
Europium beta-diketonate temperature sensors: Effects of ligands, matrix, and concentration
Upload
independentCategory
view
0download
0
PAPER www.rsc.org/materials | Journal of Materials Chemistry
Dual role of a di-urethanesil hybrid doped with europium b-diketonatecomplexes containing either water ligands or a bulky chelating ligand†
Mariana Fernandes,a Sonia S. Nobre,b Maria Cristina Goncalves,a Ana Charas,c Jorge Morgado,c
Rute A. S. Ferreira,*b Luıs D. Carlosb and Veronica de Zea Bermudez*a
Received 31st July 2008, Accepted 13th November 2008
First published as an Advance Article on the web 19th December 2008
DOI: 10.1039/b813155d
In the present work we report the unusual role played by a sol-gel derived di-urethane cross-linked
poly(oxyethylene) (POE)/siloxane (di-urethanesil, d-Ut(600)) hybrid matrix in the immobilization of
the b-diketonate aquocomplex Eu(btfac)3(H2O)2 (btfac� is 4,4,4-trifluoro-1-phenyl-2,4-butanedionate)
and in the quasi preservation in the hybrid of the 5D0 quantum efficiency q(5D0) value displayed by this
complex in the isolated state (0.12 versus 0.18, respectively). We demonstrate that the d-Ut(600)
framework acts as an inert (although optically active) support towards Eu(btfac)3(H2O)2, enabling Eu3+
sensitization by the btfac� ligands and energy transfer between the hybrid host excited states and the
lanthanide intra-4f6 levels, while the number of Eu3+-coordinated water molecules remains constant.
We also provide evidence that the incorporation of the Eu(btfac)3phen (phen is 1,10-phenantroline)
complex into the same hybrid matrix is disadvantageous from the 5D0 quantum efficiency standpoint,
because of the severe steric hindrance that emerges between the POE chains of the host structure and
the bulky phen molecules, leading to the expulsion of these ligands from the Eu3+ first coordination
sphere and to their replacement by two water molecules. As a consequence, embedding of
Eu(btfac)3phen in d-Ut(600) results in a significant reduction of the q(5D0) value of the isolated complex
(from 0.47 to 0.16). The behaviour observed in the presence of this bidentate chelating ligand correlates
well with the practically identical q(5D0) values and number of Eu3+-coordinated water molecules (ca. 2)
estimated for the two hybrids.
Introduction
Photonic materials lend themselves to technological application
in a wide variety of domains, including optical communications,
aerospace, sensing, lighting, computing and bio-medicine.
Traditionally photonic devices have been produced from semi-
conductors, glasses or polymers.
Owing to their unique luminescent features, in particular
narrow emitting bands and therefore quasi monochromatic
behaviour, lanthanide ions have been used extensively in this
context. Lanthanide ions exhibit, however, low luminescence
intensity owing to the fact that the 4f–4f transitions are parity
forbidden. One of the most successful strategies that has been
employed to overcome this drawback is the complexation of
the lanthanide ions through coordination to organic ligands
aDepartment of Chemistry, CQ-VR, University of Tras-os-Montes e AltoDouro, 5001-801 Vila Real, Portugal. E-mail: [email protected]; Fax:+351 259 350480; Tel: +351 259 350253bDepartment of Physics, CICECO, University of Aveiro, 3810-193 Aveiro,Portugal. E-mail: [email protected]; Fax: +351 234 378197; Tel: +351 234378103cInstituto de Telecomunicacoes, Department of Chemical Engineering,Instituto Superior Tecnico, 1049-001 Lisboa, Portugal
† Electronic supplementary information (ESI) available: FT-IR spectraof the Eu(btfac)3L complexes: (a) L ¼ (H2O)2 and (b) L ¼ phen; SEMmicrograph of the d-Ut(600)200Eu(btfac)3(H2O)2 di-urethanesil; XRDpatterns of the d-Ut(600)n Eu(btfac)3L di-urethanesils: A: L ¼ (H2O)2:(a, black line) n ¼ 400; (b, red line) n ¼ 200; B: L ¼ phen: (a, blackline) n ¼ 400; (b, blue line) n ¼ 200. See DOI: 10.1039/b813155d
This journal is ª The Royal Society of Chemistry 2009
(so-called ‘‘antennae’’1,2) acting as sensitisers.3–6 These ligands
absorb UV light and transfer the energy efficiently to the central
lanthanide ion (ligand-to-metal intramolecular energy transfer),
which then undergoes the corresponding radiative emitting
process.
The design of room temperature highly luminescent lantha-
nide complexes depends strongly on the efficiency of energy
transfer and on the concentration of the quenching species
present (e.g., OH groups that lead to non-radiative processes).
Apart from quantum efficiency, other critical issues, such as
solubility, volatility, photodegradability and thermal stability,
have severely limited the practical application of lanthanide
complexes. In the past few years it has been recognized that the
combination of the versatile sol-gel synthetic route7 with the
organic/inorganic hybrid concept8 allows these drawbacks to be
successfully circumvented. Firstly, the sol-gel process allows to
process, at mild conditions (moderate temperatures), homoge-
neous, transparent, easily shaped, thermally and mechanically
stable materials from a wide variety of cheap, pure and available
precursors. Secondly, the sol-gel chemistry enables the combi-
nation of organic and inorganic components, and as a conse-
quence tuning the properties of the final hybrid material. The
addition of lanthanide complexes to sol-gel derived organic/
inorganic hybrid materials is thus extremely attractive, because
the hybrid networks offer multifunctionality and tailored
features from the nanometric to the millimetric length scales.8–10
This combined strategy has resulted in the development
of several luminescent systems based on host di-urea and
J. Mater. Chem., 2009, 19, 733–742 | 733
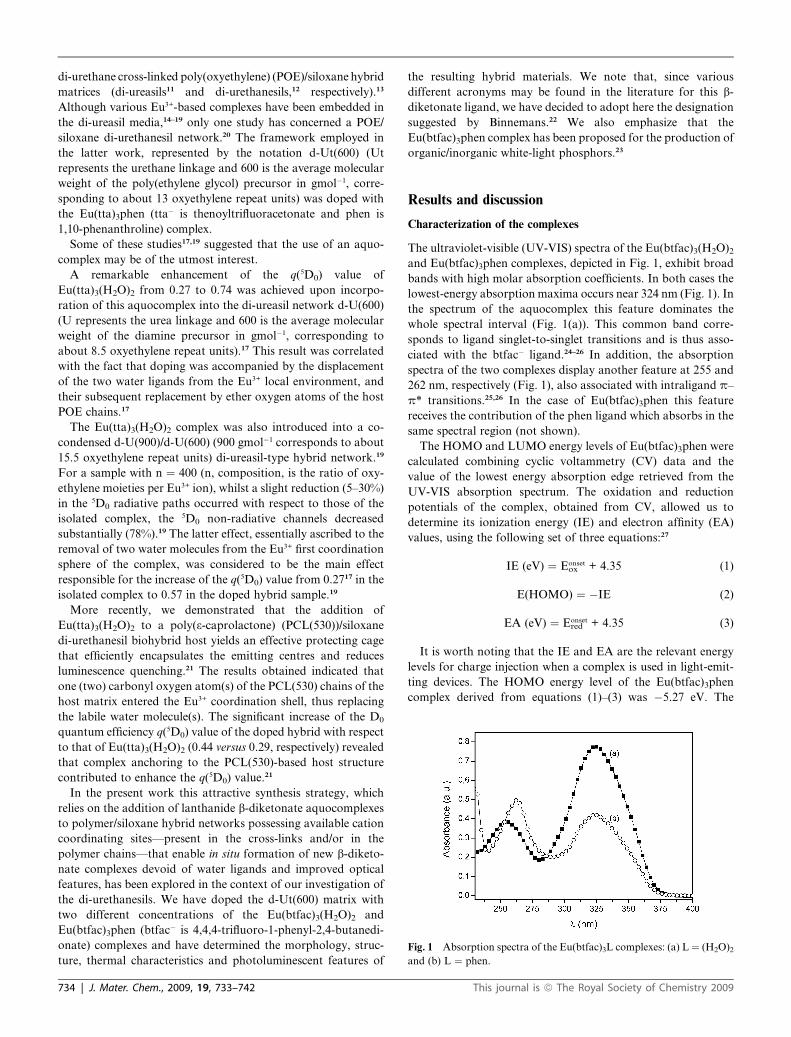
Fig. 1 Absorption spectra of the Eu(btfac)3L complexes: (a) L ¼ (H2O)2
and (b) L ¼ phen.
di-urethane cross-linked poly(oxyethylene) (POE)/siloxane hybrid
matrices (di-ureasils11 and di-urethanesils,12 respectively).13
Although various Eu3+-based complexes have been embedded in
the di-ureasil media,14–19 only one study has concerned a POE/
siloxane di-urethanesil network.20 The framework employed in
the latter work, represented by the notation d-Ut(600) (Ut
represents the urethane linkage and 600 is the average molecular
weight of the poly(ethylene glycol) precursor in gmol�1, corre-
sponding to about 13 oxyethylene repeat units) was doped with
the Eu(tta)3phen (tta� is thenoyltrifluoracetonate and phen is
1,10-phenanthroline) complex.
Some of these studies17,19 suggested that the use of an aquo-
complex may be of the utmost interest.
A remarkable enhancement of the q(5D0) value of
Eu(tta)3(H2O)2 from 0.27 to 0.74 was achieved upon incorpo-
ration of this aquocomplex into the di-ureasil network d-U(600)
(U represents the urea linkage and 600 is the average molecular
weight of the diamine precursor in gmol�1, corresponding to
about 8.5 oxyethylene repeat units).17 This result was correlated
with the fact that doping was accompanied by the displacement
of the two water ligands from the Eu3+ local environment, and
their subsequent replacement by ether oxygen atoms of the host
POE chains.17
The Eu(tta)3(H2O)2 complex was also introduced into a co-
condensed d-U(900)/d-U(600) (900 gmol�1 corresponds to about
15.5 oxyethylene repeat units) di-ureasil-type hybrid network.19
For a sample with n ¼ 400 (n, composition, is the ratio of oxy-
ethylene moieties per Eu3+ ion), whilst a slight reduction (5–30%)
in the 5D0 radiative paths occurred with respect to those of the
isolated complex, the 5D0 non-radiative channels decreased
substantially (78%).19 The latter effect, essentially ascribed to the
removal of two water molecules from the Eu3+ first coordination
sphere of the complex, was considered to be the main effect
responsible for the increase of the q(5D0) value from 0.2717 in the
isolated complex to 0.57 in the doped hybrid sample.19
More recently, we demonstrated that the addition of
Eu(tta)3(H2O)2 to a poly(3-caprolactone) (PCL(530))/siloxane
di-urethanesil biohybrid host yields an effective protecting cage
that efficiently encapsulates the emitting centres and reduces
luminescence quenching.21 The results obtained indicated that
one (two) carbonyl oxygen atom(s) of the PCL(530) chains of the
host matrix entered the Eu3+ coordination shell, thus replacing
the labile water molecule(s). The significant increase of the D0
quantum efficiency q(5D0) value of the doped hybrid with respect
to that of Eu(tta)3(H2O)2 (0.44 versus 0.29, respectively) revealed
that complex anchoring to the PCL(530)-based host structure
contributed to enhance the q(5D0) value.21
In the present work this attractive synthesis strategy, which
relies on the addition of lanthanide b-diketonate aquocomplexes
to polymer/siloxane hybrid networks possessing available cation
coordinating sites—present in the cross-links and/or in the
polymer chains—that enable in situ formation of new b-diketo-
nate complexes devoid of water ligands and improved optical
features, has been explored in the context of our investigation of
the di-urethanesils. We have doped the d-Ut(600) matrix with
two different concentrations of the Eu(btfac)3(H2O)2 and
Eu(btfac)3phen (btfac� is 4,4,4-trifluoro-1-phenyl-2,4-butanedi-
onate) complexes and have determined the morphology, struc-
ture, thermal characteristics and photoluminescent features of
734 | J. Mater. Chem., 2009, 19, 733–742
the resulting hybrid materials. We note that, since various
different acronyms may be found in the literature for this b-
diketonate ligand, we have decided to adopt here the designation
suggested by Binnemans.22 We also emphasize that the
Eu(btfac)3phen complex has been proposed for the production of
organic/inorganic white-light phosphors.23
Results and discussion
Characterization of the complexes
The ultraviolet-visible (UV-VIS) spectra of the Eu(btfac)3(H2O)2
and Eu(btfac)3phen complexes, depicted in Fig. 1, exhibit broad
bands with high molar absorption coefficients. In both cases the
lowest-energy absorption maxima occurs near 324 nm (Fig. 1). In
the spectrum of the aquocomplex this feature dominates the
whole spectral interval (Fig. 1(a)). This common band corre-
sponds to ligand singlet-to-singlet transitions and is thus asso-
ciated with the btfac� ligand.24–26 In addition, the absorption
spectra of the two complexes display another feature at 255 and
262 nm, respectively (Fig. 1), also associated with intraligand p–
p* transitions.25,26 In the case of Eu(btfac)3phen this feature
receives the contribution of the phen ligand which absorbs in the
same spectral region (not shown).
The HOMO and LUMO energy levels of Eu(btfac)3phen were
calculated combining cyclic voltammetry (CV) data and the
value of the lowest energy absorption edge retrieved from the
UV-VIS absorption spectrum. The oxidation and reduction
potentials of the complex, obtained from CV, allowed us to
determine its ionization energy (IE) and electron affinity (EA)
values, using the following set of three equations:27
IE (eV) ¼ Eonsetox + 4.35 (1)
E(HOMO) ¼ �IE (2)
EA (eV) ¼ Eonsetred + 4.35 (3)
It is worth noting that the IE and EA are the relevant energy
levels for charge injection when a complex is used in light-emit-
ting devices. The HOMO energy level of the Eu(btfac)3phen
complex derived from equations (1)–(3) was �5.27 eV. The
This journal is ª The Royal Society of Chemistry 2009
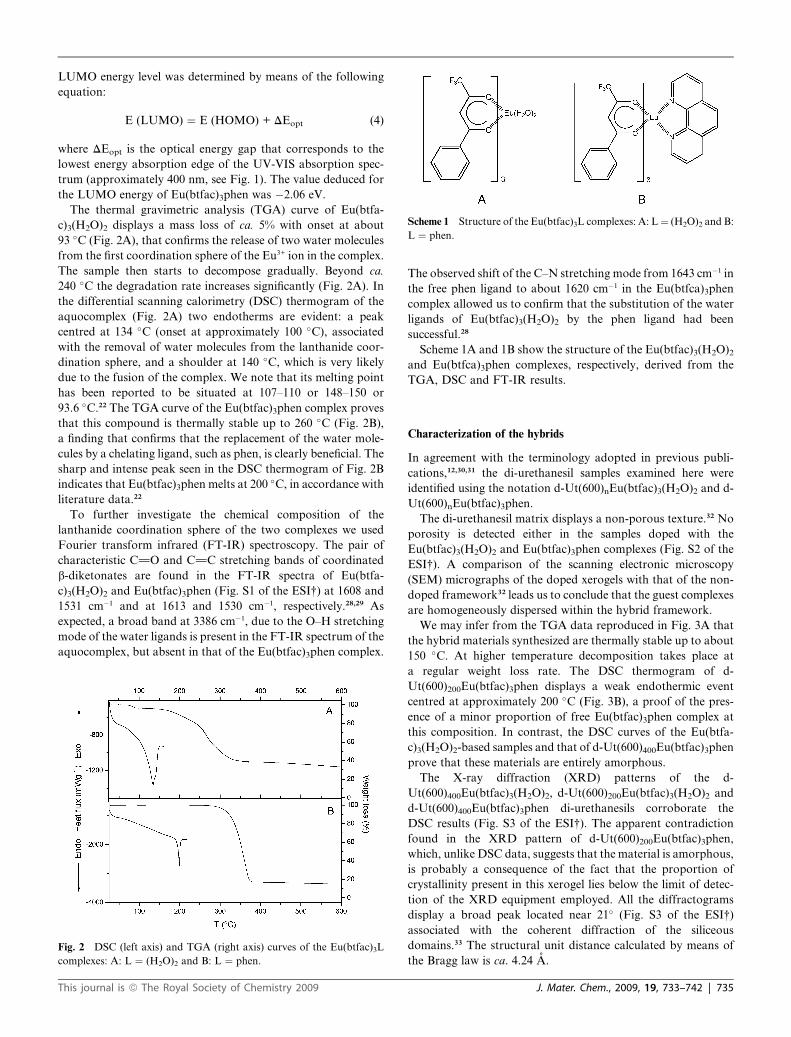
Scheme 1 Structure of the Eu(btfac)3L complexes: A: L ¼ (H2O)2 and B:
L ¼ phen.
LUMO energy level was determined by means of the following
equation:
E (LUMO) ¼ E (HOMO) + DEopt (4)
where DEopt is the optical energy gap that corresponds to the
lowest energy absorption edge of the UV-VIS absorption spec-
trum (approximately 400 nm, see Fig. 1). The value deduced for
the LUMO energy of Eu(btfac)3phen was �2.06 eV.
The thermal gravimetric analysis (TGA) curve of Eu(btfa-
c)3(H2O)2 displays a mass loss of ca. 5% with onset at about
93 �C (Fig. 2A), that confirms the release of two water molecules
from the first coordination sphere of the Eu3+ ion in the complex.
The sample then starts to decompose gradually. Beyond ca.
240 �C the degradation rate increases significantly (Fig. 2A). In
the differential scanning calorimetry (DSC) thermogram of the
aquocomplex (Fig. 2A) two endotherms are evident: a peak
centred at 134 �C (onset at approximately 100 �C), associated
with the removal of water molecules from the lanthanide coor-
dination sphere, and a shoulder at 140 �C, which is very likely
due to the fusion of the complex. We note that its melting point
has been reported to be situated at 107–110 or 148–150 or
93.6 �C.22 The TGA curve of the Eu(btfac)3phen complex proves
that this compound is thermally stable up to 260 �C (Fig. 2B),
a finding that confirms that the replacement of the water mole-
cules by a chelating ligand, such as phen, is clearly beneficial. The
sharp and intense peak seen in the DSC thermogram of Fig. 2B
indicates that Eu(btfac)3phen melts at 200 �C, in accordance with
literature data.22
To further investigate the chemical composition of the
lanthanide coordination sphere of the two complexes we used
Fourier transform infrared (FT-IR) spectroscopy. The pair of
characteristic C]O and C]C stretching bands of coordinated
b-diketonates are found in the FT-IR spectra of Eu(btfa-
c)3(H2O)2 and Eu(btfac)3phen (Fig. S1 of the ESI†) at 1608 and
1531 cm�1 and at 1613 and 1530 cm�1, respectively.28,29 As
expected, a broad band at 3386 cm�1, due to the O–H stretching
mode of the water ligands is present in the FT-IR spectrum of the
aquocomplex, but absent in that of the Eu(btfac)3phen complex.
Fig. 2 DSC (left axis) and TGA (right axis) curves of the Eu(btfac)3L
complexes: A: L ¼ (H2O)2 and B: L ¼ phen.
This journal is ª The Royal Society of Chemistry 2009
The observed shift of the C–N stretching mode from 1643 cm�1 in
the free phen ligand to about 1620 cm�1 in the Eu(btfca)3phen
complex allowed us to confirm that the substitution of the water
ligands of Eu(btfac)3(H2O)2 by the phen ligand had been
successful.28
Scheme 1A and 1B show the structure of the Eu(btfac)3(H2O)2
and Eu(btfca)3phen complexes, respectively, derived from the
TGA, DSC and FT-IR results.
Characterization of the hybrids
In agreement with the terminology adopted in previous publi-
cations,12,30,31 the di-urethanesil samples examined here were
identified using the notation d-Ut(600)nEu(btfac)3(H2O)2 and d-
Ut(600)nEu(btfac)3phen.
The di-urethanesil matrix displays a non-porous texture.32 No
porosity is detected either in the samples doped with the
Eu(btfac)3(H2O)2 and Eu(btfac)3phen complexes (Fig. S2 of the
ESI†). A comparison of the scanning electronic microscopy
(SEM) micrographs of the doped xerogels with that of the non-
doped framework32 leads us to conclude that the guest complexes
are homogeneously dispersed within the hybrid framework.
We may infer from the TGA data reproduced in Fig. 3A that
the hybrid materials synthesized are thermally stable up to about
150 �C. At higher temperature decomposition takes place at
a regular weight loss rate. The DSC thermogram of d-
Ut(600)200Eu(btfac)3phen displays a weak endothermic event
centred at approximately 200 �C (Fig. 3B), a proof of the pres-
ence of a minor proportion of free Eu(btfac)3phen complex at
this composition. In contrast, the DSC curves of the Eu(btfa-
c)3(H2O)2-based samples and that of d-Ut(600)400Eu(btfac)3phen
prove that these materials are entirely amorphous.
The X-ray diffraction (XRD) patterns of the d-
Ut(600)400Eu(btfac)3(H2O)2, d-Ut(600)200Eu(btfac)3(H2O)2 and
d-Ut(600)400Eu(btfac)3phen di-urethanesils corroborate the
DSC results (Fig. S3 of the ESI†). The apparent contradiction
found in the XRD pattern of d-Ut(600)200Eu(btfac)3phen,
which, unlike DSC data, suggests that the material is amorphous,
is probably a consequence of the fact that the proportion of
crystallinity present in this xerogel lies below the limit of detec-
tion of the XRD equipment employed. All the diffractograms
display a broad peak located near 21� (Fig. S3 of the ESI†)
associated with the coherent diffraction of the siliceous
domains.33 The structural unit distance calculated by means of
the Bragg law is ca. 4.24 A.
J. Mater. Chem., 2009, 19, 733–742 | 735
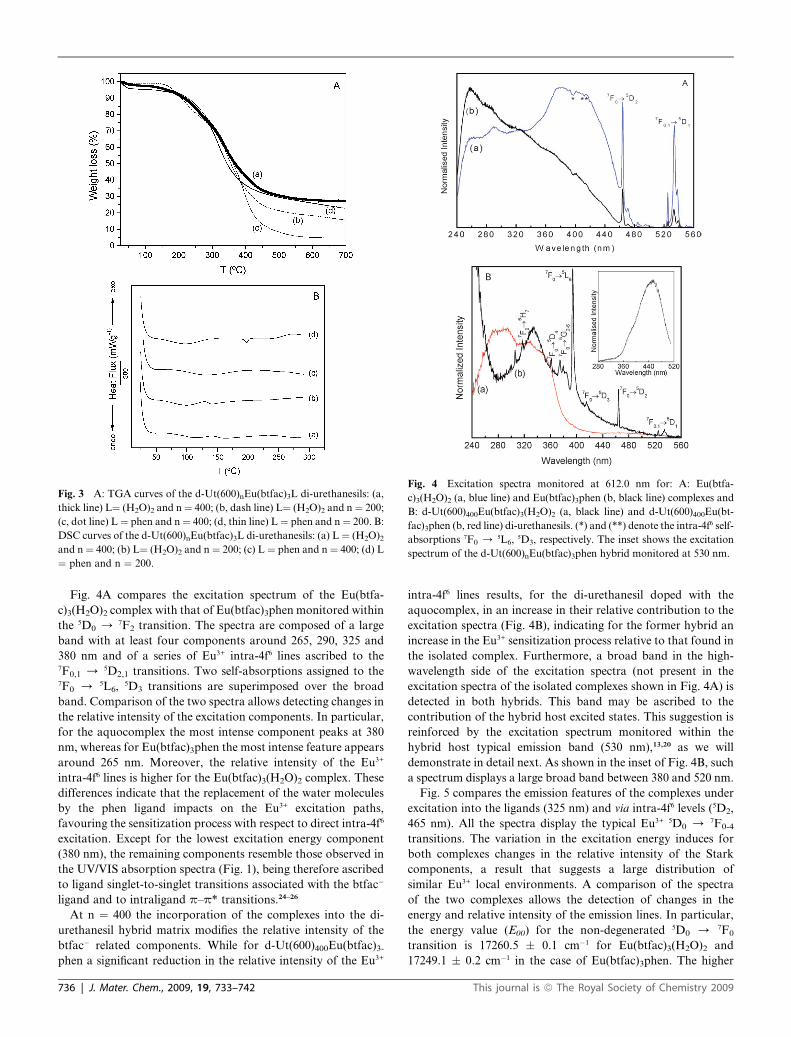
Fig. 4 Excitation spectra monitored at 612.0 nm for: A: Eu(btfa-
c)3(H2O)2 (a, blue line) and Eu(btfac)3phen (b, black line) complexes and
B: d-Ut(600)400Eu(btfac)3(H2O)2 (a, black line) and d-Ut(600)400Eu(bt-
fac)3phen (b, red line) di-urethanesils. (*) and (**) denote the intra-4f6 self-
absorptions 7F0 / 5L6, 5D3, respectively. The inset shows the excitation
spectrum of the d-Ut(600)nEu(btfac)3phen hybrid monitored at 530 nm.
Fig. 3 A: TGA curves of the d-Ut(600)nEu(btfac)3L di-urethanesils: (a,
thick line) L¼ (H2O)2 and n ¼ 400; (b, dash line) L¼ (H2O)2 and n ¼ 200;
(c, dot line) L ¼ phen and n ¼ 400; (d, thin line) L ¼ phen and n ¼ 200. B:
DSC curves of the d-Ut(600)nEu(btfac)3L di-urethanesils: (a) L ¼ (H2O)2
and n ¼ 400; (b) L¼ (H2O)2 and n ¼ 200; (c) L ¼ phen and n ¼ 400; (d) L
¼ phen and n ¼ 200.
Fig. 4A compares the excitation spectrum of the Eu(btfa-
c)3(H2O)2 complex with that of Eu(btfac)3phen monitored within
the 5D0 / 7F2 transition. The spectra are composed of a large
band with at least four components around 265, 290, 325 and
380 nm and of a series of Eu3+ intra-4f6 lines ascribed to the7F0,1 / 5D2,1 transitions. Two self-absorptions assigned to the7F0 / 5L6, 5D3 transitions are superimposed over the broad
band. Comparison of the two spectra allows detecting changes in
the relative intensity of the excitation components. In particular,
for the aquocomplex the most intense component peaks at 380
nm, whereas for Eu(btfac)3phen the most intense feature appears
around 265 nm. Moreover, the relative intensity of the Eu3+
intra-4f6 lines is higher for the Eu(btfac)3(H2O)2 complex. These
differences indicate that the replacement of the water molecules
by the phen ligand impacts on the Eu3+ excitation paths,
favouring the sensitization process with respect to direct intra-4f6
excitation. Except for the lowest excitation energy component
(380 nm), the remaining components resemble those observed in
the UV/VIS absorption spectra (Fig. 1), being therefore ascribed
to ligand singlet-to-singlet transitions associated with the btfac�
ligand and to intraligand p–p* transitions.24–26
At n ¼ 400 the incorporation of the complexes into the di-
urethanesil hybrid matrix modifies the relative intensity of the
btfac� related components. While for d-Ut(600)400Eu(btfac)3-
phen a significant reduction in the relative intensity of the Eu3+
736 | J. Mater. Chem., 2009, 19, 733–742
intra-4f6 lines results, for the di-urethanesil doped with the
aquocomplex, in an increase in their relative contribution to the
excitation spectra (Fig. 4B), indicating for the former hybrid an
increase in the Eu3+ sensitization process relative to that found in
the isolated complex. Furthermore, a broad band in the high-
wavelength side of the excitation spectra (not present in the
excitation spectra of the isolated complexes shown in Fig. 4A) is
detected in both hybrids. This band may be ascribed to the
contribution of the hybrid host excited states. This suggestion is
reinforced by the excitation spectrum monitored within the
hybrid host typical emission band (530 nm),13,20 as we will
demonstrate in detail next. As shown in the inset of Fig. 4B, such
a spectrum displays a large broad band between 380 and 520 nm.
Fig. 5 compares the emission features of the complexes under
excitation into the ligands (325 nm) and via intra-4f6 levels (5D2,
465 nm). All the spectra display the typical Eu3+ 5D0 / 7F0-4
transitions. The variation in the excitation energy induces for
both complexes changes in the relative intensity of the Stark
components, a result that suggests a large distribution of
similar Eu3+ local environments. A comparison of the spectra
of the two complexes allows the detection of changes in the
energy and relative intensity of the emission lines. In particular,
the energy value (E00) for the non-degenerated 5D0 / 7F0
transition is 17260.5 � 0.1 cm�1 for Eu(btfac)3(H2O)2 and
17249.1 � 0.2 cm�1 in the case of Eu(btfac)3phen. The higher
This journal is ª The Royal Society of Chemistry 2009
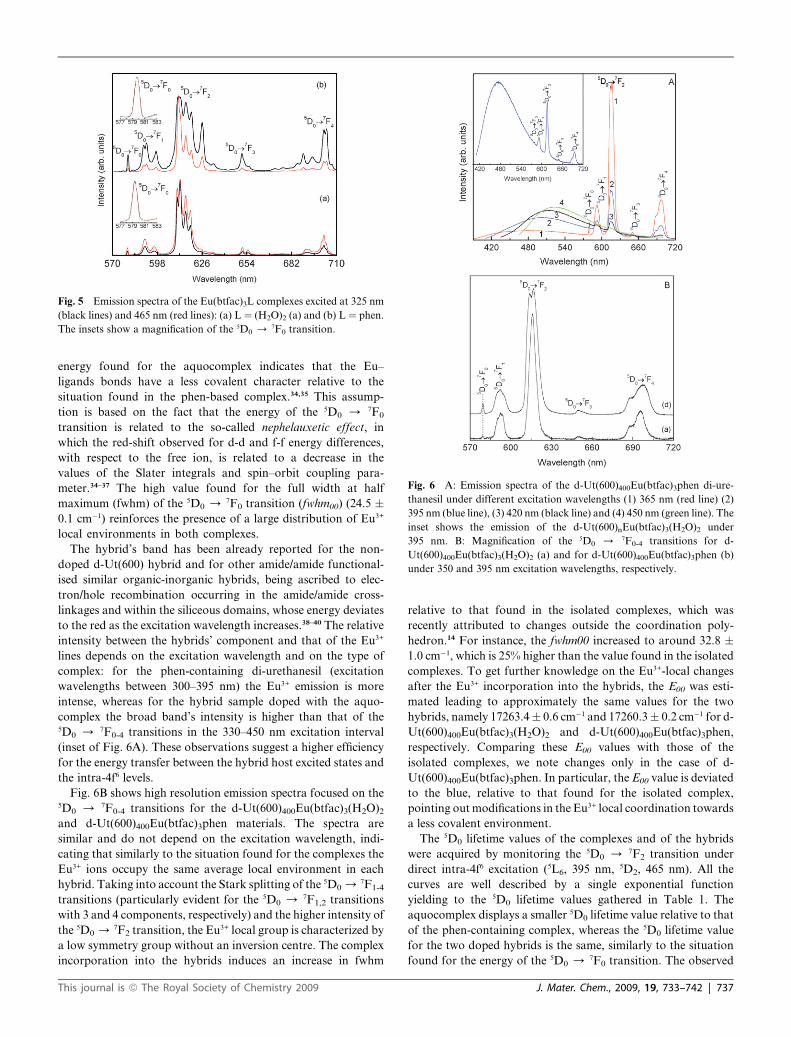
Fig. 6 A: Emission spectra of the d-Ut(600)400Eu(btfac)3phen di-ure-
thanesil under different excitation wavelengths (1) 365 nm (red line) (2)
395 nm (blue line), (3) 420 nm (black line) and (4) 450 nm (green line). The
inset shows the emission of the d-Ut(600)nEu(btfac)3(H2O)2 under
395 nm. B: Magnification of the 5D0 / 7F0-4 transitions for d-
Ut(600)400Eu(btfac)3(H2O)2 (a) and for d-Ut(600)400Eu(btfac)3phen (b)
under 350 and 395 nm excitation wavelengths, respectively.
Fig. 5 Emission spectra of the Eu(btfac)3L complexes excited at 325 nm
(black lines) and 465 nm (red lines): (a) L ¼ (H2O)2 (a) and (b) L ¼ phen.
The insets show a magnification of the 5D0 / 7F0 transition.
energy found for the aquocomplex indicates that the Eu–
ligands bonds have a less covalent character relative to the
situation found in the phen-based complex.34,35 This assump-
tion is based on the fact that the energy of the 5D0 / 7F0
transition is related to the so-called nephelauxetic effect, in
which the red-shift observed for d-d and f-f energy differences,
with respect to the free ion, is related to a decrease in the
values of the Slater integrals and spin–orbit coupling para-
meter.34–37 The high value found for the full width at half
maximum (fwhm) of the 5D0 / 7F0 transition (fwhm00) (24.5 �0.1 cm�1) reinforces the presence of a large distribution of Eu3+
local environments in both complexes.
The hybrid’s band has been already reported for the non-
doped d-Ut(600) hybrid and for other amide/amide functional-
ised similar organic-inorganic hybrids, being ascribed to elec-
tron/hole recombination occurring in the amide/amide cross-
linkages and within the siliceous domains, whose energy deviates
to the red as the excitation wavelength increases.38–40 The relative
intensity between the hybrids’ component and that of the Eu3+
lines depends on the excitation wavelength and on the type of
complex: for the phen-containing di-urethanesil (excitation
wavelengths between 300–395 nm) the Eu3+ emission is more
intense, whereas for the hybrid sample doped with the aquo-
complex the broad band’s intensity is higher than that of the5D0 / 7F0-4 transitions in the 330–450 nm excitation interval
(inset of Fig. 6A). These observations suggest a higher efficiency
for the energy transfer between the hybrid host excited states and
the intra-4f6 levels.
Fig. 6B shows high resolution emission spectra focused on the5D0 / 7F0-4 transitions for the d-Ut(600)400Eu(btfac)3(H2O)2
and d-Ut(600)400Eu(btfac)3phen materials. The spectra are
similar and do not depend on the excitation wavelength, indi-
cating that similarly to the situation found for the complexes the
Eu3+ ions occupy the same average local environment in each
hybrid. Taking into account the Stark splitting of the 5D0 /7F1-4
transitions (particularly evident for the 5D0 / 7F1,2 transitions
with 3 and 4 components, respectively) and the higher intensity of
the 5D0 /7F2 transition, the Eu3+ local group is characterized by
a low symmetry group without an inversion centre. The complex
incorporation into the hybrids induces an increase in fwhm
This journal is ª The Royal Society of Chemistry 2009
relative to that found in the isolated complexes, which was
recently attributed to changes outside the coordination poly-
hedron.14 For instance, the fwhm00 increased to around 32.8 �1.0 cm�1, which is 25% higher than the value found in the isolated
complexes. To get further knowledge on the Eu3+-local changes
after the Eu3+ incorporation into the hybrids, the E00 was esti-
mated leading to approximately the same values for the two
hybrids, namely 17263.4 � 0.6 cm�1 and 17260.3 � 0.2 cm�1 for d-
Ut(600)400Eu(btfac)3(H2O)2 and d-Ut(600)400Eu(btfac)3phen,
respectively. Comparing these E00 values with those of the
isolated complexes, we note changes only in the case of d-
Ut(600)400Eu(btfac)3phen. In particular, the E00 value is deviated
to the blue, relative to that found for the isolated complex,
pointing out modifications in the Eu3+ local coordination towards
a less covalent environment.
The 5D0 lifetime values of the complexes and of the hybrids
were acquired by monitoring the 5D0 / 7F2 transition under
direct intra-4f6 excitation (5L6, 395 nm, 5D2, 465 nm). All the
curves are well described by a single exponential function
yielding to the 5D0 lifetime values gathered in Table 1. The
aquocomplex displays a smaller 5D0 lifetime value relative to that
of the phen-containing complex, whereas the 5D0 lifetime value
for the two doped hybrids is the same, similarly to the situation
found for the energy of the 5D0 / 7F0 transition. The observed
J. Mater. Chem., 2009, 19, 733–742 | 737
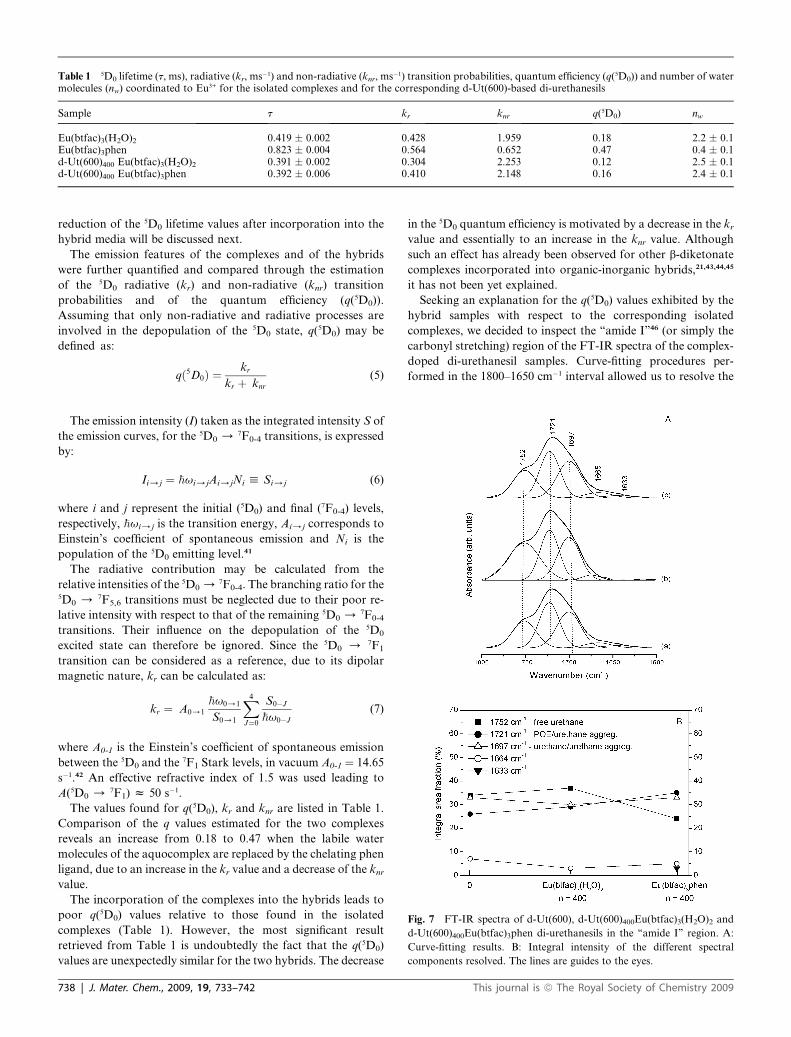
Table 1 5D0 lifetime (t, ms), radiative (kr, ms�1) and non-radiative (knr, ms�1) transition probabilities, quantum efficiency (q(5D0)) and number of watermolecules (nw) coordinated to Eu3+ for the isolated complexes and for the corresponding d-Ut(600)-based di-urethanesils
Sample t kr knr q(5D0) nw
Eu(btfac)3(H2O)2 0.419 � 0.002 0.428 1.959 0.18 2.2 � 0.1Eu(btfac)3phen 0.823 � 0.004 0.564 0.652 0.47 0.4 � 0.1d-Ut(600)400 Eu(btfac)3(H2O)2 0.391 � 0.002 0.304 2.253 0.12 2.5 � 0.1d-Ut(600)400 Eu(btfac)3phen 0.392 � 0.006 0.410 2.148 0.16 2.4 � 0.1
Fig. 7 FT-IR spectra of d-Ut(600), d-Ut(600)400Eu(btfac)3(H2O)2 and
d-Ut(600)400Eu(btfac)3phen di-urethanesils in the ‘‘amide I’’ region. A:
Curve-fitting results. B: Integral intensity of the different spectral
components resolved. The lines are guides to the eyes.
reduction of the 5D0 lifetime values after incorporation into the
hybrid media will be discussed next.
The emission features of the complexes and of the hybrids
were further quantified and compared through the estimation
of the 5D0 radiative (kr) and non-radiative (knr) transition
probabilities and of the quantum efficiency (q(5D0)).
Assuming that only non-radiative and radiative processes are
involved in the depopulation of the 5D0 state, q(5D0) may be
defined as:
qð5D0Þ ¼kr
kr þ knr(5)
The emission intensity (I) taken as the integrated intensity S of
the emission curves, for the 5D0 /7F0-4 transitions, is expressed
by:
Ii/j ¼ hui/jAi/jNi h Si/j (6)
where i and j represent the initial (5D0) and final (7F0-4) levels,
respectively, hui/j is the transition energy, Ai/j corresponds to
Einstein’s coefficient of spontaneous emission and Ni is the
population of the 5D0 emitting level.41
The radiative contribution may be calculated from the
relative intensities of the 5D0 /7F0-4. The branching ratio for the
5D0 / 7F5,6 transitions must be neglected due to their poor re-
lative intensity with respect to that of the remaining 5D0 /7F0-4
transitions. Their influence on the depopulation of the 5D0
excited state can therefore be ignored. Since the 5D0 / 7F1
transition can be considered as a reference, due to its dipolar
magnetic nature, kr can be calculated as:
kr ¼ A0/1
hu0/1
S0/1
X4
J¼0
S0�J
hu0�J
(7)
where A0-1 is the Einstein’s coefficient of spontaneous emission
between the 5D0 and the 7F1 Stark levels, in vacuum A0-1 ¼ 14.65
s�1.42 An effective refractive index of 1.5 was used leading to
A(5D0 /7F1) z 50 s�1.
The values found for q(5D0), kr and knr are listed in Table 1.
Comparison of the q values estimated for the two complexes
reveals an increase from 0.18 to 0.47 when the labile water
molecules of the aquocomplex are replaced by the chelating phen
ligand, due to an increase in the kr value and a decrease of the knrvalue.
The incorporation of the complexes into the hybrids leads to
poor q(5D0) values relative to those found in the isolated
complexes (Table 1). However, the most significant result
retrieved from Table 1 is undoubtedly the fact that the q(5D0)
values are unexpectedly similar for the two hybrids. The decrease
738 | J. Mater. Chem., 2009, 19, 733–742
in the 5D0 quantum efficiency is motivated by a decrease in the krvalue and essentially to an increase in the knr value. Although
such an effect has already been observed for other b-diketonate
complexes incorporated into organic-inorganic hybrids,21,43,44,45
it has not been yet explained.
Seeking an explanation for the q(5D0) values exhibited by the
hybrid samples with respect to the corresponding isolated
complexes, we decided to inspect the ‘‘amide I’’46 (or simply the
carbonyl stretching) region of the FT-IR spectra of the complex-
doped di-urethanesil samples. Curve-fitting procedures per-
formed in the 1800–1650 cm�1 interval allowed us to resolve the
This journal is ª The Royal Society of Chemistry 2009

band profile of the d-Ut(600) hybrid into three main distinct
components located at about 1752, 1721 and 1697 cm�1
(Fig. 7A(a)). The weak 1752 cm�1 band is ascribed to the pres-
ence of ‘‘free’’ (i.e., non-hydrogen-bonded) carbonyl groups of
the urethane linkages of d-Ut(600).47 The 1721 and 1697 cm�1
components are associated with the occurrence of hydrogen-
bonded POE/urethane and urethane/urethane aggregates,
respectively.47
The plot of Fig. 7B shows that, despite the extremely low
concentration of Eu3+ in the hybrid material with n ¼ 400 (see
Table 2 of the Experimental section), the inclusion of
Eu(btfac)3phen within d-Ut(600) alters the ‘‘amide I’’ band of
this host hybrid, leading to band redistribution. This finding may
be correlated with modifications in the chemical environment of
the urethane carbonyl groups of d-Ut(600) in the presence of this
complex. No other significant spectral changes are detected,
however, in the ‘‘amide I’’ region of d-Ut(600)400Eu(btfac)3phen,
in particular the emergence of a new Eu3+ coordination-sensitive
absorption band, usually located below 1690 cm�1, and typically
produced by the di-ureasils doped with europium complexes14,15
and mono47/di-urethanesils12,30 doped with europium salts. Close
analysis of the plot of Fig. 7B allows the inference that upon
addition of Eu(btfac)3phen, the proportion of ‘‘free’’ carbonyl
groups is reduced at the expense of the formation of POE/
urethane aggregates. Nevertheless, the fraction of urethane/
urethane aggregates remains essentially unchanged (Fig. 7B).
This means that the addition of Eu(btfac)3phen to d-Ut(600)
promotes the interaction between the POE chains and the ‘‘free’’
carbonyl groups of the hybrid matrix, but does not induce the
rupture of any hydrogen-bonded aggregates of the matrix.
Therefore the poor q(5D0) value of the d-Ut(600)400Eu(btfac)3-
phen sample (Table 1) may simply be a consequence of the
bulkiness of the phen ligand. In fact it is very likely that due to
severe steric constraints arising from the formation of the new
POE/urethane aggregates, the Eu3+-phen distance increases
significantly. In such a case, several possibilities would remain for
the emitting centres: they might either bond to water molecules
or to urethane carbonyl oxygen atoms or to ether oxygen atoms
of the POE chains. Considering that the urethane cross-links and
the polymer segments of this hybrid are deeply involved in the
process of formation of POE/urethane aggregates, the hypothesis
of the establishment of Eu3+/O]C or/and Eu3+/OCC bonds,
respectively, seems to be highly improbable. Hence, water
Table 2 Experimental details of the synthesis of the d-Ut(600)-based di-urethanesils
d-Ut(600)nEu(btfac)3L
L (H2O)2 phen
n 400 200 400 200
m(PEG) (g) 1.04 1.05 1.01 1.03v(ICPTES) (mL) 852 865 831 846v(CH3CH2OH) (mL) 815 828 795 809v(H2O) (mL) 93 95 91 93m(complex) (g) 0.04485 0.08645 0.05444 0.11081Si/Eu (molmol�1) 64 34 60 30Si/Eu (gg�1) 12 6 11 6
This journal is ª The Royal Society of Chemistry 2009
molecules will presumably replace the phen ligand in the hybrid
system to maintain the Eu3+ coordination number constant.
In contrast, the inclusion of the aquocomplex in the di-ure-
thanesil medium appears to have practically no consequences on
the hydrogen-bonded array of d-Ut(600) and on the population
of ‘‘free’’ urethane groups (Fig. 7B). Therefore, these spectral
data suggest that the Eu(btfac)3(H2O)2 complex does not interact
at all with d-Ut(600). This might explain why the q(5D0) value of
Eu(btfac)3(H2O)2 suffers practically no change upon incorpora-
tion into the d-Ut(600) structure (Table 1).
The photoluminescence data provide full support of the
conclusions retrieved from the ‘‘amide I’’ region. The number of
water molecules (nw) coordinated to Eu3+ calculated using the
empirical formula of Supkowski and Horrocks:48
nw ¼ 1.11 � [kexp � kr � 0.31] (8)
where kexp¼(t)�1 yielded nw values for the aquocomplex and for
Eu(btfac)3phen of approximately 2 and 0, respectively (Table 1),
thus in perfect agreement with the structures proposed in Scheme
1. For the corresponding hybrid samples approximately 2 water
molecules were estimated in both cases (Table 1). The latter
finding lends support to the claim that in the case of the aquo-
complex the d-Ut(600) structure acts merely as an inert support.
In addition, it may be interpreted as an indication that in the case
of the di-urethanesil doped with Eu(btfac)3phen the composition
of the coordination sphere of the Eu3+ ions is deeply modified as
a result of the significant displacement suffered by the bidentate
phen ligand away from the emitting centre and its concomitant
replacement by two water molecules. Considering the well-
known strong chelating ability of the phen ligand, this unex-
pected withdrawal from the Eu3+ chemical environment must be
related to the occurrence of severe steric effects involving the
bulky phen molecules and the POE chains.
Experimental
Materials
Europium chloride (EuCl3.6H2O, Aldrich, 99.99%), 4,4,4-trifluoro-
1-phenyl-1,3-butanedione (Hbtfac, Aldrich, 99%), 1,10-phenan-
throline (phen, Aldrich, +99%,), 3-isocyanatepropyltriethoxysilane
(ICPTES, Fluka, 95%), acetonitrile (CH3CN, Aldrich, HPLC
grade), tetrabutylammonium tetrafluoroborate (TBABF4, Aldrich
99%) and potassium bromide (KBr, Merck, spectroscopic grade)
were used as received. Poly(ethylene glycol) (PEG(600), Aldrich,
95%, MW z 600 gmol�1), ethanol (CH3CH2OH, Panreac, PA
grade) and tetrahydrofurane (THF; Merck, PA grade) were stored
over molecular sieves. High purity distilled water was used in all
experiments.
Synthesis
To synthesize Eu(btfac)3(H2O)2 (Scheme 1A)49 a mass of 0.6477 g
(2.99 mmol) of Hbtfac was dissolved in 15 mL of CH3CH2OH.
The pH of this solution was adjusted to 6–7 by adding an
appropriate amount of an aqueous NaOH solution (1 M). An
aqueous solution of EuCl3$6H2O (0.3603 g, 9.8 � 10�4 mol in
5 mL of water) was then added dropwise to the ethanolic solution
of Hbtfac. The addition of 100 mL of water on heating at 60 �C
J. Mater. Chem., 2009, 19, 733–742 | 739

for 120 minutes followed. The resulting solution was left in the
fume cupboard until precipitation. The yellow solid obtained was
filtered off, washed with water and dried in a vacuum desiccator
at room temperature. Yield: 21%. % Anal. calcd.: C: 43.21, H:
2.64; Found: C: 43.21, H: 2.49.
To synthesize Eu(btfac)3phen (Scheme 1B)50 a mass of 0.6562 g
(3 mmol) of Hbtfac and 0.1807 g, phen (1 mmol) was dissolved in
15 mL of ethanol. The pH of this solution was adjusted to 6–7 by
adding an appropriate amount of an aqueous NaOH solution
(1 M). An aqueous solution of EuCl3.6H2O (0.3575 g, 9.75 �10�4 mol in 5 mL of water) was then added dropwise to the
ethanolic solution of Hbtfac and phen on heating at 60 �C for 120
minutes. The resulting solution was left in the fume cupboard
until precipitation. The yellow solid obtained was filtered off,
washed with water and dried in a vacuum desiccator at room
temperature. Yield: 24%. % Anal. calcd: C: 51.59, H: 2.66, N:
2.87; Found: C: 51.52, H: 2.40, N: 3.01.
The doped di-urethanesil hybrid matrices were prepared by the
conventional sol-gel method, according to the two-step experi-
mental procedure described in detail previously.20 The experi-
mental details of the synthesis of the doped samples are presented
in Table 2. Transparent monolithic xerogels were thus formed. A
non-doped di-urethanesil sample was also prepared.
‡ PeakFit is a product of Jandel Corporation, 2591 Rerner Boulevard,San Rafael, CA 94901, USA
Characterization
The elemental analyses of the complexes were performed at
Centro de Apoio Cientıfico e Tecnoloxico a Investigacion
CACTI-University of Vigo (Spain).
The absorption spectra of the complexes were acquired in the
700–200 nm range with a ultraviolet/visible UV/VIS Spectronic
Genexys 2PCC spectrophotometer. The solvent employed was
CH3CH2OH.
CV studies were performed in a three-electrode cell consisting
of a platinum working electrode, a platinum counter electrode
and a saturated calomel as a reference electrode, using a scan rate
of 50 mV/s (Solartron potentiostat 1285). The redox couple
ferrocene/ferricenium ion (Fc/Fc+) was used as an external
standard. Its energy, with respect to the vacuum level, defined as
zero, was estimated to be �4.8 eV.27,51 The complexes were dis-
solved in a solution of CH3CN containing 0.2 M TBABF4. The
electrolyte was purged with nitrogen (N2) and the measurements
were made under an inert atmosphere. The oxidation and
reduction potentials were deduced from the intersection of two
curves, one corresponding to the onset potential and the other to
the residual current of the voltammogram.
Samples for thermogravimetric studies were transferred to
open platinum pans and analyzed using a TA Instruments Q50
thermobalance at a heating rate of 10 �Cmin�1 using dried N2 as
the purging gas (40 mL min�1). Prior to measurement, the
samples were vacuum-dried over phosphorus pentoxide (P2O5).
A DSC131 Setaram differential scanning calorimeter was used
to determine the thermal behaviour of the complexes and of the
di-urethanesil hybrids. A mass of 10–30 mg of the finely ground
powder sample (complex) or film (xerogel) was placed in a 30 mL
aluminium can and stored in a desiccator over P2O5 for 1 week at
room temperature under vacuum. After this drying treatment the
can was hermetically sealed and the thermograms were recorded.
Each sample was heated from 25 to 300 �C at 10 �Cmin�1.
740 | J. Mater. Chem., 2009, 19, 733–742
The purge gas used was high purity N2 supplied at a constant
35 mL min�1 flow rate.
The FT-IR spectra were acquired at room temperature on
a Unicam FT-IR spectrophotometer. The spectra were collected
in the 4000–500 cm�1 range by averaging 64 scans at a resolution
of 4 cm�1. About 2 mg of each complex was mixed with 175 mg of
KBr finely ground and pressed into pellets. Prior to recording the
spectra the complexes were stored under vacuum for about 24
hours at approximately 80 �C to reduce the levels of adsorbed
water. To evaluate broad band envelopes and to identify
underlying spectral components, the iterative least-squares
curve-fitting procedure in the PeakFit‡ software was employed.
The best fit of the experimental data was obtained by varying the
frequency, bandwidth and intensity of the bands and by using
a Gaussian shape. A linear baseline correction with a tolerance of
0.2% was employed. The standard errors of the curve-fitting
procedure were less than 0.002.
XRD measurements were performed at room temperature
with a PANalytical X’Pert Pro equipped with a X’Celerator
detector using monochromated CuKa radiation (l ¼ 1.541 A)
over the 2q range between 10 and 70�. The di-urethanesil
samples, analyzed as films, were not submitted to any thermal
pre-treatment.
To evaluate the morphology of the samples, SEM micrographs
were obtained using a SEM/ESEM-FEI Quanta 400 scanning
electron microscope at high acceleration voltage (25 kV).
The samples were coated with a thin layer of carbon by ion
sputtering.
The photoluminescence spectra were recorded at room
temperature with a modular double grating excitation spectro-
fluorimeter with a TRIAX 320 emission monochromator (Flu-
orolog-3, Jobin Yvon-Spex) coupled to a R928 Hamamatsu
photomultiplier, using the front face acquisition mode. The
excitation source was a 450W Xe arc lamp. The emission spectra
were corrected for detection and optical spectral response of the
spectrofluorimeter and the excitation spectra were corrected for
the spectral distribution of the lamp intensity using a photodiode
reference detector. The lifetime measurements were acquired
with the setup described for the luminescence spectra using
a pulsed Xe-Hg lamp (6 ms pulse at half width and 20–30 ms tail).
Conclusions
In the present work we studied the morphological, structural and
thermal properties of hybrids composed of the short chain di-
urethanesil d-Ut(600) host framework and the Eu(btfac)3(H2O)2
and Eu(btfac)3phen complexes. Xerogels with compositions
n ¼ 400 and 200 were prepared. The samples produced are
transparent, non-porous, homogeneous and thermally stable
up to about 150 �C. Except for the Eu(btfac)3phen-doped di-
urethanesil with n ¼ 200, where a minor proportion of free
complex was detected, the remaining materials are amorphous.
The ultimate goal of this investigation was to elucidate the
behaviour of the Eu(btfac)3(H2O)2 complex within the d-Ut(600)
medium and in particular to determine if, similarly to the situa-
tion observed in analogue hybrid systems previously analyzed by
This journal is ª The Royal Society of Chemistry 2009

our group (di-ureasils17,19 and di-urethanesils21), the coordinating
functionalities of the hybrid host could act as complex anchoring
sites and thus play an active role in the enhancement of the 5D0
quantum efficiency q(5D0) of the isolated aquocomplex. In the
case of d-Ut(600) this complex immobilization process was
expected to involve bonding of the Eu3+ ions to either the
carbonyl oxygen atoms of the urethane cross-linkages or the
ether oxygen atoms of the POE chains or both, and the simul-
taneous release of one(two) labile water ligand(s) of the
aquocomplex. For comparison purposes, we also examined d-
Ut(600)-based samples doped with the corresponding b-diketo-
nate complex containing a phen ligand instead of the water
ligands. The photoluminescence and FT-IR spectroscopic
analyses were focused on samples with n ¼ 400.
As a result of the lack of coordinative interaction between the
guest aquocomplex and the host hybrid matrix, the q(5D0) and nwvalues of Eu(btfac)3(H2O)2 remain practically unaltered upon
incorporation into the d-Ut(600) structure (from 0.18 to 0.12 and
from 2.2 � 0.1 to 2.5 � 0.1, respectively). These findings lead us
to suppose that the coordinating ability of the urethane carbonyl
oxygen atoms and of the ether oxygen atoms of the POE chains
of d-Ut(600) is not strong enough to displace the labile water
ligands of Eu(btfac)3(H2O)2. Consequently, d-Ut(600) plays the
mere role of support in the presence of this complex, although
ensuring the maintenance of the optical features in the isolated
state.
Doping d-Ut(600) with Eu(btfac)3phen is clearly disadvanta-
geous, as a significant reduction of the q(5D0) value of the iso-
lated complex occurs (from 0.47 to 0.16). Intense repulsive forces
originating from severe steric interactions taking place between
the POE chains and the bulky phen molecules are thought to be
responsible for the unusual displacement of the strong bidentate
chelating phen ligand from the Eu3+ first coordination shell and
its subsequent replacement by two water molecules (nw ¼ 2.4 �0.1 for d-Ut(600)400Eu(btfac)3phen). This uncommon effect
may be interpreted as an indication that in both d-Ut(600)-based
materials the local environment of the Eu3+ ions is grosso modo
the same. Not surprisingly, the q(5D0) values of both hybrids are
similar. The decrease of q(5D0) upon doping was correlated in
d-Ut(600)400Eu(btfac)(H2O)2 and d-Ut(600)400Eu(btfac)3phen
with a decrease in the radiative paths and essentially to an
increase in the non-radiative channels.
In terms of complex anchoring ability, the behaviour of
d-Ut(600) contrasts deeply with that of the PCL(530)/siloxane di-
urethanesil,21 in which the polymer carbonyl oxygen atoms
govern the complex immobilization. It also differs markedly
from that of the structurally analogous d-U(600) di-ureasil
matrix, whose POE chains dictate the in situ complex forma-
tion.17 It is thus evident that the nature of the coordinating atoms
of the cross-links and polymer chains are key parameters in the
whole process. However, the study carried out with the co-
condensed d-U(900)/d-U(600) di-ureasil-type hybrid network19
suggests that the length of the polymer chains of the hybrid
structure may also influence it.
In conclusion, the use of aquocomplexes may be attractive for
optical purposes, as long as an adequate host hybrid matrix and
appropriate ligands are employed. This choice will determine the
balance of radiative and non-radiative channels of the 5D0 level
and will consequently affect the magnitude of q(5D0).
This journal is ª The Royal Society of Chemistry 2009
Acknowledgements
The support of Fundacao para a Ciencia e Tecnologia (FCT)
(PTDC/CTM/72093/2006) is gratefully acknowledged. M. Fer-
nandes and S. Nobre thank FCT for grants (SFRH/BD/38530/
2007 and SFRH/BD/28739/2006, respectively). The authors
would like to express their gratitude to H. D. Burrows, of
the Department of Chemistry of the University of Coimbra
(Portugal), for helpful discussions.
References
1 J.-M. Lehn, Angew. Chem. Int. Ed. Engl., 1990, 29, 1304.2 G. F. de Sa, O. L. Malta, C. de Mello Donega, A. M. Simas,
R. L. Longo, P. A. Santa-Cruz and E. F. da Silva Jr., Coord. Chem.Rev., 2000, 196, 165.
3 E. B. van der Tol, H. J. van Ramesdonk, J. W. Verhoeven,F. J. Steemers, E. G. Kerver, W. Verboom and D. N. Reinhoudt,Chem. Eur. J., 1998, 4, 2315.
4 J.-C. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048.5 N. S. Baek, Y. H. Kim, S.-G. Roh, B. K. Kwak and H. K. Kim, Adv.Funct. Mater., 2006, 16, 1873.
6 M. H. V. Werts, R. H. Woudenberg, P. G. Emmerink, R. van Gassel,J. W. Hofstraat and J. W. Verhoeven, Angew. Chem. Intl. Ed., 2000,39, 4542.
7 C. J. Brinker and G. W. Scherer, in Sol-Gel Science: The Physics andChemistry of Sol-Gel Processing, Academic Press, San Diego, CA,1990.
8 P. Gomez-Romero and C. Sanchez, in Functional Hybrid Materials,Wiley Interscience, New York, 2003.
9 P. Escribano, B. Julian-Lopez, J. Planelles-Arago, E. Cordoncillo,B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23.
10 C. Sanchez, B. Julian, P. Bellevile and M. Popall, J. Mater. Chem.,2005, 15, 3559.
11 M. Armand, C. Poinsignon, J.-Y. Sanchez and V. de Zea Bermudez,U. S. Patent 5 283 310, 1993.
12 M. C. Goncalves, N. J. O. Silva, V. de Zea Bermudez, R. A. SaFerreira, L. D. Carlos, K. Dahmouche, C. V. Santilli,D. Ostrovskii, I. C. Correia Vilela and A. F. Craievich, J. Phys.Chem. B, 2005, 109, 20093.
13 L. D. Carlos, R. A. Sa Ferreira, V. de Zea Bermudez, in HybridMaterials - Synthesis, Characterization and Aplication, ed. G.Kickelbick, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,2007, (ch. 9), pp. 337–400.
14 P. P. Lima, R. A. Sa Ferreira, R. O. Freire, F. A. A. Paz, L. Fu,S. Alves Jr, L. D. Carlos and O. Malta, Chem. Phys. Chem, 2006, 7,735.
15 L. Fu, R. A. Sa Ferreira, N. J. O. Silva, A. J. Fernandes, P. Ribeiro-Claro, I. S. Goncalves, V. de Zea Bermudez and L. D. Carlos,J. Mater. Chem., 2005, 15, 3117.
16 L. Fu, R. A. Sa Ferreira, S. S. Nobre, L. D. Carlos and J. Rocha,J. Lumin., 2006, 122–123, 265.
17 C. Molina, K. Dahmouche, K. Y. Messadeq, S. J. L. Ribeiro,M. A. P. Silva, V. de Zea Bermudez and L. D. Carlos, J. Lumin.,2003, 93, 104.
18 P. P. Lima, S. A. Junior, O. L. Malta, L. D. Carlos, R. A. Sa Ferreira,R. Pavithran and M. L. P. Reddy, Eur. J. Inorg. Chem., 2006, 19,3923.
19 M. Fernandes, V. de Zea Bermudez, R. A. Sa Ferreira, L. D. Carlosand N. V. Martins, J. Lumin., 2008, 128, 205.
20 M. Fernandes, M. C. Goncalves, V. de Zea Bermudez, R. A. SaFerreira, L. D. Carlos, A. Charas and J. Morgado, J. AlloysCompds., 2008, 451, 201.
21 M. Fernandes, V. de Zea Bermudez, R. A. Sa Ferreira, L. D. Carlos,A. Charas, J. Morgado, M. M. Silva and M. J. Smith, Chem. Mater.,2007, 19, 3892.
22 K. Binnemans, Handbook on the Physics and Chemistry of RareEarths, Elsevier, Amsterdam, 2005, vol. 34, (ch. 225), pp.107–225.
23 K. M. Lee, K. W. Cheah, B. I. An, M. I. Gong and Y. L. Liu, Appl.Phys. A, 2005, 80, 337.
24 F. R. G. Silva, J. F. S. Menezes, G. B. Rocha, S. Alves, H. F. Brito,R. L. Longo and O. L. Malta, J. Alloys Compds., 2000, 303–304, 364.
J. Mater. Chem., 2009, 19, 733–742 | 741

25 H. J. Batista, A. V. M. de Andrade, R. L. Longo, A. M. Simas,G. F. de Sa and L. C. Thompson, J. Lumin., 1997, 72–74, 159.
26 G. F. de Sa, S. Alves Jr., B. J. P. da Silva and E. F. da Silva Jr., Opt.Mater., 1998, 11, 23.
27 M. Al-Ibrahim, H.-K. Roth, U. Zhokhavets, G. Gobsch andS. Sensfuss, Sol. Energy Mater. Sol. Cells, 2005, 13, 85.
28 Z. Jablonski, L. Rychlowska-Himmel and M. Dyrek, Spectrochim.Acta, 1978, 35A, 1297.
29 S.-L. Ma, W.-X. Zhu, G.-H. Huang, D.-Q. Yuan and X. Yan, J. Mol.Struct., 2003, 646, 89.
30 M. C. Goncalves, V. de Zea Bermudez, R. A. Sa Ferreira, L. D. Carlos,D. Ostrovskii and J. Rocha, Chem. Mater., 2004, 16, 2530.
31 M. C. Goncalves, V. de Zea Bermudez, R. A. Sa Ferreira,D. Ostrovskii and L. D. Carlos, Proceedings of the MaterialsResearch Society, Boston, 2005, 847, p.EE.13.1.1.
32 M. C. Goncalves, PhD Thesis, University of Tras-os-Montes e AltoDouro, Vila Real (Portugal), 2007.
33 L. D. Carlos, V. de Zea Bermudez, R. A. Sa Ferreira, L. Marques andM. Assuncao, Chem. Mater., 1999, 11, 58113.
34 O. L. Malta, H. J. Batista and L. D. Carlos, Chem. Phys., 2002,282, 21.
35 L. D. Carlos, O. L. Malta and R. Q. Albuquerque, Chem. Phys. Lett.,2005, 415, 238.
36 L. D. Carlos and A. L. L. Videira, J. Chem. Phys., 1994, 101, 8827.37 C. K. Jørgensen, Prog. Inorg. Chem., 1962, 4, 73.38 L. D. Carlos, R. A. Sa Ferreira, V. de Zea Bermudez and
S. J. L. Ribeiro, Adv. Funct. Mater., 2001, 11, 111.
742 | J. Mater. Chem., 2009, 19, 733–742
39 L. D. Carlos, R. A. Sa Ferreira, R. N. Pereira, M. Assuncao and V. deZea Bermudez, J. Chem. Phys. B., 2004, 108, 14924.
40 S. S. Nobre, P. P. Lima, L. Mafra, R. A. Sa Ferreira, R. O. Freire,L. Fu, U. Pischel, V. de Zea Bermudez, O. L. Malta andL. D. Carlos, J. Phys. Chem. C, 2007, 111, 3275.
41 L. D. Carlos, Y. Messaddeq, H. F. Brito, R. A. Sa Ferreira, V. de ZeaBermudez and S. J. L. Ribeiro, Adv. Mater., 2000, 12, 594.
42 M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem.Chem. Phys., 2002, 4, 1542.
43 P. Lenaerts, A. Storms, J. Mullens, J. D’Haen, C. Gorller-Walrand,K. Binnemans and K. Driesen, Chem. Mater., 2005, 17, 5194.
44 P. Lenaerts, C. Gorller-Walrand and K. Binnemans, J. Lumin., 2006,117, 163.
45 P. Lenaerts, K. Driesen, R. Van Deun and K. Binnemans, Chem.Mater., 2005, 17, 2148.
46 D. J. Skrovanek, S. E. Howe, P. C. Painter and M. M. Coleman,Macromolecules, 1985, 18, 1676.
47 V. de Zea Bermudez, D. Ostrovskii, M. C. Goncalves, L. D. Carlos,R. A. Sa Ferreira, L. Reis and P. Jacobsson, Phys. Chem. Chem.Phys., 2004, 6, 638.
48 R. M. Supkowski and W. de Horrocks, Inorg. Chim. Acta, 2002,340, 44.
49 R. G. Charles and R. C. Olmann, J. Inorg. Nucl. Chem., 1965, 27, 25.50 L. R. Melby, N. J. Rose, E. Abramson and J. C. Caris, J. Am. Chem.
Soc., 1964, 86, 5117.51 H. Pommerehne, W. Guss Vestweber, R. F. Mahrt, H. Bassler,
M. Porsh and J. Daub, Adv. Mater., 1995, 7, 551.
This journal is ª The Royal Society of Chemistry 2009
Copyright © 2022 FDOKUMEN




















