
Detection and Quantification of Functional Genes of Cellulose- Degrading, Fermentative, and...
11
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Apr. 2010, p. 2192–2202 Vol. 76, No. 7 0099-2240/10/$12.00 doi:10.1128/AEM.01285-09 Copyright © 2010, American Society for Microbiology. All Rights Reserved. Detection and Quantification of Functional Genes of Cellulose- Degrading, Fermentative, and Sulfate-Reducing Bacteria and Methanogenic Archaea L. P. Pereyra, 1 S. R. Hiibel, 2 † M. V. Prieto Riquelme, 3 K. F. Reardon, 2,4 and A. Pruden 1 * Department of Civil and Environmental Engineering, 1 Department of Chemical and Biological Engineering, 2 and Department of Microbiology, Immunology, and Pathology, 4 Colorado State University, Fort Collins, Colorado 80523, and Via Department of Civil and Environmental Engineering, 418 Durham Hall, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061 3 Received 3 June 2009/Accepted 21 January 2010 Cellulose degradation, fermentation, sulfate reduction, and methanogenesis are microbial processes that coexist in a variety of natural and engineered anaerobic environments. Compared to the study of 16S rRNA genes, the study of the genes encoding the enzymes responsible for these phylogenetically diverse functions is advantageous because it provides direct functional information. However, no methods are available for the broad quantification of these genes from uncultured microbes characteristic of complex environments. In this study, consensus degenerate hybrid oligonucleotide primers were designed and validated to amplify both sequenced and unsequenced glycoside hydrolase genes of cellulose-degrading bacteria, hydA genes of fermen- tative bacteria, dsrA genes of sulfate-reducing bacteria, and mcrA genes of methanogenic archaea. Specificity was verified in silico and by cloning and sequencing of PCR products obtained from an environmental sample characterized by the target functions. The primer pairs were further adapted to quantitative PCR (Q-PCR), and the method was demonstrated on samples obtained from two sulfate-reducing bioreactors treating mine drainage, one lignocellulose based and the other ethanol fed. As expected, the Q-PCR analysis revealed that the lignocellulose-based bioreactor contained higher numbers of cellulose degraders, fermenters, and methano- gens, while the ethanol-fed bioreactor was enriched in sulfate reducers. The suite of primers developed represents a significant advance over prior work, which, for the most part, has targeted only pure cultures or has suffered from low specificity. Furthermore, ensuring the suitability of the primers for Q-PCR provided broad quantitative access to genes that drive critical anaerobic catalytic processes. The gene encoding the 16S small ribosomal subunit has served as a highly suitable target for studying bacterial species. When one obtains 16S rRNA gene sequence information, it is sometimes possible to infer function from an identical match to a well-characterized pure culture. More commonly, how- ever, the similarity to pure cultures is low, and/or the highest similarities correspond to 16S rRNA gene sequences identified without isolation or phenotypic characterization. In either case, care must be taken, because distinct phenotypes [e.g., dissimilatory Fe(III) reduction, chlorate reduction] are found in microorganisms with highly similar (e.g., 99.5%) 16S rRNA gene sequences (1). In addition, 16S rRNA gene surveys of broad phylogenetic groups can be time-, labor-, and cost-in- tensive. For example, it is estimated that the 16S rRNA gene- based detection of all recognized lineages of sulfate-reducing bacteria (SRB) would require approximately 132 16S rRNA gene-targeted microarray probes (32). A more-direct approach for the study of microbes that span phylogenetic groups is to target them as a physiologically co- herent guild by using specific genetic markers (functional genes) for the functions of interest. Functional genes have been successfully targeted in bioremediation studies to inves- tigate microbial populations responsible for the degradation of various contaminants. Some examples include the use of the large alpha subunit of benzylsuccinate synthase to monitor anaerobic hydrocarbon-degrading bacteria (5), the monitoring of ars genes for the identification and quantification of arsenic- metabolizing bacteria (45), and the detection of catechol 1,2- dioxygenase in aromatic-hydrocarbon-degrading Rhodococcus spp. (48). In the field of mine drainage/metal remediation, functional genes have been used to target SRB (17, 26), but the methods have suffered both from a lack of broad specificity for SRB and from the inability to distinguish SRB from sulfur- oxidizing bacteria (SOB). A general challenge to the functional- gene approach has been the relative lack of characterization and unavailability of target sequences. As a consequence, the primer sets that are available tend to be more relevant to pure cultures than to complex environmental samples. Microbial communities in natural and engineered anaerobic environments that utilize cellulose as the primary carbon source, such as those in rumina (56), termite guts (54), decom- posing wood (7), sulfate-reducing and methanogenic sedi- ments (9, 22), wetlands (28), and sulfate-reducing bioreactors (26), are particularly challenging to characterize. 16S rRNA gene-based studies have revealed the complexity of these mi- crobial communities and their high levels of phylogenetic and functional diversity. In such anaerobic environments, mineral- * Corresponding author. Present address: Via Department of Civil and Environmental Engineering, 418 Durham Hall, Virginia Polytech- nic Institute and State University, Blacksburg, VA 24061. Phone: (540) 231-3980. Fax: (540) 231-7916. E-mail: [email protected]. † Present address: Department of Biochemistry and Molecular Bio- logy, University of Nevada, Reno, Reno, NV 89557. Published ahead of print on 5 February 2010. 2192
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Detection and Quantification of Functional Genes of Cellulose- Degrading, Fermentative, and...

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Apr. 2010, p. 2192–2202 Vol. 76, No. 70099-2240/10/$12.00 doi:10.1128/AEM.01285-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Detection and Quantification of Functional Genes of Cellulose-Degrading, Fermentative, and Sulfate-Reducing Bacteria
and Methanogenic Archaea�
L. P. Pereyra,1 S. R. Hiibel,2† M. V. Prieto Riquelme,3 K. F. Reardon,2,4 and A. Pruden1*Department of Civil and Environmental Engineering,1 Department of Chemical and Biological Engineering,2 and
Department of Microbiology, Immunology, and Pathology,4 Colorado State University, Fort Collins,Colorado 80523, and Via Department of Civil and Environmental Engineering, 418 Durham Hall,
Virginia Polytechnic Institute and State University, Blacksburg, Virginia 240613
Received 3 June 2009/Accepted 21 January 2010
Cellulose degradation, fermentation, sulfate reduction, and methanogenesis are microbial processes thatcoexist in a variety of natural and engineered anaerobic environments. Compared to the study of 16S rRNAgenes, the study of the genes encoding the enzymes responsible for these phylogenetically diverse functions isadvantageous because it provides direct functional information. However, no methods are available for thebroad quantification of these genes from uncultured microbes characteristic of complex environments. In thisstudy, consensus degenerate hybrid oligonucleotide primers were designed and validated to amplify bothsequenced and unsequenced glycoside hydrolase genes of cellulose-degrading bacteria, hydA genes of fermen-tative bacteria, dsrA genes of sulfate-reducing bacteria, and mcrA genes of methanogenic archaea. Specificitywas verified in silico and by cloning and sequencing of PCR products obtained from an environmental samplecharacterized by the target functions. The primer pairs were further adapted to quantitative PCR (Q-PCR),and the method was demonstrated on samples obtained from two sulfate-reducing bioreactors treating minedrainage, one lignocellulose based and the other ethanol fed. As expected, the Q-PCR analysis revealed that thelignocellulose-based bioreactor contained higher numbers of cellulose degraders, fermenters, and methano-gens, while the ethanol-fed bioreactor was enriched in sulfate reducers. The suite of primers developedrepresents a significant advance over prior work, which, for the most part, has targeted only pure cultures orhas suffered from low specificity. Furthermore, ensuring the suitability of the primers for Q-PCR providedbroad quantitative access to genes that drive critical anaerobic catalytic processes.
The gene encoding the 16S small ribosomal subunit hasserved as a highly suitable target for studying bacterial species.When one obtains 16S rRNA gene sequence information, it issometimes possible to infer function from an identical matchto a well-characterized pure culture. More commonly, how-ever, the similarity to pure cultures is low, and/or the highestsimilarities correspond to 16S rRNA gene sequences identifiedwithout isolation or phenotypic characterization. In eithercase, care must be taken, because distinct phenotypes [e.g.,dissimilatory Fe(III) reduction, chlorate reduction] are foundin microorganisms with highly similar (e.g., 99.5%) 16S rRNAgene sequences (1). In addition, 16S rRNA gene surveys ofbroad phylogenetic groups can be time-, labor-, and cost-in-tensive. For example, it is estimated that the 16S rRNA gene-based detection of all recognized lineages of sulfate-reducingbacteria (SRB) would require approximately 132 16S rRNAgene-targeted microarray probes (32).
A more-direct approach for the study of microbes that spanphylogenetic groups is to target them as a physiologically co-herent guild by using specific genetic markers (functional
genes) for the functions of interest. Functional genes havebeen successfully targeted in bioremediation studies to inves-tigate microbial populations responsible for the degradation ofvarious contaminants. Some examples include the use of thelarge alpha subunit of benzylsuccinate synthase to monitoranaerobic hydrocarbon-degrading bacteria (5), the monitoringof ars genes for the identification and quantification of arsenic-metabolizing bacteria (45), and the detection of catechol 1,2-dioxygenase in aromatic-hydrocarbon-degrading Rhodococcusspp. (48). In the field of mine drainage/metal remediation,functional genes have been used to target SRB (17, 26), but themethods have suffered both from a lack of broad specificity forSRB and from the inability to distinguish SRB from sulfur-oxidizing bacteria (SOB). A general challenge to the functional-gene approach has been the relative lack of characterizationand unavailability of target sequences. As a consequence, theprimer sets that are available tend to be more relevant to purecultures than to complex environmental samples.
Microbial communities in natural and engineered anaerobicenvironments that utilize cellulose as the primary carbonsource, such as those in rumina (56), termite guts (54), decom-posing wood (7), sulfate-reducing and methanogenic sedi-ments (9, 22), wetlands (28), and sulfate-reducing bioreactors(26), are particularly challenging to characterize. 16S rRNAgene-based studies have revealed the complexity of these mi-crobial communities and their high levels of phylogenetic andfunctional diversity. In such anaerobic environments, mineral-
* Corresponding author. Present address: Via Department of Civiland Environmental Engineering, 418 Durham Hall, Virginia Polytech-nic Institute and State University, Blacksburg, VA 24061. Phone: (540)231-3980. Fax: (540) 231-7916. E-mail: [email protected].
† Present address: Department of Biochemistry and Molecular Bio-logy, University of Nevada, Reno, Reno, NV 89557.
� Published ahead of print on 5 February 2010.
2192

ization of complex organic matter occurs through the con-certed action of a variety of microorganisms. Primary ferment-ers, such as cellulose degraders, break down the complexmolecules and ferment the hydrolysis products. Secondary fer-menters also ferment the hydrolysis products. When sulfate isavailable, SRB utilize the fermentation products as carbon andenergy sources. In addition, methanogens can also utilize someof the fermentation products. In many cases, functionally im-portant members, such as SRB, are present only as a smallfraction of the community (36, 38), making them difficult todetect by use of 16S rRNA gene-targeted fingerprinting meth-ods. Furthermore, the phylogenetic diversity of cellulose de-graders, fermenters, and SRB prevents their quantification us-ing a small number of 16S rRNA gene-targeted probes.
In this study, degenerate PCR primers were developed, val-idated, and demonstrated for the amplification of key functionalgroups in anaerobic environments possessing genes encoding gly-coside hydrolases of families 5 (collectively designated cel5 in thisstudy) and 48 (collectively designated cel48 in this study) (cellu-lose degradation), the alpha subunit of iron hydrogenase (hydA)(fermentation), dissimilatory sulfite reductase (dsrA) (sulfate re-duction), and methyl coenzyme M reductase (mcrA) (methano-genesis). This work is particularly novel considering that the vastmajority of existing methods are suitable only for pure cultures,especially in the cases of cel5, cel48, and hydA (21, 44, 47). Thus,the approach provides access to uncultured and unsequencedmarkers, a critical feature for the study of key anaerobic processesin complex environments. Specificity was also enhanced wherepossible, notably in the case of dsrA, for which existing primerseither do not distinguish SRB from SOB (14, 17) or have goodalignment with only a narrow range of SRB (31, 52). Finally, allprimers in this study were designed and validated for quantitativePCR (Q-PCR), in order to provide valuable quantitative func-tional information about complex anaerobic communities. Theapproach is demonstrated on mine drainage remediation systemsand is expected to be of broad value to a variety of fields, includ-ing advancing the understanding of biohydrogen production,global carbon cycling, and other important biogeochemical pro-cesses.
MATERIALS AND METHODS
Primer design. The consensus degenerate hybrid oligonucleotide primer(CODEHOP) strategy (43) was used to design all primer sets. CODEHOPs arehybrid primers that contain a relatively short 3� degenerate core and a 5� non-degenerate consensus clamp that stabilizes the hybridization of the 3� end (43).The protein sequences used to design the primers were downloaded from theNational Center for Biotechnology Information (NCBI) database (Table 1).
Using the Gibbs algorithm of the Block Maker program (23), a total of 12, 5,7, 5, and 8 blocks of conserved amino acids were found in the multiple alignmentsof the protein sequences of the cel5, cel48, hydA, dsrA, and mcrA genes presentedin Table 1, respectively. These blocks were used as input to the CODEHOPprogram for the design of primers specific to each of the target genes. Primerswere designed using all codon possibilities for the 3� degenerate core and themost frequent nucleotide in each position for the 5� consensus clamp. TheCODEHOP default parameters were used for the design of all primer sets. Foreach block, the primer with the lowest degeneracy in the 3� core was selected,and its sequence was compared in silico with the NCBI nucleotide database toconfirm target specificity. Primer sequences that did not match the desired targetwere discarded, and the primer with the next lowest degeneracy was compared tothe sequences in the NCBI database. The primers that were identified as specificwere aligned with the DNA sequences of the proteins used for the design andwere manually modified by increasing the degeneracy of the 3� region to improvethe match of the primer with the reference sequences or by decreasing the
degeneracy when it was higher in the primer than in the alignment of referencesequences. The primers were arranged in pairs that would yield an amplicon ofa size suitable for Q-PCR amplification (150 to 450 bp).
Primer validation. (i) Genomic DNA from pure cultures. Genomic DNAsfrom the following microorganisms were used as positive or negative controls forPCR: Clostridium thermocellum (ATCC 27405D), Methanococcus maripaludis(ATCC 43000D), and Desulfobacterium autotrophicum (ATCC 43914D). Purecultures of the following microorganisms were grown in the laboratory, and theirgenomic DNAs were used as positive or negative controls for PCR: Escherichiacoli, Clostridium cellulovorans (ATCC 35296), Ruminococcus flavefaciens (ATCC49949), and Fibrobacter succinogenes strain B1 (ATCC 51214).
(ii) Environmental samples. For further primer validation, samples collectedfrom two pilot-scale sulfate-reducing biochemical reactors that receive minedrainage from the National Tunnel in Black Hawk, CO (39), were selected. Thesample sources differed primarily in the carbon substrate provided. The firstreactor contained a complex lignocellulose (LC)-based substrate (wood chips[35%], limestone [20%], and corn stover [30%]). The second reactor receivedethanol (Et) as its primary substrate and was packed with limestone and azero-valent iron slag layer on the top. Both bioreactors were inoculated withhorse manure. As a negative control, an aerated (dissolved oxygen [DO], �2.0mg/liter) sequencing batch reactor (SBR) dominated by nontarget DNA wasselected. The reactor was seeded with activated sludge from a local municipalwastewater treatment plant and was fed a mixture of Bacto peptone and acetate(OAc) at an influent chemical oxygen demand of 350 mg/liter. The hydraulic andsolid residence times were 1 and 10 days, respectively.
DNA extraction. DNA was extracted from portions (approximately 5 g each)of the environmental samples using the PowerMax soil DNA isolation kit andfrom pure cultures using the UltraClean microbial DNA isolation kit (Mo Bio,Carlsbad, CA) according to the manufacturer’s protocols. The extracted DNAwas stored at �80°C for subsequent studies.
Optimization of PCR conditions. An annealing-temperature optimization wasperformed for all primer sets using pure-culture genomic DNA as the templateat the following temperatures: 52.9°C, 54.3°C, 55.8°C, 59.2°C, and 60.7°C (cel48);52.0°C, 52.8°C, 53.9°C, 55.3°C, 56.9°C, 58.5°C, 60.2°C, 61.7°C, 63.0°C, and 64.0°C(cel5); 56.0°C, 58.8°C, 62.8°C, and 65.2°C (hydA); 60.0°C and 63.0°C (dsrA); and53.6°C, 54.7°C, 56.0°C, 58.8°C, and 60.1°C (mcrA). The optimized annealingtemperature was selected based on the presence of one PCR product band of theexpected size on a 1.2% agarose gel for the positive controls and the absence ofany product in the pure-culture negative controls.
The optimized PCR master mix for the primer sets contained 1� reactionbuffer with 2 mM magnesium (5 Prime, Gaithersburg, MD); 1� PCR enhancer(5 Prime); 0.05 mM each deoxynucleoside triphosphate; 1.0 mM (mcrA), 1.5 mM(dsrA), or 2.5 mM (cel5) Mg(OAc)2; 0.15 �M (cel48, hydA, dsrA), 0.2 �M (mcrA),or 0.25 �M (cel5) each primer; 0.125 �l of formamide; 0.875 U of Taq DNApolymerase (5 Prime); an additional 0.25 U (cel5) or 0.375 U (dsrA) of iTaq DNApolymerase (Bio-Rad, Hercules, CA); 1 �l (cel48, cel5, dsrA, and mcrA) or 0.5 �l(hydA) of the DNA template; and deionized water to a final volume of 12.5 �l.The additional iTaq added to the dsrA and cel5 PCR mixtures improved theyields of otherwise weak PCR products, presumably because the polymerase ismore sensitive than Taq. The temperature program consisted of 3 min at 95°C,followed by 35 (hydA and mcrA), 40 (cel48), 45 (cel5), or 50 (dsrA) cycles of 40 sat 95°C, 30 s at the corresponding annealing temperature, 30 s at 72°C, and a finalextension of 7 min at 72°C. The annealing temperatures were 56°C, 52°C, 59°C,60°C, and 56°C for the cel48, cel5, hydA, dsrA, and mcrA primer sets, respectively.
Cloning. The DNAs from the positive-control LC bioreactor and the neg-ative-control SBR were used as the PCR templates for the primer combina-tions cel48_490F–cel48_920R, cel5_392F–cel5_754R, hydA_1290F–hydA_1538R, dsrA_290F–dsrA_660R, and mcrA_1035F–mcrA_1530R. In addition,the 16S rRNA gene was PCR amplified from DNA extracts of both the LCand Et bioreactors using primer sets 341F and 1492R (55). PCR products, ifpresent, were cloned with the TOPO TA cloning kit (Invitrogen, Carlsbad,CA) according to the manufacturer’s protocol. Inserts were PCR amplifieddirectly from colonies by using the vector-specific M13F and M13R primers.For the 16S rRNA gene clones, amplified rRNA gene restriction analysis(ARDRA) was performed visually with MspI restriction enzyme (Promega,Madison, WI)-digested inserts. The DNA corresponding to each uniqueARDRA pattern was sequenced.
Selection of restriction enzymes. The Restriction Enzyme Database(REBASE) (42) was used to select restriction endonucleases that could digest eachfunctional gene PCR product. The NEBcutter (version 2.0) tool of REBASE wasused to identify suitable restriction enzymes and the respective cleavage sites.The Hpy188III, Mbol, MnlI, NlaIII, and MboII restriction enzymes (New En-
VOL. 76, 2010 Q-PCR OF FUNCTIONAL GENES IN ANAEROBIC ENVIRONMENTS 2193
gland Biolabs, Ipswich, MA) were chosen to digest the PCR products from theprimer sets targeting the cel5, cel48, hydA, dsrA, and mcrA genes, respectively.
Restriction enzyme digestion and DNA sequencing. M13 PCR products ofcloned gene inserts were digested with the corresponding restriction enzymesaccording to the manufacturer’s recommendations, with incubation conditions of37°C for 1 h (mcrA), 1.5 h (cel5 and hydA), or 3 h (cel48 and dsrA), followed byan inactivation step of 20 min at 65°C. For each unique restriction patternidentified on 3% agarose gels, at least one clone was sequenced by the Proteo-mics and Metabolomics Facility (Colorado State University, Fort Collins, CO).The closest matches to microorganisms available in the NCBI database weredetermined using Basic Local Alignment Search Tool X (BLASTX), whichsearches the protein database using a translated nucleotide query.
Q-PCR. The primer sets were adapted to Q-PCR and were further validatedby quantifying the functional genes in samples collected from the LC and Etsulfate-reducing bioreactors. In addition, the total bacterial 16S rRNA gene wasquantified using the conditions, universal primers (BACT1369F andPROK1492R), and probe (TM1389F) described by Suzuki et al. (46). For thefunctional-gene primers, Q-PCR amplification was performed with a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) using a 12.5-�l reaction
mixture consisting of 1� Power SYBR green PCR master mix (Applied Biosys-tems); 0.15 �M (cel48, hydA, and dsrA), 0.2 �M (mcrA), or 0.25 �M (cel5) eachprimer; 1 mM (mcrA and hydA), 1.5 mM (dsrA), or 2.5 mM (cel5) Mg(OAc)2; anadditional 0.25 U (dsrA) or 0.375 U (cel5) of iTaq DNA polymerase (Bio-Rad);and 1 �l of the template. The temperature program was 10 min at 95°C, followedby 40 (cel48 and mcrA), 45 (hydA and cel5), or 50 (dsrA) cycles of 40 s at 95°C,30 s at the corresponding annealing temperature, and 30 s at 72°C. DNA extractswere diluted 1:10 and were analyzed in five replicates.
For each functional gene primer set, a triplicate four- to six-point calibrationcurve was constructed using a standard mixture of purified PCR products ob-tained with the vector-specific primers T3 and T7 from sequenced clones. For the16S rRNA gene primer set, a purified PCR product amplified from an environ-mental sample with primers 8F and 1492R (13, 55) was used as the standard. Thecalibration curves were used for absolute quantification of the target genes underthe assumption that the PCR amplification efficiencies for standards and sampleswere the same. To ensure that only samples for which this assumption was validwere analyzed, the procedure proposed by Chervoneva et al. (10) was applied.Briefly, LinRegPCR (41) was used to calculate the amplification efficiency fromthe PCR amplification curve of each replicate sample and standard. A box plot
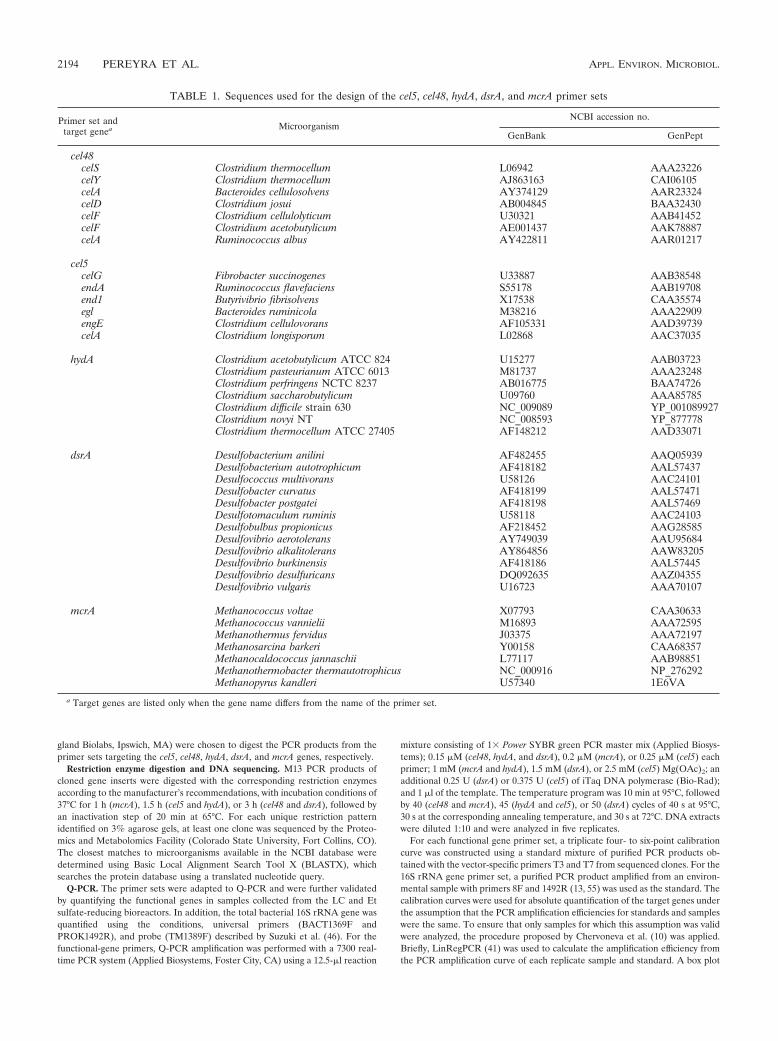
TABLE 1. Sequences used for the design of the cel5, cel48, hydA, dsrA, and mcrA primer sets
Primer set andtarget genea Microorganism
NCBI accession no.
GenBank GenPept
cel48celS Clostridium thermocellum L06942 AAA23226celY Clostridium thermocellum AJ863163 CAI06105celA Bacteroides cellulosolvens AY374129 AAR23324celD Clostridium josui AB004845 BAA32430celF Clostridium cellulolyticum U30321 AAB41452celF Clostridium acetobutylicum AE001437 AAK78887celA Ruminococcus albus AY422811 AAR01217
cel5celG Fibrobacter succinogenes U33887 AAB38548endA Ruminococcus flavefaciens S55178 AAB19708end1 Butyrivibrio fibrisolvens X17538 CAA35574egl Bacteroides ruminicola M38216 AAA22909engE Clostridium cellulovorans AF105331 AAD39739celA Clostridium longisporum L02868 AAC37035
hydA Clostridium acetobutylicum ATCC 824 U15277 AAB03723Clostridium pasteurianum ATCC 6013 M81737 AAA23248Clostridium perfringens NCTC 8237 AB016775 BAA74726Clostridium saccharobutylicum U09760 AAA85785Clostridium difficile strain 630 NC_009089 YP_001089927Clostridium novyi NT NC_008593 YP_877778Clostridium thermocellum ATCC 27405 AF148212 AAD33071
dsrA Desulfobacterium anilini AF482455 AAQ05939Desulfobacterium autotrophicum AF418182 AAL57437Desulfococcus multivorans U58126 AAC24101Desulfobacter curvatus AF418199 AAL57471Desulfobacter postgatei AF418198 AAL57469Desulfotomaculum ruminis U58118 AAC24103Desulfobulbus propionicus AF218452 AAG28585Desulfovibrio aerotolerans AY749039 AAU95684Desulfovibrio alkalitolerans AY864856 AAW83205Desulfovibrio burkinensis AF418186 AAL57445Desulfovibrio desulfuricans DQ092635 AAZ04355Desulfovibrio vulgaris U16723 AAA70107
mcrA Methanococcus voltae X07793 CAA30633Methanococcus vannielii M16893 AAA72595Methanothermus fervidus J03375 AAA72197Methanosarcina barkeri Y00158 CAA68357Methanocaldococcus jannaschii L77117 AAB98851Methanothermobacter thermautotrophicus NC_000916 NP_276292Methanopyrus kandleri U57340 1E6VA
a Target genes are listed only when the gene name differs from the name of the primer set.
2194 PEREYRA ET AL. APPL. ENVIRON. MICROBIOL.

outlier detection rule was applied to the amplification efficiency of the standardsin order to identify calibration standards that amplified with dissimilar efficien-cies, which were not used in data analysis. The Kinetic Outlier Detection method(3) was used to compare the average efficiency of the remaining standards andthe efficiency of each replicate in order to detect samples with efficiencies dis-similar from those of the standards.
Nucleotide sequence accession numbers. The cel5, cel48, hydA, dsrA, and mcrAsequence data from the LC bioreactor have been submitted to GenBank underaccession numbers GQ184058 to GQ184072, GQ184073 to GQ184082, GQ184083to GQ184095, GQ184096 to GQ184113, and GQ184114 to GQ184128, respectively.The cel48 and dsrA sequence data generated from the SBR have been submittedunder accession numbers GU563804 to GU563811 and GU563812 to GU563821,respectively.
RESULTS
Selection of genetic markers for anaerobic cellulose degra-dation, fermentation, sulfate reduction, and methanogenesis.The selection of appropriate genetic markers for the functionsof interest was the first step in designing suitable primers forquantifying cellulose degraders, fermenters, sulfate reducers,and methanogens in anaerobic environments. The goal was tocapture as much diversity among these groups as possible,including potentially uncultured members, through degenerateprimer design. It was also desired that the primers should beappropriate for Q-PCR.
Of the more than 70 glycoside hydrolase families defined onthe basis of amino acid sequence identity in their catalyticdomains (18), cellulases are found in 15 families (families 5, 6,7, 8, 9, 10, 11, 12, 26, 44, 45, 48, 51, 60, and 61) (24, 40). Thecellulase type does not correlate closely with phylogeny: mostcellulolytic microorganisms produce cellulases that belong todifferent families, and cellulases of the same family occur inwidely different microorganisms (4). However, the majority ofthe glycoside hydrolases of anaerobes belong to just threefamilies: families 5, 9, and 48 (24, 25). Families 5 and 48 wereselected in this study as targets that were particularly wellrepresented among anaerobic cellulose degraders. The major-ity of cel48 genes are found in Clostridium spp., although Bac-teroides cellulosolvens (58) and Ruminococcus albus (12) arealso known to have at least one cel48 gene each. cel48 geneshave also been found in Bacillus, Streptomyces, and Cellulomo-nas, as well as in Eukarya; however, they are highly divergentand therefore were not considered as primary targets in theprimer design. The cel5 target was selected in order to providebroader coverage of anaerobic cellulose degraders, since it isfound in a wider range of phylogenetic groups (24).
Fermentation is carried out by a broad range of species usinga variety of enzymes, some of which are also found in nonfer-menters. In nearly all fermentative bacteria, the electrons fromthe reduced electron carriers are channeled to H2 via ironhydrogenases (2). All iron hydrogenases contain a highly con-served region of 350 amino acids known as the H-cluster do-main in their alpha subunit (34, 50); this region was thereforeselected as highly suitable for primer design. Specifically, thehydA primers were designed from the two conserved proteinregions: GAGVIFGA and MACPGGCING. The second re-gion is one of the characteristic sequence signatures of theH-cluster domain of iron hydrogenases (50).
The dissimilatory reduction of sulfate to hydrogen sulfide inSRB is performed in three steps (51). First, sulfate is activatedto adenosine 5�-phosphosulfate by ATP sulfurylase. This is
followed by the partial reduction to sulfite by adenosine 5�-phosphosulfate reductase (APSR) and the final reduction tosulfide by dissimilatory sulfite reductase (DSR). The genesencoding APSR and DSR are evolutionarily conserved amongSRB, including deltaproteobacteria, clostridia, thermodesul-fobacteria, nitrospirae, and certain archaea (27, 52). However,ATP sulfurylase, APSR, and DSR appear to be involved inboth the oxidative and the reductive mode of the dissimilatorysulfur metabolism (27). This is a major challenge for the studyof SRB in systems where SOB and SRB might coexist. Sincethe dsr genes of SRB and SOB are more distantly relatedthan the corresponding aps genes (27), the dsr gene wasselected as the preferred marker for SRB. Special attentionwas given in subsequent primer design to achieving broadspecificity for SRB among the deltaproteobacteria and clos-tridia while avoiding matches for SOB.
Methanogens are a metabolically versatile group of mi-croorganisms, capable of forming methane from acetate,CO2, H2, and C1 compounds (16). A variety of enzymes areinvolved in the methanogenic pathway, and many are notexclusive to methanogenesis. Except for coenzyme M,methyl coenzyme M, and methyl coenzyme M reductase, allthe enzymes and coenzymes involved in methanogenesis arealso present in sulfate-reducing archaea (49). Similarly,methylene tetrahydromethanopterin dehydrogenase andmethenyl tetrahydromethanopterin cyclohydrolase are alsopresent in methanotrophs (11). Thus, the genes encodingmethyl coenzyme M reductase (mcrA) were selected as theideal genetic marker for methanogens, since they appear tobe unique to methanogens and are evolutionarily conserved(15).
Primer design, PCR optimization, and in silico analysis.Based on in silico specificity with minimum degeneracy usingthe CODEHOP strategy, a total of three mcrA primer sets, twoprimer sets each for cel5, cel48, and dsrA, and one hydA primerset were identified as most promising (Table 2). One of thedsrA primer sets contained a modified version of the reverseprimer RH3-dsr-R designed by Ben-Dov et al. (6) and anin-house-designed forward primer. One of the mcrA primersets (mcrA_1035F–mcrA_1530R) was designed by Luton et al.(33). The other two mcrA primer pairs each combined one ofthe Luton primers with one in-house-designed primer.
The annealing temperature and magnesium concentrationwere optimized for all primer sets (Table 2), which were thensubjected to specificity tests with positive and negative controls(Table 3). Suitability for a complex environmental sampleknown to contain all the target (as well as nontarget) groupsand PCR inhibitors was also tested. On the basis of the results,the following primer sets were selected for further validation:cel48_490F–cel48_920R, cel5_392F–cel5_754R, hydA_1290F–hydA_1538R, dsrA_290F–dsrA_660R, and mcrA_1035F–mcrA_1530R.
In order to determine the broader specificities of the primersbeyond the reference organisms used for their design (Table1), in silico analysis was performed. This was achieved by in-terrogating the NCBI Reference Sequence (RefSeq) collection(http://www.ncbi.nlm.nih.gov/RefSeq/) for each of the targetgenes except mcrA, since the mcrA primers had been describedin detail previously (33). A total of 29, 397, 202, and 14 bac-terial reference sequences were found for the cel48, cel5, hydA,
VOL. 76, 2010 Q-PCR OF FUNCTIONAL GENES IN ANAEROBIC ENVIRONMENTS 2195

and dsrA genes, respectively. Since the dsrA sequences fromthe RefSeq collection did not include sequences from all thedifferent genera that contain this gene, additional sequenceswere included in the analysis (32 in total) in order to betterrepresent the diversity. The results of the in silico analysissuggest that the hydA and dsrA primer sets have a broaderphylogenetic coverage than that for which they were directlydesigned (Table 4). The cel48 forward and reverse primersmatched mainly Clostridium sp. sequences. The reverse primeralso matched Bacillus and Anaerocellum sp. sequences. The
RefSeq collection did not include cel48 sequences from Rumi-nococcus or Bacteriodes spp. However, the Bacteroides cellulo-solvens and Ruminococcus albus sequences used for primerdesign aligned with the forward primer with 1 and 4 mis-matches and with the reverse primer with 4 and 1 mismatches,respectively. In all cases, the mismatches were not in the 3�primer region. In the case of the cel5 primer set, all referencesequences had more than 2 mismatches with the primer se-quence. This was not unexpected given the sequence diversityof the cel5 genes. Nonetheless, by cloning and DNA sequenc-
TABLE 2. Primer combinations designed by the CODEHOP strategy and selected for further validation PCR
Target and primer Sequence (5�–3�) Primer combination(s) (approximateamplicon size �bp�)a
Family 48 glycosidehydrolases
cel48_490F TNATGGTTGAAGCTCCDGAYTAYGG cel48_490F–cel48_920R (430), cel48_880F–cel48_980R (100)cel48_880F CAYTGGHTNNTGGAYGTTGAYAACTGGTA
cel48_920R CCAAANCCRTACCAGTTRTCAACRTCcel48_980R CCTGTTCACCTCTYTGRWARGTRTT
Family 5 glycosidehydrolases
cel5_392F GAGCATGGGCTGGAAYHTNGGNAA cel5_392F–cel5_525R (133), cel5_392F–cel5_754R (362)cel5_525R GAAAGGAATACGGACGGYNTTRAAHCC
cel5_754R CATCATAATCTTTGAAGTGGTTTGCAATYTGDKTCCA
Alpha subunit ironhydrogenases
hydA_1290F GGTGGAGTTATGGAAGCWGCHHT hydA_1290F–hydA_1538R (248)hydA_1538R CATCCACCWGGRCAHGCCAT
Alpha subunit dissimilatorysulfite reductase
dsrA_290F CGGCGTTGCGCATTTYCAYACVVT drsA_290F–RH3-dsr-R� (140), dsrA_290F–dsrA_660R (370)RH3-dsr-R� GTGGMGCCGTGCATGTT
dsrA_660R GCCGGACGATGCAGHTCRTCCTGRWA
Alpha subunit of methylcoenzyme M reductase
mcrA_1035F GGTGGTGTMGGATTCACACARTAYGCWACAGC mcrA_1035F–mcrA_1530R (495),mcrA_1035F–mcrA_1450R (415),mcrA_1430F–mcrA_1530R (100)
mcrA_1530R TTCATTGCRTAGTTWGGRTAGTTmcrA_1450R TTTGAAGCWCCRCAYTGGTCYTmcrA_1430F TTCTATGGTTACGACTTVCAGGACCARTGYGG
a Primers mcrA_1035F and mcrA_1530R were designed by Luton et al. (33). Primer RH3-dsr_R� is a modified version of primer RH3-dsr-R, designed by Ben-Dovet al. (6). Primer sets that were selected after specificity validation are shown in boldface.
TABLE 3. Results of PCR specificity tests with positive and negative controls
Primer combinationResult with the following positive/negative controla: Result with an
environmentalsamplebC. thermocellum C. cellulovorans R. flavefaciens F. succinogenes E. coli D. autotrophicum M. maripaludis
cel48_490F–cel48_920R � � � � � � � �cel48_880F–cel48_980R � � � � � � � �cel5_392F–cel5_525R � � � � � � � �cel5_392F–cel5_754R � � � � � � � �hydA_1290F–hydA_1538R � � � � � � � �drsA_290F–RH3-dsr-R� � � � � � � � �dsrA_290F–dsrA_660R � � � � � � � �mcrA_1035F–mcrA_1530R � � � � � � � �mcrA_1035F–mcrA_1450R � � � � � � � �mcrA_1430F–mcrA_1530R � � � � � � � �
a C. thermocellum possesses cel5, cel48, and hydA; C. cellulovorans, R. flavefaciens, and F. succinogenes possess cel5; D. autotrophicum possesses dsrA; and M.maripaludis possesses mcrA. E. coli does not possess any of the target genes.
b Substrate from a lignocellulose-based sulfate-reducing bioreactor.
2196 PEREYRA ET AL. APPL. ENVIRON. MICROBIOL.

ing of cel5 PCR products, it was confirmed that these primersamplify cel5 sequences. The primers had either no mismatchesor 1 mismatch with the cel5 sequences recovered from cloning.
Experimental validation of primer specificity. PCR productsobtained from the LC bioreactor sample were cloned, andDNA sequences were determined for 10, 15, 13, 18, and 15clones (out of 20, 30, 26, 30, and 28 per library) resulting fromthe cel48, cel5, hydA, dsrA, and mcrA primer sets, respectively.The results confirmed that the primer sets amplified only thedesired targets in all cases (Table 5). The cel48 and cel5 clonesmatched with exo- or endoglucanases belonging to families 48and 5 of glycoside hydrolases, respectively. The sequences ob-tained with the hydA primers matched the C-terminal domainof the large subunit of iron hydrogenases, which contains theH-cluster sequence used to design the primers. All of thesequences obtained with the dsrA primers matched the alphasubunit of the dissimilatory sulfite reductase of unculturedSRB. Finally, all of the mcrA sequences matched the alphasubunit of methyl coenzyme M reductase, and the majority ofthe sequences matched an uncultured archaeon belonging tothe Methanosarcinaceae. Good’s coverage estimates (19), cal-culated from the results of the restriction digest analysis, were96.6%, 90.0%, 84.6%, 93.3%, and 82.1% for the cel5, cel48,hydA, dsrA, and mcrA clones, respectively. This suggested thatthe number of clones screened was representative of the ge-netic diversity captured by each primer set.
With respect to the aerated SBR dominated by nontemplateDNA, which served as a negative control, no PCR product wasobtained from any of the primer sets, except for faint productsyielded by the cel48 and dsrA primers. Cloning and sequencingrevealed that the products did represent viable targets. dsrAclones (n 10) were most similar to sequences present in thefollowing microorganisms (with GenBank accession numbersand percentages of similarity given in parentheses): Desulfo-bulbus elongatus (AJ310430; 90%), Desulfobulbus propionicus(AF218452; 82%), Desulfobulbus rhabdoformis (AJ250473;
85%), Desulforhopalus singaporensis (AF418196; 77 to 82%),Desulfovibrio aerotolerans (AY749039; 96%), Desulfovibrioaminophilus (AY626029; 88%), and Desulfovibrio magneticusRS-1 (AP010904; 93 to 95%). cel48 clones (n 19) were allmost similar to a glycoside hydrolase present in Herpetosiphonaurantiacus (CP000875; 93 to 94%).
Adaptation to and validation of Q-PCR. The selected primersets were adapted to Q-PCR and were used for the quantifi-cation of the functional genes in the LC and Et bioreactorstreating mine drainage. All functional genes were successfullyquantified in both samples (Fig. 1A). Assuming that the num-bers of copies of functional genes present per organism aredistributed similarly between the two samples, the ratios rep-resent how many more (or less) organisms with each functionare present in the LC versus the Et bioreactor. In the LCbioreactor, the abundances of the cel48, cel5, and mcrA geneswere approximately 350 180, 580 100, and 270 70 timeshigher, respectively, than those in the ethanol-fed bioreactor.The concentration of hydA genes in the LC bioreactor was alsohigher (22 3) than that in the Et reactor. On the other hand,the Et reactor contained approximately 33 9 times moredsrA genes than the LC reactor. Based on the quantification ofthe 16S rRNA gene, the LC bioreactor contained a higherconcentration of bacterial biomass than the Et bioreactor(about 10 times higher).
Characterization by 16S rRNA gene cloning. A total of 109and 84 cloned 16S rRNA genes were analyzed by ARDRA forthe LC and Et reactors, respectively. As expected, the LCreactor was found to have higher overall bacterial diversity,with a Shannon index of 2.97, compared to 1.03 for the Etreactor. Putative functions were assigned to the bacteria rep-resented by the sequences based on information obtained fromthe highest matches in the GenBank database and a corre-sponding literature review (Table 6). Based on the putativefunctions assigned to the bacteria, the relative proportions ofcellulose degraders, fermenters, and sulfate reducers were es-timated, and these estimates were compared with the resultsobtained by Q-PCR targeting functional genes (Fig. 1B). Thetwo approaches yielded similar overall trends, with higher con-centrations of cellulose degraders and fermenters in the LCbioreactor than in the Et bioreactor, and higher concentrationsof sulfate reducers in the ethanol bioreactor. However, thevalues of the ratios cannot be directly compared, because theARDRA results were determined as ratios of the frequencies(expressed as percentages) in the clone library, and the Q-PCRresults were determined as ratios of the numbers of genecopies.
DISCUSSION
Advantages of new primers relative to existing primers. Thisstudy provides a useful new suite of tools for characterizinganaerobic sulfate-reducing and methanogenic communitiesthat utilize cellulose as the primary carbon source. The ap-proach is much less cumbersome than 16S rRNA gene profil-ing and provides direct functional information. Significant ad-vances were also made over prior functional-gene methods,which have been developed primarily for pure cultures or donot discriminate well against nonspecific targets. Even whenchallenged with samples dominated by nontarget DNA, all
TABLE 4. Results of in silico analysis of primer specificity forNCBI RefSeq database sequences
Primera Representative species with 2 orfewer mismatches
cel48_490F........................Clostridium spp.cel48_920R .......................Anaerocellum, Bacillus, and Clostridium spp.hydA_1290F......................Clostridium, Coprococcus, Dehalococcoides,
Desulfitobacterium, Geobacter, Marinitoga,Opitutus, Thermoanaerobacterium,Thermotoga, Tolumonas, and Veillonellaspp.
hydA_1538R.....................Clostridium, Desulfovibrio, Fervidobacterium,Finegoldia, Fusobacterium, Marinitoga,Petrotoga, Thermodesulfovibrio,Thermosipho, and Thermotoga spp.
dsrA_290F.........................Desulfobotulus, Desulfomicrobium,Desulfomonile, Desulforhopalus,Desulfosalina, and Desulfovibrio spp.
dsrA_660R........................Desulfobacter, Desulfobulbus, Desulfococcus,Desulfomicrobium, Desulfonema,Desulforhopalus, Desulfosalina,Desulfosarcina, Desulfotalea, andDesulfovibrio spp.
a In the case of the cel5 primer set (cel5_392F–cel5_754R), all referencesequences had more than 2 mismatches with the primer sequence.
VOL. 76, 2010 Q-PCR OF FUNCTIONAL GENES IN ANAEROBIC ENVIRONMENTS 2197

primer sets still yielded 100% specificity. The ability to detectfunctional genes corresponding to both cultured and uncul-tured organisms in complex environments is of particularvalue. With this approach, ubiquitous microbial functions, suchas cellulose degradation, fermentation, sulfate reduction, andmethanogenesis, can be studied with a smaller number ofprimer sets than that required for the 16S rRNA gene-basedapproach. All of the primer sets were also confirmed to besuitable for Q-PCR, providing valuable quantitative informa-tion about important functional groups in complex anaerobicenvironments.
The CODEHOP strategy proved to be effective for degen-erate primer design, particularly for the cel5, cel48, and hydAsequences, which, because of their lower degree of conserva-tion, were more challenging than the mcrA and dsrA se-quences. Although it was possible to identify blocks of con-served protein sequences and to design primers from them forall of the target genes, the divergence of the cel5, cel48, and
hydA sequences limited the number of primer combinationsthat could be designed and, in general, increased the degen-eracy of their 3� degenerate cores. As was intended, all primerswere capable of detecting an array of sequences even broaderthan that directly used to design the primers (Table 1). Thiswas confirmed by in silico analysis and/or experimental valida-tion. Interestingly, the similarity of the sequences from the LCbioreactor, obtained from experimental validation, to the se-quences in the NCBI database was lower than what is typicallyobserved for 16S rRNA gene sequences (�90%), particularlyfor the cel5, cel48, and hydA sequences. This is likely becausethe 16S rRNA gene has changed very slowly throughout thecourse of evolution relative to the rate of change of functionalgenes. Also, there is limited sequence information in the da-tabase relative to that for 16S rRNA.
In particular, the development of degenerate primers tar-geting cel5 and cel48 represents a significant advance over priorwork. While several sets of primers targeting various genes
TABLE 5. DNA sequencing results of clones of PCR products obtained with the cel5, cel48, hydA, dsrA, and mcrA primer sets on a samplefrom the lignocellulose-based sulfate-reducing bioreactor
Primer set and microorganism BLASTX identity (% match)a% of
screenedclones
GenPeptaccession no.
cel48Clostridium josui Exoglucanase S, glycosyl hydrolase family 48 (75) 14.3 AAC38571Clostridium acetobutylicum ATCC 824 Processive endoglucanase, glycosyl hydrolase family 48 (56) 14.3 NP_347547Clostridium cellulolyticum H10 Glycoside hydrolase, family 48 (69) 28.6 ZP_01575465Clostridium cellulovorans Exoglucanase S, glycosyl hydrolase family 48 (53–61) 42.9 AAC38571
cel5Saccharophagus degradans 2-40 Endoglucanase-like protein, glycoside hydrolase family 5
(45–59)46.7 ABD81896
Clostridium cellulovorans Endoglucanase EngB, glycoside hydrolase family 5 (43) 3.3 AAA23231Flavobacterium johnsoniae UW101 Hypothetical protein EUBSIR_0099, glycoside hydrolase
family 5 (44)6.7 ABQ03808
Eubacterium siraeum DSM 15702 Hypothetical protein EUBSIR_00991, glycoside hydrolasefamily 5 (52–53)
43.3 EDS01174
hydAClostridium botulinum B1 strain Okra �Fe� hydrogenase (72) 3.7 YP_001781395Clostridium cellulolyticum H10 Hydrogenase, Fe only (75–82) 7.4 ZP_01573883Bacteroides capillosus ATCC 29799 Hypothetical protein BACCAP 02746, iron hydrogenase
large subunit, C-terminal domain (73)7.4 ZP_02037133
Thermosinus carboxydivorans Nor1 Hydrogenase, Fe only (74) 11.1 EAX48145Pelotomaculum thermopropionicum SI Hydrogenase subunit (63) 11.1 YP_001212560Clostridium bolteae ATCC BAA-613 Hypothetical protein CLOBOL 01023, iron hydrogenase
large subunit, C-terminal domain (60–74)22.2 ZP_02083500
Clostridium thermocellum Hydrogenase-1(67–69) 37.0 AAD33071
dsrAUncultured sulfate-reducing bacterium Dissimilatory sulfite reductase alpha subunit (96) 3.3 AAQ7928Uncultured sulfate-reducing bacterium Dissimilatory sulfite reductase alpha subunit (90–91) 63.3 ACD37519Uncultured sulfate-reducing bacterium Dissimilatory sulfite reductase subunit A (82–88) 13.3 ABR45803Uncultured sulfate-reducing bacterium Dissimilatory sulfite reductase alpha subunit (89–92) 3.3 ABR09856Uncultured sulfate-reducing bacterium Dissimilatory sulfite reductase alpha subunit (96) 16.7 ABW69247
mcrAUncultured Methanosarcinaceae archaeon Methyl coenzyme M reductase alpha subunit (96) 3.6 AAN02199Uncultured euryarchaeote Methyl coenzyme M reductase alpha subunit (90–91) 3.6 AAR22542Uncultured methanogenic archaeon Methyl coenzyme M reductase alpha subunit (96) 7.1 CAN99766Uncultured archaeon Methyl coenzyme M reductase alpha subunit (97–98) 21.4 ABX75060Uncultured euryarchaeote Methyl coenzyme M reductase alpha subunit (96) 25.0 AAR24558Methanobacterium ivanovii Methyl coenzyme M reductase alpha subunit (99) 39.3 ABO93183
a Where several clones matched the same protein, the percentage of matching sequence is given as a range.
2198 PEREYRA ET AL. APPL. ENVIRON. MICROBIOL.

encoding glycoside hydrolases have been identified in the lit-erature, all were designed to target pure cultures, except forone study in which the primers were specific to the cellulases ofred claw crayfish (8). Many existing primers may, in fact, am-plify more cellulases than the single target for which they weredesigned; however, considering the broad sequence diversityamong glycoside hydrolases, a direct degenerate design ap-proach has been greatly needed. This is especially true forapplications in complex anaerobic environments utilizing cel-lulose as the primary carbon substrate.
Prior efforts to develop primers for the hydA gene have alsofocused primarily on pure cultures (37, 44, 53). In this study,the hydA primers were designed using sequences obtainedfrom several Clostridium species, and the DNA sequencingresults demonstrated that an even broader range of genera wascaptured by the primer set (Table 5). Recently, broadly specificprimers were also developed by Xing and colleagues (57) toamplify the hydA gene of H2-producing bacteria in acidophiliccommunities. The forward primer targeted the ADLTIMEEsignature sequence of the H-cluster domain to produce a PCRproduct of 500 to 600 bp. However, this fragment length is notappropriate for Q-PCR. Therefore, in our study, a new for-ward primer was designed from a conserved region closer toMACPGGCING to produce a smaller product (�248 bp),which is preferable for Q-PCR (35).
The major challenge in seeking universal primers for SRB
remains the ability to distinguish SRB from SOB while main-taining broad coverage of SRB, which span phyla and arepresent in both bacteria and archaea. Primers targeting thegenes encoding APSR have been particularly problematic inenvironmental surveys and have been observed to favor SOBover SRB when both are present (20, 26). The dsrA and dsrBgenes appear to be superior in this regard, but an in silicoanalysis (data not shown) of published primers conducted dur-ing the design phase of this study still revealed limitations.Many published dsrA or dsrB gene primer sets suffered fromeither high specificity for SOB (14, 17, 31) or low specificity forvarious SRB phyla (14, 17, 31, 52). Based on our literaturesurvey and in silico analysis, primer RH3-dsr-R, described byBen-Dov et al. (6), appeared to be the most promising andtherefore was modified for broader specificity in this study. Anin silico analysis of the dsrA primer set developed and appliedin this study indicated that there were several mismatchesbetween known dsrA genes of Thiobacillus spp. and the primers(particularly at the 3� end of the reverse primer) to preventtheir amplification. In addition, no Thiobacillus spp. were de-tected by these primers in the aerobic SBR sample. Further-more, DNA sequencing of clones of a PCR product obtainedwith the dsrA primer set from a sample of the Peerless JennyKing bioreactor, which was known to contain Thiobacillus sp.DNA (26), revealed that no Thiobacillus sp. dsrA sequenceswere amplified (data not shown).
Primers mcrA_1450R and mcrA_1430F were designed andtested in combination with the forward and reverse primersdesigned by Luton et al. (33). While the in-house-designedprimers were confirmed experimentally to be specific to mcrAgenes, the Luton combination provided better sequence diver-sity coverage (data not shown) and therefore was selected foradaptation to Q-PCR. In addition to the Luton primers, sev-eral other primer sets for mcrA gene amplification have beenpublished. However, these were not found to be suitable forQ-PCR and/or did not provide reliable information regardingthe proportion of different operational taxonomic units(OTUs) in a sample. For example, Juottonen et al. (30) con-cluded that the Luton primer combination provided betterperformance, in terms of quantitative characterization ofmethanogenic communities in peatland samples, than twoother primer sets.
Application for quantifying key functional guilds in complexanaerobic environments. Traditionally, the 16S rRNA genehas been used for the biomolecular characterization of micro-bial communities. The design of primers targeted to the 16SrRNA gene is generally effective for the study of microorgan-isms that form tight phylogenetic clusters but becomes imprac-tical when primers are used to capture the diversity of micro-organisms that span phylogenetic groups. Thus, the 16S rRNAgene-based approach is particularly problematic for character-izing complex anaerobic environments, in which cellulose de-graders, fermenters, sulfate reducers, and methanogens arekey populations with vast phylogenetic diversity. The processesdriven by these groups are of growing interest in various are-nas, including carbon cycling/climate studies, biofuel produc-tion, and bioremediation.
Mine drainage remediation is an important example of amicrobially mediated process that is poorly understood, largelydue to the complexity of the microbial communities involved.
FIG. 1. (A) Numbers of copies of the 16S rRNA, cel5, cel48, hydA,dsrA, and mcrA genes in the lignocellulose (LC)-based bioreactorrelative to those in the ethanol (Et)-fed bioreactor as determined byQ-PCR. (B) Ratios of the percentages of putative cellulose degraders,fermenters, and sulfate-reducing bacteria in the LC bioreactor to thosein the Et bioreactor as determined by ARDRA and 16S rRNA genesequencing, as well as by a subsequent literature review.
VOL. 76, 2010 Q-PCR OF FUNCTIONAL GENES IN ANAEROBIC ENVIRONMENTS 2199

Although sulfate reduction was the primary engineered func-tion, SRB were estimated to be present at only 1.9% of thetotal bacterial community of the LC bioreactor based onARDRA (Table 6). This observation is supported by severalother 16S rRNA gene surveys of similar systems (26, 29, 36,38). This indicates that for every 100 clones screened, only 2are likely to represent sulfate reducers. This is a frustratingapproach to the study of the bacteria that drive the mainfunction of interest in these bioreactors, especially if quantita-tive information is desired.
The mine drainage treatment systems investigated in thisstudy were particularly suitable for validating the primers, be-cause they differed only in the type of carbon substrate appliedto support the microbial community. The presence of ethanolwas expected to produce a microbial community enriched inSRB, because, as a product of fermentation, ethanol is not afavorable substrate for fermenters, and it can be directly uti-lized by most SRB. On the other hand, the complex lignocel-lulosic carbon source was expected to produce a more func-tionally diverse microbial community, as was confirmed byARDRA. Q-PCR utilizing the primers designed in this studyprovided quantitative support for these assumptions and wasalso in good general agreement with the 16S rRNA gene-basedcharacterization (Fig. 1). The LC bioreactor contained higher
concentrations of the cel5, cel48, hydA, and mcrA genes thanthe Et bioreactor. In contrast, the Et bioreactor containedapproximately 1.5 orders of magnitude more dsrA genes. Whilegenetic markers for cellulose degradation and fermentationwere detected in the Et bioreactor, these were likely a result ofthe residual horse manure used for inoculation.
Even though the overall trends in the results obtained fromthe 16S rRNA gene analysis and the functional gene analysiswere similar (Fig. 1), it is important to keep in mind thatassignment of bacteria to functional groups based on 16SrRNA gene sequence similarity can be highly dubious. Whilefunctional genes also are not perfect in this regard, theypresent a much higher likelihood of corresponding to the func-tion of interest.
Analysis of the 16S rRNA genes present in the sulfate-reducing bioreactors also confirmed the dominance of uncul-tured organisms in these systems (Table 6). In fact, 100% ofthe sequences obtained from the LC bioreactor presented thehighest similarities to uncultured bacteria. This underscoresthe lack of suitability of primers targeting pure cultures for thecharacterization of complex anaerobic environments. Whilethere is no guarantee that the primers designed in this studyprovide 100% coverage of the targets, especially with respectto uncultured organisms, this is the case with all primers and
TABLE 6. Summary of 16S rRNA gene sequence analysis of the lignocellulose-based versus the ethanol-fed bioreactora
Bioreactor and highest match (GenBank accession no.) % Match % of screenedclones
Putativefunctionb
Lignocellulose-based sulfate-reducing bioreactorClostridium from anoxic bulk soil, strain XB90 (AJ229234) 97 6.5 CDUncultured bacterium, clone RB323 (AB240338) 87–99 4.7 CDUncultured bacterium, clone LCFA-B03 (AB244310) 98–99 7.5 FUncultured spirochete, clone C6_2 (EF562545) 96 7.5 FUncultured bacterium, clone Eb26 (EF063613) 98 6.5 FUncultured bacterium, FukuN63 (AJ290056) 96 1.9 FUncultured bacterium, clone RA13C8 (AF407407) 99 2.8 PH/FUncultured Bacteroidetes bacterium, clone CF_5 (EF562567) 98 1.9 PH/FUncultured Bacteroidetes bacterium, clone RBE2CI-81 (EF111172) 98–99 8.4 PH/FUncultured bacterium, clone 060F03_B_SD_P93 (CR933306) 97 2.8 PH/FUncultured bacterium, clone 1013-1-CG49 (AY532557) 94–99 3.7 PH/FUncultured sulfate-reducing bacterium, clone 39B (AB069774) 99 1.9 SRUncultured bacterium, clone ELME-7 (AB288666) 96 1.9 UNKFailed sequencing 3.7
Ethanol-fed sulfate-reducing bioreactorUncultured eubacterium, AA05 (AF275916) 97 2.3 FUnidentified eubacterium, clone BSV87 (AJ229231) 99 2.3 FUncultured Acetobacterium sp. (EF420226) 99 2.3 FUncultured betaproteobacterium, clone UCT N123 (AY064177) 98 2.3 AOIron-reducing enrichment clone Cl-A9 (DQ677001) 99 6.9 PH/FUncultured Bacteroidetes bacterium (EF111172) 97 4.6 PH/FIron-reducing enrichment clone Cl-A4 (DQ676996) 99 2.3 PH/FDesulfomicrobium sp. BL (AY928663) 99 14.9 SRDesulfovibrio sp. GWE2 (AF159119) 99 14.9 SRDesulfomicrobium baculatum (AJ277896) 99 10.3 SRUncultured deltaproteobacterium (DQ676356) 99 8.0 SRDesulfovibrio putealis (AY574979) 99 5.7 SRGeobacter sp. Ply1 (EF527233) 98 4.6 SRUncultured bacterium (AB237688) 98 4.6 SRUncultured deltaproteobacterium (EF111173) 98 4.6 SRRhodopseudomonas sp. ORS 1416ri (AJ968691) 98 2.3 UNKUncultured bacterium (AY570636) 98 6.9 UNK
a The percentage of the total community was estimated by ARDRA.b CD, cellulose degradation; PH, polysaccharide hydrolysis; F, fermentation; AO, aromatics oxidation; SR, sulfate reduction; UNK, unknown.
2200 PEREYRA ET AL. APPL. ENVIRON. MICROBIOL.

probes published to date. Thus, the degenerate primers devel-oped in this study represent an important first step to broadand quantitative characterization of key functional groups inanaerobic environments.
ACKNOWLEDGMENTS
Financial support for this work was provided by NSF CBET award0651947, by the EPA Office of Research and Development Engineer-ing Technical Support Center (contract 68-C-00-136; David Reisman,Project Officer), and by the Rocky Mountain Hazardous SubstanceResearch Center through the EPA Science to Achieve Results(STAR) program (contract R 8395101-0).
The results of this study do not necessarily represent the views of theEPA or the NSF.
REFERENCES
1. Achenbach, L. A., and J. D. Coates. 2000. Disparity between bacterial phy-logeny and physiology—comparing 16S rRNA sequences to assess relation-ships can be a powerful tool, but its limitations need to be considered. ASMNews 66:714–715.
2. Adams, M. W. W., L. E. Mortenson, and J. S. Chen. 1980. Hydrogenase.Biochim. Biophys. Acta 594:105–176.
3. Bar, T., A. Stahlberg, A. Muszta, and M. Kubista. 2003. Kinetic outlierdetection (KOD) in real-time PCR. Nucleic Acids Res. 31:e105.
4. Beguin, P. 1990. Molecular biology of cellulose degradation. Annu. Rev.Microbiol. 44:219–248.
5. Beller, H. R., S. R. Kane, T. C. Legler, and P. J. J. Alvarez. 2002. A real-timepolymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ. Sci. Technol. 36:3977–3984.
6. Ben-Dov, E., A. Brenner, and A. Kushmaro. 2007. Quantification of sulfate-reducing bacteria in industrial wastewater by real-time polymerase chainreaction (PCR) using dsrA and apsA genes. Microb. Ecol. 54:439–451.
7. Borsodi, A. K., A. Micsinai, A. Rusznyak, P. Vladar, G. Kovacs, E. M. Toth,and K. Marialigeti. 2005. Diversity of alkaliphilic and alkalitolerant bacteriacultivated from decomposing reed rhizomes in a Hungarian soda lake. Mi-crob. Ecol. 50:9–18.
8. Byrne, K. A., S. A. Lehnert, S. E. Johnson, and S. S. Moore. 1999. Isolationof a cDNA encoding a putative cellulase in the red claw crayfish Cheraxquadricarinatus. Gene 239:317–324.
9. Cardenas, E., W. M. Wu, M. B. Leigh, J. Carley, S. Carroll, T. Gentry, J.Luo, D. Watson, B. Gu, M. Ginder-Vogel, P. K. Kitanidis, P. M. Jardine, J.Zhou, C. S. Criddle, T. L. Marsh, and J. A. Tiedje. 2008. Microbial commu-nities in contaminated sediments, associated with bioremediation of uraniumto submicromolar levels. Appl. Environ. Microbiol. 74:3718–3729.
10. Chervoneva, I., T. Hyslop, B. Iglewicz, L. Johns, H. R. Wolfe, S. Schulz, E.Leong, and S. Waldman. 2006. Statistical algorithm for assuring similarefficiency in standards and samples for absolute quantification by real-timereverse transcription polymerase chain reaction. Anal. Biochem. 348:198–208.
11. Chistoserdova, L., J. A. Vorholt, R. K. Thauer, and M. E. Lidstrom. 1998.C-1 transfer enzymes and coenzymes linking methylotrophic bacteria andmethanogenic Archaea. Science 281:99–102.
12. Devillard, E., D. B. Goodheart, S. K. R. Karnati, E. A. Bayer, R. Lamed, J.Miron, K. E. Nelson, and M. Morrison. 2004. Ruminococcus albus 8 mutantsdefective in cellulose degradation are deficient in two processive endocellu-lases, Cel48A and Cel9B, both of which possess a novel modular architec-ture. J. Bacteriol. 186:136–145.
13. Eden, P. A., T. M. Schmidt, R. P. Blakemore, and N. R. Pace. 1991. Phylo-genetic analysis of Aquaspirillum magnetotacticum using polymerase chainreaction-amplified 16S rRNA-specific DNA. Int. J. Syst. Bacteriol. 41:324–325.
14. Foti, M., D. Y. Sorokin, B. Lomans, M. Mussman, E. E. Zacharova, N. V.Pimenov, J. G. Kuenen, and G. Muyzer. 2007. Diversity, activity, and abun-dance of sulfate-reducing bacteria in saline and hypersaline soda lakes. Appl.Environ. Microbiol. 73:2093–2100.
15. Friedrich, M. W. 2005. Methyl-coenzyme M reductase genes: unique func-tional markers for methanogenic and anaerobic methane-oxidizing Archaea.Methods Enzymol. 397:428–442.
16. Garcia, J. L., B. K. C. Patel, and B. Ollivier. 2000. Taxonomic phylogeneticand ecological diversity of methanogenic Archaea. Anaerobe 6:205–226.
17. Geets, J., K. Vanbroekhoven, B. Borremans, J. Vangronsveld, L. Diels, andD. Van der Lelie. 2006. Column experiments to assess the effects of electrondonors on the efficiency of in situ precipitation of Zn, Cd, Co and Ni incontaminated groundwater applying the biological sulfate removal technol-ogy. Environ. Sci. Pollut. Res. 13:362–378.
18. Gilkes, N. R., B. Henrissat, D. G. Kilburn, R. C. Miller, and R. A. J. Warren.1991. Domains in microbial beta-1,4-glycanases—sequence conservation,function, and enzyme families. Microbiol. Rev. 55:303–315.
19. Good, I. J. 1953. The population frequencies of species and the estimation ofpopulation parameters. Biometrika 40:237–264.
20. Hallberg, K. B., and D. B. Johnson. 2005. Microbiology of a wetland eco-system constructed to remediate mine drainage from a heavy metal mine.Sci. Total Environ. 338:53–66.
21. Han, S. O., H. Yukawa, M. Inui, and R. H. Doi. 2003. Transcription ofClostridium cellulovorans cellulosomal cellulase and hemicellulase genes. J.Bacteriol. 185:2520–2527.
22. Heijs, S. K., R. R. Haese, P. van der Wielen, L. J. Forney, and J. D. van Elsas.2007. Use of 16S rRNA gene based clone libraries to assess microbialcommunities potentially involved in anaerobic methane oxidation in a Med-iterranean cold seep. Microb. Ecol. 53:384–398.
23. Henikoff, S., J. G. Henikoff, W. J. Alford, and S. Pietrokovski. 1995. Auto-mated construction and graphical presentation of protein blocks from un-aligned sequences. Gene 163:GC17–GC26.
24. Henrissat, B. 1991. A classification of glycosyl hydrolases based on aminoacid sequence similarities. Biochem. J. 280:309–316.
25. Henrissat, B., and A. Bairoch. 1996. Updating the sequence-based classifi-cation of glycosyl hydrolases. Biochem. J. 316:695–696.
26. Hiibel, S. R., L. P. Pereyra, L. Y. Inman, A. Tischer, D. J. Reisman, K. F.Reardon, and A. Pruden. 2008. Microbial community analysis of two field-scale sulfate-reducing bioreactors treating mine drainage. Environ. Micro-biol. 10:2087–2097.
27. Hipp, W. M., A. S. Pott, N. Thum-Schmitz, I. Faath, C. Dahl, and H. G.Truper. 1997. Towards the phylogeny of APS reductases and sirohaem sulfitereductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiol-ogy 143:2891–2902.
28. Ibekwe, A. M., C. M. Grieve, and S. R. Lyon. 2003. Characterization ofmicrobial communities and composition in constructed dairy wetland waste-water effluent. Appl. Environ. Microbiol. 69:5060–5069.
29. Johnson, D. B., and K. B. Hallberg. 2005. Biogeochemistry of the compostbioreactor components of a composite acid mine drainage passive remedia-tion system. Sci. Total Environ. 338:81–93.
30. Juottonen, H., P. E. Galand, and K. Yrjala. 2006. Detection of methanogenicArchaea in peat: comparison of PCR primers targeting the mcrA gene. Res.Microbiol. 157:914–921.
31. Karr, E. A., W. M. Sattley, M. R. Rice, D. O. Jung, M. T. Madigan, and L. A.Achenbach. 2005. Diversity and distribution of sulfate-reducing bacteria inpermanently frozen Lake Fryxell, McMurdo Dry Valleys, Antarctica. Appl.Environ. Microbiol. 71:6353–6359.
32. Loy, A., A. Lehner, N. Lee, J. Adamczyk, H. Meier, J. Ernst, K. H. Schleifer,and M. Wagner. 2002. Oligonucleotide microarray for 16S rRNA gene-baseddetection of all recognized lineages of sulfate-reducing prokaryotes in theenvironment. Appl. Environ. Microbiol. 68:5064–5081.
33. Luton, P. E., J. M. Wayne, R. J. Sharp, and P. W. Riley. 2002. The mcrA geneas an alternative to 16S rRNA in the phylogenetic analysis of methanogenpopulations in landfill. Microbiology 148:3521–3530.
34. Meyer, J. 2007. [FeFe] hydrogenases and their evolution: a genomic per-spective. Cell. Mol. Life Sci. 64:1063–1084.
35. Mohammadi, M., and P. J. R. Day. 2004. Oligonucleotides used as templatecalibrators for general application in quantitative polymerase chain reaction.Anal. Biochem. 335:299–304.
36. Morales, T. A., M. Dopson, R. Athar, and R. B. Herbert. 2005. Analysis ofbacterial diversity in acidic pond water and compost after treatment ofartificial acid mine drainage for metal removal. Biotechnol. Bioeng. 90:543–551.
37. Ozkan, M., S. G. Desai, Y. Zhang, D. M. Stevenson, J. Beane, E. A. White,M. L. Guerinot, and L. R. Lynd. 2001. Characterization of 13 newly isolatedstrains of anaerobic, cellulolytic, thermophilic bacteria. J. Ind. Microbiol.Biotechnol. 27:275–280.
38. Pereyra, L. P., S. R. Hiibel, A. Pruden, and K. F. Reardon. 2008. Comparisonof microbial community composition and activity in sulfate-reducing batchsystems remediating mine drainage. Biotechnol. Bioeng. 101:702–713.
39. Prieto, M. V., S. R. Hiibel, L. P. Pereyra, A. Pruden, K. F. Reardon, and D.Reisman. 2008. Effect of organic substrate composition on microbial com-munity structure of pilot-scale biochemical reactors treating mining influ-enced water, p. 878–891. In R. Barnhisel (ed.), Proceedings of the 2008National Meeting of the American Society of Mining and Reclamation: newopportunities to apply our science. American Society for Mining Reclama-tion, Richmond, VA.
40. Rabinovich, M. L., M. S. Melnik, and A. V. Bolobova. 2002. Microbialcellulases (review). Appl. Biochem. Microbiol. 38:305–321.
41. Ramakers, C., J. M. Ruijter, R. H. L. Deprez, and A. F. M. Moorman. 2003.Assumption-free analysis of quantitative real-time polymerase chain reaction(PCR) data. Neurosci. Lett. 339:62–66.
42. Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2007. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res.35:D269–D270.
43. Rose, T. M., E. R. Schultz, J. G. Henikoff, S. Pietrokovski, C. M. McCallum,and S. Henikoff. 1998. Consensus-degenerate hybrid oligonucleotide primersfor amplification of distantly related sequences. Nucleic Acids Res. 26:1628–1635.
VOL. 76, 2010 Q-PCR OF FUNCTIONAL GENES IN ANAEROBIC ENVIRONMENTS 2201

44. Stevenson, D. M., and P. J. Weimer. 2005. Expression of 17 genes in Clos-tridium thermocellum ATCC 27405 during fermentation of cellulose or cel-lobiose in continuous culture. Appl. Environ. Microbiol. 71:4672–4678.
45. Sun, Y. M., E. A. Polishchuk, U. Radoja, and W. R. Cullen. 2004. Identifi-cation and quantification of arsC genes in environmental samples by usingreal-time PCR. J. Microbiol. Methods 58:335–349.
46. Suzuki, M. T., L. T. Taylor, and E. F. DeLong. 2000. Quantitative analysis ofsmall-subunit rRNA genes in mixed microbial populations via 5�-nucleaseassays. Appl. Environ. Microbiol. 66:4605–4614.
47. Syutsubo, K., Y. Nagaya, S. Sakai, and A. Miya. 2005. Behavior of cellulose-degrading bacteria in thermophilic anaerobic digestion process. Water Sci.Technol. 52:79–84.
48. Tancsics, A., S. Szoboszlay, B. Kriszt, J. Kukolya, E. Baka, K. Marialigeti,and S. Revesz. 2008. Applicability of the functional gene catechol 1,2-dioxy-genase as a biomarker in the detection of BTEX-degrading Rhodococcusspecies. J. Appl. Microbiol. 105:1026–1033.
49. Thauer, R. K. 1998. Biochemistry of methanogenesis: a tribute to MarjoryStephenson. Microbiology 144:2377–2406.
50. Vignais, P. M., B. Billoud, and J. Meyer. 2001. Classification and phylogenyof hydrogenases. FEMS Microbiol. Rev. 25:455–501.
51. Wagner, M., A. Loy, M. Klein, N. Lee, N. B. Ramsing, D. A. Stahl, and M. W.Friedrich. 2005. Functional marker genes for identification of sulfate-reduc-ing prokaryotes. Methods Enzymol. 397:469–489.
52. Wagner, M., A. J. Roger, J. L. Flax, G. A. Brusseau, and D. A. Stahl. 1998.Phylogeny of dissimilatory sulfite reductases supports an early origin ofsulfate respiration. J. Bacteriol. 180:2975–2982.
53. Wang, M. Y., B. H. Olson, and J. S. Chang. 2007. Improving PCR and qPCR
detection of hydrogenase A (hydA) associated with Clostridia in pure cul-tures and environmental sludges using bovine serum albumin. Appl. Micro-biol. Biotechnol. 77:645–656.
54. Warnecke, F., P. Luginbuhl, N. Ivanova, M. Ghassemian, T. H. Richardson,J. T. Stege, M. Cayouette, A. C. McHardy, G. Djordjevic, N. Aboushadi, R.Sorek, S. G. Tringe, M. Podar, H. G. Martin, V. Kunin, D. Dalevi, J.Madejska, E. Kirton, D. Platt, E. Szeto, A. Salamov, K. Barry, N.Mikhailova, N. C. Kyrpides, E. G. Matson, E. A. Ottesen, X. N. Zhang, M.Hernandez, C. Murillo, L. G. Acosta, I. Rigoutsos, G. Tamayo, B. D. Green,C. Chang, E. M. Rubin, E. J. Mathur, D. E. Robertson, P. Hugenholtz, andJ. R. Leadbetter. 2007. Metagenomic and functional analysis of hindgutmicrobiota of a wood-feeding higher termite. Nature 450:560–565.
55. Weisburg, W. G., S. M. Barns, D. A. Pelletier, and D. J. Lane. 1991. 16Sribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173:697–703.
56. Whitford, M. F., R. J. Forster, C. E. Beard, J. H. Gong, and R. M. Teather.1998. Phylogenetic analysis of rumen bacteria by comparative sequenceanalysis of cloned 16S rRNA genes. Anaerobe 4:153–163.
57. Xing, D. F., N. Q. Ren, and B. E. Rittmann. 2008. Genetic diversity ofhydrogen-producing bacteria in an acidophilic ethanol-H2-coproducing sys-tem, analyzed using the [Fe]-hydrogenase gene. Appl. Environ. Microbiol.74:1232–1239.
58. Xu, Q., E. A. Bayer, M. Goldman, R. Kenig, Y. Shoham, and R. Lamed. 2004.Architecture of the Bacteroides cellulosolvens cellulosome: description of acell surface-anchoring scaffoldin and a family 48 cellulase. J. Bacteriol. 186:968–977.
2202 PEREYRA ET AL. APPL. ENVIRON. MICROBIOL.




















