
Le comportement des petits et moyens cabinets d'expertise comptable en matière d'utilisation des TIC
Upload
independentCategory
view
4download
0
Veterinary Microbiology 98 (2004) 3–15
Development of a monoclonal antibody based competitive-ELISAfor detection and titration of antibodies to peste des
petits ruminants (PPR) virus
R.P. Singh, B.P. Sreenivasa, P. Dhar, L.C. Shah, S.K. Bandyopadhyay∗
Division of Virology, Indian Veterinary Research Institute, Mukteswar, Nainital 263 138, Uttaranchal, India
Received 25 October 2002; received in revised form 26 June 2003; accepted 24 July 2003
Abstract
Peste des petits ruminants (PPR) is an acute febrile, viral, disease of small ruminants with great economic importance.A competitive-ELISA (c-ELISA) test was developed for detection of antibodies to PPR virus in the sera samples of goatsand sheep. The test uses monoclonal antibody to a neutralizing epitope of haemagglutinin protein of the virus. Based on thedistribution of known negative sera samples (n = 933) in respect of PPR virus antibodies in the test, a cut-off value was set as38%. This value was the result of mean of negative population added with two times the standard deviations. A total of 1668 serasamples from goat and sheep and 32 sera from cattle were screened by c-ELISA and virus neutralization test (VNT). Efficacyof c-ELISA compared very well with VNT having high relative specificity (98.4%) and sensitivity (92.4%). The sensitivity ofc-ELISA for PPR sero-surveillance could further be increased (95.4%), if the target population is non-vaccinated. c-ELISA testcorrelated well with VNT (r = 0.845) for end-point titration of PPR virus antibody in 64 goat sera samples. It could clearlyseparate infected population from uninfected in field sera. Using c-ELISA test paired sera samples from 13 goats provided aclear diagnosis of PPR virus infection. Furthermore, antibodies to PPR virus could be successfully detected during 1 year aftervaccination in four goats inoculated with an experimental PPR vaccine. Findings suggest that the c-ELISA test developed caneasily replace VNT for sero-surveillance, sero-monitoring, diagnosis from paired sera samples and end-point titration of PPRvirus antibodies.© 2003 Published by Elsevier B.V.
Keywords:Peste des petits ruminants; Antibody detection; Competitive-ELISA; Monoclonal antibody
1. Introduction
Peste des petits ruminants (PPR) is an acute febrileviral disease of small ruminants. PPR was first de-
∗ Corresponding author. Present address: Joint Director(Academic), IVRI, Izatnagar 243 122, UP, India.Tel.:/fax: +91-581-2302179.
E-mail address:[email protected] (S.K. Bandyopadhyay).
scribed in West Africa (Gargadennec and Lalanne,1942) and is characterized by necrotizing and erosivestomatitis, enteritis and pneumonia (Jones et al., 1993;Ismail et al., 1995). Its wide spread distribution hasbeen reported across the sub-Saharan Africa, Arabianpeninsula (Taylor, 1984; Lefevre and Diallo, 1990),Jordan (Lefevre et al., 1991) and Israel (Anon., 1993).PPR has become endemic in India since it was first re-ported from the south Indian state Tamil Nadu in 1987
0378-1135/$ – see front matter © 2003 Published by Elsevier B.V.doi:10.1016/j.vetmic.2003.07.007

4 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
(Shaila et al., 1989) and subsequently from other partsof the country (Shaila et al., 1990; Mondal et al., 1995;Nanda et al., 1996; Dhar et al., 1997). One uniqueoutbreak in Indian buffaloes has also been reported(Govindarajan et al., 1997). Out of the four geneticlineages of PPR viruses known to exist, lineage 4 isrestricted in Asia whereas the other three are prevalentin Africa (Shaila et al., 1996; Dhar et al., 2002).
PPR is caused by a virus belonging to Morbil-livirus genus of Paramyxoviridae and is antigenicallyclose to other morbilliviruses, closest being rinder-pest (Imagawa, 1968; Orvell and Norrby, 1974; Gibbset al., 1979; Hall et al., 1980). Both PPR and rinder-pest viruses have been found to cause similar diseasesin small ruminants (Diallo et al., 1989). Under theGlobal Rinderpest Eradication Program, many coun-tries are actively pursuing sero-surveillance program.The countries where both rinderpest and PPR arepresent or the countries which have recently acquiredthe status of provisional freedom from rinderpest,separate surveillance programs for rinderpest andPPR may be necessary. The conventional diagnos-tic techniques, e.g., Agar gel immuno-diffusion test,counter-immunoelectrophoresis, indirect ELISA can-not differentiate between rinderpest and PPR (Obi andPatrick, 1984; Obi et al., 1990). Although cross virusneutralization test (VNT) can differentiate betweenantibodies to rinderpest and PPR, it is laborious anddifficult when sample size is large.
Therefore, a diagnostic method, which is simple,rapid, specific and sensitive, is preferred over VNTfor intensive surveillance. Similar antibody detectionmethod employing monoclonal antibody raised eitheragainst haemagglutinin protein (Saliki et al., 1993;Anderson et al., 1991) or nucleoprotein (Libeau et al.,1995) have been described. The monoclonal antibod-ies used in these tests were developed using Africanlineages of PPR viruses. These tests either used gra-dient purified virus or expressed antigens. At present,only two such commercial sources are available inthe world (Anderson and McKay, 1994; Libeau et al.,1995).
The authors report here an alternative system forsero-surveillance and sero-monitoring of PPR usinga monoclonal antibody based competitive-ELISA(c-ELISA), which can be performed under field con-ditions. The test uses tissue culture propagated crudeantigen from an attenuated PPR virus and a mono-
clonal antibody, raised against the same (Sreenivasaet al., 2000).
2. Materials and methods
2.1. PPR virus, antigen and vaccine
An Indian isolate of PPR virus (Sungri isolate),adapted to grow both in vero and B95a cell lines(Sreenivasa et al., 2000), was used for VNT experi-ments. The attenuated virus (Sungri isolate) grown invero cells between passage levels of 57–62 was usedto immunize mice for hybridoma production and alsofor preparation of the antigen used in indirect ELISAand c-ELISA. The vaccine virus used in the presentstudy has been shown to be fully attenuated at 60thpassage in vero cells (Sreenivasa et al., 2000).
2.2. Monoclonal antibody
The MAb designated as 4B11 was selected froma panel of 23 monoclonal antibodies described pre-viously (Singh, 2002) on the basis of its neutral-izing ability of PPR virus infectivity, IgG isotype,strong ELISA signals against PPR virus antigen andhigh titer in culture supernatant. The MAb was di-rected to a neutralizing epitope of haemagglutinin(H) protein of PPR virus as determined by radioimmuno-precipitation assay.
2.3. Serum samples
Goat and sheep sera (n = 936) having a PPR virusneutralization titre of<1:2 were considered negativesera. These sera belong to various Indian breeds ofgoat and sheep originating from various regions ofIndia.
The sera samples (n = 733) having a PPR virusneutralization titre of≥1:8 were considered positive.Majority (n = 623) of these sera were derived fromanimals vaccinated with a live-attenuated PPR vaccineusing 1000 TCID50 of vero derived virus inoculatedby subcutaneous route. The post-vaccinate sera forthe present investigation were collected 21 days aftervaccination. With an standard dose of PPR vaccine,89% of the animals have shown sero-conversion withserum neutralization titres varying between 1:8 and

R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 5
1:256 in the sera samples collected 21 days aftervaccination. Amongst these, about 53% of animals,had VN titre between 1:32 and 1:128. Post-challengeserum from a PPR vaccinated goat was used as strongpositive control for standardization of c-ELISA.Challenge studies indicated 100% protection using10TCID50 of vaccine virus, with 50% protection(PD50) at 1 TCID50 (Sreenivasa et al., 2000).
Pre-vaccinate sera from various breeds of sheep andgoats (n = 110), already having virus neutralizationtitre of ≥1:8, possibly due to previous exposure toPPR virus infection, were also included in the study.
Paired sera samples were collected from 13 infectedgoats during and after 20 days of the outbreak. Allthese animals were confirmed for PPR by detecting theantigen using a PPR-specific immunocapture ELISA(Libeau et al., 1994). All these sera samples listedhere are available in the repository of National Mor-billivirus Reference Laboratory, Division of Virology,IVRI, Mukteswar, India.
Rinderpest positive sera were derived from rinder-pest vaccinated animals, which were found to be neg-ative (n = 32) for PPR virus antibodies in VNT(VN titre <1:8). These sera samples were availablein the repository mentioned above. The study also in-cluded goat sera samples of unknown status (n =141) received from an organized goat farm and fromsmall-herd owners for screening of antibodies to PPRvirus. The antibody status in relation to PPR were notknown for these samples. However, all the sampleswere found to be negative for rinderpest antibody ina monoclonal antibody based c-ELISA (Singh et al.,2000).
2.4. Antigen preparation
PPR virus antigen was prepared as per the pro-tocol described earlier for preparation of rinderpestantigen (Singh et al., 2000). Briefly, 48-h-old verocell monolayers were infected with PPR virus at amultiplicity of infection of 0.01. The culture washarvested at 80–90% cytopathic effect. The culturewas freeze-thawed three times and the cell debris wasclarified by centrifugation at 1000×g for 15 min. Thechilled culture supernatant was subjected to precipita-tion using 8% (w/v) PEG 6000 in the presence of 2.3%(w/v) sodium chloride. The mixture was centrifugedat 8500× g for 30 min following overnight incuba-
tion at 4◦C. The pellet was dissolved in TNE buffer(10 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.4) in10% of the original volume. PEG concentration wasrepeated twice to concentrate the material 100 times.The resulting antigen was lyophilized for later use inthe standardization of indirect ELISA and c-ELISA.
2.5. VNT and c-ELISA
Neutralizing antibodies to rinderpest virus was de-tected in a micro-assay as described byBandyopadhyayet al. (1999). For detection of antibodies to PPR virus,a similar assay was performed using either vero orB95a cells.
For c-ELISA, an attenuated PPR virus (PPRVSun-gri) was used as the coating antigen. The ELISAplates (NUNC Maxisorp) were coated with the PPRvirus antigen (50�l/well). The plates were incubatedat 37◦C for 1 h under constant orbital shaking. Un-bound antigen was washed thrice using 0.002 M PBS.Next, all the wells of the plates received 40�l ofblocking buffer (PBS having 0.2% PPR-negative goatserum and 0.1% Tween 20). 20�l of the test serasamples were added to duplicate sets of well followedby addition of 40�l of MAb in each well (exceptconjugate control wells) at a final dilution of 1:500.The plates were incubated as described earlier andwashed. Anti-mouse-HRPO conjugate (Dako) diluted1:1000 in blocking buffer was added to each well ofthe plate at 50�l/well. Plates were further incubatedfor 1 h at 37◦C under constant shaking. Substratesolution (OPD containing H2O2) was added in eachwell (50�l/well) and color reaction was developedfor 5–10 min followed by stopping with 1 M H2SO4.
2.6. Establishment of optimal MAb and serumdilutions
For optimization of working dilution of antigenand MAb in c-ELISA, a checkerboard titration wasperformed. The specific dilution of the antigen thatinduced approximately 75% absorbance (A492) of theplateau was selected. The MAb dilution was arbi-trarily selected as the dilution, which induced 100,75 and 50% absorbance of plateau as described else-where (Saliki et al., 1993; Singh et al., 2000). Thearbitrarily selected dilutions of the MAb were testedin c-ELISA for deciding the most suitable dilution of

6 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
the MAb and test sera samples to be used in the test.For this purpose sera of various immune status (neg-ative and positive) were tested in twofold dilutionsagainst arbitrarily selected three different dilutionsof monoclonal antibodies. Test sera included weakPPR-positive (n = 3) and PPR-negative samples(n = 3). The serum and MAb dilutions that gavemaximum difference in positive and negative (P/N)samples were selected for testing the samples at largerscales.
2.7. Comparative efficacy of c-ELISA with VNT
The performance of the c-ELISA was comparedwith VNT, the most reliable test for detection of mor-billivirus antibodies (Rossitter et al., 1985). The virusneutralization titers of the sera (n = 1700) and im-mune status in respect of PPR of donor animals wereknown from the records generated while evaluatingan experimental PPR vaccine developed in this lab-oratory (Sreenivasa et al., 2000). The c-ELISA wasevaluated with all these sera at a final dilution of 1:4.Diagnostic sensitivity and specificity were calculatedfrom known P/N population as per the methods de-scribed bySaliki et al. (1993)andLibeau et al. (1995)using two-sided contingency table. Correlation be-tween VNT and c-ELISA for end-point titration wasestablished using 64 goat sera samples with a knownPPR virus neutralizing antibody titre.
2.8. Statistical analysis
Percentage inhibition (PI) values in c-ELISA weregenerated using ELISA data interchange (EDI) soft-ware developed by IAEA (Jeggo and Anderson, 1992;Anon., 1994) for rinderpest sero-surveillance. The for-mula for calculation of PI values is given as:
PI = 100−(
OD of test sample
OD of monoclonal control× 100
)
The diagnostic efficacy of the assay in terms of sen-sitivity and specificity was calculated using two-sidedcontingency table (Jacobson, 1998). Sensitivity of theassay was taken as proportion of positive sample outof actual positive samples. Specificity was calculatedas proportion of negative samples out of total negativesamples.
3. Results
3.1. Selection of monoclonal antibody
The MAb for antibody detection was selectedon the basis of its virus neutralizing ability, highELISA signal and binding with haemagglutinin pro-tein in RIPA. The appropriate clone on the basisof above parameters was designated as 4B11. ThisMAb was directed against a virus neutralizing epi-tope on haemagglutinin protein of PPR virus and isof IgG1 isotype. The MAb could completely neu-tralize infectivity of 100 TCID50 of PPR virus incell culture even up to a dilution of 1:256. It alsoinduced a high signal (A492) in ELISA, which wasmore than 1.0 in indirect ELISA at a dilution of1:250.
3.2. Optimization of test reagents
For optimization of antigen and MAb dilution inc-ELISA, a checkerboard titration was performed. Thespecific dilution of the antigen was selected, whichinduced approximately 75% absorbance (A492) of theplateau. The MAb dilution on titration curve was ar-bitrarily selected as the dilution, which induced 100,75 and 50% of theA492 of plateau (Fig. 1a). A 1:500final dilution of monoclonal antibodies, which corre-sponded to 75% of maximumA492 and 1:4 final dilu-tion of field sera could clearly differentiate P/N seraand showed maximum positive–negative (P–N) differ-ential (Fig. 1b). Therefore, 1:4 dilution of the sera wasconsidered best-suited for further testing and evalua-tion of the assay.
3.3. Determination of cut-off point
After optimization of the test reagents and test pro-tocol, samples with known status as regard to PPRvirus antibody were employed for deciding cut-offvalue. For this, 936 PPR-negative sera samples fromgoat and sheep 32 rinderpest positive (but negative forPPR antibody) samples obtained from vaccinated cat-tle were tested. These samples had a mean inhibitionof 20.23% against a standard deviation of 8.92. There-fore, the cut-off value was set as 38% (mean+2S.D.)for deciding the status of sera samples in respect toPPR virus antibody (Fig. 2).
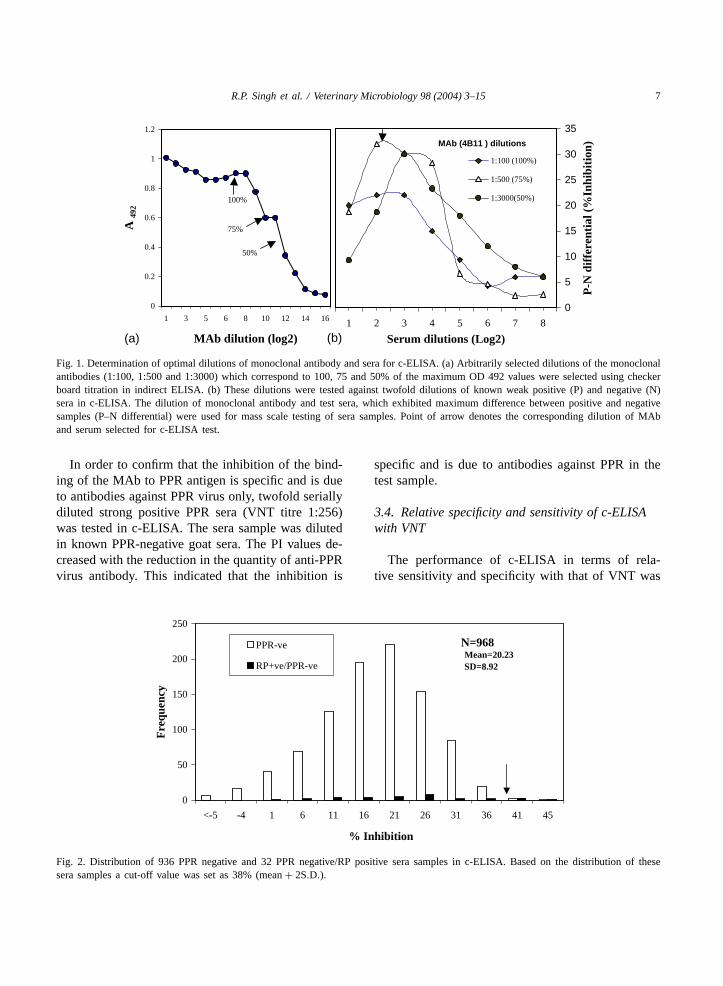
R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 7
0
0.2
0.4
0.6
0.8
1
1.2
1 3 5 6 8 10 12 14 16
MAb dilution (log2)
A 4
92
100%
75%
50%
0
5
10
15
20
25
30
35
1 2 3 4 5 6 7 8
Serum dilutions (Log2)
P-N
dif
fere
ntia
l (%
Inhi
biti
on)
1:100 (100%)
1:500 (75%)
1:3000(50%)
MAb (4B11 ) dilutions
(a) (b)
Fig. 1. Determination of optimal dilutions of monoclonal antibody and sera for c-ELISA. (a) Arbitrarily selected dilutions of the monoclonalantibodies (1:100, 1:500 and 1:3000) which correspond to 100, 75 and 50% of the maximum OD 492 values were selected using checkerboard titration in indirect ELISA. (b) These dilutions were tested against twofold dilutions of known weak positive (P) and negative (N)sera in c-ELISA. The dilution of monoclonal antibody and test sera, which exhibited maximum difference between positive and negativesamples (P–N differential) were used for mass scale testing of sera samples. Point of arrow denotes the corresponding dilution of MAband serum selected for c-ELISA test.
In order to confirm that the inhibition of the bind-ing of the MAb to PPR antigen is specific and is dueto antibodies against PPR virus only, twofold seriallydiluted strong positive PPR sera (VNT titre 1:256)was tested in c-ELISA. The sera sample was dilutedin known PPR-negative goat sera. The PI values de-creased with the reduction in the quantity of anti-PPRvirus antibody. This indicated that the inhibition is
Mean=20.23SD=8.92
0
50
100
150
200
250
<-5 -4 1 6 11 16 21 26 31 36 41 45
% Inhibition
Fre
quen
cy
PPR-ve
RP+ve/PPR-ve
N=968
Fig. 2. Distribution of 936 PPR negative and 32 PPR negative/RP positive sera samples in c-ELISA. Based on the distribution of thesesera samples a cut-off value was set as 38% (mean + 2S.D.).
specific and is due to antibodies against PPR in thetest sample.
3.4. Relative specificity and sensitivity of c-ELISAwith VNT
The performance of c-ELISA in terms of rela-tive sensitivity and specificity with that of VNT was

8 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
Table 1Relative specificity and sensitivity of c-ELISA with VNT basedon 1700 laboratory and field seraa
VNT c-ELISA
Positive Negative Total
Positive 676 57 733Negative 15 952 967
Total 691 1009 1700
a Relative specificity = 952 of 967, or 98.4%. Relativesensitivity = 676 of 733, or 92.2%.
compared using a two-sided contingency table. Theoverall specificity of c-ELISA test was 98.4% with asensitivity of 92.2% as shown in Table 1. Majority ofthe PPR-positive sera samples included in the presentstudy originated from PPR vaccinated animals whichwere collected 21 days post-immunization. A propor-tion of these samples (7.8%) were detected as falsenegative in c-ELISA (Fig. 3). When the performanceof this assay was analyzed on sera originating fromnatural infection in the field (Fig. 4), the sensitiv-ity of the test increased significantly reaching up to95.4%.
3.5. Correlation of c-ELISA with VNT for end-pointantibody titration
The MAb based c-ELISA test was also comparedwith VNT on a set of 64 sera samples for end-pointtitration of PPR virus antibodies. The test compared
Fig. 3. Distribution of 733 PPR positive sera samples in c-ELISA. Most of these sera (n = 623) were collected 21 days after inoculationof an experimental vaccine.
very well with a high correlation coefficient (r =0.845) for detection of end-point titer of PPR-positiveantibody. However, end-point values in majority of thesamples in c-ELISA were generally lower (1−3 log2)than VN titre (Fig. 5).
3.6. Suitability of the assay for sero-monitoring andsero-surveillance
Serial bleeds from three goats vaccinated with anexperimental PPR vaccine were tested in c-ELISAto study the kinetics of antibody production in theseanimals. A steady increase in the antibody was ob-served after 1 week in all the three (Fig. 6), whichcrossed the cut-off point (38%) roughly between 16and 20 days post-vaccination. Similarly, four othergoats were screened at varying interval up to a periodof 1 year post-vaccination starting from seventh day.The antibodies in these goats could be successfullydetected during an observation period of up to 1 yearpost-vaccination (Fig. 7).
The c-ELISA test was also applied for the de-tection of PPR virus antibodies from 13-paired serasamples from a confirmed out break of PPR. It couldsuccessfully detect PPR virus antibodies in all theanimals after 20 days of the reported outbreak but notin the samples collected at the time of the outbreak(Fig. 8). Further 141 goat sera samples received fromareas endemic for PPR were also tested. The testcould clearly differentiate infected from uninfectedpopulation (Fig. 9).

R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 9
Fig. 4. Distributions of 110 PPR-positive goat and sheep sera in c-ELISA originating from natural infection. Roughly 50% of these serahad a very high PI values (81–100%), indicating high titre of PPR virus antibody.
3.7. Reproducibility of assay using freeze-thawedreagents
A set of most crucial reagents i.e. antigen, mono-clonal antibody, P/N control sera were subjected torepeated cycles of freezing and thawing to evaluateits performance. The performance of the reagents didnot deteriorate even after 10 cycles of freezing andthawing.
y = 0.6583x + 0.2764R2 = 0.7148
0
1
2
3
4
5
6
7
8
9
0 1 2 3 4 5 6 7 8 9
VNT titre (log2)
c-E
LIS
A t
itre
(lo
g2)
r=0.845N=64
Fig. 5. Correlation between VNT and c-ELISA for antibody titra-tion on 64 sera samples. In general c-ELISA titers are lower(1−3 log2) than VNT titres. One point may represent several serasample.
0
10
20
30
40
50
60
70
0 6 8 10 12 14 16 18 20
Days post vaccination
% I
nhib
itio
n
Goat 295
Goat 300
Goat 306
Fig. 6. Kinetics of early antibody development as determined byc-ELISA in three goats that received an experimental PPR vaccine.Dotted line shows P/N cut-off.
4. Discussion
A diagnostic technique, which is simple, rapid, spe-cific and sensitive, is preferred for intensive surveil-lance of a disease. c-ELISA test is one such test forscreening of antibodies to various morbilliviruses(Saliki et al., 1993; Libeau et al., 1995). This testis having several advantages over VNT, as it doesnot require cell culture facility and strict sterility ofsera samples. Rapid diagnosis and screening of largenumber of sera sample is therefore, possible using theassay. Developing countries with large target popula-tion for sero-surveillance of a disease like PPR cannot

10 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
0
10
20
30
40
50
60
70
80
90
100
0 7 14 21 30 60 90 120
150
180
270
360
Days post vaccination
%In
hib
itio
n
Goat 41Goat 46Goat 50Goat 52
Fig. 7. Sero-monitoring in four goats using c-ELISA over a periodof 1 year. These goats received a single dose of an experimentalPPR vaccine. Dotted lines show negative/positive cut-off.
0
10
20
30
40
50
60
70
80
90
137 139 141 142 144 147 148 151 153 155 157 159 161
Goat numbers
%In
hibi
tion
During out break
20 days post out break
Fig. 8. PPR diagnosis using paired sera from 13 goats in c-ELISA. The sera samples were collected after confirmation of the disease inindividual animals using an internationally accepted immunocapture-ELISA kit during the PPR outbreak and 20 days after. Dotted lineshows negative/positive cut-off (38% inhibition). All the sera became positive for PPR virus antibody after 20 days of the disease outbreak in surviving goats.
afford the assays like VNT due to the lack of tech-nical expertise and facilities in the field laboratoriesand the cost factor involved. A nation-wide serolog-ical survey for PPR in India with about 200 millionsmall ruminants will require testing of approximately200,000 animals every year at the rate of 0.1% ofthe target population (Taylor et al., 1990). Intensivesero-surveillance for rinderpest virus antibodies inIndia is being pursued using a similar test. Antibodydetection systems employing monoclonal antibod-ies against haemagglutinin protein and nucleoproteinof PPR and rinderpest viruses have been described(Anderson et al., 1991; Libeau et al., 1992, 1995;Saliki et al., 1993; Singh et al., 2000). The monoclonalantibodies used in all these tests were produced usingAfrican lineage of rinderpest or PPR viruses. Thesetests either used gradient purified virus or expressedantigens. Presently, there is only one commercialsource of reagents for PPR c-ELISA test in the world(Anderson et al., 1991). c-ELISA being reported hereis an alternative system for sero-surveillance of PPRvirus antibodies, which can be performed under field
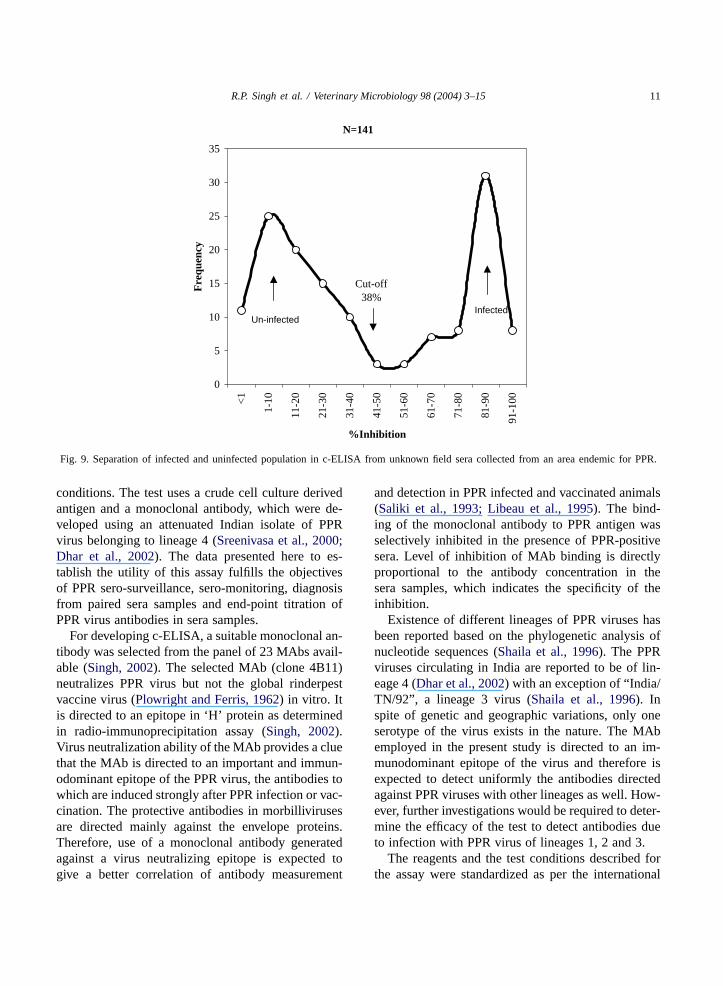
R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 11
0
5
10
15
20
25
30
35
<1
1-10
11-2
0
21-3
0
31-4
0
41-5
0
51-6
0
61-7
0
71-8
0
81-9
0
91-1
00
%Inhibition
Fre
quen
cy
N=141
Cut-off38%
Un-infectedInfected
Fig. 9. Separation of infected and uninfected population in c-ELISA from unknown field sera collected from an area endemic for PPR.
conditions. The test uses a crude cell culture derivedantigen and a monoclonal antibody, which were de-veloped using an attenuated Indian isolate of PPRvirus belonging to lineage 4 (Sreenivasa et al., 2000;Dhar et al., 2002). The data presented here to es-tablish the utility of this assay fulfills the objectivesof PPR sero-surveillance, sero-monitoring, diagnosisfrom paired sera samples and end-point titration ofPPR virus antibodies in sera samples.
For developing c-ELISA, a suitable monoclonal an-tibody was selected from the panel of 23 MAbs avail-able (Singh, 2002). The selected MAb (clone 4B11)neutralizes PPR virus but not the global rinderpestvaccine virus (Plowright and Ferris, 1962) in vitro. Itis directed to an epitope in ‘H’ protein as determinedin radio-immunoprecipitation assay (Singh, 2002).Virus neutralization ability of the MAb provides a cluethat the MAb is directed to an important and immun-odominant epitope of the PPR virus, the antibodies towhich are induced strongly after PPR infection or vac-cination. The protective antibodies in morbillivirusesare directed mainly against the envelope proteins.Therefore, use of a monoclonal antibody generatedagainst a virus neutralizing epitope is expected togive a better correlation of antibody measurement
and detection in PPR infected and vaccinated animals(Saliki et al., 1993; Libeau et al., 1995). The bind-ing of the monoclonal antibody to PPR antigen wasselectively inhibited in the presence of PPR-positivesera. Level of inhibition of MAb binding is directlyproportional to the antibody concentration in thesera samples, which indicates the specificity of theinhibition.
Existence of different lineages of PPR viruses hasbeen reported based on the phylogenetic analysis ofnucleotide sequences (Shaila et al., 1996). The PPRviruses circulating in India are reported to be of lin-eage 4 (Dhar et al., 2002) with an exception of “ India/TN/92” , a lineage 3 virus (Shaila et al., 1996). Inspite of genetic and geographic variations, only oneserotype of the virus exists in the nature. The MAbemployed in the present study is directed to an im-munodominant epitope of the virus and therefore isexpected to detect uniformly the antibodies directedagainst PPR viruses with other lineages as well. How-ever, further investigations would be required to deter-mine the efficacy of the test to detect antibodies dueto infection with PPR virus of lineages 1, 2 and 3.
The reagents and the test conditions described forthe assay were standardized as per the international

12 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
norms recommended from time to time (Jacobson,1998; Schrijver and Kramps, 1998; Wright, 1998).We have already described most of the parametersadapted in the assay for a similar test developed fordetection of antibodies to rinderpest virus (Singhet al., 2000). This test has been validated by WorldReference Laboratory of Rinderpest (John Anderson,personal communication) and accepted by Office In-ternational des Epizooties (OIE) for use in Nationalrinderpest sero-surveillance program of India. Suchtype of assays should use standard controls (con-jugate, strong positive, weak positive, negative andMAb control) to judge the quality of an assay andto ensure accuracy between the plates (Jacobson,1998). Therefore, similar controls were incorporatedin every plate to eliminate plate-to-plate variation,if any. Under field conditions the test reagents arelikely to be subjected to temperature variations be-cause of the power failures and even during routinetesting procedures. Therefore, to assure the qualityof reagents under such situations critical reagentswere freeze-thawed several times (Singh et al., 2000).Efficacy of the reagents after freezing and thawingproved that the same vial of biological can be usedseveral times without any significant loss in the per-formance of the assay. Acceptance of the plates forcontrol panel was monitored through EDI software toensure uniformity (Jeggo and Anderson, 1992; Anon.,1994). Most crucial of these reagents developed isthe antigen, which alone can affect the performanceof the assay maximally. Therefore, the method forthe preparation of the antigen was simplest possiblewith an idea for easy commercialization of the testreagents. The antigen used in the assay is a one-steppolyethylene glycol-concentrated cell-free virus, sothat large-scale production becomes easy. The anti-gen was derived from an attenuated PPR virus isolate(Sreenivasa et al., 2000) that will avoid risk of spread-ing the disease through diagnostic reagents whilebeing transported to distant places.
The objective of development of c-ELISA was toreplace VNT as a method of choice for detection ofantibodies to PPR virus. The former test is having sev-eral advantages over VNT mainly due to its simplicityand rapidity. The c-ELISA test under report had highdegree of sensitivity (92.2%) and specificity (98.4%)for disease surveillance (serological survey) and isvery convenient for large sampling frames. Further,
the performance of this assay is less affected by qual-ity of the sera samples. Sensitivity of the assay waslower for sero-monitoring of PPR antibodies usingan experimental PPR vaccine. There could be severalexplanations for the lowered sensitivity. Firstly, allthe post-vaccinate sera incorporated in the study werecollected 21 days after vaccination, wherein risingtrend of antibody development is observed during thisperiod. Some of these sera fall at the borderline forpositivity in c-ELISA. A proportion of PPR vaccinatesera, especially from sheep, were having VNT titresbetween 1:8 and 1:16. Due to slightly lower sensitiv-ity of c-ELISA test compared to VNT, some of thesesera became negative in ELISA (false negative popu-lation). It was also observed while determining corre-lation coefficient (r) for end-point antibody titration,that c-ELISA titre of sera was generally 1 − 3 log2lower than the VN titre. The test sera samples in thepresent study were used at a constant dilution of 1:4.Therefore, with a lower sensitivity (1 − 3 log2) ofthe assay, weakly positive sera in VNT could havemanaged to become negative in c-ELISA. Similarobservations were made by other workers using suchassays for detection of rinderpest and PPR virus anti-bodies (Saliki et al., 1993; Singh et al., 2000; Libeauet al., 1995). In order to overcome the problem of low-ered sensitivity of 4B11 MAb based c-ELISA, a lowcut-off (mean of negative population + 2 S.D.) waschosen. It has been suggested, for such serologicalassay that 2–3 standard deviations, if added to meanof negative population, can improve the performanceof the test (Jacobson, 1998). The authors thereforeopted to select a slightly lower cut-off to increase thesensitivity without compromising much on specificityof the assay. Even with lower cut-off, the c-ELISAtest had very high specificity (98.4%) with a sensi-tivity of 92.2% in Indian small ruminant population.Even though the monoclonal antibody used in thepresent assay is specific to PPR virus in an indirectELISA test, the present c-ELISA test detected rinder-pest virus antibodies in vaccinated animals. Amongstthe 32 sera samples which had VNT titres between1:16 and 1:256 for rinderpest virus but negative forPPR virus antibodies (VNT titres >1:8), two sampleswere detected as positive in c-ELISA. This suggeststhe involvement of certain form of steric hindrancesor interference. It may be possible that the epitope towhich 4B11 MAb is directed, lies in close proximity to

R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 13
certain other cross-reactive epitopes, which recognizePPR and rinderpest viruses both. The antibodies di-rected to the cross-reacting epitope may interfere withthe binding of antibodies on the target epitope of thepresent assay. Both the other commercially availableassays (Anderson and McKay, 1994; Libeau et al.,1995) for antibodies to PPRV, give cross-reactionswith antibodies to rinderpest as well. All these inves-tigations suggest that the presence of antibodies torinderpest virus in the target population may affect thespecificity and sensitivity of the assay. Indian smallruminant population is presently free from rinder-pest antibodies, except a proportion of vaccinatedanimals in south Indian states, where vaccinationcampaigns continued till 1996. The sera from theseanimals, however, were not included in the presentstudy.
For any infectious disease control program to beundertaken, its detailed epidemiological situation inthe country is to be studied. Such studies require ex-tensive disease surveillance (clinical and serological)in order to generate a baseline data to provide inputand justify economic implications of any such pro-gram to succeed. The presently developed c-ELISAis expected to be an ideal test to carry out serologi-cal survey of PPR virus antibody in small ruminantsof India and other South Asian countries. Further, ifmass vaccination campaign is used to control the dis-ease, sero-monitoring of antibodies in vaccinated an-imals can be carried out using the presently devel-oped c-ELISA to assess the success of vaccination.Keeping these objectives in mind, suitability of thepresent c-ELISA for sero-monitoring of antibodies toPPR vaccinated animals was investigated. The test un-der discussion could detect PPR antibodies as earlyas 16–20 days post-vaccination in three goats. Here itis important to note that the PPR virus used, was thesame as the one used for generation of monoclonalantibodies and production of ELISA. Similar findingswere also observed with a c-ELISA test developed fordetection of rinderpest virus antibodies (Singh et al.,2000). In this test, antibodies to rinderpest virus weredetected between 12 and 21 days post-vaccination us-ing RBOK strain of rinderpest virus as vaccine (JohnAnderson, personal communication). Furthermore, theassay could detect PPR virus antibodies from fourexperimentally vaccinated goats during the course ofan year-long investigation. These goats were under
long-term immunity trials. The finding conclusivelyproves that this assay could be successfully used tomonitor antibodies in PPR vaccinated animals. Thesera for this purpose could be collected any time af-ter 1 month of the vaccination with the end-point timelimit as yet undetermined. The rate of sero-conversionin vaccinated animals may be slightly higher than de-termined using the assay due to its lower sensitivity.It has been reported that, goats having a VN titre of1:4 for PPR antibody were protected against an exper-imental challenge (Sreenivasa et al., 2002). However,such goats were rarely positive for PPR virus antibodyemploying c-ELISA. Further a considerable popula-tion of PPR vaccinated sheep and few goats falling inVN titre range of 1:8–1:16 and some times even 1:32with a definite antibody protection level could not bedetected in the c-ELISA test.
India is provisionally free from rinderpest since1998 (Sinha, 1998). In the present epidemiologicalscenario, the c-ELISA, so described, could proveto be an important tool for sero-monitoring follow-ing vaccination, and sero-surveillance for PPR virusantibodies despite its slightly lower sensitivity. Theassay has been successfully used on approximately3000 additional sera samples within the laboratory.It is presently under field validation with encourag-ing results from some of the field laboratories of thecountry.
Acknowledgements
The authors are grateful to Director, Indian Vet-erinary Research Institute for providing the facilities.This study was partly funded by National AgriculturalTechnology Programme (NATP) launched by IndianCouncil of Agricultural Research.
References
Anderson, J., McKay, J.A., 1994. The identification of antibodiesagainst peste des petits ruminants virus in cattle, sheep andgoats and the possible implications to rinderpest controlprograms. Epidemiol. Infect. 112, 225–231.
Anderson, J., McKay, J.A., Butcher, R.N., 1991. The use ofmonoclonal antibodies in competition ELISA for detection ofantibodies to rinderpest and peste des petits ruminants viruses.In: The Seromonitoring of Rinderpest Throughout Africa:Phase I. IAEA, Vienna, Austria, pp. 43–53.

14 R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15
Anon., 1993. Outbreaks occurring during the month of January1993. List A diseases. Bulletin 105. Office International desEpizooties, pp. 7–10.
Anon., 1994. Recommended procedures for disease and serologicalsurveillance as part of the Global Rinderpest EradicationProgramme IAEA-TECDOC 747. IAEA, Vienna, 51 pp.
Bandyopadhyay, S.K., Singh, R.P., Chandra, U., 1999. Efficacy ofthe micro-method of the assessment of neutralizing antibodiesfollowing vaccination/infection with rinderpest virus. Indian J.Anim. Sci. 69, 82–84.
Dhar, P., Pandey, K.D., Bandyopadhyay, S.K., Negi, B.S.,Sreenivasa, B.P., Singh, R.P., Roy, R.N., 1997. Epidemilogicalobservations on peste des petits ruminants in India. In:Proceedings of the XVIII Annual Conference of IndianAssociation of Veterinary Microbiologists, Immunologists andSpecialists in Infectious Diseases and National Symposiumon Basic and Biotechnological Approaches in Animal Health,November 6–8. PAU, Ludhiana, India.
Dhar, P., Sreenivasa, B.P., Barret, T., Corteyn, M., Singh, R.P.,Bandyopadhyay, S.K., 2002. Recent epidemiology of peste despetits ruminants. Vet. Microbiol. 88, 153–159.
Diallo, A., Barrett, T., Barbron, M., Shaila, M., Subbarao, S.M.,Taylor, W.P., 1989. Differentiation of rinderpest and peste despetits ruminants viruses using specific cDNA clones. J. Virol.Meth. 23, 127–136.
Gargadennec, L., Lalanne, A., 1942. La peste des petits ruminants.Bulletin des Services Zootechniques et des Epizooties del’Afrique Occidentale Francaise 5, 16–21.
Gibbs, E.P.J., Taylor, W.P., Lawman, M.J.P., Bryant, J., 1979.Classification of peste des petits ruminants virus as fourthmember of the genus morbillivirus. Intervirology 11, 268–274.
Govindarajan, R., Koteeswaran, A., Venugopalan, A.T., Shyam, G.,Shaouna, S., Shaila, M.S., Ramachandran, S., 1997. Isolationof pestes des petits ruminants virus from an outbreak in Indianbuffalo (Bubalus bubalis). Vet. Rec. 141, 573–574.
Hall, W.W., Lamb, R.A., Chopin, P.W., 1980. The polypeptidesof canine distemper virus: synthesis in infected cells andrelatedness to the polypeptides of other morbilliviruses.Virology 100, 433–449.
Imagawa, D.T., 1968. Relationships among measles, caninedistemper and rinderpest viruses. Prog. Med. Virol. 10, 160–193.
Ismail, T.M., Yamanaka, M.K., Saliki, J.T., EL-Kholy, A.,Mebus, C., Yilma, T., 1995. Cloning and expression of thenucleoprotein of peste des petits ruminants virus in baculovirusfor use in serological diagnosis. Virology 20, 776–778.
Jacobson, R.H., 1998. Validation of serological assays for diagnosisof infectious diseases. Rev. Sci. Tech. Off. Int. Epiz. 17, 469–486.
Jeggo, M.H., Anderson, J., 1992. FAO/IAEA external qualityassurance programme for the FAO/IAEA competitive ELISAresults in 1992. In: The Sero-monitoring of RinderpestThroughout AFRICA: Phase II. International Atomic EnergyAgency, Vienna, pp. 9–21.
Jones, L., Giavedoni, L., Saliki, J.T., Brown, C., Mebus, C., Yilma,T., 1993. Protection of goats against peste des petits ruminants
with a vaccinia virus double recombinant expressing the F andH genes of rinderpest virus. Vaccine 11, 961–964.
Lefevre, P.C., Diallo, A., 1990. Peste des petits ruminants virus.Rev. Sci. Tech. Off. Int. Epiz. 9, 951–965.
Lefevre, P.C., Diallo, A., Schenkel, F., Hussein, S., Staak, G.,1991. Serological evidence of peste des petits ruminants inJordan. Vet. Rec. 128, 110.
Libeau, G., Diallo, A., Calvez, D., Lefevre, P.C., 1992. Acompetition-ELISA using anti-N monoclonal antibodies forspecific detection of rinderpest antibodies in cattle and smallruminants. Vet. Microbiol. 31, 147–160.
Libeau, G., Diallo, A., Colas, F., Guerre, L., 1994. Rapiddifferential diagnosis of rinderpest and peste des petitsruminants using an immunocapture ELISA. Vet. Rec. 134,300–304.
Libeau, G., Prehaud, C., Lancelot, R., Colas, F., Guerre, L., Bishop,D.H., Diallo, A., 1995. Development of a competitive ELISAfor detecting antibodies to the peste des petits ruminants virususing a recombinant nucleoprotein. Res. Vet. Sci. 58, 50–55.
Mondal, A.K., Chottopadhyay, A.P., Sarkar, S., Saha, G.R.,Bhowmik, M.K., 1995. Report on epizootiological andclinicopathological observations on peste des petits ruminants(PPR) of goats in West Bengal. Indian J. Anim. Health 34,145–148.
Nanda, Y.P., Chatterjee, A., Purohit, A.K., Diallo, A., Innui, K.,Sharma, R.N., Libeau, G., Thevasagayam, J.A., Bruning, A.,Kiching, R.P., Anderson, J., Barrett, T., Taylor, W.P., 1996.The isolation of peste des petits ruminants virus from NorthernIndia. Vet. Microbiol. 51, 207–216.
Obi, T.U., Patrick, D., 1984. The detection of peste des petitsruminants (PPR) virus antigen by agar gel precipitation testand counter-immunoelectrophoresis. J. Hyg. 93, 579–586.
Obi, T.U., McCullough, K.C., Taylor, W.P., 1990. The productionof peste des petits ruminants hyperimmune sera in rabbits andtheir application in virus diagnosis. J. Vet. Med. 37, 345–352.
Orvell, C., Norrby, E., 1974. Further studies on the immunologicalrelationships among measles, canine distemper and rinderpestvirus. J. Immunol. 113, 1850–1858.
Plowright, W., Ferris, R.D., 1962. Studies with rinderpest virus intissue-culture. The use of attenuated culture virus as a vaccinefor cattle. Res. Vet. Sci. 3, 172–182.
Rossitter, P.B., Jessett, D.M., Taylor, W.P., 1985. Microneutra-lisation systems for use with different strains of peste des petitsruminants virus and rinderpest virus. Trop. Anim. Health Prod.17, 75–81.
Saliki, J.T., Libeau, G., House, J.A., Mebus, C.A., Dubovi, J., 1993.A monoclonal antibody based blocking ELISA for specificdetection and titration of peste des petits ruminants antibodyin caprine and ovine sera. J. Clin. Microbiol. 31, 1075–1082.
Schrijver, R.S., Kramps, J.A., 1998. Critical factors affecting thediagnostic reliability of enzyme-linked immunosorbent assayformats. Rev. Sci. Tech. Off. Int. Epiz. 17, 550–561.
Shaila, M.S., Purushothaman, V., Bhavasar, D., Venugopal, K.,Venkatesan, R.A., 1989. Peste des petits ruminants in India.Vet. Rec. 125, 602.
Shaila, M.S., Venugopal, K., Purushothaman, V., Venkatesan,R.A., 1990. Isolation and characterization of peste des petits

R.P. Singh et al. / Veterinary Microbiology 98 (2004) 3–15 15
ruminants virus from an outbreak in Tamil Nadu sheep. Ind.Vet. J. 67, 385–386.
Shaila, M.S., Shamaki, D., Forsyth, M.A., Diallo, A., Goatley, L.,Kitching, R.P., Barrett, T., 1996. Geographic distribution ofpeste des petits ruminants viruses. Virus Res. 43, 149–153.
Singh, R.P., 2002. Production and characterization of monoclonalantibodies to peste des petits ruminants(PPR) virus. PhDThesis, Submitted to Deemed University, IVRI, Izatnagar,India.
Singh, R.P., Sreenivasa, B.P., Dhar, P., Roy, R.N., Bandyopadhyay,S.K., 2000. Development and evaluation of a monoclonalantibody based competitive enzyme-linked immunosorbentassay for the detection of the rinderpest virus antibodies. Rev.Sci. Tech. Off. Int. Epiz. 19, 754–763.
Sinha, N.K., 1998. Rinderpest in India: the delegate declares thepeninsular zone ‘provisionally free’ from the disease. Dis. Info.11, 61.
Sreenivasa, B.P., Dhar, P., Singh, R.P., Bandyopadhyay, S.K.,2000. Evaluation of an indigenously developed homologouslive-attenuated cell culture vaccine against peste des petits
ruminants infection of small ruminants. Abstract in theProceedings of the XX Annual Conference of IndianAssociation of Veterinary Microbiologists, Immunologists andSpecialists in Infectious Diseases, October, Pant nagar India,p. 84.
Sreenivasa, B.P., Dhar, P., Singh, R.P., Bandyopadhyay, S.K., 2002.Development of peste des petits ruminants (PPR) challengevirus from a field isolate. Presented at the XIV AnnualConference and National Seminar on Management of ViralDiseases with Emphasis on Global Trade and WTO Regime ofIndian Virological Society, January 18–20, 2002. IVRI, Hebbal,Bangalore, India.
Taylor, W.P., 1984. The distribution and epidemiology of pestedes petits ruminants. Prev. Vet. Med. 2, 157–166.
Taylor, W.P., Al Busaidy, S., Barrett, T., 1990. The epidemiologyof peste des petits ruminants in the Sultanate of Oman. Vet.Microbiol. 22, 341–352.
Wright, P.F., 1998. International standards for test methods andreference sera for diagnostic tests for antibody detection. Rev.Sci. Tech. Off. Int. Epiz. 17, 527–533.
Copyright © 2022 FDOKUMEN




















