
Complete genome sequence and phylogenetic analysis of hepatitis B virus isolated from turkish...
16
Virus Research 152 (2010) 137–152 Contents lists available at ScienceDirect Virus Research journal homepage: www.elsevier.com/locate/virusres Complete genome sequence and phylogenetic analysis of a recombinant Korean norovirus, CBNU1, recovered from a 2006 outbreak Sang-Im Yun a , Jin-Kyoung Kim a , Byung-Hak Song a , Ah-Yong Jeong b , Young-Mee Jee b , Chan-Hee Lee c , Soon-Young Paik d , Yongbum Koo e , Iksoo Jeon f , Sung-June Byun g , Young-Min Lee a,∗ a Department of Microbiology, College of Medicine and Medical Research Institute, Chungbuk National University, 12 Gaeshin-Dong, Heungduk-Ku, Cheongju, Chungbuk 361-763, South Korea b Division of Viral Hepatitis and Poliovirus, Center for Infectious Diseases, National Institute of Health, Seoul, South Korea c School of Life Sciences, Chungbuk National University, Cheongju, South Korea d Department of Microbiology, College of Medicine, The Catholic University of Korea, Seoul, South Korea e School of Biotechnology and Biomedical Science, Inje University, Gimhae, South Korea f Research Planning Team, National Institute of Animal Science, Suwon, South Korea g Animal Biotechnology Division, National Institute of Animal Science, Suwon, South Korea article info Article history: Received 12 March 2010 Received in revised form 17 June 2010 Accepted 18 June 2010 Available online 25 June 2010 Keywords: Norovirus CBNU1 Complete genome sequence Phylogenetic analysis abstract We have determined the complete nucleotide and deduced amino acid sequences of the RNA genome of CBNU1, a human norovirus (NoV) recovered from a 2006 outbreak in South Korea. The genome of 7547 nucleotides, excluding a 3 -poly(A) tail of 11–105 nucleotides, encodes three overlapping open read- ing frames (ORFs): ORF1 (nucleotides 5–5104), ORF2 (nucleotides 5085–6731), and ORF3 (nucleotides 6731–7495). In a comparison to 108 other currently available completely sequenced NoVs represent- ing all five genogroups (GI–GV) except GIV, the CBNU1 strain was highly similar to GII.3 NoVs. Multiple sequence alignments of the completely sequenced NoV genomes revealed five hypervariable regions throughout their genomes: two in ORF1, one in ORF2, and two in ORF3. At both the nucleotide and amino acid levels, genome-based phylogenetic analyses invariably showed that the CBNU1 strain was most closely related to three GII.3 NoVs: the American Texas/TCH04-577 and the two Japanese Saitama U18 and Saitama U201 strains; furthermore, these genome-based phylogenetic topologies corresponded most closely to those based on the ORF2 genes, as compared to those based on the ORF1 and ORF3 genes. Subsequent ORF2-based phylogenetic analyses of a selection of 126 other NoVs representing all 19 GII genotypes, in combination with genome-based Simplot analyses, showed that the CBNU1 strain was a recombinant GII.3 NoV with a breakpoint at the ORF1/ORF2 junction between two putative parent-like strains, Guangzhou/NVgz01 and Texas/TCH04-577. Overall, the CBNU1 strain represents the first Korean human NoV whose genome has been completely sequenced and for which its relationship with a large panel of genetically diverse NoVs has been extensively characterized. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Noroviruses (NoVs), formerly described as “Norwalk or Norwalk-like viruses” or “small round-structured viruses,” are currently classified as the genus Norovirus in the family Caliciviri- dae, which includes three other genera, Sapovirus, Lagovirus, and Vesivirus (Green et al., 2000; Koopmans et al., 2005; Green, 2007). Since the discovery of human NoV in 1972 from a filtrate of stool specimens derived from an 1968 outbreak in Norwalk, Ohio (Dolin et al., 1972; Kapikian et al., 1972), it has been well established as ∗ Corresponding author. Tel.: +82 43 261 2863; fax: +82 43 272 1603. E-mail address: [email protected] (Y.-M. Lee). the major etiologic agent of acute nonbacterial gastroenteritis in humans of all age groups, being responsible for a number of epi- demic and sporadic cases worldwide (Inouye et al., 2000; de Wit et al., 2001; Lopman et al., 2003; Kageyama et al., 2004; Blanton et al., 2006; Castilho et al., 2006; Medici et al., 2006; Papaventsis et al., 2007; Iturriza Gómara et al., 2008). NoV is primarily transmit- ted via a fecal-to-oral route involving ingestion of contaminated foods or water or via person-to-person contact in various settings, such as restaurants, hotels, nursing homes, schools, hospitals, and cruise ships (Glass et al., 2000b; Fankhauser et al., 2002; Green et al., 2002; Rockx et al., 2002; Widdowson et al., 2004). In addition to its multiple modes of transmission, NoV is highly contagious, with a prolonged period of environmental persistence, and it fails to induce long-lasting immunity, primarily as a result of the high level 0168-1702/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.virusres.2010.06.018
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Complete genome sequence and phylogenetic analysis of hepatitis B virus isolated from turkish...

Cn
SSa
Sb
c
d
e
f
g
a
ARRAA
KNCCP
1
NcdVSse
0d
Virus Research 152 (2010) 137–152
Contents lists available at ScienceDirect
Virus Research
journa l homepage: www.e lsev ier .com/ locate /v i rusres
omplete genome sequence and phylogenetic analysis of a recombinant Koreanorovirus, CBNU1, recovered from a 2006 outbreak
ang-Im Yuna, Jin-Kyoung Kima, Byung-Hak Songa, Ah-Yong Jeongb, Young-Mee Jeeb, Chan-Hee Leec,oon-Young Paikd, Yongbum Kooe, Iksoo Jeonf, Sung-June Byung, Young-Min Leea,∗
Department of Microbiology, College of Medicine and Medical Research Institute, Chungbuk National University, 12 Gaeshin-Dong, Heungduk-Ku, Cheongju, Chungbuk 361-763,outh KoreaDivision of Viral Hepatitis and Poliovirus, Center for Infectious Diseases, National Institute of Health, Seoul, South KoreaSchool of Life Sciences, Chungbuk National University, Cheongju, South KoreaDepartment of Microbiology, College of Medicine, The Catholic University of Korea, Seoul, South KoreaSchool of Biotechnology and Biomedical Science, Inje University, Gimhae, South KoreaResearch Planning Team, National Institute of Animal Science, Suwon, South KoreaAnimal Biotechnology Division, National Institute of Animal Science, Suwon, South Korea
r t i c l e i n f o
rticle history:eceived 12 March 2010eceived in revised form 17 June 2010ccepted 18 June 2010vailable online 25 June 2010
eywords:orovirusBNU1omplete genome sequencehylogenetic analysis
a b s t r a c t
We have determined the complete nucleotide and deduced amino acid sequences of the RNA genome ofCBNU1, a human norovirus (NoV) recovered from a 2006 outbreak in South Korea. The genome of 7547nucleotides, excluding a 3′-poly(A) tail of 11–105 nucleotides, encodes three overlapping open read-ing frames (ORFs): ORF1 (nucleotides 5–5104), ORF2 (nucleotides 5085–6731), and ORF3 (nucleotides6731–7495). In a comparison to 108 other currently available completely sequenced NoVs represent-ing all five genogroups (GI–GV) except GIV, the CBNU1 strain was highly similar to GII.3 NoVs. Multiplesequence alignments of the completely sequenced NoV genomes revealed five hypervariable regionsthroughout their genomes: two in ORF1, one in ORF2, and two in ORF3. At both the nucleotide andamino acid levels, genome-based phylogenetic analyses invariably showed that the CBNU1 strain wasmost closely related to three GII.3 NoVs: the American Texas/TCH04-577 and the two Japanese SaitamaU18 and Saitama U201 strains; furthermore, these genome-based phylogenetic topologies corresponded
most closely to those based on the ORF2 genes, as compared to those based on the ORF1 and ORF3 genes.Subsequent ORF2-based phylogenetic analyses of a selection of 126 other NoVs representing all 19 GIIgenotypes, in combination with genome-based Simplot analyses, showed that the CBNU1 strain was arecombinant GII.3 NoV with a breakpoint at the ORF1/ORF2 junction between two putative parent-likestrains, Guangzhou/NVgz01 and Texas/TCH04-577. Overall, the CBNU1 strain represents the first Koreanhuman NoV whose genome has been completely sequenced and for which its relationship with a largepanel of genetically diverse NoVs has been extensively characterized.. Introduction
Noroviruses (NoVs), formerly described as “Norwalk ororwalk-like viruses” or “small round-structured viruses,” areurrently classified as the genus Norovirus in the family Caliciviri-ae, which includes three other genera, Sapovirus, Lagovirus, and
esivirus (Green et al., 2000; Koopmans et al., 2005; Green, 2007).ince the discovery of human NoV in 1972 from a filtrate of stoolpecimens derived from an 1968 outbreak in Norwalk, Ohio (Dolint al., 1972; Kapikian et al., 1972), it has been well established as∗ Corresponding author. Tel.: +82 43 261 2863; fax: +82 43 272 1603.E-mail address: [email protected] (Y.-M. Lee).
168-1702/$ – see front matter © 2010 Elsevier B.V. All rights reserved.oi:10.1016/j.virusres.2010.06.018
© 2010 Elsevier B.V. All rights reserved.
the major etiologic agent of acute nonbacterial gastroenteritis inhumans of all age groups, being responsible for a number of epi-demic and sporadic cases worldwide (Inouye et al., 2000; de Witet al., 2001; Lopman et al., 2003; Kageyama et al., 2004; Blanton etal., 2006; Castilho et al., 2006; Medici et al., 2006; Papaventsis etal., 2007; Iturriza Gómara et al., 2008). NoV is primarily transmit-ted via a fecal-to-oral route involving ingestion of contaminatedfoods or water or via person-to-person contact in various settings,such as restaurants, hotels, nursing homes, schools, hospitals, and
cruise ships (Glass et al., 2000b; Fankhauser et al., 2002; Green etal., 2002; Rockx et al., 2002; Widdowson et al., 2004). In additionto its multiple modes of transmission, NoV is highly contagious,with a prolonged period of environmental persistence, and it fails toinduce long-lasting immunity, primarily as a result of the high level
1 esearc
oco
cogu1tiN2tdcsCPgaeeis
ii2hse2ttota2aKct3fiaigaGTla
ciTfbtswcpav
38 S.-I. Yun et al. / Virus R
f genetic and antigenic diversity of co-circulating NoVs. All theseharacteristics make it difficult to prevent the virus from spreadingnce introduced (Kuusi et al., 2002).
NoV is a non-enveloped virus of ∼27–40 nm in diameter thatontains a linear, single-stranded, positive-sense RNA genomef ∼7.5 kb, with a poly(A) tail at the 3′-end (Green, 2007). Theenome includes three open reading frames (ORFs) flanked by shortntranslated regions (UTRs) at the 5′ and 3′ ends (Jiang et al., 1990,993; Lambden et al., 1993). ORF1 encodes a nonstructural polypro-ein precursor that is cleaved by the viral 3C-like cysteine proteasento at least six mature proteins, NS1-2N-term, NS3NTPase, NS4p20,S5VPg, NS6Pro, and NS7Pol (Liu et al., 1996, 1999; Belliot et al.,003; Blakeney et al., 2003). ORF2 encodes the major capsid pro-ein, designated viral protein 1 (VP1), which folds into two majoromains: a shell (S) domain that is essential for initiation of the viralapsid assembly and a protruding (P) domain that is important fortabilization of the capsid structure (Prasad et al., 1999; Bertolotti-iarlet et al., 2002). The P domain is further divided into the P1 and2 subdomains: P2, which has the highest degree of sequence diver-ence in the genome, is involved in the binding of the virus to cellsnd antibodies (Hardy et al., 1996; Nilsson et al., 2003; Chakravartyt al., 2005; Lochridge et al., 2005; Allen et al., 2008, 2009; Xerryt al., 2008). ORF3 encodes the minor capsid protein VP2, whichs a small basic protein capable of increasing the expression andtability of VP1 (Glass et al., 2000a; Bertolotti-Ciarlet et al., 2003).
Over the past three decades, a large number of NoVs have beensolated from humans and animals. Most of these NoVs are genet-cally and antigenically diverse (Kapikian, 2000; Koopmans et al.,005; Green, 2007; Scipioni et al., 2008). NoV serotyping, however,as been hampered largely by the lack of an efficient cell cultureystem and a suitable animal infection model (Duizer et al., 2004),xcept in the case of murine NoV (Karst et al., 2003; Wobus et al.,004, 2006; Ward et al., 2007). Thus, NoV genotyping has becomehe principal approach for classifying these diverse viruses. Most ofhe previous genetic analyses have been based on partial nucleotider amino acid sequences of the genome, mainly derived from a rela-ively small region of either the ORF1 or ORF2 gene, differentiatingnumber of NoVs into their distinct lineages (Green et al., 1995,
000; Ando et al., 2000; Vinjé and Koopmans, 2000; Fankhauser etl., 2002; Katayama et al., 2002; Karst et al., 2003; Oliver et al., 2003;ageyama et al., 2004; Vinjé et al., 2004; Wang et al., 2005). A recentomprehensive study using the complete amino acid sequences ofhe ORF2 variants of 164 NoV strains, implemented with a predictedD structure-based alignment, has suggested an improved classi-cation strategy, providing a better defined genetic segregationmong the genetically diverse NoVs (Zheng et al., 2006). Accord-ng to these previous reports, NoVs are currently classified into fiveenetic groups or genogroups (GI–GV). Three genogroups (GI, GII,nd GIV) contain human NoVs, with GII also including porcine NoVs.III and GV represent bovine NoVs and murine NoVs, respectively.hese five genogroups are further subdivided into at least 31 phy-ogenetic clusters or genotypes: 8 in GI, 19 in GII, 2 in GIII, 1 in GIV,nd 1 in GV (Green, 2007).
In recent years, human NoV infection has shown to be the mostommon cause of acute viral gastroenteritis in South Korea, typ-cally peaking in the winter (Kim et al., 2005; Yoon et al., 2008).o date, a large collection of Korean strains has been recoveredrom the stool specimens of infected individuals across the nation,ut in all cases, their genomes were only partially sequenced. Inhe present study, we have determined the full-length nucleotideequence of the genomic RNA of the Korean NoV CBNU1 strain that
as responsible for a 2006 outbreak in Seoul. By comparing theomplete genome of the CBNU1 strain with those of 108 other com-letely sequenced NoVs of humans and animals that are currentlyvailable from GenBank, we have fully characterized the geneticariability of this virus at the nucleotide and predicted amino acid
h 152 (2010) 137–152
levels. Also, we have elucidated the genetic relationship of the GIICBNU1 strain to other completely sequenced, genetically diverseNoVs, not only within the same GII genogroup but also to othergenogroups. Given our observation that the genome-based phy-logeny corresponds relatively well to ORF2-based phylogeny, wehave also performed extensive ORF2-based phylogenetic analysesof a selection of 126 NoVs representing all 19 genotypes of theGII genogroup. Our results provide a comprehensive genetic rela-tionship of CBNU1, a recombinant Korean strain of human NoV,with a large group of other NoVs isolated from various geographiclocations at different times.
2. Materials and methods
2.1. Viral RNA extraction from fecal specimens
The NoV-positive stool specimen CBNU1 was collected from a∼2-year-old female child suffering from typical clinical symptomsof the viral infection during a 2006 outbreak of acute gastroen-teritis in Seoul, South Korea. A 10% (wt./vol.) stool suspension wasprepared in phosphate-buffered saline and precleared by centrifu-gation at 14,000 × g for 20 min at 4 ◦C. Aliquots (1 ml each) of thesupernatant were kept at −80 ◦C. Viral RNA was extracted from200 �l of the precleared stool suspension using TRIzol reagent(Invitrogen, Carlsbad, CA), according to the manufacturer’s instruc-tions. For consistent RNA recovery, 5 �g of glycogen (BoehringerMannheim, Indianapolis, IN) was added as a carrier to eachextraction solution before isopropanol precipitation. The RNA wasresuspended in 20 �l of RNase-free dH2O and used immediately forfurther analysis.
2.2. Synthesis and sequencing of three overlapping cDNAsrepresenting the NoV genomic RNA
Three overlapping cDNAs encompassing the full-length NoVgenomic RNA were synthesized by two-step reverse transcription(RT)-PCR from the extracted viral RNA: Fragment A, corre-sponding to nucleotides 1–3023; Fragment B, correspondingto nucleotides 1000–5104; and Fragment C, corresponding tonucleotides 3000–7547 plus the 3′-poly(A) tail. The oligonu-cleotides chosen for the RT-PCR reactions were from regions thatwere highly conserved among 35 completely sequenced GII NoVgenomes retrieved from GenBank at the initial stage of this study(Table 1). For cDNA synthesis, RT reactions were performed with10 �l of the extracted RNA in a standard 20-�l reaction mix-ture containing 10 pmol of an appropriate primer, 10 mM dNTPmix, 200 U SuperScript III reverse transcriptase (Invitrogen), 40 URNaseOUT (Invitrogen), 5 mM dithiothreitol (DTT), and the buffersupplied by the manufacturer. The three primers used in the RTreaction were FragAR for Fragment A, FragBR for Fragment B, andFragCR for Fragment C. The RT reactions involved incubation for1 h at 50 ◦C, followed by an inactivation step for 15 min at 70 ◦C. ForcDNA amplification, 10 �l of the RT reaction mixture was directlyused for PCR in a standard 100-�l reaction mixture with Pfu-derivedPyrobest DNA polymerase (Takara, Shiga, Japan), as recommendedby the manufacturer, in the presence of the following primers: Fra-gAF and FragAR for Fragment A, FragBF and FragBR for Fragment B,and FragCF and FragCR for Fragment C. The PCR program consistedof 35 cycles of 95 ◦C for 30 s, 60 ◦C for 30 s, and 72 ◦C for 4–5 min,followed by a final elongation step of 72 ◦C for 10 min. The PCRproducts of the proper sizes were gel-purified and directly used
for sequencing analysis in both directions. Two amplified cDNAs(Fragments A and B) were sequenced with internal sequencingprimers, designed from the consensus sequence of the 35 com-pletely sequenced GII NoV genomes (data not shown). Becauseof its higher degree of sequence divergence, the remaining Frag-
S.-I. Yun et al. / Virus Research 152 (2010) 137–152 139
Table 1Oligonucleotides used for cDNA synthesis and amplification of the NoV CBNU1 genomic RNA.
Oligonucleotide Sequence (5′ to 3′)a Positionb Polarity
FragAF atattaattaaGTGAATGAAGATGGCGTCTAA 1–21 SenseFragAR atacggtccgAACTGAGTCTCTCATTGTAGT 3003–3023 AntisenseFragBF atattaattaaTGAGGACCTTGCAGTGGAACTCGT 1000–1023 SenseFragBR atacggtccgTCACTCGACGCCATCTTCATT 5084–5104 AntisenseFragCF atattaattaaTAGACTACAATGAGAGACTCA 3000–3020 SenseFragCR atacggtccgTTTTTTTTTTTTTTTTTTTT poly(A) tail Antisense5raceRT GCAGCTGGTGGTTTGTGCACACCAAGTA 450–477 Antisense5racePCR/R1 CAAGACCTCGCTCCACATACAGACCG 424–449 Antisense5racePCR/R2 CATTTCCAGGATTGTTCCGGTTAGCG 375–400 Antisense3racePCR/F aataagcttACACTCAACCCAAATCACTAA 7180–7200 SensePRX ccagtgttgtggcctgcagggcgaatt Sense
ted instrain
mpop
2
5dIreSpctcitatToctT2mrab5p1psf(tiwif
2
3
PRXR gatgaattcgccctgcaggccacaaca
a NoV-specific sequences are shown in uppercase; nonviral sequences are indicab Nucleotide position refers to the complete genome sequence of the NoV CBNU1
ent C was sequenced by primer walking with the CBNU1-specificrimers, each of which was designed from the preceding roundsf sequencing. Sequence information concerning all the internalrimers used in our sequencing reactions is available upon request.
.3. Determination of the 5′-end of the NoV genomic RNA
The 5′-end of the NoV genomic RNA was determined by the′ rapid amplification of cDNA ends (5′RACE) protocol that weescribed previously (Kang et al., 2004), with slight modifications.
nitially, the first-strand cDNA corresponding to the 5′-terminalegion of the genome was reverse-transcribed from 10 �l of thextracted viral RNA in a standard 20-�l reaction mixture withuperScript III reverse transcriptase (Invitrogen) and a reverserimer 5raceRT complementary to nucleotides 450–477, under theonditions described above. The resulting RNA-cDNA hybrid wasreated with RNase H (Takara), and the remaining single-strandedDNA was then isolated with TRIzol LS reagent (Invitrogen) accord-ng to the corresponding manufacturer’s instructions. In ordero incorporate a specific primer-binding site for subsequent PCRmplification, the 3′-end of the single-stranded cDNA was ligatedo an oligonucleotide PRX that had been 5′-phosphorylated with4 polynucleotide kinase (Takara) and 3′-blocked by the additionf ddATP with terminal deoxynucleotidyl transferase (Takara). TheDNA-PRX ligation was performed for 12 h at 15 ◦C in a 40-�l reac-ion mixture containing 10 �l of the single-stranded cDNA, 40 U4 RNA ligase (New England Biolabs, Beverly, MA), 10 pmol PRX,0% PEG #6000, and the buffer supplied by the manufacturer. Fiveicroliters of the ligation reaction was directly used for the first
ound of PCR with a pair of primers, PRXR complementary to PRXnd 5racePCR/R1 complementary to nucleotides 424–449, followedy the second round of PCR with another pair of primers, PRXR andracePCR/R2 complementary to nucleotides 375–400. PCRs wereerformed with Pyrobest DNA polymerase (Takara) in a standard00-�l reaction mixture as described above, with the followingrogram: 45 cycles (for the first-round PCR) and 30 cycles (for theecond-round PCR) of 30 s at 95 ◦C, 30 s at 60 ◦C, and 1 min at 72 ◦C,ollowed by a final extension of 10 min at 72 ◦C. The PCR amplicons∼430 bp) were gel-purified, digested with PstI (incorporated intohe PRXR primer) and SpeI (preexisting at position nucleotide 238n the viral genome), and inserted into the pRS2 cloning vector,
hich had previously been digested with PstI and XbaI. Multiplendependent clones containing the insert were randomly selectedor sequencing.
.4. Determination of the 3′-end of the NoV genomic RNA
The 3′-end of the NoV genomic RNA was characterized by′RACE, which we have successfully applied to two other positive-
Antisense
lowercase.(GenBank accession no. GU980585).
sense RNA viruses, Japanese encephalitis virus (Yun et al., 2003)and porcine reproductive and respiratory syndrome virus (Kang etal., 2004). In brief, the 3′-end of the extracted viral RNA was lig-ated to a 5′-phosphorylated and 3′-blocked oligonucleotide PRX, inorder to create a specific primer-binding site for the cDNA syn-thesis and amplification. The RNA-PRX ligation was performedwith T4 RNA ligase (New England Biolabs) in a 20-�l reactionmixture including 10 �l of the extracted viral RNA, as recom-mended by the manufacturer. The PRX-ligated RNA was recoveredwith TRIzol LS reagent (Invitrogen) and then used for RT withSuperScript III reverse transcriptase (Invitrogen) in a 20-�l reac-tion mixture containing a reverse primer PRXR, as describedabove. Subsequently, one-half of the RT reaction mixture wasPCR-amplified with Pyrobest DNA polymerase (Takara) using theprimers 3racePCR/F, corresponding to nucleotides 7180–7200, andPRXR, for 45 cycles of 30 s at 95 ◦C, 30 s at 60 ◦C, and 1 min at 72 ◦C,followed by a final extension of 10 min at 72 ◦C. The PCR products(∼400–500 bp) were gel-extracted and cloned into the pGEM-3Zf(−) vector (Promega, Madison, WI) after digestion with HindIII(incorporated into the 3racePCR/F primer) and EcoRI (incorporatedinto the PRXR primer). Multiple randomly picked, independentclones were sequenced.
2.5. Multiple sequence alignments and phylogenetic analyses
A series of full-length genome-based sequence comparisons,multiple sequence alignments, and phylogenetic analyses was per-formed on a total of 109 genetically diverse NoVs, including theCBNU1 strain; a full description of their hosts, geographic ori-gins, collection dates, and GenBank accession numbers is givenin Table 2. For genotyping of the CBNU1 strain, ORF2-basedphylogenetic analyses were performed on a panel of 126 NoVs,including all 19 GII genotypes as classified in two previous reports(Wang et al., 2005; Zheng et al., 2006). Multiple sequence align-ments of the nucleotide and deduced amino acid sequenceswere obtained with Clustal X (Thompson et al., 1997). The esti-mated evolutionary distances were used for the construction ofphylogenetic trees by the neighbor-joining method (Saitou andNei, 1987). The confidence values of the internal nodes in thephylogenies were determined by performing bootstrap samplinganalysis (Felsenstein, 1985) with 1000 replicates of the constructedneighbor-joining trees. Rooted trees were generated using thegenome sequence of the human sapovirus Mc2 strain (Hansman
et al., 2004) as an outgroup. All trees were plotted using TreeViewsoftware, version 1.6.1 (Page, 1996), with minor manual adjust-ments for presentation in limited space. Recombination analyseswere performed using Simplot software, version 3.5.1 (Lole et al.,1999).
140 S.-I. Yun et al. / Virus Research 152 (2010) 137–152
Table 2Description of the NoVs used for full-length genome-based genetic analyses.
Hosta Geographicorigin
Collectiondateb
Virus GenBankaccession no.
Hosta Geographicorigin
Collectiondateb
Virus GenBankaccession no.
Hu USA 1968 Norwalk NC 001959 Hu Japan 2006 Sakai4 AB447450Hu USA 1976 Snow Mountain AY134748 Hu Japan 2006 Toyama1 AB447443Hu USA 1987 MD145-12 AY032605 Hu Japan 2006 Toyama4 AB447444Hu USA 2002 Houston/TCH186 EU310927 Hu Japan 2006 Toyama5 AB447445Hu USA 2002 Farmington Hills AY502023 Hu Japan IU YURI AB083780Hu USA 2004 Texas/TCH04-577 AB365435 Hu Japan IU YURI 32073 AB083781Hu USA 2004 MD-2004 DQ658413 Hu Japan IU GIFU’99 AB084071Hu Japan 1979 Otofuke AB187514 Hu UK 1991 Southampton L07418Hu Japan 1987 Chiba 407 AB042808 Hu UK 1993 Lordsdale X86557Hu Japan 1996 Gifu’96 AB045603 Hu UK 2002 Oxford/B2S16 AY587989Hu Japan 1997 Saitama U3 AB039776 Hu UK 2002 Oxford/B4S1 AY587988Hu Japan 1997 Saitama U4 AB039777 Hu UK 2002 Oxford/B4S2 AY587983Hu Japan 1997 Saitama U16 AB039778 Hu UK 2002 Oxford/B4S4 AY587986Hu Japan 1997 Saitama U17 AB039779 Hu UK 2002 Oxford/B4S5 AY587984Hu Japan 1997–1999 SzUG1 AB039774 Hu UK 2002 Oxford/B4S6 AY587985Hu Japan 1997–1999 Saitama U1 AB039775 Hu UK 2002 Oxford/B4S7 AY587987Hu Japan 1997–1999 Saitama U18 AB039781 Hu UK 2003 Oxford/B5S22 AY581254Hu Japan 1997–1999 Saitama U25 AB039780 Hu Germany 1997 HSS/3/97/DEU [BS5] AF093797Hu Japan 1998 Saitama U201 AB039782 Hu Germany 1997 Dresden174/pUS-NorII AY741811Hu Japan 2000 WUG1 AB081723 Hu Germany 2000 Neustrelitz260 AY772730Hu Japan 2004 MK04 DQ456824 Hu Germany 2002 Langen1061 AY485642Hu Japan 2005 Chiba/04-1050 AB220921 Hu Korea 2006 CBNU1 GU980585Hu Japan 2006 Aichi3 AB447446 Hu Vietnam 1999–2000 Vietnam 026 AF504671Hu Japan 2006 Aichi4 AB447447 Hu Thailand 2000–2001 Mc37 AY237415Hu Japan 2006 Akita1 AB447436 Hu China IU Guangzhou/NVgz01 DQ369797Hu Japan 2006 Akita2 AB447437 Sw Japan 1997 Swine43 AB126320Hu Japan 2006 Akita4 AB447438 Bo UK 1976 Newbury2 AF097917Hu Japan 2006 Akita5 AB447439 Bo UK 1994 Dumfries AY126474Hu Japan 2006 Aomori1 AB447432 Mu USA 2002 MNV-1 AY228235Hu Japan 2006 Aomori2 AB447433 Mu USA 2005 WU11 EU004663Hu Japan 2006 Aomori4 AB447434 Mu USA 2005 WU12 EU004664Hu Japan 2006 Aomori5 AB447435 Mu USA 2005 WU20 EU004665Hu Japan 2006 Ehime1 AB447453 Mu USA 2005 WU21 EU004666Hu Japan 2006 Ehime2 AB447454 Mu USA 2005 WU22 EU004667Hu Japan 2006 Ehime5 AB447455 Mu USA 2005 WU23 EU004668Hu Japan 2006 Hiroshima1 AB447451 Mu USA 2005 WU24 EU004669Hu Japan 2006 Hiroshima2 AB447452 Mu USA 2005 WU25 EU004670Hu Japan 2006 Hokkaido1 AB447427 Mu USA 2005 WU26 EU004671Hu Japan 2006 Hokkaido2 AB447428 Mu USA 2005 CR1 EU004672Hu Japan 2006 Hokkaido3 AB447429 Mu USA 2005 CR3 EU004673Hu Japan 2006 Hokkaido4 AB447430 Mu USA 2005 CR4 EU004674Hu Japan 2006 Hokkaido5 AB447431 Mu USA 2005 CR5 EU004675Hu Japan 2006 Kumamoto1 AB447459 Mu USA 2005 CR6 EU004676Hu Japan 2006 Kumamoto2 AB447460 Mu USA 2005 CR7 EU004677Hu Japan 2006 Kumamoto3 AB447461 Mu USA 2005 CR10 EU004678Hu Japan 2006 Kumamoto4 AB447462 Mu USA 2005 CR11 EU004679Hu Japan 2006 Kumamoto5 AB447463 Mu USA 2005 CR13 EU004680Hu Japan 2006 Miyagi2 AB447440 Mu USA 2005 CR15 EU004681Hu Japan 2006 Miyagi4 AB447441 Mu USA 2005 CR17 EU004682Hu Japan 2006 Miyagi5 AB447442 Mu Germany 2005 CR18 EU004683Hu Japan 2006 Saga1 AB447456 Mu Germany 2006 Berlin/04/06/DE [M21] DQ911368Hu Japan 2006 Saga4 AB447457 Mu Germany 2006 Berlin/05/06/DE [S36] EF531290Hu Japan 2006 Saga5 AB447458 Mu Germany 2006 Berlin/06/06/DE [S99] EF531291Hu Japan 2006 Sakai2 AB447448 Mu Germany 2007 Hannover1 EU854589
Hu
3
3d
wRsawsa
Hu Japan 2006 Sakai3 AB447449
a Host abbreviations: Hu, human; Sw, swine; Bo, bovine; Mu, murine.b IU, information unavailable.
. Results
.1. Molecular characterization of the full-length nucleotide andeduced amino acid sequences of the NoV CBNU1 strain
To fully characterize the NoV CBNU1 strain at the genetic level,e determined the complete nucleotide sequence of its genomicNA. The full-length genome sequence was assembled by a two-
tep strategy: (i) direct sequencing of three overlapping long cDNAmplicons encompassing the full-length genomic RNA, in such aay that all the regions were sequenced at least twice for bothtrands to eliminate sequencing artifacts, and (ii) resolving the 5′
nd 3′ ends of the genomic RNA by 5′ and 3′ RACE, respectively, both
Thailand 2000–2001 Sapovirus Mc2 AY237419
of which were followed by the cloning of their respective cDNAamplicons into a bacterial plasmid and sequencing of a large num-ber of independent clones to avoid any selection bias that mightoccur during cDNA cloning.
The complete genome sequence of CBNU1 that we assembledfrom this study was deposited into GenBank database under theaccession number GU980585. The genomic RNA of CBNU1 wascomposed of 7547 nucleotides, excluding a poly(A) tail at the 3′-end
(Fig. 1A). We found that the genome organization was the same asthat of other NoVs (Jiang et al., 1990; Lambden et al., 1993). Specif-ically, the CBNU1 genome contained an internal protein-codingregion of 7491 nucleotides that was organized into three ORFs,with ORF2 overlapping ORF1 by 20 nucleotides and ORF3 by one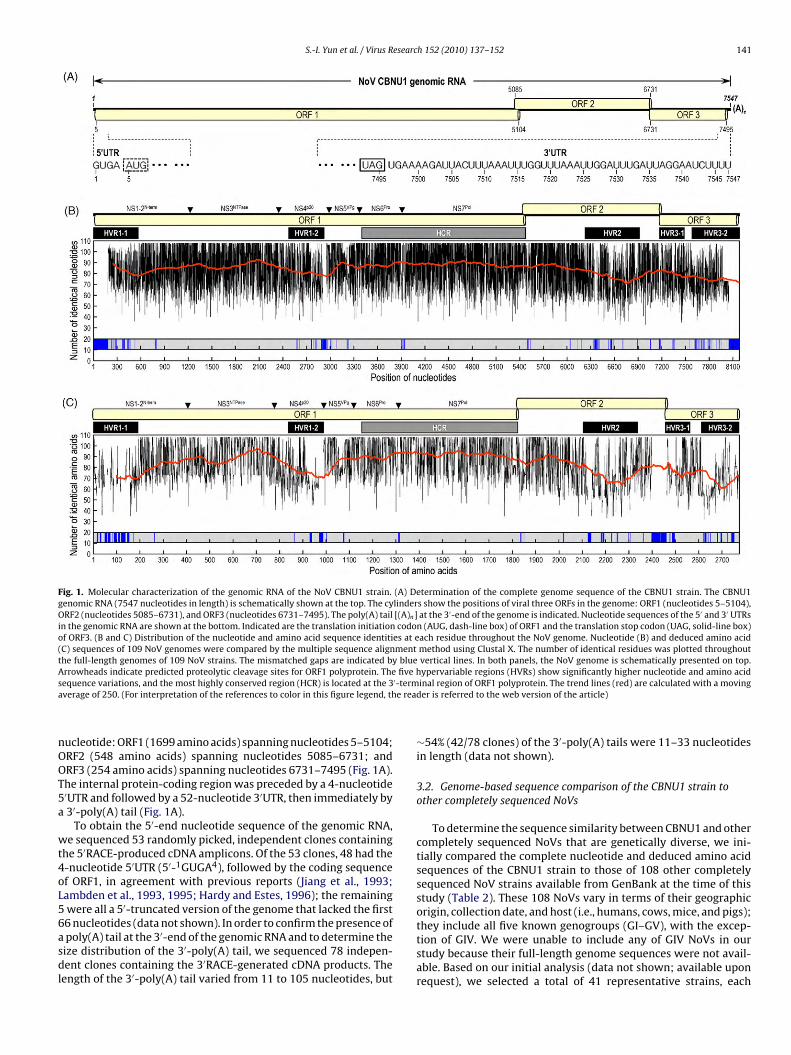
S.-I. Yun et al. / Virus Research 152 (2010) 137–152 141
Fig. 1. Molecular characterization of the genomic RNA of the NoV CBNU1 strain. (A) Determination of the complete genome sequence of the CBNU1 strain. The CBNU1genomic RNA (7547 nucleotides in length) is schematically shown at the top. The cylinders show the positions of viral three ORFs in the genome: ORF1 (nucleotides 5–5104),ORF2 (nucleotides 5085–6731), and ORF3 (nucleotides 6731–7495). The poly(A) tail [(A)n] at the 3′-end of the genome is indicated. Nucleotide sequences of the 5′ and 3′ UTRsin the genomic RNA are shown at the bottom. Indicated are the translation initiation codon (AUG, dash-line box) of ORF1 and the translation stop codon (UAG, solid-line box)of ORF3. (B and C) Distribution of the nucleotide and amino acid sequence identities at each residue throughout the NoV genome. Nucleotide (B) and deduced amino acid(C) sequences of 109 NoV genomes were compared by the multiple sequence alignment method using Clustal X. The number of identical residues was plotted throughoutthe full-length genomes of 109 NoV strains. The mismatched gaps are indicated by blue vertical lines. In both panels, the NoV genome is schematically presented on top.Arrowheads indicate predicted proteolytic cleavage sites for ORF1 polyprotein. The five hypervariable regions (HVRs) show significantly higher nucleotide and amino acids ′-terma e read
nOOT5a
wt4oL56asdl
equence variations, and the most highly conserved region (HCR) is located at the 3verage of 250. (For interpretation of the references to color in this figure legend, th
ucleotide: ORF1 (1699 amino acids) spanning nucleotides 5–5104;RF2 (548 amino acids) spanning nucleotides 5085–6731; andRF3 (254 amino acids) spanning nucleotides 6731–7495 (Fig. 1A).he internal protein-coding region was preceded by a 4-nucleotide′UTR and followed by a 52-nucleotide 3′UTR, then immediately by3′-poly(A) tail (Fig. 1A).
To obtain the 5′-end nucleotide sequence of the genomic RNA,e sequenced 53 randomly picked, independent clones containing
he 5′RACE-produced cDNA amplicons. Of the 53 clones, 48 had the-nucleotide 5′UTR (5′-1GUGA4), followed by the coding sequencef ORF1, in agreement with previous reports (Jiang et al., 1993;ambden et al., 1993, 1995; Hardy and Estes, 1996); the remainingwere all a 5′-truncated version of the genome that lacked the first
6 nucleotides (data not shown). In order to confirm the presence ofpoly(A) tail at the 3′-end of the genomic RNA and to determine theize distribution of the 3′-poly(A) tail, we sequenced 78 indepen-ent clones containing the 3′RACE-generated cDNA products. Theength of the 3′-poly(A) tail varied from 11 to 105 nucleotides, but
inal region of ORF1 polyprotein. The trend lines (red) are calculated with a movinger is referred to the web version of the article)
∼54% (42/78 clones) of the 3′-poly(A) tails were 11–33 nucleotidesin length (data not shown).
3.2. Genome-based sequence comparison of the CBNU1 strain toother completely sequenced NoVs
To determine the sequence similarity between CBNU1 and othercompletely sequenced NoVs that are genetically diverse, we ini-tially compared the complete nucleotide and deduced amino acidsequences of the CBNU1 strain to those of 108 other completelysequenced NoV strains available from GenBank at the time of thisstudy (Table 2). These 108 NoVs vary in terms of their geographicorigin, collection date, and host (i.e., humans, cows, mice, and pigs);
they include all five known genogroups (GI–GV), with the excep-tion of GIV. We were unable to include any of GIV NoVs in ourstudy because their full-length genome sequences were not avail-able. Based on our initial analysis (data not shown; available uponrequest), we selected a total of 41 representative strains, each
142 S.-I. Yun et al. / Virus Researc
Table 3Comparison of the complete genome sequence of NoV CBNU1 with those of 41completely sequenced NoVs.
Genogroup Strain Sequence identity (%)
Nucleotide Amino acida
GI
Chiba 407 53.2 51.1Otofuke 53.3 50.9Southampton 53.7 50.6HSS/3/97/DEU 53.8 50.7WUG1 53.9 51.0Norwalk 54.0 51.5SzUG1 54.1 51.8
GII
Swine43 65.8 70.1Saitama U25 66.9 71.6Saitama U4 67.1 72.9Neustrelitz260 72.9 82.1MK04 74.4 82.9Snow Mountain 77.4 85.7Langen1061 80.7 85.4Saitama U18 80.7 89.8Houston/TCH186 80.8 86.1Oxford/B5S22 80.9 85.8Oxford/B2S16 80.9 85.8Saitama U201 80.9 90.1MD-2004 80.9 85.8Saga5 81.0 85.9Aomori1 81.1 86.0Hokkaido5 81.2 86.2Sakai3 81.2 86.0Dresden174/pUS-NorII 81.6 85.8YURI 32073 81.7 86.5Lordsdale 82.1 86.5MD145-12 82.4 86.8Mc37 85.1 88.4Vietnam 026 85.2 88.4Gifu’96 86.4 89.9Saitama U1 86.6 89.7Guangzhou/NVgz01 87.1 87.9Texas/TCH04-577 87.2 94.8Chiba/04-1050 87.4 88.4Sakai2 87.5 88.4
GIIIDumfries 52.5 49.3Newbury2 52.9 49.4
GVMNV-1 51.2 45.7Berlin/04/06/DE 51.3 45.4WU12 51.5 45.6
O
oss
rs(AfNutc(ffieha
closely related to each other, but it formed a significant number
a The amino acid sequence identity is calculated for the continuousRF1/ORF2/ORF3 region.
f which was chosen from several candidates isolated from theame geographic location/time and showed a very high degree ofequence similarity at both the nucleotide and amino acid levels.
The final pairwise comparison of CBNU1 with the 41 rep-esentative NoV strains revealed a wide variety of sequenceimilarities at both the nucleotide (51.2–87.5%) and the amino acid45.4–94.8%) levels, primarily depending on genogroup (Table 3).t the nucleotide level (Table 3, nucleotide), the similarity scores
or CBNU1 and 29 GII NoVs (including 28 human NoVs and 1 swineoV) were relatively high, ranging from 65.8% to 87.5%. In partic-lar, the CBNU1 strain was highly similar to four human GII NoVs:he Japanese Sakai2 (87.5%) and Chiba/04-1050 (87.4%), the Ameri-an Texas/TCH04-577 (87.2%), and the Chinese Guangzhou/NVgz0187.1%). It was less similar to three Japanese GII NoVs: two isolatedrom humans (Saitama U4, 67.1% and Saitama U25, 66.9%) and onerom pigs (Swine43, 65.8%). Unlike the GII NoVs, however, signif-
cantly lower similarity scores were estimated with members ofach of the other three genogroups examined in our study: sevenuman GI NoVs (53.2–54.1%), two bovine GIII NoVs (52.5–52.9%),nd three murine GV NoVs (51.2–51.5%).h 152 (2010) 137–152
At the deduced amino acid level (Table 3, amino acid), the CBNU1strain also shared a spectrum of high similarity scores with 29 GIINoVs (70.1–94.8%), as observed at the nucleotide level. Specifically,the CBNU1 strain had high similarity scores with four human GIINoVs: the American Texas/TCH04-577 (94.8%) and three Japanesestrains (Saitama U201, 90.1%; Gifu’96, 89.9%; and Saitama U18,89.8%). However, it had relatively lower similarity scores withthree Japanese GII NoVs, i.e., two human NoVs (Saitama U4, 72.9%and Saitama U25, 71.6%) and one swine NoV (Swine43, 70.1%). Arange of lower similarity scores were obtained with the remain-ing NoVs of all the other three genogroups: seven human GI NoVs(50.6–51.8%), two bovine GIII NoVs (49.3–49.4%), and three murineGV NoVs (45.4–45.7%), in accord with the pattern of the nucleotidesequence similarity scores obtained in this study.
In an effort to identify any subregions of the NoV genome thatmight be highly variable or conserved, we first aligned the com-plete nucleotide and deduced amino acid sequences of all 109completely sequenced NoV genomes, including those of the CBNU1genome, and then plotted the number of nucleotide and amino acidsequences identical to their respective consensus sequences at eachresidue throughout their genomes (Fig. 1B and C). Although a largenumber of nucleotide sequence alterations were evenly scatteredthroughout the genomes, significantly higher nucleotide sequencevariations were noted in five short hypervariable regions (HVRs):(i) HVR1-1, corresponding to the 5′ half of NS1-2N-term in ORF1; (ii)HVR1-2, corresponding to NS4p20 in ORF1; (iii) HVR2, correspond-ing to the central region of ORF2 and previously designated as the P2subdomain within the NoV VP1 protein; (iv) HVR3-1, correspond-ing to the 5′-most third of ORF3; and (v) HVR3-2, corresponding tothe 3′ half of ORF3 (Fig. 1B). In agreement with these findings, mostof the nucleotide-mismatched gaps were also found exclusively inthe five HVRs (Fig. 1B). In contrast, the most highly conserved region(HCR) within the genome was located in the 3′-most third of ORF1,corresponding to the NS6Pro and NS7Pol protein-coding regions(Fig. 1B). As observed at the nucleotide level, the amino acid alter-ations and mismatched gaps were also more frequently presentin the five HVRs than in other regions of the genome (Fig. 1C).Overall, these results suggest that the five HVRs are particularlytolerant of amino acid changes that influence the viral replicationfunction.
3.3. Genome-based phylogenetic analyses of the CBNU1 strainand other completely sequenced NoVs
To better understand the genetic relationships between CBNU1and other completely sequenced NoVs, we performed full-length genome-based phylogenetic analyses using the completenucleotide and deduced amino acid sequences of 109 completelysequenced NoVs, including CBNU1, in conjunction with a sapovirusMc2 as an outgroup. A nucleotide sequence-based phylogenetictree revealed four distinct phylogenetic groups with reliable boot-strap support (Fig. 2A), which matched four genogroups, GI, GII,GIII, and GV (Zheng et al., 2006; Green, 2007). GI contained sevenhuman NoVs from USA (Norwalk), UK (Southampton), Germany(HSS/3/97/DEU), and Japan (Otofuke, WUG1, SzUG1, and Chiba 407)from 1968 to 2000; it branched into several clusters. GII included atotal of 74 strains collected during 1976–2006, and it was dividedinto two major subgroups: Subgroup A contained the Swine43,GIFU′99, and five Saitama-originated strains (Saitama U25, U16,U17, U3, and U4), all of which were isolated in Japan from 1997to 1999. Subgroup B consisted of the remaining 67 strains that are
of clusters and single branches. Within this subgroup, the CBNU1strain was most closely related to the American Texas/TCH04-577(2004) and two Japanese Saitama U18 (1997–1999) and SaitamaU201 (1998) strains. GIII contained two bovine NoVs, i.e., the British
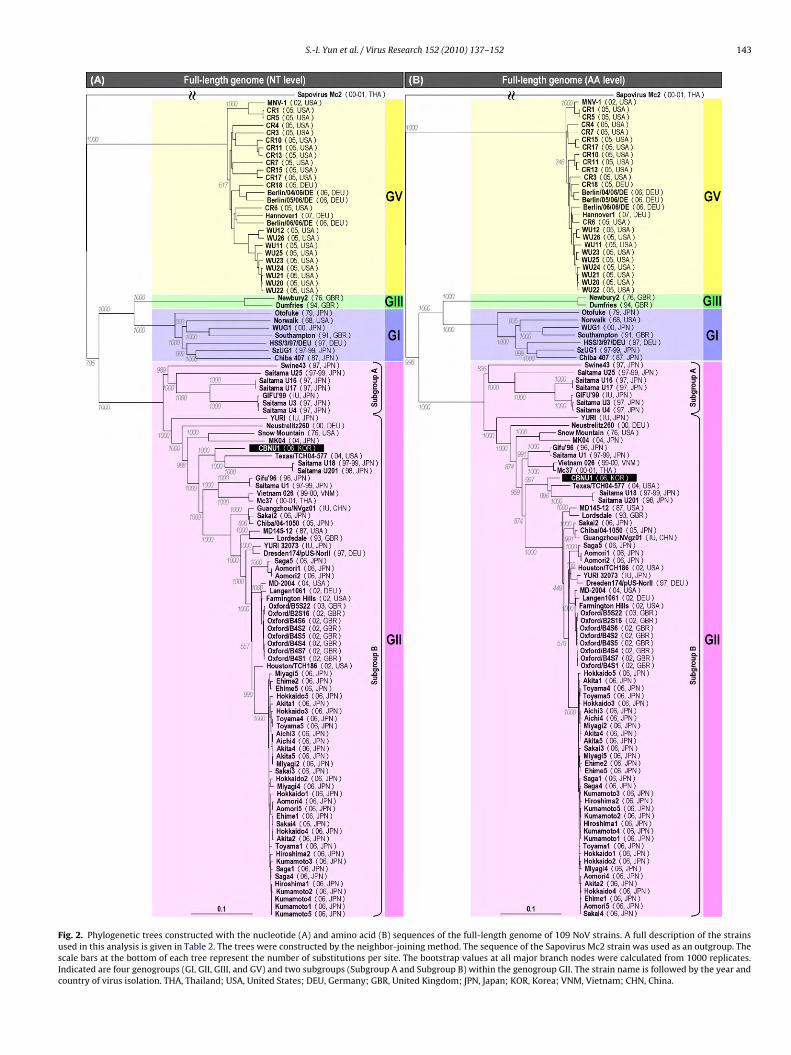
S.-I. Yun et al. / Virus Research 152 (2010) 137–152 143
Fig. 2. Phylogenetic trees constructed with the nucleotide (A) and amino acid (B) sequences of the full-length genome of 109 NoV strains. A full description of the strainsused in this analysis is given in Table 2. The trees were constructed by the neighbor-joining method. The sequence of the Sapovirus Mc2 strain was used as an outgroup. Thescale bars at the bottom of each tree represent the number of substitutions per site. The bootstrap values at all major branch nodes were calculated from 1000 replicates.Indicated are four genogroups (GI, GII, GIII, and GV) and two subgroups (Subgroup A and Subgroup B) within the genogroup GII. The strain name is followed by the year andcountry of virus isolation. THA, Thailand; USA, United States; DEU, Germany; GBR, United Kingdom; JPN, Japan; KOR, Korea; VNM, Vietnam; CHN, China.

144 S.-I. Yun et al. / Virus Research 152 (2010) 137–152
Fig. 3. Phylogenetic trees constructed with the full-length nucleotide (A) and amino acid (B) sequences of each ORF of the 109 completely sequenced NoV strains, using thesequence of the Sapovirus Mc2 strain as an outgroup. The numbers on each branch node indicate the bootstrap values for the corresponding clusters. For details of the trees,see the legend for Fig. 2.

S.-I. Yun et al. / Virus Research 152 (2010) 137–152 145
Fig. 3. (Continued ).

146 S.-I. Yun et al. / Virus Research 152 (2010) 137–152
F lete nt 145-m he doj tes th
Ns(a
tnnsgitaSnarGowatptf(
aNorsttts
FN(tbTH
ig. 4. Recombination at the ORF1/ORF2 junction of the CBNU1 genome. The compo those of three reference strains (Texas/TCH04-577, Guangzhou/NVgz01, and MD
odel in a window size of 200 bp and a step size of 20 bp with the gap strip on. Tunction, identified by maximization of �2 value, with the number of informative si
ewbury2 (1976) and Dumfries (1994) strains, whereas GV con-isted of 26 closely related murine NoVs isolated from the USAe.g., MNV-1, CR4, and WU12) or Germany (e.g., Berlin/04/06/DEnd Hannover1) in 2002–2007.
An amino acid sequence-based phylogenetic tree had an overallopology analogous to the nucleotide sequence-based phyloge-etic tree, with high bootstrap values (Fig. 2B). As seen at theucleotide level, all 109 completely sequenced NoVs were also clas-ified at the amino acid level into four phylogenetic groups (orenogroups); of the four, the GII genogroup was further subdividednto two major subgroups (Subgroup A and Subgroup B). Withinhe Subgroup B, the CBNU1 strain was again most closely relatedt the amino acid level to the Texas/TCH04-577, Saitama U18, andaitama U201 strains, forming a single minor cluster as seen at theucleotide level. This CBNU1-related cluster, however, displayedminor variation in its phylogenetic relationships to other sepa-
ate clusters defined with high bootstrap support, e.g., a cluster ofifu′96, Saitama U1, Vietnam 026, and Mc37, and another clusterf Sakai2, Chiba/04-1050, and Guangzhou/NVgz01. This variationas likely caused by the difference in the amino acid alterations
ccumulated in the genome of a particular strain. We also notedhat the GV genogroup formed a separate branch early in the viralhylogeny at both the nucleotide and amino acid levels, indicatinghat murine NoVs of the GV genogroup are significantly differentrom all other sequenced NoVs, in agreement with previous reportsKarst et al., 2003; Zheng et al., 2006).
To establish a basis for performing an extensive phylogeneticnalysis of CBNU1 with a large number of other genetically diverseoVs that have been partially sequenced, we aimed to identify anptimal subregion within the genome that would potentially mir-or the full-length genome-based phylogeny. We performed two
ets of six phylogenetic analyses, one set at the nucleotide level andhe other set at the amino acid level, using the sequence informa-ion for each viral ORF of the 109 completely sequenced NoVs; wehen compared the resulting phylogenetic topologies to those con-tructed using the complete nucleotide and amino acid sequencesig. 5. ORF2-based phylogenetic analysis used for genotyping of the CBNU1 strain. The treoV strains representative of GII.1–GII.19. The 19 genotypes are designated according t
2006). Also, indicated are two subclusters (Subcluster A and Subcluster B) formed withinhe sequence of the Sapovirus Mc2 strain as an outgroup. Numbers on each branch nodeelow indicates the number of nucleotide substitutions per site. The strain name is followHA, Thailand; MEX, Mexico; USA, United States; GBR, United Kingdom; AUS, Australia;UN, Hungary; ARG, Argentina; KOR, Korea; NZL, New Zealand; NLD, Netherlands; IU, Inf
ucleotide sequence of the genome of the CBNU1 strain (as a query) was compared12). Simplot generated a similarity plot using the Kimura-two parameter distancetted vertical line indicates the crossover point at position 5042 in the ORF1/ORF2at support clustering of the query sequence with the corresponding strains.
of the full-length genomes. At both the nucleotide (Fig. 3A) andamino acid (Fig. 3B) levels, the ORF2-based phylogenies resembledthe genome-based phylogenies in that they displayed the samefour distinct phylogenetic groups and most minor clusters of thelatter phylogenies. This result indicated that the ORF2-based phy-logenetic analysis would be useful for investigating the geneticrelationships among a large number of genetically diverse NoVs.Interestingly, the CBNU1 strain was found to be most closely relatedto the Guangzhou/NVgz01, Sakai2, and Chiba/04-1050 strains in theORF1-based phylogeny, but to the Texas/TCH04-577, Saitama U18,and Saitama U201 strains in the phylogenies based on ORF2 andORF3; this change in genetic relationship was observed at both thenucleotide (Fig. 3A) and amino acid (Fig. 3B) levels, suggesting theoccurrence of genetic recombination during NoV evolution amongdistinct viral strains.
We then looked for possible recombination events that mightoccur in the full-length genome of CBNU1. To identify the putativeparent-like strains and potential recombination sites, phyloge-netic profile analysis was performed using the Simplot program(Lole et al., 1999). Our initial analysis was performed using the34 completely sequenced NoVs that are highly similar and closelyrelated to CBNU1: e.g., YURI, Saitama U18, Saitama U201, Neustre-litz260, MK04, Texas/TCH04-577, Snow Mountain, Gifu’96, SaitamaU1, Vietnam 026, Mc37, Guangzhou/NVgz01, Sakai2, Chiba/04-1050, MD145-12, and Lordsdale. Based on this initial screen-ing, we selected three reference strains (Guangzhou/NVgz01,Texas/TCH04-577, and MD145-12) to represent the most likelyrecombination events to occur in the CBNU1 genome, because theother strains showed similarity scores lower than these three refer-ence strains or below zero percentage throughout the genome (datanot shown). When a similarity plot for the CBNU1 strain was gener-
ated with the three reference strains, a recombination breakpoint atposition 5042 of the sequence alignment was visible between twoputative parent-like strains, Guangzhou/NVgz01 and Texas/TCH04-577 (Fig. 4). This mosaicism was further confirmed by bootscanningof the same genome sequences, indicating higher levels of phy-e was constructed with the complete nucleotide sequences of ORF2 of 126 selectedo the NoV classification strategy proposed by Wang et al. (2005) and Zheng et al.the genotype GII.3. The tree was generated by the neighbor-joining method using
indicate the bootstrap values for the corresponding genetic clusters. The scale bared by the year and country of virus isolation and the GenBank accession number.
JPN, Japan; CHN, China; CAN, Canada; DEU, Germany; SWE, Sweden; FRA, France;ormation unavailable.

S.-I. Yun et al. / Virus Research 152 (2010) 137–152 147

1 esearc
ltorrt
3f
wltCGtwpa(sotaatbtUTSTnwbwSit(tGAtats
4
ht2etleiDapNm
48 S.-I. Yun et al. / Virus R
ogenetic relatedness between the CBNU1 genome sequence andhe Guangzhou/NVgz01 and Texas/TCH04-577 genome sequencen the upstream and downstream side of the recombination site,espectively (data not shown). The location and significance of theecombination site were corroborated by the highest �2 value andhe large number of informative sites, respectively (Fig. 4).
.4. ORF2-based phylogenetic analyses of the CBNU1 strain and aull spectrum of other genetically diverse GII NoVs
To investigate the genetic relationship of the CBNU1 strainith other genetically diverse GII NoVs at the sub-genogroup
evel, we carried out the extensive phylogenetic analyses usinghe nucleotide and amino acid sequences of the ORF2 gene ofBNU1 and 126 other GII NoVs representing all 19 genotypes,II.1–GII.19. The ORF2-based phylogenetic tree constructed with
he nucleotide sequences of the 127 GII NoVs, in conjunctionith a sapovirus Mc2 as an outgroup, clearly showed 19 distincthylogenetic clusters with reliable bootstrap support (Fig. 5), ingreement with a recent classification of GII NoVs into 19 genotypesWang et al., 2005; Zheng et al., 2006). Importantly, the CBNU1train belonged to genotype GII.3; for the purpose of discussionnly, this GII.3 genotype was divided into two distinct subclus-ers: Subcluster A consisted of strains from Sweden (NLV/1157-01nd NLV/2004-00), Argentina (Arg320), USA (Texas/TCH04-577),nd Germany (Herzberg 385, Oberhausen 455, and Bitburg/289);he Korean CBNU1 strain was also a member of this subcluster,eing most closely related to the Argentinian, the American, andhe three German strains. Subcluster B contained strains from theSA (MD101-2, MD134-10, New Orleans/279, Montgomery/312,owson/313, Brattleboro/321, and Lionville/247), Japan (OTH-25,aitama U201, and Saitama U18), Mexico (MX), Canada (Toronto-V24), New Zealand (Auckland), and UK (Rbh). Interestingly, weoted that two strains (Chiba/04-1050 and Guangzhou/NVgz01),hich are most closely related to the CBNU1 strain in the ORF1-
ased phylogeny, belonged to genotype GII.4; on the other hand, ase pointed out, three strains (Texas/TCH04-577, Saitama U18, and
aitama U201), which are most closely related to the CBNU1 strainn the phylogenies based on the ORF2 and ORF3, belonged to geno-ype GII.3. This finding, together with the phylogenetic profilingFig. 4), suggests that the CBNU1 strain would be an intergeno-ypic recombinant within the genogroup GII between the GII.4uangzhou/NVgz01-like and GII.3 Texas/TCH04-577-like strains.s compared to the ORF2-based phylogenetic tree constructed with
he nucleotide sequences, a virtually identical tree topology wasgain produced with the corresponding amino acid sequences, withhe CBNU1 strain being classified into genotype GII.3 (data nothown).
. Discussion
A high degree of genetic diversity has been reported amonguman NoVs circulating in different geographic locations at variousimes (Ando et al., 2000; Green et al., 2000; Vinjé and Koopmans,000; Fankhauser et al., 2002; Katayama et al., 2002; Kageyamat al., 2004; Zheng et al., 2006). This genetic diversity is linkedo the antigenic variations, which may not only alter the viru-ence and mode of viral transmission but also enable the virus toscape our immune system, changing the patterns of viral activ-ty (Noel et al., 1999; Koopmans et al., 2000; Nilsson et al., 2003;
ingle, 2004; Chakravarty et al., 2005; Koopmans, 2008; Waters etl., 2008; Verhoef et al., 2008). Thus, the availability of the com-lete genomic sequences of geographically and temporally diverseoVs is important for studies of the diagnostics, epidemiology, andolecular biology of the virus. In this work, we (i) determined theh 152 (2010) 137–152
nucleotide and predicted amino acid sequences of the full-lengthgenome of the Korean CBNU1 strain recovered from a 2006 out-break, (ii) characterized this virus at the molecular and geneticlevels by comparing it to 108 other NoV strains whose completesequences are currently available, (iii) established its genetic rela-tionship at the sub-genogroup level with the 108 other full-lengthNoV genomes, (iv) compared NoV phylogeny based on the full-length genome sequence with those based on the individual genesequences, and (v) classified the CBNU1 strain at the sub-genotypelevel by performing ORF2-based phylogenetic analyses with a largeselection of 126 other GII NoVs that have previously been groupedinto 19 genotypes (Wang et al., 2005; Zheng et al., 2006).
Based on genome sequence analysis, we found that CBNU1belonged to the GII genogroup of NoV. The genome of CBNU1consists of 7547 nucleotides preceding the 3′-poly(A) tail and is8 nucleotides shorter than that of the Lordsdale strain (Dingle etal., 1995), the representative GII NoV. On the other hand, it is 107and 161 nucleotides shorter than those of the Norwalk (Jiang etal., 1990, 1993; Matsui et al., 1991; Hardy and Estes, 1996) andSouthampton (Lambden et al., 1993, 1995) strains, respectively,both of which are representative GI NoVs. Similarly, the genomeorganization of CBNU1 corresponds to that of the Lordsdale strain,in that ORF1 and ORF2 overlap by 20 nucleotides, and ORF2 andORF3 overlap by 1 nucleotide, resulting in −2 and −1 frameshifts,respectively. In the case of the Norwalk and Southampton strains,however, ORF2 overlaps ORF1 by 17 nucleotides and ORF3 by 1nucleotide, while creating the same −2 and −1 frameshifts, respec-tively, as seen in the Lordsdale strain. The internal protein-codingregion of the CBNU1 genome is flanked by 5′ and 3′ UTRs of 4 and52 nucleotides, respectively. With respect to the size and primarysequence, the 5′UTR is well conserved in all three representa-tive strains, whereas the 3′UTR exhibits significant variability, i.e.,45 nucleotides in Lordsdale, 66 nucleotides in Norwalk, and 78nucleotides in Southampton. Also, we have shown in the presentstudy that the 3′-end of the CBNU1 genome contains a poly(A) tailof 11–105 nucleotides. In other positive-strand RNA viruses, the 5′
and 3′ terminal regions of the genomic RNA (along with 3′-poly(A)tail when the genome is polyadenylated) play an important role(s)in controlling RNA translation and replication (Dreher and Miller,2006; Liu et al., 2009). For human NoVs, however, functional anal-yses of the primary sequences and secondary structures at or nearthe 5′ and 3′ UTRs of the genome and of the presence and size ofthe 3′-poly(A) tail with regard to regulation of the viral replicationcycle have been limited by the lack of a reverse genetics system forviral genome manipulation and of a cell culture system for viruspropagation (Radford et al., 2004; Hardy, 2005).
Conventional RT-PCR, in conjunction with cDNA sequencing,has routinely been used for the molecular diagnosis of NoV infec-tion and for the genetic characterization of various NoV strains(Katayama et al., 2002; Nishida et al., 2003; Okada et al., 2005;Bull et al., 2006; Castilho et al., 2006; Ho et al., 2007; Aw et al.,2009). Also, multiplex real-time RT-PCR has been successfully usedfor the detection and quantitation of multiple NoVs simultaneously(Kageyama et al., 2003; Yan et al., 2003; Richards et al., 2004; Panget al., 2005; Hoehne and Schreier, 2006; Wolf et al., 2007; Stalset al., 2009). To search for a highly conserved region suitable formaximizing the nucleic acid sequence-based detection of genet-ically diverse NoVs, we performed multiple sequence alignmentswith the complete nucleotide and deduced amino acid sequences ofthe CBNU1 and 108 other completely sequenced human and animalNoV genomes. We found that the NS6Pro and NS7Pol protein-coding
region located at the 3′-most third of ORF1 was the most highlyconserved throughout these genomes, suggesting that this regionwould provide an optimal target sequence for a pair of primers thatreacts with a large number of genetically distinct NoVs. In addi-tion, we also identified five short stretches of hypervariable region
esearc
waraahisaIsre
nirpmsfpKssvKlarw2cSnosMsNt
2lepoptatsprHaUstiOg
Cg2
S.-I. Yun et al. / Virus R
ithin the genome: HVR1-1 and HVR1-2 in ORF1, HVR2 in ORF2,nd HVR3-1 and HVR3-2 in ORF3. These hypervariable regions wereecognized not only by the higher frequency of nucleotide andmino acid substitutions but also the accumulation of nucleotidend amino acid mismatched gaps. This finding suggests that the fiveypervariable regions might be potentially useful for differentiat-
ng the specific NoV genogroups/genotypes that are related to hostpecificity, cell tropism, pathogenicity, and/or virulence (Chen etl., 2004; Dingle, 2004; Chakravarty et al., 2005; Chen et al., 2006).n recent studies, the HVR2 region (previously known as the P2ubdomain) has been shown to be important for the antigenic andeceptor binding specificity of the NoVs (Nilsson et al., 2003; Tant al., 2003; Lochridge et al., 2005; Rohayem et al., 2005).
Since the discovery of human NoV about 40 years ago, a largeumber of genetically divergent NoVs from a variety of hosts,
ncluding humans and animals, have been isolated at an acceleratedate worldwide, but the vast majority of these NoVs have only beenartially sequenced. Previous phylogenetic analyses have been pri-arily based on these partial nucleotide or deduced amino acid
equences of the NoV genome, which have mainly been derivedrom a short conserved region of either the RNA-dependent RNAolymerase NS7Pol or the major capsid VP1 (Ando et al., 2000;oopmans, 2008). In some cases, however, because of limitedequence variation, the partial sequence-based phylogenetic analy-es might not be able to show the actual genetic relatedness amongarious NoV strains (Fankhauser et al., 2002; Katayama et al., 2002;ageyama et al., 2004). In the present study, we performed a full-
ength genome-based phylogenetic analysis of the CBNU1 strainnd 108 other completely sequenced NoVs, indicating its geneticelationships with other members of the NoV genus. Our resultsere consistent with the current classification system (Zheng et al.,
006; Green, 2007): Within the GII genogroup, the CBNU1 strainlustered with the American Texas/TCH04-577 and two Japaneseaitama U18 and Saitama U201 strains, although these strains didot show any temporal correlation. Previous phylogenetic treesf the full-length or near-full-length genome sequence were con-tructed with a panel of 18 or 60 NoV strains (Katayama et al., 2002;otomura et al., 2008). Analysis of additional full-length genome
equences will help to further elucidate the genetic diversity ofoVs, facilitating the design of a better vaccine strategy against
his pathogen.In accordance with other phylogenetic studies (Katayama et al.,
002; Motomura et al., 2008), we found that the ORF2-based phy-ogeny corresponded to the full-length genome-based phylogenyqually well for the nucleotide and amino acid sequences, as com-ared to those based on the ORF1 or ORF3. Given that the expressionf recombinant VP1 protein results in the production of virus-likearticles (VLPs) that are analogous to native NoV with respect toheir morphologic, biochemical, and antigenic properties (Jiang etl., 1992; Green et al., 1993; Prasad et al., 1994; Hutson et al., 2003),he results from our study suggest that the significant genetic diver-ity and relatedness revealed by the ORF2-based phylogeny areresumably correlated with the complex antigenic variation andelationships of these viruses (Wyatt et al., 1974; Ando et al., 2000;ansman et al., 2006). Also, we noted that the CBNU1 strain formedcluster with the Texas/TCH04-577, Saitama U18, and Saitama201 strains in the phylogenies based on the ORF2 and ORF3, as
een in the genome-based phylogeny; however, it formed a clus-er with the Chiba/04-1050, Guangzhou/NVgz01, and Sakai2 strainsn the ORF1-based phylogeny. Thus, it appears that the ORF2 andRF3 sequences, rather than the ORF1, have more weight in the
enome-based phylogeny profile.As evidenced by the ORF2-based phylogenetic analyses of theBNU1 strain and a pool of 126 NoVs, including all 19 knownenotypes of the GII genogroup (Wang et al., 2005; Zheng et al.,006), and by the results of genome-based Simplot analysis, the
h 152 (2010) 137–152 149
CBNU1 strain was shown to be a recombinant variant of GII.3 NoV,being most closely related to the Argentinian Arg320, the Amer-ican Texas/TCH04-577, and three German strains (Herzberg 385,Oberhausen 455, and Bitburg/289). Globally, GII.4 (e.g., 95/96-US)and its variant NoVs (e.g., Farmington Hills, GII.4b, and Hunter)have become the most predominant strains since the mid-1990s(Vinjé et al., 1997; Noel et al., 1999; Fankhauser et al., 2002;Lopman et al., 2004; Widdowson et al., 2004; Bull et al., 2006;Medici et al., 2006; Phan et al., 2006a; Gallimore et al., 2007;Allen et al., 2008, 2009; Sdiri-Loulizi et al., 2009; Zheng et al.,2010). A recent study has indicated that the GII.4 NoV is con-tinuously evolving by altering the surface-exposed carbohydrateligand binding domain of the VP1 protein in response to hostimmune selection; however, this antigenic drift can be toleratedby a large repertoire of histo-blood group antigens on mucosalsurfaces that are necessary for the binding, and perhaps down-stream processes, of the virus’s life cycle (Lindesmith et al., 2008;Bok et al., 2009). It is important to understand what kinds of viraland cellular factors make GII.4 and its variants the most prevalentgenotype.
In addition to mutations introduced by the RNA-dependentRNA polymerase NS7Pol, RNA recombination also contributes tothe genetic diversity and evolution of NoV (Worobey and Holmes,1999). A recombinant NoV can be defined as a single clusterwith two genetically distinct NoVs when two different regionsof the genome are used for phylogenetic analyses. This situationheld true for the CBNU1 strain, in that inconsistencies sugges-tive of a recombinant virus were observed in individual ORF-basedphylogenetic analyses. Subsequently, a nucleotide similarity anal-ysis by Simplot revealed that the intergenotypic recombinantCBNU1 strain had a high level of nucleotide conservation withthe GII.4 Guangzhou/NVgz01 strain throughout ORF1, but after theORF1/ORF2 junction, it showed a high degree of nucleotide conser-vation with the GII.3 Texas/TCH04-577 strain; the crossover pointwas mapped to position 5042 of the genome sequence alignmentthat corresponds to the junction of ORF1/ORF2. This result is con-sistent with the fact that most of the NoV recombinants had acrossover point within or around the ORF1/ORF2 junction (Bull etal., 2007). In previous studies, the Snow Mountain strain was firstreported as the naturally occurring recombinant NoV (Hardy et al.,1997); to date, a large number of recombinant NoV strains (e.g.,WUG1, MD145-12, Gifu’96, and Saitama U4) have been identified(Jiang et al., 1999; Han et al., 2004; Oliver et al., 2004; Ambert-Balayet al., 2005; Bull et al., 2005, 2007; Rohayem et al., 2005; Wanget al., 2005; Etherington et al., 2006; Phan et al., 2006a, 2006b;Tsugawa et al., 2006; Vidal et al., 2006; Waters et al., 2007; Jin etal., 2008; Nayak et al., 2008; Li et al., 2009; Martella et al., 2009;Nakamura et al., 2009; Chhabra et al., 2010; Chung et al., 2010;Dey et al., 2010). Recombination events appear to take place notonly between two different genotypes within the same genogroupbut also between two distinct genogroups (Phan et al., 2007).Within the GII genogroup, GII.4 NoVs appear to be more frequentlyinvolved in the inter- and intra-genotypic recombination, mainlybecause they are presently the most prevalent genotype worldwide(Bull et al., 2006, 2007). Also, it is possible that co-infection with twodifferent human/animal NoVs can lead to inter-genogroup recom-bination, as previously reported in a closely related calicivirus,sapovirus (Hansman et al., 2005). Given the growing number of nat-urally occurring recombinant NoVs, particularly with the crossoverpoints near or in the ORF1/ORF2 overlap, genotyping based solelyon the ORF2 sequence needs to be interpreted with caution. Ide-
ally, NoV genomes should always be compared with others foreach of the three ORFs individually. Neither partial sequencing norphylogenetic analyses based on the partial sequence, such as theNS7Pol and VP1 protein-coding regions, would be sufficient for theidentification of novel recombinant NoVs.
1 esearc
pgibtdlsaivd
A
fe(btp
R
A
A
A
A
A
B
B
B
B
B
B
B
B
B
C
C
C
50 S.-I. Yun et al. / Virus R
The results of the present study, together with previous otherhylogenetic analyses, have expanded our understanding of theenetic diversity and evolution of human NoVs and providedmportant insights into the genetic properties of the virus that maye important for controlling viral infection and spread. Althoughhe biological and/or serological significance of the virus’s geneticiversity has not yet been tested experimentally because of the
ack of an efficient cell culture system and an in vivo animal modelystem for infection, this study will help to improve the specificitynd sensitivity of nucleic acid-based diagnostic assays and facil-tate future studies aimed at understanding the genetic basis foriral pathogenesis and selecting a candidate strain(s) for vaccineevelopment.
cknowledgments
This work was supported by a grant (KRF-2007-532-C00015)rom the Korea Research Foundation, funded by the Korean Gov-rnment (MOEHRD, Basic Research Promotion Fund), and a grantCode #20070401-034-027) from the BioGreen 21 program, fundedy the Rural Development Administration, Republic of Korea. Wehank Young-Ah Kang for technical assistance with sequence com-arisons and Deborah McClellan for editorial assistance.
eferences
llen, D.J., Gray, J.J., Gallimore, C.I., Xerry, J., Iturriza-Gómara, M., 2008. Analysis ofamino acid variation in the P2 domain of the GII-4 norovirus VP1 protein revealsputative variant-specific epitopes. PLoS One 3 (1), e1485.
llen, D.J., Noad, R., Samuel, D., Gray, J.J., Roy, P., Iturriza-Gómara, M., 2009. Charac-terisation of a GII-4 norovirus variant-specific surface-exposed site involved inantibody binding. Virol. J. 6, 150.
mbert-Balay, K., Bon, F., Le Guyader, F., Pothier, P., Kohli, E., 2005. Char-acterization of new recombinant noroviruses. J. Clin. Microbiol. 43 (10),5179–5186.
ndo, T., Noel, J.S., Fankhauser, R.L., 2000. Genetic classification of “Norwalk-likeviruses”. J. Infect. Dis 181 (Suppl. 2), S336–S348.
w, T.G., Gin, K.Y., Ean Oon, L.L., Chen, E.X., Woo, C.H., 2009. Prevalence and geno-types of human noroviruses in tropical urban surface waters and clinical samplesin Singapore. Appl. Environ. Microbiol. 75 (15), 4984–4992.
elliot, G., Sosnovtsev, S.V., Mitra, T., Hammer, C., Garfield, M., Green, K.Y., 2003.In vitro proteolytic processing of the MD145 norovirus ORF1 nonstructuralpolyprotein yields stable precursors and products similar to those detected incalicivirus-infected cells. J. Virol. 77 (20), 10957–10974.
ertolotti-Ciarlet, A., Crawford, S.E., Hutson, A.M., Estes, M.K., 2003. The 3′ end ofNorwalk virus mRNA contains determinants that regulate the expression andstability of the viral capsid protein VP1: a novel function for the VP2 protein. J.Virol. 77 (21), 11603–11615.
ertolotti-Ciarlet, A., White, L.J., Chen, R., Prasad, B.V., Estes, M.K., 2002. Structuralrequirements for the assembly of Norwalk virus-like particles. J. Virol. 76 (8),4044–4055.
lakeney, S.J., Cahill, A., Reilly, P.A., 2003. Processing of Norwalk virus nonstructuralproteins by a 3C-like cysteine proteinase. Virology 308 (2), 216–224.
lanton, L.H., Adams, S.M., Beard, R.S., Wei, G., Bulens, S.N., Widdowson, M.A., Glass,R.I., Monroe, S.S., 2006. Molecular and epidemiologic trends of caliciviruses asso-ciated with outbreaks of acute gastroenteritis in the United States, 2000–2004.J. Infect. Dis 193 (3), 413–421.
ok, K., Abente, E.J., Realpe-Quintero, M., Mitra, T., Sosnovtsev, S.V., Kapikian, A.Z.,Green, K.Y., 2009. Evolutionary dynamics of GII.4 noroviruses over a 34-yearperiod. J. Virol. 83 (22), 11890–11901.
ull, R.A., Hansman, G.S., Clancy, L.E., Tanaka, M.M., Rawlinson, W.D., White, P.A.,2005. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 11 (7),1079–1085.
ull, R.A., Tanaka, M.M., White, P.A., 2007. Norovirus recombination. J. Gen. Virol. 88(Pt. 12), 3347–3359.
ull, R.A., Tu, E.T., McIver, C.J., Rawlinson, W.D., White, P.A., 2006. Emergence of anew norovirus genotype II.4 variant associated with global outbreaks of gas-troenteritis. J. Clin. Microbiol. 44 (2), 327–333.
astilho, J.G., Munford, V., Resque, H.R., Fagundes-Neto, U., Vinjé, J., Rácz, M.L., 2006.Genetic diversity of norovirus among children with gastroenteritis in São PauloState, Brazil. J. Clin. Microbiol. 44 (11), 3947–3953.
hakravarty, S., Hutson, A.M., Estes, M.K., Prasad, B.V., 2005. Evolutionary traceresidues in noroviruses: importance in receptor binding, antigenicity, virionassembly, and strain diversity. J. Virol. 79 (1), 554–568.
hen, R., Neill, J.D., Estes, M.K., Prasad, B.V., 2006. X-ray structure of a native cali-civirus: structural insights into antigenic diversity and host specificity. Proc.Natl. Acad. Sci. U.S.A. 103 (21), 8048–8053.
h 152 (2010) 137–152
Chen, R., Neill, J.D., Noel, J.S., Hutson, A.M., Glass, R.I., Estes, M.K., Prasad, B.V., 2004.Inter- and intragenus structural variations in caliciviruses and their functionalimplications. J. Virol. 78 (12), 6469–6479.
Chhabra, P., Walimbe, A.M., Chitambar, S.D., 2010. Molecular characterization ofthree novel intergenotype norovirus GII recombinant strains from western India.Virus Res. 147 (2), 242–246.
Chung, J.Y., Han, T.H., Park, S.H., Kim, S.W., Hwang, E.S., 2010. Detection ofGII-4/2006b variant and recombinant noroviruses in children with acute gas-troenteritis, South Korea. J. Med. Virol. 82 (1), 146–152.
de Wit, M.A., Koopmans, M.P., Kortbeek, L.M., Wannet, W.J., Vinjé, J., van Leus-den, F., Bartelds, A.I., van Duynhoven, Y.T., 2001. Sensor, a population-basedcohort study on gastroenteritis in the Netherlands: incidence and etiology. Am.J. Epidemiol. 154 (7), 666–674.
Dey, S.K., Phan, T.G., Mizuguchia, M., Okitsua, S., Ushijima, H., 2010. Novel recombi-nant norovirus in Japan. Virus Genes 40 (3), 362–364.
Dingle, K.E., 2004. Mutation in a Lordsdale norovirus epidemic strain as a potentialindicator of transmission routes. J. Clin. Microbiol. 42 (9), 3950–3957.
Dingle, K.E., Lambden, P.R., Caul, E.O., Clarke, I.N., 1995. Human enteric Caliciviridae:the complete genome sequence and expression of virus-like particles from agenetic group II small round structured virus. J. Gen. Virol. 76 (Pt. 9), 2349–2355.
Dolin, R., Blacklow, N.R., DuPont, H., Buscho, R.F., Wyatt, R.G., Kasel, J.A., Hornick, R.,Chanock, R.M., 1972. Biological properties of Norwalk agent of acute infectiousnonbacterial gastroenteritis. Proc. Soc. Exp. Biol. Med. 140 (2), 578–583.
Dreher, T.W., Miller, W.A., 2006. Translational control in positive strand RNA plantviruses. Virology 344 (1), 185–197.
Duizer, E., Schwab, K.J., Neill, F.H., Atmar, R.L., Koopmans, M.P., Estes, M.K., 2004.Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85 (Pt. 1), 79–87.
Etherington, G.J., Dicks, J., Roberts, I.N., 2006. High throughput sequence analysisreveals hitherto unreported recombination in the genus Norovirus. Virology 345(1), 88–95.
Fankhauser, R.L., Monroe, S.S., Noel, J.S., Humphrey, C.D., Bresee, J.S., Parashar, U.D.,Ando, T., Glass, R.I., 2002. Epidemiologic and molecular trends of “Norwalk-like viruses” associated with outbreaks of gastroenteritis in the United States. J.Infect. Dis. 186 (1), 1–7.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the boot-strap. Evolution 39 (4), 783–791.
Gallimore, C.I., Iturriza-Gomara, M., Xerry, J., Adigwe, J., Gray, J.J., 2007. Inter-seasonal diversity of norovirus genotypes: emergence and selection of virusvariants. Arch. Virol. 152 (7), 1295–1303.
Glass, P.J., White, L.J., Ball, J.M., Leparc-Goffart, I., Hardy, M.E., Estes, M.K., 2000a.Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol.74 (14), 6581–6591.
Glass, R.I., Noel, J., Ando, T., Fankhauser, R., Belliot, G., Mounts, A., Parashar, U.D.,Bresee, J.S., Monroe, S.S., 2000b. The epidemiology of enteric caliciviruses fromhumans: a reassessment using new diagnostics. J. Infect. Dis. 181 (Suppl. 2),S254–S261.
Green, K.Y., 2007. Caliciviridae: the noroviruses. In: Knipe, D.M., Howley, P.M., Grif-fin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E. (Eds.), Fields Virology,5th ed. Lippincott Williams & Wilkins, Philadelphia, PA, pp. 949–979.
Green, K.Y., Ando, T., Balayan, M.S., Berke, T., Clarke, I.N., Estes, M.K., Matson, D.O.,Nakata, S., Neill, J.D., Studdert, M.J., Thiel, H.J., 2000. Taxonomy of the cali-civiruses. J. Infect. Dis. 181 (Suppl. 2), S322–S330.
Green, K.Y., Belliot, G., Taylor, J.L., Valdesuso, J., Lew, J.F., Kapikian, A.Z., Lin, F.Y.,2002. A predominant role for Norwalk-like viruses as agents of epidemic gas-troenteritis in Maryland nursing homes for the elderly. J. Infect. Dis. 185 (2),133–146.
Green, K.Y., Lew, J.F., Jiang, X., Kapikian, A.Z., Estes, M.K., 1993. Comparison of thereactivities of baculovirus-expressed recombinant Norwalk virus capsid antigenwith those of the native Norwalk virus antigen in serologic assays and someepidemiologic observations. J. Clin. Microbiol. 31 (8), 2185–2191.
Green, S.M., Lambden, P.R., Caul, E.O., Ashley, C.R., Clarke, I.N., 1995. Capsid diversityin small round-structured viruses: molecular characterization of an antigeni-cally distinct human enteric calicivirus. Virus Res. 37 (3), 271–283.
Han, M.G., Smiley, J.R., Thomas, C., Saif, L.J., 2004. Genetic recombination betweentwo genotypes of genogroup III bovine noroviruses (BoNVs) and capsid sequencediversity among BoNVs and Nebraska-like bovine enteric caliciviruses. J. Clin.Microbiol. 42 (11), 5214–5224.
Hansman, G.S., Katayama, K., Maneekarn, N., Peerakome, S., Khamrin, P., Tonusin, S.,Okitsu, S., Nishio, O., Takeda, N., Ushijima, H., 2004. Genetic diversity of norovirusand sapovirus in hospitalized infants with sporadic cases of acute gastroenteritisin Chiang Mai, Thailand. J. Clin. Microbiol. 42 (3), 1305–1307.
Hansman, G.S., Natori, K., Shirato-Horikoshi, H., Ogawa, S., Oka, T., Katayama, K.,Tanaka, T., Miyoshi, T., Sakae, K., Kobayashi, S., Shinohara, M., Uchida, K., Sakurai,N., Shinozaki, K., Okada, M., Seto, Y., Kamata, K., Nagata, N., Tanaka, K., Miyamura,T., Takeda, N., 2006. Genetic and antigenic diversity among noroviruses. J. Gen.Virol. 87 (Pt. 4), 909–919.
Hansman, G.S., Takeda, N., Oka, T., Oseto, M., Hedlund, K.O., Katayama, K., 2005.Intergenogroup recombination in sapoviruses. Emerg. Infect. Dis. 11 (12),1916–1920.
Hardy, M.E., 2005. Norovirus protein structure and function. FEMS Microbiol. Lett.
253 (1), 1–8.Hardy, M.E., Estes, M.K., 1996. Completion of the Norwalk virus genome sequence.Virus Genes 12 (3), 287–290.
Hardy, M.E., Kramer, S.F., Treanor, J.J., Estes, M.K., 1997. Human calicivirus genogroupII capsid sequence diversity revealed by analyses of the prototype Snow Moun-tain agent. Arch. Virol. 142 (7), 1469–1479.

esearc
H
H
H
H
I
I
J
J
J
J
J
K
K
K
K
K
K
K
K
K
K
K
K
L
L
L
L
S.-I. Yun et al. / Virus R
ardy, M.E., Tanaka, T.N., Kitamoto, N., White, L.J., Ball, J.M., Jiang, X., Estes, M.K.,1996. Antigenic mapping of the recombinant Norwalk virus capsid protein usingmonoclonal antibodies. Virology 217 (1), 252–261.
o, E.C., Cheng, P.K., Lau, A.W., Wong, A.H., Lim, W.W., 2007. Atypical norovirusepidemic in Hong Kong during summer of 2006 caused by a new genogroup II/4variant. J. Clin. Microbiol. 45 (7), 2205–2211.
oehne, M., Schreier, E., 2006. Detection of Norovirus genogroup I and II by multiplexreal-time RT- PCR using a 3′-minor groove binder-DNA probe. BMC Infect. Dis.6, 69.
utson, A.M., Atmar, R.L., Marcus, D.M., Estes, M.K., 2003. Norwalk virus-like particlehemagglutination by binding to h histo-blood group antigens. J. Virol. 77 (1),405–415.
nouye, S., Yamashita, K., Yamadera, S., Yoshikawa, M., Kato, N., Okabe, N., 2000.Surveillance of viral gastroenteritis in Japan: pediatric cases and outbreak inci-dents. J. Infect. Dis. 181 (Suppl. 2), S270–S274.
turriza Gómara, M., Simpson, R., Perault, A.M., Redpath, C., Lorgelly, P., Joshi, D., Mug-ford, M., Hughes, C.A., Dalrymple, J., Desselberger, U., Gray, J., 2008. Structuredsurveillance of infantile gastroenteritis in East Anglia, UK: incidence of infectionwith common viral gastroenteric pathogens. Epidemiol. Infect. 136 (1), 23–33.
iang, X., Espul, C., Zhong, W.M., Cuello, H., Matson, D.O., 1999. Characterization ofa novel human calicivirus that may be a naturally occurring recombinant. Arch.Virol. 144 (12), 2377–2387.
iang, X., Graham, D.Y., Wang, K., Estes, M.K., 1990. Norwalk virus genome cloningand characterization. Science 250 (4987), 1580–1583.
iang, X., Wang, M., Graham, D.Y., Estes, M.K., 1992. Expression, self-assembly,and antigenicity of the Norwalk virus capsid protein. J. Virol. 66 (11),6527–6532.
iang, X., Wang, M., Wang, K., Estes, M.K., 1993. Sequence and genomic organizationof Norwalk virus. Virology 195 (1), 51–61.
in, M., Xie, H.P., Duan, Z.J., Liu, N., Zhang, Q., Wu, B.S., Li, H.Y., Cheng, W.X., Yang, S.H.,Yu, J.M., Xu, Z.Q., Cui, S.X., Zhu, L., Tan, M., Jiang, X., Fang, Z.Y., 2008. Emergenceof the GII4/2006b variant and recombinant noroviruses in China. J. Med. Virol.80 (11), 1997–2004.
ageyama, T., Kojima, S., Shinohara, M., Uchida, K., Fukushi, S., Hoshino, F.B., Takeda,N., Katayama, K., 2003. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin.Microbiol. 41 (4), 1548–1557.
ageyama, T., Shinohara, M., Uchida, K., Fukushi, S., Hoshino, F.B., Kojima, S., Takai,R., Oka, T., Takeda, N., Katayama, K., 2004. Coexistence of multiple genotypes,including newly identified genotypes, in outbreaks of gastroenteritis due toNorovirus in Japan. J. Clin. Microbiol. 42 (7), 2988–2995.
ang, S.Y., Yun, S.I., Park, H.S., Park, C.K., Choi, H.S., Lee, Y.M., 2004. Molecular char-acterization of PL97-1, the first Korean isolate of the porcine reproductive andrespiratory syndrome virus. Virus Res. 104 (2), 165–179.
apikian, A.Z., 2000. The discovery of the 27-nm Norwalk virus: an historic perspec-tive. J. Infect. Dis. 181 (Suppl. 2), S295–S302.
apikian, A.Z., Wyatt, R.G., Dolin, R., Thornhill, T.S., Kalica, A.R., Chanock, R.M., 1972.Visualization by immune electron microscopy of a 27-nm particle associatedwith acute infectious nonbacterial gastroenteritis. J. Virol. 10 (5), 1075–1081.
arst, S.M., Wobus, C.E., Lay, M., Davidson, J., Virgin, H.W., 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299 (5612),1575–1578.
atayama, K., Shirato-Horikoshi, H., Kojima, S., Kageyama, T., Oka, T., Hoshino, F.,Fukushi, S., Shinohara, M., Uchida, K., Suzuki, Y., Gojobori, T., Takeda, N., 2002.Phylogenetic analysis of the complete genome of 18 Norwalk-like viruses. Virol-ogy 299 (2), 225–239.
im, S.H., Cheon, D.S., Kim, J.H., Lee, D.H., Jheong, W.H., Heo, Y.J., Chung, H.M., Jee, Y.,Lee, J.S., 2005. Outbreaks of gastroenteritis that occurred during school excur-sions in Korea were associated with several waterborne strains of norovirus. J.Clin. Microbiol. 43 (9), 4836–4839.
oopmans, M., 2008. Progress in understanding norovirus epidemiology. Curr. Opin.Infect. Dis. 21 (5), 544–552.
oopmans, M., Vinjé, J., de Wit, M., Leenen, I., van der Poel, W., van Duynhoven,Y., 2000. Molecular epidemiology of human enteric caliciviruses in The Nether-lands. J. Infect. Dis. 181 (Suppl. 2), S262–S269.
oopmans, M.K., Green, K.Y., Ando, T., Clarke, I.N., Estes, M.K., Matson, D.O., Nakata,S., Neill, J.D., Smith, A.W., Studdert, M.J., Thiel, H.-J., 2005. Caliciviridae. In:Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A. (Eds.), VirusTaxonomy, Eighth Report of the International Committee on Taxonomy ofViruses. Academic Press, San Diego, CA, pp. 843–851.
uusi, M., Nuorti, J.P., Maunula, L., Minh Tran, N.N., Ratia, M., Karlsson, J., vonBonsdorff, C.H., 2002. A prolonged outbreak of Norwalk-like calicivirus (NLV)gastroenteritis in a rehabilitation centre due to environmental contamination.Epidemiol. Infect. 129 (1), 133–138.
ambden, P.R., Caul, E.O., Ashley, C.R., Clarke, I.N., 1993. Sequence and genome orga-nization of a human small round-structured (Norwalk-like) virus. Science 259(5094), 516–519.
ambden, P.R., Liu, B., Clarke, I.N., 1995. A conserved sequence motif at the 5′ ter-minus of the Southampton virus genome is characteristic of the Caliciviridae.Virus Genes 10 (2), 149–152.
i, H.Y., Jin, M., Zhang, Q., Liu, N., Cui, S.X., Fang, Z.Y., Duan, Z.J., 2009. Molecular char-acterization of GIIb recombinants and a novel genotype of Norovirus detectedin China. J. Infect. 59 (3), 215–218.
indesmith, L.C., Donaldson, E.F., Lobue, A.D., Cannon, J.L., Zheng, D.P., Vinje, J., Baric,R.S., 2008. Mechanisms of GII.4 norovirus persistence in human populations.PLoS Med. 5 (2), e31.
h 152 (2010) 137–152 151
Liu, B., Clarke, I.N., Lambden, P.R., 1996. Polyprotein processing in Southamptonvirus: identification of 3C-like protease cleavage sites by in vitro mutagenesis.J. Virol. 70 (4), 2605–2610.
Liu, B.L., Viljoen, G.J., Clarke, I.N., Lambden, P.R., 1999. Identification of further pro-teolytic cleavage sites in the Southampton calicivirus polyprotein by expressionof the viral protease in E. coli. J. Gen. Virol. 80 (Pt. 2), 291–296.
Liu, Y., Wimmer, E., Paul, A.V., 2009. Cis-acting RNA elements in human and animalplus-strand RNA viruses. Biochim. Biophys. Acta 1789 (9–10), 495–517.
Lochridge, V.P., Jutila, K.L., Graff, J.W., Hardy, M.E., 2005. Epitopes in the P2 domain ofnorovirus VP1 recognized by monoclonal antibodies that block cell interactions.J. Gen. Virol. 86 (Pt. 10), 2799–2806.
Lole, K.S., Bollinger, R.C., Paranjape, R.S., Gadkari, D., Kulkarni, S.S., Novak, N.G., Inger-soll, R., Sheppard, H.W., Ray, S.C., 1999. Full-length human immunodeficiencyvirus type 1 genomes from subtype C-infected seroconverters in India, withevidence of intersubtype recombination. J. Virol. 73 (1), 152–160.
Lopman, B., Vennema, H., Kohli, E., Pothier, P., Sanchez, A., Negredo, A., Buesa, J.,Schreier, E., Reacher, M., Brown, D., Gray, J., Iturriza, M., Gallimore, C., Bottiger,B., Hedlund, K.O., Torvén, M., von Bonsdorff, C.H., Maunula, L., Poljsak-Prijatelj,M., Zimsek, J., Reuter, G., Szücs, G., Melegh, B., Svennson, L., van Duijnhoven, Y.,Koopmans, M., 2004. Increase in viral gastroenteritis outbreaks in Europe andepidemic spread of new norovirus variant. Lancet 363 (9410), 682–688.
Lopman, B.A., Reacher, M.H., Van Duijnhoven, Y., Hanon, F.X., Brown, D., Koopmans,M., 2003. Viral gastroenteritis outbreaks in Europe, 1995–2000. Emerg. Infect.Dis. 9 (1), 90–96.
Martella, V., Decaro, N., Lorusso, E., Radogna, A., Moschidou, P., Amorisco, F., Lucente,M.S., Desario, C., Mari, V., Elia, G., Banyai, K., Carmichael, L.E., Buonavoglia, C.,2009. Genetic heterogeneity and recombination in canine noroviruses. J. Virol.83 (21), 1391–1396.
Matsui, S.M., Kim, J.P., Greenberg, H.B., Su, W., Sun, Q., Johnson, P.C., DuPont, H.L.,Oshiro, L.S., Reyes, G.R., 1991. The isolation and characterization of a Norwalkvirus-specific cDNA. J. Clin. Invest. 87 (4), 456–461.
Medici, M.C., Martinelli, M., Abelli, L.A., Ruggeri, F.M., Di Bartolo, I., Arcangeletti,M.C., Pinardi, F., De Conto, F., Izzi, G., Bernasconi, S., Chezzi, C., Dettori, G., 2006.Molecular epidemiology of norovirus infections in sporadic cases of viral gas-troenteritis among children in Northern Italy. J. Med. Virol. 78 (11), 1486–1492.
Motomura, K., Oka, T., Yokoyama, M., Nakamura, H., Mori, H., Ode, H., Hansman,G.S., Katayama, K., Kanda, T., Tanaka, T., Takeda, N., Sato, H., 2008. Identificationof monomorphic and divergent haplotypes in the 2006–2007 norovirus GII/4epidemic population by genomewide tracing of evolutionary history. J. Virol. 82(22), 11247–11262.
Nakamura, K., Iwai, M., Zhang, J., Obara, M., Horimoto, E., Hasegawa, S., Kurata, T.,Takizawa, T., 2009. Detection of a novel recombinant norovirus from sewagewater in Toyama prefecture, Japan. Jpn. J. Infect. Dis. 62 (5), 394–398.
Nayak, M.K., Balasubramanian, G., Sahoo, G.C., Bhattacharya, R., Vinje, J., Kobayashi,N., Sarkar, M.C., Bhattacharya, M.K., Krishnan, T., 2008. Detection of a novelintergenogroup recombinant Norovirus from Kolkata, India. Virology 377 (1),117–123.
Nilsson, M., Hedlund, K.O., Thorhagen, M., Larson, G., Johansen, K., Ekspong, A.,Svensson, L., 2003. Evolution of human calicivirus RNA in vivo: accumulation ofmutations in the protruding P2 domain of the capsid leads to structural changesand possibly a new phenotype. J. Virol. 77 (24), 13117–13124.
Nishida, T., Kimura, H., Saitoh, M., Shinohara, M., Kato, M., Fukuda, S., Munemura,T., Mikami, T., Kawamoto, A., Akiyama, M., Kato, Y., Nishi, K., Kozawa, K., Nishio,O., 2003. Detection, quantitation, and phylogenetic analysis of noroviruses inJapanese oysters. Appl. Environ. Microbiol. 69 (10), 5782–5786.
Noel, J.S., Fankhauser, R.L., Ando, T., Monroe, S.S., Glass, R.I., 1999. Identification of adistinct common strain of “Norwalk-like viruses” having a global distribution. J.Infect. Dis. 179 (6), 1334–1344.
Okada, M., Ogawa, T., Kaiho, I., Shinozaki, K., 2005. Genetic analysis of norovirusesin Chiba Prefecture, Japan, between 1999 and 2004. J. Clin. Microbiol. 43 (9),4391–4401.
Oliver, S.L., Brown, D.W., Green, J., Bridger, J.C., 2004. A chimeric bovine enteric cali-civirus: evidence for genomic recombination in genogroup III of the Norovirusgenus of the Caliciviridae. Virology 326 (2), 231–239.
Oliver, S.L., Dastjerdi, A.M., Wong, S., El-Attar, L., Gallimore, C., Brown, D.W., Green,J., Bridger, J.C., 2003. Molecular characterization of bovine enteric caliciviruses:a distinct third genogroup of noroviruses (Norwalk-like viruses) unlikely to beof risk to humans. J. Virol. 77 (4), 2789–2798.
Page, R.D., 1996. TreeView: an application to display phylogenetic trees on personalcomputers. Comput. Appl. Biosci. 12 (4), 357–358.
Pang, X.L., Preiksaitis, J.K., Lee, B., 2005. Multiplex real time RT-PCR for the detec-tion and quantitation of norovirus genogroups I and II in patients with acutegastroenteritis. J. Clin. Virol. 33 (2), 168–171.
Papaventsis, D.C., Dove, W., Cunliffe, N.A., Nakagomi, O., Combe, P., Grosjean, P., Hart,C.A., 2007. Norovirus infection in children with acute gastroenteritis, Madagas-car, 2004–2005. Emerg. Infect. Dis. 13 (6), 908–911.
Phan, T.G., Kaneshi, K., Ueda, Y., Nakaya, S., Nishimura, S., Yamamoto, A., Sugita, K.,Takanashi, S., Okitsu, S., Ushijima, H., 2007. Genetic heterogeneity, evolution,and recombination in noroviruses. J. Med. Virol. 79 (9), 1388–1400.
Phan, T.G., Kuroiwa, T., Kaneshi, K., Ueda, Y., Nakaya, S., Nishimura, S., Yamamoto,
A., Sugita, K., Nishimura, T., Yagyu, F., Okitsu, S., Müller, W.E., Maneekarn, N.,Ushijima, H., 2006a. Changing distribution of norovirus genotypes and geneticanalysis of recombinant GIIb among infants and children with diarrhea in Japan.J. Med. Virol. 78 (7), 971–978.Phan, T.G., Yan, H., Li, Y., Okitsu, S., Müller, W.E., Ushijima, H., 2006b. Novel recom-binant norovirus in China. Emerg. Infect. Dis. 12 (5), 857–858.

1 esearc
P
P
R
R
R
R
S
S
S
S
T
T
T
V
V
V
V
V
Zheng, D.P., Ando, T., Fankhauser, R.L., Beard, R.S., Glass, R.I., Monroe, S.S., 2006.Norovirus classification and proposed strain nomenclature. Virology 346 (2),
52 S.-I. Yun et al. / Virus R
rasad, B.V., Hardy, M.E., Dokland, T., Bella, J., Rossmann, M.G., Estes, M.K., 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286 (5438),287–290.
rasad, B.V., Rothnagel, R., Jiang, X., Estes, M.K., 1994. Three-dimensional structureof baculovirus-expressed Norwalk virus capsids. J. Virol. 68 (8), 5117–5125.
adford, A.D., Gaskell, R.M., Hart, C.A., 2004. Human norovirus infection and thelessons from animal caliciviruses. Curr. Opin. Infect. Dis. 17 (5), 471–478.
ichards, G.P., Watson, M.A., Fankhauser, R.L., Monroe, S.S., 2004. Genogroup I andII noroviruses detected in stool samples by real-time reverse transcription-PCRusing highly degenerate universal primers. Appl. Environ. Microbiol. 70 (12),7179–7184.
ockx, B., De Wit, M., Vennema, H., Vinjé, J., De Bruin, E., Van Duynhoven, Y., Koop-mans, M., 2002. Natural history of human calicivirus infection: a prospectivecohort study. Clin. Infect. Dis. 35 (3), 246–253.
ohayem, J., Münch, J., Rethwilm, A., 2005. Evidence of recombination in thenorovirus capsid gene. J. Virol. 79 (8), 4977–4990.
aitou, N., Nei, M., 1987. The neighbor-joining method: a new method for recon-structing phylogenetic trees. Mol. Biol. Evol. 4 (4), 406–425.
cipioni, A., Mauroy, A., Vinjé, J., Thiry, E., 2008. Animal noroviruses. Vet. J. 178 (1),32–45.
diri-Loulizi, K., Ambert-Balay, K., Gharbi-Khelifi, H., Sakly, N., Hassine, M.,Chouchane, S., Guediche, M.N., Pothier, P., Aouni, M., 2009. Molecular epidemi-ology of norovirus gastroenteritis investigated using samples collected fromchildren in Tunisia during a four-year period: detection of the norovirus variantGGII.4 Hunter as early as January 2003. J. Clin. Microbiol. 47 (2), 421–429.
tals, A., Baert, L., Botteldoorn, N., Werbrouck, H., Herman, L., Uyttendaele, M., VanCoillie, E., 2009. Multiplex real-time RT-PCR for simultaneous detection of GI/GIInoroviruses and murine norovirus 1. J. Virol. Methods 161 (2), 247–253.
an, M., Huang, P., Meller, J., Zhong, W., Farkas, T., Jiang, X., 2003. Mutations withinthe P2 domain of norovirus capsid affect binding to human histo-blood groupantigens: evidence for a binding pocket. J. Virol. 77 (23), 12562–12571.
hompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G., 1997. TheCLUSTAL X windows interface: flexible strategies for multiple sequence align-ment aided by quality analysis tools. Nucleic Acids Res. 25 (24), 4876–4882.
sugawa, T., Numata-Kinoshita, K., Honma, S., Nakata, S., Tatsumi, M., Sakai, Y.,Natori, K., Takeda, N., Kobayashi, S., Tsutsumi, H., 2006. Virological, serological,and clinical features of an outbreak of acute gastroenteritis due to recombinantgenogroup II norovirus in an infant home. J. Clin. Microbiol. 44 (1), 177–182.
erhoef, L., Depoortere, E., Boxman, I., Duizer, E., van Duynhoven, Y., Harris, J.,Johnsen, C., Kroneman, A., Le Guyader, S., Lim, W., Maunula, L., Meldal, H., Rat-cliff, R., Reuter, G., Schreier, E., Siebenga, J., Vainio, K., Varela, C., Vennema, H.,Koopmans, M., 2008. Emergence of new norovirus variants on spring cruise shipsand prediction of winter epidemics. Emerg. Infect. Dis. 14 (2), 238–243.
idal, R., Roessler, P., Solari, V., Vollaire, J., Jiang, X., Matson, D.O., Mamani, N., Prado,V., O’Ryan, M.L., 2006. Novel recombinant norovirus causing outbreaks of gas-troenteritis in Santiago, Chile. J. Clin. Microbiol. 44 (6), 2271–2275.
injé, J., Altena, S.A., Koopmans, M.P., 1997. The incidence and genetic variabilityof small round-structured viruses in outbreaks of gastroenteritis in The Nether-lands. J. Infect. Dis. 176 (5), 1374–1378.
injé, J., Hamidjaja, R.A., Sobsey, M.D., 2004. Development and application of acapsid VP1 (region D) based reverse transcription PCR assay for genotyping ofgenogroup I and II noroviruses. J. Virol. Methods 116 (2), 109–117.
injé, J., Koopmans, M.P., 2000. Simultaneous detection and genotyping of“Norwalk-like viruses” by oligonucleotide array in a reverse line blot hybridiza-tion format. J. Clin. Microbiol. 38 (7), 2595–2601.
h 152 (2010) 137–152
Wang, Q.H., Han, M.G., Cheetham, S., Souza, M., Funk, J.A., Saif, L.J., 2005.Porcine noroviruses related to human noroviruses. Emerg. Infect. Dis. 11 (12),1874–1881.
Ward, V.K., McCormick, C.J., Clarke, I.N., Salim, O., Wobus, C.E., Thackray, L.B., Virgin,H.W., Lambden, P.R., 2007. Recovery of infectious murine norovirus using polII-driven expression of full-length cDNA. Proc. Natl. Acad. Sci. U.S.A. 104 (26),11050–11055.
Waters, A., Coughlan, S., Hall, W.W., 2007. Characterisation of a novel recombinationevent in the norovirus polymerase gene. Virology 363 (1), 11–14.
Waters, A., Dunford, L., Tuite, G., Connell, J., Dooley, S., Foley, B., McKeown, P.,Hall, W.W., Coughlan, S., 2008. Significant prevalence and genetic diversity ofnorovirus infection in Irish children. Pediatr. Res. 64 (3), 312–316.
Widdowson, M.A., Cramer, E.H., Hadley, L., Bresee, J.S., Beard, R.S., Bulens, S.N.,Charles, M., Chege, W., Isakbaeva, E., Wright, J.G., Mintz, E., Forney, D., Massey, J.,Glass, R.I., Monroe, S.S., 2004. Outbreaks of acute gastroenteritis on cruise shipsand on land: identification of a predominant circulating strain of norovirus-United States, 2002. J. Infect. Dis. 190 (1), 27–36.
Wobus, C.E., Karst, S.M., Thackray, L.B., Chang, K.O., Sosnovtsev, S.V., Belliot, G., Krug,A., Mackenzie, J.M., Green, K.Y., Virgin, H.W., 2004. Replication of Norovirus incell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2(12), e432.
Wobus, C.E., Thackray, L.B., Virgin, H.W., 2006. Murine norovirus: a model systemto study norovirus biology and pathogenesis. J. Virol. 80 (11), 5104–5112.
Wolf, S., Williamson, W.M., Hewitt, J., Rivera-Aban, M., Lin, S., Ball, A., Scholes, P.,Greening, G.E., 2007. Sensitive multiplex real-time reverse transcription-PCRassay for the detection of human and animal noroviruses in clinical and envi-ronmental samples. Appl. Environ. Microbiol. 73 (17), 5464–5470.
Worobey, M., Holmes, E.C., 1999. Evolutionary aspects of recombination in RNAviruses. J. Gen. Virol. 80 (Pt. 10), 2535–2543.
Wyatt, R.G., Dolin, R., Blacklow, N.R., DuPont, H.L., Buscho, R.F., Thornhill, T.S.,Kapikian, A.Z., Chanock, R.M., 1974. Comparison of three agents of acute infec-tious nonbacterial gastroenteritis by cross-challenge in volunteers. J. Infect. Dis.129 (6), 709–714.
Xerry, J., Gallimore, C.I., Iturriza-Gómara, M., Allen, D.J., Gray, J.J., 2008. Transmis-sion events within outbreaks of gastroenteritis determined through analysisof nucleotide sequences of the P2 domain of genogroup II noroviruses. J. Clin.Microbiol. 46 (3), 947–953.
Yan, H., Yagyu, F., Okitsu, S., Nishio, O., Ushijima, H., 2003. Detection of norovirus(GI, GII), Sapovirus and astrovirus in fecal samples using reverse transcriptionsingle-round multiplex PCR. J. Virol. Methods 114 (1), 37–44.
Yoon, J.S., Lee, S.G., Hong, S.K., Lee, S.A., Jheong, W.H., Oh, S.S., Oh, M.H., Ko, G.P., Lee,C.H., Paik, S.Y., 2008. Molecular epidemiology of norovirus infections in childrenwith acute gastroenteritis in South Korea in November 2005 through November2006. J. Clin. Microbiol. 46 (4), 1474–1477.
Yun, S.I., Kim, S.Y., Rice, C.M., Lee, Y.M., 2003. Development and application ofa reverse genetics system for Japanese encephalitis virus. J. Virol. 77 (11),6450–6465.
312–323.Zheng, D.P., Widdowson, M.A., Glass, R.I., Vinjé, J., 2010. Molecular epidemiology of
genogroup II–genotype 4 noroviruses in the United States between 1994 and2006. J. Clin. Microbiol. 48 (1), 168–177.




















