
Comparison of three buffer solutions for amino acid derivatization and following analysis by liquid...
9
Journal of Chromatography A, 1245 (2012) 134–142 Contents lists available at SciVerse ScienceDirect Journal of Chromatography A j our na l ho me p ag e: www.elsevier.com/locate/chroma Comparison of three buffer solutions for amino acid derivatization and following analysis by liquid chromatography electrospray mass spectrometry Riin Rebane, Koit Herodes ∗ Institute of Chemistry, University of Tartu, Ravila 14a, 50411 Tartu, Estonia a r t i c l e i n f o Article history: Received 9 November 2011 Received in revised form 8 May 2012 Accepted 10 May 2012 Available online 17 May 2012 Keywords: Amino acids Derivatization Buffers LC–ESI-MS a b s t r a c t For reversed phase separation amino acids are usually derivatized. Several derivatization reactions are carried out at basic pH. In the present work, influence of three basic buffer solutions on liquid chromatog- raphy electrospray ionization mass-spectrometric (LC–ESI-MS) analysis of amino acid derivatives was studied. Borate buffer – the most common derivatization buffer – was found to influence ESI ionization up to 23 min retention time. For 9-fluorenylmethylmethoxycarbonyl chloride (Fmoc-Cl derivatization) carbonate buffer should be preferred as it provides higher responses. Hexafluoroisopropanol (HFIP) buffer improves chromatographic peak shapes and responses for diethyl ethoxymethylenemalonate (DEEMM) derivatives. © 2012 Elsevier B.V. All rights reserved. 1. Introduction Amino acids are compounds that are analysed widely in dif- ferent matrices, e.g. foods (wine, honey, etc.) to study their composition or determine geographic origin, and body fluids to research various metabolic systems [1–4]. Amino acids can be analysed with various analytical methods. One of the most common methods is liquid chromatography (LC). In order to improve reversed phase LC separation and detectability amino acids are derivatized. The most commonly used derivatiza- tion reagents, such as 9-fluorenylmethylmethoxycarbonyl chloride (Fmoc-Cl), o-phthaldialdehyde (OPA), phenylisothiocyanate (PITC) or 5-dimethylamino-1-naphthalenesulphonyl-chloride (DANSYL), react with the amino group of amino acids [5]. Amino acid derivatives are traditionally detected by ultravio- let (UV) or fluorescence (FL) detectors. Mass spectrometric (MS) detection with electrospray (ESI) or atmospheric pressure chemical ionization (APCI) are considerably newer techniques which provide lower detection limits and enable to confirm the identity of analytes and make the analysis of amino acids more reliable. Even though analysis of underivatized amino acids with MS detection is possi- ble [6,7], derivatization is employed for various reasons: to increase detection sensitivity, selectivity, improve chromatographic reten- tion or peak shape, facilitate sample cleanup, and to form a stable derivative for unstable analytes [8]. ∗ Corresponding author. Tel.: +372 737 6030; fax: +372 737 5264. E-mail address: [email protected] (K. Herodes). For Fmoc-Cl derivatives, besides LC-FL, liquid chromatography electrospray ionization mass spectrometry (LC–ESI-MS/MS) [9–11] has been used. LC–ESI-MS(/MS) has also been used in case of other amino acid derivatization reagents, such as OPA [12,13], 3- aminopyridyl-N-hydroxysuccinimidyl carbamate (APDS) [14] and 1,2-benzo-3,4-dihydrocarbazole-9-ethyl chloroformate (BCEOC) [15]. Abovementioned amino acid derivatization reagents and pro- cedures were originally designed for UV or FL detection. To the best of our knowledge, no work has been published studying the compatibility of respective reagents with MS detection. For various amino acid derivatization reactions pH 9 is needed. This could be provided by ammonium buffers but since ammo- nia would react with derivatization reagents, ammonium buffers are not applicable. In majority of cases borate buffer is the buffer of choice [1,2,4,9,11,13–24]. Buffers that can provide the same pH are carbonate and hexafluoroisopropanol (HFIP) buffers. Carbonate buffer has been used before in Fmoc-Cl derivatization for FL detec- tion [3,25–27]. HFIP has been used as an eluent buffer component for LC–ESI-MS analysis of oligonucleotides [28] and various basic and acidic compounds [29]. Therefore, HFIP has a potential of not interfering ESI ionization. This study was triggered by various problems with LC–ESI-MS analysis of diethyl ethoxymethylenemalonate (DEEMM) and Fmoc- Cl derivatives of amino acids. Common feature of the derivatization procedures was the use of borate buffer for attaining required basic pH. Comparison of three derivatization buffers with respect their influence on ESI-MS signal was undertaken. As model systems, two amino acid derivatization reagents – tra- ditional Fmoc-Cl [3,9,10,17–20,23,27] and relatively new DEEMM, which has gained popularity in food analysis [1,2,4,30] – were used 0021-9673/$ – see front matter © 2012 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.chroma.2012.05.039
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Comparison of three buffer solutions for amino acid derivatization and following analysis by liquid...

Ca
RI
a
ARRAA
KADBL
1
fcr
OIat(or
ldilaabdtd
0h
Journal of Chromatography A, 1245 (2012) 134– 142
Contents lists available at SciVerse ScienceDirect
Journal of Chromatography A
j our na l ho me p ag e: www.elsev ier .com/ locate /chroma
omparison of three buffer solutions for amino acid derivatization and followingnalysis by liquid chromatography electrospray mass spectrometry
iin Rebane, Koit Herodes ∗
nstitute of Chemistry, University of Tartu, Ravila 14a, 50411 Tartu, Estonia
r t i c l e i n f o
rticle history:eceived 9 November 2011eceived in revised form 8 May 2012ccepted 10 May 2012
a b s t r a c t
For reversed phase separation amino acids are usually derivatized. Several derivatization reactions arecarried out at basic pH. In the present work, influence of three basic buffer solutions on liquid chromatog-raphy electrospray ionization mass-spectrometric (LC–ESI-MS) analysis of amino acid derivatives wasstudied. Borate buffer – the most common derivatization buffer – was found to influence ESI ionization
vailable online 17 May 2012
eywords:mino acidserivatizationuffers
up to 23 min retention time. For 9-fluorenylmethylmethoxycarbonyl chloride (Fmoc-Cl derivatization)carbonate buffer should be preferred as it provides higher responses. Hexafluoroisopropanol (HFIP) bufferimproves chromatographic peak shapes and responses for diethyl ethoxymethylenemalonate (DEEMM)derivatives.
© 2012 Elsevier B.V. All rights reserved.
C–ESI-MS. Introduction
Amino acids are compounds that are analysed widely in dif-erent matrices, e.g. foods (wine, honey, etc.) to study theiromposition or determine geographic origin, and body fluids toesearch various metabolic systems [1–4].
Amino acids can be analysed with various analytical methods.ne of the most common methods is liquid chromatography (LC).
n order to improve reversed phase LC separation and detectabilitymino acids are derivatized. The most commonly used derivatiza-ion reagents, such as 9-fluorenylmethylmethoxycarbonyl chlorideFmoc-Cl), o-phthaldialdehyde (OPA), phenylisothiocyanate (PITC)r 5-dimethylamino-1-naphthalenesulphonyl-chloride (DANSYL),eact with the amino group of amino acids [5].
Amino acid derivatives are traditionally detected by ultravio-et (UV) or fluorescence (FL) detectors. Mass spectrometric (MS)etection with electrospray (ESI) or atmospheric pressure chemical
onization (APCI) are considerably newer techniques which provideower detection limits and enable to confirm the identity of analytesnd make the analysis of amino acids more reliable. Even thoughnalysis of underivatized amino acids with MS detection is possi-le [6,7], derivatization is employed for various reasons: to increaseetection sensitivity, selectivity, improve chromatographic reten-
ion or peak shape, facilitate sample cleanup, and to form a stableerivative for unstable analytes [8].∗ Corresponding author. Tel.: +372 737 6030; fax: +372 737 5264.E-mail address: [email protected] (K. Herodes).
021-9673/$ – see front matter © 2012 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.chroma.2012.05.039
For Fmoc-Cl derivatives, besides LC-FL, liquid chromatographyelectrospray ionization mass spectrometry (LC–ESI-MS/MS) [9–11]has been used. LC–ESI-MS(/MS) has also been used in case ofother amino acid derivatization reagents, such as OPA [12,13], 3-aminopyridyl-N-hydroxysuccinimidyl carbamate (APDS) [14] and1,2-benzo-3,4-dihydrocarbazole-9-ethyl chloroformate (BCEOC)[15]. Abovementioned amino acid derivatization reagents and pro-cedures were originally designed for UV or FL detection. To thebest of our knowledge, no work has been published studying thecompatibility of respective reagents with MS detection.
For various amino acid derivatization reactions pH 9 is needed.This could be provided by ammonium buffers but since ammo-nia would react with derivatization reagents, ammonium buffersare not applicable. In majority of cases borate buffer is the bufferof choice [1,2,4,9,11,13–24]. Buffers that can provide the same pHare carbonate and hexafluoroisopropanol (HFIP) buffers. Carbonatebuffer has been used before in Fmoc-Cl derivatization for FL detec-tion [3,25–27]. HFIP has been used as an eluent buffer componentfor LC–ESI-MS analysis of oligonucleotides [28] and various basicand acidic compounds [29]. Therefore, HFIP has a potential of notinterfering ESI ionization.
This study was triggered by various problems with LC–ESI-MSanalysis of diethyl ethoxymethylenemalonate (DEEMM) and Fmoc-Cl derivatives of amino acids. Common feature of the derivatizationprocedures was the use of borate buffer for attaining required basicpH. Comparison of three derivatization buffers with respect their
influence on ESI-MS signal was undertaken.As model systems, two amino acid derivatization reagents – tra-ditional Fmoc-Cl [3,9,10,17–20,23,27] and relatively new DEEMM,which has gained popularity in food analysis [1,2,4,30] – were used

omato
fbbww
2
2
JlSM((ph((ahw
psw
2
Tdsp(tCu
f(4C
R. Rebane, K. Herodes / J. Chr
or derivatization of a set of nine amino acids. Influence of borateuffer on the ionization was compared with carbonate and HFIPuffers. Results of analyses in positive and negative ion modes andith so-called waste run, where beginning of chromatographic runas diverted into waste, were compared.
. Materials and methods
.1. Materials
HPLC-grade methanol and acetonitrile were obtained from.T. Baker. Amino acid standards (l-proline, l-phenylalanine,-asparagine, l-serine, l-valine, l-methionine, l-glutamic acid,-methyl-l-cysteine) were purchased from Sigma (St. Louis,O, USA); �-alanine and Fmoc-�-alanine were from Fluka
Japan). Derivatization reagents diethyl ethoxymethylenmalonateDEEMM) and 9-fluorenylmethyl chloroformate (Fmoc-Cl) wereurchased from Fluka (Japan). Other chemicals: 1,1,1,3,3,3-exafluoro-2-propanol (Fluka, Japan); sodium hydroxideChemapol, Former Soviet Union); sodium dihydrogensulphateMerck, Germany); hydrochloric acid, orthophosphoric acid, boriccid, sodiumtetraborate, sodium bicarbonate and ammoniumydroxide were from Reakhim, Former Soviet Union. All reagentsere of analytical grade unless otherwise stated.
All aqueous solutions were prepared with ultrapure waterurified by Millipore Simplicity 185 (Millipore, USA). Amino acidtandards were dissolved in 0.1 M hydrochloric acid, and dilutedith ultrapure water to suitable concentrations.
.2. Equipment
HPLC system Agilent Series 1100 LC/MSD Trap XCT (Agilentechnologies, Santa-Clara, USA) was equipped with an in lineegasser, a binary pump, an autosampler and a column thermo-tat. For detection photodiode array detector (PDA) with 6 mmath length flow cell and electrospray interface mass spectrometerESI-MS) were used. The system was controlled with Chemsta-ion (Rev.A.10.02) and LCMSD Trap Control (Version 5.2) software.hemstation (Rev.A.10.02) and DataAnalysis (Version 3.2) weresed for UV and MS chromatograms analysis and peak integration.
Chromatographic analysis of DEEMM derivatives was per-
ormed using an analytical column Synergi Hydro-RP 80A4.6 mm × 250 mm, 4 �m) (Phenomenex, USA) with guard cartridge.0 mm × 2.0 mm, polar endcapped C18 (Phenomenex). For Fmoc-l derivatives Eclipse XDB-C18 4.6 mm × 250 mm, 5 �m analyticalFig. 1. Amino acid derivatization reaction
gr. A 1245 (2012) 134– 142 135
column with guard column (4.6 mm × 12.5 mm, 5 �m) was used(Agilent).
2.3. Preparation of buffers
Three types of buffers were prepared as follows: borate bufferwas prepared from 0.75 M boric acid solution and pH was adjustedto 9.0 with concentrated sodium hydroxide solution; carbonatebuffer was prepared from 0.6 M sodium bicarbonate solution andpH was adjusted to 9.0 with concentrated sodium hydroxide solu-tion and HFIP buffer was prepared from 0.6 M HFIP solution and pHwas adjusted to 9.0 with concentrated sodium hydroxide solution.
Borate buffer is non-volatile; both carbonate and HFIP bufferare volatile in combination with volatile base, e.g. ammoniumhydroxide. For LC–MS applications volatile ammonium hydroxidewould be a logical choice, but ammonium would interfere withderivatization reaction. From derivatization point of view, sub-stituted amines, e.g. triethylamine could be used, but those areefficiently ionized in ESI source, therefore hindering MS detection.Consequently, non-volatile sodium hydroxide was used for buffersolution preparation.
2.4. Sample preparation
Honey samples were prepared with a previously developedsample preparation procedure [2] and for tea samples, 5 g of teawas infused in 10 mL of ultrapure water (40 min at 80 ◦C). Sampleswere directly derivatized.
2.5. Derivatization procedures
Derivatization reaction for amino acids with DEEMM is pre-sented in Fig. 1a and with Fmoc-Cl in Fig. 1b.
DEEMM derivatization: to 1 mL of sample solution(0.01–0.6 mg/g in ultrapure water), 30 �L of DEEMM, 1.5 mLmethanol and 4.5 mL of borate or carbonate or HFIP buffer(0.75 M/0.6 M/0.6 M respectively, all pH 9.0) were added [1]. Thederivatized mixture was kept at room temperature protected fromdirect light. LC–MS analysis has to be carried out at least 24 h butnot more than 48 h after the derivatization [16]. Prior to LC–MSanalysis, the sample solutions were filtered through 0.45 �m
cellulose acetate syringe filter (Whatman) as in Ref. [16].Fmoc-Cl derivatization: to 300 �L of sample solution(0.01–0.6 mg/g in ultrapure water), 300 �L of borate or car-bonate or HFIP buffer (0.75 M/0.6 M/0.6 M respectively, all pH 9.0)
with (a) DEEMM and (b) Fmoc-Cl.

1 romato
avfmw(
2
pfaBmda
pefl3wc
r(
wlDmniua
aTmtwer
F(ub
36 R. Rebane, K. Herodes / J. Ch
nd 300 �L of Fmoc-Cl (5 mM in acetonitrile) were added andigorously mixed. Then the mixture was kept at room temperatureor 5 min and then 300 �L of His (8 mg/g) was added and vigorously
ixed again [24]. Prior to LC-ESI-MS analysis, the sample solutionsere filtered through 0.45 �m regenerated cellulose syringe filter
Agilent).
.6. LC/UV/MS analysis
HPLC conditions for DEEMM derivatives were as follows: mobilehase A: buffer solution (pH = 3.2; 1 mM ammonium acetate in 0.1%ormic acid); mobile phase B: acetonitrile. Gradient program wass follows: 0–12 min, 20–25%; 12–20 min 25%; 20–50 min 25–60%. The eluent flow rate was 0.9 mL min−1 and the column wasaintained at 40 ◦C and 5 �L of the sample was injected. The UV
etection wavelength was 280 nm (full spectra were acquired fordditional confirmation).
HPLC conditions for Fmoc-Cl derivatives were as follows: mobilehase A: 0.1% formic acid; mobile phase B: acetonitrile. Gradi-nt program was as follows: 0–45 min, 30–100% B. The eluentow rate was 0.8 mL min−1 and the column was maintained at0 ◦C and 10 �L of the sample was injected. The UV detectionavelength was 280 nm (full spectra were acquired for additional
onfirmation).ESI source parameters were same for both derivatization
eagents: Nebulizer gas (nitrogen) 50 psi (345 kPa), Drying gasnitrogen) flow rate 12 L min−1 and Drying gas temperature 350 ◦C.
For DEEMM and Fmoc-Cl both positive and negative ion modesere tested. Quasimolecular ions [M+H]+ and [M−H]− were ana-
ysed, respectively for positive and negative mode. In case ofEEMM derivatives sodium adduct, [M+Na]+ also appear in theass spectra and these were under the investigation but they have
o impact on the overall results. In the case of Fmoc-Cl derivativesn positive ion mode, [2M+Na]+ or [2M+H]+ appears also. This wasnder investigation and the ratios of various adducts were constantnd did not affect the results.
The derivatization buffer injected into the LC–ESI-MS systemlong with the sample, is expected to elute at the solvent front.herefore, diverting solvent front into waste could circumventatrix effect. For brevity, the experiment where initial 5 min of
he chromatographic run are diverted into waste, is called theaste run. Under regular run we mean LC–MS analysis where
ffluent enters the ESI source during the whole chromatographicun time.
ig. 2. UV chromatograms of DEEMM derivatives at 280 nm: light grey (carbonate), grey
7.4); 2, Ser (9.0); 3, Glu (12.0); 4, �-Ala (16.0); 5, Pro (21.0); 6, Me-Cys (31.7); 7, Met (34nidentified derivatization byproducts. Note that derivatization using carbonate buffer guffer solution.
gr. A 1245 (2012) 134– 142
3. Results and discussion
In the current article, two amino acid derivatization reagents arediscussed, Fmoc-Cl and DEEMM. But the problem of using boratebuffer is beyond these two derivatization reagents since boratebuffer is widely used for other amino acid derivatization procedures[14,15,21,22]. However, these were not added to the study for var-ious reasons such as the instability of OPA derivatives or the abilityto react with only primary amino acids. DEEMM and Fmoc-Cl weresuitable for testing due to their simple derivatization procedures,stable derivatives and ability to react with primary and secondaryamino acids.
In order to compare the influence of different buffers on theionization of amino acid derivatives, various amino acids were cho-sen for derivatization: Glu, Asn, Ser, �-Ala, Pro, Me-Cys, Met, Valand Phe. Amino acids were chosen to cover wide range of reten-tion times (6.4–26.1 min for Fmoc-Cl and 7.4–40.4 min for DEEMMderivatives). Concentrations of amino acids were chosen similar tothe honey samples, which had previously been analysed in the labo-ratory (0.01–1 mg/g of individual amino acid in the sample solutionprior to derivatization). In those concentration ranges UV detectionwas also possible. UV detection, as opposed to ESI-MS, does notsuffer from matrix effects. Therefore, UV-chromatograms provideinformation about the yield of a derivatization, without (possible)interference by the buffer used.
The performance of the different buffers is evaluated by meansof relative response factors (relative to the response factor ofrespective �Ala derivative derivatized in borate buffer and analysedin regular mode). Concentration used in calculating the responsefactor was the amino acid concentration (mg/g) in the injectedsample (concentration after the derivatization).
3.1. DEEMM derivatization
DEEMM derivatization procedure previously implemented foramino acid analysis in honey was applied [1,2,16]. With DEEMMderivatization, the buffer concentration in the final solution wasapproximately 0.5 M for all buffers compared.
For DEEMM derivatization, three types of buffers were consid-ered: HFIP, carbonate and borate. Up to date only borate buffer has
been used for DEEMM derivatization [1,16]. While carbonate bufferis relatively common buffer, HFIP has been used only occasionally.HFIP buffer has been demonstrated to be compatible with LC–MSeven as a component of LC eluent [28] and to provide alternative(borate) and black (HFIP). Peaks of derivatives (retention times in minutes): 1, Asn.5); 8, Val (36.0); 9, Phe (40.4); and N, derivative of ammonia. Unmarked peaks areives one extra peak at 10 min, which could be due to impurities in the carbonate

R. Rebane, K. Herodes / J. Chromatogr. A 1245 (2012) 134– 142 137
Fig. 3. Average (n = 2) ESI-MS relative response factors (relative to response factor of �Ala-DEEMM in borate buffer using regular mode) of DEEMM derivatives of amino acidsw responb
sis
Urftb
tapa
tfo
wi
3
btlentael
ith three buffers in positive ion mode for regular and waste runs. Error bars coretween respective regular and waste mode results).
electivity [29]. In the present work, HFIP was present in thenjected sample and even this small amount causes retention timehifts and changes in peaks shapes (Fig. 2).
As UV detection does not suffer from matrix effects, peak areas ofV chromatogram can be used to assess the yield of derivatization
eaction, when different buffer solutions are used. Response factorsor all amino acids in different buffers are similar demonstratinghat the yield of the derivatization reaction does not depend on theuffer type.
DEEMM derivative of Pro has considerably lower UV responsehan other amino acids for two reasons. It has lower molarbsorption coefficient than DEEMM derivatives of compounds withrimary amino group [4]. Secondly, DEEMM derivative of Pro is rel-tively less stable and is partially decomposed during the analysis.
As the concentrations of amino acid DEEMM derivatives in solu-ions of different buffers are similar, the alteration in responseactors in MS-chromatograms can be attributed to the influencef buffer compounds.
Samples derivatized using borate, carbonate or HFIP buffersere analysed using regular and waste runs in positive and negative
on mode ESI.
.1.1. DEEMM derivatives in positive ion modeIn terms of reversed phased separation, the compounds of the
uffers may usually be regarded as polar and should elute closeo the solvent front (2–5 min) (HFIP is an exception, having ratherong retention time [29]). If the initial part of the chromatographicluent is diverted into waste, the buffer components should haveo influence on ESI ionization of the analytes. Indeed, the results of
he waste run experiments show (Fig. 3) very similar responses ofll the amino acids derivatized in different buffer solutions. In sev-ral cases statistically significant differences (t-test, 95% confidenceevel) between waste and regular mode signals were obtained.d to ±1 standard deviation (*significant differences (t-test, 95% confidence level)
In the regular run, all the sample components enter the ESIsource and can cause matrix effect – ionization suppression orenhancement. As evidenced by Fig. 3, borate buffer causes signifi-cant signal enhancement in case of early eluting DEEMM derivatesof amino acids, confirmed by t-test. For example, Asn and Gluderivatives signals are up to 4 times higher with borate than withcarbonate or HFIP buffer. The effect of carbonate and HFIP buffersare similar, since the amino acid derivatives eluting in the firsthalf of chromatogram have similar responses. The late-elutingcompounds exhibit similar responses with all the tested buffersolutions.
The experiments performed also enable to understand themechanistic aspects of borate influence. One might assume thatborate elutes as a very broad peak extending to 23.3 min (reten-tion time of Met-DEEMM) where the enhancing effects cease. Ifthis was the case, then the signal enhancement would also occurin waste experiment as only the initial 5 min of the chromatogramwere diverted into waste. But enhancing effect is not observed inwaste run, and consequently borate elutes during the initial 5 minof the chromatographic run. From these observations, it can be con-cluded that in the case of regular runs, borate elutes before 5 minbut it deposits on the internal surfaces of ESI source and causesmatrix effects up to retention time 23.3 min. The exact mechanismhow deposits of borate affect ionization are yet to be explained.
In order to get more information about the mechanism, massspectra in regular and waste run were compared but no significantdifferences were identified.
3.1.2. DEEMM derivatives in negative ion mode
For negative ion mode, similar comparisons were performed asfor the positive ion mode. Waste run experiments in negative ionmode reveal, that the signals of the analytes are independent ofbuffer used for derivatization (Fig. 4), also confirmed by t-test.

138 R. Rebane, K. Herodes / J. Chromatogr. A 1245 (2012) 134– 142
Fig. 4. Average (n = 2) ESI-MS relative response factors (relative to response factor of�Ala-DEEMM in borate buffer using regular mode) of DEEMM derivatives of aminoacl
onc
tofTbt
tebr
stIam2(htt(t
3
irfmacr
i
ws
cids with three buffers in negative ion mode for regular and waste runs. Error barsorrespond to ±1 standard deviation (*significant differences (t-test, 95% confidenceevel) between respective regular and waste mode results).
In case of carbonate and HFIP buffers, similar responses arebserved in waste as well as in regular mode. Slightly higher sig-als were recorded for the analytes eluting in the beginning of thehromatogram if HFIP buffer was used for derivatization (t-test).
However, in the regular run, compared to the positive ion mode,he case is different, since up to 3 times lower responses werebserved if borate buffer was used for derivatization (t-test). Dif-erence is larger in the beginning of the chromatogram (Fig. 4).herefore, HFIP and carbonate buffers should be preferred overorate as derivatization buffers if negative ion mode ESI-MS detec-ion is used.
The mechanistic aspects of borate influence can be discussed inhe similar manner as in the positive ion mode, however, the differ-nce is, that instead of signal enhancement, suppression is causedy the deposits of borate on the internal surfaces of ESI source. Inegular run, signal is suppressed for all the amino acid derivatives.
While in search of the possible causes of this phenomenon, masspectra in the regular and waste run were investigated. It appearedhat for the borate buffer, the mass spectra exhibit clusters of peaks.n Fig. 5, two mass spectra at the retention time of Asn (7.3–7.6 min)re presented. While in case of waste run (Fig. 5b) relatively “clean”ass spectrum was obtained, in the case of regular run, around m/z
18, 288, 305, 332, 348, 402, 428 and 461 multiple peaks appearFig. 5a). Peak with m/z 301 corresponds to Asn and figure showsow excessive number of background peaks complicates interpre-ation of mass spectra and can enlarge the uncertainty of quantita-ive analysis. Waste run is simple means of avoiding those problemsFig. 5b). The influences on the mass spectra bring out the impor-ance of using waste run when analysing in negative ion mode.
.1.3. Overall comparison for DEEMM derivatizationComparing positive and negative ion mode overall, shows that
n negative ion mode, all amino acid derivatives have similaresponses (except Pro due to instability of DEEMM derivative) whileor positive ion mode, amino acid derivatives at the end of the chro-
atogram have higher responses. This is applicable to all buffersnd all modes. Higher response in the end of the chromatogramould be related to the higher organic content of the solvent, whichesults in improved ionization.
For borate buffer, it is interesting that it causes signal supression
n negative ion mode and enhancement in positive ion mode.HFIP seems to be most suitable when comparing all buffers inaste run as it offers better ionization than other buffers. In conclu-
ion, HFIP provides the best peak shape and also stable responses in
Fig. 5. Mass spectra at 7.3–7.6 min (Asn) in the negative ion mode for (a) regularrun and (b) waste run.
all modes due to the absence of non-volatile compounds that couldaccumulate on the ESI source. These two aspects makes it a betterchoice as a buffer for providing pH 9 for the DEEMM derivatization.
3.2. Fmoc-Cl derivatization
Unlike for DEEMM derivatization, for Fmoc-Cl there arenumerous derivatization procedures for amino acid analysis[3,9,10,17–20,23,27]. Moreover, Fmoc-Cl has been applied to anal-yse other compounds with amino group [11,24,25,31,32]. Thisprovides tens of various derivatization procedures where buffers,solvents, concentrations, reaction times, volumes, etc., vary. Forthis experiment, several Fmoc-Cl derivatization procedures weretested. Since Fmoc-Cl is reactive towards water, the main prob-lem is Fmoc-OH, which forms as a result and this needs to beremoved from the reaction mixture [19]. In order to avoid appear-ance of Fmoc-OH in the analysed solution extraction with pentane[18,19,32], the quenching compound (ACN–acetic acid–water,20:2:30, v/v) [33] or addition of an amino acid in excess [24] weretested as the possible derivatization procedures. Derivatizationwith excess amino acid was chosen to keep the reaction proceduresimple and similar to DEEMM derivatization. This approach is sup-ported by the experiments [20]. Concentration of 5 mM Fmoc-Cl inMeCN and reaction time 5 min at room temperature appeared tobe suitable for the analysis. The procedure is similar to the analysisof 1-deoxynojirimycin [24].
For Fmoc-Cl derivatization, the most common buffer is boratebuffer [9–11,18,20,23,24]. There are few cases of using carbonate
buffer [3,25–27] but no report of HFIP as derivatization buffer isavailable. In this work, for comparison, buffer concentrations in thefinal solutions were approximately 0.2 M for all buffers.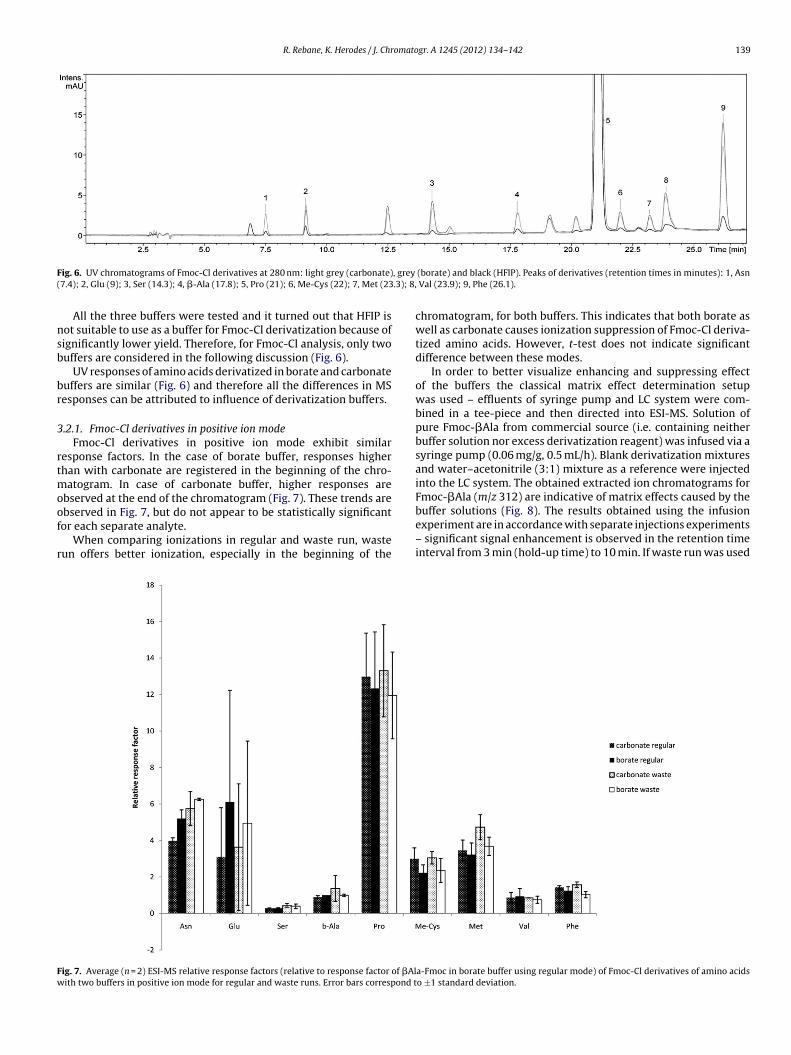
R. Rebane, K. Herodes / J. Chromatogr. A 1245 (2012) 134– 142 139
F , grey( .3); 8,
nsb
br
3
rtmoof
r
Fw
ig. 6. UV chromatograms of Fmoc-Cl derivatives at 280 nm: light grey (carbonate)7.4); 2, Glu (9); 3, Ser (14.3); 4, �-Ala (17.8); 5, Pro (21); 6, Me-Cys (22); 7, Met (23
All the three buffers were tested and it turned out that HFIP isot suitable to use as a buffer for Fmoc-Cl derivatization because ofignificantly lower yield. Therefore, for Fmoc-Cl analysis, only twouffers are considered in the following discussion (Fig. 6).
UV responses of amino acids derivatized in borate and carbonateuffers are similar (Fig. 6) and therefore all the differences in MSesponses can be attributed to influence of derivatization buffers.
.2.1. Fmoc-Cl derivatives in positive ion modeFmoc-Cl derivatives in positive ion mode exhibit similar
esponse factors. In the case of borate buffer, responses higherhan with carbonate are registered in the beginning of the chro-
atogram. In case of carbonate buffer, higher responses arebserved at the end of the chromatogram (Fig. 7). These trends are
bserved in Fig. 7, but do not appear to be statistically significantor each separate analyte.When comparing ionizations in regular and waste run, wasteun offers better ionization, especially in the beginning of the
ig. 7. Average (n = 2) ESI-MS relative response factors (relative to response factor of �Alith two buffers in positive ion mode for regular and waste runs. Error bars correspond t
(borate) and black (HFIP). Peaks of derivatives (retention times in minutes): 1, Asn Val (23.9); 9, Phe (26.1).
chromatogram, for both buffers. This indicates that both borate aswell as carbonate causes ionization suppression of Fmoc-Cl deriva-tized amino acids. However, t-test does not indicate significantdifference between these modes.
In order to better visualize enhancing and suppressing effectof the buffers the classical matrix effect determination setupwas used – effluents of syringe pump and LC system were com-bined in a tee-piece and then directed into ESI-MS. Solution ofpure Fmoc-�Ala from commercial source (i.e. containing neitherbuffer solution nor excess derivatization reagent) was infused via asyringe pump (0.06 mg/g, 0.5 mL/h). Blank derivatization mixturesand water–acetonitrile (3:1) mixture as a reference were injectedinto the LC system. The obtained extracted ion chromatograms forFmoc-�Ala (m/z 312) are indicative of matrix effects caused by the
buffer solutions (Fig. 8). The results obtained using the infusionexperiment are in accordance with separate injections experiments– significant signal enhancement is observed in the retention timeinterval from 3 min (hold-up time) to 10 min. If waste run was useda-Fmoc in borate buffer using regular mode) of Fmoc-Cl derivatives of amino acidso ±1 standard deviation.
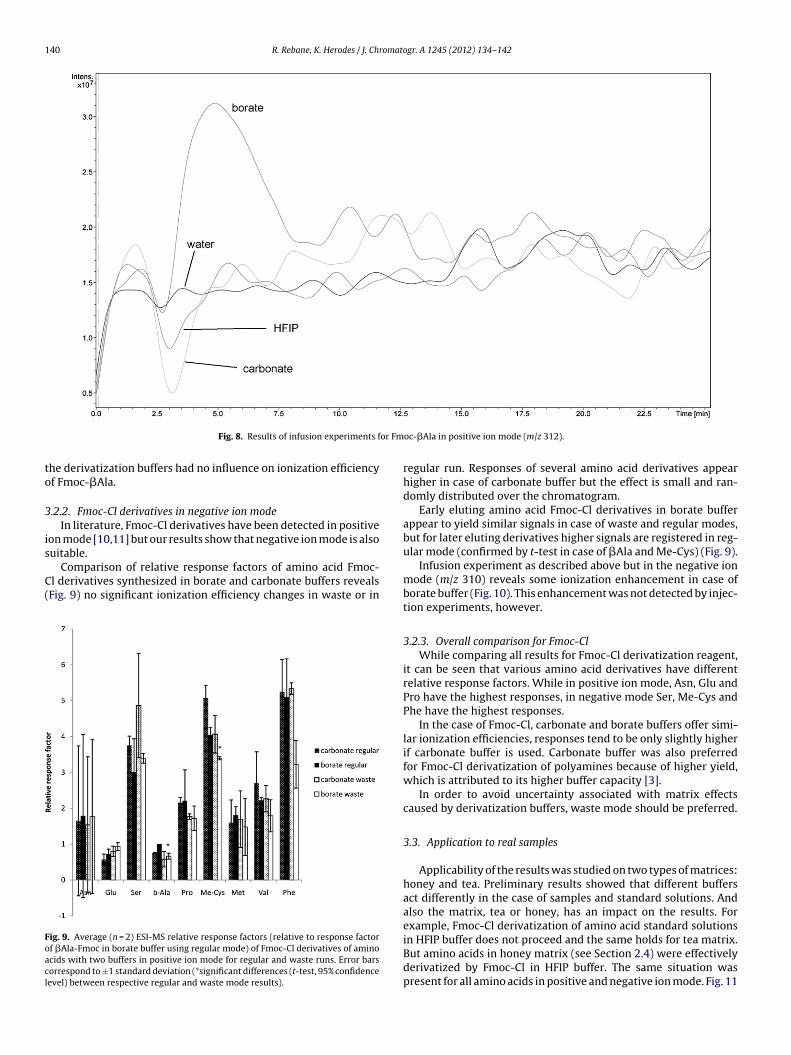
140 R. Rebane, K. Herodes / J. Chromatogr. A 1245 (2012) 134– 142
or Fm
to
3
is
C(
Foacl
Fig. 8. Results of infusion experiments f
he derivatization buffers had no influence on ionization efficiencyf Fmoc-�Ala.
.2.2. Fmoc-Cl derivatives in negative ion modeIn literature, Fmoc-Cl derivatives have been detected in positive
on mode [10,11] but our results show that negative ion mode is also
uitable.Comparison of relative response factors of amino acid Fmoc-l derivatives synthesized in borate and carbonate buffers revealsFig. 9) no significant ionization efficiency changes in waste or in
ig. 9. Average (n = 2) ESI-MS relative response factors (relative to response factorf �Ala-Fmoc in borate buffer using regular mode) of Fmoc-Cl derivatives of aminocids with two buffers in positive ion mode for regular and waste runs. Error barsorrespond to ±1 standard deviation (*significant differences (t-test, 95% confidenceevel) between respective regular and waste mode results).
oc-�Ala in positive ion mode (m/z 312).
regular run. Responses of several amino acid derivatives appearhigher in case of carbonate buffer but the effect is small and ran-domly distributed over the chromatogram.
Early eluting amino acid Fmoc-Cl derivatives in borate bufferappear to yield similar signals in case of waste and regular modes,but for later eluting derivatives higher signals are registered in reg-ular mode (confirmed by t-test in case of �Ala and Me-Cys) (Fig. 9).
Infusion experiment as described above but in the negative ionmode (m/z 310) reveals some ionization enhancement in case ofborate buffer (Fig. 10). This enhancement was not detected by injec-tion experiments, however.
3.2.3. Overall comparison for Fmoc-ClWhile comparing all results for Fmoc-Cl derivatization reagent,
it can be seen that various amino acid derivatives have differentrelative response factors. While in positive ion mode, Asn, Glu andPro have the highest responses, in negative mode Ser, Me-Cys andPhe have the highest responses.
In the case of Fmoc-Cl, carbonate and borate buffers offer simi-lar ionization efficiencies, responses tend to be only slightly higherif carbonate buffer is used. Carbonate buffer was also preferredfor Fmoc-Cl derivatization of polyamines because of higher yield,which is attributed to its higher buffer capacity [3].
In order to avoid uncertainty associated with matrix effectscaused by derivatization buffers, waste mode should be preferred.
3.3. Application to real samples
Applicability of the results was studied on two types of matrices:honey and tea. Preliminary results showed that different buffersact differently in the case of samples and standard solutions. Andalso the matrix, tea or honey, has an impact on the results. Forexample, Fmoc-Cl derivatization of amino acid standard solutions
in HFIP buffer does not proceed and the same holds for tea matrix.But amino acids in honey matrix (see Section 2.4) were effectivelyderivatized by Fmoc-Cl in HFIP buffer. The same situation waspresent for all amino acids in positive and negative ion mode. Fig. 11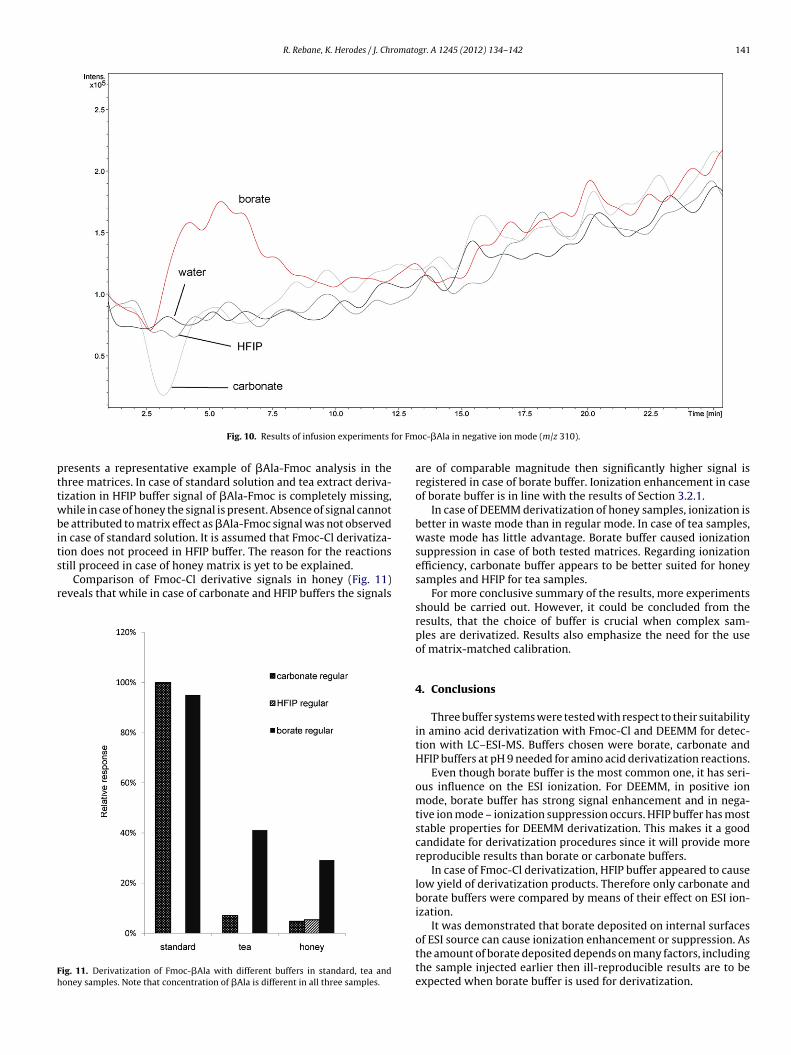
R. Rebane, K. Herodes / J. Chromatogr. A 1245 (2012) 134– 142 141
for Fm
pttwbits
r
Fh
Fig. 10. Results of infusion experiments
resents a representative example of �Ala-Fmoc analysis in thehree matrices. In case of standard solution and tea extract deriva-ization in HFIP buffer signal of �Ala-Fmoc is completely missing,hile in case of honey the signal is present. Absence of signal cannot
e attributed to matrix effect as �Ala-Fmoc signal was not observedn case of standard solution. It is assumed that Fmoc-Cl derivatiza-
ion does not proceed in HFIP buffer. The reason for the reactionstill proceed in case of honey matrix is yet to be explained.Comparison of Fmoc-Cl derivative signals in honey (Fig. 11)eveals that while in case of carbonate and HFIP buffers the signals
ig. 11. Derivatization of Fmoc-�Ala with different buffers in standard, tea andoney samples. Note that concentration of �Ala is different in all three samples.
oc-�Ala in negative ion mode (m/z 310).
are of comparable magnitude then significantly higher signal isregistered in case of borate buffer. Ionization enhancement in caseof borate buffer is in line with the results of Section 3.2.1.
In case of DEEMM derivatization of honey samples, ionization isbetter in waste mode than in regular mode. In case of tea samples,waste mode has little advantage. Borate buffer caused ionizationsuppression in case of both tested matrices. Regarding ionizationefficiency, carbonate buffer appears to be better suited for honeysamples and HFIP for tea samples.
For more conclusive summary of the results, more experimentsshould be carried out. However, it could be concluded from theresults, that the choice of buffer is crucial when complex sam-ples are derivatized. Results also emphasize the need for the useof matrix-matched calibration.
4. Conclusions
Three buffer systems were tested with respect to their suitabilityin amino acid derivatization with Fmoc-Cl and DEEMM for detec-tion with LC–ESI-MS. Buffers chosen were borate, carbonate andHFIP buffers at pH 9 needed for amino acid derivatization reactions.
Even though borate buffer is the most common one, it has seri-ous influence on the ESI ionization. For DEEMM, in positive ionmode, borate buffer has strong signal enhancement and in nega-tive ion mode – ionization suppression occurs. HFIP buffer has moststable properties for DEEMM derivatization. This makes it a goodcandidate for derivatization procedures since it will provide morereproducible results than borate or carbonate buffers.
In case of Fmoc-Cl derivatization, HFIP buffer appeared to causelow yield of derivatization products. Therefore only carbonate andborate buffers were compared by means of their effect on ESI ion-ization.
It was demonstrated that borate deposited on internal surfaces
of ESI source can cause ionization enhancement or suppression. Asthe amount of borate deposited depends on many factors, includingthe sample injected earlier then ill-reproducible results are to beexpected when borate buffer is used for derivatization.
1 romato
bTi
Mdb
A
n
R
[
[
[
[
[
[[[[[[[
[[
[
[
[[
[
[[30] J.L. Bernal, M.J. Nozal, L. Toribio, J.C. Diego, A. Ruiz, J. Sep. Sci. 28 (2005) 1039.[31] E.A. Hogendoorn, F.M. Ossendrijver, E. Dijkman, R.A. Baumann, J. Chromatogr.
42 R. Rebane, K. Herodes / J. Ch
The results of the study can be used as guidance for choosing theuffer for derivatization reaction in case of relatively clean samples.he applicability of the results to samples with complex matricess yet to be more thoroughly investigated.
Moreover, the scope of derivatization buffer suitability with ESI-S is broader than these two reagents and can be expanded to other
erivatization reagents that use borate buffer and can be detectedy LC–ESI-MS such as (OPA) [12,13], (APDS) [14] and (BCEOC) [15].
cknowledgement
This work was supported by the grant no. 8572 from the Esto-ian Science Foundation.
eferences
[1] I. Hermosín, R.M. Chicón, M.D. Cabezudo, Food Chem. 83 (2003) 263.[2] R. Rebane, K. Herodes, J. Agric. Food Chem. 56 (2008) 10716.[3] V. Lozanov, S. Petrov, V. Mitev, J. Chromatogr. A 1025 (2004) 201.[4] S. Gómez-Alonso, I. Hermosín-Gutiérrez, E. García-Romero, J. Agric. Food Chem.
55 (2007) 608.[5] T. Toyo’oka, Modern Derivatisation Methods for Separation Science, Wiley, New
York, 2002.[6] M. Piraud, C. Vianey-Saban, K. Petritis, C. Elfakir, J.-P. Steghens, A. Morla, D.
Bouchu, Rapid Commun. Mass Spectrom. 17 (2003) 1297.[7] S. Özcan, H.Z. S enyuva, J. Chromatogr. A 1135 (2006) 179.
[8] M. Jemal, Y.-Q. Xia, Curr. Drug Metab. 7 (2006) 491.[9] P.R. Uutela, A. Ketola, P. Piepponen, R. Kostiainen, Anal. Chim. Acta 633 (2009)223.10] M.G. Calabrese, G. Mamone, S. Caira, P. Ferranti, F. Addeo, Food Chem. 116
(2009) 799.
[[
gr. A 1245 (2012) 134– 142
11] P. Zavitsanos, C.K. Meng, Agilent Technologies Application, https://cp.chem.agilent.com/Library/applications/5988-4981EN.pdf (accessed 22.10.11).
12] H.M.H. van Eijk, D.R. Rooyakkers, P.B. Soeters, N.E.P. Deutz, Anal. Biochem. 271(1999) 8.
13] Y. Mengerink, D. Kuylan, F. Toth, A. Csampai, I. Molnár-Perl, J. Chromatogr. A949 (2002) 99.
14] K. Shimbo, T. Oonuki, A. Yahashi, K. Hirayama, H. Miyano, Rapid Commun. MassSpectrom. 23 (2009) 1483.
15] J. Sun, F. Li, W. Xu, G. Zhou, J. You, G. Chen, Chromatographia 70 (2009) 1627.16] R. Rebane, K. Herodes, Anal. Chim. Acta 672 (2010) 79.17] I. Molnár-Perl, J. Chromatogr. A 987 (2003) 291.18] K. Gartenmann, S. Kochhar, J. Agric. Food Chem. 47 (1999) 5068.19] D. Shangguan, Y. Zhao, H. Han, R. Zhao, G. Liu, Anal. Chem. 73 (2001) 2054.20] A. Jámbor, I. Molnár-Perl, J. Chromatogr. A 1216 (2009) 3064.21] E.H. Soufleros, E. Bouloumpasi, C. Tsarchopouls, C.G. Biliaderis, Food Chem. 80
(2003) 261.22] Á. Korös, Z. Varga, I. Molnár-Perl, J. Chromatogr. A 1203 (2008) 146.23] J. López-Cervantes, D.I. Sánchez-Machado, J.A. Rosas-Rodríguez, J. Chromatogr.
A 1105 (2006) 106.24] J.W. Kim, S.U. Kim, H.S. Lee, I. Kim, M.Y. Ahn, K.S. Ryu, J. Chromatogr. A 1002
(2003) 93.25] R. Herráez-Hernández, P. Campíns-Falcó, A. Sevillano-Cabeza, Anal. Chem. 68
(1996) 734.26] M.H. Yun, K.-i. Kwon, J. Pharm. Biomed. Anal. 40 (2006) 168.27] V. Lozanov, B. Benkova, L. Mateva, S. Petrov, E. Popov, C. Slavov, V. Mitev, J.
Chromatogr. B 860 (2007) 92.28] A. Apffel, J.A. Chakel, S. Fischer, K. Lichtenwalter, W.S. Hancock, Anal. Chem. 69
(1997) 1320.29] K. Kipper, K. Herodes, I. Leito, J. Chromatogr. A 1218 (2011) 8175.
A 833 (1999) 67.32] M. Kazachkov, P.H. Yu, J. Chromatogr. B 824 (2005) 116.33] T. Bausa, A. Blaise, F. Daumas, J.C. Cabanis, J. Chromatogr. A 707 (1995) 373.




















