
Polymorphisms in Toll-like receptor 4 ( TLR4 ) are associated with protection against leprosy
Upload
independentCategory
view
0download
0
Differential involvement of TLR2 and TLR4in host survival during pulmonary infection withChlamydia pneumoniae
Nuria Rodriguez1, Nina Wantia1, Falko Fend2, Susanne D�rr1,Hermann Wagner1 and Thomas Miethke1
1 Institute of Medical Microbiology, Immunology and Hygiene, Technical University ofMunich, Munich, Germany
2 Institute of Pathology, Technical University Munich, Munich, Germany
The relevance of TLR2 and TLR4 for recognizing Chlamydia pneumoniae in vivo duringpulmonary infection and to survive the infection was explored. We found that earlyimmune responses triggered by C. pneumoniae partially depended on TLR2, but not onTLR4. The chemokines MIP-2 and MIP-1awere not induced, while IL-12p40 levels werehigher in TLR2–/– mice compared to wild-type mice. Secretion of TNF, keratinocyte-derived chemokine and monocyte chemoattractant protein-1 was attenuated in TLR2–/–
mice, while IFN-c was increased as in wild-type mice. The pulmonary cyto- andchemokine response of TLR2–/–�TLR4d/d was similar to TLR2–/– mice. TLR2–/– andTLR2–/–�TLR4d/d mice also attracted fewer polymorphonuclear neutrophils into thelung, while TLR4d/d mice recruited them. Attenuated recruitment of polymorpho-nuclear neutrophils correlated with reduced weight loss in TLR2–/– andTLR2–/–�TLR4d/d mice and a lower chlamydial burden 3 days post infection. At9 days post infection, TLR2–/– and TLR2–/–�TLR4d/d mice produced cyto- andchemokines as efficiently as wild-type mice, indicating that the involvement of TLRin inflammation varies over time. All TLR2–/–�TLR4d/d mice succumbed to theinfection, while about 50% of TLR2–/– mice died. Taken together, the function of TLR2and TLR4 is required to survive pulmonary infection with C. pneumoniae.
Introduction
Toll-like receptors (TLR) are important for host defenseagainst microorganisms in that they recognize patho-gen-associated molecular patterns (PAMP) [1]. They areexpressed by immune cells such as monocytes/macro-
phages, dendritic cells (DC), and T cells as well asepithelial cells [2–7]. In contrast to the numerousreports on TLR-mediated recognition of PAMP, thenumber of reports demonstrating TLR as crucial tosurvive infections is limited. For example, TLR2 orMyD88–/– mice were highly susceptible to Staphylococ-cus aureus infection [8], while the survival rate ofTLR2–/– mice infected with Listeria monocytogenes wasabout 40% lower compared to wild-type (WT) mice [9].TLR2 was also important for host defenses againstCandida albicans infections since TLR2–/– mice displayeda higher rate of lethality [10]. In a chronic model ofMycobacterium bovis BCG infection, however, TLR2,TLR4 and TLR6 appeared not to be essential forsuccessful control of the infection by the host [11],
Correspondence: Thomas Miethke, Institute of Medical Mi-crobiology, Immunology and Hygiene, Technical University ofMunich, Trogerstr. 9, 81675 Munich, GermanyFax: +49-89-4140-4868e-mail: [email protected]
Received 17/6/05Revised 1/2/06
Accepted 28/2/06
[DOI 10.1002/eji.200535152]
Key words:Chemokine � Cytokine� Inflammation � Lung
� Neutrophils
Abbreviations: BMDDC: BM-derived DC � HPF: high-powerfield � IFU: inclusion-forming unit � KC: keratinocyte-derivedchemokine � PAMP: pathogen-associated molecular pattern
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1145
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

while TLR4 was required to control chronic Mycobacter-ium tuberculosis infection [12].
The relevance of TLR to recognize the intracellularbacterium Chlamydia pneumoniae in vivo is questionablefor several reasons. First, the microorganism appears tosynthesize TLR ligands of low affinity. For instance,chlamydial endotoxin stimulates macrophages via TLR4,but its activity is 100–1000-fold weaker compared toendotoxin derived from Enterobacteriaceae [13, 14].Furthermore, chlamydial DNA, a structure potentiallyrecognized via TLR9, displays a low CpG-frequency(2.99%), which is only slightly higher than that ofeukaryotic DNA (2.06%), but much lower than that ofgenomic DNA from E. coli (7.48%) or M. tuberculosis(12.73%). Perhaps, C. pneumoniae has developed –possibly due to its obligate intracellular replication – anadaptive strategy to produce very weak agonists for theTLR system.
Second, the most efficient replication of thebacterium occurs within epithelial cells of the respira-tory tract [15], where chlamydial inclusions could besequestered from detection by TLR. The cells areinfected by a metabolically inert elementary body,which develops into a metabolically active and largerreticulate body (RB). RB divide by binary fission andgenerate the chlamydial inclusion. In vitro, humanrespiratory epithelial cells appear to express TLR2, andstimulation of the cells with bacterial lipopeptide, awell-known TLR2 ligand, induces the secretion of IL-8 andhuman b defensin-2 [6]. However, intracellularlyexpressed PAMP of C. pneumoniae might not beaccessible to TLR2 since C. pneumoniae replicates insidean inclusion and potential TLR ligands may be trappedinside. In addition, respiratory epithelial cells areexposed to the bacterial flora of the upper respiratorytract and environmental stimuli; however, neitherstimuli cause inflammation. TLR-signaling must, there-fore, be tightly regulated to avoid chronic inflammationby commensal bacteria but to allow the detection ofpathogenic microorganisms.
Third, the chlamydial inclusion appears to avoidfusion with other intracellular vesicles [16]. Thus, TLR –even if they are expressed intracellularly by infectedepithelial cells – may be unable to get access to theintracellular bacteria, because they are not integratedinto the inclusion membrane.
In vitro, however, bone marrow-derived DC(BMDDC) exposed to C. pneumoniae activated thetranscription factor NF-jB and secreted proinflamma-tory cytokines like TNF and IL-12p40 [17]. Activation ofNF-jB and TNF secretion were dependent on TLR2,while secretion of IL-12p40 required the presence ofTLR2 and TLR4. Similarly, human PBMC produced TNFand IL-1 upon stimulation with C. pneumoniae in aTLR2-dependent fashion [18]. Thus, in these in vitro
systems TLR2 and to some extent TLR4 are involved inthe recognition of C. pneumoniae.
In this study we explored the relevance of TLR2 andTLR4 for the recognition of C. pneumoniae in vivo duringrespiratory infection. We also analyzed whether thepresence of these TLR is crucial to survive the infection.
Results
Influence of TLR2 and TLR4 on pulmonarychemo- and cytokine levels 3 days post infection
Chlamydia are gram-negative bacteria and purifiedendotoxin from C. trachomatis is able to stimulatemacrophages via TLR4, albeit only at high concentra-tions [14]. In addition, TLR4d/d BMDDC display a partialdefect to produce IL-12p40 upon stimulation withC. pneumoniae [17]. Therefore, we infected TLR4d/d
mice and tested whether inflammatory reactions differbetween TLR4d/d and WT mice. As Fig. 1 demonstrates,infection with C. pneumoniae stimulated the secretion ofthe cytokines TNF, IFN-c, IL-12p40 and the chemokineskeratinocyte-derived chemokine (KC), MIP-2, MCP-1and MIP-1a in WT and equally well in TLR4d/d mice.Thus, in contrast to the stimulation of BMDDC with C.pneumoniae in vitro, lack of TLR4 did not affect IL-12p40production in vivo. Since we reported earlier thatTLR2–/– BMMDC were impaired in their TNF andIL-12p40 secretion upon infection with C. pneumoniae[17], we analyzed TLR2–/– mice. In contrast to ourfindings in vitro, TLR2–/– mice displayed even higherconcentrations of IL-12p40 thanWTmice (Fig. 1). Whilepulmonary IFN-c levels were increased as in WT mice,secretion of TNF, KC and MCP-1 was attenuated inTLR2–/– mice (Fig. 1). TLR2–/– mice, however, did notup-regulate pulmonary MIP-2 and MIP-1a levels uponinfection. TLR2–/–�TLR4d/d mice essentially displayedthe same pattern of chemo- and cytokine secretion3 days post infection as TLR2–/– mice, with theexception of IFN-c and TNF. IFN-c was higher inTLR2–/–�TLR4d/d compared to WT mice (Fig. 1) andTNF was increased in TLR2–/–�TLR4d/d mice, as in WTmice. Overall these data implied that TLR2 was involvedin the recognition of C. pneumoniae in vivo, while TLR4appeared to be of minor relevance.
Pulmonary inflammation is less severe inTLR2 and TLR2–/–�TLR4d/d mice but normal inTLR4d/d mice
Since MIP-2, MIP-1a and KC attract polymorphonuclearneutrophils (PMN) and TLR2–/– mice failed or wereimpaired to produce these chemokines, we wonderedwhether in these mice PMN were less efficiently
Nuria Rodriguez et al. Eur. J. Immunol. 2006. 36: 1145–11551146
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

Figure 1. Cyto- and chemokine response ofTLR4d/d, TLR2–/– or TLR2–/–�TLR4d/d mice.TLR4d/d mice (n=6, black bar), TLR2–/– [n=7, (n=5in case of MIP-1a) black bar], or TLR2–/–�TLR4d/d
mice (TLR2/4–/–, n=6, black bar) were intrana-sally infected with C. pneumoniae (2.5 � 106 IFU/mouse). Infected C3H/HeN WT mice served ascontrols [n=6 left column, n=7 (n=5 in case ofMIP-1a) middle column, n=6 right column, graybars]. Uninfected WT or gene-deficient micewere used as negative controls (n=2, white bars).At 3 days post infection, pulmonary levels ofTNF, IL-12p40, IFN-c, KC, MCP-1, MIP-2 andMIP-1a were determined. Data are normalizedto the response of WT mice, i.e., the responsewas set to 100%. Error bars represent standarddeviation of equally treated mice. *p�0.008,#p=0.03, Mann-Whitney Rank sum test.
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1147
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

recruited to the infected lung. Analysis of the pulmonaryinfiltrate by flow cytometry revealed that the percentageof PMN as defined by GR1+/CD45+ expression [19] wasincreased in TLR2–/– or TLR2–/–�TLR4d/d mice 3 dayspost infection compared to non-infected mice (Fig. 2A,B). However, WTmice and TLR4d/d mice recruited twiceas many GR1+/CD45+ PMN (Fig. 2A, B). Recruitment ofPMN was confined to the early phase of infection sincewe were unable to detect increased PMN percentages9 days post infection (Fig. 2C). These findings corre-lated with a reduced loss of body weight in TLR2–/– orTLR2–/–�TLR4d/d mice, whereas WT or TLR4d/d micelost around 2 g of body weight within 3 days of infection
(Fig. 3). Obviously, the inflammatory reaction of micelacking TLR2 was impaired during infection withC. pneumoniae, although the microorganism wasrecognized by TLR2–/– cells since the pro-inflammatorycytokines IL-12p40 and IFN-c were induced at least tothe same level as in WT mice.
Did the bacterium benefit from impaired recognitionin TLR2–/– or TLR2–/–�TLR4d/d mice in that it replicatedto a higher extent? C. pneumoniae extensively multipliedin WT mice within 3 days, since we could recoveraround 108 inclusion forming units (IFU) from miceinitially infected with 2.5 � 106 IFU (Fig. 4A). However,in lungs of TLR2–/– and TLR2–/–�TLR4d/d mice the
Figure 2. Influence of TLR2 and TLR4 on recruitment of GR1+/CD45+ PMN at 3 days post infection with C. pneumoniae. (A)WT (C3H/HeN, WT), TLR4d/d, TLR2–/–, TLR2–/–�TLR4d/d (TLR2/4–/–) mice were infected with C. pneumoniae (2.5 � 106 IFU/mouse). At day 3,lungs were removed, digested with collagenase and cells were stained with a PE-labeled CD45 mAb and a FITC-labeled GR1 mAb.(B) Statistical analysis of granulocyte influx in WT (n=8), TLR4d/d (n=3), TLR2–/– (n=4), TLR2–/–�TLR4d/d (n=4) mice. Mock-infectedmice included 5WT, 1 TLR4d/d, 3 TLR2–/–, 2 TLR2–/–�TLR4d/dmice.Mice depicted in (A) are included in (B). *p�0.003, onewayANOVA,post-hocHolm-Sidak. (C) Percentage of granulocytes at 9 days post infectionwithC. pneumoniae (2.5 � 106 IFU/mouse) inWT (C3H/HeN, n=6) and TLR2–/–�TLR4d/d mice (n=4). Uninfected WT (C3H/HeN, n=4) and TLR2–/–�TLR4d/d mice (n=3) served as controls.
Nuria Rodriguez et al. Eur. J. Immunol. 2006. 36: 1145–11551148
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

number of C. pneumoniae IFU was lower than in WT orTLR4d/d mice 3 days post infection (Fig. 4A), a findingwhich was similar to MyD88–/– mice [19]. Comparisonof chlamydial burden and pulmonary influx of PMN inTLR2–/–, TLR2–/–�TLR4d/d, MyD88–/– and WT mice atday 3 post infection revealed that the two parameterscorrelated with each other (Fig. 4B). These datasuggested that recruitment of PMN had a beneficialinfluence on the replication of C. pneumoniae.
Involvement of TLR2 and TLR4 inC. pneumoniae-induced cytokine or chemokinesecretion varies over time
To analyze the influence of TLR2 and TLR4 oninflammatory reactions of the lung upon infection withC. pneumoniae with time, we explored pulmonary cyto-and chemokines of TLR2–/– and TLR2–/–�TLR4d/d mice9 days post infection. While these mice failed to produceMIP-2 and MIP-1a 3 days post infection, at day 9TLR2–/– and TLR2–/–�TLR4d/d mice secreted MIP-2 toa similar and MIP-1a even to a higher extent comparedto WT mice. Similarly, both gene-deficient miceproduced higher concentrations of the chemokineMCP-1 at day 9, while secretion of the chemokinewas attenuated at day 3. Pulmonary levels of thechemokine KC were still slightly lower in TLR2–/– andTLR2–/–�TLR4d/d mice 9 days post infection, while the
cytokines TNF, IL-12p40, and IFN-cwere at least as highas inWTmice (Fig. 5). Of note, IL-12p40 and IFN-cwerepresent in higher amounts in the gene-deficient micealready at day 3. In conclusion, the influence of TLR2and TLR4 on the inflammatory responses analyzed herevaried substantially over time.
Figure 3. Weight loss induced by C. pneumoniae infectiondepends on TLR2. WT (C3H/HeN, n=73 day 3, n=40 day 4, n=39day 5, n=45 day 6, n=33 day 7, n=33 day 8, n=35 day 9, blackbars), TLR4d/d (n=20 day 3, n=11 day 4, n=11 day 5, n=11 day 6,n=11 day 7, n=11 day 8, n=11 day 9, white bars), TLR2–/– (n=28day 3, n=17 day 4, n=13 day 5, n=17 day 6, n=11 day 7, n=11day 8, n=11 day 9, gray bars), TLR2–/–�TLR4d/d (TLR2/4–/–, n=22day 3, n=10 day 4, n=12 day 5, n=14 day 6, n=8 day 7, n=7 day 8,n=6 day 9, hatched bars) micewere infectedwith C. pneumoniae(2.5 � 106 IFU/mouse). At the indicated day post infection thebody weight of the mice was determined. Error bars representstandard deviation of testedmice. *p<0.05, Kruskal-Wallis one-way ANOVA on Ranks, Dunn's method.
Figure 4. (A) Kinetics of chlamydial burden on days 3, 6 and 9post infection in WT, TLR4d/d, TLR2–/– and TLR2–/–�TLR4d/d
mice. Lungs of WT (C3H/HeN, WT, n=21 day 3, n=7 day 6, n=13day 9, white bars), TLR4d/d (n=6 day 3, n=4 day 9, black bar),TLR2–/– (n=7 day 3, n=3 day 6, n=4 day 9, gray bars),TLR2–/–�TLR4d/d (TLR2/4–/–, n=8 day 3, n=3 day 6, n=4 day 9,hatched bars) mice infected with C. pneumoniae (2.5 � 106 IFU/mouse)were removed at 3, 6 and 9 days post infection,minced,and the number of IFU was determined in the supernatant oflung homogenates by culturing the supernatant with HEp2cells for 48 h. IFU were counted upon visualization withfluorescence microscopy. Error bars depict SD of equallytreated mice. *p<0.05, Kruskal-Wallis one-way ANOVA onRanks, Dunn's method. #, not determined. (B) Correlationbetween percentage of pulmonary PMN and chlamydialburden at day 3 post infection. The plot displays a linearregression analysis of the percentage of PMN found in lungs ofWT (C3H/HeN, C57BL/6), TLR4d/d, TLR2–/– backcrossed to C3H/HeN, TLR2–/– backcrossed to C57BL/6, TLR2–/–�TLR4d/d andMyD88–/– backcrossed to C57BL/6 versus pulmonary chlamydialburden of these mice, respectively.
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1149
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

Role of TLR2 and TLR4 for surviving pulmonaryinfection with C. pneumoniae
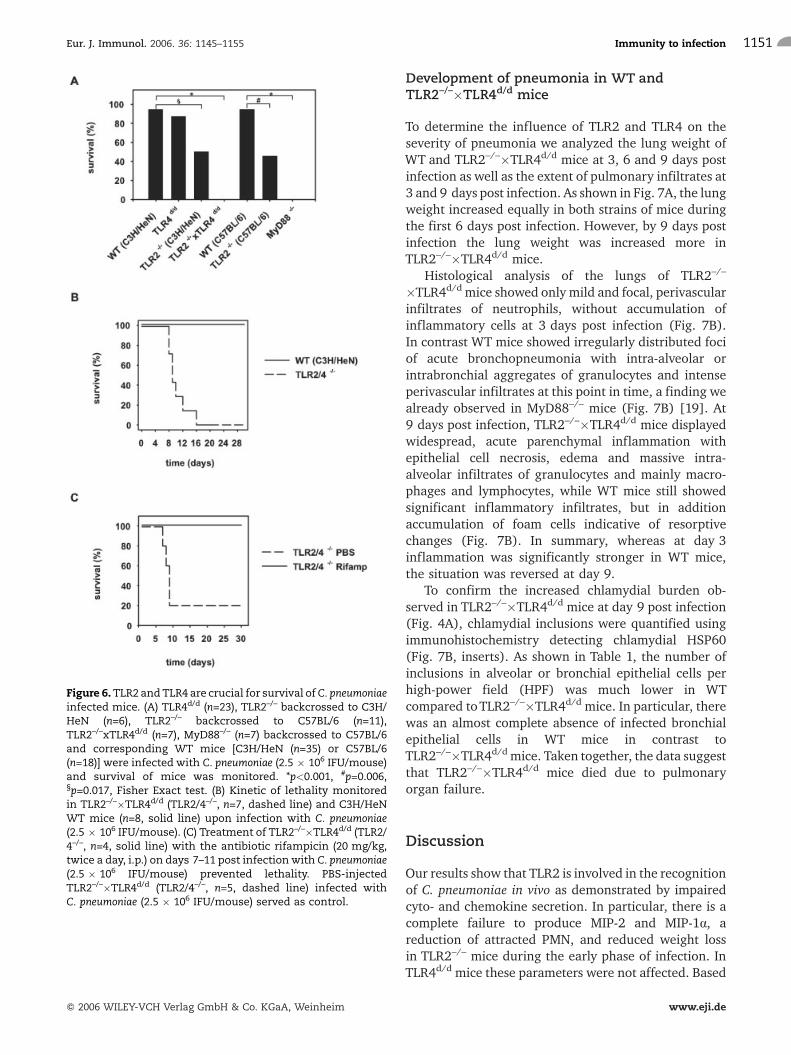
Did attenuated immune responses of TLR2–/– andTLR2–/–�TLR4d/d mice during the early phase ofinfection correlate with an impaired survival of mice?As detailed in Fig. 6A, TLR2–/– mice (backcrossed toC3H/HeN or C57BL/6 mice) survived the infection inonly 50.0% and 45.5%, respectively. In contrast, 94.3%of C3H/HeN or 94.4% of C57BL/6 WT mice survived.The survival rate of TLR4d/d mice was, with 87.0%,slightly lower than in WT mice, but the difference wasnot statistically significant. Surprisingly, all of theTLR2–/–�TLR4d/d mice died (Fig. 6A). This resultimplied that TLR4 was involved in the recognition ofC. pneumoniae even though we did not find an influenceof TLR4 on chemokine or cytokine responses at 3 dayspost infection (Fig. 1). Finally, none of the mice deficientin MyD88 survived the infection. TLR2–/–,TLR2–/–�TLR4d/d and MyD88–/– mice started to getsick at around day 6 of infection and died betweendays 7 and 16 (Fig. 6B, C and data not shown).Corresponding to their increased lethality,TLR2–/–�TLR4d/d as well as MyD88–/– mice were unableto control replication of C. pneumoniae. Thus, while WTand TLR4d/d mice lowered chlamydial burden at day 9post infection, the chlamydial load increased in TLR2–/–,TLR2–/–�TLR4d/d and MyD88–/– mice at this time point(Fig. 4 and data not shown). This was accompanied by astrong decrease in body weight of TLR2–/– andTLR2–/–�TLR4d/d mice at day 9 post infection, althoughboth strains lost less weight during the early phase ofinfection (Fig. 3). In an attempt to explore the relevanceof the increased bacterial load for the lethality observedin TLR2–/–�TLR4d/d mice, we treated these mice withthe antibiotic rifampicin between days 7 and 11 postinfection. Fig. 6C shows that all TLR2–/–�TLR4d/d micewere rescued by the antibiotic treatment, indicating thatoverwhelming infectionmight at least in part explain theincreased lethality of these mice.
·
Figure 5. Cytokine and chemokine responses in TLR2–/– andTLR2–/–�TLR4d/d mice at day 9 post infection. TLR2–/– (blackbars, n=4) and TLR2–/–�TLR4d/d mice (black bars, n=5) wereinfected with C. pneumoniae (2.5 � 106 IFU/lung) for 9 days.InfectedC3H/HeNWTmice served as controls (n=4 left column,n=5 right column, gray bars). Mock-infected WT (white bars,n=1 left column, n=2 right column), TLR2–/– (white bars, n=1)and TLR2–/–�TLR4d/d mice (TLR2/4–/–, white bars, n=2) wereused as negative controls. Pulmonary levels of TNF, IL-12p40,IFN-c, KC, MCP-1, MIP-2 andMIP-1awere determined by ELISA.Data are normalized to the response of WT mice, i.e., theresponse was set to 100%. Error bars depict SD of equallytreated mice. *p�0.032, #p=0.008, Mann-Whitney Rank sumtest.
Nuria Rodriguez et al. Eur. J. Immunol. 2006. 36: 1145–11551150
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de
Development of pneumonia in WT andTLR2–/–�TLR4d/d mice
To determine the influence of TLR2 and TLR4 on theseverity of pneumonia we analyzed the lung weight ofWT and TLR2–/–�TLR4d/d mice at 3, 6 and 9 days postinfection as well as the extent of pulmonary infiltrates at3 and 9 days post infection. As shown in Fig. 7A, the lungweight increased equally in both strains of mice duringthe first 6 days post infection. However, by 9 days postinfection the lung weight was increased more inTLR2–/–�TLR4d/d mice.
Histological analysis of the lungs of TLR2–/–
�TLR4d/d mice showed only mild and focal, perivascularinfiltrates of neutrophils, without accumulation ofinflammatory cells at 3 days post infection (Fig. 7B).In contrast WT mice showed irregularly distributed fociof acute bronchopneumonia with intra-alveolar orintrabronchial aggregates of granulocytes and intenseperivascular infiltrates at this point in time, a finding wealready observed in MyD88–/– mice (Fig. 7B) [19]. At9 days post infection, TLR2–/–�TLR4d/d mice displayedwidespread, acute parenchymal inflammation withepithelial cell necrosis, edema and massive intra-alveolar infiltrates of granulocytes and mainly macro-phages and lymphocytes, while WT mice still showedsignificant inflammatory infiltrates, but in additionaccumulation of foam cells indicative of resorptivechanges (Fig. 7B). In summary, whereas at day 3inflammation was significantly stronger in WT mice,the situation was reversed at day 9.
To confirm the increased chlamydial burden ob-served in TLR2–/–�TLR4d/d mice at day 9 post infection(Fig. 4A), chlamydial inclusions were quantified usingimmunohistochemistry detecting chlamydial HSP60(Fig. 7B, inserts). As shown in Table 1, the number ofinclusions in alveolar or bronchial epithelial cells perhigh-power field (HPF) was much lower in WTcompared toTLR2–/–�TLR4d/d mice. In particular, therewas an almost complete absence of infected bronchialepithelial cells in WT mice in contrast toTLR2–/–�TLR4d/d mice. Taken together, the data suggestthat TLR2–/–�TLR4d/d mice died due to pulmonaryorgan failure.
Discussion
Our results show that TLR2 is involved in the recognitionof C. pneumoniae in vivo as demonstrated by impairedcyto- and chemokine secretion. In particular, there is acomplete failure to produce MIP-2 and MIP-1a, areduction of attracted PMN, and reduced weight lossin TLR2–/– mice during the early phase of infection. InTLR4d/d mice these parameters were not affected. Based
Figure 6.TLR2 andTLR4 are crucial for survival ofC. pneumoniaeinfected mice. (A) TLR4d/d (n=23), TLR2–/– backcrossed to C3H/HeN (n=6), TLR2–/– backcrossed to C57BL/6 (n=11),TLR2–/–xTLR4d/d (n=7), MyD88–/– (n=7) backcrossed to C57BL/6and corresponding WT mice [C3H/HeN (n=35) or C57BL/6(n=18)] were infected with C. pneumoniae (2.5 � 106 IFU/mouse)and survival of mice was monitored. *p<0.001, #p=0.006,§p=0.017, Fisher Exact test. (B) Kinetic of lethality monitoredin TLR2–/–�TLR4d/d (TLR2/4–/–, n=7, dashed line) and C3H/HeNWT mice (n=8, solid line) upon infection with C. pneumoniae(2.5 � 106 IFU/mouse). (C) Treatment of TLR2–/–�TLR4d/d (TLR2/4–/–, n=4, solid line) with the antibiotic rifampicin (20 mg/kg,twice a day, i.p.) on days 7–11 post infection with C. pneumoniae(2.5 � 106 IFU/mouse) prevented lethality. PBS-injectedTLR2–/–�TLR4d/d (TLR2/4–/–, n=5, dashed line) infected withC. pneumoniae (2.5 � 106 IFU/mouse) served as control.
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1151
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

on these observations, we assumed that TLR4 was notrelevant for the recognition of C. pneumoniae. Also,TLR4 played only a marginal role for survival since the
infection was lethal in only 13% in the case of TLR4d/d
mice. However, while around 50% of TLR2–/– mice diedupon infectionwithC. pneumoniae, all TLR2–/–�TLR4d/d
Figure 7. Development of pneumonia in WT and TLR2–/–�TLR4d/d mice. (A) WT (C3H/HeN, n=8 day 3, n=4 day 6, n=5 day 9, whitebars) and TLR2–/–�TLR4d/dmice (n=8 day 3, n=4 day 6, n=5 day 9, black bars) were infectedwith C. pneumoniae (2.5 � 106 IFU/mouse),killed at day 3, 6 or 9 post infection and the lung weight was determined. Uninfected WT (C3H/HeN, n=7, white bar) andTLR2–/–�TLR4d/d mice (n=6, black bar) served as controls. *p=0.032, Mann-Whitney Rank sum test. (B) Histological analysis of thelungs of TLR2–/–�TLR4d/d (a, c, e, g) andWT (C3H/HeN; b, d, f, h) mice 3 and 9 days post infectionwith C. pneumoniae (2.5 � 106 IFU/mouse). At day 3 post infection the cellular pulmonary infiltratewasmore extensive inWT (n=2; b, d) compared to TLR2–/–�TLR4d/d
mice (n=2; a, c). In contrast, at day 9 post infection, WT mice (n=2; f, h) started to resolve the pulmonary infiltrate, whileTLR2–/–�TLR4d/d mice (n=2; e, g) displayed severe cellular inflammation. Hematoxylin and eosin staining, magnification �100.Inserts: immunostaining for chlamydial HSP60 showing chlamydial inclusions in bronchial epithelial cells of TLR2–/–�TLR4d/d (g)and a rare inclusion in alveolar cells of WT mice (h), magnification �400.
Table 1. Quantification of chlamydial inclusions in pulmonary epithelial cells at day 9 post infection
C3H/HeN #1(53 HPF)
C3H/HeN #2(44 HPF)
TLR2–/–xTLR4d/d #1(67 HPF)
TLR2–/–�TLR4d/d #2(59 HPF)
Inclusions inalveolar cells/HPF
0.32 0.51 1.53 1.41
Inclusions inbronchial cells/HPF
0.06 0.04 1.53 0.56
Nuria Rodriguez et al. Eur. J. Immunol. 2006. 36: 1145–11551152
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

mice succumbed to the infection, indicating that thecombined activity of TLR2 and TLR4 was crucial tocontrol the infection in vivo. All MyD88–/– mice died aswell, a finding that might have been expected sinceMyD88 interacts with all TLR. Similar findings werereported recently upon infection of MyD88–/– mice withC. pneumoniae [20]. In that study, MyD88–/– mice alsoshowed reduced signs of inflammation during the earlyphase of infection, as reported here for TLR2–/–
�TLR4d/d and elsewhere for MyD88–/– mice [19].However, in contrast to the results presented here, theinfection with C. pneumoniae was only lethal in 50% ofMyD88–/– mice. Naiki et al. [20] also reported thatTLR2–/– mice cleared C. pneumoniae from their lungs14 days post infection, whereas our results show thatchlamydial burden increased from day 6 to day 9 postinfection. The reason for these discrepancies are atpresent elusive.
The reason why the mice succumbed to the infectionis unclear. Although chlamydial burden at day 3 postinfection was lower in TLR2–/–�TLR4d/d or MyD88–/–
mice compared to WT mice, both strains were unable tocontrol chlamydial replication at later time points, andoverwhelming infection might have caused death. Thisassumption is supported by our observation thatantibiotic treatment with rifampicin started on day 7post infection rescued the mice. However, the chlamy-dial load found at day 9 post infection inTLR2–/–�TLR4d/d or MyD88–/– mice not treated withrifampicin was not higher than the one found at day 3 inWT mice. While the gene-deficient mice were severelysick at day 9 and died later on,WTmice showedwith theexception of early weight loss no signs of disease likeruffling of fur, or reduced motility at day 3 postinfection. This suggests that pathological immuneresponses might also be responsible for the lethalityobserved. For instance, at 9 days post infectionTLR2–/–�TLR4d/d mice showed high pulmonary levelsof MCP-1 and MIP-1a, chemokines known to attractmonocytes to sites of inflammation [21, 22]. Althoughalveolar monocytes were described to repair pulmonarydamage caused by infection [21], extensive attraction ofmonocytes might result in lung damage as well.
IFN-c and IL-12p40 are crucial to control chlamydialreplication in vivo, since the chlamydial load is higher inmice deficient for the IFN-c receptor or IL-12p40 or inmice treated with an IL-12p40 antibody [23–25]. It was,therefore, surprising that TLR2–/–�TLR4d/d mice suc-cumbed to infection with C. pneumoniae, although theirpulmonary IL-12p40 and IFN-c levels were at least ashigh as in WT mice. Based on the reported protectiveeffects of these cytokines we speculate that both of themwere unable to trigger their effector functions, such asinduction of inducible nitric oxide synthase (iNOS) andsubsequent release of NO in TLR2–/–�TLR4d/d mice
[24]. TLR2/4-independent release of IFN-c may beexplained by C. pneumoniae-induced secretion of IL-18,which is known to stimulate IFN-c synthesis, and wasdescribed to be produced in a TLR2/4-independentfashion [26], as reported here for IL-12p40.
Early attraction of PMN into the lungs of TLR2–/– andTLR2–/–�TLR4d/d mice was partially reduced comparedto WT or TLR4d/d mice. This can be explained by ourobservation that the former mice were impaired inraising pulmonary KC levels, and failed to secrete MIP-2and MIP-1a; these chemokines are known to attractPMN [22, 27]. In contrast, C. pneumoniae-infectedMyD88–/– mice completely failed to recruit PMN,probably because these mice were not only unable toraise pulmonary MIP-2 levels but also KC levels [19].Impaired recruitment of PMN also explains the reducedchlamydial burden found at day 3 post infection inTLR2–/– and TLR2–/–�TLR4d/d mice. As reportedrecently, PMN increase the chlamydial load in the lungof infected mice by two mechanisms. First, C. pneumo-niae infects and replicates to a limited extent in PMN[19, 28]. Second, PMN support the replication ofC. pneumoniae in HEp2 cells in vitro and in alveolarepithelial cells in vivo [19].
It is obvious from the results demonstrated here thatthe influence of TLR2 on the induction of differentinflammatory responses, in particular the production ofthe chemokines MIP-2 and MIP-1a, varies over time.Both responses, which depend on TLR2 at the earlyphase of infection with C. pneumoniae, are independentfrom TLR2 at the late stage. Also, TLR2–/–�TLR4d/dmiceare still able to recognize C. pneumoniae, and it is,therefore, conceivable that at least a third of the TLR orTLR-independent mechanisms, e.g., NOD-proteins, areinvolved in the recognition of C. pneumoniae in vivo.Since in MyD88–/– mice all parameters analyzed abovestayed at baseline with the exception of IL-12p40 [19],we consider another TLR as more likely.
In summary, this report shows that TLR, in particularTLR2 and to some extent TLR4, are of crucial importanceto control the obligate intracellular microorganismC. pneumoniae in vivo.
Materials and methods
Strains of mice
C57BL/6, C3H/HeN and C3H/HeJ mice were purchased fromHarlan Winkelmann GmbH (Borchen, Germany). Breedingpairs of MyD88–/– mice were kindly provided by Dr. S. Akira(Osaka, Japan) and mice were backcrossed eight times toC57BL/6 mice. Breeding pairs of TLR2–/– mice came fromTularik (South San Francisco, CA). These mice were back-crossed nine times to C57BL/6, nine times to C3H/HeN or sixtimes to TLR4d/d C3H/HeJ mice. Gene-deficient mice were
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1153
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

bred in our own animal facility under specific pathogen-freeconditions.
Monoclonal antibodies
PE-labeled anti-CD45 mAb (553081), FITC-labeled anti-GR1mAb (553126), and CD16/32-specific mAb (553142) to blockFc receptors were purchased from Becton Dickinson (Heidel-berg, Germany).
Infection protocol
Six-week-old mice were anesthetized with an intraperitonealinjection of Ketamin (200 lL/mouse). Subsequently, micewere intranasally infected with C. pneumoniae (CM-1, ATCCVR-1360) by applying 30 lL PBS containing 2.5 � 106 IFU ofthe microorganism. This dose causes severe non-lethalpneumoniae in WT mice and was therefore chosen in allexperiments. Animal studies were reviewed and approved bylocal authorities (Regierung von Oberbayern, file number211–2531–25/01).
Isolation of pulmonary cells
To isolate pulmonary cells, mice were killed by CO2 inhalationat 3 and 9 days post infection with C. pneumoniae. The lungswere flushed with 10 mL PBS applied through the right atriumof the heart to remove blood. Thereafter, the organwas cut intosmall pieces in a 60-mm plate and digested for 10 min withcollagenase VIII (400 U/100 lL, room temperature, C-2139,Sigma, Taufkirchen, Germany) and subsequently for another30 min (400 U collagenase/100 lL in 2 mL RPMI, 0% FCS,37�C). The digested material was filtered through a cellstrainer of 100-lm pore size (Becton Dickinson LabwareEurope, Le Pont De Claix, France) to remove debris. The cellswere incubated with ammonium chloride (0.15 M, 3 min,room temperature) to lyse erythrocytes. The remaining cellswere washed in PBS containing 3% FCS and analyzed by FACS.
Detection of chemokines and cytokines
The chemokines KC, MCP-1, MIP-2 and MIP-1a as well as thecytokines TNF, IFN-c and IL-12p40 were determined in lungsof mock and infected mice by commercially available ELISA(duo set for KC, MIP-2, MIP-1a, TNF, IFN-c, IL-12p40, R&DSystems, Wiesbaden-Nordenstadt, Germany; MCP-1, Op-tEIATM Set from Becton Dickinson, San Diego, CA). Theassays were performed as described by the manufacturer.Lungs were isolated from mice and minced to homogeneity in500 lL PBS. After centrifugation (2000 � g, 5 min) thesupernatant was analyzed for its cyto- and chemokine contentin duplicates. Data were normalized to the response observedin WT mice to compare results from different experiments forstatistical analysis.
FACS analysis
The cellular composition in lungs post infection was deter-mined by FACS. The isolated cells were preincubated with EMA(2 lg/mL, 20 min, 4�C, Molecular Probes, OR) to exclude
dead cells and anti-CD16/32 (100 lg/mL, 10 min, 4�C) toblock Fc receptors. Subsequently, cells were double stained(30 min, 4�C) with PE-labeled mAb directed against CD45(2 lg/mL) and FITC-labeled GR1 mAb (5 lg/mL). After threewash steps, the cells were fixedwith 2% paraformaldehyde andflow cytometric analysis was performed (Becton DickinsonBiosciences, San Jose, CA). All FACS data were analyzed usingFlowJo (Tree Star Inc, OR).
Bacterial quantification
To quantify C. pneumoniae in lungs of infected mice HEp2 cellswere infected with lung specimens. Briefly, lungs were mincedto homogeneity in 500 lL PBS and sucrose-phosphate-glutamate buffer was added to a final volume of 2 mL.HEp2 cells were infected with the prepared lung specimensdiluted 1:100 in culture medium (MEM-alpha, InvitrogenGmbH, Karlsruhe, Germany) in the absence of FCS butpresence of cycloheximide (1 ng/mL). After 48 h HEp2 cellswere fixed and permeabilized with methanol/acetone (1:1,5 min) and stained with FITC-labeled mAb specific forchlamydial endotoxin and counterstained with Evans blue(ACI-FITC, Progen, Biotechnik GmbH, Heidelberg, Germany).Cells were washed and embedded in glycerin/PBS (50%) andanalyzed by confocal microscopy (LSM510, Carl Zeiss JenaGmbH, G�ttingen, Germany).
Histology and immunohistochemistry
Infected WT (C3H/HeN) and TLR2–/–�TLR4d/d mice werekilled by CO2 inhalation and lungs were perfusion-fixed withbuffered 10% formalin and embedded whole in paraffin.Sections were stained with hematoxylin and eosin.
Immunohistochemistry with an mAb specific for HSP60 ofC. pneumoniae (Affinity Bioreagents, Golden, CO) at a dilutionof 1:1000 was performed on an automated immunostainer(Benchmark, Ventana Medical systems, Tucson, AZ) usingestablished protocols. In brief, slides were dewaxed, and thesubsequent procedure including heat-induced antigen retrie-val as well as blocking of nonspecific binding was performed inthe immunostainer. Detection of bound antibody wasperformedwith a biotinylated secondary antibody and alkalinephosphatase-conjugated streptavidin (Ventana), using FastRed as substrate. The same procedure with omission of theprimary antibody was performed as negative control.
For quantitative evaluation, immunoreactive inclusionbodies were counted over randomly selected HPF (�400).Inclusion bodies in the bronchial epithelium and in alveolarlining cells were evaluated separately.
Statistics
Comparison of two equally treated groups were analyzed byMann-Whitney Rank sum test. More than two equally treatedgroups were tested for significant differences with one wayANOVA, post hoc test Holm-Sidak. In case data were of unequalvariance, Kruskal-Wallis one-way ANOVA on Ranks, Dunn'smethod was applied. Fisher Exact test was used to evaluatestatistical significant differences of survival experiments.Statistical analysiswasperformedwithSigmaStat (SPSS Inc., IL).
Nuria Rodriguez et al. Eur. J. Immunol. 2006. 36: 1145–11551154
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

Acknowledgements:We thank Dr. S Akira for providingMyD88–/– mice. Thanks to Dr. C. Kirschning for criticallyreading the manuscript.
References
1 Medzhitov, R., Toll-like receptors and innate immunity. Nat. Rev. Immunol.2001. 1: 135–145.
2 Wagner, H., The immunobiology of the TLR9 subfamily. Trends Immunol.2004. 25: 381–386.
3 Iwasaki, A. and Medzhitov, R., Toll-like receptor control of the adaptiveimmune responses. Nat. Immunol. 2004. 5: 987–995.
4 Komai-Koma, M., Jones, L., Ogg, G. S., Xu, D. and Liew, F. Y., TLR2 isexpressed on activated T cells as a costimulatory receptor. Proc. Natl. Acad.Sci. USA 2004. 101: 3029–3034.
5 Sobek, V., Birkner, N., Falk, I., Wurch, A., Kirschning, C. J., Wagner, H.,Wallich, R. et al., Direct Toll-like receptor 2 mediated co-stimulation of Tcells in the mouse system as a basis for chronic inflammatory joint disease.Arthritis Res. Ther. 2004. 6: R433–R446.
6 Hertz, C. J., Wu, Q., Porter, E. M., Zhang, Y. J., Weismuller, K. H.,Godowski, P. J., Ganz, T. et al., Activation of Toll-like receptor 2 on humantracheobronchial epithelial cells induces the antimicrobial peptide humanbeta defensin-2. J. Immunol. 2003. 171: 6820–6826.
7 Monick,M.M., Yarovinsky, T. O., Powers, L. S., Butler, N. S., Carter, A. B.,Gudmundsson, G. and Hunninghake, G. W., Respiratory syncytial virusup-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol.Chem. 2003. 278: 53035–53044.
8 Takeuchi, O., Hoshino, K. and Akira, S., Cutting edge: TLR2-deficient andMyD88-deficient mice are highly susceptible to Staphylococcus aureusinfection. J. Immunol. 2000. 165: 5392–5396.
9 Torres, D., Barrier, M., Bihl, F., Quesniaux, V. J., Maillet, I., Akira, S.,Ryffel, B. and Erard, F., Toll-like receptor 2 is required for optimal control ofListeria monocytogenes infection. Infect. Immun. 2004. 72: 2131–2139.
10 Villamon, E., Gozalbo, D., Roig, P., O'Connor, J. E., Fradelizi, D. and Gil,M. L., Toll-like receptor-2 is essential in murine defenses against Candidaalbicans infections. Microbes Infect. 2004. 6: 1–7.
11 Nicolle, D., Fremond, C., Pichon, X., Bouchot, A., Maillet, I., Ryffel, B.and Quesniaux, V. J., Long-term control of Mycobacterium bovis BCGinfection in the absence of Toll-like receptors (TLR): investigation of TLR2-,TLR6-, or TLR2-TLR4-deficient mice. Infect. Immun. 2004. 72: 6994–7004.
12 Abel, B., Thieblemont, N., Quesniaux, V. J., Brown, N., Mpagi, J., Miyake,K., Bihl, F. and Ryffel, B., Toll-like receptor 4 expression is required tocontrol chronic Mycobacterium tuberculosis infection in mice. J. Immunol.2002. 169: 3155–3162.
13 Ingalls, R. R., Rice, P. A., Qureshi, N., Takayama, K., Lin, J. S. andGolenbock, D. T., The inflammatory cytokine response to Chlamydiatrachomatis infection is endotoxin mediated. Infect. Immun. 1995. 63:3125–3130.
14 Prebeck, S., Brade, H., Kirschning, C. J., da Costa, C. P., Durr, S., Wagner,H. and Miethke, T., The Gram-negative bacterium Chlamydia trachomatis
L(2) stimulates tumor necrosis factor secretion by innate immune cellsindependently of its endotoxin. Microbes Infect. 2003. 5: 463–470.
15 Yang, Z. P., Cummings, P. K., Patton, D. L. and Kuo, C. C., Ultrastructurallung pathology of experimental Chlamydia pneumoniae pneumonitis inmice. J. Infect. Dis. 1994. 170: 464–467.
16 Al Younes, H. M., Rudel, T. and Meyer, T. F., Characterization andintracellular trafficking pattern of vacuoles containing Chlamydia pneumo-niae in human epithelial cells. Cell Microbiol. 1999. 1: 237–247.
17 Prebeck, S., Kirschning, C., Durr, S., da Costa, C., Donath, B., Brand, K.,Redecke, V. et al., Predominant role of Toll-like receptor 2 versus 4 inChlamydia pneumoniae-induced activation of dendritic cells. J. Immunol.2001. 167: 3316–3323.
18 Netea, M. G., Kullberg, B. J., Galama, J. M., Stalenhoef, A. F., Dinarello,C. A. and van der Meer, J. W., Non-LPS components of Chlamydiapneumoniae stimulate cytokine production through Toll-like receptor 2-dependent pathways. Eur. J. Immunol. 2002. 32: 1188–1195.
19 Rodriguez, N., Fend, F., Jennen, L., Schiemann, M.,Wantia, N., da Costa,C. U., Durr, S. et al., Polymorphonuclear neutrophils improve replication ofChlamydia pneumoniae in vivo upon MyD88-dependent attraction. J.Immunol. 2005. 174: 4836–4844.
20 Naiki, Y., Michelsen, K. S., Schroder, N. W., Alsabeh, R., Slepenkin, A.,Zhang, W., Chen, S. et al., MyD88 is pivotal for the early inflammatoryresponse and subsequent bacterial clearance and survival in a mouse modelof Chlamydia pneumoniae pneumonia. J. Biol. Chem. 2005. 280:29242–29249.
21 Amano, H., Morimoto, K., Senba, M., Wang, H., Ishida, Y., Kumatori, A.,Yoshimine, H. et al., Essential contribution of monocyte chemoattractantprotein-1/C-C chemokine ligand-2 to resolution and repair processes inacute bacterial pneumonia. J. Immunol. 2004. 172: 398–409.
22 Menten, P., Wuyts, A. and Van Damme, J., Macrophage inflammatoryprotein-1. Cytokine Growth Factor Rev. 2002. 13: 455–481.
23 Rottenberg, M. E., Gigliotti, R. A., Gigliotti, D., Svanholm, C., Bandholtz,L. and Wigzell, H., Role of innate and adaptive immunity in the outcome ofprimary infection with Chlamydia pneumoniae, as analyzed in geneticallymodified mice. J. Immunol. 1999. 162: 2829–2836.
24 Rottenberg, M. E., Gigliotti, R. A., Gigliotti, D., Ceausu, M., Une, C.,Levitsky, V. and Wigzell, H., Regulation and role of IFN-gamma in theinnate resistance to infection with Chlamydia pneumoniae. J. Immunol.2000. 164: 4812–4818.
25 Geng, Y., Berencsi, K., Gyulai, Z., Valyi-Nagy, T., Gonczol, E. andTrinchieri, G., Roles of interleukin-12 and gamma interferon in murineChlamydia pneumoniae infection. Infect. Immun. 2000. 68: 2245–2253.
26 Netea, M. G., Kullberg, B. J., Jacobs, L. E., Verver-Jansen, T. J., van derVen-Jongekrijg, J., Galama, J. M., Stalenhoef, A. F. et al., Chlamydiapneumoniae stimulates IFN-gamma synthesis through MyD88-dependent,TLR2- and TLR4-independent induction of IL-18 release. J. Immunol. 2004.173: 1477–1482.
27 Mizgerd, J. P., Molecular mechanisms of neutrophil recruitment elicited bybacteria in the lungs. Semin. Immunol. 2002. 14: 123–132.
28 van Zandbergen, G., Gieffers, J., Kothe, H., Rupp, J., Bollinger, A., Aga,E., Klinger, M. et al., Chlamydia pneumoniae multiply in neutrophilgranulocytes and delay their spontaneous apoptosis. J. Immunol. 2004.172:1768–1776.
Eur. J. Immunol. 2006. 36: 1145–1155 Immunity to infection 1155
f 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de
Copyright © 2022 FDOKUMEN




















