
FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury
11
Regular Article FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury S. Casha, W.R. Yu, M.G. Fehlings * Spinal Program, Krembil Neuroscience Center, Toronto Western Hospital, 399 Bathurst St., Toronto, Ontario, Canada M5T 2S8 Department of Surgery, Institute of Medical Science, University of Toronto, 399 Bathurst St., Toronto, Ontario, Canada M5T 2S8 Received 1 April 2005; revised 15 July 2005; accepted 25 August 2005 Available online 3 October 2005 Abstract After spinal cord injury (SCI), apoptosis of neurons and oligodendrocytes is associated with axonal degeneration and loss of neurological function. Recent data have suggested a potential role for FAS death receptor-mediated apoptosis in the pathophysiology of SCI. In this study, we examined the effect of FAS deficiency on SCI in vitro and in vivo. FAS Lpr/lpr mutant mice and wildtype background-matched mice were subjected to a T5 – 6 clip compression SCI, and complementary studies were done in an organotypic slice culture model of SCI. Post-traumatic apoptosis in the spinal cord, which was seen in neurons and oligodendrocytes, was decreased in the FAS-deficient mice both in vivo and in vitro particularly in oligodendrocytes. FAS deficiency was also associated with improved locomotor recovery, axonal sparing and preservation of oligodendrocytes and myelin. However, FAS deficiency did not result in a significant increase in surviving neurons in the spinal cord at 6 weeks after injury, likely reflecting the importance of other cell death mechanisms for neurons. We conclude that inhibition of the FAS pathway may be a clinically attractive neuroprotective strategy directed towards oligodendroglial and axonal preservation in the treatment of SCI and neurotrauma. D 2005 Elsevier Inc. All rights reserved. Keywords: Spinal cord injury; FAS; Apoptosis; Oligodendrocyte; Neuroprotection Introduction A number of studies have linked apoptosis to spinal cord injury (SCI) (Casha and Fehlings, 2001; Crowe et al., 1997; Emery et al., 1998;Katoh et al., 1996; Li et al., 1996, 1999; Liu et al., 1997; Lou et al., 1998; Yong et al., 1998). In these reports, early apoptosis of neural cells including neurons is followed by a delayed wave of predominantly oligodendroglial programmed cell death in degenerating white matter tracts (Crowe et al., 1997; Emery et al., 1998; Li et al., 1999; Liu et al., 1997; Shuman et al., 1997). Studies of apoptosis in white matter after dorsal cordotomy or after transection suggest that glial apoptosis occurs, at least in part, as a consequence of axonal degeneration (Abe et al., 1999; Warden et al., 2001). The loss of trophic support derived by the oligodendrocyte from the axon likely results in activation of programmed cell death. However, the presence of activated microglia in contact with apoptotic oligodendrocytes after SCI suggests that this interaction may also activate cell death programs in the oligodendrocyte (Shuman et al., 1997). Secondary axonal degeneration may then follow as is seen in models of multiple sclerosis (Bjartmar et al., 1999) and in myelin-associated glycoprotein deficiency (Yin et al., 1998). It is thus logical to postulate that decreasing apoptosis in both oligodendrocytes and neurons may improve neurological outcome after SCI. FAS (CD95 or APO1) is known to interact with cytoplasmic FAS-Associated Death Domain protein (FADD) and, upon FAS ligand (FasL) binding, allows activation of caspase 8 leading to apoptosis (Siegel et al., 2000). This receptor exhibits changes in expression which may implicate it in the pathophysiology of SCI. Li and colleagues showed that white matter expression of FAS and FasL is lost within 1 day at the site of injury while increasing in segments adjacent to the injury site (Li et al., 2000a). Zurita et al. found an increase in the number of cells expressing FAS increases within 72 h of SCI (Zurita et al., 2001). We previously demonstrated that FAS expression, caspase 8 and 3 activation and apoptosis occur in a temporally 0014-4886/$ - see front matter D 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.expneurol.2005.08.020 Experimental Neurology 196 (2005) 390 – 400 www.elsevier.com/locate/yexnr * Corresponding author. Spinal Program, Krembil Neuroscience Center, Toronto Western Hospital, 399 Bathurst St., Toronto, Ontario, Canada M5T 2S8. Fax: +1 416 603 5298. E-mail address: [email protected] (M.G. Fehlings).
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury

elsevier.com/locate/yexnr
Experimental Neurology 1
Regular Article
FAS deficiency reduces apoptosis, spares axons and improves function
after spinal cord injury
S. Casha, W.R. Yu, M.G. Fehlings *
Spinal Program, Krembil Neuroscience Center, Toronto Western Hospital, 399 Bathurst St., Toronto, Ontario, Canada M5T 2S8
Department of Surgery, Institute of Medical Science, University of Toronto, 399 Bathurst St., Toronto, Ontario, Canada M5T 2S8
Received 1 April 2005; revised 15 July 2005; accepted 25 August 2005
Available online 3 October 2005
Abstract
After spinal cord injury (SCI), apoptosis of neurons and oligodendrocytes is associated with axonal degeneration and loss of neurological
function. Recent data have suggested a potential role for FAS death receptor-mediated apoptosis in the pathophysiology of SCI. In this study, we
examined the effect of FAS deficiency on SCI in vitro and in vivo. FASLpr/lpr mutant mice and wildtype background-matched mice were subjected
to a T5–6 clip compression SCI, and complementary studies were done in an organotypic slice culture model of SCI. Post-traumatic apoptosis in
the spinal cord, which was seen in neurons and oligodendrocytes, was decreased in the FAS-deficient mice both in vivo and in vitro particularly in
oligodendrocytes. FAS deficiency was also associated with improved locomotor recovery, axonal sparing and preservation of oligodendrocytes
and myelin. However, FAS deficiency did not result in a significant increase in surviving neurons in the spinal cord at 6 weeks after injury, likely
reflecting the importance of other cell death mechanisms for neurons. We conclude that inhibition of the FAS pathway may be a clinically
attractive neuroprotective strategy directed towards oligodendroglial and axonal preservation in the treatment of SCI and neurotrauma.
D 2005 Elsevier Inc. All rights reserved.
Keywords: Spinal cord injury; FAS; Apoptosis; Oligodendrocyte; Neuroprotection
Introduction
A number of studies have linked apoptosis to spinal cord
injury (SCI) (Casha and Fehlings, 2001; Crowe et al., 1997;
Emery et al., 1998;Katoh et al., 1996; Li et al., 1996, 1999; Liu
et al., 1997; Lou et al., 1998; Yong et al., 1998). In these reports,
early apoptosis of neural cells including neurons is followed by
a delayed wave of predominantly oligodendroglial programmed
cell death in degenerating white matter tracts (Crowe et al.,
1997; Emery et al., 1998; Li et al., 1999; Liu et al., 1997;
Shuman et al., 1997). Studies of apoptosis in white matter after
dorsal cordotomy or after transection suggest that glial
apoptosis occurs, at least in part, as a consequence of axonal
degeneration (Abe et al., 1999; Warden et al., 2001). The loss of
trophic support derived by the oligodendrocyte from the axon
likely results in activation of programmed cell death. However,
0014-4886/$ - see front matter D 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.expneurol.2005.08.020
* Corresponding author. Spinal Program, Krembil Neuroscience Center,
Toronto Western Hospital, 399 Bathurst St., Toronto, Ontario, Canada M5T
2S8. Fax: +1 416 603 5298.
E-mail address: [email protected] (M.G. Fehlings).
the presence of activated microglia in contact with apoptotic
oligodendrocytes after SCI suggests that this interaction may
also activate cell death programs in the oligodendrocyte
(Shuman et al., 1997). Secondary axonal degeneration may
then follow as is seen in models of multiple sclerosis (Bjartmar
et al., 1999) and in myelin-associated glycoprotein deficiency
(Yin et al., 1998). It is thus logical to postulate that decreasing
apoptosis in both oligodendrocytes and neurons may improve
neurological outcome after SCI.
FAS (CD95 or APO1) is known to interact with cytoplasmic
FAS-Associated Death Domain protein (FADD) and, upon FAS
ligand (FasL) binding, allows activation of caspase 8 leading to
apoptosis (Siegel et al., 2000). This receptor exhibits changes
in expression which may implicate it in the pathophysiology of
SCI. Li and colleagues showed that white matter expression of
FAS and FasL is lost within 1 day at the site of injury while
increasing in segments adjacent to the injury site (Li et al.,
2000a). Zurita et al. found an increase in the number of cells
expressing FAS increases within 72 h of SCI (Zurita et al.,
2001). We previously demonstrated that FAS expression,
caspase 8 and 3 activation and apoptosis occur in a temporally
96 (2005) 390 – 400
www.

S. Casha et al. / Experimental Neurology 196 (2005) 390–400 391
similar fashion after SCI (Casha and Fehlings, 2001). Demjen
et al. demonstrated that FasL neutralizing antibodies improved
neurological outcome after transaction SCI (Demjen et al.,
2004). In addition, FAS signaling has been implicated in death
of motor neurons after spinal ischemia and axotomy of the
facial nerve (Matsushita et al., 2000; Sakurai et al., 1998;
Ugolini et al., 2003). Given these observations, we hypothesize
that deficiency of FAS receptor will result in decreased
oligodendroglial cell death and improved behavioral and
histological outcome after SCI.
In this article, we compared in vivo and in vitro SCI in
C57BL/6-FASLpr/lpr mice and wildtype C57BL/6 background-
matched mice. These lpr mice are deficient in FAS expression
due to a retro-transposon insertion in the second intron of the
FAS gene leading to a premature stop codon and a non-
functional gene product (Nakatsuru et al., 1999). Phenotypical-
ly, these mice appear normal at birth but develop in a strain-
dependent manner a lymphoproliferative disorder at ages
greater than 18 weeks (Vidal et al., 1998). We used animals
with a C57Bl/6J background at 8 weeks of age to avoid this
potentially confounding feature, which develops later and less
severely in this strain (Vidal et al., 1998). In addition, an in vitro
organotypic culture model was also used to confirm findings in
the absence of systemically mediated inflammation.
Materials and methods
In vivo spinal cord injury
SCI was performed in 8-week-old female B6.MRL-Fas-lpr
mice (lpr) and C57BL/6 background-matched mice (Jackson
Laboratory, Bar Harbor, Maine) using the FEJOTAi clip
compression model at T5/6 (Joshi and Fehlings, 2002a). Under
(1.5–2%) halothane and N2O (1 L/min) anesthetic, a modified
aneurysm clip (7.8 g closing force) was applied rapidly for 1
min to the spinal cord after laminectomy. After injury, animals
were housed at 25–27-C. All protocols were in accordance
with the Canadian Council of Animal Care policies and were
approved by the animal care committee of the University
Health Network.
Organotypic slice culture model
We employed an in vitro organotypic slice culture model of
SCI (Krassioukov et al., 2002). Briefly, 350 Am slices from
T2–T10 spinal cord of 4-week-old mice were generated
(McIlwain tissue chopper, Harvard apparatus, Saint Laurent,
Quebec) and incubated in culture medium (50% MEM with
Earl’s salts and glutamine, 25% Hanks balanced salt solution,
25% horse serum, 20 mM HEPES, 6 mg/ml d-glucose, pH 7.2)
at 36-C in 5% CO2. At 14 days, the slices were injured using a
guided weight dropped onto the center of the slice (0.137 g
piston, 1.2 mm diameter, 5 mm height) (Fig. 3) 30 min after
addition of propidium iodide (10 AM) to the media. Uptake of
propidium iodide was imaged using a confocal microscope
(Bio-Rad MRC 600), and pixels with grayscale intensity >127
at 24, 48 and 72 h were counted (ImagePro plus software,
Media Cybernetics, Silver Springs, Maryland). Lpr and wild-
type counts were compared by 2-way ANOVA (n = 50 slices).
Western blotting
Five millimeters of spinal cord centered at the injury site or
the organotypic spinal cord cultures were homogenized in 10
volumes of 4-C 50 mM Tris 4 mM EDTA, pH 7.4 with 1 AMpepstatin, 100 AM leupeptin, 100 AM phenylmethylsulfonyl-
fluoride and 10 Ag/ml aprotinin. Twenty micrograms of protein
was subjected to SDS-PAGE followed by electroblotting onto
nitrocellulose. Blots were blocked (1 h) with 1% nonfat dry
milk 0.1% Tween 20 Tris-buffered saline and incubated for 1
h with primary antiserum (NF200 (Sigma, Oakville, Ontario),
MAP2 (Chemicon, Temecula, California), CNPase (Sternber-
ger Monoclonals, Lutherville, Massachusetts), caspase 8 or
caspase 3 (Santa Cruz Biotechnology, Santa Cruz, California)
and for 1 h with horseradish-peroxidase-conjugated secondary
antiserum. Immunoreactive bands were visualized by chemi-
luminescence (ECL, Amersham, Arlington Heights, Illinois),
and band density was compared (Gelpro plus software, Media
Cybernetics, Silver Springs, Maryland). Actin immunoblotting
and Coomassie Blue staining were compared to ensure equal
loading of samples (Schumacher et al., 1999). All Western
blots were performed three times using independent samples.
In situ terminal-deoxy-transferase mediated dUTP nick end
labeling (TUNEL)
Injured lpr and wildtype mice (n = 4) were perfusion-fixed
with 4% paraformaldehyde 0.1 M phosphate buffer at days 1
and 3 post-injury. Serial 10 Am paraffin spinal cord sections at
500 Am intervals were stained with hematoxylin, eosin and
Luxol Fast Blue to identify the injury epicenter. The TUNEL
assay was applied using the Apotag kit (Intergen, New York,
New York) to sections at the injury epicenter and at 1000 Am.
TUNEL-positive nuclei were counted through the entire cross-
section and compared by Student’s t test.
Wildtype mouse spinal cord sections 500 Am from the injury
epicenter 7 days after injury and sections generated from in
vitro injured spinal cord tissue were double labeled with cell-
specific antisera and TUNEL. Oligodendrocyte stained sections
were blocked for 1 h with 5% milk 0.5% BSA and incubated
with anti-CNPase antiserum (1:500) or anti-CC1 antiserum
(1:20, Calbiochem, San Diego, California) at 4-C overnight.
Neuron-stained sections were blocked for 1 h in 20% goat
serum and incubated with anti-NeuN antiserum (1:50, Chemi-
con, Temecula, California) overnight at 4-C. FITC-conjugatedsecondary antiserum was applied for 1 h followed by TUNEL
as above.
Immunohistochemistry
Wildtype mouse spinal cord sections 500 Am from the injury
epicenter 7 days after injury were double labeled with cell-
specific antisera and FAS. Sections were blocked with goat
serum and incubated overnight with anti-FAS antiserum (Santa

S. Casha et al. / Experimental Neurology 196 (2005) 390–400392
Cruz Biotechnology, Santa Cruz, CA) (1:50 in 20 mM PBS).
Sections were exposed to horseradish-peroxidase-conjugated or
fluorescent (Texas Red or FITC)-conjugated secondary antise-
rum. Aminoethyl carbazole (AEC) was used to develop the
peroxidase reaction. Oligodendrocyte-stained sections were
blocked for 1 h with 5% milk 0.5% BSA and incubated with
anti-CNPase antiserum (1:500) or anti-CC1 antiserum (1:20,
Calbiochem, San Diego, California) at 4-C overnight. Neuron-
stained sections were blocked for 1 h in 20% goat serum and
incubated with anti-NeuN antiserum (1:50, Chemicon, Teme-
cula, California) overnight at 4-C. Astrocytes were stained withanti-GFAP serum (Sigma, Oakville, Ontario) (1:200 in 20 mM
PBS) overnight at 4-C after blocking with goat serum. FITC-
conjugated or Texas-Red-conjugated secondary antiserum was
then applied for 1 h.
Sections 2000 Am from the injury epicenter at 7 days post-
injury and sham-injured controls were immunostained with anti-
CNPase antiserum or NeuN antiserum as above (n = 3/group).
A random 40� field was selected, and the number of
immunostained cells was counted. Lpr and wildtype counts
were compared by Student’s t test.
NeuN-positive neurons were also counted in paraffin cross-
sections at 500, 1000, 1500 and 2000 Am from the injury
epicenter and at 6 weeks post-injury. Counts in lpr and
wildtype animals were compared by 2-way ANOVA (variables
FAS, genome and distance n = 6/group).
In vivo behavioral scoring
Injured wildtype (n = 6) and lpr (n = 6) mice were scored
using the modified BBB locomotor behavioral score for mice
by two blinded individuals at weekly intervals following SCI
Fig. 1. Post-traumatic apoptosis in the mouse spinal cord. Immunohistochemical exa
cell-specific markers confirmed apoptosis in oligodendrocytes (A–C) and neurons (
fields double labeled with NeuN (neuronal marker) and CNPase (oligodendrocytes m
merged (scale bar, 20 Am).
(Basso et al., 1995; Joshi and Fehlings, 2002a). Scores in
wildtype and mutant mice were compared by 2-way ANOVA.
Fluorogold retrograde tracing
Six weeks after injury, the animals underwent application of
a 4% Fluorogold-soaked gelfoam pledget to the rostral surface
of a T10 transection. Five days later, the animals were
perfusion fixed with 4% paraformaldehyde, and serial 40 Amparaffin sections of the brain stem were prepared. The number
of Fluorogold containing neurons in every second section
through the vestibular, raphe, red and reticular nuclei was
counted in a blinded manner and compared by Student’s t test.
The Fluorogold application site was examined to ensure that
the area of diffusion did not approach the injury site.
Retrograde tracing in non-injured control animals and T5/6
transection control animals was also quantified.
Luxol Fast Blue staining
Serial 10 Am paraffin cross-sections of spinal cord 6 week
post-injury were stained with 0.1% Luxol Fast Blue (Sigma,
Oakville, Ontario) in 95% ethanol, 0.5% acetic acid and
destained using 0.05% LiCO3. Stained sections at 250 Amintervals around the injury epicenter were digitally imaged, and
the area of Luxol Fast Blue staining was determined as the
percent of pixels with grayscale value >127 on the inverse
image (ImagePro Plus software, Media Cybernetics, Silver
Springs, MD). Mean lpr and wildtype areas were compared by
2-way ANOVA. Pairwise comparisons were made using t test
with Bonferroni correction. Uninjured T5/6 spinal cord
sections were also compared (n = 3 mutant and n = 4 wildtype).
mination of frozen sections 500 Am from the injury epicenter with TUNEL- and
D–F) 7 days after in vivo SCI. TUNEL is shown in panels A and D. The same
arker) are shown in panels B and E respectively. In panels C and F, the fields are

S. Casha et al. / Experimental Neurology 196 (2005) 390–400 393
Statistical analyses
The specific statistical comparisons used in each experiment
are indicated in the above methods. P < 0.05 was used to reject
the null hypothesis and to indicate statistical significance. All
tests were 2-tailed.
Results
Apoptotic cell death following murine SCI
As a prelude to investigations in FAS-deficient mice, we
first confirmed the occurrence of apoptosis in this species using
the TUNEL method (Figs. 1A, D, 4A, B). Double labeling
immunohistochemistry with TUNEL- and cell-specific markers
confirmed the occurrence of apoptosis in both oligodendro-
cytes and neurons (Fig. 1). Furthermore, double labeling
immunohistochemistry with FAS and cell markers demonstrat-
ed that FAS is expressed in oligodendrocytes, neurons and
some astrocytes in the mouse (Fig. 2). TUNEL and FAS double
Fig. 2. Cellular distribution of FAS expression in wildtype mice. FAS immunohisto
labeling at 7 days post-injury focusing on a region 500 Am from the injury epicen
(arrow, D–F). FAS expression was rarely seen in astrocytes (arrow, G–I). FAS was p
in the left and center column as indicated, while the merged image is presented on
labeling confirmed the occurrence of FAS-positive apoptotic
cells after SCI (Fig. 2).
The effect of FAS deficiency on cell death after in vitro SCI
In our in vitro slice SCI model (Figs. 3A, B), we found that
the extent of cell death, as assayed by propidium iodide uptake,
was significantly lower in lpr preparations than wildtype
preparations at 48 and 72 h (2-way ANOVA: P < 0.0001 for
FAS genome and time; pairwise comparisons: P < 0.05 at 48
and 72 h) (Figs. 3C–E). Given that propidium iodide does not
distinguish apoptotic from necrotic death, we examined
caspase 8 and 3 activation in this model by Western blotting
for the p20 band of the activated caspases. Caspase 8 and 3 are
the apical and terminal caspases respectively in the FAS-
mediated apoptotic pathway. We found greater activation of
both caspases in wildtype slices than in lpr slices at 72 h post-
injury (Fig. 3F), suggesting that less programmed cell death
occurs in FAS-deficient mice after in vitro SCI and in the
absence of a systemic inflammatory response.
chemistry was combined with cell marker immunohistochemistry and TUNEL
ter. We found FAS expression in oligodendrocytes (arrow, A–C) and neurons
resent on TUNEL-positive cells (arrow, J–L). Individual labels are represented
the right (scale bar, 20 Am).

Fig. 3. Decreased cell death in injured FAS-deficient spinal cord slice preparations. (A) Organotypic slice culture preparation stained for myelin with Luxol Fast
Blue. Slices were injured by a weight drop technique ((B) the injury device positioned using a micomanipulator (1), a 0.137 g piston (3) guided by a sleeve (2)
dropped from 5 mm height on to the center of the slice). The mean area of propidium iodide uptake (pixel counts) in FAS lpr mutant slices was significantly lower (*)
than in wildtype counterparts at 48 and 72 h after injury (E) (representative propidium iodide fluorograms at 72 h are shown for (C) FAS-deficient and (D) wildtype
preparations). Densitometry of Western blots revealed significantly decreased activated caspase 8 and 3 p20 subunit (*) (F).
S. Casha et al. / Experimental Neurology 196 (2005) 390–400394
Acute cell death after in vivo SCI in FAS-deficient and wildtype
mice
In order to examine the role of FAS in a more clinically
relevant model, we next compared apoptosis in lpr and
wildtype animals after in vivo clip compression SCI. Compar-
ison of TUNEL counts (Figs. 4A–C) showed no difference
between lpr and wildtype mice at 24 h, however, there was a
significant decrease in the number of apoptotic cells in lpr mice
at 72 h (t test: P = 0.034). Similarly, by Western blotting,
caspase 3 activation was greater in wildtype animals than in lpr
animals at day 7 post-injury (t test: P = 0.002) (Fig. 5D) but
was not significantly different at days 1 and 2 post-injury or in
uninjured controls. Thus, FAS deficiency resulted in reduced
post-traumatic apoptosis in the spinal cord.
NF200 is a neuronal cytoskeletal protein that has been shown
to degrade locally in the spinal cord following injury. Its loss
reflects loss of neuronal soma or axons (Schumacher et al.,
2000). Degradation of NF200 seen in wildtype animals within
24 h of the injury was largely attenuated in the lpr mice (Fig. 5A).
MAP2 is a neuronal protein largely restricted to the neuronal
soma. On Western blotting, MAP2 signal was decreased in
wildtype mice when compared to mutant at 7 days (t test: P =
0.70 at day 1, P = 0.049 at day 7) (Fig. 5B). These data suggest
that FAS deficiency influences preservation of neuronal
elements early after injury in the spinal cord. However, counts
of NeuN-positive neurons done at 7 days (t test: P = 0.442)
(Fig. 5C) post-injury did not reveal any difference between lpr
and wildtype (similarly no difference in neuron counts was seen
at 6 weeks; ANOVA P = 0.989, data not shown). This suggests
that while FAS deficiency influences neuronal structural
proteins this does not impact neuronal survival at the injury site.
Previous work by our group and others emphasized the
importance of oligodendrocyte apoptosis after SCI (Casha and
Fehlings, 2001). We therefore examined CNPase preservation
after SCI. We observed increased CNPase signal on Western
blotting (Fig. 5D) and increased numbers of CNPase-positive
cells (t test: P = 0.027) (Fig. 5E) 7 days following SCI in lpr

Fig. 4. Reduced post-traumatic apoptosis in the spinal cord of FAS-deficient mice after in vivo SCI. Counts of TUNEL-labeled nuclei revealed no difference between
FAS mutant and wildtype animals at 24 h but were significantly decreased (*) in FAS-deficient animals at 72 h (C). TUNEL-positive nuclei were not uniformly
distributed throughout the cross-section of the spinal cord but were more numerous in the vicinity of the injury center ((A, B) TUNEL in dorsolateral white matter of
wildtype and mutant respectively at 72 h, scale bar: 50 Am). Western blotting for activated caspase 3 did not reveal a significant difference in uninjured animals or at
days 1 and 2 post-injury, however, at day 7, a significant increase (*) in caspase 3 activation was seen in wildtype animals (D). A representative Western blot for
caspase 3 in wildtype and mutant spinal cord at day 7 is illustrated in (E).
S. Casha et al. / Experimental Neurology 196 (2005) 390–400 395
mice. Oligodendrocyte counts in sham-injured wildtype mice
were not significantly different than those seen in lpr mice (t test:
P = 0.196).
In vivo behavioral and neuroanatomical comparisons of SCI in
FAS-deficient and wildtype mice
We compared spinal-cord-injured lpr mice to wildtype
background-matched mice (n = 6/group) for 6 weeks after
injury using the modified BBB locomotor behavioral score for
mice at weekly intervals (Basso et al., 1995; Joshi and Fehlings,
2002a). There were no animal mortalities in either group, and no
morbidities other than the neurological deficit caused by SCI
were seen. The behavioral recovery observed over time in the lpr
mice was significantly better than that seen in wildtype animals
(2-way ANOVA: P = 0.001 for FAS genome, P < 0.001 for time
post-injury) (Fig. 6). The mean BBB scores reached a plateau at
8 in the wildtype group (representing hindlimb lateral sweeping
or plantar placement of the hindlimbs with no weight support)
and 10 in the lpr animals (representing weight-supported plantar
steps seen in up to 50% of steps but lacking coordination with
forelimb movements).
Six weeks after injury, we re-anesthetized the animals and
administered the retrograde tracer Fluorogold caudal to the
injury. After 5 days of retrograde uptake, we counted labeled
neurons of the vestibular, raphe, red and reticular nuclei in
cross-sections through the brainstem. These bulbospinal
projections contribute significantly to recovery of hindlimb
locomotor function in mice after SCI (Joshi and Fehlings,
2002b). We found a significant increase in the number of
brainstem neurons retrogradely labeled by Fluorogold, indicat-
ing a greater preservation of descending axons across the injury
site in lpr animals (t tests: red nucleus P = 0.0006, reticular
nuclei P = 0.001, raphe nuclei P = 0.044, vestibular nuclei P =
0.014) (Fig. 7). Retrograde tracing in non-injured control
animals revealed no background difference between wildtype
and lpr mice. T5/6 transection control animals did not exhibit
any retrogradely labeled neurons in the brainstem.
At the time of perfusion fixation for Fluorogold, we removed
a 6 mm spinal cord segment centered at the injury and stained
serial paraffin cross-sections with the myelin-selective pigment
Luxol Fast Blue. We compared the area of staining within the
spinal cord images. This experiment revealed a significant
improvement in white matter myelin preservation at the injury
site in lpr animals (2-way ANOVA: P = 0.02 for FAS genome,
P < 0.001 for distance from injury epicenter) (Fig. 8). Uninjured
T5/6 spinal cord, wildtype and mutant sections showed no
difference in Luxol Fast Blue staining.
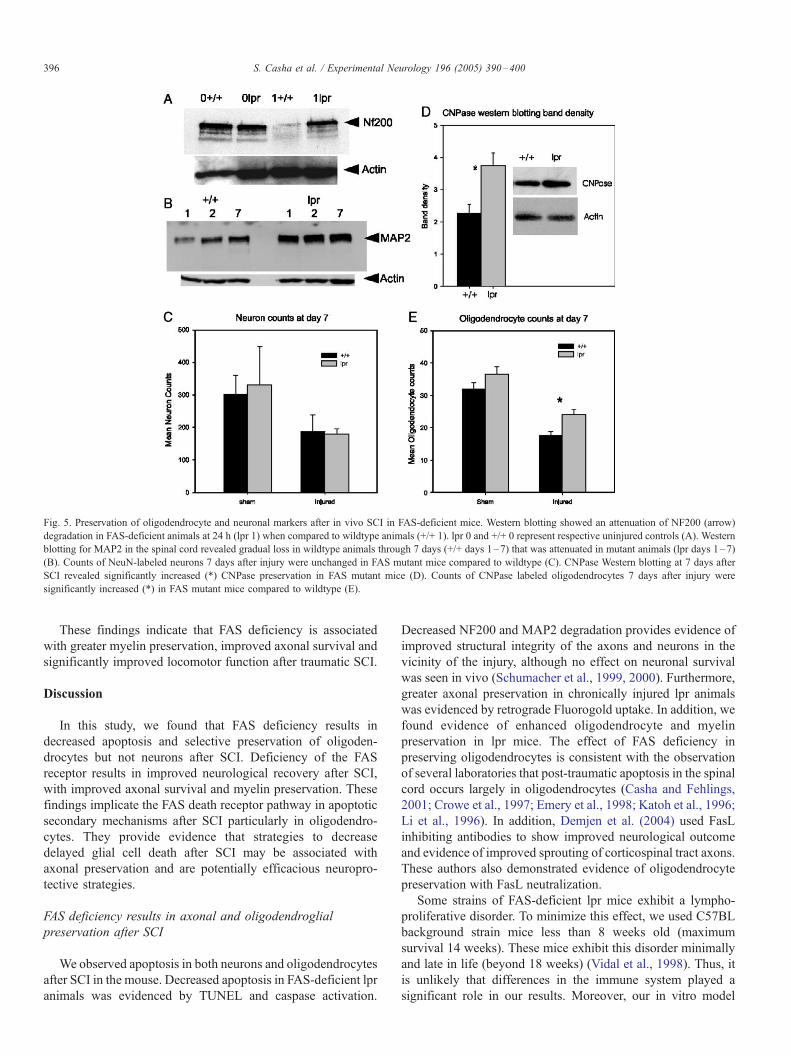
Fig. 5. Preservation of oligodendrocyte and neuronal markers after in vivo SCI in FAS-deficient mice. Western blotting showed an attenuation of NF200 (arrow)
degradation in FAS-deficient animals at 24 h (lpr 1) when compared to wildtype animals (+/+ 1). lpr 0 and +/+ 0 represent respective uninjured controls (A). Western
blotting for MAP2 in the spinal cord revealed gradual loss in wildtype animals through 7 days (+/+ days 1–7) that was attenuated in mutant animals (lpr days 1–7)
(B). Counts of NeuN-labeled neurons 7 days after injury were unchanged in FAS mutant mice compared to wildtype (C). CNPase Western blotting at 7 days after
SCI revealed significantly increased (*) CNPase preservation in FAS mutant mice (D). Counts of CNPase labeled oligodendrocytes 7 days after injury were
significantly increased (*) in FAS mutant mice compared to wildtype (E).
S. Casha et al. / Experimental Neurology 196 (2005) 390–400396
These findings indicate that FAS deficiency is associated
with greater myelin preservation, improved axonal survival and
significantly improved locomotor function after traumatic SCI.
Discussion
In this study, we found that FAS deficiency results in
decreased apoptosis and selective preservation of oligoden-
drocytes but not neurons after SCI. Deficiency of the FAS
receptor results in improved neurological recovery after SCI,
with improved axonal survival and myelin preservation. These
findings implicate the FAS death receptor pathway in apoptotic
secondary mechanisms after SCI particularly in oligodendro-
cytes. They provide evidence that strategies to decrease
delayed glial cell death after SCI may be associated with
axonal preservation and are potentially efficacious neuropro-
tective strategies.
FAS deficiency results in axonal and oligodendroglial
preservation after SCI
We observed apoptosis in both neurons and oligodendrocytes
after SCI in the mouse. Decreased apoptosis in FAS-deficient lpr
animals was evidenced by TUNEL and caspase activation.
Decreased NF200 and MAP2 degradation provides evidence of
improved structural integrity of the axons and neurons in the
vicinity of the injury, although no effect on neuronal survival
was seen in vivo (Schumacher et al., 1999, 2000). Furthermore,
greater axonal preservation in chronically injured lpr animals
was evidenced by retrograde Fluorogold uptake. In addition, we
found evidence of enhanced oligodendrocyte and myelin
preservation in lpr mice. The effect of FAS deficiency in
preserving oligodendrocytes is consistent with the observation
of several laboratories that post-traumatic apoptosis in the spinal
cord occurs largely in oligodendrocytes (Casha and Fehlings,
2001; Crowe et al., 1997; Emery et al., 1998; Katoh et al., 1996;
Li et al., 1996). In addition, Demjen et al. (2004) used FasL
inhibiting antibodies to show improved neurological outcome
and evidence of improved sprouting of corticospinal tract axons.
These authors also demonstrated evidence of oligodendrocyte
preservation with FasL neutralization.
Some strains of FAS-deficient lpr mice exhibit a lympho-
proliferative disorder. To minimize this effect, we used C57BL
background strain mice less than 8 weeks old (maximum
survival 14 weeks). These mice exhibit this disorder minimally
and late in life (beyond 18 weeks) (Vidal et al., 1998). Thus, it
is unlikely that differences in the immune system played a
significant role in our results. Moreover, our in vitro model

Fig. 7. Improved axonal preservation after SCI in FAS-deficient mice. Six
weeks after injury, a Fluorogold impregnated gelfoam pledget was applied to
the rostral surface of a T10 transection site (A1). Five days later, retrograde-
labeled neurons in the brainstem (A3) (thus having intact axons across the
injury site (A2)) were counted. Mean count T standard error of the mean is
presented (D). Differences were statistically significant at each nucleus counted
(*). Representative Fluorogold fluorograms of the red nucleus from FAS-
deficient and wildtype animals respectively are shown in panels B and C (scale
bar, 100 Am). Inset in panel A is a longitudinal section through the Fluorogold
introduction site at day 5 (scale 1 mm) demonstrating that the area of passive
diffusion of the Fluorogold extended well short of the injury site (approxi-
mately 5 mm rostral).
Fig. 6. Improved behavioral outcome after SCI in FAS-deficient mice.
Behavioral recovery following SCI determined using the BBB locomotor
score was significantly better in the lpr animals compared to wildtype (graph of
mean score T standard error of the mean vs. time post-injury). Selected
functional definitions of the BBB score relevant to the graph are presented.
S. Casha et al. / Experimental Neurology 196 (2005) 390–400 397
excluded the systemic inflammatory response suggesting that
the differences observed in mutant animals were indeed due to
the effect of FAS in the spinal cord. However, we recognize
that our in vitro preparation includes cell types other than
neurons and glia and that with time differences in cell death
and proliferation may alter the cellular composition of the slice
from that seen in vivo. Thus, while useful in confirming in vivo
findings, this model is not intended to provide strong
independent mechanistic evidence.
FAS deficiency results in improved behavioral and
neuroanatomical outcome after SCI
In our chronic in vivo study comparing lpr animals to
wildtype background-matched mice, we found behavioral
improvement, improvement in the number of intact axons
across the injury site and a decrease in white matter loss.
The beneficial outcome seen suggests that the FAS death
receptor cascade is a potentially useful target in the
treatment of SCI and that amelioration of delayed cell death
may afford significant neurological improvement. In addi-
tion, as most apoptotic cell loss occurs within 2 weeks of
injury, the therapeutic window for intervention in this
condition is longer than previous studies have explored
(Casha and Fehlings, 2001).
Yoshino et al. also compared SCI in FAS-deficient lpr
mice and wildtype controls (Yoshino et al., 2004). Similar to
our study, they demonstrated fewer TUNEL-positive cells,
improved behavioral outcome and a decrease in the
histological evidence of injury in mutant mice. However,
in contrast to our study, they observed a peak in the number
of TUNEL-positive cells at day 1 and a resolution of the
difference between mutant and wildtype by day 4. Further-
more, they identified these cells to be largely neurons. Our
data indicated little difference in neuron counts and a
predominant effect of FAS mutation in oligodendrocyte
preservation. In addition, we observed increased caspase

Fig. 8. Improved myelin preservation after SCI in FAS-deficient mice. Panels A and B are representative micrographs at the injury epicenter from a FAS-deficient
and wildtype animal respectively stained with Luxol Fast Blue (scale 50 Am). The mean area of myelin staining at the injury site was significantly greater (*) in FAS-
deficient animals -o- than in their wildtype counterparts -.- (C).
S. Casha et al. / Experimental Neurology 196 (2005) 390–400398
activation through day 7. The reason for the differences
between these two studies is unclear however may be due to
difference in the background mouse strain used or differ-
ences in the injury model. While early neuronal apoptosis
has been observed previously (Liu et al., 1997), apoptosis
predominantly affects oligodendrocytes after SCI (Casha and
Fehlings, 2001; Emery et al., 1998; Li et al., 1996; Liu et
al., 1997). Our data indicate that FAS inhibition may be
effective in decreasing this secondary injury mechanism and
improving neurological outcome.
The FAS death receptor pathway presents four sites in
particular which could serve as therapeutic targets for
pharmacotherapy development. These are FasL, the FAS
receptor, the initiator caspase 8 and the effector caspase 3.
Inhibition of the FasL and FAS receptor interaction has been
accomplished using neutralizing antibodies to these molecules
or by administering soluble FAS receptor (Demjen et al.,
2004; Schmidt et al., 2001; Silvestris et al., 2000). Inhibition
of the caspases has been accomplished through the use of
competitive and non-competitive inhibitors based on their
tetrapeptide cleavage specificity in animal models of brain
injury and ischemia (Clark et al., 2000; Gillardon et al., 1999;
Hara et al., 1997; Morita-Fujimura et al., 1999; Yakovlev et
al., 1997). Inhibition of signal transduction by manipulation
of molecules such as FADD and FLICE (caspase 8) inhibitory
protein (FLIP) has not yet been explored. Given what we
know from the FAS-deficient mouse, in vivo approaches to
inhibit the FAS pathway can be expected to have a profound
effect on the immune system possibly resulting in a
lymphoproliferative disorder and autoimmunity. This may
limit application of such strategies.
Other programmed cell death mechanisms are implicated
after SCI. These include the tumor necrosis factor death
receptor (TNFR), several caspases, several Bcl2 family
proteins, p53, cyclin D1, Cdk4 and the map kinase pathway
(Citron et al., 2000; Emery et al., 1998; Keane et al., 2001; Kim
et al., 2001; Lee et al., 2000; Li et al., 2000b; Nakahara et al.,
1999; Saito et al., 2000; Sakurai et al., 2000; Springer et al.,
1999). In addition, glutamate excitotoxicity, the inflammatory
response, free radicals and calpain activation contribute to post-
traumatic apoptosis in the spinal cord (Ghirnikar et al., 2001;
Ray et al., 2000a,b; Satake et al., 2000; Wada et al., 1999). Our
data suggest that these pathways also represent potential targets
in the treatment of SCI.
Acknowledgments
This work was funded by: The Canadian Institutes for
Health research, The Cervical Spine Research Society, The
Ontario Neurotrauma Foundation, The Christopher Reeve
Paralysis Foundation, Physicians Services Incorporated
Foundation (Ontario), Krembil Chair in Neural Repair and
Regeneration. The authors wish to thank Yang Liu and Amy
Lem for excellent technical assistance.

S. Casha et al. / Experimental Neurology 196 (2005) 390–400 399
References
Abe, Y., Yamamoto, T., Sugiyama, Y., Watanabe, T., Saito, N., Kayama, H.,
Kumagai, T., 1999. Apoptotic cells associated with Wallerian degeneration
after experimental spinal cord injury: a possible mechanism of oligoden-
droglial death. J. Neurotrauma 16, 945–952.
Basso, D.M., Beattie, M.S., Bresnahan, J.C., 1995. A sensitive and reliable
locomotor rating scale for open field testing in rats. J. Neurotrauma 12,
1–21.
Bjartmar, C., Yin, X., Trapp, B.D., 1999. Axonal pathology in myelin
disorders. J. Neurocytol. 28, 383–395.
Casha, S., Yu, W.R., Fehlings, M.G., 2001. Oligodendroglial apoptosis occurs
along degenerating axons and is associated with FAS and P75 expression
following spinal cord injury. Neuroscience 103, 203–218.
Citron, B.A., Arnold, P.M., Sebastian, C., Qin, F., Malladi, S., Ameenuddin, S.,
Landis, M.E., Festoff, B.W., 2000. Rapid upregulation of caspase-3 in rat
spinal cord after injury: mRNA, protein, and cellular localization correlates
with apoptotic cell death. Exp. Neurol. 166, 213–226.
Clark, R.S., Kochanek, P.M., Watkins, S.C., Chen, M., Dixon, C.E., Seidberg,
N.A., Melick, J., Loeffert, J.E., Nathaniel, P.D., Jin, K.L., Graham, S.H.,
2000. Caspase-3 mediated neuronal death after traumatic brain injury in
rats. J. Neurochem. 74, 740–753.
Crowe, M.J., Bresnahan, J.C., Shuman, S.L., Masters, J.N., Beattie, M.S., 1997.
Apoptosis and delayed degeneration after spinal cord injury in rats and
monkeys. Nat. Med. 3, 73–76.
Demjen, D., Klussmann, S., Kleber, S., Zuliani, C., Stieltjes, B., Metzger, C.,
Hirt, U.A., Walczak, H., Falk, W., Essig, M., Edler, L., Krammer, P.H.,
Martin-Villalba, A., 2004. Neutralization of CD95 ligand promotes
regeneration and functional recovery after spinal cord injury. Nat. Med. 7, 7.
Emery, E., Aldana, P., Bunge, M.B., Puckett, W., Srinivasan, A., Keane, R.W.,
Bethea, J., Levi, A.D., 1998. Apoptosis after traumatic human spinal cord
injury. J. Neurosurg. 89, 911–920.
Ghirnikar, R.S., Lee, Y.L., Eng, L.F., 2001. Chemokine antagonist infusion
promotes axonal sparing after spinal cord contusion injury in rat. Neurosci.
Res. 64, 582–589 (Jun 15).
Gillardon, F., Kiprianova, I., Sandkuhler, J., Hossmann, K.A., Spranger, M.,
1999. Inhibition of caspases prevents cell death of hippocampal CA1
neurons, but not impairment of hippocampal long-term potentiation
following global ischemia. Neuroscience 93, 1219–1222.
Hara, H., Friedlander, R.M., Gagliardini, V., Ayata, C., Fink, K., Huang, Z.,
Shimizu-Sasamata, M., Yuan, J., Moskowitz, M.A., 1997. Inhibition of
interleukin 1beta converting enzyme family proteases reduces ischemic
and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. U. S. A. 94,
2007–2012.
Joshi, M., Fehlings, M.G., 2002a. Development and characterization of a novel,
graded model of clip compressive spinal cord injury in the mouse: Part 1.
Clip design, behavioral outcomes, and histopathology. J. Neurotrauma 19,
175–190.
Joshi, M., Fehlings, M.G., 2002b. Development and characterization of a novel,
graded model of clip compressive spinal cord injury in the mouse: Part 2.
Quantitative neuroanatomical assessment and analysis of the relation-
ships between axonal tracts, residual tissue, and locomotor recovery.
J. Neurotrauma 19, 191–203.
Katoh, K., Ikata, T., Katoh, S., Hamada, Y., Nakauchi, K., Sano, T., Niwa, M.,
1996. Induction and its spread of apoptosis in rat spinal cord after
mechanical trauma. Neurosci. Lett. 216, 9–12.
Keane, R.W., Kraydieh, S., Lotocki, G., Bethea, J.R., Krajewski, S., Reed, J.C.,
Dietrich, W.D., 2001. Apoptotic and anti-apoptotic mechanisms following
spinal cord injury. J. Neuropathol. Exp. Neurol. 60, 422–429.
Kim, G.M., Xu, J., Xu, J., Song, S.K., Yan, P., Ku, G., Xu, X.M., Hsu, C.Y.,
2001. Tumor necrosis factor receptor deletion reduces nuclear factor-
kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and
functional recovery after traumatic spinal cord injury. J. Neurosci. 21,
6617–6625.
Krassioukov, A.V., Ackery, A., Schwartz, G., Adamchik, Y., Liu, Y., Fehlings,
M.G., 2002. An in vitro model of neurotrauma in organotypic spinal cord
cultures from adult mice. Brain Res. Brain Res. Protoc. 10, 60–68.
Lee, Y.B., Yune, T.Y., Baik, S.Y., Shin, Y.H., Du, S., Rhim, H., Lee, E.B., Kim,
Y.C., Shin, M.L., Markelonis, G.J., Oh, T.H., 2000. Role of tumor necrosis
factor-alpha in neuronal and glial apoptosis after spinal cord injury. Exp.
Neurol. 166, 190–195.
Li, G.L., Brodin, G., Farooque, M., Funa, K., Holtz, A., Wang, W.L., Olsson,
Y., 1996. Apoptosis and expression of Bcl-2 after compression trauma to rat
spinal cord. J. Neuropathol. Exp. Neurol. 55, 280–289.
Li, G.L., Farooque, M., Holtz, A., Olsson, Y., 1999. Apoptosis of
oligodendrocytes occurs for long distances away from the primary
injury after compression trauma to rat spinal cord. Acta Neuropathol.
(Berl) 98, 473–480.
Li, G.L., Farooque, M., Olsson, Y., 2000a. Changes of Fas and Fas ligand
immunoreactivity after compression trauma to rat spinal cord. Acta
Neuropathol. (Berl) 100, 75–81.
Li, X., Oudega, M., Dancausse, H.A., Levi, A.D., 2000b. The effect of
methylprednisolone on caspase-3 activation after rat spinal cord transection.
Restor. Neurol. Neurosci. 17, 203–209.
Liu, X.Z., Xu, X.M., Hu, R., Du, C., Zhang, S.X., McDonald, J.W., Dong,
H.X., Wu, Y.J., Fan, G.S., Jacquin, M.F., Hsu, C.Y., Choi, D.W., 1997.
Neuronal and glial apoptosis after traumatic spinal cord injury. J. Neurosci.
17, 5395–5406.
Lou, J., Lenke, L.G., Ludwig, F.J., O’Brien, M.F., 1998. Apoptosis as a
mechanism of neuronal cell death following acute experimental spinal cord
injury. Spinal Cord 36, 683–690.
Matsushita, K., Wu, Y., Qiu, J., Lang-Lazdunski, L., Hirt, L., Waeber, C.,
Hyman, B.T., Yuan, J., Moskowitz, M.A., 2000. Fas receptor and neuronal
cell death after spinal cord ischemia. J. Neurosci. 20, 6879–6887.
Morita-Fujimura, Y., Fujimura, M., Kawase, M., Murakami, K., Kim, G.W.,
Chan, P.H., 1999. Inhibition of interleukin-1beta converting enzyme family
proteases (caspases) reduces cold injury-induced brain trauma and DNA
fragmentation in mice. J. Cereb. Blood Flow Metab. 19, 634–642.
Nakahara, S., Yone, K., Sakou, T., Wada, S., Nagamine, T., Niiyama, T., Ichijo,
H., 1999a. Induction of apoptosis signal regulating kinase 1 (ASK1) after
spinal cord injury in rats: possible involvement of ASK1-JNK and -p38
pathways in neuronal apoptosis. J. Neuropathol. Exp. Neurol. 58, 442–450.
Nakatsuru, S., Terada, M., Nishihara, M., Kamogawa, J., Miyazaki, T., Qu,
W.M., Morimoto, K., Yazawa, C., Ogasawara, H., Abe, Y., Fukui, K.,
Ichien, G., Ito, M.R., Mori, S., Nakamura, Y., Nose, M., 1999b. Genetic
dissection of the complex pathological manifestations of collagen disease in
MRL/lpr mice. Pathol. Int. 49, 974–982.
Ray, S.K., Matzelle, D.C., Wilford, G.G., Hogan, E.L., Banik, N.L., 2000a. E-
64-d prevents both calpain upregulation and apoptosis in the lesion and
penumbra following spinal cord injury in rats. Brain Res. 867, 80–89.
Ray, S.K., Matzelle, D.D., Wilford, G.G., Hogan, E.L., Banik, N.L., 2000b.
Increased calpain expression is associated with apoptosis in rat spinal cord
injury: calpain inhibitor provides neuroprotection. Neurochem. Res. 25,
1191–1198.
Saito, N., Yamamoto, T., Watanabe, T., Abe, Y., Kumagai, T., 2000.
Implications of p53 protein expression in experimental spinal cord injury.
J. Neurotrauma 17, 173–182.
Sakurai, M., Hayashi, T., Abe, K., Sadahiro, M., Tabayashi, K., 1998. Delayed
selective motor neuron death and fas antigen induction after spinal cord
ischemia in rabbits. Brain Res. 797, 23–28.
Sakurai, M., Hayashi, T., Abe, K., Itoyama, Y., Tabayashi, K., Rosenblum,
W.I., 2000. Cyclin D1 and Cdk4 protein induction in motor neurons after
transient spinal cord ischemia in rabbits. Stroke 31, 200–207 (Jan.)
Satake, K., Matsuyama, Y., Kamiya, M., Kawakami, H., Iwata, H., Adachi,
K., Kiuchi, K., 2000. Nitric oxide via macrophage iNOS induces
apoptosis following traumatic spinal cord injury. Brain Res. Mol. Brain
Res. 85, 114–122.
Schmidt, M., Lugering, N., Lugering, A., Pauels, H.G., Schulze-Osthoff,
K., Domschke, W., Kucharzik, T., 2001. Role of the CD95/CD95 ligand
system in glucocorticoid-induced monocyte apoptosis. J. Immunol. 166,
1344–1351.
Schumacher, P.A., Eubanks, J.H., Fehlings, M.G., 1999. Increased calpain
I-mediated proteolysis, and preferential loss of dephosphorylated NF200,
following traumatic spinal cord injury. Neuroscience 91, 733–744.
Schumacher, P.A., Siman, R.G., Fehlings, M.G., 2000. Pretreatment with
calpain inhibitor CEP-4143 inhibits calpain I activation and cytoskeletal

S. Casha et al. / Experimental Neurology 196 (2005) 390–400400
degradation, improves neurological function, and enhances axonal survival
after traumatic spinal cord injury. J. Neurochem. 74, 1646–1655.
Shuman, S.L., Bresnahan, J.C., Beattie, M.S., 1997. Apoptosis of microglia and
oligodendrocytes after spinal cord contusion in rats. J. Neurosci. Res. 50,
798–808.
Siegel, R.M., Chan, F.K., Chun, H.J., Lenardo, M.J., 2000. The multifaceted
role of Fas signaling in immune cell homeostasis and autoimmunity. Nat.
Immunol. 1, 469–474.
Silvestris, F., Cafforio, P., Tucci, M., Del Prete, A., Dammacco, F., 2000.
VEINCTR-N, an immunogenic epitope of Fas (CD95/Apo-I), and soluble
Fas enhance T-cell apoptosis in vitro. II. Functional analysis and possible
implications in HIV-1 disease. Mol. Med. 6, 509–526.
Springer, J.E., Azbill, R.D., Knapp, P.E., 1999. Activation of the caspase-3
apoptotic cascade in traumatic spinal cord injury. Nat. Med. 5, 943–946.
Ugolini, G., Raoul, C., Ferri, A., Haenggeli, C., Yamamoto, Y., Salaun, D.,
Henderson, C.E., Kato, A.C., Pettmann, B., Hueber, A.O., 2003. Fas/tumor
necrosis factor receptor death signaling is required for axotomy-induced
death of motoneurons in vivo. J. Neurosci. 23, 8526–8531.
Vidal, S., Kono, D.H., Theofilopoulos, A.N., 1998. Loci predisposing to
autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. J. Clin. Invest.
101, 696–702.
Wada, S., Yone, K., Ishidou, Y., Nagamine, T., Nakahara, S., Niiyama, T.,
Sakou, T., 1999. Apoptosis following spinal cord injury in rats and
preventative effect of N-methyl-d-aspartate receptor antagonist. J. Neuro-
surg. 91, 98–104.
Warden, P., Bamber, N.I., Li, H., Esposito, A., Ahmad, K.A., Hsu, C.Y., Xu,
X.M., 2001. Delayed glial cell death following wallerian degeneration in
white matter tracts after spinal cord dorsal column cordotomy in adult rats.
Exp. Neurol. 168, 213–224.
Yakovlev, A.G., Knoblach, S.M., Fan, L., Fox, G.B., Goodnight, R., Faden,
A.I., 1997. Activation of CPP32-like caspases contributes to neuronal
apoptosis and neurological dysfunction after traumatic brain injury.
J. Neurosci. 17, 7415–7424.
Yin, X., Crawford, T.O., Griffin, J.W., Tu, P., Lee, V.M., Li, C., Roder, J.,
Trapp, B.D., 1998. Myelin-associated glycoprotein is a myelin signal that
modulates the caliber of myelinated axons. J. Neurosci. 18, 1953–1962.
Yong, C., Arnold, P.M., Zoubine, M.N., Citron, B.A., Watanabe, I., Berman,
N.E., Festoff, B.W., 1998. Apoptosis in cellular compartments of rat spinal
cord after severe contusion injury. J. Neurotrauma 15, 459–472.
Yoshino, O., Matsuno, H., Nakamura, H., Yudoh, K., Abe, Y., Sawai, T., Uzuki,
M., Yonehara, S., Kimura, T., 2004. The role of Fas-mediated apoptosis
after traumatic spinal cord injury. Spine 29, 1394–1404.
Zurita, M., Vaquero, J., Zurita, I., 2001. Presence and significance of CD-95
(Fas/APO1) expression after spinal cord injury. J. Neurosurg. 94, 257–264.




















