
Electronic and Magnetic Properties of Metal Phthalocyanines on Au(111) Surface: A First-Principles...
6
Electronic and Magnetic Properties of Metal Phthalocyanines on Au(111) Surface: A First-Principles Study Zhenpeng Hu, Bin Li, Aidi Zhao, Jinlong Yang,* and J. G. Hou* Hefei National Laboratory for Physical Sciences at Microscale, UniVersity of Science & Technology of China, Hefei, Anhui 230026, People’s Republic of China ReceiVed: May 15, 2008; ReVised Manuscript ReceiVed: June 24, 2008 A first-principles study is performed to explore the electronic and magnetic properties of several 3d transition metal phthalocyanines (including MnPc, FePc, NiPc, and CuPc) adsorbed on a Au(111) surface. Our results show that the most favorite adsorption site is the top site for MnPc molecule whereas it is the hcp site for other molecules. The electronic structures of MnPc and FePc change obviously when they are adsorbed onto the Au(111) surface, while those of NiPc and CuPc change slightly near the Fermi level due to the weak molecule-surface interactions. By analyzing the properties of d orbitals at the spatial and energy scales, we have discussed the possible Kondo effect related to these metal phthalocyanines adsorbed on the Au(111) surface. Introduction Metal phthalocyanine (M-Pc, Figure 1) molecules are man- made macrocycles conventionally used as dyes and pigments. Recently, these molecules have been found to be of much potential in nanotechnology applications such as light-emitting diodes, 1,2 field-effect transistors, 3,4 photovoltaic cells, 5,6 and single-molecule devices. 7 Various experimental techniques, such as photoemission spectroscopy, 8,9 electronic paramagnetic reso- nance, 10 nuclear magnetic resonance, 11 and gas-phase electron diffraction 12 have been used to explore the unique properties and functions of M-Pc molecules at the nanoscale. Especially, in the past two decades, scanning tunneling microscopy (STM) and spectroscopy experiments have been focused on the M-Pc molecules. 13-19 Recently, study on the Kondo effect of single M-Pc molecules adsorbed on the metal surfaces 20-22 has attracted much academic interest of the researchers. This effect is a result of the exchange interaction between the local magnetic moment in the molecule and the conducting electrons. For actual applications, such as nanodevices, the configura- tions of the M-Pc molecules adsorbed on a certain substrate or electrode and the interactions between the M-Pc molecules and the substrate/electrode are very important, since they determine the physical and chemical properties of the M-Pc molecular systems. For example, our recent STM experimental and theoretical studies show that the magnetic moment of the central ion in a CoPc molecule will disappear when the molecule is adsorbed onto an Au(111) surface, but it will be restored if eight hydrogen atoms are cut by the voltage pulse of STM tip. 22 Therefore, the first step toward a meaningful interpretation of these complex systems and developing its further applications is an accurate theoretical determination of configuration and electronic structure of the single molecule adsorbed on the metal surface. However, previous theoretical studies either are focused on the electronic structure of free molecules 23-30 or only give a few simple results of adsorbed molecules without any detail of their calculation and further analysis; 20 so far there is no systematic work about these complex nanosystems. In this paper, we employ the first-principles calculations based on density functional theory to study the effect of Au(111) surface on the electronic structures of various 3d transition metal M-Pc (M) Mn, Fe, Ni, and Cu) adsorbates and to explore how different metal ions influence the adsorption configuration, the electronic structure, and especially the magnetic properties, of these M-Pc molecules. The most favorite site of these M-Pc molecules on the Au(111) surface, the changes of the electronic structures and magnetic moments before and after adsorption, their adsorption energies, and the electronic transfers between molecules and surface are calculated and analyzed. Based on the electronic structures, we discuss the Kondo effect related to the local magnetic moment of these adsorbed M-Pc molecules. Computational Method Using the DMol 3 package, 31 we have performed the calcula- tions in local spin density approximation (LSDA) with the * To whom correspondence should be addressed. E-mail: jlyang@ ustc.edu.cn; [email protected]. Figure 1. Ball-and-stick model of metal phthalocyanine, with the metal ion sitting in the center of the molecule. Balls of different color and size represent different elements. J. Phys. Chem. C 2008, 112, 13650–13655 13650 10.1021/jp8043048 CCC: $40.75 2008 American Chemical Society Published on Web 08/09/2008
Transcript of Electronic and Magnetic Properties of Metal Phthalocyanines on Au(111) Surface: A First-Principles...

Electronic and Magnetic Properties of Metal Phthalocyanines on Au(111) Surface: AFirst-Principles Study
Zhenpeng Hu, Bin Li, Aidi Zhao, Jinlong Yang,* and J. G. Hou*Hefei National Laboratory for Physical Sciences at Microscale, UniVersity of Science & Technology of China,Hefei, Anhui 230026, People’s Republic of China
ReceiVed: May 15, 2008; ReVised Manuscript ReceiVed: June 24, 2008
A first-principles study is performed to explore the electronic and magnetic properties of several 3d transitionmetal phthalocyanines (including MnPc, FePc, NiPc, and CuPc) adsorbed on a Au(111) surface. Our resultsshow that the most favorite adsorption site is the top site for MnPc molecule whereas it is the hcp site forother molecules. The electronic structures of MnPc and FePc change obviously when they are adsorbed ontothe Au(111) surface, while those of NiPc and CuPc change slightly near the Fermi level due to the weakmolecule-surface interactions. By analyzing the properties of d orbitals at the spatial and energy scales, wehave discussed the possible Kondo effect related to these metal phthalocyanines adsorbed on the Au(111)surface.
Introduction
Metal phthalocyanine (M-Pc, Figure 1) molecules are man-made macrocycles conventionally used as dyes and pigments.Recently, these molecules have been found to be of muchpotential in nanotechnology applications such as light-emittingdiodes,1,2 field-effect transistors,3,4 photovoltaic cells,5,6 andsingle-molecule devices.7 Various experimental techniques, suchas photoemission spectroscopy,8,9 electronic paramagnetic reso-nance,10 nuclear magnetic resonance, 11 and gas-phase electrondiffraction12 have been used to explore the unique propertiesand functions of M-Pc molecules at the nanoscale. Especially,in the past two decades, scanning tunneling microscopy (STM)and spectroscopy experiments have been focused on the M-Pcmolecules.13-19 Recently, study on the Kondo effect of singleM-Pc molecules adsorbed on the metal surfaces20-22 hasattracted much academic interest of the researchers. This effectis a result of the exchange interaction between the local magneticmoment in the molecule and the conducting electrons.
For actual applications, such as nanodevices, the configura-tions of the M-Pc molecules adsorbed on a certain substrate orelectrode and the interactions between the M-Pc molecules andthe substrate/electrode are very important, since they determinethe physical and chemical properties of the M-Pc molecularsystems. For example, our recent STM experimental andtheoretical studies show that the magnetic moment of the centralion in a CoPc molecule will disappear when the molecule isadsorbed onto an Au(111) surface, but it will be restored if eighthydrogen atoms are cut by the voltage pulse of STM tip.22
Therefore, the first step toward a meaningful interpretation ofthese complex systems and developing its further applicationsis an accurate theoretical determination of configuration andelectronic structure of the single molecule adsorbed on the metalsurface. However, previous theoretical studies either are focusedon the electronic structure of free molecules23-30 or only givea few simple results of adsorbed molecules without any detailof their calculation and further analysis;20 so far there is nosystematic work about these complex nanosystems.
In this paper, we employ the first-principles calculations basedon density functional theory to study the effect of Au(111)surface on the electronic structures of various 3d transition metalM-Pc (M) Mn, Fe, Ni, and Cu) adsorbates and to explore howdifferent metal ions influence the adsorption configuration, theelectronic structure, and especially the magnetic properties, ofthese M-Pc molecules. The most favorite site of these M-Pcmolecules on the Au(111) surface, the changes of the electronicstructures and magnetic moments before and after adsorption,their adsorption energies, and the electronic transfers betweenmolecules and surface are calculated and analyzed. Based onthe electronic structures, we discuss the Kondo effect relatedto the local magnetic moment of these adsorbed M-Pc molecules.
Computational Method
Using the DMol3 package,31 we have performed the calcula-tions in local spin density approximation (LSDA) with the
* To whom correspondence should be addressed. E-mail: [email protected]; [email protected].
Figure 1. Ball-and-stick model of metal phthalocyanine, with the metalion sitting in the center of the molecule. Balls of different color andsize represent different elements.
J. Phys. Chem. C 2008, 112, 13650–1365513650
10.1021/jp8043048 CCC: $40.75 2008 American Chemical SocietyPublished on Web 08/09/2008

Vosko-Wilk-Nusair local correlation functional.32,33 The basissets are double numerical atomic orbital augmented by polariza-tion functions, which are comparable to 6-31G** sets. Thedensity functional semicore pseudopotentials are used to takethe relativistic effect into account and reduce the time ofcalculations.
In all calculations, we have employed a periodic slab modelto describe the M-Pc molecule-surface adsorption system. Thesame method has been used in our previous work22 of CoPcadsorbed on an Au(111) surface. A supercell contains total 225atoms: one M-Pc molecule with 57 atoms, as well as a threelayers metal slab with 56 Au atoms per rectangular layer. TheM-Pc molecule is on one side of the metal slab, and this slab isseparated from its periodic image by a vacuum space of 17.0Å, which is wide enough to avoid the interactions between theslabs and the M-Pc molecules in different unit cells. Althoughthe crystal structure of M-Pc indicates the molecule is notperfectly square planar,34,35 in the gas phase, its symmetry isgenerally D4h.36 Considering in the experiments the moleculesare usually dispersed by evaporation, we use a planar structureshown in Figure 1 to describe a free molecule. All gold atomsare fixed, but the molecule is fully relaxed in geometryoptimizations of the adsorption models, which are performedwith convergence criterions of 5 × 10-3 au/Å on the gradient,5 × 10-3 Å on the displacement, and 5 × 10-5 au on the energy.Medium-grid mesh points are employed for the matrix integra-tions, and only a single Γ point is used due to the numericallimitations. In our calculations, four initial configurations withM-Pc molecule adsorbed at four high-symmetry sites (Figure2a, hcp, fcc, bridge, and top sites) on the Au(111) surface havebeen considered. Additionally, we have considered severalorientations of M-Pc molecules adsorbed at the same site onthe Au(111) surface with different rotational angles to find theadsorption orientation with the lowest energy.
Results and Discussion
Adsorption Configurations. Table 1 lists our calculated totalenergies of the optimized structures for four M-Pc moleculesadsorbed on Au(111) layers with four adsorption sites, in whichthe total energy values for the most favorite adsorption site areset to zero. From Table 1, we can find that for the MnPc
molecule the top site is the most favorite adsorption site (Figure2c), which is the same as the previously reported result of MnPcon a Pb surface.20 However, for other M-Pc (M ) Fe, Ni, Cu)molecules, the configurations of the molecules adsorbed at thehcp site on the Au(111) surface (Figure 2b) have the lowesttotal energy. It can be also noticed that, for the cases of MnPcand FePc, the energy difference between two configurations withthe top and hcp adsorption sites is ∼20-30 meV, whichsuggests that these two configurations may exist simultaneouslyat room temperature. Although the same geometric configura-tions are used, our results for the case of FePc are at variancewith a previous work,21 which suggests the configurations withbridge and top adsorption sites should have much lower energy.This difference will be discussed in the next subsection. It maybe noted that our previous experimental and calculation resultsshow that the CoPc molecule is also adsorbed at the hcp siteon the Au(111) surface.22 The distances between metal ions andAu surface in the configurations with the lowest total energyare 2.81, 2.73, 3.06, and 3.04 Å for the cases of MnPc, FePc,NiPc, and CuPc molecules, respectively. These data suggest thatthe interactions between molecules and Au surface may bestronger for the cases of MnPc and FePc.
Electronic Structures. Since we are interested in themagnetic properties of the systems, spin-polarized density ofstates projected on the metal ions in the free molecules and inthe adsorbed molecules with the lowest energy configurationsare presented in Figure 3a-d to identify the influences of Ausurface on electronic structures of the adsorbates. Except forthe case of NiPc, projected densities of states (PDOS) of themetal ions in the free molecules are spin-polarized, whichindicate the existence of local magnetic moments in thesemolecules. But after the molecules have been adsorbed ontothe Au surface, the PDOS of metal ions change variously. Inthe case of MnPc molecule (Figure 3a), peaks at about +0.8and -1.8 eV disappear, and other peaks have been shifted about0.1-0.2 eV. In the case of FePc (Figure 3b), the maindifferences in PDOS of the Fe ion exist near the Fermi level:in the spin down component, the peak at +0.2 eV is absent,and the peak at +0.7 eV has been shifted to +1.0 eV; in thespin up component, all peaks are shifted nearer to the Fermilevel. These changes make the ratio of different spins near theFermi level smaller than that of the free molecule. In the caseof NiPc, there also exist peak shifting and intensity changes inPDOS of the Ni ion (Figure 3c), but its electronic structureremains non-spin-polarized. In the case of CuPc (Figure 3d),there is almost no difference between the PDOS of the freemolecule and the adsorbed one near the Fermi level, andthe changes of electronic structure can only be found withinthe energy range lower than -1.4 eV relative to EF. From the
Figure 2. (a) Four high-symmetry sites of Au(111) surface in a 1 × 1 unit cell. (b) and (c) Two configurations with the lowest total energy fordifferent M-Pc molecules.
TABLE 1: Calculated Relative Energies (meV) of SelectedConfigurations
configurations MnPc FePc NiPc CuPc
hcp 22.3 0 0 0fcc 66.4 47.0 37.8 53.3bridge 104 41.6 87.6 90.3top 0 25.3 55.4 194
Metal Phthalocyanines on Au(111) Surface J. Phys. Chem. C, Vol. 112, No. 35, 2008 13651
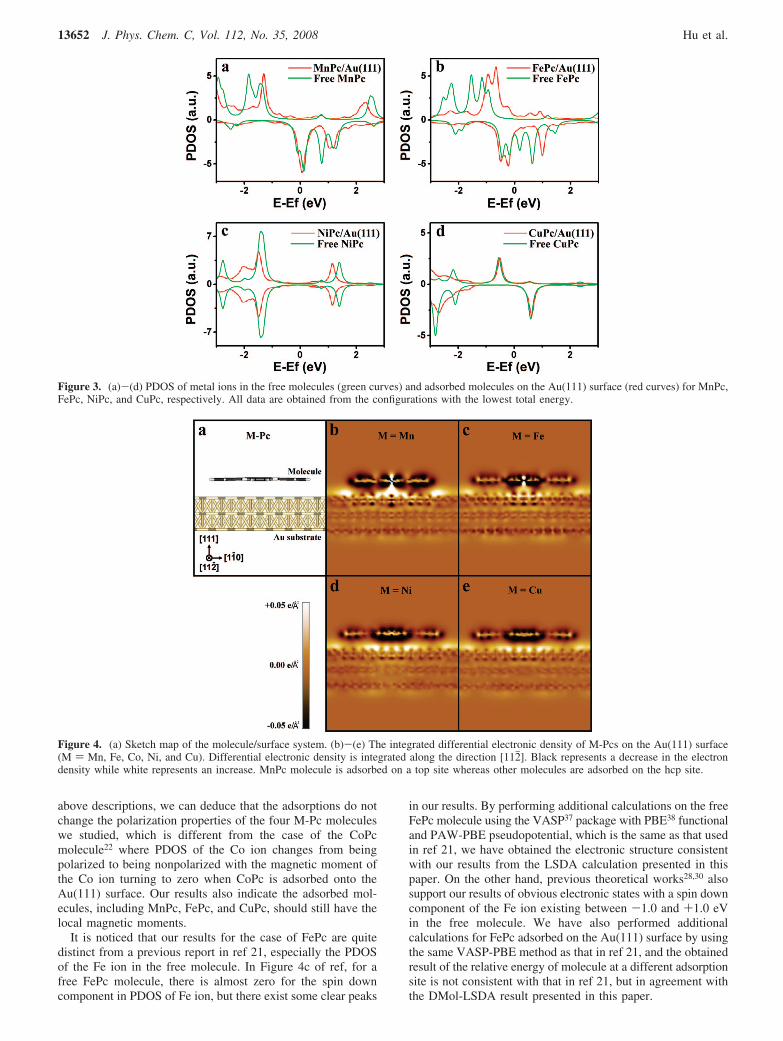
above descriptions, we can deduce that the adsorptions do notchange the polarization properties of the four M-Pc moleculeswe studied, which is different from the case of the CoPcmolecule22 where PDOS of the Co ion changes from beingpolarized to being nonpolarized with the magnetic moment ofthe Co ion turning to zero when CoPc is adsorbed onto theAu(111) surface. Our results also indicate the adsorbed mol-ecules, including MnPc, FePc, and CuPc, should still have thelocal magnetic moments.
It is noticed that our results for the case of FePc are quitedistinct from a previous report in ref 21, especially the PDOSof the Fe ion in the free molecule. In Figure 4c of ref, for afree FePc molecule, there is almost zero for the spin downcomponent in PDOS of Fe ion, but there exist some clear peaks
in our results. By performing additional calculations on the freeFePc molecule using the VASP37 package with PBE38 functionaland PAW-PBE pseudopotential, which is the same as that usedin ref 21, we have obtained the electronic structure consistentwith our results from the LSDA calculation presented in thispaper. On the other hand, previous theoretical works28,30 alsosupport our results of obvious electronic states with a spin downcomponent of the Fe ion existing between -1.0 and +1.0 eVin the free molecule. We have also performed additionalcalculations for FePc adsorbed on the Au(111) surface by usingthe same VASP-PBE method as that in ref 21, and the obtainedresult of the relative energy of molecule at a different adsorptionsite is not consistent with that in ref 21, but in agreement withthe DMol-LSDA result presented in this paper.
Figure 3. (a)-(d) PDOS of metal ions in the free molecules (green curves) and adsorbed molecules on the Au(111) surface (red curves) for MnPc,FePc, NiPc, and CuPc, respectively. All data are obtained from the configurations with the lowest total energy.
Figure 4. (a) Sketch map of the molecule/surface system. (b)-(e) The integrated differential electronic density of M-Pcs on the Au(111) surface(M ) Mn, Fe, Co, Ni, and Cu). Differential electronic density is integrated along the direction [112j]. Black represents a decrease in the electrondensity while white represents an increase. MnPc molecule is adsorbed on a top site whereas other molecules are adsorbed on the hcp site.
13652 J. Phys. Chem. C, Vol. 112, No. 35, 2008 Hu et al.

Adsorption Energy and Charge Transfer. In order todescribe the strength of molecule-surface interaction, we havecalculated the adsorption energies (Ead) and charge transferswhen different M-Pc molecules are adsorbed onto the Au(111)surface. The adsorption energy is defined by the terms of theenergies of the various species as follows:
Ead )E(sub ⁄ mol)- {E(sub)+E(mol)}
where E(sub/mol) is the total energy of the molecule-surfaceadsorption system and E(sub) and E(mol) are energies of thesurface and a free molecule, respectively. Our calculationsobtained the adsorption energies of -3.58, -3.67, -3.25, and-3.27 eV for the cases of MnPc, FePc, NiPc, and CuPc,respectively. Here, negative values mean the adsorptions of thesemolecules are exothermic processes. More interestingly, thesedata are nearly linear with the distances between metal ionsand Au surface, suggesting a trend that the molecule-surfaceinteraction will be stronger with M-Pc molecule closer to Ausurface.
To get the quantity of charge transfer, we adopt a methoddescribed in a theoretical study about the C60 molecule on metallicsurfaces,39 in which an xy-integrated differential electron densitycurve against the z coordinate is plotted to determine the boundarybetween the C60 molecule and the surface. Here the differentialelectron density can show the change in electron density that occursin the molecule and the surface when the molecule is adsorbedonto the surface. Our calculation results of the differential electrondensities of different molecules are presented in Figure 4. Figure4a is a sketch map showing the spatial positions of the surfaceand M-Pc molecule. The z axis is along [111] direction of Ausurface while y axis is along [112j] direction. Panels b-e in Figure4 show the differential electron densities integrated over the ydirection for the cases of four M-Pc molecules. For all fouradsorption cases of M-Pc molecules, there are obvious chargetransfers between metal ions and surface at the molecule-surfaceinterface, and the most significant differences between themare seen to occur at the central part of the M-Pc molecule, wherethe metal ion resides. There exist some regions of increase in theelectron density around the metal ions for the cases of MnPc andFePc (Figure 4b and c). However, for the cases of NiPc and CuPcmolecules (Figure 4d and e), there are only regions of decrease inthe electron density around the metal ions. This phenomenon isconsistent with the argument that MnPc and FePc having aunoccupied low-lying bonding orbital are much easier to get oneelectron to form anions than NiPc and CuPc having a high-lyingantibonding orbital.27,28 It may be also suggested that, for the casesof MnPc and FePc, there is a complex redistribution of electronsaround the metal ions because of the stronger molecule-surfaceinteraction. According to our calculations, the total amounts ofelectron transfer from M-Pc molecules to Au surfaces are 0.68,0.56, 0.73, and 0.70 e for the cases of MnPc, FePc, NiPc, andCuPc, respectively. We find that the electron transfer from FePcto the Au surface is obviously lower than those from other threeM-Pc molecules, which may be related to the greatest changes ofPDOS of metal ion in the FePc molecule after its adsorption asdiscussed before.
Magnetic Property and Kondo Effect. Spin-polarized elec-tronic structures of the metal ions can lead to a local magneticmoment in the M-Pc molecules. Our calculated results of electronicmagnetic moment from Mulliken analysis have been presented inTable 2. Whether being adsorbed on the Au surface or not, MnPc,FePc, and CuPc molecules have the local magnetic moments, withvariance only in the changes of quantity after the adsorption. Themagnetic moment of the MnPc molecule changes in a small amount
of 0.22 µB after its adsorption onto the Au(111) surface, but forFePc, the value is 0.98 µB, which is almost half of the magneticmoment in a free FePc molecule, while the magnetic moment ofCuPc keeps a value of 0.54 µB. All these changes are consistentwith the changes of PDOS and charge transfers discussed above.The magnetic moment of the NiPc molecule is always zero beforeand after being adsorbed onto the Au surface due to the nonpo-larized electronic structure. Specially, a free CoPc molecule has amagnetic moment of 1.10 µB, but the magnetic moment has beenquenched when the CoPc molecule is adsorbed onto the Au surface,and after cutting eight hydrogen atoms by STM tip CoPc moleculerecovers its magnetic moment, as shown by our previous research.22
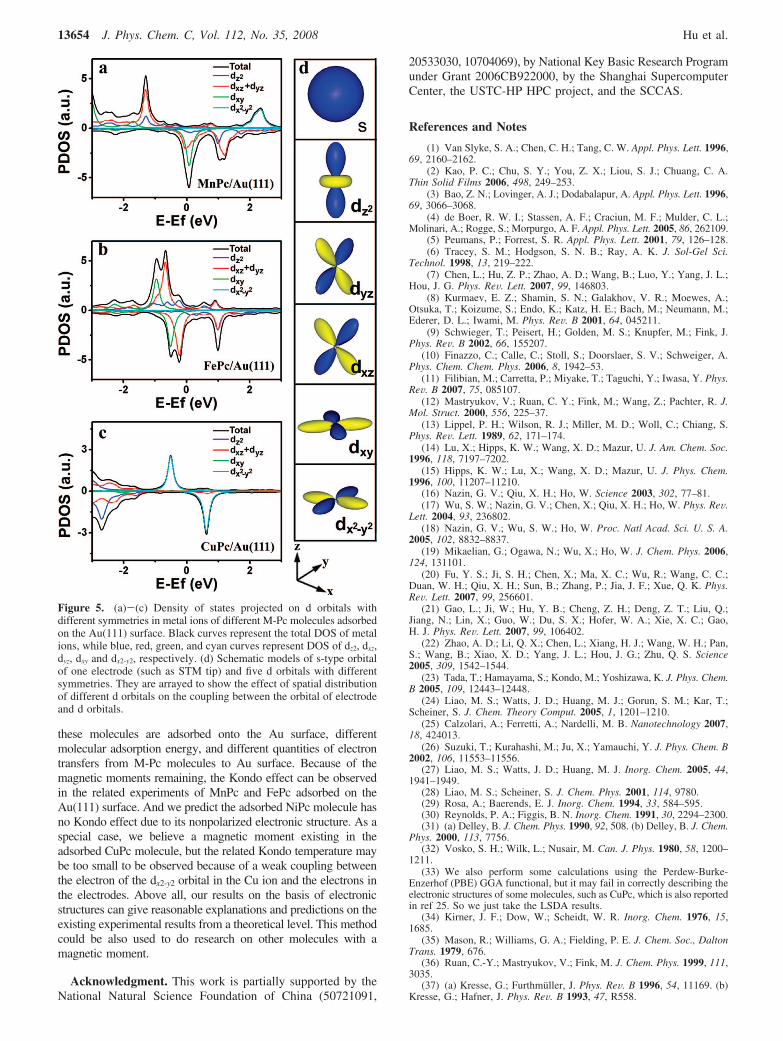
Generally, the existence of a local magnetic moment is animportant precondition of the Kondo effect observed in the STMexperiments. The Kondo effect has been observed when the MnPcmolecule is adsorbed on the Pb surface,20 and a similar result hasalso been reported for the case of FePc adsorbed on the Ausurface.21 Although the CuPc molecule has a magnetic moment,there is no report on the observation of the Kondo effect until now.To understand these experimental results, we present the densityof states projected on different symmetries of d orbital in the metalions for the cases of MnPc, FePc, and CuPc (Figure 5). The dxz,dyz, and dxy orbitals dominate the PDOS of adsorbed MnPc andFePc molecules (Figure 5a and b) near the Fermi level, and thedz2 orbital has only a very small component, which is differentfrom the calculation results in ref 21 But for the case of CuPcmoeclule (Figure 5c), the dx2-y2 orbital dominates the PDOS of CuPcmolecule near the Fermi level, which is consistent with the resultfrom a recent study combining experiment and simulation.40 Thedxz and dyz orbitals have stronger z-direction component which isperpendicular to Au surface, while the dx2-y2 orbital has morecomponents in the molecular plane parallel to the surface. Thus,the electrons of dxz and dyz orbitals can couple much more withthe electron in two electrodes (such as STM tip and Au substratein an STM experiment) than those of dx2-y 2orbital (see Figure 5d).As a result, the Kondo temperature of CuPc molecule may be toosmall41,42 to be observed under current experimental conditions.In our previous report for the case of CoPc, the PDOS of thed-CoPc molecule (with eight hydrogen atoms being cut) near theFermi level is dominated by the dz2 orbital that has the strongestcoupling with the states of STM tip and the Au surface, so theKondo effect can be observed.22 Furthermore, we predict that theKondo effect could be also observed due to the spin-polarizedelectronic structure near the Fermi level when a MnPc moleculeis adsorbed on the Au(111) surface, and there could be no Kondoeffect for the NiPc molecule adsorbed on the Au surface due to itsnonpolarized electronic structure.
Summary
The calculated results presented in this work demonstrate thatM-Pc (M ) Fe, Ni, Cu) molecules would like to be adsorbed onthe hcp site of the Au(111) surface, whereas the top site is mostfavored for the MnPc molecule. The interactions between M-Pcmolecules and the Au surface are different and may be related tovarious d orbital occupations of those metal ions. These discrep-ancies result in different changes in PDOS of metal ions when
TABLE 2: Electronic Magnetic Moment (µB) of Molecules
M-Pc magnetic moment
free adsorbedM ) Mn 3.17 2.95M ) Fe 2.03 1.05M ) Ni 0.00 0.00M ) Cu 0.54 0.54
Metal Phthalocyanines on Au(111) Surface J. Phys. Chem. C, Vol. 112, No. 35, 2008 13653
these molecules are adsorbed onto the Au surface, differentmolecular adsorption energy, and different quantities of electrontransfers from M-Pc molecules to Au surface. Because of themagnetic moments remaining, the Kondo effect can be observedin the related experiments of MnPc and FePc adsorbed on theAu(111) surface. And we predict the adsorbed NiPc molecule hasno Kondo effect due to its nonpolarized electronic structure. As aspecial case, we believe a magnetic moment existing in theadsorbed CuPc molecule, but the related Kondo temperature maybe too small to be observed because of a weak coupling betweenthe electron of the dx2-y2 orbital in the Cu ion and the electrons inthe electrodes. Above all, our results on the basis of electronicstructures can give reasonable explanations and predictions on theexisting experimental results from a theoretical level. This methodcould be also used to do research on other molecules with amagnetic moment.
Acknowledgment. This work is partially supported by theNational Natural Science Foundation of China (50721091,
20533030, 10704069), by National Key Basic Research Programunder Grant 2006CB922000, by the Shanghai SupercomputerCenter, the USTC-HP HPC project, and the SCCAS.
References and Notes
(1) Van Slyke, S. A.; Chen, C. H.; Tang, C. W. Appl. Phys. Lett. 1996,69, 2160–2162.
(2) Kao, P. C.; Chu, S. Y.; You, Z. X.; Liou, S. J.; Chuang, C. A.Thin Solid Films 2006, 498, 249–253.
(3) Bao, Z. N.; Lovinger, A. J.; Dodabalapur, A. Appl. Phys. Lett. 1996,69, 3066–3068.
(4) de Boer, R. W. I.; Stassen, A. F.; Craciun, M. F.; Mulder, C. L.;Molinari, A.; Rogge, S.; Morpurgo, A. F. Appl. Phys. Lett. 2005, 86, 262109.
(5) Peumans, P.; Forrest, S. R. Appl. Phys. Lett. 2001, 79, 126–128.(6) Tracey, S. M.; Hodgson, S. N. B.; Ray, A. K. J. Sol-Gel Sci.
Technol. 1998, 13, 219–222.(7) Chen, L.; Hu, Z. P.; Zhao, A. D.; Wang, B.; Luo, Y.; Yang, J. L.;
Hou, J. G. Phys. ReV. Lett. 2007, 99, 146803.(8) Kurmaev, E. Z.; Shamin, S. N.; Galakhov, V. R.; Moewes, A.;
Otsuka, T.; Koizume, S.; Endo, K.; Katz, H. E.; Bach, M.; Neumann, M.;Ederer, D. L.; Iwami, M. Phys. ReV. B 2001, 64, 045211.
(9) Schwieger, T.; Peisert, H.; Golden, M. S.; Knupfer, M.; Fink, J.Phys. ReV. B 2002, 66, 155207.
(10) Finazzo, C.; Calle, C.; Stoll, S.; Doorslaer, S. V.; Schweiger, A.Phys. Chem. Chem. Phys. 2006, 8, 1942–53.
(11) Filibian, M.; Carretta, P.; Miyake, T.; Taguchi, Y.; Iwasa, Y. Phys.ReV. B 2007, 75, 085107.
(12) Mastryukov, V.; Ruan, C. Y.; Fink, M.; Wang, Z.; Pachter, R. J.Mol. Struct. 2000, 556, 225–37.
(13) Lippel, P. H.; Wilson, R. J.; Miller, M. D.; Woll, C.; Chiang, S.Phys. ReV. Lett. 1989, 62, 171–174.
(14) Lu, X.; Hipps, K. W.; Wang, X. D.; Mazur, U. J. Am. Chem. Soc.1996, 118, 7197–7202.
(15) Hipps, K. W.; Lu, X.; Wang, X. D.; Mazur, U. J. Phys. Chem.1996, 100, 11207–11210.
(16) Nazin, G. V.; Qiu, X. H.; Ho, W. Science 2003, 302, 77–81.(17) Wu, S. W.; Nazin, G. V.; Chen, X.; Qiu, X. H.; Ho, W. Phys. ReV.
Lett. 2004, 93, 236802.(18) Nazin, G. V.; Wu, S. W.; Ho, W. Proc. Natl Acad. Sci. U. S. A.
2005, 102, 8832–8837.(19) Mikaelian, G.; Ogawa, N.; Wu, X.; Ho, W. J. Chem. Phys. 2006,
124, 131101.(20) Fu, Y. S.; Ji, S. H.; Chen, X.; Ma, X. C.; Wu, R.; Wang, C. C.;
Duan, W. H.; Qiu, X. H.; Sun, B.; Zhang, P.; Jia, J. F.; Xue, Q. K. Phys.ReV. Lett. 2007, 99, 256601.
(21) Gao, L.; Ji, W.; Hu, Y. B.; Cheng, Z. H.; Deng, Z. T.; Liu, Q.;Jiang, N.; Lin, X.; Guo, W.; Du, S. X.; Hofer, W. A.; Xie, X. C.; Gao,H. J. Phys. ReV. Lett. 2007, 99, 106402.
(22) Zhao, A. D.; Li, Q. X.; Chen, L.; Xiang, H. J.; Wang, W. H.; Pan,S.; Wang, B.; Xiao, X. D.; Yang, J. L.; Hou, J. G.; Zhu, Q. S. Science2005, 309, 1542–1544.
(23) Tada, T.; Hamayama, S.; Kondo, M.; Yoshizawa, K. J. Phys. Chem.B 2005, 109, 12443–12448.
(24) Liao, M. S.; Watts, J. D.; Huang, M. J.; Gorun, S. M.; Kar, T.;Scheiner, S. J. Chem. Theory Comput. 2005, 1, 1201–1210.
(25) Calzolari, A.; Ferretti, A.; Nardelli, M. B. Nanotechnology 2007,18, 424013.
(26) Suzuki, T.; Kurahashi, M.; Ju, X.; Yamauchi, Y. J. Phys. Chem. B2002, 106, 11553–11556.
(27) Liao, M. S.; Watts, J. D.; Huang, M. J. Inorg. Chem. 2005, 44,1941–1949.
(28) Liao, M. S.; Scheiner, S. J. Chem. Phys. 2001, 114, 9780.(29) Rosa, A.; Baerends, E. J. Inorg. Chem. 1994, 33, 584–595.(30) Reynolds, P. A.; Figgis, B. N. Inorg. Chem. 1991, 30, 2294–2300.(31) (a) Delley, B. J. Chem. Phys. 1990, 92, 508. (b) Delley, B. J. Chem.
Phys. 2000, 113, 7756.(32) Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200–
1211.(33) We also perform some calculations using the Perdew-Burke-
Enzerhof (PBE) GGA functional, but it may fail in correctly describing theelectronic structures of some molecules, such as CuPc, which is also reportedin ref 25. So we just take the LSDA results.
(34) Kirner, J. F.; Dow, W.; Scheidt, W. R. Inorg. Chem. 1976, 15,1685.
(35) Mason, R.; Williams, G. A.; Fielding, P. E. J. Chem. Soc., DaltonTrans. 1979, 676.
(36) Ruan, C.-Y.; Mastryukov, V.; Fink, M. J. Chem. Phys. 1999, 111,3035.
(37) (a) Kresse, G.; Furthmuller, J. Phys. ReV. B 1996, 54, 11169. (b)Kresse, G.; Hafner, J. Phys. ReV. B 1993, 47, R558.
Figure 5. (a)-(c) Density of states projected on d orbitals withdifferent symmetries in metal ions of different M-Pc molecules adsorbedon the Au(111) surface. Black curves represent the total DOS of metalions, while blue, red, green, and cyan curves represent DOS of dz2, dxz,dyz, dxy and dx2-y2, respectively. (d) Schematic models of s-type orbitalof one electrode (such as STM tip) and five d orbitals with differentsymmetries. They are arrayed to show the effect of spatial distributionof different d orbitals on the coupling between the orbital of electrodeand d orbitals.
13654 J. Phys. Chem. C, Vol. 112, No. 35, 2008 Hu et al.

(38) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. ReV. Lett. 1996, 77,3865.
(39) Lu, X. H.; Grobis, M.; Khoo, K. H.; Louie, S. G.; Crommie, M. F.Phys. ReV. B 2004, 70, 115418.
(40) Evangelista, F.; Carravetta, V.; Stefani, G.; Jansik, B.; Alagia, M.;Stranges, S.; Ruocco, A. J. Chem. Phys. 2007, 126, 124709.
(41) Madhavan, V.; Chen, W.; Jamneala, T.; Crommie, M. F.; Wingreen,N. S. Science 1998, 280, 567–569.
(42) Madhavan, V.; Chen, W.; Jamneala, T.; Crommie, M. F. Phys. ReV.B 2001, 64, 165412.
JP8043048
Metal Phthalocyanines on Au(111) Surface J. Phys. Chem. C, Vol. 112, No. 35, 2008 13655




















