
Effect of DNA methylation on molecular diversity of watermelon heirlooms and stability of...
11
Effect of DNA methylation on molecular diversity of watermelon heirlooms and stability of methylation specific polymorphisms across the genealogies Padma Nimmakayala • Gopinath Vajja • Rene ´e A. Gist • Yan R. Tomason • Amnon Levi • Umesh K. Reddy Received: 6 May 2010 / Accepted: 14 September 2010 / Published online: 24 September 2010 Ó Springer Science+Business Media B.V. 2010 Abstract American watermelon heirlooms are phe- notypically diverse in terms of their growth habits, fruit traits and responses to biotic and abiotic stress. Wide ranging DNA marker tools resolved narrow molecular diversity among these collections. The current research explored additional insights such as extent of diversity at the methylation level among the watermelon cultivars. DNA profiles were generated using Methylation-sensi- tive AFLP assay for 47 watermelon heirlooms. Results indicated that methylation specific diversity (43%) in US watermelon heirlooms is higher than the diversity (19.8%) estimated by several investigators using con- ventional DNA markers. In tree topologies of Neighbor- Joining (NJ) phenograms, the clustering pattern of principal component analyses of separate data sets obtained from the methylation specific isoschizomers MspI and HpaII resolved the diversity associated with methylation. Methylation-induced clustering was fur- ther verified using model-based population structure analysis. Our study clearly revealed the extent of methylations that are shared between parental heirlooms and progeny heirlooms, when tracked in known gene- alogies and breeding histories of heirlooms. Methyla- tion sites that were not carried over and de novo methylations in the progeny heirlooms were fewer, when compared to the methylations that are stable. Keywords mAFLP Watermelon heirlooms Methylation Epigenetic diversity Linkage disequilibrium Introduction Watermelon is the fifth most economically important vegetable crop and is grown in 44 states in the United States. There is wide phenotypic diversity in terms of growth habits, fruit traits and resistance to biotic and abiotic stress (Levi et al. 2004). After using wide ranging DNA marker tools, Levi et al. (2001, 2004), concluded that there is a very narrow genetic diversity at the DNA sequence level. In order to exploit the cultivar phenotypic potential to the fullest extent, insights into other sources of variation such as epigenetic and functional diversity might be needed to understand the interplay between wide phenotypic diversity and narrow DNA sequence diversity. Knowledge of DNA methylation profiles in cultivated watermelon will contribute to our current understand- ing of molecular divergence of watermelons. Umesh K. Reddy contributed equally to this work. P. Nimmakayala G. Vajja R. A. Gist Y. R. Tomason U. K. Reddy (&) Gus R. Douglass Institute and Department of Biology, West Virginia State University, Institute, WV 25112-1000, USA e-mail: [email protected] A. Levi US Vegetable Laboratory, USDA, ARS, 2875 Savannah Highway, Charleston, SC 29414, USA 123 Euphytica (2011) 177:79–89 DOI 10.1007/s10681-010-0259-z
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Effect of DNA methylation on molecular diversity of watermelon heirlooms and stability of...

Effect of DNA methylation on molecular diversityof watermelon heirlooms and stability of methylationspecific polymorphisms across the genealogies
Padma Nimmakayala • Gopinath Vajja •
Renee A. Gist • Yan R. Tomason • Amnon Levi •
Umesh K. Reddy
Received: 6 May 2010 / Accepted: 14 September 2010 / Published online: 24 September 2010
� Springer Science+Business Media B.V. 2010
Abstract American watermelon heirlooms are phe-
notypically diverse in terms of their growth habits, fruit
traits and responses to biotic and abiotic stress. Wide
ranging DNA marker tools resolved narrow molecular
diversity among these collections. The current research
explored additional insights such as extent of diversity at
the methylation level among the watermelon cultivars.
DNA profiles were generated using Methylation-sensi-
tive AFLP assay for 47 watermelon heirlooms. Results
indicated that methylation specific diversity (43%) in
US watermelon heirlooms is higher than the diversity
(19.8%) estimated by several investigators using con-
ventional DNA markers. In tree topologies of Neighbor-
Joining (NJ) phenograms, the clustering pattern of
principal component analyses of separate data sets
obtained from the methylation specific isoschizomers
MspI and HpaII resolved the diversity associated with
methylation. Methylation-induced clustering was fur-
ther verified using model-based population structure
analysis. Our study clearly revealed the extent of
methylations that are shared between parental heirlooms
and progeny heirlooms, when tracked in known gene-
alogies and breeding histories of heirlooms. Methyla-
tion sites that were not carried over and de novo
methylations in the progeny heirlooms were fewer,
when compared to the methylations that are stable.
Keywords mAFLP � Watermelon heirlooms �Methylation � Epigenetic diversity �Linkage disequilibrium
Introduction
Watermelon is the fifth most economically important
vegetable crop and is grown in 44 states in the United
States. There is wide phenotypic diversity in terms of
growth habits, fruit traits and resistance to biotic and
abiotic stress (Levi et al. 2004). After using wide
ranging DNA marker tools, Levi et al. (2001, 2004),
concluded that there is a very narrow genetic
diversity at the DNA sequence level. In order to
exploit the cultivar phenotypic potential to the fullest
extent, insights into other sources of variation such as
epigenetic and functional diversity might be needed
to understand the interplay between wide phenotypic
diversity and narrow DNA sequence diversity.
Knowledge of DNA methylation profiles in cultivated
watermelon will contribute to our current understand-
ing of molecular divergence of watermelons.
Umesh K. Reddy contributed equally to this work.
P. Nimmakayala � G. Vajja � R. A. Gist �Y. R. Tomason � U. K. Reddy (&)
Gus R. Douglass Institute and Department of Biology,
West Virginia State University, Institute,
WV 25112-1000, USA
e-mail: [email protected]
A. Levi
US Vegetable Laboratory, USDA, ARS, 2875 Savannah
Highway, Charleston, SC 29414, USA
123
Euphytica (2011) 177:79–89
DOI 10.1007/s10681-010-0259-z

Methylation is a major epigenetic process that has
great impact on genetic variation in plant species
(Grant 1971; Wendel 2000). DNA methylation is a
common phenomenon in plants and is very important
in gene regulation in a number of biological
processes, and together with histone modifications
represent a self-propagating epigenetic network (He-
slop-Harrison 2000; Attwood et al. 2002). In plant
genomes, a large amount of CpG sequence was found
in regions upstream of the start codon and compar-
atively less in non-coding regions (Shimizu et al.
1997; Messeguer et al. 1991). Moreover, in Arabid-
opsis, the genome-wide high resolution mapping of
DNA methylation has revealed that over one-third of
the expressed genes contain methylations within
transcribed regions (Zhang et al. 2006).
Measuring sequence variations by methods such as
RAPD, RFLP, AFLP and variable number of tandem
repeats (VNTR) amplification (Karp et al. 1996) will
only give an estimate of variation in DNA sequences.
The amount of epigenetic variation that is stored by
the genomes cannot be adequately described by
conventional DNA techniques, and a detailed study
of epigenetic variation would be a major step forward
in understanding the dynamics of genetic diversity.
Xu et al. (2000) reported a step wise protocol and the
results pertaining to methylation-specific AFLP
(mAFLP). Keyte et al. (2006) carried out mAFLP
on 20 diverse hirsutum lines and two barbadense
cotton cultivars, and showed extensive methylation
specific polymorphism (MP) among the cultivars.
Keyte et al. (2006) also noted that their MP data
distinctly correlated with functional genes, which
provided additional clues as to how methylation
might have further expanded existing genetic diver-
sity. Madlung et al. (2002) assayed selfed synthetic
allotetraploids using methylation-sensitive amplified
polymorphism (MSAP) and showed that there was
variation in methylation patterns.
At the level of DNA sequence, alterations such as
deletions, point mutations and chromosomal rear-
rangements have been the main foci for decades in
explaining how genomes and populations evolved
and further diversified. There is no dispute that such
alterations are fundamental to generate variation,
both at neutral and non-neutral levels, upon which
selection intensified. Interestingly, there is evidence
that the mutational frequency of methyl-cytosine is
substantially greater than unmodified cytosines
(Rideout et al. 1990). The objective of this research
is to understand the dynamics of genetic diversity
among American watermelon [Citrullus lanatus
(Thunb.) Matsum. & Nakai] cultivars at the methyl-
ation level, and to estimate epigenetic diversity based
on methylation profiles to compare with previously
published DNA level diversity (Levi et al. 2004).
Materials and methods
Forty-seven heirloom watermelon cultivars (Table 1)
and a wild species accession Citrullus colocynthis
were grown under uniform conditions in a growth
chamber. DNA was extracted from the leaf tissues of
three 21 days old plants using the method described
in the QIAGEN DNeasy Plant Mini Kit.
Methylation-sensitive AFLP assays on all the DNAs
were carried out using standard protocols (Xu et al. 2000;
Keyte et al. 2006 and Liu et al. 2001). Adapter sequences
of HpaII and MspI were used from the protocol of Xu
et al. (2000). DNA samples from each cultivar were
digested with EcoRI then with HpaII, and for another set,
all the samples were digested with EcoRI then with MspI.
The ligation reaction containing 10 pmol of EcoRI
adaptors, 100 pmol of HpaII/MspI adaptors with 0.5
units of T4 DNA ligase and 1 ll of T4 DNA ligase buffer
was added to 20 ll of digested reaction. The adaptors
were ligated overnight at room temperature. The adaptor
ligated reaction was diluted 1:10 using 19 TE buffer.
Pre-selective amplification was performed using a single
selective base at the 30 end of each of the HpaII and
EcoRI primers. Selective amplification was carried out
using EcoRI labeled primers using a 4300 LI-COR DNA
analyzer. Adapter sequences of HpaII and MspI were
adopted from the protocol of Xu et al. (2000). The primer
combinations were EcoRI ? AAC/HpaII/MspI ? AC
(EAAC/HAC), EcoRI ? ACT/HpaII/MspI ? CT
(EACT/HCT), EcoRI ? ACC/HpaII/MspI ? CT
(EACC/HCT), EcoRI ? ACA/HpaII/MspI ? CA
(EACA/HCA), EcoRI ? ACA/HpaII/MspI ? AC
(EACA/HAC), EcoRI ? ACG/HpaII/MspI ? AC
(EACG/HAC), EcoRI ? AGC/HpaII/MspI ? AC
(EAGC/HAC), EcoRI ? AGC/HpaII/MspI ? CA
(EAGC/HCA), EcoRI ? ACA/HpaII/MspI ? CC
(EACA/HCC), EcoRI ? AAG/HpaII/MspI ? GC
(EAAG/HGC), EcoRI ? AAC/HpaII/MspI ? GC
(EAAC/HGC) and EcoRI ? ACG/HpaII/MspI ? CA
(EACG/HCA).
80 Euphytica (2011) 177:79–89
123

Table 1 List of the U.S.
watermelon heirloom
collections
Cultivar Source of
seeds
Year
released
Breeding parentage
All Sweet Sunseeds 1972 [Miles 9 Peacock] 9 Charleston gray
Astrakansk – – –
AU-Golden Producer Hollar 1993 A selection from AU-Producer
AU-Jubliant Hollar 1985 Jubliee 9 PI271778
AU-Producer Hollar 1985 Crimson Sweet 9 PI189225
Black Diamond Sunseeds – –
Black Stone Hollar – Black Diamond, Fairfax
Calhoun Gray Sunseeds – –
Calhoun Sweet Sunseeds – –
Charleston Gray NSL-5267 1954 Africa 8, Iowa Belle, Garrison, Hawkesbury
Coles Early Sunseeds 1982 –
Congo Syngenta 1949 [African 9 Iowa Belle] 9 Garrison
Crimson Sweet Hollar 1963 [Miles 9 Peacock] 9 Charleston Gray
Dixilee Hollar 1979 Texas W5, Wilt resistant Paecock, Fairfax
Dixi Queen Sunseeds 1890 F1 Hybrid
Dunbarton NSL-6637 1953 [African 9 Iowa Belle] 9 Garrison
Fairfax Sunseeds 1952 Garrison, African, Iwoa Belle, Hawkesbury
Family Fun Syngenta 1973 F1 Hybrid
Garrison NSL-2053 – –
Garrisonian Willhite 1957 Africa 8, Iowa Belle, Garrison, Hawkesbury
Georgia Rattlesnake Seed Savers – –
Golden Honey Hollar 1954 Mixed strain from Japan seed, Co.
Golden Midget NSL-5288 1959 New Hampshire Midget 9 Pumpkin Rind
Hawkesbury Syngenta 1936 –
Iopride Syngenta – –
Ironsides NSL-7369 1950 [Leesburg 9 Hawkesbury] 9 Garrison
Jubliee Hollar 1963 Africa 8, Iowa Belle, Garrison, Hawkesbury
King and Queen Hollar – –
Kleckely’s Sweet Seed Savers – –
Klondike Syngenta 1959 –
Klondike Sweet – – –
Leesburg NSL-7368 1936 Selection From Kleckley Sweet
Melitoplesky – – –
Mickylee Hollar 1986 Texas W5, Fairfax, Summit, Graybelle
Miles NSL-6688 1948 Dixi Queen 9 Klondike R-7
Minilee Hollar 1986 Texas W5, Fairfax, Summit, Graybelle
New Hamshire Midget Syngenta 1951 –
Northern Sweet Syngenta 1938 –
Parker Willhite – F1 Hybrid
Peacock Hollar 1939 –
Picnic Syngenta 1972 Peacock type
Prince Charles Hollar 1978 F1 Hybrid ….
Sangria – – –
Stone Mountain Hollar 1924 –
Sugar Baby Sunseeds 1955 Tough sweets selection
Summit – – –
Sweet Princess – – –
Euphytica (2011) 177:79–89 81
123

Data scoring and analysis
Typically, AFLP bands are grouped into three types.
Type 1 are those in which bands are present (or
absent) in both the HpaII and MspI gels. Presumably,
no methylation events occurred in these bands. The
isoschizomers HpaII and MspI recognize the same
tetranucleotide sequence (50-CCGG-30), but display
different sensitivity to DNA methylation. Type 2 are
those in which bands are present in MspI based
amplification and absent in HpaII based amplification.
These bands are due to 50-CmCGG-30 cytosine meth-
ylation. Type 3 are those in which bands are present in
HpaII and absent in MspI based amplification. These
bands indicate the presence of hemi-methylated
50-CmCGG-30 or 50-mCCGG-30 sites, which can be
digested by HpaII but not by MspI (Walder et al.
1983). Genetic similarities based on Jaccard’s coef-
ficients (Jaccard 1908) were calculated using the
SIMQUAL program of the numerical taxonomy
multivariate analysis system (NTSYS-pc) version
2.0 software package (Rohlf 1994). The resulting
genetic similarity indices were used to generate a tree
using the neighbor joining method (Saitou and Nei
1987). PAUP *4.0 was used to generate 1,000 boot-
strap replicates for testing the reliability of the dataset
and to draw a consensus tree. Principal component
analysis (PCA) based on the genetic similarity
matrices was performed using DCENTER and EIGEN
algorithms of NTSYS-pc. The program STRUC-
TURE (version 2.2) utilizes a model-based (Bayesian)
clustering algorithm (Pritchard et al. 2000). Assump-
tions were made to form 2–6 subpopulations (K). In
this method, individual genotypes were assigned to
unknown clusters with the assumption that Hardy–
Weinberg equilibrium or linkage equilibrium exists
within clusters, but is absent between clusters and all
clusters have distinct allele frequencies (Pritchard
et al. 2000). For each subpopulation, the program was
run three times with a Burn in period of 100,000 and
MCMC reps of 200,000.
Evaluation of linkage disequilibrium (LD)
among the methylated sites
LD among the methylated sites was estimated using
the software TASSEL. Pair wise LD (r2, P and D0)were estimated. The loci pairs were considered to be
in significant LD if P \ 0.01 or r2 [ 0.5. As the
methylation sites were not known for its chromo-
somal location, intra/inter-chromosomal LD could
not be estimated.
Results
Methylation profiles in watermelon heirlooms
DNA methylation profiles were generated using MSA
assays for 47 watermelon heirlooms and one single
accession of the species Citrullus colocynthis (Table 1).
A CCGG site for a particular heirloom was classified as
‘‘methylated’’ if a band was uniquely present in the MspI
or HpaII lane but not in both (Fig. 1). To facilitate
comparison, two lanes containing amplicons of both
isoschizomers (EcoRI/HpaII and EcoRI/MspI) of a
single heirloom were loaded next to each other in the
gels. A total of twelve primer combinations were used in
both isoschizomer orientations. The primer combina-
tions EAAC/HAC, EACT/HCT, EACC/HCT, EACA/
HCA, EACA/HAC, EACG/HAC, EAGC/HAC,
EAGC/HCA, EACA/HCC, EAAG/HGC, EAAC/
HGC, and EACG/HCA amplified a total number of
17, 33, 22, 21, 16, 32, 25, 29, 8, 21, 23, and 47 bands
respectively. Among the heirlooms, Peacock had 8
MspI-specific bands, whereas the heirlooms Sweet
Princess, Hawkesbury and Calhoun Gray each had 66
MspI-specific bands. For the HpaII, the heirlooms
Family Fun and Dunbarton had the least (35) and the
highest (69) number of bands. The heirloom Peacock
and the wild accession PI386016 of C.colocynthis had
the least number of total bands (HpaII ? MspI), 52 and
69 respectively. The heirloom Au-Golden Producer had
the highest number (125) of total bands. The heirlooms
Miles (120), All Sweet (121), Golden Honey (122),
Stone Mountain (124), and Northern Sweet (124) also
had a very high number of total bands. In the current
study, analysis was performed by separating the meth-
ylation datasets of HpaII and MspI, as the methylations
generated by MspI are from fully methylated sites and
the profiles of HpaII, are derivatives of hemi-
methylation.
Estimation of epigenetic diversity based
on methylation sites
The methylated bands that are polymorphic (MP) were
used to construct Neighbor-Joining (NJ) phenograms
82 Euphytica (2011) 177:79–89
123

to explore whether the patterns of genetic relationships
were consistent with those determined previously
(Levi et al. 2004). The first and the second analyses
included specific MPs of MspI and HpaII, respec-
tively, to compare the tree topologies of complete
methylation (MspI) versus hemi-methylation (HpaII).
Both the trees were rooted using C.colocynthis as the
outgroup. When estimated based on the MspI-specific
MPs, the range of genetic diversity among various
groups was within 16–43%. In contrast, GDs between
the groups ranged between 17–36% when estimated
based on MPs specific to HpaII. Subsequently, the
major clusters of the combined NJ phenogram were
supported with Bootstrap Values (BVs) in the range of
52–94%. The NJ phenogram made from HpaII-
specific MPs resolved into two clear clusters (Fig. 2).
However, the main split was not supported by BVs,
but internally the sub-clustering in cluster II showed
robust support with a BV range of 52–95%. The MspI-
specific MPs resolved into three different clusters, with
a BV range of 56–83% (Fig. 3). Interestingly, some of
the heirlooms that got clustered into two major groups
in the NJ tree constructed from HpaII-specific MPs
were seen hitchhiking into a third cluster in the NJ tree
of MspI.
To corroborate results of the diversity analysis, we
also carried out a PCA using the MPs of HpaII. The
first three eigenvectors (Vector I = 33.61, Vector
II = 9.34, Vector III = 6.21) that were used to
construct the PCA cumulatively absorbed variance
of 49.17, indicating good support of the analysis. This
PCA resolved into two clusters as in the NJ
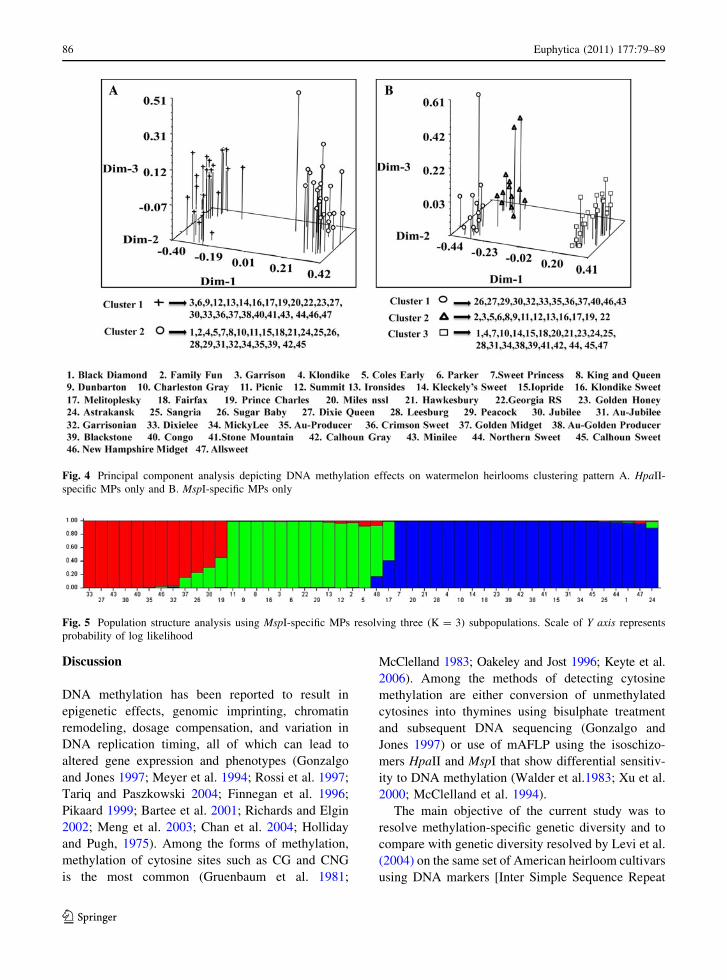
phenogram of HpaII-specific MPs. The clustering
pattern of the heirlooms can be noted in Fig. 4a. The
PCA constructed using the first three eigenvectors
(Vector I = 22.31, Vector II = 12.29, Vector
III = 8.49) derived from similarity indices of MspI-
specific MPs resolved into three distinct clusters
(Fig. 4b). The first cluster included the heirlooms
Sugar Baby, Dixie Queen, Peacock, Jubilee, Garriso-
nian, Dixilee, Au-Producer, Crimson Sweet, Golden
Midget, Congo, New Hampshire Midget, and
Minilee. The second cluster contained heirlooms
Family Fun, Garrison, Coles Early, Parker, King, and
Queen, Dunbarton, Picnic, Summit, Ironsides, Klon-
dike Sweet, Melitoplesky, Prince Charles, and Geor-
gia Rattlesnake. The heirlooms in cluster I and cluster
III of MspI-specific PCA were similar of cluster I and
II, respectively, of HpaII-specific PCA. The most
intriguing result of the current study is that the second
(middle) cluster of MspI-specific PCA contains
heirlooms from the first and second cluster PCA of
HpaII, thus confirming the shifting of heirlooms into
a new cluster due to methylations of MspI sites. It
clearly showed that the HpaII-specific hemi-methyl-
ated products could not resolve into the third cluster,
which was newly formed by using the fully methyl-
ated MPs of MspI. To further support this finding, a
model based population structure program was used
assuming (K = 2 to K = 6) using MPs of MspI to
resolve the clustering pattern induced by DNA
methylation effects. The analysis of K = 3 had a
highest mean alpha value of 0.13, which supports the
formation of three subpopulations in the current
study. Further, the analysis K = 3 had three clusters
with FSTs (population differentiation estimates) of
0.44, 0.63, and 0.29 respectively, indicating the
extent of divergence within individual clusters. The
analysis that involved higher K assumptions (more
than K = 3) was not informative and had a smaller
mean alpha. Allele frequency divergence (AFD) was
1.44 between the first and second cluster, and was
Fig. 1 DNA methylation profiles in a set of US watermelon heirlooms by methylation sensitive AFLP technique (H;HpaII, M;MspI)
Euphytica (2011) 177:79–89 83
123

0.84 between the first and third clusters. AFD
between the second and third clusters was also 1.44.
The first cluster of K = 3 analysis (Fig. 5) is in
total agreement with the first cluster of PCA of MspI
(Fig. 4b) except in the case of heirloom Peacock. The
cultivars that formed the second (middle) cluster in
K = 3 analysis were same as the middle cluster(II) in
the PCA of MspI with minor changes in the case of the
heirlooms Melitoplesky (#17)and Prince Charles
(#19) that showed shared ancestry between adjacent
clusters. In addition to population structure, LD is
another important feature that deals with the non-
random association of alleles at different loci within a
population. LD parameters r2 and D0 were estimated
among the methylation sites and significant LDs
(P \ 0.01) are presented in Fig. 6. Our results showed
that 71 MP pairs had strong LD that is r2 above 0.5.
Average r2 for all the genome-wide methylation
polymorphisms (6724 MP pairs) was 0.08 indicating
genome wide distribution of methylation.
Inheritance and stability of methylation specific
sites across generations
Based on breeding histories and pedigree records, we
selected four sets of heirlooms that had parental
heirlooms and derivative progenies. We compared
methylation sites present in parents and the extent of
methylation that transmitted across generations to
progeny and further impacted the varietal evolution.
From the MPs dataset, the proportion of methylations
that are not transmitted or lost in the progeny and the
Fig. 2 Phenogram obtained with HpaII-specific methylation polymorphisms. The numbers adjacent to some nodes indicate bootstrap
confidence values (1,000 bootstrap replicates)
84 Euphytica (2011) 177:79–89
123

extent of unique methylations accumulated in the
progeny heirloom could be identified. Details of
stably inherited MPs, methylations that are lost or not
carried (nc), and de novo methylation sites or unique
sites (u) are presented in Fig. 7. Our genealogy
analysis of MPs clearly indicated the proportions of
shared MPs, unique or de novo methylations and the
methylations that are lost in varietal evolution or
history.
Heirloom Ironside is a derivative of a three way
cross [Leesburg (l) 9 Hawkesbury (h)] 9 Garrison
(Table 1). A majority of MPs (70%) were stably
inherited by the heirloom Ironside. A set of 30% of
MPs were not transmitted or lost in the process of
varietal evolution of Ironside. 0.7% of the total MPs
in Ironside were de novo in nature. In breeding
histories of the heirlooms Crimson Sweet and All
Sweet, which were selected from the recombinants
of a cross [Miles (m) 9 Peacock (p)] 9 Charleston
Gray, only 17% of MPs could not be traced when
compared to the MPs that were present in the
parental genomes. A set of 6% MPs were collec-
tively de novo or unique in the heirlooms Crimson
Sweet and All Sweet. In individual breeding histo-
ries of Blackstone and Miles, a majority of *90%
of MPs was stably inherited and only a small set of
MPs were de novo or unique (Fig. 7). Our results
clearly indicate that the proportions of unique or de
novo methylations and of methylations that are
present in parental heirlooms and not inherited or
lost are lower than the proportion of methylations
that are stably inherited.
Fig. 3 Phenogram obtained with MspI-specific methylation polymorphisms. The numbers adjacent to some nodes indicate bootstrap
confidence values (1,000 bootstrap replicates)
Euphytica (2011) 177:79–89 85
123
Discussion
DNA methylation has been reported to result in
epigenetic effects, genomic imprinting, chromatin
remodeling, dosage compensation, and variation in
DNA replication timing, all of which can lead to
altered gene expression and phenotypes (Gonzalgo
and Jones 1997; Meyer et al. 1994; Rossi et al. 1997;
Tariq and Paszkowski 2004; Finnegan et al. 1996;
Pikaard 1999; Bartee et al. 2001; Richards and Elgin
2002; Meng et al. 2003; Chan et al. 2004; Holliday
and Pugh, 1975). Among the forms of methylation,
methylation of cytosine sites such as CG and CNG
is the most common (Gruenbaum et al. 1981;
McClelland 1983; Oakeley and Jost 1996; Keyte et al.
2006). Among the methods of detecting cytosine
methylation are either conversion of unmethylated
cytosines into thymines using bisulphate treatment
and subsequent DNA sequencing (Gonzalgo and
Jones 1997) or use of mAFLP using the isoschizo-
mers HpaII and MspI that show differential sensitiv-
ity to DNA methylation (Walder et al.1983; Xu et al.
2000; McClelland et al. 1994).
The main objective of the current study was to
resolve methylation-specific genetic diversity and to
compare with genetic diversity resolved by Levi et al.
(2004) on the same set of American heirloom cultivars
using DNA markers [Inter Simple Sequence Repeat
Fig. 4 Principal component analysis depicting DNA methylation effects on watermelon heirlooms clustering pattern A. HpaII-
specific MPs only and B. MspI-specific MPs only
Fig. 5 Population structure analysis using MspI-specific MPs resolving three (K = 3) subpopulations. Scale of Y axis represents
probability of log likelihood
86 Euphytica (2011) 177:79–89
123

makers (ISSR) and AFLP markers]. In the current
study, epigenetic diversity was noted to be 16–43%
among the cultivated watermelons, in contrast to
3.2–19.8% of genetic diversity estimated using con-
ventional DNA markers by Levi et al. This study
revealed that the diversity at the methylation level is
three times higher than the genetic diversity revealed
by DNA markers on the same set of heirloom DNAs.
Our results showed that DNA methylation is wide-
spread within the cultivated watermelon germplasm
and the proportion of unique or de novo methylations
and the methylations that are not carried over or lost
are proportionately smaller, when compared to the
methylations that are stable and shared among the
parental heirlooms and their progeny.
Ellul et al. (2007), Wehner et al. (2008) and Parris
(1949) reported extensive phenotypic variation for
fruit characters such as shape, color, size, rind
thickness, flesh color, flesh texture, flesh flavor, sugar
content, size, shape and color of the seeds, as well as
plant characteristics such as vigor, earliness, prolif-
icacy, resistance to diseases, insects and/or mechan-
ical injury, stress tolerance, fruit setting tendency and
ecological adaptation. Systematic analyses involving
transmission genetics and QTL location will provide
direct evidence on how these methylation specific
polymorphisms impact phenotypic diversity.
Fig. 6 Significant LD among 426 MP pairs
Fig. 7 Pattern of inheritance and stability of methylated specific polymorphism across the genealogies and breeding histories
Euphytica (2011) 177:79–89 87
123

There are comparable studies from other plants
such as in Arabidopsis (Cervera et al. 2002; Riddle
and Richards 2002), rice (Ashikawa 2001; Wang et al.
2004; Sakthivel et al. 2010), Pisum (Knox and Ellis
2001) and cotton (Keyte et al. 2006) suggesting that
the MPs are widespread among plants and can serve as
epigenetic markers for use in plant breeding. Data on
the influence of DNA methylation on QTLs and the
potential use of epigenetic markers for QTL analysis
is on hand for humans (Gibbs et al. 2010). Similar
mechanisms may be the secret for the high degree of
phenotypic variation known to be present among the
American heirloom watermelon cultivars despite their
derivation from a narrow genetic base. The explora-
tion of cultivated watermelon diversity is truly
dynamic and requires investigations at all levels of
the molecular spectrum, from sequence variations to
epigenetic to functional analyses. It seems likely that
additional insights will continue to emerge from the
interplay between structural, functional genomics, and
epigenetics, along with the phenotypic potential and
selection dynamics of these heirlooms in plant
breeding programs.
The information on epigenetic diversity in culti-
vated watermelon revealed in the current study will
further contribute to our understanding of phenotypic
plasticity of watermelon heirlooms across ecological
zones. Higher and significant LD among the meth-
ylation sites may indicate the existence of some
common methylation control mechanism among
these sites. This is the first study that undertook LD
analysis among methylation sites. When we used the
MP sites in the current study for LD-based associ-
ation mapping of various phenotypic trait variation in
these heirlooms, a set of MP sites were identified that
are significantly linked to the QTLs with wide
ranging effects of various fruit traits in watermelon
heirlooms (Data unpublished).
Results pertaining to identification of shared and
de novo methylations across genealogies helped to
understand their pace in varietal evolution, or the
level of stability of methylations within the same
generation and subsequent generations. These results
provide clear evidence that methylation patterns are
highly heritable across watermelon breeding histories
and hence will be of great interest to watermelon
breeders. More generally, our demonstration of stable
inheritance of numerous epi-alleles over many gen-
erations highlights the need to integrate epigenetic
information into genetic studies. In support of our
results, Peng and Zhang (2009) confirmed that plants
are known to accumulate and memorize the methyl-
ation modifications and further faithfully transmit
them to progeny. The findings of Johannes et al.
(2009) recently provided the first rationale to identify
epi-allelic variants that contributed to heritable
variation in complex traits.
Conclusions
A natural extension of our study would be to examine
the effects of methylation on gene expression.
Although PCA results presented here suggest patterns
of methylation diversity and hitchhiking, mapping of
these patterns could only identify how these methy-
lations alter phenotypic variation. Our immediate
endeavor is to further validation by isolating individ-
ual MPs through extensive gel elution and sequencing
for methylated PCR that uses bisulfite-treated DNAs.
Acknowledgments The authors are grateful to Drs. Vickie
Woolf, Clint Magill and Gerry Hankins for their critical
comments. Funding support is provided by USDA-CSREES
Research (Grant #2007-38814-18472).
References
Ashikawa I (2001) Surveying CpG methylation at 50-CCGG in
the genomes of rice cultivars. Plant Mol Biol 45:31–39
Attwood JT, Young RL, Richardson BC (2002) DNA meth-
ylation and the regulation of gene transcription. Cell Mol
Life Sci 59:241–257
Bartee L, Malagnac F, Bender J (2001) Arabidopsis cmt3 chro-
momethylase mutations block non-CG methylation and
silencing of an endogenous gene. Genes Dev 15:1753–1758
Cervera MT, Ruiz-Garcıa L, Martınez-Zapater JM (2002)
Analysis of DNA methylation in Arabidopsis thalianabased on methylation-sensitive AFLP markers. Mol Genet
Genomics 268:543–552
Chan SWL, Zilberman D, Xie Z, Johansen LK, Carrington JC,
Jacobsen SE (2004) RNA silencing genes control de novo
DNA methylation. Science 303:1336
Ellul P, Lelivelt C, Naval MM, Nogueral FJ, Sanchez S, Atares
A, Moreno V, Corella P, Dirks R (2007) Watermelon bio-
technology, In: agriculture and forestry, transgenic crops,
vol 60. Springer Verlag publication, Berlin pp 129–165
Finnegan EJ, Peacock WJ, Dennis S (1996) Reduced DNA
methylation in Arabidopsis thaliana results in abnormal
plant development. Proc Natl Acad Sci USA 93:8449–8454
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls
MA et al (2010) Abundant quantitative trait loci exist for
DNA methylation and gene expression in human brain.
88 Euphytica (2011) 177:79–89
123

PLoS Genet 6(5):e1000952. doi:10.1371/journal.pgen.
1000952
Gonzalgo ML, Jones PA (1997) Rapid quantification of
methylation at specific sites using methylation-sensitive
single nucleotide primer extension (Ms-SNuPE). Nucleic
Acids Res 25:2529–2531
Grant V (1971) Plant Speciation. Columbia University Press,
New York
Gruenbaum Y, Naveh-Many T, Cedar H, Razin A (1981)
Sequence specificity of methylation in higher plant DNA.
Nature 292:860–862
Heslop-Harrison JS (2000) Comparative genome organization
in plants: from sequence and markers to chromatin and
chromosomes. Plant Cell 12:617–635
Holliday R, Pugh JE (1975) DNA modification mechanisms and
gene activity during development. Science 187:226–232
Jaccard P (1908) Nouvelles recherches sur la distribution flo-
rale. Bul Soc Vaudoise Sci Nat 44:223–270
Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V,
Simon M et al (2009) Assessing the impact of transgen-
erational epigenetic variation on complex traits. PLoS
Genet 5(6):e1000530. doi:10.1371/journal.pgen.1000530
Karp A, Seberg O, Buiatti M (1996) Molecular techniques in the
assessment of biological diversity. Ann Bot 78:143–149
Keyte Al, Percifield R, Liu B, Wendel JF (2006) Infraspecific
DNA methylation polymorphism in cotton (Gossypiumhirsutum L.). J Hered 97:445–446
Knox MR, Ellis THN (2001) Stability and inheritance of
methylation states at PstI sites in Pisum. Mol Genet
Genomics 265:497–507
Levi A, Thomas CE, Wehner TC, Zhang X (2001) Low genetic
diversity indicates the need to broaden the genetic base of
cultivated watermelon. J Am Soc Hort Sci 36:1096–1101
Levi A, Thomas CE, Newman M, Reddy UK, Zhang X, Xu Y
(2004) ISSR and AFLP markers differ among American
watermelon cultivars with limited genetic diversity. J Am
Soc Hort Sci 129:553–558
Liu B, Brubaker CL, Mergeai G, Cronn RC, Wendel JF (2001)
Polyploid formation in cotton is not accompanied by rapid
genomic changes. Genome 44:321–330
Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J
(2002) Remodeling of DNA methylation and phenotypic
and transcriptional changes in synthetic Arabidopsis al-lotetraploids. Plant Physiol 129:733–746
McClelland M (1983) The frequency and distribution of
methylatable DNA sequences in leguminous plant protein
coding genes. J Mol Evol 19:346–354
McClelland M, Nelson M, Raschke E (1994) Effect of site-
specific modification on restriction endonucleases and
DNA modification methyltransferases. Nucleic Acids Res
22:3640–3659
Meng L, Bregitzer P, Zhang S, Lemaux PG (2003) Methylation
of the exon/intron region in the ubi1 promoter complex
correlates with transgene silencing in barley. Plant Mol
Biol 53:327–340
Messeguer R, Ganal MW, Stevens JC, Tanksley SD (1991)
Characterization of the level, target sites and inheritance
of cytosine methylation in tomato nuclear DNA. Plant
Mol Biol 16:753–770
Meyer P, Niedenhof I, Lohuis M (1994) Evidence for cytosine
methylation of non-symmetrical sequences in transgenic
Petunia hybrida. EMBO J 13:2084–2088
Oakeley EJ, Jost JP (1996) Nonsymmetrical cytosine methyl-
ation in tobacco pollen DNA. Plant Mol Biol 31:927–930
Parris GK (1949) Watermelon breeding. Econ Bot 3:193–212
Peng H, Zhang J (2009) Plant genomic DNA methylation in
response to stresses: potential applications and challenges
in plant breeding. Prog Nat Sci 19:1037–1045
Pikaard CS (1999) Nucleolar dominance and silencing of
transcription. Trends Plant Sci 4:478–483
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000)
Association mapping in structured populations. Am J
Hum Genet 67:170–181
Richards EJ, Elgin SC (2002) Epigenetic codes for hetero-
chromatin formation and silencing: rounding up the usual
suspects. Cell 108:489–500
Riddle NC, Richards EJ (2002) The control of natural variation in
cytosine methylation in Arabidopsis. Genetics 162:355–363
Rideout WG 3rd, Coetzee GA, Olumi AF, Jones PA (1990) 5-
Methylcytosine as an endogenous mutagen in the human
LDL receptor and p53 genes. Science 249:1288–1290
Rohlf FJ (1994) NTSYS-pc: Numerical taxonomy and multivari-
ate analysis system, ver. 1.80. Exeter Software, Setauket
Rossi V, Motto M, Pelligrini L (1997) Analysis of the meth-
ylation pattern of the maize Opaque-2 (O2) promoter and
in vitro binding studies indicate that the O2 B-zip protein
and other endosperm factors can bind to methylated target
sequences. J Biol Chem 272:13758–13765
Saitou W, Nei M (1987) The neighbor-joining method: a new
method for reconstructing phylogenetic trees. Molec Biol
Evol 4:406–425
Sakthivel K, Girishkumar K, Ramkumar G, Shenoy VV, Ka-
jjidoni T, Salimath PM (2010) Alterations in inheritance
pattern and level of cytosine DNA methylation, and their
relationship with heterosis in rice. Euphytica 175:303–314
Shimizu TS, Takahashi K, Tomita M (1997) CpG distribution
patterns in methylated and non-methylated species. Gene
205:103–107
Tariq M, Paszkowski J (2004) DNA and histone methylation in
plants. Trends Genet 20:244–251
Walder RY, Langtimm C, Chatterjee J, Walder JA (1983)
Cloning of the MspI modification enzyme. The site of
modification and its effects on cleavage by MspI and
HpaII. J Biol Chem 258:1235–1241
Wang JL, Tian A, Madlung H, Lee S, Chen M (2004) Sto-
chastic and epigenetic changes of gene expression in
Arabidopsis polyploids. Genetics 167:1961–1973
Wehner TC (2008) Watermelon. In: Prohens J, Nuez F (eds)
Handbook of plant breeding; vegetables I: Asteraceae,
Brassicaceae, Chenopodiaceae, and Cucurbitaceae.
Springer, New York, pp 381–418
Wendel JF (2000) Genome evolution in polyploid. Plant Mol
Biol 42:225–249
Xu M, Li X, Korban S (2000) AFLP-based detection of DNA
methylation. Plant Mol Biol 18:361–368
Zhang X et al (2006) Genome-wide high-resolution mapping
and functional analysis of DNA methylation in Arabid-opsis. Cell 126:1189–1201
Euphytica (2011) 177:79–89 89
123




















