
Copper(II) complexes of thioarylazo-pentanedione: Structures, magnetism, redox properties and...
10
Copper(II) complexes of thioarylazo-pentanedione: Structures, magnetism, redox properties and correlation with DFT calculations Mrinal Kanti Paira a , Tapan Kumar Mondal a , Elena López-Torres b , Joan Ribas c , Chittaranjan Sinha a,⇑ a Inorganic Chemistry Section, Department of Chemistry, Jadavpur University, Kolkata 700 032, India b Departamento de Química Inorgánica, c/Francisco Tomás y Valiente, 7. Universidad, Autónoma de Madrid, Cantoblanco, 28049-Madrid, Spain c Departament de Química Inorgànica, Universitat de Barcelona, Diagonal, 6487, 08028-Barcelona, Spain article info Article history: Received 16 November 2009 Accepted 7 August 2010 Available online 24 August 2010 Keywords: Azoacetylacetonate Copper(II) complex Bridged dimer Antiferromagetic coupling Spin density abstract Copper(II) complexes of 3-((2-(alkylthio)phenylazo)-2,4-pentanedione, tridentate O, N, S donor ligands, are described in this work. Chloride bridged copper(II) polymers (1) and thiocyanato bridged copper(II) dimmers (2) are characterized by a single crystal X-ray diffraction study. The complexes show antiferro- magnetic interactions, with J = 0.5 ± 0.1 cm 1 (1a) and 25.8 ± 0.5 cm 1 (2b), which implies stronger coupling in the –SCN-bridging compound. The spectra, redox and magnetism are explained by DFT studies. Ó 2010 Elsevier Ltd. All rights reserved. 1. Introduction Copper is second to iron in its usefulness in life and society. The metal and its compounds are used in every sphere of life. The po- tential role played by copper ions, present in the active sites of many metalloproteins having the CuN 2 S 2 chromophore [1–7], has stimulated the design of new ligand frames having N, S donor sets and their copper complexes as models for providing a better understanding of biological systems [8–11]. The copper(II)-N,S chelates have antineoplastic activities [12–20] and act as efficient photosensitizers to cleave DNA [21,22]. The magnetic properties of copper complexes, as discrete complexes and bridging dimers, trimers, etc., are of great importance because of their potential applications [23–25]. As a consequence, there has been continued interest in the investigation of copper(II) complexes with the ½Cu II 2 ðl-XÞ 2 (X = Cl , Br ,N 3 , SCN , etc.) core which display differ- ent structures with a variety of Cu–X bond distances and Cu–X–Cu bridge angles, depending on the type of terminal donor atoms and other structure varying parameters [26,27]. The metal complexes of O, N and S donor ligands has expanded enormously [28–33] and embraces very wide and diversified sub- jects comprising vast areas of organometallic compounds and cat- alysts, various aspects of bioinorganic chemistry, metal clusters, supramolecular complexes, etc. In the development of the coordi- nation chemistry of N-donor centers, imine, –N@C– and azo groups, –N@N– have assumed leading roles. The coordination chemistry of transition metals with azo ligands is of interest due to the observation of several interesting properties [34–38]. Azo compounds are used as dyes and pigments. They exhibit antifungal and antibacterial properties. The metal complexes of azo dyes show more efficient activity than the organic molecules alone, due to charge sharing and the availability of a binding site about the metal ion, and have been put forward as photo-stable, weather stable pigments [34–36]. Recently, some metal complexes of dyes have found applications in photoelectronic devices, optical recording media, light emitting diodes, field effect transistors, photovoltaic cells (PVCs) [37,38,39–42], etc. The azo function is photochromatic, induces easy charge transfer and exhibits intense colour in the visible region. Compounds containing the azo group are also pH-responsive and redox active [43–49,50]. In analytical chemistry, azo compounds have been used as acid–base and metal- lochromic indicators [51–55]. Some of them play a role as efficient and selective spectrophotometric reagents, colorants in textile industry and have been used in solid phase extraction processes [56–59]. The p-acidity and metal binding ability of the azo nitro- gen have drawn interest to the exploration of the chemistry of me- tal complexes incorporating azo ligands. Thus, the synthesis of ligands incorporating the azo function in different backbones is an inspiring field of research. We have been engaged for last dec- ade in the design of azo-conjugated ligands and characterization of their metal complexes [50,56–59,60,61,62–65,66]. Acetylace- tone has an active –CH 2 – group and undergoes electrophilic substitution by the diazonium ion, Ar-N@N–, to synthesise azo 0277-5387/$ - see front matter Ó 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.poly.2010.08.024 ⇑ Corresponding author. E-mail address: [email protected] (C. Sinha). Polyhedron 29 (2010) 3147–3156 Contents lists available at ScienceDirect Polyhedron journal homepage: www.elsevier.com/locate/poly
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Copper(II) complexes of thioarylazo-pentanedione: Structures, magnetism, redox properties and...

Polyhedron 29 (2010) 3147–3156
Contents lists available at ScienceDirect
Polyhedron
journal homepage: www.elsevier .com/locate /poly
Copper(II) complexes of thioarylazo-pentanedione: Structures, magnetism,redox properties and correlation with DFT calculations
Mrinal Kanti Paira a, Tapan Kumar Mondal a, Elena López-Torres b, Joan Ribas c, Chittaranjan Sinha a,⇑a Inorganic Chemistry Section, Department of Chemistry, Jadavpur University, Kolkata 700 032, Indiab Departamento de Química Inorgánica, c/Francisco Tomás y Valiente, 7. Universidad, Autónoma de Madrid, Cantoblanco, 28049-Madrid, Spainc Departament de Química Inorgànica, Universitat de Barcelona, Diagonal, 6487, 08028-Barcelona, Spain
a r t i c l e i n f o
Article history:Received 16 November 2009Accepted 7 August 2010Available online 24 August 2010
Keywords:AzoacetylacetonateCopper(II) complexBridged dimerAntiferromagetic couplingSpin density
0277-5387/$ - see front matter � 2010 Elsevier Ltd. Adoi:10.1016/j.poly.2010.08.024
⇑ Corresponding author.E-mail address: [email protected] (C. Sinha).
a b s t r a c t
Copper(II) complexes of 3-((2-(alkylthio)phenylazo)-2,4-pentanedione, tridentate O, N, S donor ligands,are described in this work. Chloride bridged copper(II) polymers (1) and thiocyanato bridged copper(II)dimmers (2) are characterized by a single crystal X-ray diffraction study. The complexes show antiferro-magnetic interactions, with J = �0.5 ± 0.1 cm�1 (1a) and �25.8 ± 0.5 cm�1 (2b), which implies strongercoupling in the –SCN-bridging compound. The spectra, redox and magnetism are explained by DFTstudies.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Copper is second to iron in its usefulness in life and society. Themetal and its compounds are used in every sphere of life. The po-tential role played by copper ions, present in the active sites ofmany metalloproteins having the CuN2S2 chromophore [1–7], hasstimulated the design of new ligand frames having N, S donor setsand their copper complexes as models for providing a betterunderstanding of biological systems [8–11]. The copper(II)-N,Schelates have antineoplastic activities [12–20] and act as efficientphotosensitizers to cleave DNA [21,22]. The magnetic propertiesof copper complexes, as discrete complexes and bridging dimers,trimers, etc., are of great importance because of their potentialapplications [23–25]. As a consequence, there has been continuedinterest in the investigation of copper(II) complexes with the½CuII
2ðl-XÞ2� (X = Cl�, Br�, N3�, SCN�, etc.) core which display differ-
ent structures with a variety of Cu–X bond distances and Cu–X–Cubridge angles, depending on the type of terminal donor atoms andother structure varying parameters [26,27].
The metal complexes of O, N and S donor ligands has expandedenormously [28–33] and embraces very wide and diversified sub-jects comprising vast areas of organometallic compounds and cat-alysts, various aspects of bioinorganic chemistry, metal clusters,supramolecular complexes, etc. In the development of the coordi-nation chemistry of N-donor centers, imine, –N@C– and azo
ll rights reserved.
groups, –N@N– have assumed leading roles. The coordinationchemistry of transition metals with azo ligands is of interest dueto the observation of several interesting properties [34–38]. Azocompounds are used as dyes and pigments. They exhibit antifungaland antibacterial properties. The metal complexes of azo dyesshow more efficient activity than the organic molecules alone,due to charge sharing and the availability of a binding site aboutthe metal ion, and have been put forward as photo-stable, weatherstable pigments [34–36]. Recently, some metal complexes of dyeshave found applications in photoelectronic devices, opticalrecording media, light emitting diodes, field effect transistors,photovoltaic cells (PVCs) [37,38,39–42], etc. The azo function isphotochromatic, induces easy charge transfer and exhibits intensecolour in the visible region. Compounds containing the azo groupare also pH-responsive and redox active [43–49,50]. In analyticalchemistry, azo compounds have been used as acid–base and metal-lochromic indicators [51–55]. Some of them play a role as efficientand selective spectrophotometric reagents, colorants in textileindustry and have been used in solid phase extraction processes[56–59]. The p-acidity and metal binding ability of the azo nitro-gen have drawn interest to the exploration of the chemistry of me-tal complexes incorporating azo ligands. Thus, the synthesis ofligands incorporating the azo function in different backbones isan inspiring field of research. We have been engaged for last dec-ade in the design of azo-conjugated ligands and characterizationof their metal complexes [50,56–59,60,61,62–65,66]. Acetylace-tone has an active –CH2– group and undergoes electrophilicsubstitution by the diazonium ion, Ar-N@N–, to synthesise azo

3148 M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156
compounds. Because of the potential application of acetylacetona-to metal chelates, chemists are trying to synthesize newer deriva-tives and their complexes. Arylazo appended acetylacetone hasbeen known in the literature since 1925 [67–69]. However, thio-arylazo (R–S–C6H4–N@N–) functionalized acetylacetone is hithertounknown. We have undertaken a programme to synthesise, 3-((2-(alkylthio)phenylazo)-2,4-pentanedione, and then to synthesisecopper(II) complexes with this compound as a ligand. In somecases the structures were confirmed by a X-ray diffraction study.Variable temperature magnetic measurements were carried outto explain the magnetic interactions present in the complexes.The electronic, redox and magnetic properties are explained byDFT computation using an optimized geometry for the complexes.
2. Experimental
2.1. Materials and measurements
Acetylacetone (Hacac), 2-aminothiophenol, iodomethane (MeI),iodoethane (EtI), CuCl2, 2H2O and NH4SCN were purchased from E.Merck, India. All other chemicals used were of A.R. quality andwere used as received. The organic solvents were purified anddried by standard methods [70]. The 2-(alkylthio)anilines wereprepared by a reported procedure [71].
UV–Vis spectra were recorded using a Perkin–Elmer Lambda 25UV–Vis spectrophotometer and infrared spectra were obtainedfrom a Perkin–Elmer Spectrum RX1 instrument. Microanalyseswere collected on a Perkin–Elmer 2400 CHN elemental analyser.1H NMR spectra were recorded on a Brucker 300 MHz FT-NMR.The room temperature magnetic moments were measured usinga Magnetic Susceptibility Balance, Sherwood Scientific Cambridge,UK. Molar conductances (KM) were measured in a Systronics con-ductivity meter 304 model using ca. 10�3 M solutions in acetoni-trile. Electrochemical measurements were performed usingcomputer-controlled PAR model 250 VersaStat electrochemicalinstruments with Pt-disk electrodes. All measurements were car-ried out under a nitrogen environment at 298 K with reference toSCE in acetonitrile using [nBu4N][ClO4] as the supporting electro-lyte. The reported potentials are uncorrected for the junction po-tential. EPR spectra were measured in MeCN–CH2Cl2 solution atroom temperature (298 K) and at liquid nitrogen temperature(77 K) using a Bruker EPR spectrometer model EMX 10/12, X-bandER 4119 HS cylindrical resonator.
2.1.1. Preparation of the ligands2.1.1.1. 3-(2-(Methylthio)phenylazo)-2,4-pentanedione, HL1. Into a50 ml ethanol solution of 2-(amino)thiophenol (2.9 g, 0.023 mol),metallic sodium (0.534 g, 0.023 mol) was slowly added under coldconditions, stirring with a magnetic stirrer. Stirring was continuedfor 1 h, maintaining the cold conditions, whereupon the colourchanged from yellow to orange. Then 1.45 ml (0.023 mol) of MeIwas added under the same conditions, and the stirring was contin-ued for 30 min. The mixture was then refluxed for 2 h at 65–70 �Cin a water bath, and the colour of the solution changed to red. Thewhole mixture was then poured into a large excess of water, agummy mass separated out, and it was extracted in benzene andwashed with water. The benzene was then removed by a rotaryevaporator. The gummy mass was then dissolved 1:1 in HCl, andNaNO2 solution was added dropwise to it at 0–5 �C. This solutionwas then added to a Na2CO3 solution of acetylacetone (2.34 ml,0.023 mol) dropwise. A yellowish orange precipitate was obtained;it was filtered and washed with cold water, dried over CaCl2 desic-cators. Yield 4.80 g (82.6%), m.p. 121 �C. Microanalytical data: Anal.Calc. for C12H14N2O2S: C, 57.58; H, 5.64; N, 11.19. Found: C, 57.44;H, 5.67; N, 11.06%. IR data (KBr disc) (m, cm�1): 1671(s), 1626(m),
1506, 1357, 1319. 1H NMR data in CDCl3 (d, ppm): 15.05 (OH, s),7.78 (11-H, d, J = 8.1 Hz), 7.49 (8-H, d, J = 7.2 Hz), 7.35 (9-H, t,J = 7.7 Hz), 7.16 (10-H, t, J = 7.2 Hz), 2.63 (1-CH3, s), 2.51 (5-CH3,s), 2.47 (12-CH3, s). 13C NMR data in CDCl3 (d, ppm): 197.1 (2-C),196.9 (4-C), 122.1–142.7 (ArC, 6C), 115.1 (3-C), 30.2 (1-C), 26.3(5-C), 24.3 (12-C). kmax (e � 10�3, M�1 cm�1): 397 (18.3); 380(21.3); 275 (8.82); 253 (15.0) in CHCl3.
2.1.1.2. 3-(2-(Ethylthio)phenylazo)-2,4-pentanedione, HL2. 2-(Ami-no)-thioethylphenol was first synthesized following the abovementioned method using 3.0 g (0.024 mol) 2-(amino)thiophenoland 1.92 ml (0.024 mol) of EtI followed by coupling with 2.44 ml(0.024 mol) acetylacetone. A yellowish orange precipitate was ob-tained; it was filtered and washed with cold water, dried overCaCl2 desiccators. Yield 4.95 g (78.1%), m.p. 101 �C. Microanalyticaldata: Anal. Calc. for C13H16N2O2S: C, 59.05; H, 6.10; N, 10.59.Found: C, 59.20; H, 6.08; N, 10.43%. IR data (KBr disc) (m, cm�1):1673(s), 1627(m), 1507, 1358, 1321. 1H NMR data in CDCl3 (d,ppm): 15.07 (OH, s), 7.80 (11-H, d, J = 8.2 Hz), 7.53 (8-H, d,J = 7.6 Hz), 7.40 (9-H, t, J = 7.7 Hz), 7.15 (10-H, t, J = 7.5 Hz), 2.80(12-CH2, q, J = 14.6 Hz), 2.64 (1-CH3, s), 2.52 (5-H, s), 1.25 (13-CH3, t, J = 7.3 Hz). 13C NMR data in CDCl3 (d, ppm): 197.3(2-C),197.1 (4-C), 122.7–143.0 (ArC, 6C), 115.3 (3-C), 31.5 (12-C), 30.0(1-C), 26.6 (5-C), 14.7 (13-C). kmax (e � 10�3, M�1 cm�1): 396(14.3); 374 (17.6); 277 (5.63); 253 (12.6) in CHCl3.
2.1.2. Preparation of 1/n[Cu(L1)Cl]n (1a)HL1 (92.6 mg, 0.37 mmol) was dissolved in methanol (20 ml)
and was added to a methanolic solution of CuCl2�2H2O (65.2 mg,0.38 mmol) with stirring at room temperature and the stirringwas continued for 1 h. The colour of the solution changed from or-ange yellow to blue green. The solution was then filtered and keptfor crystallization after the addition of a few drops of DMF. Blockshape crystals appeared on the inner wall of the beaker and werecollected by filtration, washed with MeOH–water (1:1, v/v) anddried over CaCl2 in a desiccator. Yield, 98 mg (76%). Microanalyticaldata: Anal. Calc. for C12H13N2O2SClCu: C, 41.38; H, 3.76; N, 8.04.Found: C, 41.29; H, 3.85; N, 8.00%. IR data (KBr disc) (m, cm�1):1657, 1364, 305. kmax (e � 10�3, M�1 cm�1): 608 (0.420); 405(10.418); 290 (8.039); 268 (10.981) in CHCl3.
2.1.3. Preparation of 1/n[Cu(L2)Cl]n (1b)The complex 1b was prepared following the same procedure as
1a using 100.3 mg (0.38 mmol) of HL2 and 65.2 mg (0.38 mmol) ofCuCl2�2H2O. Yield, 101 mg (73%). Microanalytical data: Anal. Calc.for C13H15N2O2SClCu: C, 43.09; H, 4.17; N, 7.73. Found: C, 43.00;H, 4.20; N, 7.80%. IR data (KBr disc) (m, cm�1): 1652, 1355, 308. kmax
(e � 10�3, M�1 cm�1): 607 (0.529); 406 (14.493); 289 (11.024); 267(15.109) in CHCl3.
2.1.4. Preparation of [Cu(L1)(SCN)]2 (2a)To a CuCl2�2H2O solution (85.3 mg, 0.50 mmol) in methanol
(15 ml), HL1 (121.3 mg, 0.49 mmol) was added in same solvent(15 ml MeOH) and the mixture was stirred for 30 min. ThenNH4SCN (938.3 mg, 0.50 mmol) in MeOH solution was added andthe stirring was continued for another 2.5 h, whereupon the colourof the solution changed to greenish yellow. The solution was fil-tered and kept for crystallization, after the addition of few dropsof DMF, by slow evaporation in air. A crystalline product was ob-tained within 3 days. The crystals were collected and washed withMeOH–water (1:1, v/v) and dried over CaCl2 in a desiccator. Yield127 mg (71%). Microanalytical data: Anal. Calc. for C26H26N6O4S4-
Cu2: C, 42.09; H, 3.53; N, 11.33. Found: C, 42.00; H, 3.50; N,11.28%. IR data (KBr disc) (m, cm�1): 2110, 1659, 1356. kmax
(e � 10�3, M�1 cm�1): 599 (0.337); 420 (15.40), 374 (16.816);327 (5.711); 278 (6.380) in CHCl3.

M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156 3149
2.1.5. Preparation of [Cu(L2)(SCN)]2 (2b)Complex 2b was prepared following the procedure of 2a using
132.0 mg (0.50 mmol) of HL2, 85.3 mg (0.50 mmol) CuCl2�2H2Oand 938.3 mg (0.50 mmol) of NH4SCN in MeOH. Yield 135 mg(72%). Microanalytical data: Anal. Calc. for C28H30N6O4S4Cu2: C,43.68; H, 3.93; N, 10.91. Found: C, 43.65; H, 3.95; N, 11.00%. IR data(KBr disc) (m, cm�1): 2114, 1660, 1356. kmax (e � 10�3, M�1 cm�1):616 (0.630); 407 (13.217); 295 (10.760); 270 (13.041) in CHCl3.
2.2. Crystal structure determination
The crystals, [Cu(L1)Cl]n (1a) (0.15 � 0.20 � 0.30 mm), [Cu(L2)-(NCS)]2 (2b) (0.15 � 0.30 � 0.30 mm) were grown by slow evapo-ration of the reaction mixture in methanol over a week. Data werecollected (Table 1) using a Bruker AXS Kappa Apex-II diffractome-ter equipped with an Apex-II CCD area detector using a graphitemonochromator (Mo Ka radiation, k = 0.71 073 Å). Diffractionswere recorded with 2h in the range 4.62 6 2h 6 57.40� for 1a and2.96 6 2h 6 57.40� for 2b. Reflection data were recorded usingthe w and x scan techniques. Data were corrected for Lp and anempirical absorption correction in the hkl range: �14 6 h 6 14;�23 6 k 6 23; �10 6 l 6 10 for 1a and �17 6 h 6 17; �19 6 k 619; �19 6 l 6 19 for 2b. The substantial redundancy in data allowsempirical absorption corrections (SADABS) [72] to be applied usingmultiple measurements of symmetry-equivalent reflections. Theraw intensity data frames were integrated with the SAINT program,which also applied corrections for Lorentz and polarization effects.[73] The software package SHELXTL version 6.10 was used for spacegroup determination, structure solution and refinement. The struc-tures were solved by direct methods (SHELXS-97), [74] completedwith difference Fourier syntheses, and refined with full-matrixleast squares using SHELXL-97 minimizing xðF2
0 � F2c ). Weighted R
factors (Rw) and all goodness of fit S are based on F2; conventionalR factors (R) are based on F [75]. All non-hydrogen atoms were re-fined with anisotropic displacement parameters and hydrogenatoms were located by difference maps, although they were posi-tioned geometrically after each cycle of refinement. All scattering
Table 1Crystallographic data of [Cu(L1)Cl]n (1a) and [Cu(L2)(SCN)]2 (2b).
Crystal parameter (1a) (2b)
Empirical formula C12H13ClCuN2O2S C28H30Cu2N6O4S4
Formula weight 348.29 769.90Crystal system monoclinic triclinicSpace group P2(1)/c �PUnit cell dimensionsa (Å) 10.7980(4) 12.9475(11)b (Å) 17.6826(7) 14.1966(12)c (Å) 7.4096(3) 14.2019(11)a (�) 90.00 76.541(4)b (�) 109.201(2) 88.561(4)c (�) 90.00 66.165(4)V (Å3) 1336.06(9) 2315.3(3)k (Å) 0.71073 0.71073qcalcd (Mg m�3) 1.732 1.657Z 4 3F(0 0 0) 708 1182l (mm�1) 1.988 1.694T (K) 100(2) 296Total data 14 853 103 922Unique data 3432 11 880Refined parameters 224 604Ra 0.0232 0.0470wRb 0.0608 0.1431GOF 1.057 1.09
a R = R||F0| � |Fc||/R|F0|.b wR2 = [RwðF2
0 � F2c Þ
2=P
wðF20Þ
2�1=2, w = 1/[r2(F0)2 + (0.0258P)2 + 0.9848P] for[Cu(L1)Cl]n (1a); w = 1/[r2(F0)2 + (0.0682P)2 + 6.9845P] for [Cu(L2)(SCN)]2 (2b);where P ¼ ðF2
0 þ 2F2c Þ=3.
factors and anomalous dispersions factors are contained in theSHELXTL 6.10 program library.
2.3. Variable temperature magnetic measurements
The magnetic behavior of [Cu(L1)Cl]n (1a) and [Cu(L2)(SCN)]2
(2b) were measured on a SQUID susceptometer in the temperaturerange 2–300 K and with a magnetic field value of 0.1 T. Diamagne-tism was corrected from Pascal Tables. Magnetization measure-ments were made in the same SQUID at 2 K.
2.4. Computation
Full geometry optimizations of the complexes were carried outusing the density functional theory method at the (U)B3LYP level[76–78]. All elements except Cu were assigned the 6-31G(d) basisset. LanL2DZ with an effective core potential for a Cu atom was used.The vibrational frequency calculations were performed to ensurethat the optimized geometries represent the local minima and thatthere are only positive eigen values. All calculations were performedwith GAUSSIAN03 program package [79] with the aid of the GaussViewvisualization program [80]. Vertical electronic excitations based onB3LYP optimized geometries were computed using the time-depen-dent density functional theory (TD-DFT) formalism [81–83] in chlo-roform using a conductor-like polarizable continuum model (CPCM)[84–86]. GaussSum [87] was used to calculate the fractional contri-butions of various groups to each molecular orbital.
3. Results and discussion
3.1. Synthesis and formulation
The 3-(2-(alkylthio)-2-phenylazo)-2,4-pentanediones (R = Me,HL1; Et, HL2) are synthesized by coupling 2-(alkylthio)phenyldiazo-nium chloride with acetylacetone in basic medium (Na2CO3
solution, pH 7) (Scheme 1) and are purified by repeated crystallisa-tion from an aqueous ethanol (1:1, v/v) mixture. The purity of theproduct was checked by elemental analyses.
The reaction between a methanolic solution of CuCl2�2H2O andHL in a 1:1 mole ratio has isolated shining dark green crystals of
S
N
N
O
OH
HL1 HL2
12
34
5
6
7
8
9
10
11
12
S
N
N
O
OH
12
34
5
6
7
8
9
10
11
1213
S
N
O
S
N
N
O
O
R
Cu
SCN
S
N
N
OCH3
CH3
Cu
O
Cl
R
Cl
2; R = Me ( a), Et (b)
2
1
n
Scheme 1. The ligands and the complexes.
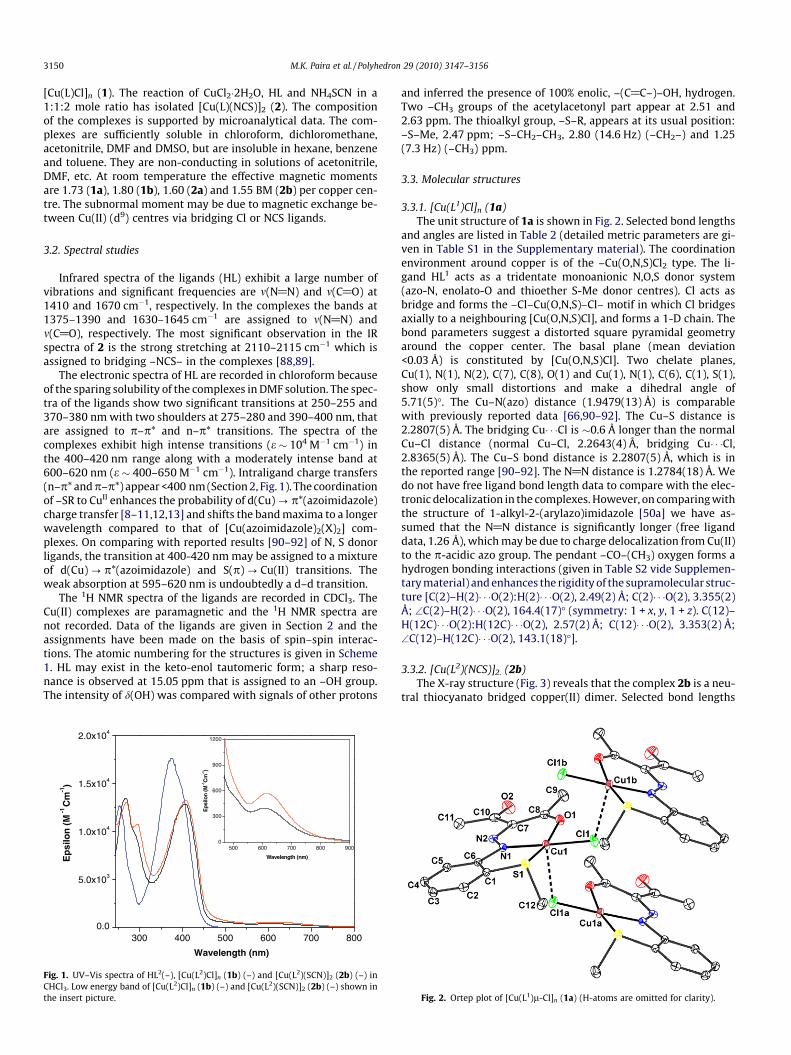
3150 M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156
[Cu(L)Cl]n (1). The reaction of CuCl2�2H2O, HL and NH4SCN in a1:1:2 mole ratio has isolated [Cu(L)(NCS)]2 (2). The compositionof the complexes is supported by microanalytical data. The com-plexes are sufficiently soluble in chloroform, dichloromethane,acetonitrile, DMF and DMSO, but are insoluble in hexane, benzeneand toluene. They are non-conducting in solutions of acetonitrile,DMF, etc. At room temperature the effective magnetic momentsare 1.73 (1a), 1.80 (1b), 1.60 (2a) and 1.55 BM (2b) per copper cen-tre. The subnormal moment may be due to magnetic exchange be-tween Cu(II) (d9) centres via bridging Cl or NCS ligands.
3.2. Spectral studies
Infrared spectra of the ligands (HL) exhibit a large number ofvibrations and significant frequencies are m(N@N) and m(C@O) at1410 and 1670 cm�1, respectively. In the complexes the bands at1375–1390 and 1630–1645 cm�1 are assigned to m(N@N) andm(C@O), respectively. The most significant observation in the IRspectra of 2 is the strong stretching at 2110–2115 cm�1 which isassigned to bridging –NCS– in the complexes [88,89].
The electronic spectra of HL are recorded in chloroform becauseof the sparing solubility of the complexes in DMF solution. The spec-tra of the ligands show two significant transitions at 250–255 and370–380 nm with two shoulders at 275–280 and 390–400 nm, thatare assigned to p–p* and n–p* transitions. The spectra of thecomplexes exhibit high intense transitions (e � 104 M�1 cm�1) inthe 400–420 nm range along with a moderately intense band at600–620 nm (e � 400–650 M�1 cm�1). Intraligand charge transfers(n–p* andp–p*) appear <400 nm (Section 2, Fig. 1). The coordinationof –SR to CuII enhances the probability of d(Cu) ? p*(azoimidazole)charge transfer [8–11,12,13] and shifts the band maxima to a longerwavelength compared to that of [Cu(azoimidazole)2(X)2] com-plexes. On comparing with reported results [90–92] of N, S donorligands, the transition at 400-420 nm may be assigned to a mixtureof d(Cu) ? p*(azoimidazole) and S(p) ? Cu(II) transitions. Theweak absorption at 595–620 nm is undoubtedly a d–d transition.
The 1H NMR spectra of the ligands are recorded in CDCl3. TheCu(II) complexes are paramagnetic and the 1H NMR spectra arenot recorded. Data of the ligands are given in Section 2 and theassignments have been made on the basis of spin–spin interac-tions. The atomic numbering for the structures is given in Scheme1. HL may exist in the keto-enol tautomeric form; a sharp reso-nance is observed at 15.05 ppm that is assigned to an –OH group.The intensity of d(OH) was compared with signals of other protons
300 400 500 600 700 8000.0
5.0x103
1.0x104
1.5x104
2.0x104
500 600 700 800 9000
300
600
900
1200
Eps
ilon
(M-1C
m-1)
Wavelength (nm)Ep
silo
n (
M-1
Cm
-1)
Wavelength (nm)
Fig. 1. UV–Vis spectra of HL2(–), [Cu(L2)Cl]n (1b) (–) and [Cu(L2)(SCN)]2 (2b) (–) inCHCl3. Low energy band of [Cu(L2)Cl]n (1b) (–) and [Cu(L2)(SCN)]2 (2b) (–) shown inthe insert picture.
and inferred the presence of 100% enolic, –(C@C–)–OH, hydrogen.Two –CH3 groups of the acetylacetonyl part appear at 2.51 and2.63 ppm. The thioalkyl group, –S–R, appears at its usual position:–S–Me, 2.47 ppm; –S–CH2–CH3, 2.80 (14.6 Hz) (–CH2–) and 1.25(7.3 Hz) (–CH3) ppm.
3.3. Molecular structures
3.3.1. [Cu(L1)Cl]n (1a)The unit structure of 1a is shown in Fig. 2. Selected bond lengths
and angles are listed in Table 2 (detailed metric parameters are gi-ven in Table S1 in the Supplementary material). The coordinationenvironment around copper is of the –Cu(O,N,S)Cl2 type. The li-gand HL1 acts as a tridentate monoanionic N,O,S donor system(azo-N, enolato-O and thioether S-Me donor centres). Cl acts asbridge and forms the –Cl–Cu(O,N,S)–Cl– motif in which Cl bridgesaxially to a neighbouring [Cu(O,N,S)Cl], and forms a 1-D chain. Thebond parameters suggest a distorted square pyramidal geometryaround the copper center. The basal plane (mean deviation<0.03 Å) is constituted by [Cu(O,N,S)Cl]. Two chelate planes,Cu(1), N(1), N(2), C(7), C(8), O(1) and Cu(1), N(1), C(6), C(1), S(1),show only small distortions and make a dihedral angle of5.71(5)�. The Cu–N(azo) distance (1.9479(13) Å) is comparablewith previously reported data [66,90–92]. The Cu–S distance is2.2807(5) Å. The bridging Cu� � �Cl is �0.6 Å longer than the normalCu–Cl distance (normal Cu–Cl, 2.2643(4) Å, bridging Cu� � �Cl,2.8365(5) Å). The Cu–S bond distance is 2.2807(5) Å, which is inthe reported range [90–92]. The N@N distance is 1.2784(18) Å. Wedo not have free ligand bond length data to compare with the elec-tronic delocalization in the complexes. However, on comparing withthe structure of 1-alkyl-2-(arylazo)imidazole [50a] we have as-sumed that the N@N distance is significantly longer (free liganddata, 1.26 Å), which may be due to charge delocalization from Cu(II)to the p-acidic azo group. The pendant –CO–(CH3) oxygen forms ahydrogen bonding interactions (given in Table S2 vide Supplemen-tary material) and enhances the rigidity of the supramolecular struc-ture [C(2)–H(2)� � �O(2):H(2)� � �O(2), 2.49(2) Å; C(2)� � �O(2), 3.355(2)Å; \C(2)–H(2)� � �O(2), 164.4(17)� (symmetry: 1 + x, y, 1 + z). C(12)–H(12C)� � �O(2):H(12C)� � �O(2), 2.57(2) Å; C(12)� � �O(2), 3.353(2) Å;\C(12)–H(12C)� � �O(2), 143.1(18)�].
3.3.2. [Cu(L2)(NCS)]2. (2b)The X-ray structure (Fig. 3) reveals that the complex 2b is a neu-
tral thiocyanato bridged copper(II) dimer. Selected bond lengths
Fig. 2. Ortep plot of [Cu(L1)l-Cl]n (1a) (H-atoms are omitted for clarity).

Table 2Bond distances and bond angles.
Bond lengths (Å) Bond angles (�)
X-ray DFT X-ray DFT
[Cu(L1)Cl]n (1a)Cu(1)–Cl(1) 2.2643(4) 2.278 Cl(1)–Cu(1)–S(1) 88.06(2) 90.43Cu(1)–S(1) 2.2807(5) 2.326 Cl(1)–Cu(1)–O(1) 93.40(4) 94.40Cu(1)–O(1) 1.9271(13) 1.930 Cl(1)–Cu(1)–N(1) 174.10(4) 168.2Cu(1)–N(1) 1.9479(13) 1.954 Cl(1)–Cu(1)–Cl(1b) 103.08(1) –Cu(1)–Cl(1b) 2.8365(5) – S(1)–Cu(1)–O(1) 167.50(4) 168.5N(1)–N(2) 1.2784(18) 1.289 S(1)–Cu(1)–N(1) 87.06(4) 87.65
O(1)–Cu(1)–N(1) 90.65(5) 89.67
[Cu(L2)(SCN)2]2 (2b)Cu(1)–S(1) 2.3588(9) 2.49003 S(1)–Cu(1) –S(2) 101.42(3) 98.85229Cu(1)–S(2) 2.6468(10) 2.80135 S(1)–Cu(1) –O(1) 150.23(8) 152.32424Cu(1)–O(1) 1.956(2) 2.03427 S(1)–Cu(1) –N(1) 85.70(8) 85.62552Cu(1)–N(1) 1.933(3) 1.90819 S(1)–Cu(1) –N(6) 93.58(9) 92.21789Cu(1)–N(6) 1.932(3) 1.84951 S(2)–Cu(1) –O(1) 107.78(8) 107.88309Cu(2)–S(3) 2.3230(9) 2.46138 S(2)–Cu(1) –N(1) 88.67(8) 87.26765Cu(2)–S(4) 2.8088(9) 2.92318 S(2)–Cu(1) –N(6) 94.36(8) 96.31118Cu(2)–O(3) 1.938(2) 2.00338 O(1)–Cu(1) –N(1) 89.24(11) 88.57363Cu(2)–N(3) 1.952(3) 1.85792 O(1)–Cu(1) –N(6) 89.94(12) 91.84358Cu(2)–N(4) 1.934(3) 1.90798 N(1)–Cu(1) –N(6) 176.96(11) 176.07654N(1)–N(2) 1.282(3) 1.28718 S(3)–Cu(2) –S(4) 85.21(3) 86.14518N(4)–N(5) 1.289(4) 1.28691 S(3)–Cu(2) –O(3) 169.68(8) 168.92936
S(3)–Cu(2) –N(3) 92.70(8) 91.63577S(3)–Cu(2) –N(4) 87.04(8) 86.69494S(4)–Cu(2)–O(3) 104.60(7) 104.16889S(4)–Cu(2)–N(3) 93.97(8) 96.66019S(4)–Cu(2)–N(4) 86.58(8) 89.37554
M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156 3151
and angles are given in Table 2 (detailed metric parameters are gi-ven in Table S1 in the Supplementary material). The copper lies in adistorted square pyramid with CuON2S2 donor sets. There are twodimers in the unit cell. The dimer is generated by two –NCS– bridges.The bridge angles are N(3)–Cu(2)–S(4), 93.96(8)�, and N(6)–Cu(1)–S(2), 94.35(9)�. The –NCS– unit is almost linear, as is revealed fromthe angles N(3)–C(14)–S(2), 179.1(3)� and N(6)–C(28)–S(4),178.0(3)�. The chelate angles are \O(1)–Cu(1)–N(1), 89.23(10)�and \N(1)–Cu(1)–S(1), 85.70(8)�. The two chelate planes, Cu(1),N(1), N(2), C(7), C(8), O(1) and Cu(1), N(1), C(1), C(6), S(1), are notplanar and make a dihedral angle of 19.37�. The distorted squareplane is constituted by CuON2S, on which the bridged thiocyanato-S lies perpendicular (�94�). There are two N donor centres: N(azo)and N(thiocyanato), and the Cu–N distances lie within the range1.889–1.953 Å. The two S donor centres show different Cu–S dis-tances: the chelated thioether-S shows lower Cu–S distances(2.32–2.36 Å) than that of Cu–S(thiocyanate) (2.64–2.81 Å). TheN@N distances are N(1)–N(2), 1.282(3); N(4)–N(5), 1.289(3);N(7)–N(8), 1.306(3) Å. The significant elongation of the N@N bond
Fig. 3. ORTEP plot of [Cu(L2)(SCN)]2 (2b) (H-atoms are omitted for clarity).
length implies some amount of charge delocalization, d(Cu) ? p*(azo) (which is supported from spectral observation, vide supra).The pendant –CO–(CH3) forms hydrogen bonding interactions(given in Table S3 vide Supplementary material): C(23)–H(23B)� � �O(4):H(23B)� � �O(4), 2.4211 Å; C(23)� � �O(4), 2.761(5) Å;\C(23)–H(23B)� � �O(4), 100.43� (symmetry: x, y, 1 + z). C(26)–H(26B)� � �O(4):H(26B)� � �O(4), 2.4711 Å; C(26)� � �O(4), 3.222(4) Å;\C(26)–H(26B)� � �O(4), 133.99� (symmetry: �x, �y, 1 � z) C(40)–H(40A)� � �O(2):H(40A)� � �O(2), 2.3870 Å; C(40)� � �O(2), 3.352(4) Å;\C(40)–H(40A)� � �O(2), 173.15� (symmetry: x, y, �1 + z).
3.4. Electrochemistry
The voltammogram of the complexes shows a redox response at�0.3 V (Table 3, Fig. 4). The free ligand does not show any oxida-tion while irreversible reductive responses appear at <�1.5 V,which is referred to electron addition to the azo function. Thequasireversible response at 0.3 V (DEP > 100 mV) may be ascribedto the Cu(II)/Cu(I) response [66]. On scanning at <�0.7 V potential,an upsurge of current at av. �0.3 V is observed. The high currentdensity may be due to reoxidation of adsorbed Cu (metallic) onthe electrode surface which has been produced after reduction ofCu(I) at negative potential. The peak-to-peak separation of the re-dox couple positive to the reference electrode is dependent on thescan rate and increases from 140 mV at 30 mV s�1 to 600 mV at1000 mV s�1. At slow scan rates (30–100 mV s�1), DEP remains al-most constant, as do the EPa and the EPc values. This corresponds toa low heterogeneous electron-transfer rate constant which hasbeen influenced by the applied potential. In general, the electro-chemical reduction of copper(II) complexes is associated with achange in coordination geometry. The solution structure of thecopper(II) complexes show a square pyramidal or trigonal bipyra-midal geometry, which upon reduction rearranges fast to a tetra-hedral geometry via bond rupture and bond formation. The twoquasireversible couples at �0.8 and �1.3 V are assigned to theprocesses [-N=N-] / [-N N-] - and N-] - / [-N-N-][-N , respec-tively (Table 3).
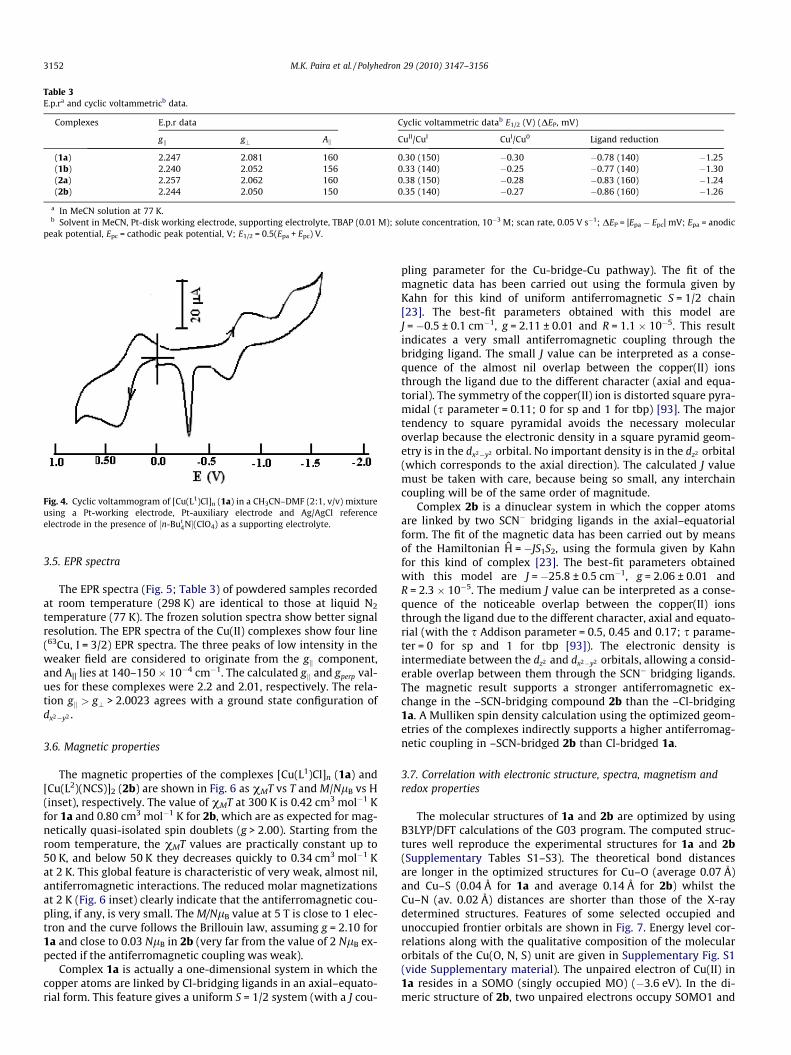
Table 3E.p.ra and cyclic voltammetricb data.
Complexes E.p.r data Cyclic voltammetric datab E1/2 (V) (DEP, mV)
gk g? Ak CuII/CuI CuI/Cu0 Ligand reduction
(1a) 2.247 2.081 160 0.30 (150) �0.30 �0.78 (140) �1.25(1b) 2.240 2.052 156 0.33 (140) �0.25 �0.77 (140) �1.30(2a) 2.257 2.062 160 0.38 (150) �0.28 �0.83 (160) �1.24(2b) 2.244 2.050 150 0.35 (140) �0.27 �0.86 (160) �1.26
a In MeCN solution at 77 K.b Solvent in MeCN, Pt-disk working electrode, supporting electrolyte, TBAP (0.01 M); solute concentration, 10�3 M; scan rate, 0.05 V s�1; DEP = |Epa � Epc| mV; Epa = anodic
peak potential, Epc = cathodic peak potential, V; E1/2 = 0.5(Epa + Epc) V.
Fig. 4. Cyclic voltammogram of [Cu(L1)Cl]n (1a) in a CH3CN–DMF (2:1, v/v) mixtureusing a Pt-working electrode, Pt-auxiliary electrode and Ag/AgCl referenceelectrode in the presence of ½n-But
4N�(ClO4) as a supporting electrolyte.
3152 M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156
3.5. EPR spectra
The EPR spectra (Fig. 5; Table 3) of powdered samples recordedat room temperature (298 K) are identical to those at liquid N2
temperature (77 K). The frozen solution spectra show better signalresolution. The EPR spectra of the Cu(II) complexes show four line(63Cu, I = 3/2) EPR spectra. The three peaks of low intensity in theweaker field are considered to originate from the gk component,and A|| lies at 140–150 � 10�4 cm�1. The calculated gk and gperp val-ues for these complexes were 2.2 and 2.01, respectively. The rela-tion gk > g? > 2.0023 agrees with a ground state configuration ofdx2�y2 .
3.6. Magnetic properties
The magnetic properties of the complexes [Cu(L1)Cl]n (1a) and[Cu(L2)(NCS)]2 (2b) are shown in Fig. 6 as vMT vs T and M/NlB vs H(inset), respectively. The value of vMT at 300 K is 0.42 cm3 mol�1 Kfor 1a and 0.80 cm3 mol�1 K for 2b, which are as expected for mag-netically quasi-isolated spin doublets (g > 2.00). Starting from theroom temperature, the vMT values are practically constant up to50 K, and below 50 K they decreases quickly to 0.34 cm3 mol�1 Kat 2 K. This global feature is characteristic of very weak, almost nil,antiferromagnetic interactions. The reduced molar magnetizationsat 2 K (Fig. 6 inset) clearly indicate that the antiferromagnetic cou-pling, if any, is very small. The M/NlB value at 5 T is close to 1 elec-tron and the curve follows the Brillouin law, assuming g = 2.10 for1a and close to 0.03 NlB in 2b (very far from the value of 2 NlB ex-pected if the antiferromagnetic coupling was weak).
Complex 1a is actually a one-dimensional system in which thecopper atoms are linked by Cl-bridging ligands in an axial–equato-rial form. This feature gives a uniform S = 1/2 system (with a J cou-
pling parameter for the Cu-bridge-Cu pathway). The fit of themagnetic data has been carried out using the formula given byKahn for this kind of uniform antiferromagnetic S = 1/2 chain[23]. The best-fit parameters obtained with this model areJ = �0.5 ± 0.1 cm�1, g = 2.11 ± 0.01 and R = 1.1 � 10�5. This resultindicates a very small antiferromagnetic coupling through thebridging ligand. The small J value can be interpreted as a conse-quence of the almost nil overlap between the copper(II) ionsthrough the ligand due to the different character (axial and equa-torial). The symmetry of the copper(II) ion is distorted square pyra-midal (s parameter = 0.11; 0 for sp and 1 for tbp) [93]. The majortendency to square pyramidal avoids the necessary molecularoverlap because the electronic density in a square pyramid geom-etry is in the dx2�y2 orbital. No important density is in the dz2 orbital(which corresponds to the axial direction). The calculated J valuemust be taken with care, because being so small, any interchaincoupling will be of the same order of magnitude.
Complex 2b is a dinuclear system in which the copper atomsare linked by two SCN� bridging ligands in the axial–equatorialform. The fit of the magnetic data has been carried out by meansof the Hamiltonian H = �JS1S2, using the formula given by Kahnfor this kind of complex [23]. The best-fit parameters obtainedwith this model are J = �25.8 ± 0.5 cm�1, g = 2.06 ± 0.01 andR = 2.3 � 10�5. The medium J value can be interpreted as a conse-quence of the noticeable overlap between the copper(II) ionsthrough the ligand due to the different character, axial and equato-rial (with the s Addison parameter = 0.5, 0.45 and 0.17; s parame-ter = 0 for sp and 1 for tbp [93]). The electronic density isintermediate between the dz2 and dx2�y2 orbitals, allowing a consid-erable overlap between them through the SCN� bridging ligands.The magnetic result supports a stronger antiferromagnetic ex-change in the –SCN-bridging compound 2b than the –Cl-bridging1a. A Mulliken spin density calculation using the optimized geom-etries of the complexes indirectly supports a higher antiferromag-netic coupling in –SCN-bridged 2b than Cl-bridged 1a.
3.7. Correlation with electronic structure, spectra, magnetism andredox properties
The molecular structures of 1a and 2b are optimized by usingB3LYP/DFT calculations of the G03 program. The computed struc-tures well reproduce the experimental structures for 1a and 2b(Supplementary Tables S1–S3). The theoretical bond distancesare longer in the optimized structures for Cu–O (average 0.07 Å)and Cu–S (0.04 Å for 1a and average 0.14 Å for 2b) whilst theCu–N (av. 0.02 Å) distances are shorter than those of the X-raydetermined structures. Features of some selected occupied andunoccupied frontier orbitals are shown in Fig. 7. Energy level cor-relations along with the qualitative composition of the molecularorbitals of the Cu(O, N, S) unit are given in Supplementary Fig. S1(vide Supplementary material). The unpaired electron of Cu(II) in1a resides in a SOMO (singly occupied MO) (�3.6 eV). In the di-meric structure of 2b, two unpaired electrons occupy SOMO1 and
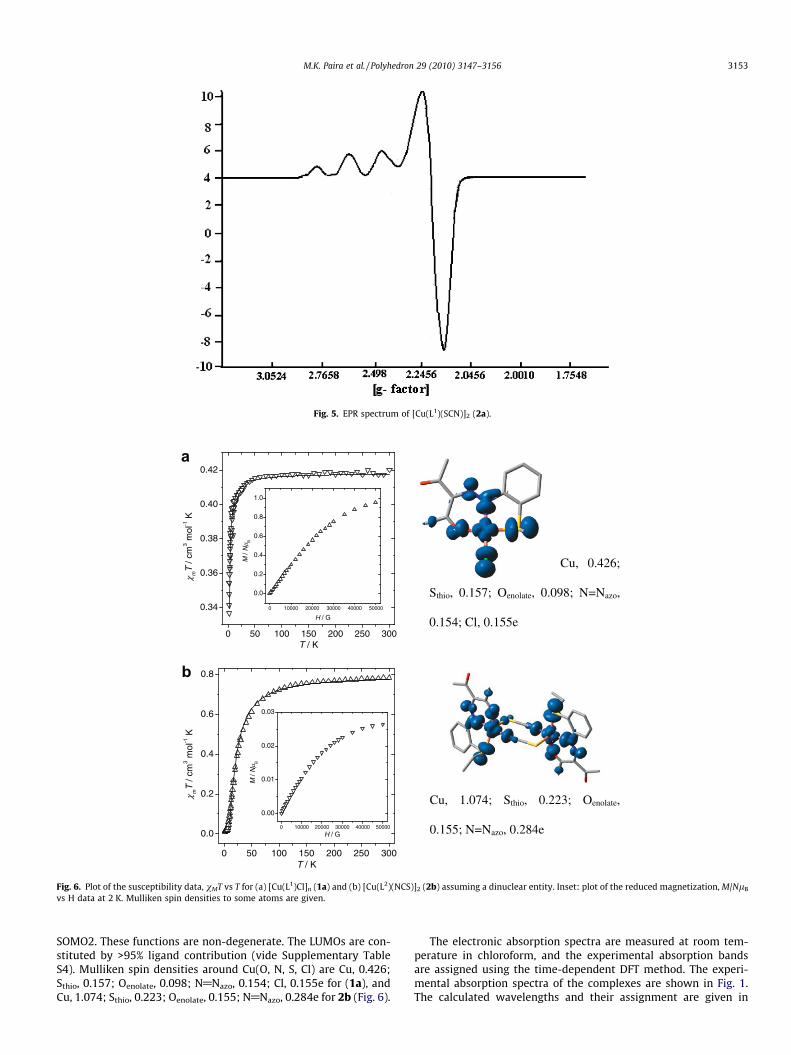
Fig. 5. EPR spectrum of [Cu(L1)(SCN)]2 (2a).
Cu, 0.426;
Sthio, 0.157; Oenolate, 0.098; N=Nazo,
0.154; Cl, 0.155e
Cu, 1.074; Sthio, 0.223; Oenolate,
0.155; N=Nazo, 0.284e 0.0
0.2
0.4
0.6
0.8
0 10000 20000 30000 40000 50000
0.00
0.01
0.02
0.03
χ mT
/ cm
3 mol
-1 K
T / K
M /
Nμ B
H / G
0 50 100 150 200 250 300
0 50 100 150 200 250 300
0.34
0.36
0.38
0.40
0.42
0 10000 20000 30000 40000 50000
0.0
0.2
0.4
0.6
0.8
1.0
χ mT
/ cm
3 mol
-1 K
T / K
M /
Nμ B
H / G
a
b
Fig. 6. Plot of the susceptibility data, vMT vs T for (a) [Cu(L1)Cl]n (1a) and (b) [Cu(L2)(NCS)]2 (2b) assuming a dinuclear entity. Inset: plot of the reduced magnetization, M/NlB
vs H data at 2 K. Mulliken spin densities to some atoms are given.
M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156 3153
SOMO2. These functions are non-degenerate. The LUMOs are con-stituted by >95% ligand contribution (vide Supplementary TableS4). Mulliken spin densities around Cu(O, N, S, Cl) are Cu, 0.426;Sthio, 0.157; Oenolate, 0.098; N@Nazo, 0.154; Cl, 0.155e for (1a), andCu, 1.074; Sthio, 0.223; Oenolate, 0.155; N@Nazo, 0.284e for 2b (Fig. 6).
The electronic absorption spectra are measured at room tem-perature in chloroform, and the experimental absorption bandsare assigned using the time-dependent DFT method. The experi-mental absorption spectra of the complexes are shown in Fig. 1.The calculated wavelengths and their assignment are given in
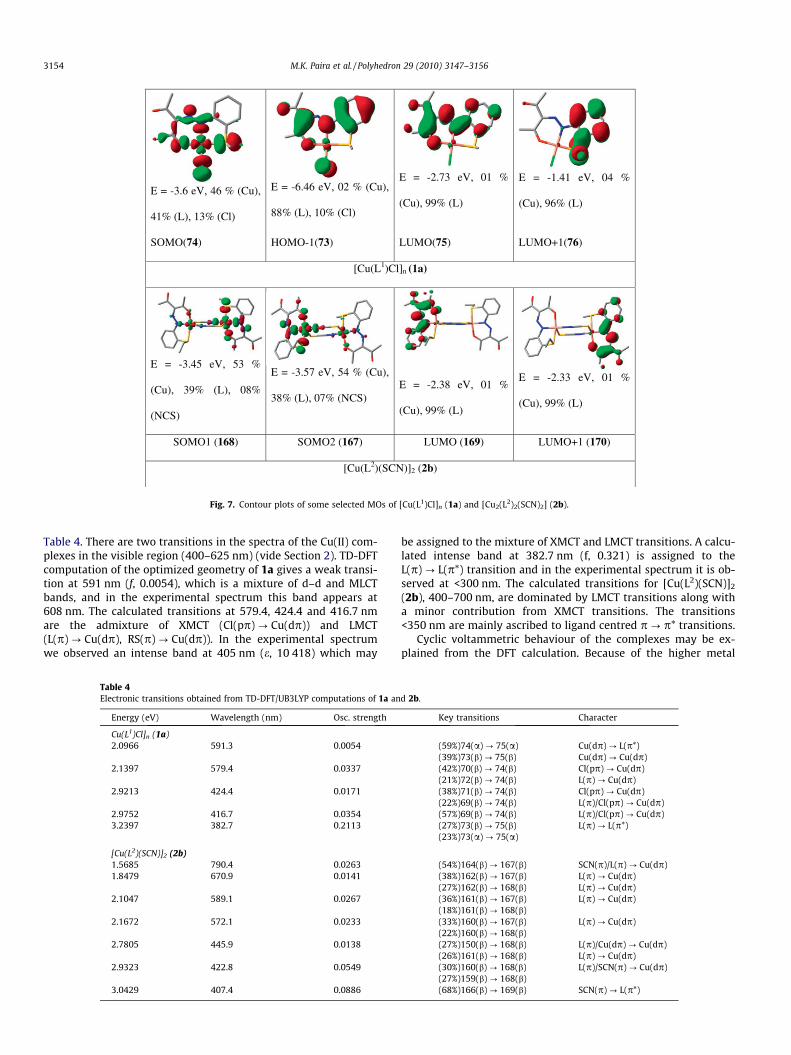
E = -3.6 eV, 46 % (Cu),
41% (L), 13% (Cl)
E = -6.46 eV, 02 % (Cu),
88% (L), 10% (Cl)
E = -2.73 eV, 01 %
(Cu), 99% (L)
E = -1.41 eV, 04 %
(Cu), 96% (L)
SOMO(74) HOMO-1(73) LUMO(75) LUMO+1(76)
[Cu(L1)Cl]n (1a)
E = -3.45 eV, 53 %
(Cu), 39% (L), 08%
(NCS)
E = -3.57 eV, 54 % (Cu),
38% (L), 07% (NCS) E = -2.38 eV, 01 %
(Cu), 99% (L)
E = -2.33 eV, 01 %
(Cu), 99% (L)
SOMO1 (168) SOMO2 (167) LUMO (169) LUMO+1 (170)
[Cu(L2)(SCN)]2 (2b)
Fig. 7. Contour plots of some selected MOs of [Cu(L1)Cl]n (1a) and [Cu2(L2)2(SCN)2] (2b).
3154 M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156
Table 4. There are two transitions in the spectra of the Cu(II) com-plexes in the visible region (400–625 nm) (vide Section 2). TD-DFTcomputation of the optimized geometry of 1a gives a weak transi-tion at 591 nm (f, 0.0054), which is a mixture of d–d and MLCTbands, and in the experimental spectrum this band appears at608 nm. The calculated transitions at 579.4, 424.4 and 416.7 nmare the admixture of XMCT (Cl(pp) ? Cu(dp)) and LMCT(L(p) ? Cu(dp), RS(p) ? Cu(dp)). In the experimental spectrumwe observed an intense band at 405 nm (e, 10 418) which may
Table 4Electronic transitions obtained from TD-DFT/UB3LYP computations of 1a an
Energy (eV) Wavelength (nm) Osc. strength
Cu(L1)Cl]n (1a)2.0966 591.3 0.0054
2.1397 579.4 0.0337
2.9213 424.4 0.0171
2.9752 416.7 0.03543.2397 382.7 0.2113
[Cu(L2)(SCN)]2 (2b)1.5685 790.4 0.02631.8479 670.9 0.0141
2.1047 589.1 0.0267
2.1672 572.1 0.0233
2.7805 445.9 0.0138
2.9323 422.8 0.0549
3.0429 407.4 0.0886
be assigned to the mixture of XMCT and LMCT transitions. A calcu-lated intense band at 382.7 nm (f, 0.321) is assigned to theL(p) ? L(p*) transition and in the experimental spectrum it is ob-served at <300 nm. The calculated transitions for [Cu(L2)(SCN)]2
(2b), 400–700 nm, are dominated by LMCT transitions along witha minor contribution from XMCT transitions. The transitions<350 nm are mainly ascribed to ligand centred p ? p* transitions.
Cyclic voltammetric behaviour of the complexes may be ex-plained from the DFT calculation. Because of the higher metal
d 2b.
Key transitions Character
(59%)74(a) ? 75(a)(39%)73(b) ? 75(b)
Cu(dp) ? L(p*)Cu(dp) ? Cu(dp)
(42%)70(b) ? 74(b)(21%)72(b) ? 74(b)
Cl(pp) ? Cu(dp)L(p) ? Cu(dp)
(38%)71(b) ? 74(b)(22%)69(b) ? 74(b)
Cl(pp) ? Cu(dp)L(p)/Cl(pp) ? Cu(dp)
(57%)69(b) ? 74(b) L(p)/Cl(pp) ? Cu(dp)(27%)73(b) ? 75(b)(23%)73(a) ? 75(a)
L(p) ? L(p*)
(54%)164(b) ? 167(b) SCN(p)/L(p) ? Cu(dp)(38%)162(b) ? 167(b)(27%)162(b) ? 168(b)
L(p) ? Cu(dp)L(p) ? Cu(dp)
(36%)161(b) ? 167(b)(18%)161(b) ? 168(b)
L(p) ? Cu(dp)
(33%)160(b) ? 167(b)(22%)160(b) ? 168(b)
L(p) ? Cu(dp)
(27%)150(b) ? 168(b)(26%)161(b) ? 168(b)
L(p)/Cu(dp) ? Cu(dp)L(p) ? Cu(dp)
(30%)160(b) ? 168(b)(27%)159(b) ? 168(b)
L(p)/SCN(p) ? Cu(dp)
(68%)166(b) ? 169(b) SCN(p) ? L(p*)

M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156 3155
(Cu) function in the SOMO/HOMO-1, the observed oxidation is cor-rectly ascribed to the Cu(II)/Cu(I) redox couple. Unoccupied MOsare significantly dominated by the ligand function (>95%), thusreduction may refer to electron accommodation at the azo domi-nated orbital of the ligand. So the assignment of azo reductionsis justified.
The spin density calculation has explained the degree of delo-calization of the unpaired electron over the metal centres in thebridged Cu–X–Cu unit. The coordinating atoms of the ligand partic-ipate in the singly occupied molecular orbital (SOMO), which leadsto the enhancement of the electron density of the donor atoms.Interestingly, the spin density is mainly distributed amongst thecopper dx2�y2 magnetic orbital and the Cl/NCS bridging betweentwo Cu centres (Cu(1)� � �Cu(2)) in the dinuclear entity. It is to benoted that the calculated exchange coupling constant gives anantiferromagnetic interaction (J) �0.14 cm�1 (1a) and 18.65 cm�1
(2b). The overlap between the magnetic orbitals of the copperatoms is taking place via the bonding between the dx2�y2 magneticorbital of copper and the hybrid orbital of the bridging atom. Thus,the spin delocalization on the bridging group corroborates the anti-ferromagnetic interaction. The calculated spin density (Fig. 6) onthe bridging atoms is much higher in 2b than 1a and may explainthe high degree of delocalization in the –SCN-bridged complex (2b)than the Cl- (1a) bridged compound.
4. Conclusion
3-(2-(Alkylthio)-2-phenylazo)-2,4-pentanedione (HL) coordi-nates to Cu(II) as an O, N, S, donor ligand and generates a Cl-bridged polymer or a –SCN-bridged dimer. Electrochemistry showsa quasireversible Cu(II)/Cu(I) redox couple along with a Cu(I)/Cu(0)response. The solution spectra show a high intense metal-to-azo-imine charge transfer. The complexes show antiferromagnetic cou-pling with J = �0.5 ± 0.1 cm�1 for Cl-bridging (1a) and �25.8 ±0.5 cm�1 for –SCN-bridging (2b). A higher degree of delocalizationin the –SCN-bridged dimer than the Cl-bridged polymer may be ex-plained by spin density calculation using optimized geometries ofthe complexes. The spectra, magnetism and redox properties areexplained using DFT computation on the optimized geometries.
Acknowledgements
Financial support from the CAS program, University GrantsCommission and Department of Science & Technology, New Delhiare gratefully acknowledged. One of us, MP, thanks CSIR for a fel-lowship. J.R. acknowledges the financial support from the SpanishGovernment (Grant CTQ2006/03949).
Appendix A. Supplementary material
CCDC 753060 and 753061 contain the supplementary crystallo-graphic data for [Cu(L1)Cl]n (1a) and [Cu(L1)Cl]n (1a). These datacan be obtained free of charge via http://www.ccdc.cam.ac.uk/con-ts/retrieving.html, or from the Cambridge Crystallographic DataCentre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223336 033; or e-mail: [email protected]. Supplementary dataassociated with this article can be found, in the online version, atdoi:10.1016/j.poly.2010.08.024.
References
[1] E.I. Solomon, R.K. Szilagyi, S.D. George, L. Basumallick, Chem. Rev. 104 (2004)419.
[2] D.B. Rorabacher, Chem. Rev. 104 (2004) 651.[3] H.W. Yim, L.M. Tran, E.D. Dobbin, D. Rabinovich, L.M. Liable-Sands, C.D.
Incarvito, K.-C. Larn, A.L. Rheingold, Inorg. Chem. 38 (1999) 2211.
[4] P. Ge, B.S. Haggerty, A.L. Rheingold, C.G. Riordan, J. Am. Chem. Soc. 116 (1994)8406.
[5] A.J. Barton, J. Connolly, W. Levason, A. Mendia-Jalon, S.D. Orchard, G. Reid,Polyhedron 19 (2000) 1373.
[6] P.L. Holland, W.B. Tolman, J. Am. Chem. Soc. 122 (2000) 6331.[7] J.A. Graden, M.C. Posewitz, J.R. Simon, G.N. George, I.J. Pickering, D.R. Winge,
Biochemistry 35 (1996) 14583.[8] D.A. Robb, in: R. Lontie (Ed.), Copper Proteins and Copper Enzymes, vol. 2, CRC
Press, Bocaraton, 1984, p. 207.[9] K.D. Karlin, Z, Tyeklar. Bioinorganic Chemistry of Copper. Chapmann & Hall,
New York, 1993.[10] W. Kaim, B. Schwederski, Bioinorganic Chemistry: Inorganic Elements in the
Chemistry of Life, John Wiley & Sons, Chichester-New York-Brisbane-Toronto-Singapore, 1994. pp. 22 and 196.
[11] P.M. Bush, J.P. Whitehead, C.C. Pink, E.C. Gramm, J.L. Eglin, S.P. Watton, L.E.Pence, Inorg. Chem. 40 (2001) 1871.
[12] E.I. Solomon, P. Chen, M. Metz, S.-K. Lee, A.E. Palmer, Angew. Chem., Int. Ed. 40(2001) 4570.
[13] R. Balamurugan, M. Palaniandavar, S.R. Gopalan, G.U. Kulkarni, Inorg. Chim.Acta 357 (2004) 919.
[14] M. Vidyanathan, R. Balamurugan, U. Sivagnanam, M. Palaniandavar, J. Chem.Soc., Dalton Trans. (2001) 3498 (and references cited therein).
[15] B.C. Westerby, K.L. Juntunen, G.H. Leggett, V.B. Pett, M.J. Koenigbauer, M.D.Purgett, M.J. Taschner, L.A. Ochrymowycz, D.B. Rorabacher, Inorg. Chem. 30(1991) 2109.
[16] K.K. Nanda, A.W. Addison, R.J. Butcher, M.R. McDevitt, T.N. Rao, E. Sinn, Inorg.Chem. 36 (1997) 134.
[17] S. Torelli, C. Belle, C. Philouze, J.-L. Pierre, W. Rammal, E. Saint-Aman, Eur. J.Inorg. Chem. (2003) 2452.
[18] E.A. Ambundo, L.A. Ochrymowycz, D.B. Rorabacher, Inorg. Chem. 40 (2001)5133.
[19] L.Q. Hatcher, D.-H. Lee, M.A. Vance, A.E. Milligan, R. Sarangi, K.O. Hodgson, B.Hedman, E.I. Solomon, K.D. Karlin, Inorg. Chem. 45 (2006) 10055.
[20] P.J. Blower, J.S. Lewis, J. Zweit, Nucl. Med. Biol. 23 (1996) 957.[21] A.R. Cowley, J.R. Dilworth, P.S. Donnelly, E. Labisbal, A. Sousa, J. Am. Chem. Soc.
124 (2002) 5270.[22] S. Dhar, P.A.N. Reddy, M. Nethaji, S. Mahadevan, M.K. Saha, A.R. Chakravorty,
Inorg. Chem. 41 (2002) 3469.[23] S. Dhar, D. Senapati, P.K. Das, P. Chattopadhyay, M. Nethaji, A.R. Chakravarty, J.
Am. Chem. Soc. 125 (2003) 12118.[24] O. Kahn, Molecular Magnetism, VCH, New York, 1993.[25] O. Kahn, Acc. Chem. Res. 33 (2000) 647;
(b) M. Verdaguer, Polyhedron 20 (2001) 1115;(c) D. Gatteschi, R. Sessoli, Angew. Chem., Int. Ed. 42 (2003) 268;(d) H. Oshio, M. Nakano, Chem. Eur. J. 11 (2005) 5178;(e) J.S. Miller, J. Chem. Soc., Dalton Trans. (2006) 2742.
[26] J. Ribas, A. Escuer, M. Monfort, R. Vicente, R. Cortés, L. Lezama, T. Rojo, Coord.Chem. Rev. 193–195 (1999) 1027;(b) M. Ohba, H. Õkawa, Coord. Chem. Rev. 198 (2000) 313;(c) J.S. Miller, J.L. Manson, Acc. Chem. Res. 34 (2001) 563;(d) L.M.C. Beltran, J.R. Long, Acc. Chem. Res. 38 (2005) 325;(e) R. Lescouëzec, L.M. Toma, J. Vaissermann, M. Verdaguer, F.S. Delgado, C.Ruiz-Pérez, F. Lloret, M. Julve, Coord. Chem. Rev. 249 (2005) 2691.
[27] R. Mukherjee, Coord. Chem. Rev. 203 (2000) 151;(b) R. Mukherjee, in: J.A. McCleverty, T.J. Meyer (Eds.), ComprehensiveCoordination Chemistry-II: From Biology to Nanotechnology, vol. 6, ElsevierPergamon, Amsterdam, 2004, pp. 747–910;(c) S. Mandal, F. Lloret, R. Mukherjee, Inorg. Chim. Acta 362 (2009) 27.
[28] D.J. Hodgson, Prog. Inorg. Chem. 19 (1976) 173;(b) S.G.N. Roundhill, D.M. Roundhill, D.R. Bloomquist, C. Landee, R.D. Willett,D.M. Dooley, H.B. Gray, Inorg. Chem. 18 (1979) 831;(c) W.E. Marsh, W.E. Hatfield, D.J. Hodgson, Inorg. Chem. 21 (1982) 2679;(d) W.E. Marsh, D.S. Eggleston, W.E. Hatfield, D.J. Hodgson, Inorg. Chim. Acta70 (1983) 137;(e) W.E. Marsh, K.C. Patel, W.E. Hatfield, D.J. Hodgson, Inorg. Chem. 22 (1983)511;(f) C.P. Landee, R.E. Greeney, Inorg. Chem. 25 (1986) 3771;(g) T. Rojo, M.I. Arriortua, J. Ruiz, J. Darriet, G. Villeneuve, D. Beltran-Porter, J.Chem. Soc., Dalton Trans. (1987) 285;(h) F. Tuna, L. Patron, Y. Journaux, M. Andruh, W. Plass, J.-C. Trombe, J. Chem.Soc., Dalton Trans. (1999) 539;(i) M. Rodriguez, A. Llobet, M. Corbella, A.E. Martell, J. Reibenspies, Inorg.Chem. 38 (1999) 2328;(j) M. Rodriguez, A. Llobet, M. Corbella, Polyhedron 19 (2000) 2483;(k) P. Kapoor, A. Pathak, R. Kapoor, P. Venugopalan, M. Corbella, M. Rodriguez,J. Robles, A. Llobet, Inorg. Chem. 41 (2002) 6153;(l) P. DeHoog, P. Gamez, O. Roubeau, M. Lutz, W.L. Driessen, A.L. Spek, J.Reedijk, New J. Chem. 27 (2003) 18;(m) W.A. Alves, R.H. de Almeida Santos, A. Paduan-Filho, C.C. Becerra, A.C.Borin, A.M. Da Costa Ferreira, Inorg. Chim. Acta 357 (2004) 2269;(n) S.-L. Ma, X.-X. Sun, S. Gao, C.-M. Qi, H.-B. Huang, W.-X. Zhu, Eur. J. Inorg.Chem. 12 (2007) 846 (and references cited therein).
[29] V.W.-W. Yam, V.C.-Y. Lau, K.-K. Cheung, J. Chem. Soc., Chem. Commun. (1995)259.
[30] S. Frantz, J. Fiedler, I. Hartenbach, T. Schleid, W. Kaim, J. Organomet. Chem. 689(2004) 3031.

3156 M.K. Paira et al. / Polyhedron 29 (2010) 3147–3156
[31] A.C. Cope, R.W. Siekman, J. Am. Chem. Soc. 87 (1965) 3272.[32] (a) S. Ganguly, S. Chattopadhyay, C. Sinha, A. Chakravorty, Inorg. Chem. 39
(2000) 2954;(b) S. Ganguly, V. Manivannan, A. Chakravorty, J. Chem. Soc., Dalton Trans.(1998) 461;(c) S. Karmakar, S.B. Choudhury, S. Ganguly, A. Chakravorty, J. Chem. Soc.,Dalton Trans. 12 (1997) 585 (and references cited therein).
[33] (a) R. Acharyya, S.-M. Peng, G.-H. Lee, S. Bhattacharya, Inorg. Chem. 42 (2003)7378;(b) S. Nag, P. Gupta, R.J. Butcher, S. Bhattacharya, Inorg. Chem. 43 (2004) 4814;(c) P. Gupta, R.J. Butcher, S. Bhattacharya, Inorg. Chem. 42 (2003) 5405;(d) R. Aharyya, F. Basuli, R.-Z. Wang, T.C.W. Mak, S. Bhattacharya, Inorg. Chem.43 (2004) 704 (and references cited therein).
[35] (a) A.H. Velders, K. van der Schilden, A.C.G. Hotze, J. Reedijk, H. Kooijman, A.L.Spek, J. Chem. Soc., Dalton Trans. (2004) 448;(b) M. Shivakumar, K. Pramanik, I. Bhattacharyya, A. Chakravorty, Inorg.Chem. 39 (2000) 4332;(c) S. Banerjee, S. Bhattacharyya, B.K. Dirghangi, M. Menon, A. Chakravorty,Inorg. Chem. 39 (2000) 6;(d) C. Das, A. Saha, C.H. Hung, G.-H. Lee, S.-M. Peng, S. Goswami, Inorg. Chem.42 (2003) 198.
[35] G. Pandey, K.K. Narang, Synth. React. Inorg. Metal-Org. Nano-Metal Chem. 34(2005) 397.
[36] Z. Zheng, K. Shetty, J. Agric. Food Chem. 48 (2000) 932.[37] I. Nor, R. Ene, V. Hurduc, M. Bercea, J. Optoelectron. Adv. Mater. 10 (2008) 683.[38] K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C.
Kasada, A. Shinpo, S. Suga, J. Phys. Chem. B 109 (2005) 15476.[39] Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo, K. Hara, J. Phys. Chem. C 111
(2007) 7224.[40] C.D. Dimitrakopoulos, P.R.L. Malenfant, Adv. Mater. 14 (2002) 99.[41] J. Veres, S. Ogier, G. Lloyd, D. de Leeuw, Chem. Mater. 16 (2004) 4543.[42] C.J. Brabec, N.S. Sariciftci, J.C. Hummelen, Adv. Funct. Mater. 11 (2001) 15.[43] K.M. Coakley, M.D. McGehee, Chem. Mater. 16 (2004) 4533.[44] H. Rau, in: H. Dürr, H. Bounas-Laurent (Eds.), Photochromism, Molecules and
Systems, Elsevier, Amsterdam, 1990, p. 165.[45] H. Nishihara, Bull. Chem. Soc. Jpn. 77 (2004) 407.[46] N. Tamai, H. Miyasaka, Chem. Rev. 100 (2000) 1875.[47] S. Yagai, T. Karatsu, A. Kitamura, Chem. Eur. J. 11 (2005) 4054.[48] M. Irie, Science 268 (1995**) (1873).[49] S. Kawata, Y. Kawata, Chem. Rev. 100 (2000) 1777.[50] (a) J. Otsuki, K. Suwa, K. Narutaki, C. Sinha, I. Yoshikawa, K. Araki, J. Phys.
Chem. A 109 (2005) 8064;(b) K.K. Sarker, B.G. Chand, K. Suwa, J. Cheng, T.-H. Lu, J. Otsuki, C. Sinha, Inorg.Chem. 46 (2007) 670;(c) J. Otsuki, K. Suwa, K.K. Sarker, C. Sinha, J. Phys. Chem. A 111 (2007) 1403;(d) K.K. Sarker, D. Sardar, K. Suwa, J. Otsuki, C. Sinha, Inorg. Chem. 46 (2007)8291;(e) P. Pratihar, T.K. Mondal, A.K. Patra, C. Sinha, Inorg. Chem. 48 (2009) 2760.
[51] I. Vogel, A Text Book of Quantitative Inorganic Analysis, ELBS, Longman, 1975.[52] K. Brajter, E. Dbek-Zotorzyska, Analyst 113 (1988) 1571.[53] H. Lee, J.S. Kim, M.Y. Suh, W. Lee, Anal. Chim. Acta 339 (1997) 303.[54] P.K. Tewari, A.K. Singh, Talanta 53 (2001) 823.[55] M.M. Kenawy, M.A.H. Hafez, M.A. Akl, R.R. Lashein, Anal. Sci. 16 (2000) 493.[56] W. Lee, S.E. Lee, C.H. Lee, Y.S. Kim, Y.I. Lee, Microchem. J. 70 (2001) 195.[57] P. Chattopadhyay, C. Sinha, D.K. Pal, Fresenius J. Anal. Chem. 357 (1997) 368.
[58] D. Das, A.K. Das, C. Sinha, Talanta 48 (1999) 1013.[59] D. Das, A.K. Das, C. Sinha, Anal. Lett. 32 (1999) 567.[60] B.C. Mondal, D. Das, A.K. Das, J. Indian Chem. Soc. 48 (2001) 89.[61] R. Roy, P. Chattopadhyay, C. Sinha, S. Chattopadhyay, Polyhedron 15 (1996)
3361.[62] C.K. Pal, S. Chattopadhyay, C. Sinha, D. Bandyopadhyay, A. Chakravorty,
Polyhedron 13 (1994) 999.[63] P.K. Santra, D. Das, T.K. Misra, R. Roy, C. Sinha, S.-M. Peng, Polyhedron 18
(1999) 1909.[64] P.K. Santra, T.K. Misra, D. Das, C. Sinha, A.M.Z. Slawin, J.D. Woollins, Polyhedron
18 (1999) 2869.[65] S. Senapoti, U.S. Ray, P.K. Santra, C. Sinha, J.D. Woollins, A.M.Z. Slawin,
Polyhedron 21 (2002) 753.[66] P.K. Santra, P. Byabartta, S. Chattopadhyay, L.R. Falvello, C. Sinha, Eur. J. Inorg.
Chem (2002) 1124.[67] D. Banerjee, U.S. Ray, Polyhedron 25 (1299) (2006).[68] C. Bulow, W. Spengler, Ber. 58B (1925) 1375.[69] H.V. Patel, P.S. Fernandes, Indian J. Chem., Sect. B 28 (1989) 167.[70] L. Mishra, A.K. Yadaw, R.S. Phadke, C.S. Choi, K. Araki, Metal-Based Drugs 8
(2001) 65.[71] A.I. Vogel, A Text Book of Practical Organic Chemistry, second ed., Longman,
London, 1959.[72] P. Chattopadhyay, C. Sinha, Polyhedron 13 (1994) 2689.[73] G.M. Sheldrick, SADABS Version 2.03, Program for Empirical Absorption
Corrections, Universität Göttingen, Göttingen, Germany, 1997–2001.[74] G.M. Sheldrick, SAINT+NT (Version 6.04) SAX Area-Detector Integration
Program, Bruker AXS, Madison, WI, 1997–2001.[75] G.M. Sheldrick, SHELXTL (Version 6.10) Structure Determination Package, Bruker
AXS, Madison, WI, 2000.[76] G.M. Sheldrick, Acta Crystallogr., Sect. A 46 (1990) 467.[77] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.[78] D. Andrae, U. Häußermann, M. Dolg, H. Stoll, H. Preuß, Theor. Chim. Acta 77
(1990) 123.[79] P. Fuentealba, H. Preuss, H. Stoll, L.V. Szentpaly, Chem. Phys. Lett. 95 (1983)
617.[80] M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., GAUSSIAN 03, Revision D.01,
Gaussian, Inc., Wallingford CT, 2004.[81] GaussView3.0, Gaussian, Pittsburgh, PA.[82] R. Bauernschmitt, R. Ahlrichs, Chem. Phys. Lett. 256 (1996) 454.[83] R.E. Stratmann, G.E. Scuseria, M.J. Frisch, J. Chem. Phys. 109 (1998) 8218.[84] M.E. Casida, C. Jamorski, K.C. Casida, D.R. Salahub, J. Chem. Phys. 108 (1998)
4439.[85] V. Barone, M. Cossi, J. Phys. Chem. A 102 (1998) 1995.[86] M. Cossi, V. Barone, J. Chem. Phys. 115 (2001) 4708.[87] M. Cossi, N. Rega, G. Scalmani, V. Barone, J. Comput. Chem. 24 (2003) 669.[88] N.M. O’Boyle, A.L. Tenderholt, K.M. Langner, J. Comput. Chem. 29 (2008) 839.[89] S. Youngme, J. Phatchimkun, U. Suksangpanya, C. Pakawatchai, G.A. van
Albada, M. Quesada, J. Reedijk, Inorg. Chem. Commun. 9 (2006) 242.[90] P.K. Dhara, S. Pramanik, T.-H. Lu, M.G.B. Drew, P. Chattopadhyay, Polyhedron
23 (2004) 2457.[91] S. Torelli, C. Belle, C. Philouze, J.–L. Pierre, W. Rammal, E. Saint-Aman, Eur. J.
Inorg. Chem. (2003) 2452.[92] B. Adhikari, C.R. Lucas, Inorg. Chem. 33 (1994) 1376.[93] A.W. Addison, T.N. Rao, J. Reedijk, J. van Rijn, G.C. Verschoor, J. Chem. Soc.,
Dalton Trans. (1984) 1349.




















