
Analysis of hybridoma mutants defective in synthesis of immunoglobulin M

Essential global role of CDC14 in DNA synthesisrevealed by chromosome underreplicationunrecognized by checkpoints in cdc14 mutantsStanimir Duleva,c, Christelle de Rentyb, Rajvi Mehtaa, Ivan Minkovc, Etienne Schwobb, and Alexander Strunnikova,1
aNational Institutes of Health, National Institute of Child Health and Human Development, Bethesda, Maryland, 20892; bInstitute of Molecular Genetics,Centre National de la Recherche Scientifique Unite Mixte de Recherche 5535, University Montpellier 2, 34293, France; and cUniversity of Plovdiv, Plovdiv4000, Bulgaria
Edited by Richard D. Kolodner, University of California at San Diego School of Medicine, La Jolla, CA, and approved June 30, 2009 (received for reviewMarch 18, 2009)
The CDC14 family of multifunctional evolutionarily conserved phos-phatases includes major regulators of mitosis in eukaryotes and ofDNA damage response in humans. The CDC14 function is also crucialfor accurate chromosome segregation, which is exemplified by itsabsolute requirement in yeast for the anaphase segregation ofnucleolar organizers; however the nature of this essential pathway isnot understood. Upon investigation of the rDNA nondisjunctionphenomenon, it was found that cdc14 mutants fail to completereplication of this locus. Moreover, other late-replicating genomicregions (10% of the genome) are also underreplicated in cdc14mutants undergoing anaphase. This selective genome-wide replica-tion defect is due to dosage insufficiency of replication factors in thenucleus, which stems from two defects, both contingent on thereduced CDC14 function in the preceding mitosis. First, a constitutivenuclear import defect results in a drastic dosage decrease for thosereplication proteins that are regulated by nuclear transport. Particu-larly, essential RPA subunits display both lower mRNA and proteinlevels, as well as abnormal cytoplasmic localization. Second, thereduced transcription of MBF and SBF-controlled genes in G1 leads tothe reduction in protein levels of many proteins involved in DNAreplication. The failure to complete replication of late replicons is theprimary reason for chromosome nondisjunction upon CDC14 dysfunc-tion. As the genome-wide slow-down of DNA replication does nottrigger checkpoints [Lengronne A, Schwob E (2002) Mol Cell 9:1067–1078], CDC14 mutations pose an overwhelming challenge to genomestability, both generating chromosome damage and undermining thecheckpoint control mechanisms.
DNA replication � genome � rDNA � RFA2 � SWI6 � importins
Successful chromosome segregation in mitosis requires the co-ordinated work of cellular regulatory circuits, which transmit
cell cycle signals to chromatin and spindle proteins. One of suchfactors is the Cdc14 protein, an evolutionary conserved proteinphosphatase (1–3). While in human cells a CDC14 ortholog hasbeen recently shown to play a key role in DNA damage response (4),studies on S. cerevisiae were mostly focused on Cdc14p roles inanaphase regulation and in the exit from mitosis. The scope ofCdc14p activity in budding yeast is believed to be limited toanaphase, because Cdc14p is sequestered in the nucleolus (5) inapparently inactive form (6) at other cell cycle stages. Therefore,while Cdc14 can potentially dephosphorylate many substrates (7, 8),the most studied physiological pathways are the anaphase pathways(FEAR and MEN), which are both dependent on the two sequen-tial bursts of Cdc14 release (1, 9). The cdc14 mutations cause amitotic exit block, but also display defects in nucleolar (10) andtelomeric (11) segregation. The mechanisms of chromosome seg-regation defects (11–15) in cdc14 mutants are generally poorlyunderstood. While condensin mutations phenocopy the cdc14rDNA nondisjunction (11, 16) and Cdc14p is required for conden-sin loading to rDNA (14), it is unlikely that condensin deficiency isthe primary cause of chromosome missegregation in cdc14 mutants.
Indeed the interference of rDNA transcription with condensinbinding (17) and the elevated levels of mitotic rDNA transcriptionin cdc14 cells (18–20) suggest that the role of Cdc14 in condensinloading is indirect. Incidentally, some role of CDC14 in DNAreplication was demonstrated genetically (21), and many replicationfactors could be direct substrates of Cdc14p (7). Cdc14 is alsoknown to organize prereplication complex formation and the G1transcriptional program, which controls expression of cyclins andreplication factors. While bulk DNA replication is complete atcdc14 arrest (22), the rDNA locus is sensitive to collision oftranscription with DNA replication (23, 24), which could be relatedto the specific increase of rDNA nondisjunction in cdc14 mutants.
Here we demonstrate that chromosome nondisjunction in cdc14mutants is largely caused by stretches of unreplicated DNA. Weshow that the compounding deregulation of both G1 transcriptionand nuclear import of replication factors in cdc14 is the mostprobable mechanism responsible for the DNA underreplication inthis mutant. This phenotype encompasses two cell cycles due to theconstitutive (hypomorphic) defect of the mutant Cdc14 protein,which likely affects multiple targets relevant to DNA replication.However, because DNA replication is not stalled in the mutants, theDNA replication checkpoint is not triggered, demonstrating that ahypomorphic mutation in a single gene can significantly compro-mise genome stability by generating genome-wide chromosomelesions that are invisible to checkpoint control mechanisms.
ResultsrDNA Is Underreplicated in cdc14. As the nature of the linkagebetween nondisjoined sister chromatids in the cdc14 mutant an-aphase remains unknown, we tested whether rDNA replication isdefective in cdc14 mutants. Due to its extended replicon size (25)and largely unidirectional replication, the rDNA locus must beparticularly sensitive to DNA underreplication, which might pro-duce irresolvable sister chromatids links (Fig. 1A). First, we used aplasmid loss assay (21) to examine the activity of rDNA ARS incdc14 mutants. The effect of cdc14 mutation was quite dramatic onplasmids carrying rDNA-derived origins: both colony size andtransformation efficiency were markedly reduced, and theminichromosome stability was below 5% (Fig. 1B). This suggeststhat the cdc14-1 allele is a strong hypomorph for rDNA originfunction. In contrast, the early-firing ARS1 or its weakened ars1�derivative showed a comparable stability in wild type and cdc14
Author contributions: S.D., E.S., and A.S. designed research; S.D., C.d.R., R.M., E.S., and A.S.performed research; C.d.R. and E.S. contributed new reagents/analytic tools; S.D., I.M., E.S.,and A.S. analyzed data; and A.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.
1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0900190106/DCSupplemental.
14466–14471 � PNAS � August 25, 2009 � vol. 106 � no. 34 www.pnas.org�cgi�doi�10.1073�pnas.0900190106
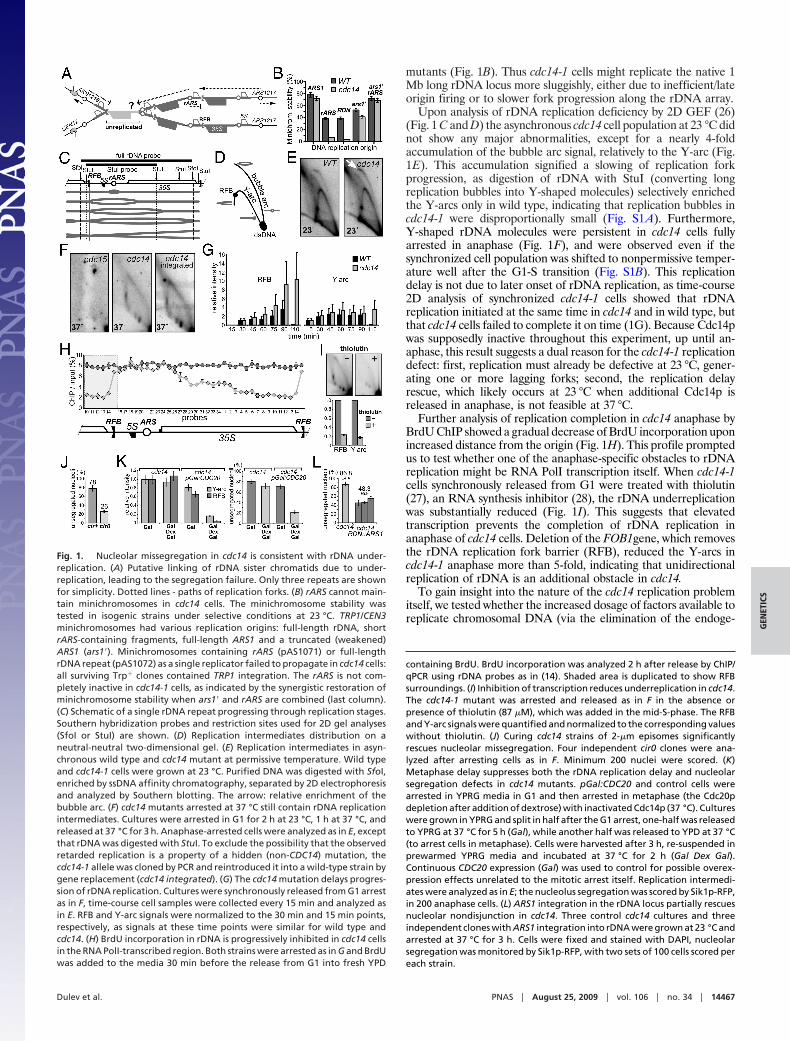
mutants (Fig. 1B). Thus cdc14-1 cells might replicate the native 1Mb long rDNA locus more sluggishly, either due to inefficient/lateorigin firing or to slower fork progression along the rDNA array.
Upon analysis of rDNA replication deficiency by 2D GEF (26)(Fig. 1 C and D) the asynchronous cdc14 cell population at 23 °C didnot show any major abnormalities, except for a nearly 4-foldaccumulation of the bubble arc signal, relatively to the Y-arc (Fig.1E). This accumulation signified a slowing of replication forkprogression, as digestion of rDNA with StuI (converting longreplication bubbles into Y-shaped molecules) selectively enrichedthe Y-arcs only in wild type, indicating that replication bubbles incdc14-1 were disproportionally small (Fig. S1A). Furthermore,Y-shaped rDNA molecules were persistent in cdc14 cells fullyarrested in anaphase (Fig. 1F), and were observed even if thesynchronized cell population was shifted to nonpermissive temper-ature well after the G1-S transition (Fig. S1B). This replicationdelay is not due to later onset of rDNA replication, as time-course2D analysis of synchronized cdc14-1 cells showed that rDNAreplication initiated at the same time in cdc14 and in wild type, butthat cdc14 cells failed to complete it on time (1G). Because Cdc14pwas supposedly inactive throughout this experiment, up until an-aphase, this result suggests a dual reason for the cdc14-1 replicationdefect: first, replication must already be defective at 23 °C, gener-ating one or more lagging forks; second, the replication delayrescue, which likely occurs at 23 °C when additional Cdc14p isreleased in anaphase, is not feasible at 37 °C.
Further analysis of replication completion in cdc14 anaphase byBrdU ChIP showed a gradual decrease of BrdU incorporation uponincreased distance from the origin (Fig. 1H). This profile promptedus to test whether one of the anaphase-specific obstacles to rDNAreplication might be RNA PolI transcription itself. When cdc14-1cells synchronously released from G1 were treated with thiolutin(27), an RNA synthesis inhibitor (28), the rDNA underreplicationwas substantially reduced (Fig. 1I). This suggests that elevatedtranscription prevents the completion of rDNA replication inanaphase of cdc14 cells. Deletion of the FOB1gene, which removesthe rDNA replication fork barrier (RFB), reduced the Y-arcs incdc14-1 anaphase more than 5-fold, indicating that unidirectionalreplication of rDNA is an additional obstacle in cdc14.
To gain insight into the nature of the cdc14 replication problemitself, we tested whether the increased dosage of factors available toreplicate chromosomal DNA (via the elimination of the endoge-
containing BrdU. BrdU incorporation was analyzed 2 h after release by ChIP/qPCR using rDNA probes as in (14). Shaded area is duplicated to show RFBsurroundings. (I) Inhibition of transcription reduces underreplication in cdc14.The cdc14-1 mutant was arrested and released as in F in the absence orpresence of thiolutin (87 �M), which was added in the mid-S-phase. The RFBand Y-arc signals were quantified and normalized to the corresponding valueswithout thiolutin. (J) Curing cdc14 strains of 2-�m episomes significantlyrescues nucleolar missegregation. Four independent cir0 clones were ana-lyzed after arresting cells as in F. Minimum 200 nuclei were scored. (K)Metaphase delay suppresses both the rDNA replication delay and nucleolarsegregation defects in cdc14 mutants. pGal:CDC20 and control cells werearrested in YPRG media in G1 and then arrested in metaphase (the Cdc20pdepletion after addition of dextrose) with inactivated Cdc14p (37 °C). Cultureswere grown in YPRG and split in half after the G1 arrest, one-half was releasedto YPRG at 37 °C for 5 h (Gal), while another half was released to YPD at 37 °C(to arrest cells in metaphase). Cells were harvested after 3 h, re-suspended inprewarmed YPRG media and incubated at 37 °C for 2 h (Gal Dex Gal).Continuous CDC20 expression (Gal) was used to control for possible overex-pression effects unrelated to the mitotic arrest itself. Replication intermedi-ates were analyzed as in E; the nucleolus segregation was scored by Sik1p-RFP,in 200 anaphase cells. (L) ARS1 integration in the rDNA locus partially rescuesnucleolar nondisjunction in cdc14. Three control cdc14 cultures and threeindependent clones with ARS1 integration into rDNA were grown at 23 °C andarrested at 37 °C for 3 h. Cells were fixed and stained with DAPI, nucleolarsegregation was monitored by Sik1p-RFP, with two sets of 100 cells scored pereach strain.
Fig. 1. Nucleolar missegregation in cdc14 is consistent with rDNA under-replication. (A) Putative linking of rDNA sister chromatids due to under-replication, leading to the segregation failure. Only three repeats are shownfor simplicity. Dotted lines - paths of replication forks. (B) rARS cannot main-tain minichromosomes in cdc14 cells. The minichromosome stability wastested in isogenic strains under selective conditions at 23 °C. TRP1/CEN3minichromosomes had various replication origins: full-length rDNA, shortrARS-containing fragments, full-length ARS1 and a truncated (weakened)ARS1 (ars1�). Minichromosomes containing rARS (pAS1071) or full-lengthrDNA repeat (pAS1072) as a single replicator failed to propagate in cdc14 cells:all surviving Trp� clones contained TRP1 integration. The rARS is not com-pletely inactive in cdc14-1 cells, as indicated by the synergistic restoration ofminichromosome stability when ars1� and rARS are combined (last column).(C) Schematic of a single rDNA repeat progressing through replication stages.Southern hybridization probes and restriction sites used for 2D gel analyses(SfoI or StuI) are shown. (D) Replication intermediates distribution on aneutral-neutral two-dimensional gel. (E) Replication intermediates in asyn-chronous wild type and cdc14 mutant at permissive temperature. Wild typeand cdc14-1 cells were grown at 23 °C. Purified DNA was digested with SfoI,enriched by ssDNA affinity chromatography, separated by 2D electrophoresisand analyzed by Southern blotting. The arrow: relative enrichment of thebubble arc. (F) cdc14 mutants arrested at 37 °C still contain rDNA replicationintermediates. Cultures were arrested in G1 for 2 h at 23 °C, 1 h at 37 °C, andreleased at 37 °C for 3 h. Anaphase-arrested cells were analyzed as in E, exceptthat rDNA was digested with StuI. To exclude the possibility that the observedretarded replication is a property of a hidden (non-CDC14) mutation, thecdc14-1 allele was cloned by PCR and reintroduced it into a wild-type strain bygene replacement (cdc14 integrated). (G) The cdc14 mutation delays progres-sion of rDNA replication. Cultures were synchronously released from G1 arrestas in F, time-course cell samples were collected every 15 min and analyzed asin E. RFB and Y-arc signals were normalized to the 30 min and 15 min points,respectively, as signals at these time points were similar for wild type andcdc14. (H) BrdU incorporation in rDNA is progressively inhibited in cdc14 cellsin the RNA PolI-transcribed region. Both strains were arrested as in G and BrdUwas added to the media 30 min before the release from G1 into fresh YPD
Dulev et al. PNAS � August 25, 2009 � vol. 106 � no. 34 � 14467
GEN
ETIC
S

nous 2-�m plasmid replicons) would suppress rDNA missegrega-tion in cdc14. Indeed, such curing of cdc14 strains significantlysuppressed the nucleolar nondisjunction phenotype (Fig. 1J). Fur-thermore, if cdc14 replication forks were slowed down, but notstalled, DNA replication might be rescued by delaying anaphaseentry. We therefore generated such a delay by the depletion ofCdc20p, which does not interfere with DNA replication machineryor spindle formation. Transient, but significantly longer than de-scribed previously (13), pGAL:CDC20 shut-off resulted in bothsignificant reduction of replication intermediates and in prominentrescue of the cdc14 nucleolar segregation defect (Fig. 1K). Thesedata are consistent with 2D gel analysis above and concur with theidea that delayed replication might be responsible, at least in part,for the cdc14 rDNA segregation phenotype. In rDNA, this delaymay be exacerbated by sparse positioning of active origins, becauseintegration of ARS1 into the rDNA locus partially rescues itssegregation (Fig. 1L). Thus, DNA replication forks in CDC14-deficient cells are likely active, but have reduced speed and/orefficiency.
Genome-Wide Defects in Chromosome Replication in cdc14. As Cdc14mutations are known to interfere with the function of non-rDNAorigins (21), we compared the cdc14 rDNA replication delay to thewhole genome. A time-course analysis of replication by DNAmolecular combing (25) (Fig. 2A and B), revealed genome-wideunderreplication in cdc14. In wild-type, the fraction of unreplicatedDNA fibers dropped from 24% at 60 min to the background 2% at90 min, as expected from the FACS data (Fig. 2A). In cdc14-1 cellshowever, there were still 18% unreplicated fibers at 120 min afterrelease, that is, 60 min after the entire culture had apparently (byFACS) completed S phase. This indicated that DNA replicationwas significantly delayed in cdc14-1 mutant cells at 37 °C, althoughprobably not fully blocked as this number fell from 18 to 10% duringthe following 30 min (Fig. 2D). Surprisingly, the replication com-pletion delay was only marginally more pronounced for rDNA,where 12% of the fibers remained unreplicated 150 min afterrelease from G1, that is, when cells are well arrested in anaphase(Fig. 2C). These data are consistent with Fig. 1 results and concurwith the idea that delayed replication might be responsible, at leastin part, for the chromosome segregation defects of cdc14-1 cells.
To better characterize which chromosomal regions are under-replicated, we first probed telomeric sites that were shown tomissegregate in cdc14 mutants (11). BrdU incorporation followedby ChIP/qPCR indicated that the two tested telomeres (TEL15R,TEL9L) were still underreplicated in cdc14 mutants 2 h afterrelease from G1 (nocodazole-arrested cells) (Fig. 2E). This under-replication was specific to cdc14-1 cells and not seen at early-replicating regions (around ARS305 or ARS309). Furthermore,using the Sir3p-mRFP telomeric marker, we showed that telomeresegregation was severely disorganized in cdc14 mutants undergoinganaphase: more than 60% of cells had incomplete telomere sepa-ration and up to one-third of the total population showed clearlyidentifiable signs of asymmetric segregation (Fig. 2F). Next weanalyzed the entire chromosomes III, IV, and V by BrdU ChIP-chipto get a more global picture of the cdc14 underrepplication defect.This revealed several distinct zones where the BrdU incorporationratio (cdc14 to wild type) was less than 0.5 (Fig. 2H and Fig. S2).A sample of such zones was validated by qPCR/ChIP, which showedan average 70% reduction of BrdU incorporation in cdc14 com-pared to wild type (Fig. 2I). Furthermore, from the analysis of three
Fig. 2. cdc14-specific DNA under-replication is not limited to rDNA. (A) DNAcontent (FACS) in a time-course experiment. Cultures were grown at 23 °C,arrested in G1 with �-factor, shifted to 37 °C for 30 min in YPD � BrdU and thenreleased at 37 °C in YPD � BrdU for combing analyses in (B–D). (B) Represen-tative image of combed DNA fibers showing fully replicated molecules andunreplicated molecules (red only) detected both with anti-ssDNA (red) andanti-BrdU (green) antibodies. Analysis was performed both for total genomicDNA and specifically for rDNA after cutting genomic DNA with restrictionenzymes (BamHI and XhoI) that cleave every 5–10 kb, but leave the 1.5-MbrDNA array intact. (C and D) Percent of DNA fibers (�100 kb) largely unrep-licated after DNA combing. DNA was analyzed at the indicated time for bothtotal genomic DNA and rDNA. Plotting the percentage of unreplicated DNAfibers (lacking BrdU signal for longer than 100 kb) with respect to time afterrelease shows the progression and completion of S phase. (E) BrdU incorpo-ration is reduced at telomeres in cdc14. Strains, arrest and labeling proceduresare as in A. BrdU incorporation into DNA was analyzed (2 h after release into37 °C/nocodazole from G1) by ChIP/qPCR using the probes covering the telo-meric regions of two chromosomes (15R and 9L). Neither probe set is uniquefor these locations, usually corresponding to 2–6 sites at other telomeres. Theearly-replicating regions of chromosome III were analyzed similarly, as acontrol. (F) Telomere segregation is impaired in cdc14. Strains carryingSIR3:mRFP gene replacement were arrested as in Fig. 1F, stained with DAPI(green), and scored for Sir3p-mRFP signal position (red). Unlike wild type,cdc14 had blocked telomere segregation (arrows and higher magnification),and prominent additional signs of asymmetric segregation (asterisk). (G)Telomere missegregation in cdc14-3 is suppressed by the loss of two-�mepisomes. Four independent cir0 clones were analyzed after arresting cells asin Fig. 1F. Minimum 200 nuclei were scored. TEL5R-adjacent chromosomal tagswere as in (11). (H) Late-replicating regions have reduced BrdU incorporationin cdc14. ChIP-chip analysis of BrdU incorporation is shown for chromosome IVusing the arrest/labeling as in A. Four experiments (SI Microarray Hybridiza-tion Data) were averaged for each strain and plotted as a cdc14 to wild typeratio. Red circles (i.e., individual microarray features) show the relative BrdU
incorporation in cdc14. The black curve: 10-feature running average. Bluecircles: replication timing of microarray features and origin positions (Lower)from (63). Arrows: locations of validation regions (in H). (I) cdc14 cells delayBrdU incorporation in late-replicating regions. ChIP/qPCR validation of ChIP-chip results in G was done as in E, with five regions (numbered according to G)where three non-overlapping 300-bp PCR probes were selected.
14468 � www.pnas.org�cgi�doi�10.1073�pnas.0900190106 Dulev et al.

chromosomes, it became evident that delayed BrdU incorporationcoincides with late-replicating regions (Fig. 2H and Fig. S2). Thus,cdc14 underreplication affects the whole genome, specifically re-gions containing late-firing origins and/or replicating late during Sphase.
DNA Checkpoint Is Not Activated by Underreplication in cdc14. Strik-ingly, chromosome underreplication in cdc14-1 mutants does notactivate the DNA replication checkpoint, since these cells arrest inanaphase/telophase. One possibility is that the checkpoint itselfcould be inactivated in cdc14 mutants, at least partially. This isunlikely, however, as cdc14 cells released from G1 at 37 °C in thepresence of hydroxyurea (Fig. 3A) or MMS (Fig. 3B) showed themetaphase arrest kinetics similar to wild type. The phosphorylationlevels of Rad53p (dependent on Mec1p or Tel1p) in cdc14 werecomparable to cdc15 (control) in response to both stalled forks(hydroxyurea) and DNA damage (MMS) (Fig. 3C). The Mec1p/Tel1p-dependent phosphorylation of H2A at the serine residue 129was also unaltered in cdc14 (Fig. 3D). Thus, an alternative expla-nation is likely, that is, slowed replication in cdc14 is carried out byreplication forks that are structurally normal, and therefore cannottrigger checkpoints (29, 30). This appears to be different from clb5mutants, which do not fire late origins (31) but do activate a Rad53response (32).
G1 Transcription and Nuclear Import Defects in cdc14 Impair Chro-mosome Replication. The pathway(s) responsible for cdc14 replica-tion insufficiency can be potentially revealed through the identifi-cation of Cdc14-dependent events in the previous cell cycle.Incidentally, dephosphorylation of Swi6p (Ser-160) by Cdc14 isneeded for its nuclear import in telophase (33) and formation of theSBF and MBF transciption complexes (34), which activate the G1expression of a wide range of genes essential for S phase. Indeed,we found that several key SWI6-controlled genes involved in DNAreplication and other processes showed reproducibly lower tran-script levels in cdc14 in G1 (at 23 °C), as well as 10 and 20 min intoS phase (at 37 °C) (Fig. 4A, 10-min time point is shown). Swi6poverexpression (i.e., dosage increase of dephosphorylated Swi6p)also notably improved rDNA segregation in cdc14 (Fig. 4B). Thus,due to insufficient Swi6p amount in the nucleus (33), the replicationmachinery as a whole is expressed at a lower level in cdc14 mutants.
Interestingly, the expression of RFA2, under direct MBFcontrol (34), was reduced more (Fig. 4A) than RFA1, which isactivated by the downstream factor Yhp1p (35). We investigatedthe RPA example further, and found that Rfa1 protein levelswere comparable in cdc14 and cdc15 strains (Fig. 4C). However,the Rfa2p level was somewhat decreased in cdc14-1 in G1, andespecially in mitosis (nocodazole and 37 °C arrests) (Fig. 4C).DNA damaging treatments, however, enabled the increase ofRfa2p levels even at 37 °C (Fig. 4C), probably due to theactivation of the DNA-damage induced transcription of RFA2(36, 37), further indicating that DNA checkpoints are functionalbut not triggered in cdc14 mutants.
As both Rfa1p and Rfa2p are known to be regulated by nuclearimport (38, 39), similarly to Swi6p, we analyzed their localization incdc14 cells. The RPA2 subunit is phosphorylated in mammals (40,41) and, in response to DNA damage (42), in yeast (by the nuclearkinase Mec1). However, in cdc14 cells, Rfa2p is not phosphorylatedat this site, even at 23 °C (Fig. 4D), indicating that Rfa2p may beconstitutively mislocalized. Indeed, Rfa2p-GFP was abnormallyenriched in the cytoplasm in arrested cdc14 cells, unlike in cdc15(Fig. 4E) and wild type. This defect is also evident at 23 °C (Fig.S3A), but can be alleviated by the transient expression of wild typeCdc14p activity in anaphase (Fig. S3B).
To ensure that GFP does not interfere with Rfa2p import weseparated nuclear and cytoplasmic fractions from untagged strains:both Rfa1p and Rfa2p were underrepresented in the nucleus andabundant in cytoplasm in cdc14 mutants (Fig. 4F). Furthermore,
Rfa2p was not able to interact with the Kap60/Kap95 complexknown to mediate the cell-cycle dependent import of Rfa2p into thenucleus (38, 39). As the RPA example in Fig. 4 shows, themechanism of replication insufficiency in cdc14 mutants has a dualnature: the constitutively decreased G1-specific transcription ofMBF/SBF-controlled genes and the insufficient/deregulated nu-clear import of at least some replication proteins from anaphasethrough G1-S transition. Furthermore, additional data suggest thatthe replication protein insufficiency in the nucleus, as well asnuclear transport imbalance, associated with cdc14 lesions areglobal defects. First, when we overexpressed the RPA holocomplex(43), it did not suppress cdc14 phenotypes; second, nucleo-cytoplasmic distribution of many proteins is changed in cdc14 cells(Fig. S4 and Table S2).
DiscussionUnderreplication Blocks rDNA Segregation in cdc14 Mutants. Incom-plete chromosome replication is rarely considered as a mechanismresponsible for chromosome nondisjunction during cell division(44), because cells with underreplicated DNA should not enteranaphase due to the activity of DNA integrity checkpoints (45, 46).However, the rDNA locus in budding yeast stands out in thisrespect, as it has persistent DSBs (47), which apparently do nottrigger a checkpoint response, because nucleolar chromatin isshielded from DNA damage checkpoint signaling (48–51). Weshow that in the cdc14 mutant the bulk of rDNA nondisjunction canbe attributed to incomplete DNA replication (Fig. 1). The inabilityof cdc14 mutant to down-regulate transcription in mitosis (20) alsocontributes to underreplication specifically at rDNA (Fig. 1I).
Fig. 3. cdc14 mutants are proficient in DNA-damage checkpoints. (A and B)cdc14 mutants are properly arrested in response to DNA damage. Isogenic strainswere grown to log phase at 23 °C, arrested in G1, and released at 37 °C for 2 h (A)with hydroxyurea (HU, 0.2 M) or (B) with MMS (0.01%). Cell were stained withDAPI and counted. (C) Phosphorylation of Rad53p is properly induced in cdc14mutants in response to DNA damage. Cells were grown at 23 °C, arrested in G1and released at 37 °C with either hydroxyurea (HU, 0.2 M) or MMS (0.01%), for3 h. Checkpoint induction was monitored by western blotting with anti-Rad53antibodies. (D) H2A is phosphorylated in cdc14 mutants in response to DSB.Cultures were grown in YPD to log phase at 23 °C, arrested in G1 and released inYPD at 37 °C for 3 h without any drug (37°), with hydroxyurea (HU, 0.2 M), withMMS (0.01%) or with nocodazole (Nz). The DSB presence was determined bywestern blotting with anti-phospho-S129 H2A antibody (64).
Dulev et al. PNAS � August 25, 2009 � vol. 106 � no. 34 � 14469
GEN
ETIC
S

However, the underreplication-related chromosome nondisjunc-tion in cdc14 mutants is not rDNA-specific (Fig. 2) and is apparentlydue to a complex defect, which interferes with duplication of thelate-replicating regions of the genome. Experiments in Fig. 1Kindicate, however, that replication forks are still active duringmitosis in cdc14 mutants, as an extended metaphase delay enablesboth the completion of rDNA replication and the improvement ofnucleolar segregation.
Mechanisms of Underreplication in cdc14. The molecular combingexperiments have shown that the cdc14 underreplication phenotypeis only slightly more penetrant for rDNA than other parts of thegenome (Fig. 2 C and D). This initial observation was extensively
confirmed by other approaches (Fig. 2 E–I). Thus, chromosome loss(12) in cdc14 can be explained by the presence of unreplicatedregions in chromosomes. The asymmetric and blocked segregationof telomeres (Fig. 2 F and G) is consistent with such an assessment.
The specific sensitivity of late replicating regions to the Cdc14function (Fig. 2 H and I) let us to hypothesize that the late DNAreplication ‘‘bottleneck’’ in cdc14 mutant cells is not a defect in asingle specific replication protein, but is a cumulative quantitativeinsufficiency of replication machinery (Fig. 1J). This low dosage ofreplication machinery apparently enables initiation of early originsbut prevents effective replication from the late origins, as replica-tion factors are exhausted by the commitment to early forks. cdc14mutants have at least two defects in the amount and balance ofreplication proteins (Fig. S5). First, the SBF/MBF-dependent pro-duction of the optimal supply of replication factors is constitutivelydecreased due to insufficient Swi6p import into the nucleus (Fig. 4A–C). Second, cdc14 mutants show defects in the nuclear importboth at 23 °C and 37 °C, which is illustrated with a particular caseof Rfa1p and Rfa2p (Fig. 4 D–G). Thus, the impaired functions ofCdc14p likely most drastically affect replication proteins that areregulated by both Swi6p and nuclear entry. The execution point ofthis Cdc14p activity (i.e., activation of replication-specific nuclearimport pathways) is likely in anaphase (Fig. S3B), but the resultingdefect of the impaired nuclear import in cdc14 mutants has a longspan: from anaphase (e.g., Rfa2p re-import) to G1-S transition(e.g., importing of the newly made pool of Rfa1p and Rfa2p, Fig.S3A and Fig. 4F).
The spectrum of Cdc14 phosphatase substrates in anaphase,which are relevant for the following G1 and S-phase, remains to beidentified. It is conceivable that several cargo proteins regulated bynuclear import have to be directly dephosphorylated to enter thenucleus, by analogy with Swi6p (Fig. S5). Indeed, preliminary datafrom 2D-electrophoresis indicate that the spectrum of phosphor-ylated proteins in cdc14 cytoplasm (Pro-Q staining) is significantlydistinct from cdc15 mutants. However, the prolonged cell-cyclespan of the import defect, including the G1-S transition (whenCdc14p is inactive), suggests a possibility that the import machineryitself is also a substrate of the Cdc14 phosphatase. Indeed, a numberof nuclear pore components interacting with Kap60p are CDKsubstrates (52), that is, potential Cdc14p targets; mutations in theNup84 subcomplex are synthetically lethal with FEAR pathwaymutations (53).
Dual Replication/Checkpoint Insufficiency in cdc14 Mutants. The factthat DNA under-replication does not trigger DNA damage check-points in cdc14 mutants may either indicate that (a) cdc14 repli-cation forks are not recognized as abnormal by checkpoints or that(b) the checkpoint system itself is impaired. However, a severeimpairment of checkpoints is unlikely, because critical DNAdamage-induced branches of the ATM/ATR signaling pathway areunaffected (Fig. 3). At the same time, the loss of Mec1p-mediatedphosphorylation of S-122 in Rfa2p (Fig. 4D) indirectly indicates thatsome, more obscure, aspects of ATM/ATR activities may becompromised. However, the requirement of Mec1p for replicationof late-replicating regions is attributed to Mec1’s role in the forkprogression itself (54, 55) (through direct phosphorylation ofreplication machinery), not due to a defect in ATR signaling. Thus,it is compelling to hypothesize that in cdc14 mutants failure torecognize underreplication is not due to a checkpoint system defect,but rather is a function of the slow replication speed itself, as wasshown for DNA polymerase epsilon mutants (56, 57).
In conclusion, cdc14 mutants present a case of a lesion, which isextremely challenging to the cell and thus can potentially result ina rapid destabilization of the genome. Namely, a single pointmutation creates profound chromosomal damage (underreplica-tion), as well as eludes its detection by checkpoints. Genomestability mechanisms are found to be compromised in many cancercell types, and thus are believed to contribute to multistep onco-
Fig. 4. Both transcription and nuclear import defects lead to replicationdeficiency in cdc14. (A) Genes under control of Swi6p have partially decreasedtranscription in cdc14 mutants. Cells were arrested in G1 at 23 °C for 3 h, shiftedat 37 °C for 30 min, and then released from G1 for 10 min. Expression levels weredeterminedbyqRT-PCR.All signalsarenormalizedtoTUB2 transcript, rDNANTS1- negative control. (B) Swi6p overexpression partially rescues the nucleolus seg-regation defect in cdc14. The cdc14 strain with Sik1p-RFP was transformed witheither the pRS426gal vector (mock) or the pGAL:GST:SWI6 plasmid [derived fromthe library in (65)]. The cells were grown at 23 °C in plasmid-selective raffinosemedia, arrested with alpha factor in YPRaf for 2 h at 23 °C, galactose was addedfor 1 h/37 °C in the presence of alpha factor, and the cells were the released intoYPRG/37 °C for 3 h. (C) RPA subunit imbalance in cdc14 mutants. Cells werearrested in G1 at 23 °C and released at 37 °C for 3 h untreated (37°), withhydroxyurea (HU, 0.2 M), with MMS (0.01%) or with nocodazole (NZ). Alpha-factor arrested cells were incubated at 37° for 1 h. Rfa2p detection: anti-GFPantibodies, Rfa1p detection: by specific antibodies. (D) Rfa2p phosphorylation isaltered in cdc14 mutants even when expression is induced to wild-type level.Strains were arrested in G1 at 23 °C and then released to 37 °C or, in the presenceof hydroxyurea (HU, 0.2 M), to 23 °C. Rfa2p detection as in E. The Mec1-dependentRfa2pSer-122phosphorylationwasdetectedwithaspecificantibody.Cdc28p levels (anti-PSTAIRE antibody): the loading control. (E) Rfa2p-GFP islargely delocalized from the nucleus in arrested cdc14 cells. Cultures were grownin YPD to log phase at 23 °C and shifted to 37 °C for 3 h. (F) Rfa1p and Rfa2paccumulate in cytoplasm in cdc14 mutants. Both cultures were arrested at 23 °Cfor 3 h, shifted at 37 °C for 30 min and released at 37 °C, samples were collectedat indicated time points. Localization of Rfa1p and Rfa2p in nuclear (nuc) andcytoplasmic (cyt) fractions was analyzed by western blotting with specific poly-clonal antibodies. Hmo1: chromatin and loading control. (G) Rfa2p does notinteract with Kap60p-Kap95p importin complex in cdc14. Cells were arrested at23 °C for 3 h, shifted at 37 °C for 30 min and split in half, with one half releasedfrom G1 at 37 °C. Rfa2p was immunoprecipitated from both G1 and anaphasecells, and analyzed with anti-Rfa2, anti-Kap60, and anti-Kap95 antibodies.
14470 � www.pnas.org�cgi�doi�10.1073�pnas.0900190106 Dulev et al.

genic transformations. Thus, hypomorphic mutations in CDC14genes in mammals can potentially generate a wide spectrum ofgenome instability signatures seen in cancers. Strikingly, a recentstudy points out that expression of CDC14B is decreased in a varietyof human tumors (4).
MethodsAdditional methods, including DNA combing (58), are detailed in SI Methods. Allyeast strains (Table S1) handling was done according to standard protocols. ARSvariant plasmids were constructed based on the pAS231 backbone, which con-tained truncated ars1� and was generated by inserting the AluI-SalI fragment ofCEN3 into pRS404 digested with Ecl136 and XhoI. All other variants were con-structed by replacing the HincII-PstI ars1� fragment. Cloning of the cdc14-1 allelewas done from a mutant strain by PCR. The TRP1 marker was inserted betweenthe engineered SpeI and BglII restriction sites within the CDC14 terminator,giving the pAS1063 targeting construct.
For 2D DNA analysis, DNA was purified by CTAB method, as in (59), anddigested with SfoI or StuI. In some experiments, ssDNA was enriched by affinity
chromatography using BND-cellulose as in (23). Neutral-neutral 2D gel electro-phoreses was as described by (47), with minor modifications.
For analysis of BrdU incorporation by ChIP, yeast strains were labeled essen-tiallyas inDNAcombingexperiment:after2harrestat37 °C, cellswerefixedwith4 °C PBS with 0.1% sodium azide, and total DNA was extracted with QIAGENgenomic DNA kit. DNA was precipitated as in (60), except that replicated regionswere captured using Protein G Dynabeads (Dynal) prebound with anti-BrdUantibody (Millipore). Immunoprecipitated material was analyzed by qPCR. ForChIP-CHIP experiments, precipitated DNA was further purified and amplified asin (61), 10 �g of DNA was sheared into 100-bp fragments by partial DNaseIdigestion and end-labeled with biotin-N6-ddATP by recombinant calf terminaltransferase. Probes were hybridized to the Affymetrix SC3456a pseudotilingarray(chromosomes III, IV,V,andpartialVI) (62).Sampleswerehybridizedfor16hat 42 °C in a GeneChip hybri-oven 320 (Affymetrix), washed and stained on aGeneChip fluidic station 450 (FS450�0001 protocol, Affymetrix).
ACKNOWLEDGMENTS. We thank A. Amon, S. Brill, M. Nomura, E. Schiebel, I.Dawid, and G. Brush for research materials; R. Woodgate, M. Lichten, and D.Gordenin for critical comments; and N. Sghir for help on DNA combingexperiments. E.S. acknowledges Association pour la Recherche sur le Cancer(ARC) and Institut National du Cancer (INCa) for financial support.
1. Stegmeier F, Amon A (2004) Closing mitosis: The functions of the Cdc14 phosphataseand its regulation. Annu Rev Genet 38:203–232.
2. Trautmann S, et al. (2004) The S. pombe Cdc14-like phosphatase Clp1p regulateschromosome biorientation and interacts with Aurora kinase. Dev Cell 7:755–762.
3. Krasinska L, et al. (2007) Regulation of multiple cell cycle events by Cdc14 homologuesin vertebrates. Exp Cell Res 313:1225–1239.
4. Bassermann F, et al. (2008) The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134:256–267.
5. Visintin R, et al. (1999) Cfi1 prevents premature exit from mitosis by anchoring Cdc14phosphatase in the nucleolus. Nature 398:818–823.
6. Traverso EE, et al. (2001) Characterization of the Net1 cell cycle-dependent regulatorof the Cdc14 phosphatase from budding yeast. J Biol Chem 276:21924–21931.
7. Bloom J, Cross FR (2007) Novel role for Cdc14 sequestration: Cdc14 dephosphorylatesfactors that promote DNA replication. Mol Cell Biol 27:842–853.
8. Holt LJ, et al. (2007) Evolution of Ime2 phosphorylation sites on Cdk1 substrates provides amechanism to limit the effects of the phosphatase Cdc14 in meiosis. Mol Cell 25:689–702.
9. Sullivan M, Uhlmann F (2003) A non-proteolytic function of separase links the onset ofanaphase to mitotic exit. Nat Cell Biol 5:249–254.
10. Granot D, Snyder M (1991) Segregation of the nucleolus during mitosis in budding andfission yeast. Cell Motil Cytoskeleton 20:47–54.
11. D’Amours D, et al. (2004) Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell 117:455–469.
12. Palmer R, et al. (1990) Mitotic transmission of artificial chromosomes in cdc mutants ofthe yeast, S cerevisiae. Genetics 125:763–774.
13. Sullivan M, et al. (2004) Cdc14 phosphatase induces rDNA condensation and resolvescohesin-independent cohesion during budding yeast anaphase. Cell 117:471–482.
14. WangBD,etal. (2004)Cdc14p/FEARpathwaycontrols segregationofnucleolus inS.cerevisiaeby facilitating condensin targeting to rDNA chromatin in anaphase. Cell Cycle 3:960–967.
15. Torres-Rosell J ,et al. (2004) Nucleolar segregation lags behind the rest of the genomeand requires Cdc14p activation by the FEAR network. Cell Cycle 3:496–502.
16. Freeman L, et al. (2000) The condensin complex governs chromosome condensationand mitotic transmission of rDNA. J Cell Biol 149:811–824.
17. Wang BD, et al. (2006) Condensin function in mitotic nucleolar segregation is regulatedby rDNA transcription. Cell Cycle 5:2260–2267.
18. Tomson BN, et al. (2006) Ribosomal DNA transcription-dependent processes interferewith chromosome segregation. Mol Cell Biol 26:6239–6247.
19. Machin F, et al. (2006) Transcription of ribosomal genes can cause nondisjunction. J CellBiol 173:893–903.
20. Clemente-Blanco A, et al. (2009) Cdc14 inhibits transcription by RNA polymerase Iduring anaphase. Nature 458:219–222.
21. Hogan E, Koshland D (1992) Addition of extra origins of replication to a minichromo-some suppresses its mitotic loss in cdc6 and cdc14 mutants of S. cerevisiae. Proc NatlAcad Sci USA 3098–3102.
22. Fitzpatrick PJ, et al. (1998) DNA replication is completed in S. cerevisiae cells that lackfunctional Cdc14, a dual-specificity protein phosphatase. Mol Gen Genet 258:437–441.
23. Muller M, et al. (2000) Replication of yeast rDNA initiates downstream of transcrip-tionally active genes. Mol Cell 5:767–777.
24. Takeuchi Y, et al. (2003) Transcription-dependent recombination and the role of forkcollision in yeast rDNA. Genes Dev 17:1497–1506.
25. Pasero P, et al. (2002) Single-molecule analysis reveals clustering and epigeneticregulation of replication origins at the yeast rDNA locus. Genes Dev 16:2479–2484.
26. Brewer BJ, et al. (1988) Analysis of replication intermediates by two-dimensionalagarose gel electrophoresis. Methods Enzymol 6:229–234.
27. Khachatourians GG, Tipper DJ (1974) Inhibition of messenger ribonucleic acid synthesisin Escherichia coli by thiolutin. J Bacteriol 119:795–804.
28. Grigull J, et al. (2004) Genome-wide analysis of mRNA stability using transcriptioninhibitors and microarrays reveals posttranscriptional control of ribosome biogenesisfactors. Mol Cell Biol 24:5534–5547.
29. Lengronne A, Schwob E (2002) The yeast CDK inhibitor Sic1 prevents genomic insta-bility by promoting replication origin licensing in late G(1). Mol Cell 9:1067–1078.
30. Shimada K, et al. (2002) ORC and the intra-S-phase checkpoint: A threshold regulatesRad53p activation in S phase. Genes Dev 16:3236–3252.
31. McCune HJ, et al. (2008) The temporal program of chromosome replication: Genome-wide rReplication in clb5{Delta} S. cerevisiae. Genetics 180:1833–1847.
32. Gibson DG, et al. (2004) Diminished S-phase cyclin-dependent kinase function elicits vitalRad53-dependent checkpoint responses in S. cerevisiae. Mol Cell Biol 24:10208–10222.
33. Geymonat M, et al. (2004) Clb6/Cdc28 and Cdc14 regulate phosphorylation status andcellular localization of Swi6. Mol Cell Biol 24:2277–2285.
34. Iyer VR, et al. (2001) Genomic binding sites of the yeast cell-cycle transcription factorsSBF and MBF. Nature 409:533–538.
35. Horak CE, et al. (2002) Complex transcriptional circuitry at the G1/S transition in S.cerevisiae. Genes Dev 16:3017–3033.
36. Benton MG, et al. (2006) Analyzing the dose-dependence of the S. cerevisiae global transcrip-tional response to methyl methanesulfonate and ionizing radiation. BMC Genomics 7:305.
37. Fry RC, et al. (2006) The DNA-damage signature in S. cerevisiae is associated withsingle-strand breaks in DNA. BMC Genomics 7:313.
38. Yoshida K, Blobel G (2001) The karyopherin Kap142p/Msn5p mediates nuclear importand nuclear export of different cargo proteins. J Cell Biol 152:729–740.
39. Belanger KD, et al. (2004) The karyopherin Msn5/Kap142 requires Nup82 for nuclearexport and performs a function distinct from translocation in RPA protein import. J BiolChem 279:43530–43539.
40. Bartrand AJ, et al. (2004) DNA stimulates Mec1-mediated phosphorylation of replica-tion protein A. J Biol Chem 279:26762–26767.
41. Brush GS, Kelly TJ (2000) Phosphorylation of the replication protein A large subunit inthe S. cerevisiae checkpoint response. Nucleic Acids Res 28:3725–3732.
42. Mallory JC, et al. (2003) Amino acid changes in Xrs2p, Dun1p, and Rfa2p that removethe preferred targets of the ATM family of protein kinases do not affect DNA repair ortelomere length in S cerevisiae DNA Repair (Amst) 2:1041–1064.
43. Maniar HS, et al. (1997) Roles of replication protein-A subunits 2 and 3 in DNAreplication fork movement in S. cerevisiae. Genetics 145:891–902.
44. Belyaeva ES, et al. (2006) DNA underreplication in intercalary heterochromatin regionsin polytene chromosomes of Drosophila melanogaster correlates with the formationof partial chromosomal aberrations and ectopic pairing. Chromosoma 115:355–366.
45. Sanchez Y, et al. (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1in yeast cell cycle checkpoint pathways. Science 271:357–360.
46. Krishnan V, et al. (2004) DNA replication checkpoint prevents precocious chromosomesegregation by regulating spindle behavior. Mol Cell 16:687–700.
47. Burkhalter MD, Sogo JM (2004) rDNA enhancer affects replication initiation andmitotic recombination: Fob1 mediates nucleolytic processing independently of repli-cation. Mol Cell 15:409–421.
48. Torres-Rosell J, et al. (2007) Anaphase onset before complete DNA replication withintact checkpoint responses. Science 315:1411–1415.
49. Ozenberger BA, Roeder GS (1991) A unique pathway of double-strand break repairoperates in tandemly repeated genes. Mol Cell Biol 11:1222–1231.
50. Gangloff S, et al. (1996) Gene conversion plays the major role in controlling the stabilityof large tandem repeats in yeast. EMBO J 15:1715–1725.
51. Torres-Rosell J, et al. (2007) The Smc5-Smc6 complex and SUMO modification of Rad52regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9:923–931.
52. Ubersax JA, et al. (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425:859–864.53. Ye P, et al. (2005) Gene function prediction from congruent synthetic lethal interac-
tions in yeast. Mol Syst Biol 1:2005 0026.54. Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphory-
lation in response to DNA replication stress activates Rad53. Genes Dev 17:1755–1767.55. Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting
breaks in replication slow zones. Science 297:602–606.56. Navas TA, et al. (1995) DNA polymerase epsilon links the DNA replication machinery to
the S phase checkpoint. Cell 80:29–39.57. Dua R, et al. (1998) Role of the putative zinc finger domain of S. cerevisiae DNA
polymerase epsilon in DNA replication and the S/M checkpoint pathway. J Biol Chem273:30046–30055.
58. Lengronne A, et al. (2001) Monitoring S phase progression globally and locally usingBrdU incorporation in TK(�) yeast strains. Nucleic Acids Res 29:1433–1442.
59. Allers T, Lichten M (2000) A method for preparing genomic DNA that restrains branchmigration of Holliday junctions. Nucleic Acids Res 28:e6.
60. Katou Y, et al. (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stablereplication-pausing complex. Nature 424:1078–1083.
61. Winzeler EA, et al. (1998) Direct allelic variation scanning of the yeast genome. Science281:1194–1197.
62. Lengronne A, et al. (2004) Cohesin relocation from sites of chromosomal loading toplaces of convergent transcription. Nature 430:573–578.
63. Raghuraman MK, et al. (2001) Replication dynamics of the yeast genome. Science294:115–121.
64. Redon C, et al. (2003) Yeast histone 2A serine 129 is essential for the efficient repair ofcheckpoint-blind DNA damage. EMBO Rep 4:678–684.
65. Ptacek J, et al. (2005) Global analysis of protein phosphorylation in yeast. Nature438:679–684.
Dulev et al. PNAS � August 25, 2009 � vol. 106 � no. 34 � 14471
GEN
ETIC
S
Copyright © 2022 FDOKUMEN




















