
Effects of the reaction parameters on the properties of thermosensitive poly(N-isopropylacrylamide)...
15
Effects of the Reaction Parameters on the Properties of Thermosensitive Poly(N-isopropylacrylamide) Microspheres Prepared by Precipitation and Dispersion Polymerization HANA MACKOVA ´ , DANIEL HORA ´ K Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Square 2, 162 06 Prague 6, Czech Republic Received 6 September 2005; accepted 25 October 2005 DOI: 10.1002/pola.21223 Published online in Wiley InterScience (www.interscience.wiley.com). ABSTRACT: Poly(N-isopropylacrylamide) (PNIPAAm)-based microspheres were pre- pared by precipitation and dispersion polymerization. The effects of several reaction parameters, such as the type and concentration of the crosslinker (N,N 0 -methylene- bisacrylamide or ethylene dimethacrylate), medium polarity, concentration of the monomer and initiator, and polymerization temperature, on the properties were examined. The hydrogel microspheres were characterized in terms of their chemical structure, size and size distribution, and morphological and temperature-induced swelling properties. A decrease in the particle size was observed with increasing polarity of the reaction medium or increasing concentration of poly(N-vinylpyrroli- done) as a stabilizer in the dispersion polymerization. The higher the content was of the crosslinking agent, the lower the swelling ratio was. Too much crosslinker gave unstable dispersions. Although the solvency of the precipitation polymerization mix- ture controlled the PNIPAAm microsphere size in the range of 0.2–1 lm, a micro- meter range was obtained in the Shellvis 50 and Kraton G 1650 stabilized dispersion polymerizations of N-isopropylacrylamide in toluene/heptane. Typically, the particles had fairly narrow size distributions. Copolymerization with the functional glycidyl methacrylate monomer afforded microspheres with reactive oxirane groups. V V C 2005 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 968–982, 2006 Keywords: colloids; dispersion polymerization; dynamic light scattering; micro- spheres; N-isopropylacrylamide; precipitation polymerization; swelling; thermosensitive INTRODUCTION Intelligent (environmentally sensitive) hydrogels have been the subject of extensive research and development since the 1990s because their vol- ume changes (swelling or shrinking transition) can be triggered by various external stimuli, such as the temperature, 1 solvent composition, 2 hydro- static pressure, 3 electric fields, and antigen and radiation forces. A prominent role is played here especially by temperature-sensitive gels, an im- portant representative of which is nonionic poly (N-isopropylacrylamide) (PNIPAAm). 4,5 One of the reasons that it has been so widely studied is that PNIPAAm can shrink and/or swell in water in response to changes in the temperature and even undergo a large volume change at a temperature of 32 8C, which is called the volume-phase-tran- sition temperature (VPTT) in crosslinked poly- mers or lower critical solution temperature (LCST) Correspondence to: D. Hora ´ k (E-mail: [email protected]) Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 44, 968–982 (2006) V V C 2005 Wiley Periodicals, Inc. 968
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Effects of the reaction parameters on the properties of thermosensitive poly(N-isopropylacrylamide)...

Effects of the Reaction Parameters on the Propertiesof Thermosensitive Poly(N-isopropylacrylamide)Microspheres Prepared by Precipitationand Dispersion Polymerization
HANA MACKOVA, DANIEL HORAK
Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic,Heyrovsky Square 2, 162 06 Prague 6, Czech Republic
Received 6 September 2005; accepted 25 October 2005DOI: 10.1002/pola.21223Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: Poly(N-isopropylacrylamide) (PNIPAAm)-based microspheres were pre-pared by precipitation and dispersion polymerization. The effects of several reactionparameters, such as the type and concentration of the crosslinker (N,N0-methylene-bisacrylamide or ethylene dimethacrylate), medium polarity, concentration of themonomer and initiator, and polymerization temperature, on the properties wereexamined. The hydrogel microspheres were characterized in terms of their chemicalstructure, size and size distribution, and morphological and temperature-inducedswelling properties. A decrease in the particle size was observed with increasingpolarity of the reaction medium or increasing concentration of poly(N-vinylpyrroli-done) as a stabilizer in the dispersion polymerization. The higher the content was ofthe crosslinking agent, the lower the swelling ratio was. Too much crosslinker gaveunstable dispersions. Although the solvency of the precipitation polymerization mix-ture controlled the PNIPAAm microsphere size in the range of 0.2–1 lm, a micro-meter range was obtained in the Shellvis 50 and Kraton G 1650 stabilized dispersionpolymerizations of N-isopropylacrylamide in toluene/heptane. Typically, the particleshad fairly narrow size distributions. Copolymerization with the functional glycidylmethacrylate monomer afforded microspheres with reactive oxirane groups. VVC 2005
Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 968–982, 2006
Keywords: colloids; dispersion polymerization; dynamic light scattering; micro-spheres; N-isopropylacrylamide; precipitation polymerization; swelling; thermosensitive
INTRODUCTION
Intelligent (environmentally sensitive) hydrogelshave been the subject of extensive research anddevelopment since the 1990s because their vol-ume changes (swelling or shrinking transition)can be triggered by various external stimuli, suchas the temperature,1 solvent composition,2 hydro-
static pressure,3 electric fields, and antigen andradiation forces. A prominent role is played hereespecially by temperature-sensitive gels, an im-portant representative of which is nonionic poly(N-isopropylacrylamide) (PNIPAAm).4,5 One of thereasons that it has been so widely studied is thatPNIPAAm can shrink and/or swell in water inresponse to changes in the temperature and evenundergo a large volume change at a temperatureof �32 8C, which is called the volume-phase-tran-sition temperature (VPTT) in crosslinked poly-mers or lower critical solution temperature (LCST)
Correspondence to: D. Horak (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 44, 968–982 (2006)VVC 2005 Wiley Periodicals, Inc.
968

in linear polymers. Upon a slight change in thetemperature, it undergoes a transition from ahydrophilic structure to a hydrophobic structure.Water can be expelled from the gel matrix as aresult of the disruption of hydrogen bonds andhydrophobic interactions of isopropyl groups ofneighboring polymer chains. At the same time,the particles can undergo a morphological transi-tion from a loosely swollen network to rigidspheres. In other words, at temperatures belowthe LCST, water is a good solvent, and thespheres are highly swollen. At elevated tempera-tures, the solvent quality changes, and above theLCST, water is a nonsolvent; this leads to the col-lapse of the particles. The environmental sensi-tivity is mainly ascribed to the strong hydropho-bicity of the isopropyl groups located in sidechains. At lower temperatures (e.g., < 32 8C),water molecules around the isopropyl groups areordered and form an iceberg structure6 to mini-mize the number of contacts with the hydropho-bic groups. A number of other factors, such as thestructure and concentration of the crosslinkingagent used, incorporation of comonomers, proper-ties of solvents and surfactants, and addition ofsalts, also have a strong influence on the phasebehavior of the gels.7 Crosslinking or copolymer-ization with hydrophobic monomers often weak-ens or even eliminates the thermal sensitivity ofPNIPAAm-based hydrogels. In fact, the introduc-tion of hydrophobic compounds generally lowersthe transition temperature by reducing the poly-mer solvency in the aqueous phase, whereas theincorporation of hydrophilic compounds increasesthe LCST of the corresponding copolymers by theenhancement of the polymer solubility in water.Interesting temperature- and pH-responsive inter-penetrating networks were obtained from N-iso-propylacrylamide (NIPAAm) and sodium acrylate.8
The temperature of about 32 8C is close to thetemperature of the human body (37 8C), andhence such PNIPAAm hydrogels have been eval-uated in temperature-regulated drug deliverysystems9–11 and separation processes.12 In thesesystems, the substrates in PNIPAAm hydrogelsare squeezed out at the target tissue in the bodyby the shrinking of the gels in response to tem-perature. Moreover, thermosensitive materialsmay yield surfaces that control biological interac-tions such as bioadhesion.13,14 On the PNIPAAm-containing surface, cells adhere, spread, andproliferate at 37 8C, and below the LCST, the cul-tured cell monolayers spontaneously detach (har-vest) as single cell sheets from the hydrophilic
surfaces without enzymatic digestion.15 The uni-que thermoresponsive property of PNIPAAm andits copolymers makes it not only particularly rele-vant as a novel method for drug delivery and tis-sue engineering but also promising for injectablepolymer implants.16 Thermosensitive biodegrad-able hydrogels composed of NIPAAm, poly(L-lacticacid), and dextran show great potential for bio-medical applications.17 Another possible applica-tion of thermoresponsive gels consists of their useas smart actuators and on–off environmentalstimulation switches, for which a fast response isneeded. PNIPAAm chains then act as a tempera-ture sensor18 and as a valve controlling the filtra-tion characteristics of the membrane.
A small dimension is necessary for the stimuli-responsive hydrogels for drug delivery systems totransverse certain organs. Hence, it is necessaryto synthesize the gels in the form of microspheres,the small size of which minimizes any potentialirritant reaction at the injection site. Moreover,reducing the size of PNIPAAm hydrogels mayimprove their response rate in comparison withconventional bulky PNIPAAm hydrogels, whichhave rather slow response rates. A uniform mi-crosphere particle size is equally important fordrug delivery systems because the distribution ofthe microspheres in the body and their interac-tion with biological cells are greatly affected bythe particle size.19 If monodisperse microspheresare available, physical/chemical properties areuniform, and the drug release kinetics can bemanipulated; this makes it easier to formulatemore sophisticated intelligent drug delivery sys-tems. Despite plentiful excellent literature dataon the design of thermosensitive PNIPAAmmicrospheres,7,20–22 the effects of various reactionparameters, such as the type and concentrationof the crosslinking agent, medium polarity, mono-mer and initiator concentration, incorporation ofreactive oxirane groups, and polymerization tem-perature, on the particle size and monodispersityhave not yet been examined in detail.
EXPERIMENTAL
Materials
NIPAAm (Aldrich, Milwaukee, WI) and N,N0-methylenebisacrylamide (MBAAm; Aldrich) wereused as received. Ammonium persulfate (APS)was from Lachema (Brno, Czech Republic), and2,20-azobisisobutyronitrile (AIBN) was obtained
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 969

from Fluka (Buchs, Switzerland) and recrystal-lized from ethanol. Ethylene dimethacrylate(EDMA; Rohm GmbH, Darmstadt, Germany) wasdistilled; heptane and toluene were from Lache-ma. Poly(N-vinylpyrrolidone) [PVP; weight-averagemolecular weight (Mw) ¼ 40,000; K30, Fluka],Shellvis 50 [poly(hydrogenated isoprene-block-styrene); Mw ¼ 180,000, 25 wt % polystyrene(PSt)], and Kraton G 1650 [poly(styrene-block-hy-drogenated butadiene-block-styrene); Mw ¼ 74,000,number-average molecular weight ¼ 70,000, PSt/polybutadiene (PBu) ¼ 29/71 w/w] were stabil-izers from Shell (Houston, TX). Other solventsand reagents were from Aldrich.
Synthesis of the PNIPAAm Hydrogel Microspheres
PNIPAAm particles were prepared by precipita-tion polymerization in a single step. In a typicalexperiment, after the dissolution of 1.52 g ofNIPAAm, 0.08 g of MBAAm, and 14 mg of APS in80 mL of distilled water, the mixture was bubbledwith nitrogen for 10 min and heated to 70 8C tostart the polymerization. The reaction proceededunder stirring (400 rpm) for 8 h at 70 8C. Aftercooling, the particles were thoroughly dialyzed(molecular weight cutoff ¼ 14,000; Visking mem-brane, Carl Roth GmbH, Germany) for 14 daysagainst distilled water to remove residual mono-mers and initiator. Finally, the particles werequickly frozen in liquid nitrogen and freeze-driedin vacuo at �42 8C in a Lyovac GT 2 freezer (Ley-bold, Cologne, Germany) for at least 48 h.
Analogously, the dispersion polymerization ofNIPAAm in water was stabilized with PVP. Thedispersion polymerization of NIPAAm (1 g) in tolu-ene/heptane (27 g) was stabilized with Shellvis 50and Kraton G 1650 (always 0.27 g) and initiatedwith AIBN (0.01 g).
Characterization
The hydrodynamic particle sizes were measuredwith quasielastic light scattering on a model ZEN3600 Zetasizer nanoinstrument (Malvern Instru-ments, Malvern, United Kingdom). The swellingratio was (Dh/Dhc)
3, where Dh and Dhc are thehydrodynamic diameters of the normal and col-lapsed particles, measured at room temperatureand at 37 8C, respectively. The particle size in thedry state and the particle size distribution werethen analyzed by scanning electron microscopy(SEM; JSM 6400, JEOL). The number-averagediameter (Dn), weight-average diameter (Dw),and uniformity [polydispersity index (PDI) ¼ Dw/
Dn] were calculated with Atlas software (TescanDigital Microscopy Imaging, Brno, Czech Repub-lic) by the counting of at least 500 individual par-ticles from SEM microphotographs. Dn and Dw
can be expressed as follows:
Dn ¼X
niDi=X
ni ð1Þ
Dw ¼X
niD4i =
XniD
3i ð2Þ
where n and D are the number and the diameterof the particles, respectively.
The IR spectra of the polymers were recordedon a PerkinElmer Paragon 1000PC Fouriertransform infrared spectrometer. The oxiranegroup content was determined on the basis of thepeak area at 910 cm�1. The composition of thecopolymers [including the incorporation of gly-cidyl methacrylate (GMA)] was determined witha PerkinElmer 2400 CHN elemental analyzer.The amount of NIPAAm in the microspheres wascalculated from the N content.
RESULTS AND DISCUSSION
PNIPAAm Microspheres by PrecipitationPolymerization
Particle Size and Size Distribution
Both the particle size and size distribution affectthe efficiency of their utilization in a given appli-cation. They were determined by two independ-ent methods: quasielastic light scattering andSEM. SEM analyses showed that monodisperselatexes were produced (PDI < 1.05). Such a nar-row particle size distribution indicates a veryshort nucleation period leading to a constant par-ticle number throughout the polymerization pro-cess.23 The diameters obtained by electron micros-copy were smaller than those by dynamic lightscattering measured at 25 8C (Fig. 1) because ofthe preparation conditions of SEM samples,which led to a loss of water and hence to shrink-age of the microspheres. In contrast, quasielasticlight scattering measures the hydrodynamic dia-meter (geometrical diameter for hard spheres) ofthe particles in water. Because such particles arehighly water-swollen at 25 8C, the hydrodynamicparticle size was larger than the particle sizemeasured by SEM. The diameter of the particlesshrunken at 37 8C generally coincided with thediameter of dry particles measured by SEM.There are, however, many parameters controlling
970 MACKOVA AND HORAK
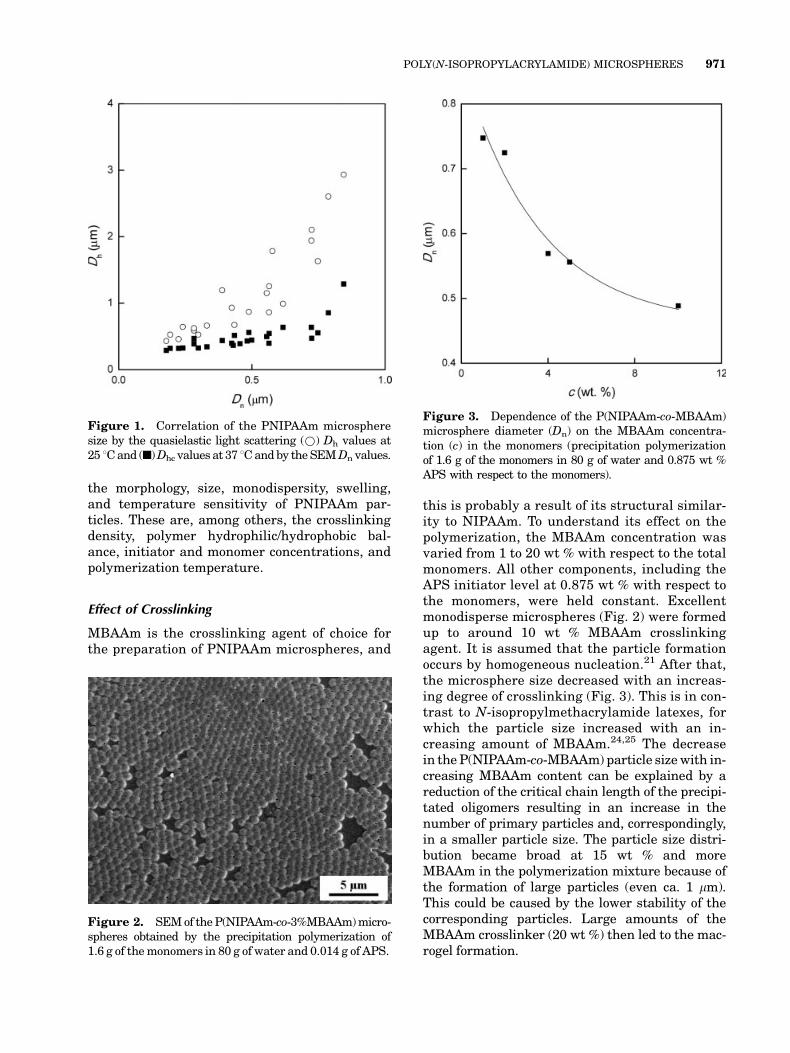
the morphology, size, monodispersity, swelling,and temperature sensitivity of PNIPAAm par-ticles. These are, among others, the crosslinkingdensity, polymer hydrophilic/hydrophobic bal-ance, initiator and monomer concentrations, andpolymerization temperature.
Effect of Crosslinking
MBAAm is the crosslinking agent of choice forthe preparation of PNIPAAm microspheres, and
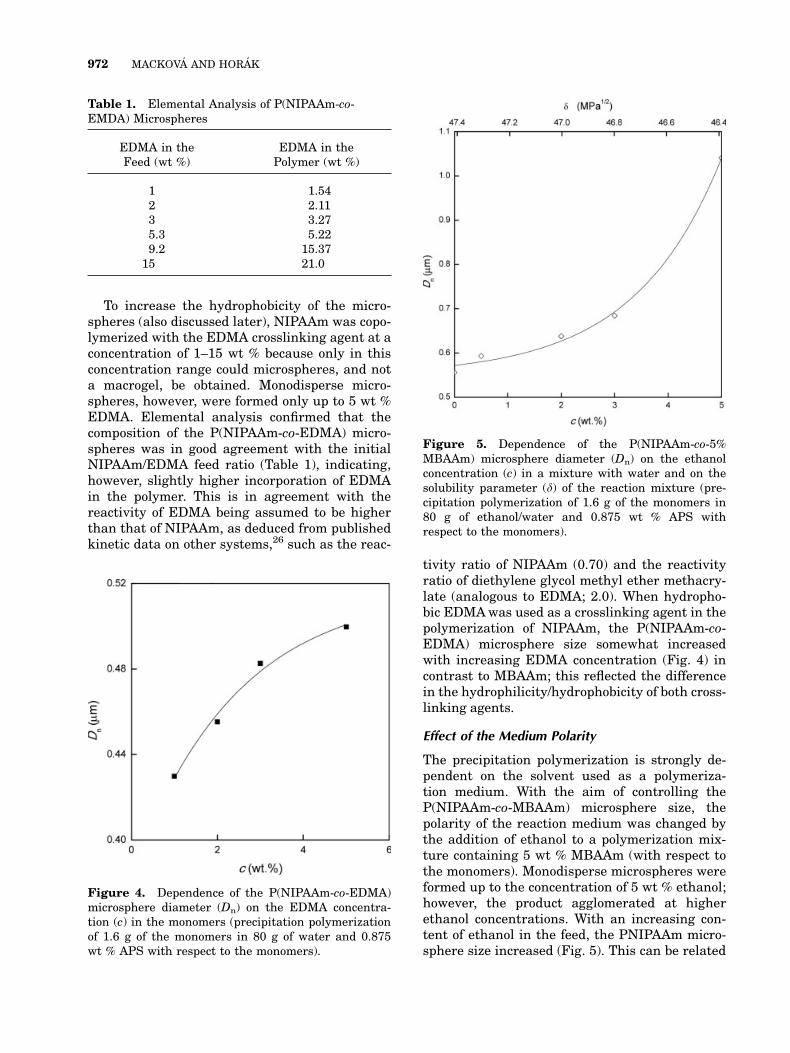
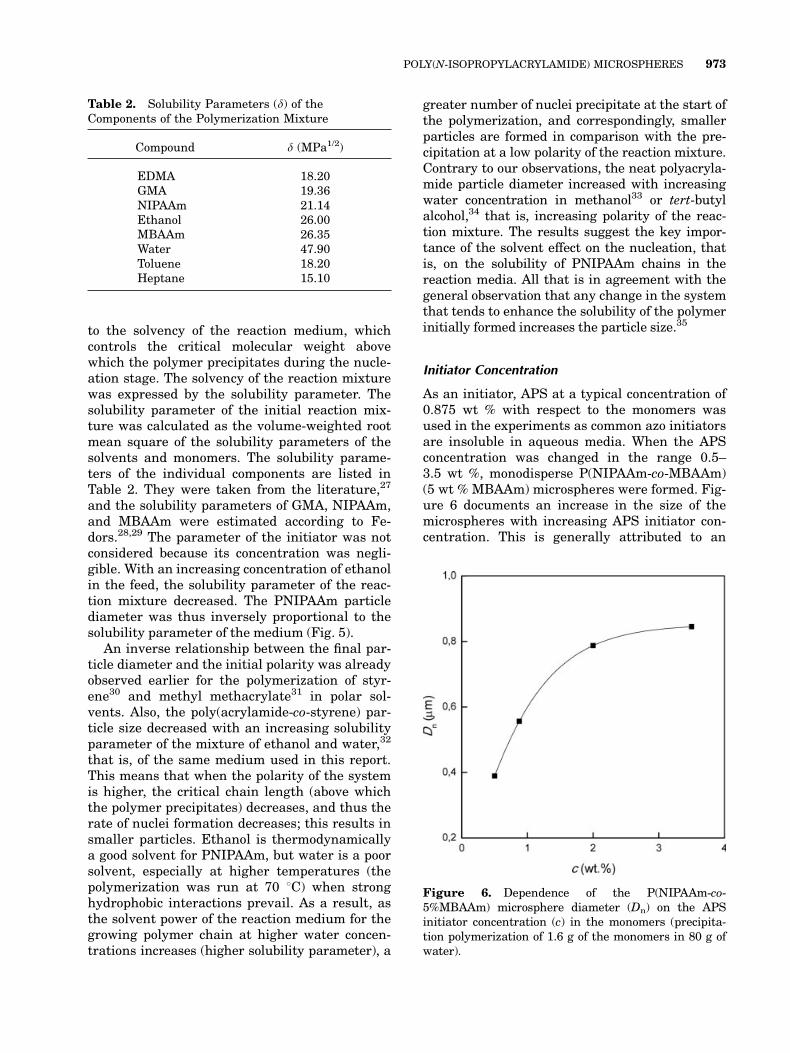
this is probably a result of its structural similar-ity to NIPAAm. To understand its effect on thepolymerization, the MBAAm concentration wasvaried from 1 to 20 wt % with respect to the totalmonomers. All other components, including theAPS initiator level at 0.875 wt % with respect tothe monomers, were held constant. Excellentmonodisperse microspheres (Fig. 2) were formedup to around 10 wt % MBAAm crosslinkingagent. It is assumed that the particle formationoccurs by homogeneous nucleation.21 After that,the microsphere size decreased with an increas-ing degree of crosslinking (Fig. 3). This is in con-trast to N-isopropylmethacrylamide latexes, forwhich the particle size increased with an in-creasing amount of MBAAm.24,25 The decreasein the P(NIPAAm-co-MBAAm) particle sizewith in-creasing MBAAm content can be explained by areduction of the critical chain length of the precipi-tated oligomers resulting in an increase in thenumber of primary particles and, correspondingly,in a smaller particle size. The particle size distri-bution became broad at 15 wt % and moreMBAAm in the polymerization mixture because ofthe formation of large particles (even ca. 1 lm).This could be caused by the lower stability of thecorresponding particles. Large amounts of theMBAAm crosslinker (20 wt %) then led to the mac-rogel formation.
Figure 1. Correlation of the PNIPAAm microspheresize by the quasielastic light scattering (*) Dh values at25 8Cand (n)Dhc values at 37 8Candby theSEMDn values.
Figure 2. SEMof theP(NIPAAm-co-3%MBAAm)micro-spheres obtained by the precipitation polymerization of1.6 g of themonomers in 80 g of water and 0.014 g of APS.
Figure 3. Dependence of the P(NIPAAm-co-MBAAm)microsphere diameter (Dn) on the MBAAm concentra-tion (c) in the monomers (precipitation polymerizationof 1.6 g of the monomers in 80 g of water and 0.875 wt %APS with respect to the monomers).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 971
To increase the hydrophobicity of the micro-spheres (also discussed later), NIPAAm was copo-lymerized with the EDMA crosslinking agent at aconcentration of 1–15 wt % because only in thisconcentration range could microspheres, and nota macrogel, be obtained. Monodisperse micro-spheres, however, were formed only up to 5 wt %EDMA. Elemental analysis confirmed that thecomposition of the P(NIPAAm-co-EDMA) micro-spheres was in good agreement with the initialNIPAAm/EDMA feed ratio (Table 1), indicating,however, slightly higher incorporation of EDMAin the polymer. This is in agreement with thereactivity of EDMA being assumed to be higherthan that of NIPAAm, as deduced from publishedkinetic data on other systems,26 such as the reac-
tivity ratio of NIPAAm (0.70) and the reactivityratio of diethylene glycol methyl ether methacry-late (analogous to EDMA; 2.0). When hydropho-bic EDMAwas used as a crosslinking agent in thepolymerization of NIPAAm, the P(NIPAAm-co-EDMA) microsphere size somewhat increasedwith increasing EDMA concentration (Fig. 4) incontrast to MBAAm; this reflected the differencein the hydrophilicity/hydrophobicity of both cross-linking agents.
Effect of the Medium Polarity
The precipitation polymerization is strongly de-pendent on the solvent used as a polymeriza-tion medium. With the aim of controlling theP(NIPAAm-co-MBAAm) microsphere size, thepolarity of the reaction medium was changed bythe addition of ethanol to a polymerization mix-ture containing 5 wt % MBAAm (with respect tothe monomers). Monodisperse microspheres wereformed up to the concentration of 5 wt % ethanol;however, the product agglomerated at higherethanol concentrations. With an increasing con-tent of ethanol in the feed, the PNIPAAm micro-sphere size increased (Fig. 5). This can be related
Figure 4. Dependence of the P(NIPAAm-co-EDMA)microsphere diameter (Dn) on the EDMA concentra-tion (c) in the monomers (precipitation polymerizationof 1.6 g of the monomers in 80 g of water and 0.875wt % APS with respect to the monomers).
Figure 5. Dependence of the P(NIPAAm-co-5%MBAAm) microsphere diameter (Dn) on the ethanolconcentration (c) in a mixture with water and on thesolubility parameter (d) of the reaction mixture (pre-cipitation polymerization of 1.6 g of the monomers in80 g of ethanol/water and 0.875 wt % APS withrespect to the monomers).
Table 1. Elemental Analysis of P(NIPAAm-co-EMDA) Microspheres
EDMA in theFeed (wt %)
EDMA in thePolymer (wt %)
1 1.542 2.113 3.275.3 5.229.2 15.37
15 21.0
972 MACKOVA AND HORAK
to the solvency of the reaction medium, whichcontrols the critical molecular weight abovewhich the polymer precipitates during the nucle-ation stage. The solvency of the reaction mixturewas expressed by the solubility parameter. Thesolubility parameter of the initial reaction mix-ture was calculated as the volume-weighted rootmean square of the solubility parameters of thesolvents and monomers. The solubility parame-ters of the individual components are listed inTable 2. They were taken from the literature,27
and the solubility parameters of GMA, NIPAAm,and MBAAm were estimated according to Fe-dors.28,29 The parameter of the initiator was notconsidered because its concentration was negli-gible. With an increasing concentration of ethanolin the feed, the solubility parameter of the reac-tion mixture decreased. The PNIPAAm particlediameter was thus inversely proportional to thesolubility parameter of the medium (Fig. 5).
An inverse relationship between the final par-ticle diameter and the initial polarity was alreadyobserved earlier for the polymerization of styr-ene30 and methyl methacrylate31 in polar sol-vents. Also, the poly(acrylamide-co-styrene) par-ticle size decreased with an increasing solubilityparameter of the mixture of ethanol and water,32
that is, of the same medium used in this report.This means that when the polarity of the systemis higher, the critical chain length (above whichthe polymer precipitates) decreases, and thus therate of nuclei formation decreases; this results insmaller particles. Ethanol is thermodynamicallya good solvent for PNIPAAm, but water is a poorsolvent, especially at higher temperatures (thepolymerization was run at 70 8C) when stronghydrophobic interactions prevail. As a result, asthe solvent power of the reaction medium for thegrowing polymer chain at higher water concen-trations increases (higher solubility parameter), a
greater number of nuclei precipitate at the start ofthe polymerization, and correspondingly, smallerparticles are formed in comparison with the pre-cipitation at a low polarity of the reaction mixture.Contrary to our observations, the neat polyacryla-mide particle diameter increased with increasingwater concentration in methanol33 or tert-butylalcohol,34 that is, increasing polarity of the reac-tion mixture. The results suggest the key impor-tance of the solvent effect on the nucleation, thatis, on the solubility of PNIPAAm chains in thereaction media. All that is in agreement with thegeneral observation that any change in the systemthat tends to enhance the solubility of the polymerinitially formed increases the particle size.35
Initiator Concentration
As an initiator, APS at a typical concentration of0.875 wt % with respect to the monomers wasused in the experiments as common azo initiatorsare insoluble in aqueous media. When the APSconcentration was changed in the range 0.5–3.5 wt %, monodisperse P(NIPAAm-co-MBAAm)(5 wt % MBAAm) microspheres were formed. Fig-ure 6 documents an increase in the size of themicrospheres with increasing APS initiator con-centration. This is generally attributed to an
Table 2. Solubility Parameters (d) of theComponents of the Polymerization Mixture
Compound d (MPa1/2)
EDMA 18.20GMA 19.36NIPAAm 21.14Ethanol 26.00MBAAm 26.35Water 47.90Toluene 18.20Heptane 15.10
Figure 6. Dependence of the P(NIPAAm-co-5%MBAAm) microsphere diameter (Dn) on the APSinitiator concentration (c) in the monomers (precipita-tion polymerization of 1.6 g of the monomers in 80 g ofwater).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 973
increase in the instantaneous concentration ofthe growing oligomeric radicals with increasinginitiator concentration. This in turn increasedthe rate of the oligomer association and the coag-ulation rate of unstable nuclei to form larger per-manent particle nuclei.36 Stronger coagulationthen led to larger but fewer final particles.29,37
The increase in the particle size with increasinginitiator concentration is in conformity withmany dispersion polymerizations.29,37,38 In othersystems, however, the initiator concentrationincrease caused the final particle size to decrease,as long as the initiator concentration was lowerthan the critical coagulation concentration of pri-mary particles.22
Effect of the Monomer Concentration
The standard monomer concentration in water inthis report was 2 wt %. It could be increased to3 wt % to get microspheres; however, a broad sizedistribution was produced because of extendednucleation. The particles showed a tendency tocoagulate at a monomer concentration higherthan 3 wt %. This is in agreement with the resultsof Stover’s group on precipitation polymerization,who reported that stable spherical particles were
obtained only with up to 5 vol % divinylbenzene(DVB)39 and with up to a 2 vol % monomer mix-ture consisting of (chloromethyl)styrene and DVBadded to the polymerization medium.40 Abovethese concentrations, coagulated particles wereobtained. The size of the PNIPAAm microspheresincreased with increasing monomer loading(Fig. 7), and this is in agreement with resultspublished earlier.41 The effect of the monomerconcentration on the size of the polymer particlesand their distribution is mainly due to thechanges in the initial solvency of the reactantmixture (the monomer is a good solvent for thepolymer), which may affect the nucleation in thepolymerization process. Extended nucleation pro-duces fewer primary particles, which in turn areof larger size.
Effect of the Polymerization Temperature
A typical polymerization temperature in thisreport was 70 8C, that is, sufficiently above theLCST of the corresponding polymer and highenough to favor precipitation of the oligomersoriginating in the water phase. The polymeriza-tion temperature had a pronounced effect on thequality of the resulting product. Although partialparticle agglomeration occurred at 60 8C, excel-lent monodispersity of the microspheres wasachieved at polymerization temperatures in therange of 70–90 8C (Fig. 8). Increasing the temper-ature apparently led to an increase in the decom-position rate of the initiator, resulting in the
Figure 7. Dependence of the P(NIPAAm-co-5%MBAAm) microsphere diameter (Dn) on the monomerconcentration (c; precipitation polymerization in 80 gof water and 0.875 wt % APS with respect to themonomers).
Figure 8. P(NIPAAm-co-5%MBAAm) microspheres ob-tained by the precipitation polymerization of 1.6 g ofthe monomers in 80 g of water at 80 8C and 0.014 g ofAPS.
974 MACKOVA AND HORAK
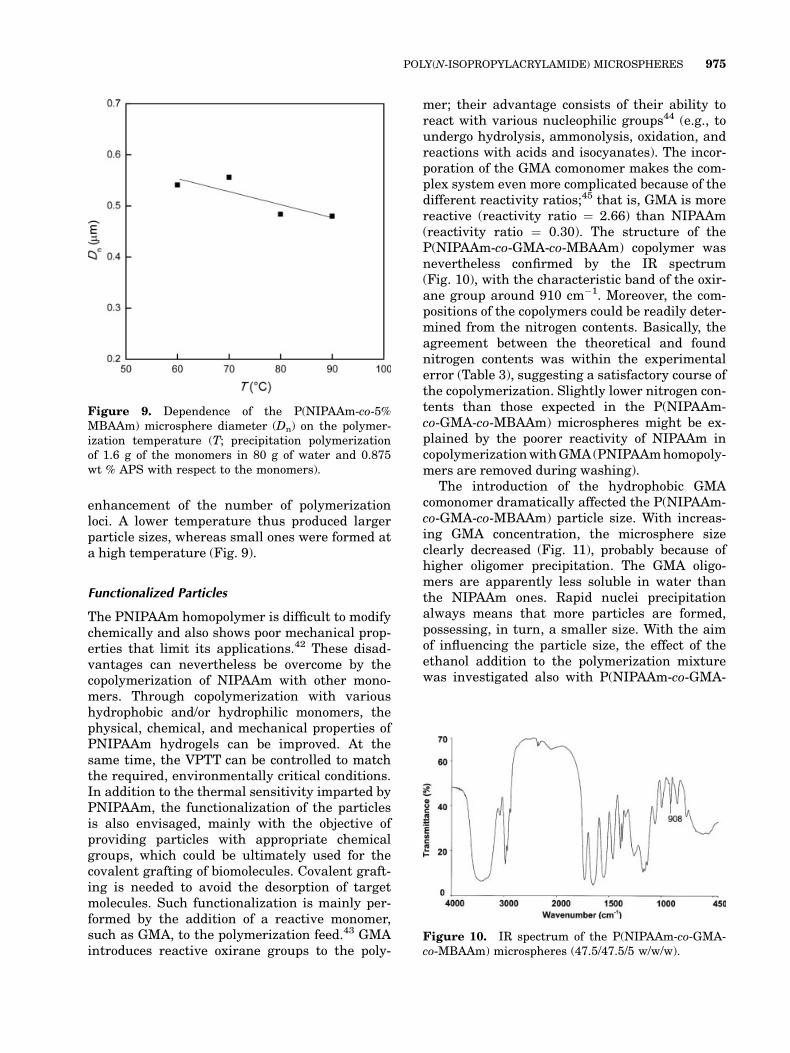
enhancement of the number of polymerizationloci. A lower temperature thus produced largerparticle sizes, whereas small ones were formed ata high temperature (Fig. 9).
Functionalized Particles
The PNIPAAm homopolymer is difficult to modifychemically and also shows poor mechanical prop-erties that limit its applications.42 These disad-vantages can nevertheless be overcome by thecopolymerization of NIPAAm with other mono-mers. Through copolymerization with varioushydrophobic and/or hydrophilic monomers, thephysical, chemical, and mechanical properties ofPNIPAAm hydrogels can be improved. At thesame time, the VPTT can be controlled to matchthe required, environmentally critical conditions.In addition to the thermal sensitivity imparted byPNIPAAm, the functionalization of the particlesis also envisaged, mainly with the objective ofproviding particles with appropriate chemicalgroups, which could be ultimately used for thecovalent grafting of biomolecules. Covalent graft-ing is needed to avoid the desorption of targetmolecules. Such functionalization is mainly per-formed by the addition of a reactive monomer,such as GMA, to the polymerization feed.43 GMAintroduces reactive oxirane groups to the poly-
mer; their advantage consists of their ability toreact with various nucleophilic groups44 (e.g., toundergo hydrolysis, ammonolysis, oxidation, andreactions with acids and isocyanates). The incor-poration of the GMA comonomer makes the com-plex system even more complicated because of thedifferent reactivity ratios;45 that is, GMA is morereactive (reactivity ratio ¼ 2.66) than NIPAAm(reactivity ratio ¼ 0.30). The structure of theP(NIPAAm-co-GMA-co-MBAAm) copolymer wasnevertheless confirmed by the IR spectrum(Fig. 10), with the characteristic band of the oxir-ane group around 910 cm�1. Moreover, the com-positions of the copolymers could be readily deter-mined from the nitrogen contents. Basically, theagreement between the theoretical and foundnitrogen contents was within the experimentalerror (Table 3), suggesting a satisfactory course ofthe copolymerization. Slightly lower nitrogen con-tents than those expected in the P(NIPAAm-co-GMA-co-MBAAm) microspheres might be ex-plained by the poorer reactivity of NIPAAm incopolymerizationwithGMA(PNIPAAmhomopoly-mers are removed during washing).
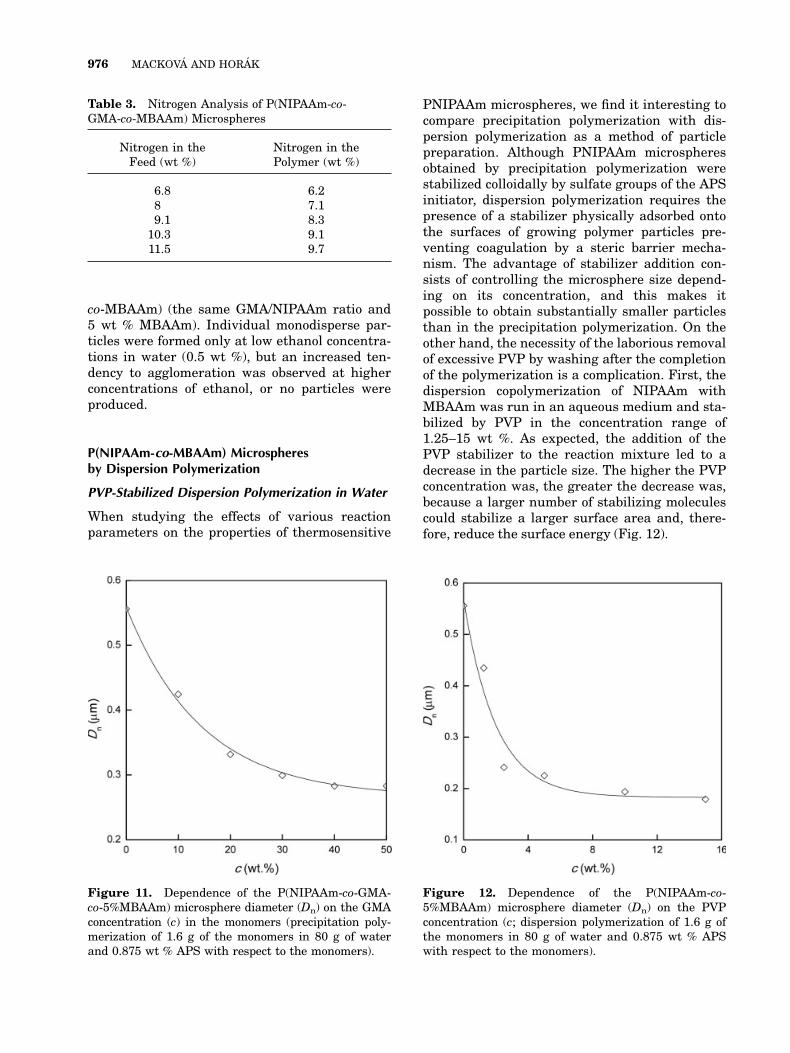
The introduction of the hydrophobic GMAcomonomer dramatically affected the P(NIPAAm-co-GMA-co-MBAAm) particle size. With increas-ing GMA concentration, the microsphere sizeclearly decreased (Fig. 11), probably because ofhigher oligomer precipitation. The GMA oligo-mers are apparently less soluble in water thanthe NIPAAm ones. Rapid nuclei precipitationalways means that more particles are formed,possessing, in turn, a smaller size. With the aimof influencing the particle size, the effect of theethanol addition to the polymerization mixturewas investigated also with P(NIPAAm-co-GMA-
Figure 9. Dependence of the P(NIPAAm-co-5%MBAAm) microsphere diameter (Dn) on the polymer-ization temperature (T; precipitation polymerizationof 1.6 g of the monomers in 80 g of water and 0.875wt % APS with respect to the monomers).
Figure 10. IR spectrum of the P(NIPAAm-co-GMA-co-MBAAm) microspheres (47.5/47.5/5 w/w/w).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 975
co-MBAAm) (the same GMA/NIPAAm ratio and5 wt % MBAAm). Individual monodisperse par-ticles were formed only at low ethanol concentra-tions in water (0.5 wt %), but an increased ten-dency to agglomeration was observed at higherconcentrations of ethanol, or no particles wereproduced.
P(NIPAAm-co-MBAAm) Microspheresby Dispersion Polymerization
PVP-Stabilized Dispersion Polymerization in Water
When studying the effects of various reactionparameters on the properties of thermosensitive
PNIPAAm microspheres, we find it interesting tocompare precipitation polymerization with dis-persion polymerization as a method of particlepreparation. Although PNIPAAm microspheresobtained by precipitation polymerization werestabilized colloidally by sulfate groups of the APSinitiator, dispersion polymerization requires thepresence of a stabilizer physically adsorbed ontothe surfaces of growing polymer particles pre-venting coagulation by a steric barrier mecha-nism. The advantage of stabilizer addition con-sists of controlling the microsphere size depend-ing on its concentration, and this makes itpossible to obtain substantially smaller particlesthan in the precipitation polymerization. On theother hand, the necessity of the laborious removalof excessive PVP by washing after the completionof the polymerization is a complication. First, thedispersion copolymerization of NIPAAm withMBAAm was run in an aqueous medium and sta-bilized by PVP in the concentration range of1.25–15 wt %. As expected, the addition of thePVP stabilizer to the reaction mixture led to adecrease in the particle size. The higher the PVPconcentration was, the greater the decrease was,because a larger number of stabilizing moleculescould stabilize a larger surface area and, there-fore, reduce the surface energy (Fig. 12).
Table 3. Nitrogen Analysis of P(NIPAAm-co-GMA-co-MBAAm) Microspheres
Nitrogen in theFeed (wt %)
Nitrogen in thePolymer (wt %)
6.8 6.28 7.19.1 8.3
10.3 9.111.5 9.7
Figure 11. Dependence of the P(NIPAAm-co-GMA-co-5%MBAAm) microsphere diameter (Dn) on the GMAconcentration (c) in the monomers (precipitation poly-merization of 1.6 g of the monomers in 80 g of waterand 0.875 wt % APS with respect to the monomers).
Figure 12. Dependence of the P(NIPAAm-co-5%MBAAm) microsphere diameter (Dn) on the PVPconcentration (c; dispersion polymerization of 1.6 g ofthe monomers in 80 g of water and 0.875 wt % APSwith respect to the monomers).
976 MACKOVA AND HORAK
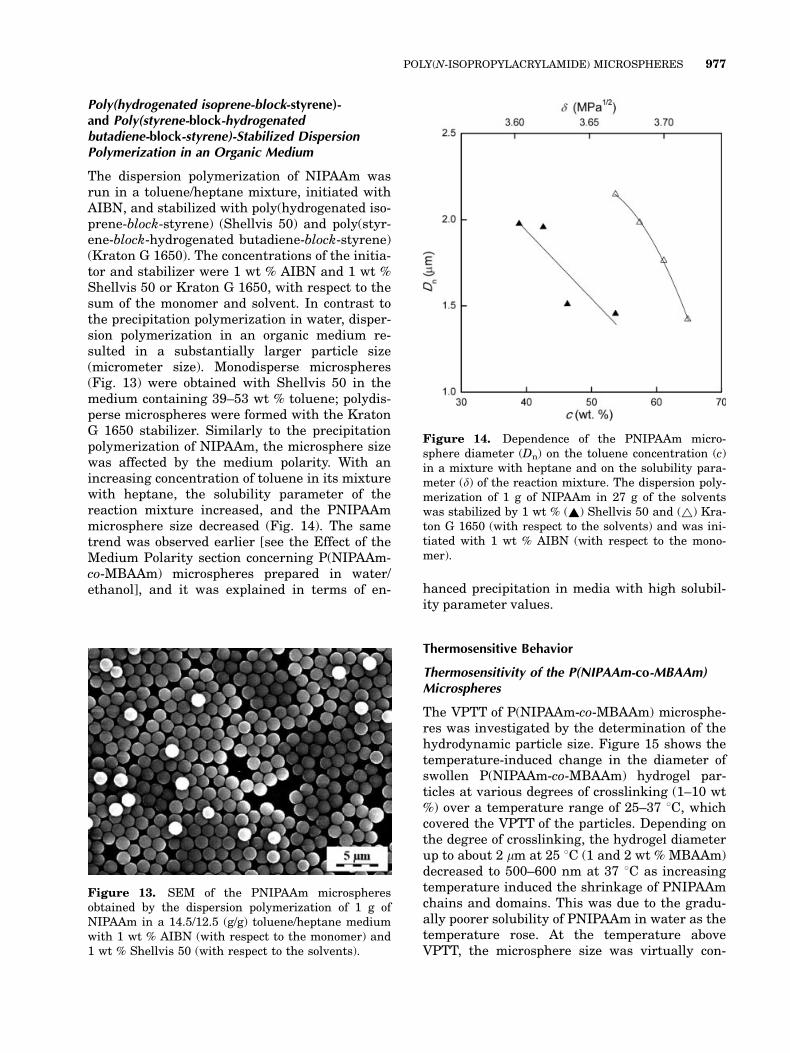
Poly(hydrogenated isoprene-block-styrene)-and Poly(styrene-block-hydrogenatedbutadiene-block-styrene)-Stabilized DispersionPolymerization in an Organic Medium
The dispersion polymerization of NIPAAm wasrun in a toluene/heptane mixture, initiated withAIBN, and stabilized with poly(hydrogenated iso-prene-block-styrene) (Shellvis 50) and poly(styr-ene-block-hydrogenated butadiene-block-styrene)(Kraton G 1650). The concentrations of the initia-tor and stabilizer were 1 wt % AIBN and 1 wt %Shellvis 50 or Kraton G 1650, with respect to thesum of the monomer and solvent. In contrast tothe precipitation polymerization in water, disper-sion polymerization in an organic medium re-sulted in a substantially larger particle size(micrometer size). Monodisperse microspheres(Fig. 13) were obtained with Shellvis 50 in themedium containing 39–53 wt % toluene; polydis-perse microspheres were formed with the KratonG 1650 stabilizer. Similarly to the precipitationpolymerization of NIPAAm, the microsphere sizewas affected by the medium polarity. With anincreasing concentration of toluene in its mixturewith heptane, the solubility parameter of thereaction mixture increased, and the PNIPAAmmicrosphere size decreased (Fig. 14). The sametrend was observed earlier [see the Effect of theMedium Polarity section concerning P(NIPAAm-co-MBAAm) microspheres prepared in water/ethanol], and it was explained in terms of en- hanced precipitation in media with high solubil-
ity parameter values.
Thermosensitive Behavior
Thermosensitivity of the P(NIPAAm-co-MBAAm)Microspheres
The VPTT of P(NIPAAm-co-MBAAm) microsphe-res was investigated by the determination of thehydrodynamic particle size. Figure 15 shows thetemperature-induced change in the diameter ofswollen P(NIPAAm-co-MBAAm) hydrogel par-ticles at various degrees of crosslinking (1–10 wt%) over a temperature range of 25–37 8C, whichcovered the VPTT of the particles. Depending onthe degree of crosslinking, the hydrogel diameterup to about 2 lm at 25 8C (1 and 2 wt % MBAAm)decreased to 500–600 nm at 37 8C as increasingtemperature induced the shrinkage of PNIPAAmchains and domains. This was due to the gradu-ally poorer solubility of PNIPAAm in water as thetemperature rose. At the temperature aboveVPTT, the microsphere size was virtually con-
Figure 13. SEM of the PNIPAAm microspheresobtained by the dispersion polymerization of 1 g ofNIPAAm in a 14.5/12.5 (g/g) toluene/heptane mediumwith 1 wt % AIBN (with respect to the monomer) and1 wt % Shellvis 50 (with respect to the solvents).
Figure 14. Dependence of the PNIPAAm micro-sphere diameter (Dn) on the toluene concentration (c)in a mixture with heptane and on the solubility para-meter (d) of the reaction mixture. The dispersion poly-merization of 1 g of NIPAAm in 27 g of the solventswas stabilized by 1 wt % (~) Shellvis 50 and (~) Kra-ton G 1650 (with respect to the solvents) and was ini-tiated with 1 wt % AIBN (with respect to the mono-mer).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 977
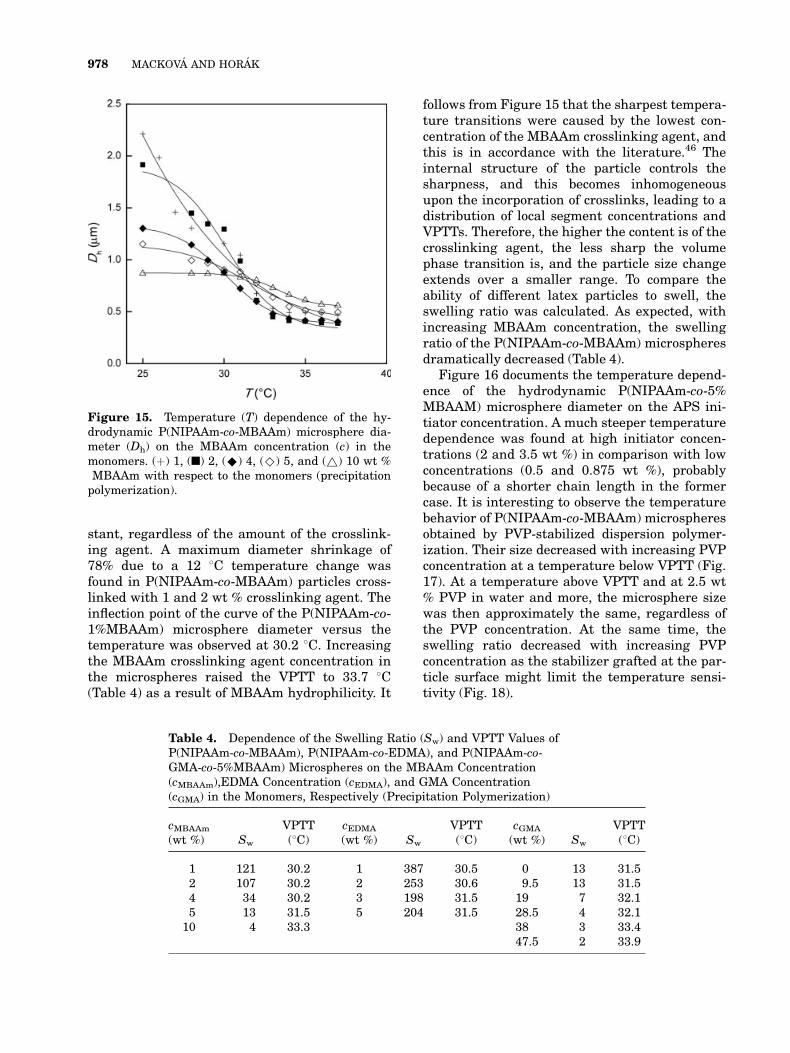
stant, regardless of the amount of the crosslink-ing agent. A maximum diameter shrinkage of78% due to a 12 8C temperature change wasfound in P(NIPAAm-co-MBAAm) particles cross-linked with 1 and 2 wt % crosslinking agent. Theinflection point of the curve of the P(NIPAAm-co-1%MBAAm) microsphere diameter versus thetemperature was observed at 30.2 8C. Increasingthe MBAAm crosslinking agent concentration inthe microspheres raised the VPTT to 33.7 8C(Table 4) as a result of MBAAm hydrophilicity. It
follows from Figure 15 that the sharpest tempera-ture transitions were caused by the lowest con-centration of the MBAAm crosslinking agent, andthis is in accordance with the literature.46 Theinternal structure of the particle controls thesharpness, and this becomes inhomogeneousupon the incorporation of crosslinks, leading to adistribution of local segment concentrations andVPTTs. Therefore, the higher the content is of thecrosslinking agent, the less sharp the volumephase transition is, and the particle size changeextends over a smaller range. To compare theability of different latex particles to swell, theswelling ratio was calculated. As expected, withincreasing MBAAm concentration, the swellingratio of the P(NIPAAm-co-MBAAm) microspheresdramatically decreased (Table 4).
Figure 16 documents the temperature depend-ence of the hydrodynamic P(NIPAAm-co-5%MBAAM) microsphere diameter on the APS ini-tiator concentration. A much steeper temperaturedependence was found at high initiator concen-trations (2 and 3.5 wt %) in comparison with lowconcentrations (0.5 and 0.875 wt %), probablybecause of a shorter chain length in the formercase. It is interesting to observe the temperaturebehavior of P(NIPAAm-co-MBAAm) microspheresobtained by PVP-stabilized dispersion polymer-ization. Their size decreased with increasing PVPconcentration at a temperature below VPTT (Fig.17). At a temperature above VPTT and at 2.5 wt% PVP in water and more, the microsphere sizewas then approximately the same, regardless ofthe PVP concentration. At the same time, theswelling ratio decreased with increasing PVPconcentration as the stabilizer grafted at the par-ticle surface might limit the temperature sensi-tivity (Fig. 18).
Figure 15. Temperature (T) dependence of the hy-drodynamic P(NIPAAm-co-MBAAm) microsphere dia-meter (Dh) on the MBAAm concentration (c) in themonomers. (þ) 1, (n) 2, (^) 4, (^) 5, and (~) 10 wt %MBAAm with respect to the monomers (precipitationpolymerization).
Table 4. Dependence of the Swelling Ratio (Sw) and VPTT Values ofP(NIPAAm-co-MBAAm), P(NIPAAm-co-EDMA), and P(NIPAAm-co-GMA-co-5%MBAAm) Microspheres on the MBAAm Concentration(cMBAAm),EDMA Concentration (cEDMA), and GMA Concentration(cGMA) in the Monomers, Respectively (Precipitation Polymerization)
cMBAAm
(wt %) Sw
VPTT(8C)
cEDMA
(wt %) Sw
VPTT(8C)
cGMA
(wt %) Sw
VPTT(8C)
1 121 30.2 1 387 30.5 0 13 31.52 107 30.2 2 253 30.6 9.5 13 31.54 34 30.2 3 198 31.5 19 7 32.15 13 31.5 5 204 31.5 28.5 4 32.1
10 4 33.3 38 3 33.447.5 2 33.9
978 MACKOVA AND HORAK
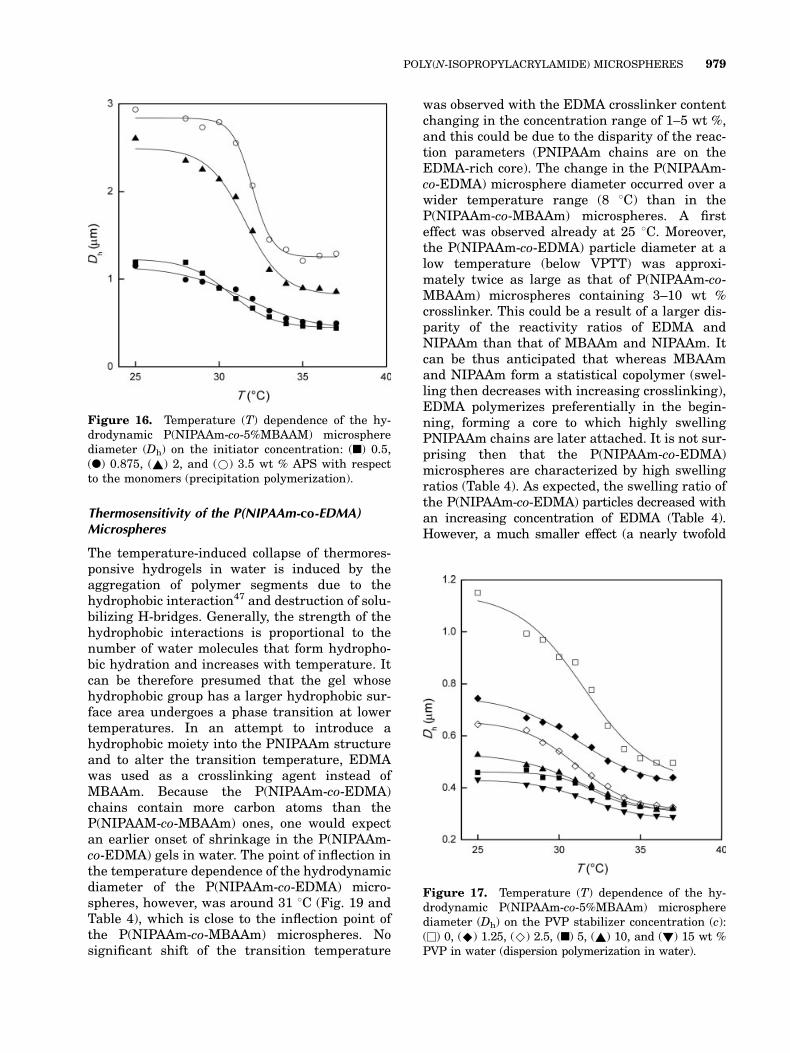
Thermosensitivity of the P(NIPAAm-co-EDMA)Microspheres
The temperature-induced collapse of thermores-ponsive hydrogels in water is induced by theaggregation of polymer segments due to thehydrophobic interaction47 and destruction of solu-bilizing H-bridges. Generally, the strength of thehydrophobic interactions is proportional to thenumber of water molecules that form hydropho-bic hydration and increases with temperature. Itcan be therefore presumed that the gel whosehydrophobic group has a larger hydrophobic sur-face area undergoes a phase transition at lowertemperatures. In an attempt to introduce ahydrophobic moiety into the PNIPAAm structureand to alter the transition temperature, EDMAwas used as a crosslinking agent instead ofMBAAm. Because the P(NIPAAm-co-EDMA)chains contain more carbon atoms than theP(NIPAAM-co-MBAAm) ones, one would expectan earlier onset of shrinkage in the P(NIPAAm-co-EDMA) gels in water. The point of inflection inthe temperature dependence of the hydrodynamicdiameter of the P(NIPAAm-co-EDMA) micro-spheres, however, was around 31 8C (Fig. 19 andTable 4), which is close to the inflection point ofthe P(NIPAAm-co-MBAAm) microspheres. Nosignificant shift of the transition temperature
was observed with the EDMA crosslinker contentchanging in the concentration range of 1–5 wt %,and this could be due to the disparity of the reac-tion parameters (PNIPAAm chains are on theEDMA-rich core). The change in the P(NIPAAm-co-EDMA) microsphere diameter occurred over awider temperature range (8 8C) than in theP(NIPAAm-co-MBAAm) microspheres. A firsteffect was observed already at 25 8C. Moreover,the P(NIPAAm-co-EDMA) particle diameter at alow temperature (below VPTT) was approxi-mately twice as large as that of P(NIPAAm-co-MBAAm) microspheres containing 3–10 wt %crosslinker. This could be a result of a larger dis-parity of the reactivity ratios of EDMA andNIPAAm than that of MBAAm and NIPAAm. Itcan be thus anticipated that whereas MBAAmand NIPAAm form a statistical copolymer (swel-ling then decreases with increasing crosslinking),EDMA polymerizes preferentially in the begin-ning, forming a core to which highly swellingPNIPAAm chains are later attached. It is not sur-prising then that the P(NIPAAm-co-EDMA)microspheres are characterized by high swellingratios (Table 4). As expected, the swelling ratio ofthe P(NIPAAm-co-EDMA) particles decreased withan increasing concentration of EDMA (Table 4).However, a much smaller effect (a nearly twofold
Figure 16. Temperature (T) dependence of the hy-drodynamic P(NIPAAm-co-5%MBAAM) microspherediameter (Dh) on the initiator concentration: (n) 0.5,(l) 0.875, (~) 2, and (*) 3.5 wt % APS with respectto the monomers (precipitation polymerization).
Figure 17. Temperature (T) dependence of the hy-drodynamic P(NIPAAm-co-5%MBAAm) microspherediameter (Dh) on the PVP stabilizer concentration (c):(u) 0, (^) 1.25, (^) 2.5, (n) 5, (~) 10, and (!) 15 wt %PVP in water (dispersion polymerization in water).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 979
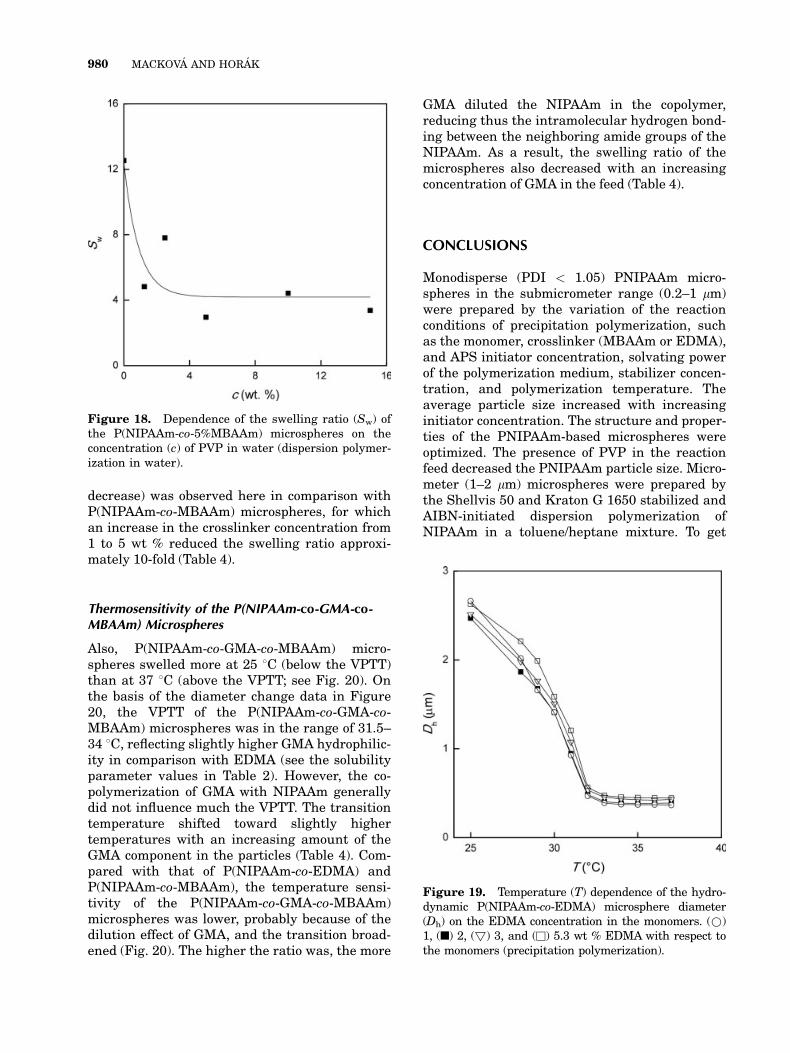
decrease) was observed here in comparison withP(NIPAAm-co-MBAAm) microspheres, for whichan increase in the crosslinker concentration from1 to 5 wt % reduced the swelling ratio approxi-mately 10-fold (Table 4).
Thermosensitivity of the P(NIPAAm-co-GMA-co-MBAAm) Microspheres
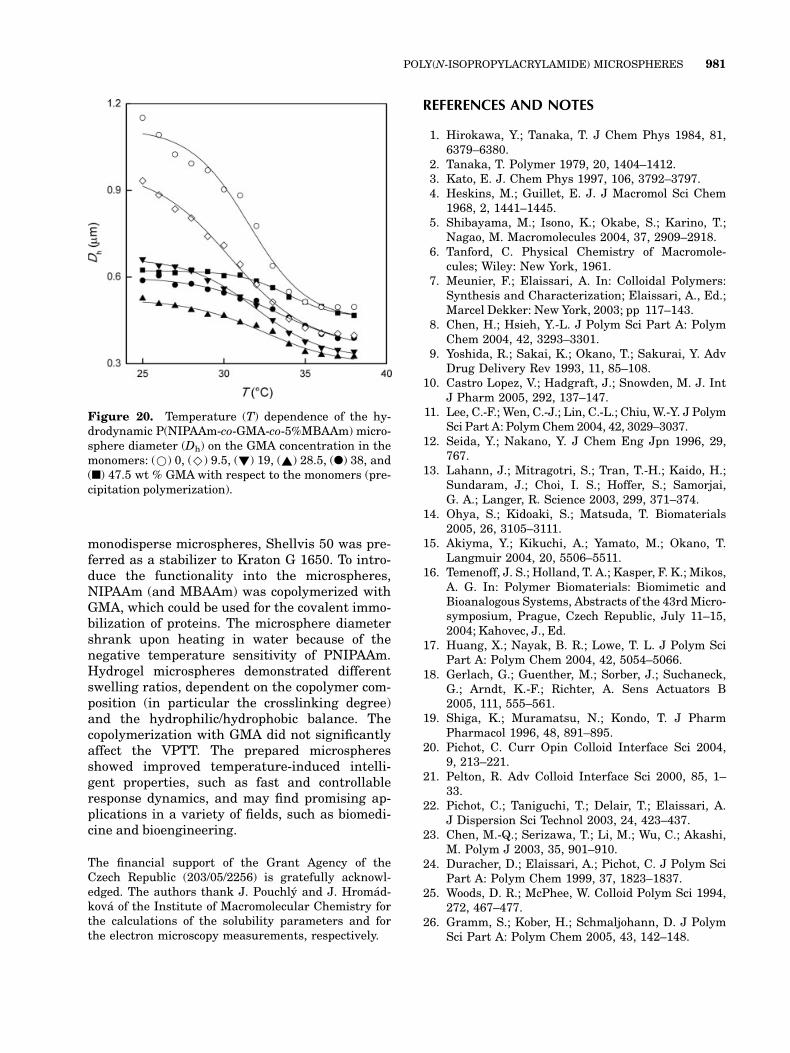
Also, P(NIPAAm-co-GMA-co-MBAAm) micro-spheres swelled more at 25 8C (below the VPTT)than at 37 8C (above the VPTT; see Fig. 20). Onthe basis of the diameter change data in Figure20, the VPTT of the P(NIPAAm-co-GMA-co-MBAAm) microspheres was in the range of 31.5–34 8C, reflecting slightly higher GMA hydrophilic-ity in comparison with EDMA (see the solubilityparameter values in Table 2). However, the co-polymerization of GMA with NIPAAm generallydid not influence much the VPTT. The transitiontemperature shifted toward slightly highertemperatures with an increasing amount of theGMA component in the particles (Table 4). Com-pared with that of P(NIPAAm-co-EDMA) andP(NIPAAm-co-MBAAm), the temperature sensi-tivity of the P(NIPAAm-co-GMA-co-MBAAm)microspheres was lower, probably because of thedilution effect of GMA, and the transition broad-ened (Fig. 20). The higher the ratio was, the more
GMA diluted the NIPAAm in the copolymer,reducing thus the intramolecular hydrogen bond-ing between the neighboring amide groups of theNIPAAm. As a result, the swelling ratio of themicrospheres also decreased with an increasingconcentration of GMA in the feed (Table 4).
CONCLUSIONS
Monodisperse (PDI < 1.05) PNIPAAm micro-spheres in the submicrometer range (0.2–1 lm)were prepared by the variation of the reactionconditions of precipitation polymerization, suchas the monomer, crosslinker (MBAAm or EDMA),and APS initiator concentration, solvating powerof the polymerization medium, stabilizer concen-tration, and polymerization temperature. Theaverage particle size increased with increasinginitiator concentration. The structure and proper-ties of the PNIPAAm-based microspheres wereoptimized. The presence of PVP in the reactionfeed decreased the PNIPAAm particle size. Micro-meter (1–2 lm) microspheres were prepared bythe Shellvis 50 and Kraton G 1650 stabilized andAIBN-initiated dispersion polymerization ofNIPAAm in a toluene/heptane mixture. To get
Figure 18. Dependence of the swelling ratio (Sw) ofthe P(NIPAAm-co-5%MBAAm) microspheres on theconcentration (c) of PVP in water (dispersion polymer-ization in water).
Figure 19. Temperature (T) dependence of the hydro-dynamic P(NIPAAm-co-EDMA) microsphere diameter(Dh) on the EDMA concentration in the monomers. (*)1, (n) 2, (!) 3, and (u) 5.3 wt % EDMA with respect tothe monomers (precipitation polymerization).
980 MACKOVA AND HORAK
monodisperse microspheres, Shellvis 50 was pre-ferred as a stabilizer to Kraton G 1650. To intro-duce the functionality into the microspheres,NIPAAm (and MBAAm) was copolymerized withGMA, which could be used for the covalent immo-bilization of proteins. The microsphere diametershrank upon heating in water because of thenegative temperature sensitivity of PNIPAAm.Hydrogel microspheres demonstrated differentswelling ratios, dependent on the copolymer com-position (in particular the crosslinking degree)and the hydrophilic/hydrophobic balance. Thecopolymerization with GMA did not significantlyaffect the VPTT. The prepared microspheresshowed improved temperature-induced intelli-gent properties, such as fast and controllableresponse dynamics, and may find promising ap-plications in a variety of fields, such as biomedi-cine and bioengineering.
The financial support of the Grant Agency of theCzech Republic (203/05/2256) is gratefully acknowl-edged. The authors thank J. Pouchly and J. Hromad-kova of the Institute of Macromolecular Chemistry forthe calculations of the solubility parameters and forthe electron microscopy measurements, respectively.
REFERENCES AND NOTES
1. Hirokawa, Y.; Tanaka, T. J Chem Phys 1984, 81,6379–6380.
2. Tanaka, T. Polymer 1979, 20, 1404–1412.3. Kato, E. J. Chem Phys 1997, 106, 3792–3797.4. Heskins, M.; Guillet, E. J. J Macromol Sci Chem
1968, 2, 1441–1445.5. Shibayama, M.; Isono, K.; Okabe, S.; Karino, T.;
Nagao, M. Macromolecules 2004, 37, 2909–2918.6. Tanford, C. Physical Chemistry of Macromole-
cules; Wiley: New York, 1961.7. Meunier, F.; Elaissari, A. In: Colloidal Polymers:
Synthesis and Characterization; Elaissari, A., Ed.;Marcel Dekker: New York, 2003; pp 117–143.
8. Chen, H.; Hsieh, Y.-L. J Polym Sci Part A: PolymChem 2004, 42, 3293–3301.
9. Yoshida, R.; Sakai, K.; Okano, T.; Sakurai, Y. AdvDrug Delivery Rev 1993, 11, 85–108.
10. Castro Lopez, V.; Hadgraft, J.; Snowden, M. J. IntJ Pharm 2005, 292, 137–147.
11. Lee, C.-F.; Wen, C.-J.; Lin, C.-L.; Chiu, W.-Y. J PolymSci Part A: PolymChem 2004, 42, 3029–3037.
12. Seida, Y.; Nakano, Y. J Chem Eng Jpn 1996, 29,767.
13. Lahann, J.; Mitragotri, S.; Tran, T.-H.; Kaido, H.;Sundaram, J.; Choi, I. S.; Hoffer, S.; Samorjai,G. A.; Langer, R. Science 2003, 299, 371–374.
14. Ohya, S.; Kidoaki, S.; Matsuda, T. Biomaterials2005, 26, 3105–3111.
15. Akiyma, Y.; Kikuchi, A.; Yamato, M.; Okano, T.Langmuir 2004, 20, 5506–5511.
16. Temenoff, J. S.; Holland, T. A.; Kasper, F. K.; Mikos,A. G. In: Polymer Biomaterials: Biomimetic andBioanalogous Systems, Abstracts of the 43rdMicro-symposium, Prague, Czech Republic, July 11–15,2004; Kahovec, J., Ed.
17. Huang, X.; Nayak, B. R.; Lowe, T. L. J Polym SciPart A: Polym Chem 2004, 42, 5054–5066.
18. Gerlach, G.; Guenther, M.; Sorber, J.; Suchaneck,G.; Arndt, K.-F.; Richter, A. Sens Actuators B2005, 111, 555–561.
19. Shiga, K.; Muramatsu, N.; Kondo, T. J PharmPharmacol 1996, 48, 891–895.
20. Pichot, C. Curr Opin Colloid Interface Sci 2004,9, 213–221.
21. Pelton, R. Adv Colloid Interface Sci 2000, 85, 1–33.
22. Pichot, C.; Taniguchi, T.; Delair, T.; Elaissari, A.J Dispersion Sci Technol 2003, 24, 423–437.
23. Chen, M.-Q.; Serizawa, T.; Li, M.; Wu, C.; Akashi,M. Polym J 2003, 35, 901–910.
24. Duracher, D.; Elaissari, A.; Pichot, C. J Polym SciPart A: Polym Chem 1999, 37, 1823–1837.
25. Woods, D. R.; McPhee, W. Colloid Polym Sci 1994,272, 467–477.
26. Gramm, S.; Kober, H.; Schmaljohann, D. J PolymSci Part A: Polym Chem 2005, 43, 142–148.
Figure 20. Temperature (T) dependence of the hy-drodynamic P(NIPAAm-co-GMA-co-5%MBAAm) micro-sphere diameter (Dh) on the GMA concentration in themonomers: (*) 0, (^) 9.5, (!) 19, (~) 28.5, (l) 38, and(n) 47.5 wt % GMAwith respect to the monomers (pre-cipitation polymerization).
POLY(N-ISOPROPYLACRYLAMIDE) MICROSPHERES 981

27. In Polymer Handbook, 4th ed.; Brandrup, J.; Im-mergut, E. H.; Grulke, E. A., Eds.; Wiley: New York,1999; Chapter 7, pp 684–686.
28. Fedors, R. F. Polym Eng Sci 1974, 14, 147–154.
29. Fedors, R. F. Polym Eng Sci 1974, 14, 472.30. Ober, C. K.; Lok, K. P. Macromolecules 1987, 20,
268–273.31. Shen, S.; Sudol, E. D.; El-Aasser, M. S. J Polym
Sci Part A: Polym Chem 1993, 31, 1393–1402.
32. Tao, Z.; Yang, W.; Zhou, H.; Wang, C.; Fu, S. Col-loid Polym Sci 2000, 278, 509–516.
33. Lee, K.-C.; Lee, S.-E. Macromol Res 2002, 10,140–144.
34. Ray, B.; Mandal, B. M. Langmuir 1997, 13, 2191–2196.
35. Ugelstad, J.; Berge, A.; Ellingsen, T.; Schmid, R.;Nilsen, T.-N.; Mørk, P. C.; Stenstad, P.; Hornes,E.; Olsvik, O. Prog Polym Sci 1992, 17, 87–161.
36. Liu, X.-X.; Ding, X.-B.; Zheng, Z.-H.; Peng, Y.-X.;Long, X.-P.; Wang, X.-C.; Chan, A. S. C.; Yip,C. W. J Appl Polym Sci 2003, 90, 1879–1884.
37. Tseng, C. M.; Lu, Y. Y.; El-Aasser, M. S.; Vanderh-off, J. W. J Polym Sci Part A: Polym Chem 1986,24, 2995–3007.
38. Paine, A. J.; Luymes, W.; McNulty, J. Macromole-cules 1990, 23, 3104–3109.
39. Li, K.; Stover, H. D. H. J Polym Sci Part A: PolymChem 1993, 31, 3257–3263.
40. Li, W. H.; Stover, H. D. H. J Polym Sci Part A:Polym Chem 1999, 37, 2295–2303.
41. Bai, F.; Yang, X.; Huang, W. Macromolecules2004, 37, 9746–9752.
42. Gutowska, A.; Bae, Y. H.; Jacobs, H.; Feijen, J.;Kim, S. W. Macromolecules 1994, 27, 4167–4175.
43. Horak, D.; Benedyk, N. J Polym Sci Part A:Polym Chem 2004, 42, 5827–5837.
44. Epoxy Resins: Chemistry and Technology; May,A. C., Ed.; Marcel Dekker: New York, 1988.
45. Virtanen, J.; Tenku, H. J Polym Sci Part A: PolymChem 2001, 39, 3716–3725.
46. Saunders, B. R. In: Colloidal Polymers: Synthesisand Characterization; Elaissari, A., Ed.; MarcelDekker: New York, 2003; pp 419–437.
47. Otake, K.; Inomata, H.; Konno, M.; Saito, S. Macro-molecules 1990, 23, 283–289.
982 MACKOVA AND HORAK
















![Drug Delivery System Based on Covalently Bonded Poly[N-Isopropylacrylamide-co-2-Hydroxyethylacrylate]-Based Nanoparticle Networks](https://static.fdokumen.com/doc/165x107/6340d5f6e0dac3b265042228/drug-delivery-system-based-on-covalently-bonded-polyn-isopropylacrylamide-co-2-hydroxyethylacrylate-based.jpg)



