
Early diagenesis and organic matter preservation — a molecular stable carbon isotope perspective
15
ELSEVIER Chemical Geology 114 (1994) 365-379 CHEMICAL GEOLOGY ISOTOPE (;EOS('IE.VCE Early diagenesis and organic matter preservation --- a molecular stable carbon isotope perspective Stephen A. Macko a, Michael H. Engel b, Yaorong Qian b "Department of Environmental Sciences, Universityof Virginia, Charlottesville, VA 22903, USA bSchool Geologyand Geophysics, Universityof Oklahoma, Norman, OK 73019, USA (Received June 22, 1993; revision accepted November 6, 1993) Abstract Through a new development in stable isotope capability, gas chromatography coupled to a stable isotope ratio mass spectrometer (GC-C-IRMS), the reaction pathways of organic matter diagenesis can potentially be traced by analysis of the isotopic compositions of individual molecular components. In this sludy, changes in bulk stable carbon and nitrogen isotope compositions, amino acid contents and carbon isotope compositions of individual amino acids are reported for seagrass and sediment degradation experiments using isotopically resolvable organic substrates. Seagrass showed slight enrichments in d lSN ( + 0.4%o) with little change in d13C following 4 weeks of decomposition. During that period, the identifiable amino acid content decreased by .~ 50% for each amino acid. Mixtures of marine sediment with the same seagrass showed enrichments in 15N ( + 2.3%0) with associated deple- tions in carbon ( - 2.8%o) isotopic compositions over the same time span. These changes are attributed to hydrol- ysis, deamination, decarboxylation and condensation reactions. Control experiments on the sediments without added fresh seagrass showed no change in isotopic content. At the molecular level, using GC-C-IRMS, certain amino acids in the seagrass (e.g., Glu, Ala, Val, Gly) are seen to decrease in t3C content during decomposition whereas others remained constant (e.g., Leu, Ile, Pro) or became increasingly enriched in 13C (Asp). The molec- ular isotope approach indicates that the process of early diagenesis, leading to the eventual residual materials which are preserved, is significantly more complex than simple breakdown and loss. A portion of the organic matter consequently preserved in organic-rich deposits can be attributed to new production during early diagenetic stages. 1. Introduction Detailed chemical and isotopic characteriza- tion offers the potential for the discovery of a particular source of an organic material or inter- pretation of its diagenetic history. Although many studies have reported the changes which occur in organic molecules through diagenesis, it has re- mained difficult to identify whether the compo- nent being quantified is indigenous or a product of reactions following deposition of the organic matter. Such detailed chemical characteriza- tions have enabled investigators to suggest, for example, specific algal or bacterial sources for the isolated material (Klok, 1984; Sigleo and Macko, 1985) or processes such as organic polymeriza- tion (Nissenbaum and Kaplan, 1972; Henrichs and Williams, 1985). However, even with de- tailed analyses, the conclusions are often, at best, based on correlations in the data or geological stratigraphy (Drozdova, 1973; Orem and Hatcher, 1987), lacking direct evidence of the 0009-2541/94/$07.00 © 1994 Elsevier Science B.V. All rights reserved SSDI 0009-2541 ( 93 ) E0259-V
Transcript of Early diagenesis and organic matter preservation — a molecular stable carbon isotope perspective

ELSEVIER Chemical Geology 114 (1994) 365-379
CHEMICAL GEOLOGY
ISOTOPE (;EOS('IE.VCE
Early diagenesis and organic matter preservation --- a molecular stable carbon isotope perspective
Stephen A. Macko a, Michael H. Engel b, Yaorong Qian b
"Department of Environmental Sciences, University of Virginia, Charlottesville, VA 22903, USA bSchool Geology and Geophysics, University of Oklahoma, Norman, OK 73019, USA
(Received June 22, 1993; revision accepted November 6, 1993)
Abstract
Through a new development in stable isotope capability, gas chromatography coupled to a stable isotope ratio mass spectrometer (GC-C-IRMS), the reaction pathways of organic matter diagenesis can potentially be traced by analysis of the isotopic compositions of individual molecular components. In this sludy, changes in bulk stable carbon and nitrogen isotope compositions, amino acid contents and carbon isotope compositions of individual amino acids are reported for seagrass and sediment degradation experiments using isotopically resolvable organic substrates. Seagrass showed slight enrichments in d lSN ( + 0.4%o) with little change in d13C following 4 weeks of decomposition. During that period, the identifiable amino acid content decreased by .~ 50% for each amino acid. Mixtures of marine sediment with the same seagrass showed enrichments in 15N ( + 2.3%0) with associated deple- tions in carbon ( - 2.8%o) isotopic compositions over the same time span. These changes are attributed to hydrol- ysis, deamination, decarboxylation and condensation reactions. Control experiments on the sediments without added fresh seagrass showed no change in isotopic content. At the molecular level, using GC-C-IRMS, certain amino acids in the seagrass (e.g., Glu, Ala, Val, Gly) are seen to decrease in t3C content during decomposition whereas others remained constant (e.g., Leu, Ile, Pro) or became increasingly enriched in 13C (Asp). The molec- ular isotope approach indicates that the process of early diagenesis, leading to the eventual residual materials which are preserved, is significantly more complex than simple breakdown and loss. A portion of the organic matter consequently preserved in organic-rich deposits can be attributed to new production during early diagenetic stages.
1. Introduction
Detailed chemical and isotopic characteriza- tion offers the potential for the discovery of a particular source of an organic material or inter- pretation of its diagenetic history. Although many studies have reported the changes which occur in organic molecules through diagenesis, it has re- mained difficult to identify whether the compo- nent being quantified is indigenous or a product of reactions following deposition of the organic
matter. Such detailed chemical characteriza- tions have enabled investigators to suggest, for example, specific algal or bacterial sources for the isolated material (Klok, 1984; Sigleo and Macko, 1985) or processes such as organic polymeriza- tion (Nissenbaum and Kaplan, 1972; Henrichs and Williams, 1985). However, even with de- tailed analyses, the conclusions are often, at best, based on correlations in the data or geological stratigraphy (Drozdova, 1973; Orem and Hatcher, 1987), lacking direct evidence of the
0009-2541/94/$07.00 © 1994 Elsevier Science B.V. All rights reserved SSDI 0009-2541 ( 93 ) E0259-V

366 S.A. Macko et aL / Chemical Geology 114 (1994) 365-379
specific source or diagenetic pathways of the molecules studied (Degens and Mopper, 1976; Mopper, 1977; Burdige and Martens, 1988 ).
Stable isotope compositions (~13C, ~SN) of the bulk organic material have improved the perspective of the overall diagenetic process, but also have limited resolution on the multitude of reactions occurring during diagenesis. Geopoly- mers tend to be depleted in ~3C relative to the organic matter from which they form (Nissen- baum et al., 1972; Nissenbaum, 1974; Galimov, 1980). Abelson and Hoering (1961) reported that the carboxyl groups of amino acids are more enriched in ~3C relative to the remaining carbon constituents in these molecules. It has been spec- ulated that the loss of ~3C-enriched carboxyl groups during early diagenesis could in part ac- count for the depletion in 13C observed in resid- ual organic materials (Nissenbaum et al., 1972; Nissenbaum, 1974; Galimov, 1980).
The diagenesis of organic matter in sediments is thought to proceed via a series of chemical and microbially mediated reactions that result in losses from the system (Martens et al., 1992) or in the formation ofgeomacromolecules which in turn are transformed into kerogen (Nissenbaum and Schallinger, 1974; Welte, 1974). The recog- nition of what is preserved in a sedimentary de- posit, beyond establishment of the refractory or labile nature of the component, has been an ex- ceedingly arduous task. This is especially true if one considers that compounds with exactly the same chemical structure as those found in the original material that was deposited may in fact be produced by processes or organisms during diagenesis. The possibility of resolving such dif- ferent sources for a component could lie in the assessment of the isotopic composition of the compound, if the original source material is iso- topically distinguishable from that which is newly produced. If differences do exist, compound spe- cific isotope analysis could furnish a way of not only identifying the different sources, but also quantifying them through mass-balance equations.
Recently, there has been developed an in- creased capability for such an approach through the use of advanced analytical methods in con-
junction with a technique often used in the anal- ysis of bulk organic materials: stable isotopic compositions. The initial studies have yielded unique information regarding the source and history of the compounds isotopically character- ized (Summons and Powell, 1986; Hayes et al., 1987; Macko et al., 1987). Through the recent technological advances, gas chromatographic (GC) effluents can be combusted (C) and the resulting carbon dioxide directly introduced into the stable isotope ratio mass spectrometer (IRMS). This modification, GC-C-IRMS, al- lows for rapid analysis of the carbon isotopes of components in a mixture as well as increased sensitivity, on the order of 0.5 nmol of each com- pound (Freedman et al., 1988). Gas chromato- graphic-based systems are presently constrained by the volatility of the components investigated. Compounds which do not have this constraint include hydrocarbons (Hayes et al., 1990), the analyses of which have already clearly demon- strated the power of such technology and mea- surements in the assessments of source (Prahl et al., 1992) and the history of organic materials (Kohnen et al., 1992; Sinninghe Damst6 et al., 1993; Wakeham et al., 1993).
Multifunctional molecules, which include amino acids, carbohydrates and fatty acids are nonvolatile, and thus require derivatization prior to gas chromatographic analysis to increase vol- atility. Through derivatization, additional car- bon is thus added to the parent compound. This addition, as well as fractionations associated with derivatization procedures involving bond rup- ture and formation in, for example, esterifica- tion and acylation (Silfer et al., 1991 ) need to be corrected for in order to ascertain the original isotopic compositions of the compound. The original carbon isotopic compositions of indi- vidual amino acids and their stereoisomers (Sil- fer et al., 1991 ), carbohydrates (Macko et al., 1992) and fatty acids (Abrajano et al., 1992) have been computed, however, through analysis of standards prepared in a similar fashion. The isotopic compositions of individual amino acids have been studied, with pathways for nitrogen metabolism (Macko et al., 1987), comparative biochemistry in fossil materials (Hare and Es-

S.A. Macko et al. /Chemical Geology 114 (1994) 365-3 79 367
tep, 1983) and indigeneity in fossils (Engel and Macko, 1984, 1986; Serban et al., 1988) and ex- traterrestrial materials (Engel et al., 1990) being elucidated.
Presented here is a new perspective on the pro- cess of early diagenesis of organic matter through the investigation of changes in both the concen- trations and the stable isotope abundances of in- dividual amino acids. This paper reports the re- suits of a laboratory simulation of early diagenesis using natural, isotopically resolvable substrates. Samples from these experiments were analyzed for variations in bulk organic carbon and nitrogen isotopic composition, the amino acid contents and their isotopic compositions. Through the isotopic label, the sources and his- tory of amino acids can potentially be traced during the decomposition of organic matter. Further, production of non-indigenous amino acids, as suggested through more depleted iso- topic compositions, may be associated with bac- terial action.
2. Methods
Samples of a seagrass, Halodule wrightii, from the Laguna Madre, Texas, U.S.A., were washed of any surface epiphytic growth, freeze-dried and ground to a fine powder. A surficial, carbonate- rich sediment sample from Baffin Bay, Texas, was freeze-dried and ground to a powder. These sub- strates for the experiment were chosen because previous isotope analyses on the organic fraction had shown that these two materials were isotop- ically distinguishable (Table 1, week 0).
Fifteen individual 2-cm diameter by 20 cm in length Winogradsky columns were prepared us- ing the following substrates (five columns each of the three substrates): the seagrass mixed with filtered (0.45 pro) seawater with a salinity of ~ 20%o (H series of samples); the sediment slur- ried with the same seawater sample (M series of samples); a combination (C series) of the sea- grass and the sediment on a 1 g:50 g dry weight basis which was also slurried with the filtered seawater. Winogradsky columns are a standard microbiological tool used to enrich for a wide va-
riety of procaryotes, especially phototrophic an- aerobes (Brock and Madigan, 1991 ). The sepa- rate columns were stopped on a weekly basis by freezing the column.
Samples of the plant materials and sediments from the columns were characterized with re- spect to their amino acid compositions after hy- drolysis with quartz-distilled 6 N HC1 in tubes that were sealed under N2 and heated at 100 °C for 24 hr. An aliquot of the acid was diluted with quartz-distilled water for injection into an amino acid analyzer. Separation and quantification of the amino acids was done by high-performance liquid chromatography (HPLC) using ion ex- change with post column derivatization with or- tho-phthaldialdehyde (OPA) and fluorescence detection (Hare, 1972 ). A cation-exchange 5-/lm resin column was used with stepwise isocratic elution (St. John Assoc., Beltsville, Maryland, U.S.A.). Replicate analyses on these samples typically showed concentration differences of less than 10%, and often better than 5%.
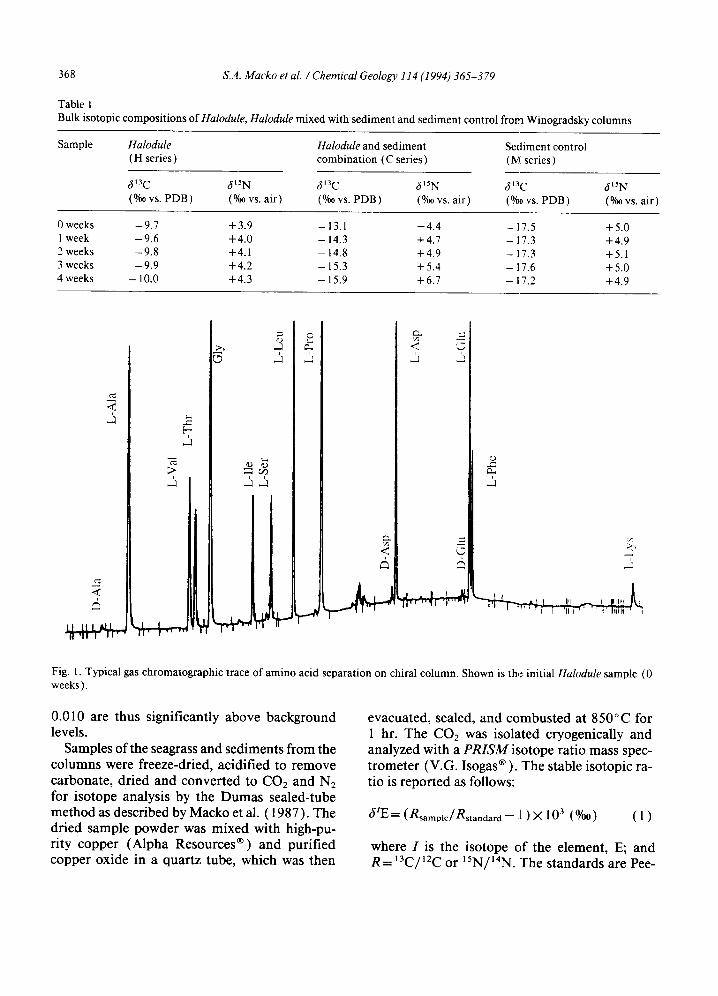
For stereoisomer characterization of the amino acids, a portion of the acid hydrolyzate was passed through Metricel ® DM-450 (0.45 pm) filters, and desalted on a Biorad ® AG50-X8 (50- 100 mesh, hydrogen form) cation-exchange col- umn. An aliquot of the desalted extract was de- rivatized to N,O-trifluoroacetyl (TFA) isopro- pyl esters (Engel and Hare, 1985 ) to facilitate the separation of the stereoisomers by gas chro- matography (GC). Separation was achieved us- ing a Hewlett Packard ® .5890 GC equipped with a 50 m×250/~m (i.d.) fused silica capillary col- umn coated with an optically active stationary phase (Chirasil-Val III® ), and detected with a nitrogen/phosphorous detector with ratio esti- mates made from the integration values ob- tained for the peaks with a Hewlett Packard ® 3396A integrator (Fig. 1 ). The GC analyses for the D /L values have errors derived from repli- cate analyses of better than ___0.004. Whereas hydrolysis of natural samples can induce slight racemization, standard L-enantiomers which had been hydrolyzed and derivatized through the above procedure have no detectable D-enan- tiomers present. The D / L values for the natural samples in this present study which are above
368 S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
Table 1
Bulk isotopic compositions of Halodule, Halodule mixed with sediment and sediment control from Winogradsky columns
Sample Halodule Halodule and sediment Sediment control (H series) combination (C series) (M series)
~13 C ¢~15 N 513C 815 N 8J3C 815N
(%0 vs. PDB) (%0 vs. air) (%0 vs. PDB) (%0 vs. air) (%0 vs. PDB) (%0 vs. air)
0 weeks - 9 . 7 +3 .9 - 13.1 +4 .4 - 17.5 +5 .0 1 week - 9 . 6 +4 .0 - 14.3 +4 .7 - 17.3 +4 .9 2 weeks - 9 . 8 +4.1 - 14.8 + 4.9 - 17.3 + 5.1 3 weeks - 9 . 9 +4.2 - 15.3 +5 .4 - 17.6 +5 .0 4 weeks - 10.0 + 4.3 - 15.9 + 6.7 - 17.2 + 4.9
<
<
c5
E-
>
09
=
r~
=t:
11
Fig. 1. Typical gas chromatographic trace of amino acid separation on chiral column. Shown is the initial Halodule sample (0 weeks).
0.010 are thus significantly above background levels.
Samples of the seagrass and sediments from the columns were freeze-dried, acidified to remove carbonate, dried and converted to CO2 and N2 for isotope analysis by the Dumas sealed-tube method as described by Macko et al. (1987). The dried sample powder was mixed with high-pu- rity copper (Alpha Resources ® ) and purified copper oxide in a quartz tube, which was then
evacuated, sealed, and combusted at 850°C for 1 hr. The CO2 was isolated cryogenically and analyzed with a PRISM isotope ratio mass spec- trometer (V.G. Isogas ® ). The stable isotopic ra- tio is reported as follows:
~ I E = (Rsample/Rstandard - - 1 ) X 10 3 ( % o ) ( 1 )
where I is the isotope of the element, E; and R = 13C/12C or 15N/14N. The standards are Pee-

S.A. Macko et al. / Chemical Geology 114 (1994) 365-3 79 369
dee Belemnite limestone (PDB) for carbon and atmospheric N2 (air) for nitrogen, which are as- signed 0NE-values of 0.00/00. The reproducibility of the measurement is better than _+ 0.1%o for 013C and 0~SN. In the laboratory, the samples are commonly measured against tanks of carbon dioxide and nitrogen gases which have been cal- ibrated against NBS 22 and atmospheric N2.
Stable isotope compositions of the above amino acid TFA isopropyl esters were also as- sessed using the combined G C - C - I R M S system. In our laboratory, this system consists of a Hew- lett Packard ® 5890 gas chromatograph inter- faced to the PRISM stable isotope ratio mass spec- trometer through a combustion furnace at 850 °C and a water trap at - 100°C (Macko and Engel, 1991; Silfer et al., 1991 ). Separation of the amino acid TFA isopropyl esters for GC-C- IRMS analysis is identical to that described above, and the isotope compositions are reported as in the case of bulk measurements. Back-calculated iso- topic compositions of the individual amino acids (after correcting for the effects of carbon addi- tion and kinetic isotope effects during derivati- zation) have standard deviations which are 0.25+_0.06%o, based on replicate analyses of standard and natural samples (Silfer et al., 199 l, 1994).
3 . R e s u l t s
The columns containing the Halodule and the mixture of the Halodule with the sediment were reducing by the termination of the experiment, with both C4 and H4 having noticeable hydro- gen sulfide. The sediment and Halodule mixture were both black, consistent with colonization by sulfate-reducing bacteria, although no attempt was made to quantify or identify the strains that may have been present. It is also emphasized that differences were observed between parallel col- umns in the amount of coloration and thus mi- crobial colonization. These differences likely were the result of variations in the exposure of the columns to light, the homogeneity of the ini- tial substrates and variations in the population of organisms added to the column initially. Such
differences likely account for a portion of the ob- served fluctuations in later chemical and iso- topic measurements.
3.1. Bulk stable isotopes
The isotopic composition of the Halodule in the Winogradsky columns exhibited little change over the course of the experiment, increasing slightly in 0~SN from +3.9 to +4.4%0 and de- creasing in 013C from - 9 . 7 to -10.0%o. These changes are within the variation expected among natural samples. The sediment control samples also showed little variation and had no discern- ible trend in bulk isotopic content, averaging - 17.4_+ 0.16%0 for carbon and + 5.0 +_ 0.08%0 for nitrogen. The seagrass which was mixed with the sediment into a slurry showed significant changes in isotopic composition, having enrich- ments in 15N (+2.3%o) with associated deple- tions in carbon ( -2 .8%o) isotopic compositions over the 4-week time span (Table 1 ).
3.2. Amino acid concentrations
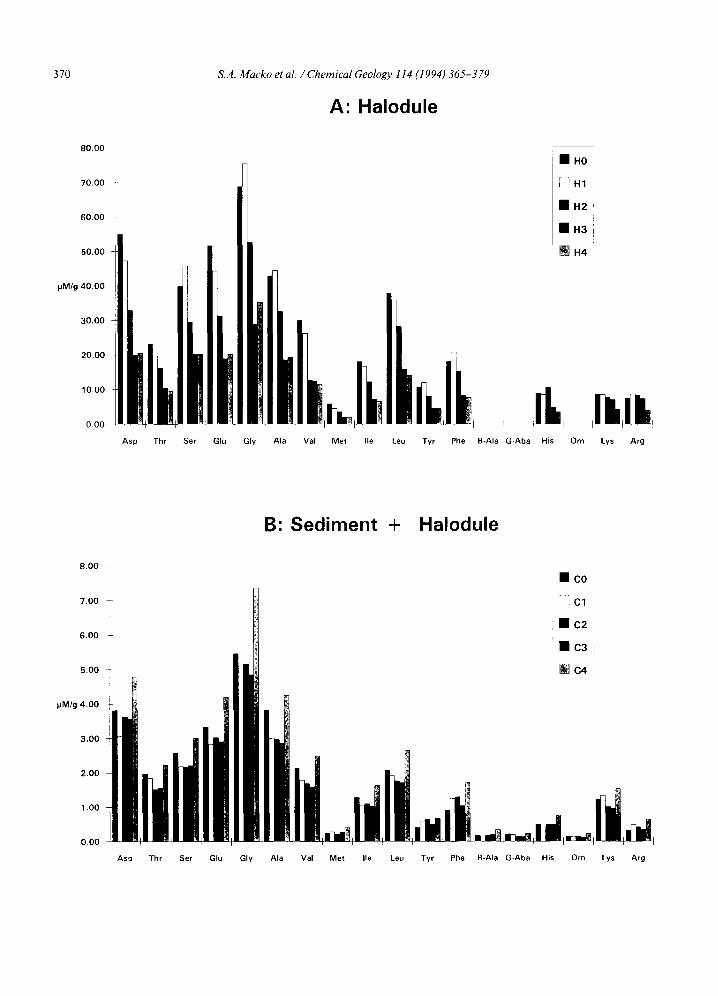
The seagrass lost ~50% of the identifiable amino acids over the time period of decomposi- tion on both an individual and total basis (Fig. 2A). In both the sediment control and in the combination seagrass-sediment decomposition columns, the amino acid levels observed were es- sentially the same (within the margin of replica- tion for these natural samples) as those found in the initial sediment (Fig. 2B and C), with excep- tion of the final mixed substrate (C4). In that column, the levels of Gly, Glu, Ala, Val and Asp were as much as one-third higher than at the start of the experiment, and are above the variation expected for sample replication. The amino acids added with the Halodule to the sediment slurry were essentially masked by the presence of indig- enous amino acids in the sediment. Ornithine, fl- alanine and 7-aminobutyric acid were detected in the sediment and in the sediment-seagrass combination, but were never observed at mea- surable levels in the isolated Halodule experiment.
3 7 0 S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
A: Halodule
80.00
• HO
70.00 i E~ H1
• H2 60.00
• H3
50.00 [ ] H4
pM/g 40.00
30.00
20.00
10.00
0.00 +
Asp Thr Ser Glu Gly Ala Val Met lie Leu Tyr Phe B-Ala G-Aba His Orn Lys Arg
lIJ
B" Sediment + Halodule
8.00 -
7.00
6.00
• co
ii)ci
• c2
• c~
° 5.00 C4
,uM/g 4.00 J
3.00
2.00
1.00
0.00
Asp Thr Ser Glu Gly Ala Val Met lie Leu Tyr Phe B-Ala G-Aba His Orn Lys Arg

S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
C" Sediment 371
6.00
= I i
5.00 -~
4.00
pM/g 3.00
2.00
1.00
0.00
• M0
~ ] M 1
• M2
• M3
• M4
Asp Thr Set Glu Gly Ala Val Met lie Leu Tyr Phe 6-Ala G-Aba His Orn Lys Arg
Fig. 2. Amino acid distributions of materials from Winogradsky columns (/tmol g-~ dry weight basis): (A) Halodule; (B) sediment and Halodule slurry; and (C) sediment slurry.
Table 2 Relative amounts of D-amino acid to L-amino acid for those amino acids with D-isomers above baseline detection (BD) for Halodule, Halodule mixed with sediment and sediment controls (values greater than 0.010 are significantly above BD)
Sample Ala Asp Glu Phe
Halodule (H series): 0 weeks 0.019 0.044 0.021 BD 1 week 0.007 0.043 0.019 BD 2 weeks 0.020 0.050 0.046 0.031 3 weeks 0.030 0.100 0.086 0.083 4 weeks 0.017 0.055 0.042 BD
Halodule and sediment combination (C series): 0 weeks 0.053 0.095 0.066 BD 1 week 0.062 0.108 0.117 BD 2 weeks 0.066 0.115 0.114 0.026 3 weeks 0.065 0.116 0.112 0.026 4 weeks 0.065 0.112 0.130 0.025
Sediment control (M series): 0 weeks 0.075 0.112 0.064 0.025 1 week 0.084 0.125 0.082 0.035 2 weeks 0.091 0.132 0.085 0.029 3 weeks 0.086 0.125 0.110 0.020 4 weeks 0.086 0.126 0.071 0.020
372 S.A. Macko et al. / C h e m i c a l G e o l o g y 114 (1994) 365-379
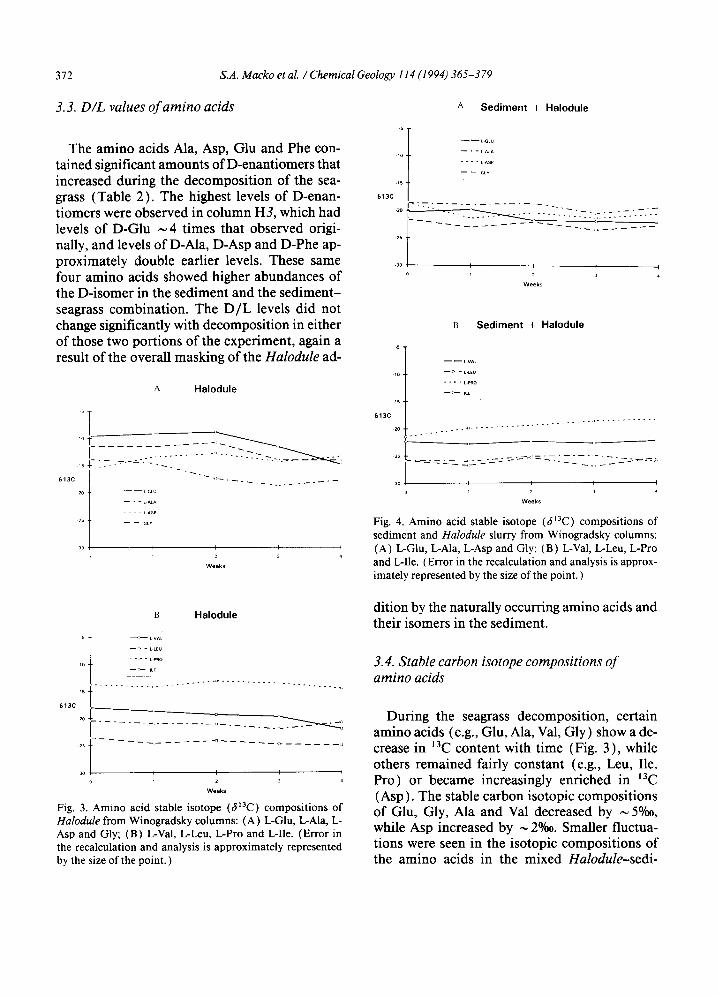
3.3. D/L values of amino acids
The amino acids Ala, Asp, Glu and Phe con- tained significant amounts of D-enantiomers that increased during the decomposition of the sea- grass (Table 2). The highest levels of D-enan- tiomers were observed in column H3, which had levels of D-Glu ~ 4 times that observed origi- nally, and levels of D-Ala, D-Asp and D-Phe ap- proximately double earlier levels. These same four amino acids showed higher abundances of the D-isomer in the sediment and the sediment- seagrass combination. The D/L levels did not change significantly with decomposition in either of those two portions of the experiment, again a result of the overall masking of the Halodule ad-
A Halodule
1 5
6 1 3 C
20
10 - " ~
. . . . . . . . . . . . . . . . . . . . . . ~ . " : - - ~ . ~ - - ~ ---_- . . . . - . - . . ~ .
- - - - t GLU
- - " - L . a L a
I I
2 3
Weeks
A Sediment + Halodule
~ - - L GLU
- - ' - L A t ~
. . . . . t ASP
- - ~ GLV
- lS
b 1 3 C
~o ~ : ---- 7 - - ~ L 2 5 . . . . . . . . . . . . . . . . . . . . . . . . . . . " - ~-~'~-:-L . . . . . . . . . . ::7-: . . . . . . . . . . . .
-3o I I ~ I
~ :~ 4
Weeks
B Sediment + Halodule
. s .
~ L .VAL
. 10 , - - c > - L4=EU
" "~ " t - ~O
- - ~ ILE
1 S
~ 1 3 C
-2O . . . . . . . ~' . . . . . . . . . . . . . " . . . . . .
3o I I I I
1 2 ~ 4
Weeks
Fig. 4. Amino acid stable isotope (c~3C) compositions of sediment and H a l o d u l e slurry from Winogradsky columns: (A) L-GIu, L-AIa, L-Asp and Gly; (B) L-Val, L-Leu, L-Pro and L-Ile. (Error in the recalculation and analysis is approx- imately represented by the size of the point. )
B Halodule
it ...... - - ~ - L.LEU
" "~ - t -PeO 1
- - 2 ILE
~S ~ - . . . . . . . . . . . . . . . . . . . . . . . . ~ . . . . . . . . . . . ~ . . . . . . . . . . .
! 8 1 3 C
3o I I I I
0 1 2 3 4
W e e k s
Fig. 3. Amino acid stable isotope (&~3C) compositions of H a l o d u l e from Winogradsky columns: (A) I.,-Glu, L-Ala, L- Asp and Gly; (B) L-Val, L-Leu, L-Pro and L-Ile. (Error in the recalculation and analysis is approximately represented by the size of the point. )
dition by the naturally occurring amino acids and their isomers in the sediment.
3.4. Stable carbon isotope compositions of amino acids
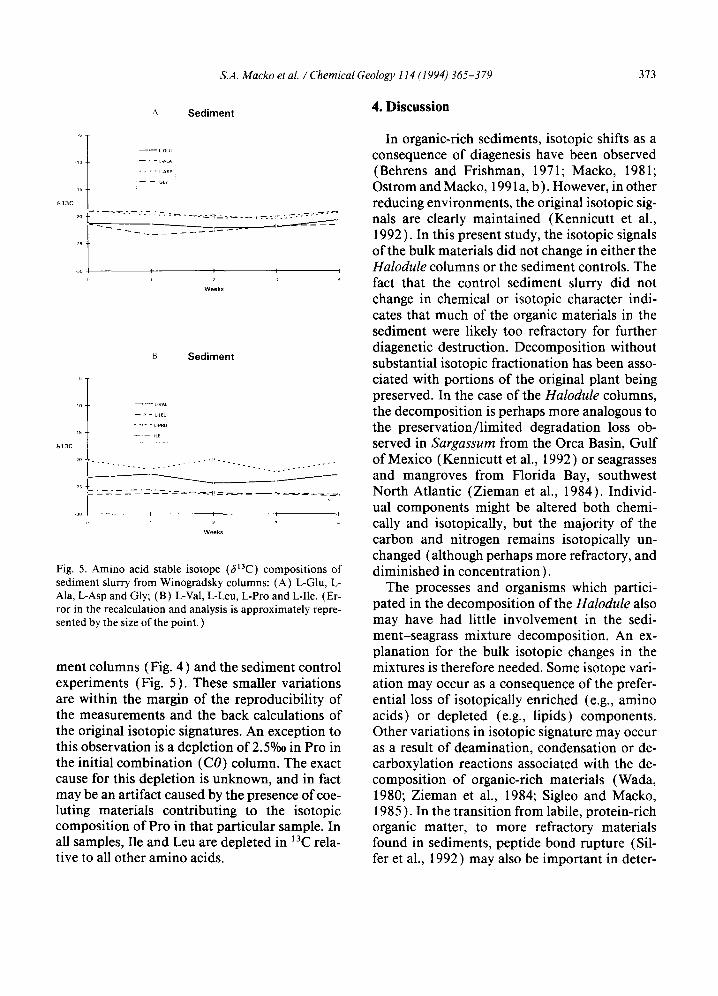
During the seagrass decomposition, certain amino acids (e.g., Glu, Ala, Val, Gly ) show a de- crease in 13C content with time (Fig. 3), while others remained fairly constant (e.g., Leu, Ile, Pro) or became increasingly enriched in laC (Asp). The stable carbon isotopic compositions of Glu, Gly, Ala and Val decreased by ~ 5°/oo, while Asp increased by ~ 2%o. Smaller fluctua- tions were seen in the isotopic compositions of the amino acids in the mixed Halodule-sedi-
S.A. Macko et al. / Chemical Geology 114 (1994) 365-3 79 373
A Sediment 4. Discussion
5 J ----LGLU - - " - t A ~
.... LASP - - - - G L Y ~5
&13C I
I I I I
2
Weeks
B Sediment
l o
~5
&13C
20
| 25
3o I I
, 1 2
W e e k s
~ t . V A L
- - ' : " - L t E U
. . . . . L P a O
- - - - ILE
. . . ° "
Fig. 5. Amino acid stable isotope (6~3C) compositions of sediment slurry from Winogradsky columns: (A) L-GIu, L- Ala, L-Asp and Gly; (B) L-Val, L-Leu, L-Pro and L-lie. (Er- ror in the recalculation and analysis is approximately repre- sented by the size of the point. )
ment columns (Fig. 4) and the sediment control experiments (Fig. 5). These smaller variations are within the margin of the reproducibility of the measurements and the back calculations of the original isotopic signatures. An exception to this observation is a depletion of 2.5%0 in Pro in the initial combination (CO) column. The exact cause for this depletion is unknown, and in fact may be an artifact caused by the presence ofcoe- luting materials contributing to the isotopic composition of Pro in that particular sample. In all samples, Ile and Leu are depleted in ~3C rela- tive to all other amino acids.
In organic-rich sediments, isotopic shifts as a consequence of diagenesis have been observed (Behrens and Frishman, 1971; Macko, 1981; Ostrom and Macko, 1991 a, b). However, in other reducing environments, the original isotopic sig- nals are clearly maintained (Kennicutt et al., 1992 ). In this present study, the isotopic signals of the bulk materials did not change in either the Halodule columns or the sediment controls. The fact that the control sediment slurry did not change in chemical or isotopic character indi- cates that much of the organic materials in the sediment were likely too refractory for further diagenetic destruction. Decomposition without substantial isotopic fractionation has been asso- ciated with portions of the original plant being preserved. In the case of the Halodule columns, the decomposition is perhaps more analogous to the preservation/limited degradation loss ob- served in Sargassum from the Orca Basin, Gulf of Mexico (Kennicutt et al., 1992) or seagrasses and mangroves from Florida Bay, southwest North Atlantic (Zieman et al., 1984). Individ- ual components might be altered both chemi- cally and isotopically, but the majority of the carbon and nitrogen remains isotopically un- changed (although perhaps more refractory, and diminished in concentration ).
The processes and organisms which partici- pated in the decomposition of the Halodule also may have had little involvement in the sedi- ment-seagrass mixture decomposition. An ex- planation for the bulk isotopic changes in the mixtures is therefore needed. Some isotope vari- ation may occur as a consequence of the prefer- ential loss of isotopically enriched (e.g., amino acids) or depleted (e.g., lipids) components. Other variations in isotopic signature may occur as a result of deamination, condensation or de- carboxylation reactions associated with the de- composition of organic-rich materials (Wade, 1980; Zieman et al., 1984; Sigleo and Macko, 1985 ). In the transition from labile, protein-rich organic matter, to more refractory materials found in sediments, peptide bond rupture (Sil- fer et al., 1992 ) may also be important in deter-

374 S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
mining the final isotopic compositions of the or- ganic nitrogen and carbon. An enrichment in the
t 3C of algal mat organic matter was reported to be associated with the loss of depleted lipid car- bon (Behrens and Frishman, 1971; Macko, 1981 ). Loss of carbohydrate carbon has been suggested to explain isotopic depletions in sap- ropelic peats, relative to the original plant sources (Spiker and Hatcher, 1984, 1987), and also could account for the depletions observed in the mixed columns.
Nitrogen isotope enrichments during diagene- sis of a size similar to those reported here for the combination columns have been observed pre- viously. An enrichment in nitrogen isotopes with a decline in the amino acid content is indicative of diagenesis, possibly the result of peptide bond hydrolysis, followed by bacterial utilization of the free amino acids or shorter-chain peptides. Iso- topic fractionations of the ~SN of amino nitrogen that were similar in size (fl~ 1.004) and direc- tion to those described here have been reported in a laboratory study on the peptide hydrolysis of glycylglycine (Silfer et al., 1992 ) and in natu- ral samples of particulate organic materials (Saino and Hattori, 1980; Wada, 1980). Thus, in the mixed columns, losses of carbon and ni- trogen accompanied by bond rupture and for- mation can alter the isotopic compositions, yet the overall bulk chemistry (in this case observa- tions were limited to the hydrolyzable amino acid) may not change substantially. Significant changes in other portions of the substrate (e.g., carbohydrates ) or the nature of the material (e.g., becoming more refractory) are possible expla- nations for the isotopic signal changes.
The abundances of the D-amino acids in these experiments (up to 8.6% of the glutamic acid and 10.0% of the aspartic acid) indicate that micro- biological input and not partial racemization of the original plant material is the source of these compounds. Simply not enough time elapsed during the course of the experiment for substan- tial natural racemization to occur at laboratory temperatures, and the effects from hydrolysis and derivatization are minimal (see Section 2 ). Fur- ther, other amino acids beyond the four noted would have shown heightened levels of the D-en-
antiomers. Zieman et al. (1984) noted similar levels of these D-amino acids in mangrove sedi- ments found in Florida. These D-amino acids are commonly found in bacterial cell walls (Lehn- inger, 1975 ) or in the free state inside microbial cells (S.A. Macko and M.L. Fogel, unpublished data, 1983).
Increases observed in the concentrations of the amino acids, constrained by correlative interpre- tations and sample inhomogeneity, may be the result of microbial activity and production. Pro- duction of new amino acids of the same chemi- cal nature as ones originally present in the sam- ple, during a period of utilization and destruction of the same types of compounds, cannot be iden- tified using normal concentration based obser- vations. For example, Glu which is undergoing destruction via deamination, decarboxylation, etc., and thus declining in concentration, is chemically identical to Glu which may originate by an alternate path of production. Chemically, there is no method of distinguishing or quanti- fying the two sources of the Glu. Compounds as- sociated with new production during microbial decomposition appear to be depleted in ~3C rel- ative to the initial plant matter (Macko et al., 1990, 1991 ). It is suggested that the amino acids which show significant depletions in 13C in fact represent materials which have substantial con- tribution from new production in the sediment- Halodule columns. Three of the amino acids (Glu, Ala, Asp) which show the most pro- nounced changes in isotopic composition also exhibit the largest contributions of D-isomers, which further suggests microbial inputs.
It is somewhat surprising that ornithine, fl- alanine and 7-amino butyric acid were not ob- served in the Halodule decomposition, given the fact that these amino acids have often been sug- gested to be a product of decomposition of other amino acids. Clearly, the stage of degradation of the initial substrate plays a role in the amounts of these breakdown products present. Demon- stration of the linkage between the breakdown product amino acids and their precursors in nat- ural samples may in fact be possible through the use of GC-C-IRMS and natural isotopic labels of the parent compounds. Further, the isotopic

S.A. Macko et al. / Chemical Geology 114 (1994,) 365-379 375
depletions observed in the 13C in Ile and Leu are likely products of their biosynthetic pathways, and their association with long-chain carbon synthesis (Macko et al., 1987). The isotopic compositions of individual components may therefore be useful in comparative biochemical assessment in natural systems.
It is normally difficult to resolve indigenous plant material which is a residue of the decay process, from material which has been added through microbial (or fungal) growth. These or- ganisms may produce significant amounts of or- ganic material during the decay of primary pro- duced materials. The amino acid content of bacterial cells (dry weight) may be/> 55% (Brock and Madigan, 1991 ). If one takes the observed changes in the amino acid content of the samples from the Halodule decomposition columns to- gether with the isotopic composition changes seen in the individual amino acids in those sam- pies, an attempt at quantifying new production during the diagenesis of the seagrass could be made through a mass balance of the isotopes and amino acids using:
¢~13C M :Fot~13Co q- FN~I3CN (2)
where M is the measured component; O is the original material; and N is the newly produced material. The sum of the mole fractions (F) con- tributed by each of the sources in this two-mem- bered mixture must equal unity ( 1 = Fo + FN ). The four amino acids, Glu, Gly, Ala and Val, represent ~ 50% of the total molar abundances of amino acids in the Halodule decomposition. Each of these amino acids also declined by ~ 50% during the course of the experiment. Further, the ~13C of these four amino acids declined by an average of 5%0. If one assumes a typical fraction- ation of 10%o (Macko and Estep, 1984; Macko et al., 1987) in the conversion of natural sub- strates to microbial cells, then 50% of these four amino acids, measured in the final column (H4), were not originally present at the start of the ex- periment. If the fractionation is larger, the rela- tive contribution of the original material dimin- ishes (a 15%0 fractionation decreases the contribution to 33%). It can be shown that the relative contribution is independent of the ab-
solute isotopic composition of the starting ma- terial. The relative amount of the contribution of new production to the total of the amino acids measured in H4 can also be calculated to be 25% based on 50% molar fraction mentioned above. This estimate is also somewhat conservative be- cause other amino acids would also be produced, and were not included in the calculation since their isotopic signals were not observed to de- crease significantly. The D / L isomer values of these newly produced amino acids would also in- crease by a factor of 2 in the above example. The rates of loss for the amino acids from the original substrate would be 50% higher than estimates based purely on concentration differences. This example also emphasizes the need for controlled microbial culture experiments to evaluate the magnitudes of isotopic fractionation on natural substrates. Obviously, allowing these experi- ments to continue for longer durations of months to years might alter the chemical and isotopic compositions further, thus even more clearly distinguishing among components which result from decomposition and microbial production. It is apparent, however that through the isotopic analysis of the individual amino acids, a better characterization of the complex processes in- volved in organic preservation may be possible.
5. Summary
Through isotopic analysis on individual amino acids, a unique perspective on the mechanisms of organic preservation and diagenesis is possi- ble. Substantial changes during early diagenesis were observed in bulk isotopic compositions and in both the concentration and isotopic composi- tions of individual amino acids. The effects of microbiological action on the transformation of organic matter in natural environments, partic- ularly on the natural biopolymers of amino acids, fatty acids and carbohydrates, may be better understood through isotopic assessment of indi- vidual components. The production of new, la- bile compounds during the processes of organic matter alteration and preservation, attendant to incorporation of more refractory components is
376 S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
apparently a significant portion of the pathway to the eventual preservation of organic matter. Beyond clearer understanding of the diagenetic processes affecting organic materials, is the pos- sibility of establishing sources for unusual amino acids observed in water columns (Henrichs and Farrington, 1979, 1980), organic deposits (Cas- agrande and Given, 1974, 1980) and sediments (Whelan, 1977; Whelan and Emeis, 1992). Through further application of this molecular/ isotope approach, these processes could be better understood and addressed.
Acknowledgements
Acknowledgement is made to the Donors of the Petroleum Research Fund, administered by the American Chemical Society for partial support of the work (S.A.M.). Other support was pro- vided by the National Science Foundation, Earth Sciences Division (EAR-9118011 and EAR- 9005489) (M.H.E. and S.A.M.). We would like to express our appreciation to C. Lee and two anonymous reviewers for their detailed and in- sightful comments.
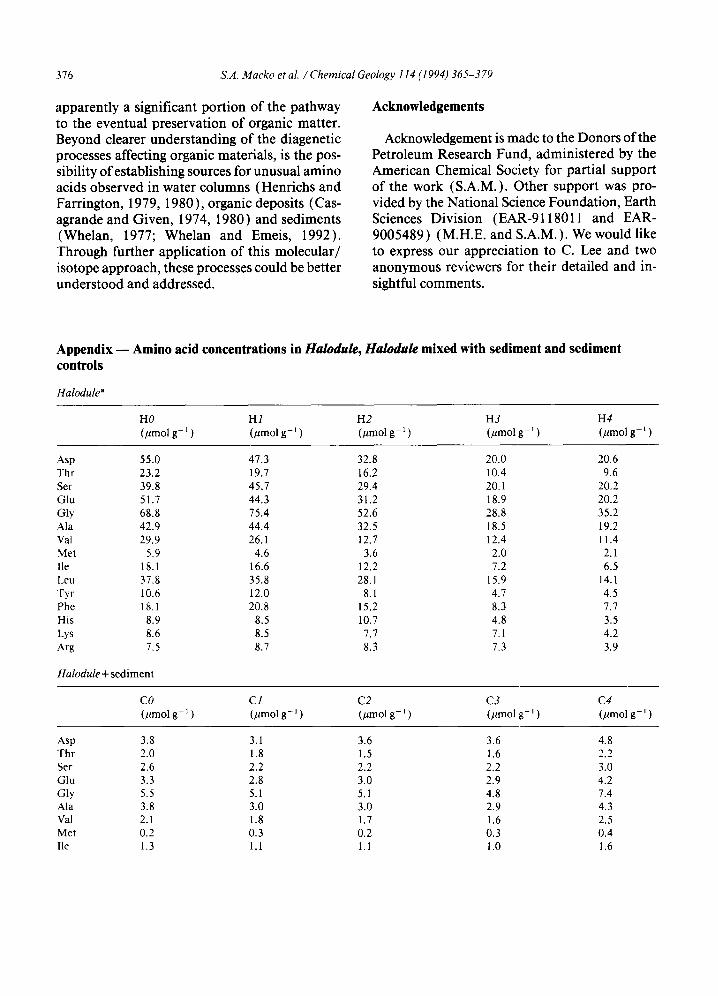
Appendix - - Amino acid concentrations in Halodule, Halodule mixed with sediment and sediment controls
Halodule a
HO HI H2 H3 H4 (#mol g- ~ ) (/lmol g- ~ ) (pmol g- 1 ) (#tool g- ~ ) (#mol g- ~ )
Asp 55.0 47.3 32.8 20.0 20.6 Thr 23.2 19.7 16.2 10.4 9.6 Ser 39.8 45.7 29.4 20. I 20.2 Glu 51.7 44.3 31.2 18.9 20.2 Gly 68.8 75.4 52.6 28.8 35.2 Ala 42.9 44.4 32.5 18.5 19.2 Val 29.9 26.1 12.7 12.4 11.4 Met 5.9 4.6 3.6 2.0 2.1 lle 18.1 16.6 12.2 7.2 6.5 Leu 37.8 35.8 28.l 15.9 14.1 Tyr 10.6 12.0 8.l 4.7 4.5 Phe 18.1 20.8 15.2 8.3 7.7 His 8.9 8.5 10.7 4.8 3.5 Lys 8.6 8.5 7.7 7.1 4.2 Arg 7.5 8.7 8.3 7.3 3.9
Halodule + sediment
CO C1 C2 C3 C4 (pmol g- 1 ) (/~mol g- 1 ) (#mol g- 1 ) (pmol g- l ) (/tmol g- l )
Asp 3.8 3.1 3.6 3.6 4.8 Thr 2.0 1.8 1.5 1.6 2.2 Ser 2.6 2.2 2.2 2.2 3.0 Glu 3.3 2.8 3.0 2.9 4.2 Gly 5.5 5.1 5.1 4.8 7.4 Ala 3.8 3.0 3.0 2.9 4.3 Val 2.1 1.8 1.7 1.6 2.5 Met 0.2 0.3 0.2 0.3 0.4 Ile 1.3 1.1 1.1 1.0 1.6
S.A. Macko et aL /Chemical Geology 114 (1994) 365-379 377
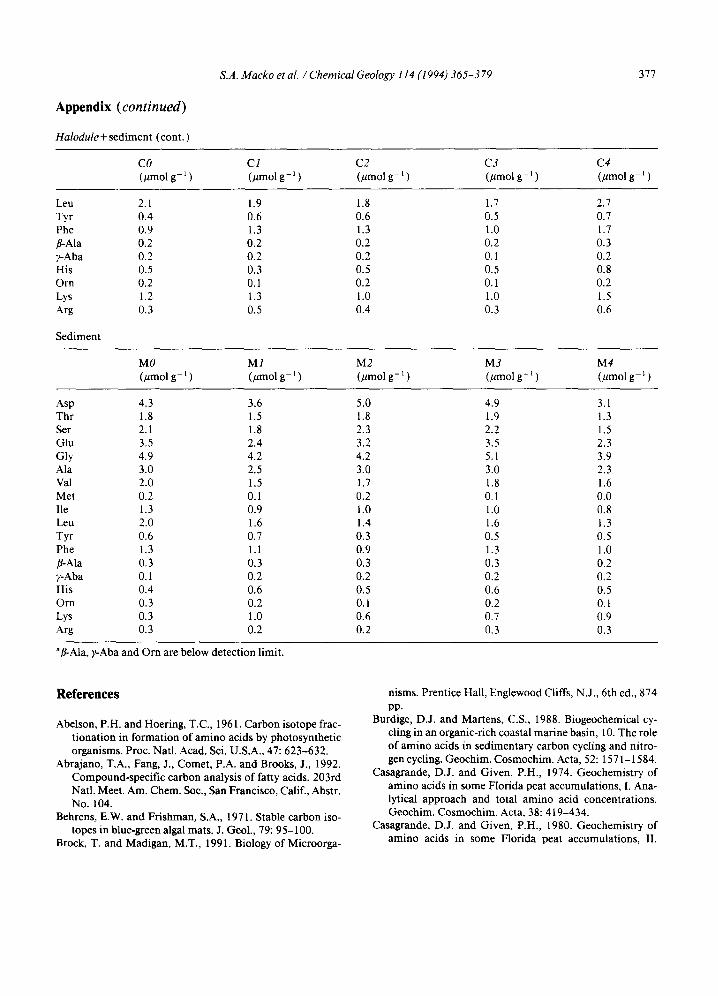
Appendix (continued)
Halodule+ sediment (cont.
CO C1 C2 C3 C4 (#molg -* (~molg -~ ) (#molg -* ) (~molg -~ ) (~molg -* )
Leu 2.1 1.9 1.8 1.7 2.7 Tyr 0.4 0.6 0.6 0.5 0.7 Phe 0.9 1.3 1.3 1.0 1.7 ~Ala 0.2 0.2 0.2 0.2 0.3 ~Aba 0.2 0.2 0.2 0.1 0.2 His 0.5 0.3 0.5 0.5 0.8 Orn 0.2 0.1 0.2 0.1 0.2 Lys 1.2 1.3 1.0 1.0 1.5 Arg 0.3 0.5 0.4 0.3 0.6
Sediment
M0 M1 M2 M3 M4 (#mol g- ~ (#mol g- 1 ) (pmol g- ~ ) (~mol g- ~ ) (/zmol g- ~ )
Asp 4.3 3.6 5.0 4.9 3.1 Thr 1.8 1.5 1.8 1.9 1.3 Ser 2.1 1.8 2.3 2.2 1.5 Glu 3.5 2.4 3.2 3.5 2.3 Gly 4.9 4.2 4.2 5.1 3.9 Ala 3.0 2.5 3.0 3.0 2.3 Val 2.0 1.5 1.7 1.8 1.6 Met 0.2 0.1 0.2 0.1 0.0 lie 1.3 0.9 1.0 1.0 0.8 ~ u 2.0 1.6 1.4 1.6 1.3 Tyr 0.6 0.7 0.3 0.5 0.5 Phe 1.3 1.1 0.9 1.3 1.0 ~Ala 0.3 0.3 0.3 0.3 0.2 ~Aba 0.1 0.2 0.2 0.2 0.2 His 0.4 0.6 0.5 0.6 0.5 Orn 0.3 0.2 0.1 0.2 0.1 ~ s 0.3 1.0 0.6 0.7 0.9 Arg 0.3 0.2 0.2 0.3 0.3
afl-Ala, y-Aba and Orn are below detection limit.
References
Abelson, P.H. and Hoering, T.C., 1961. Carbon isotope frac- tionation in formation of amino acids by photosynthetic organisms. Proc. Natl. Acad. Sci. U.S.A., 47: 623-632.
Abrajano, T.A., Fang, J., Comet, P.A. and Brooks, J., 1992. Compound-specific carbon analysis of fatty acids. 203rd Natl. Meet. Am. Chem. Soc., San Francisco, Calif., Abstr. No. 104.
Behrens, E.W. and Frishman, S.A., 1971. Stable carbon iso- topes in blue-green algal mats. J. Geol., 79: 95-100.
Brock, T. and Madigan, M.T., 1991. Biology of Microorga-
nisms. Prentice Hall, Englewood Cliffs, N.J., 6th ed., 874 PP.
Burdige, D.J. and Martens, C.S., 1988. Biogeochemical cy- cling in an organic-rich coastal marine basin, 10. The role of amino acids in sedimentary carbon cycling and nitro- gen cycling. Geochim. Cosmochim. Acta, 52:1571-1584.
Casagrande, D.J. and Given,, P.H., 1974. Geochemistry of amino acids in some Florida peat accumulations, I. Ana- lytical approach and total amino acid concentrations. Geochim. Cosmochim. Acta, 38:419-434.
Casagrande, D.J. and Given, P.H., 1980. Geochemistry of amino acids in some Florida peat accumulations, II.

378 S.A. Macko et al. / Chemical Geology 114 (1994) 365-379
Amino acid distributions. Geochim. Cosmochim. Acta, 44: 1493-1507.
Degens, E.T. and Mopper, K., 1976. Factors controlling the distribution and early diagenesis of organic material in marine sediments. In: J.P. Riley and R. Chester (Edi- tors), Chemical Oceanography, Vol. 6. Academic Press, London, pp. 59-113.
Drozdova, T.V., 1973. Geochemistry of amino acids and car- bohydrates in old and recent sediments. In: B. Tissot and F. Bienner (Editors), Advances in Organic Geochemistry 1973. Technip, Paris, pp. 285-292.
Engel, M.H. and Hare, P.E., 1985. Gas-liquid chromato- graphic separation of amino acids and their derivatives. In: G.C. Barrett (Editor), Chemistry and Biochemistry of the Amino Acids. Chapman and Hall, New York, N.Y., pp. 462-479.
Engel, M.H. and Macko, S.A., 1984. Separation of amino acid enantiomers for stable nitrogen and carbon isotopic anal- yses. Anal. Chem., 56: 2598-2600.
Engel, M.H. and Macko, S.A., 1986. Stable isotope evalua- tion of the origins of amino acids in fossils. Nature (Lon- don), 323: 531-533.
Engel, M.H., Macko, S.A. and Silfer, J.A., 1990. Carbon iso- tope composition of individual amino acids in the Mur- chison meteorite. Nature (London), 348: 47-49.
Freedman, P.A., Gillyon, E.C.P. and Jumeau, E.J., 1988. De- sign and application of a new instrument for GC-isotope ratio MS. Am. Lab., June 1988, pp. 114-119.
Galimov, E.M., 1980. C~3/C ~2 in kerogen. In: B. Durand (Editor), Kerogen-Insoluble Organic Matter from Sedi- mentary Rocks. Technip, Paris, pp. 271-299.
Hare, P.E., 1972. Ion exchange chromatography in lunar or- ganic analyses. Space Life Sci., 3: 354-359.
Hare, P.E. and Estep, M.L., 1983. Carbon and nitrogen iso- topic composition of amino acids in modern and fossil collagens. Carnegie Inst. Washington Yearbk., 82: 410- 414.
Hayes, J.M., Takigiku, R., Ocampo, R., Callot, H.J. and Al- brecht, P., 1987. Isotopic compositions and probable origins of organic molecules in the Eocene Messel shale. Nature (London), 329:48-51.
Hayes, J.M., Freeman, K.H., Popp, B.N and Hoham, C.H., 1990. Compound-specific isotope analysis: a novel tool for reconstruction of ancient biogeochemical processes. Org. Geochem., 16:1115-1128.
Henrichs, S.M. and Farrington, J.W., 1979. Amino acids in interstitial waters of marine sediments. Nature (Lon- don), 279: 319-322.
Henrichs, S.M. and Farrington, J.W., 1980. Amino acids in interstitial waters of marine sediments: A comparison of results from varied sedimentary environments. In: A.G. Douglas and J.R. Maxwell (Editors), Advances in Or- ganic Geochemistry 1979. Pergamon, Oxford, pp. 435- 443.
Henrichs, S.M. and Williams, P.M., 1985. Dissolved and particulate amino acids and carbohydrates in the sea sur- face microlayer. Mar. Chem., 17: 141-163.
Kennicutt, M,C., Macko, S.A., Harvey, H.R. and Bidigare, R.R., 1992. Preservation of Sargassum under anoxic con- ditions: molecular and isotopic evidence. In: J.K. Whelan and J.W. Farrington (Editors), Productivity, Accumula- tion and Preservation of Organic Matter: Recent and An- cient Sediments. Columbia University Press, New York, N.Y., pp. 123-141.
Klok, J., 1984. Composition and origin of complex organic matter in recent marine sediments. Ph.D. Thesis, Delft University, Delft, 111 pp.
Kohnen, M.E.L., Schouten, S., Sinninghe Damstr, J.S., de Leeuw, J.W., Merritt, D.A. and Hayes, J.M., 1992. Rec- ognition of paleochemical by a combined molecular sul- fur isotope geochemical approach. Science, 256: 358-362.
Lehninger, A.L., 1975. Biochemistry. Worth, New York, N.Y., 1104 pp.
Macko, S.A., 1981. Stable nitrogen isotope ratios as tracers of organic geochemical processes. Ph.D. Thesis, Univer- sity of Texas at Austin, Austin. Texas, 181 pp.
Macko, S.A. and Engel, M,H., 1991. Assessment ofindigene- ity in fossil organic matter: amino acids and stable iso- topes. Philos. Trans. R. Soc. London, Ser. B, 333: 367- 374.
Macko, S.A. and Estep, M.L.F., ][ 984. Microbial alteration of stable nitrogen and carbon isotopic compositions of or- ganic substrates. Org. Geochem., 6: 787-790.
Macko, S.A., Estep, M.F., Engel, M.H. and Hare, P.E., 1986. Kinetic fractionation of stable nitrogen isotopes during amino acid transamination. Geochim. Cosmochim. Acta., 50: 2143-2146.
Macko, S.A., Estep, M.L.F., Hare, P.E. and Hoering, T.C., 1987. Isotopic fractionation of nitrogen and carbon in the synthesis of amino acids by microorganisms. Chem. Geol. (Isot. Geosci. Sect.), 65: 79-92.
Macko, S.A., Helleur, R., Hartley, G. and Jackman, P., 1990. Diagenesis of organic matter - - a study using stable iso- topes of individual carbohydrates. Org. Geochem., 16: 1129-1137.
Macko, S.A., Engel, M.H., Hartley, G., Hatcher, P., Helleur, R., Jackman, P. and Silfer, J.A., 1991. Isotopic composi- tions of individual carbohydrates as indicators of early diagenesis of organic matter. In: J.A. Curiale, R. Alex- ander and P.W. Brooks (Editors), Organic Geochemistry of Hydrocarbon Basins. Chem. Geol., 93:147-161 (spe- cial issue).
Macko, S.A., Leskey, T., Ryan, M. and Engel, M.H., 1992. Carbon isotopic analysis of individual carbohydrates by GC/IRMS. 203rd Natl. Meet. Am. Chem. Soc., San Fran- cisco, Calif., Abstr. No. 107.
Martens, C., Haddad, R.I. and Chanton, J.P., 1992. Organic matter accumulation, remineralization, and burial in an anoxic coastal sediment. In: J.K. Whelan and J.W. Far- rington (Editors), Productivity, Accumulation and Pres- ervation of Organic Matter: Recent and Ancient Sedi- ments. Columbia University Press, New York, N.Y., pp. 82-99.
Mopper, K., 1977. Sugars and uronic acids in sediment and

S.A. Macko et al. /Chemical Geology 114 (1994) 365-379 379
water from the Black Sea and North Sea with emphasis on analytical techniques. Mar. Chem., 5: 585-603.
Nissenbaum, A., 1974. The organic geochemistry of marine and terrestrial humic substances: Implications of carbon and hydrogen isotope studies. In: B. Tissot and F. Bienner (Editors), Advances in Organic Geochemistry 1973. Technip, Paris, pp. 39-52.
Nissenbaum, A. and Kaplan, I.R., 1972. Chemical and iso- topic evidence for the in situ origin of marine humic sub- stances. Limnol. Oceanogr., 17: 570-582.
Nissenbaum, A. and Schallinger, K.M., 1974. The distribu- tion of the stable carbon isotope (13C/12C) in fractions of soil organic matter. Geoderma, 11: 137-145.
Nissenbaum, A., Presley, B.J. and Kaplan, I.R., 1972. Early diagenesis in a reducing fjord, Saanich Inlet, British Co- lumbia, I. Chemical and isotopic changes in major com- ponents of interstitial water. Geochim. Cosmochim. Acta, 36: 1007-1027.
Orem, W.H. and Hatcher, P.G., 1987. Early diagenesis of or- ganic matter in a sawgrass peat from the Everglades, Flor- ida. Int. J. Coal. Geol., 8: 33-54.
Ostrom, N.E. and Macko, S.A., 1991a. Sources, cycling and distribution of water column particulate and sedimentary organic matter in northern Newfoundland tjords and bays: a stable isotope study. In: J.K. Whelan and J.W. Farting- ton (Editors), Productivity, Accumulation and Preser- vation of Organic Matter: Recent and Ancient Sediments. Columbia University Press, New York, N.Y., pp. 55-81.
Ostrom, N.E. and Macko, S.A., 1991b. Late Wisconsinan to present sedimentation of organic matter off northern Newfoundland in response to climatological events. Cont. ShelfRes., 1 l: 1285-1296.
Prahl, F.G, Hayes, J.M. and Xie, T.M., 1992. Diploptene: An indicator of terrigenous organic carbon in Washington coastal sediments. Limnol. Oceanogr., 37: 1290-1300.
Saino, T. and Hattori, A., 1980. ~SN natural abundance in oceanic suspended particulate matter. Nature (London), 283: 752-754.
Serban, A., Engel, M.H. and Macko, S.A., 1988. The distri- bution, stereochemistry and stable isotopic composition of amino acid constituents of fossil and modern mollusk shells. Org. Geochem., 13: 1123-1129.
Sigleo, A,C. and Macko, S.A., 1985. Stable isotope and amino acid composition of estuarine dissolved colloidal mate- rial. In: A. Sigleo and A. Hattori (Editors), Marine and Estuarine Geochemistry. Lewis, Chelsea, Mich., pp. 29- 46.
Silfer, J.A., Engel, M.H., Macko, S.A. and Jumeau, E.J., 1991. Stable carbon isotope analysis of amino acid enantiomers by conventional isotope ratio mass spectrometry and combined gas chromatography-isotope ratio mass spec- trometry. Anal. Chem., 63: 370-374.
Silfer, J.A., Engel, M.H. and Macko, S.A., 1992. Kinetic frac-
tionation of stable carbon and nitrogen isotopes during peptide bond hydrolysis: experimental evidence and geo- chemical implications. In: S.A. Macko and M.H. Engel (Guest-Editors), Isotope Fractionations in Organic Mat- ter. Chem. Geol. (Isot. Geosci. Sect. ), 101:211-221 (special issue).
Silfer, J.A., Qian, Y., Macko, S.A. and Engel, M.H., 1994. Stable carbon isotope compositions of individual amino acid enantiomers by GC/C/IRMS. Org. Geocbem. (in press).
Sinninghe Damstr, J.S., Wakeham, S.G., Kohnen, M.E.L., Hayes, J.M. and de Leeuw, J.W., 1993. A 6,000 year sed- imentary molecular record of the chemocline excursions in the Black Sea. Nature (London), 363: 827-829.
Spiker, E.C. and Hatcher, P.G., 1984. Carbon isotope frac- tionation of sapropelic organic matter during early dia- genesis. Org. Geochem., 5: 283-290.
Spiker, E.C. and Hatcher, P.G., 1987. The effects of early diagenesis on the chemical and stable carbon isotopic composition of wood. Geochim. Cosmochim. Acta, 51: 1385-1391.
Summons, R.E. and Powell, T.G., 1986. Chlorobiaceae in Paleozoic seas revealed by biological markers, isotopes and geology. Nature (London), 319: 763-765.
Treibs, A., 1934. Chlorophyll-- und H~iminderivate in bitu- minrsen Gesteinen, Erdrlen, Erdwachsen und Asphalten. Ann. Chem., 510: 42-62.
Wada, E., 1980. Nitrogen isotope fractionation and its signif- icance in biogeochemical processes occurring in marine environments. In: E.D. Goldberg and Y. Horibe (Edi- tors), Isotope Marine Chemistry. Uchida Rokakuho, To- kyo, pp. 375-398.
Wakeham, S.G., Freeman, K.H., Pease, T.K. and Hayes, J.M., 1993. A photoautotrophic source for lycopane in marine water columns. Geochim. Cosmochim. Acta, 57:159-165.
Welte, D.H., 1974. Recent advances in organic geochemistry of humic substances and kcrogen - - A review. In: B. Tis- sot and F. Bienner (Editors), Advances in Organic Geo- chemistry 1973. Technip, Paris, pp. 39-52.
Whelan, J.K., 1977. Amino acids in a surface sediment core of the Atlantic abyssal plain. Geochim. Cosmochim. Acta, 41: 803-810.
Whelan, J.K. and Emeis, K.C., 1992. Sedimentation and preservation of amino compounds and carbohydrates in marine sediments. In: J.K. Whelan and J.W. Farrington (Editors), Productivity, Accumulation and Preservation of Organic Matter: Recent and Ancient Sediments. Co- lumbia University Press, New York, N.Y., pp. 176-200.
Zieman, J.C., Macko, S.A. and Mills, A.L., 1984. Role of sea- grasses and mangroves in estuarine food webs: temporal and spatial changes in stable isotope composition and amino acid content during decomposition. Bull. Mar. Sci., 35: 380-392.



















