Examination of 2DE in the Human Proteome Organisation Brain Proteome Project pilot studies with the...
18
RESEARCH ARTICLE Examination of 2-DE in the Human Proteome Organisation Brain Proteome Project pilot studies with the new RAIN gel matching technique Andrew W. Dowsey 1 , Jane English 2 , Kyla Pennington 2 , David Cotter 2 , Kai Stuehler 3 , Katrin Marcus 3 , Helmut E. Meyer 3 , Michael J. Dunn 4 and Guang-Zhong Yang 1 1 Royal Society / Wolfson Foundation Medical Image Computing Laboratory, Department of Computing, Imperial College London, UK 2 Department of Psychiatry, Royal College of Surgeons in Ireland, Dublin, Ireland 3 Medizinisches Proteom-Center, Ruhr-Universität Bochum, Bochum, Germany 4 Proteome Research Centre, Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Ireland The Human Proteome Organisation (HUPO) Brain Proteome Project (BPP) pilot studies have generated over 200 2-D gels from eight participating laboratories. This data includes 67 single- channel and 60 DIGE gels comparing 30 whole frozen C57/BL6 female mouse brains, ten each at embryonic day 16, postnatal day 7 (juvenile) and postnatal day 54–56 (adult); and ten single- channel and three DIGE gels comparing human epilepsy surgery of the temporal front lobe with a corresponding post-mortem specimen. The samples were generated centrally and distributed to the participating laboratories, but otherwise no restrictions were placed on sample prepara- tion, running and staining protocols, nor on the 2-D gel analysis packages used. Spots were characterised by MS and the annotated gel images published on a ProteinScape web server. In order to examine the resultant differential expression and protein identifications, we have reprocessed a large subset of the gels using the newly developed RAIN (Robust Automated Image Normalisation) 2-D gel matching algorithm. Traditional approaches use symbolic repre- sentation of spots at the very early stages of the analysis, which introduces persistent errors due to inaccuracies in spot modelling and matching. With RAIN, image intensity distributions, rather than selected features, are used, where smooth geometric deformation and expression bias are modelled using multi-resolution image registration and bias-field correction. The method includes a new approach of volume-invariant warping which ensures the volume of protein expression under transformation is preserved. An image-based statistical expression analysis phase is then proposed, where small insignificant expression changes over one gel pair can be revealed when reinforced by the same consistent changes in others. Results of the proposed method as applied to the HUPO BPP data show significant intra-labo- ratory improvements in matching accuracy over a previous state-of-the-art technique, Multi-res- olution Image Registration (MIR), and the commercial Progenesis PG240 package. Received: February 25, 2006 Revised: May 9, 2006 Accepted: June 22, 2006 Keywords: 2-DE / Gel matching / Human Proteome Organisation Brain Proteome Project / Image registration 5030 Proteomics 2006, 6, 5030–5047 Correspondence: Professor Guang-Zhong Yang, Royal Society / Wolfson Foundation Medical Image Computing Laboratory, Department of Computing, 180 Queen’s Gate, Imperial College London, London SW7 2AZ, UK E-mail: [email protected] Fax: 144-0-20-7581-8024 Abbreviations: BPP , Brain Proteome Project; HUPO, Human Pro- teome Organisation; MIR, multi-resolution image registration; PSI, Proteomics Standard Initiative; RAIN, robust automated image normalisation; SEA, statistical expression analysis DOI 10.1002/pmic.200600152 © 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Transcript of Examination of 2DE in the Human Proteome Organisation Brain Proteome Project pilot studies with the...

RESEARCH ARTICLE
Examination of 2-DE in the Human Proteome
Organisation Brain Proteome Project pilot studies
with the new RAIN gel matching technique
Andrew W. Dowsey1, Jane English2, Kyla Pennington2, David Cotter2, Kai Stuehler3,Katrin Marcus3, Helmut E. Meyer3, Michael J. Dunn4 and Guang-Zhong Yang1
1 Royal Society / Wolfson Foundation Medical Image Computing Laboratory, Department of Computing,Imperial College London, UK
2 Department of Psychiatry, Royal College of Surgeons in Ireland, Dublin, Ireland3 Medizinisches Proteom-Center, Ruhr-Universität Bochum, Bochum, Germany4 Proteome Research Centre, Conway Institute of Biomolecular and Biomedical Research,
University College Dublin, Ireland
The Human Proteome Organisation (HUPO) Brain Proteome Project (BPP) pilot studies havegenerated over 200 2-D gels from eight participating laboratories. This data includes 67 single-channel and 60 DIGE gels comparing 30 whole frozen C57/BL6 female mouse brains, ten each atembryonic day 16, postnatal day 7 (juvenile) and postnatal day 54–56 (adult); and ten single-channel and three DIGE gels comparing human epilepsy surgery of the temporal front lobe witha corresponding post-mortem specimen. The samples were generated centrally and distributedto the participating laboratories, but otherwise no restrictions were placed on sample prepara-tion, running and staining protocols, nor on the 2-D gel analysis packages used. Spots werecharacterised by MS and the annotated gel images published on a ProteinScape web server.In order to examine the resultant differential expression and protein identifications, we havereprocessed a large subset of the gels using the newly developed RAIN (Robust AutomatedImage Normalisation) 2-D gel matching algorithm. Traditional approaches use symbolic repre-sentation of spots at the very early stages of the analysis, which introduces persistent errors dueto inaccuracies in spot modelling and matching. With RAIN, image intensity distributions,rather than selected features, are used, where smooth geometric deformation and expressionbias are modelled using multi-resolution image registration and bias-field correction. Themethod includes a new approach of volume-invariant warping which ensures the volume ofprotein expression under transformation is preserved. An image-based statistical expressionanalysis phase is then proposed, where small insignificant expression changes over one gel paircan be revealed when reinforced by the same consistent changes in others.Results of the proposed method as applied to the HUPO BPP data show significant intra-labo-ratory improvements in matching accuracy over a previous state-of-the-art technique, Multi-res-olution Image Registration (MIR), and the commercial Progenesis PG240 package.
Received: February 25, 2006Revised: May 9, 2006
Accepted: June 22, 2006
Keywords:
2-DE / Gel matching / Human Proteome Organisation Brain Proteome Project / Imageregistration
5030 Proteomics 2006, 6, 5030–5047
Correspondence: Professor Guang-Zhong Yang, Royal Society /Wolfson Foundation Medical Image Computing Laboratory,Department of Computing, 180 Queen’s Gate, Imperial CollegeLondon, London SW7 2AZ, UKE-mail: [email protected]: 144-0-20-7581-8024
Abbreviations: BPP, Brain Proteome Project; HUPO, Human Pro-teome Organisation; MIR, multi-resolution image registration;PSI, Proteomics Standard Initiative; RAIN, robust automatedimage normalisation; SEA, statistical expression analysis
DOI 10.1002/pmic.200600152
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5031
1 Introduction
In January 2004, the Human Proteome Organisation(HUPO) Brain Proteome Project (BPP) [1] launched two pilotstudies to explore the standardisation of techniques neededto obtain reliable results on the phenotyping of patient andmouse models and complete characterisation of brain tissuethat is to be performed in the main phase. Participants har-nessed a wide range of quantitative 2-D and non-2-D gelbased proteomics technologies in order to discover the pro-tein complement and differential expression in two experi-ments: a) Analysis of whole dry-ice frozen brain of C57/Bl6mice at three development stages of growth (embryonic day16, postnatal day 7 and postnatal days 54–56) was conductedto assess the quality of different proteome analysis tech-niques and to compare proteome and transciptome withmRNA profiling. No pooling was allowed and analysis wasperformed on five to ten different samples for each stage;b) A further study was also launched to compare the pro-teome of human brain from an autopsy sample with thatfrom an epilepsy biopsy in order to assess post-mortem pro-tein stability. Both projects had the remit of feeding the brainproteome with reliable data. ProteinScape was used as thestandardised data collection software and web server.
To provide homogeneity, the samples for each experi-ment were provided centrally and distributed to the partici-pants. No other restrictions were placed on the experimentalprotocol, so the 2-D gel data provided by eight participatinginstitutions varied in sample preparation, first- and second-dimension running conditions, staining protocol and imageacquisition instrumentation – no laboratory used the samecombination of IPG strip pH gradient, SDS-PAGE gel con-centration and gel size. Furthermore, no standardisation wasenforced on the 2-DE software analysis package used or on
their parameters, such as the significance level for spotdetection, matching and differential expression. A total of266 images from 140 2-D gels were generated for differentialexpression analysis. Statistics on the number of gels pro-vided by each laboratory and the variability in protocols arelisted in Table 1.
In order to revalidate the 2-DE complement of the HUPOBPP pilot studies and assess the possibilities for inter-labo-ratory matching without stringent protocol standardisation,we have analysed a significant subset of the 2-D gels withour new fully-automatic gel matching algorithm RobustAdvanced Image Normalisation (RAIN). RAIN is an image-based registration and bias-correction technique that correctsboth geometric and intensity variations between 2-D gels. Noprior spot detection is required, and after processing thesample gel image is warped to align directly onto the refer-ence gel image. Before proceeding to the details of this study,we begin by introducing the challenges in 2-DE bioinfor-matics and the limitations of conventional spot-detection andmatching techniques.
1.1 2-D gel matching
2-DE remains the preferred core technology for the majorityof applied proteomic experiments due to its ability to indicatePTMs that result in alterations in protein pH and/or Mr [2, 3],as well as its capacity to simultaneously separate thousandsof proteins. At present, however, several experimental factorshinder the separation power of 2-DE. Briefly:
(i) Co-migrating proteins can make their separate reso-lution intractable.
(ii) Spots can often exhibit severe tails in the Mr dimen-sion.
Table 1. List of laboratories providing 2-D gels to the HUPO BPP pilot studies, the various protocols used and the resulting gel imagesgenerated. For the human brain experiment: B = epilepsy biopsy tissue, A = post-mortem autopsy tissue, B-A = Cy2 pooled sam-ple images. For the mouse brain experiment: E = embryonic day 16, J = juvenile postnatal day 7, A = adult postnatal day 54–56, E-
A, E-J and J-A = respective Cy2 pooled sample images. Note that figures represent images, not gels, e.g. a human brain DIGE gelis listed as 1 biopsy image, 1 autopsy issue and 1 pooled sample image
Lab IEF SDS-PAGE
Detection Human gel images Mouse gel images Sum
B A B-A E J A E-A E-J J-A
1 IPG 24 cm pH 3–11NLKlose system [35]
11%15%
SilverSilver
2 2 54
55
88
2217
2 IPG 24 cm pH 4–7IPG 24 cm pH 6–9
12%12%
DIGE DIGE 1 1 1 1010
1010
1010
55
55
55
4845
4 Klose system 15% Silver 3 3 3 96 IPG 18 cm pH 3–10NL 12% SYPRORuby 4 4 8
10 IPG 18 cm pH 4–7IPG 18 cm pH 6–9
12%12%
DIGEDIGE
2 2 2 1010
1010
1010
55
55
55
5145
11 IPG 18 cm pH 3–10NL 12.5% Silver 5 5 5 1712 IPG 18 cm pH 3–10 12.5% Silver 2 2 414 IPG 24 cm pH 3–10 12.5% Silver 1 1 2
Sum 8 8 3 57 62 68 20 20 20 266
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5032 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
(iii) The dynamic range of 2-DE is large [4], with thefaintest spots hard to discriminate from noise.
(iv) Geometric deformations in the electrophoretic diffu-sion process make it difficult to match spots between gels.
(v) Contrast varies from gel to gel due to stain exposure[5], sample loading errors and protein losses at differentstages of gel running.
(vi) Staining techniques are well known to be far fromstoichiometric [6].
(vii) The intensity of the background can vary across theimage [7].
Therefore, multiple technical replicates must be run foreach sample in order to establish statistical significance ofdifferential expression between them.
A comprehensive review of bioinformatics in 2-DE isavailable in [8], which includes references to commercialpackages and academic research in the area. In general, thetraditional image processing pipeline for 2-DE consists of:
(i) Gel image pre-processing, e.g. noise reduction, back-ground subtraction.
(ii) Spot detection, modelling and expression quantifica-tion.
(iii) Spot matching to implicitly remove geometric defor-mation.
(iv) Identification of differential expression (statisticaloutliers).
(v) Creation of federated 2-DE databases.In conventional spot matching, point pattern matching
is used to find the closest spot correspondence between apoint pattern (source spot list) and a target point set (refer-ence spot list). These methods need to be robust to outlierscaused both by failed spot detection and true differentialexpression, as well as the geometric distortion present. Awide variety of methods have been developed includingiterative closet point, the alignment method, geometrichashing and robust pattern matching. However, only robustpattern matching methods interleave image warping directly[9]. With feature matching, a large number of candidatetransforms need to be evaluated due to combinational explo-sion when each arc (drawn between two points) in the pointpattern is mapped onto every arc in the target point set. Toreduce the search space, arc reduction techniques such asDelaunay triangulation [10] are regularly used.
It is now believed that the inaccuracies of the feature-based approach are largely due to insufficient data. Whencomplex spot matching fails, a manual landmark warpingphase is traditionally pre-pended to compensate for severegeometric differences. Even then, significant manual post-validation of the spot correspondences is required. Recently,Nonlinear Dynamics (Newcastle, UK) have recently releasedtheir TT900 S2S package which provides a graphical userinterface for smooth landmark-based image warping, and anoutput facility to export the matched images into the user’spreferred proteomics analysis package. Whilst this techniqueis entirely manual, entailing 20–30 min work per gel pair, theimproved results underline the benefits gained by reducing
the role of spot matching with a view to eventually eliminat-ing it altogether. If one focuses on the raw pixel values,numerous features such as spot shape, streaks, smears, spottails and background structure are available which are other-wise lost in the spot detection phase.
Z3 [11] was one of the latest automatic algorithms toincorporate pixel level information, by comparing rectan-gular image regions each containing a few spots. Morerecently, MIR (Multi-resolution Image Registration) [12], andthen Gustafsson et al. [13], have presented classical imageregistration techniques which have no recourse to detectingspots. In image registration, geometric and intensity distor-tion attributed to experimental uncertainty (e.g. protein dif-fusion) is eliminated, whilst intrinsic differences (e.g.changes in protein expression) are kept. The source image Is
is brought into alignment with the reference image Ir, con-strained by M, the space of allowable transformations (map-pings) on Is, and guided by the similarity measure functionsim, which is maximal when the images are optimallyaligned (Eq. 1):
arg maxm2M
sim Is �my; Irð Þ (1)
The maximum is found by an optimisation procedure on theparameters Y of the mapping function m, which transformseach pixel location in the sample image space to a corre-sponding location in the reference image space. The natureof M is important, as there is a trade-off between finding thetransformation that leads to the highest similarity, whilstensuring that it is realistic. Therefore, in MIR a multi-reso-lution approach is used, where low-resolution images areused to find an initial match with a single bilinear mapping,and then finer and finer images are analysed as the bilinearmapping is subdivided to become more deformable. Nor-malised cross-correlation is used as the similarity measure.In a comparative study between MIR and Z3, MIR tooksimilar time and scored better 29 out of 30 times in a testwhere an expert quantified visually the number of mis-registered spots. More recently, Woodward et al. [14] haveutilised wavelets, an alternate formalism of multi-resolutionanalysis where only a sub-band of image frequencies (rangeof deformations) are kept at each iteration.
These algorithms have highlighted the possibility offully-automated gel matching. However, we can note thatmuch more is possible to improve the matching process andcorrect for other systematic errors in the 2-DE process:
(i) Users find it impossible to perform accurate proteinexpression quantification on warped gels. This is because nocurrent image-based registration algorithm outputs a realis-tically smooth warp, nor ensures that the volume of proteinin the image remains consistent. Commercial packagesmitigate this problem by performing protein quantificationon the original images only.
(ii) Intensity inhomogeneities between gels are wellknown, but are assumed to follow a Gaussian distributionand suppressed with statistical techniques.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5033
(iii) There is an extensive range of protocols in the 2-DEfield, but no image-based technique has proved to be robustenough to cope with the variety of gels that are created. Forexample, limited work has been attempted to match gelstreated with different staining or labelling protocols, or gelsfrom different laboratories, which is of interest to large col-laborative efforts such as the HUPO BPP. The dynamicrange and background variability of DIGE is much greaterthan in silver stained gels but no technique has been vali-dated against such gels.
In practice, these questions must be answered beforeassurance can be placed on a totally automated high-throughput solution, especially those related to statisticalapproaches based on multiple gel runs and image miningtechniques through the use of parallel processing. We believethat a combination of image-based registration, bias-fieldmodelling and high-throughput statistical expression analy-sis moves someway to reaching this goal.
1.1.1 Bias-field modelling
The variability in spot volumes between even technicalreplicates is well known and documented [15, 16]. No current2-DE software package attempts to correct gels for theexpression bias caused by pipetting, gel electrophoresis andstaining errors, relying instead on extrapolation of the back-ground variability to the spot areas, which can add significantsoftware-induced variance [17]. Since Rabilloud [3], little re-search has been performed on the nature of this bias-fielduntil Gustafsson et al. [13] reported a spatial dependence inprotein quantification error over externally Studentised resi-duals in three representative gel sets [18]. With an absence of2-DE formation models to extract the bias-field on an indi-vidual gel basis, analysis must be performed relative to asecond gel. However, Gustafsson et al. [13] argue that somebiological conditions could be concealed by normalisingspatial trends in data using between treatment group com-parisons. For example, a higher degree of proteolysis candecrease the volumes of spots corresponding to full-sizeproteins, whilst enlarging spot volumes corresponding totheir degradation fragments that are positioned furtherdown the second dimension in the gel. To counter this claim,it can be argued that bias-field modelling for image registra-tion is better posed than bias-field modelling for expressionquantification:
(i) For image registration, the bias-field modelling can bemore aggressive as techniques which remove biological var-iation between gels increase the similarity between corre-sponding spots, and so aids in their matching. The resultingbias-fields would have to be removed from the final result.
(ii) For expression quantification, differential expression(outliers) will always cause errors in fitting techniques thatassume normally distributed data, such as bias-field correc-tion. Therefore, the bias-field correction of each spot must benormalised by considering the expression levels in the sur-rounding protein pattern, so that real differential expression
is not removed unintentionally. This can be performed byrestricting the bias-field to vary smoothly, whilst differentialexpression causes much more abrupt changes in intensitybetween gels.
1.1.2 High-throughput statistical expression
analysis (SEA)
Recent studies on the accuracy of different spot detectionalgorithms in main software packages in the field haveshown marked difference in their respective performance [7,19], which highlights the major obstacles in realising the fullpotential of the differential powers of 2-DE. In practice, ifpair-wise statistical analyses were performed simultaneouslyon whole sets of pre-aligned images, then small insignificantexpression changes over one pair could become significantwhen reinforced by the same consistent changes in the oth-ers. This permits the creation of a database of probabilisticbaselines, or ‘norms’, which characterise confidence levelsfor protein expression under different experimental settings.Such evidence will build up a statistical formation model of2-DE, which would then be mined to discover intrinsictrends, thus increasing the acuity of new results and provid-ing valuable feedback to 2-DE scientists on the sensitivity oftheir experiments. With the proposed SEA, the richness ininformation and its associated uncertainties are fully cap-tured in a model independent manner. To this end, the fol-lowing issues must be addressed:
(i) Normalisation: i.e. by removing geometric and inten-sity inhomogeneities through image registration and bias-field correction. There will be no loss of data – the gels willremain in image format.
(ii) Representation: differentially expressed proteins willbe extracted from the gels by image mining – comparingthem with statistical norms from a large-scale proteomicsdatabase. Data must be stored in an intermediate repre-sentation that draws a compromise between tractable com-putation and maximal information retention.(iii) Interfacing: standards have only now been established bythe HUPO Proteomics Standards Initiative (PSI) [20] (http://psidev.sourceforge.net/), to ensure that data generated at thislevel can be fully and automatically integrated with the rele-vant upstream (e.g. experimental, sample, separation) anddownstream (e.g. MS) data pipelines.
(iv) Computation: in order to cope with the large volumeof data and complexity in analysis brought by the processingof large gel sets and a corresponding proteomics repository,distributed parallel computation is required.
We have recently developed the high-throughput pro-Turbo framework [21] for task farming massive batches of2-D gels. Spare capacity is harvested from idle officemachines when their users are away. The next step towardsan automated pipeline is to tackle the normalisation issue asa prerequisite for SEA image mining. The RAIN algorithmhas been developed to meet this goal [22], which is sum-marised below. The remainder of this paper then presents
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5034 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
the validation of RAIN against MIR on gel sets generatedfrom the HUPO BPP pilot studies, and against ProgenesisPG240 on a large gel set from the Royal College of Surgeonsin Ireland (Dublin), together with insights for future collab-orative 2-D gel generation in the main HUPO BPP projectphase.
2 Materials and methods
The objective of RAIN is to remove the systematic geometricand intensity distortions in the 2-DE process, so gel imagescan be directly analysed. Specifically, RAIN is an image-based technique to simultaneously register and bias-fieldcorrect 2-D gel pairs with a smooth volume-invariant multi-resolution B-spline transformation. The method involves theuse of a sum of squared residuals similarity metric and theLimited Memory BFGS optimiser, which shall be explainedin this section. The novel approach used includes processingof expression intensities to normalise out systematic inten-sity inhomogeneity between gels, in order to ensure accuratepost-quantification. It is expected that protein expression inthe output images will subsequently be accurately quantifiedwith SEA or conventional techniques.
2.1 B-spline transformations for geometric and
bias-field modelling
B-splines were introduced in 1946 by Schoenberg [23] asconnected piecewise polynomials of order n. At each con-nection a smoothness constraint imposes continuity of thespline and its n-1 derivatives, so a k segment spline is definedby only k1n parameters. B-splines have been chosen fortheir efficiency in optimising geometrical transformationsfor image registration, since each coefficient has a locality ofeffect of n11 segments, or less at the ends. Furthermore,with the Oslo algorithm [24] one can sub-divide a B-splinecurve without altering its path, in order to increase thenumber of parameters and therefore its deformability. Formodelling geometric deformation and bias-fields we requirepiecewise surfaces, not segment-wise curves, and asB-splines are linearly separable this is a simple extension.Note, however, that two B-spline surfaces are required, inorder to model the smooth deformation in both x and y axes.
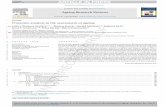
Since most conventional warping algorithms aredesigned to correct for optical distortion, not the physicaldistortion present in 2-DE, they introduce protein over-expression when the surface stretches and under-expressionwhen it contracts. Figure 1 gives an example on a syntheticGaussian spot stretched by a conventional warp and a vol-ume-invariant warp. To preserve volume invariance, theintensity of each warped pixel must be weighted by itschange in density. This can be achieved by considering theJacobian, which is the set of partial derivatives of the map-ping with respect to the coordinate axes [25]. Each derivativecorresponds to the change in density along the respective
Figure 1. Volume-invariant warping of asynthetic protein spot. (a) Original spot.(b) As stretched by a typical imagewarping algorithm. The spot intensityremains constant and so the volumeincreases. (c) Stretched by a volume-invariant warp that normalises intensityby the determinant of the Jacobian ofthe transformation mapping.
axis, so the determinant of the Jacobian represents the totalchange in volume. If the intensities of the warped image arenormalised by the determinant at each pixel, the integratedoptical density (IOD) of each protein spot remains constanteven after warping. In order for the normalisation to be rea-listically smooth, the B-spline surfaces must be at least 3rd
order. This ensures that the partial derivatives are 2nd orderand so the change in density is also continuous. Previousimage-based techniques employ 1st order transformations,such as piecewise bilinear mappings and Delaunay triangu-lation, that are discontinuous at piece boundaries and sodistort spots badly if they lie across those boundaries. Fur-thermore, volume-invariant warping is not advisable as thechange in density is constant for each piece and so the dis-continuity between pieces is even more noticeable.
It has been shown previously that the bias-field correc-tion technique of Lai and Fang [26] is suitable for extractingspatial bias from lists of spot matches in 2-DE [8]. However,this technique is a feature-based approach which requiresgood manual verification of the spot lists. In RAIN, bias-fieldcorrection is integrated into the image registration phase bymodelling multiplicative and additive bias-fields by separate3rd order B-spline surfaces. A multiplicative bias-field bU
represents a smooth variation in contrast over the image,whilst an additive bias-field bV provides the same for imagebrightness (Eq. 2):
IU;Vðx; yÞ ¼ Iðx; yÞ þ bVðx; yÞð Þ � ebUðx;yÞ (2)
The additive bias-field and multiplicative bias field each actin turn on the pixel intensity at location (x,y) in image I. WithB-spline surface modelling bV and bU are constrained tosmoothly vary over the image to account for expressioninhomogeneities without correcting for true differentialexpression. The goal is to find the bias-field parameters,which when applied to the sample image, maximise thesimilarity between the sample and reference images.
In image-based 2-D gel matching, matching errors arecomputed by differences in pixel intensity between sampleand reference images. The difference in intensity between astrongly expressed spot and the background intensity isgreater than that of a weakly expressed spot; thereforestrongly expressed spots predominate in the error calcula-
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5035
tion. To minimise this effect, the study is conducted in thelog domain [13]. This has the effect of enhancing feint spots,since a ratio between two intensity values in the image do-main becomes a constant difference in the log domain. In2-DE, the weaker the expression, the more significant theeffects of noise. Log-images enhance this noise as well as theweak expression, so a weighting factor a is necessary, whichblends between a pure log-image at a = 0, to the originalimage at a = ?. The equation Jr for sampling the referenceimage Ir is therefore given by Eq. (3):
Jr ¼ lnðIrðx; yÞ þ aÞ (3)
Equivalently, the equation Js for sampling the sample imageIs, including both bias-fields and volume-invariant geometrictransformation, is defined by Eq. (4):
Js;C ¼ ln det b0X; b0Y
� ��� �� � Is bX; bYð Þ þ bVð Þ � ebU þ a� �
(4)
where image Is is under geometric B-spline surface transfor-mation in x and y, (bX, bY), multiplicative bias-field correctionbU, additive bias-field correction bV, and normalised into avolume invariant warp by the Jacobian determinantdet b0X; b
0Y
� ��� ��. In order to find the optimum warp and bias-fields, the total set of B-spline parameters C = {X,Y,U,V}must be searched until the optimum is found. This requiresan optimisation strategy using a similarity metric objectivefunction, which is described in the next section.
Since B-splines are continuous, sampling Js at each pixelwill almost always lead to Is being sampled between pixels. Aresampler must be applied to make Is continuous. ForRAIN’s final match warp, a 3rd order B-spline interpolator isused. A piecewise B-spline surface, with number of piecesequal to the sample image’s width6height, is fitted to thesample image by a fast singular value decomposition tech-nique. Sampling then proceeds directly from the B-splinesurface.
2.2 Finding the optimal match
The goal of image registration is to ensure the devised simi-larity measure and search strategy find the optimal match, inthe wide range of 2-D gel protocols in use and in the widestvariety of cases. Whilst it is acceptable that an automatedapproach can be batched overnight, performance is stillimportant due to the great number of comparisons thatresults from the image mining of large gel sets. It is there-fore imperative that the registration strategy, similaritymeasure and optimisation procedure are tailored to the tar-get application.
As in MIR, a multi-resolution approach is adopted tocope with misalignment that ranges from global changes tosmall imperfections. Multi-resolution has numerous advan-tages as long as the increase in image resolution at eachstage is paired well with a complementary increase thecomplexity of the deformation corrected for. In this way,
local minima that would cause the optimiser to converge toan incorrect transformation will be smoothed away instead.However, the resolutions chosen must not be too low, sincethe true optimum will also be smoothed away to anapproximation. It is also important for the bias-fields to beconstrained in this way, so that their optimisation is notmisled by the residual geometric deformation. The Haarbasis [27] is used to create the multi-resolution image pyr-amids. The reference and sample images are first resized to204862048, and then detail is removed iteratively by mer-ging each square of four pixels into a single pixel, by takingthe average. Each output image therefore has a resolution ofhalf the width and half the height, and so a pyramid ofimages is created bottom-up from resolution 102461024 to16616, as illustrated in Figure 2(a). When RAIN matchesthe images, it proceeds in reverse, determining the optimalparameters of single-piece B-spline surfaces X, Y, U and V atpyramid level 16616, and then at each stage sub-dividingthe B-spline surfaces whilst increasing the pyramid level, sofinally at 102461024 resolution the B-spline surfaces have32632 pieces.
A large number of candidate transformations are eval-uated by RAIN, so to increase performance tri-linear inter-polation is used to resample Is in Eq. (4) for these candidates.In tri-linear interpolation, the change in size of the pixel isfirst evaluated, just as in volume-invariant warping. The twopyramid levels with pixels nearest in size are selected, andbilinear interpolation performed on each. Another linearinterpolation is then performed between the two results. Oneadvantage of this approach is that the image gradients arekept conservatively smooth to help the convergence of theoptimiser.
Since intensity level correspondence between images isdealt with explicitly with bias-field modelling, the implicitglobal contrast and brightness compensation provided by thenormalised cross-correlation in MIR is not necessary.Instead, we calculate the much less computationally expen-sive sum of squared residuals (Eq. 5):
sim Js;C; Jr
� �¼ �
X
x;yð Þ2Ir
Js;C x; yð Þ � Jr x; yð Þ� �2
(5)
The choice of algorithm to optimise the parameters C is acritical step in image registration, and depends on the pres-ence of local minima. Since the aim of the multi-resolutionanalysis is to remove all local minima, we can employ a gra-dient descent algorithm which searches the parameter spaceby always moving in a downward direction. In Eq. (5), this isanalogous to ensuring sim increases at every iteration. Withquasi-Newton methods, the Jacobian must be supplied (see[22] for a derivation), but the matrix of 2nd order partial deri-vatives (the Hessian) can be gradually built up by using gra-dient information from some or all of the previous iterations.The BFGS Hessian update has been shown to be the methodof choice in most circumstances [28]. B-spline locality can beeasily exploited, e.g. for a 3rd order B-spline each parameter
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5036 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
Figure 2. (a) An image pyramid of shifted-log images. The first four levels of overlaid gels are shown, with pixel dimensions from 16616 to128 6128. The shifted log transform, Equation (3), has a = 0.1. (b) Synthetic warps generated for the robustness analysis. The transfor-mation parameters were perturbed by a normally distributed random process with SDs of 0.05, 0.15, 0.25 and 0.35 of B-spline patch size(see Table 2).
only affects 16 pieces (and less near surface boundaries). Forfurther performance gains, we have chosen the limited-memory BFGS method [28], in which only the significantcomponent of the Hessian is recalculated.
To combine the optimisation of the bias-fields with thatof the image registration, it is important to note that a goodbias-field correction improves convergence of the registra-tion, but conversely a good registration also improves bias-field estimation. For this reason, iterations of bias-field cor-rection and image registration were interleaved. In areas oflittle image gradient (sparse or no spots), the convergencecan be significantly affected by numerical round-off errors.To avoid this, we threshold the derivatives of the similaritymeasure, so that the warp will only deform in directions withsufficient evidence. A global correction stage was also added,so that simple translation displacements and a global changein gel contrast and brightness is corrected for on 16616images before B-spline modelling. The optimisation thenterminates after 6 B-spline levels, since the fraction of spotsmatched at level 7 but not at level 6 did not warrant increasedrunning time from an average of 7.5 min per gel pair.
Due to the nature of DIGE gels, an additional processingstrategy is required. DIGE has the benefit that matching dif-ferentially expressed gels can be performed on the non-dif-ferentially expressed Cy2 pooled components. The Cy3 andCy5 sample images are then warped with the transformationgenerated between the Cy2 sample and reference. Eachimage needs to be bias-field corrected individually, so opti-mal bias-fields were found through the RAIN process foreach of the Cy2, Cy3 and Cy5 sample images, and Cy3 andCy5 references images, against the reference Cy2 image. It isalso noted that images produced by high dynamic range flu-
orescence scanners write the square root of actual pixelintensity to the image file, since in this way lower intensityspots can be more accurately represented. This phenomenonhad to be taken into account for volume-invariant warping tofunction correctly.
2.2.1 Examination of algorithm robustness
It cannot be stressed enough that in optimisation the choiceof internal parameter values are critical when gradient des-cent techniques are used. Each choice affects the nature ofthe objective function, such as its curvature, the presence oflocal minima that cause convergence to incorrect end result,and the accuracy of the true optimum. In our exploration ofthe technical aspects of RAIN [22], the analyses were per-formed as follows:
(i) Examination of the objective function over an exten-sive range of gels for different B-spline configurations andlog-image weighting factors. The optimal parameter valuesare illustrated in Figure 2(a).
(ii) Validation of the robustness of interleaved optimisa-tion of geometric deformation modelling and bias-fieldmodelling on both real and synthetically distorted gel sets.
(iii) Investigation of the upper limits of deformation andbias that can be corrected for using RAIN, by processing gelsunder progressively increasing synthetic distortion. Resultsare given in Table 2 and examples of synthetic warps shownin Figure 2(b).
Illustration of the RAIN gel matching process proceedsas follows. Disparities between an example gel pair areshown in Figure 3. Hierarchical matching with RAIN is thenshown in Figure 4 whilst Figure 5 illustrates the derived bias-
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5037
Table 2. Robustness analysis. The geometric deformation and bias-field B-spline parameters were perturbed by anormally distributed random process with SD between 0 150 and 0 350 of B-spline patch size. For fiverepresentative gels and five random processes, the mean squared error (MSE) between image intensitiesand average mismatched spots (Mismatch) were quantified after RAIN correction. It can be seen thatRAIN is able to robustly match spots whilst the standard deviation �0.25. See Figure 2(b) for examplewarps
SD 0.100 0.125 0.150 0.175 0.200 0.225 0.250 0.275 0.300
MSE 0.022 0.022 0.022 0.022 0.023 0.024 0.025 0.026 0.028Mismatch 0.1 0.1 0.2 0.5 1.5 2.8 7.3 25.3 36.5
Figure 3. The deformities between two human skin cell gels areclearly shown when the reference (top-right) in green is overlaidwith the sample (bottom-right) in magenta. Note the higher con-trast in the sample gel.
fields for the first four stages. The final image in Figure 4 hasthe full unconstrained bias-field applied, which will masksubtle differential expression. Figure 6 shows the end resultwithout bias-field correction, where real differential expres-sion is masked by intensity inhomogeneities, and with a464 smooth bias-field applied. It is evident that a suitablecompromise is achieved, though substantial validation willhave to be carried out in future before confidence can beplaced in propagating the bias-field information downstreamto SEA or conventional spot analysis.
2.3 Electrophoresis
The brain tissue from the epilepsy surgery was provided byAlbert Becker (University of Bonn). The biopsy specimenwas obtained from a patient (male, age 44) with chronicpharmacoresistant temporal lobe epilepsy [29], who under-went surgical treatment in the Epilepsy Surgery Program atthe University of Bonn Medical Center. A pre-surgical evalu-ation using a combination of non-invasive and invasive pro-cedures [30] revealed that seizures originated in the mesialtemporal lobe. Surgical removal of the hippocampus andtemporal lobe was clinically indicated. All procedures were
conducted in accordance with the Declaration of Helsinkiand approved by the ethics committee of the University ofBonn Medical Centre. Informed written consent wasobtained from the patient. A standardised neuropathologicalevaluation of the hippocampus revealed Ammon’s hornsclerosis, i.e. segmental neuronal cell loss in CA1 as well asCA3/4, gliosis, and axonal reorganization [31]. Temporal lobespecimen showed heterotopic neurons in the white matter.
Post-mortem tissues came from Brain-Net Europethrough Hans Kretzschmar (Ludwig-Maximilians Uni-versity, Munich). The brain originated from a 60 year oldCaucasian male (postmortem interval: 11 h). Pathologicalcharacterisation revealed: Neuropathological diagnosis:Braak&Braak: 1–2; Cerad: 0; Lewy bodys (Mckeiths): 0; Clin-ical diagnosis: Prostate carcinoma, no other markers of neu-rodegeneration Cause of death: Peritonitis. Samples weretaken from the area corresponding to the biopsy sample (su-perior temporal gyrus).
The mouse brain samples were donated by Gert Lubec(Medical University of Vienna). The C57/Bl6 mice weretreated identically under standard breeding conditions;brains were removed after decapitation and immediatelyfrozen on dry ice. Analysis included three different develop-mental stages: embryonic day 16, postnatal day 7; and 8weeks. Detailed intra-laboratory validation was performed onthe mouse brain samples from Laboratories 1 and 2, and the2-D electrophoresis is described below. Mouse and humaninter-laboratory comparison between all eight laboratories isalso considered, but for conciseness the electrophoresis isnot presented in depth; see Table 1 for an overview.
Laboratory 1 supplied two sets of silver stained gels andvarious test gels run with varying protocols, e.g. CBB stain.The first set of 2-D gels (four embryonic day 16, five juvenilepostnatal day 7 and five adult 8 week mouse brain samples)was generated using the standard IPG 2-DE method [32]. IEFwas performed on the IPGphor system (GE Healthcare)according to the manufacturer’s instructions. After rehydra-tion of the IPG-strips (essentially according to[33], 8 M urea,2 M thiourea, 2% CHAPS, 0.8% Servalyte, 100 mM DTT) thesamples were loaded via cup loading (80 mg of protein).Focusing was performed at max. 207C for 110 kVh as follows:6 h at 100 V/step, 1 h at 300 V/gradient, 3 h at 300 V/step, 6 hat 1000 V/gradient, 1.5 h at 6000 V/gradient, 16 h at 6000
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5038 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
Figure 4. The RAIN algorithmmatching the two gels in Fig-ure 3. After the initial search forthe optimal translation compo-nent, a single B-spline surfacepatch is fitted. Finer and finerdeformations are then modelledwith B-spline surfaces of 262 to32632 pieces. This is necessarysince distortions this fine arestill evident in the gels, e.g. seethe spot highlighted in red,which is only matched at the laststage.
V/step. Following focusing, the IPG-strips equilibratedaccording to [32] (15 min in equilibration buffer A: pH 8.8,6 M urea, 50 mM Tris, 2% SDS w/v, 30% glycerin w/v,65 mM DTT, 15 min in equilibration buffer B: pH 8.8, 6 Murea, 50 mM Tris, 2% SDS w/v, 30% glycerin w/v, 280 mMiodoacetamide). The IPG-strips were transferred on the topof an 11% 25 cm620 cm SDS-PAGE gel and sealed withagarose sealing solution (50 mM Tris-HCl, 0.1% SDS w/v,
192 mM glycine, 0.3% agarose w/v, bromphenol blue). TheSDS-PAGE was carried out vertically in a EttanDALTTMapparatus (GE Healtcare), according to the manufacturer’sinstructions, with a current of 20 W per gel at 207C. Silverpost-staining was performed after gel scanning using a MS-compatible protocol, as described in [34].
The other set from laboratory 1 (4 embryonal, 3 juvenileand 4 adult) was created using the method described in [35].
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5039
Figure 5. Multiplicative (top row) and additive (bottom row) bias-field correction from 161 to 868 pieces of the process in Fig-ure 4. A green bias field represents a shift x towards the sample,whilst a magenta bias-field represents a shift towards the refer-ence. The multiplicative bias-field shows Is?ex = Ir, where22�x�2. The weaker additive bias-field shows Is 1 x = Ir, where21/4� x � 1/4. The algorithm can be seen correcting for thesample gel’s stronger contrast around the margins of the gel. Thebias fields appear to become unrealistic after 464 pieces (boxedin red), but still help convergence dramatically.
Carrier ampholyte based IEF was performed in a self-madeIEF chambers using tube gels (20 cm60.9 mm). After run-ning a 21.25 h voltage gradient, the ejected tube gels wereincubated in equilibration buffer (125 mM Tris, 40% glycerolw/v, 3% SDS w/v, 65 mM DTT, pH 6.8) for 10 min. The sec-ond dimension was performed in a Desaphor VA 300 systemusing polyacrylamide gels (15.2% total acrylamide, 1.3%bisacrylamide). The IEF tube gels were placed onto the poly-acrylamide gels (20 cm630 cm60.7 mm) and fixed using1.0% agarose w/v containing 0.01% bromophenol blue dyew/v (Riedel deHaen, Seelze, Germany). For protein identifi-cation the preparative sized gel system (IEF:20 cm61.5 mm, SDS-PAGE: 20 cm630 cm61.5 mm) wasapplied under identical conditions. Silver staining was per-formed as before.
In laboratory 2 mouse brain samples were lysed in600 mL (embryonic day 16) or 1 mL (postnantal day 7, 8weeks) of lysis buffer (9.5 M urea, 2% Chaps, 20 mM Tris
(pH 8.0) and analysed using the DIGE system (Table 1). Aninternal standard for the mouse study was prepared withequal fractions of all mouse samples and bulk labelled with400 pmol Cy2 per 50 ìg of protein. Fifty microgrammes ofeach sample protein was labelled with 400 pmol of either Cy3or Cy5, performing a so-called dye swap, making sure eachdevelopmental stage had an equal amount of Cy3 and Cy5labelled samples. Cy2, Cy3, and Cy5 labelled samples weremultiplexed appropriately and applied to both 24 cm pH 4–7IPG and pH 6–9 strips by in-gel rehydration and IEF wascarried out using an Ettan IPGphor II IEF system at 207C anda maximum of 50 mA/strip and 3500 V for 75 kVh, followedby 8000 V for 1 h. After equilibration buffer, the strips wererun in the second dimension on 12% SDS-PAGE gels usingan Ettan Dalt system overnight at 157C and 1 W/gel. Gelswere scanned in with a Typhoon scanner.
Objective evaluation against Progenesis PG240 (Non-linear Dynamics, Newcastle, UK) was performed on a subsetof a 105 2-D gel study of post-mortem brain tissue carried outas described previously [36]. Briefly, total proteins were solu-bilised from samples of grey matter taken from the dorso-lateral prefrontal cortex in schizophrenia, bipolar disorderand control subject groups (35 subjects per group) in lysisbuffer (9.5 M urea, 1% DTT, 2% CHAPS and 0.8% Pharma-lyte, pH 3–10). Each sample (100 mg protein) was then sepa-rated in the first dimension by IEF on 18 cm pH 4–7 IPG,using in-gel rehydration method [37]. The strips werefocused at 0.05 mA/IPG strip for 68 kVh at 207C using aMultiphor II system. After IEF, the strips were equilibratedand 2-D SDS-PAGE was performed using 12% SDS-PAGEgels using a Hoefer DALT system. The 2-D separation wascarried out overnight at 20 mA/gel at 87C and was stopped asthe Bromophenol blue dye front just left the bottom of thegels. Following 2-DE, the gels were fixed in 50% methanol,10% acetic acid and silver stained using the Owl 2-D SilverStaining Kit (Insight Biotechnology, Wembley, UK). Thestained gels were scanned at 100 mm resolution using a Mo-lecular Dynamics Personal SI laser densitometer (GEHealthcare, Amersham Biosciences).
Figure 6. The effect of bias-fieldcorrection. The final match fromFigure 4, (a) without bias-fieldcorrection, and (b) with the 464bias-field correction boxed inred in Figure 5. Subtle differ-ential expression is more clearlyvisible.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5040 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
We also looked at two silver stained experiments thatVeeser et al. [12] published to act as gold standards to com-pare future image registration algorithms against (http://www.doc.ic.ac.uk/vip/2d-gel/index.htm/).
2.4 Validation strategy
For intra-laboratory subjective validation, an expert was giventhe task of quantifying the amount of spot mismatches be-tween gels processed by MIR and RAIN. Expert scrutiny isnecessary in order to identify true positives from false posi-tives. The threshold criterion used was that 75% of the spotshape must overlap for a match. This is in line with the cri-teria used for assessment of MIR against Z3 (Veeser,unpublished). In order to conduct a fair test, the analysis wasperformed in a double blind manner. Despite the increaseddifficulty for the user, RAIN’s bias field correction wasturned off or else the user could have easily differentiatedbetween MIR and RAIN processed pairs. Both intra andinter-sample 2-D gel sets were examined, to assess eachalgorithm’s robustness to differentially expressed spots. Thescoring method used (Table 3) was devised [12] to relate theamount of spot mismatches to user validation time. Forinter-laboratory validation, comparison was unnecessarysince matching was too hard for MIR.
In the objective validation, 14 control gels and 14 samplegels were pre-warped using RAIN. To quantify the effect ofRAIN on the 2-DE bioinformatics process, two groups werecompared, the original unaligned gels and the RAIN pre-warped gels, and the same control gel was assigned as thereference in both cases. The two groups were imported intothe same experiment in Progenesis PG240 using the experi-ment wizard. Spots were automatically detected by the soft-ware and subsequently automatically warped and matched tothe reference gel. Statistical analysis was then carried outusing the average gel for each set, to compare:
(i) Original control gels vs original sample gels(ii) RAIN pre-warped control gels vs RAIN pre-warped
sample gelsStatistically significant differential expression was found
and manually validated, and the corresponding spot matcheswere scrutinised in both groups for mismatches.
Table 3. Scores used to assess the performance of RAIN againstMIR
Score Criteria
5 Spots mismatched=04 0,spots mismatched�53 5,spots mismatched�102 10,spots mismatched�201 20,spots mismatched, but affecting less than
a quarter of the gel0 20,spots mismatched, but affecting more than
a quarter of the gel
3 Results
Subjective validation was performed between technicalreplicates (intra-set assessment–Table 4) and between con-trol and sample gels (inter-set assessment–Table 5.)
Mean and SD is shown for the score and number of spotmismatches per gel, excluding outlier results such as totalfailures and problem regions, which would bias the dis-tribution. For both statistics in all the comparisons, the Sha-piro-Wilk test rejected the null hypothesis that the differencebetween RAIN and MIR was normally distributed. The Wil-coxon signed rank test was therefore deemed more suitablethan the paired t-test, and with this test at the 95% con-fidence interval it was found that RAIN significantly out-performed MIR with higher scores and a lower number ofspot mismatches in each comparison listed in Tables 4 and 5.Four of the most challenging gel pairs are shown in Figure 7and the results of their matching with MIR and RAIN are il-lustrated in Figure 8.
Intra-set assessment was performed between 36 pairs inthe psychiatric disorder experiment (labelled Psy Silver), 36pairs in the laboratory 1 HUPO BPP silver sets (HUPO BPPSilver) and 36 pairs in the laboratory 2 HUPO BPP DIGE set(HUPO BPP DIGE). Technical replicates were not availablein the Veeser experiments. The results show RAIN matchessubstantially more spots without introducing extensive false-positives. In the DIGE experiment, MIR failed to converge atall in 6 gel pairs due to the significantly higher backgroundbias in DIGE. With the scoring scheme RAIN consistentlyscored 3 or higher, whilst MIR often scored 0 or 1, mis-matching more than 20 spots, and so resulting in consider-able user editing.
Inter-set assessment was performed between three setsof psychiatric disorder gels (24 pairs–Psy Silver), the threestages of mouse brain growth (24 pairs–HUPO BPP Silver)and the ten pairs in the Veeser easy and medium groups.Inter-assessment is not applicable to DIGE gels since thematching is always performed against the Cy2 pooled sam-ple. Both MIR and RAIN’s matching performance decreasesunder differential expression, but RAIN is more robust asthe images become less and less similar. The laboratory 1HUPO BPP gels exhibit large disparities, especially betweenthe embryonic and juvenile/adult stages, as shown in Fig-ure 7(c). MIR proved problematic on these gels, divergingthree times and mis-matching many spots. This notablyoccurred in heavily streaked or smeared regions, whereasRAIN’s additive bias field equalised the disparity. The PsySilver gels showed less dissimilarity overall, but differencesbetween the schizophrenia and control gels were still signif-icant. Two of the medium difficulty Veeser gels exhibit prob-lem regions due to issues in the first dimension IPG strip.These regions were included in the analysis, yet RAIN onlymismatched 2 spots over the 10 pairs once the regions wereexcluded from the results. This highlights the robustness ofRAIN to problem regions. MIR performed relatively poorly,mismatching an average of 4.8 spots per gel.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5041
Table 4. Subjective intra-assessment. Thirty six gel pairs were matched with MIR and RAIN for the mental disorderexperiment (Psy Silver), and the mouse brain experiment from HUPO BPP laboratory 1 (HUPO BPP Silver)and laboratory 2 (HUPO BPP DIGE)
MismatchedSpots
Psy Silver HUPO BPP Silver HUPO BPP DIGE
MIR RAIN MIR RAIN MIR RAIN
Mean 15.50 1.17 24.44 5.44 22.00a) 4.60Std Dev 2.40 1.36 9.09 3.07 9.33a) 2.38
Score MIR RAIN MIR RAIN MIR RAIN
Mean 2.33 4.33 1.44 3.67 1.00 3.60Std Dev 0.48 0.48 0.69 0.68 0.91 0.50
a) Excluding 6 divergences
Table 5. Subjective inter-assessment. Twenty four gel pairs were matched with MIR and RAIN for the mental dis-order experiment (Psy Silver) and laboratory 1 mouse brain experiment (HUPO BPP Silver), plus 10 pairsfrom [12] (Veeser Silver)
MismatchedSpots
Psy Silver HUPO BPP Silver Veeser Silver
MIR RAIN MIR RAIN MIR RAIN
Mean 17.67 6.33 31.00a) 9.83 4.80b) 0.20b)
Std Dev 10.97 6.84 13.50a) 4.58 3.49b) 0.42b)
Score MIR RAIN MIR RAIN MIR RAIN
Mean 2.17 3.50 0.50 2.83 3.80b) 4.80b)
Std Dev 1.24 1.14 0.78 0.70 0.79b) 0.42b)
a) Excluding three divergencesb) Excluding four problem regions
Figure 7. A selection of difficult HUPO BPP gels analysed. (a) Laboratory 1 IPG gels, silver stained, from different adult mouse brains. (b)Laboratory 1 gels prepared with the Klose system. The samples are from the same mouse brain, comparing silver stain (top) with CBB(bottom). (c) Laboratory 1 IPG method between juvenile (top) and embryonic mouse brain (bottom). (d) Laboratory 2 DIGE gels. Fluores-cence imaging has a higher dynamic range than silver stain, so these images are presented histogram equalised to make the low abun-dance spots visible.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5042 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
In the objective validation, after automatic warping andmatching of the 28 gels, Progenesis PG240 had matchedan average of 812 spots per gel in the original group andan average of 1020 spots per gel in the RAIN pre-warpedgroup. Comparison for one gel pair is given in Figure 9.Once again, these differences were not normally distribut-ed, so the Wilcoxon signed rank test was chosen. At the95% confidence interval significantly more spots werematched in the RAIN pre-warped group, and by visualinspection the RAIN group provided a much more accu-rate match. Since verification of every spot was intractable,Progenesis was employed to find significant differentialexpression between the schizophrenia and control sets foreach group. Seventy-three differentially expressed spots(p,0.05) were found in the original group and 57 in theRAIN pre-warped group, of which only nine were found inboth groups.
Table 6 shows the results of manually quantifying thenumber of gels in which these spots were mismatched byProgenesis. The fact that the spots appeared in an average of19.6 gels in the original set and an average of 21.3 gels in theRAIN pre-warped group was not found to be significant.However, in the RAIN pre-warped group all 9 spots were
Table 6. Objective intra-assessment using Progenesis PG240 toextract differential expression between 14 control and14 schizophrenia gels of dorsolateral prefrontal cortex.The original gel images (Original) and RAIN pre-warped images (RAIN) were processed through Pro-genesis and nine spots (p,0.05) were present in bothgroups. The spots were then checked manually. Thenumber of gels each spot match was present in, andthe number of these gels the spot match was incorrectin, are shown
No. of gels with positivematch
No. of gels with false positivematch
Original RAIN Original RAIN
28 28 3 014 22 3 017 16 2 011 14 3 028 28 2 018 23 1 027 28 1 026 26 3 07 7 1 0
Figure 8. Subjective validation of MIR (top) and RAIN (bottom, shown bias-corrected) with the gels from Figure 7. (a) Both algorithms fail inthe problem region at the top, but MIR drags surrounding spots out of alignment. The central mass of protein also disrupted peripherymatches with MIR. (b) MIR is unable to match the spots on the left of the gel due to the streaking in the silver stained gel. RAIN is able tosuppress the streaking artefacts by increasing the additive bias in this area. Also, the spots in the bottom left region are very under-expressed on the CBB gel, and so are too feint for MIR to match. RAIN does a much better job by amplifying the multiplicative bias in thisarea. (c) There is a massive increase in expression from embryonic to juvenile mouse brain. Both MIR and RAIN find it harder to matchspots in the wake of misleading differential expression, though MIR is also weaker in peripheral regions. (d) MIR finds matching very dif-ficult since the bias levels between the two gels are too great. MIR was not validated against DIGE gels in [12]. RAIN is able to bias-correctwell, and matches the majority of the spots.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5043
Figure 9. Example of a gel pair which was unable to be warped in automatic mode by Progenesis PG240 and as a result the reference gelcould only be matched to a) 833 spots in the sample gel, in comparison with b) 1100 in the RAIN pre-warped gel.
matched over all 28 gels, yet in the original group Progenesismismatched each spot an average of 2.1 times, which waseasily verified as significant.
Whilst it is possible to use RAIN to compare sampleswith different staining protocols, as shown in Figure 8(b),inter-laboratory comparison proved much tougher. Fig-ure 10(a–b) shows gels created by laboratories 12 and 14 onthe human epilepsy biopsy sample. Both gels are silverstained and run with the same parameters, except that IPGstrip length was 18 cm in laboratory 12 and 24 cm in labora-tory 14. Whilst RAIN was able to discern a match, the re-sultant warp was much distorted, which illustrates the largegeometric variability in gels between laboratories. In fact,comparing the gels between any other pair of laboratories ledto too much dissimilarity for RAIN to match. An example isgiven in Figure 10(c). RAIN’s matching ability is built on theassumption that gradient information in the two images arerelated by some underlying function, but in these cases theassumption does not hold. In order to perform inter-labora-tory comparisons in future, more information must be har-nessed.
4 Discussion
In this paper, we have presented the progress towards a fully-automated high-throughput bioinformatics solution for2-DE through the use of image-based registration, bias fieldmodelling and distributed cluster image computing. Wehave shown that RAIN is a substantial improvement over the
previous state-of-art MIR, due to the incorporation of asmooth 3rd order mapping, bias-field correction, volumeinvariant warping and the capacity to match DIGE gels. Thenew method can handle both artefacts and intensity inho-mogeities, and remains robust as deformation severityincreases. Whilst RAIN is comparatively slow (,7.5 min), itis fully automatic so can be run in the background or batchedovernight. MIR analysis can be performed interactively sinceit only takes around 3 s per match. However, it is fast becausethe transformation is simple and the number of optimisableparameters is low.
In order to get meaningful data from 2-D gels importedinto Progenesis PG240, considerable manual warping andmatching is required. Often spots matched automatically byProgenesis are mismatched to the wrong spots in the refer-ence gel, and so the user must go over the warping andmatch manually in order to ensure correct matching of themajority of protein spots. However, using RAIN pre-warping,this error in mismatching was reduced considerably. Fur-thermore, investigation of the nine proteins found to be dif-ferentially expressed between the two groups were mis-matched in none of the RAIN pre-warped gels. We areendeavouring to perform objective validation of this type on avariety of sample types, and once the infrastructure is inplace during the HUPO BPP main phase, it is hoped thatHUPO BPP members will conduct this analysis in theirlaboratories whilst utilising a wide variety of 2-DE protocols.
Inter-laboratory comparison was found to be intractablein the majority of circumstances without extra information.In fact, we do have such information in the form of protein
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5044 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
Figure 10. a) Gels created bylaboratories 12 (top) and 14(bottom) on the human epilepsybiopsy sample. Both gels aresilver stained and run with thesame parameters, except thatthe IPG strip length was 18 cm inlaboratory 12 and 24 cm in labo-ratory 14. b) Whilst RAIN wasable to discern a match (top), theresultant warp was much dis-torted (bottom), which illus-trates the significant variability.c) Example of intractable inter-laboratory comparison. pH 6–9DIGE mouse brain sample gelsfor laboratory 2 (top) and labo-ratory 10 (bottom), histogramequalised.
identification lists that each laboratory has obtained throughspot excision and characterisation with MS. If we can find co-identified spots between laboratories, then these can bebrought into alignment as a precursor to gel matching. Forexample, by comparing the subset of proteins identified inboth the IPG and Klose system 2-D gel sets from laboratory1, it is clear that substantially more proteins, many repre-senting multiple post-translationally modified forms, wereidentified in the Klose system set. If these gel spots wereannotated with their mass spectra, an algorithm could bedeveloped to find the most realistic warp between the twogels that also provided a statistically significant alignment ofco-identified protein isoforms.
This is a target for future research, but at present 4 matchvectors were identified manually (the offset between proteinspot location p in the Klose system 2-D gel to the correspond-ing spot location q in the IPG 2-D gel). A weighted combina-tion of thin plate splines [26] centred about each match vectorwas then generated to give an interpolation function whichpasses through the points exactly, whilst maximising thesmoothness of the warp (the integral of the squares of thesecond derivatives). In effect, the IPG gel is warped so that the4 spots align directly onto the corresponding spots in theKlose system 2-D gel. For a good alignment of the whole gel,the match vectors should be spread out evenly. If this is not thecase, further manual landmarks can be placed by eye, analo-gous to the methodology of the TT900 package, and thewarping updated. Once an acceptable match is found, spots ofinterest can then be manually revalidated between labora-tories by considering their neighbouring protein patterns.
However, it is still clear from the large variety of iso-forms and residual geometric variation that revalidation willbe a long and difficult proposition. It is therefore apparent
that if cross-comparison of 2-D gels is desired, inter-labora-tory analyses should be based on more standardised proto-cols. Unfortunately, protocol standardisation restricts eachindividual laboratory’s specialist expertise and limits thecoverage of proteome mapping, so in certain cases greaterheterogeneity should be allowed instead, with attentiongiven to improved intra-laboratory verification and valida-tion.
Feature-based matching with point-pattern matchingtechniques was the original solution in 2-DE. These methodshave the opportunity to match larger distortions than image-based approaches, since the deformation corrected by thesealgorithms is usually unconstrained, and the pattern profilesdo not need to overlap. Unfortunately, unconstrainedmatching can lead to a higher false-positive rate, so to reducemismatches, image warping constraints had to be aug-mented [10]. Typically, feature-based approaches performbetter in situations where they can distinguish between cor-rect and incorrect features. In 2-DE, this translates to streaksand smearing. Feature-based approaches are weak in com-plex areas and for low abundance proteins, i.e. where idealspot models break-down. Image-based techniques are farmore robust in these areas since they make no assumptionsabout the data. We have shown with RAIN that by usingminimal assumptions unrelated to spot models (e.g. the na-ture of the bias-field), image-based algorithms can exceed theperformance of feature-based approaches. Furthermore,thanks to the volume-invariant warping, image-basedapproaches now do not affect expression post-quantification.However, feature-based algorithms are still in active re-search, and more robust spot models [38] and matchingtechniques may emerge in future. There is consequently adesperate need for a gold-standard validation process to
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5045
evaluate new techniques, as individual papers can only vali-date against one or two other packages or algorithms, whichrenders cross-validation infeasible.
We believe that the existing spot matching approachesare based on incomplete information, and 2-DE gel analysismethodology should instead be built from the bottom-up. Atthe bottom level general information should be used, i.e.intensity profiles. The RAIN algorithm provides a uniqueopportunity for incorporating all possible generic informa-tion into the matching of 2-D gels. Only when the usefulnessof this data is exhausted should we then move upwards tousing more and more domain-specific knowledge, such asmodels of spot shape. In RAIN, we intend to incorporate thisby regressing the results of SEA spot surface analysis back tothe image registration phase in a feedback loop. In this way,gel matching and differential expression modelling can beiteratively improved until both are optimal. By taking thismethodology further, once feature-based models haveexhausted their use, attention will turn to the use of expertinformation, i.e. domain models. This can come from theuse of machine-based learning and its associated caveats, orin our opinion, from the compilation and analysis of statis-tical norms built from vast quantities of 2-D gels, to for-mulate a statistical atlas of 2-DE gel formation.
Our results on the multiplicative bias-field correlate wellwith the observations of spatial-dependence found by [18].The additive bias-field was only intended to correct for resi-dual errors, but has also been found to be very useful toequalise gels that suffer large differences in streaking orsmearing artefacts. Bias-field correction substantially aidsthe optimal convergence of the image registration process.This is even the case when the bias-fields begin to appearunrealistic, which occurs once they are constrained less asthe warping corrects for finer deformations. It is clear that itsmodelling is perhaps too simplistic with regard to theunderlying biological process. A study will have to be per-formed to deduce which of the following models is true:
(i) The nonlinear non-stoichiometric response of stain tobinding matter is the reason why background and proteinexpression appear to follow two different linear models.
(ii) The background and protein expression surfaces doin fact follow different, possibly linear, models.
For the latter, improved bias-field estimation can onlyfollow from either spot detection or reusing downstreamSEA spot surface analysis in re-registration with a feedbackloop. If the former is true, then we can model the non-linear bias-fields implicitly using a quadratic or cubic bias-field model. A physical explanation for the bias-field, relat-ed to a model of gel formation, would be of value in fullyunderstanding the systematic errors we are correcting forhere.
Also the SEA approach could significantly improve theresolution and sensitivity of 2-DE analyses by using imageinformation from sets of technical replicates for differentialexpression quantification. We believe that several areas of2-DE bioinformatics can be improved upon with SEA:
4.1 Spot detection
Technical replicates by their nature should exhibit the samespot profiles, apart from experimental uncertainty. Therefore,if a spot is found in one gel, it must have counterparts in theother technical replicates, even though in some they may be soweakly expressed that they are individually undetectable fromnoise with conventional techniques. By basing the analysesstatistically on the technical replicates, evidence of a protein onone gel would reinforce the evidence on other gels. Evidencefor proteins exhibiting low abundance on all gels, which wouldbe missed by using conventional techniques, would accumu-late with SEA. With all gels taken as a whole, evidence for thatspot could be significant. This type of analysis was rarely con-ducted previously [39] because the gel sets must be pre-matched in the image domain. This correlates with our view-point, since we believe that whole image information shouldbe kept as far into the pipeline as possible. In other words, spotsegmentation should be the very last task performed.
4.2 Spot segmentation and quantification
Traditional techniques treat spot quantification between setsof technical replicates in a statistical manner, but the preced-ing segmentation process makes concrete decisions based onindividual intensity profiles. We suggest that spot modellingshould be an implicit, rather than an explicit process. Thequestion should be: what part of this pixel is protein, and whatpart is background, given its surrounding neighbourhood?The response should then be: given the space of possible spotshapes, what is the probability that the neighbourhood lieswithin that space, and what is the protein level associatedwith the maximum likelihood outcome of spot shape, giventhat the background is modelled by a smooth deformationsurface (such as B-splines)? By formulating the problem assuch, the information in the technical replicates can beincorporated to strengthen the final result.
4.3 Background subtraction
Rather than the initial pre-processing step, background sub-traction should be a consequence of spot modelling. Back-ground can be separated into components based on whetherit is random noise, or contaminants present in all the tech-nical replicates. Once the set has been matched and spotmodelled, the shared contaminant field is extracted and anyresidual error can be discounted. In this way, backgrounderrors are removed even before we introduce a second set ofgels to analyse for differential expression.
5 Concluding remarks
To conclude, rather than look for alternative methods of pro-tein separation, we have chosen to base our research intoimproving the elucidation of information that is ‘locked’ into
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

5046 A. W. Dowsey et al. Proteomics 2006, 6, 5030–5047
2-D gels, with a view towards creating a fully automatedpipeline. The goal of our ProteomeGRID project (http://www.proteomegrid.org/) is to interface RAIN and proTurbowith a SEA-based differential expression analysis engine, andto integrate an exemplar PSI-standard database imple-mentation. This will realise our main goal of providing acentralised and standardised bioinformatics resource for theproteomics community. Gels annotated with PSI metadatawill be entered into the database and then calibrated fordirect comparison with RAIN. The standardised gels arethen mined for differential expression, drawing on the sta-tistical norms for that tissue type, protocol and laboratoryfrom the integrated proteomics database. The statisticalnorms are updated with the results, whilst the differentialspot locations are excised from the gel and the proteinsidentified by the usual techniques. Dissemination and vali-dation of the results will be available through a web basedinterface.
Maintaining statistical norms will allow, for the firsttime, the investigation of a statistical atlas of gel formation.Annotations incorporated into the norms adhere to the sameprinciples, so gels matched in the system will be auto-matically probabilistically annotated. By categorising thenorms on the metadata dictionary and annotations, the atlascould be queried to answer questions such as:
(i) Which sample preparation/staining/running proto-cols have resulted in the highest confidence levels of expres-sion and differential expression quantification?
(ii) At what levels of abundance do certain staining pro-tocols fail to produce a confident quantification?
(iii) For a particular protocol, how many technical repli-cates does one require to be 95% confident that expressionquantification will be 95% confident?
(iv) In which sample type does biomarker protein A mostconfidently appear?
(v) Under what conditions is protein A under- or over-regulated?
Therefore, the power of a statistical atlas of 2-D gel for-mation lies in the data mining of subtle trends and relation-ships in protein patterns over vast quantities of 2-D gels thatcould indicate new links in the underlying biological pro-cesses of function and disease.
HUPO BPP is supported by the German Ministry for Edu-cation and Research with grants 0313318B and 01GR0440. Thematerial from MJD’s laboratory is based upon works supportedby the Science Foundation Ireland under Grant No. 04/RP1/B499 to MJD. The Proteome Research Centre at UCD has beensupported by the Higher Education Authority (HEA) in Ireland.AWD is supported by the Medical Research Council (UK) andthe British Heart Foundation, and GZY by the Royal Society andWolfson Foundation. We are greatly indebted to all the labora-tories and participants of the HUPO BPP for their advice andhelp in organising this study. ProteinScape was donated by Bru-ker Daltronics.
6 References
[1] Hamacher, M., Klose, J., Rossier, J., Marcus, K. et al., Prote-omics 2004, 4, 1932–1934.
[2] Gorg, A., Weiss, W., Dunn, M. J., Proteomics 2004, 4, 3665–3685.
[3] Rabilloud, T., Proteomics 2002, 2, 3–10.
[4] Anderson, N. L., Anderson, N. G., Mol. Cell. Proteomics2002, 1, 845–867.
[5] Ireland, W. P., Sulston, K. W., Summan, M., Electrophoresis2002, 23, 1652–1658.
[6] Herbert, B. R., Sanchez, J.-C., Bini, L., in: Wilkins, M. R., Wil-liams, K. L., Appel, R. D., Hochstrasser, D. F. (Eds), ProteomeResearch: New Frontiers in Functional Genomics, Springer-Verlag, Heidelberg 1997, pp. 13–34.
[7] Nishihara, J. C., Champion, K. M., Electrophoresis 2002, 23,2203–2215.
[8] Dowsey, A. W., Dunn, M. J., Yang, G. Z., Proteomics 2003, 3,1567–1596.
[9] Pedersen, L., Ersbøll, B., 12th Scandinavian Conference onImage Analysis, Bergen, Norway, 2001, 118–125.
[10] Efrat, A., Hoffmann, F., Kriegel, K., Schultz, C. et al., J. Com-put. Biol. 2002, 9, 299–315.
[11] Smilansky, Z., Electrophoresis 2001, 22, 1616–1626.
[12] Veeser, S., Dunn, M. J., Yang, G. Z., Proteomics 2001, 1, 856–870.
[13] Gustafsson, J. S., Blomberg, A., Rudemo, M., Electrophore-sis 2002, 23, 1731–1744.
[14] Woodward, A. M., Rowland, J. J., Kell, D. B., Analyst 2004,129, 542–552.
[15] Almeida, J. S., Stanislaus, R., Krug, E., Arthur, J. M., Prote-omics 2005, 5, 1242–1249.
[16] Choe, L. H., Lee, K. H., Electrophoresis 2003, 24, 3500–3507.
[17] Wheelock, A. M., Buckpitt, A. R., Electrophoresis 2005, 26,4508–4520.
[18] Gustafsson, J. S., Ceasar, R., Glasbey, C. A., Blomberg, A. etal., Proteomics 2004, 4, 3791–3799.
[19] Rogers, M., Graham, J., Tonge, R. P., Proteomics 2003, 3,879–886.
[20] Orchard, S., Hermjakob, H., Apweiler, R., Proteomics 2003, 3,1374–1376.
[21] Dowsey, A. W., Dunn, M. J., Yang, G. Z., Proteomics 2004, 4,3800–3812.
[22] Dowsey, A. W., PhD Thesis, Department of Computing,Imperial College London, 2005.
[23] Schoenberg, I. J., Quart. Appl. Math. 1946, 4, 45–99.
[24] Cohen, E., Lyche, T., Riesenfeld, R., Comput. Graph. ImageProcess. 1980, 14, 87–111.
[25] Shen, D., Davatzikos, C., Neuroimage 2003, 18, 28–41.
[26] Lai, S. H., Fang, M., Med. Image Anal. 1999, 3, 409–424.
[27] Strang, G., Bull. Am. Math. Soc. 1993, 28, 288–305.
[28] Nocedal, J., Math. Comp. 1980, 35, 773–782.
[29] Elger, C. E., Schramm, J., Radiologe 1993, 33, 165–171.
[30] Kral, T., Clusmann, H., Urbach, J., Schramm, J. et al., Zen-tralbl. Neurochir. 2002, 63, 106–110.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2006, 6, 5030–5047 Bioinformatics 5047
[31] Blumcke, I., Zuschratter, W., Schewe, J. C., Suter, B. et al., J.Comp. Neurol. 1999, 414, 437–453.
[32] Gorg, A., Postel, W., Gunther, S., Electrophoresis 1988, 9,531–546.
[33] Marcus, K., Immler, D., Sternberger, J., Meyer, H. E., Elec-trophoresis 2000, 21, 2622–2636.
[34] Nesterenko, M. V., Tilley, M., Upton, S. J., J. Biochem. Bio-phys. Methods 1994, 28, 239–242.
[35] Klose, J., Kobalz, U., Electrophoresis 1995, 16, 1034–1059.
[36] Beasley, C. L., Pennington, K., Behan, A., Wait, R. et al., Pro-teomics 2006, 6, 3414–3425.
[37] Rabilloud, T., Valette, C., Lawrence, J. J., Electrophoresis1994, 15, 1552–1558.
[38] Rogers, M., Graham, J., Tonge, R. P., Proteomics 2003, 3,887–896.
[39] Baker, M., Busse, H., Vogt, M., Medical Imaging 2000: ImageProcessing, San Diego, SPIE, Bellingham 2000, 3979, 426–436.
© 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com