
Effect of Ischemia on the In Vivo Release of Striatal Dopamine, Glutamate, and ?-Aminobutyric Acid...
10
Journal of Neurochemistry Raven Press, Ltd., New York 0 1988 International Society for Neurochemistry Effect of Ischemia on the In Vivo Release of Striatal Dopamine, Glutamate, and y-Aminobutyric Acid Studied by Intracerebral Microdialysis Mordecai Y.-T. Globus, Raul Busto, W. Dalton Dietrich, Elena Martinez, Isabel Valdes, and Myron D. Ginsberg Cerebral Vascular Disease Research Center, Department of Neurology, University of Miami School of Medicine, Miami, Florida, U.S.A. Abstract: We have previously described a marked attenuation of postischemic striatal neuronal death by prior substantia nigra (SN) lesioning. The present study was carried out to evaluate whether the protective effect of the lesion involves changes in the degree of local cerebral blood flow (ICBF) reduction, energy metabolite depletion, or alterations in the extracellular release of striatal dopamine (DA), glutamate (Glu), or y-aminobutyric acid (GABA). Control and SN-le- sioned rats were subjected to 20 min of forebrain ischemia by four-vessel occlusion combined with systemic hypotension. Levels of lCBF, as measured by the autoradiographic method, and energy metabolites were uniformly reduced in both the ipsi- and contralateral striata at the end of the ischemic period, a finding implying that the lesion did not affect the severity of the ischemic insult itself. Extracellular neurotransmitter levels were measured by microdialysis; the perfusate was col- lected before, during, and after ischemia. An -500-fold in- crease in DA content, a 7-fold increase in Glu content, and a 5-fold increase in GABA content were observed during ischemia in nonlesioned animals. These levels gradually re- turned to baseline by 30 min of reperfusion. In SN-lesioned rats, the release of DA was completely prevented, the release of GABA was not affected, and the release of Glu was partially attenuated. However, excessive extracellular Glu concentra- tions were still attained, which are potentially toxic. This, taken together with the previous neuropathological findings, suggests that excessive release of DA is important for the development of ischemic cell damage in the striatum. Key Words: Dopamine-Glutamate-Striatum-Cerebral isch- emia-Microdialysis. Globus M. Y.-T. et al. Effect of isch- emia on the in vivo release of striatal dopamine, glutamate, and y-aminobutyric acid studied by intracerebral microdi- alysis. J. Neurochem. 51, 1455-1464 (1988). Neuropathologistshave long noted that certain brain regions and specific neuronal types are especially vul- nerable to ischemic insults (Brierley, 1976). Among the neurons that are particularly susceptible to transient ischemia are the CA 1 pyramidal cells of the hippocam- pus and the small to medium-sized neurons of the dor- solateral striatum (Pulsinelli et al., 1982; Ginsberg et al., 1985). Although the selective vulnerability of these neurons is well established morphologically, its patho- genesis remains obscure. Recent studies have described elevations of the extracellular concentration of gluta- mate (Glu) and 7-aminobutyric acid (GABA) in the hippocampus following transient ischemia (Benveniste et al., 1984; Hagberg et al., 1985), and others have demonstrated that ischemia-induced cell death in the hippocampus is dependent on the integrity of its glu- tamatergic input (Wieloch et al., 1985~; Johansen et al., 1986; Onodera et al., 1986). These results suggest that the release of excitatory neurotransmitters is in- volved in the development of ischemic cell damage in the hippocampus. The striatum, a region highly vulnerable to ischemia, is richly innervated both by the corticostriatal gluta- matergic pathway and by nigrostriatal dopaminergic projections, which have been shown to interact with each other (Cheramy et al., 1986; Chiodo and Berger, Received March 11, 1988; revised manuscript received May 9, 1988; accepted May 12, 1988. Address correspondence and reprint requests to Dr. M. Y.-T. Glo- bus at Cerebral Vascular Disease Research Center, Department of Neurology ( D 4 - 9 , University of Miami School of Medicine, P.O. Box 016960, Miami, FL 33101, U.S.A. Abbreviations used: DA, doparnine; DOPAC, 3,4-dihydroxyphe- nylacetic acid; GABA, y-arninobutyric acid; Glu, glutamate; HVA, homovanillic acid;lCBF, local cerebral blood flow; NMDA, N-methyl- D-aspartate; PCA, perchloric acid; PCr, phosphocreatine; SN, sub- stantia nigra. 1455
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Effect of Ischemia on the In Vivo Release of Striatal Dopamine, Glutamate, and ?-Aminobutyric Acid...

Journal of Neurochemistry Raven Press, Ltd., New York 0 1988 International Society for Neurochemistry
Effect of Ischemia on the In Vivo Release of Striatal Dopamine, Glutamate, and y-Aminobutyric Acid Studied
by Intracerebral Microdialysis
Mordecai Y.-T. Globus, Raul Busto, W. Dalton Dietrich, Elena Martinez, Isabel Valdes, and Myron D. Ginsberg
Cerebral Vascular Disease Research Center, Department of Neurology, University of Miami School of Medicine, Miami, Florida, U.S.A.
Abstract: We have previously described a marked attenuation of postischemic striatal neuronal death by prior substantia nigra (SN) lesioning. The present study was carried out to evaluate whether the protective effect of the lesion involves changes in the degree of local cerebral blood flow (ICBF) reduction, energy metabolite depletion, or alterations in the extracellular release of striatal dopamine (DA), glutamate (Glu), or y-aminobutyric acid (GABA). Control and SN-le- sioned rats were subjected to 20 min of forebrain ischemia by four-vessel occlusion combined with systemic hypotension. Levels of lCBF, as measured by the autoradiographic method, and energy metabolites were uniformly reduced in both the ipsi- and contralateral striata at the end of the ischemic period, a finding implying that the lesion did not affect the severity of the ischemic insult itself. Extracellular neurotransmitter levels were measured by microdialysis; the perfusate was col- lected before, during, and after ischemia. An -500-fold in-
crease in DA content, a 7-fold increase in Glu content, and a 5-fold increase in GABA content were observed during ischemia in nonlesioned animals. These levels gradually re- turned to baseline by 30 min of reperfusion. In SN-lesioned rats, the release of DA was completely prevented, the release of GABA was not affected, and the release of Glu was partially attenuated. However, excessive extracellular Glu concentra- tions were still attained, which are potentially toxic. This, taken together with the previous neuropathological findings, suggests that excessive release of DA is important for the development of ischemic cell damage in the striatum. Key Words: Dopamine-Glutamate-Striatum-Cerebral isch- emia-Microdialysis. Globus M. Y.-T. et al. Effect of isch- emia on the in vivo release of striatal dopamine, glutamate, and y-aminobutyric acid studied by intracerebral microdi- alysis. J. Neurochem. 51, 1455-1464 (1988).
Neuropathologists have long noted that certain brain regions and specific neuronal types are especially vul- nerable to ischemic insults (Brierley, 1976). Among the neurons that are particularly susceptible to transient ischemia are the CA 1 pyramidal cells of the hippocam- pus and the small to medium-sized neurons of the dor- solateral striatum (Pulsinelli et al., 1982; Ginsberg et al., 1985). Although the selective vulnerability of these neurons is well established morphologically, its patho- genesis remains obscure. Recent studies have described elevations of the extracellular concentration of gluta- mate (Glu) and 7-aminobutyric acid (GABA) in the hippocampus following transient ischemia (Benveniste
et al., 1984; Hagberg et al., 1985), and others have demonstrated that ischemia-induced cell death in the hippocampus is dependent on the integrity of its glu- tamatergic input (Wieloch et al., 1985~; Johansen et al., 1986; Onodera et al., 1986). These results suggest that the release of excitatory neurotransmitters is in- volved in the development of ischemic cell damage in the hippocampus.
The striatum, a region highly vulnerable to ischemia, is richly innervated both by the corticostriatal gluta- matergic pathway and by nigrostriatal dopaminergic projections, which have been shown to interact with each other (Cheramy et al., 1986; Chiodo and Berger,
Received March 11, 1988; revised manuscript received May 9, 1988; accepted May 12, 1988.
Address correspondence and reprint requests to Dr. M. Y.-T. Glo- bus at Cerebral Vascular Disease Research Center, Department of Neurology (D4-9, University of Miami School of Medicine, P.O. Box 016960, Miami, FL 33101, U.S.A.
Abbreviations used: DA, doparnine; DOPAC, 3,4-dihydroxyphe- nylacetic acid; GABA, y-arninobutyric acid; Glu, glutamate; HVA, homovanillic acid; lCBF, local cerebral blood flow; NMDA, N-methyl- D-aspartate; PCA, perchloric acid; PCr, phosphocreatine; SN, sub- stantia nigra.
1455

1456 M. Y.-T. GLOBUS ET AL.
1986; Kornhuber and Kornhuber, 1986; Romo et al., 1986). We have recently found that prior lesioning of the substantia nigra (SN) exerts a strong protective ef- fect on postischemic striatal neuronal death in a rat model of transient ischemia (Globus et al., 1987~). Al- though these results tend to implicate dopamine (DA) in the genesis of ischemic striatal injury, one cannot exclude the possibility that the protective effect of such a lesion might be attributable to its influence on the release of other putative neurotransmitters, such as Glu or GABA, during ischemia and recirculation.
The present study used a newly developed technique of regional brain microdialysis (Ungerstedt, 1984) to measure extracellular levels of striatal DA and its me- tabolites, of Glu, and of GABA during ischemia and recirculation in control rats and in animals with uni- lateral SN lesions. Local cerebral blood flow (1CBF) and metabolic studies were carried out to document the severity of the ischemic insult.
MATERIALS AND METHODS
SN lesion Male Wistar rats (weighing 280-300 g) were kept under
diurnal light conditions with free access to food and water for at least 7 days before the morning of the operation. They were then anesthetized with Equitensin, and unilateral SN lesions were made by stereotaxic infusion of 3 pl of 6-hy- droxydopamine solution containing 6 pg of 6-hydroxydo- pamine base, as described previously (Hank et al., 1982). Eleven days after SN lesioning, rats were injected with apo- morphine, 0.5 mg/kg s.c., and observed for rotational be- havior for 20 min. Only rats that exhibited consistent con- tralateral turning, a behavior indicating >90% depletion of DA in the ipsilateral striatum (Hefti et al., 1980), were used in our experiments.
Brain dialysis procedure and transient forebrain ischemia
Two weeks after SN lesioning, rats were subjected to 20 rnin of global ischemia by four-vessel occlusion combined with systemic hypotension. The procedure consisted of bi- lateral temporary ligation of the common carotid arteries in rats with prior permanent occlusion of the vertebral arteries. One day before ischemia, both vertebral arteries were per- manently electrocoagulated under halothane (3%) and a mixture of nitrous oxide (70%) and oxygen (30%), as described previously (Pulsinelli and Brierley, 1979). On the following day, both femoral arteries and one femoral vein were can- d a t e d , and ligatures consisting of a loop of PE-I0 polyeth- ylene tubing contained within a double-bore Silastic tubing were placed around each common carotid artery.
After the rats were intubated and mechanically ventilated, they were transferred to a stereotaxic frame (David Kopf) for microdialysis probe implantation. The skull was exposed, and a hole was drilled, with the animal under halothane anesthesia, according to the coordinates for the striatum (1 mm anterior and 2.5 mm lateral to the bregma). A micro- dialysis probe (4 mm dialysis membrane; Carnegie Medicin, Sweden), mounted on a probe clip and camer, was then low- ered into the striatum (5.5 mm ventral to the dura) and sub- sequently perfused with Ringer's solution at a flow rate of 2 pl/min by means of a microinfusion pump (Carnegie Med-
icin). Halothane administration was discontinued, and the animals were maintained on nitrous oxide and oxygen and immobilized by pancuronium bromide, 0.6 mg/kg i.v.; ad- ditional doses of 0.3 mg/kg were administered at 0.5-h in- tervals throughout the experiment. Arterial P,co~ and P,02 were kept within the normal range.
Following a 1.5-h stabilization period, the ischemic insult was initiated. Before the ischemic insult, blood was withdrawn to adjust the preischemic blood pressure to 90 mm Hg. Ce- rebral ischemia was then produced for 20 rnin by tightening the carotid ligatures bilaterally and maintaining the mean blood pressure at 80 mm Hg by gradual withdrawal of ad- ditional blood or by reinjecting heparinized blood as needed. Rectal and head (temporalis muscle) temperatures were monitored with thermistor probes and were kept at 36-37°C throughout the experiment by warming lamps (Busto et al., 1987). To permit postischemic reperfusion, carotid ligatures were removed, and the blood, kept at 36-37"C, was reinfused to restore the mean arterial pressure to 100-120 mm Hg. Samples of the microdialysis perfusate, representing extra- cellular fluid, were collected in an ice bath at 1 0-min intervals, beginning 30 rnin before ischemia, during ischemia, and dur- ing the first 30 rnin and at the end of 1 and 2 h of recirculation. All samples were immediately frozen and stored at -80°C until analysis.
Biochemical analysis of striatal perfusate Each collecting tube contained 10 pl of 0. I M perchloric
acid (PCA) (to prevent air oxidation of the monoamines) and 0.5 pM 3,4-dihydroxybenzylamine as an internal standard. At a perfusion rate of 2 pllmin, each 10-min period yielded a sample volume of 30 pl, of which 10 pl was analyzed for DA and its metabolites, and the remaining 20 p1 was neu- tralized with 0.1 M KOH for Glu and GABA assessment.
Quantijcation of monoamines. Analysis of DA, 3,4-di- hydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) concentrations in the perfusate was performed by HPLC with electrochemical coulometric detection (Kilpatrick et al., 1986). Perfusate samples were injected directly into a solvent delivery module (model 5700; ESA), and mono- amines were separated on a 250- X 4.6-mm reversed-phase analytical column (Ultrasphere ODS 5 pm; Beckman). The mobile phase consisted of 0.09 M sodium acetate, 0.035 M citric acid, 130 pM EDTA, 50 pM octane sulfonate, and 13.5% (vol/vol) methanol. The coulometric detectors (ESA) consisted of a control module (model 5 loo), conditioning cell (model 502 I), and a dual-electrode analytical cell (model 50 1 1). For optimal performance, current-voltage curves were measured for each species analyzed. Based on these curves, the values of applied potential used in our study were 0.10 V at detector 1 and -0.40 V at detector 2; the conditioning cell potential was 0.60 V. Basal levels of DA in the perfusate were three to five times higher than the limit of detection.
Glu and GABA assay. Glu was analyzed by combining the following enzymatic reactions. Two microliters of perfusate was first reacted with 50 pl of enzymatic reagent, which con- tained glutamate dehydrogenase (EC 1.4. I .3), and with NAD+ for 45 min to form NADH, as described by Folbergrovi et al. (1 972). Microtubes that contained blank (Ringer's solu- tion, 3,4-dihydroxybenzylamine, and PCA) or standard were analyzed in parallel with the samples. The reaction was ter- minated by adding 0.5 M NaOH and heating the samples at 80°C for 20 min. After the reaction was stopped, 10 p1 of 0.5 MHCl was added, and a 5-pl aliquot was used for NADH enzymatic cycling reactions in the presence of alcohol de-
J . Neurochem., Vol. 51. Nu. 5. 1988

CEREBRAL ISCHEMIA AND EXTRACELLULAR NEUROTRANSMITTERS 1457
hydrogenase (EC 1.1.1.1) and malate dehydrogenase (EC 1.1.1.37), as described by Kato et al. (1973). At the end of the reactions, the fluorescence of NADH was measured.
Microdetermination of GABA levels was carried out by a combination of the enzymatic assay for GABA and the en- zymatic cycling technique, which have been described in de- tail (Otsuka et al., 197 1). Two-microliter samples of perfusate, blank, and standard were each reacted with 26 pl of the en- zymatic reagent, which contained GABA-glutamate trans- aminase (EC 2.6.1.19) and succinate semialdehyde dehydro- genase (EC 1.2.1.16), in a sealed container and heated at 38°C. The reaction was stopped after 1 h by addition of 5 pl of 0.5 MNaOH and heating the mixture at 60°C for 20 min. Five microliters of aliquot was then incubated at 38°C with 100 pl of the cycling reagent, which contained glutamate de- hydrogenase and glucose-6-phosphodehydrogenase (EC 1.1.1.49). After 90 min, the cycling reaction was terminated by heating the mixture for 5 rnin at 100°C. To each tube, 1 ml of 6-phosphogluconate reagent was then added, and the baseline reading was taken. The samples were then reacted with 6-phosphogluconate dehydrogenase (EC 1.1.1.44) for 30 min, and the fluorescence of NADH was then measured.
Metabolic and lCBF measurements To determine whether the SN lesion altered the severity
of the ischemic insult, metabolic and lCBF measurements were camed out at the end of the ischemic period in the striata both ipsi- and contralateral to the SN lesion. Striatal metabolites were also evaluated at the end of 15 and 30 min of recirculation, and lCBF was measured in other forebrain structures.
Metabolic measurements. In animals prepared for metab- olite assay, the brain was frozen transcalvarially with liquid nitrogen over a 5-min period at the end ofthe ischemic insult or the recirculation period. During the freezing process, me- chanical ventilation was continued, and the mean arterial blood pressure was maintained at 80 mm Hg (Ponth et al., 1973). The frozen head was then trimmed under continuous imgation with liquid nitrogen on a band saw to remove ex- tracranial tissue, and coronal brain slices were stored under liquid nitrogen. For striatal sampling, coronal sections were transferred to a glove box refrigerated at -30°C. After a 15- rnin temperature equilibration period, an 18-gauge needle was used to punch cores of striatal tissue. Each tissue core, weighing 5-10 mg, was prepared for analysis as described previously (Busto and Ginsberg, 1985). Assays for creatine, phosphocreatine (PCr), ATP, ADP, AMP, lactate, and glucose were performed by a direct fluorometric technique (Lowry and Passonneau, 1972). Pyruvate was quantified by cycling (Kato and Lowry, 1973), and glycogen by using amylo-a- 1,4-a- 1,6-glucosidase (Passonneau and Lauderdale, 1974).
Measurements of ICBF. The lCBF was measured by the autoradiographic method (Sakurada et al., 1978). In brief, at the end of the ischemic period, rats were infused with 30 pCi of iodo[14C]antipyrine at a constant rate during a 45-s period while multiple blood samples were drawn for whole-blood I4C activity measurements. Animals were killed by decapi- tation, and their brains were quickly removed and frozen. Coronal brain sections (20 pm thick) were cut at -20°C in a cryostat and, together with calibrated [‘4C]methyl meth- acrylate standards, were exposed to Kodak SB-5 film for 10 days. The resulting autoradiograms were digitized on a ro- tating-drum scanning densitometer (Optronics) interfaced to a PDP 1 1 /44 minicomputer. Optical density information was transformed to color-coded quantitative representations of
lCBF by means of the usual operational equation (Sakurada et al., 1978). Measurements of lCBF were performed in dif- ferent forebrain structures by means of interactive region-of- interest routines developed in our laboratory, using a square cursor of user-adjustable size positioned over individual co- ronal sections. The sections selected for analysis corresponded to the levels of frontal cortex; anterior, middle, and posterior caudate nucleus; and anterior and posterior hippocampus. Measurements were made from a total of 11 regions from each cerebral hemisphere, with at least three serial sections being used for replicate measurements at each level.
RESULTS
In rats with a unilateral SN lesion analyzed for brain metabolites, profound depletion of energy metabolites of a similar degree was observed at the end of the isch- emic insult in both the ipsi- and contralateral striata (Table 1). PCr and ATP levels were nearly depleted, whereas AMP and creatine concentrations were in- creased, but ADP levels did not change significantly. Similar degrees of glucose and glycogen content re- duction and lactate accumulation were observed in the two striata. The pattern of recovery of these metabolites during recirculation was also similar bilaterally (Table 1). ATP levels gradually increased with time but did not reach control values by 30 rnin of reperfusion. PCr, creatine, and AMP levels gradually recovered by 30 rnin of recirculation, whereas the ADP level was un- changed. Glycogen content gradually increased but did not reach the control level by 30 rnin of reperfusion. Glucose content, on the other hand, recovered quickly, exceeding control levels at 30 min, whereas lactate lev- els gradually returned to normal. These results were expected after severe ischemia and indicate that a uni- lateral SN lesion did not have an effect on the severity of striatal energy depletion induced by the ischemic insult or on the process of early postischemic metabolite recovery.
To corroborate our conclusion from the metabolic analysis, lCBF was assessed autoradiographically in nonischemic rats with an SN lesion and in SN-lesioned animals at the end of the ischemic period (Table 2). In control, nonischemic rats with an SN lesion, no sig- nificant side-to-side differences in lCBF values were found in any brain region analyzed. In SN-lesioned animals with ischemia, blood flow was severely reduced at the end of the ischemic period, to 1-4% of control, in all forebrain regions analyzed, with no side-to-side differences. These results confirm that the severity of the ischemic insult was uniform in both striata.
Unilateral striatal microdialysis sampling was carried out during ischemia and recirculation in nonlesioned rats (n = 10) and from the denervated striatum of SN- lesioned animals (n = 7). The time course of change in DA release in nonlesioned and SN-lesioned rats is illustrated in Fig. 1. Basal levels of endogenous DA were stable in three consecutive perfusate samples col- lected from both the denervated and nondenervated striata before ischemia. Basal DA levels were similar
J. Neurochem., Vol. 51, No. 5, 1988

1458 M. Y.-T. GLOBUS ET AL.
TABLE 1. Levels of striatal metabolites in rats with a unilateral SN lesion under control conditions and at the end of ischemia with and without 15 or 30 rnin of recirculation
Recirculation
Control (n = 7) End of ischemia (n = 4) 15 rnin (n = 5) 30 rnin (n = 5)
Ipsilateral Contralateral Ipsilateral Contralateral Ipsilateral Contralateral Iusilateral Contralateral
PCr Cr ATP ADP AMP Glucose Glycogen Lac Pyr
5.98 f 0.12 5.68 f 0.12 3.02 f 0.1 1 0.20 f 0.02 0.02 f 0.01 1.05 f 0.03 1.38 f 0.12 1.42 f 0.10 0.09 f 0.01
5.74 f 0.14 5.60 f 0.06 2.99 f 0.1 1 0.23 f 0.02 0.02 f 0.01 1.08 t 0.10 1.29 f 0.14 1.38 f 0.09 0.09 f 0.01
0.31 f 0.09" I 1.30 f 0.55" 0.04 f 0.04" 0.28 f 0.02 1.06 f 0.20" 0.02 f 0.OlC 0.14 f 0.05"
12.80 f 0.75" 0.07 f 0.01
0.39 k 0.17' I 1 .SO f 0.62" 0.06 f 0.06' 0.22 f 0.04 1.10 f 0.20" 0.07 f 0.02' 0.12 f 0.02"
13.50 f 0.77" 0.1 1 f 0.03
5.24 f 0.13' 6.07 f 0.44 2.01 f 0.16' 0.18 f 0.02 0.07 f 0.02 3.07 f 0.54" 0.43 f 0.1 1' 7.71 f 1.06" 0.20 f 0.04'
4.99 f 0.09' 6.77 f 0.29 1.86 f 0.18" 0.15 f 0.02 0.09 f 0.02 2.24 f 0.47 0.37 f 0.08" 7.83 f 0.97" 0.17 f 0.03
5.44 f 0.27 5.79 f 0.40 1.86 f 0.04" 0.20 f 0.02 0.12 f 0.04 3.25 f 0.79" 0.54 f 0.06" 3.70 f 0.47 0.16 f 0.03
5.67 f 0.23 5.36 f 0.54 1.87 f 0.11" 0.20 f 0.03 0.10 f 0.04 3.76 f 0.75" 0.53 f 0.04" 3.76 f 0.61 0.16 f 0.04
Data are mean f SEM values (in pmol/g wet weight). Cr, creatine; Lac, lactate; Pyr, pyruvate. Values significantly different from the corresponding control value are indicated: "p < 0.01 (by two-way analysis of variance), 'p < 0.05 (by
two-way analysis of variance), "p < 0.001 (by Student's t test).
in the denervated and nondenervated striata. In nonle- sioned rats, a massive increase in striatal DA concen- tration was detected in the perfusate within 10 min of ischemia (mean basal levels = 0.003 pM; mean isch- emic levels = 1.6 pM). This increased release persisted during the 20 rnin of ischemia, and the levels gradually returned to baseline values by 30 rnin of reperfusion. In SN-lesioned animals, levels of DA were low and unchanged throughout the ischemia and recirculation periods. These results indicate that a massive release of striatal DA occurs during ischemia and early recir- culation, which is completely inhibited by prior SN lesioning. The ischemic and postischemic alterations in DOPAC and HVA perfusate concentrations in nonlesioned and SN-lesioned rats are depicted in Figs. 2 and 3, respectively. Whereas in the nonlesioned rats
the basal levels of DOPAC and HVA were much higher than those of the parent amine, in SN-lesioned animals they were barely detectable. After 20 rnin of ischemia, a significant decrease in DOPAC and HVA concen- trations was observed in the perfusate of the nonle- sioned animals (by 63 and 59%, respectively); on re- perfusion, these levels gradually returned to baseline. In SN-lesioned animals, DOPAC and HVA perfusate levels remained low and hardly detectable during the ischemia and recirculation periods.
The time course of changes in Glu and GABA release in nonlesioned and SN-lesioned animals during isch- emia and recirculation is presented in Figs. 4 and 5, respectively. In nonlesioned animals, a gradual extra- cellular increase in levels of both amino acids was ob- served in the striatal perfusate; levels were maximal at
TABLE 2. Regional cerebral bloodflow values in rats with a unilateral SN lesion under control conditions and at the end of ischemia
Control (n = 7) End of ischemia
Structure Ipsilateral Contralateral Ipsilateral Contralateral
Cingulate cortex Frontal cortex Nucleus accumbens Donolateral striatum Medioventral striatum Globus pallidus Motor cortex Somatosensory cortex Anterior thalamus Anterior hippocampus Posterior hippocampus
1.19 f 0.16 0.99 f 0.13 1.09 f 0.15 1.00 f 0.13 0.91 f 0.10 0.62 f 0.08 1.17 f 0.17 1.04 f 0.08 0.99 f 0.09 0.62 f 0.08 0.70 f 0.09
1.19 f 0.20 1.02 f 0.15 1.20 f 0.24 1.03 f 0.10 0.97 f 0. I 1 0.56 f 0.04 1.12 f 0.15 1.13 k 0.10 1.00 f 0.14 0.63 f 0.07 0.70 f 0.09
0.01 f 0.00 0.01 * 0.00 0.02 f 0.01 0.02 f 0.00 0.01 f 0.00 0.02 * 0.01 0.01 f 0.00 0.02 f 0.01 0.03 f 0.02 0.01 f 0.00 0.02 f 0.01
~~
0.01 f 0.00 0.01 f 0.00 0.02 f 0.01 0.02 f 0.01 0.01 f 0.00 0.02 f 0.00 0.01 f 0.00 0.02 f 0.01 0.02 f 0.02 0.01 f 0.00 0.02 f 0.01
The lCBF values (ml/g/min) are expressed as mean t SD. Statistical significance was assessed by two-way analysis of variance. All flow values at the end of ischemia
were significantly lower than the corresponding control values (p < 0.01). No significant difference was found between ipsilateral and contralateral sides.
J. Neurochem., Vol. 51, No. 5, 1988
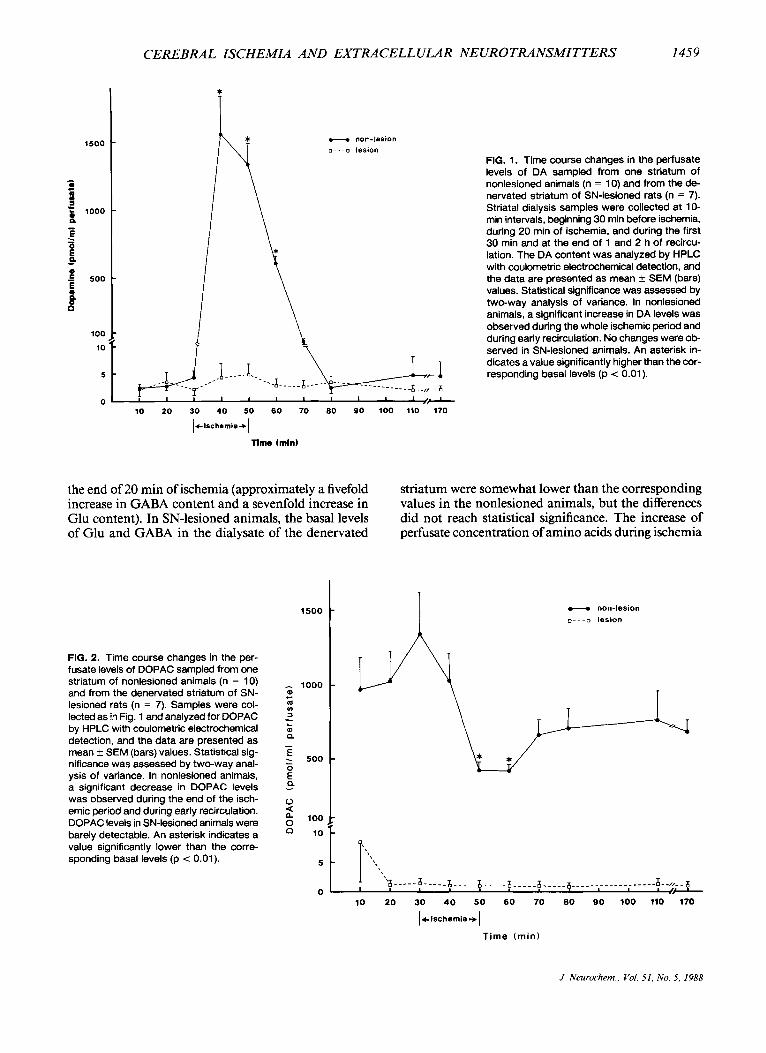
CEREBRAL ISCHEMIA AND EXTRACELLULAR NEUROTRANSMITTERS 1459
1500
- 1000
m m c
v) f
m - L a
E . 500
- - 0
k
g 100
- 0 a
1500
0 - I 5 1000
P i E a
E
0
c
n
- f 500
g
100
10
5
0
-
-
-
-
*
- non-lesion o---o lesion
FIG. 1. Time course changes in the perfusate levels of DA sampled from one striatum of nonlesioned animals (n = 10) and from the de- nervated striatum of SN-lesioned rats (n = 7). Striatal dialysis samples were collected at 10- min intervals, beginning 30 min before ischemia, during 20 min of ischemia. and during the first 30 min and at the end of 1 and 2 h of recircu- lation. The DA content was analyzed by HPLC with coulometric electrochemical detection, and the data are presented as mean f SEM (bars) values. Statistical significance was assessed by two-way analysis of variance. In nonlesioned animals, a significant increase in DA levels was observed during the whole ischemic period and during early recirculation. No changes were ob- served in SN-lesioned animals. An asterisk in- dicates a value significantly higher than the cor- responding basal levels (p < 0.01).
10 20 30 40 50 60 70 80 SO 100 110 170
lelschsrnis+)
nme (min)
the end of 20 min of ischemia (approximately a fivefold increase in GABA content and a sevenfold increase in Glu content). In SN-lesioned animals, the basal levels of Glu and GABA in the dialysate of the denervated
striatum were somewhat lower than the corresponding values in the nonlesioned animals, but the differences did not reach statistical significance. The increase of perfusate concentration of amino acids during ischemia
FIG. 2. Time course changes in the per- fusate levels of DOPAC sampled from one striatum of nonlesioned animals (n = 10) and from the denervated striatum of SN- lesioned rats (n = 7). Samples were col- lected as in Fig. 1 and analyzed for DOPAC by HPLC with coulometric electrochemical detection, and the data are presented as mean f SEM (bars) values. Statistical sig- nificance was assessed by two-way anal- ysis of variance. In nonlesioned animals, a significant decrease in DOPAC levels was observed during the end of the isch- emic period and during early recirculation. DOPAC levels in SN-lesioned animals were barely detectable. An asterisk indicates a value significantly lower than the corre- sponding basal levels (p < 0.01).
t. non- les ion D-- -L) l e s i o n
I 'a----- &-----A ___.. __.._ & _ _ _ _ _ & ___._ 6 _ . _ _ _ _ _ _ _ _ _ _ _ _ - - - - A - -,,-. &
0 ' ' /+ 10 20 30 40 50 60 70 00 SO 100 110 170
1-Ischemia-, 1 Time (rnin)
J. Neurochem.. Vol. 51. No. 5. 1988

I460 M. Y.-T. GLOBUS ET AL.
1000
- c m u) 3 r 500 m
E 0 - . - 0
100 - u lo > I
5
0
T t. non-lesion o-- -D lesion
_ _ _ _ z - _ _ _ _ 6- __.. _ _ _ __._.-.-- -- - - - !- -,,- - & f+
- - - - - " - _ _ _ _ _ _ - - - - 10 20 30 40 50 60 70 80 90 100 110 170
Ic Ischemia+l
Time (min)
was similar in pattern in the nonlesioned and lesioned animals but was of lesser magnitude in the denervated striatum. However, by the end of 20 min of ischemia, only the Glu level was significantly lower in the de- nervated striatum as compared with the nondenervated striaturn (30 vs. 50 pM, respectively). In all experi- mental animals, perfusate levels of both Glu and GABA gradually returned to baseline by 20 min of reperfusion.
DISCUSSION
Both the degree of lCBF reduction and the extent of striatal tissue energy depletion indicate that a high- grade cerebral ischemic insult was produced in our modified four-vessel occlusion model. Control of head temperature during ischemia was found to be a critical factor in this ischemia model, because we have shown that small variations in intraischemic brain tempera-
FIG. 4. Time course changes in the per- fusate levels of Glu sampled from one striatum of nonlesioned animals (n = 10) and from the denervated striatum of SN- lesioned rats (n = 7). Samples were col- lected as in Fig. 1 and analyzed for Glu by an enzymatic assay, and the data are pre- sented as mean f SEM (bars) values. Statistical significance was assessed by two-way analysis of variance. In both nonlesioned and lesioned animals, a sig- nificant increase in Glu levels was ob- served during the end of the ischemic pe- riod and during early recirculation. An as- terisk indicates a value significantly higher than the corresponding basal levels (p < 0.01). The letter a indicates a value sig- nificantly lower than the corresponding level in the nonlesioned animals (p < 0.01).
60 - al L
t ii
E
a r
40 - :
E - al w m 2 0 E m a B L
0
FIG. 3. Time course changes in the perfusate levels of HVA sampled from one striatum of nonlesioned animals (n = 10) and from the denervated striatum of SN-lesioned rats (n = 7). Samples were collected as in Fig. 1 and analyzed for HVA by HPLC with coulometric electrochemical detection, and the data are presented as mean +- SEM (bars) values. Sta- tistical significance was assessed by two-way analysis of variance. In nonlesioned animals, a significant decrease in HVA levels was ob- served during the end of the ischemic period and during early recirculation. HVA levels in SN-lesioned animals were barely detectable. An asterisk indicates a value significantly lower than the corresponding basal levels (p < 0.01).
ture markedly influence the extent of ischemic injury (Busto et al., 1987). The uniformity in flow reduction and energy depletion in both striata, ipsi- and contra- lateral to the SN lesion, implies that SN lesioning did not affect the severity of the ischemic insult itself. Nev- ertheless, this insult has been shown to give rise to con- sistently severe ischemic cell damage in the striaturn contralateral to the SN lesion but only mild changes on the ipsilateral side (Globus et al., 1987a). Thus, striatal DA denervation confers a protective effect. The aim of the present study was to evaluate the effect of SN lesioning on the extracellular release of several pu- tative neurotransmitters that could be involved in the mediation of the ischemic insult in the striatum.
In control nonlesioned animals, global forebrain ischemia evoked a profound increase in the extracel- lular concentrations of striatal DA accompanied by a decrease in DOPAC and HVA levels. Extracellular
* T
t. non-lesion o - - - ~ lesion
.. 10 20 30 40 50 60 70 80 90 100 110 170
lclsehemia+)
Time (mln)
J . Neurochem., Vol. 51, No. 5. 1988

CEREBRAL ISCHEMIA AND EXTRA CELLULAR NEUROTRANSMITTERS 1461
i
FIG. 5. Time course changes in the perfusate levels of GABA sampled from one striaturn of nonlesioned animals (n = 10) and from the de- neNated striaturn of SN-lesioned rats (n = 7). Samples were collected as in Fig. 1 and ana- lyzed for GABA by an enzymatic assay, and the data are presented as mean * SEM (bars) values. Statistical significance was assessed by two-way analysis of variance. In both nonle- sioned and lesioned animals, a significant in- crease in GABA levels was observed during the end of the ischemic period and during early re- circulation. No significant differences were ob- served between the lesioned and nonlesioned animals. An asterisk indicates a value signifi- cantly higher than the corresponding basal lev- els (p -= 0.01).
I I I I 1 I I ' f- 10 20 30 40 SO 60 70 80 90 100 110 170
I*.lschernia *I Tlmo (mln)
levels of both Glu and GABA also increased, but to a far lesser extent in percentage terms. It is conceivable that this dramatic increase in the neurotransmitters quantified could have resulted, in part, from ingress through a damaged blood-brain bamer. However, no acute barrier changes have been documented in the transient ischemia model we used (Petito et al., 1982). Therefore, it is likely that the changes in extracellular levels resulted from leakage from intracellular com- partments. Under normal conditions, extracellular neurotransmitter levels are controlled by a dynamic, energy-requiring efflux-influx cycle of transport. Nor- mal levels of ATP and normal ATP metabolism are required to keep DA compartmentalized inside syn- aptic terminals and to maintain the normal reuptake process, which is also dependent on transmembrane ionic gradients (Iversen, 197 1). Similarly, Glu released at glutamatergic nerve terminals during synaptic ac- tivity is generally regarded as being taken up by an energy-requiring, high-affinity transport process into glial cells (Fonnum, 1984). During ischemia, tissue en- ergy stores are depleted, leading to inhibition of these energy-dependent transport processes and of Na+,K+- ATPase (Pulsinelli and Duffy, 1983). Thus, increased transmitter efflux due to neuronal depolarization, cou- pled with inhibition of reuptake, provides an expla- nation for the increased extracellular content of DA and the amino acids observed in the present study. The delay in appearance of peak extracellular levels of Glu and GABA suggests that reuptake mechanisms might initially compensate for the depolarization-evoked re- lease, because the compartment responsible for amino
acid uptake-namely, glial cells-is fairly resistant to ischemia (Jenkins et al., 1979). The decrease in DOPAC and HVA content probably reflects decreased reuptake of DA into the nerve terminals (Pastuszko et al., 1982), as well as a decrease in monoamine oxidase activity (Cvejic et al., 1980). During recirculation, striatal tissue levels of DOPAC and HVA have been shown to in- crease significantly (Globus et al., 19876).
It has been hypothesized that excessive release of excitatory neurotransmitters, such as Glu, and in- creased synaptic activity play causative roles in the mediation of postischemic neuronal cell death (Roth- man, 1983; Rothman and Olney, 1986). The results of our study show that in contrast to DA, whose in- crease in level during ischemia was completely inhib- ited by SN lesioning, release of Glu was only partially prevented. One may, therefore, argue that the protec- tive effect of SN lesioning may result from the possi- bility that Glu levels on the side of the nigral lesion do not reach toxic levels for a sufficient interval. However, in preliminary in vitro studies, we have found that the recovery factor of the dialysis membrane for Glu is 25%. Thus, the extracellular concentration of Glu in the denervated striatum during ischemia is estimated to be 120 pM. Because a Glu concentration of 50- 1,000 pM has been shown to cause irreversible cell loss in vitro (Choi et al., 1987), an extracellular concentra- tion of 120 pA4 is potentially toxic. This, taken together with the neuropathological findings (Globus et al., 1987a), suggests that the protective effect of SN lesion- ing is related to the inhibition of excessive DA and not Glu release during ischemia and recirculation. Addi-
J. Neurochem.. Vul 51, No. 5. 1988

1462 M. Y.-T. GLOBUS ET AL.
tional evidence supports the role of catecholamines in the mediation of ischemic cell death. Depletion of cat- echolamines by pretreatment with a-methyl-ptyrosine exerts a strong protective effect on postischemic damage to nerve terminals in the gerbil (Weinberger et al., 1985), and hypoglycemic striatal neuronal injury is re- duced by a lesion of the nigrostriatal dopaminergic sys- tem (Lindvall et al., 1986).
Several lines of evidence support a direct deleterious effect of DA. We have observed that, after transient ischemia, relative glucose hypermetabolism combined with hypoperfusion occurs in the striatum, resulting in marked metabolism/flow uncoupling (Ginsberg et al., 1985). This early restoration of metabolic activity is suppressed in the DA-depleted striatum, an observation confirming that it is associated with increased dopa- minergic activity (Globus et al., 19873). Thus, the del- eterious effect of DA release on the ischemic striatum may be mediated by a mechanism involving enhanced metabolic activity in dopaminergic terminals or other neuronal components. The oxidation of the excessive DA that is released during ischemia could also be par- tially responsible for the production of free radicals during reperfusion (Slivka and Cohen, 1985) and, through such a mechanism, contribute to the matu- ration of the ischemic damage in the striatum.
Another possibility that must be contemplated is that the injurious effect of DA is mediated indirectly by modulating the excitotoxic effect of Glu. Several recent in vitro and in vivo studies have implicated activation of the N-methyla-aspartate (NMDA) excitatory amino acid receptor subtype in the pathophysiology of exci- totoxin-induced ischemic brain damage (Rothman and Olney, 1987). NMDA antagonists prevent anoxia from causing irreversible loss of normal electrophysiological properties in the CA 1 region of rat hippocampal slices (Clark and Rothman, 1987; Rothman et al., 1987). In rats subjected to forebrain ischemia, intrahippocampal injection of a competitive NMDA antagonist attenuates hippocampal damage at the injection site (Simon et al., 1984). The noncompetitive NMDA antagonist MK-80 1 has been shown to reduce the extent of isch- emic cell damage in models of global and focal ischemia (Foster et al., 1987; Gill et al., 1987; Park et al., 1987). The NMDA receptor channel allows current flow only when two conditions are met: (a) presynaptic release of a neurotransmitter, such as Glu, that binds to the receptor and (b) a postsynaptic cell sufficiently depo- larized to relieve the voltage-dependent Mg2+ blockade of the channel (Cotman and Iversen, 1987). Therefore, other types of receptors and regulatory systems, which are coupled to NMDA receptors, can enhance NMDA- induced responses through depolarization of the cell membrane. Potentiation of NMDA sensitivity has been reported with glycine (Johnson and Ascher, 1987), and recently it has been suggested that catecholamines can function as modulators of the ability of motor neurons to respond to excitatory amino acids (Wohlberg et al.,
1987). Therefore, it is possible that the mechanism by which DA endangers striatal neurons in the aftermath of ischemia involves activation of the voltage-depen- dent NMDA channel, thereby enabling the NMDA- mediated excitotoxic effect of Glu. Others have shown that cortical ablation and local injection of a Glu an- tagonist into the striatum ameliorate neuronal damage following hypoglycemia (Wieloch, 1985; Wieloch et al., 19853). Thus, excessive release of both DA and Glu during ischemia may be necessary for the devel- opment of ischemic injury in the striatum. This hy- pothesis is supported by studies showing that electrical stimulation of the SN or iontophoretic application of DA into the striatum potentiates the excitatory re- sponse of certain neurons in the dorsolateral striatum to cortical electric stimulation (Hirata et al., 1984) and that DA enhances excitatory amino acid-gated con- ductances in cultured retinal horizontal cells (Knapp and Dowling, 1987).
In summary, our results indicate that the protective effect of SN lesioning on the development of ischemic striatal injury is related to inhibition of extracellular release of DA during ischemia, a conclusion implicating DA in ischemic neuronal damage. These results, taken together with others (Blomqvist et al., 1985; Von Lubitz et al., 1987), suggest that neuronal death in ischemia may be modulated by the interplay of various neuro- transmitters and neuromodulators. Thus, specific pharmacological modification, aimed at multiple sites, might prove to be an appropriate therapeutic strategy in the treatment of brain ischemia.
Acknowledgment: This study was supported by US. Public Health Service grants NS-05820 and NS-22603 and by a Grant-in-Aid of the American Heart Association, with funds contributed in part by the Greater Miami Chapter. Dr. M. D. Ginsberg is the recipient of a Jacob Javits Neuroscience Investigator Award. Dr. W. D. Dietrich is an Established In- vestigator of the American Heart Association.
REFERENCES Benveniste H., Drejer J., Schousboe A., and Diemer N. H. (1984)
Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 43,
Blomqvist P., Lindvall O., and Wieloch T. (1985) Lesions of the locus coeruleus system aggravate ischemic damage in the rat brain. Neurosci. Lett. 58, 353-358.
Brierley J. (1 976) Cerebral hypoxia, in Greenfield’s Neuropathology (Blackwood W. and Corsellis A., eds), pp. 43-85. Edward Arnold, London.
Busto R. and Ginsberg M. D. (1985) Graded focal cerebral ischemia in the rat by unilateral carotid occlusion and elevated intracranial pressure: hemodynamic and biochemical characterization. Stroke
Busto R., Dietrich W. D., Globus M. Y.-T., Valdes I., Scheinberg P., and Ginsberg M. D. (1987) Small differences in intra-ischemic brain temperature critically determine the extent of ischemic neuronal injury. J. Cereb. Blood Flow Metab. 7,729-738.
1369-1374.
16,466-476.
J Neurochem., Vol. 51, No. 5, 1988

CEREBRAL ISCHEMIA AND EXTRACELLULAR NEUROTRANSMITTERS 1463
Cheramy A,, Romo R., Godeheu G., Baruch P., and Glowinski J. (1 986) In vivo presynaptic control of dopamine release in the cat caudate nucleus: facilitatory or inhibitory influence of L- glutamate. Neuroscience 19, 1081-1090.
Chiodo L. A. and Berger T. W. (1986) Interactions between dopamine and amino acid-induced excitation and inhibition in the striatum. Brain Res. 375, 198-203.
Choi D. W., Maulucci-Gedde M., and Kriegstein A. R. (1987) Glu- tamate neurotoxicity in cortical cell culture. J. Neurosci. 7,357- 368.
Clark G. D. and Rothman S. M. (1987) Blockade of excitatory amino acid receptors protects anoxic hippocampal slices. Neuroscience
Cotman C. W. and Iversen L. L. (1987) Excitatory amino acids in the brain: focus on NMDA receptors. Trends Neurosci. 10,263- 265.
Cvejic V., Micic D. V., Djuricic B. M., Mrsulja B. J., and Mrsulja B. B. (1980) Monoamines and related enzymes in cerebral cortex and basal ganglia following transient ischemia in gerbils. Acta Neuropathol. (Berl.) 51, 7 1 -77.
Folbergrovl J., MacMillan V., and Siesjo B. K. (1972) The effect of hypercapnic acidosis upon some glycolytic and Krebs cycle-as- sociated intermediates in the rat brain. J. Neurochem. 19,2507- 2517.
Fonnum F. (1984) Glutamate: a neurotransmitter in mammalian brain. J. Neurochem. 42, 1-1 1.
Foster A. C., Gill R., Kemp J. A., and Woodruff G. N. (1987) Systemic administration of MK-80 1 prevents N-methyl-Baspartate-in- duced neuronal degeneration in rat brain. Neurosci. Lett. 76,
Gill R., Foster A. C., and Woodruff G. N. (1987) Systemic admin- istration of MK-80 1 protects against ischemia-induced hippo- campal neurodegeneration in the gerbil. J. Neurosci. 7, 3343- 3349.
Ginsberg M. D., Graham D. I., and Busto R. (1985) Regional glucose utilization and blood flow following graded forebrain ischemia in the rat: correlation with neuropathology. Ann. Neurol. 18,
Globus M. Y.-T., Ginsberg M. D., Dietrich W. D., Busto R., and Scheinberg P. (1987~) Substantia nigra lesion protects against ischemic damage in the striaturn. Neurosci. Lett. 80, 25 1-256.
Globus M. Y.-T., Ginsberg M. D., Hank S. I., Busto R., and Dietrich W. D. (1987b) Role of dopamine in ischemic striatal injury: metabolic evidence. Neurology 37, 17 12- 17 19.
Hagberg H., Lehmann A., Sandberg M., Nystrom B., Jacobson I., and Hamberger A. (1985) Ischemia-induced shift of inhibitory and excitatory amino acids from intra- to extracellular com- partments. J. Cereb. Blood Flow Metab. 5,413-419.
Harik S. I., LaManna J. C., Snyder S., Wetherbee J. R., and Rosenthal M. (1982) Abnormalities of cerebral oxidative metabolism in animal models of Parkinson disease. Neurology 32, 382-389.
Hefti F., Melamed E., and Wurtman R. J. (1980) Partial lesions of the dopaminergic nigrostriatal system in rat brain: biochemical characterization. Brain Res. 195, 123-137.
Hirata K., Yim C. Y., and Mogenson G. J. (1984) Excitatory input from sensory motor cortex to neostriatum and its modification by conditioning stimulation of the substantia nigra. Brain Res.
Iversen L. L. (1971) Catecholamine uptake processes, in Biogenic Amines and Physiological Membrane Drug Therapy (Biel J. and Abood L. G., eds), pp. 259-327. Marcel Dekker, New York.
Jenkins L. W., Povlishock J. T., Becker D. P., Miller J. D., and Sullivan H. G. (1979) Complete cerebral ischemia. An ultra- structural study. Acta Neuropathol. (Berl.) 48, 1 13-125.
Johansen F. F., Jorgensen M. B., and Diemer N. H. (1986) Ischemic CA- 1 pyramidal cell loss is prevented by preischemic colchicine destruction of dentate gyms granule cells. Brain Res. 377,344- 347.
Johnson J. W. and Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529- 531.
21,665-67 1.
307-3 1 1.
470-48 1.
321, 1-8.
Kato T. and Lowry 0. H. (1973) Enzymes of energy-converting sys- tems in individual mammalian nerve cell bodies. J. Neurochem.
Kato T., Berger S. J., Carter J. A., and Lowry 0. H. (1973) An en- zymatic cycling method for nicotinamide-adenine dinucleotide with malic and alcohol dehydrogenases. Anal. Biochem. 53,86- 97.
Kilpatrick I. C., Jones M. W., and Phillipson 0. T. (1986) A semiau- tomated analysis method for catecholamines, indoleamines, and some prominent metabolites in microdissected regions of the nervous system: an isocratic HPLC technique employing cou- lometric detection and minimal sample preparation. J. Neuro- chem. 46, 1865-1876.
Knapp A. G. and Dowling J. E. (1987) Dopamine enhances excitatory amino acid-gated conductances in cultured retinal horizontal cells. Nature 325,437-439.
Kornhuber J. and Kornhuber M. E. (1986) Presynaptic dopaminergic modulation of cortical input to the striatum. Life Sci. 39,669- 674.
Lindvall O., Auer R. N., and Siesjo B. K. (1986) Selective lesions of mesostriatal dopamine neurons ameliorate hypoglycemic dam- age in the caudate-putamen. Exp. Brain Res. 63, 382-386.
Lowry 0. H. and Passonneau J. V. (1972) A Flexible System ofEn- zymatic Analysis. Academic Press, New York.
Onodera H., Sat0 G., and Kogure K. (1986) Lesions to Schaffer col- laterals prevent ischemic death of CA 1 pyramidal cells. Neurosci. Lett. 68, 169-174.
Otsuka M., Obata K., Miyata Y., and Tanaka U. (197 1) Measurement of gamma-aminobutyric acid in isolated nerve cells of cat central nervous system. J. Neurochem. 18,287-295.
Park C., Nehls D. G., Ozyurt E., Graham D. I., and McCulloch J. (1987) Ischemic brain damage is reduced by systemic admin- istration of the N-methyl-Baspartate (NMDA) antagonist, MK- 801. (Abstr) SOC. Neurosci. Abstr. 13, 1029.
Passonneau J. V. and Lauderdale V. R. (1974) A comparison ofthree methods of glycogen measurements in tissues. Anal. Biochem.
Pastuszko A., Wilson D. F., and Erecinska M. (1982) Neurotrans- mitter metabolism in rat brain synaptosomes: effect of anoxia and pH. J. Neurochem. 38, 1657-1667.
Petito C. K., Pulsinelli W. A., Jacobson G., and Plum F. (1982) Edema and vascular permeability in cerebral ischemia: com- parison between ischemic neuronal damage and infarction. J. Neuropathol. Exp. Neurol. 41, 423-426.
Pon th U., Ratcheson R. A., Salford L. G., and Siesjo B. K. (1973) Optimal freezing conditions for cerebral metabolites in rats. J. Neurochem. 21, 1127-1 138.
Pulsinelli W. A. and Brierley J. B. (1979) A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 10,267- 272.
Pulsinelli W. A. and Duffy T. E. (1983) Regional energy balance in rat brain after transient forebrain ischemia. J. Neurochem. 40,
Pulsinelli W. A,, Brierley J. B., and Plum F. (1982) Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 11, 491-498.
Romo R., Cheramy A., Godeheu G., and Glowinski J. (1986) In vivo presynaptic control of dopamine release in the cat caudate nucleus: further evidence for the implication of corticostriatal glutamatergic neurons. Neuroscience 19, 1091-1099.
Rothman S. M. (1983) Synaptic activity mediates death of hypoxic neurons. Science 220,536-537.
Rothman S . M. and Olney J. W. (1986) Glutamate and the patho- physiology of hypoxic-ischemic brain damage. Ann. Neurol. 19, 105-1 1 I .
Rothman S. M. and Olney J. W. (1987) Excitotoxicity and the NMDA receptor. Trends Neurosci. 10, 299-302.
Rothman S. M., Thurston J. H., Hauhart R. E., Clark G. D., and Solomon J. S. (1987) Ketamine protects hippocampal neurons from anoxia in vitro. Neuroscience 21, 673-678.
Sakurada O., Kennedy C., Jehle J., Brown J. D., Carbin G. L., and
20, 151-163.
60,405-412.
1500-1503.
J. Neurochem., Vol. 51, No. 5 , 1988

1464 M. Y.-T. GLOBUS ET AL.
Sokoloff L. (1978) Measurement of local cerebral blood flow with iodo(14C)antipyrine. Am. J. Physiol. 234, H59-H66.
Simon R. P., Swan J. H., Griffiths T., and Meldrum B. S. (1984) Blockade of N-methyh-aspartate receptors may protect against ischemic damage in the brain. Science 226,850-852.
Slivka A. and Cohen G. (1985) Hydroxyl radical attack on dopamine. J. Biol. Chem. 260, 15466-15472.
Ungerstedt U. (1984) Measurement of neurotransmitter release by intracranial dialysis, in Measurement of Neurotransmitter Release In Vivo (Marsden C. A., ed), pp. 81-105. John Wiley and Sons, New York.
Von Lubitz D. K. J. E., Cohan S. L., and Redmond D. J. (1987) Postischemic application of cyclohexyl adenosine (CHA) in ger- bils: enhancement of survival. (Abstr) SOC. Neurosci. Abstr. 13, 1495.
Weinberger J., Nieves-Rosa J., and Cohen G. (1985) Nerve terminal
damage in cerebral ischemia: protective effect of alpha-methyl- para-tyrosine. Stroke 16, 864-870.
Wieloch T. (1 985) Hypoglycemia-induced neuronal damage pre- vented by an N-methyl-Baspartate antagonist. Science 230,68 1- 683.
Wieloch T., Lindvall O., Blomqvist P., and Gage F. H. (19854 Ev- idence for amelioration of ischemic neuronal damage in the hippocampal formation by lesions of the perforant path. Neurol. Res. 7,24-26.
Wieloch T., Engelsen B., Westerberg E., and Auer R. (198%) Lesions of the glutamatergic cortico-striatal projections in the rat ame- liorate hypoglycemic brain damage in the striatum. Neurosci. Lett. 58, 25-30.
Wohlberg C. J., Hackman J. C., and Davidoff R. A. (1987) Epi- nephrine and norepinephrine modulate neuronal responses to excitatory amino acids and agonists in frog spinal cord. Synapse 1,202-207.
J. Neuroehern., Vol. 51. No. 5. 1988




















