
Electrochemical behavior of c-type cytochromes at clay-modified carbon electrodes: a model for the...
13
www.elsevier.nl/locate/jelechem Journal of Electroanalytical Chemistry 493 (2000) 37 – 49 Electrochemical behavior of c -type cytochromes at clay-modified carbon electrodes: a model for the interaction between proteins and soils Y. Sallez, P. Bianco, E. Lojou * Unite ´ de Bioe ´nerge ´tique et Inge ´nierie des Prote ´ines, IFR 1, CNRS 31, ch. J. Aiguier, 13402 Marseille Cedex 20, France Received 4 April 2000; received in revised form 10 July 2000; accepted 11 July 2000 Abstract Voltammetric studies of c -type cytochromes at clay-modified pyrolytic graphite electrodes have been undertaken as providing a good model for investigating interactions of proteins with soils. Different clays have been investigated according to their global charge, composition and hydrophobicity. The electrochemical behavior of either mitochondrial or bacterial cytochromes at these modified electrodes has been used to define a general rule for the nature of interactions between cytochromes and clays. The influence of various parameters such as pI values of proteins, pH, ionic strength, composition of the clay coating, on the electrochemical response is reported. It is demonstrated that while electrostatic interactions are responsible for the electrochemical promotion of cytochrome c, hydrophobic factors play a key role in the electron transfer process on multiheme cytochromes at clay-modified electrodes. Finally, the consequences of these interactions on the metal reductase activity developed by bacterial polyheme c -type cytochromes have been discussed. © 2000 Elsevier Science B.V. All rights reserved. Keywords: Clay; Modified-electrode; Voltammetry; Cytochromes 1. Introduction Soils are complex microporous media, which have large specific surface areas in the solid phase. Linked to the presence of clays, oxyhydroxides and organic mat- ter, the main functional groups in soil systems are able to interact with positively charged species, including metallic and organic cations. Interactions are intricate phenomena involving ion exchange and/or specific ad- sorption properties. Entrapments in structural voids or in micelle-like microdomains are not excluded. Undoubtedly, proteins in soils are also candidates for adsorption on charged surfaces. This is the case for enzymes secreted by microorganisms, which play an important role in the mineralization of the soil organic matter [1]. These extracellular enzymes are able to cleave the various high molar mass macromolecules into elementary monomer constituents such as amino acids and sugars, and inorganic ions such as orthophos- phate and sulfate. Adsorption of enzymes by soil col- loids leads to a decrease in the mobility of the enzymes in the soil and a modification of their catalytic activity. A shift towards alkaline pH of the optimum catalytic activity has been recognized as a general trend when the enzyme is adsorbed on a negatively charged surface such as a clay mineral [2]. Microbial metal reduction is another field where the adsorption of microorganisms on soil components plays an important role. This process has been recognized to have a prominent effect on the geochemistry of soils and sediments [3]. Indeed, it concerns the most abun- dant metal in the environment, namely iron. Ferric iron is essentially insoluble at neutral pHs, and amorphous iron oxide is the predominant form of Fe(III) in anaer- obic environments. Recent investigations have shown that sulfur and sulfate reducing bacteria develop an iron reductase activity in anaerobiosis. Thus, the main question has been the way in which the electrons flow outside from the bacterial cell to the metal oxide. A first hypothesis is based on close contact between the * Corresponding author. Tel.: +33-491-164443; fax: +33-491- 164578. E-mail address: [email protected] (E. Lojou). 0022-0728/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved. PII:S0022-0728(00)00280-1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Electrochemical behavior of c-type cytochromes at clay-modified carbon electrodes: a model for the...

www.elsevier.nl/locate/jelechem
Journal of Electroanalytical Chemistry 493 (2000) 37–49
Electrochemical behavior of c-type cytochromes at clay-modifiedcarbon electrodes: a model for the interaction between proteins
and soils
Y. Sallez, P. Bianco, E. Lojou *Unite de Bioenergetique et Ingenierie des Proteines, IFR 1, CNRS 31, ch. J. Aiguier, 13402 Marseille Cedex 20, France
Received 4 April 2000; received in revised form 10 July 2000; accepted 11 July 2000
Abstract
Voltammetric studies of c-type cytochromes at clay-modified pyrolytic graphite electrodes have been undertaken as providinga good model for investigating interactions of proteins with soils. Different clays have been investigated according to their globalcharge, composition and hydrophobicity. The electrochemical behavior of either mitochondrial or bacterial cytochromes at thesemodified electrodes has been used to define a general rule for the nature of interactions between cytochromes and clays. Theinfluence of various parameters such as pI values of proteins, pH, ionic strength, composition of the clay coating, on theelectrochemical response is reported. It is demonstrated that while electrostatic interactions are responsible for the electrochemicalpromotion of cytochrome c, hydrophobic factors play a key role in the electron transfer process on multiheme cytochromes atclay-modified electrodes. Finally, the consequences of these interactions on the metal reductase activity developed by bacterialpolyheme c-type cytochromes have been discussed. © 2000 Elsevier Science B.V. All rights reserved.
Keywords: Clay; Modified-electrode; Voltammetry; Cytochromes
1. Introduction
Soils are complex microporous media, which havelarge specific surface areas in the solid phase. Linked tothe presence of clays, oxyhydroxides and organic mat-ter, the main functional groups in soil systems are ableto interact with positively charged species, includingmetallic and organic cations. Interactions are intricatephenomena involving ion exchange and/or specific ad-sorption properties. Entrapments in structural voids orin micelle-like microdomains are not excluded.
Undoubtedly, proteins in soils are also candidates foradsorption on charged surfaces. This is the case forenzymes secreted by microorganisms, which play animportant role in the mineralization of the soil organicmatter [1]. These extracellular enzymes are able tocleave the various high molar mass macromoleculesinto elementary monomer constituents such as amino
acids and sugars, and inorganic ions such as orthophos-phate and sulfate. Adsorption of enzymes by soil col-loids leads to a decrease in the mobility of the enzymesin the soil and a modification of their catalytic activity.A shift towards alkaline pH of the optimum catalyticactivity has been recognized as a general trend when theenzyme is adsorbed on a negatively charged surfacesuch as a clay mineral [2].
Microbial metal reduction is another field where theadsorption of microorganisms on soil components playsan important role. This process has been recognized tohave a prominent effect on the geochemistry of soilsand sediments [3]. Indeed, it concerns the most abun-dant metal in the environment, namely iron. Ferric ironis essentially insoluble at neutral pHs, and amorphousiron oxide is the predominant form of Fe(III) in anaer-obic environments. Recent investigations have shownthat sulfur and sulfate reducing bacteria develop aniron reductase activity in anaerobiosis. Thus, the mainquestion has been the way in which the electrons flowoutside from the bacterial cell to the metal oxide. Afirst hypothesis is based on close contact between the
* Corresponding author. Tel.: +33-491-164443; fax: +33-491-164578.
E-mail address: [email protected] (E. Lojou).
0022-0728/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved.PII: S 0 022 -0728 (00 )00280 -1

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4938
cell and Fe(III) minerals [4]. It must be rememberedthat growth under anoxic conditions induces produc-tion of c-type cytochromes in the outer membrane ofthe bacterial cell. It is thus reasonable to envisage theinvolvement of these cytochromes, or other electroncarriers that accept electrons from them, in the electrontransfer chain to metal oxides. Actually, from studieson Fe(III) reduction, it was demonstrated that theperiplasmic polyheme cytochromes c3, which are ubi-quitous in sulfur and sulfate reducing bacteria, weredirectly involved in such a process [5].
Modified electrodes offer a simple and convenienttechnology for investigating the interactions betweenvarious substrates and soil components. In particular, ithas been shown in our laboratory [6] that the electrodesurface can be advantageously modified by clay-suspen-sion in view of developing novel electrochemical clay-based sensors. In this case, the clay suspension wasentrapped at the electrode surface via a dialysis mem-brane. These clay-modified electrodes have been provedto be suitable for the detection of various cations ofenvironmental interest [7].
Concerning proteins, very few reports [8] describe theelectrochemistry of cytochromes at clay-modified elec-trodes. In the present paper, an exhaustive study on theelectrochemical behavior of different monoheme orpolyheme c-type cytochromes investigated as proteinmodels is developed at graphite electrodes coated byclay films. Various mineral surfaces have been chosenaccording to their respective charge and hydrophobicitycharacteristics (see Table 1): kaolinite, two clays be-longing to the smectite group, montmorillonite andtalc, and a mineral surface (ochre) composed of amixture of kaolinite and oxyhydroxide mineral.Goethite, an iron oxyhydroxide mineral, has also beenstudied for additional understanding of the phenomena.Mitochondrial cytochrome c is a good candidate forsuch an approach, because of extensive investigationson its biological and biochemical properties. Cy-tochrome c belongs to the respiratory chain. Struc-turally, it contains one heme as the prosthetic groupthat can exchange one electron with partner proteins.From an electrochemical point of view, the response atvarious working electrodes is observed provided that
strict conditions (presence of anionic species or pro-moters, specific treatment of the surface, etc.) are ful-filled (see for example [14–19]). In this work, the natureof the interaction between cytochrome c and mineralsurfaces is discussed according to the experimentalparameters: pH, ionic strength, nature of the clay,composition of the electrode coating, etc. In a secondstep, the voltammetric response at clay-modified elec-trodes of bacterial polyheme c-type cytochromes, whichbehave as fast reversible electrochemical systems at barePG electrodes, is reported for the first time. Further-more, a complete study of the electrochemical behaviorof these cytochromes at a goethite-modified electrodegives new insight into the interactions between mineralsurfaces and polyheme cytochromes. The consequencesof these specific interactions on further metal reducingactivity developed by bacterial cytochromes are finallyanalyzed.
2. Experimental
2.1. Materials
Stock suspensions of clays were prepared by dispers-ing the commercial material in deionized water. Thesuspension was stirred for 2 h, and then stocked at+4°C when not in use. Sodium-montmorillonite (SWy-2) and kaolinite (KGa-1b) were purchased from theSource Clay Minerals Repository (University of Mis-souri, Columbia, MO) and talc from ICN Biochemi-cals. Artificial goethite was prepared using thetechnique of Schwertmann et al. [20], by adding KOHto a solution of Fe(NO3)3 and leaving the mixture for 2weeks at 20°C. Oxy red ochre (named ‘ochre’ in thefollowing manuscript) was purchased from Conserva-toire des Ocres et Pigments Appliques, Usine Mathieu,Roussillon, France.
Hexaammineruthenium(III) chloride (Ru(NH3)63+),
tetraammine copper(II) sulfate (Cu(NH3)42+) from
Strem Chemicals, 1,1%-dimethyl-4,4%-bipyridiniumdichloride (methylviologen, MV2+), hexacyanofer-rate(III) (Fe(CN)6
3−) and cobalt(III) sepulchratetrichloride (Co(sep)3+) from Fluka, 1,1%-ethylene-2,2%-
Table 1Composition and properties of the mineral surfaces used in this work
Clay Composition Charge Remarks Reference
Tetrahedral silica sheets (s)+octahedralKaolinite Negative at all pH values (isomorphic [9]alumina layers (g) substitutions: Si(IV) by Al(III))
Negative at all pH values (isomorphic Hydrophobic [10]Montmorillonite s–g–s stackingNa+ form substitutions: Al(III) by Mg(II))
[10]Talc Mg3Si4O10(OH)2 HydrophobicNo chargeGoethite [11,12]aFeOOH HydrophilicpH dependent pHpzc 8
Kaolinite+goethiteOchre [13]

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 39
bipyridinium dibromide (diquat DQ2+) from Riedel deHaen were used without further purification. All otherchemicals were reagent grade. Horse heart cytochromec was purchased from ICN Biochemicals. Cytochromesc3 and c553 from Desulfo6ibrio 6ulgaris Hildenborough(DvH) [21,22], c3 (Mr 26 000) from Desulfomicrobiumnor6egium (Dn) [23], c3 from Desulfo6ibrio gigas [21]and c7 from Desulfuromonas acetoxidans (Da) [24] wereprepared and purified as described previously.
All solutions were prepared from distilled, deionizedwater. Unless stated otherwise, the supporting elec-trolyte was 10 mM Tris–chloride buffer, pH 7.6.
2.2. Apparatus and procedure
2.2.1. ElectrochemistryAn EG&G 273A potentiostat controlled by a Pro-
linea 4/66 microcomputer with EG&G PARC 270/250software was used for cyclic voltammetry (CV). Athree-electrode system consisting of a MetrohmAg � AgCl � NaCl sat reference electrode, a platinumwire auxiliary electrode and the working electrode (seebelow) was used throughout. Unless otherwise stated,all potentials are referred to the Ag � AgCl � NaCl satelectrode. Prior to each voltammetric experiment, thesolutions were deoxygenated by bubbling high-puritynitrogen. All experiments were carried out at roomtemperature (23°C).
Working electrodes were constructed from 4 mmrods of pyrolytic graphite (PG–A=0.13 cm2) from LeCarbone Lorraine (Paris) housed in epoxy sheaths andcut with the disk face parallel to the edge plane. First,the electrode surface was polished with 0.05 mm alu-mina slurry. A small volume (2 ml) of clay suspensionwas deposited on the freshly polished surface. Thecoating was then allowed to dry gently under a heatgun before experiment. Unless specified, the concentra-tion of the clay was 3% w/v. The film thickness hasbeen shown to be directly proportional to the totalmass of clay added [25]. Based on published data [25]the thickness of the dry clay-coatings can be estimatedto be in the 2–5 mm range.
2.2.2. UV–6is spectroscopyUV–vis absorption spectroscopy was done with a
Cary 5 spectrophotometer from Varian. Mixtures of200 mM cytochrome c in 10 mM Tris–chloride buffer,pH 7.6 with clay suspensions (1 or 2% w/v) were firstrealized. Dry films of cytochrome c–clay mixture wereprepared by depositing a volume of the mixtures ontoindium tin oxide coated slides (ITO slides were a kindgift from Professor F.M. Hawkridge, Virginia Com-monwealth University, Richmond, USA). The dropswere then gently dried in air before spectroscopicmeasurements.
3. Results and discussion
3.1. Characterization of the clay-modified electrode inthe presence of small electroacti6e species
As the CV current observed at a clay-modified elec-trode depends on the porosity of the clay film and onthe number of sites responsible for the partitioning ofthe complex into the film, two distinct classes of soluteswere investigated. Fe(CN)6
3− was chosen at first as anelectrochemically active probe ion for an indirect mea-sure of the porosity of the film [25]. Indeed, the anioniccharacter of this complex precludes strong interactionwith the negatively charged clays, namely montmoril-lonite, kaolinite, and ochre. Secondly, Ru(NH3)6
3+
cations are known to incorporate into clay films such asmontmorillonite films [26], via long range electrostaticinteractions with the negatively charged clay surface.Thus, repetitive cyclic voltammograms were performedin 100 mM Ru(NH3)6
3+ using the four selected mineralsurfaces: kaolinite, ochre, talc, and goethite. The result-ing data were compared to Ru(NH3)6
3+ incorporationin montmorillonite films.
Typical CV curves obtained in 100 mM Fe(CN)63− in
10 mM Tris–chloride buffer, pH 7.6 either at a bare orat a montmorillonite-modified PG electrode are shownin Fig. 1. Peak shaped CVs are observed in both cases,in agreement with previous results [25], but the totalsurface of the underlying electrode is restricted in thecase of a clay-modified electrode as demonstrated by alowering in peak heights. The ratio of the peak currentobtained at the modified to the bare electrode leads toan estimation of the void space of the film, whichincludes both porosity and tortuosity of the coating. Rtakes the values of 0.55 for a 1% montmorillonite-modified electrode, suggesting that the total film porespace is at the most 55%. The same observations havebeen made at kaolinite and ochre-modified electrodes in100 mM Fe(CN)6
3− solution in 10 mM Tris–chloridebuffer, pH 7.6. Based on R values, a 55% void spacehas been reached with suspensions made from 3%kaolinite and 3% ochre. Talc and goethite, whichpresent neutral or positive surfaces at pH 7.6, respec-tively, exhibit R values of 0.65 and 0.90, respectively.
As described previously in the case of montmoril-lonite films [25], Ru(NH3)6
3+ cations readily concen-trated in kaolinite-coated electrodes. This leads tocurrents larger than at the bare electrode. Since kaolin-ite possesses a negative charge as a consequence ofisomorphic substitutions, this result is consistent withan electrostatic driven exchange mechanism. When theconcentration of the kaolinite in the suspension wasincreased, a marked increase in the enhancement ratioRe (defined as the ratio of the constant CV cathodicpeak current values at the modified electrode to thatmeasured at the electrode in the absence of clay) was

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4940
Fig. 1. Cyclic voltammetry of 100 mM Fe(CN)63− in 10 mM Tris–chloride buffer solution, pH 7.6; scan rate: 20 mV s−1; (fine line) cyclic
voltammogram at the bare PG electrode and (solid line) steady-state voltammogram at the montmorillonite-modified PG electrode (deposit of 2ml of 1% montmorillonite suspension).
Table 2Enhancement ratios (Re) for 100 mM Ru(NH3)6
3+ incorporation in various mineral coatings a
Ochre Montmorillonite Talc GoethiteClay Kaolinite
3 1 3% w/v 11 3 3 3Re 2 3.3 1.2 3.4 7.3 3.1 1.4 1.3
a Tris–chloride 10 mM pH 7.6, CV scan rate: 20 mV s−1.
recorded, denoting more and more favorable incorpora-tion. The maximum value in CV peak currents wasobserved when using 3% w/v suspension as coatingmaterial. Under these conditions the value of Re was3.3. As the clay concentration increased beyond 5%w/v, the time needed to reach the steady-state becamelonger and longer. Furthermore, mechanical holdingbecame hazardous when the thickness of the coatingwas increased.
Enhancement ratios for Ru(NH3)63+ uptake in the
various mineral surfaces are gathered in Table 2. As ageneral feature, it can be seen that negatively chargedclays, namely kaolinite, ochre and montmorillonite, areable to concentrate Ru(NH3)6
3+ in their film structure.On the contrary, due to their global surface charge, talcand goethite exhibit enhancement ratios very close tounity.
The Ru(NH3)63+ incorporation dependence on ochre
concentration is similar to that described for kaolinite.Thus, the presence of goethite in ochre does not seemto affect the cation uptake process greatly. This resultagrees with previous data devoted to adsorption phe-nomena on both ‘clean’ and ‘dirty’ (coated with Feoxyhydroxides) kaolinite [27], which leads to the con-clusion that identical substrate adsorption mechanisms
are involved with both types of clays. It must benoticed that a smaller amount of montmorillonite com-pared to kaolinite or ochre is needed to obtain a highenhancement ratio. By measuring the activity of sweetalmond b-D-glucosidase adsorbed on clay suspension, itwas shown that montmorillonite adsorbs 55 times moreenzymes than kaolinite [2]. This was related to thedifference in specific area, 800 and 14 m2 g−1 formontmorillonite and kaolinite, respectively, which canexplain the results obtained in this work.
Other species, including cations of environmentalinterest were studied at the ochre-modified electrode.This is particularly attractive since ochre reflects in situconditions where clay minerals are coated at least par-tially with mixtures of polymeric oxides and hydrox-ides. The respective enhancement ratios are given in
Table 3Enhancement ratios (Re) for the incorporation of various 100 mMcationic species in ochre coatings (from 3% suspension) a
Ru(NH3)63+ Co(sep)3+ MV2+ DQ2+Species Cu(NH3)4
2+
10.51053.4Re 2
a Tris–chloride 10 mM pH 7.6, 20 mV s−1.

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 41
Fig. 2. Cyclic voltammetry of 200 mM cytochrome c in 10 mM Tris–chloride buffer solution, pH 7.6; scan rate: 20 mV s−1; (A) (---) cyclicvoltammogram at the bare PG electrode and (—) repetitive voltammograms at the ochre-modified PG electrode (deposit of 2 ml of 3% ochresuspension); (B) dependence of the steady-state cathodic peak current recorded at the ochre-modified PG electrode on the scan rate.
Table 3. For the three metal complexes investigated, thefollowing sequence for the enhancement ratio is ob-served: Cu(NH3)4
2+BRu(NH3)63+BCo(sep)3+. The
lowest Re is attained in the case of Cu(NH3)42+ that
bears the lowest positive charge. These points confirmthe marked role played by electrostatic interactions inthe incorporation process. Very high enhancement ra-tios are observed for the two dipyridylium herbicidesMV2+ and DQ2+. As previously noted in anotherwork [7], this can have severe consequences on thefurther gradual change of such cations in the naturalenvironment.
3.2. Interaction between heme proteins and clays
3.2.1. Interaction between mitochondrial cytochrome cand clays
3.2.1.1. UV–6is spectroscopy of mixtures of cytochromec and clays. The position of the Soret absorption bandof heme iron provides information about cytochrome cconformation. It was especially demonstrated that de-naturation of cytochrome c upon increasing tempera-ture yields a blue shift of the Soret band [28].
In this work, it has been shown that spectra of eithercytochrome c solution in 10 mM Tris–chloride bufferpH 7.6 or dry film cast from cytochrome c solution ontransparent ITO glasses exhibit a Soret band at 408 nm.
Mixtures of cytochrome c solutions with three differ-ent clay dispersions, i.e. montmorillonite, talc and
ochre, were prepared. These particular clays were cho-sen because of their difference in charge, hydrophobic-ity and composition. As a first feature, it wasinteresting to observe that montmorillonite settled withtime thus dragging the cytochrome c towards the bot-tom of the UV–vis cell. As a consequence, within a fewminutes, the characteristic UV–vis spectrum of cy-tochrome c became completely flat. On the contrary,both talc and ochre settled leaving cytochrome c in thesupernatant. These results suggest a much strongeradsorption of cytochrome c on montmorillonite com-pared to ochre or talc.
Dry films cast from cytochrome c–clay suspensionson transparent ITO glasses were then examined. Usingcytochrome c-ochre dry film, the UV–vis spectrum ischaracterized by a Soret band centered at 407 nm,suggesting that in ochre films cytochrome c retains itsnative state. The UV–vis spectrum of cytochrome c-montmorillonite film exhibits a Soret band at 401 nm.This blue shift indicates that adsorption of cytochromec in dry films of montmorillonite is accompanied by achange in protein conformation.
3.2.1.2. Electrochemical beha6ior of cytochrome c atochre-modified electrodes. Fig. 2A compares the CVcurves for 200 mM cytochrome c at ochre-coated (solidline) and bare (dotted line) PG electrodes in 10 mMTris–chloride buffer, pH 7.6. Whereas no redox wave isobserved at the bare electrode, well-peaked CV curvesdevelop in the presence of an ochre coating at the

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4942
surface of the electrode at Epc= +0.03 V, Epa= +0.12V, in agreement with the literature data [29]. It is thussuggested that cytochrome c molecules are arranged ina manner favorable for electron exchange at the claysurface. Very likely, the presence of negatively chargedsites within the clay yields a promoting effect towardscytochrome c as a result of favorable electrostatic inter-actions with the positively charged residues of theprotein (cytochrome c has an isoelectric point of 10.5).The same promoting effect has been already noticedusing poly(ester-sulfonic acid) polymers as electrodemodifiers [17].
The steady-state is reached after 4 min of cyclingwhen using a coating from 3% ochre-suspension. Underthese conditions, the cathodic peak current is propor-tional to the square root of the potential scan rate inthe 2–200 mV s−1 range (Fig. 2B), indicative of thepredominance of a diffusion controlled process. Thepeak separation between the anodic and the cathodicpeak potentials is 90 mV at 20 mV s−1, suggesting aone-electron quasi-reversible process.
From film transfer experiments, it can be concludedthat cytochrome c is not strongly adsorbed on the ochrecoating. Rapid and substantial leaching of transferredcytochrome c occurs within the first few seconds follow-ing the immersion of the modified electrode. Actually,the peak current recorded immediately after the transferof the cytochrome-charged coated electrode is only 30%of the steady state current recorded in the cytochrome
solution. After 10 min cycling only 3% of the cy-tochrome remains in the coating.
3.2.1.3. Effect of pH and ionic strength on the electro-chemical beha6ior of cytochrome c at an ochre-modifiedelectrode. If electrostatic interactions are the majorfactor that governs the incorporation process of cy-tochrome c, as is the case for metal cation uptake, itcan be expected that the level of incorporated speciesmight be strongly affected by variations in pH valuesand ionic strength of the solution.
The dependence of the steady-state peak current onpH for cytochrome c at an ochre-modified electrode inthe 5–10 pH range is presented in Fig. 3A. The peakcurrent–pH curve is characterized by a maximum atpH 7.5 and a sharp decrease for pH\8.5. The decreasein cytochrome c incorporation at acidic pH can berelated to the competitive adsorption of protons on thenegative clay sites. This specific adsorption obviouslyweakens the electrostatic forces between the clay andthe protein. At alkaline pH, the current–pH profilereminds us of that observed in previous work at thegold electrode activated by 4,4%-bipyridine [30]. Fromthe study of the effect of pH on cytochrome c elec-troactivity [30], it was demonstrated that the electroac-tive and electroinactive forms of cytochrome c atalkaline pH yielded a marked decrease in peak currentsabove pH 7.
Fig. 3. Cyclic voltammetry of 200 mM cytochrome c in 10 mM Tris–chloride buffer solution, pH 7.6, at the ochre-modified PG electrode (depositof 2 ml of 3% ochre suspension); scan rate: 20 mV s−1; (A) dependence of the steady-state cathodic peak current on the pH; (B) dependence ofthe steady-state cathodic peak current on the ionic strength.

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 43
Fig. 4. Cyclic voltammetry of 200 mM cytochrome c in 10 mM Tris–chloride buffer solution, pH 7.6, at various clay-modified PG electrodes; scanrate: 20 mV s−1; (A) 3% talc; (B) 3% goethite: (---) first cycle and (—) 10th cycle; (C) 1% montmorillonite; and (D) 3% kaolinite.
A gradual decrease in Ipc when NaCl concentration isincreased in the 0–500 mM range is observed forcytochrome c electrochemical behavior at an ochre-modified electrode (Fig. 3B). The profile of the steady-state peak current when NaCl is added into solutionprobably results from two concomitant processes. Atfirst, such a decrease is expected for an incorporationuptake governed by electrostatic interactions. But pre-vious studies have demonstrated that increasing NaClconcentration decreases the interlayer space in clayfilms such as montmorillonite films [31]. As an examplethe distance between two negatively charged clay plateswas shown to diminish from 100 to 10 A, when theNaCl concentration was increased from 10 to 300 mM.Cytochrome c can be described as a globular proteinwhose diameter is 40 A, [32]. Thus, it is expected thatthe protein can no longer penetrate the film when theNaCl concentration is increased.
3.2.1.4. Influence of the nature of the clay on the electro-chemical response for cytochrome c. A comparativestudy of the effect of the different mineral surfaces onthe direct electrochemistry of cytochrome c has beenperformed. CV curves are presented in Fig. 4 for 200mM cytochrome c in 10 mM Tris–chloride buffer, pH7.6. Two distinct types of behavior are obtained accord-ing to the net surface charge of the clays.
Poorly defined steady-state electrochemical signalsare detected at talc- and goethite-modified electrodeswhatever the concentration of the clay in the 0.1–5%
w/v range (4A, B, respectively). The hydrophobic char-acter of the talc surface coupled to the neutral surfacecharge is supposed t o be responsible for unproductiveinteraction between talc and cytochrome c. Contrary totalc, however, the many hydroxyl groups in the goethitestructure can easily form hydrogen bonds with eithercarboxyl or amino groups of cytochrome c moleculesand thus constitute a hydrophilic environment neces-sary for direct electron transfer from/to cytochrome, asdemonstrated in previous work [29]. This is surely thecase in the first step of cytochrome c incorporation.Indeed, a peak shaped voltammogram is obtained forthe first cycle (dotted line in Fig. 4B). However it mustbe noticed that a modification of the electrochemicalresponse upon voltammetric cycling is recorded at thegoethite-modified electrode. The peak shaped CV curveprogressively vanishes to yield the poorly definedsteady-state profile (solid line in Fig. 4B). This peculiarbehavior can be related to the net charge of goethite atpH 7.6 which is not favorable to positive electrostaticinteractions with cytochrome c (see Table 1). It mustthen be assumed that coulombic interactions play anincreasing role in the control of the overall process.
On the contrary, persistent peak shaped CV curvesare obtained at montmorillonite and kaolinite modified-electrodes (4C, D, respectively). As already mentionedfor ochre material, these results suggest that favorableelectrostatic interactions between the negatively chargedclays and the positively charged protein are responsiblefor the electrochemical promotion of cytochrome c.

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4944
In the uptake process, the thickness of the coating isexpected to have an effect on the electrochemistry ofcytochrome c within the clay since it affects the percent-age of exchangeable sites and the total void space. Fig.5 shows the dependence of cathodic peak current (Ipc)for 200 mM cytochrome c in 10 mM Tris–chloridebuffer, pH 7.6, on the percentage of clays in the suspen-sion. As expected, the two kaolinite-containing clays,i.e. ochre and pure kaolinite, exhibit the same profile:an increase in Ipc at the lowest concentrations, a maxi-mum at about 10% w/v clay percentage, followed by adecrease in Ipc for the highest clay concentrations. Therising part of the curve reflects the increasing number offavorable sites for cytochrome c incorporation. For aclay percentage greater than 10% w/v, it is reasonableto assume that the thickness of the coating induces ahigh tortuosity factor which is unfavorable to transportof cytochrome c molecules within the film. In contrast,although the same profile is observed with montmoril-lonite, it is noticeable that the maximum in Ipc isobtained for coatings made with only 1% w/v montmo-rillonite concentration in the suspension. This result isvery much like that obtained for cationic species in theabove section, where the high specific area of montmo-
rillonite was supposed to take this particularity intoaccount.
Table 4 compares the electrochemical data for cy-tochrome c at the three negative clays tested in thisstudy that permit direct electrochemistry of cytochromec molecules. Concentration of the various clays hasbeen chosen on the basis of Fe(CN)6
3− electrochemicalbehavior, in order to obtain films of equivalent voidvolumes (see Section 3.1), and the best electrochemicalsignal. The cathodic peak current values are in thefollowing range: montmorilloniteBkaoliniteBochre.Analysis of the complete data shows that ochre is thematerial that allows the highest incorporation rate andthe fastest electron transfer rate to be achieved. Previ-ous studies devoted to swollen polyelectrolytes [17,33]have considered the coated film as a two phase domain:the ‘Donnan’ domain in the immediate neighborhoodof the fixed charged groups, and the remainder volumeconsisting of pores filled by the solution of interest. Inthe case of the diffusion of cytochrome c within theochre film, film transfer experiments have shown thatprotein molecules were rapidly released from the claycoating. It can thus be assumed that the overall processis predominantly governed by diffusion of cytochromec through the pores of the clay film. Thus, based on thevalue of the diffusion coefficient of cytochrome c insolution (1.2×10−6 cm2 s−1) [34], the heterogeneousrate constant has been calculated. From the peak sepa-ration data using the relation given in Ref. [35] a valueof 1×10−3 cm s−1 has been estimated, which is oneorder less than that estimated at a bare graphite elec-trode [35].
Results in Table 4 underline that CV peak separationgreatly increases at a montmorillonite-modified elec-trode. As an example, it reaches 175 mV at a sweep rateof 20 mV s−1. This decrease in the electron transferrate is accompanied by an increase in protein retentionwithin the clay coating. Indeed, whereas only 30% ofthe cytochrome c remains in the ochre structure imme-diately after transfer in pure electrolyte, 70% of cy-tochrome c is recovered in the same experiment usingthe montmorillonite coating. It can therefore be con-cluded that cytochrome c is more tightly bound tomontmorillonite than to ochre or kaolinite. This type ofadsorption is favorable to retention of the protein inthe clay structure but decreases the kinetics of chargetransfer by favoring denaturation of cytochrome c, asnoticed in other work [36]. This is strongly supportedby the results obtained from UV–vis experiments inthis work.
3.2.2. Electrochemical beha6ior of bacterial c-typepolyheme cytochromes at clay-modified electrodes
Three different polyheme c-type cytochromes havebeen tested at modified clay-electrodes, DvH cy-tochrome c3, Da cytochrome c7 and Dn cytochrome c3
Fig. 5. Dependence of the steady-state cathodic peak current on thepercentage of the clay in the suspension for 200 mM cytochrome c in10 mM Tris–chloride buffer solution, pH 7.6; scan rate: 20 mV s−1;() ochre, () kaolinite, and ( ) montmorillonite.
Table 4Peak characteristics for the electrochemical signals of cytochrome c atvarious negatively charged clay-modified electrodes
DEp/mVEpa/mVClay Epc/mVIpc/nA
5883% Ochre +30 +120 901153% Kaolinite 484 +133+18
+1581% Montmorillonite 175331 −17

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 45
Fig. 6. Steady-state cyclic voltammograms of polyheme cytochromes at a montmorillonite-modified PG electrode in 10 mM Tris–chloride buffersolution, pH 7.6; scan rate: 20 mV s−1; (A) 100 mM DvH cytochrome c3 at (---) 2% montmorillonite-modified electrode and (—) 0.5%montmorillonite-modified electrode; (B) 200 mM Da cytochrome c7 at (---) bare and (—) 2% montmorillonite-modified electrode; (C) 100 mM Dncytochrome c3 (Mr 26 000) at (---) bare and (—) 0.5% montmorillonite-modified electrode.
(Mr 26 000), whose isoelectric points are 10.2, 7.8 and4.8, respectively. These bacterial cytochromes exchangeelectrons directly and reversibly with naked PG elec-trodes in the absence of any dissolved promoting spe-cies [37]. If electrostatic interactions are the drivingforce for the electron transfer between proteins and claymaterial, different electrochemical behavior is expecteddepending essentially on the net charge of both clayand cytochrome.
3.2.2.1. Kaolinite-modified PG electrode. CV curvesrecorded at the negatively charged kaolinite-modifiedelectrode for solutions of polyheme cytochromes in 10mM Tris–chloride buffer pH 7.6, exhibit well peakshaped waves. As observed for cytochrome c solution,peak currents are significantly higher at the clay-modified electrode than at the bare PG electrode. Peakpotential values are nearly the same whether the elec-trode is modified or not.
By comparing steady-state CV curves recorded foreach polyheme cytochrome at the kaolinite-modifiedelectrode (clay concentration in the suspension was 3%w/v), it clearly appears that the more favorable theelectrostatic interactions between the negatively chargedclay and the protein, the higher is the enhancementratio. Actually, the enhancement ratios are 2.5, 1.6 and1.4 for the positively charged DvH cytochrome c3,neutral Da cytochrome c7 and negatively charged Dncytochrome c3 (Mr 26 000), respectively. It is noticeable
however that electron transfer is observed even in theabsence of favorable interactions as is the case for Dncytochrome c3 (Mr 26 000).
3.2.2.2. Montmorillonite-modified PG electrode. Al-though favorable electrostatic interactions are fulfilled,electron transfer on positively charged DvH cy-tochrome c3 is achieved only for montmorillonite con-centrations below 0.5% w/v (Fig. 6A, solid line). Usingthis particular montmorillonite concentration, the en-hancement ratio takes a low value of 1.5. More concen-trated montmorillonite dispersions do not allow anelectrochemical signal for DvH cytochrome c3 to beobserved (Fig. 6A, dashed line). This result concurswith that given for cytochrome c electrochemistry,where a maximum in cytochrome c promotion wasrealized with diluted montmorillonite suspension com-pared to ochre or kaolinite.
Besides, no redox signals of Da cytochrome c7 or Dncytochrome c3 (Mr 26 000) (6B, C, respectively) areobserved, even at the most diluted montmorillonitecoating. This is clearly denoted in Fig. 6C where noelectrochemical wave develops in Dn cytochrome c3 (Mr
26 000) at 0.5% montmorillonite-modified electrode(Fig. 6C solid line), contrary to the electrochemicaltwo-wave signal observed at the bare PG electrode (Fig.6C dashed line).
Two concomitant interaction forces have to be con-sidered for taking the complete phenomenon into ac-

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4946
count. Differences in electrochemical behavior of multi-heme cytochromes at diluted montmorillonite-modifiedPG electrodes depending on the pI values of the cy-tochromes confirm the involvement of electrostaticforces in the electrochemical process. But interventionof hydrophobic forces is mandatory in order to under-stand why the positively charged DvH cytochrome c3
develops a low Re in the hydrophobic montmorillonite-modified electrode. In this case, strong adsorption andpossible reconformation of bacterial polyheme cy-tochrome within the montmorillonite structure is sup-posed to forbid the electron transfer.
3.2.2.3. Talc-modified PG electrode. Talc material alsopresents a hydrophobic surface but no overall charge.Thus, it constitutes a good mineral surface for deter-mining the predominance of either electrostatic or hy-
drophobic forces in the interactions between proteinsand clays. At the talc-modified electrode, the threepolyheme cytochromes behave as fast reversible electro-chemical systems. From the CV curves (not shownhere), the enhancement ratios are found to be very closeto unity, however: 1.3, 1.1 and 1.1 for DvH cytochromec3, Da cytochrome c7 and Dn cytochrome c3 (Mr
26 000), respectively. The fact that talc does not forbidthe electron transfer on polyheme cytochromes al-though it possesses a hydrophobic surface like mont-morillonite, demonstrates that electrostatic interactionsmust play the most important role in the electrochemi-cal process.
3.2.2.4. Goethite-modified PG electrode. Goethitecoated-electrodes were used to study different c-typecytochrome solutions in 10 mM Tris–chloride buffer,pH 7.6, i.e. DvH cytochrome c3 (pI 10.5), Da cy-tochrome c7 (pI 7.8), Dn cytochrome c3 (Mr 26 000) (pI4.8) (see 7A–C, respectively), and Dg cytochrome c3 (pI5) (CV curve not shown). Each CV curve is character-ized by greatly enhanced cathodic currents at themodified electrode, though the proteins have differentglobal charges. The enhancement ratios (or ratio of thelimiting cathodic current at the modified electrode tothe peak current at the bare electrode in the peculiarcases of DvH cytochrome c3 and Dn cytochrome c3 (Mr
26 000)) are very high, reaching values of 7.0, 5.5 and6.0 for DvH cytochrome c3, Da cytochrome c7 and Dncytochrome c3 (Mr 26 000), respectively. In the case ofthe acidic Dg cytochrome c3, an even higher value of Re
is attained (Re=10). As illustrated by the inset to Fig.7A in the particular case of DvH cytochrome c3, Re
increases with increasing percentage of goethite in thesuspension until a plateau is reached. A study of thepH-dependence of CV peak currents at the goethite-modified electrode was performed in Da cytochrome c7
solution. Although the global charge of Da cytochromec7 is changed in the 5–10 pH range, no variation inpeak currents is detected.
Thus, it appears that interaction between the proteinand the mineral occurs even if the protein and thesorbent have the same charge. Even more, the magni-tude of the interaction seems to be independent of thecharge of the protein as indicated from the peak cur-rent–pH evolution. This suggests that factors otherthan coulombic interactions are responsible for theenhanced cathodic current at the goethite-modifiedelectrode.
A first explanation for the behavior of polyhemec-type cytochromes having different pI values atgoethite-modified electrodes could originate in the hy-drophilic character of the mineral surface. Other hy-drophilic surfaces were demonstrated previously asbeing capable of adsorbing significant amounts ofprotein despite repulsive electrostatic interactions. In
Fig. 7. Steady-state cyclic voltammograms of polyheme cytochromesat (---) bare and (—) 3% goethite-modified PG electrodes in 10 mMTris–chloride buffer solution, pH 7.6; scan rate: 20 mV s−1; (A) 100mM DvH cytochrome c3, inset: dependence of Re on the percentage ofgoethite; (B) 200 mM Da cytochrome c7; (C) 100 mM Dn cytochromec3 (Mr 26 000).

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 47
Fig. 8. Steady-state cyclic voltammograms of polyheme cytochromesat (---) bare and (—) 3% ochre-modified PG electrode in 10 mMTris–chloride buffer solution, pH 7.6; scan rate: 20 mV s−1; (A) 100mM DvH cytochrome c3; (B) 200 mM Da cytochrome c7; (C) 100 mMDn cytochrome c3 (Mr 26 000).
cytochrome interaction has to be put forward. At thegoethite–modified electrode, Da cytochrome c7 behavesas a reversible electrochemical system. Both cathodicand anodic peak currents are enhanced and Ipc/Ipa isclose to unity as is the case at the bare PG electrode(Fig. 7B). CV curves recorded in DvH c3 and Dncytochrome c3 (Mr 26 000), however exhibit large ca-thodic currents while weak anodic currents are ob-served on the reverse scan (7A, C). Dg cytochrome c3
(curve not given) exhibits very similar behavior. Fromthis feature, it can be concluded that some catalyticcontribution is implied in the electrochemical process.Such a phenomenon is not unexpected in this casebecause of the presence of iron atoms in goethite. It hasbeen established in previous work [40] that polyhemecytochromes were able to reduce suspensions of metaloxides, such as MnO2, V2O5 or FeOOH catalytically. Inthis work, catalytic reduction of a dry coating ofgoethite is achieved by the cytochromes that present thelowest redox heme potentials. Da cytochrome c7 whichpossesses active heme centers with redox potentials 100mV higher than DvH cytochrome c3, Dn cytochromepreviously or Dg cytochrome c3, is not able to reducegoethite. Additional experiments on the electrochemicalbehavior of monoheme DvH cytochrome c553 at thegoethite-modified electrode confirm that the redox po-tential of cytochrome is the driving force of the cata-lytic process. Actually, DvH cytochrome c553 behaves asa fast electrochemical system at a bare PG electrodewith a redox potential of −220 mV versus Ag � AgCl.At the goethite-modified electrode, no catalytic processis observed in the CV curves, but enhanced cathodicand anodic currents resulting from the positive interac-tions between the clay and the protein.
3.2.2.5. Ochre-modified PG electrode. The electrochemi-cal behavior of polyheme cytochromes at ochre-modified electrodes reflects the presence of bothkaolinite and goethite in the natural clay. Effectively,enhanced currents are observed with Re values of 3.8, 3and 1.8 for DvH cytochrome c3, Da cytochrome c7 andDn cytochrome c3 (Mr 26 000), respectively (8A–C).Moreover, in the case of DvH cytochrome c3, thecatalytic contribution to the overall cathodic current isobserved (Fig. 8A). This is not the case for Dn cy-tochrome c3 (Mr 26 000), which suffers from unfavor-able electrostatic interaction with the negativelycharged kaolinite component (Fig. 8C). Da cytochromec7 behaves similarly either at the goethite or at theochre-modified electrode. The Re value is weaker forthe ochre material because of the lower percentage ofhydrophilic goethite in the mixed clay (Fig. 8B). Fromthe linearity of the cathodic peak current with squareroot of the scan rate in the range 5–50 mV s−1, it isshown that the process is diffusion limited.
particular, hematite (Fe2O3) was reported to bind hu-man plasma albumin, although both the sorbent andprotein carry a positive charge [38]. Otherwise, thedirect electrochemistry of cytochrome c and myoglobinwas shown to be possible at hydrophilic surface elec-trodes that were complementary to the binding surfaceof proteins. The surface was thought to serve to bindthe protein reversibly and orient it favorably [15,39].Thus, in this work, it can be reasonably concluded thatthe hydrophilic character of goethite is responsible forthe efficient interaction between the mineral and poly-heme cytochromes.
From the shape of the CV curves in Fig. 7, anotherfactor that plays a key role in the goethite-polyheme
Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–4948
3.2.3. Interaction between bacterial polyhemecytochromes and clays: consequences for the metalreductase acti6ity
Previous work [5] has demonstrated the ability ofpolyheme c-type cytochromes from various strains tocatalyze the reduction of a soluble iron(III) complex.New paths for the use of sulfur and sulfate reducingbacteria in the bioremediation of metal-contaminatedwaters and waste streams have been opened using sucha property. Recent work reports the application of thisbacterial process to the removal of Fe(III) impuritiesfrom porcelain [41]. The question is to know whetherthis activity can be retained when bacteria are en-trapped in clay matrixes.
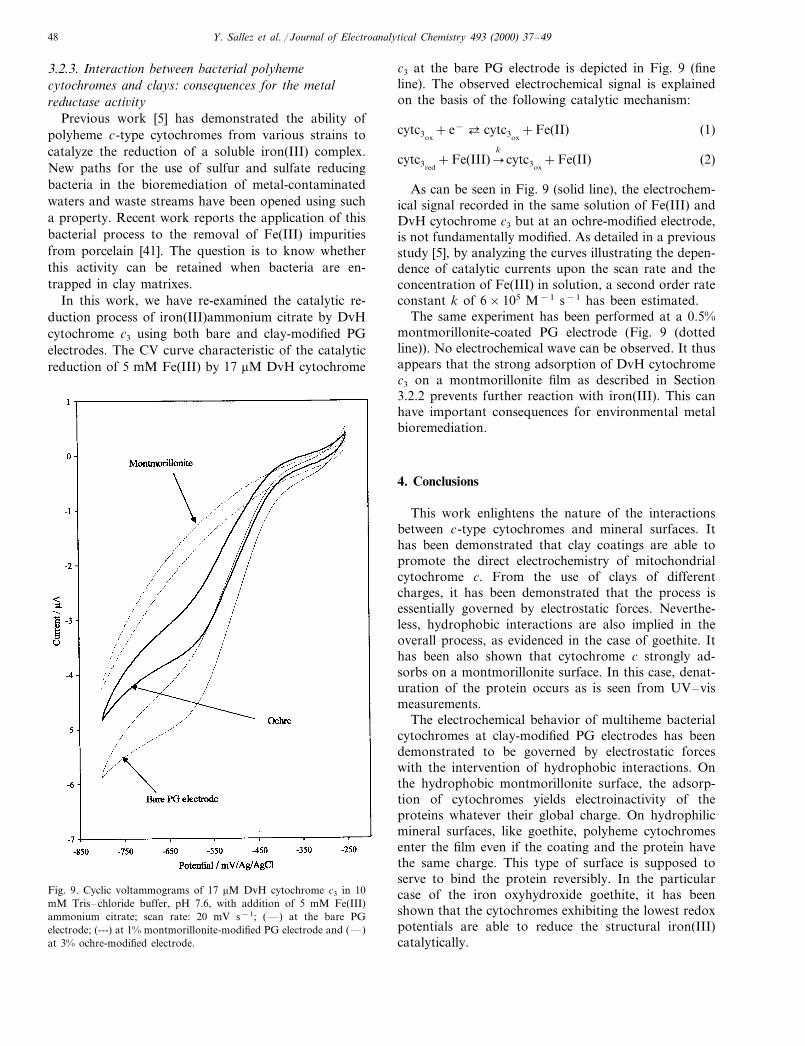
In this work, we have re-examined the catalytic re-duction process of iron(III)ammonium citrate by DvHcytochrome c3 using both bare and clay-modified PGelectrodes. The CV curve characteristic of the catalyticreduction of 5 mM Fe(III) by 17 mM DvH cytochrome
c3 at the bare PG electrode is depicted in Fig. 9 (fineline). The observed electrochemical signal is explainedon the basis of the following catalytic mechanism:
cytc3ox+e− ? cytc3ox
+Fe(II) (1)
cytc3red+Fe(III)�
kcytc3ox
+Fe(II) (2)
As can be seen in Fig. 9 (solid line), the electrochem-ical signal recorded in the same solution of Fe(III) andDvH cytochrome c3 but at an ochre-modified electrode,is not fundamentally modified. As detailed in a previousstudy [5], by analyzing the curves illustrating the depen-dence of catalytic currents upon the scan rate and theconcentration of Fe(III) in solution, a second order rateconstant k of 6×105 M−1 s−1 has been estimated.
The same experiment has been performed at a 0.5%montmorillonite-coated PG electrode (Fig. 9 (dottedline)). No electrochemical wave can be observed. It thusappears that the strong adsorption of DvH cytochromec3 on a montmorillonite film as described in Section3.2.2 prevents further reaction with iron(III). This canhave important consequences for environmental metalbioremediation.
4. Conclusions
This work enlightens the nature of the interactionsbetween c-type cytochromes and mineral surfaces. Ithas been demonstrated that clay coatings are able topromote the direct electrochemistry of mitochondrialcytochrome c. From the use of clays of differentcharges, it has been demonstrated that the process isessentially governed by electrostatic forces. Neverthe-less, hydrophobic interactions are also implied in theoverall process, as evidenced in the case of goethite. Ithas been also shown that cytochrome c strongly ad-sorbs on a montmorillonite surface. In this case, denat-uration of the protein occurs as is seen from UV–vismeasurements.
The electrochemical behavior of multiheme bacterialcytochromes at clay-modified PG electrodes has beendemonstrated to be governed by electrostatic forceswith the intervention of hydrophobic interactions. Onthe hydrophobic montmorillonite surface, the adsorp-tion of cytochromes yields electroinactivity of theproteins whatever their global charge. On hydrophilicmineral surfaces, like goethite, polyheme cytochromesenter the film even if the coating and the protein havethe same charge. This type of surface is supposed toserve to bind the protein reversibly. In the particularcase of the iron oxyhydroxide goethite, it has beenshown that the cytochromes exhibiting the lowest redoxpotentials are able to reduce the structural iron(III)catalytically.
Fig. 9. Cyclic voltammograms of 17 mM DvH cytochrome c3 in 10mM Tris–chloride buffer, pH 7.6, with addition of 5 mM Fe(III)ammonium citrate; scan rate: 20 mV s−1; (—) at the bare PGelectrode; (---) at 1% montmorillonite-modified PG electrode and (—)at 3% ochre-modified electrode.

Y. Sallez et al. / Journal of Electroanalytical Chemistry 493 (2000) 37–49 49
The clay–cytochrome interactions govern the metalreductase activity developed by bacterial c-type cy-tochromes. Actually, in the case of strong adsorption ofthe protein, the activity is lost. This is, in particular, thecase for DvH cytochrome c3 on montmorillonite whichis not able to reduce the soluble complex of iron(III)investigated here.
Acknowledgements
We thank Dr H. Quiquampoix for kindly supplyingthe goethite sample. We are indebted to Dr M. Bruschifor the gift of purified polyheme cytochrome samples.
References
[1] R.G. Burns, Soil Biol. Biochem. 14 (1982) 423.[2] H. Quiquampoix, Biochimie 69 (1987) 753.[3] M.L. Coleman, D.B. Hedrick, D.R. Lovley, D.C. White, K. Pye,
Nature 361 (1993) 436.[4] D.R. Lovley, Microbiol. Rev. 55 (1991) 259.[5] E. Lojou, P. Bianco, M. Bruschi, Electrochim. Acta 43 (1998)
2005.[6] P. Bianco, N. Agrhoud, Electroanalysis 9 (1997) 602.[7] M. Pecorari, P. Bianco, Electroanalysis 10 (1998) 181.[8] C. Lei, F. Lisdat, U. Wollenberger, F. Scheller, Electroanalysis
11 (1999) 274.[9] M.J. Angove, B.B. Johnson, J.D. Wells, Colloids Surf. 126
(1997) 137.[10] U. Hofmann, K. Endell, D. Wilm, Z. Kristallogr. 86 (1933) 340.[11] L. Sigg, W. Stumm, Colloids Surf. 2 (1981) 101.[12] D.P. Rodda, B.B. Johnson, J.D. Wells, J. Colloid Interface Sci.
161 (1993) 57.[13] S. Bec, Ocres, Luberon Images et Signes, Edisud, 1997.[14] M.J. Eddowes, H.A.O. Hill, J. Chem. Soc. Chem. Commun.
(1977) 3154.[15] H.A.O. Hill, L.H. Guo, G. McLendon, in: R.A. Scott (Ed.),
Cytochrome c : A Multidisciplinary Approach, University Sci-ences Books, CA, 1996 (Chapter 7).
[16] K.R. Brown, A.P. Fox, M.J. Natan, J. Am. Chem. Soc. 118(1996) 1154.
[17] E. Lojou, P. Luciano, S. Nitsche, P. Bianco, Electrochim. Acta44 (1999) 3341.
[18] Q. Huang, Z. Lu, J.F. Rusling, Langmuir 12 (1996) 5472.[19] P. Bianco, A. Taye, J. Haladjian, J. Electroanal. Chem. 377
(1994) 299.[20] U. Schwertmann, P. Cambier, E. Murat, Clays Clay Minerals 33
(1985) 369.[21] J. Le Gall, G. Mazza, N. Dragoni, Biochem. Biophys. Acta 99
(1965) 30.[22] P. Bianco, J. Haladjian, R. Pilard, M. Bruschi, J. Electroanal.
Chem. 136 (1982) 291.[23] F. Guerlesquin, G. Bovier-Lapierre, M. Bruschi, Biochem. Bio-
phys. Res. Commun. 106 (1982) 291.[24] I. Probst, M. Bruschi, N. Pfennig, J. Le Gall, Biochem. Biophys.
Acta 460 (1977) 58.[25] P. Subramanlan, A. Fitch, Environ. Sci. Technol. 26 (1992) 1775.[26] A. Fitch, J. Song, J. Stein, Clays Clay Minerals 44 (1996) 370.[27] P. Fusi, G.G. Ristori, L. Calamai, G. Stotzky, Soil Biol.
Biochem. 21 (1989) 911.[28] K.L. Hanrahan, S.M. MacDonald, S.G. Roscoe, Electrochim.
Acta 41 (1996) 2469.[29] F.A. Armstrong, P.A. Cox, H.A.O. Hill, V.J. Lowe, P.N. Oliver,
J. Electroanal. Chem. 217 (1987) 331.[30] J. Haladjian, R. Pilard, P. Bianco, P.A. Serre, Bioelectrochem.
Bioenerg. 9 (1982) 91.[31] A. Fitch, S.A. Lee, J. Electroanal. Chem. 344 (1993) 45.[32] N. Mandel, G. Mandel, L.T. Benes, J. Rosenberg, G. Carlson,
R.E. Dickerson, J. Biol. Chem. 252 (1977) 4619.[33] F.C. Anson, T. Ohsaka, J.M. Saveant, J. Am. Chem. Soc. 105
(1983) 4883.[34] E.F. Bowden, J.F. Hawkridge, E.E. Chlebowski, C. Bancroft,
H.N. Thorpe, J. Blount, J. Am. Chem. Soc. 104 (1982) 7641.[35] (a) E. Laviron, J. Electroanal. Chem. 101 (1979) 19. (b) H.A.O.
Hill, D. Whitford, J. Electroanal. Chem. 235 (1987) 153.[36] A. Szucs, M. Novak, J. Electroanal. Chem. 383 (1995) 75.[37] P. Bianco, J. Haladjian Biochim. 76 (1994) 605.[38] P.G. Koutsoukos, W. Norde, J. Zyklemai, J. Colloid Interface
Sci. 95 (1983) 385.[39] M. Tominaga, T. Kumagai, S. Takita, I. Taniguchi, Chem. Lett.
(1993) 1771.[40] E. Lojou, P. Bianco, M. Bruschi, J. Electroanal. Chem. 452
(1998) 167.[41] E.Y. Lee, K. Cho, H.W. Ryu, Y.K. Chang, J. Biosci. Bioenerg.
87 (1999) 397.
.




















