
Exploring the Structural and Electronic Properties of Pt/Ceria- Modified TiO 2 and Its...
9
Exploring the Structural and Electronic Properties of Pt/Ceria- Modified TiO 2 and Its Photocatalytic Activity for Water Splitting under Visible Light Shankhamala Kundu, † Jim Ciston, § Sanjaya D. Senanayake, † Dario A. Arena, ‡ Etsuko Fujita, † Dario Stacchiola, † Laura Barrio, ⊥ Rufino M. Navarro, ⊥ Jose L. G. Fierro, ⊥ and Jose ́ A. Rodriguez* , † † Chemistry Department, and ‡ National Synchrotron Light Source, Brookhaven National Laboratory, Upton, New York 11973, United States § National Center for Electron Microscopy, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States ⊥ Instituto de Cata ́ lisis y Petroleoquímica, CSIC, Cantoblanco, 28049 Madrid, Spain ABSTRACT: In the past few years there has been intensive research focused on doping TiO 2 with other metal oxides in order to synthesize a mixed metal oxide based semiconductor with modified band gap which can split water efficiently in visible light. In this study we modified titania−anatase powders through deposition of ceria nanoparticles on them. The formation of a mixed metal oxide at the CeO x /TiO 2 interface was observed using transmission electron microscopy and near-edge X-ray absorption spectroscopy. UV−vis spectra show that this mixed metal oxide can absorb photons in the visible region due to the presence of stabilized Ce 3+ in the mixed oxide phase. The addition of 0.5 wt % Pt imposes a significant chemical change on the ceria nanoparticles by substantially enhancing the concentration of Ce 3+ and makes possible photocatalytic activity by providing an electron trap. When irradiated with visible light, 0.5 wt % Pt loaded on CeO 2 -modified TiO 2 generates oxygen almost seven times more efficiently than a standard WO 3 catalyst. A correlation was found between the concentration of Ce 3+ in Pt/CeO x /TiO 2 and its photocatalytic activity. ■ INTRODUCTION The conversion of solar energy into hydrogen via the water- splitting process assisted by semiconductor photocatalysts is one of the more interesting ways of achieving clean and renewable energy systems. Many types of semiconductors, with over 130 materials including oxides, nitrides, sulfides, carbides, and phosphides, have been reported to act as efficient photocatalysts for water splitting. 1−3 Water splitting was first demonstrated using a TiO 2 photoelectrode with some external bias; 4 however, powdered TiO 2 photocatalyst does not efficiently split water under visible light. This is mainly due to the wide band gap of TiO 2 (3.2 eV) which allows us to use only a small fraction of the solar spectrum 4 (UV fraction, 3−4% of total solar spectrum). Intensive studies have been carried out to improve the visible light sensitivity of Ti oxide-based catalysts. Among the various approaches undertaken in the search for more efficient and active TiO 2 photocatalysts, the following can be mentioned: 1,5,6 (i) nanodesign to control the size, morphology, and defects of photocatalysts, (ii) modifying the band gap energy by substitutional doping (cations or anions), (iii) surface modification of TiO 2 by deposition of cocatalysts to reduce the activation energy for gas evolution and/or decrease the electron−hole recombination, (iv) developing multicomponent photocatalyst by forming solid solutions, and (v) sensitization. In principle, the mixing of two different metal oxides could improve the performance of the involved oxides by producing different physical and chemical properties with respect to the individual components. 7−9 Despite that bulk ceria and titania do not adopt similar crystal structures, 7,10−12 cerium ions are particularly interesting to mix with TiO 2 because the Ti 4+ ions can be replaced by cerium ions modifying the physicochemical properties (redox capability, thermal sintering, etc.) of TiO 2. 12−14 However, the characterization of the interfacial and surface structures of this type of system, i.e., CeO 2 /TiO 2 or TiO 2 /CeO 2 , remains as a challenge because of the complexity of the structures that can be generated. 8 Recently there have been studies focused on the properties of CeO 2 −TiO 2 powders with the aim of using this mixed metal oxide in catalysis and photocatalysis. 15−18 Lopez et al. studied the structure and morphology of the CeO 2 −TiO 2 mixed oxide (10 wt % CeO 2 −90 wt % TiO 2 ) calcined at different temperatures and found that the presence of cerium conferred an increased thermal stability to the TiO 2 materials against particle sintering and pore collapse. 15 Furthermore, it has been reported that ceria-modified TiO 2 shows activity for the Received: May 8, 2012 Revised: June 7, 2012 Published: June 12, 2012 Article pubs.acs.org/JPCC © 2012 American Chemical Society 14062 dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−14070
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Exploring the Structural and Electronic Properties of Pt/Ceria- Modified TiO 2 and Its...

Exploring the Structural and Electronic Properties of Pt/Ceria-Modified TiO2 and Its Photocatalytic Activity for Water Splittingunder Visible LightShankhamala Kundu, † Jim Ciston,§ Sanjaya D. Senanayake, † Dario A. Arena,‡ Etsuko Fujita, †
Dario Stacchiola, † Laura Barrio,⊥ Rufino M. Navarro,⊥ Jose L. G. Fierro,⊥ and Jose A. Rodriguez*, †
†Chemistry Department, and ‡National Synchrotron Light Source, Brookhaven National Laboratory, Upton, New York 11973,United States§National Center for Electron Microscopy, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States⊥Instituto de Catalisis y Petroleoquímica, CSIC, Cantoblanco, 28049 Madrid, Spain
ABSTRACT: In the past few years there has been intensiveresearch focused on doping TiO2 with other metal oxides inorder to synthesize a mixed metal oxide based semiconductorwith modified band gap which can split water efficiently invisible light. In this study we modified titania−anatase powdersthrough deposition of ceria nanoparticles on them. Theformation of a mixed metal oxide at the CeOx/TiO2 interfacewas observed using transmission electron microscopy andnear-edge X-ray absorption spectroscopy. UV−vis spectrashow that this mixed metal oxide can absorb photons in thevisible region due to the presence of stabilized Ce3+ in the mixed oxide phase. The addition of 0.5 wt % Pt imposes a significantchemical change on the ceria nanoparticles by substantially enhancing the concentration of Ce3+ and makes possiblephotocatalytic activity by providing an electron trap. When irradiated with visible light, 0.5 wt % Pt loaded on CeO2-modifiedTiO2 generates oxygen almost seven times more efficiently than a standard WO3 catalyst. A correlation was found between theconcentration of Ce3+ in Pt/CeOx/TiO2 and its photocatalytic activity.
■ INTRODUCTIONThe conversion of solar energy into hydrogen via the water-splitting process assisted by semiconductor photocatalysts isone of the more interesting ways of achieving clean andrenewable energy systems. Many types of semiconductors, withover 130 materials including oxides, nitrides, sulfides, carbides,and phosphides, have been reported to act as efficientphotocatalysts for water splitting.1−3 Water splitting was firstdemonstrated using a TiO2 photoelectrode with some externalbias;4 however, powdered TiO2 photocatalyst does notefficiently split water under visible light. This is mainly dueto the wide band gap of TiO2 (3.2 eV) which allows us to useonly a small fraction of the solar spectrum4 (UV fraction, 3−4%of total solar spectrum). Intensive studies have been carried outto improve the visible light sensitivity of Ti oxide-basedcatalysts. Among the various approaches undertaken in thesearch for more efficient and active TiO2 photocatalysts, thefollowing can be mentioned:1,5,6 (i) nanodesign to control thesize, morphology, and defects of photocatalysts, (ii) modifyingthe band gap energy by substitutional doping (cations oranions), (iii) surface modification of TiO2 by deposition ofcocatalysts to reduce the activation energy for gas evolutionand/or decrease the electron−hole recombination, (iv)developing multicomponent photocatalyst by forming solidsolutions, and (v) sensitization.
In principle, the mixing of two different metal oxides couldimprove the performance of the involved oxides by producingdifferent physical and chemical properties with respect to theindividual components.7−9 Despite that bulk ceria and titaniado not adopt similar crystal structures,7,10−12 cerium ions areparticularly interesting to mix with TiO2 because the Ti
4+ ionscan be replaced by cerium ions modifying the physicochemicalproperties (redox capability, thermal sintering, etc.) ofTiO2.
12−14 However, the characterization of the interfacialand surface structures of this type of system, i.e., CeO2/TiO2 orTiO2/CeO2, remains as a challenge because of the complexityof the structures that can be generated.8
Recently there have been studies focused on the properties ofCeO2−TiO2 powders with the aim of using this mixed metaloxide in catalysis and photocatalysis.15−18 Lopez et al. studiedthe structure and morphology of the CeO2−TiO2 mixed oxide(10 wt % CeO2−90 wt % TiO2) calcined at differenttemperatures and found that the presence of cerium conferredan increased thermal stability to the TiO2 materials againstparticle sintering and pore collapse.15 Furthermore, it has beenreported that ceria-modified TiO2 shows activity for the
Received: May 8, 2012Revised: June 7, 2012Published: June 12, 2012
Article
pubs.acs.org/JPCC
© 2012 American Chemical Society 14062 dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−14070

photochemical degradation of methylene blue under visible-light irradiation.16,17 The authors of this study speculate thatthe CeO2 species may be confined in the anatase nanocrystalframework. Such interaction between the frameworks of CeO2and TiO2 could be the key for the enhancement ofphotoactivity in the visible-light region. Theoretical calculationsdone by Catlow et al. suggest that the presence of reducedcerium cations is necessary in order to enhance the photo-catalytic activity of TiO2.
18 However, until now experimentalstudies have not fully explored the correlation between thephotocatalytic activities of CeO2-modified TiO2 with thepresence of Ce3+ and a possible mechanism behind it. RecentlyPrimo et al. reported that a small amount of gold supported oncerium nanoparticles generates oxygen from water moreefficiently than the standard WO3 under visible light irradiation,indicating that ceria can be an useful material in photocatlysis.19
Considering the importance in developing novel photocatalystswith visible-light activity,20 our objective here is to explore thepossibility of using photocatalysts prepared by depositingnanoparticles of CeO2 on TiO2, thus designing a semi-conductor with a modified band gap which can perform thechallenging goal of splitting water using visible light.As mentioned above, the third way to enhance the activity of
TiO2 photocatalyst is through the addition of small amounts ofnoble metals such as Pt and Rh/Ru.1,5,20 Such enhancement inactivity has been explained in terms of a photoelectrochemicalmechanism in which the electrons generated by light irradiationof the semiconductor transfer to the loaded metal particles,while the holes remain in the semiconductors, resulting in adecrease in the electron−hole recombination. Additionally, thepresence of noble metals on the metal oxide surfaces couldimpose chemical changes on the cerium and/or titaniananoparticles that could have influence on the optoelectronicproperties of the photocatalytic systems.With this background, in the present paper, we have
investigated the structural and electronic properties of ceriananoparticles modified on titania combining transmissionelectron microscopy (TEM) and different variations of near-edge X-ray absorption fine structure (NEXAFS). Our resultsdemonstrate that the interface between a CeO2 nanoparticleand the TiO2 support is partially disordered and contains Ce3+
cations. UV−vis measurements were conducted to study thephoton absorption capability of this mixed metal oxide in theUV and visible region. The presence of the CeOx nanoparticleson the TiO2 surface enhanced the absorption of light in thevisible range substantially, but it did not produce an efficientcatalyst for the splitting of water. When a very small amount ofPt was loaded on the ceria-modified titania support, complexinteractions led to an increase in the amount of Ce3+ present,and we obtained a system that generated oxygen 10 times moreefficiently than Pt/TiO2 during the photocatalytic splitting ofwater at λ ≥ 400 nm under our experimental conditions.
■ EXPERIMENTAL SECTIONTiO2 powders (titanium(IV) oxide, catalyst support Alfa Aesar)were stabilized by thermal treatment at 773 K for 4 h beforebeing used for cerium deposition.14,21 The Ce-modified TiO2samples (Ce−TiO2) were prepared by wet impregnation of thethermally stabilized TiO2 powders using an aqueous solution ofcerium nitrate (Alfa Aesar, Reacton 99.5%) with two differentloading of Ce, 6 and 15 wt %. The ceria loadings usedcorrespond to half and complete theoretical monolayercoverage, respectively. The theoretical monolayer coverage of
ceria for the TiO2 support was calculated taking into accountthe dispersion capacity of ceria on TiO2 (7.23 Ce4+ ions/nm2
TiO2) published in literature. After cerium impregnation, thepowders were calcined under air at 873 K for 8 h to obtain theCe-modified TiO2 photocatalysts. Below the two sampleprepared by this methodology are labeled as 6 wt % CeO2/TiO2 (in short 6CeTi) and 15 wt % CeO2/TiO2 (in short15CeTi). The supported-Pt photocatalysts (0.5 wt % Pt) wereprepared by impregnating each of the CeO2/TiO2 supportsunder stirring at 353 K for 2 h using aqueous solutions ofH2PtCl6 metal precursor (Johnson−Matthey, 40.78 wt % Pt)).After Pt loading, the samples were dried at 393 K for 12 h, andsubsequently they were calcined in air at 773 K to remove thechlorine.14 Pt loaded on two different ceria-modified TiO2supports are labeled as Pt6CeTi and Pt15CeTi for short.TEM experiments were performed on the spherical and
chromatic aberration-corrected TEAM I microscope at theNational Center for Electron Microscopy at Lawrence BerkeleyNational Laboratory (http://ncem.lbl.gov/TEAM-project/).Images were obtained in both scanning TEM (STEM) andconventional high-resolution TEM (HREM) geometries.Electron energy loss spectroscopy (EELS) was collected usinga Gatan Tridiem energy filter with a probe convergence of 30mrad, collection angle of 35 mrad, and 0.2 eV/pixel sampling.In order to minimize damage to the beam-sensitive TiO2 phase,an accelerating voltage of 80 kV was used, and STEM probecurrent was limited to 40 pA. TEM images were simulatedusing the MacTempas software package (totalresolution.com).XRD patterns for the CeO2/TiO2 and Pt/CeO2/TiO2
samples were collected at beamline X7B of the NationalSynchrotron Light Source (NSLS) as described in refs 14 and21. XANES spectra of Ce L3 absorption edge were collected atbeamline X19A of the National Synchrotron Light Source(NSLS) in the ‘‘fluorescence-yield mode’’ using a passivatedimplanted planar silicon (PIPS) detector cooled withcirculating water. The XANES data was then analyzed usingthe Athena program.22 The NEXAFS experiments with soft X-rays were performed at the end station of the U4B beamline atthe NSLS. The end station is equipped with a partial electronyield detector and a partial fluorescence yield detector whichallowed us to simultaneously measure fluorescence and electronyields. Partial electron yield intensities were measured with asingle channeltron with a front-end bias of −250 V to reducesignals from secondary electrons. All NEXAFS data reportedhere were collected with the photon beam fixed at 55° from thesurface normal of the sample. All NEXAFS scans werenormalized by the incident beam flux, monitored by an Aumesh located upstream of the sample. The NEXAFS spectrawere measured as a function of the incident X-ray photonenergy in the vicinity of the titanium L-edge (445−490 eV), theoxygen K-edge (520−590 eV), and the cerium M-edge (870−930 eV) regions. The energy resolution in the NEXAFSexperiments was about 0.2−0.4 eV depending on the edge.UV−vis spectroscopy measurements were performed in
diffuse reflection mode using UV−vis/NIR spectrophotometerPerkinElmer Lambda 950 located in the center of functionalnanomaterials at Brookhaven National Laboratory. The photo-catalytic experiments were carried out in a 30 mL closed quartzreactor. The head space of the reactor was connected to a gas-tight valve, allowing the measurement of the evolved gas. In thephotocatalytic reactions the photocatalyst powder (45 mg) wasdispersed in 20 mL of water containing 0.01 M of AgNO3. Inthe overall water splitting, water becomes reduced to hydrogen
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014063

and simultaneously oxidized to oxygen. Of these twosemireactions, hydrogen generation is considered to be thesimpler step compared to oxygen generation. In principle, toproduce hydrogen the protons present in the water acceptelectrons trapped by noble metals that act as hydrogenevolution centers. In contrast, formation of oxygen fromwater is conceptually more challenging since it involves severalintermediate steps, requiring transfer of four positive holes andthe formation of O−O bonds.19,23,24 For that reason in ourstudy we choose the AgNO3 as sacrificial reagent in order toprobe the oxygen evolution as a measure of efficiency of thephotocatalyst. The suspension was purged with an argon flowfor at least 60 min before irradiation in order to removedissolved air. Then it was irradiated for 4 h using a 150 Wxenon lamp with a visible light cutoff filter (λ > 400 nm). Thestationary temperature of the reactor was kept at 15 °C by acontinuous water circulation. The formation of oxygen wasconfirmed by injecting 0.5 mL of the reactor head space gas in agas chromatograph operating at isothermal conditions (40 °C)using argon as the carrier gas.
■ RESULTS AND DISCUSSIONStructure and Morphology of Ceria Nanoparticles on
TiO2. A. Structural Characterization. Table 1 summarizes thesamples studied along this work, their chemical composition asdetermined by TXRF analysis, and their BET surface area, aswell as the cell dimension of CeO2 fluorite and TiO2 anatasephases as determined by Rietveld refinement of XRD patterns.
Diffraction patterns of the CeO2/TiO2 samples weredominated by the expected lines for TiO2−anatase14,21 withvery small features for ceria in a fluorite structure. After addingPt to the CeO2/TiO2 systems, there were no extra diffractionslines. This is due to the low concentration of Pt in the samples,but it could also be a sign of the high dispersion of Pt species(size <1 nm), since no periodic structures are observed.B. Morphological Characterization. Figure 1a shows a
TEM image for a Pt/15 wt % CeO2/TiO2 system. The TiO2support nanoparticles are monocrystalline and present anaverage size of 10−15 nm. They exhibit low index [−1 1 2] and[3 3 1] facets according to an atomistic model. The bright spotsas indicated by the arrows represent CeO2 nanoparticles withan average diameter of about 4−5 nm. CeO2 particles aregenerally located at a TiO2 corner site, and they have atruncated pyramid shape as shown in the HRTEM image inFigure 1b. Pt nanoparticles are barely visible. A more magnifiedTEM image (not shown here) indicates that the Pt particles arequite small and monodispersed with a size of 0.53 ± 0.06 nm.The HRTEM image shows the orientation relationships
between the cerium islands and the TiO2 support particles tobe CeO2 fluorite (0 0 1) || TiO2 anatase (−1 1 2) and CeO2fluorite [1 1 0] || TiO2 anatase [3 1 1]. The orientation
relationships were confirmed with TEM image simulations.CeO2 nanoparticles formed a highly ordered cubic fluoritestructure of about 2−3 nm height above the interface of theCeO2 nanoparticle and TiO2 support. However, at the interfacewe can observe the presence of a more distorted structureindicating the intermixing of two metal oxides. Previously, usingcrystal simulation, Lopez et al. have suggested that ceriumaffected the crystallographic and sintering characteristics ofCeO2−TiO2 compounds due to the progressive segregation ofcerium on the surface of TiO2 globules as the temperature wasraised and by possible substitutions of Ti4+ by Ce4+ specieswithin the TiO2 anatase structure,10 thus supporting ourobservation of intermixing two metal oxides. However, whetherthe intermixing of the two oxides induces any change in theoxidation state of cerium has yet to be determined.Electron energy loss spectroscopy (EELS) was employed to
measure the Ce3+/Ce4+ ratio with high spatial resolution withinand around the ceria nanoparticles. The Ce valence state wasquantified using the second derivative method of Fortner etal.25 with Ce +3 reference data taken from Yang et al.26 Themost sensitive signals with a clear dependency on cationvalence are the Ti L-edges, which probe d-electron occupancyin Ti, and the Ce M-edges, which probe f-electron occupancy inCe. These edges were measured in a spatially resolved wayusing EELS line scans. The red line in the image inset of Figure2 shows the EELS line scan that was performed along with theSTEM imaging. Ti L2- and L3-edges were integrated to get theTi signal, and Ce M4- and M5-edges were used to obtain the Cesignal. The first two points of the Ce valence plot with respectto the probing depth, when there is only Ce signal present,show Ce4+ oxidation state. The first significant change in the
Table 1. Summary of Structural Characterization Results14,21
TXRF TiO2 phase CeO2 phase
sample % Ce % Pt BET (m2/g) a = b c
TiO2 − − 82 3.786 9.512 −6CeTi 6.6 − 100 3.786 9.513 −15CeTi 14.9 − 101 3.786 9.514 5.415Pt6CeTi 6.6 0.5 96 3.785 9.511 −Pt15CeTi 14.9 0.5 93 3.786 9.514 5.413
Figure 1. (a) HAADF STEM image of Pt/15 wt % CeO2/TiO2catalyst; (b) HRTEM image of a typical CeO2 nanoparticle depositedon TiO2.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014064
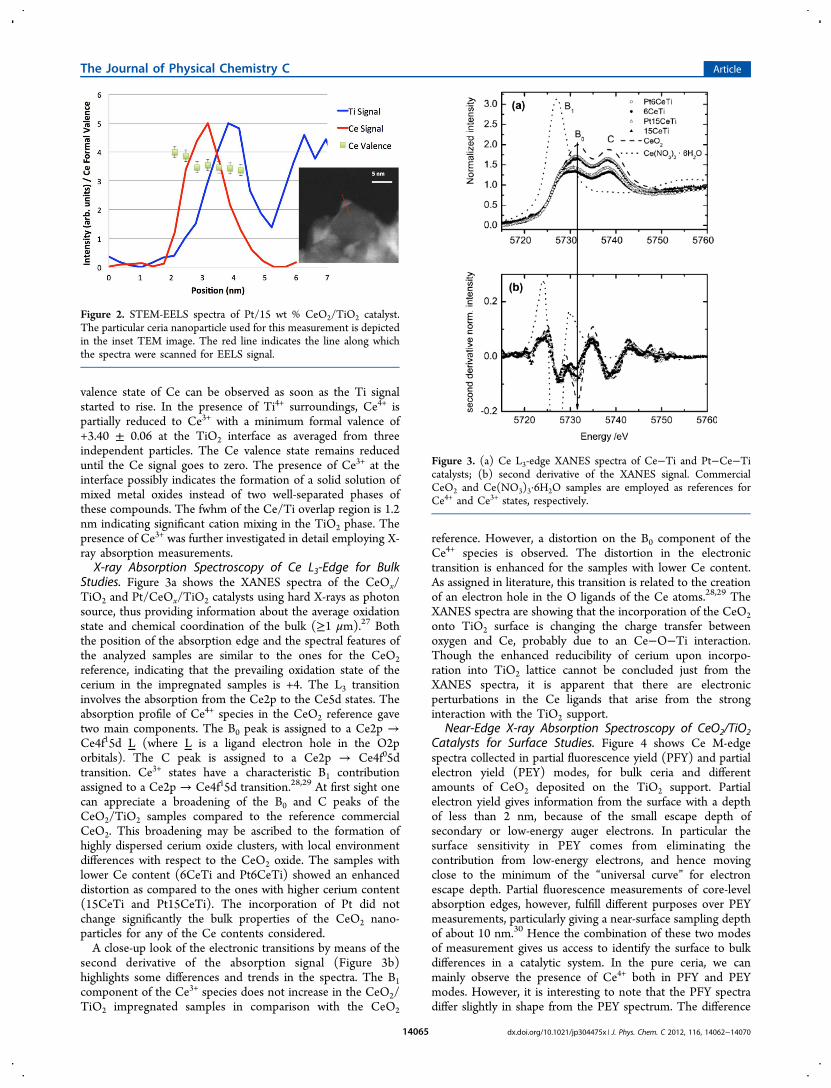
valence state of Ce can be observed as soon as the Ti signalstarted to rise. In the presence of Ti4+ surroundings, Ce4+ ispartially reduced to Ce3+ with a minimum formal valence of+3.40 ± 0.06 at the TiO2 interface as averaged from threeindependent particles. The Ce valence state remains reduceduntil the Ce signal goes to zero. The presence of Ce3+ at theinterface possibly indicates the formation of a solid solution ofmixed metal oxides instead of two well-separated phases ofthese compounds. The fwhm of the Ce/Ti overlap region is 1.2nm indicating significant cation mixing in the TiO2 phase. Thepresence of Ce3+ was further investigated in detail employing X-ray absorption measurements.X-ray Absorption Spectroscopy of Ce L3-Edge for Bulk
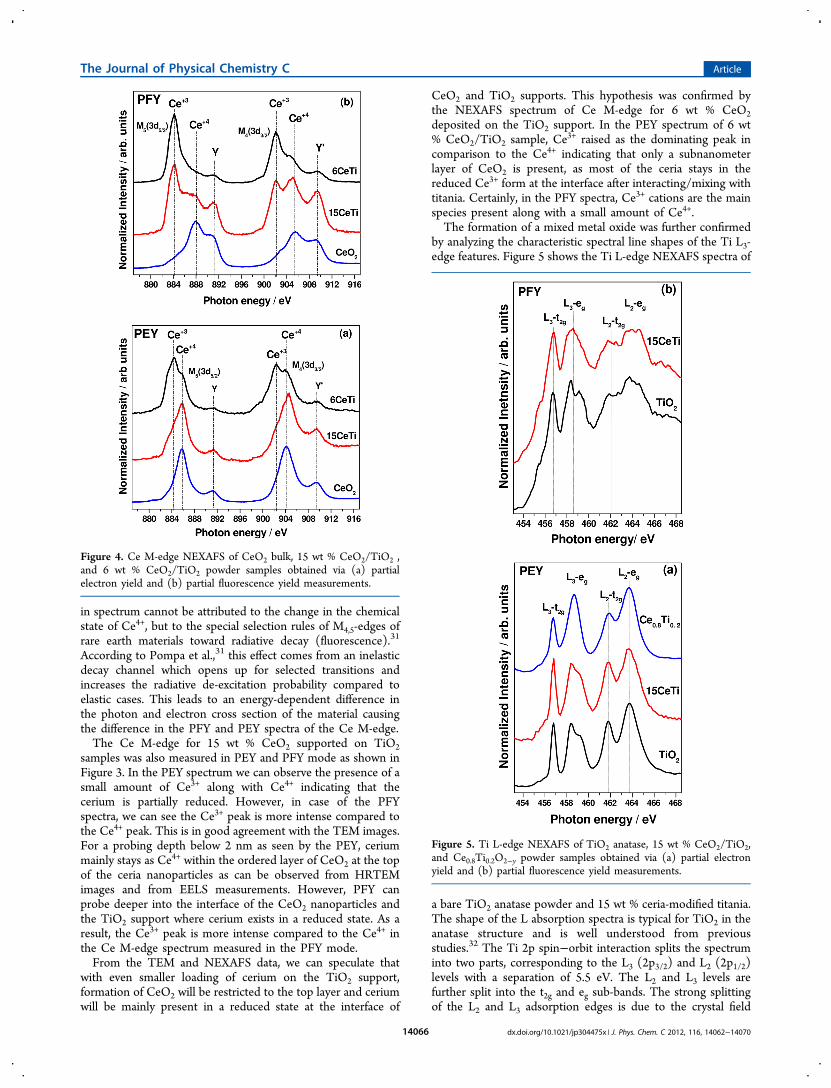
Studies. Figure 3a shows the XANES spectra of the CeOx/TiO2 and Pt/CeOx/TiO2 catalysts using hard X-rays as photonsource, thus providing information about the average oxidationstate and chemical coordination of the bulk (≥1 μm).27 Boththe position of the absorption edge and the spectral features ofthe analyzed samples are similar to the ones for the CeO2reference, indicating that the prevailing oxidation state of thecerium in the impregnated samples is +4. The L3 transitioninvolves the absorption from the Ce2p to the Ce5d states. Theabsorption profile of Ce4+ species in the CeO2 reference gavetwo main components. The B0 peak is assigned to a Ce2p →Ce4f15d L (where L is a ligand electron hole in the O2porbitals). The C peak is assigned to a Ce2p → Ce4f05dtransition. Ce3+ states have a characteristic B1 contributionassigned to a Ce2p → Ce4f15d transition.28,29 At first sight onecan appreciate a broadening of the B0 and C peaks of theCeO2/TiO2 samples compared to the reference commercialCeO2. This broadening may be ascribed to the formation ofhighly dispersed cerium oxide clusters, with local environmentdifferences with respect to the CeO2 oxide. The samples withlower Ce content (6CeTi and Pt6CeTi) showed an enhanceddistortion as compared to the ones with higher cerium content(15CeTi and Pt15CeTi). The incorporation of Pt did notchange significantly the bulk properties of the CeO2 nano-particles for any of the Ce contents considered.A close-up look of the electronic transitions by means of the
second derivative of the absorption signal (Figure 3b)highlights some differences and trends in the spectra. The B1component of the Ce3+ species does not increase in the CeO2/TiO2 impregnated samples in comparison with the CeO2
reference. However, a distortion on the B0 component of theCe4+ species is observed. The distortion in the electronictransition is enhanced for the samples with lower Ce content.As assigned in literature, this transition is related to the creationof an electron hole in the O ligands of the Ce atoms.28,29 TheXANES spectra are showing that the incorporation of the CeO2onto TiO2 surface is changing the charge transfer betweenoxygen and Ce, probably due to an Ce−O−Ti interaction.Though the enhanced reducibility of cerium upon incorpo-ration into TiO2 lattice cannot be concluded just from theXANES spectra, it is apparent that there are electronicperturbations in the Ce ligands that arise from the stronginteraction with the TiO2 support.
Near-Edge X-ray Absorption Spectroscopy of CeO2/TiO2Catalysts for Surface Studies. Figure 4 shows Ce M-edgespectra collected in partial fluorescence yield (PFY) and partialelectron yield (PEY) modes, for bulk ceria and differentamounts of CeO2 deposited on the TiO2 support. Partialelectron yield gives information from the surface with a depthof less than 2 nm, because of the small escape depth ofsecondary or low-energy auger electrons. In particular thesurface sensitivity in PEY comes from eliminating thecontribution from low-energy electrons, and hence movingclose to the minimum of the “universal curve” for electronescape depth. Partial fluorescence measurements of core-levelabsorption edges, however, fulfill different purposes over PEYmeasurements, particularly giving a near-surface sampling depthof about 10 nm.30 Hence the combination of these two modesof measurement gives us access to identify the surface to bulkdifferences in a catalytic system. In the pure ceria, we canmainly observe the presence of Ce4+ both in PFY and PEYmodes. However, it is interesting to note that the PFY spectradiffer slightly in shape from the PEY spectrum. The difference
Figure 2. STEM-EELS spectra of Pt/15 wt % CeO2/TiO2 catalyst.The particular ceria nanoparticle used for this measurement is depictedin the inset TEM image. The red line indicates the line along whichthe spectra were scanned for EELS signal.
Figure 3. (a) Ce L3-edge XANES spectra of Ce−Ti and Pt−Ce−Ticatalysts; (b) second derivative of the XANES signal. CommercialCeO2 and Ce(NO3)3·6H2O samples are employed as references forCe4+ and Ce3+ states, respectively.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014065
in spectrum cannot be attributed to the change in the chemicalstate of Ce4+, but to the special selection rules of M4,5-edges ofrare earth materials toward radiative decay (fluorescence).31
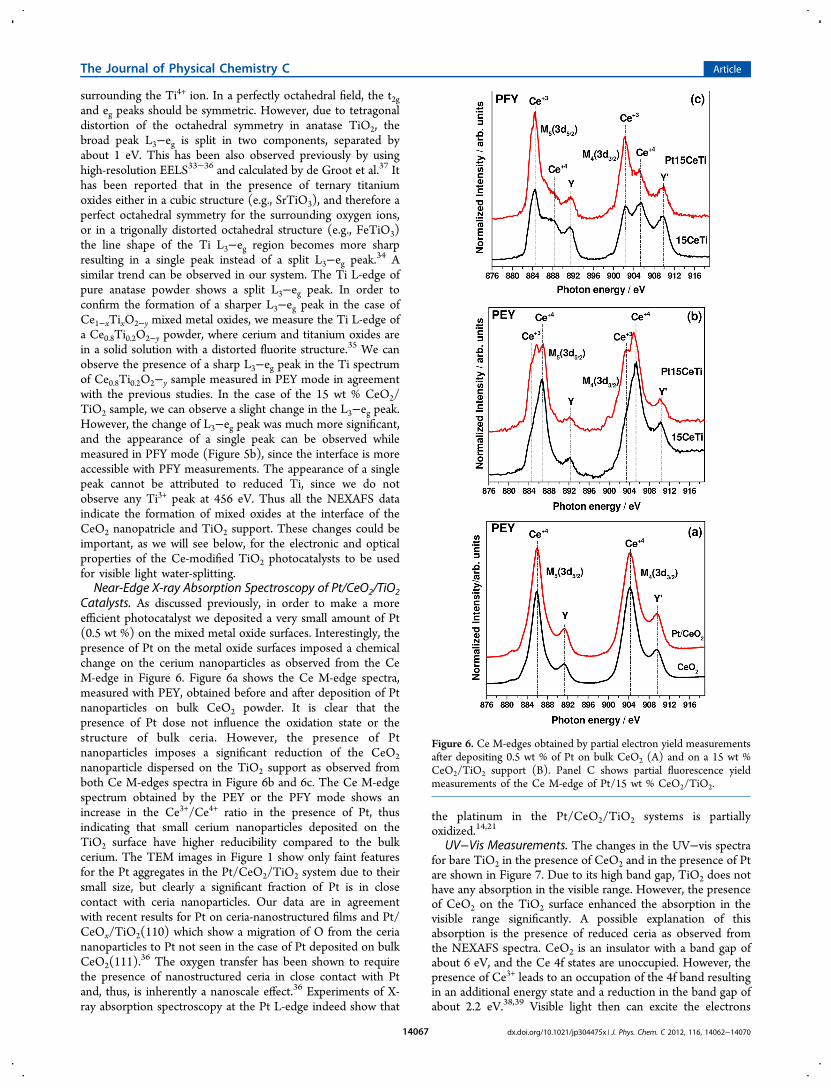
According to Pompa et al.,31 this effect comes from an inelasticdecay channel which opens up for selected transitions andincreases the radiative de-excitation probability compared toelastic cases. This leads to an energy-dependent difference inthe photon and electron cross section of the material causingthe difference in the PFY and PEY spectra of the Ce M-edge.The Ce M-edge for 15 wt % CeO2 supported on TiO2
samples was also measured in PEY and PFY mode as shown inFigure 3. In the PEY spectrum we can observe the presence of asmall amount of Ce3+ along with Ce4+ indicating that thecerium is partially reduced. However, in case of the PFYspectra, we can see the Ce3+ peak is more intense compared tothe Ce4+ peak. This is in good agreement with the TEM images.For a probing depth below 2 nm as seen by the PEY, ceriummainly stays as Ce4+ within the ordered layer of CeO2 at the topof the ceria nanoparticles as can be observed from HRTEMimages and from EELS measurements. However, PFY canprobe deeper into the interface of the CeO2 nanoparticles andthe TiO2 support where cerium exists in a reduced state. As aresult, the Ce3+ peak is more intense compared to the Ce4+ inthe Ce M-edge spectrum measured in the PFY mode.From the TEM and NEXAFS data, we can speculate that
with even smaller loading of cerium on the TiO2 support,formation of CeO2 will be restricted to the top layer and ceriumwill be mainly present in a reduced state at the interface of
CeO2 and TiO2 supports. This hypothesis was confirmed bythe NEXAFS spectrum of Ce M-edge for 6 wt % CeO2deposited on the TiO2 support. In the PEY spectrum of 6 wt% CeO2/TiO2 sample, Ce
3+ raised as the dominating peak incomparison to the Ce4+ indicating that only a subnanometerlayer of CeO2 is present, as most of the ceria stays in thereduced Ce3+ form at the interface after interacting/mixing withtitania. Certainly, in the PFY spectra, Ce3+ cations are the mainspecies present along with a small amount of Ce4+.The formation of a mixed metal oxide was further confirmed
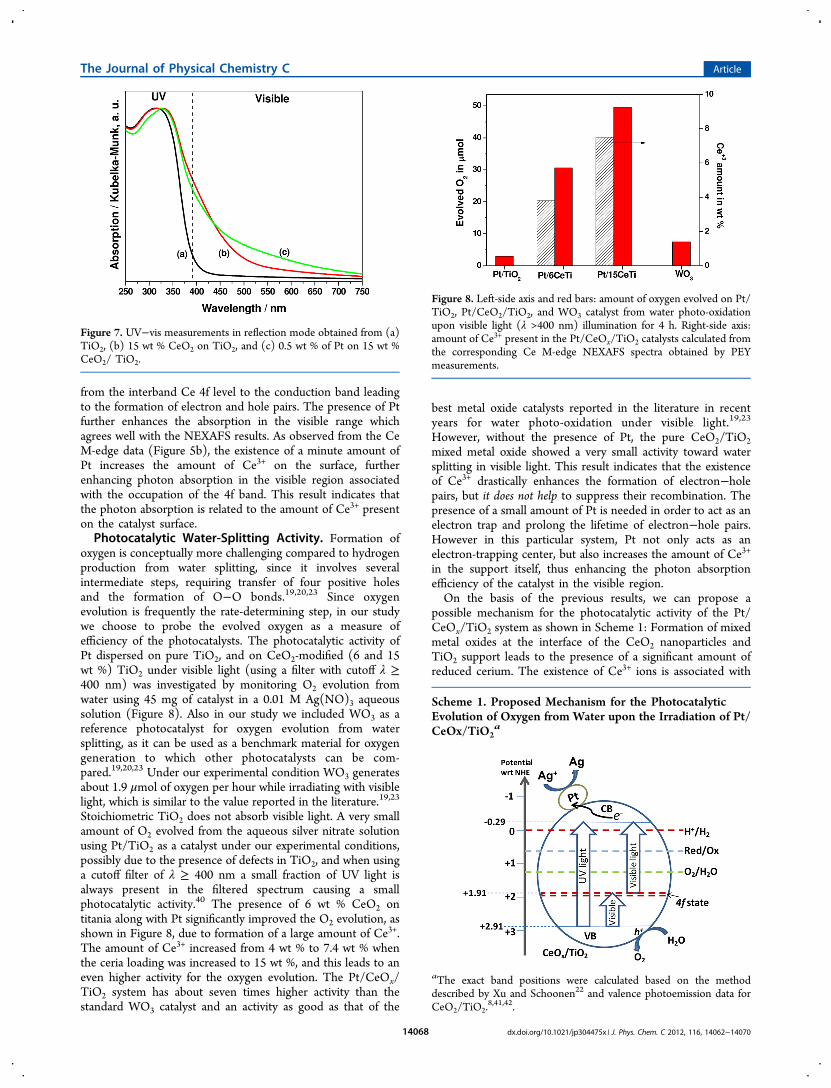
by analyzing the characteristic spectral line shapes of the Ti L3-edge features. Figure 5 shows the Ti L-edge NEXAFS spectra of
a bare TiO2 anatase powder and 15 wt % ceria-modified titania.The shape of the L absorption spectra is typical for TiO2 in theanatase structure and is well understood from previousstudies.32 The Ti 2p spin−orbit interaction splits the spectruminto two parts, corresponding to the L3 (2p3/2) and L2 (2p1/2)levels with a separation of 5.5 eV. The L2 and L3 levels arefurther split into the t2g and eg sub-bands. The strong splittingof the L2 and L3 adsorption edges is due to the crystal field
Figure 4. Ce M-edge NEXAFS of CeO2 bulk, 15 wt % CeO2/TiO2 ,and 6 wt % CeO2/TiO2 powder samples obtained via (a) partialelectron yield and (b) partial fluorescence yield measurements.
Figure 5. Ti L-edge NEXAFS of TiO2 anatase, 15 wt % CeO2/TiO2,and Ce0.8Ti0.2O2−y powder samples obtained via (a) partial electronyield and (b) partial fluorescence yield measurements.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014066
surrounding the Ti4+ ion. In a perfectly octahedral field, the t2gand eg peaks should be symmetric. However, due to tetragonaldistortion of the octahedral symmetry in anatase TiO2, thebroad peak L3−eg is split in two components, separated byabout 1 eV. This has been also observed previously by usinghigh-resolution EELS33−36 and calculated by de Groot et al.37 Ithas been reported that in the presence of ternary titaniumoxides either in a cubic structure (e.g., SrTiO3), and therefore aperfect octahedral symmetry for the surrounding oxygen ions,or in a trigonally distorted octahedral structure (e.g., FeTiO3)the line shape of the Ti L3−eg region becomes more sharpresulting in a single peak instead of a split L3−eg peak.34 Asimilar trend can be observed in our system. The Ti L-edge ofpure anatase powder shows a split L3−eg peak. In order toconfirm the formation of a sharper L3−eg peak in the case ofCe1−xTixO2−y mixed metal oxides, we measure the Ti L-edge ofa Ce0.8Ti0.2O2−y powder, where cerium and titanium oxides arein a solid solution with a distorted fluorite structure.35 We canobserve the presence of a sharp L3−eg peak in the Ti spectrumof Ce0.8Ti0.2O2−y sample measured in PEY mode in agreementwith the previous studies. In the case of the 15 wt % CeO2/TiO2 sample, we can observe a slight change in the L3−eg peak.However, the change of L3−eg peak was much more significant,and the appearance of a single peak can be observed whilemeasured in PFY mode (Figure 5b), since the interface is moreaccessible with PFY measurements. The appearance of a singlepeak cannot be attributed to reduced Ti, since we do notobserve any Ti3+ peak at 456 eV. Thus all the NEXAFS dataindicate the formation of mixed oxides at the interface of theCeO2 nanopatricle and TiO2 support. These changes could beimportant, as we will see below, for the electronic and opticalproperties of the Ce-modified TiO2 photocatalysts to be usedfor visible light water-splitting.Near-Edge X-ray Absorption Spectroscopy of Pt/CeO2/TiO2
Catalysts. As discussed previously, in order to make a moreefficient photocatalyst we deposited a very small amount of Pt(0.5 wt %) on the mixed metal oxide surfaces. Interestingly, thepresence of Pt on the metal oxide surfaces imposed a chemicalchange on the cerium nanoparticles as observed from the CeM-edge in Figure 6. Figure 6a shows the Ce M-edge spectra,measured with PEY, obtained before and after deposition of Ptnanoparticles on bulk CeO2 powder. It is clear that thepresence of Pt dose not influence the oxidation state or thestructure of bulk ceria. However, the presence of Ptnanoparticles imposes a significant reduction of the CeO2nanoparticle dispersed on the TiO2 support as observed fromboth Ce M-edges spectra in Figure 6b and 6c. The Ce M-edgespectrum obtained by the PEY or the PFY mode shows anincrease in the Ce3+/Ce4+ ratio in the presence of Pt, thusindicating that small cerium nanoparticles deposited on theTiO2 surface have higher reducibility compared to the bulkcerium. The TEM images in Figure 1 show only faint featuresfor the Pt aggregates in the Pt/CeO2/TiO2 system due to theirsmall size, but clearly a significant fraction of Pt is in closecontact with ceria nanoparticles. Our data are in agreementwith recent results for Pt on ceria-nanostructured films and Pt/CeOx/TiO2(110) which show a migration of O from the ceriananoparticles to Pt not seen in the case of Pt deposited on bulkCeO2(111).
36 The oxygen transfer has been shown to requirethe presence of nanostructured ceria in close contact with Ptand, thus, is inherently a nanoscale effect.36 Experiments of X-ray absorption spectroscopy at the Pt L-edge indeed show that
the platinum in the Pt/CeO2/TiO2 systems is partiallyoxidized.14,21
UV−Vis Measurements. The changes in the UV−vis spectrafor bare TiO2 in the presence of CeO2 and in the presence of Ptare shown in Figure 7. Due to its high band gap, TiO2 does nothave any absorption in the visible range. However, the presenceof CeO2 on the TiO2 surface enhanced the absorption in thevisible range significantly. A possible explanation of thisabsorption is the presence of reduced ceria as observed fromthe NEXAFS spectra. CeO2 is an insulator with a band gap ofabout 6 eV, and the Ce 4f states are unoccupied. However, thepresence of Ce3+ leads to an occupation of the 4f band resultingin an additional energy state and a reduction in the band gap ofabout 2.2 eV.38,39 Visible light then can excite the electrons
Figure 6. Ce M-edges obtained by partial electron yield measurementsafter depositing 0.5 wt % of Pt on bulk CeO2 (A) and on a 15 wt %CeO2/TiO2 support (B). Panel C shows partial fluorescence yieldmeasurements of the Ce M-edge of Pt/15 wt % CeO2/TiO2.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014067
from the interband Ce 4f level to the conduction band leadingto the formation of electron and hole pairs. The presence of Ptfurther enhances the absorption in the visible range whichagrees well with the NEXAFS results. As observed from the CeM-edge data (Figure 5b), the existence of a minute amount ofPt increases the amount of Ce3+ on the surface, furtherenhancing photon absorption in the visible region associatedwith the occupation of the 4f band. This result indicates thatthe photon absorption is related to the amount of Ce3+ presenton the catalyst surface.Photocatalytic Water-Splitting Activity. Formation of
oxygen is conceptually more challenging compared to hydrogenproduction from water splitting, since it involves severalintermediate steps, requiring transfer of four positive holesand the formation of O−O bonds.19,20,23 Since oxygenevolution is frequently the rate-determining step, in our studywe choose to probe the evolved oxygen as a measure ofefficiency of the photocatalysts. The photocatalytic activity ofPt dispersed on pure TiO2, and on CeO2-modified (6 and 15wt %) TiO2 under visible light (using a filter with cutoff λ ≥400 nm) was investigated by monitoring O2 evolution fromwater using 45 mg of catalyst in a 0.01 M Ag(NO)3 aqueoussolution (Figure 8). Also in our study we included WO3 as areference photocatalyst for oxygen evolution from watersplitting, as it can be used as a benchmark material for oxygengeneration to which other photocatalysts can be com-pared.19,20,23 Under our experimental condition WO3 generatesabout 1.9 μmol of oxygen per hour while irradiating with visiblelight, which is similar to the value reported in the literature.19,23
Stoichiometric TiO2 does not absorb visible light. A very smallamount of O2 evolved from the aqueous silver nitrate solutionusing Pt/TiO2 as a catalyst under our experimental conditions,possibly due to the presence of defects in TiO2, and when usinga cutoff filter of λ ≥ 400 nm a small fraction of UV light isalways present in the filtered spectrum causing a smallphotocatalytic activity.40 The presence of 6 wt % CeO2 ontitania along with Pt significantly improved the O2 evolution, asshown in Figure 8, due to formation of a large amount of Ce3+.The amount of Ce3+ increased from 4 wt % to 7.4 wt % whenthe ceria loading was increased to 15 wt %, and this leads to aneven higher activity for the oxygen evolution. The Pt/CeOx/TiO2 system has about seven times higher activity than thestandard WO3 catalyst and an activity as good as that of the
best metal oxide catalysts reported in the literature in recentyears for water photo-oxidation under visible light.19,23
However, without the presence of Pt, the pure CeO2/TiO2mixed metal oxide showed a very small activity toward watersplitting in visible light. This result indicates that the existenceof Ce3+ drastically enhances the formation of electron−holepairs, but it does not help to suppress their recombination. Thepresence of a small amount of Pt is needed in order to act as anelectron trap and prolong the lifetime of electron−hole pairs.However in this particular system, Pt not only acts as anelectron-trapping center, but also increases the amount of Ce3+
in the support itself, thus enhancing the photon absorptionefficiency of the catalyst in the visible region.On the basis of the previous results, we can propose a
possible mechanism for the photocatalytic activity of the Pt/CeOx/TiO2 system as shown in Scheme 1: Formation of mixedmetal oxides at the interface of the CeO2 nanoparticles andTiO2 support leads to the presence of a significant amount ofreduced cerium. The existence of Ce3+ ions is associated with
Figure 7. UV−vis measurements in reflection mode obtained from (a)TiO2, (b) 15 wt % CeO2 on TiO2, and (c) 0.5 wt % of Pt on 15 wt %CeO2/ TiO2.
Figure 8. Left-side axis and red bars: amount of oxygen evolved on Pt/TiO2, Pt/CeO2/TiO2, and WO3 catalyst from water photo-oxidationupon visible light (λ >400 nm) illumination for 4 h. Right-side axis:amount of Ce3+ present in the Pt/CeOx/TiO2 catalysts calculated fromthe corresponding Ce M-edge NEXAFS spectra obtained by PEYmeasurements.
Scheme 1. Proposed Mechanism for the PhotocatalyticEvolution of Oxygen from Water upon the Irradiation of Pt/CeOx/TiO2
a
aThe exact band positions were calculated based on the methoddescribed by Xu and Schoonen22 and valence photoemission data forCeO2/TiO2.
8,41,42.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014068

an extra energy state, belonging to the partially occupied 4flevel, between the conduction and valence bands ofTiO2.
13,41,42 This generates a reduced band gap of about 2.2eV in the mixed metal oxide phase. Visible light now can excitethe electrons from the valence band to the intermediate Ce 4fband or from the Ce 4f band to the conduction band, thusforming electron−hole pairs. Electrons then get trapped by Ptnanoparticles and reduce the silver nitrate present in thesolution. On the other hand, the holes will oxidize the water tooxygen. Previous experimental and theoretical studies haveexamined the adsorption of water on CeOx/TiO2(110) surfaceswhere there is a significant amount of Ce3+ sites, showing thefacile dissociation of O−H bonds.41,42 In DFT calculationsthere is a large reduction in the activation energy for thedissociation of water when going from pure TiO2(110) toCeOx/TiO2(110), with the activation energy for O−H bondcleavage on the mixed metal oxide being only 0.04 eV.41,42
Thus, the Pt/CeOx/TiO2(110) photocatalyst operates in acomplex way where the Pt and ceria nanoparticles each havedouble roles. The Pt nanoparticles help in the formation ofCe3+ centers and slow down the rate of electron−holerecombination. On the other hand, the Ce3+ centers in theceria nanoparticles dissociate water and facilitate the formationof electron−hole pairs with visible light, with the holeseventually interacting with adsorbed OH or O to form O2molecules. The coupling of the titania and ceria is essential forstabilizing the Ce3+ sites that enhance the formation ofelectron−hole pairs with visible light.The behavior of Pt/CeO2/TiO2 illustrates the advantages of
using a multifunctional configuration in which the photocatalystexposes metal nanoparticles and chemically active oxidenanoparticles to the reacting water. Thus, the water moleculescan interact with specific sites of the oxide nanoparticles, metalsites, and metal−oxide interfaces. Nanoparticles of VOx, WOx,RuOx, and CeOx in contact with a titania support exhibitunique electronic properties and create electronic states withinthe titania band gap.42−46
■ CONCLUSIONSIn summary, the deposition of CeOx nanoparticles on TiO2proves to be an efficient way to modify the chemical andelectronic properties of both metal oxides as observed fromNEXAFS and UV−vis measurements. Although Ce2O3 is not astable oxide, our study shows that it is possible to stabilize Ce3+
cations within a mixed oxide interface, which then can absorbphotons in the visible region. Pt/CeOx/TiO2 proved to beexcellent for the photocatalytic splitting of water in the visibleregion being about seven times more active than a WO3standard. Our findings open up the way to design photo-catalysts taking advantage of the unique interactions that occurin a mixed metal oxide at the nanometer level.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSL.B. acknowledges support from E.U. FP7-People-2007-4-IOF-219674. Dr. G. Zhou is gratefully acknowledged for providingthe Ce0.8Ti0.2O2 mixed oxide sample. The authors also thank
Dr. David Grill for his help with the photocatalytic activitymeasurements. The work at BNL was financed by the U.S.Department of Energy (DOE), Office of Basic Energy Science(BES) under Contract DE-AC02-98CH10086. The authorsalso acknowledge support of the National Center for ElectronMicroscopy, Lawrence Berkeley National Laboratory, which issupported by the DOE BES under Contract DE-AC02-05CH11231. The work at ICP-CSIC was financed by MCIIN(ENE2010-21198-C04-01).
■ REFERENCES(1) Kudo, A.; Miseki, Y. Chem. Soc. Rev. 2009, 38, 253−278.(2) Osterloh, F. E. Chem. Mater. 2008, 20, 35−54.(3) Kudo, A. Catal. Surv. Asia 2003, 7, 31−38.(4) Fujishima, A.; Honda, K. Nature 1972, 238, 37−38.(5) Chen, X.; Mao, S. S. Chem. Rev. 2007, 107, 2891−2959.(6) Navarro, R. M.; Alvarez-Galvan, M. C.; del Valle, F.; Villoria de laMano, J. A.; Fierro, J. L. G. ChemSusChem 2009, 2, 471−485.(7) Garcıa, M. F.; Arias, A. M.; Hanson, J. C.; Rodriguez, J. A. Chem.Rev. 2004, 104, 4063−4104.(8) Rodriguez, J. A.; Stacchiola, D. Phys. Chem. Chem. Phys. 2010, 12,9557−9565.(9) Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Science 2003,301, 935−938.(10) Linsebigler, A. L.; Lu, G.; Yates, J. T., Jr. Chem. Rev. 1995, 95,735−758.(11) Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Science2001, 293, 269−272.(12) Trovarelli, A. Catal. Rev. Sci. Eng. 1996, 38, 439−520.(13) Park, J. B.; Graciani, J.; Evans, J.; Stacchiola, D.; Ma, S.; Liu, P.;Nambu, A.; Sanz, J. F.; Hrbek, J.; Rodriguez, J. A. Proc. Natl. Acad. Sci.U.S.A. 2009, 106, 4975−4980.(14) Gonzalez, I. D.; Navarro, R. M.; Wenb, W.; Marinkovic, N.;Rodriguez, J. A.; Rosa, F. J.; Fierro, L. G. Catal. Today 2010, 149,372−379.(15) Lopez, T.; Rojas, F.; Alexander-Katz, R.; Galindo, F.; Balankin,A.; Buljan, A. J. Solid State Chem. 2004, 177, 1873−1881.(16) Li, G.; Zhang, D.; Yu, J. C. Phys. Chem. Chem. Phys. 2009, 11,3775−3782.(17) Xiea, J.; Jianga, D.; Chena, M.; Lia, D.; Zhua, J.; Lua, X.; Yan, X.Colloids Surf., A 2010, 372, 107−114.(18) Catlow, C. R. A.; Guo, Z. X.; Miskufova, M.; Shevlin, S. A.;Smith, A. G. H.; Sokol, A. A.; Walsh, A.; Wilson, D. J.; Woodley, S. M.Philos. Trans. R. Soc. A 2010, 368, 3379−3456.(19) Primo, A.; Marino, T.; Corma, A.; Molinari, R.; García, H. J. Am.Chem. Soc. 2011, 13, 6930−6933.(20) Kubacka, A.; Fernandez-García, M.; Colon, G. Chem. Rev. 2012,12, 1555−1614.(21) Barrio, L.; Zhou, G.; Gonzalez, I. D.; Estrella, M.; Hanson, J.;Rodriguez, J. A.; Navarro, R. M.; Fierro, J. L. G. Phys. Chem. Chem.Phys. 2012, in press.(22) Xu, Y.; Schoonen, M. A. Am. Mineral. 2000, 85, 543−556.(23) Silva, C. G.; Bouizi, Y.; Fornes, V.; Garcia, H. J. Am. Chem. Soc.2009, 131, 13833−13839.(24) Ravel, B.; Newville, M. J. Synchrotron Radiat. 2005, 12, 537.(25) Fortner, J. A.; Buck, E. C.; Ellison, A. J. G.; Bates, J. K.Ultramicroscopy 1997, 67, 77−81.(26) Yang, G.; Mobus, G.; Hand, R. J. Micron 2006, 37, 433−441.(27) Mamontova, E.; Egamia, T.; Dmowskia, W.; Kao, C. C. J. Phys.Chem. Solids 2001, 62, 819−823.(28) Zhang, J.; Ju, X.; Wu, Z. Y.; Liiu, T.; Hu, T. D.; Xie, Y. N. Z.;Zhang, L. Chem. Mater. 2001, 13, 4192−4197.(29) Soldatov, A. V.; Ivanchenko, T. S.; Della Longa, S.; Kotani, A.;Iwamoto, Y.; Bianconi, A. Phys. Rev. B 1994, 50, 5074.(30) Kucheyev, S. O.; Clapsaddle, B. J.; Wang, Y. M.; van Buuren, T.;Hamza, A. V. Phys. Rev. B 2007, 76, 235420−5.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014069

(31) Pompa, M.; Flank, A. M.; Lagarde, P.; Rife, J. C.; Stekhin, L.;Stekhin, I.; Nakazawa, M.; Ogasawara, H.; Kotani, A. Phys. Rev. B1997, 56, 2267−2272.(32) Turchini, S.; Delaunay, R.; Lagarde, P.; Vogel, J.; Sacchi, S. J.Electron Spectrosc. 1995, 71, 31−37.(33) Lusvardi, V. S.; Barteau, M. A.; Chen, J. G.; Fruhberger, J.; B.;Teplyakov, A. Surf. Sci. 1998, 397, 237−250.(34) Brydson, R.; Sauer, H.; Engel, W.; Thomas, J. M.; Zeitler, E.;Kosugi, N.; Kuroda, H. J. Phys.: Condens. Matter 1989, 1, 797−812.(35) Zhou, G.; Shah, P. R.; Kim, T.; Fornasiero, P.; Gorte, R. J. Catal.Today 2007, 123, 86.(36) (a) Vayssilov, G. N.; Lykhach, Y.; Migani, A.; Satudt, T.;Petrova, G. P.; Tsud, N.; Skala, T.; Bruix, A.; Illas, F.; Prince, K. C.;Matolin, V.; Neyman, K. M.; Libuda, J. Nat. Mater. 2011, 10, 310−315.(b) Bruix, A.; Rodriguez, J. A.; Ramirez, P. J.; Senanayake, S. D.; Evans,J.; Park, J. B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. J. Am. Chem.Soc. 2012, 134, 8968−8974.(37) de Groot, F. M. F.; Fuggle, J. C.; Thole, B. T.; Sawatzky, G. A.Phys. Rev. B 1990, 41, 928−937.(38) Skorodumova, N. V.; Ahuja, R.; Simak, S. I.; Abrikosov, I. A.;Johansson, B.; Lundqvist, B. I. Phys. Rev. B 2001, 64, 115108−9.(39) Andersson, D. A.; Simak, L.; Johansson, B.; Abrikosov, I. A.;Skorodumova, N. V. Phys. Rev. B 2007, 75, 035109−035113.(40) Kitano, M.; Masato, T.; Masaya, M.; Thomas, J. M.; Anpo, M.Catal. Today 2007, 120, 133−138.(41) Park, J. B.; Graciani, J.; Evans, J.; Stacchiola, D.; Senanayake, S.D.; Barrio, L.; Liu, P.; Sanz, J. F.; Hrbek, J.; Rodriguez, J. A. J. Am.Chem. Soc. 2010, 132, 356−363.(42) Graciani, J.; Plata, J. J.; Sanz, J. F.; Liu, P.; Rodriguez, J. A. J.Chem. Phys. 2010, 132, 104703.(43) Lee, S.; Zajac, G. W.; Goodman, D. W. Top. Catal. 2006, 38,127−131.(44) Zhang, Z.; Henrich, V. E. Surf. Sci. 1992, 277, 263−272.(45) Yang, F.; Kundu, S.; Vidal, A. B.; Graciani, J.; Ramirez, P. J.;Senanayake, S. D.; Stacchiola, D.; Evans, J.; Liu, P.; Sanz, J. F.;Rodriguez, J. A. Angew. Chem., Int. Ed. 2011, 50, 10198−10202.(46) Kundu, S.; Vidal, A. B.; Yang, F.; Ramirez, P. J.; Senanayake, S.D.; Stacchiola, D.; Evans, J.; Liu, P.; Rodriguez, J. A. J. Phys. Chem. C2012, 116, 4767−4773.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp304475x | J. Phys. Chem. C 2012, 116, 14062−1407014070




















