
CXCL10 blockade protects mice from cyclophosphamide-induced cystitis

Endothelin receptor blockade combined with phosphodiesterase-5 inhibitionincreases right ventricular mitochondrial capacity in pulmonaryarterial hypertension
Koen T. B. Mouchaers,1 Ingrid Schalij,1 Amanda M. G. Versteilen,2 Awal M. Hadi,1
Geerten P. van Nieuw Amerongen,2 Victor W. M. van Hinsbergh,2 Pieter E. Postmus,1
Willem J. van der Laarse,2 and Anton Vonk-Noordegraaf1
Departments of 1Pulmonary Diseases and 2Physiology, Institute for Cardiovascular Research, Vrije Universiteit UniversityMedical Center, Amsterdam, The Netherlands
Submitted 14 August 2008; accepted in final form 13 April 2009
Mouchaers KT, Schalij I, Versteilen AM, Hadi AM, van NieuwAmerongen GP, van Hinsbergh VW, Postmus PE, van der LaarseWJ, and Vonk-Noordegraaf A. Endothelin receptor blockade combinedwith phosphodiesterase-5 inhibition increases right ventricular mito-chondrial capacity in pulmonary arterial hypertension. Am J PhysiolHeart Circ Physiol 297: H200–H207, 2009. First published April 24,2009; doi:10.1152/ajpheart.00893.2008.—Pulmonary arterial hyper-tension (PAH) is often treated with endothelin (ET) receptor blockadeor phosphodiesterase-5 (PDE5) inhibition. Little is known about thespecific effects on right ventricular (RV) function and metabolism.We determined the effects of single and combination treatment withBosentan [an ET type A (ETA)/type B (ETB) receptor blocker] andSildenafil (a PDE5 inhibitor) on RV function and oxidative metabo-lism in monocrotaline (MCT)-induced PAH. Fourteen days after MCTinjection, male Wistar rats were orally treated for 10 days withBosentan, Sildenafil, or both. RV catheterization and echocardiogra-phy showed that MCT clearly induced PAH. This was evidenced byincreased RV systolic pressure, reduced cardiac output, increasedpulmonary vascular resistance (PVR), and reduced RV fractionalshortening. Quantitative histochemistry showed marked RV hypertro-phy and fibrosis. Monotreatment with Bosentan or Sildenafil had noeffect on RV systolic pressure or cardiac function, but RV fibrosis wasreduced and RV capillarization increased. Combination treatment didnot reduce RV systolic pressure, but significantly lowered PVR, andnormalized cardiac output, RV fractional shortening, and fibrosis.Only combination treatment increased the mitochondrial capacity ofthe RV, as reflected by increased succinate dehydrogenase and cyto-chrome c oxidase activities, associated with an activation of PKG, asindicated by increased VASP phosphorylation. Moreover, significantinteractions were found between Bosentan and Sildenafil on PVR,cardiac output, RV contractility, PKG activity, and mitochondrialcapacity. These data indicate that the combination of Bosentan andSildenafil may beneficially contribute to RV adaptation in PAH, notonly by reducing PVR but also by acting on the mitochondria in theheart.
Bosentan; Sildenafil; mitochondria; oxidative capacity; chronic heartfailure
PULMONARY ARTERIAL HYPERTENSION (PAH) is a severe progres-sive disease of the small lung vasculature leading to increasedpulmonary vascular resistance (PVR). The right ventricle (RV)hypertrophies to withstand the rise in PVR and maintaincardiac output (CO) (16, 48). Subsequently, RV contractiledysfunction occurs, and failure may develop (8, 51). Many
factors influence the capacity of the RV to adapt to PAH, suchas the degree of RV wall stress, myocardial ischemia, ormicrovascular endothelial dysfunction. However, the exactsequence of events leading the RV toward failure, and theeffects of treatments to counteract this process remain uncer-tain (51).
Various treatments for PAH have been developed over time,which primarily act as vasodilators of the pulmonary vascula-ture. These treatments include anticoagulant therapy, prostacy-clin infusion, endothelin (ET) receptor blockade, and phospho-diesterase-5 (PDE5) inhibition (9, 25). ET receptor blockadeby Bosentan and PDE5 inhibition by Sildenafil are both com-mon strategies in PAH treatment (9, 25, 43). The ET system ishighly active in PAH and causes sustained vasoconstriction ofpulmonary arteries. It increases the mitogenic activity ofsmooth muscle cells and fibroblasts in the pulmonary vesselwall, thereby decreasing the lumen of pulmonary vessels, alsocontributing to increased pulmonary vascular resistance (PVR)(24, 40). In addition, it is known that ET receptor expression inthe myocardium of the RV increases upon PAH (47). Simi-larly, PDE5 is overactive in the pulmonary vasculature of PAHpatients, where it prevents normal relaxation of smooth musclecells in the vessel wall by excessive degradation of cGMP.PDE5 inhibition causes relaxation of pulmonary vessels andalso exerts antiproliferative effects on pulmonary arterysmooth muscle cells (24, 53). In addition, it has been shownthat PDE5 is highly expressed in the RV of PAH patients andrats, and that acute inhibition increases RV contractility (35).
Although these reports have shed light on the etiology andtreatment of the arterial pathology and pulmonary hyperten-sion, limited information is available regarding counteractingthe (mal)adaptation of the heart in PAH (34, 46). Central in this(mal)adaptation is the hypertrophy of RV cardiomyocytes thatis needed to generate sufficient power to perfuse the lungvascular bed. However, the increased demand of power outputof the RV in PAH requires a rise in O2 delivery to RVcardiomyocytes to maintain sufficient ATP synthesis. Theincreased O2 consumption and diffusion distances for O2 inhypertrophied cardiomyocytes, in combination with the re-duced capillary density, can lead to a mismatch between O2
supply and demand (15, 54). In turn, this can cause hypoxia inindividual cardiomyocytes and may lead to cellular damage,contributing to RV failure (45, 51).
Bosentan and Sildenafil have been extensively studied inregard to their effects on pulmonary vessels. However, knowl-edge about their specific effects on the RV is still limited. We
Address for reprint requests and other correspondence: A. Vonk-Noordegraaf,Dept. of Pulmonology, C5-P, Vrije Universiteit Univ. Medical Center, PO Box7057, Amsterdam 1007 MB, The Netherlands (e-mail: [email protected]).
Am J Physiol Heart Circ Physiol 297: H200–H207, 2009.First published April 24, 2009; doi:10.1152/ajpheart.00893.2008.
0363-6135/09 $8.00 Copyright © 2009 the American Physiological Society http://www.ajpheart.orgH200

hypothesized that treatment with Bosentan, Sildenafil, or theircombination affects cardiac function and determinants for RVcontractility and oxidative metabolism in a rat reversal modelof monocrotaline (MCT)-induced PAH. To this end, we stud-ied RV contractility using echocardiography and measured RVcardiomyocyte hypertrophy as a measure for O2 diffusiondistances. In addition, we studied the RV myoglobin concen-tration as a parameter for intracellular O2 diffusion and buff-ering and RV capillary density as a determinant of O2 supplycapacity. Furthermore, we investigated two mitochondrial en-zymes as measures of mitochondrial capacity, changes incardiomyocyte PKG activity, and the formation of fibrosis inthe RV.
MATERIALS AND METHODS
See the online Supplement Material for additional details onreagents and methods.1
Study design. PAH was induced in 28 male Wistar rats by a singleinjection of MCT (MCT rats; 40 mg/kg sc, Sigma-Aldrich, Zwijndrecht,The Netherlands). After 14 days, PAH rats were randomly assigned toreceive oral treatment for 10 days with Bosentan (100mg �kg�1 �day�1, n � 7, Actelion Pharmaceuticals, Allschwil, Swit-zerland), Sildenafil (1 mg �kg�1 �day�1, n � 7, Pfizer), or theircombination (same dosages, n � 7; Fig. 1). On day 25, rats wereeuthanized by heart excision under isoflurane anesthesia (�3.5%). Allexperiments were approved by the Institutional Animal Care and UseCommittee and conformed with Helsinki conventions for the use andcare of animals.
Echocardiographic and hemodynamic measurements. On days 0,14, and 25, M-mode echocardiographic images were made of the RVand left ventricle (LV) at midpapillary level and the heart rate (HR)was recorded using an Aloka SSD4000 ultrasonographic system(Biomedic, Almere, The Netherlands; Fig. 1). RV and LV end-systolic diameters (ESDs) and end-diastolic diameters (EDDs) ofthree cardiac cycles in four different recordings were measured usingScion Image (version 4.0.3.2, Scioncorp). Averaged values of theLVEDD and LVESD were used to calculate stroke volume (SV) andCO by calculating end-systolic and end-diastolic volumes assumingthe LV as a prolate spheroid (14, 42). In addition, RV and LVfractional shortening (FS) values were calculated. Before death, theRV systolic pressure (RVSP) was measured using a Millar pressurecatheter (Millar) by direct insertion through the RV walls afterthoracotomy. Data were obtained using a PowerLab setup (AD In-struments, New South Wales, Australia). Systemic blood pressure wasmonitored using a fluid-filled catheter inserted into the left carotidartery. PVR was estimated according to Poiseuille’s law as previouslydescribed (7, 21).
Quantitative histochemistry of myocardial tissue. Cryosections ofmyocardial tissue (5 �m) were incubated for succinate dehydrogenase(SDH) activity (15), and adjacent sections were incubated for cyto-chrome c oxidase activity (39). SDH and cytochrome c oxidaseactivities were measured in 40 cardiomyocytes of the RV and LV.SDH activity determined in this way is proportional to the maximumrate of O2 consumption in cardiomyocytes (15). Cytochrome c oxi-dase activity was determined as a confirmation for SDH activity.Adjacent sections were stained with hematoxylin and eosin. Mean RVand LV cardiomyocyte cross-sectional area (CSA) of 40 cardiomyo-cytes were measured randomly. The myoglobin concentration wasdetermined in both ventricles by calibrated histochemistry (47a). Thenumber of capillaries per cardiomyocyte was determined using col-lagen type IV staining (15). Picrosirius red-stained sections were used
to determine the amount of fibrosis using polarization filters. Cyto-chrome c release was determined as an indicator of mitochondrialmembrane integrity (47b).
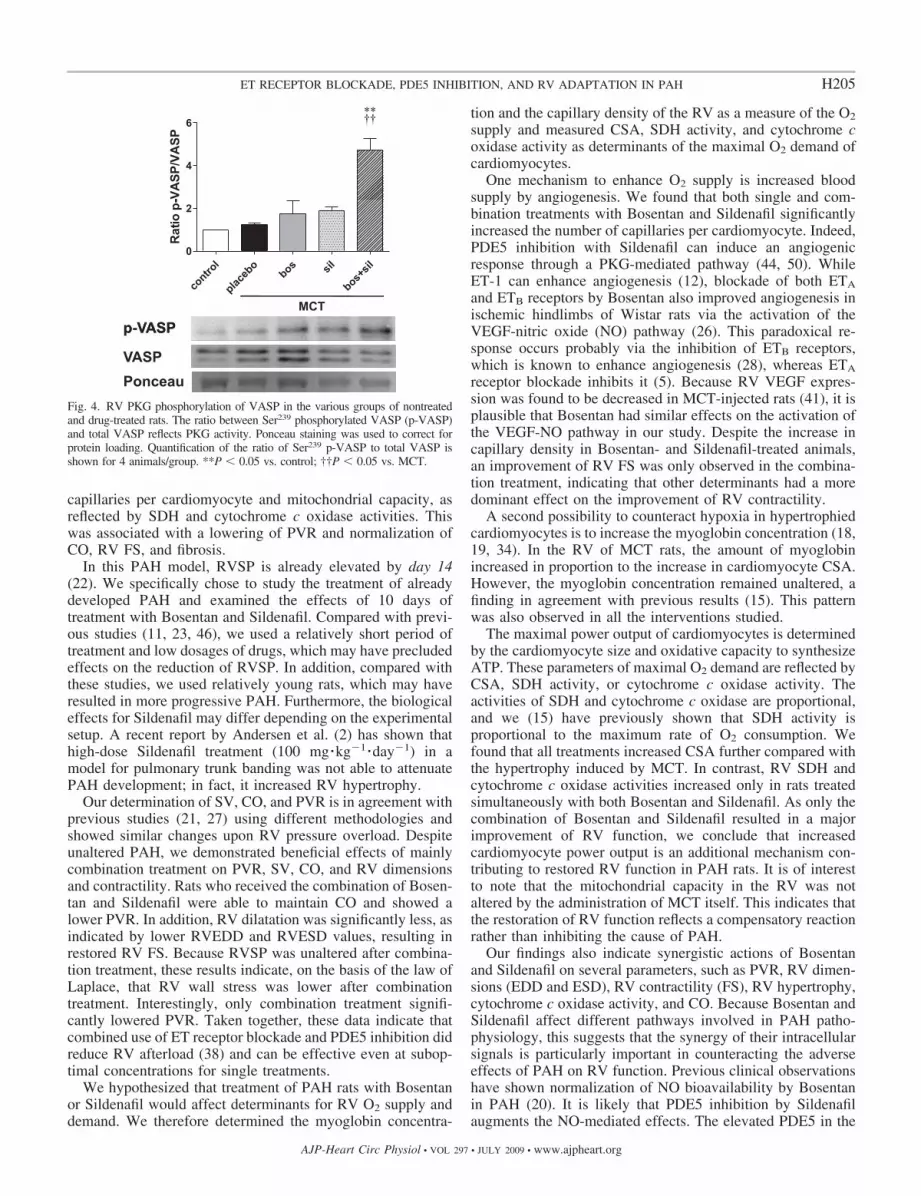
RV PKG activity. The RV activity of PKG was studied by mea-suring the Ser239 phosphorylation of VASP, a myocardial PKG-1target (29), using immunological Western blot analysis. Densitometry(over total VASP expression) was performed and is presented aspreviously described (33, 35, 49). Myocardial tissue of the RV washomogenized. Detection of VASP (C17) and phosphorylated VASP(Ser239) (both 1:200, Santa Cruz Biotechnology) was performed.VASP phosphorylation is known to cause a migrational shift onSDS-PAGE, which causes a separation of VASP over two bands (6).Both bands together served as total VASP. Before the experiments,we performed a signal linearity control for protein loading (Pon-ceau).We used horseradish peroxidase-conjugated anti-rabbit second-ary antibody (1:1,000, DAKO Cytomation). Horseradish peroxidasewas detected using the ECL plus Western blot detection system(Amersham Biosciences). Relative density was determined as previ-ously described (49). Sample densities were expressed as a ratiobetween phospho-VASP and total VASP, where MCT and MCT �treatment samples are expressed relative to the ratio of the controlsamples, which was set to 1.
Statistical analysis. Mean values � SE are given. Since compari-sons in this study were planned, Student’s t-test was used to determinedifferences between means. Two-way ANOVA was used to detectinteractions between Bosentan and Sildenafil treatments. P valuesof �0.05 were considered to be statistically significant.
RESULTS
Effects of treatment on hemodynamics, body mass, and lungmass. Table 1 shows hemodynamic parameters and body andlung masses of the various groups of animals studied. MCTclearly induced PAH, which was clearly demonstrated by thehigher RVSP on day 25 than in control animals. Neither singletreatment with either Bosentan or Sildenafil nor simultaneoustreatment with both drugs significantly reduced MCT-inducedRVSP significantly. Nontreated MCT rats and both groups ofsingle drug-treated MCT rats had lower systemic blood pres-sures than control animals.
Body masses of all PAH rats, including treated rats, weresignificantly lower on day 25 compared with their controlcounterparts (Table 1). In addition, lung wet masses weresignificantly increased in MCT-, Bosentan � MCT-, and Sil-denafil � MCT-treated rats, whereas the combination treat-ment normalized lung mass.
Effects of treatment on RV and LV dimensions and contrac-tility, CO, SV, and PVR. To evaluate the effects of MCT-induced PAH and treatment by Bosentan and Sildenafil on theheart, functional echocardiography was performed in anesthe-sized rats. Representative pictures of M-mode echocardio-graphic recordings of the RV are shown in Fig. 1. Echocardio-graphic data are shown in Table 1. After 25 days, MCT ratsshowed marked dilation of the RV, as demonstrated by in-creased RVEDDs and RVESDs. RV FS significantly de-creased. RVEDD and RVESD values of Bosentan � MCT-and Sildenafil � MCT-treated rats were not different fromthose of untreated MCT rats, but the calculation of RV FSrevealed a further decrease. Combined Bosentan and Sildenafiltreatment of MCT rats resulted in less RV dilation, as evi-denced by smaller RVEDD and RVESD values, resulting innormalized RV FS. LVEDD and LVESD significantly de-creased upon MCT exposure and further decreased uponBosentan monotreatment, which was not observed in Silde-
1 Supplemental Material for this article is available online at the AmericanJournal of Physiology-Heart and Circulatory Physiology website.
H201ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org

nafil-treated rats. Treatment with the combination of bothdrugs normalized these parameters. LV FS was similar in allgroups except for Bosentan-treated rats, which revealed asignificant increase. The calculation of LV SV showed asignificant reduction in nontreated MCT rats compared withcontrols. Single treatment of MCT rats with Bosentan orSildenafil did not improve this. Correspondingly, the CO waslower in nontreated or single drug-treated MCT rats. MCT ratsthat received combination treatment showed a normalization ofSV and CO. HR was reduced in nontreated MCT rats. Bosen-tan further decreased HR, but HR was unaffected in Sildenafil-and Sildenafil � Bosentan-treated rats. MCT induced a markedincrease of PVR, which was not lowered upon Bosentan orSildenafil treatment, whereas the combination treatment signif-icantly lowered PVR. On days 0 and 14, no differences inRVEDD, RVESD, LVEDD, LVESD, RV FS, CO, SV, or HRwere observed between the groups (see Table E1 in the onlineSupplemental Material). These data show that the changes in
cardiac performance in rat PAH as determined by echocardi-ography correspond well with clinical observations and areimproved by combined treatment with Bosentan and Sildenafil.
RV cardiomyocyte adaptation to PAH: hypertrophy, myo-globin, capillary density, fibrosis, and mitochondrial capacity.Subsequently, we evaluated, by quantitative histochemistry,whether the adaptation of metabolic parameters (energy supplyand demand) and their structural organization occurred in RVcardiomyocytes of PAH rats and whether this was influencedby Sildenafil and Bosentan. No mitochondrial damage wasobserved in control, nontreated, or any of the treated MCT rats,as indicated by the absence of cytochrome c release in cardio-myocytes (data not shown). Typical examples of sections ofmyocardial tissue stained for hematoxylin and eosin, myoglo-bin, collagen type IV, Picrosirius red, SDH, and cytochrome coxidase are shown in Fig. 2, whereas Fig. 3 shows the quan-tifications of these data for the various groups of animals. MCTclearly induced RV hypertrophy, indicated by an increase in
Table 1. Hemodynamic and echocardiographic data for the RV and LV, body mass, and lung mass of rats from the control,untreated MCT, MCT � Bosentan, MCT � Sildenafil, and combination treatment groups
ControlUntreated (Placebo)
MCT MCT � Bosentan MCT � SildenafilMCT � Bosentan
� Sildenafil
P Value of theInteraction ofBosentan and
Sildenafil
RV systolic pressure, mmHg 28�2‡ 62�7* 64�5* 55�5* 55�10* NSSystemic pressure, mmHg 89�3 70�4* 67�7* 74�5* 76�7 NSRVEDD, mm 2.48�0.05‡ 4.18�0.52* 5.24�0.50* 4.91�0.44* 3.37�0.55 0.0003RVESD, mm 1.07�0.06‡ 2.39�0.38* 3.52�0.38* 3.16�0.27* 1.60�0.21* 0.0003RV FS, % 57�3‡ 44�3* 33�3*‡ 35�2*‡ 52�2‡ �0.0001LVEDD, mm 6.75�0.17‡ 5.86�0.22* 5.43�0.21* 5.99�0.45 6.65�0.18‡ 0.043LVESD, mm 2.97�0.17‡ 2.32�0.20* 1.86�0.15† 2.39�0.26 2.63�0.26 NSLV FS, % 56�2 61�2 66�2† 60�4 61�3 NSStroke volume, �l 296�21 201�21 164�18 228�44 234�19 NSCardiac output, ml/min 116�10‡ 71�10* 47�6† 74�14* 112�8‡ 0.0048Pulmonary vascular resistance, dyn � s �cm�5 15,005�780‡ 46,224�8,451* 69,276�8,913† 60,350�5,983† 22,732�2,687*‡ 0.0005Heart rate, beats/min 392�13‡ 345�15* 291�11*‡ 334�16* 387�9‡ 0.015Body mass, g 363�9§ 311�12† 297�11† 305�12† 330�9* NSLung mass, g 1.69�0.11‡ 2.06�0.06* 2.21�0.11† 2.25�0.13† 1.96�0.13 0.048
Values are means � SE; n �5–7 rats/group (also see the online Supplemental Material). RV, right ventricle; LV, left ventricle, MCT, monocrotaline; RVEDDand LVEDD, RV and LV end-diastolic diameter, respectively; RVESD and LVESD, RV and LV end-systolic diameter, respectively; FS, fractional shortening;NS, not significant. *P � 0.05 and †P � 0.005 vs. control; ‡P � 0.05 and §P � 0.005 vs. untreated MCT rats.
Fig. 1. Top: study design. Pulmonary arterial hypertension(PAH) was induced in male Wistar rats by an injection ofmonocrotaline (MCT; 40 mg/kg sc). Fourteen days after MCTtreatment, rats were orally treated with Bosentan (Bos), Silde-nafil (Sil), or both drugs for 10 days. On days 0, 14, and 25,echocardiography was performed to monitor right ventricular(RV) and left ventricular (LV) function. On day 25, right heartcatheterization was performed to measure RV systolic pressure(RVSP), and, subsequently, hearts were excised for quantita-tive histochemistry. Bottom: representative echocardiographicimages of the RV obtained in the M-mode, which used tocalculate RV fractional shortening. Vertical bars show the RVend-diastolic diameter (EDD) and end-systolic diameter(ESD). MCT rats showed RV dilation, as indicated by theincreased RVEDD and RVESD. This was not reversed bysingle treatments. Rats that received the combination treatmentshowed less dilation (also see Table 1).
H202 ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org

mean RV cardiomyocyte CSA. Bosentan, Sildenafil, and alsothe combination treatment further increased cardiomyocytehypertrophy (Figs. 2A and 3A). The myoglobin concentrationdid not differ between cardiomyocytes of control, nontreated,and drug-treated MCT rats (Figs. 2B and 3B). Nontreated MCTrats showed a trend toward an increase in capillary density. Thenumber of capillaries per cardiomyocyte increased signifi-cantly in MCT rats exposed to both single drug and combina-tion treatments compared with control rats (Figs. 2C and 3C).In addition, MCT increased the formation of fibrosis in the RV,which reduced significantly upon single treatments with eitherBosentan or Sildenafil and normalized upon combination treat-ment (Figs. 2D and 3D). The mean RV cardiomyocyte SDHactivity of control, nontreated MCT, or single drug-treatedMCT rats was similar, whereas the combination treatment
significantly increased SDH activity (Figs. 2E and 3E). Cyto-chrome c oxidase activity revealed a similar pattern (Figs. 2Fand 3F). SDH and cytochrome c oxidase activities were pro-portional (P � 0.0001, r2 � 0.66).
For comparison, mean LV cardiomyocyte CSA, myoglobinconcentrations, and capillary densities were similar in allgroups (382 � 13 �m2, 0.74 � 0.03 mM, and 1.60 � 0.03capillaries/cell, respectively, n � 31). However, LV fibrosissignificantly increased in nontreated MCT rats compared withcontrol rats (2.9 � 0.8% vs. 1.1 � 0.4%, respectively, P �0.0002) and was only lowered by the combined treatment ofBosentan and Sildenafil (to 1.9 � 0.3%, P � 0.012 vs. MCTrats). In addition, SDH activity increased in Sildenafil- andcombination-treated rats [absorbance (A) at 660 nm � 0.172 �0.006, n � 14; pooled value for the two groups compared with
Fig. 2. Typical examples of myocardial sections from thevarious groups of rats (see Fig. 1) used for quantification.A: hematoxylin and eosin staining of the RV myocardium,which was used to determine the mean RV cardiomyocytecross-sectional area (CSA). Arrows show representative cardi-omyocytes. B: RV staining for myoglobin, which was used todetermine the myoglobin concentration in the RV. C: collagentype IV staining of the RV myocardium, which was used todetermine the capillary density of the RV. Arrows showrepresentative capillaries. D: Picrosirius red staining, whichwas used to quantify the amount of RV fibrosis. Arrows showcollagen fibers. E: succinate dehydrogenase (SDH) activity.F: cytochrome c oxidase (CYTOX) activity. Mitochondrialenzyme activities were determined by microdensitometry. Sec-tions for the LV are not shown.
H203ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org

pooled control, nontreated, and Bosentan treated MCT rats,which were similar: A660nm � 0.133 � 0.005, P � 0.0001, n �17], which was also observed for cytochrome c oxidase activity(A436nm� 0.223 � 0.004, n � 14; pooled value for the twogroups compared with pooled control, nontreated, and Bosen-tan treated MCT rats, which were similar: A436nm � 0.193 �0.005, P � 0.0001, n � 17).
Effect of treatment on RV PKG activity. Subsequently, westudied whether the effects of treatment on the RV could beexplained by a change in cGMP. To this end, the activity ofPKG in the RV was measured by determining the degree ofSer239 phosphorylation of VASP. Western blot analysisshowed that the RV of MCT rats revealed no difference in RVPKG activity compared with control rats. This shows that PAHinduction by MCT itself does not alter PKG activity in RVcardiomyocytes. However, while single treatment with Bosen-
tan or Sildenafil had no significant effect on VASP phosphor-ylation, the combination of both drugs significantly increasedPKG activity by 4.6-fold (Fig. 4).
Interactions between Bosentan and Sildenafil. Two-wayANOVA detected significant interactions between Bosentanand Sildenafil treatment. In addition to those shown Table 1,interactions were found for mean RV CSA (P � 0.034), Ser239
phosphorylation of VASP (P � 0.023), and cytochrome coxidase activity (P � 0.0035).
DISCUSSION
The main finding of our study is that, in this model ofMCT-induced PAH, the combination of orally administeredBosentan and Sildenafil, but not single treatments, improvedthe RV adaptation to severe PAH with respect to the number of
Fig. 3. Cardiomyocyte adaptation to PAH. A: MCTclearly induced RV cardiomyocyte hypertrophy com-pared with control, which further developed despitetreatment. B: no differences in the myoglobin con-centration was found in control, nontreated, Bosen-tan-, and/or Sildenafil-treated MCT rats. C: both sin-gle and combination treatments induced a significantincrease in the capillary-to-cardiomyocyte ratio.D: RV fibrosis was increased in nontreated MCT ratscompared with control rats. All treatments reducedRV fibrosis significantly, with the most pronouncedeffect seen upon combination treatment. E: comparedwith control, SDH activity was not changed in non-treated MCT rats or upon single treatment, whereasadditional combination drug treatment significantlyincreased SDH activity. F: cytochrome c oxidaseactivity revealed the same pattern as SDH activity.Values are means � SE. *P � 0.05 and **P � 0.005vs. control rats; †P � 0.05 and ††P � 0.005 vs. MCTrats.
H204 ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org
capillaries per cardiomyocyte and mitochondrial capacity, asreflected by SDH and cytochrome c oxidase activities. Thiswas associated with a lowering of PVR and normalization ofCO, RV FS, and fibrosis.
In this PAH model, RVSP is already elevated by day 14(22). We specifically chose to study the treatment of alreadydeveloped PAH and examined the effects of 10 days oftreatment with Bosentan and Sildenafil. Compared with previ-ous studies (11, 23, 46), we used a relatively short period oftreatment and low dosages of drugs, which may have precludedeffects on the reduction of RVSP. In addition, compared withthese studies, we used relatively young rats, which may haveresulted in more progressive PAH. Furthermore, the biologicaleffects for Sildenafil may differ depending on the experimentalsetup. A recent report by Andersen et al. (2) has shown thathigh-dose Sildenafil treatment (100 mg �kg�1 �day�1) in amodel for pulmonary trunk banding was not able to attenuatePAH development; in fact, it increased RV hypertrophy.
Our determination of SV, CO, and PVR is in agreement withprevious studies (21, 27) using different methodologies andshowed similar changes upon RV pressure overload. Despiteunaltered PAH, we demonstrated beneficial effects of mainlycombination treatment on PVR, SV, CO, and RV dimensionsand contractility. Rats who received the combination of Bosen-tan and Sildenafil were able to maintain CO and showed alower PVR. In addition, RV dilatation was significantly less, asindicated by lower RVEDD and RVESD values, resulting inrestored RV FS. Because RVSP was unaltered after combina-tion treatment, these results indicate, on the basis of the law ofLaplace, that RV wall stress was lower after combinationtreatment. Interestingly, only combination treatment signifi-cantly lowered PVR. Taken together, these data indicate thatcombined use of ET receptor blockade and PDE5 inhibition didreduce RV afterload (38) and can be effective even at subop-timal concentrations for single treatments.
We hypothesized that treatment of PAH rats with Bosentanor Sildenafil would affect determinants for RV O2 supply anddemand. We therefore determined the myoglobin concentra-
tion and the capillary density of the RV as a measure of the O2
supply and measured CSA, SDH activity, and cytochrome coxidase activity as determinants of the maximal O2 demand ofcardiomyocytes.
One mechanism to enhance O2 supply is increased bloodsupply by angiogenesis. We found that both single and com-bination treatments with Bosentan and Sildenafil significantlyincreased the number of capillaries per cardiomyocyte. Indeed,PDE5 inhibition with Sildenafil can induce an angiogenicresponse through a PKG-mediated pathway (44, 50). WhileET-1 can enhance angiogenesis (12), blockade of both ETA
and ETB receptors by Bosentan also improved angiogenesis inischemic hindlimbs of Wistar rats via the activation of theVEGF-nitric oxide (NO) pathway (26). This paradoxical re-sponse occurs probably via the inhibition of ETB receptors,which is known to enhance angiogenesis (28), whereas ETA
receptor blockade inhibits it (5). Because RV VEGF expres-sion was found to be decreased in MCT-injected rats (41), it isplausible that Bosentan had similar effects on the activation ofthe VEGF-NO pathway in our study. Despite the increase incapillary density in Bosentan- and Sildenafil-treated animals,an improvement of RV FS was only observed in the combina-tion treatment, indicating that other determinants had a moredominant effect on the improvement of RV contractility.
A second possibility to counteract hypoxia in hypertrophiedcardiomyocytes is to increase the myoglobin concentration (18,19, 34). In the RV of MCT rats, the amount of myoglobinincreased in proportion to the increase in cardiomyocyte CSA.However, the myoglobin concentration remained unaltered, afinding in agreement with previous results (15). This patternwas also observed in all the interventions studied.
The maximal power output of cardiomyocytes is determinedby the cardiomyocyte size and oxidative capacity to synthesizeATP. These parameters of maximal O2 demand are reflected byCSA, SDH activity, or cytochrome c oxidase activity. Theactivities of SDH and cytochrome c oxidase are proportional,and we (15) have previously shown that SDH activity isproportional to the maximum rate of O2 consumption. Wefound that all treatments increased CSA further compared withthe hypertrophy induced by MCT. In contrast, RV SDH andcytochrome c oxidase activities increased only in rats treatedsimultaneously with both Bosentan and Sildenafil. As only thecombination of Bosentan and Sildenafil resulted in a majorimprovement of RV function, we conclude that increasedcardiomyocyte power output is an additional mechanism con-tributing to restored RV function in PAH rats. It is of interestto note that the mitochondrial capacity in the RV was notaltered by the administration of MCT itself. This indicates thatthe restoration of RV function reflects a compensatory reactionrather than inhibiting the cause of PAH.
Our findings also indicate synergistic actions of Bosentanand Sildenafil on several parameters, such as PVR, RV dimen-sions (EDD and ESD), RV contractility (FS), RV hypertrophy,cytochrome c oxidase activity, and CO. Because Bosentan andSildenafil affect different pathways involved in PAH patho-physiology, this suggests that the synergy of their intracellularsignals is particularly important in counteracting the adverseeffects of PAH on RV function. Previous clinical observationshave shown normalization of NO bioavailability by Bosentanin PAH (20). It is likely that PDE5 inhibition by Sildenafilaugments the NO-mediated effects. The elevated PDE5 in the
Fig. 4. RV PKG phosphorylation of VASP in the various groups of nontreatedand drug-treated rats. The ratio between Ser239 phosphorylated VASP (p-VASP)and total VASP reflects PKG activity. Ponceau staining was used to correct forprotein loading. Quantification of the ratio of Ser239 p-VASP to total VASP isshown for 4 animals/group. **P � 0.05 vs. control; ††P � 0.05 vs. MCT.
H205ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org

RV of PAH patients and MCT rats (35) would locally reducecGMP generated by NO-stimulated guanylate cyclase. Suchsynergy of Bosentan and Sildenafil on NO/cGMP-mediatedprocesses may also underlie the observed improvement ofmitochondrial density reflected by SDH and cytochrome coxidase activities. Indeed, it has been shown that NO stimu-lates mitochondrial biogenesis through the activation of themitochondrial transcription factors peroxisome proliferator-activated receptor-� coactivator-1, nuclear respiratory fac-tor-1, and mitochondrial transcription factor A in a cGMP-dependent manner (10, 36, 37). We therefore investigatedwhether the combination of Bosentan and Sildenafil would beable to augment the NO-cGMP pathway by investigating theRV Ser239 phosphorylation of VASP. Indeed, we found anincrease in RV VASP phosphorylation only with the combi-nation of Bosentan and Sildenafil and not with the singletreatments. These data confirm the synergism and indicate thatan increase in mitochondrial capacity is likely an effect of theactivation of PKG through the rescue of the NO-cGMP path-way.
The observed effects in our study were predominant for theRV, as treatments did not affect LV histological characteristicsexcept for SDH and cytochrome c oxidase activities in bothgroups that received Sildenafil. Recent studies have shown thatSildenafil is able to reduce the production of ROS by mito-chondria (17) and that it exerts cardioprotective effects throughopening of Ca2�-sensitive, ATP-sensitive K� channels (13,52). Whether this is related to an increase in oxidative capacityremains to be investigated. Fibrosis was also reduced uponsingle and combination treatments in both ventricles. Althoughit is generally believed that MCT only affects the RV, evidenceis increasing indicating that the LV is also affected by MCTthrough the activation of various neurohumoral factors (1, 31).In addition, we observed altered LV dimensions as indicatedby reduced LVEDD and LVESD, which was also observed inBosentan-treated rats. This suggests that the LV of PAH ratswas also subjected to mechanical changes secondary to thepressure-overloaded RV, such as impaired LV filling (30, 32).
In conclusion, we demonstrated that the combination of ETreceptor blockade by Bosentan and PDE5 inhibition by Silde-nafil, at dosages that are ineffective in single treatments, is ableto increase RV mitochondrial enzyme activity in a progressivemodel of MCT-induced PAH, likely through the activation ofPKG by enforcement of the NO-cGMP pathway. This wasassociated with a lowering of PVR, a restoration of RVcontractility, a normalization of RV fibrosis, and a significantincrease in RV capillarization, resulting in the maintenance ofnormal CO. Our data confirm the synergism between Bosentanand Sildenafil and indicate that combination treatment withboth drugs may act at the mitochondrial level in the heart.
ACKNOWLEDGMENTS
The authors thank Prof. Dr. N. Westerhof for the expert advice anddiscussions.
GRANTS
This work was supported by the Institute for Cardiovascular Research of theVrije Universiteit University Medical Center (Amsterdam, The Netherlands).G. P. van Nieuw Amerongen was supported by The Netherlands HeartFoundation Grant T2003-0032; A. Vonk-Noordegraaf was supported by DutchScientific Organization Grant VIDI 917.96.306.
REFERENCES
1. Akhavein F, St Michel EJ, Seifert E, Rohlicek CV. Decreased leftventricular function, myocarditis, and coronary arteriolar medial thicken-ing following monocrotaline administration in adult rats. J Appl Physiol103: 287–295, 2007.
2. Andersen A, Nielsen JM, Peters CD, Schou UK, Sloth E, Nielsen-Kudsk JE. Effects of phosphodiesterase-5 inhibition by sildenafil in thepressure overloaded right heart. Eur J Heart Fail 10: 1158–1165, 2008.
5. Bhargava S, Stummeyer T, Hotz B, Hines OJ, Reber HA, Buhr HJ,Hotz HG. Selective inhibition of endothelin receptor A as an anti-angiogenic and anti-proliferative strategy for human pancreatic cancer. JGastrointest Surg 9: 703–709, 2005.
6. Blume C, Benz PM, Walter U, Ha J, Kemp BE, Renne T. AMP-activated protein kinase impairs endothelial actin cytoskeleton assemblyby phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem282: 4601–4612, 2007.
7. Chemla D, Castelain V, Humbert M, Hebert JL, Simonneau G,Lecarpentier Y, Herve P. New formula for predicting mean pulmonaryartery pressure using systolic pulmonary artery pressure. Chest 126:1313–1317, 2004.
8. Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonaryhypertension. Coron Artery Dis 16: 13–18, 2005.
9. Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol51: 1527–1538, 2008.
10. Clementi E, Nisoli E. Nitric oxide and mitochondrial biogenesis: a key tolong-term regulation of cellular metabolism. Comp Biochem Physiol AMol Integr Physiol 142: 102–110, 2005.
11. Clozel M, Hess P, Rey M, Iglarz M, Binkert C, Qiu C. Bosentan,sildenafil, and their combination in the monocrotaline model of pulmonaryhypertension in rats. Exp Biol Med (Maywood) 231: 967–973, 2006.
12. Cruz A, Parnot C, Ribatti D, Corvol P, Gasc JM. Endothelin-1, aregulator of angiogenesis in the chick chorioallantoic membrane. J VascRes 38: 536–545, 2001.
13. Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafilpreconditions adult cardiac myocytes against necrosis and apoptosis.Essential role of nitric oxide signaling. J Biol Chem 280: 12944–12955,2005.
14. de Simone G, Wallerson DC, Volpe M, Devereux RB. Echocardio-graphic measurement of left ventricular mass and volume in normotensiveand hypertensive rats. Necropsy validation. Am J Hypertens 3: 688–696,1990.
15. Des Tombe AL, van Beek-Harmsen BJ, Lee-De Groot MB, Van DerLaarse WJ. Calibrated histochemistry applied to oxygen supply anddemand in hypertrophied rat myocardium. Microsc Res Tech 58: 412–420,2002.
16. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med351: 1655–1665, 2004.
17. Fernandes MA, Marques RJ, Vicente JA, Santos MS, Monteiro P,Moreno AJ, Custodio JB. Sildenafil citrate concentrations not affectingoxidative phosphorylation depress H2O2 generation by rat heart mitochon-dria. Mol Cell Biochem 309: 77–85, 2008.
18. Garry DJ, Kanatous SB, Mammen PP. Emerging roles for myoglobin inthe heart. Trends Cardiovasc Med 13: 111–116, 2003.
19. Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, GrangeRW, Bassel-Duby R, Williams RS. Mice without myoglobin. Nature395: 905–908, 1998.
20. Girgis RE, Champion HC, Diette GB, Johns RA, Permutt S, SylvesterJT. Decreased exhaled nitric oxide in pulmonary arterial hypertension:response to bosentan therapy. Am J Respir Crit Care Med 172: 352–357,2005.
21. Handoko ML, Schalij I, Kramer K, Sebkhi A, Postmus PE, Van DerLaarse WJ, Paulus WJ, Vonk-Noordegraaf A. A refined radio-teleme-try technique to monitor right ventricle or pulmonary artery pressures inrats: a useful tool in pulmonary hypertension research. Pflugers Arch 455:951–959, 2008.
22. Henkens IR, Mouchaers KT, Vliegen HW, van der Laarse WJ,Swenne CA, Maan AC, Draisma HH, Schalij I, van der Wall EE,Schalij MJ, Vonk-Noordegraaf A. Early changes in rat hearts withdeveloping pulmonary arterial hypertension can be detected with 3-dimen-sional electrocardiography. Am J Physiol Heart Circ Physiol 293: H1300–H1307, 2007.
H206 ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org

23. Hill NS, Warburton RR, Pietras L, Klinger JR. Nonspecific endothelin-receptor antagonist blunts monocrotaline-induced pulmonary hypertensionin rats. J Appl Physiol 83: 1209–1215, 1997.
24. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR,Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF,Rabinovitch M. Cellular and molecular pathobiology of pulmonary arte-rial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004.
25. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterialhypertension. N Engl J Med 351: 1425–1436, 2004.
26. Iglarz M, Silvestre JS, Duriez M, Henrion D, Levy BI. Chronicblockade of endothelin receptors improves ischemia-induced angiogenesisin rat hindlimbs through activation of vascular endothelial growth fac-tor-no pathway. Arterioscler Thromb Vasc Biol 21: 1598–1603, 2001.
27. Jungebluth P, Ostertag H, Macchiarini P. An experimental animalmodel of postobstructive pulmonary hypertension. J Surg Res 147: 75–78,2008.
28. Lahav R, Suva ML, Rimoldi D, Patterson PH, Stamenkovic I. Endo-thelin receptor B inhibition triggers apoptosis and enhances angiogenesisin melanomas. Cancer Res 64: 8945–8953, 2004.
29. Li Z, Xi X, Gu M, Feil R, Ye RD, Eigenthaler M, Hofmann F, Du X.A stimulatory role for cGMP-dependent protein kinase in platelet activa-tion. Cell 112: 77–86, 2003.
30. Louie EK, Rich S, Brundage BH. Doppler echocardiographic assessmentof impaired left ventricular filling in patients with right ventricular pres-sure overload due to primary pulmonary hypertension. J Am Coll Cardiol8: 1298–1306, 1986.
31. Lourenco AP, Roncon-Albuquerque R Jr, Bras-Silva C, Faria B,Wieland J, Henriques-Coelho T, Correia-Pinto J and Leite-MoreiraAF. Myocardial dysfunction and neurohumoral activation without remod-eling in left ventricle of monocrotaline-induced pulmonary hypertensiverats. Am J Physiol Heart Circ Physiol 291: H1587–H1594, 2006.
32. Mahmud E, Raisinghani A, Hassankhani A, Sadeghi HM, StrachanGM, Auger W, DeMaria AN, Blanchard DG. Correlation of leftventricular diastolic filling characteristics with right ventricular overloadand pulmonary artery pressure in chronic thromboembolic pulmonaryhypertension. J Am Coll Cardiol 40: 318–324, 2002.
33. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, HarryG, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targetingsurvivin selectively induces pulmonary vascular apoptosis and reversespulmonary arterial hypertension. J Clin Invest 115: 1479–1491, 2005.
34. Murray JD. On the role of myoglobin in muscle respiration. J Theor Biol47: 115–126, 1974.
35. Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A,St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR,Michelakis ED. Phosphodiesterase type 5 is highly expressed in thehypertrophied human right ventricle, and acute inhibition of phosphodi-esterase type 5 improves contractility. Circulation 116: 238–248, 2007.
36. Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J CellSci 119: 2855–2862, 2006.
37. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C,Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO.Mitochondrial biogenesis in mammals: the role of endogenous nitricoxide. Science 299: 896–899, 2003.
38. Norton JM. Toward consistent definitions for preload and afterload. AdvPhysiol Educ 25: 53–61, 2001.
39. Old SL, Johnson MA. Methods of microphotometric assay of succinatedehydrogenase and cytochrome c oxidase activities for use on humanskeletal muscle. Histochem J 21: 545–555, 1989.
40. Pak O, Aldashev A, Welsh D, Peacock A. The effects of hypoxia on thecells of the pulmonary vasculature. Eur Respir J 30: 364–372, 2007.
41. Partovian C, Adnot S, Eddahibi S, Teiger E, Levame M, Dreyfus P,Raffestin B, Frelin C. Heart and lung VEGF mRNA expression in ratswith monocrotaline- or hypoxia-induced pulmonary hypertension. Am JPhysiol Heart Circ Physiol 275: H1948–H1956, 1998.
42. Pombo JF, Troy BL, Russell RO Jr. Left ventricular volumes andejection fraction by echocardiography. Circulation 43: 480–490, 1971.
43. Puri A, McGoon MD, Kushwaha SS. Pulmonary arterial hypertension:current therapeutic strategies. Nat Clin Pract Cardiovasc Med 4: 319–329,2007.
44. Pyriochou A, Zhou Z, Koika V, Petrou C, Cordopatis P, Sessa WC,Papapetropoulos A. The phosphodiesterase 5 inhibitor sildenafil stimu-lates angiogenesis through a protein kinase G/MAPK pathway. J CellPhysiol 211: 197–204, 2007.
45. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y,Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T,Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-inducedinhibition of Hif-1 causes cardiac dysfunction during pressure overload.Nature 446: 444–448, 2007.
46. Schermuly RT, Kreisselmeier KP, Ghofrani HA, Yilmaz H, ButrousG, Ermert L, Ermert M, Weissmann N, Rose F, Guenther A,Walmrath D, Seeger W, Grimminger F. Chronic sildenafil treatmentinhibits monocrotaline-induced pulmonary hypertension in rats. Am JRespir Crit Care Med 169: 39–45, 2004.
47. Ueno M, Miyauchi T, Sakai S, Kobayashi T, Goto K, Yamaguchi I.Effects of physiological or pathological pressure load in vivo on myocar-dial expression of ET-1 and receptors. Am J Physiol Regul Integr CompPhysiol 277: R1321–R1330, 1999.
47a.van Beek-Harmsen BJ, Bekedam MA, Feenstra HM, Visser FC, VanDer Laarse WJ. Determination of myoglobin concentration and oxidativecapacity in cryostat sections of human and rat skeletal muscle fibres and ratcardiomyocytes. Histochem Cell Biol 121: 335–342, 2004.
47b.van Beek-Harmsen BJ, Van Der Laarse WJ. Immunohistochemicaldetermination of cytosolic cytochrome C concentration in cardiomyocytes.J Histochem Cytochem 53: 803–807, 2005.
48. van Wolferen SA, Grunberg K, Vonk-Noordegraaf A. Diagnosis andmanagement of pulmonary hypertension over the past 100 years. RespirMed 101: 389–398, 2007.
49. Versteilen AM, Korstjens IJ, Musters RJ, Groeneveld AB, Sipkema P.Rho kinase regulates renal blood flow by modulating eNOS activity inischemia-reperfusion of the rat kidney. Am J Physiol Renal Physiol 291:F606–F611, 2006.
50. Vidavalur R, Penumathsa SV, Zhan L, Thirunavukkarasu M, MaulikN. Sildenafil induces angiogenic response in human coronary arteriolarendothelial cells through the expression of thioredoxin, hemeoxygenaseand vascular endothelial growth factor. Vasc Pharmacol 45: 91–95, 2006.
51. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD,Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ,Gladwin M, Denholm EM, Gail DB. Right ventricular function andfailure: report of a National Heart, Lung, and Blood Institute workinggroup on cellular and molecular mechanisms of right heart failure. Cir-culation 114: 1883–1891, 2006.
52. Wang X, Fisher PW, Xi L, Kukreja RC. Essential role of mitochondrialCa2�-activated and ATP-sensitive K� channels in sildenafil-induced latecardioprotection. J Mol Cell Cardiol 44: 105–113, 2008.
53. Wharton J, Strange JW, Moller GM, Growcott EJ, Ren X, FranklynAP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodies-terase type 5 inhibition in human pulmonary artery cells. Am J Respir CritCare Med 172: 105–113, 2005.
54. Zong P, Tune JD, Downey HF. Mechanisms of oxygen demand/supplybalance in the right ventricle. Exp Biol Med (Maywood) 230: 507–519,2005.
H207ET RECEPTOR BLOCKADE, PDE5 INHIBITION, AND RV ADAPTATION IN PAH
AJP-Heart Circ Physiol • VOL 297 • JULY 2009 • www.ajpheart.org
Copyright © 2022 FDOKUMEN




















