
Does tautomeric equilibrium exist in ortho-nitrosonaphthols?
10
Does tautomeric equilibrium exist in ortho-nitrosonaphthols? Galya Ivanova, Venelin Enchev * Institute of Organic Chemistry, Bulgarian Academy of Sciences, Acad G Bonchev str, 1113 So®a, Bulgaria Received 2 October 2000; in ®nal form 9 January 2001 Abstract The structure and conformational equilibrium of the monooximes of 1,2-naphthoquinone were studied by solid and liquid state NMR spectroscopy and non-empirical quantum-chemical calculations. According to the experimental data and the ab initio HF/6-31G** and MP4SDTQ)/6-31G**//6-31G** levels) calculations the compounds studied exist in the gas phase and in solution as oxime tautomers only. The relative stabilities of the above compounds in chloroform and dimethylsulfoxide solution are calculated within the polarizable continuum model. Solvent eects are found to change the relative stability of the syn- and anti-isomers of 1,2-naphthoquinone-2-oxime. The presence of syn- and anti- oxime isomers of 1,2-naphthoquinone-2-oxime and two rotameric forms of syn-1,2-naphthoquinone-1-oxime in solu- tion is proved by NMR spectroscopy. Ó 2001 Elsevier Science B.V. All rights reserved. Keywords: NMR; Solvent; Ab initio calculations; Oxime; Nitroso 1. Introduction The nitroso±oxime tautomerism of 1-nitroso- 2-naphthol and 2-nitroso-1-naphthol has been treated in several papers [1±8]. By means of UV [1], IR [2], and NMR [3±5] spectroscopy it has been shown that 1-nitroso-2-naphthol exists in solution in oxime form only. According to X-ray data [6] in the solid state the same compound exists as an syn- oxime tautomer. However, the existence of both tautomeric forms of 2-nitroso-1-naphthol in chlo- roform [4] and dimethylsulfoxide adapted with 5% dioxane or acetic acid [3,4] has been suggested, based on 1 H NMR data. To our knowledge there are only two theoretical studies on the nitroso± oxime tautomerism of two ortho isomers of the nitrosonaphthol [7,8]. Pilipenko and Savranskii [7] have reported that the p-electron energies of quinone monooxime tautomers of 1-nitroso-2- naphthol and 2-nitroso-1-naphthol, calculated by means of the semiempirical PPP method, are lower than those of their respective nitroso tautomers. A more systematic theoretical study concerning the tautomerism of ortho-nitrosonaphthols has been reported by Krzan et al. [8]. Using HF/6-31G ab initio calculations the authors have shown that the nitroso tautomers of 1-nitroso-2-naphthol and 2-nitroso-1-naphthol are more stable than the ox- ime forms by 0.5 and 0.01 kcal/mol, respectively. However, these results are in contradiction with the experimental data published before [1±6]. The aim of the present study is the reinvesti- gation of the structure and relative stabilities of the tautomeric forms of 1-nitroso-2-naphthol and 2- nitroso-1-naphthol in the gas phase, solution non- polar and polar solvents) and solid state by a Chemical Physics 264 2001) 235±244 www.elsevier.nl/locate/chemphys * Corresponding author. Fax: +359-2-700225. E-mail address: [email protected] V. Enchev). 0301-0104/01/$ - see front matter Ó 2001 Elsevier Science B.V. All rights reserved. PII:S0301-010401)00245-2
Transcript of Does tautomeric equilibrium exist in ortho-nitrosonaphthols?

Does tautomeric equilibrium exist in ortho-nitrosonaphthols?
Galya Ivanova, Venelin Enchev *
Institute of Organic Chemistry, Bulgarian Academy of Sciences, Acad G Bonchev str, 1113 So®a, Bulgaria
Received 2 October 2000; in ®nal form 9 January 2001
Abstract
The structure and conformational equilibrium of the monooximes of 1,2-naphthoquinone were studied by solid and
liquid state NMR spectroscopy and non-empirical quantum-chemical calculations. According to the experimental data
and the ab initio HF/6-31G** and MP4 SDTQ)/6-31G**//6-31G** levels) calculations the compounds studied exist in
the gas phase and in solution as oxime tautomers only. The relative stabilities of the above compounds in chloroform
and dimethylsulfoxide solution are calculated within the polarizable continuum model. Solvent e�ects are found to
change the relative stability of the syn- and anti-isomers of 1,2-naphthoquinone-2-oxime. The presence of syn- and anti-
oxime isomers of 1,2-naphthoquinone-2-oxime and two rotameric forms of syn-1,2-naphthoquinone-1-oxime in solu-
tion is proved by NMR spectroscopy. Ó 2001 Elsevier Science B.V. All rights reserved.
Keywords: NMR; Solvent; Ab initio calculations; Oxime; Nitroso
1. Introduction
The nitroso±oxime tautomerism of 1-nitroso-
2-naphthol and 2-nitroso-1-naphthol has been
treated in several papers [1±8]. By means of UV [1],
IR [2], and NMR [3±5] spectroscopy it has been
shown that 1-nitroso-2-naphthol exists in solution
in oxime form only. According to X-ray data [6] in
the solid state the same compound exists as an syn-
oxime tautomer. However, the existence of both
tautomeric forms of 2-nitroso-1-naphthol in chlo-
roform [4] and dimethylsulfoxide adapted with 5%
dioxane or acetic acid [3,4] has been suggested,
based on 1H NMR data. To our knowledge there
are only two theoretical studies on the nitroso±
oxime tautomerism of two ortho isomers of the
nitrosonaphthol [7,8]. Pilipenko and Savranskii
[7] have reported that the p-electron energies of
quinone monooxime tautomers of 1-nitroso-2-
naphthol and 2-nitroso-1-naphthol, calculated by
means of the semiempirical PPP method, are lower
than those of their respective nitroso tautomers. A
more systematic theoretical study concerning the
tautomerism of ortho-nitrosonaphthols has been
reported by Krzan et al. [8]. Using HF/6-31G
ab initio calculations the authors have shown that
the nitroso tautomers of 1-nitroso-2-naphthol and
2-nitroso-1-naphthol are more stable than the ox-
ime forms by 0.5 and 0.01 kcal/mol, respectively.
However, these results are in contradiction with
the experimental data published before [1±6].
The aim of the present study is the reinvesti-
gation of the structure and relative stabilities of the
tautomeric forms of 1-nitroso-2-naphthol and 2-
nitroso-1-naphthol in the gas phase, solution non-
polar and polar solvents) and solid state by a
Chemical Physics 264 2001) 235±244
www.elsevier.nl/locate/chemphys
* Corresponding author. Fax: +359-2-700225.
E-mail address: [email protected] V. Enchev).
0301-0104/01/$ - see front matter Ó 2001 Elsevier Science B.V. All rights reserved.
PII: S0301-0104 01 )00245-2

combination of NMR and ab initio quantum-
chemical methods. The solvent e�ect on the rela-
tive stabilities of the possible tautomeric, isomeric
and rotameric forms of both nitrosonaphthols is
estimated using the polarizable continuum model
PCM).
2. Computational and spectroscopic methods
2.1. Computational details
Ab initio calculations were performed with 3-
21G [9] and 6-31G** [10,11] basis sets at Hartree±
Fock level using the GAMESS package [12]. The
geometry optimizations of the structures investi-
gated Figs. 1 and 2) were done without geomet-
rical constrains. The mean gradient threshold was
1 � 10ÿ4 hartree/Bohr. The transition structures
for tautomeric �A1 ! B1� and rotameric �A1 !A2� conversions were located at 6-31G** level.
Numerical harmonic vibrational frequency calcu-
lations were carried out to characterize the nature
of all stationary points at 6-31G** level and to
estimate the zero-point vibrational energy ZPE).
The ZPE values were scaled by 0.893 [13] to ac-
count for the overestimation of vibrational fre-
quencies. The scaled ZPE corrections were in-
cluded in the relative energy values. All transition
structures were characterized by only one imagi-
nary vibrational frequency. The force calculations
at the equilibrium structures of the selected species
yielded only real frequencies.
To further correct for electron correlation,
single-point calculations were carried out at the
MP4 SDTQ)/6-31G**//6-31G** level of theory.
To account for solvent e�ects on the relative
stabilities of selected species of the compounds
investigated, the PCM implemented in GAMESS
was used. The method computes the free energy of
solvation as the sum of electrostatic, cavitation
and van der Waals components. Speci®c compu-
tational details regarding the PCM formalism are
extensively described in reviews [14,15]. In our
investigations the electrostatic and cavitation
contributions were taken into account. The cavi-
tation component was evaluated following Pier-
ottiÕs theory [16]. It has been reported that the
solvent e�ect on the geometrical parameters is
small [17,18]. Hence for the sake of economy, the
structures investigated were not reoptimized in
solution.
Fig. 1. Oxime A and nitroso B tautomeric forms of 1-nitroso-2-naphthol 1.
236 G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244

2.2. NMR measurements
The NMR spectra were obtained on a Bruker
Avance DRX-250 spectrometer, operating at
250.13 and 62.90 MHz for 1H and 13C, respec-
tively, using a dual 5 mm probe head. The chem-
ical shifts were referenced to tetramethylsilane
TMS). The measurements in CDCl3, DMSO-d6
and DMSO-d6 � 5% CD3COOD solutions were
carried out at ambient temperature 300 K).
The following standard Bruker library pulse
programs were used for all experiments: zg, dept,
zgig, cosydftp, invbtp, inv4lplrnd. Standard 1D
experiments with 30° pulses, 1 s relaxation delays,
16K time domain points, zero-®lled to 64K for the
protons and 32K for the carbons were performed.
Hard pulses with 90° pulse widths of 11.8 ls for
the protons and 6.4 ls for the carbons at a power
level of 3 dB below the maximum output were
used. The distortionless enhancement by polar-
ization transfer DEPT) spectra were obtained
under the same conditions as the 13C-NMR spec-
tra, and s � �2 1JCH�ÿ1
� 3:45 ls was used. The13C-NMR spectra in DMSO-d6 were also recorded
using an inverse-gated sequence. The 2D 1H±1H
phase-sensitive double-quantum ®ltered COSY
DQCOSY) spectra were typically performed with
a spectral width �600 Hz, relaxation delay 2 s,
number of increments 256 or 512 and FT size
2K � 1K. The 2D 1H±13C heteronuclear multiple
quantum coherence HMQC) and 1H±13C hetero-
nuclear multiple bond connectivity HMBC) ex-
periments were carried out with a spectral width of
�2000 Hz for 1H and 7000 Hz for 13C, relaxation
delay 1.5 s, FT size 1K � 512W for the HMQC
and 2K � 256W for HMBC.13C cross polarization magic angle spinning
total suppression of side bands CPMAS TOSS)
and 13C cross polarization magic angle spinning
non-quaternary suppression total suppression of
side bands CPMAS NQS TOSS) NMR spectra
were obtained on a Bruker MSL 300 spectrometer
operating at 75.5 MHz for 13C. 13C CPMAS TOSS
and 13C CPMAS NQS TOSS experiments were
performed at magic angle spinning MAS) rates 5
kHz, using cylindrical zircon rotor with Kel-F cap,
a contact time 2 ms and repetition rate 5 s.
Commercially available Fluka) 1-nitroso-2-
naphthol and 2-nitroso-1-naphthol were used.
3. Results and discussion
1-Nitroso-2-naphthol 1) and 2-nitroso-1-naph-
thol 2) may exist in two tautomeric forms, oxime
A and nitroso B. The possible tautomeric, isomeric
and rotameric structures of these compounds are
presented in Figs. 1 and 2. The structural forms of
A are designated as A1 for the syn-isomer, A3 for
the anti-isomer, and A2 and A4 for their rotamers,
Fig. 2. Oxime A and nitroso B tautomeric forms of 2-nitroso-1-naphthol 2.
G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244 237
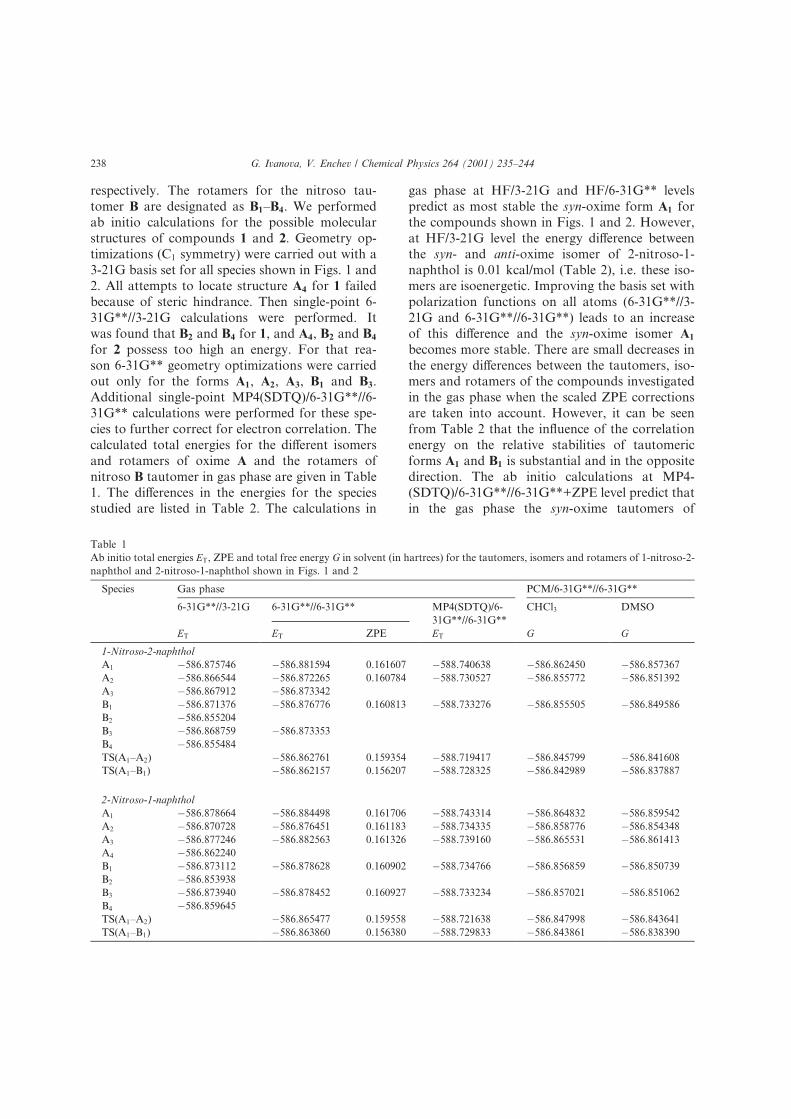
respectively. The rotamers for the nitroso tau-
tomer B are designated as B1±B4. We performed
ab initio calculations for the possible molecular
structures of compounds 1 and 2. Geometry op-
timizations C1 symmetry) were carried out with a
3-21G basis set for all species shown in Figs. 1 and
2. All attempts to locate structure A4 for 1 failed
because of steric hindrance. Then single-point 6-
31G**//3-21G calculations were performed. It
was found that B2 and B4 for 1, and A4, B2 and B4
for 2 possess too high an energy. For that rea-
son 6-31G** geometry optimizations were carried
out only for the forms A1, A2, A3, B1 and B3.
Additional single-point MP4 SDTQ)/6-31G**//6-
31G** calculations were performed for these spe-
cies to further correct for electron correlation. The
calculated total energies for the di�erent isomers
and rotamers of oxime A and the rotamers of
nitroso B tautomer in gas phase are given in Table
1. The di�erences in the energies for the species
studied are listed in Table 2. The calculations in
gas phase at HF/3-21G and HF/6-31G** levels
predict as most stable the syn-oxime form A1 for
the compounds shown in Figs. 1 and 2. However,
at HF/3-21G level the energy di�erence between
the syn- and anti-oxime isomer of 2-nitroso-1-
naphthol is 0.01 kcal/mol Table 2), i.e. these iso-
mers are isoenergetic. Improving the basis set with
polarization functions on all atoms 6-31G**//3-
21G and 6-31G**//6-31G**) leads to an increase
of this di�erence and the syn-oxime isomer A1
becomes more stable. There are small decreases in
the energy di�erences between the tautomers, iso-
mers and rotamers of the compounds investigated
in the gas phase when the scaled ZPE corrections
are taken into account. However, it can be seen
from Table 2 that the in¯uence of the correlation
energy on the relative stabilities of tautomeric
forms A1 and B1 is substantial and in the opposite
direction. The ab initio calculations at MP4-
SDTQ)/6-31G**//6-31G**+ZPE level predict that
in the gas phase the syn-oxime tautomers of
Table 1
Ab initio total energies ET, ZPE and total free energy G in solvent in hartrees) for the tautomers, isomers and rotamers of 1-nitroso-2-
naphthol and 2-nitroso-1-naphthol shown in Figs. 1 and 2
Species Gas phase PCM/6-31G**//6-31G**
6-31G**//3-21G 6-31G**//6-31G** MP4 SDTQ)/6-
31G**//6-31G**
CHCl3 DMSO
ET ET ZPE ET G G
1-Nitroso-2-naphthol
A1 ÿ586.875746 ÿ586.881594 0.161607 ÿ588.740638 ÿ586.862450 ÿ586.857367
A2 ÿ586.866544 ÿ586.872265 0.160784 ÿ588.730527 ÿ586.855772 ÿ586.851392
A3 ÿ586.867912 ÿ586.873342
B1 ÿ586.871376 ÿ586.876776 0.160813 ÿ588.733276 ÿ586.855505 ÿ586.849586
B2 ÿ586.855204
B3 ÿ586.868759 ÿ586.873353
B4 ÿ586.855484
TS A1±A2) ÿ586.862761 0.159354 ÿ588.719417 ÿ586.845799 ÿ586.841608
TS A1±B1) ÿ586.862157 0.156207 ÿ588.728325 ÿ586.842989 ÿ586.837887
2-Nitroso-1-naphthol
A1 ÿ586.878664 ÿ586.884498 0.161706 ÿ588.743314 ÿ586.864832 ÿ586.859542
A2 ÿ586.870728 ÿ586.876451 0.161183 ÿ588.734335 ÿ586.858776 ÿ586.854348
A3 ÿ586.877246 ÿ586.882563 0.161326 ÿ588.739160 ÿ586.865531 ÿ586.861413
A4 ÿ586.862240
B1 ÿ586.873112 ÿ586.878628 0.160902 ÿ588.734766 ÿ586.856859 ÿ586.850739
B2 ÿ586.853938
B3 ÿ586.873940 ÿ586.878452 0.160927 ÿ588.733234 ÿ586.857021 ÿ586.851062
B4 ÿ586.859645
TS A1±A2) ÿ586.865477 0.159558 ÿ588.721638 ÿ586.847998 ÿ586.843641
TS A1±B1) ÿ586.863860 0.156380 ÿ588.729833 ÿ586.843861 ÿ586.838390
238 G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244

compounds 1 and 2 are more stable than the
nitroso ones by 4.17 and 4.91 kcal/mol, respec-
tively.
The structures of 1-nitroso-2-naphthol and 2-
nitroso-1-naphthol in solid state and in solution
were established on the basis of NMR spectral
investigations. The complete assignments of the 1H
NMR and 13C NMR spectra of 1 and 2 in CDCl3,
DMSO-d6 and DMSO-d6 � acetic acid have been
achieved on the basis of DQ ®ltered COSY, 1H/13C
HMQC and 1H/13C HMBC experiments.
3.1. 1-Nitroso-2-naphthol
The assignments of the proton spectra of 1 in
the three solvents are in agreement with the liter-
ature data except for protons H5, H6 and H7,
which are reverse [1±5] and the chemical shifts are
now in the right order Table 3). The carbon NMR
data for compound 1 in CDCl3, DMSO-d6 and
DMSO-d6 � CD3COOD solutions at 300 K are
presented in Table 4. The 13C NMR data are in
agreement with published data [4,19] and con®rm
existence of tautomer A only. However, we ob-
serve a doubling of the resonance peaks for all CH
carbons in the carbon spectrum in chloroform
solution but the presence of only one form in
DMSO solution. We assume the existence of two
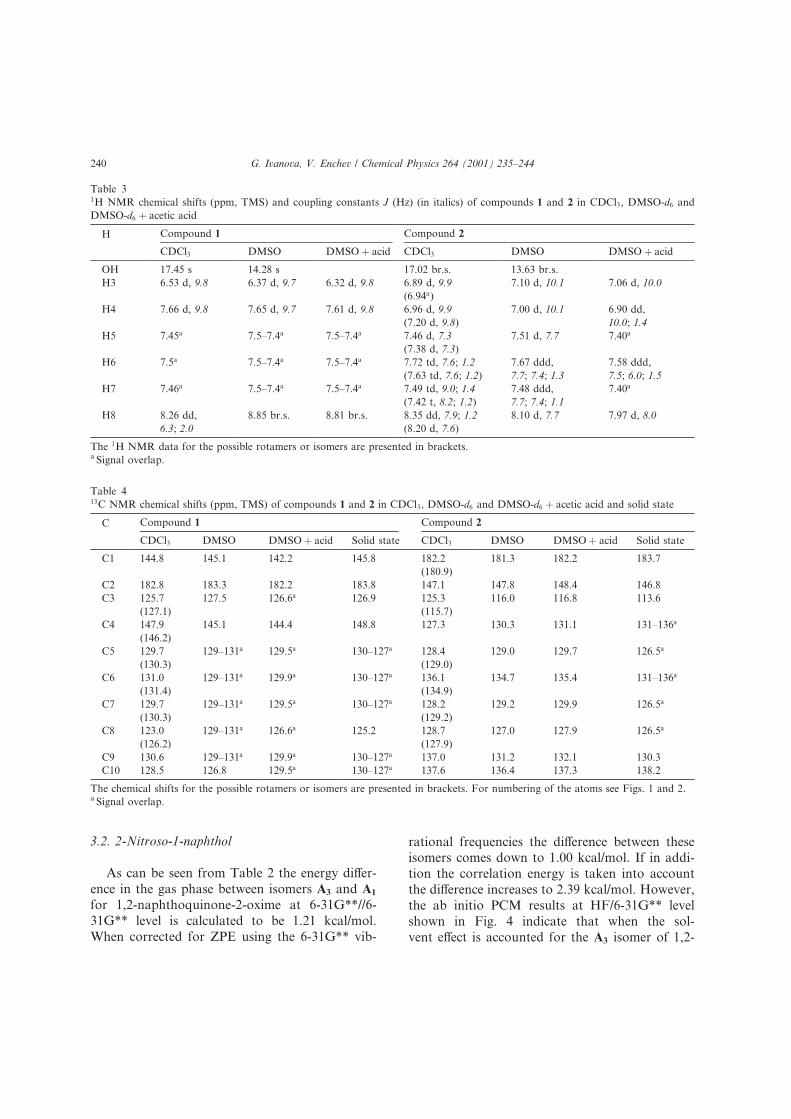
rotamers of the oxime form of 1 in chloroform.
According to ab initio PCM calculations the en-
ergy di�erence between rotamers A1 and A2 as well
as the rotational barrier between them decrease
with the increasing of the solvent polarity from
chloroform to dimethylsulfoxide Fig. 3). This dif-
ference in the rotational barrier heights could ex-
plain the presence of the resonance peaks of the
rotameric forms, A1 and A2 observed by us in the
carbon NMR spectrum in chloroform solution.
The ab initio calculated bond lengths of syn-1,2-
naphthoquinone-1-oxime A1 given in Table 5 are
in agreement with available X-ray data [6]. A
comparison of the calculated and experimental
data shows that the di�erences in the C±C bond
lengths do not exceed 0.018 �A. There are more
substantial changes in the C±O, C±N and N±O
bond lengths, which are shorter by 0.041, 0.044
and 0.042 �A, respectively. The experimental and
calculated data show that an intramolecular hy-
drogen bond is formed Table 5). The calculations
predict a longer by 0.073 �A) interatomic distance
between the oxygen atoms O12 and O13 partici-
pating in the intramolecular hydrogen bond.
Table 2
Calculated relative stabilities in kcal/mol) for all possible species of 1-nitroso-2-naphthol and 2-nitroso-1-naphthol, shown in Figs. 1
and 2, at di�erent computational levels in the gas phase and in solution CHCl3 and DMSO)
Computational level A1 A2 A3 A4 B1 B2 B3 B4
1-Nitroso-2-naphthol
3-21G//3-21G 0.00 6.22 4.19 5.43 19.84 8.62 18.53
6-31G**//3-21G 0.00 5.77 4.92 2.74 12.89 4.38 12.71
6-31G**//6-31G** 0.00 5.88 5.18 3.02 5.17
6-31G**//6-31G**+ZPE 0.00 5.39 2.58
MP4 SDTQ)/6-31G**//6-31G**+ZPE 0.00 5.88 4.17
PCM/6-31G**//6-31G**+ZPEa 0.00 3.73 3.91
PCM/6-31G**//6-31G**+ZPEb 0.00 3.29 4.44
2-Nitroso-1-naphthol
3-21G//3-21G 0.00 5.15 0.01 13.10 6.17 23.55 8.29 19.26
6-31G**//3-21G 0.00 4.98 0.89 10.30 3.48 15.52 2.96 11.93
6-31G**//6-31G** 0.00 5.05 1.21 3.68 3.79
6-31G**//6-31G**+ZPE 0.00 4.76 1.00 3.23 3.36
MP4 SDTQ)/6-31G**//6-31G**+ZPE 0.00 5.34 2.39 4.91 5.89
PCM/6-31G**//6-31G**+ZPEa 0.67 4.16 0.00 5.20 5.12
PCM/6-31G**//6-31G**+ZPEb 1.41 4.35 0.00 6.46 6.27
a In CHCl3.b In DMSO.
G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244 239
3.2. 2-Nitroso-1-naphthol
As can be seen from Table 2 the energy di�er-
ence in the gas phase between isomers A3 and A1
for 1,2-naphthoquinone-2-oxime at 6-31G**//6-
31G** level is calculated to be 1.21 kcal/mol.
When corrected for ZPE using the 6-31G** vib-
rational frequencies the di�erence between these
isomers comes down to 1.00 kcal/mol. If in addi-
tion the correlation energy is taken into account
the di�erence increases to 2.39 kcal/mol. However,
the ab initio PCM results at HF/6-31G** level
shown in Fig. 4 indicate that when the sol-
vent e�ect is accounted for the A3 isomer of 1,2-
Table 31H NMR chemical shifts ppm, TMS) and coupling constants J Hz) in italics) of compounds 1 and 2 in CDCl3, DMSO-d6 and
DMSO-d6 � acetic acid
H Compound 1 Compound 2
CDCl3 DMSO DMSO � acid CDCl3 DMSO DMSO � acid
OH 17.45 s 14.28 s 17.02 br.s. 13.63 br.s.
H3 6.53 d, 9.8 6.37 d, 9.7 6.32 d, 9.8 6.89 d, 9.9 7.10 d, 10.1 7.06 d, 10.0
6.94a)
H4 7.66 d, 9.8 7.65 d, 9.7 7.61 d, 9.8 6.96 d, 9.9 7.00 d, 10.1 6.90 dd,
7.20 d, 9.8) 10.0; 1.4
H5 7.45a 7.5±7.4a 7.5±7.4a 7.46 d, 7.3 7.51 d, 7.7 7.40a
7.38 d, 7.3)
H6 7.5a 7.5±7.4a 7.5±7.4a 7.72 td, 7.6; 1.2 7.67 ddd, 7.58 ddd,
7.63 td, 7.6; 1.2) 7.7; 7.4; 1.3 7.5; 6.0; 1.5
H7 7.46a 7.5±7.4a 7.5±7.4a 7.49 td, 9.0; 1.4 7.48 ddd, 7.40a
7.42 t, 8.2; 1.2) 7.7; 7.4; 1.1
H8 8.26 dd, 8.85 br.s. 8.81 br.s. 8.35 dd, 7.9; 1.2 8.10 d, 7.7 7.97 d, 8.0
6.3; 2.0 8.20 d, 7.6)
The 1H NMR data for the possible rotamers or isomers are presented in brackets.a Signal overlap.
Table 413C NMR chemical shifts ppm, TMS) of compounds 1 and 2 in CDCl3, DMSO-d6 and DMSO-d6 � acetic acid and solid state
C Compound 1 Compound 2
CDCl3 DMSO DMSO � acid Solid state CDCl3 DMSO DMSO � acid Solid state
C1 144.8 145.1 142.2 145.8 182.2 181.3 182.2 183.7
180.9)
C2 182.8 183.3 182.2 183.8 147.1 147.8 148.4 146.8
C3 125.7 127.5 126.6a 126.9 125.3 116.0 116.8 113.6
127.1) 115.7)
C4 147.9 145.1 144.4 148.8 127.3 130.3 131.1 131±136a
146.2)
C5 129.7 129±131a 129.5a 130±127a 128.4 129.0 129.7 126.5a
130.3) 129.0)
C6 131.0 129±131a 129.9a 130±127a 136.1 134.7 135.4 131±136a
131.4) 134.9)
C7 129.7 129±131a 129.5a 130±127a 128.2 129.2 129.9 126.5a
130.3) 129.2)
C8 123.0 129±131a 126.6a 125.2 128.7 127.0 127.9 126.5a
126.2) 127.9)
C9 130.6 129±131a 129.9a 130±127a 137.0 131.2 132.1 130.3
C10 128.5 126.8 129.5a 130±127a 137.6 136.4 137.3 138.2
The chemical shifts for the possible rotamers or isomers are presented in brackets. For numbering of the atoms see Figs. 1 and 2.a Signal overlap.
240 G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244
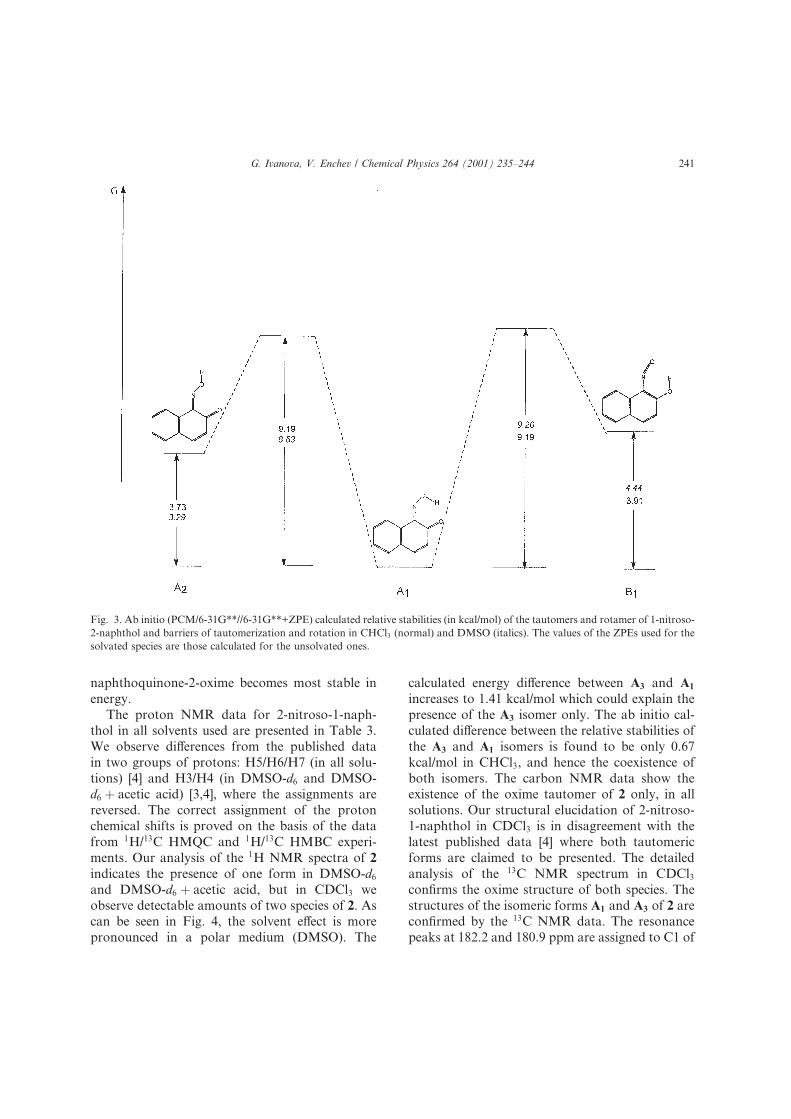
naphthoquinone-2-oxime becomes most stable in
energy.
The proton NMR data for 2-nitroso-1-naph-
thol in all solvents used are presented in Table 3.
We observe di�erences from the published data
in two groups of protons: H5/H6/H7 in all solu-
tions) [4] and H3/H4 in DMSO-d6 and DMSO-
d6 � acetic acid) [3,4], where the assignments are
reversed. The correct assignment of the proton
chemical shifts is proved on the basis of the data
from 1H/13C HMQC and 1H/13C HMBC experi-
ments. Our analysis of the 1H NMR spectra of 2
indicates the presence of one form in DMSO-d6
and DMSO-d6 � acetic acid, but in CDCl3 we
observe detectable amounts of two species of 2. As
can be seen in Fig. 4, the solvent e�ect is more
pronounced in a polar medium DMSO). The
calculated energy di�erence between A3 and A1
increases to 1.41 kcal/mol which could explain the
presence of the A3 isomer only. The ab initio cal-
culated di�erence between the relative stabilities of
the A3 and A1 isomers is found to be only 0.67
kcal/mol in CHCl3, and hence the coexistence of
both isomers. The carbon NMR data show the
existence of the oxime tautomer of 2 only, in all
solutions. Our structural elucidation of 2-nitroso-
1-naphthol in CDCl3 is in disagreement with the
latest published data [4] where both tautomeric
forms are claimed to be presented. The detailed
analysis of the 13C NMR spectrum in CDCl3con®rms the oxime structure of both species. The
structures of the isomeric forms A1 and A3 of 2 are
con®rmed by the 13C NMR data. The resonance
peaks at 182.2 and 180.9 ppm are assigned to C1 of
Fig. 3. Ab initio PCM/6-31G**//6-31G**+ZPE) calculated relative stabilities in kcal/mol) of the tautomers and rotamer of 1-nitroso-
2-naphthol and barriers of tautomerization and rotation in CHCl3 normal) and DMSO italics). The values of the ZPEs used for the
solvated species are those calculated for the unsolvated ones.
G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244 241

isomers A1 and A3, respectively. The calculations
predict an interatomic distance of 2.6028 �A Table
5) between the oxygen atoms O12 and O13, i.e.
there is an intramolecular hydrogen bond. The
existence of an intramolecular hydrogen bond in
the syn-isomer A1 leads to a down®eld chemical
shift of C1 by 1.3 ppm. We also observe changes in
the chemical shift of C3 b-carbon chemical shift)
associated with the cis±trans position of the oxime
group at C2 [20,21]. A low®eld shift of the reso-
nance peak of C3 of the syn-isomer A1 but up®eld
shift of C3 of the anti-isomer A3 are detected
Table 4). The resonance at 125.3 ppm is assigned
to C3 of the syn-isomer A1 but that at 115.7 ppm
to C3 of the anti-isomer A3.
3.3. Solid state 13C NMR data
The solid state 13C NMR data indicate the pres-
ence of compounds 1 and 2 in their oxime forms
only Table 4). The resonance assignment of the13C CPMAS NMR spectrum of 2 and more spe-
Table 5
Ab initio 6-31G** calculated interatomic distances in �A) for the oxime forms A1 of compound 1 and A1 and A3 of compound 2 in gas
phase
Distances Compound 1 ± syn-oxime A1
C1±C2 1.4999 1.485)
C2±C3 1.4623 1.444)
C3±C4 1.3310 1.328)
C4±C10 1.4622 1.451)
C5±C10 1.3935 1.397)
C9±C10 1.3970 1.411)
C5±C6 1.3784 1.377)
C6±C7 1.3890 1.394)
C7±C8 1.3810 1.385)
C8±C9 1.3916 1.390)
C1±C9 1.4862 1.477)
C2±O13 1.2089 1.250)
C1±N11 1.2705 1.315)
N11±O12 1.3216 1.364)
O12±H 0.9570 0.726)
O13±H 1.7474 1.886)
O12±O13 2.5826 2.510)
Compound 2
syn-oxime A1 anti-oxime A3
C1±C2 1.4966 1.5131
C2±C3 1.4652 1.4684
C3±C4 1.3260 1.3265
C4±C10 1.4668 1.4687
C5±C10 1.3923 1.3900
C9±C10 1.4003 1.3980
C5±C6 1.3814 1.3837
C6±C7 1.3899 1.3868
C7±C8 1.3791 1.3830
C8±C9 1.3921 1.3878
C1±C9 1.4774 1.4911
C1±O11 1.2084 1.1924
C2±N12 1.2730 1.2617
N12±O13 1.3215 1.3534
O13±H 0.9562 0.9435
O11±H 1.7711 4.4058
O11±O13 2.6028 4.0333
242 G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244

ci®cally the chemical shift of C3 indicates the exis-
tence of the anti-1,2-naphthoquinone-2-oxime A3.
4. Concluding remarks
On the basis of NMR spectroscopy and ab initio
quantum-chemical calculations, only the oxime
tautomeric form of 1-nitroso-2-naphthol and 2-
nitroso-1-naphthol is found to exist. The syn-1,2-
naphthoquinone-1-oxime A1 is the most stable one
in the gas phase, solution and solid state. Two
rotameric forms of this tautomer are observed in
solution by us for the ®rst time. It can be seen from
the Table 6 that the barrier height of rotation
around the C±O bond A1 ! A2 process) in the gas
phase increases when the electron correlation at
MP4 level) is taken into account but the increase
is not substantial. However, there is a decrease
of the barrier height of rotation in solution, in
comparison with the gas phase see Table 6 and
Figs. 3 and 4). Increasing the solvent polarity leads
to a lowering of the rotational barrier Figs. 3
and 4).
Fig. 4. Ab initio PCM/6-31G**//6-31G**+ZPE) calculated relative stabilities in kcal/mol) of the tautomers, isomer and rotamer of 2-
nitroso-1-naphthol and barriers of tautomerization and rotation in CHCl3 normal) and DMSO italics). The values of the ZPEs used
for the solvated species are those calculated for the unsolvated ones.
Table 6
Calculated barriers in kcal/mol) of tautomerization A1 $ B1 and of rotation A1 $ A2 in the gas phase
Computational level A1 ! B1 B1 ! A1 m# A1 ! A2 A2 ! A1 m
#
1-Nitroso-2-naphthol
6-31G**//6-31G** 12.20 9.18 1625i 11.82 5.97 370i
6-31G**//6-31G**+ZPE 9.17 6.59 10.56 5.17
MP4 SDTQ)/6-31G**//6-31G**+ZPE 4.70 0.53 12.05 6.17
2-Nitroso-1-naphthol
6-31G**//6-31G** 12.95 9.27 1636i 11.94 6.89 402i
6-31G**//6-31G**+ZPE 9.96 6.23 10.73 5.97
MP4 SDTQ)/6-31G**//6-31G**+ZPE 5.48 0.57 12.40 7.06
Imaginary frequencies m# are scaled by 0.893.
G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244 243

The calculations at a 6-31G**//6-31G** and
MP4 SDTQ)/6-31G**//6-31G**+ZPE level pre-
dict 1,2-naphthoquinone-2-oxime to exists as syn-
oxime isomer A1 in the gas phase. However, our
experimental and theoretical results prove the
presence of syn- and anti-oxime isomers, A1 and
A3, in solution. We have observed a similar co-
existence of syn- and anti-isomers of acenaph-
thenedionemonoxime [20]. In the solid state 13C
CPMAS NMR spectrum, resonance peaks of the
anti-oxime isomer A3 are observed.
According to the literature data [3,4], both
tautomeric forms of 2-nitroso-1-naphthol are
present in solution. However, in the 13C NMR
spectra of 2 in solution, the characteristic reso-
nance signal for an aromatic carbon with attached
hydroxy group in the range 155±170 ppm [20,22±
24] has not been observed. Tautomers A1 and
B1 are interconvertible by intramolecular proton
transfer. It can be seen from Table 6 that the in-
clusion of electron correlation in the calculations
strongly in¯uences the tautomerization barrier.
The barriers of tautomerization in the gas phase
for compounds 1 and 2 at the MP4 SDTQ)/6-
31G**//6-31G**+ZPE level are predicted to be
4.70 and 5.48 kcal/mol, respectively. In the gas
phase, the barrier of tautomerization is found to
be lower than that of rotation Table 6) and maybe
in this case tautomerization could occur. In solu-
tion, however, the barrier of tautomerization cal-
culated at HF/6-31G** level is found to be higher
than the rotation one Figs. 3 and 4) and this could
explain the absence of tautomer B1. It can be seen
from Figs. 3 and 4 that solvent polarity has almost
no in¯uence on the barrier height of tautomeriza-
tion between A1 and B1.
Acknowledgements
We thank Dr. A.E. Aliev, University College,
London, UK, for recording the solid state 13C
NMR spectra.
References
[1] A. Buraway, M. Cais, J.T. Chamberlain, F. Liversedge,
A.R. Thomson, J. Chem. Soc. 1955) 3727.
[2] D. Hadzi, J. Chem. Soc. 1956) 2725.
[3] T. Shono, Y. Hayashi, K. Shinra, Bull. Chem. Soc. Jpn. 44
1971) 3179.
[4] C.F.G.C. Geraldes, M.I.F. Silva, Opt. Pura Apl. 21 1988)
71.
[5] D. Herbison-Evans, R.E. Richards, Mol. Phys. 8 1964)
19.
[6] H. Saarinen, J. Korvenranta, Finn. Chem. Lett. 1978)
233.
[7] A.T. Pilipenko, L.I. Savranskii, Dokl. Akad. Nauk SSSR
197 1971) 1092.
[8] A. Krzan, D.R. Crist, V. Horak, J. Mol. Struct. THEO-
CHEM 528 2000) 237.
[9] J.S. Binkley, J.A. Pople, W.J. Hehre, J. Am. Chem. Soc.
102 1980) 939.
[10] M.M. Frankl, W.J. Pietro, W.J. Hehre, J.S. Binkley, M.S.
Gordon, D.J. De Frees, J.A. Pople, J. Chem. Phys. 77
1982) 3654.
[11] P.C. Hariharan, J.A. Pople, Theor. Chim. Acta 28 1973)
213.
[12] M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert,
M.S. Gordon, J.H. Jensen, S. Koseki, N. Matsunaga, K.A.
Nguyen, S. Su, T.L. Windus, M. Dupuis, J.A. Montgom-
ery, J. Comput. Chem. 14 1993) 1347.
[13] J.A. Pople, M. Head-Gordon, D.J. Fox, K. Raghavachari,
L.A. Curtiss, J. Chem. Phys. 90 1989) 5622.
[14] J. Tomasi, M. Persico, Chem. Rev. 94 1994) 2027.
[15] C. Amovilli, V. Barone, R. Cammi, E. Cances, M. Cossi,
B. Mennucci, C.S. Pomelli, J. Tomasi, Adv. Quant. Chem.
32 1999) 227.
[16] R.A. Pierotti, Chem. Rev. 76 1976) 717.
[17] F.J. Lique, J.M. Lopez-Bes, J. Cemeli, M. Aroztegui, M.
Orozco, Theor. Chem. Acc. 96 1997) 105.
[18] B. Champagne, B. Mennucci, M. Cossi, R. Cammi,
J. Tomasi, Chem. Phys. 238 1998) 153.
[19] P.E. Hansen, S. Bolvig, Magn. Res. Chem. 35 1997)
520.
[20] V. Enchev, G. Ivanova, A. Ugrinov, G.D. Neykov, J. Mol.
Struct. 508 1999) 149.
[21] G.E. Hawkes, K. Herwing, J.D. Roberts, J. Org. Chem. 39
1974) 1017.
[22] Z. Rozwadowski, T. Dziembowska, Magn. Res. Chem. 37
1999) 274.
[23] L.A. Fedorov, M.S. Zhukov, N.Y. Korsakova, Y.M.
Dedkov, A.N. Ermatov, Izv. Akad. Nauk SSSR, Ser.
Khim 1984) 1763.
[24] P.E. Hansen, Org. Magn. Res. 12 1979) 109.
244 G. Ivanova, V. Enchev / Chemical Physics 264 2001) 235±244




















