mitochon-rial membrane depolarization induced by nonoxidativethanol metabolites was largely prevented by prior load-ng of cells with the calcium chelator BAPTA (FigureB), demonstrating the loss of membrane potential thatccurs without BAPTA to be predominantly due tolobal, sustained increases of [Ca2�]C that overload mi-ochondria. Nevertheless, with prior loading of BAPTA,ADH levels still fell following extracellular (100mol/L) palmitoleic acid (Figure 7B), suggesting mito-
hondrial impairment from futile cycling and inhibitionf respiration. Consistent with this, low-dose palmitoleiccid did not alter mitochondrial membrane potential butid induce a more modest, global, sustained increase ofCa2�]C from calcium-ATPase pump failure (Figure 7C).hus, uncoupling of oxidative phosphorylation through
nhibition of respiration remains the most probable pri-ary means whereby fatty acids reduce pancreatic acinaritochondrial ATP production.28–30 Fatty acids combineith cytosolic CoASH to form acyl-CoA (“activated fatty
cid”), the acyl group of which is temporarily bound toarnitine and transferred across the inner mitochondrialembrane by carnitine palmitoyltransferases I and II, to
ombine with mitochondrial CoASH, and again formcyl-CoA. Acyl-CoA then enters the �-oxidation path-ay to produce acetyl-CoA for the tricarboxylic acid
ycle that produces NADH and FADH2 to create theroton gradient that drives subsequent ATP production.itochondrial uncoupling proteins dissipate energy
hrough fatty acid export prior to �-oxidation, protect-ng mitochondria from high concentrations of fatty acids,otably long-chain fatty acids,28–30 and superoxide.42
xcessive fatty acids may overload these mechanisms,onsume CoASH as acyl-CoA, and so reduce the avail-bility of CoASH for later steps in �-oxidation and thericarboxylic acid cycle, inhibiting respiration.30 Addi-ionally, the combination of fatty acids with cytosolicoASH requires ATP, not replaced by futile cycling.30
he initial impairment of mitochondrial respiration in-uced by nonoxidative ethanol metabolites is likely toontribute to the subsequent complete mitochondrialepolarization induced by the sustained, toxic increases
f [Ca2�]C that lead to cell death.
apastspepAbhitwcwaiatctcdbifiaoia
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
792 CRIDDLE ET AL GASTROENTEROLOGY Vol. 130, No. 3
The formation of fatty acid ethyl ester requires fattycid to be present as a cofactor to induce alcoholicancreatitis. Hyperlipidemia, a high-fat diet, and fastingll increase the relative role of fatty acids as metabolicubstrates,30,43 elevating cellular fatty acid concentra-ions. Fatty acid toxicity, through the mechanisms de-cribed, may also be the cause of acute pancreatitis inatients with hyperlipidemia, which is prevented byffective lipid-regulation44; also, alcohol is a recognizedrecipitant of pancreatitis in this group of patients.45
lthough the sparse epidemiologic data on the contri-ution of a high fat intake to the pathogenesis of alco-olic pancreatitis are conflicting,46,47 ethanol alone isnsufficient for the induction of experimental pancreati-is, whereas ethanol combined with a high-fat diet is aell-documented model.9,10 The lack of effect of ethanol
ontrasts with that of long-chain fatty acid infusion,hich results in a severe form of acute pancreatitis char-
cterized by marked ATP depletion,48 which our exper-ments suggest is central to the pathogenesis of bothlcoholic and hyperlipidemic acute pancreatitis. In addi-ion, alcohol and/or a high fat intake exacerbate bothaerulein and bile-induced experimental acute pancreati-is.11–13,49,50 Furthermore, the intermittent excessiveonsumption of alcohol that is often seen in patientseveloping alcoholic pancreatitis51 may be accompaniedy little simultaneous food intake, again leading to anncrease in fatty acids as metabolic substrates.30 Thendings reported here suggest that dietary and otherppropriate measures that reduce the relative availabilityf fatty acids as respiratory substrates could be beneficialn those at risk of pancreatitis from ethanol and itsssociated metabolites.
References1. Ammann RW, Heitz PU, Kloppel G. Course of alcoholic chronic
pancreatitis: a prospective clinicomorphological long-term study.Gastroenterology 1996;111:224–231.
2. Laposata EA, Lange LG. Presence of nonoxidative ethanol metab-olism in human organs commonly damaged by ethanol abuse.Science 1986;231:497–499.
3. Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN,Faller LD, Pandol SJ. Ethanol metabolism and transcription factoractivation in pancreatic acinar cells in rats. Gastroenterology2002;122:106–118.
4. Diczfalusy MA, Bjorkhem I, Einarsson C, Hillebrant CG, AlexsonSE. Characterization of enzymes involved in formation of ethylesters of long-chain fatty acids in humans. J Lipid Res 2001;42:1025–1032.
5. Werner J, Laposata M, Fernandez-del Castillo C, Saghir M, IozzoRV, Lewandrowski KB, Warshaw AL. Pancreatic injury in ratsinduced by fatty acid ethyl ester, a nonoxidative metabolite ofalcohol. Gastroenterology 1997;113:286–294.
6. Werner J, Saghir M, Warshaw AL, Lewandrowski KB, Laposata M,Iozzo RV, Carter EA, Schatz RJ, Fernandez-Del Castillo C. Alco-
holic pancreatitis in rats: injury from nonoxidative metabolites ofethanol. Am J Physiol Gastrointest Liver Physiol2002;283:G65–G73.
7. Deng X, Wang L, Elm MS, Gabazadeh D, Diorio GJ, Eagon PK,Whitcomb DC. Chronic alcohol consumption accelerates fibrosisin response to cerulein-induced pancreatitis in rats. Am J Pathol2005;166:93–106.
8. Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, PetersenOH, Sutton R. Ethanol toxicity in pancreatic acinar cells: media-tion by nonoxidative fatty acid metabolites. Proc Natl Acad SciU S A 2004;101:10738–10743.
9. Tsukamoto H, Towner SJ, Yu GS, French SW. Potentiation ofethanol-induced pancreatic injury by dietary fat. Induction ofchronic pancreatitis by alcohol in rats. Am J Pathol 1988;131:246–257.
0. Kono H, Nakagami M, Rusyn I, Connor HD, Stefanovic B, BrennerDA, Mason RP, Arteel GE, Thurman RG. Development of ananimal model of chronic alcohol-induced pancreatitis in the rat.Am J Physiol Gastrointest Liver Physiol 2001;280:G1178–G1186.
1. Katz M, Carangelo R, Miller LJ, Gorelick F. Effect of ethanol oncholecystokinin-stimulated zymogen conversion in pancreatic aci-nar cells. Am J Physiol 1996;270:G171–G175.
2. Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y,Solomon TE, Gukovskaya AS, Tsukamoto H. Ethanol diet in-creases the sensitivity of rats to pancreatitis induced by cho-lecystokinin octapeptide. Gastroenterology 1999;117:706–716.
3. Lu Z, Karne S, Kolodecik T, Gorelick FS. Alcohols enhancecaerulein-induced zymogen activation in pancreatic acinar cells.Am J Physiol Gastrointest Liver Physiol 2002;282:G501–G507.
4. Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, SuttonR, Petersen OH. Calcium-dependent enzyme activation and vac-uole formation in the apical granular region of pancreatic acinarcells. Proc Natl Acad Sci U S A 2000;97:13126–13131.
5. Park MK, Petersen OH, Tepikin AV. The endoplasmic reticulum asone continuous Ca2� pool: visualization of rapid Ca2� move-ments and equilibration. EMBO J 2000;19:5729–5739.
6. Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH,Tepikin A. Correlation of NADH and Ca2� signals in mouse pan-creatic acinar cells. J Physiol 2002;539:41–52.
7. Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR.The relationship between mitochondrial state, ATP hydrolysis,[Mg2�]i and [Ca2�]i studied in isolated rat cardiomyocytes.J Physiol 1996;496:111–128.
8. Inoue M, Fujishiro N, Imanaga I, Sakamoto Y. Role of ATP de-crease in secretion induced by mitochondrial dysfunction in guin-ea-pig adrenal chromaffin cells. J Physiol 2002;539:145–155.
9. Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Pe-tersen OH, Watson AJ. Menadione-induced apoptosis: roles ofcytosolic Ca2� elevations and the mitochondrial permeabilitytransition pore. J Cell Sci 2002;115:485–497.
0. Wakui M, Osipchuk YV, Petersen OH. Receptor-activated cyto-plasmic Ca2� spiking mediated by inositol trisphosphate is dueto Ca2�-induced Ca2� release. Cell 1990;63:1025–1032.
1. Thorn P, Lawrie AM, Smith PM, Gallacher DV, Petersen OH. Localand global cytosolic Ca2� oscillations in exocrine cells evoked byagonists and inositol trisphosphate. Cell 1993;74:661–668.
2. Park MK, Lomax RB, Tepikin AV, Petersen OH. Local uncaging ofcaged Ca2� reveals distribution of Ca2�-activated Cl� channelsin pancreatic acinar cells. Proc Natl Acad Sci U S A 2001;98:10948–10953.
3. Petersen OH. Ca2� signalling and Ca2�-activated ion channels inexocrine acinar cells. Cell Calcium 2005;38:171–200.
4. Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. Thepharmacology of intracellular Ca2�-release channels. Trends
Pharmacol Sci 1994;15:145–149.
2
2
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
4
4
4
4
4
5
5
sUe
fW
March 2006 FATTY ACID ETHYL ESTER PANCREATIC CALCIUM TOXICITY 793
5. Toescu EC, O’Neill SC, Petersen OH, Eisner DA. Caffeine inhibitsthe agonist-evoked cytosolic Ca2� signal in mouse pancreaticacinar cells by blocking inositol trisphosphate production. J BiolChem 1992;267:23467–23470.
6. Lange LG, Sobel BE. Mitochondrial dysfunction induced by fattyacid ethyl esters, myocardial metabolites of ethanol. J Clin Invest1983;72:724–731.
7. Penzo D, Tagliapietra C, Colonna R, Petronilli V, Bernardi P.Effects of fatty acids on mitochondria: implications for cell death.Biochim Biophys Acta 2002;1555:160–165.
8. Jaburek M, Varecha M, Gimeno RE, Dembski M, Jezek P, ZhangM, Burn P, Tartaglia LA, Garlid KD. Transport function and regu-lation of mitochondrial uncoupling proteins 2 and 3. J Biol Chem1999;274:26003–26007.
9. Hunt MC, Nousiainen SE, Huttunen MK, Orii KE, Svensson LT,Alexson SE. Peroxisome proliferator-induced long chain acyl-CoAthioesterases comprise a highly conserved novel multi-gene fam-ily involved in lipid metabolism. J Biol Chem 1999;274:34317–34326.
0. Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-DoulcierAM, Bouillaud F, Ricquier D. The biology of mitochondrial uncou-pling proteins. Diabetes 2004;53(Suppl 1):S130–S135.
1. Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. CellCalcium 2005;38:261–272.
2. Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K,Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J,Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secre-tion underlying energy metabolism. Science 2005;309:2232–2234.
3. Ashby MC, Craske M, Park MK, Gerasimenko OV, Burgoyne RD,Petersen OH, Tepikin AV. Localized Ca2� uncaging reveals polar-ized distribution of Ca2�-sensitive Ca2� release sites: mecha-nism of unidirectional Ca2� waves. J Cell Biol 2002;158:283–292.
4. Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, WuytackF, Racymaekers L, Muallem S. Polarized expression of Ca2�
channels in pancreatic and salivary gland cells. Correlation withinitiation and propagation of [Ca2�]i waves. J Biol Chem 1997;272:15765–15770.
5. Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A. Ca2�
dynamics in the lumen of the endoplasmic reticulum in sensoryneurons: direct visualization of Ca2�-induced Ca2� release trig-gered by physiological Ca2� entry. EMBO J 2002;21:622–630.
6. Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, Pe-tersen OH. Two different but converging messenger pathways tointracellular Ca2� release: the roles of NAADP, cADPR and IP3.EMBO J 2000;19:2549–2557.
7. Osipchuk YV, Wakui M, Yule DI, Gallacher DV, Petersen OH.Cytoplasmic Ca2� oscillations evoked by receptor stimulation,G-protein activation, internal application of inositol trisphosphateor Ca2�: simultaneous microfluorimetry and Ca2� dependent Cl�
current recording in single pancreatic acinar cells. EMBO J 1990;9:697–704.
8. Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A. Bile
acids induce calcium signals in mouse pancreatic acinar cells:implications for bile-induced pancreatic pathology. J Physiol2002;540:49–55.
9. Lau BW, Colella M, Ruder WC, Ranieri M, Curci S, Hofer AM.Deoxycholic acid activates protein kinase C and phospholipase Cvia increased Ca2� entry at plasma membrane. Gastroenterology2005;128:695–707.
0. Morton C, Klatsky AL, Udaltsova N. Smoking, coffee, and pancre-atitis. Am J Gastroenterol 2004;99:731–738.
1. Pfutzer RH, Tadic SD, Li HS, Thompson BS, Zhang JY, Ford ME,Eagon PK, Whitcomb DC. Pancreatic cholesterol esterase,ES-10, and fatty acid ethyl ester synthase III gene expressionare increased in the pancreas and liver but not in the brain orheart with long-term ethanol feeding in rats. Pancreas 2002;25:101–106.
2. Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S,Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S,Clapham JC, Brand MD. Superoxide activates mitochondrial un-coupling proteins. Nature 2002;415:96–99.
3. Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia, andthe metabolic syndrome. Am J Cardiol 1998;81:18B–25B.
4. Yadav D, Pitchumoni CS. Issues in hyperlipidemic pancreatitis.J Clin Gastroenterol 2003;36:54–62.
5. Dickson AP, O’Neill J, Imrie CW. Hyperlipidaemia, alcohol abuseand acute pancreatitis. Br J Surg 1984;71:685–688.
6. Durbec JP, Sarles H. Multicenter survey of the etiology of pan-creatic diseases. Relationship between the relative risk of devel-oping chronic pancreaitis and alcohol, protein and lipid consump-tion. Digestion 1978;18:337–350.
7. Wilson JS, Bernstein L, McDonald C, Tait A, McNeil D, Pirola RC.Diet and drinking habits in relation to the development of alco-holic pancreatitis. Gut 1985;26:882–887.
8. Nordback IH, Clemens JA, Chacko VP, Olson JL, Cameron JL.Changes in high-energy phosphate metabolism and cell morphol-ogy in four models of acute experimental pancreatitis. Ann Surg1991;213:341–349.
9. Ramo OJ. Antecedent long-term ethanol consumption in combi-nation with different diets alters the severity of experimentalacute pancreatitis in rats. Gut 1987;28:64–69.
0. Hofbauer B, Friess H, Weber A, Baczako K, Kisling P, Schilling M,Uhl W, Dervenis C, Buchler MW. Hyperlipaemia intensifies thecourse of acute oedematous and acute necrotising pancreatitisin the rat. Gut 1996;38:753–758.
1. Stigendal L, Olsson R. Alcohol consumption pattern and serumlipids in alcoholic cirrhosis and pancreatitis. A comparative study.Scand J Gastroenterol 1984;19:582–587.
Received July 1, 2005. Accepted December 7, 2005.Address requests for reprints to: Robert Sutton, DPhil, FRCS, Divi-
ion of Surgery and Oncology, University of Liverpool, Royal Liverpoolniversity Hospital, Daulby Street, Liverpool L69 3BX, United Kingdom.-mail: [email protected]; fax: (44) 151 706 5826.Supported by Programme, Cooperative, and Component grants
rom the Medical Research Council (United Kingdom) and an Ameliearing Clinical Research Fellowship from CORE (to J.M.).
O.H.P. is a Medical Research Council professor.Copyright © 2022 FDOKUMEN