
Department für Chemie - Abstracts
126
Department für Chemie Institut für Chemie nachwachsender Rohstoffe Departmentleiterin: Ao.Univ.Prof. Dipl.-Ing. Dr.nat.techn. Erika Staudacher Betreuer: Univ.Prof. Dipl.-Chem. Dr.rer.nat. Dr.h.c. Thomas Rosenau LIGNOFOAMS: POTENTIAL FOR 100% BIO-BASED MATRICES AND RESINS LIGNINSCHÄUME: POTENZIAL FÜR 100% BIO- BASIERTE MATRIZEN UND HARZE Dissertation zur Erlangung des Doktorgrades an der Universität für Bodenkultur Wien Eingereicht von Vebi Mimini, MSc Wien, April 2019
-
Upload
khangminh22 -
Category
Documents
-
view
11 -
download
0
Transcript of Department für Chemie - Abstracts

Department für Chemie
Institut für Chemie nachwachsender Rohstoffe
Departmentleiterin:
Ao.Univ.Prof. Dipl.-Ing. Dr.nat.techn. Erika Staudacher
Betreuer:
Univ.Prof. Dipl.-Chem. Dr.rer.nat. Dr.h.c. Thomas Rosenau
LIGNOFOAMS: POTENTIAL FOR 100% BIO-BASED MATRICES AND RESINS LIGNINSCHÄUME: POTENZIAL FÜR 100% BIO-BASIERTE MATRIZEN UND HARZE
Dissertation zur Erlangung des Doktorgrades an der Universität für Bodenkultur Wien
Eingereicht von
Vebi Mimini, MSc
Wien, April 2019

Danksagung
Meinen Betreuern Antje und Rosi möchte ich ganz herzlich für die allseitige und exemplarische Unterstützung bedanken. Dankeschön, dass Ihr mich in eure „Familie“ – die NAWARO-Gruppe – aufgenommen habt und mir die Möglichkeit gegeben habt, meine Dissertation in einem sehr spannenden Gebiet zu schreiben. Eure Betreuung, euer Rat, und die einzigartige Motivationsweise haben mich auch abseits der Wissenschaft enorm im Alltag weitergebracht. Danke!
Ein großes Dankeschön geht an meine Familie und speziell an meine Frau Shahe, die mich in jeglicher Form kontinuierlich und bedingungslos unterstützt hat. Danke, dass du dich immer mit größter Sorgfalt um unsere beiden Töchter Eliza und Amelia gekümmert hast, um mir die nötige Zeit zu verschaffen, die Arbeit konzentriert durchzuführen, vor allem manchmal auch an Wochenenden. Hierbei möchte ich mich auch gleich bei meinen zwei Töchtern Eliza und Amelia entschuldigen, dass euer Papa anstelle vom Spielplatz in dieser Zeit manchmal auch im Labor war oder Manuskripte geschrieben hat.
Des Weiteren möchte ich mich bei der Wood K Plus, der Lenzing AG und der FFG für die Administration und großzügige finanzielle Förderung des Projektes und die Ermöglichung der Konferenzreisen bedanken. Danke Robert, Karin, Robert B., Ireen, und ein Dankeschön auch im Andenken an Vasken (der leider viel zu früh plötzlich und unerwartet verstorben ist). Ohne euch hätte diese aufregende Arbeit nicht durchgeführt werden können, vor allem nicht so gründlich und erfolgreich.
Meinen KollegInnen der NAWARO-Gruppe, besonderes Hassan und Hubert, danke ich für die wertvollen Diskussionen, prachtvollen Ideen und für die Betreuung bei den Laborexperimenten. Vielen Dank auch an alle Co-Autoren und Institute für die Mithilfe bei der Gestaltung unsere gemeinsamen Publikationen.
Danken möchte ich außerdem meinen Verwandten und Bekannten, die mich im Rahmen der ganzen Studienzeit begleitet und moralisch unterstützt haben.
DANKE!
FALEMINDERIT!

Kurzfassung
Lignin, oder genauer technisches Lignin, ist trotz vielfältiger Vorteile, wie hohem Kohlenstoffgehalt, phenolische Struktur-Einheiten, große Verfügbarkeit usw., leider immer noch das am häufigsten ungenutzte Material, welches aus nachwachsenden Rohstoffen stammt. Die vorliegende Arbeit befasst sich mit Lignosulfonat (LS), einem aus dem Sulfitverfahren gewonnenen technischen Lignin, und hat als Ziel, wichtige chemische Faktoren herauszufinden, welche LS zu einem wertvollen Material (z.B. in Form von Schäumen) transformieren können.
Die Literaturrecherche über den Stand der Technik von Lignin-basierten Schäumen („Lignofoams“), als erstes Ziel dieser Arbeit, zeigte, dass die Verwendung von Ligninen im große, technischen Maßstab mit bestehenden technologischen Ansätzen und chemischen Formulierungen nicht möglich ist (Publikation I). Die Inhomogenität der Schwarzlauge und die intrinsische, komplexe Struktur des darin enthaltenen Lignins sind maßgebliche Faktoren und diesbezüglich als Hauptfaktoren anzusehen.
Die Isolierung und Aufreinigung von LS mittels des XAD-7-Verfahrens sowie eine umfassende Charakterisierung von LS und Sulfit Schwarzlauge (SSL) haben dazu beigetragen, die grundlegenden Charakteristika und Unterschiede zwischen LS and SSL genauer zu verstehen. Die Bestimmung und Skalierung des produktivsten Verhältnisses von Adsorbat zu Adsorbens ohne signifikanten Verlust der Qualität des gereinigten LS zeigte, dass das XAD-7-Verfahren eine sehr vielversprechende Alternative zur Reinigung von LS auch im großen Maßstab darstellt.
Die Extrusion von gereinigtem LS zusammen mit Polymilchsäure (PLA) ohne Modifikation und Zugabe weiterer Reagenzien oder Kompatibilisatoren, zeigte eine beschleunigte Kristallisation
Figure 1: Reale Nutzungmöglichkeiten für Lignosulfonat

von PLA und zusätzlich Porenbildung in LS-PLA-Gemischen (Publikation II). Diese Feststellung wirft ein neues Licht auf gereinigtes LS als Nukleierungsmittel mit einer Perspektive für (Bio)-Polymerschäume.
Unter Verwendung der wichtigen Eigenschaft von LS, der Wasserlöslichkeit, wurde das Schäumen mittels eines Mixers als geeignete Technik zur Herstellung von Nassschäumen für SSL und gereinigtes LS etabliert. Weiteres wurde festgestellt, dass die Konzentration und die Reinheit von LS die kritischsten Faktoren sind, welche die resultierende Morphologie und Eigenschaften der erhaltenen nassen und getrockneten LS-Schaumstoffe beeinflussen.
In einem weiteren Ansatz dieser Arbeit wurden Polyurethanmaterialien (PU) auf LS-Basis mit ungiftigen Reagenzien und durch umweltfreundliche Verfahren erfolgreich synthetisiert (Publikation III). Die Modifizierung des gereinigten LS mit biobasierten cyclischen Carbonaten (ausgehend von Glycerin) und die anschließende Umsetzung mit Diaminen zeigten die Möglichkeit der Entwicklung von PU auf nicht-Isocyanat Basis unter Verwendung von technischem LS als Ausgangsmaterial.
Neben der oben beschriebenen Hauptarbeit wurde in einem Nebenprojekt ein neuer Syntheseweg für Coniferylalkohol und dem Derivat Coniferylthiol vorgestellt (Publikation IV). Während die erste Substanz häufig als Modellverbindung in der Ligninchemie verwendet wird, kann die letztere zur Untersuchung der Schwefelchemie in Ligninen bzw. in LS verwendet werden.

Abstract
Lignin, or more specifically technical lignin, is despite diverse advantageous, such as high carbon content, phenolic units, huge availability, etc., unfortunately until today the most underutilized renewable-based resource. The present thesis is concerned with lignosulfonate (LS), a type of technical lignin obtained after sulfite pulping of wood, with the goal to bring in light the most important factors which can transform it to a valuable material.
Reviewing the state of the art of lignin-based foams (lignofoams) as a first objective of this work revealed that bulk utilization of lignins with existing technological methods and chemical formulations is infeasible (Publication I). The inhomogeneity of the liquor and the lignin´s intrinsic and complex structure were shown to play the major role thereof.
Isolation and purification of LS as well as comprehensive characterization of LS and sulfite spent liquor (SSL) contributed to a better understanding of the fundamental insights and differences between each other. Determination and upscaling of the most productive adsorbate-to-adsorbent ratio without losing the quality performance of the purified LS demonstrated the XAD-7 method as a very promising alternative for purification of LS also on a larger scale.
Extrusion of the purified LS together with polylactic acid (PLA) without modification and addition of any further reagents or compatibilizers revealed an accelerated crystallization of PLA and pore formation in LS-PLA blends (Publication II). This finding
Figure 2: Real utilisation possibilities for lignosulfonate

points towards a potential application of purified LS as nucleating agent with a perspective in (bio)polymer foaming.
Using the unique characteristic of LS, namely its water solubility, frothing was established as the suitable technique for the production of wet foams for SSL and purified LS. Concentration and purity of LS were found to be the most critical factors that influence the resulting morphology and properties of the obtained wet and dried LS foams.
In a further approach of this work, LS-based polyurethane (PU) materials were successfully synthetized by non-toxic reagents and through environmentally friendly processes (Publication III). Modification of the purified LS with bio-based (glycerin-derived) cyclic carbonate and subsequent reaction with a diamine derivative showed the feasibility of developing non-isocyanate-derived PU by utilizing technical LS as starting material.
In addition to the main work described above a new synthesis pathway for coniferyl alcohol and its derivative coniferyl thiol is presented as a result of a side-project (Publication IV). While the first substance is frequently used as a model compound in lignin chemistry, the second can be used to investigate sulfur chemistry in technical lignins, and LS, respectively.

Keywords
Bio-composite, coniferyl alcohol, coniferyl thiol, cyclic carbonate, extrusion, foaming, insulation, kraft lignin, lignin, lignofoams, lignosulfonate, lignosulfonate nucleation, organosolv lignin, polylactic acid, polyurethane, technical lignin utilization, 3D-printing

List of abbreviations
ATR-FTIR Attenuated total reflection Fourier-transform infrared spectroscopy
BL Black liquor
BPO Benzoyl peroxide
CBA Chemical blowing agent
CC Cyclic carbonate
DBU 1,8-Diazabicyclo[5.4.0]undec-7-en
DMAP 4-Dimethylaminopyridine
DMC Dimethyl carbonate
DMF N,N-Dimethylformamide
DSC Differential scanning calorimetry
EA Elemental analysis
EPS Expanded polystyrene
EXS Extruded polystyrene
FT-IR Fourier transform infrared spectroscopy
GC Glycerol 1,2-carbonate
GCC Glycerol cyclic carbonate
GC-MS Gas chromatography coupled to mass spectrometry
GPC Gel permeation chromatography
HMBC Heteronuclear Multiple Bond Correlation
HMDA 1,6-Hexamethylenediamine
H-PDMS-H Poly(dimethylsiloxane)
HSQC Heteronuclear Single Quantum Coherence)
ICP-MS Inductively coupled plasma mass spectrometry
KL Kraft lignin
LS Lignosulfonate
LSA Lignosulfonic acid
MVR Melt volume rate
MMD Molar mass distribution
Mw Molar mass
NMR Nuclear magnetic resonance
ODP Ozone-depletion potential
OH Hydroxyl group
OSL Organosolv lignin
PBA Physical blowing agent
PBD Polybutadiene glycol
PBS Polybutylene succinate
PC Polycarbonate
PEG Polyethylene glycol
PETG Polyethylene terephthalate
PF Phenol-formaldehyde
Ph Phenol
PHB Polyhydroxybutyrate
PHMS Poly(hydroxymethylsiloxane)

PIR Polyisocyanurate
PLA Polylactic acid
PO Polypropylene oxide
PP Polypropylene
PPD Polypropylene glycol
pQM para-Quinone methide
PS Polystyrene
PU Polyurethane
PVA Polyvinyl alcohol
PVC Polyvinyl chloride
RPU Rigid polyurethane
SEM Scanning electron microscopy
SL Spent liquor
SSL Sulfite spent liquor
SS-NMR Solid state nuclear magnetic resonance
TDS Total dissolved solid
TGA Thermogravimetric analysis
TLC Thin-layer chromatography
TMDP 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane
VA Vanillyl alcohol

Table of Contents 1 Aim of the study .......................................................................................................... 11
2 Introduction ................................................................................................................. 13
2.1 Lignin ..................................................................................................................... 13
2.2 Pulping, the source of technical lignin ..................................................................... 13
2.2.1 Sulfite pulping, lignosulfonate (LS) .................................................................. 14
2.3 Obstacles in utilization of technical lignins .............................................................. 16
2.4 Analysis of technical lignins and LS ........................................................................ 17
2.4.1 Analytical techniques for the determination of functional groups in LS ............. 17
2.4.2 Molar mass (Mw) and molar mass distribution (MMD) of LS ............................. 19
3 Isolation of LS and lignosulfonic acid (LSA) from sulfite spent liquor (SSL).......... 21
3.1 Upscaling of the XAD-7 method for isolation of LS from SSL.................................. 22
3.2 Upscaling: Comparison of small-scale and large-scale batches ............................. 24
3.2.1 EXPERIMENTAL............................................................................................. 25
4 Foaming of Lignosulfonate ......................................................................................... 26
4.1 Influence of diverse parameters on LS and SSL wet-foaming................................. 27
4.1.1 EXPERIMENTAL............................................................................................. 29
4.1.2 Preparation of LS and SSL wet-foams ............................................................. 29
5 Compatibility of LS with PLA (Publication II) ............................................................ 30
6 Non-isocyanate urethanes based on LS (Publication III) .......................................... 31
7 References ................................................................................................................... 32
8 Publications ................................................................................................................. 34
8.1 Lignin-based foams as insulations materials: a review (Publication I) ..................... 35
8.2 Compatibility of Kraft Lignin, Organosolv Lignin and Lignosulfonate with PLA in 3D Printing (Publication II) ...................................................................................................... 51
8.3 Lignosulfonate-based polyurethane materials via cyclic carbonates - preparation and characterization (Publication III) ................................................................................. 71
9 Related Publication ................................................................................................... 103
9.1 Gram-scale economical synthesis of trans-coniferyl alcohol and its corresponding thiol (Publication IV) ........................................................................................................ 103
10 Appendix ................................................................................................................ 111
10.1 Additional publication ........................................................................................... 111
10.1.1 Novel carbamoyl type quinine and quinidine based chiral anion exchangers implementing alkyne–azide cycloaddition immobilization chemistry (Publication V) ..... 111
10.2 Conference contributions ...................................................................................... 123
10.2.1 Oral presentations ......................................................................................... 123
10.2.2 Poster presentation ....................................................................................... 123
11 Curriculum vitae..................................................................................................... 126

1 Aim of the study
The aim of the present work is the establishment of fundamental insights for LS that will enlighten and determine its valorization prospective. The main focus lies in the investigation of the key factors, such as purity, chemical constitution and modification of LS, which directly influence the production of novel valuable LS-based porous materials. Reviewing the state of the art of foamed lignin-based polymers (lignofoams), comprehensive characterization of LS and SSL, as well as the examination of their compatibility as native compounds with other bio-based copolymers are the first milestones to be achieved. Inhomogeneity and the poor reactivity of LS are principal drawbacks that seriously limit its bulk utilization. However, these drawbacks could be overcome through the isolation and purification of LS from SSL, and through increasing its reactivity by converting or introducing new functional groups. Subsequently, the obtained results are expected to show the feasibility of developing alternative materials by utilizing technical lignosulfonate as the starting source instead of fossil-derived materials.
11

12

2 Introduction
2.1 Lignin
Lignin is – apart from cellulose and hemicellulose – one of the three main wood components. It acts as composite linker between cellulose and hemicellulose in the plants, gives strength and rigidity to the cell walls and is responsible for water imparts and microorganism attacks (Weng and Chapple 2010). Lignin can be isolated from botanical sources (e.g. for research purposes), but generally it is obtained as a byproduct from pulping processes or papermaking during the production of cellulose, pulp or fibers from wood. The 3D irregular aromatic structure and the high molar mass makes lignin a biopolymer with a complex chemical constitution (Balakshin et al. 2008, Fengel and Wegener 1989) different from those of polysaccharides and other biopolymers such as proteins or DNA. Lignin consists largely of phenol rings and propane-chains equipped with diverse functional groups, namely hydroxyls, methoxyls, carboxyls, and carbonyl groups as well as – in case of lignosulfonate – additionally, sulfonic acid groups. Depending on the type of wood, grass or agricultural crop, the phenylpropane monomers occur in three different forms and ratios: coniferyl alcohol (guaiacyl lignin, G, or gymnosperm), syringyl alcohol (sinapyl lignin, S, or angiosperm) and p-coumaryl alcohol (p-hydroxyphenyl, H, or graminaceous) (Figure 3).
CH
CH2OH
CH
OH
OCH3
CH
CH2OH
CH
OH
OCH3H3CO
CH
CH2OH
CH
OH
(I) (II) (III)
CH2
CH3
CH2
R
OCH3
2
34
5
61
R= OH, OCH3, Lig
Figure 3: Main reaction sites of the phenyl propane unit (left) and the building monomers of lignin: (I) coniferyl alcohol, (II) sinapyl alcohol, (III) p-coumaryl alcohol (adapted and modified from Fengel and Wegener 1989).
The complexity of the 3D lignin structure attributes to many bonding-patterns possible between individual phenylpropane units (C9), which are linked by various ether and C-C bonds (Balakshin et al. 2014, Ralph et al. 2004). During the biosynthesis of lignin from monomeric phenylpropane units through dehydrogenative enzyme-catalyzed polymerization, lignin is not formed by a genetically prescribed, regular mechanism, but by random coupling of lignol radicals to a non-linear polymer (Fengel and Wegener 1989). Therefore, the final structure of lignin is determined primarily by the reactivity and quantity of the building units involved in the radical polymerization and the chemical modifications during the pulping process or other isolation techniques.
2.2 Pulping, the source of technical lignin
In general, the pulping process can be divided into mechanical, chemical and semi-mechanical pulping (a combination of mechanical and chemical). While during the mechanical pulping lignin is not removed, in semi-mechanical pulping also a substantial portion of the lignin remains. In contrast to those, the chemical pulping leads to near total
13

removal of lignin and non-fibrous wood compounds. The chemical separation of lignin from the cellulose fibers and their derivatives as well as hemicelluloses – also called delignification – can be divided into sulfur (Kraft lignin KL and Lignosulfonates LS) and non-sulfur processes (Soda lignin and Organosolv lignin OSL). The sulfur-containing pulping processes are technically the most dominant chemical pulping methods, whereby it has to be distinguished between alkaline and acidic processes. The pulp or papers produced from the chemical pulping additionally have a higher quality than the fibers obtained from other pulping methods; however, they still need to be bleached without damaging the cellulose fibers in order to remove residual lignin. The main byproduct, which remains after pulping, is liquor that – depending on the employed process – is either called black liquor (BL) for Kraft pulping or (sulfite) spent liquor (SSL) in case of sulfite pulping. In the alkaline/Kraft process, which is also called sulfate pulping due to the sodium sulfate obtained, the active substance is sodium sulfide (Na2S). For details on the Kraft process is referred to Sjöström 1993 and Biermann 1996. In the following the sulfite pulping process is described in more detail, since for the present thesis LS and SSL were used as starting materials.
2.2.1 Sulfite pulping, lignosulfonate (LS)
In the sulfite process, the wood chips are digested in aqueous sulfite solution of a sulfite or bisulfite salt (HSO3
- or SO32-) at 140 – 170 °C for 3 – 7 h (Figure 4). During the digestion
process the physical cleavage between lignin and polysaccharides, and the sulfonation of lignin by attack of the negatively charged sulfite or bisulfite ions on the lignin structure occurs, resulting in highly charged lignin and thus solubilization in aqueous media (Lora 2008). The nucleophilic attack at the lignin structure occurs at a benzylium cation as the main intermediate. Under acidic conditions, the oxygen of OH or OR-group in α-position – in both phenolic and non-phenolic lignin units – is protonated. Then, the protonated oxygen undergoes a cleavage reaction as water, alcohol or phenol forming the benzylium cation, which is next attacked from sulfite or bisulfite ions (Figure 5). Under these conditions the most common β-O-4 linkages are stable, and as a result lignin is dissolved in liquor only by α-substituent elimination and without
Figure 4: Production of lignosulfonate. Figure adapted and reprinted with permission from Elsevier.
14

OCH3
CH
OR1
CH
OR
R2
CH2OH
OCH3
CH+
OR1
CH R2
CH2OH
OCH3
CH
OR1
CH
SO3H
R2
CH2OH
+ HSO3-
+ H+
- ROH
R= H, alkyl, aryl; R 1= H, lignin; R 2= lignin
Figure 5: Reaction scheme demonstrating the formation of lignosulfonate in case of the sulfite pulping process. Figure adapted and reprinted with permission from Elsevier.
substantial degradation. Additional aldol condensation takes place forming stable C-C bonds that leads to less fragmentation and higher molar mass (Mw) of LS compared to alkaline systems. LS have an average Mw between 4.000 and 30.000 g/mol (see Section: Molar mass (Mw) and molar mass distribution (MMD) of LS).
The amount of sulfur incorporated in lignin is typically 4-8% (Fengel and Wegener 1989). Depending on the counter-ion used during the pulping process, the sulfonic acid group is accompanied with a cation (Na+, NH4
+, Ca2+, Mg2+) and appears in form of a salt. Protonation of the sulfonic acid group removes the cations and yields the lignosulfonic acid (LSA). As can be seen in Figure 6, LS has an amphiphilic structure due to the presence of hydrophobic phenolic units and the hydrophilic sulfonic acid groups. The sulfonate groups are mainly positioned on the surface of the highly crosslinked LS with high content of polyaromatic chains, which are disorderly coiled (Rezanowich and Goring, 1960). The sulfonate groups make LS a polyelectrolyte with a large number of negatively charged sulfonic acid groups with diverse unique characteristics, such as high surface activity, stabilizing and dispersing properties and most importantly, enabling the solubility of LS in water.
Mg2+
Ca2+
Na+
R=H, LS
OCH3
O
S
O
OO
-
R
O
OLS
LS
NH4+
Figure 6: Idealized model of ligninosulfonate. The right figure is adapted and reprinted with permission from Elsevier.
15

2.3 Obstacles in utilization of technical lignins
Although technical lignin comprises various valuable properties, such as high carbon content, phenolic units, thermal stability etc., and simultaneously the benefit that is available in huge quantity (worldwide more than 70 Mt year-1), lignin is still underutilized. Valorization as value-added product in adhesives (Mansouri and Salvadó 2006), dispersants (Yang et al. 2015), plasticizers in concrete (Mullick 1996), stabilizers, superabsorbents (Mansouri and Salvadó 2006), surfactants etc., accounts for less than 3% of the total volume. The rest is used in low-performance sectors or simply combusted for the generation of energy and heat after the recovery of particular chemicals, which is especially true in case of the Kraft process.
The main obstacles which seriously limit its application potentials are the inhomogeneity of BL/SSL and the low reactivity of the complex lignin structure (Fatehi and Chen 2016). The composition of the liquor after pulping is mainly based on organic impurities (free sugars, hemicelluloses and organic acids) or inorganic ingredients (especially chemicals and high salt content from pulping processes) (Saeed et al. 2011), which, among others, are low-molar mass compounds compared to the macromolecular nature of LS/lignin. During the synthesis of novel lignin/LS based materials directly from BL/SSL, e.g. porous materials, gels, blends etc., the small and easily dissolvable compounds can react comparably quickly with the co-reactants as in contrast to the high molar mass compounds. Consequently an improvement or control of the desired physicochemical characteristics of the end product, such as mechanical stability, heat conductivity, flexibility, density, stability against humidity and fire etc., cannot be carried out selectively. Additionally, the small substances which are not covalently bound to the matrix can diffuse and thus achieve a number of unpredictable effects that limit the use of the lignin-based composites.
In order to design market-compliant products (comprising mechanical stability, homogeneity, elasticity, emissions, leaching resistance, toxicity), isolation and purification of lignin/LS from other liquor residues, as well as characterization and modification of target functional groups such as aromatic/aliphatic -OH, are expected to overcome these restrictions.
16

2.4 Analysis of technical lignins and LS
Chemical characterization of lignins/LS is the basis for their proper utilization. It provides information about the pulping process, constitution, purity grade, reactivity, etc., which are of great interest and importance in terms of further chemical modification and processing. To enlighten the chemical composition and structural features, the characterization was performed using different superior analytical techniques convenient for the SSL and the respective LS. Besides some preliminary investigations of SSL, like pH or total dissolved solids (TDS), the following methods were used for the analysis:
Elemental analysis (EA) for the determination of organic matter and sulfur content,
Methanolysis (by GC-MS) for free sugar and hemicellulose composition and concentration,
Thermogravimetric analysis (TGA) and Differential Scanning Calorimetry (DSC) to follow the thermal degradation and determination of ash content,
Fourier transform infrared spectroscopy (FT-IR) for structural analysis,
Inductively coupled plasma mass spectrometry (ICP-MS) for determination of most common metals and
Scanning electron microscopy (SEM) for surface imaging and particle size determination.
The following further techniques were applied additionally for LS characterization:
Conductometric titration for sulfonic acid group determination (-SO3H),
Gas chromatography and mass spectrometry (GC-MS) for methoxyl group (-OCH3) and -SO3H determination,
Gel permeations chromatography (GPC) for molar mass (Mw) and molar mass distribution (MMD), and
Nuclear magnetic resonance spectroscopy methods (NMR) like 1H, 13C, 2D HSQC, HMBC and 31P for structure elucidation and hydroxyl group determination.
Lignin-PLA blends were additionally analyzed by Melt volume rate (MVR); Flexural and Impact strength (Publication II).
Synthesized LS-PU insoluble materials were characterized by solid-state NMR (SS-NMR, Publication III).
Some of the methods relevant for LS analysis are described in more detail below.
2.4.1 Analytical techniques for the determination of functional groups in LS
2.4.1.1 Determination of sulfonic acid groups ( SO3H) in LS
The sulfonic acid groups are characteristic functional groups of LS, which originate from the sulfite pulping process (see Section: Sulfite pulping, lignosulfonate (LS). The sulfonic acid groups and the degree of sulfonation are decisive for the solubility extent of LS in water and - more importantly - they directly influence the properties and the characteristics of the LS-based products during the production and end-use application. So for e.g. LSs with higher SO3H content used during the present PhD work (e.g. 20%) foamed quickly and built stable wet-foams compared to LSs with lower SO3H content (e.g. 12%). The degree of sulfonation is additionally responsible for the polyelectrolyte character and the negatively charged surface of LS. It is proportional to material hydrophilicity and to surface activity (Ge et al. 2014).
The sulfonic acid group content can be determined indirectly e.g. via elemental analysis, X-ray fluorescence spectrometry, combustion/ion chromatography, or directly e.g. via conductometric titration (Stephen and Carlton 1992), UV spectrometry or via headspace gas shromatography coupled with mass spectrometry (Korntner et al. 2018). In case of the indirect methods the sulfonic acid groups content is determined by measuring the sulfur content and calculating the sulfonic acid groups under the assumption that all sulfur is
17
organic sulfur originating from the sulfonic acid groups. The direct methods on the other hand are specific analysis methods directly focusing on the sulfonic acid groups in LS and are thus more precise. An additional advantage of the direct methods, e.g. GC-MS headspace analysis, is the easy sample preparation, relatively low sample amount needed and higher screening throughput.
2.4.1.2 Determination of hydroxyl groups ( OH) in LS
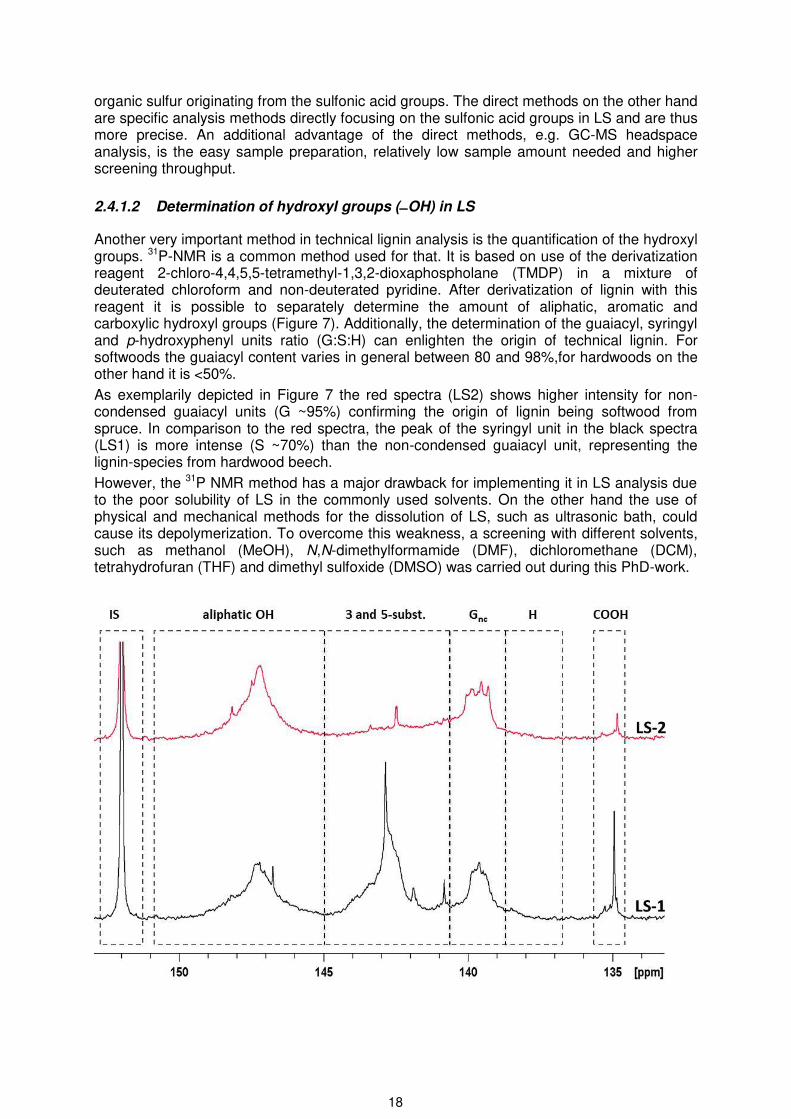
Another very important method in technical lignin analysis is the quantification of the hydroxyl groups. 31P-NMR is a common method used for that. It is based on use of the derivatization reagent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) in a mixture of deuterated chloroform and non-deuterated pyridine. After derivatization of lignin with this reagent it is possible to separately determine the amount of aliphatic, aromatic and carboxylic hydroxyl groups (Figure 7). Additionally, the determination of the guaiacyl, syringyl and p-hydroxyphenyl units ratio (G:S:H) can enlighten the origin of technical lignin. For softwoods the guaiacyl content varies in general between 80 and 98%,for hardwoods on the other hand it is <50%.
As exemplarily depicted in Figure 7 the red spectra (LS2) shows higher intensity for non-condensed guaiacyl units (G ~95%) confirming the origin of lignin being softwood from spruce. In comparison to the red spectra, the peak of the syringyl unit in the black spectra (LS1) is more intense (S ~70%) than the non-condensed guaiacyl unit, representing the lignin-species from hardwood beech.
However, the 31P NMR method has a major drawback for implementing it in LS analysis due to the poor solubility of LS in the commonly used solvents. On the other hand the use of physical and mechanical methods for the dissolution of LS, such as ultrasonic bath, could cause its depolymerization. To overcome this weakness, a screening with different solvents, such as methanol (MeOH), N,N-dimethylformamide (DMF), dichloromethane (DCM), tetrahydrofuran (THF) and dimethyl sulfoxide (DMSO) was carried out during this PhD-work.
18

Figure 7: 31
P NMR spectra of two different magnesium-LSs (LS1 and LS2) recorded in a solvent mixture of deuterated DMF and non-deuterated pyridine. 3- and 5-subst. range from 140.7– 145 ppm representing the condensed syringyl and guaiacyl units; Gnc: non-condensed guaiacyl units; H: p-hydroxyphenyl units; and COOH: carboxyl units.
The highest solubility of LS was observed in DMF. The compatibility of DMF with LS regarding the OH-group determination was evaluated following the same conditions of the traditional method (for more details see Publication III, Section: Analytical characterization). As from 31P NMR spectra in Figure 7 can be seen, DMF does not react with the phosphitylation reagent TMDP. In addition, the hydroxyl content of the samples measured in DMF was up to 40% higher than the respective measurement in CDCl3. A similar approach with DMF as a suitable solvent for OH determination was meanwhile published by Stücker et al. (2018).
2.4.2 Molar mass (Mw) and molar mass distribution (MMD) of LS
Besides the functional groups, Mw is one of the main important parameters for the characterization of lignin/LS. Its characteristics, such as MMD and polydispersity index, determine the functional properties of lignin/LS (Zinovyev et al. 2016, Sulaeva et al. 2017; 2018). Depending on the delignification procedure and wood type, the Mw of lignins various significantly, e.g. 1000 – 11.000 g/mol for Organosolv; 2000 - 7500 g/mol for Kraft lignin or 4000 - 64000 g/mol for LS (Calvo-Flores et al. 2015). LS has a higher molar mass due to the applied acidic pulping process, in which condensation reactions take place (see Section: Sulfite pulping, lignosulfonate (LS). Also the LS originating from softwood has a higher Mw than LS originating from hardwood. However, although the origin of lignin/LS; the pulping or extraction conditions and the technique used for the isolation of lignins/LS from black liquor/SSL are primary factors influencing directly the molar weight, a proper isolation and determination technique is equally significant and is just as important. In general, Mw-analysis of lignin/LS is hindered due to heterogeneity of the structural and chemical characteristics of lignin/LS and lignin/LS fractions (Sulaeva et al. 2015). In case of LS the analysis is even more complicated due to the respective cationic counter ions of the sulfonic acid groups and due to its low solubility in organic eluents.
An approach, which converts LS into the respective lignosulfonic acid form (LSA) before analysis and simultaneously separates any impurities by yielding protonated LS (lignosulfonic acid LSA) was reported by Sulaeva et al. (2017). It overcomes to a high extent the limitations in conventional techniques, such as size exclusion chromatography (SEC) or gel permeation chromatography (GPC) systems without the necessity of derivatization steps before. This method for Mw determination with DMSO/LiBr as a solvent, was used for both LS types used in this work (Figure 8). For more details on how LS is converted to LSA see Section: Isolation of LS and lignosulfonic acid (LSA) from sulfite spent liquor (SSL).
19
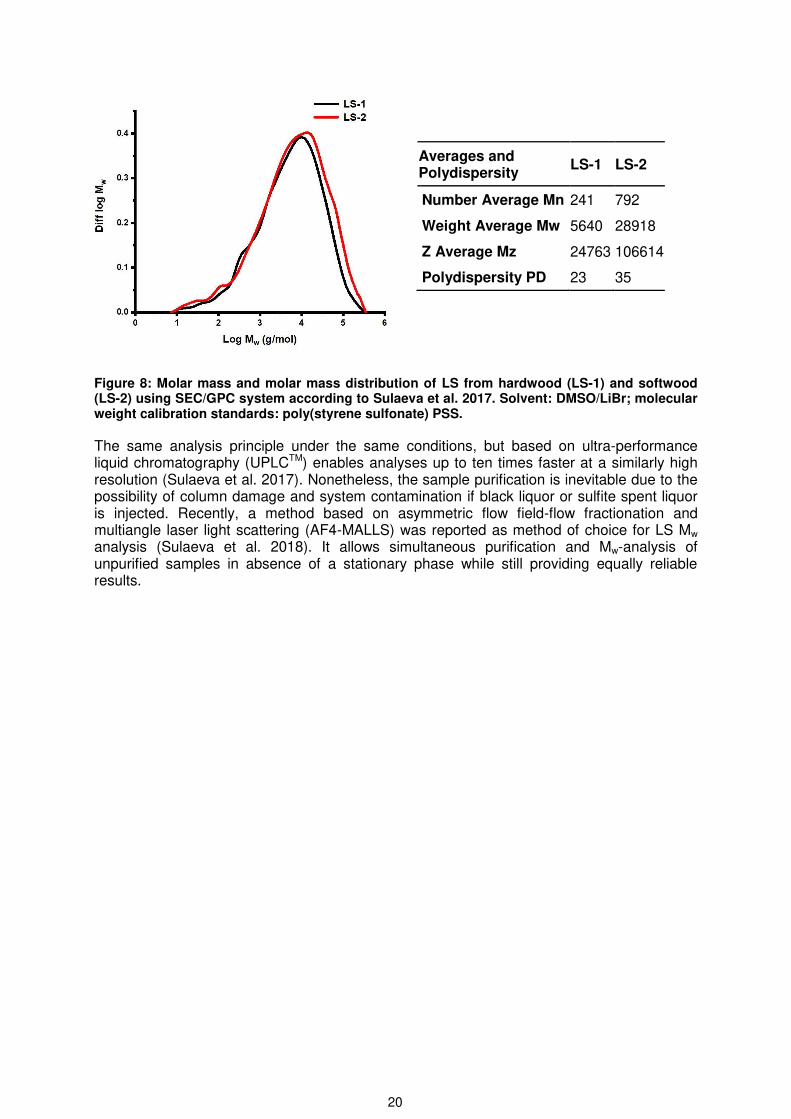
Averages and Polydispersity
LS-1 LS-2
Number Average Mn 241 792
Weight Average Mw 5640 28918
Z Average Mz 24763 106614
Polydispersity PD 23 35
Figure 8: Molar mass and molar mass distribution of LS from hardwood (LS-1) and softwood (LS-2) using SEC/GPC system according to Sulaeva et al. 2017. Solvent: DMSO/LiBr; molecular weight calibration standards: poly(styrene sulfonate) PSS.
The same analysis principle under the same conditions, but based on ultra-performance liquid chromatography (UPLCTM) enables analyses up to ten times faster at a similarly high resolution (Sulaeva et al. 2017). Nonetheless, the sample purification is inevitable due to the possibility of column damage and system contamination if black liquor or sulfite spent liquor is injected. Recently, a method based on asymmetric flow field-flow fractionation and multiangle laser light scattering (AF4-MALLS) was reported as method of choice for LS Mw analysis (Sulaeva et al. 2018). It allows simultaneous purification and Mw-analysis of unpurified samples in absence of a stationary phase while still providing equally reliable results.
20

3 Isolation of LS and lignosulfonic acid (LSA) from sulfite spent liquor (SSL)
For the isolation and purification of LS diverse methods are proposed in the literature or used on laboratory scale, such as amine extraction, dialysis, electrolysis, ion exchange resins etc. (Fatehi and Chen 2016). The common method, which is also the most utilized on industrial scale, is ultrafiltration. However, the majority of these methods is accompanied with several drawbacks, e.g. membrane instability (ultrafiltration), the recovery of organic solvents (amine extraction), the long isolation time (dialysis), the high energy costs and the fouling of the electrodes (electrolysis), etc. (Fatehi and Chen 2016). Even though the ultrafiltration became more important in the last decades through the application of improved membranes, typical problems discussed are flux, membrane fouling and the formation of a gel layer (Ringena et al. 2005).
Additionally, the sole isolation of pure LS by ultrafiltration is without separately collection of membranes almost inevitable due to the similar molar mass of polysaccharide residues. However, methods based on adsorption processes have shown to be promising alternatives towards convenient and easy isolation of LS. The high adsorption capacities and the selectivity of polyacryl- or polyaromatic-based resins as well as the low operational costs render methods based on adsorption realistic alternatives with a high replacement potential. Sumerskii et al. (2015) established a method for the isolation and purification of LS on analytical scale based on macroporous poly(methyl methacrylate) beads (Amberlite® XAD-7 resin). As an optional step LS can be protonated e.g. before treatment with the XAD-7 resin. Ion-exchanger resins, such as DOWEX® 50WX8 or strong acids, such as HCl, H2SO4 or carboxylic acids, protonate the sulfonate groups leading to lignosulfonic acid (LSA).
Figure 9: Isolation protocol of lignosulfonate from spent sulfite liquor (left); protonation of lignosulfonate by DOWEX-50WX8 cation-exchange resin (right).
21

As can be seen in Figure 9, the protonated LS is suspended together with the respective resin, and, after adsorption of LS, the resin is washed with deionized water to remove any impurities. Subsequently, LS is desorbed with alcohol, concentrated by evaporation of the eluent and lyophilized to yield LS or LSA, respectively, in high purity.
3.1 Upscaling of the XAD-7 method for isolation of LS from SSL
Isolation of pure LS in gram amounts or more with traditional methods such as ultrafiltration is challenging due to the long separation time and high costs. The previously established method of purifying LS for analytical purposes from Sumerskii et al. (2015) was extended and optimized for large scale application. In order to find the most productive protocol for preparative purposes in terms of quality and quantity, different adsorbate-to-adsorbent ratios on small (g) and large scale (kg) were carried out and compared to each other (Table 1).
It should be noted that in case of the preparative-scale approaches LS was protonated beforehand through addition of hydrochloric acid and after adsorption purified by continuous washing with deionized water for 15-25 minutes.
As expected, the gravimetric analysis revealed the maximum yield of LS (29% of total dissolved solids, TDS = 60%, Figure 10) at the highest relative amount of XAD-7 resin (XAD-7:SSL = 10:1). A double volume increase of SSL (XAD-7:SSL = 5:1) does not increase the percentage yield of LS, but contrary, the amount of isolated LS decreases about 2%, which indicates either the adsorption capacity of XAD-7 is reached or loss of lignosulfonates through longer washing time has occurred. Further reducing of resin amount with respect to liquor decreased the yield of isolated LS, however, the 2:1 XAD-7:SSL ratio still yielded acceptable results (25% of TDS), e.g. by comparison with adsorption capacity at the same proportion (1:1), which is the lowest (17% of TDS).
According to the literature, the range of adsorption capacity of XAD-7 resin varies greatly between 100-300 mg g-1 (Sumerskii et al. 2015). Theoretically, 2500 g XAD-7 have an isolation capacity of 250 – 750 g of LS.
In the present case, where the liquor has a TDS of 60% and a maximum lignin content of 40% with respect to TDS, the amount of SSL that can be treated with 2500 g resin without overloading it is between 1 and 3 kg SSL. However, although the amount of the liquor in the first two batches was below 1 kg, in both cases the maximum yield of 40% of the TDS was not reached. The low isolated quantity may be a consequence of the high hydrophilic character of LS, which – during continuous washing with deionized water (approx. 20 minutes) – desorbs and solubilizes.
Table 1: Large scale isolation of LS by different adsorbate/adsorbent ratios. The XAD-7 amount was kept constant.
# XAD-7 : SSL [g/g] ratio
1 2500 : 250 10 : 1
2 2500 : 500 5 : 1
3 2500 : 1250 2 : 1
4 1500 : 1500 1 : 1
22

Figure 10: Yield of isolated LS calculated from total dissolved solids (TDS) and SSL and free sugars and hemicelluloses content by different adsorbate-to-adsorbent ratios.
The purity of isolated LS was analyzed by determining the free sugar and hemicellulose content as determined after methanolysis followed by GC-MS analysis (Figure 10 secondary axis), as well as by determining the most frequent metals (ICP-MS, Table 2). From Figure 10 it is clearly visible, that LS from 1:1 XAD-7:SSL ratio contained more than the double amount of free sugars and hemicelluloses compared to the respective other three ratios. On the other hand, the metal contents between the four ratios do not show substantial differences. The total metal (cation) amount is between 0.30% and 0.35% for the isolated LS and 4.8% for the SSL, respectively. Taking into consideration the quality and quantity of the isolated LS, the 2:1 XAD-7:SSL ratio was established as the boundary line between purity and adsorption capacity. Down-scaling of this ratio from 2.5 kg XAD-7 and 1.25 kg SSL to 50 g XAD-7 and 25 g SSL revealed almost the same yield of isolated LS (23 – 25% of TDS) without losing the quality performance of the purified LS (not shown here).
Table 2: Metal ions and phosphorous content in SSL and respective LS isolation approaches.
XAD-7 : SSL ratio
Metals [mg kg-1] SSL 10:1 5:1 2:1 1:1
Mg 42710 2578 2626 2480 2523
K 3369 4
Ca 1063 41
Mn 532 267 286 263 276
Fe 395 196 248 208 223
Ni 6.9 6 5 3 3
Cu 0.6 3 10 1 1
Zn 16 8 280 8 9
P 354
∑ [mg kg-1] 48446 3099 3456 2968 3037
∑ [%] 4.8 0.31 0.35 0.30 0.30
23
3.2 Upscaling: Comparison of small-scale and large-scale batches
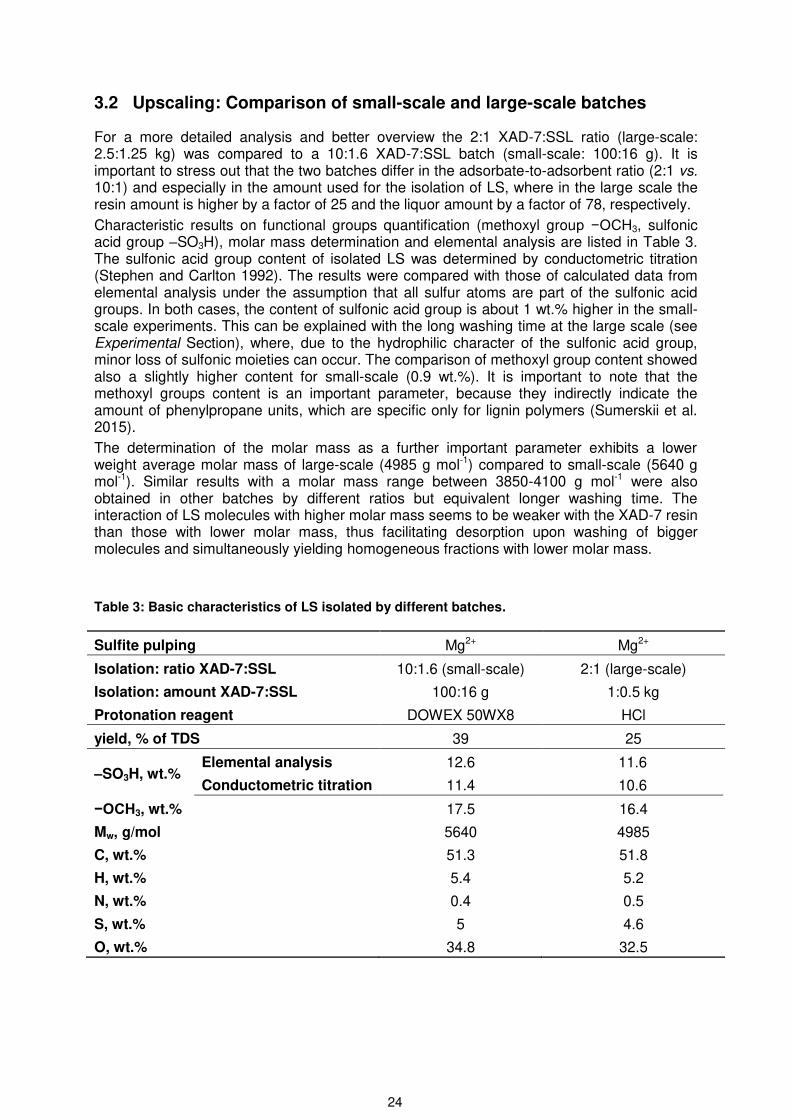
For a more detailed analysis and better overview the 2:1 XAD-7:SSL ratio (large-scale: 2.5:1.25 kg) was compared to a 10:1.6 XAD-7:SSL batch (small-scale: 100:16 g). It is important to stress out that the two batches differ in the adsorbate-to-adsorbent ratio (2:1 vs. 10:1) and especially in the amount used for the isolation of LS, where in the large scale the resin amount is higher by a factor of 25 and the liquor amount by a factor of 78, respectively.
Characteristic results on functional groups quantification (methoxyl group −OCH3, sulfonic acid group –SO3H), molar mass determination and elemental analysis are listed in Table 3. The sulfonic acid group content of isolated LS was determined by conductometric titration (Stephen and Carlton 1992). The results were compared with those of calculated data from elemental analysis under the assumption that all sulfur atoms are part of the sulfonic acid groups. In both cases, the content of sulfonic acid group is about 1 wt.% higher in the small-scale experiments. This can be explained with the long washing time at the large scale (see Experimental Section), where, due to the hydrophilic character of the sulfonic acid group, minor loss of sulfonic moieties can occur. The comparison of methoxyl group content showed also a slightly higher content for small-scale (0.9 wt.%). It is important to note that the methoxyl groups content is an important parameter, because they indirectly indicate the amount of phenylpropane units, which are specific only for lignin polymers (Sumerskii et al. 2015).
The determination of the molar mass as a further important parameter exhibits a lower weight average molar mass of large-scale (4985 g mol-1) compared to small-scale (5640 g mol-1). Similar results with a molar mass range between 3850-4100 g mol-1 were also obtained in other batches by different ratios but equivalent longer washing time. The interaction of LS molecules with higher molar mass seems to be weaker with the XAD-7 resin than those with lower molar mass, thus facilitating desorption upon washing of bigger molecules and simultaneously yielding homogeneous fractions with lower molar mass.
Table 3: Basic characteristics of LS isolated by different batches.
Sulfite pulping Mg2+ Mg2+
Isolation: ratio XAD-7:SSL 10:1.6 (small-scale) 2:1 (large-scale)
Isolation: amount XAD-7:SSL 100:16 g 1:0.5 kg
Protonation reagent DOWEX 50WX8 HCl
yield, % of TDS 39 25
–SO3H, wt.% Elemental analysis 12.6 11.6
Conductometric titration 11.4 10.6
−OCH3, wt.% 17.5 16.4
Mw, g/mol 5640 4985
C, wt.% 51.3 51.8
H, wt.% 5.4 5.2
N, wt.% 0.4 0.5
S, wt.% 5 4.6
O, wt.% 34.8 32.5
24

3.2.1 EXPERIMENTAL
3.2.1.1 Materials and Chemicals
Magnesium-SSL was obtained from Lenzing AG. Amberlite® XAD-7 HP (20-60 mesh) was purchased from Acros Organics (Geel, Belgium). Cation-exchange resin DOWEX® 50WX8 (50-100 mesh), ethanol, sodium hydroxide (NaOH) and hydrochloric acid (HCl) were purchased from Sigma-Aldrich GmbH, Schnelldorf, Germany.
Before use, XAD-7 resin was purified in a 5 L custom designed Soxhlet Extractor to remove micro-particles and low molar mass contaminants such as monomers and dimers. The resin was extracted with ethanol for 48 h (until 15 extraction cycles). After extraction it was washed with 0.1 M NaOH and 0.1 M HCl and stored until needed under slightly acidic conditions (0.01 M HCl).
Total dissolved solid content (TDS) was determined by gravimetric analysis using thermal energy (volatilization) and freeze-drying (lyophilization).
3.2.1.2 Isolation and purification of LS by adsorption on XAD-7
Isolation and purification of LS for the upscaling experiments were carried out according to Sumerskii et al. (2015) with a modified protocol. Protonation of Magnesium-SSL was carried out with 1 M HCl and pH was adjusted to <1. The protonated LS was poured into a predetermined amount of XAD-7 resin and shaken overnight at room temperature. Non-adsorbed compounds were removed by vacuum filtration through a custom-designed filter (10×2 cm) in an adapted 5 L Schott bottle. A modified bottle cap with a filter and a pipe with holes (designed for 5 L Schott bottle cap) enabled continuous washing of the resin/adsorbed LS with deionized H2O for 15–25 minutes. Thereby the impurities were removed semi-automatically. The remaining water was removed by vacuum filtration and the adsorbed LS was desorbed through addition of technical ethanol (4 times). The filtrates were combined and ethanol was evaporated under reduced pressure. The remainder was dissolved in deionized water and frozen at -80 °C. After freeze-drying, the LS (now present as lignosulfonic acid LSA) was further dried in a vacuum oven at 40 °C until it reached constant weight.
The isolation of the small-scale batches was done according to literature protocol (Sumerskii et al. 2015) without any modification.
3.2.1.3 Characterization
The free sugar and hemicellulose contents analyses were performed by a modified acidic methanolysis protocol (Sundberg et al. 1996). Methoxyl-groups were analyzed according to Sumerskii et al. (2017). The molar mass and molar mass distribution analyses were determined according to literature procedure (Sulaeva et al. 2017). For details on the three mentioned methods also see Publication II, section Characterization. Sulfonic acid group contents were determined through conductometric titration (Stephen and Carlton 1992) and also calculated from the sulfur content as measured by elemental analysis. Elemental analysis was carried out using an EURO EA 3000 CHNS-O instrument from HEKAtech (Wegberg, Germany). Additional ICP-MS measurements were performed using Perkin Elmer 9000 DRCe.
25

4 Foaming of Lignosulfonate
Foaming is a dynamic and complex phenomenon governed by scientific principles and engineering parameters (Lee and Ramesh 2004). The two major foaming methodologies which are widely used in traditional polymeric foams are soluble foaming or physical foaming (e.g. polystyrene) and reactive foaming or chemical foaming (e.g. polyurethane). Both techniques involve the fundamental mechanisms for a typical foaming process: gas implementation, gas expansion and foam stabilization (Figure 11).
Besides the foaming technologies, the materials used for the foaming process are crucial. These materials, which consist – among others – of the main components polymer and gas, need to fulfill the foaming conditions, e.g. to solubilize, nucleate or stabilize foaming within certain parameters. Nevertheless, for a successful production of polymeric foams, the technique and the material have to be compatible with each other. In general, materials that are extensively utilized for polymeric foams are commonly derived from fossil sources or petrochemicals, or derived from environmentally unfriendly, highly toxic starting materials, e.g. isocyanates in case of polyurethanes (Mimini et al. 2018). Another ecological hazard about commercial foams is the composition of the used blowing agents, which are often environmentally harmful, such as nowadays banned chlorofluorcarbons.
Greener starting materials based on biopolymers and renewable resources, such as cellulose, starch, polylactic acid, recycled paper, etc., are believed to be an alternative to current fossil-derived materials applied in polymeric foams. However, their production on a large scale is – due to the cost of starting material or processes – economically challenging compared to commercial fossil-based polymeric foams. The abovementioned obstacles may be avoided to a certain degree by a long term sustainable alternative material, such as technical lignin, a biopolymer delivered in huge quantities as a byproduct from pulping and papermaking processes. While technical lignins, e.g. Kraft lignin, can be used in rigid or flexible polymeric foams, LS has the highest potential towards wet foaming. The reason thereof is the constitution of LS that – apart the common functional groups present in other technical lignin types – additionally contains sulfonic acid groups (−SO3H). The sulfonic acid groups have hydrophilic character and high surface activity. By contact with water the acidic groups solubilize first, prompting the dissolution of the residual LS, respectively. On the other hand, the high content of the hydrophobic aromatic moieties tend to distance from water molecules, revealing at the same time surfactant-like properties for the amphiphilic LS.
Figure 11: Steps in foaming process. Figure adapted from Lee and Ramesh (2004).
26

Figure 12: Frothing of pure lignosulfonate: a) pure water evidently does not foam upon mixing; b) addition of lignosulfonate solution with a syringe; c) mixing of the water-lignosulfonate solution results in significant volume increase; d) metastable wet LS-water foam.
By introduction of a gas to the mixture, bubbles are formed, which grow within certain parameters, such as concentration, temperature etc., and build a wet foam. This specific assumption of foaming LS was tested for purified LS and SSL with diverse methods, such as by gas introduction, disperser (Ultra-Turrax®) and frothing (mixer). The gas (N2 or CO2, respectively) was introduced through an adapted syringe outlet in a controlled manner. For the dispersion and frothing falcon tubes or plastic containers were used, and ambient air was used as respective gas for the last two methods. The respective best entries of each method were frozen by liquid N2 or at -80°C and freeze-dried. Parameters taken into consideration for foams investigation were pH, concentration, volume, speed, temperature, and time. While the first method yielded foams which were almost impossible to stabilize against coalescence, the foams produced by dispersion were very dense.
On the other hand, frothing, e.g. by a simple household milk frother, showed to be the method of choice for gas introduction into solubilized LS or SSL. As demonstrated in the Figure 12, by addition of LS and mixing of the water-LS solution, the volume increased significantly. The resulted water-foam is metastable and coalesces after a few minutes. However, besides the foaming techniques, it was found that the performance of the foams (wet or dried) highly depends on the applied conditions.
4.1 Influence of diverse parameters on LS and SSL wet-foaming
The variation of the diverse parameters such as pH, total volume, frothing speed, concentration of lignosulfonate, temperature and time, revealed the concentration and the purity of LS as being the most influencing factors on foam building and foam stabilization.
27

These fundamental parameters related to the wet-foam properties of LSA and SSL (e.g. high foam volume, homogenous bubbles) are shown in Table 4. At first, entries of LSA (a-c) and SSL (a-c) with similar lignin concertation (5 wt.%) but at different pH were compared with each other.
Mg(OH)2 as a weak base reached maximum pH 4, on the other hand, sodium hydroxide alkalized LS and SSL showed in general better performance compared to acidic entries, however, all foams collapsed within a few seconds.
LSA (d-i) with pH 8 and pH 10 and with different concentrations (0.8, 1.7 and 3.3 wt.%, respectively), revealed that lower LS concentrations (e.g. 0.8 wt.%) lead to brighter wet-foams with large pore sizes, while higher LS concentrations (3.3 wt.%) lead to darker wet-foams with smaller pore sizes but with lower total volume (Figure 13). As the concentration between 2 – 2.5 wt.% LS was established as the most suitable, the last entries (j-l) for both LSA and SSL were carried out with 2.2 wt.% LS in basic and acidic medium.
Table 4: Different entries for wet-foaming of LS and SSL by frothing method.
starting material [mg] dissolved in total volume
[mL] pH
lignosulfonate
concentration [%]
LSA*
a 1000 H2O 20 1.28 5
b 1000 H2O + NaOH 20 10 5
c 1000 H2O + Mg(OH)2 20 1.55 5
d 160 H2O + NaOH 20 8.5 0.8
e 160 H2O + NaOH 20 10 0.8
f 340 H2O + NaOH 20 8.6 1.7
g 340 H2O + NaOH 20 10.1 1.7
h 660 H2O + NaOH 20 8.3 3.3
i 660 H2O + NaOH 20 10 3.3
j 330 H2O 15 2.27 2.2
k 330 H2O + NaOH 15 9.08 2.2
l 330 H2O + Mg(OH)2 15 2.40 2.2
SSL**
a 1000 H2O 20 3.86 5
b 1000 H2O + NaOH 20 10 5
c 1000 H2O + Mg(OH)2 20 3.9 5
j 825 H2O 15 3.94 2.2***
k 825 H2O + NaOH 15 9.12 2.2***
l 825 H2O + Mg(OH)2 15 4.0 2.2***
*purified through 2:1 XAD-7:SSL, **freeze-dried, ***calculated based on TDS, 40% of TDS is LS
28

Figure 13: Freeze-dried LS foams (3× magnified) produced by frothing: left 0.8 wt.% LS, right 3.3 wt.% LS.
In general, the yellowish SSL foams look visually compacter than the browner LS foams, however, after freeze-drying the volume of the material reduces spontaneously at room temperature and the porous structure of SSL foams gets lost. This behavior and additionally an adhesive property onto glass or plastic of the dried SSL foams can be attributed to the free sugars and hemicelluloses content in SSL. One noteworthy disadvantage in this respect is the strength of bad odor, which may occur due to the presence of inorganic ingredients, such as metals and salt from pulping processes.
In summary there is no need for additional chemicals or blowing agents for building of LS based wet-foams by frothing (mixer). Water as liquid medium and air as gas for nucleation are very suitable. The increase of foam volume by a factor of 5 at room temperature is reachable within less than a minute. Additionally, the foams derived from the purified LS do not show any bad odor and are stable at room temperature.
In general, foaming of LS in such a simple and versatile manner shows big potential in utilizing LS as a surfactant or dispersing agent for water-based applications, such as oil recovery, concrete or for detergency purposes. Also an application as a fire extinguisher agent could be possible, especially by modifying LS with fire resistant materials, such as silicates, or by using CO2 gas as foaming agent instead of air. However, for utilization as a rigid or flexible foam material, there is a need of crosslinking or reinforcing of the obtained LS homogeneous cellular structure.
4.1.1 EXPERIMENTAL
4.1.2 Preparation of LS and SSL wet-foams
Different predetermined amounts of purified LS or dried SSL powders were added to a predetermined amount of pure water or alkaline water solutions. The pH of the solutions was adjusted using NaOH or Mg(OH)2. Homogenization and powder deagglomeration was carried out by shaking of the water-lignosulfonate solution e.g. as by gas introduction method, or by dispersing/mixing, respectively. In the first method, foaming was carried out in syringes (12 – 60 mL) with an adapted outlet and gas (N2 and CO2) was introduced in a controlled manner. Air as gas, Ultra-Turrax® (IKA magic LAB or T8) disperser and milk frother (Xavax, 14.000 U/min) were used for wet-foaming in falcon tubes (50 mL) or plastic containers (100 mL). Subsequently the wet-foams were frozen by liquid N2 or at -80°C overnight and freeze-dried. Liquid N2 was used for freezing of less stable foams because it avoided forming of a thin layer in the bottom of the vessel.
29

5 Compatibility of LS with PLA (Publication II)
The demand for renewable alternatives in material sciences is constantly growing. Compared to conventional petroleum-based composites, the bio-originated materials are required mostly in medical applications, food packaging, automotive industry, 3D printing, construction etc. However, the use of renewable resources and especially native polymers is – compared to current petrochemical-based materials – often hampered by inferior physicochemical properties and high costs. A combination of lignin, which is available in large quantities and simultaneously counted as reinforcement agent, and polylactic acid (PLA), is most probably perceived as a sustainable alternative towards this purpose. In the present study, three unmodified different lignins, such as Kraft lignin, Organosolv lignin, and LS, were compounded with PLA (Figure 14). The focus was their compatibility with PLA and the comparison of the lignin-PLA samples with respect to thermogravimetric, morphological and mechanical properties.
Figure 14: Production of lignosulfonate-PLA filaments and 3D-printed bars.
Organosolv-PLA blends showed higher thermal resistance and higher flexural strength than other counterparts. An interesting behavior was observed for LS-PLA blends in differential scanning calorimetry (DSC), whereby the presence of LS increased the PLA crystallinity. Scanning electron microscopy (SEM) images showed a porous structure of LS-PLA blends, confirming the observed action of LS as a nucleating agent.
30

6 Non-isocyanate urethanes based on LS (Publication III)
Polyurethanes (PUs) are well known materials which occupy a large part of the market as foam (insulation) materials, high performance adhesives, surface coating etc. Their main drawback is the origin and release of toxic chemicals, such as isocyanate and aromatic amines (anilines). In this study the key objective was the synthesis of bio-based PU materials with LS as starting compound (Figure 15). Glycerol 1,2-carbonate (GC) and dimethyl carbonate (DMC) – both glycerol derivatives – were used to equip LS with cyclic carbonate moieties, which subsequently yield the desired carbamate functional group after the reaction with a diamine, such as 1,6-hexamethylenediamine. During oxypropylations with GC the formation of a cyclic carbonate was assumed, however, it was almost impossible to proof in case of LS. The same behavior – as reported in literature – was supposed to occur in other lignins, such as kraft-, organsolv- and soda-lignins. The question of whether the cyclic carbonate is bound or not to lignin/LS – and especially without a transesterification step with DMC – has been answered unambiguously with advanced analytical techniques, such as 2D HMBC NMR technique, and corroborated by use of the model compound vanillyl alcohol (VA). As a consequence, the production of LS-PUs via cyclic carbonates was simplified and accelerated.
Figure 15: Synthesis of lignosulfonate-based polyurethane (PU) from the pulping byproduct sulfite spent liquor (SSL).
The obtained materials - synthesized from non-toxic reagents using environmentally friendly processes - show a high potential of the most underutilized renewable-based resource technical lignin as a starting material for bio-based PU composites. Additionally, taking into account the unique characteristic of LS, such as the highly negatively charged surface, LS-PU materials and their application can be of particular interest.
31

7 References
Balakshin, M.Y, Capanema, E.A., and Berlin, A. (2014) Isolation and analysis of lignin-carbohydrate complexes, Preparation with traditional and advanced Methods: A review. In: Studies in Natural Products Chemistry. Eds. Atta-ur-Rahman. Elsevier. The Netherlands. 42. pp. 83-115.
Balakshin, M.Y., Capanema, E.A., Chang, H.-M. (2009) Recent Advances in the Isolation and Analysis of Lignins and Lignin–Carbohydrate Complexes. In: Characterization of Lignocellulosic Materials. Hu, T.Q. Blackwell Publishing Ltd., online. pp. 148-166.
Biermann, C.J. (1996) Pulping Fundamentals. In: Handbook of Pulping and Papermaking (2nd Edition). Ed. Biermann, C.J. Academic Press, San Diego. pp. 55-100
Calvo-Flores, F. G., Dobado, J. A., Isac-García, J., Martín-Martínez, F. J. (2015) Functional and Spectroscopic Characterization of Lignins. In: Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications
Fatehi, P., Chen, J. (2016) Extraction of Technical Lignins from Pulping Spent Liquors, Challenges and Opportunities. In: Production of Biofuels and Chemicals from Lignin. Eds. Fang, Z., Smith, J.R.L. Springer Singapore, Singapore. pp. 35-54.
Fengel, D., Wegener, G. Wood: Chemistry, Ultrastructure, Reactions. de Gruyter, Berlin; New York, 1984.
Ge, Yuanyuan, Li, Dingwei, LI, Zhili (2014) Effects of Lignosulfonate Structure on the Surface Activity and Wettability to a Hydrophobic Powder. BioResources, 9(4), 7119-7127.
Korntner, P., Schedl, A., Sumerskii, I., Zweckmair, T., Mahler, A.K., Rosenau, T., Potthast, A. (2018) Sulfonic Acid Group Determination in Lignosulfonates by Headspace Gas Chromatography. ACS Sustain. Chem. Eng. 6:6240-6246.
Lee, S.T., Ramesh, N.S. Polymeric Foams: Mechanisms and Materials. CRC Press, New York, 2004.
Lora, J. (2008) Industrial Commercial Lignins: Sources, Properties and Applications In: Monomers, Polymers and Composites from Renewable Resources. Eds. Belgacem, M. N. and Gandini, Elsevier, The Netherlands. pp. 226-230.
Mansouri, N.-E.E., Salvadó, J. (2006) Structural characterization of technical lignins for the production of adhesives: Application to lignosulfonate, kraft, soda-anthraquinone, organosolv and ethanol process lignins. Ind. Crop. Prod. 24(1):8-16.
Mimini, V., Kabrelian, V., Fackler, K., Hettegger, H., Potthast, A., Rosenau, T. (2018) Lignin-based foams as insulation materials: a review. Holzforschung. 73(1):117.
Mullick, A.K. (1996) 7 - Use of lignin-based products in concrete. In: Waste Materials Used in Concrete Manufacturing. Ed. Chandra, S. William Andrew Publishing, West-wood, New York. pp. 352-429.
Ralph, J., Lundquist, K., Brunow, G., Lu, F., Kim, H., Schatz, P. F., and Marita, J. M., (2004). Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 3: pp. 29-60.
Rezanowich, A., Goring, D.A.I. (1960) Polyelectrolyte expansion of a lignin sulfonate microgel. J. Colloi Interf. Sci. 15(5):452-471.
Ringena, O., Saake, B., Lehnen, R. (2005) Isolation and fractionation of lignosulfonates by amine extraction and ultrafiltration: A comparative study. Holzforschung. 59(4):405.
Saeed, A., P. Fatehi, and Y. Ni, (2011) Chitosan as a flocculant for pre-hydrolysis liquor of kraft-based dissolving pulp production process. Carbohyd. Polymers. 86(4). pp. 1630-1636.
Sjöström, E. (1993) Wood pulping. In: Wood Chemistry, Fundamentals and Applications (2nd Edition). Ed. Sjöström, E. Academic Press, San Diego. pp. 114-164
32

Stücker, A., Podschun, J., Saake, B., Lehnen, R. (2018) A novel quantitative 31P NMR spectroscopic analysis of hydroxyl groups in lignosulfonic acids. Analytical Methods. 10:3481-3488.
Sulaeva, I., Sumerskii, I., Bacher, M., Zinovyev, G., Henniges, U., Rosenau, T., Potthast, A. (2015) Comparing Different Approaches to Measure Molar Mass of Lignin: SEC, DOSY and AsFlFFF. ACS National Meeting & Exposition, Conference: 249th, Denver, CO, United States.
Sulaeva, I., Vejdovszky, P., Henniges, U., Mahler, A. K., Rosenau T., Potthast, A. (2018) Molar Mass Characterization of Crude Lignosulfonates by Asymmetric Flow Field-Flow Fractionation. ACS Sustainable Chem. Eng., 2019, 7 (1), pp 216–223.
Sulaeva, I., Zinovyev, G., Plankeele, J.-M., Sumerskii, I., Rosenau, T., Potthast, A. (2017) Fast Track to Molar-Mass Distributions of Technical Lignins. ChemSusChem. 10(3):629-635.
Sumerskii, I., Korntner, P., Zinovyev, G., Rosenau, T., Potthast, A. (2015) Fast track for quantitative isolation of lignosulfonates from spent sulfite liquors. RSC Adv. 5:92732-92742.
Sumerskii, I., Zweckmair, T., Hettegger, H., Zinovyev, G., Bacher, M., Rosenau, T., Potthast, A. (2017) A fast track for the accurate determination of methoxyl and ethoxyl groups in lignin. RSC Adv. 7:22974-22982.
Sundberg, A., Sundberg, K., Lillandt, C., Holmbom, B. (1996) Determination of hemicelluloses and pectins in wood and pulp fibres by acid methanolysis and gas chromatography. Nord. Pulp Pap Res J. 11, 216-219, 226.
Stephen, Y., L. and Carlton W., D. (1992) Methods in Lignin Chemistry, 1st Edition, Springer-Verlag, Berlin, Heidelberg, Germany.
Yang, D., Li, H., Qin, Y., Zhong, R., Bai, M., Qiu, X. (2015) Structure and Properties of Sodium Lignosulfonate with Different Molecular Weight Used as Dye Dispersant. J. Disper. Sci. Technol. 36:532-539.
Weng, J.-K., and Chapple, C. (2010) The origin and evolution of lignin biosynthesis. New Phytol. 187:273-285.
Zinovyev, G., Sumerskii, I., Korntner, P., Sulaeva, I., Rosenau, T., Potthast, A. (2017) Molar mass-dependent profiles of functional groups and carbohydrates in kraft lignin. J. Wood Chem. Techn., 37:171-183.
33

8 Publications
Reprints of the publications are attached below. The respective author is underlined.
Publication I
Mimini, V., Kabrelian, V., Fackler, K., Hettegger, H., Potthast, A., Rosenau, T. (2018) Lignin-based foams as insulation materials: a review. Holzforschung. 73(1):117.
Publication II
Mimini, V., Sykacek, E., Hashim, S. N. A. S., Holzweber, J., Hettegger, H., Fackler, K., Potthast, A., Mundigler, N., Rosenau, T. (2019) Compatibility of Kraft Lignin, Organosolv Lignin and Lignosulfonate with PLA in 3D Printing. Journal of Wood Chemistry and Technology, 39: 14-30.
Publication III
Mimini, V., Amer, H., Hettegger, H., Bacher, M., Gebauer, I., Bischof, R., Fackler, K., Potthast, A., Rosenau, T. (2019) Lignosulfonate-based polyurethane materials via cyclic carbonates - preparation and characterization. Holzforschung
Related Publication:
Publication IV
Amer, H., Mimini, V., Schild, D., Rinner, U., Bacher, M., Potthast, A., and Rosenau, T. (2019)
Gram-scale economical synthesis of selectively protected trans-coniferyl alcohol and its
corresponding thiol. Holzforschung
34

35
8.1 Lignin-based foams as insulations materials: a review (Publication I)

36

Holzforschung 2019; 73(1): 117–130
Vebi Mimini, Vasken Kabreliana, Karin Fackler, Hubert Hettegger, Antje Potthast and Thomas Rosenau*
Lignin-based foams as insulation materials: a review
https://doi.org/10.1515/hf-2018-0111Received May 13, 2018; accepted October 26, 2018; previously published online December 8, 2018
Abstract: The bulk use of renewable polymers is cur-rently largely limited to cellulose and, less significantly, hemicelluloses. Technical lignins are only applied in novel materials to a rather limited extent, although bulk lignin utilization is a worldwide research object. Native lignins, which belong to the second or third most abun-dant biopoly mers of terrestrial plants, are mostly used in the form of technical lignins from wood pulping pro-cesses; they are employed in low-performance sectors or simply burnt for the generation of energy. Technical lignins are available in huge quantities and have a large application potential, mainly in areas where their aro-matic nature is of relevance. This review presents the state of the art of foamed lignin-based polymers (lignofoams) as high-performance insulation materials. In the focus of this presentation are the fundamental foaming principles and influential agents that have an improvement poten-tial concerning the matrix interactions between technical lignins (including lignosulfonates) and a copolymer in foam composites. The different approaches for foam prep-aration are critically compared. In general, the reviewed papers disclose that the lignin part in foams should be less
than 37%. There are significant difficulties to improve the properties of lignofoams, and thus intensive research is needed to find better formulations and new technologies.
Keywords: foaming, insulation, lignin, lignin utilization, lignofoams, lignosulfonate, technical lignin
Introduction
Renewable resources, such as lignocellulosic biomass, can partly substitute fossil materials for the produc-tion of established or new materials. This approach con-tributes to the solution of environmental problems, and serves national security and rural economic development (Brown and Brown 2014). Biomass-derived carbohydrates are broadly consumed but the lignin moiety of biomass is underutilized in high-value and high-volume applications. Technical lignins are aromatic polymers with high carbon contents of around 60–63% and have a high thermal sta-bility. As they originate from different raw materials and pulping processes, their structures and application poten-tials are also diverse. Kraft lignin and lignosulfonates (LSs) contain sulfur in contrast to soda lignins, organo-solv lignins or hydrolysis lignins. The covalently bound sulfur is difficult to remove and thus such lignins cannot be applied in nutrition. Sulfur hinders transition metal-catalyzed reactions and may cause odor problems (Vishtal and Kraslawski 2011). Kraft lignins are used as binders, fer-tilizers or resins and are alternative to polyols due to their high content of hydroxyl groups. The sulfonic acid groups (-SO
3−) in LS provide water solubility and a high surface
activity. They are well suited for emulsion stabilizers, plas-ticizers, dispersants, surfactants, etc. LSs are capable of trapping air in concrete, and they have a potential applica-tion in wet foams. Organosolv lignins, on the other hand, are sulfur-free, and have lower molecular weights and higher homogeneity than kraft lignins and LSs. Their more alkylated nature permits application in formulations of inks, varnishes and paints (Belgacem et al. 2003).
From the 70 Mt year−1 technical lignin production worldwide, only less than 2% is used as material (Cateto et al. 2013; Agarwal et al. 2014). This is due to their hetero-geneous structure and the presence of various inorganic
aDeceased*Corresponding author: Thomas Rosenau, Division of Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Straße 24, Tulln A-3430, Austria; and Johan Gadolin Process Chemistry Centre, Åbo Akademi University, Porthansgatan 3, Åbo/Turku FI-20500, Finland, e-mail: [email protected] Mimini: Division of Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Straße 24, Tulln A-3430, Austria; and Wood Kplus – Kompetenzzentrum Holz GmbH, Altenberger Straße 69, Linz A-4040, AustriaVasken Kabrelian: Wood Kplus – Kompetenzzentrum Holz GmbH, Altenberger Straße 69, Linz A-4040, AustriaKarin Fackler: Lenzing AG, Werkstraße 1, Lenzing A-4860, AustriaHubert Hettegger and Antje Potthast: Division of Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Straße 24, Tulln A-3430, Austria
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4237

118 V. Mimini et al.: Lignin-based foams as insulation materials
and organic impurities like ash, silicates and carbohy-drates (Vishtal and Kraslawski 2011). Generally, technical lignin is burned to produce energy for the pulping process, but up to 50% of the lignin could be diverged from the pro-duction process for other utilizations without endanger-ing the energy balance of a plant (Fisher and Fong 2014).
Insulation materials are commonly derived from pet-rochemicals [mainly polystyrene (PS; Figure 1)] or from inorganic natural resources, which have a high energy demand for production, such as glass and rock wools (Asdrubali et al. 2015). Large amounts of technical lignin could be utilized for foam production. Lignin application for this purpose is not a trivial matter because of their relatively low compatibility with other polymers. To over-come some problems, several process steps are conceiv-able, such as adjusting the molar mass, separating sugars, extractives and inorganic compounds, targeted modifica-tion reactions for reactivity improvement via introduction of new active sites, functionalizing of hydroxyl groups, or copolymerizing with other suitable polymers.
In this review, the compatibility of technical lignins with other polymers are focused on with regard to foam pro-duction. The application of blowing and crosslinking agents and various chemical modification methods was scrutinized in the context of lignin-based insulation materials.
Polymeric foams
The first cellular polymer that reached the market was sponge rubber produced from natural rubber latex
(Schidrowitz and Goldsbrough 1915). Since then, the inter-est in porous materials has continually grown as polymer foaming materials have unique properties, such as low weight, low heat transfer, excellent strength-to-weight ratio, low dielectric constant, superior thermal and acous-tic insulating potential, and higher flexibility (Lee and Ramesh 2004; Frisch 2006). The incorporation of micro-bubbles into the polymer matrix increases its volume and simultaneously reduces the polymer amount required. This lowers production costs per unit volume compared to non-foamed materials and needs fewer resources. In general, foamed polymers can be flexible or rigid (Ikem et al. 2010). Flexible foams are expected to gain a market share in the future due to their unique properties, such as easy handling and shaping. The increasing trend is illustrated based on the forecasted polyurethane (PU) foams market in Germany (Figure 2). Depending on the cell morphology, there are open- or closed-cell foams. The latter are more suitable as thermal insulation materials due to their low permeability (Lee et al. 2005), while the open-cell foams are much better in sound absorption (Álvarez-Láinez et al. 2014; Bohnke et al. 2014; Mostafa and Abdolreza 2015). In terms of cell size, the porous materials are classified into microporous (<2 nm), mesoporous (between 2 and 50 nm) and macroporous (≥50 nm) foams (Sing et al. 1984). However, both mesoporous and microporous materials are often designated in the literature as “nanoporous”.
The most common polymeric foams are PS, expanded PS (EPS) or extruded PS (EXS), PU, polyisocyanurate (PIR), polyvinyl chloride (PVC), polyimide, phenolic and polyolefin foams (Figure 1). The application field is broad, such as in household appliances, packaging, transport and furniture, just to mention a few, but the polymeric foams are mostly used as insulation materials in buildings
Figure 1: The European thermal insulation market by insulant type in 2014.The total value for the thermal insulation products in Europe was just below 234.6 M m3 in 2014. Figure adapted and reprinted with permission from IAL Consultants.
6000.0
5000.0
4000.0
3000.0
2000.0
1000.0
–
Rigid foamCoatingsElastomers
Flexible foamAdhesives and sealantsOthers
2010
2011
2012
2013
2014
2015
2016
2017
2018
2019
2020
2021
2022
2023
2024
Figure 2: German polyurethane (PU) market revenue by product, 2010–2024 (in million USD).Figure adapted and reprinted with permission from http://www.grandviewresearch.com.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4238
V. Mimini et al.: Lignin-based foams as insulation materials 119
and for construction (Hingmann 2017). Their global market demand is very high, and it is expected to increase continuously (Figures 3 and 4).
Fundamental principles in polymer
foaming
Polymer foams can be produced by means of chemi-cal, physical or mechanical techniques, such as thermal decomposition (Kim et al. 2001; Zhai et al. 2010), sintering of particles (Wong et al. 2009), polymerization of emul-sion templates and ultraviolet-polymerizable particles (Lee et al. 2011), leaching (Hwa et al. 1964; Engelmann 1978), phase separation (Hwa et al. 1964; Mannella et al. 2015), extrusion, molding (Zhang et al. 2015) and froth-ing (Szczurek et al. 2014; Merle et al. 2016a,b). Blowing agents are the key materials for polymer foaming. They decompose or vaporize easily and develop gases or vapors in large quantities (Štěpek and Daoust 1983). They are
subdivided into chemical blowing agents (CBAs) and physical blowing agents (PBAs). CBAs are organic sol-vents or inorganic salts for example, ethanol; isocyanates and water; carbonates and bicarbonates and/or nitrogen-based compounds like ammonium carbonate, ammo-nium carbamate, nitrites and azo-hydrazine (Singh 2001). These agents release gases such as N
2, NH
3, CO and CO
2,
which form holes/bubbles in the matrix leading to cel-lular structures (Štěpek and Daoust 1983). This irrevers-ible process is significantly influenced by particle size and the concentration of the chemical agent (Petchwat-tana and Covavisaruch 2011). PBAs are compounds such as n-pentane, isobutane and methyl formate or gases like CO
2 and N
2 that are not chemically produced during the
foaming process, but released e.g. by evaporation, and that do not react with the polymer (Štěpek and Daoust 1983). During physical foaming, the respective inert gas dissolves in the polymer matrix through pressurization and is in a supersaturated state. Subsequently, at a certain temperature, the gas begins to expand and forms bubble nuclei (Lee et al. 2006; Sauceau et al. 2011). PBAs, as sig-nificantly lower-cost agents, deliver low-density materials with a highly homogenous foam structure (Singh 2001).
The type of blowing agents is relevant to foam quality and also in terms of environmental safety issues. World-wide environmental agreements like the Montreal Protocol (1987) and the Kyoto Protocol (1997) strictly define the use of certain blowing agents and ban those with ozone-deple-tion potential (ODP); for example, the chlorofluorocarbons. Carbon dioxide, as an environmentally compatible, non-toxic and highly soluble agent, is frequently the first choice. It is releasable during production processes. Another advantage of CO
2 is its large diffusivity with zero surface
tension, and it is suitable as a supercritical fluid (Han et al. 2007). On the other hand, it is not easy to handle, and it must be purified, dried and liquefied before use.
Lignofoams: lignin-based foam
materials
Lignofoams are polymeric lignin-based biomaterials com-posed of an internal porous/cellular structure. They have a large production potential on an industrial scale and are applicable in the construction/housing sector as an insula-tion material. Lignofoams are made of renewable materials and can be designated as eco-friendly materials. However, to the best of our knowledge, no commercially available foams have been produced so far with technical lignin as the main component with or without modification.
Figure 3: Market for foams: estimated assessable market by segment.Figure reproduced from Hingmann (2017).
5000.04500.04000.03500.03000.02500.02000.01500.01000.0500.0
2013 2014
Packaging
Funrniture and bedding
Others
Building and constructions
Automotive
2015 2016 2017 2018 2019 2020 2021 2022 2023 2024
–
Figure 4: US polymer foam market volume by application, 2013–2024 (in kilotons).Figure adapted and reprinted with permission from http://www.grandviewresearch.com.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4239

120 V. Mimini et al.: Lignin-based foams as insulation materials
PU-lignin foams
The introduction of lignin in PU foams is currently one of the most studied topics. The multipurpose properties of PU foams make them the most produced foams in the world (Obaid et al. 2016). They are suitable for several applications, such as in packaging, automotive and trans-portation industries, cushioning, machinery, foundries and as insulation. Nonetheless, the isocyanate starting compounds are graded as environmentally unfriendly, toxic and difficult to produce due to safety issues, which is a considerable disadvantage. PUs are made from polyols and isocyanates, both derived from fossil sources (Obaid et al. 2016). Lignin as a polyol containing multiple hydroxyl groups is a suitable alternative for conventional polyols in the production of PU foams (Rials and Glasser 1986; Cateto et al. 2011; Obaid et al. 2016). PU foams can be divided into flexible and rigid foams. Li and Ragauskas (2012) and Cateto et al. (2013) synthesized rigid PU foams (RPU) based on different lignin-based polyols obtained after oxypropylation. This approach converts solid tech-nical lignin (e.g. kraft lignin) into a liquid polyol, which provides a better control of polyol properties and allows for incorporating higher amounts of lignin in PU foams (Li and Ragauskas 2012). Additionally, Li and Ragauskas (2012) reported that lignin polyol-based foams have better mechanical properties than commercial RPU, and also lignin polyols can be used solely for RPU production without another polyol or chain extenders (Figure 5).
Jeong et al. (2013) investigated the effect of lignin in PU foams aiming at flexible PU foams in combina-tion with polyethylene glycol (PEG). An increasing lignin fraction in the blend led to a higher compressive modulus of the materials, i.e. lignin gives hardness and PEG gives softness to the foam. Probably, the aromatic groups of lignin are responsible for the higher stiffness of the polymer chains (Yoshida et al. 1987, 1990). On the other hand, PEG is incorporated into lignin at the molecular level and operates as a chain extender leading to more ductility (Obaid et al. 2016). That chain extend-ers improve the foams’ flexibility was also observed by others. Such commonly used extenders are PEG, poly-propylene glycol (PBD glycol; Cinelli et al. 2013; Ber-nardini et al. 2015), castor oil (CO; Cinelli et al. 2013), polybutadiene glycol (PBD glycol; Saraf et al. 1985), butanediol (Pan and Saddler 2013), propylene carbon-ate (PC; Kühnel et al. 2015) and polypropylene oxide (PO; Mahmood et al. 2016).
Phenolic resin – lignin foams
Foaming of phenolic resins [phenol-formaldehyde (PF) or phenol-formaldehyde-urea resins] is also a very appealing approach. Highly temperature-resistant materials with low thermal conductivity (such as phenolic resins) are required in thermal insulation applications. Many studies on phenolic resins have already been conducted that replaced phenol with technical lignin, in some cases by up to 40% (Ghorbani et al. 2016, 2017). However, a complete replacement is almost impossible due to the low reactiv-ity of lignin toward formaldehyde (Obaid et al. 2016). Hu et al. (2012) have reported on the replacement of phenol with LS in phenolic foams in a ratio Ph:LS = 1:5 in alkaline media. n-Pentane was the most suitable foaming agent, sulfuric acid served as a catalyst and Tween 80 was the surfactant. The LS-modified phenolic foams possess lower density (Figure 6) and better thermal insulation properties compared to their neat phenolic counterparts.
Starch-lignin foams
Lignin is a candidate for modifying starch foam in pack-aging applications due to its low water absorption (Obaid et al. 2016). Stevens et al. (2010) described the first suc-cessful starch-kraft lignin foam prepared by a technique similar to compression molding. Up to 20% starch substi-tution by lignin, the density and morphology of the foam are unchanged. Lignin stabilizes the starch structure and,
0.20 0%
10%
30%
60%
100%
Only
Lignin polyol
0.15
0.10
0.05
0.000 5 10 15 20
Strain (%)
Str
ess (
MP
a)
Figure 5: Mechanical properties of kraft lignin-based PU foams by compression testing.Both the stress and the strain increase with 10 and 30% lignin polyol content. The optimum properties are obtained with foams prepared with only lignin polyol without the addition of any other commercial polyol. Adapted from Li et al. (2012), reprinted with permission from Taylor & Francis.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4240

V. Mimini et al.: Lignin-based foams as insulation materials 121
as assumed, decreases the water absorption (Figure 7). Compared to foamed PS, the starch-lignin foams showed larger moduli of elasticity, similar flexural strengths and smaller strains at maximum stress. However, a wide utili-zation of starch-lignin foams is limited due to their highly hydrophilic character.
PS-lignin foams
PS foams – found as rigid PS and EPS or EXS – are poly-mers produced from styrene as a liquid hydrocarbon monomer originating from petroleum. PSs are one of the most common polymeric foams with a versatile applica-tion potential, such as insulation, construction, protective
and food packaging, and in automotive and transportation industries (Figure 1). Their advantages are high expansion (up to approximately 95% air), light weight, inexpen-sive production and outstanding insulation properties. However, the recycling of PS foams and their fossil-based origin are problematic. Additionally, the foams are fragile and brittle and have lower quality in terms of thermal conductivity. A quality improvement by incorporation with other polymers and/or inorganic or organic fillers is usually recommended. A combination with lignin seems to be advantageous as PS is produced by radical polym-erization, while lignin has radical scavenging properties. Sequential mass-suspension polymerization, atom-trans-fer radical polymerization and melt blending are the most employed processes in this context. Victor et al. (2018) attempted to replace PS with up to 20% kraft lignin or previously esterified kraft lignin via the sequential mass-suspension polymerization process. Kraft lignin was cata-lytically esterified by 1-methylimidazole with methacrylic anhydride as the esterification agent, which ensures organic phase homogeneity by increasing its solubility in organic solvents. Benzoyl peroxide (BPO) was used as an initiator and polyvinyl alcohol (PVA) as a suspend-ing agent. According to the authors, the esterified lignin enables free-radical copolymerization with styrene, avoid-ing the lixiviation problem of lignin from the thermoplastic PS matrix during both the polymerization reaction and the polymer processing. Scanning electron microscopy (SEM) reveals smaller particle size of the modified lignin after substitution of hydroxyl groups with ester groups, but the T
g was increasing, which was attributable to the higher
ester group concentration, which reduces the mobility of lignin chains. Furthermore, the PS-esterified lignin com-posites showed higher average molar masses, viscosity and porosity. However, after the polymerization of lignin with styrene, oxidation of lignin was observed. This unde-sirable effect was minimized by increasing the amount of BPO and PVA. Lignin acts as a free-radical scavenger and
Figure 6: SEM micrograph of phenolic resin foams.On the left, conventional phenolic foam (3.0 mm section); in the middle, modified phenolic foam (3.0 mm section); and on the right, magnification of modified phenolic foam (30.0 µm section). Adapted and reprinted with permission from Hu et al. (2012).
0.08
0.06
0.04mt (
g c
m–
2)
0.02
0.000 10 20 30 40 50 60
3N
1
t1/2 (s1/2)
Figure 7: Water absorption for starch (sample 1) and starch-lignin (sample 3N) described by an empirical power law model (mt = ktn with mt for mass of absorbed water per unit area at time).The slopes increase linearly at longer times with lignin as a water absorption inhibitor. Adapted and reprinted with permission from Stevens et al. (2010).
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4241
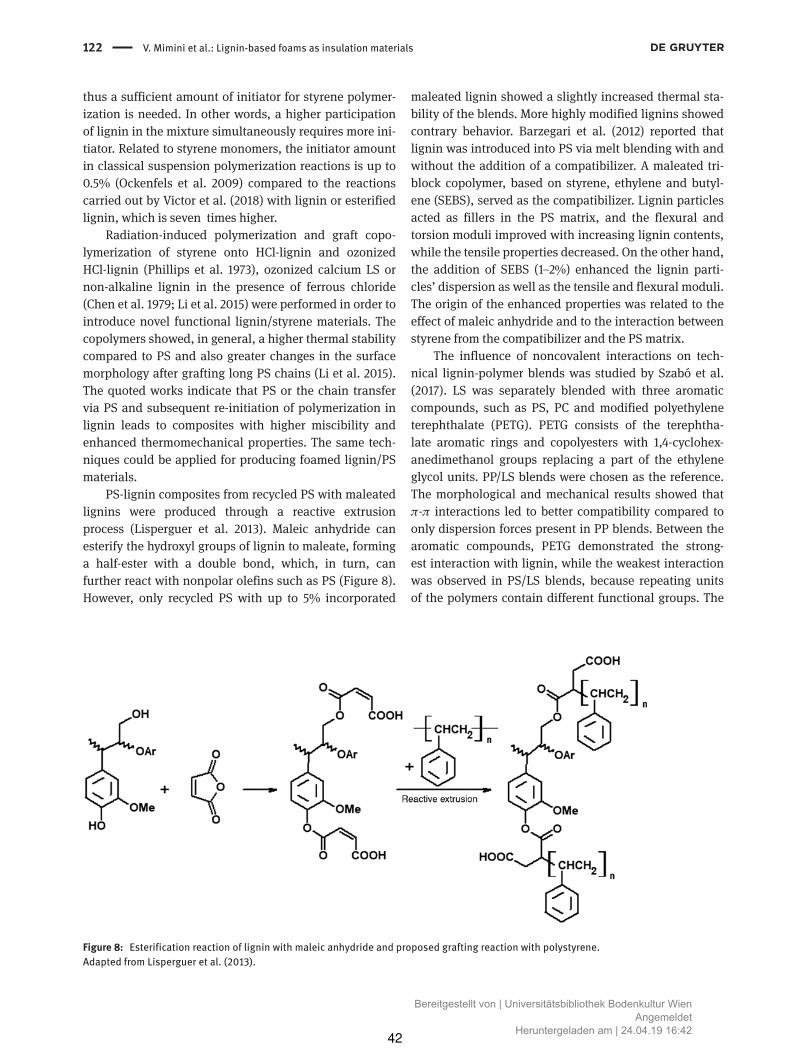
122 V. Mimini et al.: Lignin-based foams as insulation materials
thus a sufficient amount of initiator for styrene polymer-ization is needed. In other words, a higher participation of lignin in the mixture simultaneously requires more ini-tiator. Related to styrene monomers, the initiator amount in classical suspension polymerization reactions is up to 0.5% (Ockenfels et al. 2009) compared to the reactions carried out by Victor et al. (2018) with lignin or esterified lignin, which is seven times higher.
Radiation-induced polymerization and graft copo-lymerization of styrene onto HCl-lignin and ozonized HCl-lignin (Phillips et al. 1973), ozonized calcium LS or non-alkaline lignin in the presence of ferrous chloride (Chen et al. 1979; Li et al. 2015) were performed in order to introduce novel functional lignin/styrene materials. The copolymers showed, in general, a higher thermal stability compared to PS and also greater changes in the surface morphology after grafting long PS chains (Li et al. 2015). The quoted works indicate that PS or the chain transfer via PS and subsequent re-initiation of polymerization in lignin leads to composites with higher miscibility and enhanced thermomechanical properties. The same tech-niques could be applied for producing foamed lignin/PS materials.
PS-lignin composites from recycled PS with maleated lignins were produced through a reactive extrusion process (Lisperguer et al. 2013). Maleic anhydride can esterify the hydroxyl groups of lignin to maleate, forming a half-ester with a double bond, which, in turn, can further react with nonpolar olefins such as PS (Figure 8). However, only recycled PS with up to 5% incorporated
maleated lignin showed a slightly increased thermal sta-bility of the blends. More highly modified lignins showed contrary behavior. Barzegari et al. (2012) reported that lignin was introduced into PS via melt blending with and without the addition of a compatibilizer. A maleated tri-block copolymer, based on styrene, ethylene and butyl-ene (SEBS), served as the compatibilizer. Lignin particles acted as fillers in the PS matrix, and the flexural and torsion moduli improved with increasing lignin contents, while the tensile properties decreased. On the other hand, the addition of SEBS (1–2%) enhanced the lignin parti-cles’ dispersion as well as the tensile and flexural moduli. The origin of the enhanced properties was related to the effect of maleic anhydride and to the interaction between styrene from the compatibilizer and the PS matrix.
The influence of noncovalent interactions on tech-nical lignin-polymer blends was studied by Szabó et al. (2017). LS was separately blended with three aromatic compounds, such as PS, PC and modified polyethylene terephthalate (PETG). PETG consists of the terephtha-late aromatic rings and copolyesters with 1,4-cyclohex-anedimethanol groups replacing a part of the ethylene glycol units. PP/LS blends were chosen as the reference. The morphological and mechanical results showed that π-π interactions led to better compatibility compared to only dispersion forces present in PP blends. Between the aromatic compounds, PETG demonstrated the strong-est interaction with lignin, while the weakest interaction was observed in PS/LS blends, because repeating units of the polymers contain different functional groups. The
Figure 8: Esterification reaction of lignin with maleic anhydride and proposed grafting reaction with polystyrene.Adapted from Lisperguer et al. (2013).
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4242

V. Mimini et al.: Lignin-based foams as insulation materials 123
π-π interactions can arise between the aromatic rings of LS and the aromatic rings of PS, PC or PETG. Hydro-gen bonding can also be effective between the carbonyl oxygen of polyesters (two C=O groups per a repeating unit) or PC (one C=O group per repeating unit) and acidic lignin hydroxyl protons. Hydrogen bonding and/or π-π interac-tions increase the miscibility of technical lignins with dif-ferent polymers, but these interactions are weak compared to the strong interaction among lignin molecules (Szabó et al. 2017). A stronger interaction or covalent bonding could increase the miscibility of lignin/polymer blends and thus improve the thermomechanical properties.
Silicone-lignin foams
Zhang et al. (2015) reported the development of closed-cell lignin-silicone foams based on suitable mixtures of hydrosilanes. Based on the Piers-Rubinsztajn reaction (Brook et al. 2010), the authors applied a simple two-step process for the production of flexible lignin-silicone com-posite elastomers: (1) extrusion at room temperature to obtain a consistent mixture and (2) compression molding of the extruded precursor lignin-silicone at elevated tem-peratures to get a foamed elastomer. The lignin content was varied between 25 and 55%. For adequate mechanical
properties and compact closed-cell foams, the optimal lignin content was 42%. Depending on the proportion of the catalyst, lignin particles and hydrosilane composi-tion, three different foam materials were obtained: Type A, foams with large cells that are easily breakable; Type B, foams with uniform morphology and flexible behav-ior; and Type C, foams with fewer cells and rigid behavior compared to Type A and Type B (Figure 9). The foams of Type B showed a homogeneous cell structure and satis-factory properties. The formulation of B1 consisted of 25% lignin, 70% hydride-terminated poly(dimethylsiloxane) (H-PDMS-H) and 5% poly(hydromethylsiloxane) (PHMS). The formulation of B2 contained 41% lignin, 50% H-PDMS-H and 8% PHMS. Both types were prepared at 90°C in the presence of B(C
6F
5)
3 (∼2050 ppm) as a catalyst.
The released gases, primarily H2 from -OH groups and CH
4
from Ar-OMe groups, can act as in situ blowing agents for foaming during molding. This metal-free, organo-catalyzed approach reduces lignin model compounds to phenols and primary alcohols by Lewis acid B (C
6F
5)
3 as
a selective catalyst by the reductive cleavage of β-O-4 and α-O-4 linkages via hydrosilylation (Feghali and Cantat 2014). Kai et al. (2016) found that the technical lignin in hybrid functional silicone elastomers is completely modi-fied and not only surface-modified. It should be empha-sized that lignin is degraded to small molecules carrying
Figure 9: Illustrations of lignin-silicone foam structures and their resulting mechanical properties.Tensile modulus increases with crosslinking density. Type A structures are foams either with large trapped (A1) or collapsed voids (A2, closer to an elastomer than a foam). Type A1 foams are easy to break with extension or bending due to their large defects. Type B structures are foams with uniform morphology; these foams are flexible with tunable toughness. Type C structures are foams with excessive crosslinking density and fewer voids. Type C1 foams are tough, while Type C2 are brittle. Adapted from Zhang et al. (2015), reprinted with permission from Green Chemistry.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4243

124 V. Mimini et al.: Lignin-based foams as insulation materials
silyl ethers (Figure 10). Silicone segments are responsible for the formation of longer chain segments that results in elastomer behavior, which means that silyl groups cross-link between each other and not – as initially supposed – under the participation of lignin fragments. This can be gathered from the optimized formulation (Type B), where a better foam network was obtained by an input of higher amounts of PHMS, H-PDMS-H or a mixture of H-PDMS-H with different molecular weights. However, the processing method by Brook et al. (2010) provides utilization of lignin in silicones. On the other hand, upscaling in industry will still be a challenge due to the high cost of silicones and low hydrolytic stability of the respective silane ethers.
Lignin-furan foams
Based on the previous findings of tannin-based foams (Tondi and Pizzi 2009), Tondi et al. (2016) used spent liquor (SL) from a magnesium bisulfite pulping process for lignin foams manufactured with 24–37% LS participation. The influence of the components ratio and temperature during the production processes were studied in detail. In addition to SL, furfuryl alcohol served as a co-monomer
(19–30%), ethanol as a blowing agent (5–9%) and sulfu-ric acid as a catalyst (4–8%). Wood particles and additives such as LS powder, PVA and mimosa tannins (5–13%) were added for adjusting viscosity. A higher ratio of the co-monomer to lignin in the formulation reduces the vis-cosity and, as a result, increases the crosslinking degree with shorter hardening times. On the one hand, a rise in the catalyst amount or temperature led to a shorter hard-ening time as well and, on the other hand, caused an inhomogeneous cell distribution. The additives increased the viscosity accompanied with a slower hardening time and homogeneous cell dimensions and distribution, and also formed a denser product (Figure 11). The increase in density was attributed to additives, such as wood particles (Tondi et al. 2016) or inorganic compounds, which do not participate chemically in the actual foam formation.
The applicability of SL without any purification is a big advantage, especially in terms of production costs. However, except for the lignin function, the role of other accompanying compounds in the SL such as sugars and particularly inorganic residues during the foaming pro-cesses is not yet understood. They could have positive and negative effects. Some of the sugars, like xylose or xylose-based oligomers/polymers, convert under strongly acidic
Figure 10: Reductive cleavage of lignin via hydrosilylation.Adapted from Zhang et al. (2015), reprinted with permission from Green Chemistry.
Figure 11: Visual appearance of the foams produced with (a) and without (b) wood particles.Adapted and reprinted with permission from Tondi et al. (2016).
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4244

V. Mimini et al.: Lignin-based foams as insulation materials 125
conditions to furfural, which in turn acts as a crosslink-ing agent forming lignin-furfural condensation products in situ (Dongre et al. 2015). Future research should clarify the foam-scaffold development as well as the true role of the residual carbohydrates and added furfuryl alcohol.
Tannin-lignin foams
Tannin foams have been intensely explored due to their high chemical stability, high flame resistance and low thermal conductivity (Tondi and Pizzi 2009; Sanchez-Martin et al. 2013; Szczurek et al. 2014). These bio-based foams are produced by a well-known and easily imple-mented mechanical frothing process that is inter alia used for PU foam preparation (Barron and Dunlap 1974; Gribble et al. 2004). Nonetheless, the high costs and low availability of raw tannin materials limit its industrial-scale utilization. A realistic alternative for petroleum-based insulation foams could be the combination of tannins with other biomaterials such as lignin. Merle et al. (2016a,b) presented bio-based, microporous and polymeric materials prepared from hydrolyzable and con-densed tannins (from oak/chestnut and mimose, respec-tively) with LS. The foams were produced mechanically by aerating the mixture in the presence of Tween 80 as a surfactant, a crosslinker (hexamine) and – depending on the formulation – a hardener (glyoxal). The obtained solid, self-standing and open-cell foams showed morpho-logical behavior close to phenolic resin and PU foams. Investigations, concerning the reactions of different main components with each other or their possible role as fillers in the formulations, revealed that (except for the mimosa tannin) the other building blocks (LS and two hydrolyzable tannins) do not react with hexamine. It was
observed that the formulations consisting of the mimosa tannin and lignin or the combination thereof are stable and do not crack after curing. The lignins associated with hydrolyzable tannins gave solid foams. The addi-tion of condensed tannins increased the compression modulus, providing high-resistance foams. As shown in Table 1, Merle et al. (2016a,b) observed better results in the presence of glyoxal together with mixtures of mimosa tannins, chestnut and sulfite SL.
However, in a post-study of the same group, it was highlighted that, actually, an increase in the amount of glyoxal does not really increase the mechanical proper-ties (Merle et al. 2016a,b). Moreover, its amount should be minimized due to economic and environmental reasons. A comparison of the optimized results with petroleum-derived products revealed a lower perfor-mance for tannin-lignin foams, especially in terms of density and thermal conductivity. The authors suggested that the surfactant concentration should be increased and the hexamine should be added progressively in a low total amount during the foaming process to reduce foam density.
Lignin for reinforcement in foamed
composites
One of the functions of native lignin in lignocellulosics is increasing the pressure resistance and hardness (Davis et al. 2016; Engelmann and Ganster 2016). This property is also helpful if technical lignins are to be used as a rein-forcement in foamed composites. Thakur et al. (2014) reviewed the performance of technical lignin in this field. The great potential of lignins as a reinforcement agent
Table 1: Formulation and mechanical properties of the different foams.
Sample Specimens
M1 M2 M3 M4
Mimosa (%) 15 15 13.5 13.5Oak (%) 15 0 13.5 0Chestnut (%) 0 15 0 13.5Lignosulfonate (%) 16.5 16.5 16 16H2O (%) 47 47 52.5 52.5Hexamine (%) 3 3 3 3Glyoxal (%) 2.5 2.5 0 0Tween 80 (%) 1 1 1.5 1.5Compressive modulus (MPa) 21.4 ± 0.6 22.9 ± 2.2 6.3 ± 1.4 3.5 ± 0.3Therm. conduct. (W · m−1 × K−1) 0.038 ± 0.0005 0.042 ± 0.001 0.035 ± 0.001 0.037 ± 0.001
Adapted from Merle et al. (2016a,b) and reprinted with permission from Elsevier.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4245

126 V. Mimini et al.: Lignin-based foams as insulation materials
in foam-based composites was confirmed in the context of lignin/phenol foams (Del Saz-Orozco et al. 2012) and lignin-reinforced soybean oil in PU foams (Luo et al. 2013). Del Saz-Orozco et al. (2012) prepared phenolic foams with lignin nanoparticles as reinforcement agents (up to 8.5%), n-pentane as a blowing agent and phenol-4-sulfonic acid as an acid catalyst. The incorporation of LS nanoparticles in phenolic foams led to lower density, and LS drastically improved the mechanical properties, such as compres-sive modulus and compressive strength, of the resulting composites.
Luo et al. (2013) studied the morphological, mechani-cal, thermal and density behavior of lignin-reinforced bio-foams. Different formulations of bio-foam (BioPU) pre-pared from soybean oil-based polyol and Jeffol A-630 were reinforced by lignin at concentrations of 10 and 15%. The composite densities and thermal stabilities increased as a function of the lignin content, and the mechanical
properties increased up to 10% lignin content. Higher lignin concentrations were detrimental and prevented a proper crystallization of urethane (Luo et al. 2013; Thakur et al. 2014). SEM images (Figure 12) show more uniform cell structures in composites with lignin than in a lignin-free bio-foam.
Lignin-based insulation materials
with modified lignins
Technical lignins can be utilized in composites without and with previous modification but it can only be incorpo-rated in limited percentages into foams, mainly because of structural incompatibility. Technical lignins have a broad molar mass distribution (Fengel and Wegener 1984; Balakshin et al. 2009) and the internal functional groups
Figure 12: SEM images of neat bio-foam and bio-foam composites (100 ×; scale bar, 1 mm): (a) BioPULignin0, (b) BioPULignin5, (c) BioPULignin10 and (d) BioPULignin15 the digits in the sample names giving the percentage of added lignin.Adapted from Luo et al. (2013), reprinted with permission from Industrial Crops and Products.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4246

V. Mimini et al.: Lignin-based foams as insulation materials 127
of large molecules are not accessible due to steric hin-drance, which limits the utilization possibilities (Obaid et al. 2016). Technical lignins show molar mass-depend-ent reaction profiles, and there is a relationship between structure-property and application (Zinovyev et al. 2017). Chemical modification is one of the remedies. Laurichesse and Avérous (2014) classified the most significant chemi-cal modifications into three categories: (1) fragmentation, (2) introduction of new chemical active sites and (3) func-tionalization of OH groups.
Lignin fragmentation can be effectuated via thermo-chemical, biochemical or chemical processes, such as by pyrolysis (thermolysis), chemical or enzymatic oxidation, hydrogenolysis, gasification, hydrolysis and microbial conversion (Amen-Chen et al. 2011; Pandey and Kim 2011; Laurichesse and Avérous 2014; Negrao et al. 2015). An interesting approach is the fragmentation through a mul-tifunctional catalyst, such as Fe
3O
4@Nb
2O
5@Co@Re with
different metal loadings between 2 and 7% (Opris et al. 2017). Magnetic nanoparticles lead to a selective hydrog-enolysis of the C-O bonds and hydrocracking of the C-C bonds. Molecular fragments between 200 and 1000 Da were obtained, while fragments with a molecular weight between 400 and 600 Da were water-soluble. Low mole-cular weight fragments are highly branched and are more
processable toward foams than linear polymers. The cata-lyst can be recovered and reused without loss of activity or selectivity. However, the high price of the catalyst and difficulties in the fractionation have hitherto impeded a large-scale utilization.
Technical lignins can be modified by the introduction of chemically active sites via hydroxyalkylation, amination or nitration (Figure 13). The additional functional groups do not impede the reactivity of the native ones. The hydroxy-alkylation is the best investigated approach mainly for the synthesis of PF resins. It increases accessibility and availa-bility of reactive hydroxyl groups. These lignopolyols are of great interest in isocyanate chemistry for PU foam produc-tion as well as in crosslinking with poly amines (Huo et al. 2012). Nitrated lignin was used in the production of inter-penetrating polymer networks (Huang and Zhang 2002). The reduction of nitrolignin leads to aminated structures, which can further react, e.g. with polyglycidyl ethers, as a crosslinking agent (Czaplicki et al. 2004) resulting in foam products based on epoxy and amine components. The phenolic hydroxyl (OH
phen) groups are the most reac-
tive ones and there are several pathways to modify them toward a multifunctional lignin macromolecule. Lignin modification can be effectuated by esterification, pheno-lation, hydroxypropylation, reactions with isocyanates,
Figure 13: Modification possibilities of lignin.Introduction of new functional groups into aromatic rings (green), functionalization of hydroxyl groups (red) and reactions that – depending on the conditions – occur at an aromatic or aliphatic hydroxyl group.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4247

128 V. Mimini et al.: Lignin-based foams as insulation materials
etherification (oxypropylation), alkylation and silylation (Laurichesse and Avérous 2014; Kai et al. 2016). Oxypro-pylation, as the most frequently applied method, lique-fies technical lignin and increases its solubility in organic solvents. Such lignopolyols are the starting products for PU foams (Nadji et al. 2005; Cateto et al. 2013). The origi-nal phenolic OH groups of lignins can be “extended” and simultaneously converted to diols, thereby doubling the number of hydroxyl groups and their reactivity.
Demethylation replaces the methyl group of methoxy-substituted aromatic rings with hydrogen (Chung and Washburn 2012). This can be done under alkaline condi-tions and in molten sulfur, in which two methyl groups from lignin are cleaved off as dimethylsulfide, which is further oxidized to dimethyl sulfoxide. Demethylation can also be performed with hydroiodic or hydrobromic acid. Depending on the reaction and the reaction conditions, the amount of phenolic and aliphatic OH groups can be increased up to 28%.
Conclusions
Although technical lignin has a high potential to replace petroleum-derived insulation materials, there are only a few foam composites so far that include technical lignins. Moreover, the quantity of lignins incorporated in such foams is less than 37%. The industrialization and bulk uti-lization of lignin in foam formations as insulation mate-rials is not yet feasible based on the currently available chemical formulations and technological approaches. This is mainly due to technical lignin’s intrinsic structure, high complexity, low reactivity and the steric hindrance of their reactive functional groups. The organic and inor-ganic impurities in industrial lignins also deteriorate their industrial application. Pretreatment steps for purification, drying and multi-step synthesis are costly. Lignin frag-mentation to defined molar mass fractions would facilitate economically viable foam production. Chemical modi-fication of lignins by means of eco-friendly and low-cost compounds (e.g. polyols), can increase the accessibility of their functional groups and would act simultaneously as crosslinkers that provide mechanically stabile foams.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.Research funding: Financial support to WOOD Kplus was provided by the Austrian government, the provinces of lower Austria, upper Austria, and Carinthia as well as by Lenzing AG. We also express our gratitude to the
University of Natural Resources and Life Sciences, Vienna (BOKU University), the Johannes Kepler University, Linz, and Lenzing AG for their in-kind contributions.Employment or leadership: None declared.Honorarium: None declared.Conflict of interest statement: No conflicts of interest declared.
References
Agarwal, K., Prasad, M., Sharma, R., Setua, D.K. (2014) Novel bio-degradable lignin reinforced NBR composites. Int. J. Energy Eng. 4:47–62.
Álvarez-Láinez, M., Rodríguez-Pérez, M.A., de Saja, J.A. (2014) Acoustic absorption coefficient of open-cell polyolefin-based foams. Mater. Lett. 121:26–30.
Amen-Chen, C., Pakdel, H., Roy, C. (2001) Production of monomeric phenols by thermochemical conversion of biomass: a review. Bioresour. Technol. 79:277–299.
Asdrubali, F., D’Alessandro, F., Schiavoni, S. (2015) A review of unconventional sustainable building insulation materials. Sustain. Mater. Technol. 4:1–17.
Balakshin, M.Y., Capanema, E.A., Chang, H.-M. (2009) Recent advances in the isolation and analysis of lignins and lignin–carbohydrate complexes. In: Characterization of lignocellulosic materials. Ed. Hu, T.Q. Blackwell Publishing Ltd., Oxford. pp. 148–166.
Barron, B., Dunlap, J. (1974) Air frothed polyurethane foams. US3821130A.
Barzegari, M.R., Alemdar, A., Zhang, Y., Rodrigue, D. (2012) Mechan-ical and rheological behavior of highly filled polystyrene with lignin. Polym. Compos. 33:353–361.
Belgacem, M.N., Blayo, A., Gandini, A. (2003) Organosolv lignin as a filler in inks, varnishes and paints. Ind. Crops Prod. 18:145–153.
Bernardini, J., Anguillesi, I., Coltelli, M.-B., Cinelli, P., Lazzeri, A. (2015) Optimizing the lignin based synthesis of flexible polyu-rethane foams employing reactive liquefying agents. Polym. Int. 64:1235–1244.
Bohnke, L., Beaujean, J., Albach, R., Li, J., Ling, S. (2014) Rigid polyurethane foams with high acoustic absorption. US20160046758A1.
Brook, M.A., Grande, J.B., Ganachaud, F. (2010) New synthetic strate-gies for structured silicones using B(C6F5)3. In: Silicon Polymers. Ed: Muzafarov, A.M. Springer, Berlin, Heidelberg. pp. 161–183.
Brown, R.C., Brown, T.R. Biorenewable Resources: Engineering New Products from Agriculture. John Wiley & Sons, Inc., New Jersey. 2014.
Cateto, C.A., Barreiro, M.F., Rodrigues, A.E., Belgacem, M.N. (2011) Kinetic study of the formation of lignin-based polyurethanes in bulk. Reactive Func. Polym. 71:863–869.
Cateto, C.A., Barreiro, M.F., Ottati, C., Lopretti, M., Rodrigues, A.E., Belgacem, M.N. (2013) Lignin-based rigid polyurethane foams with improved biodegradation. J. Cell. Plast. 50:81–95.
Chen, R., Kokta, B.V., Valade, J.L. (1979) Graft copolymerization of lignosulfonate and styrene. J. Appl. Polym. Sci. 24: 1609–1618.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4248

V. Mimini et al.: Lignin-based foams as insulation materials 129
Chung, H., Washburn, N.R. (2012) Improved lignin polyurethane properties with Lewis acid treatment. ACS Appl. Mater. Inter-faces 4:2840–2846.
Cinelli, P., Anguillesi, I., Lazzeri, A. (2013) Green synthesis of flex-ible polyurethane foams from liquefied lignin. Europ. Polym. J. 49:1174–1184.
Czaplicki, M., Kosal, D., Madaus, K. (2004) Two component (epoxy/amine) structural foam-in-place material. US20050020703A1.
Davis, K., Rover, M., Brown, R., Bai, X., Wen, Z., Jarboe, L. (2016) Recovery and utilization of lignin monomers as part of the biorefinery approach. Energies 9:808.
Del Saz-Orozco, B., Oliet, M., Alonso, M.V., Rojo, E., Rodríguez, F. (2012) Formulation optimization of unreinforced and lignin nanoparticle-reinforced phenolic foams using an analysis of variance approach. Compos. Sci. Technol. 72:667–674.
Dongre, P., Driscoll, M., Amidon, T., Bujanovic, B. (2015) Lignin-furfural based adhesives. Energies 8:7897–7914.
Engelmann, W.H. (1978) Foam injection leaching process for frag-mented ore. US4080419A.
Engelmann, G., Ganster, J. (2016) Lignin reinforcement in thermo-sets composites. In: Lignin in Polymer Composites. Eds. Faruk, O., Sain, M. William Andrew Publishing, Norwich, NY. pp. 119–151.
Feghali, E., Cantat, T. (2014) Unprecedented organocatalytic reduc-tion of lignin model compounds to phenols and primary alco-hols using hydrosilanes. Chem. Commun. (Camb) 50:862–5.
Fengel, D., Wegener, G. Wood: Chemistry, Ultrastructure, Reactions. De Gruyter, Berlin, New York, 1984.
Fisher, A.B., Fong, S.S. (2014) Lignin biodegradation and industrial implications. AIMS Bioeng. 1:92–112.
Frisch, K.C. (2006) History of science and technology of polymeric foams. J. Macromol. Sci. A: Chemistry 15:1089–1112.
Ghorbani, M., Liebner, F., Van Herwijnen, H.W.G., Pfungen, L., Krahofer, M., Budjav, E., Konnerth, J. (2016) Lignin phenol formaldehyde resoles: the impact of lignin type on adhesive properties. BioResources 11:6727–6741.
Ghorbani, M., Konnerth, J., Budjav, E., Silva, A., Zinovyev, G., van Herwijnen, H., Edler, M., Griesser, T., Liebner, F. (2017) Ammox-idized fenton-activated pine kraft lignin accelerates synthesis and curing of resole resins. J. Polym. 9:43.
Gribble, M.Y., Kennedy, J.G., White, D.P., Van Bellegem, P.C.J.M., Autenrieth, R.E. (2004) Process for applying polyurethane dispersion-based foam to article. US 20040109992A1.
Han, X., Shen, J., Huang, H., Tomasko, D.L., Lee, L.J. (2007) CO2 foaming based on polystyrene/poly(methyl methacrylate) blend and nanoclay. Polym. Eng. Sci. 47:103–111.
Hingmann, R. (2017) Thermoplastic foams: an exciting story of versatile cellular materials for multifaceted applications and markets. SPE Foams 2017, Conference on Advances in Foam Materials & Technology. Bayreuth, Germany.
Hu, L.H., Zhou, Y.H., Zhang, M., Liu, R.J. (2012) Characterization and properties of a lignosulfonate-based phenolic foam. BioResources 7:554–564.
Huang, J., Zhang, L. (2002) Structure and properties of regenerated cellulose films coated with polyurethane-nitrolignin graft-IPNs coating. J. Appl. Polym. Sci. 86:1799–1806.
Huo, S.P., Nie, M.C., Kong, Z.W., Wu, G.M., Chen, J. (2012) Crosslink-ing kinetics of the formation of lignin-aminated polyol-based polyurethane foam. J. Appl. Polym. Sci. 125:152–157.
Hwa, C.C.L., McNeil, D.W. (1964) Method for leaching a polyurethane foam. US3125541A.
Ikem, V.O., Menner, A., Bismarck, A. (2010) High-porosity macropo-rous polymers synthesized from titania-particle-stabilized medium and high internal phase emulsions. Langmuir 26:8836–8841.
Jeong, H., Park, J., Kim, S., Lee, J., Ahn, N. (2013) Compressive viscoelastic properties of softwood kraft lignin-based flexible polyurethane foams. Fibers Polym. 14:1301–1310.
Kai, D., Tan, M.J., Chee, P.L., Chua, Y.K., Yap, Y.L., Loh, X.J. (2016) Towards lignin-based functional materials in a sustainable world. Green Chem. 18:1175–1200.
Kim, D.J., Kim, S.W., Kang, K.J., Seo, K.H. (2001) Foaming of aliphatic polyester using chemical blowing agent. J. Appl. Polym. Sci. 81:2443–2454.
Kühnel, I., Podschun, J., Saake, B., Lehnen, R. (2015) Synthesis of lignin polyols via oxyalkylation with propylene carbonate. Holzforschung 69:531–538.
Kyoto Protocol to the United Nations Framework Convention on Climate Change, Kyoto (1997, in force 2005), Vol. 2303 United Nations, Treaty Series, p. 162.
Laurichesse, S., Avérous, L. (2014) Chemical modification of lignins: towards biobased polymers. Prog. Polym. Sci. 39:1266–1290.
Lee, S.T., Ramesh, N.S. Polymeric Foams: Mechanisms and Materi-als. CRC Press, New York, 2004.
Lee, L., Zeng, C., Cao, X., Han, X., Shen, J., Xu, G. (2005) Polymer nanocomposite foams. Compos. Sci. Technol. 65:2344–2363.
Lee, S.T., Park, C.B., Ramesh, N.S. Polymeric Foams: Science and Technology. CRC Press, Taylor and Francis Group, New York, 2006.
Lee, K.-Y., Wong, L.L.C., Blaker, J.J., Hodgkinson, J.M., Bismarck, A. (2011) Bio-based macroporous polymer nanocomposites made by mechanical frothing of acrylated epoxidised soybean oil. Green Chem. 13:3117.
Li, Y., Ragauskas, A.J. (2012) Kraft lignin-based rigid polyurethane foam. J. Wood Chem. Technol. 32:210–224.
Li, H., Zhang, Q., Gao, P., Wang, L.-L. (2015) Preparation and characterization of graft copolymer from dealkaline lignin and styrene. J. Appl. Polym. Sci. 132:41900.
Lisperguer, J., Núñez, C., Pérez-Guerrero, P. (2013) Structure and thermal properties of maleated lignin-recycled polystyrene composites. J. Chil. Chem. Soc. 58:1937–1940.
Luo, X., Mohanty, A., Misra, M. (2013) Lignin as a reactive reinforc-ing filler for water-blown rigid biofoam composites from soy oil-based polyurethane. Ind. Crops Prod. 47:13–19.
Mahmood, N., Yuan, Z., Schmidt, J., Tymchyshyn, M., Xu, C. (2016) Hydrolytic liquefaction of hydrolysis lignin for the prepara-tion of bio-based rigid polyurethane foam. Green Chem. 18:2385–2398.
Mannella, G.A., Conoscenti, G., Carfì Pavia, F., La Carrubba, V., Bru-cato, V. (2015) Preparation of polymeric foams with a pore size gradient via thermally induced phase separation (TIPS). Mater. Lett. 160:31–33.
Merle, J., Birot, M., Deleuze, H., Mitterer, C., Carré, H., Bouhtoury, F.C.-E. (2016a) New biobased foams from wood byproducts. Mater. Design 91:186–192.
Merle, J., Trinsoutrot, P., Charrier-El Bouhtoury, F. (2016b) Optimiza-tion of the formulation for the synthesis of bio-based foams. Eur. Polym. J. 84:577–588.
Montreal Protocol on Substances that Deplete the Ozone Layer, Montreal (1987, in force 1989), Vol. 1522 United Nations, Treaty Series, p. 3.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4249

130 V. Mimini et al.: Lignin-based foams as insulation materials
Mostafa, A., Abdolreza, O. (2015) Improving sound absorption of polyurethane foams by the incorporation of nano-particles. The 22th International Congress on Sound and Vibration, Florence, Italy.
Nadji, H., Bruzzèse, C., Belgacem, M.N., Benaboura, A., Gandini, A. (2005) Oxypropylation of lignins and preparation of rigid polyurethane foams from the ensuing polyols. Macromol. Mater. Eng. 290:1009–1016.
Negrao, D.R., Sain, M., Leao, A.L., Sameni, J., Jeng, R., de Jesus, J.P.F., Monteiro, R.T.R. (2015) Fragmentation of lignin from organosolv black liquor by white rot fungi. BioResources 10:1553–1573.
Obaid, N., Kortschot, M.T., Sain, M. (2016) Lignin-based foaming materials. In: Lignin in Polymer Composites. Eds. Faruk, O., Sain, M. William Andrew/Elsevier, Oxford, UK. pp. 217–232.
Ockenfels, H.-M., Uter, W., Lessmann, H., Schnuch, A., Geier, J. (2009) Patch testing with benzoyl peroxide: reaction profile and interpretation of positive patch test reactions. Contact Dermatitis 61:209–216.
Opris, C., Cojocaru, B., Gheorghe, N., Tudorache, M., Coman, S.M., Parvulescu, V.I., Duraki, B., Krumeich, F., van Bokhoven, J.A. (2017) Lignin fragmentation onto multifunctional Fe3O4@Nb2O5@Co@Re catalysts: the role of the composition and deposition route of rhenium. ACS Catal. 7:3257–3267.
Pan, X., Saddler, J.N. (2013) Effect of replacing polyol by organosolv and kraft lignin on the property and structure of rigid polyure-thane foam. Biotechnol. Biofuels 6:12.
Pandey, M.P., Kim, C.S. (2011) Lignin depolymerization and conversion: a review of thermochemical methods. Chem. Eng. Technol. 34:29–41.
Petchwattana, N., Covavisaruch, S. (2011) Influences of particle sizes and contents of chemical blowing agents on foaming wood plastic composites prepared from poly(vinyl chloride) and rice hull. Mater. Des. 32:2844–2850.
Phillips, R.B., Brown, W., Stannett, V. (1973) The graft copolym-erization of styrene and lignin. III. Chain transfer reactions of lignin and lignin model compounds. J. Appl. Polym. Sci. 17:443–451.
Rials, T.G., Glasser, W.G. (1986) Engineering plastics from lignin – XIII. Effect of lignin structure on polyurethane network forma-tion. Holzforschung 40:353–360.
Sanchez-Martin, J., Beltran-Heredia, J., Delgado-Regana, A., Rodri-guez-Gonzales, M.A., Rubio-Alonso, F. (2013) Optimization of tannin rigid foam as adsorbents for wastewater treatment. Ind. Crops Prod. 49:507–514.
Saraf, V.P., Glasser, W.G., Wilkes, G.L. (1985) Engineering plas-tics from lignin. VII. Structure property relationships of poly(butadiene glycol)-containing polyurethane networks. J. Appl. Polym. Sci. 30:3809–3823.
Sauceau, M., Fages, J., Common, A., Nikitine, C., Rodier, E. (2011) New challenges in polymer foaming: a review of extrusion processes assisted by supercritical carbon dioxide. Prog. Polym. Sci. 36:749–766.
Schidrowitz, P., Goldsbrough, H.A. (1915) British patent No. 1,111.Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A.,
Rouquerol, J., Siemieniewska, T. (1984) Reporting physisorp-
tion data for gas-solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 57:603–619.
Singh, S.N. Blowing Agents for Polyurethane Foams. Rapra Techno-logy Ltd., online, 2001.
Štěpek, J., Daoust, H. (1983) Chemical and physical blowing agents. In: Additives for Plastics. Springer New York, New York, pp. 112–123.
Stevens, E.S., Klamczynski, A., Glenn, G.M. (2010) Starch-lignin foams. Express Polym. Lett. 4:311–320.
Szabó, G., Romhányi, V., Kun, D., Renner, K., Pukánszky, B. (2017) Competitive interactions in aromatic polymer/lignosulfonate blends. ACS Sustain. Chem. Eng. 5:410–419.
Szczurek, A., Fierro, V., Pizzi, A., Stauber, M., Celzard, A. (2014) A new method for preparing tannin-based foams. Ind. Crops Prod. 54:40–53.
Thakur, V.K., Thakur, M.K., Raghavan, P., Kessler, M.R. (2014) Progress in green polymer composites from lignin for multi-functional applications: a review. ACS Sustain. Chem. Eng. 2:1072–1092.
Tondi, G., Pizzi, A. (2009) Tannin-based rigid foams: characteriza-tion and modification. Ind. Crops Prod. 29:356–363.
Tondi, G., Link, M., Kolbitsch, C., Gavino, J., Luckeneder, P., Petutschnigg, A., Herchl, R., Van Doorslaer, C. (2016) Lignin-based foams: production process and characterization. BioRe-sources 11:2972–2986.
Victor, P.A., Gonçalves, S.B., Machado, F. (2018) Styrene/lignin-based polymeric composites obtained through a sequential mass-suspension polymerization process. J. Polym. Environ. 26:1755–1774.
Vishtal, A., Kraslawski, A. (2011) Challenges in industrial applica-tions of technical lignins. BioResources 6:3547–3568.
Wong, J.C.H., Tervoort, E., Busato, S., Gonzenbach, U.T., Studart, A.R., Ermanni, P., Gauckler, L.J. (2009) Macroporous polymers from particle-stabilized foams. J. Mater. Chem. 19:5129.
Yoshida, H., Morck, R., Kringstad, K.P., Hatakeyama, H. (1987) Kraft lignin in polyurethanes I. Mechanical properties of polyu-rethanes from a kraft lignin-polyether triol-polymeric MDI System. J. Appl. Polym. Sci. 34:1187–1198.
Yoshida, H., Morck, R., Kringstad, K.P., Hatakeyama, H. (1990) Kraft lignin in polyurethanes II. Effects of the molecular weight of kraft lignin on the properties of polyurethanes from a kraft lignin-polyether triol-polymeric MDI system. J. Appl. Polym. Sci. 40:1819–1832.
Zhai, W., Leung, S.N., Wang, L., Naguib, H.E., Park, C.B. (2010) Preparation of microcellular poly(ethylene-co-octene) rubber foam with supercritical carbon dioxide. J. Appl. Polym. Sci. 116:1997–2004.
Zhang, J., Fleury, E., Brook, M.A. (2015) Foamed lignin–silicone bio-composites by extrusion and then compression molding. Green Chem. 17:4647–4656.
Zinovyev, G., Sumerskii, I., Korntner, P., Sulaeva, I., Rosenau, T., Potthast, A. (2017) Molar mass-dependent profiles of func-tional groups and carbohydrates in kraft lignin. J. Wood Chem. Technol. 37:171–183.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:4250

51
8.2 Compatibility of Kraft Lignin, Organosolv Lignin and Lignosulfonate
with PLA in 3D Printing (Publication II)

52

COMPATIBILITY OF KRAFT LIGNIN, ORGANOSOLV LIGNIN ANDLIGNOSULFONATE WITH PLA IN 3D PRINTING
Vebi Mimini,1,2� Eva Sykacek,3� Sharifah Nurul Ain Syed Hashim,1 Julian Holzweber,3
Hubert Hettegger,1 Karin Fackler,4 Antje Potthast,1 Norbert Mundigler,3 andThomas Rosenau1,5
1Department of Chemistry, Division of Chemistry of Renewable Resources, University ofNatural Resources and Life Sciences Vienna, Tulln, Austria2Wood Kplus – Kompetenzzentrum Holz GmbH, Linz, Austria3Department of Agrobiotechnology, Institute of Natural Materials Technology, University ofNatural Resources and Life Sciences Vienna, Tulln, Austria4Lenzing AG, Lenzing, Austria5Johan Gadolin Process Chemistry Centre, Abo Akademi University, Abo/Turku, Finland
The compatibility of three different types of lignin, such as Kraft lignin, Organosolv lignin,and lignosulfonate, with PLA, in particular without the addition of any further reagents orcompatibilizers, is investigated. The unmodified lignins were thoroughly characterized toestablish their interaction potential in lignin-PLA composite formation. The obtained dou-ble-extruded binary systems containing 5, 10, and 15wt % lignin in PLA were characterizedin terms of chemical conversion (31P NMR), thermal behavior (TGA, DSC), and cellularmorphology (SEM). Additionally, the flexural and impact properties as well as the melt vol-ume rate (MVR) of the extruded materials, in the form of 3D-printed bars, were analyzed,and the effect of lignin addition to PLA studied. The results revealed poor mechanical prop-erties for the Kraft lignin-PLA blends, but higher compatibility of Organosolv lignin-PLAblends. Lignosulfonate-PLA blends showed a promising behavior for 3D-printing. A specialnucleation effect, potentially useful in foaming applications, was observed in this study.
KEYWORDS. Bio-composite; extrusion; Kraft lignin; lignosulfonate; nucleation; Organosolvlignin; polylactic acid; 3D-printing
INTRODUCTION
To this day non-biodegradable materialscontribute to the progressive increase ofenvironmental pollution, which is one of thegreatest ecological issues that the world iscurrently facing. Permanent plastics, whichare mostly derived from petroleum sources,are used almost ubiquitously in packaging,automotive industry, transportation, construc-tion, etc. However, due to their fossil originthey are becoming more and more
unattractive and suspicious with regard tohealth hazards, such as for example plasticsused in children’s toys, or in medical applica-tions.[1–4] For the welfare of the environmentand our health – and even in approaches toimprove national security and rural economicdevelopment[5,6] – the use of biodegradablematerials or bioplastics obtained from renew-able sources is a sustainable alternative solu-tion.[7,8] The term bioplastic is used forpolymers which are produced from a bio-
Address correspondence to Thomas Rosenau, Department of Chemistry, Division of Chemistry of RenewableResources, University of Natural Resources and Life Sciences Vienna, Konrad-Lorenz-Strasze 24, 3430 Tulln, Austria.E-mail: [email protected]
�These authors contributed equally to this work.Color versions of one or more of the figures in the article can be found online at www.tandfonline.com/lwct.
14
Journal of Wood Chemistry and Technology, 39: 14–30
Copyright # 2019 Taylor & Francis Group, LLC
ISSN: 0277-3813 print/1532-2319 online
DOI: 10.1080/02773813.2018.1488875
53

logical resource as well as for biodegradableplastics which are either manufactured fromfossils or from biomass. Currently, various bio-plastics are commercially available which ful-fill these requirements, such as polylactic acid(PLA), polyhydroxybutyrate (PHB), or polybu-tylene succinate (PBS).[9] PLA, today thestandard example for successful bioplastics, isbiodegradable and biocompatible, and it isproduced via bacterial fermentation of carbo-hydrates such as starch extracted from corn,or sugars extracted from sugar beet, whey,molasses, etc. or chemical synthesis.[10,11]
Despite its obvious benefits, it has someinferior characteristics such as moisture sensi-tivity, low glass transition temperature, lowelongation at break, it cannot be widely usedas a single polymer in hot packaging or inapplications that need materials with certaindeformation properties.[11] The current highcosts of the polymer are the overwhelmingobstacle limiting its bulk utilization.
A very promising biomaterial for combin-ation with the above-mentioned biopolymers– especially with the aim of overcomingabove-mentioned disadvantages – is lignin. Asan industrial by-product, it has high potentialnot just due to its high availability with anproduction output of 70 million tons perannum on a global scale,[12] but much moreowing to its inherent structure containinghigh carbon content, aromatic rings and vari-ous functional groups available for functional-ization, such as hydroxyl, carbonyl, andcarboxyl, and in case of lignosulfonate, sul-fonic acid groups. Depending on the pulpingprocess, in which lignin is separated from thecellulose fibers and partly from hemicellulo-ses, it needs to be distinguished between sul-fur-containing Kraft lignin and lignosulfonateand non-sulfur processes such as Organosolvand soda lignin. Together with the woodtype, the pulping process is responsible forthe final structure of lignin. The more struc-turally homogenous it is, the more favorableand interesting it becomes for utilization innovel biomaterials. From a chemical point ofview, the polyester-type PLA contains highly
polar motifs that are available for interactionwith hydroxyl groups of lignin by hydrogenbonds and electrostatic interactions.[13]
Compared to non-polar polymers, the polarpolyesters are more reactive toward lig-nins.[14] Another advantage of a lignin-PLAcombination would be the economic aspect,reducing the high costs of the polyester whenused as a single component.
Much research has been done in lignin-PLA blending. In almost all cases, lignin wasmodified for greater compatibility, or thereaction between lignin and PLA was cata-lyzed.[15–17] However, from a chemical pointof view the reasons for the poor compatibilitybetween unmodified lignins and PLA withoutaddition of any further reagents were eithernot or only little illuminated.
The aim of this paper is to study the com-patibility of Kraft lignin (KL), Organosolv lignin(OSL), and lignosulfonate (LS) with PLA inabsence of any solvent, compatibilizers orother reagents added. Another aim of thisstudy is the examination of lignin-PLAextruded materials in 3D printing. Specialattention was paid to analyze the chemicalinteraction, thermal stability, and the mech-anical properties of the obtained PLA-ligninbiocomposites, finding out the optimal lignintype and establishing appropriate conditionsfor compounding it with PLA. These resultsare correlated with the chemical features oflignin in an attempt to establish structure –property relationships which must be seen asprerequisites to any new innovative PLA-lig-nin composite applications.
MATERIALS AND METHODS
Raw Materials
PLA (NatureWorks IngeoTM Biopolymer4043D) was purchased from RESINEX AustriaGmbH, Allhaming, Austria. Pine Kraft ligninIndulin ATTM was from Mead WestvacoCorp, Richmond, VA. Beech Organosolv lig-nin was purchased from Fraunhofer Centerfor Chemical Biotechnological Processes CBP,Leuna, Germany. Beech lignosulfonate was
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 15
54

obtained as liquor. Before its use it was sepa-rated and purified (see below).
Materials Preparation
Prior to extrusion the equilibrium watercontents of the lignins were determined usinga Mettler Toledo moisture analyzer HS153resulting in 4.53% water in the Kraft lignin, aswell as 3.39% and 4.52% in the Organosolvlignin and lignosulfonate, respectively. Thedifferent lignins were dried in order to pre-vent hydrolysis of the PLA during the extru-sion process. After 4 days drying at 85 �C thewater content of all lignins was decreasedbeneath 0.5%. Prior to thermal processing,the PLA was dried in a Heraeus circulatingair oven at 80 �C overnight. For the com-pounding with PLA concentrations of 5, 10,and 15wt % of each lignin type were chosen.Of each composition five batches of 200 gwere manually mixed and poured in sealableplastic bags to reduce water uptake dur-ing processing.
Isolation of Lignosulfonate byAdsorption onto XAD-7
While Kraft lignin and Organosolv ligninwere used as purchased, the lignosulfonatewas isolated and purified from respective sul-fite spent liquor (SSL) by XAD-7, an anionexchange resin with high absorption capacity.To obtain salt-free sulfonated lignin, the SSLwas first treated with 1 M aqueous hydro-chloric acid.[18] The pH of the protonatedSSL was adjusted below 1. Before the add-ition of protonated SSL to wet XAD-7 resin,the resin was washed by Soxhlet extractionwith ethanol and sequentially washed with0.1 M NaOH and 0.1 M HCl. The adsorptionof SSL onto XAD-7 resin was carried outovernight through shaking at a XAD-7/SSLratio of 2:1. Subsequently, the adsorbedlignosulfonate was washed sequentially withacidified and deionized water. The washingsteps were repeated 3 times to removeresidual sugar and inorganic contents. Theremaining adsorbate was treated 4 times withethanol by shaking for 1 h at 45 �C to desorb
the LSA from XAD-7 resin. The filtrates werecombined and evaporated under reducedpressure to quantitatively remove ethanol.The remainder was dissolved in deionizedwater, frozen overnight at �80 �C, andfreeze-dried. Subsequently, the isolated LSnow present as lignosulfonic acid (LSA)[19]
was dried in a vacuum drying oven at 40 �Cuntil it reached constant weight.
Extrusion
For better miscibility, lignin-PLA blendswere double-extruded, i.e. the lignins werecompounded with PLA, and the obtained fil-aments were cut to granules and extrudedagain. Compounding and filament extrusionwere conducted by means of counter rotat-ing, parallel extruder Collin ZK 25 (Dr. CollinGmbH, Ebersberg, Germany). The screw hasa diameter of 25mm, an aspect ratio of 18and was equipped with one smear head.During compounding the barrel temperatureswere adjusted to 160 �C at the entry, 175and 170 �C at the subsequent heating zonesand 165 �C at the die. The single batches ofthe mixtures were filled in the hopper toachieve highest charging of the screw whichwas maintained at 50 rpm in all trials. Theresulting strands of approximately 3.5mmdiameter were cut to granules by use of aRieter Primo 100S blade granulator. Prior tofilament extrusion the granules including neatPLA4043D as reference were dried at 80 �Covernight. Lower barrel temperatures of150 �C (feeding zone), 165 �C and 150 �Cwere used in filament extrusion to increasemelt pressure and hence shaping of the2.8mm strands. The hopper was maintainedin full state and a screw speed of 30 rpm wasused. The filaments were cooled with pres-surized air.
3 D-Printing Tests
The test bars with dimensions of4� 10�80mm3 were printed with therespective extruded materials by use of aReprap X400 3D-printer (RepRap GmbH,Feldkirchen, Germany) which was equipped
16 MIMINI ET AL.
55

with a strand die of 0.3mm. The print bedwas tempered at 60 �C and the extruder tem-perature was maintained at 210 �C for allcompositions including the PLA reference.
CHARACTERIZATION
Molecular Weight and MolecularWeight Distribution: GPC
The molecular weight and molecularweight distribution analyses were performedaccording to the literature[20] by using anUltiMateV
R
3000 Standard LC System,equipped with Kontron 420 HPLC pump andpulse damper, in which 280 nm UV andShodex RI-101 detectors were used. ThreePL GPC column of 300� 7.5mm were cali-brated by polystyrene sulfonates as polymerstandards of known molecular mass.Approximately 10mg of the lignin samplewere dissolved in DMSO/LiBr (0.5% w/v),allowed to shake overnight, and filtered using0.45 mm PHENEX PTFE filters prior to injec-tion. Chromatography parameters: flow rate:0.5mL/min�1, injection volume: 10 mL, col-umn temperature: 40 �C. Data were eval-uated using Chromeleon 6.8 software.
Methoxy Group Determination:Headspace GC-MS
The methoxy group determination wasexecuted according to Sumerskii et al.[21] Inshort: the respective lignin sample (�5mg)and the internal standard 4-methoxybenzoicacid-d3 (MBA, �5mg) were added to a head-space vial provided with a magnetic stirringbar. Aqueous hydroiodic acid (1mL, 57%)was added to the mixture and the vial wasclosed with a cap and heated at 130 �Cunder stirring. After 1 h the sample wasallowed to cool to room temperature. ExcessHI was neutralized. Headspace GC-MS analy-ses were performed on an Agilent 5890N gaschromatograph (DB-5ms column30 m� 0.25mm) coupled with an Agilent5975B inert XL mass selective detector(MSD). Helium was used as a carrier gas witha constant column flow of 1mL/min. The
method was calibrated by responses frommethyl iodide and deuterated methyl iodideobtained from vanillin and known amounts ofthe internal standard MBA.
Free Sugar and HemicelluloseDetermination: Methanolysis GC-MS
The free sugar and hemicellulose contentswere determined by a modified acidic meth-anolysis protocol.[22] Ten milligrams of samplewere soaked in 2mL of 2 M acidic methanoland mixed with a vortex mixer for 2min. Thesoaked samples were ultrasonicated for2min, heated in an oven (100 �C, 5 h), andcooled to room temperature. One hundredmicroliters of anhydrous pyridine and sorbitolstandard solutions were added, vortexed, andevaporated under a nitrogen stream. Theevaporated samples were freeze-dried over-night. Derivatization started with the additionof 100 mL anhydrous pyridine to the freeze-dried samples. 4-Dimethylaminopyridine(DMAP/pyridine 1.5mg/mL) and N,O-bis(tri-methylsilyl)trifluoroacetamide (BSTFA with10% trimethylsilyl chloride) were added tothe samples prior to heating at 70 �C for 2 h.Ethyl acetate was added prior to analysis withGC-FID (Agilent Technologies model 7890B).Parameters: sample injection volume: 1 mL,split ratio of 10:1. A HP1 (Agilent 19091Z-413) methyl siloxane column(30 m� 320 mm� 0.25 mm) using He as acarrier gas at a flow rate of 2mL/min wasused for analysis. The oven temperature wasprogramed at 140 �C for 1min, ramped at4 �C/min to 210 �C and then heated at 30 �C/min to 260 �C with a hold time of 5min. Thetemperatures of the injector and detectorwere maintained at 260 �C and 280 �C,respectively. The FID temperature was fixedat 320 �C with a He flow rate of 30mL/min.
Sulfonic Acid Group Determination
Sulfonic acid group contents of isolatedlignosulfonate was determined through con-ductometric titration.[19] The results werecomparable with those of calculated datafrom elemental analysis.
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 17
56

Hydroxy Group Determination:31P-NMR
Hydroxy groups in lignins were analyzedaccording to Korntner et al.[23] The samplesand internal standard N-hydroxy-5-norbor-nene-2,3-dicarboxylic acid imide were dis-solved in 800 mL of a mixture of chloroform/pyridine (1:1.6 v/v) and allowed to shakeovernight. Approximately 100 mL of relaxationreagent (see below) and 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane wereadded to the dissolved samples which wereshaken for 1 h. The relaxation reagent wasprepared by adding 5mg of chromium(III)acetylacetonate to 1mL of a chloroform/pyri-dine (v/v: 1:1.6) mixture.
Thermogravimetric Analysis: TGA
Thermal analyses were performed using aNETZSCH TG 209F1 instrument. The sampleswere heated from 25 �C up to 900 �C undernitrogen atmosphere at a 10 K/min heatingrate. The temperature was held at 80 �C for10min to remove any possible moisture.Approximately 5–12mg of each sample wereweighed into an alumina ceramic crucible,with a thermal stability up to 2000 �C,and then placed onto the balance of theTGA instrument. Before use the equipmenthad undergone three times nitrogengas evacuation.
Differential Scanning Calorimetry: DSC
DSC was performed using a NETZSCHDSC 200F3 instrument. The samples wereheated from 25 �C up to 250 �C under nitro-gen atmosphere at a 10 K/min heating rate.After heating, the samples were cooled downto 25 �C with liquid nitrogen at a rate of 2 K/min. Approximately 5–12mg of each samplewere weighed in the alumina pierced lid panand then placed onto the balance of the DSCequipment. Both TGA and DSC data wereplotted using NETZSCH Proteus 6.1 ThermalAnalysis software.
Scanning Electron Microscopy (SEM)
Double-extruded samples were studiedwith SEM on a FEI INSPECT S50 instrument(Hillsboro, OR). Gold was used in a Leica EMSCD005 sputter coater for sputtering of thesamples (8 nm layer thickness).
Determination of Melt Volume Rateand Mechanical Tests
Melt volume rates (MVR) of the compos-ite granules and the neat PLA reference werecarried out according to ISO 1133 (190 �C,2.16 kg) on a Instron Ceast MF20 melt flowtester. Flexural tests were carried out after14 days storage of the printed test bars in anorm climate (23 �C, 50% r.h.) according toISO 178 on a Frank universal testingmachine 81816.
Data Analyses
Spearman’s rank correlations were calcu-lated with the program Statgraphics Centurionto find possible relationships betweenmolecular weight properties of the differentlignins, filler concentration, and mechanicalproperties of the lignin-PLA composites.
RESULTS AND DISCUSSION
Chemical Analysis of Neat Compoundsand Lignin-PLA Composites
In order to assess purity grade and poten-tial reactivity of lignins, various pre-character-ization techniques were performed. Sugarconcentration, differences in molecularweight (MW), and functional groups, such asmethoxyl-, carboxyl-, carbonyl-, and sulfonicacid groups as in the case of LS, are signifi-cant parameters for data interpretation interms of potential chemical interaction andreaction selectivity, lignin modification andfurther processing. Initial analysis was con-ducted by elemental analysis (see Table 1).
LS shows a 15% lower carbon contentcompared to OSL and KL. Taking into consid-eration the percentage amount of all meas-ured elements (C, H, N, S), the proportion ofoxygen in LS is �12% higher than in OSL
18 MIMINI ET AL.
57

and KL. Together with sulfur, which indicatesthe type of pulping process, the high amountof oxygen and sulfur is due to the sulfonicacid groups, which are typical for LS. Thefree sugar and hemicellulose content, asmeasured by methanolysis GC-MS accordingto Sundberg et al.,[22] is lower for the isolatedLS compared to KL and OSL. This is causedby the efficient XAD-7 isolation method usedfor LS purification.[18] In general, lower sugarcontent is a characteristic of more homoge-neous lignins. The determination of molecularweight and molecular weight distributionthrough GPC[24] revealed higher mean valuesfor KL and LS. By contrast, OSL containsmore methoxy groups than LS and KL. LowerMW allows for higher structural flexibilityaccompanied with better functional groupaccessibility.
For the evaluation of the interactionintensity of lignins with PLA upon extrusion,the lignin-PLA copolymers were phosphity-lated. The 31P NMR results were comparedto those of the phosphitylated neat com-pounds (Table 2 and Figure 1). To ensure acorrect comparison, the content of granulesfor the 31 P NMR measurement was adapted,so that the content of lignin is the same in all
samples (e.g. to set 6mg of lignin in all sam-ples, 40mg of 15wt % and 120mg of 5wt %composite were required). For accurate spec-tra interpretation, the integration approach byBalakshin and Capanema[25] was used.
The phosphite esters of aliphatic(151.2–145 ppm), aromatic (syringyl, guaiacyl,and p-hydroxyphenyl, 145–136.7 ppm) aswell as carboxylic hydroxyl groups(135.7–134.6 ppm) of KL and OSL are shownin Figure 1. Not readily soluble LS-PLA blendsshowed only weak NMR signals and are notpresented. The resonances at 146.7 and135.7 ppm are assigned to PLA and134.4 ppm to CDCl3, respectively. FromTable 2, it is clear that the total amount of–OH groups is higher in KL than in OSL(6.07mmol/g and 5.09mmol/g, respectively).After compounding, the 5wt % lignin loadedKL/OSL-PLA blends reveal in both casesapproximately a decrease in –OH groups bya factor of 10. The absence of any OHgroups at 5wt % lignin loading (except theminor signal for guaiacyl in case of 5wt % lig-nin KL-PLA, see Figure 1) and the presenceof PLA signals at 146.7 ppm indicate thatmost –OH groups of both lignin types havereacted. At 5 and 10wt % KL lignin loading,
TABLE 1. Characteristics of the used lignins.a
Lignin type C (%) H (%) N (%) S (%) Sugar (mg/mg) Mw (g/mol) Dispersity –OMe (%) –SO3H (%)
LS 44.8 5.7 0.5 4.6 11.0 3994 4.65 17 11OSL 61.3 6.28 0.29 <0.02 19.1 1249 2.80 21 –KL 59.9 5.5 1.2 1.7 18.5 3889 6.54 13 –
aKL: Kraft lignin; OSL: Organosolv lignin; and LS: lignosulfonate.
TABLE 2. Hydroxyl group content of the starting components and the obtained composites.a
Lignin type Aliphatic –OH, (mmol/g) Aromatic –OH, (mmol/g) Carboxylic –OH, (mmol/g) Total�OH, (mmol/g)
Organosolv lignin 1.56 3.42 0.11 5.09OSL-PLA: 5wt % lignin 0.19 0.32 – 0.51OSL-PLA: 10wt % lignin 0.60 2.32 – 2.92OSL-PLA: 15wt % lignin 0.77 3.14 – 3.91Kraft lignin 2.17 3.46 0.44 6.07KL-PLA: 5wt % lignin 0.19 0.46 – 0.65KL-PLA: 10wt % lignin 0.45 0.72 – 1.17KL-PLA: 15wt % lignin 0.66 1.58 – 2.24
aComparison of Kraft lignin (KL), Organosolv lignin (OSL) and their copolymers. Due to the low solubility of LS in the CDCl3/pyridinesolvent system, data from LS-PLA composites are not given.
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 19
58

the syringyl –OH groups in the KL-PLA com-posites reacted completely. At higher lignincontents, the total –OH groups increasedproportionally with lignin loading. The situ-ation is different for OSL blends, for whichlignin-PLA compatibility is reached at 10wt %lignin. These mixtures showed more than 2times higher –OH group contents comparedto that of the KL blends for the same loading(OSL-PLA 10wt % lignin =2.92mmol/g; KL-PLA 10wt % lignin =1.17mmol/g).
These differences are attributed to thechemical composition of KL and OSL. NeatKL contained more total –OH groups com-pared to neat OSL and reacted better withPLA (Table 2). As shown in Scheme 1 sche-matically, the terminal COOH group of PLAcan react with phenolic and aliphatic alcoholgroups of unmodified lignins. Chemical bond-ing occurs during the extrusion process, inwhich under high pressure and temperature(10 bars, 160–175 �C) non-catalyzed esterifi-cation between lignin and PLA can takeplace.[26] On the other hand, also free radicalgeneration may occur that can effect covalentbonding due to grafting-type reactions.[27] It is
known that lignin acts as a free radical scav-enger due to its aromatic structure and multi-functional side groups,[28] thus stabilizing thesolvent-free system. It must also be consideredthat the water, which is released during theinitial condensation reaction between PLAand lignin and other elimination processes, ishighly reactive under the extrusion conditionsand will readily effect hydrolysis of PLA, gen-erating new anchor groups for PLA-ligninbonding and thus leading to changes in themolecular weight fractions of lignin-PLA com-posites. Nevertheless, from 31 P NMR spectrait is difficult to verify such dynamic chemicalchanges, while the fact that a reactionoccurred between the PLA and lignin compo-nents can be unambiguously stated from thelowered amounts of free hydroxyl groups. Ingeneral, high molecular weight and high poly-dispersity lignins are accompanied with lowerflexibility and lower internal accessibility,while more highly branched lignins, such asKL, provide broader surface area with highfunctional group accessibility (KL = 6.54, OSL= 2.8). Additionally, the high content ofmethoxy groups of OSL (21%; 7% more than
PLA
OSL
KL
a)
b)
c)
a)
b)
c)
Int. Std. Aliph. 3 and 5-subst. Gnc H COOH
135140145150 [ppm]
FIGURE 1. 31P NMR spectra of polylactic acid (PLA, green line); Kraft lignin (KL, black line); Organosolv lignin (OSL, red line). (a–c)The lignin-PLA copolymers with 5, 10, and 15wt % lignin loading. The range of ‘3- and 5-substituted’ groups contains syringyl andcondensed guaiacyl units. Gnc: guaiacyl non-condensed units; H: p-hydroxyphenyl units; COOH: carboxyl units. For betterillustration the intensity of PLA spectra was 5 times increased.
20 MIMINI ET AL.
59

KL) might generates repulsive interactions dueto slight sterical hindrance toward PLA chainswhich can result in an overall distancing oflignin from PLA.
Thermal Behavior of Lignin-PLA Composites
The high thermal stability of lignin makesit of high interest for composites because it isexpected to enhance their net ther-mal properties.
The intermolecular interactions of the 3Dlignin structure, such as hydrogen bonding,electrostatic and p-p interactions, with suit-able co-components create a strong networkand lower flexibility than in the individualcomponents, resulting in higher thermalresistance. In addition to intra- and intermo-lecular interactions, the degree of covalentcrosslinking and the molecular weight of lig-nin are important parameters influencing thethermal behavior, e.g. glass transition tem-perature Tg.
As shown in Figure 2, the lignins ther-mally decomposed over a broad temperaturerange and they have more than one decom-position step. While PLA starts to quantita-tively decompose at around 300 �C, the
lignins start to lose weight earlier(110–170 �C), with OSL being the most ther-moresistant lignin at this early stage (170 �C).The multistep decomposition occurs due tothe structural heterogeneity of lignins – thevarious functional groups and structural ele-ments start to cleave off at different tempera-tures reducing the total mass.[29,30]
Up to 400 �C, the weight of PLAdecreases by 99%; that of OSL and LS by35% and that of KL by 26%. Before inductionof oxygen at 700 �C, OSL and LS lose 83%and 88% of their weight, respectively. KL ismore stable at this temperature with less than69% weight loss. These results are in accord-ance with M€uller-Hagedorn et al.[31] whostated that coniferous lignin (Figure 1,138.7–140.7 ppm) is thermally more stablethan hardwood lignin (Figure1, 140.7–145 ppm).
The thermograms of lignin-PLA compo-sites are shown in Figure 2(a–c). The respect-ive mass losses at 400 �C are given in Table 3.Except for the 5wt % KL-PLA composite,which has the highest weight loss at 400 �C(98%) and behaves comparable to neat PLA(99%), all other composites reveal lower masschanges. In general, the decomposition
O
O
O
H
Lig-O H
H
OCH3
+O
OH
OH
CH3
OCH3
O
160 - 175 °C
n
O
OH CH3
O
CH3
OO
OCH3
O
OLig-O
O
OH
CH3
O
CH3
O
O
OHCH3
O
CH3
O
n
n
n
SCHEME 1. Example reaction demonstrating covalent linking of PLA and lignin component.
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 21
60

degree of LS-PLA composites is higher thanthat of OSL and KL composites at the sameratio. Furthermore, the LS-PLA compositeshave not only the highest weight loss, butthey also decompose at up to 15 �C lowertemperatures than their counterparts, transfer-ring the behavior of pure LS, as the ligninwith the lowest thermoresistance, to the com-posite case. The introduction of LS into thePLA matrix accelerated the decomposition ofPLA and thus caused weaker PLA thermalproperties (Figure 2(a–c)). On the other hand,OSL and KL composites show clear differen-ces at 10 and 15wt % lignin (Figure 2(b,c)), inwhich coincidentally the reduced mass
TABLE 3. Thermal properties of PLA, lignins, and theircomposites.a
Lignin/PLA TGA, weight loss at 400 �C (%) Tg (�C) Tm (�C)
0/100 99 61 155.5LS 5/10 95 56.0 150.9
10/90 95 57.4 147.715/85 90 57.4 148.8100/0 35 – –
OSL 5/10 94 58.7 151.910/90 91 59.3 152.015/85 85 58.3 150.8100/0 35 – –
KL 5/10 98 60.02 154.210/90 90 59.4 153.315/85 86 57.4 148.8100/0 26 – –
aTg: glass transition temperature; Tm: melting temperature.
100 200 300 400 500 600 700 800 900
0
20
40
60
80
100
TG
/%
Temperature /°C
Temperature /°C
PLA
KL
OSL
LS
O2
Mass Change -99 %
Mass Change: -69.1 %
Mass Change: -83.3 %
Mass Change: -88.1%
320 340 360 380 400
0
20
40
60
80
100
320 340 360 380 400 320 340 360 380 400
TG
/%
(a)
10°C
(b) (c)
18%
FIGURE 2. TGA curves of PLA (green line); Kraft lignin (KL, black line); Organosolv lignin (OSL, red line) and Lignosulfonate (blueline). (a–c) The lignin-PLA copolymers with 5, 10, and 15wt % lignin loading, respectively. Up to 700 �C, the measurements werecarried out in nitrogen atmosphere, above that, oxygen was introduced.
22 MIMINI ET AL.
61
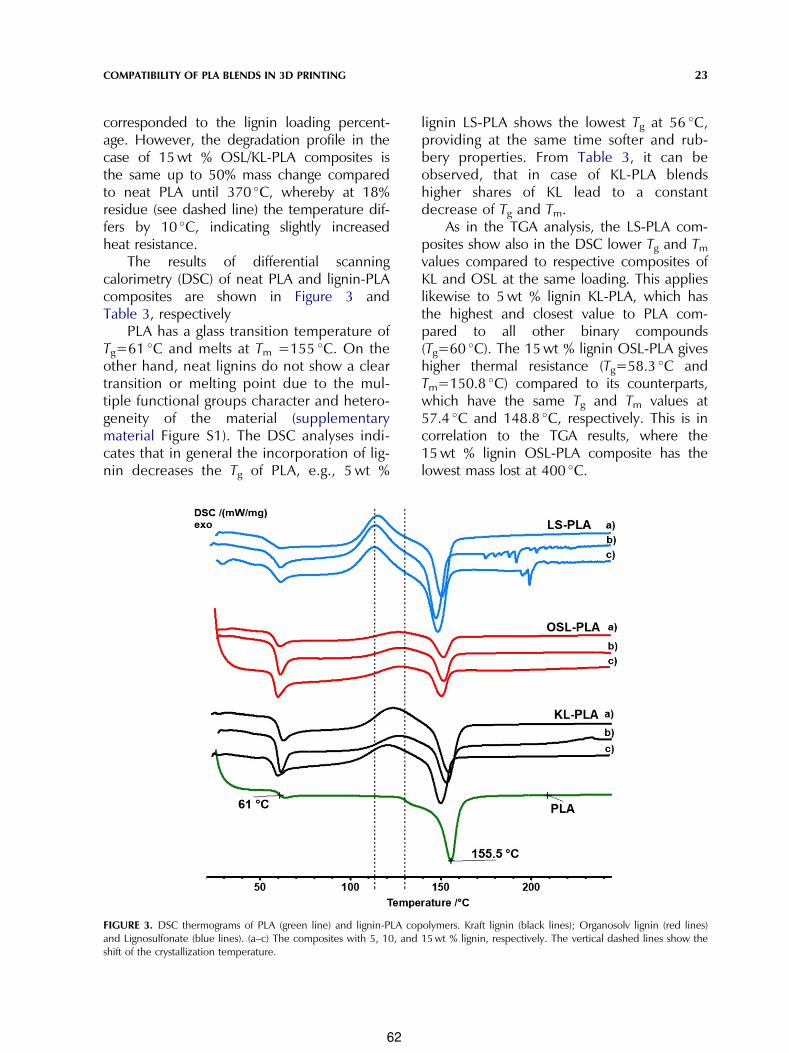
corresponded to the lignin loading percent-age. However, the degradation profile in thecase of 15wt % OSL/KL-PLA composites isthe same up to 50% mass change comparedto neat PLA until 370 �C, whereby at 18%residue (see dashed line) the temperature dif-fers by 10 �C, indicating slightly increasedheat resistance.
The results of differential scanningcalorimetry (DSC) of neat PLA and lignin-PLAcomposites are shown in Figure 3 andTable 3, respectively
PLA has a glass transition temperature ofTg=61 �C and melts at Tm =155 �C. On theother hand, neat lignins do not show a cleartransition or melting point due to the mul-tiple functional groups character and hetero-geneity of the material (supplementarymaterial Figure S1). The DSC analyses indi-cates that in general the incorporation of lig-nin decreases the Tg of PLA, e.g., 5 wt %
lignin LS-PLA shows the lowest Tg at 56 �C,providing at the same time softer and rub-bery properties. From Table 3, it can beobserved, that in case of KL-PLA blendshigher shares of KL lead to a constantdecrease of Tg and Tm.
As in the TGA analysis, the LS-PLA com-posites show also in the DSC lower Tg and Tmvalues compared to respective composites ofKL and OSL at the same loading. This applieslikewise to 5wt % lignin KL-PLA, which hasthe highest and closest value to PLA com-pared to all other binary compounds(Tg=60 �C). The 15wt % lignin OSL-PLA giveshigher thermal resistance (Tg=58.3 �C andTm=150.8 �C) compared to its counterparts,which have the same Tg and Tm values at57.4 �C and 148.8 �C, respectively. This is incorrelation to the TGA results, where the15wt % lignin OSL-PLA composite has thelowest mass lost at 400 �C.
FIGURE 3. DSC thermograms of PLA (green line) and lignin-PLA copolymers. Kraft lignin (black lines); Organosolv lignin (red lines)and Lignosulfonate (blue lines). (a–c) The composites with 5, 10, and 15wt % lignin, respectively. The vertical dashed lines show theshift of the crystallization temperature.
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 23
62

In Figure 3, a further difference betweenbinary blends and PLA can be observed inthe crystallization properties. While OSL-PLAcomposites do not show any prominentchanges, the KL-PLA and LS-PLA blends showreduction of the cold crystallization tempera-ture of PLA from 130 to �120 �C for KL-PLAblends and to �115 �C for LS-PLA blends.Crystallization can be affected by many fac-tors, such as spatial confinement, nucleationon sample boundaries, temperature gradients,melt flow, etc.,[32] but in general it is a resultof interplay between the kinetics of crystal-lization and potential for crystallization.[33]
The introduction of an additional phase inthe PLA matrix, such as lignin, can accelerateor decelerate the crystallization by assisting orhindering the chain mobility. Additionally, asecond phase can influence the crystallizationkinetics by providing a higher surface area,which can reduce the free energy and as aresult improve nucleation.[32,33] In the case ofLS-PLA blends, the introduction of a secondphase shows an acceleration of the cold crys-tallization temperature with sharper peakscompared to KL-PLA blends, which is indi-cated by broader peaks and higher crystalliza-tion temperatures. The shift of the coldcrystallization temperature is higher in 10 wt% lignin LS-PLA blends. When the content ofLS becomes 15wt %, the crystallization tem-perature does not decrease significantly, indi-cating a saturation of the PLA matrix.
Melt Volume Rate of PLA and Lignin-PLA Composites
Hydrolysis is one of the main characteris-tics of PLA, which affects several materialproperties and hence service life.[34]
Polymeric ester groups can be easily hydro-lyzed, in particular under the drastic condi-tions of melt extrusion. This reaction can beeither acid-catalyzed or base-catalyzed.[35]
Under acidic conditions, the regenerated car-boxylic acid groups act autocatalytically, pro-moting further chain cleavage. As aconsequence of this degradation, products ofautocatalytic random chain scissions are
found.[36,37] Besides hydrolysis also other deg-radation types of the polymer occur duringmelt processing caused by shear stress, heatand catalyst residues,[38] mainly belonging tothe group of elimination reactions. A simpleand abundantly used method to detectchanges in molecular weight of PLA and com-parable polymers is the measurement of themelt volume rate (MVR).[39]
Independent of the kind of polymer, allstudies reveal significant increases in MVRwith decreasing molecular weight. TheMVRs of the KL-PLA composites are signifi-cantly above the values obtained for therespective OSL and LS composites (seeFigure 4). The obviously stronger promotionof chain scission by use of KL could becaused by initial differences in the pH-value,[35] by the stronger hygroscopicity ofthis lignin type and by by-products releasedat higher processing temperatures.[40]
Interestingly, the strongly acidic sulfonategroups in LS did not have a significant nega-tive impact on PLA chain cleavage. Thedecrease in PLA molecular weight propertiesof the PLA-Kraft lignin composites as indi-cated by increasing MVRs are in consistencewith the study of Cicala et al.[40] whichreveals accelerated degradation of PLAinduced by incorporation of lignin.Apparently, the influence of reactions dur-ing processing, such as the release ofbyproducts, water, possible acids, etc., ismore decisive than functional groups in thelignin components present fromthe beginning.
0 5 10 150
2
4
6
8
10
12
14
16
18
neat PLA KL OSL LS
Me
lt V
olu
me
Ra
te in
cm
³/1
0m
in
lignin loading in [%]
FIGURE 4. Melt volume rates of neat PLA and lignin-PLA composites.
24 MIMINI ET AL.
63

Flexural and Impact Strength
No improvement in the flexural strengthof PLA is achieved by incorporation of thedifferent lignins in different concentrations(Figure 5). The reduction in strength uponincorporation of lignin is attributed to weakinterfaces which impede the stress transferfrom PLA to the lignin reinforcement (seebelow SEM results). The highest strength val-ues are obtained with 5% reinforcement ofOSL. Generally, OSL yields the higheststrength at each addition level. The lowestreinforcing effect is obtained by use of LS.
The significant decrease (rSp= �0.84;p=.01) in flexural strength with increasingmolecular weight Mn of the lignin typesmight indicate disrupted interaction betweenthe PLA chains. The deterioration of strengthof PLA upon incorporation of lignin in con-centrations above 5wt % is in agreementwith the results in the literature.[9,16,41]
Contrary results are reported by Sun et al.,[42]
who achieved a strong matrix/filler interfacialinteraction by blending lignin-rubber-PDLA(lig-rubber-D) and commercial PLLAin chloroform.
The impact strength of PLA is reduced byblending with all of the tested lignin types.The slightly better value of 5wt % KL-PLA(15.3 kJ/m2) seems to be less significant, seealso the error bars in Figure 5. The impactstrength is correlated with the filler load(rSp=�0.81; p=.01). The partially significantdecrease in impact strength of PLA by blend-ing with lignin suggests a higher rigidity of all
lignin types compared to neat PLA matrix.[43]
Also, the voids and air pockets, as shown inFigure 6, diminish impact strength by intro-ducing flaws in the composite.
Scanning Electron Microscopy (SEM) ofLS Composites
The morphology of lignin-PLA compositeswas examined by SEM. Due to the high num-ber of combinations, only the micrographs ofneat lignins (Figure 6, KL, OSL, and LS), andof LS-PLA combinations (Figure 6, LS1–LS6)are displayed.
As illustrated in Figure 6, the particles ofKL are spherical compared to LS particleswhich are irregular with a sharp shape. Bothshow a large scatter in particle size whichranges from 10 to 200 mm (SEM at 500 mm).OSL consists of fine spherical particles (SEM at100 mm) which are up to 200 times smallerthan KL and LS particles. The spherical formof the particles is a prerequisite to isotropicfiller reinforcement, and on the other side, thefiller size affects the surface area to volumeratio.[33] Micrographs (100 mm) of LS-PLAcomposites with 5, 10, and 15wt % LS andtherefore reflect differences in the PLA-lignincross-linking and thus in the thermomechani-cal properties. This can be observed in theTGA and DSC results, with the OSL-PLA andKL-PLA composites showing better thermalresistance than the LS-PLA composites (Table3), and by flexural and impact strength, withOSL-PLA composites giving better results(Figure 5). Additionally, the lower melt volumerate of OSL-PLA composites indicates a better
0 5 10 150
20
40
60
80
100
120
neat PLA KL OSL LS
Fle
xu
ral str
en
gth
in
MP
a
lignin loading in [%]
0 5 10 150
2
4
6
8
10
12
14
16
18
neat PLA KL OSL LS
Imp
act
str
en
gth
in
kJ/m
2
lignin loading in [%]
FIGURE 5. Flexural and impact strength of neat PLA and lignin-PLA composites.
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 25
64

fit of the small and spherical particles of OSLinto the PLA matrix (Figure 4).
In the SEM images taken at 200 mm(marked as LS1, LS2, and LS3) representing5, 10, and 15wt % LS loading) irregularbubbles can be observed at the surface ofthe LS-PLA composites compared to theircounterparts, which showed smooth surfaceswith a surrounding PLA matrix. The bubblesat 5 wt % LS are smaller and sparse andwith increasing LS concentration expand
and multiply. The cross section of the LS-PLA blends illustrated through micrographsat higher magnification (100 mm) revealscavities between PLA and the LS particles(Figure 6, LS4–LS6). The pores are withinthe micron range and are quite evident inLS5 (10wt % LS), which further confirms thereduced flexural and impact strength.However, with increasing content of LS(LS6, 15wt % LS), the pores become morehomogeneous and smaller.
FIGURE 6. SEM images of neat lignins; KL (Kraft lignin) and LS (Lignosulfonate) at 500 mm; OSL (Organosolv) at 100 mm. LS1–LS3are surface micrographs at 200 mm from LS-PLA-composites with 5, 10, and 15wt % LS, respectively. LS4–LS6 show cross sections.
26 MIMINI ET AL.
65

The LS behavior as a nucleating agentwas recognized in the thermal studies as aresult of an increase in the PLA crystallinity(Figure 3). In addition to the factors statedfrom DSC, which influence crystallinity andtherefore nucleation, such as chain mobility,surface area, amount of the second phase,etc., further co-factors of the second phase,such as particle shape, surface, and size, arereported to have a negligible effect in thenucleation of PLA.[33,44] However, excludingOSL which has ‘incomparable’ particles to LSdue to smaller size and different form, the KLparticles show the same size as LS with differ-ing morphological shape, but no nucleationin KL-PLA composites was detected.Furthermore, considering the almost sameMw of KL and LS, the nucleation effect of LSmay be caused by differences in functionalgroups. As mentioned above, compared toOSL and KL, LS contains sulfonic acid groupsmaking it a polymer with net negative chargeand thus giving diverse advantages, such ashigh surface activity, stabilizing and dispersingproperties and most importantly, enabling thesolubility of LS in water. The sulfonic acidgroups, however, impart a degree of hygro-scopicity,[45,46] whereby by introduction ofheat, as by extrusion, the moisture vaporizesand is released as gas. However, to under-stand and figure out the PLA nucleationproperties of LS, further detailed investiga-tions are required, in particular post adsorp-tion effects of LS by high temperatureand pressure.
CONCLUSION
PLA-Lignin composites were successfullyproduced by double extrusion followed by3D-printing. Physicochemical and thermome-chanical characterization was carried outbefore and after extrusion as well as after 3D-printing to examine the behavior and thepotential of unmodified lignins in the bioplas-tic field.
31P NMR analysis revealed a higheramount of total OH groups for neat KL andlower for KL composites than OSL and OSL-
composites, which indicates that the OHgroups of the chemically reactive KL havereacted to a higher degree upon compound-ing. However, except the 5wt % KL-PLAblends which showed higher impact strength,the other KL-PLA composites provided infer-ior mechanical properties compared to OSLand LS composites.
The OSL-PLA blends yielded better ther-mal resistance, lower melt volume rate andthe highest flexural strength values comparedto their counterparts. The high compatibilityis attributed to the low molecular weight,lower dispersity of OSL, and its small (lessthan 10 mm) and spherical particles, whichcan effortlessly blend into the PLA matrixleading to better interfacial interactions.
The LS-PLA composites showed a higherdecomposition degree, however, lower glasstransition, crystallization temperature, meltingpoint as well as flexural strength valuesrespective to composites of KL and OSL atthe same loading. Due to the high strength ofPLA, the highest reduction by incorporationof 15% lignosulfonate still yields quite accept-able values around 80MPa which shouldallow implementation of these composites indiverse applications, such as 3D-prints oroffice supplies. The accelerated crystallizationof PLA in presence of LS, as detected byDSC, as well as the generated pores observedby SEM micrographs, shine a new light onpurified LS as nucleating agent with a per-spective in (bio)polymer foaming.
ACKNOWLEDGMENTS
Financial support to WOOD Kplus isgratefully acknowledged.
FUNDING
Austrian government; the provinces ofLower Austria, Upper Austria, and Carinthia;industrial partners of WOOD Kplus;€Osterreichische Forschungsf€orderungsgesells-chaft (FFG).
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 27
66

SUPPLEMENTAL MATERIAL
Supplemental data for this article can beaccessed on the publisher’s website at http://dx.doi.org/10.1080/02773813.2018.1488875
REFERENCES
[1] Flaris, V.; Singh, G. RecentDevelopments in Biopolymers. Journal ofVinyl and Additive Technology 2009, 15(1),1–11. doi:10.1002/vnl.20171.
[2] Lim, L. T.; Auras, R.; Rubino, M.Processing Technologies for Poly(Lactic Acid).Progress in Polymer Science 2008, 33(8),820–852. doi:10.1016/j.progpolymsci.2008.05.004.3. North, E. J.; Halden, R. U. Plastics and
Environmental Health: The Road Ahead.Reviews on Environmental Health 2013,28(1), 1–8. doi:10.1515/reveh-2012-0030.4. Song, J. H.; Murphy, R. J.; Narayan, R.;
Davies, G. B. H. Biodegradable andCompostable Alternatives to ConventionalPlastics. Philosophical Transactions of theRoyal Society B: Biological Sciences 2009,364(1526), 2127–2139.5. Brown, R. C.; Brown, T. R. (2013).
Biorenewable Resources: Engineering NewProducts from Agriculture; Wiley-Blackwell:Hoboken, NJ, USA.6. Thakur, V. K.; Thakur, M. K.; Gupta,
R. K. Graft Copolymers of Natural Fibers forGreen Composites. Carbohydrate Polymers2014, 104(Supplement C), 87–93.doi:10.1016/j.carbpol.2014.01.016.7. Lora, J. H.; Glasser, W. G. Recent
Industrial Applications of Lignin: ASustainable Alternative to NonrenewableMaterials. Journal of Polymers and theEnvironment 2002, 10(1/2), 39–48.doi:10.1023/a:1021070006895.8. Voicu, S. I.; Condruz, R. M.; Mitran, V.;
Cimpean, A.; Miculescu, F.; Andronescu, C.;Miculescu, M.; Thakur, V. K. Sericin CovalentImmobilization onto Cellulose AcetateMembrane for Biomedical Applications. ACSSustainable Chemistry & Engineering 2016,
4(3), 1765–1774. doi:10.1021/acssusche-meng.5b01756.
[9] Li, J.; He, Y.; Inoue, Y. Thermal andMechanical Properties of BiodegradableBlends of Poly(L-Lactic Acid) and Lignin.Polymer International 2003, 52(6), 949–955.doi:10.1002/pi.1137.10. Li, Y.; Susan Sun, X. Mechanical and
Thermal Properties of Biocomposites FromPoly(Lactic Acid) and DDGS. Journal ofApplied Polymer Science 2011, 121(1),589–597. doi:10.1002/app.33681.11. Murariu, M.; Dubois, P. PLA
Composites: From Production to Properties.Advanced Drug Delivery Reviews 2016,107(Supplement C), 17–46. doi:10.1016/j.addr.2016.04.00312. Lora, J. Chapter 10 – Industrial
Commercial Lignins: Sources, Properties andApplications A2 – Belgacem, Mohamed Naceur.In Monomers, Polymers and Composites fromRenewable Resources; Gandini, A., Ed.; Elsevier:Amsterdam, 2008, 225–241.13. Chen, R.; Abdelwahab, M. A.; Misra,
M.; Mohanty, A. K. Biobased Ternary Blendsof Lignin, Poly(Lactic Acid), andPoly(Butylene Adipate-Co-Terephthalate): TheEffect of Lignin Heterogeneity on BlendMorphology and Compatibility. Journal ofPolymers and the Environment 2014, 22(4),439–448. doi:10.1007/s10924-014-0704-5.14. Doherty, W. O. S.; Mousavioun, P.;
Fellows, C. M. Value-Adding to CellulosicEthanol: Lignin Polymers. Industrial Crops andProducts 2011, 33(2), 259–276. doi:10.1016/j.indcrop.2010.10.022.15. Arifur, R. M.; Diego, D. S.; Gloria, S.;
Giorgio, R.; Maurizio, P.; Thanh, P. V.;Andrea, L. Biocomposites Based on Ligninand Plasticized Poly(L-Lactic Acid). Journal ofApplied Polymer Science 2013, 129(1),202–214. doi:10.1002/app.38705.16. Gordobil, O.; Eg€u�es, I.; Llano-Ponte, R.;
Labidi, J. Physicochemical Properties of PLALignin Blends. Polymer Degradationand Stability 2014, 108(Supplement C),330–338. doi:10.1016/j.polymdegradstab.2014.01.002.
28 MIMINI ET AL.
67

17. Kim, S.; Oh, S.; Lee, J.; Ahn, N.; Roh,H.; Cho, J.; Chun, B.; Park, J. Effect ofAlkyl-Chain-Modified Lignin in the PLA Matrix.Fibers and Polymers 2014, 15(12), 2458–2465.doi:10.1007/s12221-014-2458-z.18. Sumerskii, I.; Korntner, P.; Zinovyev,
G.; Rosenau, T.; Potthast, A. Fast Track forQuantitative Isolation of Lignosulfonates fromSpent Sulfite Liquors. RSC Advances 2015,5(112), 92732–92742. doi:10.1039/C5RA14080C.19. Lin Stephen, Y.; Carlton, W. D. (1992.).
Methods in Lignin Chemistry. Springer Verlag:Berlin, New York. doi:10.1007/978-3-642-74065-7.20. Sulaeva, I.; Zinovyev, G.; Plankeele, J.-
M.; Sumerskii, I.; Rosenau, T.; Potthast, A.Fast Track to Molar-Mass Distributions ofTechnical Lignins. ChemSusChem 2017,10(3), 629–635. doi:10.1002/cssc.201601517.21. Sumerskii, I.; Zweckmair, T.; Hettegger,
H.; Zinovyev, G.; Bacher, M.; Rosenau, T.;Potthast, A. A fast track for the accuratedetermination of methoxyl and ethoxylgroups in lignin. RSC Advances 2017, 7(37),22974–22982. doi:10.1039/C7RA00690J.22. Sundberg, A.; Sundberg, K.; Lillandt, C.;
Holmbom, B. Determination ofHemicelluloses and Pectins in Wood andPulp Fibres by Acid Methanolysis and gaschromatography. Nordic Pulp and Paper1996, 11, 216–219.23. Korntner, P.; Sumerskii, I.; Bacher, M.;
Rosenau, T.; Potthast, A. Characterization ofTechnical Lignins by NMR Spectroscopy:Optimization of Functional Group Analysis by31P NMR Spectroscopy. Holzforschung 2015,69, 807–814.24. Zinovyev, G.; Sumerskii, I.; Korntner,
P.; Sulaeva, I.; Rosenau, T.; Potthast, A.Molar Mass-Dependent Profiles of FunctionalGroups and Carbohydrates in Kraft Lignin.Journal of Wood Chemistry and Technology2017, 37(3), 171–183. doi:10.1080/02773813.2016.125310325. Balakshin, M.; Capanema, E. On the
Quantification of Lignin Hydroxyl Groups
with 31P and 13C NMR Spectroscopy.Journal of Wood Chemistry and Technology2015, 35(3), 220–237. doi:10.1080/02773813.2014.92832826. Shiramizu, M. L.; Salciccioli, M. S.;
Neeraj, C. T. In World Intellectual PropertyOrganization Report. WIPO: Stockholm,2017.27. Evtuguin, D. V.; Neto, C. P.; Silvestre,
A. J. D. Condensation reactions of lignin dur-ing oxygen delignification under acidic condi-tions. Journal of Wood Chemistry andTechnology 1997, 17(1-2), 41–55.doi:10.1080/02773819708003117.28. Mohammad Jawaid, O. Y. A.; Sapuan,
S. M. Green Biocomposites: Manufacturingand Properties. Springer: Berlin, New York,2017.29. Al�en, R.; Kuoppala, E.; Oesch, P.
Formation of the Main DegradationCompound Groups from Wood and itsComponents During Pyrolysis. Journal ofAnalytical and Applied Pyrolysis 1996,36(2), 137–148. doi:10.1016/0165-2370(96)00932-1.30. Rodrigues, J.; Graca, J.; Pereira, H.
Influence of Tree Eccentric Growth onSyringyl/Guaiacyl Ratio in EucalyptusGlobulus Wood Lignin Assessed by AnalyticalPyrolysis. Journal of Analytical and AppliedPyrolysis 2001, 58-59(Supplement C),481–489. doi:10.1016/S0165-2370(00)00121-2.31. M€uller-Hagedorn, M.; Bockhorn, H.;
Krebs, L.; M€uller, U. A Comparative KineticStudy on the Pyrolysis of Three DifferentWood Species. Journal of Analytical andApplied Pyrolysis 2003, 68-69, 231–249.doi:10.1016/S0165-2370(03)00065-2.32. Piorkowska, E.; Galeski, A. Overall
Crystallization Kinetics Handbook of PolymerCrystallization; John Wiley & Sons, Inc.:Hoboken, NJ, 2013, 215–236.33. Anwer, M. A. S.; Naguib, H. E.; Celzard,
A.; Fierro, V. Comparison of the Thermal,Dynamic Mechanical and MorphologicalProperties of PLA-Lignin & PLA-TanninParticulate Green Composites. Composites
COMPATIBILITY OF PLA BLENDS IN 3D PRINTING 29
68

Part B: Engineering 2015, 82, 92–99.doi:10.1016/j.compositesb.2015.08.028.34. Huang, Y.; Zhang, C.; Pan, Y.; Zhou, Y.;
Jiang, L.; Dan, Y. Effect of NR on the HydrolyticDegradation of PLA. Polymer Degradation andStability 2013, 98(5), 943–950. doi:10.1016/j.polymdegradstab.2013.02.01835. Gorrasi, G.; Pantani, R. Effect of PLA
Grades and Morphologies on HydrolyticDegradation at Composting Temperature:Assessment of Structural Modification andKinetic Parameters. Polymer Degradation andStability 2013, 98(5), 1006–1014.doi:10.1016/j.polymdegradstab.2013.02.005.36. Antheunis, H.; van der Meer, J.-C.; de
Geus, M.; Heise, A.; Koning, C. E.Autocatalytic Equation Describing the Changein Molecular Weight During HydrolyticDegradation of Aliphatic Polyesters.Biomacromolecules 2010, 11(4), 1118–1124.doi:10.1021/bm100125b.37. Li, S. Hydrolytic Degradation
Characteristics of Aliphatic Polyesters Derivedfrom Lactic and Glycolic acids. Journal ofBiomedical Materials Research 1999, 48(3),342–353. doi:10.1002/(SICI)1097-4636(1999)48:3< 342::AID-JBM20>3.0.CO;2-7.38. _Zenkiewicz, M.; Richert, J.; Rytlewski,
P.; Moraczewski, K.; Stepczy�nska, M.;Karasiewicz, T. Characterisation of Multi-Extruded Poly(Lactic Acid). Polymer Testing2009, 28(4), 412–418. doi:10.1016/j.polymertesting.2009.01.012.39. Signori, F.; Coltelli, M.-B.; Bronco, S.
Thermal Degradation of Poly(Lactic Acid)(PLA) and Poly(Butylene Adipate-Co-Terephthalate) (PBAT) and Their Blends UponMelt Processing. Polymer Degradation andStability 2009, 94(1), 74. doi:10.1016/j.polymdegradstab.2008.10.004.
40. Cicala, G.; Saccullo, G.; Blanco, I.;Samal, S.; Battiato, S.; Dattilo, S.; Saake, B.Polylactide/Lignin Blends: Effects OfProcessing Conditions on Structure andThermo-Mechanical Properties. Journal ofThermal Analysis and Calorimetry 2017,130(1), 515–524.41. Domenek, S.; Louaifi, A.; Guinault, A.;
Baumberger, S. Potential of Lignins asAntioxidant Additive in ActiveBiodegradable Packaging Materials. Journalof Polymers and the Environment 2013,21(3), 692–701. doi:10.1007/s10924-013-0570-642. Sun, Y.; Yang, L.; Lu, X.; He, C.
Biodegradable and Renewable Poly(Lactide)-Lignin Composites: Synthesis, Interface andToughening Mechanism. Journal of MaterialsChemistry A 2015, 3(7), 3699–3709.doi:10.1039/C4TA05991C43. Ouyang, W.; Huang, Y.; Luo, H.;
Wang, D. Poly(Lactic Acid) Blended withCellulolytic Enzyme Lignin: Mechanical andThermal Properties and MorphologyEvaluation. Journal of Polymers and theEnvironment 2012, 20(1), 1–9. doi:10.1007/s10924-011-0359-444. Liao, R.; Yang, B.; Yu, W.; Zhou, C.
Isothermal Cold Crystallization Kinetics ofPolylactide/Nucleating Agents. Journal ofApplied Polymer Science 2007, 104(1),310–317. doi:10.1002/app.2573345. Pizzi, A. WOOD Adhesives. Chemistry
and Technology 1989, 2, 417.46. Feldman, D. Lignin: Properties and
Materials. Journal of Polymer Science:Polymer Letters Edition 1990,28(5), 184–185. doi:10.1002/pol.1990.140280511.
30 MIMINI ET AL.
69

70

71
8.3 Lignosulfonate-based polyurethane materials via cyclic carbonates -
preparation and characterization (Publication III)

72

For Review O
nly
Lignosulfonate-based polyurethane materials via cyclic carbonates: preparation and characterization
Vebi Mimini1,2, Hassan Amer1,3, Hubert Hettegger1, Markus Bacher1, Ireen Gebauer2, Robert Bischof4, Karin Fackler4, Antje Potthast1 and Thomas Rosenau1,5,*
1 Division of Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Straße 24, A-3430 Tulln, Austria
2 Wood Kplus - Kompetenzzentrum Holz GmbH, Altenberger Straße 69, A-4040 Linz, Austria
3 Department of Natural and Microbial Products Chemistry, National Research Centre, 33 Al Bohous St., Dokki, Giza 12622, Egypt
4 Lenzing AG, Werkstraße 1, A-4860 Lenzing, Austria
5 Johan Gadolin Process Chemistry Centre, Åbo Akademi University, Porthansgatan 3, Åbo/Turku FI-20500, Finland
*Corresponding author: e-mail: [email protected]
V. Mimini et al.: Lignosulfonate-based polyurethane materials
Received December 14, 2018; accepted March 28, 2019
Page 1 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
73

For Review O
nly
Abstract: Usage of lignin and its derivatives as chemical and carbon source, i.e. in
processes other than burning, is one of the most active fields in renewable resource
chemistry today. In this work, the synthesis of lignosulfonate-based polyurethane
materials from non-toxic reagents and through environmentally friendly processes is
presented. Lignosulfonate (LS), modified with bio-based (glycerin-derived) cyclic
carbonate moieties, was reacted with 1,6-hexamethylenediamine to form
characteristic polyurethane (PU) material. For mechanistic studies and reaction
optimization, cyclic carbonates and 1,2-diol derivatives of vanillyl alcohol, as a
simplifying lignin model compound, were employed. A LS-bound cyclic carbonate can
be formed in one pot without a transesterification step, which simplifies the route
towards non-isocyanate lignin-based PU materials. ATR-FTIR spectra showed typical
linkages of cyclic carbonates and 1,2-diols on lignosulfonate. Further analytical
characterization, in both the model compound and the LS polymer case, was
provided by liquid state NMR spectroscopy (1D, 2D and 31P) and 13C solid state
NMR. The production of PU materials from sulfonated lignin and glycerol carbonate,
synthesized through a non-isocyanate reaction pathway, confirms the good potential
of LS utilization in the development of PU composites based on renewable
resources.
Keywords: cyclic carbonate, dimethyl carbonate, glycerol carbonate, lignin
utilization, lignosulfonate, lignosulfonate glycerol cyclic carbonate, non-isocyanate
polyurethane, polyurethane
Page 2 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
74

For Review O
nly
Introduction
Conventionally, industrial synthesis of PUs starts from diol and diisocyanate
compounds which in turn come from amines and phosgene (Król 2007, Akindoyo et
al. 2016). Both, isocyanates and phosgene are highly toxic and dangerous
compounds. The world´s most horrific industrial disaster, the Bhopal catastrophe in
1984 with nearly 4000 victims, has been related to (methyl) isocyanate chemistry. In
the light of such danger, the additional disadvantages of sensitivity to humidity and
difficult general handling appear to shrink to merely minor annoyances. Alternative,
sustainable non-isocyanate PUs have consequently been the targets of research in
the last decade (Scheme 1). Among all studied routes, either transurethanization /
polycondensation between a bis-carbamate and a diol, or the polyaddition between
cyclic carbonates and amines are considered as the most promising pathways to
overcome the disadvantages of isocyanate and phosgene chemistry (Scheme 1, A
and B; Kreye et al. 2013, Maisonneuve et al. 2015). The first pathway requires
expensive catalysts, leads to lower molecular weight bis-carbamates and offers
generally lower reactivity for polymerization (Maisonneuve et al. 2015), which seems
to disfavor this approach. The cyclic carbonates of the second alternative are
synthesized through fixation of CO2 by epoxides (oxiranes) which, however, must be
separately generated (Scheme 1, B1; Guan et al. 2011). Straight implementation of
this methodology into renewable materials is not possible because biopolymers, such
as lignins, technical lignins or LS, obviously do not contain epoxide building blocks
per se. The addition of the three-membered cyclic ether ring to the biopolymer is
typically achieved by reaction with epichlorohydrin (Lee et al. 2002, Nouailhas et al.
2011), once again a hazardous and toxic material, although less dangerous than
most isocyanates. In the next step, the CO2 insertion requires high pressure (20−40
Page 3 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
75

For Review O
nly
bar) or special catalysts and equipment, therefore preparation of cyclic carbonates
via CO2 and oxiranes remains industrially and economically challenging.
An environmentally friendly approach towards cyclic carbonates is based on the
transesterification of 1,2-diols (e.g. ethylene glycol, 1,2-propanediol, and glycerol)
with dialkyl carbonates (dimethyl or diethyl carbonate), thus avoiding the above-
mentioned obstacles (Kihara et al. 1993, Shaikh et al. 1996, Takagaki et al. 2010,
Schäffner et al. 2010, Pyo et al. 2011). This reaction is entropically driven by the
formation of the 1,3-dioxolan-2-one heterocycle (cyclic carbonate). This mild and eco-
friendly approach was recently implemented in derivatization of biopolymers, such as
organosolv-, kraft-, and soda lignin, by Kühnel et al. (2018). Technical lignins, i.e.
lignins retrieved from processing liquors of commercial pulping operations, are a
suitable alternative for conventional polyols for isocyanate/PU composites due to
their high amounts of hydroxyl groups (Cateto et al. 2011, Cateto et al. 2014, Obaid
et al. 2016), and they thus offer a high potential for the production of renewable-
based PU composites. The reactive hydroxyl groups of lignins can be modified, e.g.
esterified, etherified, silylated etc., under mild conditions (Laurichesse et al. 2014).
While Saake’s and Lehnen’s group already oxyalkylated organosolv, kraft, and soda
lignin through cyclic organic carbonates (Kühnel et al. 2017), and additionally
successfully equipped organosolv lignin with cyclic carbonate functionalities (Kühnel
et al. 2018), to the best of our knowledge, there are no examples reported in
literature about the synthesis of LS cyclic carbonates.
In this report, we present a non-isocyanate, non-toxic and environmentally
compatible approach to LS-based PU materials. Mechanism and byproduct formation
were also studied by means of model compound conversions, the reaction of glycerol
carbonate with vanillyl alcohol.
Page 4 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
76

For Review O
nly
Materials and methods
Starting materials: Beech lignosulfonate was obtained from Lenzing AG, Austria.
Before use it was purified according to Sumerskii et al. (2015) with a modification
(see below). Amberlite XAD-7 HP (20-60 mesh) was purchased from Acros Organics
(Geel, Belgium). Glycerol 1,2-carbonate (GC, purity 90%) was purchased from abcr
GmbH (Karlsruhe, Germany). Dimethyl carbonate (DMC, purity 99%) and the catalyst
1,8-diazabicyclo[5.4.0]undec-7-en (DBU, purity 98%) were purchased from
Honeywell FlukaTM Chemicals (Seelze, Germany). 4-Hydroxy-3-methoxybenzyl
alcohol (vanillyl alcohol, VA, purity 98%) was purchased from Alfa Aesar (Karlsruhe,
Germany). 1,6-Hexamethylenediamine (HMDA, purity 98%) and all other chemicals
and solvents were purchased from Sigma-Aldrich (Schnelldorf, Germany). Prior to
use, the solvents were dried over 4 Å molecular sieves.
Purification of lignosulfonate by adsorption onto XAD-7 resin: Sulfite spent
liquor (SSL, 100 g) was diluted with 1 M aqueous HCl (30 mL) and the pH was
adjusted to <1. The protonated SSL was mixed with XAD-7 resin (mass ratio SSL /
XAD-7, 1:2) and shaken overnight. Non-adsorbed compounds were removed by
washing with acidified (pH 2) and deionized water until neutral. Subsequently,
lignosulfonic acid (LSA) was desorbed from XAD-7 with ethanol at 45°C. After
evaporation of ethanol under reduced pressure, LSA was lyophilized and dried in a
vacuum oven at 40°C.
Page 5 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
77

For Review O
nly
Analytical characterization: The 1D and 2D nuclear magnetic resonance analyses
(NMR) were performed on a Bruker Avance II 400 instrument (Bruker, Rheinstetten,
Germany) with a 1H resonance frequency at 400.13 MHz, 13C at 100.61 MHz, and
31P at 162.08 MHz. The samples were dissolved in CDCl3 or MeOD and spectra were
recorded at room temperature. 31P NMR analyses for the determination of the total
hydroxyl group contents were performed following a modified protocol (Korntner et al.
2015): 30 mg of each LSA sample was dissolved in anhydrous DMF (700 µL).
Pyridine (100 µL), a relaxation reagent (100 µL, prepared with 5 mg of chromium(III)
acetylacetonate in 1 mL of DMF/pyridine (7:1, v/v) mixture) and 2-chloro-4,4,5,5-
tetramethyl-1,3,2-dioxaphospholane (TMDP, 100 µL) were added and the mixture
shaken for 1 h. NMR data were processed using Bruker TopSpin 3.5 pl 7 software.
13C solid-state NMR (ss-NMR) spectra were recorded on a Bruker Avance III HD 400
instrument (Bruker, Rheinstetten, Germany) with a 13C resonance frequency of
100.68 MHz. Attenuated Total Reflection Infrared Spectroscopy (ATR-FTIR) was
carried out using a PerkinElmer Frontier (Universal ATR Sampling Accessory; Perkin-
Elmer, Waltham, MA, USA) FT-IR spectrometer. Thin-layer chromatography (TLC)
was performed on silica gel 60 F254 coated glass plates (Merck, Darmstadt,
Germany).
Syntheses:
Etherification of lignosulfonate with glycerol carbonate: LSA was etherified and
transesterified according to a modified literature protocol for lignins (Kühnel et al.
2018). Isolated LSA (10 g, 25.6 mmol) was suspended in glycerol carbonate (33 mL,
0.39 mol). A 250 mL three-neck round-bottom flask was flushed with argon and DBU
(0.52 mL, 3.48 mmol) was added. The reaction mixture was heated at 170°C with
Page 6 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
78

For Review O
nly
stirring under argon overnight. Absolute ethanol (150 mL) was added to stop the
reaction, and the mixture was stirred at 40°C for 1 h. The residue was filtered,
washed several times with absolute ethanol to remove any residual glycerol
carbonate byproducts, centrifuged and subsequently dried under vacuum at 40°C to
afford a reddish-brown powder of partially cyclic carbonated lignosulfonate-glycerol
(LS-GCC) mixed with lignosulfonate 1,2-diol (LS-diol). Yield: 5.4 g.
Further cyclic carbonation of LS-diol/LS-GCC lignosulfonate with dimethyl carbonate:
The isolated reddish-brown LS-diol/LS-GCC mixture (3.5 g) was transferred to a
three-neck round bottom flask and mixed with dimethyl carbonate (21.8 mL,
260 mmol) under an inert argon atmosphere. DBU (0.4 mL, 2.68 mmol) was added
and the reaction mixture was stirred at 75°C for 12 h. The mixture was centrifuged,
washed several times with absolute ethanol and subsequently dried under vacuum at
40°C to afford LS-GCC in the form of a reddish-brown powder. Yield: 3.2 g.
Synthesis of non-isocyanate lignosulfonate-based polyurethane: LS-GCC
(500 mg) was suspended in 1,6-hexamethylenediamine (5 mL, 36.1 mmol) and
stirred overnight at 70°C. The reaction mixture was washed with ethyl acetate to
remove the non-reacted reagents, during which a black-brown precipitate formed.
The soft precipitate was dried, powdered and washed several times with ethanol to
remove traces of diamine to afford LS-PU (490 mg) as a brown solid.
Page 7 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
79

For Review O
nly
Synthesis of 3-O-(3-methoxy-4-acetoxy-benzyl)-propane-1,2-carbonate (VA-
GCC) and 3-O-(3-methoxy-4-acetoxy-benzyl)-1,2-diacetoxypropane (VA-diol):
Vanillyl alcohol (1 g, 6.49 mmol) and glycerol 1,2-carbonate (1.1 mL, 12.97 mmol)
were dissolved in dioxane (20 mL) under inert atmosphere. DBU (97 µL, 0.65 mmol)
was added and the reaction mixture was stirred overnight at 110°C. The reaction
progress was monitored by TLC (heptane / ethyl acetate, v/v=1:3). The reaction
mixture was concentrated under vacuum. For purification, the crude product was
dissolved in dry pyridine (4 mL). Acetic anhydride (2 mL, 21.16 mmol) and a catalytic
amount of DMAP (10 mg, 82 µmol) were added and the reaction mixture stirred
overnight at room temperature. Methanol (4 mL) was added to quench the excess
acetic anhydride. The reaction mixture was co-evaporated with toluene to remove the
residual pyridine. The concentrated yellow-brown oily syrup was purified by column
chromatography (heptane / ethyl acetate, v/v=1:2) to afford VA-GCC (59%) as a
white solid and VA-diol (23%) as colorless syrup.
3-O-(3-Methoxy-4-acetoxy-benzyl)-propane-1,2-carbonate (VA-GCC): For atom
numbering see Scheme 4. 1H NMR (CDCl3, [ppm]): 7.00 (d, 1H, J = 8.0 Hz, H-5),
6.96 (d, 1H, J = 1.8 Hz H-2), 6.85 (dd, 1H, J = 8.0, 1.8 Hz, H-6), 4.82 (m, 1H, H-9),
4.60 (d, 1H, J = 12.6 Hz, H-7a), 4.56 (d, 1H, J = 12.6 Hz, H-7b), 4.49 (t, 1H, J = 8.2
Hz, H-10a), 4.40 (dd, 1H, J = 8.2, 6.0 Hz, H-10b), 3.85 (s, 3H, 3-OMe), 3.74 (dd, J =
11.1, 3.8 Hz, H-8a), 3.63 (dd, J = 11.1, 3.7 Hz, H-8b), 2.31 (s, 3H, 3-OAc-CH3).
13C NMR (CDCl3, [ppm]): 168.91 (4-Ac-CO), 154.87 (C-11), 151.18 (C-3), 139.28
(C-4), 136.10 (C-1), 122.56 (C-5), 119.49 (C-6), 111.44 (C-2), 74.99 (C-9), 73.00 (C-
7), 68.88 (C-8), 66.06 (C-10), 65.81 (OMe), 20.49 (Ac-CH3).
Page 8 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
80

For Review O
nly
3-O-(3-Methoxy-4-acetoxy-benzyl)-1,2-diacetoxypropane (VA-diol): For atom
numbering see Scheme 4. 1H NMR (CDCl3, [ppm]): 6.96 (d, 1H, J = 8.0 Hz, H-5),
6.92 (d, 1H, J = 1.7 Hz H-2), 6.83 (dd, 1H, J = 8.0, 1.7 Hz, H-6), 5.20 (m, 1H, H-9),
4.50 (d, 1H, J = 12.3 Hz, H-7a), 4.45 (d, 1H, J = 12.3 Hz, H-7b), 4.31 (dd, 1H, J =
11.9, 3.9 Hz, H-10a), 4.15 (dd, 1H, J = 11.9, 6.3 Hz, H-10b), 3.80 (s, 3H, 3-OMe),
3.57 (m, 2H, H-8a, b), 2.26 (s, 3H, 4-OAc-CH3), 2.04 (s, 3H, 9-OAc-CH3), 2.00 (s, 3H,
10-OAc-CH3).
13C NMR (CDCl3, [ppm]): 170.45 (10-Ac-CO), 170.11 (9-Ac-CO), 168.83 (4-Ac-CO),
151.07 (C-3), 139.18 (C-4), 136.57 (C-1), 122.48 (C-5), 119.59 (C-6), 111.47 (C-2),
72.79 (C-7), 70.07 (C-9), 68.08 (C-8), 62.62 (C-10), 55.74 (OMe), 20.84 (9-OAc-
CH3), 20.55 (10-OAc-CH3), 20.48 (4-OAc-CH3).
Results and discussion
Synthesis of lignosulfonate-glycerol cyclic carbonates (LS-GCC)
As demonstrated in Scheme 2, the conversion of OH functional groups to cyclic
carbonates occurs in a two-step reaction, so-called etherification or oxypropylation
followed by transesterification. Oxypropylation, a common reaction for lignin
modification, increases the number and accessibility of aliphatic OH groups
compared to the sterically more hindered (technical) lignin or LS (Li et al. 2012,
Cateto et al. 2013). Further carbonation of the initially resulting diols leads to cyclic
carbonates. Both reactions (oxypropylation and carbonation) are water-sensitive and
are usually carried out in dry organic solvents. As the coreactant glycerol carbonate
is a liquid, it was advantageously used simultaneously as the reaction medium to
achieve a homogenous suspension of lignosulfonate. After completion of the
reaction, the insoluble material was filtered and washed several times with absolute
Page 9 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
81

For Review O
nly
ethanol to remove any residual glycerol carbonate byproducts and residual DBU
catalyst to furnish a solid product (LS-diol/LS-GCC). While LS-diol is the actual
primary product of LS-oxypropylation with glycerol carbonate, the co-product LS-
GCC is formed by subsequent carbonation of already formed LS-diol with excess
glycerol carbonate. Apparently the carbonation of LS-diol could not be driven to
completion just by increasing the excess of glycerol carbonate. LS-diol/LS-GCC was
further carbonated with dimethyl carbonate to afford LS-GCC. This reaction, driven
by entropic favorization of the cyclic carbonate structure, causes conversion of
accessible LS-diol moieties into LS-GCC. Structural changes of the LSA and
products were analytically followed (see e.g. the ATR-FTIR in Figure 1). The peak
intensities close to 2780-3030 cm-1 increased in LS-diol/GCC and esterified
lignosulfonate due to additional CH2 and CH groups of the glyceryl moiety.
Interestingly, the spectrum of the initial oxypropylation product showed bands at 3400
and 1792 cm-1, which originate from the OH of the oxypropyl group (LS-diol) and the
C=O of oxypropyl moieties with cyclic carbonate groups, respectively. These data are
in agreements with those reported by Kühnel et al. (2017) and confirm the coupling of
glycerol carbonate with LS to produce not only LS-diol, but a mixture of LS-diol and
LS-GCC. Further exhaustive carbonation of the LS-diol/LS-GCC mixture with
dimethyl carbonate resulted in a dramatic decrease of the hydroxyl band intensity at
3400 cm-1 confirming complete conversion of the LS-diol and a relative increase of
the peak intensity at 1792 cm-1 (cyclic carbonate in LS-GCC). In addition, the
shoulder appearing at 1749 cm-1 in the LS-GCC spectrum originates from linear
carbonate side-products (R-O-CO-OMe).
For further confirmation of etherification of LS by glycerol carbonate followed by
carbonation with dimethyl carbonate, respectively, the total hydroxyl group content
was determined by 31P NMR according to the common phosphitylation protocol. The
Page 10 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
82

For Review O
nly
corresponding spectra are shown in Figure 2, the resonances at 152 ppm and
132 ppm being related to the internal standard (IS) and the hydrolysis product of the
phosphitylation reagent TMDP, respectively. Aliphatic phosphite esters, aromatic
phosphite esters and carboxylic phosphite anhydride appear in the regions of 151-
145 ppm, 145-136 ppm and 136-134 ppm, respectively. The spectra show a
complete disappearance of aromatic OH groups in the LS-diol and the LS-GCC,
confirming complete reaction (oxypropylation) at these positions. New peaks appear
in the aliphatic region and the overall intensity of aliphatic OH groups increases
significantly. This confirmed that the actual goal of the oxypropylation, increase and
introduction of reactive aliphatic hydroxyl groups, was met, in accordance with data
reported by Kühnel et al. 2018.
Evaluation of the total hydroxyl group content (3.48 mmol/g in LSA) showed an
increase of 54% of free hydroxyl groups (5.36 mmol/g) by reaction of LSA with
glycerol carbonate (note that also cyclic carbonates are introduces which are not
monitored by the 31P NMR method). Further carbonation of LS-diol/LS-GCC with
dimethyl carbonate, as discussed in the next section, decreased the free OH groups
to 1.92 mmol/g of free hydroxyl groups in the final product (LS-GCC).
Reaction of lignosulfonate-glycerol cyclic carbonate (LS-GCC) with diamines
The production of lignosulfonate polyurethane materials (LS-PU) by reaction of LS-
GCC with diamines is illustrated in Scheme 3. The actual urethane formation occurs
through reaction of a primary amine with the cyclic carbonate under ring-opening,
while the utilization of a diamine ensures crosslinking and thus the polymerization
character of the process. Upon washing with ethyl acetate, the initially formed
viscous product precipitates and agglomerates to a rubbery round-shaped material.
Page 11 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
83

For Review O
nly
The chemical characterization of LS-PU was first performed by FTIR spectroscopy
(Figure 3). For spectral comparison, conventional PU from methylene diphenyl
diisocyanate and polyethylene glycol was used. The peaks at 1792 cm-1 and
1749 cm-1 in LS-GCC (Figure 3, blue line), which originate from cyclic carbonate
carbonyl groups, disappear completely as expected in the spectrum of LS-PU,
proving the complete conversion of those moieties. On the other hand, a new
carbonyl group signal appeared at 1704 cm-1 (green line), indicating the successful
formation of carbamate linkages. The same signal was observed in the PU control as
well (purple line, 1714 cm-1). The increase in intensity of the two peaks at 3050-
2800 cm-1 in the spectrum of LS-PU (green line) demonstrates the incorporation of
methylene groups from 1,6-hexamethylenediamine. Also the appearance of new
hydroxyl groups, formed upon amination opening of the cyclic carbonates, can be
observed in the OH region (3050-3700 cm-1).
Due to the poor solubility of the lignosulfonate polyurethanes (LS-PU), their chemical
characterization was done by 13C solid state NMR, again with comparison to the
corresponding conventional PU (Figure 4). In the spectra of LSA the sharp peak at
56 ppm is related to the LS´s methoxy groups (Nimz 1974, Hatfield et al. 1987). The
second prominent signal at around 147 ppm was attributed to phenolic carbon C-4 in
S units and G units. After the oxypropylation of LSA, the majority of C-4 intensity, as
shown in LS-diol/GCC spectrum, was shifted to 152 ppm, which confirmed the
substitution occurring on the phenolic OH groups. At the same time two new
characteristic peaks of secondary and primary carbons, related to the glycerol
moiety, appeared at around 71 ppm and 63 ppm, respectively.
Upon further carbonation of the LS-diol/LS-GCC mixture, the 5-membered cyclic
carbonate is formed, giving a broad signal appearing at 155 ppm (blue line). The
Page 12 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
84

For Review O
nly
reaction of LS-GCC with 1,6-hexamethylenediamine successfully afforded the
desired LS-PU product, with the two signals at 41 and 27 ppm coming from the
methylene and aminomethylene moieties. The signal at 157 ppm is typical of the
urethane group, appearing at 154 ppm in the conventional PU (purple line).
Carbonation reaction of a model compound
Due to solubility issues and the three-dimensional structural complexity of the
modified lignosulfonates (LS, LS-diol/LS-GCC and LS-GCC), spectral analyses
(ATR-FTIR and NMR) are limited, so that additional support from the reaction of a
model compound was sought. Vanillyl alcohol was used as a strongly simplifying
lignin model compound, and the primary reaction with glycerol carbonate as well as
further carbonation was studied in depth (Scheme 4).
In the literature, vanillyl alcohol has been applied in many studies as a lignin model
compound due to similar structural properties to lignin units (Rothenberg et al. 1966,
Cheng et al. 2017, Zhao et al. 2017). Besides the aromatic methoxyl-group, vanillyl
alcohol contains phenolic and aliphatic OH-groups with different nucleophilicity. Our
first attempt was the solvent-free reaction of vanillyl alcohol and glycerol carbonate
carried out in inert atmosphere at 110°C, which showed a fast color change from
yellowish to yellow-brown. The viscosity was also increased, indicating self-
polymerization of glycerol carbonate. Therefore, dioxane was used as solvent and
reaction medium to minimize the amount of resulting byproducts and polymerization.
The crude product of this reaction was purified by column chromatography after
acetylation to afford VA-diol and VA-GCC. As illustrated in Figure 5 the FTIR spectra
of VA-diol showed – in comparison to untreated vanillyl alcohol – disappearance of
OH peak at 3050-3700 cm-1 and an increase of the absorption band intensity of the
Page 13 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
85

For Review O
nly
CH2 / CH region near 2780-3000 cm-1. In addition, the presence of acetyl groups was
confirmed by the appearance of a sharp band at 1740 cm-1. The spectrum of VA-
GCC showed a sharp peak at 1789 cm-1, which is typical of C=O in cyclic
carbonates.
Further characterization of VA-diol and VA-GCC was carried out by 1D- and 2D-NMR
spectroscopy. As shown in Figure 6, HMBC spectra (long range H-C connectivities
over 2 or more bonds) of VA-GCC show long-range crosspeaks from H9 at
4.82 ppm as well as from H-10 ( 4.50 and 4.40 ppm) to a carbonate carbon at
ca. 155 ppm, confirming the presence of the cyclic carbonate. Moreover, long-range
crosspeaks from H-7 to C-8 and H-8 to C-7 and the presence of an acetyl group on
C-4 also proved that the etherification occurred only at the benzylic position of vanillyl
alcohol and not at the phenolic hydroxyl group. The 1H chemical shift of the acetyl´s
methyl group was detected at 2.3 ppm, which is typical of aromatic acetic acid esters.
The 13C NMR data of VA-CC was in agreement with recent literature data (Salanti et
al. 2016).
The 1H NMR spectrum of the acetylated VA-diol showed the phenolic acetyl group (
2.26 ppm) as well as two additional acetyl groups at 2.04 and 2.00 ppm. In the
HMBC spectra, long-range crosspeaks of the corresponding acetyl carbonyl carbons
at ca. 170 ppm to H-9 and H-10 of the glycerol sidechain and crosspeaks from H-7
to C-8 and H-8 to C-7 unambiguously confirmed that the etherification resulted in the
diol-containing side chain (Figure 7). Previously, acetylated VA-diol had been
synthesized by another synthetic route by Leary et al. (1983); the 13C NMR data of
that study and ours being in good agreement.
In conclusion, vanillyl alcohol reacted with glycerol carbonate at its aliphatic hydroxyl
group, affording a mixture of oxypropylated 1,2-diol and oxypropylated cyclic
Page 14 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
86

For Review O
nly
carbonate (ratio 1:3). However, in spite of the high reactivity of the phenolic OH and
the ease of its deprotonation to the corresponding phenolate in the presence of an
auxiliary base (in our case: DBU), we did not find any reaction at the phenolic site,
contrary to our first expectations. This might be due to ready para-quinone methide
(pQM) intermediate formation under base-catalysis. The same process occurs
through an intra-molecular elimination of water (H+ from the phenolic OH and OH
from the benzylic position) at higher temperatures (Higuchi et al. 2001, Li et al. 2017).
The pQM formation is additionally promoted in the presence of a base (Higuchi et al.
2001). The resulting para-quinone methide intermediate, which can also be
described by its zwitterionic, aromatic resonance structure carrying a phenolate
oxygen (anion) and a benzylic methylene cation, can either add water to re-form the
starting material vanillyl alcohol or a hydroxyl/alkoxyl group from glycerol carbonate
to give the observed VA-diol / VA-GCC. Due to its zwitterionic character, the
corresponding pQM is significantly more reactive than the uncharged vanillyl alcohol
and undergoes nucleophilic attack much faster.
Experiments with homovanillyl alcohol, an homologue to vanillyl alcohol with an
additional methylene group – intended to substantiate the suggested pQM
involvement – showed substitution of both types of hydroxyl groups with glycerol
carbonate (spectra not shown). This confirms that the unusual benzylic
regioselectivity in the vanillyl alcohol case is caused by pQM involvement, while
compounds unable to form such pQM intermediates, such as homovanillyl alcohol,
show the expected reactivity of both phenolic and aliphatic hydroxyl groups, similar to
lignin and LS. Besides the oxypropylated diol and oxypropylated cyclic carbonate
derivatives of homovanillyl alcohol, we observed other side products, such as
oligoglyceryl moieties “capped” with vanillyl alcohol. Formation of these byproducts,
which are inseparable due to their different chain lengths, depends on the solvent
Page 15 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
87

For Review O
nly
amount used and on the vanillyl alcohol / glycerol carbonate ratio. These experiments
demonstrated that vanillyl alcohol was properly chosen with regard to studies of the
oxypropylation reaction, but it was not an appropriate model compound as far as a
similar reactivity to LS was concerned.
As briefly mentioned above, excess glycerol carbonate caused its polymerization to
polyglycerol at 110°C. The same situation is likely to be responsible also for yield
losses in the reaction with lignosulfonate. About 55 wt% LS-GCC/LS-diol powder was
isolated in the oxypropylation, and the rest was difficult to recover, likely because of
etherification with condensed oligoglycerol or polygylcerol carbonate to afford so-
called “LS-polyol” or “liquified LS”. To overcome those undesired polymerization
reactions and the related isolation problems and yield losses, the oxypropylation
should be carried out in suitable solvents, such as dioxane or tetrahydrofuran, and
addition of the glycerol carbonate is to be carried out in small portions, as a starting
point for further optimization.
Conclusion
Polyurethanes based on the bio-based and renewable raw material LS were
successfully synthesized without involvement of questionable isocyanate
intermediates. LS was equipped with glycerol-derived cyclic carbonate moieties that
reacted with an alkyl diamine to the desired polyurethane. The process was mirrored
in a model compound reaction between vanillyl alcohol as a simple lignin model
compound and glycerol carbonate.
To conclude, the outcome of this study shows the feasibility of developing non-
isocyanate-derived PU by utilizing technical LS as the starting material. It is evident
Page 16 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
88

For Review O
nly
that the production of lignin-based PU with optimized or even tailored
physicochemical properties requires much additional research activities in future
which would by far exceed the scope of this account. Nevertheless, with the
presented route, it was shown for the first time that also LS, which are often treated
somewhat stepmotherly in comparison to the well-research kraft or organosolv
lignins, might also serve as the base for PU materials. Due to the special LS
properties – SO3H content, water solubility, relatively high hydrophilicity – the derived
LS-PU obviously have different material properties and application areas, since they
offer qualities that conventional lignin-derived PUs lack, such as water and solvent
compatibility, hydrogel formation, and swelling. LS-PU properties will be the topic of a
follow-up account.
Research funding: Financial support to WOOD Kplus was provided by the Austrian
government, the provinces of lower Austria, upper Austria, and Carinthia, as well as
by Lenzing AG. We also express our gratitude to the University of Natural Resources
and Life Sciences Vienna (BOKU Vienna), the Johannes Kepler University Linz, and
Lenzing AG for their in-kind contributions.
Page 17 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
89

For Review O
nly
References
Akindoyo, J.O., Beg, M.D.H., Ghazali, S., Islam, M.R., Jeyaratnam, N., Yuvaraj, A.R. (2016) Polyurethane types, synthesis and applications – a review. RSC Adv. 6:114453-114482.
Cateto, C.A., Barreiro, M.F., Rodrigues, A.E., Belgacem, M.N. (2011) Kinetic study of the formation of lignin-based polyurethanes in bulk. Reactive and Func. Polym. 71:863-869.
Cateto, C.A., Barreiro, M.F., Ottati, C., Lopretti, M., Rodrigues, A.E., Belgacem, M.N. (2013) Lignin-based rigid polyurethane foams with improved biodegradation. J. Cell. Plast. 50:81-95.
Cheng, C., Wang, J., Shen, D., Xue, J., Guan, S., Gu, S., Luo, K. (2017) Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review. J. Polym. 9:240.
Guan, J., Song, Y., Lin, Y., Yin, X., Zuo, M., Zhao, Y., Tao, X., Zheng, Q. (2011) Progress in Study of Non-Isocyanate Polyurethane. Ind. Eng. Chem. Res. 50:6517-6527.
Hatfield, G.R., Maciel, G.E., Erbatur, O., Erbatur, G. (1987) Qualitative and quantitative analysis of solid lignin samples by carbon-13 nuclear magnetic resonance spectrometry. Anal. Chem. 59:172-179.
Higuchi, M., Yoshimatsu, T., Urakawa, T., Morita, M. (2001) Kinetics and Mechanisms of the Condensation Reactions of Phenolic Resins II. Base-Catalyzed Self-Condensation of 4-Hydroxymethylphenol. Polym. J. 33:799-806.
Kihara, N., Endo, T. (1993) Synthesis and properties of poly(hydroxyurethane)s. J. Polym. Sci. Part A: Polym. Chem. 31:2765-2773.
Korntner, P., Sumerskii, I., Bacher, M., Rosenau, T., Potthast, A. (2015) Characterization of technical lignins by NMR spectroscopy: optimization of functional group analysis by 31P NMR spectroscopy. Holzforschung 69(6): 807-814.
Kreye, O., Mutlu, H., Meier, M.A.R. (2013) Sustainable routes to polyurethane precursors. Green Chem. 15:1431-1455.
Król, P. (2007) Synthesis methods, chemical structures and phase structures of linear polyurethanes. Properties and applications of linear polyurethanes in polyurethane elastomers, copolymers and ionomers. Prog. Mater. Sci. 52:915-1015.
Kühnel, I., Saake, B., Lehnen, R. (2017) Comparison of different cyclic organic carbonates in the oxyalkylation of various types of lignin. Reactive Func. Polym. 120:83-91.
Kühnel, I., Saake, B., Lehnen, R. (2018) A New Environmentally Friendly Approach to Lignin-Based Cyclic Carbonates. Macromol. Chem. Phys. 219:1700613.
Leary, G. J., Sawtell, D.A., Wong, H. (1983) The Formation of Model Lignin -Carbohydrate Compounds in Aqueous Solution. Holzforschung 37:11-16.
Laurichesse, S., Avérous, L. (2014) Chemical modification of lignins: towards biobased polymers. Prog. Polym. Sci. 39:1266-1290.
Lee, B.M., Kang, H.-C., Park, J.-m., Yoon, J.H. (2002) Method of synthesizing glycidyl ether compounds in the absence of water and organic solvents, Korea Research Institute of Chemical Technology. US6392064B2.
Page 18 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
90

For Review O
nly
Li, T., Cao, M., Liang, J., Xie, X., Du, G. (2017) Theoretical Confirmation of the Quinone Methide Hypothesis for the Condensation Reactions in Phenol-Formaldehyde Resin Synthesis. J. Polym. 9:45.
Li, Y., Ragauskas, A.J. (2012) Kraft Lignin-Based Rigid Polyurethane Foam. J. Wood Chem. Technol. 32:210-224.
Maisonneuve, L., Lamarzelle, O., Rix, E., Grau, E., Cramail, H. (2015) Isocyanate-Free Routes to Polyurethanes and Poly(hydroxy Urethane)s. Chem. Rev. 115:12407-12439.
Nimz, H. (1974) Beech Lignin—Proposal of a Constitutional Scheme. Angew. Chem. Int. Edit. 13:313-321.
Nouailhas, H., Aouf, C., Le Guerneve, C., Caillol, S., Boutevin, B., Fulcrand, H. (2011) Synthesis and properties of biobased epoxy resins. part 1. Glycidylation of flavonoids by epichlorohydrin. J. Polym. Sci. Part A: Polym. Chem. 49:2261-2270.
Obaid, N., Kortschot, M.T., Sain, M. (2016) Lignin-based foaming materials. In: Lignin in Polymer Composites. Eds. Faruk, O., Sain, M. William Andrew/Elsevier, Oxford, UK. pp. 217-232.
Pyo, S.-H., Persson, P., Mollaahmad, M.A., Sörensen, K., Lundmark, S., Hatti-Kaul, R. (2011) Cyclic carbonates as monomers for phosgene- and isocyanate-free polyurethanes and polycarbonates. Pure Appl. Chem. 84:637-661.
Rothenberg, S., Luner, P. (1966) Formation of Colored Compounds in the Reaction of Lignin Model Compounds with Alkali and Oxygen. In: Lignin Structure and Reactions. Eds. Marton., Washington, D.C., USA. pp. 90-109.
Salanti, A., Zoia, L., Orlandi, M. (2016) Chemical modifications of lignin for the preparation of macromers containing cyclic carbonates. Green Chem. 18:4063-4072.
Schäffner, B., Schäffner, F., Verevkin, S.P., Börner, A. (2010) Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 110:4554-4581.
Shaikh, A.-A.G., Sivaram, S. (1996) Organic Carbonates. Chem. Rev. 96:951-976.Sumerskii, I., Korntner, P., Zinovyev, G., Rosenau, T., Potthast, A. (2015) Fast track
for quantitative isolation of lignosulfonates from spent sulfite liquors. RSC Adv. 5:92732-92742.
Takagaki, A., Iwatani, K., Nishimura, S., Ebitani, K. (2010) Synthesis of glycerol carbonate from glycerol and dialkyl carbonates using hydrotalcite as a reusable heterogeneous base catalyst. Green Chem. 12:578-581.
Zhao, X., Zhang, Y., Wei, L., Hu, H., Huang, Z., Yang, M., Huang, A., Wu, J., Feng, Z. (2017) Esterification mechanism of lignin with different catalysts based on lignin model compounds by mechanical activation-assisted solid-phase synthesis. RSC Adv. 7:52382-52390.
Page 19 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
91

For Review O
nly
Scheme 1: Alternative synthesis routes for sustainable non-isocyanate polyurethanes. A)
transurethanization between a bis-carbamate and a diol; B) polyaddition between cyclic carbonates and
amines; and B1) synthesis of a cyclic carbonate by CO2 fixation in epoxides.
1031x483mm (120 x 120 DPI)
Page 20 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
92

For Review O
nly
Scheme 2: Synthesis route to lignosulfonate-glycerol cyclic carbonates (LS-GCC). a) etherification of
lignosulfonate acid (LSA) with glycerol carbonate (GC); b) transesterification of the mixture LS-diol/GCC
with dimethyl carbonate (DMC).
236x91mm (120 x 120 DPI)
Page 21 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
93

For Review O
nly
Scheme 3: Reaction of lignosulfonate-glycerol cyclic carbonate (LS-GCC) with diamines to lignosulfonate-
based non-isocyanate polyurethane materials (LS-PU). a) 1,6-hexamethylenediamine (HMDA), stirring
overnight, 70°C; b) part of synthesized LS-PU material 10x magnified.
174x76mm (120 x 120 DPI)
Page 22 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
94
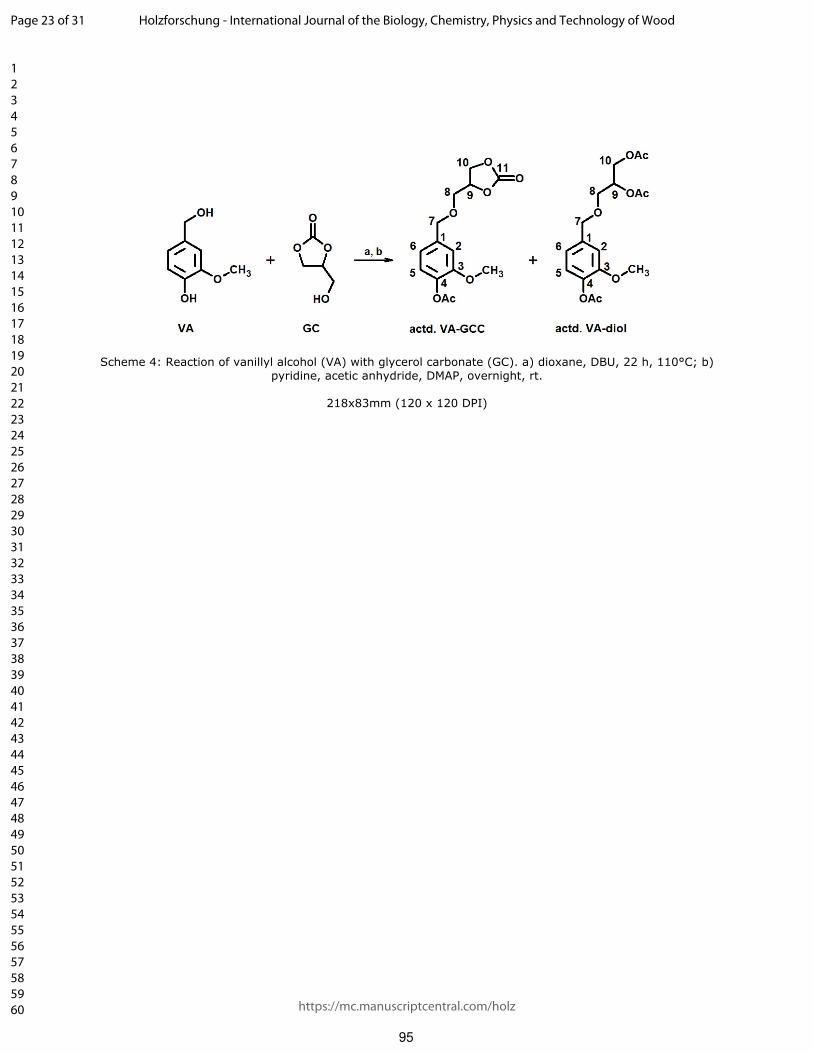
For Review O
nly
Scheme 4: Reaction of vanillyl alcohol (VA) with glycerol carbonate (GC). a) dioxane, DBU, 22 h, 110°C; b)
pyridine, acetic anhydride, DMAP, overnight, rt.
218x83mm (120 x 120 DPI)
Page 23 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
95

For Review O
nly
Figure 1: FTIR spectra of lignosulfonic acid (LSA, black) and its derivatives: mixture of lignosulfonate 1,2-
diol / lignosulfonate-glycerol cyclic carbonate (LS-diol/GCC, red) and lignosulfonate-glycerol cyclic carbonate
(LS-GCC, blue). Spectra were normalized at 1500 cm-1.
234x170mm (120 x 120 DPI)
Page 24 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
96
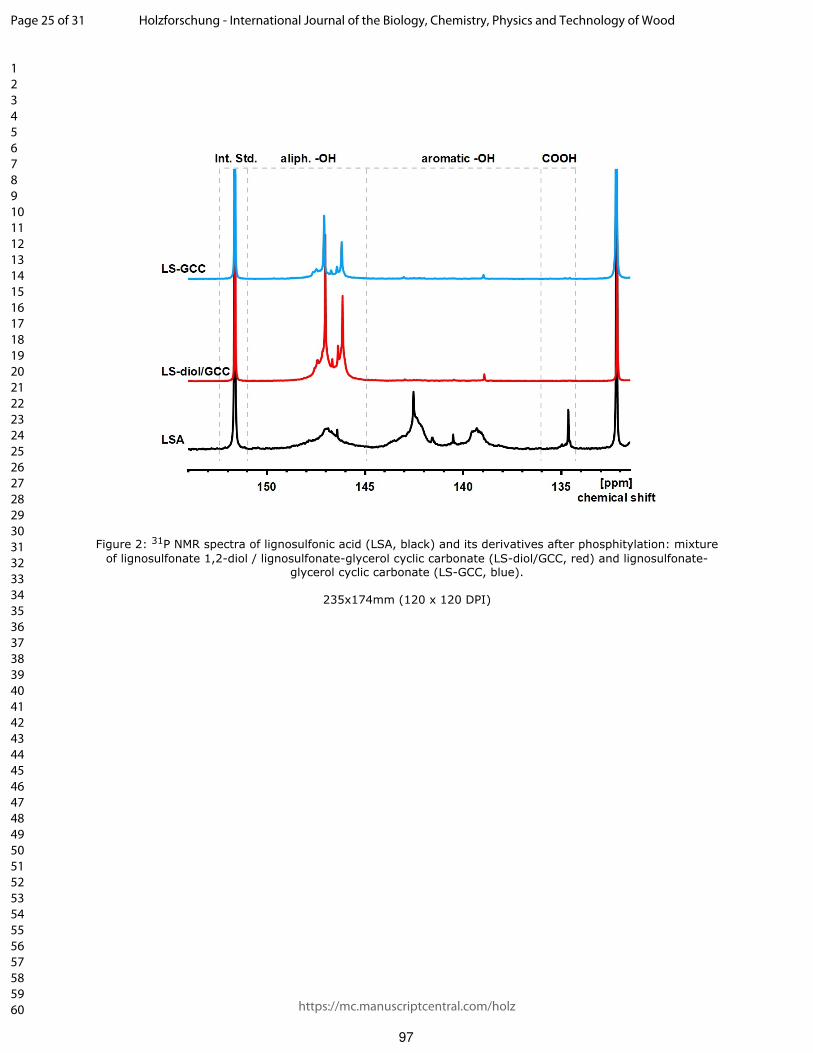
For Review O
nly
Figure 2: 31P NMR spectra of lignosulfonic acid (LSA, black) and its derivatives after phosphitylation: mixture
of lignosulfonate 1,2-diol / lignosulfonate-glycerol cyclic carbonate (LS-diol/GCC, red) and lignosulfonate-
glycerol cyclic carbonate (LS-GCC, blue).
235x174mm (120 x 120 DPI)
Page 25 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
97

For Review O
nly
Figure 3: FTIR spectra of lignosulfonate-glycerol cyclic carbonate (LS-GCC, blue), lignosulfonate
polyurethane (LS-PU, green) and conventional polyurethane (conv. PU, purple).
233x172mm (120 x 120 DPI)
Page 26 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
98

For Review O
nly
Figure 4: 13C solid-state NMR spectra of lignosulfonic acid (LSA, black), mixture of lignosulfonate 1,2-diol /
lignosulfonate-glycerol cyclic carbonate (LS-diol/GCC, red), lignosulfonate-glycerol cyclic carbonate (LS-
GCC, blue), lignosulfonate polyurethane (LS-PU, green) and conventional polyurethane (conv. PU, purple).
234x175mm (120 x 120 DPI)
Page 27 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
99

For Review O
nly
Figure 5: FTIR spectra of vanillyl alcohol (VA, black) and its acetylated derivatives (indicated and
abbreviated as actd., respectively): vanillyl 1,2-diol (VA-diol, red) and vanillyl-glycerol cyclic carbonate (VA-
GCC, blue). Spectra are normalized at 1500 cm-1.
234x174mm (120 x 120 DPI)
Page 28 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
100

For Review O
nly
Figure 6: 1H, 13C HMBC spectrum of acetylated vanillyl-glycerol cyclic carbonate (VA-GCC).
262x229mm (120 x 120 DPI)
Page 29 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
101

For Review O
nly
Figure 7: 1H, 13C HMBC spectrum of acetylated vanillyl-glycerol 1,2-diol (VA-diol).
273x229mm (120 x 120 DPI)
Page 30 of 31
https://mc.manuscriptcentral.com/holz
Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
102

103
9 Related Publication
9.1 Gram-scale economical synthesis of trans-coniferyl alcohol and its
corresponding thiol (Publication IV)

104

Holzforschung 2019; aop
Hassan Amer*, Vebi Mimini, Dominik Schild, Uwe Rinner, Markus Bacher, Antje Potthast and Thomas Rosenau*
Gram-scale economical synthesis of trans-coniferyl alcohol and its corresponding thiol
https://doi.org/10.1515/hf-2018-0297Received December 13, 2018; accepted February 19, 2019; previously published online xx
Abstract: Coniferyl alcohol is considered to be a potent
antioxidant and a precursor of several bioactive products.
In addition, it is a frequently used as a model compound
in lignin chemistry. Coniferyl thiol is used analogously to
study the sulfur chemistry in technical lignins. Coniferyl
alcohol was synthesized in a large scale from commer-
cially available ferulic acid by a mixed anhydride reduc-
tion method which affords high yields (84%) under
very mild conditions and allows using sodium borohy-
dride. The nucleophilic substitution of 4-O-acetylated
coniferyl alcohol (3) with thioacetic acid in the presence
of dimethylformamide (DMF) dineopentylacetal afforded
4-O-acetylated coniferyl thioacetate (5) in a 70% yield,
which, in a 72% yield, was deprotected to the respective
thiol (6). Both coniferyl alcohol and coniferyl thiol were
comprehensively analytically characterized [one-dimen-
sional (1D) and two-dimensional (2D) nuclear magnetic
resonance (NMR) spectroscopy]. The presented approach
renders the two model substances readily available on a
gram scale and according to low-risk, environmentally
compatible protocols.
Keywords: coniferyl alcohol, coniferyl thiol, lignin, tech-
nical lignin
Introduction
The antioxidative activity of polyphenols is commonly
attributed to their hydroxyl groups, but it is not the only
factor defining their potency. In the case of coniferyl
alcohol and its corresponding thiol (coniferyl thiol),
there is a single phenolic hydroxyl group at an aromatic
ring in para-position to a conjugated allyl side chain.
This para-substitution and the conjugated double
bond allow the corresponding phenoxyl radical to be
highly delocalized. This delocalization is a fundamen-
tal feature in the biosynthesis of lignin (Boerjan et al.
2003; Weng and Chapple 2010). The compounds are also
thought to be beneficial to human health by exerting
various biological effects, such as free radical scaveng-
ing, metal chelation, modulation of enzymatic activity
and alteration of signal transduction pathways (Singh
and Aggarwa 1995; Stocker 1999; Xu and Simon Cho
1999; Cho and Xu 2000).
Phenolic allylic alcohols are precursors of several
bioactive products, such as flavonoids, lignans and cou-
marins (Prakash Chaturvedula et al. 2004; El-Gamal
et al. 2005; Ochi et al. 2005). They are also important as
key intermediates in the chemical synthesis of many bio-
logically active natural product analogs (Yamamoto et al.
2003; Wan et al. 2005, 2006).
Several multi-step synthetic approaches have been
developed to prepare coniferyl alcohol from various com-
mercially available starting materials, such as ferulic acid
(Wang et al. 2009), guaiacol (Kobota et al. 2011), eugenol
and isoeugenol (Zanarotti 1985), and coniferaldehyde
*Corresponding authors: Hassan Amer, Department of Chemistry, Division of Chemistry of Renewables, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Str. 24, A-3430 Tulln, Austria; IMC Fachhochschule Krems Gmbh, Department of Life Sciences, Campus Krems, Trakt G, A-3500 Krems, Austria; and Department of Natural and Microbial Products Chemistry, National Research Centre, P.O. 12622, Dokki, Giza, Egypt, e-mail: [email protected], [email protected]; and Thomas Rosenau, Department of Chemistry, Division of Chemistry of Renewables, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Str. 24, A-3430 Tulln, Austria; and Johan Gadolin Process Chemistry Centre, Åbo Akademi University, Porthansgatan 3, Åbo/Turku FI-20500, Finland, e-mail: [email protected] Vebi Mimini: Department of Chemistry, Division of Chemistry of Renewables, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Str. 24, A-3430 Tulln, Austria; and Wood Kplus – Kompetenzzentrum Holz GmbH, Altenberger Straße 69, A-4040 Linz, AustriaDominik Schild and Uwe Rinner: IMC Fachhochschule Krems Gmbh, Department of Life Sciences, Campus Krems, Trakt G, A-3500 Krems, AustriaMarkus Bacher and Antje Potthast: Department of Chemistry, Division of Chemistry of Renewables, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Str. 24, A-3430 Tulln, Austria
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46105

2 H. Amer et al.: Gram-scale synthesis of coniferyl alcohol and its corresponding thiol
(Lu and Ralph 1998). Among these methods, the reduc-
tion of ferulic acid derivatives with different reducing
agents, mostly LiAlH4 derivatives, is the most prominent
approach. However, the reduction of ferulic esters suffers
from the formation of undesirable and hard-to-separate
side products, complicated workup, and sometimes low
yields, which taken together would not make an appro-
priate method for large-scale preparation. One important
negative aspect with regard to handling and upscalabil-
ity is the use of aluminum hydride reductants with their
inherent dangers of flammability and hydrogen evolu-
tion. Coniferyl alcohol has been prepared on a 20-g scale
by the selective reduction of ethyl ferulate by diisobutyl
aluminum hydride (DIBAL-H) (Quideau and Ralph 1992).
Normally, cinnamic alcohols prepared according to
such reduction methods are contaminated with varying
amounts of side products, which are hard to separate, at
least without further derivatization (acetylation). Because
of that, all of the aforementioned methods are not eco-
nomically feasible: the price of coniferyl alcohol still
remains 80 times higher than that of ferulic acid. This is
not only due to the reagent costs per se, but also due to the
requirements for workup and purification which must be
taken into account.
Allyl thiols, the major metabolites of garlic, have
received increased attention because of their therapeutic
effects. Coniferyl thiol, as substituted allyl thiol, is con-
sidered to be a bioactive product and a key intermediate
for the synthesis of multifunctional sulfur-containing
polyphenolic derivatives. Also, it is a free radical scaven-
ger and a reductant due to the reducing ability of its thiol
group (Jiang et al. 2018).
There are rather few reports on the synthesis of
coniferyl thiols. Lindeberg prepared coniferyl thioacetate
from the corresponding coniferyl bromide with potassium
thioacetate in a 68% yield (Lindeberg 1980). Coniferyl
thioacetate was detected during kraft pulping of pine
chips (after peracetylation) and confirmed by 1H and 13C
nuclear magnetic resonance (NMR) (Robert et al. 1984).
Later, coniferyl thioacetate has been synthesized in high
yields from selectively protected coniferyl alcohol and
thioacetic acid in the presence of triphenyl phosphine and
diisopropyl azodicarboxylate (Zhao and Sinnott 2000).
Based on a recent study (Panzella et al. 2018), we
report a one-pot, conventional mixed anhydride reduc-
tion method for the direct chemoselective reduction of
4-O-acetylated ferulic acid with sodium borohydride,
which produces the corresponding coniferyl alcohol in
high yields on a large gram scale. Further reaction with
thioacetic acid followed by complete deprotection affords
the corresponding coniferyl thiol.
Materials and methods
Ferulic acid was purchased from Sigma-Aldrich Co. (Schnelldorf,
Germany) and used without further purification. Reagent-grade sol-
vents were used for all extraction and workup procedures. Commercial
chemicals, including solvents, were of the highest purity available and
used without further purification unless otherwise noted. All reactions
were carried out in flame-dried glassware under an argon atmosphere,
unless otherwise specified. Thin-layer chromatography (TLC) was
performed on silica gel 60 F254 pre-coated glass plates (Merck, Darm-
stadt, Germany). Flash column chromatography was performed on
silica gel 60 from Merck. NMR spectra were run in perdeuterated sol-
vents, on a Bruker Avance II 400 instrument (Rheinstetten, Germany)
with a resonance frequency of 400.13 MHz for 1H and 100.62 MHz for 13C. The chemical shift values are given in δ parts per million (ppm) val-
ues relative to tetramethylsilane (TMS), and the coupling constants in
Hz. Complete resonance assignments were done based on one-dimen-
sional (1D) and two-dimensional (2D) NMR methods.
trans-3-(4-Acetoxy-3-methoxyphenyl)-2-propenoic acid (2): trans-
Ferulic acid (1, 0.26 mol, 50 g) was dissolved in pyridine (100 ml) and
stirred at 4°C under argon for 1 h. Acetic anhydride (50 ml) was added
drop-wise at the same temperature. The reaction mixture was stirred
at room temperature overnight under an argon atmosphere. The mix-
ture was poured onto crushed ice and neutralized with the drop-wise
addition of ice-cold concentrated sulfuric acid. The residue was sepa-
rated by filtration, washed with cold water and dried under vacuum
to yield 2 in an 89% yield (54 g) as a white solid.1H NMR (DMSO-d
6): δ 12.41 (br.s, 1H, OH); 7.57 (d, 1H, 3J = 16.0 Hz,
H-α), 7.47 (d, 1H, 4J = 1.8 Hz, H-2), 7.25 (dd, 1H, 3J = 8.1 Hz, 4J = 1.8 Hz,
H-6), 7.11 (d, 1H, 3J = 8.1 Hz, H-5), 6.58 (d, 1H, 3J = 16.0 Hz, H-β), 3.81 (s,
3H, OCH3), 2.25 (s, 3H, CH
3 in OAc).
13C NMR (DMSO-d6): δ 168.5 (C=O in OAc), 167.7 (C-γ), 151.3 (C-3),
143.5 (C-α), 140.9 (C-4), 133.4 (C-1), 123.3 (C-5), 121.5 (C-6), 119.7 (C-β),
111.9 (C-2), 56.1 (OCH3), 20.5 (CH
3 in OAc).
trans-3-(4-Acetoxy-3-methoxyphenyl)-2-propenol (3): To a solu-
tion of carboxylic acid 2 (50 g, 0.21 mol) in EtOAc (500 ml) were
added Et3N (45 ml) and ethyl chloroformate (25 ml, 0.26 mol) at 4°C.
After stirring overnight at 4°C, the mixture was cooled to −10°C and
NaBH4 (25 g, 0.66 mol) suspended in 25 ml of DMF was added drop-
wise to the solution. The mixture was stirred at 4°C overnight and
the excess of NaBH4 was quenched with cold water (200 ml). The
organic layer was washed with ice-cold 5 M HCl (50 ml), to neutralize
the reaction mixture, and with brine. The organic extract was filtered
through a piece of cotton to remove the residual water and concen-
trated in vacuo. The residue was purified by column chromatography
(EtOAc/n-heptane, v/v = 1:1) to yield compound 3 (42 g, 89%) as a pale
yellow syrup.1H NMR (CDCl
3): δ 6.98 (d, 1H, 4J = 1.6 Hz, H-2), 6.98 (d, 1H,
3J = 8.1 Hz, H-5), 6.95 (dd, 1H, 3J = 8.1 Hz, 4J = 1.6 Hz, H-6), 6.58 (dt, 1H, 3J = 15.9 Hz, 4J = 1.6 Hz, H-α), 6.31 (dt, 1H, 3J = 15.9 Hz, 3J = 5.7 Hz, H-β),
4.32 (dd, 2H, 3J = 5.7 Hz, 4J = 1.6 Hz, H-γ), 3.84 (s, 3H, OCH3), 2.31 (s,
3H, CH3 in OAc). 13C NMR (CDCl
3): δ 169 (C=O in OAc), 151.1 (C-3), 139.4
(C-4), 135.8 (C-1), 130.4 (C-α), 128.9 (C-β), 122.8 (C-5), 119.1 (C-6), 110.5
(C-2), 63.5 (C-γ), 55.8 (OCH3), 20.6 (CH
3 in OAc).
trans-3-(4-Hydroxy-3-methoxyphenyl)-2-propenol (4): To a solu-
tion of 4-O-acetyl coniferyl alcohol 3 (1.0 g, 4.28 mmol) in DMF
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46106

H. Amer et al.: Gram-scale synthesis of coniferyl alcohol and its corresponding thiol 3
(10 ml), hydrazine acetate (1.0 g, 2 equiv.) was added. The solution
was stirred overnight at room temperature. The reaction mixture was
diluted with toluene and concentrated in vacuo. The crude product
was chromatographed on silica gel (n-heptane/EtOAc, v/v = 1:1) to
yield coniferyl alcohol 4 (700 mg, 86%). The deprotection of acetate
3 should be carried out shortly before usage of the free coniferyl alco-
hol, as the former is far more stable upon storage.1H NMR (CDCl
3): δ 6.92 (d, 1H, 4J = 1.8 Hz, H-2), 6.90 (dd, 1H,
3J = 8.2 Hz, 4J = 1.8 Hz, H-6), 6.86 (d, 1H, 3J = 8.2 Hz, H-5), 6.54 (dt, 1H, 3J = 15.8 Hz, 4J = 1.4, H-α), 6.22 (dt, 1H, 3J = 15.8 Hz, 3J = 5.9 Hz, H-β), 5.63
(s, 1H, OHph.
), 4.30 (d, 2H, 3J = 5.9 Hz, H-γ), 3.91 (s, 3H, OCH3), 1.42 (br.s,
1H, γ-OH). 13C NMR (CDCl3): δ 146.6 (C-3), 145.6 (C-4), 131.4 (C-α), 129.2
(C-1), 126.1 (C-β), 120.3 (C-6), 114.4 (C-5), 108.3 (C-2), 63.8 (C-γ), 55.8
(OCH3).
trans-3-(4-Acetoxy-3-methoxyphenyl)-2-propenyl thioacetate (5):
A mixture of dimethylformamide (DMF) dineopentylacetal (15.1 ml,
54.2 mmol) and thioacetic acid (3.8 ml, 53.9 mmol) in toluene (300 ml)
was added to a solution of 3 (4.0 g, 18 mmol) in toluene (100 ml) at
room temperature under argon with stirring. The mixture was heated
at 70°C, with stirring overnight. The reaction mixture was concen-
trated in vacuo to give a brown residue. Column chromatography on
silica gel (n-heptane/EtOAc, v/v = 2:1) gave compound 5 (3.7 g, 70%)
as a brownish-yellow solid.1H NMR (CDCl
3): δ 6.92 (d, 1H, 3J = 8.2 Hz, H-5), 6.90 (d, 1H,
4J = 1.9 Hz, H-2), 6.86 (dd, 1H, 3J = 8.2 Hz, 4J = 1.9 Hz, H-6), 6.47 (br.d,
1H, 3J = 15.6 Hz, H-α), 6.07 (dt, 1H, 3J = 15.6 Hz, 3J = 7.4 Hz, H-β), 3.76 (s,
3H, OCH3), 3.64 (dd, 2H, 3J = 7.4 Hz, 4J = 1.2 Hz, H-γ), 2.30 (s, 3H, CH
3 in
SAc), 2.24 (s, 3H, CH3 in OAc).
13C NMR (CDCl3): δ 194.5 (C=O in SAc), 168.5 (C=O in OAc), 150.8
(C-3), 139.1 (C-4), 135.3 (C-1), 132.1 (C-α), 124.5 (C-β), 122.5 (C-5), 118.7 (C-6),
109.7 (C-2), 55.4 (OCH3), 31.2 (C-γ), 30.1 (CH
3 in SAc), 20.2 (CH
3 in OAc).
trans-3-(4-Hydroxy-3-methoxyphenyl)-2-propenethiol (6): Coniferyl
thioacetate 5 (500 mg, 1.78 mmol) was dissolved in tetrahydrofuran
(THF) (5 ml). Cyclohexene (1 ml), ethanol (1 ml) and hydrazine
hydrate (700 µl, ∼14 mmol) were added. The solution was stirred at
room temperature for 4 h and diluted with toluene (50 ml). On con-
centration, it gave a brownish-yellow solid that was purified by chro-
matography on silica gel (n-heptane/EtOAc, v/v = 2:1) to yield thiol 6
(250 mg, 72%) as a yellow syrup.1H NMR (CDCl
3): δ 6.89 (bs, 1H, H-2), 6.8 (d, J = 1.1 Hz, 2H, H-5,
H-6), 6.41 (dt, 1H, 3J = 15.6 Hz, 4J = 1.3 Hz, H-α), 6.15 (dt, 1H, 3J = 15.6 Hz, 3J = 7.4 Hz, H-β), 5.61 (s, 1H, OH
ph), 3.91 (s, 3H, OCH
3), 3.33 (dt, 2H,
3J = 7.4 Hz, 4J = 1.3 Hz, H-γ), 1.51 (t, 1H, 3J = 7.6 Hz, SH).
13C NMR (CDCl3): δ 146.6 (C-3), 145.7 (C-4), 130.7 (C-α), 129.3 (C-1),
126.5 (C-β), 120.2 (C-5), 114.4 (C-6), 108.2 (C-2), 55.9 (OCH3), 27.3 (C-γ).
Results and discussion
The target compounds coniferyl alcohol (4) and coniferyl
thiol (6) were synthesized according to the route outlined
in Scheme 1. Commercially available and relatively inex-
pensive ferulic acid was used as the starting material. All
intermediates and the products were fully analytically
characterized by NMR (see the Materials and methods
section). The synthesis began with the protection of (E)-
ferulic acid (1) by conventional acetylation with acetic
anhydride in pyridine to afford 4-O-acetylated ferulic acid
(2) quantitatively.
The 1H spectrum of 2 shows a singlet at 2.15 ppm char-
acteristic of the methyl group in phenolic acetates (see
Table 1). The NMR data were fully in accordance with
those reported previously (Hosoda et al. 2001). For reduc-
tion, 2 was converted to a mixed carboxylic-carbonic
anhydride with ethyl chloroformate in the presence of
trimethylamine at a low temperature. The intermediate
mixed anhydride is formed in situ and does not need to be
isolated. Sodium borohydride is able to reduce the mixed
anhydride without affecting the olefinic double bond. This
mild reduction approach provided the 4-O-Ac protected
coniferyl alcohol as the only product (3). Multigram quan-
tities of 4-O-Ac- ferulic acid were reduced by this one-pot
reaction in an excellent 89% yield without any difficulty.
One of the practical advantages – besides the lack of side
products as found upon the reduction of ferulic acid esters
and the high yield – is that no aluminum hydride-based
reductants were required which are hazardous and hard to
handle on a larger scale. Another benefit is the commercial
availability and the low cost of ferulic acid compared with
the somewhat more exotic and definitely more expensive
coniferaldehyde as the starting material (Lu and Ralph
SH
OH
OMeOMe
OR
SR’OH
OMe
OR
OHO
OMe
OR
5 R = R’ = Ac 63 R = Ac
4 R = H2 R = Ac
1 R = H
i
ii
iii
iv v
Scheme 1: Reagents and reaction conditions: (i) Ac2O/pyridine/RT, 89%; (ii) ethyl chloroformate/NaBH4/DMF, 89%; (iii) hydrazine acetate/DMF, 86%; (iv) AcSH/DMF dineopentyl acetal/PhMe/80°C, 70%; (v) NH2NH2/THF:cyclohexene (5:1), 72%.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46107

4 H. Amer et al.: Gram-scale synthesis of coniferyl alcohol and its corresponding thiol
1998). Attempts to crystallize 4-O-acetylated coniferyl
alcohol from methylene chloride/diethyl ether were unsuc-
cessful, but succeeded with the deprotected compound 4
(Lu and Ralph 1998).
Spectroscopic evidence for the reduction of 2 was
provided by the characteristic signal of the γ-protons at δ
4.32 ppm in the 1H NMR spectrum of product 3 (Table 1).
Furthermore, the presence of resonances at δ 6.58 and 6.31
associated with the α- and β-protons, respectively, con-
firmed the selectivity of the reduction, not affecting the
olefinic double bond. 1H and 13C chemical shift values for
compound 3 were in good agreement with those reported
(Zhu et al. 2013).
The phenolic acetate was completely cleaved in the
presence of hydrazine acetate, a buffered, good nucleo-
phile, to afford coniferyl alcohol 4 in an 86% yield. The
NMR spectral data (Tables 1 and 2) of coniferyl alcohol
4 are in full agreement with published data (Nakamura
and Higuchi 1976). As coniferyl alcohol is not stable upon
storage at room temperature under air, it is recommended
to store it in its stable, protected form of acetate 3, and
deprotect only the amounts needed short before usage.
Based on natural thiol-containing antioxidant com-
pounds, it was suggested that a sulfhydryl group in addi-
tion to a phenolic hydroxyl group and conjugated double
bonds can increase the antioxidant capacity (Devasa-
gayam et al. 1991; Jiang et al. 2018).
Tab
le 1
: 1 H
-NM
R da
ta o
f com
poun
ds 2
–6
.
Pro
ton
2
3
4
5
6
δ
J [H
z]δ
J
[Hz]
δ
J [H
z]δ
J
[Hz]
δ
J [H
z]
2
7.47
(d)
1.
8 6.
98 (d
)
1.6
6.92
(d)
1.
8 6.
90 (d
)
1.9
6.89
(bs)
5
7.
11 (d
)
8.1
6.98
(d)
8.
1 6.
86 (d
)
8.2
6.91
(d)
8.
2 6.
8 (d
)
1.1
6
7.25
(dd)
8.
1; 1
.8
6.95
(dd)
8.
1; 1
.6
6.90
(dd)
8.
2; 1
.8
6.86
(dd)
8.
2; 1
.9
6.8
(d)
1.
1α
7.
57 (d
)
16.0
6.
58 (d
t)
15.9
; 1.5
6.
54 (d
t)
15.8
; 1.4
6.
47 (b
d)
15.6
6.
41 (d
t)
15.6
; 1.3
β
6.58
(d)
16
.0
6.31
(dt)
15
.9; 5
.7
6.22
(dt)
15
.8; 5
.9
6.07
(dt)
7.
4; 7
.3; 1
5.6
6.15
(dt)
15
.6; 7
.2γ
4.
32 (d
d)
5.7;
1.5
4.
30 (d
)
5.9
3.64
(dd)
7.
4; 1
.2
3.33
(dt)
7.
4; 1
.3-O
CH
3
3.81
(s)
3.84
(s)
3.91
(s)
3.76
(s)
3.91
(s)
O
COC
H3 (p
he
no
l.)
2.15
(s)
2.31
(s)
2.24
(s)
S
COC
H3 (a
lip
h.)
2.
30 (s
)
-OH
12
.41
(br.
s)
5.
63 (s
)1.
42 (b
r.s)
5.61
(s)
-SH
1.
51 (t
)
7.6
NM
R, n
ucle
ar m
agne
tic
reso
nanc
e.
Table 2: 13C NMR data of compounds 2–6.
Carbon 2 3 4 5 6
1 133.4 135.8 129.2 135.3 129.32 111.9 110.5 108.3 109.7 108.23 151.3 151.1 145.6 150.8 146.64 140.9 139.4 146.6 139.1 145.55 123.3 122.8 114.4 122.5 114.46 121.5 119.1 120.3 118.7 120.2α 143.5 130.4 131.4 132.1 130.7β 119.7 128.9 126.1 124.5 126.5γ 167.7 63.5 63.8 31.2 27.3-OCH3 56.1 55.8 55.8 55.4 55.9OCOCH
3 (phenol.)
20.5 20.6 20.2OCOCH3
(phenol.)168.5 169.0 168.5
SCOCH3 (aliph.)
30.1SCOCH3
(aliph.)194.5
All assignments are based on 1D and 2D NMR (HMBC, HSQC) experiments. Compound 2 was measured in DMSO-d6, and the other compounds in CDCl3.1D, One-dimensional; 2D, two-dimensional; DMSO, dimethyl sulfoxide; HMBC, heteronuclear multiple bond correlation; HSQC, heteronuclear single quantum coherence; NMR, nuclear magnetic resonance.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46108

H. Amer et al.: Gram-scale synthesis of coniferyl alcohol and its corresponding thiol 5
The second part of our study was thus concerned with
coniferyl thiol. The introduction of the thioacetate group
into the 4-O-Ac protected coniferyl alcohol was accom-
plished by reaction with thioacetic acid in the presence of
N,N-DMF dineopentyl acetal. The primary alcohol is acti-
vated by N,N-DMF dineopentyl acetal toward subsequent
displacement with the strongly nucleophilic thioacetic
acid. The yields of thioester 5 are moderate to good at 70%.
The 1H NMR spectrum of 5 shows an additional singlet at
2.30 ppm corresponding to the thioacetate methyl group,
besides the phenolic acetate at 2.24 ppm, for the compiled
NMR data, which are in full agreement with the literature
(Lindeberg 1980), see Tables 1 and 2.
The final step of the synthetic route was the simul-
taneous deacetylation of both the thiol and the phenolic
hydroxyl moieties with hydrazine hydrate to give coniferyl
thiol 6 in a 72% yield. The 1H spectrum of the product in
CDCl3 shows a singlet at 5.61 ppm and a triplet at 1.51 ppm
corresponding to the phenolic hydroxyl proton and thiol
proton, respectively. The thiol-bearing γ-methylene group
resonated at 3.33 ppm/27.3 ppm (1H/13C). NMR data fully
agreed with the literature (Zhao and Sinnott 2000).
Conclusion
We have described an easily scalable approach for the
preparation of coniferyl alcohol and coniferyl thiol, start-
ing from the inexpensive and commercially available
ferulic acid. By a mixed anhydride reduction method, it
avoids intermediate ferulic esters and their notorious
byproduct formation, as well as aluminum hydride rea-
gents that are hard to handle in larger amounts.
This method is attractive due to the ready availabil-
ity and low price of the starting material and reducing
agent and the high overall yields. We hope that the pro-
cedure will be accepted soon by the community of lignin
researchers and will find wide usage.
Author contributions: All the authors have accepted
responsibility for the entire content of this submitted
manuscript and approved submission.
Research funding: Financial support from the Austrian
Research Promotion Agency (FFG) and Amt der NÖ
Landesregierung, Abteilung Wirtschaft, Tourismus and
Technologie Landhausplatz 1, Haus 14, 3109 St. Pölten,
Austria is gratefully acknowledged.
Employment or leadership: None declared.
Honorarium: None declared.
References
Boerjan, W., Ralph, J., Baucher, M. (2003) Lignin biosynthesis. Annu. Rev. Plant Biol. 54:519–546.
Cho, B., Xu, S. (2000) Effects of allyl mercaptan and various allium-derived compounds on cholesterol synthesis and secretion in hep-G2 cells. Comp. Biochem. Physiol. C Toxicol. Pharmacol. C 126:195–201.
Devasagayam, T.P., Sundquist, A.R., Mascio, P.D., Kaiser, S., Sies, H. (1991) Activity of thiols as singlet molecular oxygen quenchers. J. Photochem. Photobiol. B: Biol. 1:105–116.
El-Gamal, A., Wang, S.-K., Dai, C.-F., Chen, C.I.-G., Duh, C.-Y. (2005) Prenylbicyclogermacrane diterpenoids from the formosan soft coral Nephthea pacifica. J. Nat. Prod. 68:74–77.
Hosoda, A., Nomura, E., Mizuno, K., Taniguchi, H. (2001) Preparation of a (±)-1,6-Di-O-feruloyl-myo-inositol derivative: an efficient method for introduction of ferulic acid to 1,6-vicinal hydroxyl groups of myo-inositol. J. Org. Chem. 66:7199–7201.
Jiang, J., Zang, S., Li, D., Wang, K., Tian, S., Yu, A., Zhang, Z. (2018) Determination of antioxidant capacity of thiol-containing com-pounds by electron spin resonance spectroscopy based on Cu2+ ion reduction. Talanta 184:23–28.
Kobota, K., Mimura, M., Sigawara, M., Watanabe, T. (2011) Synthesis of stable isotope-labeled precursors for the biosyntheses of capsaicinoids, capsinoids, and capsiconinoids. Biosci. Biotech-nol. Biochem. 75:1611–1614.
Lindeberg, O. (1980) Synthesis of coniferyl and dihydroconiferyl derivatives using radical bromination with N-bromosuccinimide as the key step. Acta Chem. Scand. B 34:15–20.
Lu, F., Ralph, J. (1998) Highly selective syntheses of coniferyl and sinapyl alcohols. J. Agric. Food Chem. 46:1794–1796.
Nakamura, Y., Higuchi, T. (1976) A new synthesis of coniferyl alde-hyde and alcohol. Wood Res. 59/60:101–105.
Ochi, T., Shibata, H., Higuti, T., Kodama, K., Kusumi, T., Takaishi, Y. (2005) Anti-Helicobacter pylori compounds from Santalum
album. J. Nat. Prod. 68:819–824.Panzella, L., DellaGreca, M., Longobardo, L. (2018) A facile
preparation of hydroxycinnamyl alcohols with simultaneous protection of phenol groups as carbonate. ChemistrySelect 3:10637–10640.
Prakash Chaturvedula, V.S., Hecht, M.S., Gao, Z., Jones, S.H., Feng, X., Kingston, D.G. (2004) New neolignans that inhibit DNA polymerase β lyase. J. Nat. Prod. 67:964–967.
Quideau, S., Ralph, J. (1992) Facile large-scale synthesis of coniferyl, sinapyl, and p-coumaryl alcohol. J. Agric. Food Chem. 40:1108–1110.
Robert, D., Bardet, M., Gellerstedt, G., Lindfors, E.-L. (1984) Structural changes in lignin during kraft cooking. Part 3. On the structure of dissolved lignins. J. Wood Chem. Technol. 4:239–263.
Singh, S., Aggarwa, B.B. (1995) Activation of transcription factor NF-jB is suppressed by curcumin (diferuloylmethane). J. Biol. Chem. 270:24995–25000.
Stocker, R. (1999) Dietary and pharmacological antioxidants in atherosclerosis. Curr. Opin. Lipidol. 10:589–597.
Wan, S., Landis-Piwowar, K., Kuhn, D., Chen, D., Dou, Q. (2005) Structure-activity study of epi-gallocatechin gallate (EGCG) analogs as proteasome inhibitors. Bioorg. Med. Chem. 13:2177–2185.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46109

6 H. Amer et al.: Gram-scale synthesis of coniferyl alcohol and its corresponding thiol
Wan, S., Dou, Q., Chan, T. (2006) Regiospecific and enantioselective synthesis of methylated metabolites of tea catechins. Tetrahe-dron 62:5897–5904.
Wang, X., Li, X., Xue, J., Zhao, Y., Zhang, Y. (2009) A novel and efficient procedure for the preparation of allylic alcohols from α,β-unsaturated carboxylic esters using LiAlH4/BnCl. Tetrahe-dron Lett. 50:413–415.
Weng, J.-K., Chapple, C. (2010) The origin and evolution of lignin. New Phytol. 187:273–285.
Xu, S., Simon Cho, B. (1999) Allyl mercaptan, a major metabolite of garlic compounds, reduces cellular cholesterol synthesis and its secretion in Hep-G2 cells. J. Nutr. Biochem. 10:654–659.
Yamamoto, K., Garbaccio, R.M., Stachel, S., Solit, D., Chiosis, G., Rosen, N., Danishefsky, S. (2003) Total synthesis
as a resource in the discovery of potentially valuable antitumor agents: cycloproparadicicol. J. Angew. Chem. Int. Ed. 42:1280–1284.
Zanarotti, A. (1985) Synthesis and reactivity of vinyl quinone meth-ides. J. Org. Chem. 50:941–945.
Zhao, Y., Sinnott, M.L. (2000) A new affinity ligand for the isolation of a single ‘feruloyl esterase’ (FAE-III) from Aspergillus niger. Bioorg. Med. Chem. 8:917–924.
Zhu, Y., Regner, M., Lu, F., Kim, H., Mohammadi, A., Pearson, T.J., Ralph, J. (2013) Preparation of monolignol γ-acetate, γ-p-hydroxycinnamate, and γ-p-hydroxybenzoate conjugates: selective deacylation of phenolic acetates with hydrazine acetate. RSC Adv. 3: 21964–21970.
Bereitgestellt von | Universitätsbibliothek Bodenkultur Wien
Angemeldet
Heruntergeladen am | 24.04.19 16:46110

111
10 Appendix
10.1 Additional publication
10.1.1 Novel carbamoyl type quinine and quinidine based chiral anion exchangers implementing alkyne–azide cycloaddition immobilization chemistry (Publication V)

112

Journal of Chromatography A, 1337 (2014) 85–94
Contents lists available at ScienceDirect
Journal of Chromatography A
j o ur na l ho me page: www.elsev ier .com/ locate /chroma
Novel carbamoyl type quinine and quinidine based chiral anion
exchangers implementing alkyne–azide cycloaddition
immobilization chemistry
Hubert Hettegger a, Michal Kohoutb, Vebi Mimini a, Wolfgang Lindner a,∗
a Institute of Analytical Chemistry, University of Vienna, Waehringer Strasse 38, 1090 Vienna, Austriab Department of Organic Chemistry, Institute of Chemical Technology Prague, Technická 5, 16628 Prague, Czech Republic
a r t i c l e i n f o
Article history:
Received 31 December 2013
Received in revised form 10 February 2014
Accepted 11 February 2014
Available online 18 February 2014
Keywords:
Chiral anion exchange chromatography
Cinchona alkaloids
Click chemistry
Copper-catalysis
Enantiomer separation
Liquid chromatography
a b s t r a c t
The synthesis and chromatographic evaluation of a series of new Cinchona derived chiral weak anion
exchangers is presented. Huisgen Cu(I) mediated alkyne–azide cycloaddition, so-called click chemistry,
was used as an immobilization strategy. In this way it was possible to immobilize about 90% of offered
selector via 1,2,3-triazole linker, which displays a more efficient way of binding the selector to modified
silica compared to common radical mediated thiol-ene addition. Problems associated with potential
radical scavenging properties of chiral selectors thereby could be circumvented. The evaluation of the
synthesized chiral stationary phases regarding chromatographic behavior was carried out using polar
organic mode mobile phase composition and a set of representative chiral organic acids. Different loading
densities revealed an optimum selector density of about 310 �mol/g chiral stationary phase with respect
to resolution and selectivity. A decrease of performance was observed for higher loading, indicating
mutual spatial influence of selector units leading to sterical hindrance. In addition, we observed that
the effect of free azide groups on retention is negligible and the overall chromatographic behavior is
comparable to other Cinchona derived chiral stationary phases.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
The separation of chiral compounds is an important topic
both in analytical science and on industrial scale, including the
separation of bioactive compounds [1–3], flavors and fragrances
[4,5], environmental pollutants [6–8], food contaminants [9],
purity determination of pharmaceutical products [10,11] and drug
development [12]. Besides HPLC, various other methods such as
supercritical fluid chromatography [13], capillary electrophoresis
[14], gas chromatography [15], simulated moving bed technology
[16], enzymatic resolution [17] and crystallization [18] are rou-
tinely used for the enantiomer separation of enantiomers either
on analytical or preparative scale.
Chiral stationary phases (CSPs) for the separation of chiral
compounds using liquid chromatography are most commonly
based on modified silica materials comprising either immobi-
lized low-molecular mass chiral selectors (e.g. Cinchona-based ion
exchangers) [19], macromolecular [20] (e.g. biopolymers or syn-
thetic polymers) or macrocylic selectors such as cyclodextrins [21],
∗ Corresponding author. Tel.: +43 1 4277 52014; fax: +43 1 4277 9523.
E-mail address: [email protected] (W. Lindner).
antibiotics [22] or chiral crown ethers [23]. The broad family of CSPs
has enabled separation of almost any racemic mixture of choice,
ranging from neutral lipophilic to highly polar hydrophilic com-
pounds. For the latter especially ion exchange CSPs have been found
favorable [24,25]. Besides anion and cation exchange materials one
can also distinguish zwitterion ion exchange-type CSPs [26–29].
Separation of chiral organic acids is feasible on chiral anion
exchange materials. These are based on selectors containing
ionizable primary, secondary or tertiary amino group as well
as permanently charged quaternary amines [30], whereas the
last represents the strongest anion exchanger type. In case of
Cinchona-based materials retention is primarily driven by long-
range electrostatic forces related to the protonated quinuclidine
group with a deprotonated acidic group and the formation of an ion-
pair. Chiral recognition is enabled by formation of a diastereomeric
pair supported by additional directed interactions like intermolec-
ular hydrogen bonds, �–�-interactions, Van der Waals and steric
influences [31,32]. A chiral recognition model for carbamoylated
Cinchona-based selectors has been recently reviewed by Laemmer-
hofer and Lindner [24].
Commercially available chiral anion exchange materials
(Chiralpak® QN-AX and QD-AX), which are used in this study as
reference materials, are immobilized via a radical addition reaction
http://dx.doi.org/10.1016/j.chroma.2014.02.026
0021-9673/© 2014 Elsevier B.V. All rights reserved.
113

86 H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94
3R
4S
1S
89
N
NO
MeO
HNH
O
R
R
ON
NN
SiO
OMe
MeO
3R
4S
1S
89
NNN
SiO
OMe
MeO
N
NO
MeO
HNH
O
O
a)
b)
3R
4S
1S
89
c)
N
NO
MeO
HNH
O S SiO
OMe
MeO
Fig. 1. Structures of the CSPs comprising selectors A–F immobilized onto AzPrSi via
click chemistry. The structures of commercially available QN-AX and QD-AX CSPs are
also shown. a) CSP-1 + CSP-2 R = Cl (8S,9R); CSP-3 to CSP-6 R = Cl (8R,9S); CSP-7 + CSP-
8 R = OMe (8S,9R); CSP-9 + CSP-10 R = OMe (8R,9S); CSP-13 to CSP-15 R = Cl (8R,9S);
b) CSP-11 (8S,9R); CSP-12 (8R,9S). For selector density on silica see Table 1. c) QN-
AX (8S,9R), QD-AX (8R,9S). Selector density of QN-AX and QD-AX: app. 340 �mol/g
silica.
concept of a thiol group. Generally, mercaptopropyl-modified sil-
ica gel is often used as a backbone material for the immobilization
of chiral low molecular mass selectors comprising a double bond
such as Cinchona alkaloids quinine and quinidine [33]. Since this
type of radical mediated immobilization reaction does not work
for selectors with radical-quenching activity, an alternative way for
immobilization was evaluated in this study – Huisgen 1,3-dipolar
cycloaddition, the so-called click chemistry [34], which was already
applied for the immobilization of Cinchona based chiral selectors
by Kacprzak et al. [35].
In this reaction 1,2,3-triazoles are formed from azides and ter-
minal alkyne moieties using Cu(I) as a catalyst. Besides copper
catalysis, also ruthenium(II)-based complexes can be used for this
type of reaction [36,37]. Copper can be used as Cu(I) salts such as
CuI, which is however air sensitive, or as CuSO4 precatalyst in com-
bination with a reducing agent such as ascorbic acid, whereas Cu(I)
is formed in situ [38].
The novel chiral ion exchange materials presented in this work
are low-molecular mass selectors based on diverse carbamoylated
dihydroquinine derivatives. Because the selectivity of quinine-
based chiral ion exchange materials can be modified relatively
easily by different substitution pattern on the hydroxy-group at
position C9 (see Fig. 1) of the quinine/quinidine building block
[39], this concept was applied using different isocyanates for car-
bamoylation. In contrast to the work of e.g. Kacprzak et al. [31],
immobilization took place via a carbamoyl linker and not by the
vinyl moiety at the quinuclidine ring, which changes the geom-
etry of the bound selector with respect to the silica surface of the
backbone material. By this strategy the overall selectivity of the CSP
may be altered (see also Fig. 1). Subsequently the CSPs prepared in
this way were evaluated in terms of their chiral separation power.
2. Experimental
2.1. Materials and methods
NMR-spectra were recorded on a Bruker DRX 400 spectrom-
eter (Karlsruhe, Germany) operating at 400 MHz. Either CDCl3 or
CD3OD (both 99.8%, Deutero GmbH, Kastellaun, Germany) was used
as a solvent and the solvent signals were used as a reference. The
raw data were processed with SpinWorks 2.5 software. The FTIR-
measurements were carried out on a Bruker Tensor 27 Diamond
ATR spectrometer (Ettlingen, Germany) with Opus 4.2 software.
Mass spectrometric measurements of the various selectors were
performed using a 4000 QqLIT mass spectrometer equipped with
an ESI ion source from Applied Biosystems (Foster City, USA). All
mass spectra were measured in positive ionization mode. For data
processing Analyst 1.5 software was used. Elemental analyses were
operated on a EURO EA 3000 CHNS-O instrument from HEKAt-
ech (Wegberg, Germany). Determination of the chlorine content
was performed using potentiometric titration with a Mettler DL
21 titrator (Greifensee, Switzerland). Melting points were deter-
mined using heating stage Leica VM TG (Bensheim, Germany). Flash
column chromatography was carried out using Normasil 60 Sil-
ica Gel from VWR. Daisogel SP-120-5-P from Daiso (Japan) was
used as a basis for silica derivatization (spherical particles with
a mean particle size of 5 �m, mean pore diameter 120 A, particle
size distribution ≤1.25, pore volume 1.0 mL/g, specific surface area
300 m2/g).
Analytical grade solvents for synthesis were purchased from
DonauChem, Fluka, Merck, ROTH, Sigma-Aldrich and VWR.
Reagents and catalysts were obtained from ABCR, Acros Organics,
Buchler, Fluka, Merck, Sigma-Aldrich, TCI and VWR. As the bulk
mobile phase and additives the following chemicals were used:
MeOH (HPLC grade quality, 99.8%, VWR), AcOH (≥99%, Sigma-
Aldrich) and NH4OAc (≥97%, p.a., Fluka). For comparative purposes
commercially available Chiralpak® QN-AX and QD-AX columns
were used (150 × 4 mm ID, 5 �m), because these two quinine
and quinidine derived carbamoylated reference materials show
high capabilities in terms of resolution of chiral acidic compounds
[32,40–43].
2.2. Chromatography
The chromatographic screening of the columns was carried out
on a 1290 series Infinity HPLC system from Agilent Technologies
(Waldbronn, Germany) equipped with a column compartment for
six columns and a diode array detector (DAD). The concentration
of analytes was approximately 1 mg/mL in MeOH and the detec-
tion wavelength was 254 nm. The injected volume was set to 5 �L.
The columns were thermostated at 25 ◦C. Elution was performed in
the isocratic mode with a flow rate of 1.0 mL/min. The composition
of the polar organic eluent was MeOH/AcOH/NH4OAc = 99/1/0.25
(v/v/w). The mobile phase was degassed by sonication prior to use.
Acetone (50 �L/mL in MeOH) was used as a non-retained void vol-
ume marker. Data processing was carried out with ChemStation
chromatographic data software from Agilent and Excel spreadsheet
software from Microsoft Corporation.
2.3. Analytes
The retention on Cinchona alkaloids based CSPs is primarily
driven by an ion pairing (exchange) mechanism [33]. Due to the
large number of CSPs to be screened only a relatively small set
of organic acid type analytes (see Fig. 2) was chosen for the eval-
uation. The test compounds were either commercially available,
114

H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94 87
NH
O
OH
O
Bz-Leu
NH
O
OH
O
O2N
NO2
DNB-Leu
N
O
OH
O
O2N
NO2
DNB-N-Me-Leu
NH
O
OH
O
Cl
Cl
DCB-Le u
NH
O
OH
O
CF3
F3C
BTFMB-Leu
NH
O
OHO
O
Z-Phe
NH
O
OHO
O
BOC-Ph e
NH
O
OH
O
Bz-Phe
N
OOHO
O
FMOC-Aze
O
O
OH
Cl Cl
Dich lorprop
PI-2-25 -1
NHO
O
PO
OHO
PI-2-34 -1
NHO
O
PO
OHO
PI-2-56 -2
NHO
O
PO
OHO
Fig. 2. Chemical structures of the chiral test analytes used for the evaluation of the CSPs.
synthesized earlier in our working group or gifts of other working
groups [44,45]. For the determination of the elution order single
enantiomers were used if available.
2.4. Synthesis
The numbering of the compounds refers to Figs. 3 and 4.
2.4.1. 3,5-Dichloro-4-propargyloxybenzoyl azide (3)
To a suspension of 3,5-dichloro-4-propargyloxybenzoic acid 1
[46] (7.30 g, 29.8 mmol) in oxalyl chloride (50 mL), catalytic amount
of DMF was added and the suspension was stirred at 30 ◦C for
45 min. The unreacted oxalyl chloride was distilled off. The crude
product was diluted with petroleum ether (120 mL) and activated
carbon (200 mg) was added. The suspension was heated under
reflux for 3 min, filtered and evaporated to dryness. The acid chlo-
ride was directly used in the subsequent azide–halide exchange.
To a solution of 3,5-dichloro-4-propargyloxybenzoyl chloride
(7.16 g, 27.2 mmol) in acetone (70 mL) at 0 ◦C, a solution of NaN3
(5.48 g, 84.3 mmol) in H2O (30 mL) was added dropwise under
vigorous stirring and the reaction mixture was stirred for 1 h at
0 ◦C. The white precipitate was filtered and washed with cold H2O
115

88 H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94
Fig. 3. Synthetic procedures for preparation of selectors A–D. Reaction conditions: a) (COCl)2 , DMF (catalyst), 30 ◦C, 45 min; b) NaN3 , acetone, water, 0 ◦C, 1 h; c) toluene,
reflux, N2 atmosphere, 3 h; d) 10,11-dihydroquinine, toluene, dibutyltin dilaurate (catalyst), N2 atmosphere, reflux, 18 h; e) 10,11-dihydroquinidine, toluene, dibutyltin
dilaurate (catalyst), N2 atmosphere, reflux, 18 h.
(2 × 50 mL). The filtration cake was dissolved in EtOAc (150 mL),
washed with H2O and brine (each 40 mL) and dried with anhy-
drous MgSO4. The solvent was evaporated and the product was
dried under reduced pressure at room temperature. It was obtained
6.43 g (23.8 mmol, 88%) of 3,5-dichloro-4-propargyloxybenzoyl
azide as a white solid, m.p. 96–97 ◦C. 1H-NMR [CDCl3]: ı = 7.91 (s,
2H), 4.81 (d, 2H), 2.47 (t, 1H). FTIR (cm−1): 3297, 2166, 1686.
Analogously, 3,5-dimethoxy-4-propargyloxybenzoyl azide (4)
was prepared, yielding 8.28 g (3.2 mmol, 75%) of a white solid, m.p.
108–109 ◦C. 1H-NMR [CDCl3]: ı = 7.22 (s, 2H), 4.75 (d, 2H), 3.84 (s,
6H), 2.37 (t, 1H). FTIR (cm−1): 3284, 2944, 2145, 1685.
2.4.2. 10,11-Dihydroquinine
9-[3,5-dichloro-4-(prop-2-yn-1-yloxy)phenyl]carbamate (A)
3,5-Dichloro-4-propargyloxybenzoyl azide 3 (2.83 g, 8.8 mmol)
was dissolved in freshly azeotropically dried toluene (50 mL) and
subsequently refluxed for 3 h to accomplish Curtius rearrangement.
A sample of the mixture showed an intensive IR-absorption at
2263 cm−1, indicating the successful conversion into the respective
isocyanate, while absorption band of the azide group at 2166 cm−1
disappeared completely. The reaction solution was cooled to room
temperature and directly used without further purification.
Commercially available 10,11-dihydroquinine (1.85 g,
5.7 mmol) was dissolved in toluene (80 mL) and half of the
solvent was distilled off using a Dean-Stark trap. After flushing
with nitrogen for 10 min and cooling below the boiling point
of toluene the previously prepared solution of 3,5-dichloro-4-
propargyloxyphenyl isocyanate 5 in dry toluene was added.
Dibutyltin dilaurate as a catalyst was added (40 �L) and the solu-
tion was refluxed for 18 h. The reaction mixture was evaporated
to dryness and the crude product was purified by flash column
chromatography (eluent: CH2Cl2/MeOH, 10/1) yielding 2.10 g
(3.7 mmol, 65%) of the selector A as a greenish solidified foam.1H-NMR [CDCl3]: ı = 8.60 (d, 1H), 7.91 (d, 1H), 7.41 (d, 1H), 7.30 (d,
1H), 7.20 (s, 2H), 7.19 (d, 1H), 6.46 (d, 1H), 4.64 (d, 2H), 3.89 (s, 3H),
3.27 (q, 1H), 3.08–2.89 (broad, 2H), 2.56 (m, 1H), 2.43 (t, 1H), 2.29
(d, 1H), 1.83–1.71 (broad, 2H), 1.69–1.59 (broad, 1H), 1.51–1.32
(broad, 3H), 1.32–1.21 (broad, 2H), 0.80 (t, 3H). FTIR (cm−1): 3298,
2930, 2868, 1730. MS [ESI, positive]: 1158 [2M + Na]+, 568 [M + H]+,
284 [M + 2H]2+.
In the similar way as for the selector A, 10,11-Dihydroquinidine
9-[3,5-dichloro-4-(prop-2-yn-1-yloxy)phenyl]carbamate (B)
was prepared from 10,11-dihydroquinidine and 3,5-dichloro-
4-propargyloxyphenyl isocyanate 5. Purification by column
chromatography (eluent: CH2Cl2/MeOH, 10/1) afforded selector
B (2.07 g, 3.6 mmol, 56%) as a brownish solidified foam. 1H-NMR
[CDCl3]: ı = 8.68 (d, 1H), 7.99 (d, 1H), 7.45 (d, 1H), 7.37 (s, 2H), 7.34
(d, 1H), 7.30 (d, 1H), 6.53 (d, 1H), 4.71 (d, 2H), 3.95 (s, 3H), 3.28
(q, 1H), 2.93–2.85 (broad, 1H), 2.82–2.54 (broad, 3H), 2.50 (t, 1H),
1.80–1.72 (broad, 2H), 1.61–1.53 (broad, 2H), 1.50–1.38 (broad,
Fig. 4. Synthetic procedures for preparation of selectors E and F. Reaction conditions: a) Propargyl bromide, NaH, THF, N2 atmosphere, −10 ◦C 2 h, RT 18 h; b) NaOH, MeOH,
reflux, 4 h; c) (COCl)2 , DMF (catalyst), reflux, 2 h; d) NaN3 , acetone, water, 0 ◦C, 2 h; e) toluene, reflux, N2 atmosphere, 3 h; f) 10,11-dihydroquinine, toluene, dibutyltin dilaurate
(catalyst), N2 atmosphere, reflux, 18 h; g) 10,11-dihydroquinidine, toluene, dibutyltin dilaurate (catalyst), N2 atmosphere, reflux, 18 h.
116

H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94 89
2H), 1.27–1.21 (broad, 2H), 0.90 (t, 3H). FTIR (cm−1): 3296, 2932,
2870, 1728. MS [ESI, positive]: 568 [M + H]+, 284 [M + 2H]2+.
Analogously, using the synthetic procedure described above
for the selector A, 10,11-Dihydroquinine 9-[3,5-dimethoxy-4-
(prop-2-yn-1-yloxy)phenyl]carbamate (C) was prepared from
10,11-dihydroquinine and 3,5-dimethoxy-4-propargyloxyphenyl
isocyanate 6. After purification by column chromatography (elu-
ent: CH2Cl2/MeOH, 10/1), the selector C was obtained as a brownish
solidified foam (3.60 g, 0.64 mmol, 52%). 1H-NMR [CDCl3]: ı = 8.72
(d, 1H), 8.01 (d, 1H), 7.49 (d, 1H), 7.37 (d, 1H), 7.35 (d, 1H), 7.34 (s,
2H), 6.52 (d, 1H), 4.64 (d, 2H), 3.96 (s, 3H), 3.78 (s, 6H), 3.35 (q, 1H),
3.18–2.98 (b, 2H), 2.64 (m, 1H), 2.4 (t, 1H), 2.34 (d, 1H), 1.87–1.77
(b, 2H), 1.77–1.67 (b, 1H), 1.61–1.52 (b, 1H), 1.52–1.38 (b, 2H), 1.33
(m, 1H), 1.23 (t, 1H), 0.86 (t, 3H). FTIR (cm−1): 3290, 2932, 1723.
MS [ESI, positive]: 583 [M + Na]+, 561 [M + H]+, 281 [M + 2H]2+.
In the same way, 10,11-Dihydroquinidine 9-[3,5-dimethoxy-
4-(prop-2-yn-1-yloxy)phenyl]carbamate (D) was prepared. It was
obtained 2.98 g (0.54 mmol, 42%) of a brownish solidified foam. 1H-
NMR [CDCl3]: ı = 8.70 (d, 1H), 8.01 (d, 1H), 7.69 (d, 1H), 7.65 (d, 1H),
7.48 (d, 1H), 7.36 (s, 2H), 6.56 (d, 1H), 4.60 (d, 2H), 3.97 (s, 3H), 3.76
(s, 6H), 3.60 (s, 1H), 3.40–3.17 (b, 2H), 3.10–2.83 (b, 1H), 2.83–2.66
(b, 1H), 2.39 (t, 1H), 1.86 (s, 1H), 1.77 (s, 1H), 1.70–1.44 (b, 3H), 1.39
(t, 1H), 1.24 (m, 1H), 1.23 (t, 1H), 0.86 (t, 3H). FTIR (cm−1): 3289,
2932, 1727. MS [ESI, positive]: 561 [M + H]+, 281 [M + 2H]2+.
2.4.3. 2,2-Dimethyl-3-propargyloxypropanoyl azide (7)
To a suspension of NaH (60% in mineral oil, 10 g, 250.0 mmol)
in dry THF (100 mL) cooled to −10 ◦C, a solution of methyl 3-
hydroxypivalate (25 g, 189.2 mmol) in dry THF (100 mL) was added
dropwise in the inert nitrogen atmosphere. The reaction mixture
was allowed to equilibrate for 1 h. Propargyl bromide solution (80%
in toluene, 31 mL, 278.3 mmol) was added dropwise during con-
stant cooling in an inert nitrogen atmosphere. After 2 h the cooling
bath was removed and stirring was continued for 18 h at room
temperature. The reaction was decomposed with MeOH (150 mL)
and diluted with ethyl acetate (200 mL) and H2O (400 mL). The lay-
ers were separated and the water layer was extracted with EtOAc
(3 × 200 mL). The combined organic solution was washed with H2O
(100 mL), brine (50 mL) and dried with anhydrous MgSO4. The sol-
vent was removed and the liquid product was separated from
mineral oil by liquid phase separation. Drying was achieved by
high vacuum. It was obtained 21.33 g (125.3 mmol, 66%) of methyl
3-propargyloxypivalate [47].
To a solution of methyl 3-propargyloxypivalate (21.33 g,
125.3 mmol) in MeOH (200 mL), a solution of NaOH (20 g,
500 mmol) in H2O (40 mL) was added slowly. The reaction mix-
ture was refluxed for 4 h and after cooling to room temperature
poured into H2O (200 mL) and acidified with aqueous HCl to pH ∼ 2.
The mixture was extracted with dichloromethane (3 × 75 mL).
The combined organic solution was washed with H2O (50 mL),
brine (50 mL) and dried with anhydrous MgSO4. The solvent was
removed and the product was dried under high vacuum at 50 ◦C for
18 h, affording 16.10 g (103 mmol, 82%) of 3-propargyloxypivalic
acid as a yellowish oil. 1H-NMR [CDCl3]: ı = 4.19 (d, 2H), 3.57 (s,
2H), 2.44 (t, 1H), 1.25 (s, 6H). FTIR (cm−1): 3276, 2918, 2117, 1691.
3-Propargyloxypivalic acid (13.8 g, 88.4 mmol) was dissolved in
oxalyl chloride (60 mL), a catalytic amount of DMF (one drop) was
added and the reaction mixture was refluxed for 2 h. The unreacted
oxalyl chloride was distilled off, the crude acid chloride (15.2 g,
87.0 mmol, 98%) was dried under vacuum at room temperature and
directly used for azide–halide exchange.
To a solution of acid chloride in acetone (150 mL), a solution
of NaN3 (17.0 g, 261.5 mmol) in H2O (60 mL) was added dropwise
during vigorous stirring at 0 ◦C. After 2 h the mixture was extracted
with ethyl acetate (3 × 100 mL). The combined organic solution was
washed with H2O (60 mL) and brine (60 mL), and dried with anhy-
drous MgSO4. Evaporation of the solvent at room temperature and
drying under vacuum yielded 14.0 g (77.3 mmol, 89%) of azide 7 as
a brownish liquid. 1H-NMR [CDCl3]: ı = 4.12 (d, 2H), 3.48 (s, 2H),
2.42 (t, 1H), 1.17 (s, 6H). FTIR (cm−1): 3290, 2978, 2135, 1709.
2.4.4. 10,11-Dihydroquinine
9-[2-methyl-1-(prop-2-yn-1-yloxy)propan-2-yl]carbamate (E)
Carbamoylation was carried out in the same way as described
above for selector A by Curtius rearrangement of 7 in dry
toluene and the subsequent addition of 10,11-dihydroquinine. The
selector was purified via flash column chromatography (eluent:
EtOAc/MeOH, 4/1). Yield: 4.1 g (8.5 mmol, 72%) of selector E as a
brownish solid. 1H-NMR [CDCl3]: ı = 8.66 (d, 1H), 7.92 (d, 1H), 7.40
(s, 1H), 7.29 (d, 1H), 7.27 (d, 1H), 6.39 (d, 1H), 5.03 (s, 1H), 4.02 (d,
2H), 3.89 (s, 3H), 3.40–3.31 (b, 2H), 3.25–3.16 (b, 1H), 3.14–2.94 (b,
2H), 2.59 (m, 1H), 2.33–2.28 (b, 1H), 1.76–1.59 (b, 2H), 1.57–1.46
(b, 2H), 1.46–1.31 (b, 2H), 1.21 (d, 2H), 1.18 (s, 6H), 0.77 (t, 3H). FTIR
(cm−1): 3292, 2936, 1722. MS [ESI, positive]: 480 [M + H]+.
2.4.5. 10,11-Dihydroquinidine
9-[2-methyl-1-(prop-2-yn-1-yloxy)propan-2-yl]carbamate (F)
The synthesis of selector F was carried out in the same way as
described above for the respective dihydroquinine type selector E.
Yield: 4.3 g (8.9 mmol, 75%) of a brownish solid. 1H-NMR [CDCl3]:
ı = 8.66 (d, 1H), 7.92 (d, 1H), 7.40 (s, 1H), 7.30 (d, 1H), 7.27 (d, 1H),
6.45 (s, 1H), 4.99 (s, 1H), 4.04 (d, 2H), 3.91 (s, 3H), 3.40–3.36 (b, 2H),
3.22–3.12 (b, 1H), 2.98–2.57 (b, 3H), 2.35 (s, 1H), 1.87–1.61 (b, 2H),
1.55–1.28 (b, 4H), 1.24 (s, 2H), 1.22 (s, 6H), 0.86 (t, 3H). FTIR (cm−1):
3292, 2936, 1717. MS [ESI, positive]: 480 [M + H]+.
2.5. Immobilization and preparation of the stationary phases
All synthesized selectors were immobilized onto azidopropyl-
modified silica gel (AzPrSi) which was prepared according to a
previously described protocol of Kacprzak et al. [48]. Azide load-
ing on the silica surface was determined by elemental analysis (EA)
[w-%]: C: 3.64, H: 0.92, N: 2.64, S: <0.01, Cl: 0.18. This corresponds
to the azide loading of 630 �mol/g silica. The immobilization pro-
cedure was similar to the previously described one by Kacprzak
et al. [31]. Briefly: In a typical immobilization procedure AzPrSi
(2.50 g) was suspended in ACN (60 mL) in a sealable glass bottle
and the desired amounts of the respective selector (see Table 1),
N,N-diisopropylethylamine (1.00 mL) and CuI as a catalyst (50 mg)
were added. The suspension was degassed with N2 and tightly
sealed. The glass bottle was mounted on an overhead shaker and
allowed to rotate for 72 h at room temperature. The modified silica
was filtered and washed with ACN, MeOH, aqueous ethylenedi-
aminetetraacetic acid (EDTA) solution (2%, w/v), MeOH/H2O = 1/1
(v/v), MeOH/AcOH = 10/1 (v/v) and MeOH (each 150 mL). The sil-
ica material was allowed to dry at 60 ◦C for 4 h followed by 60 ◦C
under vacuum for 18 h. The amount of immobilized selector was
determined by EA (see Table 1). The CSPs were packed in house
in 150 × 4 mm ID stainless steel columns. Packing was carried out
by a conventional slurry packing method using isopropanol/acetic
acid, 10/1 (v/v) as the slurry solvent and MeOH as the packing sol-
vent at a pressure of approximately 650 bars. After packing, the
columns were washed with aqueous EDTA solution (2%, w/v) to
remove remaining CuI, followed by rinsing with H2O and MeOH
(each 50 mL, flow rate: 0.5 mL/min).
CSP-13 was prepared by endcapping of remaining azide groups
of CSP-3. Therefore CSP-3 (2.50 g) was subjected to another
immobilization step following the protocol described above using
propargyl alcohol (100 �L, 690 �mol/g CSP) instead of a selector.
EA [w-%]: C: 8.98, H: 1.33, N: 2.72, S: <0.02, Cl: 1.16.
117

90 H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94
Table 1
Immobilization and elemental analysis results; ec = endcapping after immobilization of the selector. CSP-13 was endcapped with propargyl alcohol, CSP-15 using 1-pentyne.
CSP-15 was almost a 1:1 homogeneous physical mixture of CSP-6 and pure AzPrSi.
CSP Selector (type) Offered
selector
[mg]
Offered
selector
[�mol/g]
C [w-%] H [w-%] N [w-%] S [w-%] Cl [w-%] Immobilized
selector
[�mol/g]
Immobilized
selector
[�mol/m2]
Loading
efficiency [%]
CSP-1 A (DHQN) 327 231 10.04 1.41 3.42 <0.02 1.52 210 0.70 91
CSP-2 A (DHQN) 1483 1043 19.20 2.16 4.24 <0.02 3.38 480 1.60 n.d.
CSP-3 B (DHQD) 213 150 7.75 1.24 2.82 <0.02 1.24 150 0.50 100
CSP-4 B (DHQD) 261 250 9.90 1.38 3.11 <0.02 1.83 230 0.77 92
CSP-5 B (DHQD) 512 360 12.96 1.65 3.28 <0.02 2.23 310 1.03 86
CSP-6 B (DHQD) 1442 1013 19.04 2.19 4.18 <0.02 3.48 490 1.63 n.d.
CSP-7 C (DHQN) 326 234 10.64 1.52 3.11 <0.02 n.d. 200 0.67 85
CSP-8 C (DHQN) 1340 531 20.01 2.41 3.86 <0.02 n.d. 450 1.50 85
CSP-9 D (DHQD) 326 234 8.58 1.38 2.92 <0.02 n.d. 160 0.53 68
CSP-10 D (DHQD) 1340 531 19.81 2.51 3.89 <0.02 n.d. 440 1.47 83
CSP-11 E (DHQN) 300 250 9.20 1.47 3.08 <0.02 n.d. 180 0.60 72
CSP-12 F (DHQD) 300 250 9.89 1.53 3.15 <0.02 n.d. 200 0.67 80
CSP-13 B (DHQD, ec) n.d. n.d. 8.98 1.33 2.72 <0.02 1.16 150 0.50 n.d.
CSP-14 B (DHQD, ec) n.d. n.d. 9.86 1.57 2.82 <0.02 1.18 150 0.50 n.d.
CSP-15 B (DHQD, phys. mix) n.d. n.d. 11.14 1.54 3.34 <0.02 1.69 240 0.80 n.d.
CSP-14 was prepared in the same way as CSP-13 using 1-
pentyne instead of propargyl alcohol for endcapping. EA [w-%]: C:
9.86, H: 1.57, N: 2.82, S: <0.02, Cl: 1.18.
CSP-15 represents a homogenous physical mixture of CSP-
6 (1.23 g) and AzPrSi (1.28 g) resulting in a selector loading of
240 �mol/g. EA [w-%]: C: 11.14, H: 1.54, N: 3.34, S: <0.02, Cl: 1.69.
One column was packed with pure AzPrSi as a non-chiral
stationary phase for comparative purposes and to elucidate the
retention characteristics of the free azide groups for chiral acids.
3. Results and discussion
3.1. Synthetic aspects
In this contribution, the synthesis of a number of different chiral
selectors, their characterization and immobilization onto modified
silica via so-called click chemistry is presented.
Enantioseparation strongly depends on the choice of an appro-
priate mobile phase and especially the chiral selector as a stationary
phase. The retention mechanism e.g. for commercially available
Cinchona derived anion exchangers Chiralpak® QN-AX and QD-
AX is primarily driven by the formation of an ion-pair between
an anionic analyte and the quinuclidine ring of the selector
[24,49]. This is enabled via protonation of the tertiary amine in
the quinuclidine ring under slightly acidic mobile phase condi-
tions. The principally non-directed, long range Coulomb force as
primary attraction is accompanied by various non-covalent inter-
molecular interactions, among them directed hydrogen bonding,
�–�-stacking, van der Waals interactions and steric influences
[32,40]. The protonated quinuclidine group can, however, also act
via a hydrogen-supported ion-pairing site, which makes it directed.
The synthesized selectors represent similar selector structures
(see Fig. 1), which possess similar primary interaction sites as
the commercially available anion exchangers mentioned above.
However, their bonding geometry toward the liquid bulk phase is
modified. QN-AX and QD-AX comprise tert-butyl carbamoylated
quinine and quinidine immobilized via radical mediated thiol-ene
addition at C11 of the quinuclidine ring (both about 340 �mol/g
selector loading). This is sometimes also called thiol click chem-
istry. In contrast, immobilization in this study was carried out via
alkyne–azide cycloaddition using spacers that were attached to the
Cinchona derivatives at C9-OH. The structure of selectors E and F is
therefore almost identical to QN-AX and QD-AX selectors, respec-
tively. The other four selectors A–D possess a 3,5-disubstituted
aromatic ring, which may provide additional �–�-interaction sites
with analytes.
Although covalent immobilization of quinine/quinidine based
ion exchange selectors, and materials in general, can be car-
ried out via radical mediated thiol-ene immobilization onto
mercaptopropyl-modified silica gel [28], we gave preference to
the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (often
called also azide click chemistry). Using allyl functionality instead of
propargyl as an immobilization anchor employing thiol-ene immo-
bilization yielded a selector loading below 150 �mol/g, in particular
for dichloro-substituted aromatic linkers (data not shown). The
low immobilization efficiency could be based on radical scavenging
properties of dichloro-substituted aromatic systems as shown by
Li et al. [50]. Click chemistry circumvents the drawback of the rad-
ical reaction and benefits from high reaction yields even at room
temperature.
The Cinchona-based selectors (A–F) were synthesized by
carbamoylation of dihydroquinine and, its in terms of chro-
matographic behavior pseudoenantiomer, dihydroquinidine
[40,49,51,52] using in situ generated isocyanates formed from the
corresponding acyl azides (see Figs. 3 and 4). After flash column
chromatographic purification, the selectors were obtained in
42–75% yield. The selectors were characterized by NMR, FTIR and
LC-MS and found to have sufficient purity for the subsequent
heterogeneous click chemistry immobilization. The loading effi-
ciency was 84 ± 9% in average, determined by EA. At a low level
of offered selector (150 �mol/g in case of CSP-3) the total amount
was immobilized onto the modified silica as it is claimed that this
chemistry concept works stoichiometrically. The loading efficiency
decreased almost linearly with higher amount of offered selector
(see Table 1), providing that the selector has been offered in deficit
with respect to the azide loading of AzPrSi (630 �mol/g silica). The
maximum practical loadability achieved with excess of selector B
was 490 �mol/g (Table 1). The stoichiometric maximum of surface
capacity (630 �mol/g) was not reached even with a considerable
excess of selector, which reflects the impact of steric hindrance.
A similar effect was observed by Marshall and Mottola [53]. At
selector densities higher than 490 �mol/g the spatially demanding
selectors showed saturation phenomena. This observation renders
the preparation of highly loaded silica surfaces elusive.
3.2. CSP evaluation
3.2.1. Influence of free azide groups and endcapping
Since the surface of the CSPs consists of chiral selector and
unreacted azide groups, there could be a significant influence of
the free azide moieties on retention and resolution of analytes
[54]. The azide group itself is highly polarized and therefore it can
118

H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94 91
potentially interact with polar or polarizable groups of analytes.
In order to evaluate the impact of free azide groups, one column
was packed with pure AzPrSi (630 �mol/g). With standard mobile
phase composition the average absolute k-value was about 0.1.
There was of course no enantioselectivity observed. As a result it
can be claimed that the influence of the remaining free azide groups
is negligible.
CSP-14 and CSP-15 (both of 150 �mol/g loading of DHQD type
selector B) represent endcapped material. The remaining azide
groups (430 �mol –N3 per gram silica) on the modified silica mate-
rial from CSP-3 were allowed to react with propargyl alcohol or
1-pentyne, respectively, according to the established synthetic
protocol. Evaluation of the prepared columns showed almost no
influence on the ˛-values (for all chromatographic results see
Table 2). Improved selectivity ( = 1.05) was found only for DNB-
N-Me-Leu, which could be partially separated on the 1-pentyne
endcapped material. It is reasonable to assume that modification
of the mobile phase could help to further increase the separa-
tion factor. Although the alpha values remained almost unchanged,
resolution increased for the separable analytes in the order non-
endcapped < propargyl alcohol endcapped < 1-pentyne endcapped
CSP.
3.2.2. Reversal of the elution order
A reversal of the elution order on quinine and quinidine
based CSPs was observed for every single separated racemic
analyte, without any exception. This behavior reflects the pseu-
doenantiomeric nature of the selector building blocks, namely
dihydroquinine type (8S,9R) and dihydroquinidine type (8R,9S). The
reversal of elution order for these two pseudoenantiomeric types
of chiral scaffolds on the enantioselectivity was reported in several
other publications [40,41,49,51,52]. In case of N-protected amino
acids the elution order was always D before L for DHQN type CSPs.
The exactly opposite behavior of DHQD type CSPs indicates an equal
separation mechanism for N-protected amino acid derivatives.
3.2.3. Influence of building blocks regarding selectivity and
resolution
CSPs 1, 4, 11 and 12 are well suited for the comparison of DHQN
and DHQD type CSPs since they all contain approximately the same
amount of selector immobilized onto the silica surface. CSP-1 and
CSP-11 are of DHQN type containing 210 �mol/g (selector A) and
180 �mol/g (selector E). The same applies for CSP-4 and CSP-12,
which are of DHQD type containing 230 �mol/g (selector B) and
200 �mol/g (selector F), respectively.
An increase in selectivity was observed for DHQD type CSPs. This
can be illustrated on the separation of Bz-Phe on CSP-1 and CSP-4.
The alpha value increased from 1.39 to 1.50 (+8%), while resolu-
tion increased from 4.22 to 5.64 (+34%). Another example is the
aminophosphonate analyte PI-2-25-1. No separation took place in
case of CSP-1. CSP-4 however showed base line separation of the
enantiomers with = 1.22 and R = 2.70. Taking all the other ana-
lytes into consideration, although it is still a limited number, one
can conclude that the DHQD type chiral stationary phases seem to
be generally more suitable for the separation of chiral acids, pro-
vided that the selector loading onto silica is roughly the same. Such
behavior is in agreement to the commercial materials QN-AX and
QD-AX used in this study (see Table 2).
3.2.4. Influence of carbamoyl substitution pattern
For a comparison of the impact of different aryl substitu-
tion patterns on the carbamoyl moiety, CSP-3 and CSP-9 can be
used. In general, both the structures of the selectors, immobi-
lization strategy and the amount of the immobilized selectors
(150 and 160 �mol/g, respectively) were comparable. While CSP-
3 contains a 3,5-dichloro substituted ring, CSP-9 possesses a
3,5-dimethoxy substitution. The two analytes DNB-N-Me-Leu and
the amino phosphonate PI-2-56-2 could not be separated using
the 3,5-dichloro substituted selector of CSP-3. An electron donat-
ing (3,5-dimethoxy) substitution of the carbamoyl residue as in
case of CSP-9 was proven to be beneficial because at least a partial
separation of these two analytes was observed. Similar behavior
was found also for other analytes such as DCB-Leu, Z-Phe and
FMOC-Aze. The separation factors (+5%) as well as the resolution
values (average increase of 50%) were generally higher in case
of CSP-9.
In comparison to the arylcarbamoylated selector in case of CSP-7
(DHQN-type 3,5-dimethoxy substitution, 200 �mol/g), the struc-
ture of the selector in CSP-11 comprises an aliphatic tert-butyl
moiety (DHQN-type, 180 �mol/g). This selector is basically imitat-
ing the structure of the commercially available QN-AX CSP, except
for the immobilization strategy and thus the selector bonding
geometry. The comparison shows that both selectivity and espe-
cially resolution were increased in case of the thiol clicked tert-butyl
type selector. This was particularly pronounced in case of Bz-Leu,
DNB-Leu and BOC-Phe.
Dichlorprop could not be separated on CSP-7, while in case of
CSP-11 baseline separation was observed (R = 2.33). These results
show the potential of tert-butyl carbamoylated quinine/quinidine-
based chiral anion exchange-type materials compared to other
carbamoyl derivatives of cinchonanes. A direct comparison of
the novel tert-butyl type selector immobilized via Huisgen
alkyne–azide cycloaddition and the commercially available one,
which was immobilized using radical mediated thiol-ene addition,
is given below.
3.2.5. Impact of different attachment modes on the chiral
separation power of tert-butyl type selectors
As it can be seen in Fig. 1, the core structures of the selectors of
CSP-12 and the commercially available QD-AX were basically the
same. The QD-AX selector was immobilized via the allyl moiety
at the quinuclidine ring using radical mediated thiol-ene immobi-
lization strategy yielding a selector density of 340 �mol/g silica.
The novel selector CSP-12 synthesized in this work comprised
a modified carbamoyl residue and was thereby suitable for click
immobilization onto AzPrSi. The differences in chromatographic
behavior, which may be related to variation of flexibility and spatial
demand of the carbamoyl part of the selector, are summarized in
the following. However, it should be noted that the selector loading
of the two CSPs mentioned above (CSP-12 and QD-AX) was signif-
icantly different (200 �mol/g in case of CSP-12) and therefore a
direct comparison is rather difficult.
Interestingly, the alpha value of DNB-Leu was found almost
double for the CSP-12 in comparison to QD-AX. In contrast, DNB-
N-Me-Leu was not separated on CSP-12 while QD-AX provided at
least partial separation of this compound (R = 0.78). Remarkable is
the fact that all three N-protected aminophosphonates were bet-
ter resolved on the novel tert-butyl materials using quinine as a
building block. The quinidine-based materials showed contrasting
behavior – with the exception of the analyte PI-2-56-2.
This observation indicates that the chiral recognition mecha-
nism for the new selectors is consistent with the process described
for other 9-O-immobilized selectors [55]. It also becomes evi-
dent that the extension of the carbamoyl part and introduction
of 1,2,3-triazole linker does grosso modo not significantly influ-
ence interactions of the tested small molecule type analytes with
the selectors. For larger analytes the situation may be more
different. The conformation of the binding pocket formed by
the quinoline aromatic unit, quinuclidine ring and carbamoyl
moiety, is not influenced by spatially demanding linker unit. More-
over, the vicinity of this unit and silica surface may support
119
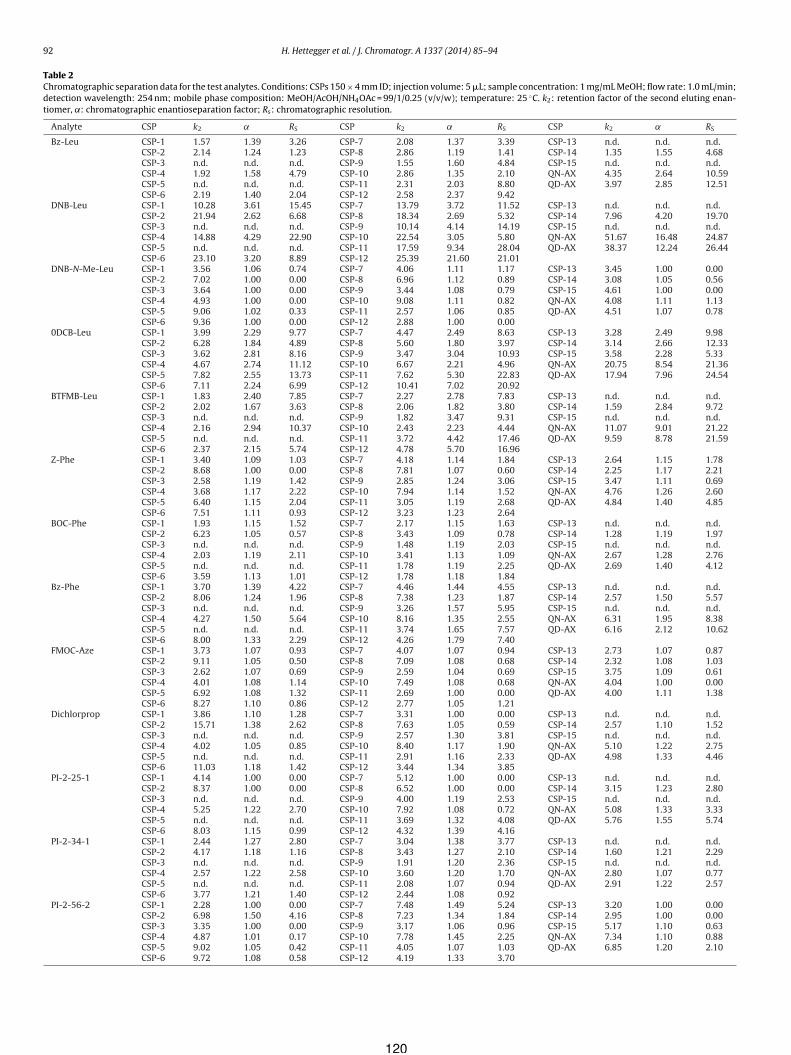
92 H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94
Table 2
Chromatographic separation data for the test analytes. Conditions: CSPs 150 × 4 mm ID; injection volume: 5 �L; sample concentration: 1 mg/mL MeOH; flow rate: 1.0 mL/min;
detection wavelength: 254 nm; mobile phase composition: MeOH/AcOH/NH4OAc = 99/1/0.25 (v/v/w); temperature: 25 ◦C. k2: retention factor of the second eluting enan-
tiomer, ˛: chromatographic enantioseparation factor; Rs: chromatographic resolution.
Analyte CSP k2 ˛ RS CSP k2 ˛ RS CSP k2 RS
Bz-Leu CSP-1 1.57 1.39 3.26 CSP-7 2.08 1.37 3.39 CSP-13 n.d. n.d. n.d.CSP-2 2.14 1.24 1.23 CSP-8 2.86 1.19 1.41 CSP-14 1.35 1.55 4.68
CSP-3 n.d. n.d. n.d. CSP-9 1.55 1.60 4.84 CSP-15 n.d. n.d. n.d.
CSP-4 1.92 1.58 4.79 CSP-10 2.86 1.35 2.10 QN-AX 4.35 2.64 10.59
CSP-5 n.d. n.d. n.d. CSP-11 2.31 2.03 8.80 QD-AX 3.97 2.85 12.51
CSP-6 2.19 1.40 2.04 CSP-12 2.58 2.37 9.42
DNB-Leu CSP-1 10.28 3.61 15.45 CSP-7 13.79 3.72 11.52 CSP-13 n.d. n.d. n.d.CSP-2 21.94 2.62 6.68 CSP-8 18.34 2.69 5.32 CSP-14 7.96 4.20 19.70
CSP-3 n.d. n.d. n.d. CSP-9 10.14 4.14 14.19 CSP-15 n.d. n.d. n.d.
CSP-4 14.88 4.29 22.90 CSP-10 22.54 3.05 5.80 QN-AX 51.67 16.48 24.87
CSP-5 n.d. n.d. n.d. CSP-11 17.59 9.34 28.04 QD-AX 38.37 12.24 26.44CSP-6 23.10 3.20 8.89 CSP-12 25.39 21.60 21.01
DNB-N-Me-Leu CSP-1 3.56 1.06 0.74 CSP-7 4.06 1.11 1.17 CSP-13 3.45 1.00 0.00CSP-2 7.02 1.00 0.00 CSP-8 6.96 1.12 0.89 CSP-14 3.08 1.05 0.56
CSP-3 3.64 1.00 0.00 CSP-9 3.44 1.08 0.79 CSP-15 4.61 1.00 0.00
CSP-4 4.93 1.00 0.00 CSP-10 9.08 1.11 0.82 QN-AX 4.08 1.11 1.13CSP-5 9.06 1.02 0.33 CSP-11 2.57 1.06 0.85 QD-AX 4.51 1.07 0.78
CSP-6 9.36 1.00 0.00 CSP-12 2.88 1.00 0.00
0DCB-Leu CSP-1 3.99 2.29 9.77 CSP-7 4.47 2.49 8.63 CSP-13 3.28 2.49 9.98
CSP-2 6.28 1.84 4.89 CSP-8 5.60 1.80 3.97 CSP-14 3.14 2.66 12.33CSP-3 3.62 2.81 8.16 CSP-9 3.47 3.04 10.93 CSP-15 3.58 2.28 5.33CSP-4 4.67 2.74 11.12 CSP-10 6.67 2.21 4.96 QN-AX 20.75 8.54 21.36
CSP-5 7.82 2.55 13.73 CSP-11 7.62 5.30 22.83 QD-AX 17.94 7.96 24.54
CSP-6 7.11 2.24 6.99 CSP-12 10.41 7.02 20.92
BTFMB-Leu CSP-1 1.83 2.40 7.85 CSP-7 2.27 2.78 7.83 CSP-13 n.d. n.d. n.d.
CSP-2 2.02 1.67 3.63 CSP-8 2.06 1.82 3.80 CSP-14 1.59 2.84 9.72
CSP-3 n.d. n.d. n.d. CSP-9 1.82 3.47 9.31 CSP-15 n.d. n.d. n.d.CSP-4 2.16 2.94 10.37 CSP-10 2.43 2.23 4.44 QN-AX 11.07 9.01 21.22CSP-5 n.d. n.d. n.d. CSP-11 3.72 4.42 17.46 QD-AX 9.59 8.78 21.59
CSP-6 2.37 2.15 5.74 CSP-12 4.78 5.70 16.96Z-Phe CSP-1 3.40 1.09 1.03 CSP-7 4.18 1.14 1.84 CSP-13 2.64 1.15 1.78
CSP-2 8.68 1.00 0.00 CSP-8 7.81 1.07 0.60 CSP-14 2.25 1.17 2.21CSP-3 2.58 1.19 1.42 CSP-9 2.85 1.24 3.06 CSP-15 3.47 1.11 0.69
CSP-4 3.68 1.17 2.22 CSP-10 7.94 1.14 1.52 QN-AX 4.76 1.26 2.60CSP-5 6.40 1.15 2.04 CSP-11 3.05 1.19 2.68 QD-AX 4.84 1.40 4.85CSP-6 7.51 1.11 0.93 CSP-12 3.23 1.23 2.64
BOC-Phe CSP-1 1.93 1.15 1.52 CSP-7 2.17 1.15 1.63 CSP-13 n.d. n.d. n.d.CSP-2 6.23 1.05 0.57 CSP-8 3.43 1.09 0.78 CSP-14 1.28 1.19 1.97CSP-3 n.d. n.d. n.d. CSP-9 1.48 1.19 2.03 CSP-15 n.d. n.d. n.d.
CSP-4 2.03 1.19 2.11 CSP-10 3.41 1.13 1.09 QN-AX 2.67 1.28 2.76
CSP-5 n.d. n.d. n.d. CSP-11 1.78 1.19 2.25 QD-AX 2.69 1.40 4.12CSP-6 3.59 1.13 1.01 CSP-12 1.78 1.18 1.84
Bz-Phe CSP-1 3.70 1.39 4.22 CSP-7 4.46 1.44 4.55 CSP-13 n.d. n.d. n.d.
CSP-2 8.06 1.24 1.96 CSP-8 7.38 1.23 1.87 CSP-14 2.57 1.50 5.57
CSP-3 n.d. n.d. n.d. CSP-9 3.26 1.57 5.95 CSP-15 n.d. n.d. n.d.CSP-4 4.27 1.50 5.64 CSP-10 8.16 1.35 2.55 QN-AX 6.31 1.95 8.38
CSP-5 n.d. n.d. n.d. CSP-11 3.74 1.65 7.57 QD-AX 6.16 2.12 10.62CSP-6 8.00 1.33 2.29 CSP-12 4.26 1.79 7.40
FMOC-Aze CSP-1 3.73 1.07 0.93 CSP-7 4.07 1.07 0.94 CSP-13 2.73 1.07 0.87
CSP-2 9.11 1.05 0.50 CSP-8 7.09 1.08 0.68 CSP-14 2.32 1.08 1.03
CSP-3 2.62 1.07 0.69 CSP-9 2.59 1.04 0.69 CSP-15 3.75 1.09 0.61
CSP-4 4.01 1.08 1.14 CSP-10 7.49 1.08 0.68 QN-AX 4.04 1.00 0.00CSP-5 6.92 1.08 1.32 CSP-11 2.69 1.00 0.00 QD-AX 4.00 1.11 1.38
CSP-6 8.27 1.10 0.86 CSP-12 2.77 1.05 1.21Dichlorprop CSP-1 3.86 1.10 1.28 CSP-7 3.31 1.00 0.00 CSP-13 n.d. n.d. n.d.
CSP-2 15.71 1.38 2.62 CSP-8 7.63 1.05 0.59 CSP-14 2.57 1.10 1.52CSP-3 n.d. n.d. n.d. CSP-9 2.57 1.30 3.81 CSP-15 n.d. n.d. n.d.
CSP-4 4.02 1.05 0.85 CSP-10 8.40 1.17 1.90 QN-AX 5.10 1.22 2.75CSP-5 n.d. n.d. n.d. CSP-11 2.91 1.16 2.33 QD-AX 4.98 1.33 4.46
CSP-6 11.03 1.18 1.42 CSP-12 3.44 1.34 3.85PI-2-25-1 CSP-1 4.14 1.00 0.00 CSP-7 5.12 1.00 0.00 CSP-13 n.d. n.d. n.d.
CSP-2 8.37 1.00 0.00 CSP-8 6.52 1.00 0.00 CSP-14 3.15 1.23 2.80
CSP-3 n.d. n.d. n.d. CSP-9 4.00 1.19 2.53 CSP-15 n.d. n.d. n.d.CSP-4 5.25 1.22 2.70 CSP-10 7.92 1.08 0.72 QN-AX 5.08 1.33 3.33CSP-5 n.d. n.d. n.d. CSP-11 3.69 1.32 4.08 QD-AX 5.76 1.55 5.74
CSP-6 8.03 1.15 0.99 CSP-12 4.32 1.39 4.16PI-2-34-1 CSP-1 2.44 1.27 2.80 CSP-7 3.04 1.38 3.77 CSP-13 n.d. n.d. n.d.
CSP-2 4.17 1.18 1.16 CSP-8 3.43 1.27 2.10 CSP-14 1.60 1.21 2.29CSP-3 n.d. n.d. n.d. CSP-9 1.91 1.20 2.36 CSP-15 n.d. n.d. n.d.CSP-4 2.57 1.22 2.58 CSP-10 3.60 1.20 1.70 QN-AX 2.80 1.07 0.77
CSP-5 n.d. n.d. n.d. CSP-11 2.08 1.07 0.94 QD-AX 2.91 1.22 2.57CSP-6 3.77 1.21 1.40 CSP-12 2.44 1.08 0.92
PI-2-56-2 CSP-1 2.28 1.00 0.00 CSP-7 7.48 1.49 5.24 CSP-13 3.20 1.00 0.00
CSP-2 6.98 1.50 4.16 CSP-8 7.23 1.34 1.84 CSP-14 2.95 1.00 0.00
CSP-3 3.35 1.00 0.00 CSP-9 3.17 1.06 0.96 CSP-15 5.17 1.10 0.63CSP-4 4.87 1.01 0.17 CSP-10 7.78 1.45 2.25 QN-AX 7.34 1.10 0.88
CSP-5 9.02 1.05 0.42 CSP-11 4.05 1.07 1.03 QD-AX 6.85 1.20 2.10CSP-6 9.72 1.08 0.58 CSP-12 4.19 1.33 3.70
120

H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94 93
Fig. 5. Retention characteristics of racemic DCB-Leu separated on selector B type
CSPs 3–6 comprising loading densities of 150, 230, 310 and 490 �mol/g, respectively.
Chromatographic conditions: CSPs 150 × 4 mm ID; injection volume: 5 �L; sam-
ple concentration: 1 mg/mL MeOH; flow rate: 1.0 mL/min; detection wavelength:
254 nm; mobile phase composition: MeOH/AcOH/NH4OAc = 99/1/0.25 (v/v/w); tem-
perature: 25 ◦C. k1: retention factor of the first eluting enantiomer, k2: retention
factor of the second eluting enantiomer, ˛: chromatographic enantioseparation
factor; Rs: chromatographic resolution.
preferences toward one enantiomer by blocking one side of the
selector.
3.2.6. Impact of different selector loading
Since four columns with different selector loading comprising
the same selector B were prepared we could study the effect of
selector loading on selectivity and resolution. The selector den-
sities in an ascending order (from CSP-3 to CSP-6) are 150, 230,
310 and 490 �mol selector B per gram silica. The retention times
and respective k-values of each single enantiomer were enhanced
with increasing selector loading, which reflects the expectation (see
Fig. 5 using DCB-Leu as an illustrative example). The higher the
selector density, the more interaction sites are available resulting in
longer retention times. The retention of the first peak follows grosso
modo the stoichiometric displacement model [56] and it is assumed
that the retention is more based on the non-stereoselective ion-
pairing event. The retention of the second peak on the other hand
relates to the additional stereoselective interaction increments.
The increase of retention and stereoselectivity is not necessarily
paralleling; in contrary, if the selector density is overcrowded we
observe unfavorable effects, and if the selector content is too low
we also lose enantioselectivity. We assume at this point a statis-
tical even distribution of the immobilized selector on the surface
and not so much of an “island type” selector distribution.
In general, selectivity seems to be dependent on the type of
the analyte. For example, the alpha value of DCB-Leu dropped lin-
early with higher selector densities (from 2.81 to 2.24, R2 = 0.987),
however for the aminophosphonate PI-2-56-2 it slightly increased
(from 1.00 to 1.08) which resulted in partial separation of PI-2-56-2
on highly loaded CSPs.
The trend found for resolution was different and it increased
with increasing selector loading. At a selector loading of 310 �mol/g
resolution reached a maximum. For the highest selector loading
a rapid loss of resolution was observed, which indicates a partial
blockage of the binding sites due to high selector loading. The very
high loading (490 �mol/g) leads to a limited conformational free-
dom of the immobilized selectors, and thus hindered interactions
with the respective analytes. We have not stepwise optimized the
selector loading in the interval between 310 and 490 �mol/g, there-
fore a selector concentration of about 310 �mol/g is considered in
this case to be the optimum coverage. This value is similar to the
coverage of the commercially available reference QN-AX and QD-
AX columns. Because the new selectors are slightly bigger, due to
the 1,2,3-triazole linker unit, it is assumed that the amount of the
selector is in the optimum range.
The big advantage of the synthetic procedure presented in this
study is the controllability of selector loading via the azide click
chemistry without facing restrictions of radical scavenging selec-
tors in case of thiol-ene click chemistry. Thus the controllability of
loading using radical mediated thiol-ene chemistry can be poorer.
However, there are examples described in the literature that also
the thiol-ene click chemistry in binding appropriate selectors to
thiol activated silica can work nearly stoichiometrically [54]. Via
azide click chemistry such restriction can be circumvented in any
case, since in average about 90% of offered selector can be immo-
bilized, representing a more economic reaction rate as well. As
deducted from these studies a stepwise optimization of the selec-
tor density on the silica surface in the course of CSP developments
is advised.
4. Conclusion
A series of novel weak anion exchange chiral stationary phases
based on 3,5-dichlorophenyl-, 3,5-dimethoxyphenyl- and tert-
butyl-carbamoylated Cinchona type selectors immobilized via Cu(I)
mediated Huisgen alkyne–azide cycloaddition has been designed
and evaluated in terms of chromatographic behavior. The imple-
mentation of azide click chemistry circumvents the drawbacks
regarding immobilization associated with potential radical scav-
enging properties of chiral selectors. Furthermore it offers the
possibility to control the optimum selector density of the respective
CSP. The effect of free azide groups on retention is negligible, while
the overall chromatographic behavior in terms of elution order,
enantioselectivity and resolution is comparable to other Cinchona
derived chiral anion exchangers. By means of the established syn-
thetic protocols and immobilization strategies in combination with
different carbamoyl residues further investigations in the fields of
enantioseparation and selector density optimization are possible.
References
[1] E.R. Francotte, J. Chromatogr. A 666 (1994) 565.
[2] E.R. Francotte, A. Junker-Buchheit, J. Chromatogr. Biomed. Appl. 576 (1992) 1.
[3] E.R. Francotte, J. Chromatogr. A 906 (2001) 379.
[4] C. Bicchi, A. D’Amato, P. Rubiolo, J. Chromatogr. A 843 (1999) 99.
[5] T.J. Betts, J. Chromatogr. A 936 (2001) 33.
[6] W. Vetter, V. Schurig, J. Chromatogr. A 774 (1997) 143.
[7] J.W. Cochran, G.M. Frame, J. Chromatogr. A 843 (1999) 323.[8] K. Bester, Anal. Bioanal. Chem. 376 (2003) 302.
[9] F.E. Ahmed, TrAC Trends Anal. Chem. 22 (2003) 170.[10] N.W. Smith, M.B. Evans, Chromatographia 38 (1994) 649.
[11] M.R. Euerby, C.M. Johnson, K.D. Bartle, P. Myers, S.C.P. Roulin, Anal. Commun.33 (1996) 403.
[12] N.M. Maier, P. Franco, W. Lindner, J. Chromatogr. A 906 (2001) 3.
[13] S. Han, K. Row, Chin. J. Chem. Eng. 13 (2005) 741.[14] M. Laemmerhofer, W. Lindner, J. Chromatogr. A 829 (1998) 115.
[15] L. He, T.E. Beesley, J. Liq. Chromatogr. Relat. Technol. 28 (2005) 1075.[16] A.E. Ribeiro, P.S. Gomes, L.S. Pais, A.E. Rodrigues, Sep. Sci. Technol. 46 (2011)
1726.[17] S.S. Ribeiro, C. Raminelli, A.L.M. Porto, J. Fluorine Chem. 154 (2013) 53.
[18] H. Lorenz, A. Perlberg, D. Sapoundjiev, M.P. Elsner, A. Seidel-Morgenstern,
Chem. Eng. Process. 45 (2006) 863.
[19] R. Hellinger, J. Horak, W. Lindner, Anal. Bioanal. Chem. 405 (2013) 8105.[20] T. Miyabe, H. Iida, A. Ohnishi, E. Yashima, Chem. Sci. 3 (2012) 863.
[21] X.U. Xue-Feng, G.U.O. Zhi-Mou, Xin-Miao. LIANG, Separation of ben-zoxazine enantiomers on �-cyclodextrin bonded chiral stationaryphases[J], Chinese Journal of Chromatography 30 (11) (2012) 1188–1193,http://dx.doi.org/10.3724/SP.J.1123.2012.02011.
[22] T.A. Gondova, J. Petrovaj, M. Sucha, D.W. Armstrong, J. Liq. Chromatogr. Relat.Technol. 34 (2011) 2304.
[23] S. Park, S.J. Kim, M.H. Hyun, Bull. Korean Chem. Soc. 33 (2012) 3497.
[24] M. Laemmerhofer, J. Chromatogr. A. 1217 (2010) 814.[25] W.H. Pirkle, T.C. Pochapsky, Chem. Rev. 89 (1989) 347.
121

94 H. Hettegger et al. / J. Chromatogr. A 1337 (2014) 85–94
[26] C.V. Hoffmann, M. Laemmerhofer, W. Lindner, J. Chromatogr. A. 1161 (2007)
242.[27] A.F.G. Gargano, M. Kohout, P. Macikova, M. Laemmerhofer, W. Lindner, Anal.
Bioanal. Chem. 405 (2013) 8027.
[28] C.V. Hoffmann, R. Pell, M. Lammerhofer, W. Lindner, Anal. Chem. 80 (2008)
8780.[29] C.V. Hoffmann, R. Reischl, N.M. Maier, M. Laemmerhofer, W. Lindner, J. Chro-
matogr. A. 1216 (2009) 1147.[30] C.V. Hoffmann, R. Pell, M. Laemmerhofer, W. Lindner, Anal. Chem. (Washington,
DC, U.S.) 80 (2008) 8780.[31] K.M. Kacprzak, W. Lindner, J. Sep. Sci. 34 (2011) 2391.
[32] A. Mandl, L. Nicoletti, M. Lammerhofer, W. Lindner, J. Chromatogr. A 858 (1999)
1.[33] W.R. Oberleitner, N.M. Maier, W. Lindner, J. Chromatogr. A 960 (2002) 97.
[34] V.V. Rostovtsev, L.G. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed. 41(2002) 2596.
[35] K.M. Kacprzak, N.M. Maier, W. Lindner, J. Chromatogr. A. 1218 (2011) 1452.
[36] C.W. Tornoe, C. Christensen, M. Meldal, J. Org. Chem. 67 (2002) 3057.[37] B.C. Boren, S. Narayan, L.K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia, V.V. Fokin,
J. Am. Chem. Soc. 130 (2008) 8923.[38] M. Barra, O. Roy, M. Traikia, C. Taillefumier, Org. Biomol. Chem. 8 (2010) 2941.
[39] S. Wernisch, R. Pell, W. Lindner, J. Sep. Sci. 35 (2012) 1560.
[40] M. Laemmerhofer, W. Lindner, J. Chromatogr. A 741 (1996) 33.[41] N.M. Maier, L. Nicoletti, M. Lammerhofer, W. Lindner, Chirality 11 (1999) 522.
[42] M. Laemmerhofer, P. Di Eugenio, I. Molnar, W. Lindner, J. Chromatogr. B Biomed.Appl. 689 (1997) 123.
[43] V. Piette, M. Lammerhofer, K. Bischoff, W. Lindner, Chirality 9 (1997) 157.[44] J. Picha, M. Budesinsky, I. Hanclova, M. Sanda, P. Fiedler, V. Vanek, J. Jiracek,
Tetrahedron 65 (2009) 6090.[45] E. Gavioli, N.M. Maier, K. Haupt, K. Mosbach, W. Lindner, Anal. Chem. 77 (2005)
5009.
[46] D. Wolrab, P. Fruehauf, M. Kohout, W. Lindner, J. Sep. Sci. 36 (2013) 2826.[47] M. Ichikawa, M. Ohtsuka, H. Ohki, N. Haginoya, M. Itoh, K. Sugita, H. Usui, M.
Suzuki, K. Terayama, A. Kanda, Bioorg. Med. Chem. 20 (2012) 3072.[48] K.M. Kacprzak, N.M. Maier, W. Lindner, Tetrahedron Lett. 47 (2006) 8721.
[49] N.M. Maier, Chromatogr. Today 2009 (2009) 3.[50] G.-X. Li, Z.-Q. Liu, X.-Y. Luo, Eur. J. Med. Chem. 45 (2010) 1821.
[51] G.D.H. Dijkstra, R.M. Kellogg, H. Wynberg, J.S. Svendsen, I. Marko, K.B. Sharpless,J. Am. Chem. Soc. 111 (1989) 8069.
[52] C. Czerwenka, M. Laemmerhofer, N.M. Maier, K. Rissanen, W. Lindner, Anal.Chem. 74 (2002) 5658.
[53] M.A. Marshall, H.A. Mottola, Anal. Chem. 57 (1985) 375.[54] P. Levkin, N.M. Maier, W. Lindner, V. Schurig, J. Chromatogr. A. 1269 (2012) 270.
[55] K.H. Krawinkler, E. Gavioli, N.M. Maier, W. Lindner, Chromatographia 58 (2003)
555.[56] M. Laemmerhofer, W. Lindner, in: E. Grushka, N. Grinberg (Eds.), Advances in
Chromatography, CRC Press, Boca Raton, FL, 2008, p. 1.
122

10.2 Conference contributions
The respective presenter is underlined.
10.2.1 Oral presentations
Mimini, V; Hettegger, H; Amer, H; Fackler, K; Potthast, A; Rosenau, T (2018): Lignosulfonate shaping: Purification, modification and crosslinking towards lignin-based function materials. 3rd International Conference on Advanced Functional Materials, San Francisco, USA, AUG 3-5, 2018. In: ICAFM2018 (Eds.), Key Engineering Materials - 3rd International Conference on Advanced Functional Materials, Book of Abstracts , p.27; ISBN: 1662-9795.
Mimini, V; Hashim, SNAS; Sumerskii, I; Zinovyev, G; Fackler, K; Potthast, A; Rosenau, T (2017): Isolation and characterisation of lignosulfonate obtained from sulfite spent liquor by adsorption on XAD-7 resin. International Conference: Renewable Plant Resources - Chemistry, Technology, Medicine, Saint Petersburg, RUSSIA, SEP 18-22, 2017. In: Saint Petersburg State Forest Technical University, 2017 International Conference: Renewable Plant Resources - Chemistry, Technology, Medicine. Book of Abstracts, p.61.
10.2.2 Poster presentation
Mimini, V; Hashim, SNAS; Sumerskii, I; Gebauer, I; Fackler, K; Potthast, A; Rosenau, T (2018): Upscaling of the XAD-7 method for isolation of lignosulfonates from sulfite spent liquor. 15th European Workshop on Lignocellulosics and Pulp (EWLP), Aveiro, PORTUGAL, JUN 26-29, 2018. In: University of Aveiro (Eds.), 15th European Workshop on Lignocellulosics and Pulp (EWLP), Conference Proceedings, pp. 43-45.
Mimini, V; Kabrelian, V; Fackler K; Potthast, A; Rosenau, T (2016): Lignofoam: Isolation and Characterization of Lignosulfonate. 4th Doc Day 2016, Tulln, OCT 18, 2016. In: University of Natural Resources and Life Sciences (BOKU), Kompetenzzentrum Holz GmbH, 4th DocDay- Book of Abstracts, p.55
123

124

125

11 Curriculum vitae
Personal Details
Name: Vebi Mimini
Date of birth: April 11th, 1982
Place of birth: Struga, North Macedonia
Academic credentials
10/2015 – 06/2019 Dissertation, Institute for Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences (BOKU), Vienna; Subject of the PhD Thesis: “Lignofoams: Potential for 100% bio-based matrices and resins”. Supervisor: Univ.-Prof. Dipl.-Chem. Dr.rer.nat. Dr.h.c. Thomas Rosenau
07/2012 – 02/2013 Project staff at the Department of Analytical Chemistry, University of Vienna, Group of Prof. Wolfgang Lindner
10/2012 – 07/2015 MSc (Chemistry), Department of Analytical Chemistry, University of Vienna; Subject of the Master Thesis: “Herstellung und Evaluierung von chiralen stationären Phasen (CSPs) auf Basis von Chinin/Chinidin sowie von 9-epi- Chinin/Chinidin” Supervisor: Em.o.Univ.-Prof. Dr. Wolfgang Lindner
03/2008 – 07/2012 BSc (Chemistry), Institute of Analytical Chemistry, University of Vienna; Subject of the Bachelor Thesis: “Herstellung von Propargyloxyphenyl-Carbamaten aus Chinchonanen, deren Immobilisierung auf dem Kieselgel und Evaluierung der zugehörigen chiralen stationären Phasen (CSPs)” Supervisor: Em.o.Univ.-Prof. Dr. Wolfgang Lindner
126




















